Note: Descriptions are shown in the official language in which they were submitted.
CA 02312935 2000-06-02
wo ~n~s~ rcT~r~o
-1-
This invention relates to a series of substituted a-aminosulphonyl-
acetohydroxamic acids which are inhibitors of zinc-dependent metalloprotease
enzymes. In particular, the compounds are inhibitors of certain members of the
matrix metalloprotease (MMP) family.
Matrix metalloproteases (MMPs) constitute a family of structurally similar
zinc-containing metalloproteases, which are- involved in the remodelling and
degradation of extracellular matrix proteins, both as part of normal
physiological
processes and in pathological conditions. Since they have high destructive
potential, MMPs are usually under close regulation and failure to maintain MMP
regulation may be a component of a number of diseases and pathological
conditions, including atheroscierotic plaque rupture, heart failure,
restenosis,
~5 periodontal disease, tissue ulceration, wound repair, cancer metastasis,
tumour
angiogenesis, age-related macular degeneration, fibrotic disease, rheumatoid
arthritis, osteoarthritis and inflammatory diseases dependent on migratory
inflammatory cells.
Another important function of certain MMPs is to activate various
2o enzymes, including other MMPs, by cleaving the pro-domains from their
protease domains. Thus some MMPs act to regulate the activities of other
MMPs, so that over-production of one MMP may lead to excessive proteolysis
of extracellular matrix by another. Moreover, MMPs have different substrate
preferences (shown in the following Table for selected family members) and
25 different functions within normal and pathological conditions. For recent
reviews of MMPs, see Current Pharmaceutical Design, 1996, ~, 624 and Exp.
Opin. Ther. Patents, 1996; ~, 1305.
CA 02312935 2000-06-02
wo 99n~ss~ Pcr~r9sro~o
_2_
Enzyme C)ther Names Preferred Substrates
r
MMP-1 collagenase-1; interstitial collagens I, II, III,
VII, X;
collagenase gelatins
MMP-2 gelatinase A; 72kDa gelatinasegelatins; collagens IV,
V, VII,
X; elastin; fibronectin;
activates pro-MMP-13
MMP-3 stromelysin-1 proteoglycans; laminin;
fibronectin; gelatins
MMP-8 collagenase-2; neutrophil collagens I, II, III
collagenase
MMP-9 gelatinase B; 92kDa gelatinasegelatins; collagens IV,
V;
elastin
MMP-13 collagenase-3 collagens 1, II, III;
gelatins
MMP-14 MT-MMP-1 activates pro-MMP-2 &
13;
gelatins
Excessive production of MMP-3 is thought to be responsible for
s pathological tissue breakdown which underlies a number of diseases and
conditions. For example, MMP-3 has been found in the synovium and cartilage
of osteoarthritis and fieumatoid arthritis patients, thus implicating MMP-3 in
the
joint damage caused by these diseases: see Biochemistry, 1989, Z$, 8691 and
Biochem. J., 1989, 2~$, 115. MMP-13 is also thought to play an important role
o in the pathology of osteoarthritis and rheumatoid arthritis: see Lab.
Invest.,
1997, ~, 717 and Arthritis Rheum., 1997, 4Q, 1391. The compounds of the
present invention inhibit both MMP-3 and MMP-13 and thus may be of utility in
treating these diseases.
CA 02312935 2000-06-02
wo ~n~s~ rcr~o
-3-
The over-expression of MMP-3 is also thought to be responsible for
much of the tissue damage and chronicity of chronic wounds, such as venous
ulcers, diabetic ulcers and pressure sores: see Brit. J. Dermatology, 1996, ~,
52.
Furthermore, the production of MMP-3 may also cause tissue damage in
conditions where there is ulceration of the colon (as in ulcerative colitis
and
Crohn's disease: see J. Immunol., 1997 ~$, 1582 and J. Clin. Pathol., 1994,
~, 113) or of the duodenum (see Am. J. Pathol., 1996, 148, 519).
Moreover, MMP-3 may also be involved in skin diseases such as
dystrophic epidermolysis bullosa (see Arch. Dermatol. Res., 1995, ~, 428)
and dermatitis herpetiformis (see J. Invest. Dermatology, 1995, ~, 184).
Finally, rupture of atherosclerotic plaques by MMP-3 may lead to cardiac
or cerebral infarction: see Circulation, 1997, ~, 396. Thus, MMP-3 inhibitors
~5 may find utility in the prevention of heart attack and stroke.
Studies of human cancers have shown that MMP-2 is activated on the
invasive tumour cell surface (see J. BioLChem., 1993, ?,~$, 14033) and BB-94,
a non-selective peptidic hydroxamate MMP inhibitor, has been reported to
decrease the tumour burden and prolong the survival of mice carrying human
20 ovarian carcinoma xenografts (see Cancer Res., 1993, ~, 2087). Certain
compounds of the present invention inhibit MMP-2 and therefore may be useful
in the treatment of cancer metastasis and tumour angiogenesis.
Various series of MMP inhibitors have appeared in the patent literature.
For example, a-arylsulphonamido-substituted acetohydroxamic acids are
25 disclosed in EP-A-0606046, WO A-9627583 and WO-A-9719068, whilst EP A-
0780386 discloses certain related suiphone-substituted hydroxamic acids.
The compounds of the present invention are inhibitors of some of the
members of the MMP family. In particular, they are potent inhibitors of MMP-3
and MMP-13, with certain compounds exhibiting varying degrees of selectivity
30 over other MMPs, such as MMP-1, MMP-2 and MMP-9. Certain of the
compounds are potent MMP-2 inhibitors.
CA 02312935 2000-06-02
wo ~n9s6~ rcr~~s/o~eo
-4-
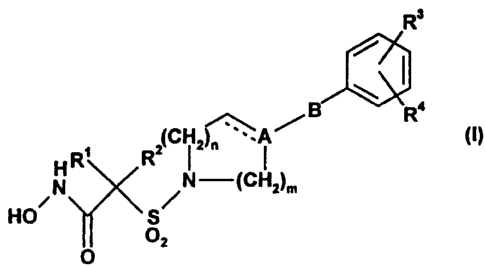
Thus, according to the present invention, there is provided a compound
of formula (I):
Ra
g R4
R~ R=(C i z1- ~ ~
/N /N-(CHa)m
HO 'S
~2
O
or a pharmaceutically or veterinarily acceptable salt thereof, or a
pharmaceutically or veterinarily acceptable solvate (including hydrate) of
either
entity,
wherein the broken line represents an optional bond;
A is C or CH;
o B is CHZ, O or absent;
R' and R2 are each independently selected from hydrogen, C~ to
C6 alkyl optionally substituted with C~ to C4 alkoxy or phenyl, and
C1 to CB alkenyl; or, together with the carbon atom to which they
are attached, form a C3 to Cs cycloalkyl group which optionally
~5 incorporates a heteroatom linkage selected from O, SO, S02 and
NR6 or which is optionally benzo-fused;
R3 is hydrogen, halo, R' or OR';
R4 is hydrogen, C1 to C4 alkyl, C~ to C4 alkoxy, trifluoromethyl or
halo;
2o R6 is hydrogen or C~ to C,, alkyl;
R' is a monocyclic or bicyclic ring system selected from phenyl,
thienyl, furyl, pyridinyl, pyrimidinyl, naphthyl, indanyl, benzothienyl,
benzofuranyl, 2,3-dihydrobenzofuranyl, indolyl, quinolinyl,
isoquinolinyl, benzodioxolyl, benzimidazolyl, benzoxazolyl,
25 benzothiazolyl and benzodioxanyl, any of which ring systems
CA 02312935 2000-06-02
wo ~n~~ Pc~rn~~sross4o
-5-
is optionally substituted with one or two substituents selected from
C~ to C4 alkyl optionally substituted with C~ to C4 alkoxy or
hydroxy, C~-C4 alkoxy optionally substituted with C~ to C4 alkoxy
or hydroxy, C~ to C,, alkylthio, trifluoromethyl, trifluoromethoxy,
halo and cyano;
m is 1 or 2;
and n is 0, 1 or 2;
with the proviso that B is not O when A is C.
In the above definition, unless otherwise indicated, alkyl, alkoxy, alkylthio
and alkenyl groups having three or more carbon atoms may be straight chain or
branched chain. Halo means fluoro, chloro, bromo or iodo.
The compounds of formula (I) may contain one or more chiral centres
and therefore can exist as stereoisomers, i.e. as enantiomers or
~5 diastereoisomers, as well as mixtures thereof. The invention includes both
the
individual stereoisomers of the compounds of formula (I) and any mixture
thereof. Separation of diastereoisomers may be achieved by conventional
techniques, e.g. by fractional crystallisation or chromatography (including
HPLC) of a diastereoisomeric mixture of a compound of fom~ula (I) or a
suitable
2o salt or derivative thereof. An individual enantiomer of a compound of
formula (I)
may be prepared from a corresponding optically pure intermediate or by
resolution, either by HPLC of the racemate using a suitable chiral support or,
where appropriate, by fractional crystallisation of the diastereoisomeric
salts
formed by reaction of the racemate with a suitable optically active base or
acid.
25 Furthermore, compound of formula (I) which contain alkenyl groups can
exist as cis-stereoisomers or traps-stereoisomers. Again, the invention
includes both the separated individual stereoisomers as well as mixtures
thereof.
CA 02312935 2000-06-02
wo ~n~~ pc~r~ro
-6.
Also included in the invention are radiolabelled derivatives of compounds
of formula (1) which are suitable for biological studies.
Compounds of formulae (I) may provide pharmaceutically or veterinarily
acceptable base salts, in particular non-toxic alkali metal salts, with bases.
Examples include the sodium and potassium salts. The pharmaceutically or
veterinarily acceptable salts of the compounds of formula (I) which contain a
basic centre are, for example, non toxic acid addition salts formed with
inorganic acids such as hydrochloric, hydrobromic, sulphuric and phosphoric
acid, with organo-carboxylic acids, or with organo-sulphonic acids.
A preferred group of compounds of formula (I) is that wherein B is
absent; R' is hydrogen, C~ to C4 alkyl optionally substituted with methoxy or
phenyl, or C~ to C5 alkenyl; R2 is hydrogen or C~ to C4 alkyl; or R' and R2,
together with the carbon atom to which they are attached, form a C4 to C5
cycloalkyl group which optionally incorporates a heteroatom linkage selected
from O and NR6 or which is optionally benzo-fused; R3 is selected from 4
phenyl, 4-pyridinyl, 4-(indan-5-yl), 4-(2,3-dihydrobenzofuran-5-yl), 4-
(quinolin-3-
yl), 4-(benzodioxol-5-yl) and 4-(benzimidazol-5-yl), any of which is
optionally
substituted with one or two substituents selected from C~ to C3 alkyl
optionally
substituted with methoxy or hydroxy, C~ to C3 alkoxy optionally substituted
with
methoxy or hydroxy, methylthio, trifluoromethyl, trifluoromethoxy, fluoro,
chloro
and cyano; R4 is hydrogen, methyl, ethyl, methoxy, trifluoromethyl, fluoro or
chloro; Rs is methyl; m is 2; and n is 1.
A more preferred group of compounds of formula (I) is that wherein R' is
2s hydrogen, methyl, ethyl, 2-methylprop-1-yl, but-1-yt, 2-methoxyethyl,
benzyl, 3-
phenylprop-1-yl, allyl, 2-methylallyl, 3,3-dimethylallyl; R2 is hydrogen,
methyl or
ethyl; or R' and R2, together with the carbon atom to which they are attached,
form a cyclobutyl, cyctopentyl, tetrahydropyran-4,4-diyl, 1-methylpiperidin-
4,4-
diyl or indan-2,2-diyl group; R3 is 4-phenyl, 4-(2-methylphenyl), 4-(3-
CA 02312935 2000-06-02
wo ~n~~ Prr~r9so
_7_
methylphenyl), 4-(3-ethylphenyl), 4-[3-(prop-2-yl)phenyl], 4-(3,5-
dimethytphenyl), 4-(3-methoxymethylphenyl), 4-(3-hydroxymethylphenyl), 4-(2-
methoxyphenyl), 4-(3-methoxyphenyl), 4-(3-ethoxyphenyl), 4-(4-ethoxyphenyl),
4-[3-(prop-1-oxy)phenyl], 4-[3-(prop-2-oxy)phenyl], 4-[4-(prop-2-oxy)phenyl],
4-
(3,4-dimethoxyphenyl), 4-[3-(2-methoxyethoxy)phenyl], 4-[3-(2-
hydroxyethoxy)phenyl], 4-(3-methylthiophenyl), 4-(3-trifluoromethylphenyl), 4-
(3-trifluoromethoxyphenyl), 4-(2-fluorophenyl), 4-(3-chloro-4-fluorophenyl), 4-
(3-
cyanophenyl), 4-(pyridin-2-yl), 4-(pyridin-3-yl), 4-(pyridin-4-yl), 4-(6-
ethoxypyridin-2-yl), 4-{5-ethoxypyridin-3-yl), 4-(indan-5-yl), 4-(2,3-
dihydrobenzofuran-5-yl), 4-(quinolin-3-yl), 4-{benzodioxol-5-yl), 4-(2,2-
dimethylbenzodioxol-5-yl) and 4-(1,2-dimethylbenzimidazol-5-yl); and Ra is
hydrogen, 2-methyl, 3-methyl, 3-ethyl, 3-methoxy, 3-trifluoromethyl, 3-fluoro
or
3-chloro.
~5 A particularly preferred group of compounds of formula (I) is that wherein
R' and R2 are both hydrogen or methyl or, together with the carbon atom to
which they are attached, form a cyclobutyl, cyclopentyl, tetrahydropyran-4,4-
diyl
or 1-methylpiperidin-4,4-diyl group; R3 is 4-phenyl, 4-(3-methoxyphenyl), 4-(3-
ethoxyphenyl), 4-[3-(2-methoxyethoxy)phenyl], 4-[3-(2-hydroxyethoxy)phenyl]
2o or 4-(6-ethoxypyridin-2-yl); and R4 is 3-methyl or 3-methoxy.
Especially preferred individual compounds of the invention include
N-hydroxy-2-{4-[4-(3-ethoxyphenyl)-3-methylphenyl]-1,2, 3,6-
tetrahydropyridin-1-ylsuiphonyl?acetamide;
N-hyd roxy-2-{4-[4-(3-ethoxyphenyl)-3-methylphenyl]-1, 2, 3,6-
25 tetrahydropyridin-1-ylsulphonyl}-2-methylpropanamide;
N-hydroxy-2-{4-[4-(3-ethoxyphenyl)-3-methylphenyl]piperidin-1-
ylsulphonyl)-2-methylpropanamide;
N-hydroxy-1-{4-[4-(3-methoxyphenyl}-3-methylphenyl]piperidin-'1-
ylsulphonyl}cyclopentanecarboxamide;
CA 02312935 2000-06-02
wo 99nx~~ r~r~rsaros~o
N-hydroxy-1-{4-[4-(3-methoxyphenyl)-3-methylphenyl]piperidin-1-
ylsulphonyl}cyclobutanecarboxamide;
N-hydroxy-2-{4-[4-(3-ethoxyphenyl)-3-methoxyphenyl]piperidin-1-
yisulphonyl}-2-methytpropanamide;
N-hydroxy-2-{4-[4-(6-ethoxypyridin-2-yl)-3-methytphenyt]piperidin-1-
ylsulphonyl)-2-methylpropanamide;
N-hydroxy-2-{4-[4-(3-[2-methoxyethoxy]phenyl)-3-methylphenyl]-
piperidin-1-ylsuiphonyl)-2-methylpropanamide; and
to N-hydroxy-2-{4-(4-(3-[2~~hydroxyethoxy]phenyl)-3-methylphenyl]piperidine
-1-ylsulphonyl}-2-methylpropanamide.
In a further aspect, the present invention provides processes for the
preparation of a compound of formula (I), or a pharmaceutically or
veterinarity
~5 acceptable salt thereof, or a pharmaceutically or veterinarily acceptable
solvate
(including hydrate) of either entity, as illustrated below.
It will be appreciated by persons skilled in the art that, within certain of
the processes described, the order of the synthetic steps employed may be
varied and will depend 'mnteyatia on factors such as the nature of other
2o functional groups present in a particular substrate, the availability of
key
intermediates and the protecting group strategy (if any) to be adopted.
Clearly,
such factors will also influence the choice of reagent for use in the said
synthetic steps.
Illustrative of protecting group strategies are the synthetic routes to
25 Example 64, in which an O-benzyl protected hydroxamate is formed prior to
the
required Suzuki reaction step, and to Example 66, in which alcohol protection
using a t-butyldtphenylsilyl group is employed.
CA 02312935 2000-06-02
wo 99nx~~ Pcr~r~sio~oo
_g_
It will also be appreciated that various standard substituent or functional
group interconversions and transformations within certain compounds of
formula (1) will provide other compounds of formula (I). An example is the
conversion of the tetrahydropyridine derivative (Example 28) to the piperidine
derivative (Example 29) by hydrogenation.
The following processes are illustrative of the general synthetic
procedures which may be adopted in order to obtain the compounds of the
invention.
~o A compound of formula (I) may be prepared directly from an ester of
formula (II):
Rs
R~
R, Rs(C i Z)~ i ~ (It)
Ra0 /N-(CHZ)m
'S
Oz
O
wherein R5 is C~ to C3 alkyl, and the broken line, A,B, R', R2, R3, R", m and
n
~ 5 are as previously defined for formula (I), or y1~ the intermediacy of the
corresponding carboxylic acid of formula (II) wherein R5 is hydrogen.
When prepared directly from an ester of formula (II), the reaction may be
carried out by treatment of the ester with up to a 3 fold excess of
hydroxylamine
in a suitable solvent at from about room temperature to about 85°C. The
2o hydroxylamine is conveniently generated jp, ~d from its hydrochloride salt
by
conducting the reaction in the presence of a molar equivalent amount of a
suitable base such as an alkali metal carbonate or bicarbonate, e.g. potassium
carbonate. Preferably the solvent is methanol , optionally combined with
tetrahydrofuran or dichloromethane as co-solvent, and the reaction temperature
25 is from about 65 to 70°C.
CA 02312935 2000-06-02
wo 99nsss~ pc~r~r9sro~o
-10-
Alternatively, the ester may be converted by conventional hydrolysis to
the corresponding carboxylic acid which is then transformed to the required
hydroxamic acid of formula (I).
s Preferably the hydrolysis is effected under basic conditions using up to
about a 6-fold excess of an alkali metal hydroxide in aqueous solution,
optionally in the presence of a co-solvent, at from about room temperature to
about 85°C. Typically the co-solvent is selected from methanol, 1,4-
dioxan, a
mixture of methanol and tetrahydrofuran and a mixture of methanol and 1,4-
o dioxan and the reaction temperature is from about 40 to about 70°C.
The subsequent coupling step may be achieved using conventional
amide-bond forming techniques, e.g. yj,~ the aryl chloride derivative and
hydroxylamine hydrochloride in the presence of an excess of a tertiary amine
such as triethylamine or pyridine to act as acid-scavenger, optionally in the
s presence of a catalyst such as 4-dimethylaminopyridine, in a suitable
solvent
such as dichloromethane, at from about 0°C to about room temperature.
For
convenience, pyridine may also be used as the solvent.
In particular, any one of a host of amino acid coupling variations 'may be
used. For example, the acid of formula (II) wherein R5 is hydrogen may be
2o activated using a carbodiimide such as 1,3-dicyclohexylcarbodiimide or 1-
ethyl-
3-(3-dimethylaminoprop-1-yl)carbodiimide optionally in the presence of 1-
hydroxybenzotriazole and/or a catalyst such as 4-dimethylaminopyridine, or by
using a halotrisaminophosphonium salt such as bromottis(pyrrolidino)-
phosphonium hexafluorophosphate. Either type of coupling is conducted in a
2s suitable solvent such as dichloromethane or dimethylformamide, optionally
in
the presence of a tertiary amine such as N-methylmorpholine or N-
ethyldiisopropylamine (for example when either the hydroxylamine or the
CA 02312935 2000-06-02
wo ~an9s6~ Pcr~r~a~os~oo
-11-
activating reagent is presented in the form of an acid addition salt), at from
about 0°C to about room temperature. Typically, from 1.1 to 2.0
molecular
equivalents of the activating reagent and from 1.0 to 4.0 molecular
equivalents
s of any tertiary amine present are employed.
A preferred reagent for mediating the coupling reaction is O-(7-
azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU).
Preferably a solution of the acid and from 1.0 to 1.2 molecular
equivalents of N-ethyldiisopropylamine in a suitable solvent such as anhydrous
dimethylformamide or anhydrous 1-methylpyrrolidin-2-one, under nitrogen, is
treated with up to a 50% excess of HATU at about room temperature followed,
after about 15 to 30 minutes, with up to about a 3-fold excess of
hydroxylamine
hydrochloride and up to about a 4 fold excess of N-ethyidiisopropylamine,
optionally in the same solvent, at the same temperature.
15 An ester of formula (II) may be prepared from an amine of formula (III):
R'
4
(CHz)~A~ R (111)
I~N-(CHZ)m
wherein the broken line, A,B, R3, R4, m and n are as previously defined for
formula (II), by sulphonylation with a compound of formula (I~:.
Rz
R (m
R60
SOzZ
20 O
wherein Z is halo, R5 is C~ to C3 alkyl and R' and R2 are as previously
defined
for formula (II). Preferably, Z is chloro.
CA 02312935 2000-06-02
wo ~n~6s~ Pcr~~os~o
-12-
When Rg is hydrogen, it will normally be advantageous to protect this
secondary amino linkage with a conventional amine protecting group.
The reaction may be effected in the presence of up to a 50% excess of
s an appropriate base in a suitable solvent at from about 0°C to about
room
temperature. For example, when both R' and R2 are hydrogen, an appropriate
base is 1,8-diazabicyclo[5.4.0]undec-7-ene and a suitable solvent is
dichloromethane.
Alternatively, the anion of (III) may be generated initially using up to a
20°!° excess of a strong base in a suitable solvent, under
nitrogen, and then the
sulphonylation with from 1.0 to 1.2 molecular equivalents of (I~ effected.
Conveniently, such a coupling may be carried out at room temperature
with N,O-bis(trimethylsilyl)acetamide as base and anhydrous tetrahydrofuran as
solvent.
Further routes to the preparation of an ester of formula (II), wherein R3 is
R', rely on exploitation of either a Suzuki reaction or a Stille reaction with
an
ester of formula (II) wherein R3 (but not R~) is either bromo or iodo.
Thus, in the Suzuki reaction, the latter ester is treated with from 1.0 to
1.5 molecular equivalents of a boronic acid of formula R'B(OH)2, in the
2o presence of from 2.0 to 3.0 molecular equivalents of an alkali metal
fluoride,
about 0.1 molecular equivalents of a triarylphosphine and about 0.05 molecular
equivalents of a palladium catalyst in a suitable solvent, under nitrogen, at
from
about 65 to about 100°C. Typically, the fluoride is cesium fluoride,
the
phosphine is tri-o-tolylphosphine, the catalyst is tris(dibenzylideneacetone)-
2s dipalladium(0) and the solvent is degassed 1,2-dimethoxyethane optionally
with
1-methylpyrrolidin-2-one as co-solvent.
CA 02312935 2000-06-02
wo ~n~6~ Pcr~~siostao
-13-
In the Sttlle reaction, the aforementioned ester starting material of
formula (II) is treated with from 1.0 to 2.0 molecular equivalents of a
suitable
trialkylstannane derivative of formula R'Sn(alkyl)3 wherein alkyl is, for
example,
n-butyl, in the presence of from 2.0 to 3.0 molecular equivalents of a
tertiary
base, from 0.3 to 0.6 molecular equivalents of a triarylphosphine and from
0.05
to 0.2 molecular equivalents of a palladium catalyst in a suitable solvent,
under
nitrogen, at from about 65 to about 100°C. Typically, the base is
triethylamine,
the phosphine is tri-o-tolylphosphine, the catalyst is palladium(II) acetate
optionally in the presence of tetrakis(triphenylphosphine)palladium(0) and the
solvent is anhydrous acetonitrile.
Certain esters of formula (II) wherein at least one of R' and R2 is other
than hydrogen may be conveniently obtained from the a-carbanion of an ester
of formula (II) wherein at least one of R' and R2 is hydrogen by conventional
C-
~5 alkylation procedures using an alkylating agent of formula NA) or NB):
RX X-W Y
NA) NB>
2o wherein R is as previously defined for R' or R2 but is not hydrogen, X and
Y
may be the same or different and are suitable leaving groups, and W is a C2 to
C5 alkylene group which optionally incorporates a heteroatom linkage selected
from O, SO, S02 and NRs or which is optionally benzo-fused. When R6 is to be
hydrogen in a compound of formula (I), then a conventional amine protecting
25 group strategy may be of advantage during this alkylation procedure.
A suitable leaving group may be selected from halo (e.g. chloro, bromo
or iodo), C~-C4 alkanesulphonyloxy, trifluoromethanesulphonyloxy and
arylsulphonyloxy (e.g. benzenesulphonyloxy or p-toluenesulphonyloxy).
CA 02312935 2000-06-02
WO 99/Z9667 PCT/EP98/0664~
-14-
Preferably, X and Y are selected from bromo, iodo and p-
toluenesulphonyloxy.
The carbanion may be generated using an appropriate base in a suitable
solvent. Typical base-solvent combinations may be selected from lithium,
sodium or potassium hydride, lithium, sodium or potassium
bis(trimethylsilyl)amide, lithium diisopropylamide and butyllithium, together
with
toluene, ether, 1,2-dimethoxyethane, tetrahydrofuran, 1,4-dioxan,
dimethylformamide, N,N-dimethylacetamide, 1-methylpyrrolidin-2-one and any
o mixture thereof.
Preferably the base is sodium hydride and the solvent is anhydrous
dimethylformamide, optionally with anhydrous tetrahydrofuran as co-solvent, or
anhydrous 1-methylpyrrolidin-2-one. For monoalkylation, up to about a 10%
excess of base is employed whilst, for dialkylation, from about 2 to about 3
~5 molar equivalents are generally appropriate.
Typically, the carbanion is generated at about room temperature, under
nitrogen, and subsequently treated with up to about a 30% excess of the
required alkylating agent at the same temperature.
Clearly, when dialkylation is required and R' and R2 are different, the
2o substituents may be introduced in tandem in a one-pot reaction° or
in separate
steps.
A particularly convenient, alternative alkylation method involves
treatment of the substrate with the required alkylating agent in the presence
of
from 3.0 to 3.5 molecular equivalents of anhydrous potassium carbonate in
25 anhydrous dimethyl sulphoxide or anhydrous 1,2-dimethoxyethane, under
nitrogen, at about room temperature.
CA 02312935 2000-06-02
WO 99/29667 PCT/EP9t3/06640
-15-
Clearly, an alternative variation for preparing a compound of formula (II)
is to introduce R' and/or R2 into a suitable bromo or iodo intermediate beforg
further elaboration yj~, for example, a Suzuki or Stille reaction.
An amine of formula (III) may be obtained by standard chemical
procedures. For example, when B is absent, m is 2 and n is 1, a suitably N-
protected piperidin-4-one of formula (VI):
0
PN (Vt)
wherein P is a conventional amine protecting group, is reacted with a
carbanion
1o derivative of a compound of formula (VII):
Rs
Z ~ ~ 4 ~tt)
R
wherein Z is as previously defined for formula (IV) and R3 and R4 are as
previously defined for formula (III), to provide a compound of fomnula (VIII):
(vut)
Preferably, Z is chloro, bromo or iodo.
Conveniently (VII) is converted to an aryllithium or aryl Grignard
derivative whilst, of the plethora of amine protecting groups available, P is
typically t-butoxycarbonyl (Boc) or benzyl.
CA 02312935 2000-06-02
wo ~n~s~ pc~r~r9s~o~o
-16-
When P is Boc, (VIII) may be transformed directly to a compound of
formula (1t1), wherein the broken line represents a bond, A is C, B is absent,
m
is 2, n is 1 and R3 and R° are as previously defined for formula (III),
using
trifluoroacetic acid optionally in a suitable solvent such as dichloromethane
at
about room temperature. Alternatively, when P is benzyl, (Vllt) may be
converted in two steps to the same compound of formula (III). For example, in
the first step, dehydration may be effected in refluxing toluene using p-
tofuenesulphonic acid and a Dean-Stark apparatus. N-Deprotection of the
resulting alkene (1,2,3,6-tetrahydropyridine derivative), in the second step,
may
be achieved using 1-chloroethyl chloroformate in refluxing toluene followed by
treatment of the reaction mixture, at room temperature, with either methanol
or
ethanol.
This unsaturated piperidine may be converted to a compound of formula
(III) wherein the broken line does ~ represent a bond, A is CH, B is absent, m
is 2, n is 1 and Ra and R4 are as previously defined for formula (III) under
conventional catalytic, or catalytic transfer, hydrogenation conditions.
Alternatively, these hydrogenation conditions may be employed to convert the
previously described N-benryl alkene (1,2,3,6-tetrahydropyridine derivative)
to
2o the same piperidine derivative, directly in one step. Furthermore, this
fully
saturated piperidine is also available in one step from (VIII) when P is Boc
by
standard ionic hydrogenation using, for example, triethylsilane and
trifluoroacetic acid in dichloromethane.
Other amines of formula (III), when neither commercially available nor
subsequently described, can be obtained either by analogy with the processes
described in the Preparations section or by conventional synthetic procedures,
in accordance with standard textbooks on organic chemistry or literature
precedent, from readily accessible starting materials using appropriate
reagents
and reaction conditions.
CA 02312935 2003-04-04
69387-288
-17-
Moreover, persons skilled in the art will be aware of variations of, and
alternatives to, those processes described hereinafter in the Examples and
Preparations sections which allow the compounds defined by formula (I) to be
obtained.
The pharmaceutically and veterinarily acceptable base salts of the
compounds of formula (I) may also be prepared in a conventional manner. For
example a solution of the hydroxamic acid is treated with the appropriate
base,
either neat or in a suitable solvent, and the resulting salt isolated either
by
1o filtration or by evaporation under vacuum of the reaction solvent.
Pharmaceutically and veterinarily acceptable acid addition salts can be
obtained in an analogous manner by treating a solution of a basic compound of
formula (1) with the appropriate acid. Both types of salt may be formed or
interconverted using ion-exchange resin techniques.
The biological activities of the compounds of the present invention were
determined by the following test methods, which are based on the ability of
the
compounds to inhibit the cleavage of various fluorogenic peptides by MMPs 1,
2, 3, 9, 13 and 14.
2o The assays for MMPs 2, 3, 9 and 14 are based upon the original
protocol described in FEBS, 1992, ~C9 , 263, with the minor modifications
described below.
Inhibition of MMP-1
Enz~rme Preparation
Catalytic domain MMP-1 was prepared in Pfizer Central Research
laboratories. A stock solution of MMP-1 (1 ~iM) was activiated by the addition
of aminophenylmercuric acetate (APMA), at a final concentration of 1 mM, for
20
minutes at 37°C. MMP-1 was then diluted in Tris-HCI assay buffer (50mM
Tris,
200mM NaCI, 5mM CaCl2, 20~M ZnS04 and 0.05% Brij 35, pH 7.5) to a
3o concentration of 10nM. The final concentration of enzyme used in the assay
was 1 nM.
*Trade-mark
CA 02312935 2003-04-04
69387-288
-18-
~.strate
The fluorogenic substrate used in this assay was Dnp-Pro-~i-cyclohexyl-
Ala-Gly-Cys(Me)-His-Ala-Lys-(N-Me-Ala)-NH2 as originally described in Anal.
s Biochem., 1993, ~2., 58. The final substrate concentration used in the assay
was 10uM.
Determination of Enzyme Inhibition
The test compound was dissolved in dimethyl sulphoxide and diluted
with assay buffer so that no more than 1 % dimethyl sulphoxide was present.
Test compound and enzyme were added to each well of a 96 well plate and
allowed to equilibrate for 15 minutes at 37°C in an orbital shaker
prior to the
addition of substrate. Plates were then incubated for 1 hour at 37°G
prior to
determination of fluorescence (substrate cleavage) using a fluorimeter
(Fluostar; BMG LabTechnologies, Aylesbury, UK) at an excitation wavelength
~5 of 355 nm and emission wavelength of 440 nm. The potency of inhibition was
measured from the amount of substrate cleavage obtained using a range of test
compound concentrations and, from the resulting dose-response curve, an IC5o
value (the concentration of inhibitor required to inhibit 50% of the enzyme
activity) was calculated.
20 Inhibition of MMP-2 MMP-3 and MMP-9
~nzy,tme Preparation
Catalytic domains MMP-2, MMP-3 and MMP-9 were prepared in Pfizer
Central Research laboratories. A stock solution of MMP-2, MMP-3 or MMP-9
(1~M) was activated by the addition of APMA. For MMP-2 and MMP-9, a final
25 concentration of 1 mM APMA was added, followed by incubation for 1 hour at
37°C. MMP-3 was activated by the addition of 2mM APMA, followed by
incubation for 3 hours at 37°C_ The enzymes were then diluted in Tris-
HCI
assay buffer (100mM Tris, 100mM NaCI, 10mM CaCl2 and 0.16% Brij 35, pH
7.5) to a concentration of 10nM. The final concentration of enzyme used in the
so assays was 1 nM_
*Trade-mark
CA 02312935 2003-04-04
69387-288
-19-
substrate
The fluorogenic substrate used in this screen was Mca-Arg-Pro-Lys-Pro-
Tyr-Ala-Nva-Trp-Met-~ys(Dnp)-NH2 (Bachem Ltd., Essex, UK) as originally
described in J.BioLChem., 1994, X69, 20952. This substrate was selected
because it has a balanced hydrolysis rate against MMPs 2, 3 and 9 (k~t/km of
54,000, 59,400 and 55,300 s~' M-' respectively). The final substrate
concentration used in the assay was SpM.
Determination of Enz~rme Inhibition
The test compound was dissolved in dimethyl sulphoxide and diluted
with assay buffer so that no more than 1 % dimethyi sulphoxide was present.
Test compound and enzyme were added to each well of a 96 well plate and
allowed to equilibrate for 15 minutes at 37°C in an orbital shaker
prior to the
addition of substrate. Plates were then incubated for 1 hour at 37°C,
prior to
~5 determination of fluorescence using a fluorimeter (Fluostar; BMG
LabTechnologies, Aylesbury, UK) at an excitation wavelength of 328nm and
emission wavelength of 393nm. The potency of inhibition was measured from
the amount of substrate cleavage obtained using a range of test compound
concentrations and, from the resulting dose-response curve, an ICSO value (the
20 concentration of inhibitor required to inhibit 50% of the enzyme activity)
was
calculated.
Inhibition of MMP-13
Enz~ mr a Preparation
25 Human recombinant MMP-13 was prepared by PanVera Corporation
(Madison, Wisconsin) and characterised at Pfizer Central Research
laboratories. A 1.9 mg/rnf stock solution was activated with 2mM APMA for 2
hours at 37°C. MMP-13 was then diluted in assay buffer (50mM Tris,
200mM
NaCI, 5mM CaCl2, 20pM ZnCl2 and 0.02% Brij 35, pH 7.5) to a concentration of
30 5.3nM. The final concentration of enzyme used in the assay was 1.3nM.
*Trade-mark
CA 02312935 2003-04-04
69387-288 .
-20-
Substrate
The tluorogenic substrate used in this screen was Dnp-Pro-Cha-Gly-
Cys(Me)-His-Ala-Lys(NMA)-NH2. The final substrate concentration used in the
s assay was 10pM.
Determination of Enz,~Lme Inhibition
The test compound was dissolved in dimethyl sulphoxide and diluted
with assay buffer so that no more than 1 % dimethyl sulphoxide was present.
Test compound and enzyme were added to each well of a 96 well plate. The
1o addition of substrate to each well initiated the reaction. Fluorescence
intensity
was determined using a 96 well plate fluorimeter (Cytofluor II; PerSeptive
Biosystems, Inc., Framingham, MA) at an excitation wavelength of 360nm and
emission wavelength of 460nm. The potency of inhibition was measured from
the amount of substrate cleavage obtained using a range of test compound
~s concentrations and, from the resulting dose-response curve, an ICSO value
(the
concentration of inhibitor required to inhibit 50% of the enzyme activity) was
calculated.
Inhibition of MMP-14
Enzyme Preparation
2o Catalytic domain MMP-14 was prepared in Pfizer Central Research
laboratories. A lOpM enzyme stock solution was activated for 20 minutes at
25°C following the addition of 5pglml of trypsin (Sigma, Dorset, UK).
The
trypsin activity was then neutralised by the addition of 50pglml of soyabean
trypsin inhibitor (Sigma, Dorset, UK), prior to dilution of this enzyme stock
25 solution in Tris-HCI assay buffer (100mM Tris, 100nM NaCI, 10mM CaCl2,
0.16% Brij 35, pH 7.5) to a concentration of 10nM. The final concentration of
enzyme used in the assay was 1 nM.
*Trade-mark
CA 02312935 2000-06-02
WO 99/29667 PCT/EP98/06640
-21-
The fluorogenic substrate used in this screen was Mca-Pro-Leu-Gly-Leu-
Dpa-Ala-Arg-NH2 (Sachem Ltd., Essex, UK) as described in J.BioLChem.,
1996, ~, 17119.
This was performed as described for MMPs 2, 3 and 9.
In human therapy, the compounds of formula (I), their pharmaceutically
acceptat~le salts, and pharmaceutically acceptable solvates of either entity,
can
be administered alone, but will generally be administered in admixture with a
pharmaceutical carrier selected with regard to the intended route of
administration and standard pharmaceutical practice.
They may be administered orally in the form of tablets containing such
~ 5 excipients as starch or lactose, or in capsules or ovules either alone or
in
admixture with excipients, or in the form of elixirs, solution or suspensions
containing flavouring or colouring agents. They can also be injected, for
example intravenously, intramuscularly or subcutaneously, and are best used in
the form of a sterile aqueous solution which may contain other substances, for
2o example enough salts or monosaccarides to make the solution isotonic with
blood. For other routes of parenteral administration, such as buccal or
sublingual, they may be administered in the form of tablets or lozenges which
can be formulated in a conventional manner.
In addition, the compounds and their salts may be administered topically
25 in the form of sterile creams, gels, suspensions, lotions, ointments,
dusting
powders, sprays, drug-incorporated dressings or yj~ a skin patch. For example,
they can be incorporated into a cream consisting of an aqueous or oily
emulsion of polyethylene glycols or liquid paraffin, or they can be
incorporated
into an ointment consisting of a white wax soft paraffin base, or as a
hydrogel
CA 02312935 2000-06-02
wo ~n~ss~ rc~r~r9aross~o
-22-
with cellulose or polyacrylate derivatives or other viscosity modifiers, or as
a dry
powder or liquid spray or aerosol with butane/propane, HFA or CFC
propellants, or as a drug-incorporated dressing either as a tulle dressing,
with
s white soft paraffn or polyethylene glycol impregnated gauze dressings or
with
hydrogel, hydrocolloid, alginate or film dressings. Moreover, the compounds
and salts may be administered intraocularly as an eye drop with appropriate
buffers, viscosity mod~ers (e.g. cellulose derivatives), preservatives (e.g.
benzalkonium chlorides (BZK)) and agents to adjust tonicity (e.g. sodium
o chloride).
All such formulations may also contain stabilisers and preservatives.
Depending on the route of administration to human patients, the daily
dosage level of the compounds of formula (I) and their salts may be from 0.001
to 20 mg/kg, in single or divided doses. Thus, for example, tablets or
capsules
s could contain ftom 0.02 to 500 mg of active compound for administration
singly
or two or more at a time as appropriate.
The physician in any event will determine the actual dosage which will be
most suitable for an individual patient and it will vary with the age, weight
and
response of the particular patient. The above dosages are exemplary of the
2o average case; there can of course be individual instances where higher or
lower dosage ranges are merited and such are within the scope of this
invention.
For veterinary use, a compound of formula (I), or a veterinarily
acceptable salt thereof, or a veterinarily acceptable solvate of either
entity, is
2s administered as a suitably acceptable formulation in accordance with normal
veterinary practice and the veterinary surgeon will determine the dosing
regimen and route of administration which will be most appropriate for a
particular animal.
CA 02312935 2000-06-02
wo ~n9s~s~ rcT~~~os~oo
-23-
Thus the invention provides a pharmaceutical composition comprising a
compound of formula (I), or a pharmaceutically acceptable salt thereof, or a
pharmaceutically acceptable solvate of either entity, together with a
pharmaceutically acceptable diluent or carrier.
It further provides a veterinary formulation comprising a compound of
formula (I), or a veterinarily acceptable salt thereof, or a veterinarily
acceptable
solvate of either entity, together with a veterinarily acceptable diluent or
carrier.
The invention also provides a compound of formula (I), or a
pharmaceutically acceptable salt ther~;of, or a pharmaceutically acceptable
solvate of either entity, or a pharmaceutical composition containing any of
the
foregoing, for use as a human medicament.
In addition, it provides a compound of formula (I), or a veterinarily
acceptable salt thereof, or a veterinarily acceptable solvate of either
entity, or a
~5 veterinary formulation containing any of the foregoing, for use as an
animal
medicament.
In yet another aspect, the invention provides the use of a compound of
formula (1), or a pharmaceutically acceptable salt thereof, or a
pharmaceutically acceptable solvate of either entity, for the manufacture of a
2o human medicament for the curative or prophylactic treatment of a medical
condition for which a MMP inhibitor is indicated.
It also provides the use of a compound of formula (I), or a veterinarily
acceptable salt thereof, or a veterinarily acceptable solvate of either
entity, for
the manufacture of an animal medicament for the curative or prophylactic
25 treatment of a medical condition for which a MMP inhibitor is indicated.
Moreover, the invention provides the use of a compound of formula (I),
or a pharmaceutically acceptable salt thereof, or a pharmaceutically
acceptable
solvate containing either entity, for the manufacture of a human medicament
for
the curative or prophylactic treatment of atherosclerotic plaque rupture,
CA 02312935 2003-04-04
69387-288
- 2 4 - . ..,. . ,
myocardial infarction, heart failure, restenosis, stroke,
periodontal disease, tissue ulceration, wound repair, skin,
diseases, cancer metastasis, tumour angiogenesis, age- ,
related macular degeneration, fibrotic disease, rheumatoid
arthritis, osteoarthritis and inflammatory diseases
dependent on migratory inflammatory cells.
It also provides the use of a compound of
formula (I), or a veterinarily acceptable salt thereof, or a
veterinarily acceptable solvate containing either entity,
for the manufacture of an animal medicament for the curative
or prophylactic treatment of atherosclerotic plaque rupture,
myocardial infarction, heart failure, restenosis, stroke,
periodontal disease, tissue ulceration, wound repair, skin
diseases, cancer metastatis, tumour angiogenesis, age-
related macular degeneration, fibrotic disease, rheumatoid
arthritis, osteoarthritis and inflammatory diseases
dependent on migratory inflammatory cells.
The invention also provides a commercial package
comprising: (a) a first dosage form comprising a compound
of formula (I), or a pharmaceutically or veterinarily
acceptable salt thereof, or a pharmaceutically or
veterinarily acceptable solvate of either entity, together
with a pharmaceutically or veterinarily acceptable diluent
or carrier; and (b) a written matter describing instructions
for the use thereof for treating or preventing a medical
condition for which a MMP inhibitor is indicated in a mammal
(including a human being).
It also provides a commercial package comprising:
(a) a first dosage form comprising a compound of
formula (I), or a pharmaceutically or veterinarily
acceptable salt thereof, or a pharmaceutically or
CA 02312935 2003-04-04
69387-288
- 24a - , ,, ,, .,.
veterinarily acceptable solvate of either entity, together
with a pharmaceutically or veterinarily acceptable diluent,
or carrier; and (b) a written matter describing instructions
for the use thereof for treating or preventing
atherosclerotic plaque rupture, myocardial infarction, heart
failure, restenosis, stroke, periodontal disease, tissue
ulceration, wound repair, skin diseases, cancer metastasis,
tumour angiogenesis, age-related macular degeneration,
fibrotic disease, rheumatoid arthritis, osteoarthritis and
inflammatory diseases dependent on migratory inflammatory
cells in a mammal (including a human being).
Additionally, the invention provides a method of
treating or preventing a medical condition for which a MMP
inhibitor is indicated, in a mammal (including a human
being), which comprises administering to said mammal a
therapeutically effective amount of a compound of
formula (I), or a pharmaceutically or veterinarily
acceptable salt thereof, or a pharmaceutically or
veterinarily acceptable solvate of either entity, or a
pharmaceutical composition or veterinary formulation
containing any of the foregoing.
Still further, the invention provides a method of
treating or preventing atherosclerotic plaque rupture,
myocardial infarction, heart failure, restenosis, stroke,
periodontal disease, tissue ulceration, wound repair, skin
diseases, cancer metastasis, tumour angiogenesis, age
related macular degeneration, fibrotic disease, rheumatoid
arthritis, osteoarthritis and inflammatory diseases
dependent on migratory inflammatory cells, in a mammal
(including a human being), which comprises administering to
said mammal a therapeutically effective amount of a compound
of formula (I), or a pharmaceutically or
CA 02312935 2000-06-02
wo ~n~s~ rc~r~r9sross~eo
-25-
veterinarily acceptable salt thereof, or a pharmaceutically or veterinarily
acceptable solvate of either entity, or a pharmaceutical composition or
veterinary formulation containing any of the foregoing.
The invention also includes any novel intermediates described herein, for
example those of formula (11).
The syntheses of the compound of the invention and of the
intermediates for use therein are illustrated by the following Examples and
Preparations.
Room temperature means 20 to 25°C.
Flash chromatography refers to column chromatography on silica gel
(Kieselgel 60, 230-400 mesh).
Melting points are uncorrected.
'H Nuclear magnetic resonance (NMR) spectra were recorded using a
Broker AC300, a Varian Unity Inova-300 or a Varian Unity tnova-400
spectrometer and were in all cases consistent with the proposed structures.
Characteristic chemical shifts (8) are given in parts-per-million downfield
from
tetramethylsilane using conventional abbreviations for designation of major
peaks: e.g. s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br,
broad.
2o Mass spectra were recorded using a Finnigan Mat. TSQ 7000 or a
Fisons Intruments Trio 1000 mass spectrometer. l_RMS means low resolution
mass spectrum and the calculated and observed ions quoted refer to the
isotopic composition of lowest mass.
CA 02312935 2000-06-02
WO 99I29b67 PCT/EP98/06640
-26
EXAMPLE 1
Potassium carbonate (207mg, 1.5 mmol) was added to a stirred mixture
of the title compound of Preparation 8 (186 mg, 0.5 mmol), hydroxylamine
hydrochloride (104 mg, 1.5 mmol), tetrahydrofuran (2.m1) and methanol (3 ml).
The reaction mixture was heated under reflux for 20 hours, allowed to cool,
diluted with water (15 ml} and ethyl acetate (10 ml) and acidified with
concentrated hydrochloric acid. This nuxture was then briefly heated at
100°C,
allowed to cool and filtered. The material thus obtained was washed
sequentially with water and ethyl acetate, then dried under vacuum to provide
the title compound (125 mg, 67%) as a colourless solid, m.p. 216-218°C.
Found: C,61.05; H, 5.35; N, 7.41. C~gH2pN204S requires C, 61.27; H, 5.41; N,
7.52%. & {DMSOd6): 2.62 (m,2H), 3.50 (m,2H), 3.92 (s,2H), 3.98 (m,2H), 6.24
{brs, 1 H), 7.35 (m,1 H), 7.41-7.48 (m,2H), 7.52-7.58 (m,2H), 7.62-7.73
(m,4H),
9.22 (s,1 H), 10.82 (s,1 H).
LRMS (APCI): 373 (M+H)+.
2o E?~ld~L~2
N-HydrQ~r-2-[~~-~ -methyrl-4-Rhenyrli~henyrl)~-1.2.3.6-tetrahyrdroRyridin-1-
Obtained as a colourless solid (58%), m.p. 190-191°C, from the
title
compound of Preparation 9, using the procedure of Example 1. Found:
C,62.05; H,5.74; N,7.12. C~H~N204S requires C,62.16; H,5.74; N,7.25%.
S(DMSO~): 2.23 {s,3H), 2.60 (m,2H), 3.47 (t,2H), 3.90 {s,2H), 3.95 (m,2H),
6.20 (brs, 1 H), 7.17 (d,1 H), 7.30-7.48 (m,7H), 9.20 (s,1 H), 10.80 (s,1 H).
LRMS (Thermospray}: 388(M+H}''.
CA 02312935 2000-06-02
wo ~n~ss~ pcr~o
-27-
Obtained as a colourless solid (62%), m.p. 200-201°C, from the
title
compound of Preparation 10, using the procedure of Example 1. Found:
C,60.96; H,5.86; N,6.97. C~gH22NZO4S requires C,60.94; H,5.92; N,7.48%.
S(DMSO~): 1.63 (m,2H), 1.83 (m,2H), 2.66 (m,1H), 2.98 (t,2H), 3.70 (m,2H),
3.83 (s,2H), 7.30 (m,3H), 7.40 (t,2H), 7.54-7.60 (m,4H), 9.18 (s,1 H), 10.75
(s,1 H).
LRMS (Theimospray): 375 (M+H)''.
N-Hyd roxv-Z=(4-p h~nyl-1.2. 3.6-tetra hyrd rohyrrid in-1-yrlsu I
Rhonyrllacetam ide
Obtained as a colourless solid (76%), m.p. 175-176°C, from the
title
~5 compound of Preparation 11, using the procedure of Example 1. Found:
C,52.41; H,5.39; N,9.35. C~3H~sN204S requries C,52.69; H,5.44; N,9.45%.
8(DMSO~): 2.58 (m,2H), 3.46 (t,2H), 3.90 (s,2H), 3.95 (m,2H), 6.18 (brs, 1 H),
7.23-7.48 (m,5H), 9.20 (s,1 H), 10.80 (s,1 H).
2o E~FLE 5
Obtained as a colourless solid (44%), m.p. 185-187°C, from the
title
compound of Preparation 12, using the procedure of Example 1. Found:
C,52.08; H,6.04; N,9.23. C~3H~8N204S requires C,52.33; H,6.08; N,9.39%.
25 8(DMSOdg): 1.62 (m,2H), 1.82 (m,2H), 2.62 (m,1H), 2.98 (t,2H), 3.70 (m,2H),
3.84 (s,2H), 7.15-7.33 (m,SH), 9.20 (s,1 H), 10.78 (s,1 H).
CA 02312935 2000-06-02
wo ~n~~ Pcr~r9sios~o
-28-
Obtained as a colourless solid (35%), m.p. 132-135°C, from the
title
compound of Preparation 13, using the procedure of Example 1. Found:
C,53.66; H,6.43; N,8.82. C~4H2pN2O4S requires C,53:83; H,6.45; N,8.97%.
8(DMSO~): 1.13 (m,2H), 1.58 (m,3H), 2.49 (d,2H), 2:75 (t,2H), 3.50 (d,2H),
3.73 {s,2H), 7.10 (m,3H), 7.22 (m,2H), 9.10 (s,1H), 10.70 (s,lH).
EXAMPLE 7
O-{7-Azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronium
hexafluorophosphate (100 mg, 0.26 mmol) was added to a stirred solution of
~5 the title compound of Preparation 15 (70 mg, 0.18 mmol} and N-
ethyldiisopropylamine (0.03 ml, 0.18 mmol) in anhydrous dimethylformamide
(1 ml}, under nitrogen, at room temperature. After 15 minutes, a solution of
hydroxylamine hydrochloride (37 mg, 0.53 mmol) and N-ethyldiisopropylamine
{0.12 rnl, 0.7 mmol) in anhydrous dimethylformamide (0.5 ml) was added and
2o the reaction mixture stirred for 20 hours, then partitioned between ethyl
acetate
and aqueous phosphate buffer (pH 7). The organic phase was separated,
washed with water, dried (MgS04) and evaporated under reduced pressure,
then the residue purified by flash chromatography, using
dichloromethane:methanol (97:3) as eluant, ~o give the title compound (55 mg)
25 as a colourless, amorphous solid. S(DMSOds): 2.50-2.80 (m,4H), 3.50 (m,2H),
3.80 (dd,1 H), 4.00 (m,2H), 5.03-5.18 (m,2H), 5.62 (m,1 H), 6.23 {brs, 1 H),
7.37
(m,1H), 7.45 (m,2H), 7.53 (m,2H), 7.67 (m,4H), 9.22 {s,1H), 10.85 (s,1H).
LRMS (Thermospray): 413 {M+H)+.
CA 02312935 2000-06-02
wo ~n~s~ rcT~r9s~o
-29-
Obtained as a solid (13%) from the title compound of Preparation 17,
using the procedure of Example 7, but with an elution gradient of
dichloromethane:methanol (100:0 to 90:10) for the chromatographic
purification.
. step. 8(CDCI3): 2.25 (s,3H), 2.62 (m,2H), 2.82 (m,2H), 3.62 (m,2H), 3.80
(dd,1 H), 4.10 (m,2H), 5.10-5.22 (m,2H), 5.75 (m,1 H), 6.03 (brs, 1 H), 7.20-
7.43
(m,BHj.
LRMS (APCI): 427 (M+H)+.
y~~_Iphon,yrljc~entanamide
Obtained as a colourless solid (35%), m.p. 150°C (decomp.), from
the
title compound of Preparation 18, using the procedure of Example 1.
8(DMSO~): 1.50 (m,2H), 1.80 (m,lH), 2.00 (m,1H), 2.54 (m,4H), 3.45 (m,2H),
3.75 (dd,1 H), 3.98 (m,2H), 6.10 (brs,1 H), 7.10-7.54 (m,1 OH), 7.63 (m,4H),
9.10
(s,1H), 10.88 (s,1H).
l_RMS (Thermospray): 492 (M+H)'.
1~1~,~~,'lhhonyll~ronan~ amide
Obtained as an amorphous solid (41 %) from the title compound of
Preparation 19, using the procedure of Example 7. S(DMSOds): 1.50 (s,6H),
2.66 (m,2H), 3.50 (m,2H), 4.00 (m,2H), 6.10 (brs,1H), 7.30-7.70 (m,9H), 9.00
(s,1 H), 10.78 (s,1 H).
CA 02312935 2000-06-02
WO 99129667 PCT/EP98/06640
-30-
Obtained as a solid (44%) from the title compound of Preparation 21,
using the procedure of Example 7, but with dichloromethane:methanol (99:1) as
eluant for the chromatographic purification step. 8(CDC13): 1.70 (m,2H), 1.86
(m,2H), 2.27 (s,3H), 2.37 (m,2H), 2.48 (m,2H), 2.62 (m,2H), 3.60 (t,2H), 4.05
(m,2H), 6.02 (brs,1H), 7.20-7.43 {m,BH).
Obtained as a solid (56%) from the title compound of Preparation 23,
using the procedure of Example 7. 8(DMSOds): 0.90 (m,6H), 1.95-2.13 (m,4H),
2.52 (m,2H), 3.48 (m,2H), 3.98 (m,2H), 6.10 (brs,1 H), 7.35 (m,1 H), 7.44
(m,2H),
7.52 (m,2H), 7.64 {m,4H), 9.03 (brs,1 H), 10.70 (brs,1 H).
,~!-.l~ix~yr-ZtR.~)~-L~(~.ah~nyl~.hgny~)-1.2.3.6-tetrahyrdropyrridin-1-
Obtained as a colourless solid (72%), m.p. 186-189°C, from the
title
compound of Preparation 25, using the procedure of Example 7, but with 1-
methylpyrrolidin-2-one as reaction solvent and with crystallisation from
diisopropyl ether:ethyl acetate, rather than flash chromatography, as the
purification technique. Found: C,63.03; H,6.60; N,6.43. C23H28N2O4S; 0.50
H20 requires C,63.13; H,6.68; N,6.40%. S(DMSO~): 0.83 (t,3H), 1.10-1.35
(m,4H), 1.78 (m,1 H), 1.98 (m,1 H), 2.55 (m,2H), 3.50 (m,2H), 3.70 (dd,1 H),
3.98
CA 02312935 2000-06-02
wo 99n~s~s~ Pcr~~smo
-31-
(m,2H), 6.12 (brs, 1 H), 7.32 (m,1 H), 7.44 (m,2H), 7.52 (m,2H), 7.64 (m,4H),
9.20 (brs, 1 H), 10.85 (brs,1 H).
LRMS (APCI): 429 (M+H)~.
Obtained as a colourless solid (33%), m.p. 170-171°C, from the
title
compound of Preparation 27, using the procedure of Example 7, but with an
elution gradient of dichloromethane:methanol (100:0 to 98:2) for the
chromatographic purification step. S(DMSOds): 1.65 (s,3H), 2.40-2.80 (m,4H),
3.52 (m,2H), 3.90 (dd,1 H), 4.00 (m,2H), 4.70 (s,1 H), 4.78 (s,1 H), 6.23
(brs,1 H),
7.35 (m,1 H), 7.44 (m,2H), 7.52 (d,2H), 7.65 (m,4H), 9.22 (s,1 H), 10.85 (s,1
H).
~5 LRMS (APCI): 427 (M+H)+.
2o Obtained as a colourless gum (20%) from the title compound of
Preparation 29, using the procedure of Example 7, but with an elution gradient
of dichloromethane:methanol (100:0 to 98:2 to 95:5) for the chromatographic
purification step. b (CDCI3): 1.60 (s,3H), 2.28 (s,3H), 2.64 (m,3H), 3.00 (m,1
H),
3.62 (m,2H), 4.10 (m,2H), 5.21 (m,2H), 5.70 (m,1 H), 6.03 (brs,1 H), 7.20-7.44
25 (m,BH).
LRMS (APCI): 441 (M+H)+.
CA 02312935 2000-06-02
WO 99I~9667 PCT/EP98/06640
-32
~~~pLE 16
N-Hyrdroxyr-2-[3-(4-ph, envlohenoxvliazetidin-1-ylsulphonvllaceta_mide
Obtained as a colourless solid (66%) from the title compound of
Preparation 32, using the procedure of EXAMPLE 1. 8(DMSO~): 4.03 (s,2H),
4.05 (dd,2H), 5.09 (m,1 H), 6.94 (d,2H), 7.30 (m,1 H), 7.40 (m,2H), 7.60
(m,4H),
9.25 (s,1 H), 10.80 (brs,1 H).
LRMS (Thermospray): 364 (M+H)+.
EXAMPLE 17
~yrdro r-2-~~ f~3-ethox~«nyrl)-3-methyr~~y'~[~-1,~,~,6-tetra~yrdropyrridin-
Potassium carbonate (406 mg, 3 mmol) was added to a stirred mixture of
the title compound of Preparation 41 (429 mg, 1 mmol), hydroxylamine
hydrochloride (212 mg, 3 mmol) and methanol (20 ml). The reaction mixture
was heated under reflux for about 6 hours, allowed to cool and partitioned
between ethyl acetate and 1 M hydrochloric acid. The separated organic phase
was dried (MgS04) and evaporated under reduced pressure, then the residue
triturated with diisopropyl ether and crystallised from ethyl acetate to yield
the
title compound (148 mg, 34%) as a colourless solid, m.p. 151-153°C.
Found:
C,61.01; H, 6.04; N, 6.48. C~H2gN2O5S requires C, 61.38; H, 6.09; N, 6.51 %.
8 (DMSO~): 1.33 (t,3H), 2.23 (s,3H), 2.60 (m,2H), 3.46 (t,2H), 3.91 (s,2H),
3.96 (m,2H), 4.03 (q,2H), 6.10 (brs,1 H), 6.80-6.95 (m,3H), 7.17 (d,1 H), 7.28-
7.38 (m,3H), 9.20 (brs,1 H), 10.8 (brs,1 H).
LRMS (APCI): 431 (M+H)+.
CA 02312935 2000-06-02
wo 99n9ss~ pcr~r~sro~o
-33
EXAMPLE 18
s Potassium carbonate (95 mg, 0.7 mmol) was added to a stirred mixture
of the title compound of Preparation 45 (170 mg, 0.4 mmol), hydroxylamine
hydrochloride (49 mg, 0.7 mmol) and methanol (3 ml). The reaction mixture
was heated under reflux for about 2 hours, allowed to cool, diluted with
phosphate buffer (15 ml) and extracted with ethyl acetate (2 x 15 ml). The
o combined organic phases were dried (MgS04) and evaporated under reduced
pressure, then the residue was triturated with ethyl acetate to furnish the
title
compound (50 mg, 30%) as a colourless solid, m.p. 175-177°C. Found:
C,58.84; H, 5.51; N, 6.70. C2oH~N205S; 0.10 CHZCI2 requires C, 58.75; H,
5.45; N, 6.82%. b (DMSO~): 2.63 (m,2H), 3.50 (t,2H), 3.80 (s,3H), 3.93 (s,2H),
~ 5 3.99 (s,2H), 6.27 (s,1 H), 7.10 (d,1 H), 7.16 (s,1 H), 7.27 (d,1 H), 7.31
(d,1 H), 7.41
(t,2H), 7.47 (d,2H), 9.23 (brs,1 H), 10.80 (brs,1 H).
LRMS (Thermospray): 420 (M+NH4)+.
2o ~[yrdroxy[~~3=..m_ethoxy-4-henyrlphenyrl)~-1.2.3.6-tetrahyrdrohyrridin-1-
O-(7-Azabenzotriazol-1-yl)-1,1, 3, 3-tetramethyluroniurn
hexafluorophosphate (274 mg, 0.72 mmol) was added to a stirred solution of
the title compound of Preparation 50 (200 mg, 0.48 mmol) and N-
2s ethyldiisopropylamine (0.08 ml, 0.48 mmol) in anhydrous dimethylformamide
(4 ml), under nitrogen, at room temperature. After 15 minutes, hydroxylamine
hydrochloride (100 mg, 1.44 mmol) and N-ethyldiisopropylamine (0.33 ml, 1.9
mmol) were added and the reaction mixture stin-ed for about 3 hours, then
partitioned between ethyl acetate and aqueous phosphate buffer (pH 7). The
CA 02312935 2000-06-02
wo ~n~~ rc~'~cr9sros~ao
_34_
organic phase was separated, washed with water, dried (MgS04) and
evaporated under reduced pressure, then the residue was triturated with
diisopropyl ether to give the title compound (71 mg, 36%) as a colourless,
amorphous solid, m.p. 156-158°C. Found: C,60.80; H, 6.17; N, 6.25.
CnH~N205S; 0.10 H20 requires C, 61.12; H, 6.11; N, 6.48%. S(DMSO~);
1.51 {s,6H), 2.57 (m,2H), 3.43 (t,2H), 3.80 {s,3H), 4.03 (m,2H), 6.25
(brs,1H),
7.09 (d,1 H), 7.13 (s,1 H), 7.26 (d,1 H), 7.31 (d,1 H), 7.39 (t,2H), 7.46
(d,2H), 9.24
(brs;1 H), 10.79 (brs,1 H).
LRMS (APCI): 431 (M+H)+.
Obtained as a colourless solid (40%), m.p. 184-188°C, from the
title
compound of Preparation 47, using the procedure of Example 1. Found:
C,58.39; H,4.90; N,6.84. C~9H~9FN204S requires C,58.45; H,4.91; N,7.17%.
8(DMSO~): 2.61 {m,2H), 3.47 (t,2H), 3.94 (s,2H), 4.00 (s;2H), 6.35 (brs,1H),
7.33-7.60 (m,BH), 9.23 (brs,1 H), 10.80 (brs,1 H).
2o LRMS (Thermospray): 408 (M+NH4)+.
Obtained as a colourless solid (76%), m.p. 168-170°C, from the
title
compound of Preparation 56, using the procedure of Example 1. 8(DMSO~):
1.34 (t,3H), 2.61 (m,2H), 3.49 {t,2H), 3.94 (s,2H), 3.98 (s,2H), 4.10 (q,2H),
6.25
(brs,1 H), 6.91 {d,1 H), 7.17 (s,1 H), 7.22 {d,1 H), 7.33 (t,1 H), 7.52
{d,2H), 7.66
(d,2H), 9.22 (brs,1 H), 10.80 (brs,1 H).
so LRMS (Thermospray): 434 (M+NH4)+.
CA 02312935 2000-06-02
wo ~nx~s~ Pc~r~msro~o
-35-
Obtained as a colourless solid (76%), m.p. 162-165°C, from the
title
compound of Preparation 61, using the procedure of Example 1. Found:
C,60.26; H, 5.86; N, 6.43. CZ~H24N2O5S requires C, 60.56; H, 5.81; N, 6.73%.
8(DMSOds): 2.23 (s,3H), 2.60 (m,2H), 3.47 (t,2H), 3.77 (s,3H), 3.93 (s,2H),
3.97
(s,2H), 6.20 (brs,1H), 6.83-6.94(m,3H), 7.18 (d,1H), 7.27-7.39 (m, 3H), 9.22
(brs,1 H), 10.80 (brs,1 H).
LRMS (APCI): 416 (M)+.
~~n~,phonyl?a~~etamide
Obtained as a colourless solid (58%), m.p. 151-154°C, from the
title
compound of Preparation 63, using the procedure of Example 1. Found:
C,62.75; H, 6.24; N, 6.26. C22H2sN2O4S; 0.50 H20 requires C, 62.39; H, 6.43;
N, 6.61 %. 8(DMSOds): 1.20 (t,3H), 2.24 (s,3H), 2.61 (m,4H), 3.47 (t,2H), 3.92
(s,2H), 3.97 (s,2H), 6.20 (brs,1H), 7.10-7.23 (m,4H), 7.27-7.38 (m,3H), 9.22
(brs,1 H), 10.81 (brs,1 H).
LRMS (APCI): 414 (M)''.
~(~(yrdroxrr-4-~(4 j4-(,,,~-Methoxy~~~,~1-3-methy~p~,p,»[~-1.2.3.6-
tetrahlrdrp,~vridin-1 ~,rlsu'~ ho rllt, etrahyrdrQpyjan-4-carboxamide
Obtained as a pale yellow solid (47%), m.p. 160-170°C, from the
title
compound of Preparation 65, using the procedure of Example 19. 8(DMSOds):
1.93 (m,2H), 2.23 (s,3H), 2.39 (m,2H), 2.43 (m,2H), 3.20 (t,2H), 3.49 (m,2H),
CA 02312935 2000-06-02
wo ~nx~~ rcr~r~sroc~4o
-36-
3.77 (s,3H), 3.86 (m,2H), 4.00 (m,2H), 6.16 (brs,1 H), 6.83-6.94 (m,3H), 7.17
(d,1 H), 7.36 (m,3H), 9.21 (brs,1 H), 11.00 (brs,1 H).
LRMS (APCI) 487 (M+H)''.
Obtained as a white solid (82%), m.p. 200-202°C, from the title
1o compound of Pr~:paration 67, using the procedure of Example 19, except that
the residue was crystallised from methanol. Found: C,60.02; H, 6.78; N, 5.45.
~'25H32N206S~ CH30H requires C, 59.98; H, 6.97; N, 5.38%. 8(DMSOds): 1.60
(m,2H), 1.78 (m,2H), 1.90 (m,2H), 2.20 (s,3H), 2.38 (m,2H), 2.64 (m,1 H), 3.04
(t,2H), 3.20 (t,2H), 3.70 (m,2H), 3.77 (s,3H), 3.86 (m,2H), 6.87 (m,3H), 7.13
(m,3H), 7.33 (t,1 H), 9.16 (brs,1 H), 10.97 (brs,1 H).
LRMS (APCI) 489 (M+H)''.
2o yrlsulyhonyrl~~2-methyrl~ronanamide
Obtained as a white solid (47%), m.p. 161-163°C, from the title
compound of Preparation 74, using the procedure of Example 19, except that
the residue was purled by flash chromatography using
dichloromethane:methanol:concentrated aqueous ammonia solution (90:10:1 )
as eluant: Found: C,59:39; H, 6.58; N, 6.13. C23H~N2O6S requires C, 59.72;
H, 6.54; N, 6.06%. 8(DMSO~): 1.49 (s,6H), 1.64 (m;2H), 1.81 (m,2H), 2.70
(m,1H), 3.06 (t,2H); 3.75 (s,BH), 6.87 (m,2H), 6.98 (m,3H), 7.20 (d,lH), 7.27
(t,1 I-t), 8.99 (brs,1 H), 10.75 (brs,1 H).
LRMS {Thermospray): 480 (M+NH4)+.
CA 02312935 2000-06-02
WO 99/2966? PCT/EP98/06640
-37-
Obtained as a white solid (39%), m.p. 134-136°C, from the title
compound of Preparation 77, using the procedure of Example 26. Found:
C,60.60; H, 6.80; N, 5.82. C24H32N2~6S requires C, 60.48; H, 6.77; N, 5.88%.
8(DMSOds): 1.32 (t,3H), 1.49 (s,6H), 1.66 {m,2H), 1.81 (m,2H), 2.70 (m,1 H),
3.07 (t,2H), 3.76 (s,SH), 4.02 (q,2H), 6.85 (m,2H), 6.98 (m,3H), 7.20 (d,iH},
0 7.27 (t,1 H), 9.00 (brs,1 H), 10.76 (brs,1 H).
t_RMS (Thermospray): 494 (M+NH4)'.
1v,. isi honyj}tetrahyrdroRyl~n-4~carboxamide
Obtained as a colourless solid (87%), m.p. 152-154°C, from the
title
compound of Preparation 79, using the procedure of Example 19. Found:
C,61.99; H, 6.47; N, 5.54. C~Hg2N2OgS requires C, 62.38; H, 6.44; N, 5.60%.
8(DMSO~): 1.33 (t,3H), 1.93 (m,2H), 2.24 (s,3H), 2.40 (d,2H), 2.52 (m,2H),
3.21 (dd,2H), 3.50 (m,2H), 3.88 (m,2H), 3.98-4.10 (m,4H}, 6.18 (brs,1H}; 6.80-
6.95 (m,3H), 7.18 (d,1H), 7.26-7.37 (m,3H), 9.22 (brs,1H), 11.05 (brs,1H).
LRMS (APCI): 501 (M+H}+.
~il~y~~L4 !L4-G~en~L~~-3-methy!lRh~111~R1 ~ in-1-
Palladium on barium sulfate (5%, 5 mg) was added to a stirred solution
of the tile compound of Example 28 (50 mg, 0.1 rnmol) in a mixture of 1,2-
CA 02312935 2000-06-02
wo ~n~s~ rcr~~s~~a
-38-
dimethoxyethane (1 ml) and methanol (3 ml), then the reaction mixture
hydrogenated at 345 kPa (50 psi) pressure for about 20 hours. A further
portion
of palladium on barium sulfate (5%, 5 mg) was added and hydrogenation
continued for an additional 20 hours. The catalyst was removed by filtration,
the solvent evaporated under reduced pressure and the residue crystallised
from ether hexane to afford the title compound (34 mg, 66%) as a colourless
solid, m.p. 165-167°C. Found: C,60.81; H, 6.76; N, 5.35. C26H~NZO6S;
0.50
H20 requires C, 61.03; H, 6.89; N, 5.47%. 8 {DMSO~): 1.35 {t,3H), 1.60
(m,2H), 1.78 (m,2H), 1.92 (m,2H), 2.20 (s,3H), 2.40 (d,2H), 2.62 (m,1H), 3.03
(dd,2H), 3.20 (dd,2H), 3.73 (m,2H), 3.86 (m,2H), 4.04 (q,2H), 6.80-6.92
{m,3H),
7.07-7.17 (m,3H), 7.30 (t,1 H), 9.18 (brs,1 H), 11.0 (brs,1 H).
LRMS (Thermospray): 520 (M+NH4)+.
EXAMPLE 30
1-yrlsulphonyrl)~-2-methyrl~nan,. amide
Obtained as a colourless solid (33%), m.p. 116-118°C, from the title
compound
of Preparation 81, using the procedure of Example 19. Found: C,62.52; H,
6.47; N, 6.00. C24H3pN2O5S requires C, 62.86; H, 6.59; N, 6.11 %. S(DMSO~):
1.34 (t,3H), 1.52 (s,6H), 2.23 (s,3H), 2.53 (m,2H), 3.50 (m,2H), 4.00-4.10
(m,4H), 6.18 (brs,1H), 6.80-6.95 (m,3H), 7.18 (d,1H), 7.28-7.38 (m,3H), 9.03
(brs,1 H), 10.8 (brs,1 H).
LRMS (Thermospray): 459 (M+H)+.
CA 02312935 2000-06-02
wo ~n~~ rc~rn~~ar~o
-39-
Obtained as a colourless solid (48%), m.p. 179-180°C (crystallised
from
diisopropyl ether), from the title compound of Preparation 82, using the
procedure of Example 17. Found: C,60.72.; H,6.49; N,6.36. C~H2gN2O5S
requires C,61.09; H,6.53; N,6.48%. 8(DMSOds): 1.31 (t,3H), 1.68 (m,2H), 1.86
(m,2H), 2.20 (s,3H), 2.65 (m,1H), 3.00 (m,2H), 3.72 (m,2H), 3.86 (s,2H), 4.03
(q.2H), 6.80-6.90 (m,3H), 7.10 (s,2H), 7.17 (s,1 H), 7.30 (t,1 H), 9.20 (brs,1
H),
10.8 (brs,1 H).
t_RMS (APCI): 433 (M+H)+.
~5 N-Hyrdroxy(4-~4-(3~etho~,~henyrly-3-methyrj~g~yr_Ijp,1 ' in-1-ylsulr~honvll-
2-
Obtained as a colourless solid (53%), m.p. 172-174°C (crystallised
from
diisopropyl ether), from the title compound of Preparation 84, using the
procedure of Example 19. Found: C,62.20; H,6.99; N,6.02. C24Hs2N20sS
2o requires C,62.58; H,7.00; N,6.08%. 8(DMSOds): 1.31 (t,3H), 1.50 (s,6H),
1.61
(m,2H), 1.79 (m,2H), 2.20 (s,3H), 2.65 (m,1H), 3.05 (m,2H), 3.75 (m,2H), 4.03
(q,2H), 6.80-6.90 (m,3H), 7.08-7.18 (m,3H), 7.30 (dd,1H), 8.98 (brs,1H), 10.75
(brs,1 H).
LRMS (APCI): 461 (M+H)''.
CA 02312935 2000-06-02
wo ~n~as~ rcr~~srot~o
-40-
Obtained as a colourless amorphous solid (50%) from the title compound
of Preparation 85, using the procedure of Example 17. 8(DMSOds): 2.33
(s,3H), 2.60 (m,2H), 3.46 (t,2H), 3.91 (s,2H), 3.97 (m,2H), 6.12 (brs,1H),
7.30-
7.40 (m,4H), 7.50 (d,1 H), 7.84 (dd,1 H), 8.63 (d,1 H), 9.20 (brs,1 H), 10.8
(brs,1 H).
~o LRMS (APCI): 388 (M+H)''.
Obtained as a colourless solid (57%), m.p. 132-9 36°C, from the
title
compound of Preparation 86, using the procedure of Example 31. 8(DMSOds):
2.23 (s,3H), 2.60 (m,2H), 3.46 (t,2H), 3.91 (s,2H), 3.97 (m,2H), 6.12
(brs,1H),
7.21 (d,1 H), 7.35 (d,1 H), 7.40 (s,1 H), 7.45 (dd,1 H), 7.78 (d,1 H); 8.55
(m,2H),
9.20 (brs,1 H), 10.8 (brs,1 H).
2o LRMS (APCI): 388 (iul+H)~.
Obtained as a colourless solid (20%), m.p. 165-167° C, from the
title
compound of Preparation 87, using the procedure of Example 31. 8(DMSOds):
2.26 (s,3H), 2.60 (m,2H), 3.47 (t,2H), 3.92 (s,2H), 3.97 (m,2H), 6.12
(brs,1H),
7.22 (d,1 H), 7.35-7.42 (m,4H), 8.62 (d,2H), 9.20 (brs,1 H), 10.8 (brs,1 H).
LRMS (APCI): 388 (M+H)+.
CA 02312935 2000-06-02
WO 99/29667 PCT/EP98/06640
-41-
Obtained as a colourless solid (30%), m.p. 184-187° C, from the
title
compound of Preparation 91, using the procedure of Example 19, except that
the residue was purified by flash chromatography using dichloromethane:
ethanol ( 98:2) as eluant. 8(DMSOds): 1.30 (t,3H), 1.48 (s,6H), 1.63 (m,2H),
1.79 (m,2H), 2.35 (s,3H), 2.67 (m,lH), 3.05 (t,2H), 3.75 (d,2H), 4.30 (q,2H),
6.72 (d,1 H}, 7.05 (d,1 H), 7.15 (m,2H), 7.34 (a,1 H), 7.73 (t,1 H), 9.00
(brs,1 H),
10.75 (brs,1 H).
HRMS (positive ion electrospray): 462.206 (M+H)'.
ydroxv-4-f4-~4-oThe, nw henyl)~-1.2.3.x-tetrahyr~py ' in-1-
yl,~~ honyl~tetrahyrdropyrran-4-carboxamide
Obtained as a colourless solid (68%), m.p. 191-193°C, from the
title
compound of Preparation 93, using the procedure of Example 19, but with
crystallisation of the residue from methanol. 8(DMSOds): 1.93 (m,2H), 2.40
(d,2H), 2.55 (m,2H), 3.20 (t,2H), 3.48 (m,2H), 3.85 (m,2H), 4.00 (m,2H), 6.11
(brs,1H), 7.35 (t,1H), 7.44 (m,2H), 7.52 (d,2H), 7.65 (m,4H), 9.22 (brs,1H),
11.05 (brs,1H).
LRMS (APCI): 443 (M+H)+.
F_XAMPLE 38
1-~!lQhg~y,~tetra ~,rdroRvran-4-carboxamide
Obtained as a colourless solid (36%), m.p. 159-161°C, from the
title
compound of Preparation 95, using the procedure of Example 19, but with
3o crystallisation of the residue from dichloromethane-diisopropyl ether.
CA 02312935 2000-06-02
wo ~n~ss~ pc'r~~s~osc~o
-42-
8(DMSO,~): 1.35 (t,3H), 1.94 (m,2H), 2.22 (s,3H), 2.38 (d,2H), 2.50 (brs,2H),
3.20 (t,2H), 3.50 (brs,2H), 3.87 (dd,2H), 3.98 (brs,2H), 4.04 (q,2H), 6.15
(brs,1 H), 6.96 (d,2H), 7.13 (d,1 H), 7.22 (d,2H), 7.28 (d,1 H), 7.33 (s,1 H),
9.20
(brs,1 H), 11.05 (brs,1 H).
LRMS (Thermospray): 515 (M+NH4)''.
~. to rahy~o~yridin-1 a Isu ~ony~JEacetamide
Obtained as a colourless solid (63%), m.p. 174-176°C (crystallised
from
methanol-diisopropyl ether), from the title compound of Preparation 97, using
the procedure of Example 17. Found: C,60.35; H,5.75; N,6.70. C2~H24N2O5S
requires C,60.56; H,5.81; N,6.73%. 8(DMSO~): 2.22 (s,3H), 2.60 (m,2H), 3.47
~5 (t,2H), 3.93 (s,2H), 3.97 (s,2H), 4.53 (d,2H), 5.19 (t,1H exchangeable),
6.20
(brs,1 H), 7.15-7.42 (m,7H), 9.20 (brs,1 H), 10.80 (brs,1 H).
LRMS (APCI): 417 (M+H)+.
2o ~'I-H dr.~~~yJ~~~I~_mp yL(auinolin-3-yl~lphenyll 1.2.3.6-
Obtained as a colourless solid (51 %), m.p. 158-160°C, from the
title
compound of Preparation 100, using the procedure of Example 19. 8(DMSO~):
1.50 (s,6H), 2.32 (s,3H), 2.57 (m,2H), 3.53 (m,2H), 4.03 (m,2H), 6.23
(brs,1H),
25 7.34-7.48 (m,3H), 7.63 (t,1 H), 7.79 (t,1 H), 8.04 (t,2H), 8.37 (s,1 H),
8.91 (s,1 H),
9.04 (brs,1 H), 10.8 (brs,1 H).
LRMS (APCI): 466 (M+H)+.
CA 02312935 2000-06-02
wo ~n~ss~ Pcr~r9sio~ao
-43
F~,~pLE 41
N-Hyrdro~X; 2~(4-[3-methyl-4.-(3-methyrlthiophenyrlynhenyrl]-1.2.3:6
tetra yrdrooyrridin-1-yrlsulpho_nyrlJlacetamide
Obtained as a colourless solid (17%), from the title compound of
Preparation 102, using the procedure of Example 1. 8(DMSOd6): 2.23 (s,3H),
2.49 (s,3H), 2.60 (m,2H), 3.47 (t,2H), 3.91 (s,2H); 3.97 (s,2H), 6.22
(brs,1H),
7.08 (d,1 H), ?.17 (m,2H), 7.24 (d,1 H), 7.34 (m, 3H), 9.22 (brs,1 H), 10.80
(brs,1 H).
1o LRMS (APCI): 432 (M)+.
Obtained as a colourless solid (28%), m.p. 151-153°C (crystallised
from
ethyl acetate-diisopropyl ether), from the title compound of Preparation 104,
using the procedure of Example 17. Found: C,60.16; H,6.00; N,6.28.
C22H~N20~S; 0.10 H20 requires C,60.11; H,6.19; N,6.37%. S(DMSOds): 2.22
(s,3H), 2.60 (m,2H), 3.30 (s,3H), 3.47 (t,2H), 3.93 (s,2H), 3.97 (s,2H), 4.04
(s,2H), 6.20 (brs,1 H), 7.17 (d,1 H), 7.21-7.42 (m,6H), 9.20 {brs,1 H), 10.80
(brs,1 H).
LRMS (Thermospray): 432 (M+H)+.
N-EjyrdroxVr-2.~-f4-(3 j -meikh-oxyet_ hoxyj,Rh_e_nyrl)~-3-methyrlphenyrlj-
1.2.3.6-
tetrah~LoR~ ride in-1-ylsulphon,yrj}acetamide
Obtained as a colourless solid (64%), m.p. 158-160°C (crystallised
from
ethyl acetate), from the title compound of Preparation 107, using the
procedure
of Example 17. Found: C,59.78; H,6.10; N,6.01. C23H28NZO6S requires
CA 02312935 2000-06-02
WO 99/29667 PCT/EP98J06640
-44-
C,59.98; H,6.13; N,6.08%. 8(DMSO~): 2.24 (s,3H), 2.60 (m,2H), 3.30 (s,3H),
3.47 (t,2H), 3.66 (m,2H), 3.92 {s,2H), 3.97 (m,2H), 4.12 (t,2H), 6.20 (brs,1
H),
6.89 (m,3H), 7.18 (d,1 H), 7.34 (m,3H), 9.20 {brs,1 H), 10.80 (brs,1 H).
LRMS (Thermospray): 460 (M)+.
o Obtained as a colourless solid (56%), m.N. 158-162°C, from the title
compound of Preparation 112, using the procedure of Example 17. Found:
C,57.01; H,5.47; N,5.26. C22H2gN2O5S; 0.60 CH2CI2 requires C,56.61; H,5.30;
N,5.84%. 8(DMSO~): 2.23 (s,3H), 2.58 (m,2H), 3.20 (t,2H), 3.44 (t,2H}, 3.92
(s,2H), 3.96 (s,2H), 4.55 (t,2H), 6.20 (brs,1 H), 6.78 (d,1 H), 7.01 (d,1 H},
7.12
~ 5 (d,1 H), 7.17 (s,1 H), 7:27 (d,1 H), 7.33 (s,1 H), 9.20 (brs,1 H), 10.80
(brs,1 H).
LRMS (APCI): 428 (M+H)'.
2o tetrahyrdroayrridin-1-yrlsul;,nhon~~}acetamide
Obtained as a colourless solid (50%}, m.p. 168-170°C (crystallised
from
diisopropyl ether), from the title compound of Preparation 116, using the
procedure of Example 17, except that tetrahydrofuran was used as a co-solvent
for the reaction. Found: C,54.96; H,4.73; N,5.97. C2~H2~F~N2O4S; 0.25 HZO
25 requires C,54.96; H,4.72; N,6.10%. 8(DMSOds): 2.24 (s,3H), 2.60 {m,2H),
3.47
(t,2H), 3.93 (s,2H), 3.97 (m,2H), 6.22 (brs,1 H), 7.23 (d,1 H), 7.36 (d,1 H},
7.41
(s,1 H), 7.68 (m,4H), 9.20 {brs,1 H), 10.80 (brs,1 H).
LRMS (APCI): 455 (M+H)+.
CA 02312935 2000-06-02
wo ~n~ss~ Pc~r~~s~o~o
-45-
Obtained as a colourless solid (13%), m.p. 176-179°C, from the
title
compound of Preparation 119, using the procedure of Example 17, except that
dichloromethane was used as a co-solvent for the reaction. Found: C,57.92;
H,5.62; N,6.97. C~9H~Nz05S; 0.20 HZO requires C,57.91; H,5.73; N,7.11%.
8(DMSO~): 1.63 (m,2H), 1.84 (m,2H), 2.63 (m,1 H), 2.98 (t,2H), 3.69 (m,2H),
3.85 (s,2H), 6.96 (m,4H), 7.10 (t,1H), 7.27 (d,2H), 7.36 (t,2H), 9.20
(brs,1H),
10.80 (brs,1 H).
LRMS {Thermospray): 392 (M+H)''.
~ Ir sul~onyl~2-methyl~ronanamide
Obtained as a colourless solid (42%), m.p. 155-156°C, from the
title
compound of Preparation 122, using the procedure of Example 19, except that
the residue was purified by flash chromatography, using
dichloromethane:methanol (97:3) as eluant, prior to crystallisation from
2o dichloromethane-diisopropyl ether. 8(DMSO~): 1.50 (s,6H), 1.60 (m,2H), 1.80
(m,2H), 2.20 (s,3H), 2.65 (m,1H), 3.05 (t,2H), 3.30 (s,3H), 3.62 (t,2H), 3.74
(d,2H), 4.10 (t,2H), 6.80-6.92 (m,3H), 7.10-7.17 (m,3H), 7.30 (t,1H), 9.02
(brs,1 H), 10.7 (brs,1 H).
LRMS (Electrospray): 513 (M+Na)''.
CA 02312935 2000-06-02
wo ~n~s~ pc~r~t~ros~o
-46-
Obtained as a colourless solid (39%), m.p. 135-136°C, from the
title
compound of Preparation 124, using the procedure of Example 19, except that
the residue was purified by r'lash chromatography using dichloromethane:
methanol (97:3) as eluant, prior to crystallisation from diisopropyl ether.
8(DMSOds): 2.10 (m,2H), 2.23 (s,3H), 2.57 (m,2H),3.20 (s,4H), 3.36 (m,1 H),
3.48 (m,2H), 3.87 (dd,1 H), 3.98 (m,2H), 6.20 (brs,1 H), 7.16 (d,1 H), 7.32
(m,SH),
7.43 (m,2H), 9.22 (brs,1 H), 10.95 (brs,1 H).
LRMS (Thermospray): 444 (M)+.
~,yrdroxy-4-[4-.(3-methyl-4-2henyluhenyrl)-1.2.3.6-tetrahyrdro~yrridin-1-
~ Ir sulphony~ltetrahyrdropyrran-4-carboxamide
Obtained as a colourless solid (91%), m.p. 188-190°C, from the
title
compound of Preparation 126, using the procedure of Example 38. Found:
C,61.89; H,6.15; N,5.94. C24HZgN2O5S; 0.50 H20 requires C,61.91; H,6.28;
2o N,6.02%. 8(DMSOds): 1.95 (m,2H), 2.22 (s,3H), 2.39 (d,2H), 2.50 (m,2H),
3.20
(t,2H), 3.48 (brs,2H), 3.87 (dd,2H), 4.00 (brs,2H), 6.17 (brs,1 H), 7.16 (d,1
H),
7.31 (m,SH), 7.42 (m,2H).9.22 (brs,1H), 11.05 (brs,1H).
LRMS (APCI): 457 (M+H)+.
CA 02312935 2000-06-02
wo ~n~~ rc~r~r~s~os~o
-47-
Obtained as a colourless solid (63%), m.p. 199-202°C, from the
title
compound of Preparation 130, using the procedure of Example 19, but with
crystallisation of the residue from diisopropyl ether. Found: C,66.25; H,6.18;
N,5.18. C~H32N20~S; 0.30 H20 requires C,61.21; H,6.25; N,5.33%.
8(DMSO~): 1.55 (m,2H), 1.76 (m,2H), 2.20 (s,3H), 2.54 (m,1H), 2.89 (t,2H),
0 3.48 (m,2H), 3.77 (m,7H), 6.87 (m,3H), 7.07-7.3~ (m,BH), 9.10 (brs,1H),
11.05
(brs,1 H).
yrlss,~(phonyr~~yrclobutanecarboxamide
Obtained as a colourless solid (52%), m.p. 157-160°C, from the
title
compound of Preparation 132, using the procedure of Example 50. Found:
C,62.79; H,fi.60; N,5.93. C24H3oN2O5S requires C,62.86; H,6.59; N,6.11 %.
8(DMSOds): 1.60 (m,2H), 1.78 (m,3H), 1.93 (m,1H), 2.20 (s,3H), 2.57 (m,SH),
2.97 (t,2H), 3.72 (m,2H), 3.77 (s,3H), 6:87 (m,3H), 7.11 (s,2H), 7.15 (s,1H),
7.36 (t,1 H), 9.10 (brs,1 H), 10.92 (brs,1 H).
LRMS (Thermospray): 459 (M+H)'.
Obtained as a colourless solid (13%), from the title compound of
Preparation 134, using the procedure of Example 19, except that the residue
was purified by flash chromatography using dichloromethane:methanol:
CA 02312935 2000-06-02
WO 99/2966? PCf/EP98/06~640
-48-
concentrated aqueous ammonia solution {90:10:1 ) as eluant. 8(CDC13): 1.79
(m,2H), 1.90 (m,2H), 2.16 (m,2H), 2.27 (s,3H), 2.30 (s,3H), 2.33 (m,4H), 2.64
(m,1H), 2.91 (brd,2H), 3.10 (t,2H), 3.83 (s,3H), 3.91 (m,2H), 6.88 (m,3H),
7.08
(m,2H), 7.18 (d,1 H), 7.30 (t,1 H).
LRMS (Thermospray): 502 (M+H)'.
yrIsuITyrljpropanamide
Obtained as a colourless solid (31 %); m.p. 202-205°C, from the
title
compound of Preparation 136, using,the procedure of Example 19, except that
the residue was triturated with dichloromethane. Found: C,66.05; H,5.82;
N,6.15. C~H~N204S; 0.50 H2O requires C,66.22; H,5.77; N,5.94%.
8(DMSO~): 2.60 (m,2H), 3.15 (m,2H), 3.57 (m,2H), 4.03 (m,3H), 6.07 (brs,1H),
7.16-7.36 (m,6H), 7.45 (m,2H), 7.57 (m,2H), 7.65 (m,4H), 9.17 (brs,1 H), 10.70
(brs,1 H).
LRMS (APCI): 463 (M+H)+.
EXAMPLE 54
yrlsu hony~]indane-2-carboxamide
Obtained as a colourless solid (27%), m.p. 159-161 °C, from the
title
compound of Preparation 138, using the procedure of Example 19, except that
the residue was purified by flash chromatography using
dichloromethane:methano! (98:2) as eluant, followed by trituration with
diisopropyl ether. 8(DMSO~): 2.55 (m,2H), 3.41 (m,2H), 3.53 (d,2H), 3.77
(d,2H), 3.96 (m,2H), 6.18 (brs,1H), 7.16 (m,2H), 7.23 (m,2H), 7.34-7.53
(m,SH),
7.65 (m,4H), 9.15 (brs,1 H), 11.10 (brs,1 H).
3o LRMS (APCI): 463 (M+H)~.
CA 02312935 2000-06-02
wo ~n~s~ pcr~~sro~oo
-49-
Obtained as a colourless solid (60%), m.p. 181-183°C (crystallised
from
ether), from the title compound of Preparation 142, using the procedure of
Example 17, except that tetrahydrofuran was used as a co-solvent for the
reaction. Found: C,54.65; H,4.61; N,6.13. CZOH2oCIFN2O4S requires C,54.73;
H,4.59; N,6.38%. 8(DMSO~): 2.24 (s,3H), 2.60 (m,2H), 3.47 (t,2H), 3.90
o (s,2H), 3.97 (m,2H), 6.22 (brs,1H), 7.20 (d,1H), 7.30-7.40 (m,3H), 7.45
(t,1H),
7.56 (m,1 H), 9.20 (brs,1 H), 10.80 (brs,1 H).
LRMS (APCI): 439 {M+H)+.
Jy-Hydroxy-2-{4-~4-(1.3-benzodioxol-5-~~)~- -methyrlphenyl)-1.2.3.6-
Obtained as a colourless solid (69%), m.p. 165-167°C, from the
title
compound of Preparation 146, using the procedure of Example 45. Found:
C,58.60; H,5.10; N,6.01. CZ~H22N2O6S requires C,58.59; H,5.15; N,6.51%.
8(DMSO~): 2.22 (s,3H), 2.59 (m,2H), 3.46 (t,2H), 3.90 {s,2H), 3.96 (m,2H),
6.04 (s,2H), 6.20 (brs,1 H), 6.76 (d,1 H), 6.89 (s,1 H), 6.95 (d,1 H), 7.15
(d,1 H),
7.28 (d,1 H), 7.35 (s,1 H), 9.20 (brs,1 H), 10.80 (brs,1 H).
LRMS (APCI): 431 (M+H)+.
CA 02312935 2000-06-02
wo ~n~~ rc~r~r9s~o
-50
~AMPLE 57
Obtained as a colourless solid (32%), m.p. 160-163°C, from the
title
compound of Preparation 150, using the procedure of Example 17, except that
tetrahydrofuran was used as a co-solvent for the reaction. 8(DMSO~): 2.12
(s,3H), 2.60 (m,2H), 3.48 (t,2H), 3.84 (s,2H), 3.98 (m,2H), 6.21 (brs,1H),
7.17
(d,1 H), 7.23-7.35 (m,4H), 7.40 (s,1 H), 7.42 (m,1 H).
1o LRMS (APCI): 405 (M+H)+.
~- I[yrdroxyr-2-(4-(~-f,3.4-dimethoxy~he_~yrl)-3-methyrl~heny (]-1.2.3.6
tetrahvdro~ ridr in-1-yr~~ulphonv acetamide
Obtained as a colourless solid (61%), m.p. 172-174°C, from the
title
compound of Preparation 151, using the procedure of Example 57. Found:
C,59.36; H,6.08; N,5.87. C~HZgN206S requires C,59.18; H,5.87; N,6.27%.
8(DMSOds): 2.25 (s,3H), 2.60 (m,2H), 3.48 (t,2H), 3.75 (s,3H), 3.78 (s,3H),
3.90
(s,2H), 3.96 (m,2H), 6.19 (brs,1 H), 6:82 (d,1 H), 6.88 (s,1 H), 7.00 (d,1 H),
7.19
(d,1 H), 7.29 (d,1 H), 7.36 (s,1 H), 9.20 (brs,1 H), 10.80 (brs,1 H).
LRMS (APCI): 447 (M+H)+.
N-H~ droocrr_~~~-(~,(indan-~yy-3-methylphenyrl)-1,2.3.6-tetrahyrdrooyrridin-1-
yrlsul~ahonyrl~Eacetamide
Obtained as a colourless solid (58%), m.p. 149-152°C, from the
title
compound of Preparation 152, using the procedure of Example 17. 8(DMSO~):
CA 02312935 2000-06-02
wo ~n~s~ rcr~r9aio~o
-51-
2.03 (m,2H), 2.22 (s,3H), 2.59 (m,2H), 2.90 (m,4H), 3.47 (m,2H), 3.90 (s,2H),
3.96 (m,2H), 6.19 (brs,1H), 7.05 (d,1H), 7.15 (m,2H), ?.23-7.30 (m,2H), 7.36
(s,1 H), 9.20 (brs,1 H), 10.80 (brs,1 H).
LRMS (APCI): 427 (M+H)~.
1o Obtained as a colourless solid (13%), m.p. 154°C (crystallised from
dichloromethane-diisopropyl ether), from the title compound of Preparation
153,
using the procedure of Example 17. b{DMSOds): 2.23 (s,3H), 2.59 (m,2H),
3.47 {t,2H), 3.90 (s,2H), 3.96 (m,2H), 6.22 (brs,1 H), 7.20 (d,1 H), 7.30-7.40
(m,SH), 7.58 (t,1 H), 9.20 (brs,1 H), 10.80 (brs,1 H).
~5 LRMS (Thermospray): 488 (M+NH4)+.
2o Obtained as a colourless solid (43%), m.p. 143-145°C, from the title
compound of Preparation 157, using the procedure of Example 17. Found:
C,54.63; H,4.35; N,5.90. C2oH~9F3N20~S requires C,54.54; H,4.35; N,6:36%.
8(DMSO~): 2.66 (m,2H), 3.50 (m,2H), 3.93 (s,2H), 4.00 (m,2H), 6.38 (brs,1H),
7.30 (m,2H), 7.35-7.47 (m,4H), 7.79 (d,1 H), 7.82 (s,1 H), 9.20 (brs,1 H),
10.80
25 (brs,1 H).
LRMS (Thermospray): 458 (M+NH4)+.
CA 02312935 2000-06-02
wo ~n96s~ pc~r~msro~o
-52
F~AMPLI~62
N-Hydroxv-2-{4-(4-(2.2-dimethyl-1.3-benzodioxol-5-yrl)-3-meth~~~p henvll
Rj,peridin-1-yrlsu~hopyrly-~ hvj~ron~onamide
Obtained as a colourless solid (52%), m.p. 184-186°C, from the
title
compound of Preparation 161, using the procedure of Example 19. 8(DMSOds):
1.47 (s,6H), 1.60 (m,2H), 1.65 (s,6H), 1.78 (m,2H), 2.20 (s,3H), 2.65 (m,1H),
3.04 (m,2H), 3.73 (m,2H), 6.68 (d,1 H), 6.77 (s,1 H), 6.83 (d,1 H), 7.07
(s,2H),
7.12 (s,1 H), 9.00 (brs,1 H), 10.75 (brs,1 H).
1 o LRMS (APCI): 489 (M+H)+.
~5 Obtained as a colourless solid (22%), m.p. 213-215°C, from the title
compound of Preparation 165, using the procedure of Example 19. 8(DMSOds):
1.48 (s,6H), 1.62 (m,2H), 1.80 (m,2H), 2.20 (s;3H), 2.52 (s,3H), 2.67 (m,1H),
3.08 (t,2H), 3.73 (s,3H), 3.75 (m,2H), 7.08-7.15 (m,4H), 7.39 (s,1H), 7.47
{d,1H), 9.00 (brs,1H), 11.75 (brs,1H).
2o LRMS (Thermospray): 485 (M+H)'.
25 Palladium on barium sulfate (5%, 10 mg) was added to a stirred solution
of the title compound of Preparation 168 {70 mg, 0.13 mmol) in methanol (2
ml),
then the reaction mixture hydrogenated at 345 kPa (50 psi) pressure for about
20 hours. A further portion of palladium on barium sulfate (5%; 10 mg) was
added and hydrogenation continued for an additional 4 days. The catalyst was
CA 02312935 2000-06-02
WO 99/29667 PCT/EP98/06640
-53-
removed by filtration, the solvent evaporated under reduced pressure and the
residue flash chromatographed, using methanol:dichloromethane (5:95) as
eluant, to give the title compound (13 mg, 23%) as a colourless amorphous
solid. 8 {CDCI3): 1.63 (s,6H), 1.63-1.95 (m,4H), 2.26 (s,3H), 2.67 {m,1H),
3.10
{m,2H), 3.96 (m,2H), 7.04-7.20 (m,3H), 7.50-7.70 (m,4H)
IR (KBr) 2240 crri' for cyano.
~y~~yr-2-~4-(4-(5-etho rpyrridin-3-yrl)-3-meth~~nhenyrl],pjperidin-1-
Obtained as a colourless foam (62%), from the title compound of
Preparation 172, using the procedure of Example 19, except that the residue
was purified by flash chromatography, using dichloromethane:ethanol ( 95:5) as
~5 eluant. 8{DMSO~): 1.32 (t,3H), 1.48 {s,6H), 1.61 {m,2H), 1.79 (m,2H), 2.21
(s,3H), 2.67 (m,1H), 3.05 (t,2H), 3.75 (d,2H), 4.13 (q,2H), 7.15-7.20 (m,3H),
7.30 (s,1 H), 8.10 (s,1 H), 8.23 (s,1 H), 8.98 {brs,1 H), 10.75 (brs,1 H).
LRMS (Thermospray): 462 (M+H)+.
2o EXAMPLE 66
O-(7-Azabenzotriazol-1-yl)-1,1,3, 3-tetramethyluronium
hexafluorophosphate (510 mg, 1.15 mmol) was added to a stirred solution of
25 the title compound of Preparation 178 (530 mg, 0.76 mmol) and N-
ethyldiisopropylamine (0.20 ml, 0.9 mmol) in anhydrous dimethylformamide (4
ml), under nitrogen, at room temperature. After 15 minutes, hydroxylamine
hydrochloride {188 mg, 2.2 mmol) and N-ethyldiisopropylamine (0.6 ml, 2.7
mmol) were added and the reaction mixture stirred for about 16 hours, then
CA 02312935 2000-06-02
WO 99129667 PCT/EP98/06640
partitioned between ethyl acetate and aqueous phosphate buffer (pH 7). The
organic phase was separated, washed with water, dried (MgS04) and
evaporated under reduced pressure. The residue was dissolved in anhydrous
tetrahydrofuran (15 ml), tetra-n-butylammonium fluoride (1.4 m! of a 1.0M
solution in tetrahydrofuran; 1.4 mmol) added and the resulting solution
stirred at
room temperature for 1.5 hours, then partitioned between ethyl acetate and
aqueous phosphate buffer (pH 7). The organic phase was separated, washed
with water, dried (MgS04) and evaporated under reduced pressure.
o Purification of the residue by flash chromatography, using
dichloromethane:methanol: concentrated aqueous ammonia solution (90:10:1 )
as eluant, followed by crystallisation from methanol-water, provided the
product
as a colourless solid (155mg, 43%), m.p. 147-150°C. Found: C,60.26;
H,6.81;
N,5.76. C24H32N20sS requires C,60.48; H,6.77; N,5.88%. 8(DMSOds): 1.49
~s {s,6H), 1.63 (m,2H), 1.80 (m,2H), 2.21 (s,3H), 2.67 (m,1H), 3.08 (t,2H),
3.74
(m,4H), 4.02 {t,2H), 4.83 (t,1H), 6.82-6.92 (m,3H), 7.10 (s,2H), 7.16 (s,1H),
7.30
(t,1 H), 9.00 {brs,1 H), 10.75 (brs,1 H).
CA 02312935 2000-06-02
wo ~nx~~ Pcr~r9s~os~o
-55-
A 2.5M solution of n-butyllithium in hexane (8 ml, 20 mmol) was added
over about 10 minutes to a stirred mixture of 4-bromobiphenyl (4.66 g, 20
mmol), anhydrous ether (100 ml) and anhydrous tetrahydrofuran (10 ml), under
nitrogen, at about -75°C. After a further 1 hour, a solution of t-butyl
4-
oxopiperidin-1-carboxylate (3.98 g, 20 mmol) in anhydrous tetrahydrofuran (10
ml) was added at such a rate that the reaction temperature was maintained
o below -60°C.
The reaction mixture was stirred at about -75°C for 3 hours and
quenched with aqueous ammonium chloride solution, then the organic phase
was separated, washed with water, dried (MgS04) and evaporated under
reduced pressure. Crystallisation of the residue from diisopropyl ether gave
the
~5 title compound (4.29 g) as a colourless solid, m.p. 144-146°C.
S(CDC13): 1.50
(s,9H), 1.78 (d,2H), 2.06 (m,2H), 3.28 (dd,2H), 4.06 (m,2H), 7.35 (t,1H), 7.43
(m,2H), 7.50-7.65 (m,6H).
t_RMS (APCI): 354 {M+H)+.
20 PREPARATION 2
Trifluoroacetic acid (20 ml) was added to a stirred solution of the title
compound of Preparation 1 (4.2 g, 11.9 mmol) in dichloromethane (20 ml) at
room temperature. After a further 3 hours, the reaction mixture was evaporated
25 under reduced pressure and the residue basified with 1 M aqueous sodium
hydroxide solution. The resulting mixture was extracted with dichloromethane,
then the combined extracts washed with water, dried (MgS04) and evaporated
under reduced pressure to yield the title compound (2.79 g) as a colourless
CA 02312935 2000-06-02
WO 99/29667 PCT/EP98/06640
-56-
solid. S(CDCI3): 1.53 (s,1H), 2.50 (m,2H), 3.14 (t,2H), 3.58 (m,2H), 6.20
(brs,1 H), 7.34 (t,1 H), 7.45 {m,4H), 7.60 {m,4H).
LRMS (Thermospray): 236 (M+H)+.
1-Benzyl-4-hyrd_ roxrr-4-(4-~yrlphenyrl)~,peridine
A 1.6M solution of n-butyllithium in hexane {39 ml, 63 mmol) was added
to a stirred solution of 4-bromobiphenyl (11.7 g, 50 mmol) in anhydrous
o tetrahydrofuran (50 ml), under nitrogen, at about -50°C, whilst
ensuring that the
reaction temperature was kept below -40°C. After a further 1 hour, a
solution of
1-benzyl-4-oxopiperidine (10.4 g, 55 mmol) in anhydrous tetrahydrofuran (30
ml) was added at such a rate that the reaction temperature was maintained
below -40°C. The cooling bath was then removed and, after a further 1
hour,
~5 the reaction mixture was partitioned between dichloromethane (400 ml) and
brine (200 ml). The organic phase was separated, washed with water, dried
(MgS04) and evaporated under reduced pressure, then the residue crystallised
from ethyl acetate to provide the title compound (13.9 g) as a colourless
solid.
8(DMSO,~): 1.80 (d,2H), 2.52 {m,2H), 3.24 (m,4H), 4.33 (d,2H), 7.28-7.75
20 (m,14H), 11.30 (brs, 1 H).
1-Ben,Zyl-4-(4-phenyrlphe,_n_~,yrl)~-1.2.3.6-tetrah~Qy 'mdine
A solution of the title compound of Preparation 3 (13.8 g, 40.2 mmol) and
25 p-toluenesulphonic acid (15.3 g, 80.4 mmol) in toluene (100 ml) was heated
under ceflux in a Dean-Stark apparatus until water removal was complete {~. 2
hours), then allowed to cool and diluted with water {200 ml). The resulting
mixture was basified with concentrated aqueous ammonia solution and
CA 02312935 2000-06-02
wo ~n~w~ pcr~r9srosseo
-57-
extracted with dichloromethane (4 x 200 ml), then the combined extracts dried
(MgS04) and evaporated under reduced pressure to furnish the title compound
(10.6 g) as an off white solid. 8(CDCI3): 2.57 (m,2H), 2.70 (m,2H), 3.18
(m,2H), 3.62 (s,2H), 6.10 (brs, 1 H), 7.20-7.60 (m,14H).
A stirred mixture of the title compound of Preparation 4 (5.07 g, 15.6
o mmol), ammonium formaie (4 g, 62 mmol), palladium hydroxide on carbon (500
mg) and methanol (50 ml) was heated under reflux for 4.5 hours, allowed to
cool and filtered. The filtrate was evaporated under reduced pressure and the
residue partitioned between 2M aqueous sodium hydroxide solution and
dichloromethane. The organic phase was separated and combined with
dichloromethane extracts .(3 x 100 ml) of the aqueous phase, then the
combined dichloromethane solutions dried (MgS04) and evaporated under
pressure to afford the title compound (3.5 g) as an off white solid, m.p. 104-
107°C. 8(CDCI3): 1.50 (brs,1H), 1.63 (m,2H), 1.83 (m,2H), 2.62 (m,1H),
2.75
(m,2H), 3.19 (d,2H), 7.25 (m,3H), 7.40 (m,2H), 7.53 (m,4H).
~-Bn yrl 4-hyrdroxy-4-(,yi-methyrl-4-~enyrlphenyrl)R~eridine-1-carboxrrlate
Obtained as a colourless solid (60%), m.p. 142-144°C, from 4-bromo-
2-
methylbipheny! (J.Amer.Chem.Soc., 1926, ~$, 1372) and t-butyl 4-oxopiperidin-
1-carboxylate, using the procedure of Preparation 1. 8(CDCI3): 1.48 (s,9H),
1.78 (m,2H), 2.04 (m,2H), 2.30 (s,3H), 3.28 (m,2H), 4.05 (m,2H), 7.20-7.42
(m,8H).
t_RMS (Thermospray): 468 (M+H)+.
CA 02312935 2000-06-02
wo ~n9~~ Pc~r~~s~os~o
-58
pREPARATION 7
~(~~-Meth henyrlphenyrl)-1.2.3.6-tetrah~ d~ro,;pyrridine
Obtained as a colourless solid (90%) from the title compound of
Preparation 6, using the procedure of Preparation 2. 8(CDCI~): 1.85 (s,1 H),
2.28 (s,3H), 2.50 (m,2H), 3.14 (t,2H), 3.57 (m,2H), 6.18 (brs, 1 H), 7.20-7.42
(m,BH).
LRMS (APCI): 250 (M+H)''.
PREPARATION 8
A solution of methyl chlorosulphonylacetate (0.35 g, 2 mmol) in
dichloromethane (2 ml) was added dropwise to a stirred solution of the title
compound of Preparation 2 (470 mg, 2 mmol) and' 1,8-diazabicyclo
~5 [5.4.Ojundec-7-ene (0.3 ml, 2 mmol) in dichloromethane (8 ml) at about
0°C, the
cooling bath removed and the reaction mixture stirred at room temperature for
4
hours, then diluted with dichloromethane. The resulting mixture was washed
with 0.1 M hydrochloric acid, dried (MgS04) and evaporated under reduced
pressure. The residue was purified by flash chromatography, using
2o dichloromethane as eluant, followed by crystallisation from diisopropyl
ether, to
give the title compound (250 mg) as a colourless solid, m.p. 182-183°C.
Found: C,64.32; H,5.59; N,3.77. C2pH2~NO4S requires C,64.66; H,5.70;
N,3.77%. 8(CDCI3): 2.66 (m,2H), 3.62 (t,2H), 3.78 (s,3H), 3.99 (s,2H), 4.08
(m,2H), 6.08 (brs, 1 H), 7.32 (m,1 H), 7.38-7.44 (m,4H), 7.53-7.60 (m,4H).
25 LRMS (APCI): 372 (M+H)+.
CA 02312935 2000-06-02
wo ~n~s~ pcr~~os~o
-59-
Obtained as a colourless solid (30%), m.p. 104-105°C, from the
title
compound of Preparation 7 and methyl chlorosulphonylacetate, using the
procedure of Preparation 8. Found: C,fi5.18; H,6.03; N,3.59. C2~H23NO4S
requires C,65.43; H,6.01; N,3.63%. 8(CDCI3): 2.28 (s,3H), 2.68 (m,2H), 3.64
(t,2H), 3.81 (s,3H), 4.02 (s,2H), 4.10 (m,2H), 6.08 (brs,1H), 7.20-7.47
(m,BH).
LRMS (Thermospray): 386 (M+H)+.
Obtained as a colourless solid (27%), m.p. 169-170°C, from the
title
~ 5 compound of Preparation 5 and methyl chlorosulphonylacetate, using the
procedure of Preparation 8. Found: C,63.99; H,6.18; N,3.69. C2oH23NO4S
requires C,64.32; H, 6.21; N,3.75. 8(CDCI3): 1.83 (m,2H), 1.95 (m,2H), 2.68
(m,1 H), 3.00 (t,2H), 3.80 (s,3H), 3.95 (s,2H), 3.97 (m,2H), 7.20-7.35 (m,3H),
7.40 (t,2H), 7.50-7.60 (m,4H).
Obtained as a colourless solid (20%), m.p. 93-94°C, from 4-phenyl-
1,2,3,6-tetrahydropyridine and methyl chlorosulphonylacetate, using the
procedure of Preparation 8. Found: C,56.86; H,5.79; N,4.76, C~4H~~N04S
requires C,56.93; H,5.80; N,4.74%. 8(CDCI3): 2.62 (m,2H), 3.60 (t,2H), 3.78
(s,3H), 3.99 (s,2H), 4.05 (m,2H), 6.00 (brs,lH), 7.22-7.35 (m,SH).
CA 02312935 2000-06-02
wo ~n~s~ pcr~r9sios~o
-60-
Obtained as a colourless solid (35%), m.p. 98-100°C, from 4-
phenylpiperidine and methyl chlorosulphonyfacetate, using the procedure of
Preparation 8. Found: C,56.43; H,6.41; N,4.64. C~4H~9N04S requires C,56.55;
H,6.44; N,4.71 %. 8(CDCI3): 1.80 (m,2H), 1.90 (m,2H), 2.60 (m,1 H), 2.97
(m,2H), 3.80 (s,3H), 3.92 (s,2H), 3.93 (m,2H), 7.15-7.33 (m,SH).
PREPARATION 13
Obtained as an amorphous solid (24%) from 4-benzyipiperidine and
methyl chlorosulphonylacetate, using the procedure of Preparation 8, but with
an elution gradient of dichloromethane:methanol (100:0 to 95:5) for the
chromatographic purification step. 8(CDCI3): 1.30 (m,2H), 1.62 (m,1H), 1.70
(m,2H), 2.54 (d,2H), 2.78 (t,2H), 3.73 (s,3H), 3.76 (m,2H), 3.88 (s,2H), 7.08
(d,2H), 7.17 (t,lH); 7.24 (m,2H).
LRMS (APCI): 312 (M+H)+.
PREPA BTjON 14
60% Sodium hydride in a mineral oil dispersion (21 mg, 0.53 mmol) was
added to a stirred solution of the title compound of Preparation 8 (180 mg,
0.48
mmol) in a mixture of anhydrous tetrahydrofu~an (1 ml) and anhydrous
dimethylformamide (1 ml), under nitrogen, at room temperature. After 30
minutes, allyl bromide (0.05 ml, 0.53 mmol) was added and stirring continued
CA 02312935 2000-06-02
wo ~n~s~ pcr~r9siossoo
-61-
for a further 2 hours, then the resulting mixture was partitioned between
ethyl
acetate and aqueous phosphate buffer (pH 7). The organic phase was
separated, washed with water, dried (MgS04) and evaporated under reduced
pressure, then the residue triturated with diisopropyl ether to yield the
title
compound (170 mg) as a colourless solid. b (CDCI3): 2.60-2.85 (rn,4H), 3.55-
3.77 (m,2H), 3.79 (s,3H), 4.03 (dd,1 H), 4.12 (m,2H), 5.10-5.22 (m,2H), 5.74
(m,1 H), 6.08 (brs, 1 H), 7.36 (m,1 H), 7.43 (m,4H), 7.60 (m,4H).
LRMS (APCI): 411 (M+H)+.
1 M Aqueous sodium hydroxide solution (1.2 ml, 1.2 mmol) was added to
~5 a stirred solution of the title compound of Preparation 14 (160 mg, 0.39
mmoi)
in a mixture of tetrahydrofuran (5 ml) and methanol (10 ml). The resuming
solution was heated at 50°C for 3 hours, then evaporated under reduced
pressure and the residue dissolved in water. This aqueous solution was
acid~ed with concentrated hydrochloric acid and the resulting emulsion
2o extracted with ethyl acetate. The combined extracts were dried (MgS04) and
evaporated under reduced pressure, then the residue purified by flash
chromatography, using an elution gradient of ethyl acetate:methanol:glacial
acetic acid (100:0:0 to 97:3:0 to 96:3:1), followed by trituration with
hexane, to
provide the title compound (90 mg) as a colourless, amorphous solid. 8(CDCI3):
25 2.60-2.87 (m,4H), 3.60-3.72 (m,2H), 4.05 (dd,1 H), 4.12 (s,2H), 5.10-5.23
(m,2H), 5.79 (m,1H), 6.06 (brs,1H), 7.30-7.43 (m,SH), 7.50-7.60 (m,4H):
LRMS (Thermospray): 415 (M+NH~)+.
CA 02312935 2000-06-02
WO 99/Z9667 PCT/EP98~06640
-62-
Obtained as a solid (67%) from the title compound of Preparation 9 and
ally) bromide, using the procedure of Preparation 14, but with flash
chromatography, employing an elution gradient of hexane:ethyl acetate (100:0
to 80:2)), as the pur~cation step. 8(CDCI3): 2.30 (s,3H), 2.68 (m,2H), 2.85
(m,2H), 3.56 {m,1 H), 3.70 (m,1 H), 3.80 (s,3H), 4.03 (dd,1 H), 4.10 (m,2H),
5.10-
5.22 (m,2H), 5.73 (m,1 H), 6.06 (brs,1 H), 7.20-7.45 (m,BH~.
LRMS (Thermospray): 426 (M+H)''.
yjsulnhonyrl~~e~-~,~ic acid
Obtained as an amorphous solid {46%) from the title compound of
Preparation 16, using the procedure of Preparation 15. 8(CDCI3): 2.30 (s,3H),
2.66 {m,2H), 2.87 (m,2H), 3.60 (m,1 H), 3.70 (m,1 H), 4.03 (dd,1 H), 4.12
(m,2H),
5.13-5.25 (m,2H), 5.78 (m,1 H), 6.04 {brs,1 H), 7.20-7.43 (m,BH).
2o LRMS (Thermospray): 368 (M+H-C02)+.
Obtained as a colourless solid (65%), m.p. 146-148°C, from the
title
compound of Preparation 8 and 1-bromo-3-phenylpropane, using the procedure
of Preparation 14. 8(CDCi3): 1.70 {m,2H), 2.15 (m,2H), 2.64 {m,4H), 3.52
(m,1 H), 3.63 (m,1 H), 3.79 (s,3H), 3.98 (dd,1 H), 4.06 {m,2H), 6.05 (m,1 H),
7.10-
7.50 (m,10H), 7.60 (m,4H).
3o LRMS (Thermospray): 490 (M+H)'.
CA 02312935 2000-06-02
WO 99/29667 PGT/EP98/46640
-63
PREPARATION 19
60% Sodium hydride in a mineral oii dispersion (48 mg, 1.2 mmol) was
added to a stirred solution of the title compound of Preparation 8 (150 mg,
0.4
mmol) in a mixture of anhydrous tetrahydrofuran (3 ml) and anhydrous
dimethylformamide (1 ml), under nitrogen, at room temperature. After 30
minutes, methyl p-toluenesulphonate (220 mg, 1.2 mmol) was added and
o stirring continued for a further 20 hours, then the resulting mixture was
partitioned between ethyl acetate and water. The aqueous phase was acidified
with 2M hydrochloric acid and extracted with dichloromethane (3 x 50 ml). The
combined extracts were dried {MgS04) and evaporated under reduced
pressure, then the residue purified by flash chromatography, using
~5 dichloromethane:methanol:glacial acetic acid {89:10:1) as eluant, to
furnish the
title compound (60 mg) as a pale yellow, amorphous solid. b{CDCI3): 1.70
(s,6H), 2.63 (m,2H), 3.67 (m,2H), 4.17 (m,2H), 6.08 (brs,1H), 7.25-7.70
(m,9H).
60% Sodium hydride in a mineral oil dispersion (43 mg, 1.07 mmol) was
added to a stirred solution of the title compound of Preparation 9 (380 mg,
0.99
mmol) in anhydrous dimethylformamide (5 ml), under nitrogen, at room
temperature. After 30 minutes, 1,4-diiodobutane (0.14 m1, 1.06 mmol) was
added and stirring continued for 18 hours, then more 60% sodium hydride
dispersion (43 mg, 1.07 mmol) was added and stirring continued for a further 4
hours. The resulting mixture was partitioned between ethyl acetate and water,
then the organic phase separated, washed with water, dried (MgS04) and
so evaporated under reduced pressure. The residue was purified by flash
chromatography, using hexane:ethyl acetate (9:1 ) as eluant; to afford the
title
CA 02312935 2000-06-02
wo ~n~ss~ pcr~wmo
compound (340 mg) as an amorphous solid. S(CDCI3): 1.64 (m,2H), 1.88
(m,2H), 2.28 (s,3H), 2.37 (m,2H), 2.52 (m,2H), 2.63 (m,2H), 3.60 (t,2H), 3.80
(s,3H), 4.10 (m,2H), 6.03 (brs,1H), 7.20-7.43 (m,BH).
LRMS (Thermospray): 440 (M+H)+.
~o Obtained as a solid (54%) from the title compound of Preparation 20,
using the procedure of Preparation 15, but with an elution gradient of
hexane:ethyl acetate (75:25 to 0:100) for the chromatographic purification
step.
8(CDCI3): 1.65 (m,2H), 1.88 (m,2H), 2.27 (s,3H), 2.39 (m,2H), 2.50 (m,2H),
2.63 (m,2H), 3.64 (t,2H), 4.12 (m,2H), 6.03 (brs,1H), 7.20-7.43 (m,BH).
LRMS (Thermospray): 426 (M+H)+.
This was conducted as for Preparation 19 and on the same molar scale,
using the title compound of Preparation 8 and ethyl iodide. However, in this
case, no concomitant ester hydrolysis was apparent and therefore the required
product was isolated from the org ai nic phase during work-up and purified by
flash chromatography, using dichloromethane as eluant, to give the title
compound (125 mg) as a white amorphous solid. b(CDCl3): 1.04 (t,6H), 2.08-
2.26 (m,4H), 2.66 (m,2H), 3.60 (m,2H), 3.80 (s,3H), 4.10 (m,2H), 6.08
(brs,1H),
7.35 (m,1H), 7.43 (m,4H), 7.60 (m,4H).
CA 02312935 2000-06-02
wo ~n~~ Pcr~r9sross4o
-65
PREPARATION 23
1 M Aqueous sodium hydroxide solution (1.5 ml, 1.5 mmol) was added to
a stirred solution of the title compound of Preparation 22 (120 mg, 0.28 mmol}
in a mixture of tetrahydrofuran (5 ml) and methanol (2 ml). The resulting
mixture was heated under reflux for 70 hours, allowed to coo! to room
temperature, acidified with 2M hydrochloric acid and extracted with
1o dichloromethane (3 x 20 ml). The combined extracts were dried (MgS04) and
evaporated under reduced pressure to yield the title compound (95 mg) as a
pale yellow, amorphous solid. 8(CDCI3): 1.10 (t,6H), 2.10-2.28 (m,4H), 2.64
(m,2H), 3.65 (m,2H), 4.15 (m,2H), 6.08 (brs,1H), 7.35 (m,1H), 7.43 (m,4H),
7.58
(m,4H}.
This was conducted essentially as for Preparation 14, using the title
2o compound of Preparation 8 and n-butyl iodide, but employing 1.0 mol.equiv.
of
sodium hydride, 1-methylpyrrolidin-2-one as solvent and an alkylation reaction
time of 70 hours, to provide the title compound (78%) as a colourless solid,
m.p.
152-154°C. Found: C, 67.09; H, 6.76; N, 3.22. C24H~N04S requires C,
fi7.42;
H, 6.84; N, 3.28%. 8(CDCI3): 0.90 (t,3H), 1.30 (m,4H), 2.05 (m,lH), 2.15
(m,1 H), 2.60-2.75 (m,2H), 3.55 (m,1 H), 3.70 (m,1 H), 3.80 (s,3H), 3.95 (dd,1
H),
4.10 (m,2H), 6.08 (brs, 1 H), 7.35 (m,1 H), 7.43 (m,4H}, 7.58 (m,4H).
LRMS (Thermospray): 428 (M+H)+.
CA 02312935 2000-06-02
wo ~n~ss~ pcr~o
-66
PREPARATION 25
1 M Aqueous sodium hydroxide solution (1.5 ml, 1.5 mmol) was added to
a stirred solution of the title compound of Preparation 24 (220 mg, 0.51 mmol)
in a mixture of 1,4-dioxan (3 ml) and methanol (10 ml). The resulting mixture
was heated under reflux for 45 minutes, diluted with water and acidified with
concentrated hydrochloric acid, then the precipitate collected and dried under
o vacuum to furnish the title compound (200 mg) as a colourless, crystalline
solid,
m.p. 180-182°C. Found: C, 65.64; H, 6.42; N, 3.30. C23H2~NO4S; 0.50 H20
requires C, 65.38; H, 6.68; N, 3.32%. 8(DMSOds): 0.83 (t,3H), 1.27 (m,4H),
1.81 (m,1 H), 1.93 (m,1 H), 2.58 (m,2H), 3.50 (m,2H), 4.00 (m,2H), 4.05 (dd,1
H),
6.22 (brs, 1 H), 7.33 (m,1 H), 7.43 (m,2H), 7.52 (m,2H), 7.64 (m,4H), 13.30
(brs,1 H).
LRMS (Thermospray): 387 (M+NH4-C02)''.
Met rl4-methyrl-2y~?,,S)-j4-(4-phenyrlnhenyrl)-1,2,3.6-tetrahvdroRyridin-1-
2o yr,~,ul hp onvlicent-4-enoate
Obtained as a solid (79%), m.p. 149-151 °C after crystallisation
from
diisopropyl ether, from the title compound of Preparation 8 and 3-bromo-2-
methylprop-1-ene, using the procedure of Preparation 14. 8(CDCl3): 1.77
(s,3H), 2.60-2.75 (m,3H), 2.80 (dd,1 H), 3.57 (m,1 H), 3.70 (m,1 H), 3.78
(s,3H),
4.10 (m,2H), 4.18 (dd,1 H), 4.75 (s,1 H), 4.82 (s,1 H), 6.10 (brs,1 H), 7.35
(m,1 H),
7.44 (m,4H), 7.60 (m,4H).
LRMS (Thermospray): 426 (M+H)''.
CA 02312935 2000-06-02
wo ~n~~
-67
PREPARATION 27
Obtained as a colourless solid (94%), m.p. 153-155°C, from the
title
compound of Preparation, 26, using the procedure of Preparation 15. 8(CDCI3):
1.79 (s,3H}, 2.65 (m,2H}, 2.75 (dd,1 H), 2.90 (dd,1 H}, 3.60 (m,1 H), 3.70
(m,1 H),
4.13 (m,2H), 4.20 (dd,1 H), 4.80 (s,1 H), 4.88 (s,1 H), fi.08 (s,1 H), 7.35
(m,1 H),
7.44 (m,4H), 7.60 (m,4H).
LRMS (Thermospray): 368 (M+H-COz)+.
60% Sodium hydride in a mineral oil dispersion (70 mg, 1.75 mmol) was
added to a stirred solution of the title compound of Preparation 9 (600 mg,
1.56
mmol) in anhydrous dimethylformamide (5 ml), under nitrogen, at room
temperature. After 20 minutes, ally) bromide (0.145 ml, 1.72 mmol) was added
and stirring continued for 2 hours, then more 60% sodium hydride dispersion
(70 mg, 1.75 mmol) was added followed, after 20 minutes, by methyl iodide
(0.11 ml, 1.72 mmol). The reaction mixture was stirred for a further 20 hours,
then partitioned between ethyl acetate and water. The organic phase was
separated, dried (MgSO,,) and evaporated under reduced pressure, then the
residue purified by flash chromatography, using hexane:ethyl acetate
(92.5:7.5)
as eluant, to afford the title compound (263 mg) as a colourless gum.
8(CDCI3):
1.62 (s,3H), 2.30 (s,3H), 2.59 (m,1 H), 2.66 (m,2H), 3.14 (m,1 H), 3.62
(m,2H),
3.80 (s,3H), 4.10 (m,2H), 5.20 (m,2H), 5.62 (m,1 H), 6.08 (brs,1 H), 7.20-7.44
(m,BH).
LRMS (Thermospray): 440 (M+H)+.
CA 02312935 2000-06-02
wo ~n~ss~ rcr~~rsros~o
-68
pJf3_E ,PARATION 29
Obtained as a colourless gum (90%) from the title compound of
Preparation 28, using the procedure of Preparation 15, but with a reaction
duration of 24 hours. 8(CDCI3): 1.63 (s,3H), 2.30 (s,3H), 2.62 (m,3H), 3.13
(m,1 H), 3.68 (m,2H), 4.18 (m,2H), 5.22 (m,2H), 5.70 (m,1 H), 6.07 (brs,1 H),
7.17-7.45 (m,BH).
o LRMS (Thermospray): 382 (M+H-C02)+, 426 (M+H)+.
_P$EPARATIONy~O
1-Benzhyrdtyl-.~-(4-pheny heno t,~;~zetidine
Potassium carbonate (2.39 g, 17.4 mmol) was added to a stirred mixture
of 1-benzhydryl-3-methanesulphonyloxyazetidine (J.Org.Chem., 1972, ~,
3953; 5 g, 15.8 mmol), 4-phenylphenol (2.95 g, 17.4 mmol) and anhydrous
dimethylfocmamide (65 ml), then the resulting mixture heated under reflux for
4
hours, allowed to cool and partitioned between ethyl acetate and water. The
organic phase was separated, washed with saturated brine, dried (MgS04) and
2o evaporated under reduced pressure, then the residue purified by flash
chromatography, using hexane:ethyl acetate (95:5) as eluant, to give the title
compound (1.32 g) as a colourless, amorphous solid. 8(CDCI3): 3.17 (dd,2H),
3.75 (dd,2H), 4.43 (s,2H), 4.83 (m,1 H), 6.81 (d,1 H), 7.16-7.55 (m,17H).
L.RMS (Thermospray): 392 (M+H)+.
A stirred mixture of the title compound of Preparation 30 (1.03 g, 2.63
mmol), 10% palladium hydroxide on activated carbon (100 mg), ethanol (100
3o m1), ethyl acetate (20 ml) and glacial acetic acid (10 ml) was hydrogenated
at
CA 02312935 2000-06-02
WO 99/Z9667 PCT/EP98/~6640
-69-
345 kPa (50 psi) and 50°C for 20 hours, then filtered. The filter pad
was
washed with ethanol and the combined washings and filtrate evaporated under
reduced pressure, then the residue was based with 2M aqueous sodium
hydroxide solution. This mixture was extracted with ethyl acetate and the
combined extracts dried (MgS04) and evaporated under reduced pressure to
yield the title compound containing 1 mol.equiv. of diphenylmethane* (456 mg)
as a colourless, amorphous solid. 8(CDCI3): 1.75 (brs,1H), 3.83 (m,2H), 3.94
(m,2H), 3.98 (s,2H)*, 6.82 (d,2H), 7.20-7.55 (m,17H)*.
o LRMS (Thermospray): 226 (M+H)+.
Obtained as a colourless, amorphous solid (22%) from the title
~5 compound of Preparation 31 and methyl chlorosulphonylacetate, using the
procedure of Preparation 8. 8(CDC13): 3.81 (s,3H), 4.07 (s,2H), 4.23 (dd,2H),
4.42 (dd,2H), 4.98 (m,1 H), 6.80 (d,2H), 7.34 (m,1 H), 7.42 (m,2H), 7.54
(m,4H).
LRMS (APCI): 362 (M+H)+.
20 PREPARATION 33
e. A 2.5M solution of n-butyllithium in hexane (3.8 ml, 9.4 mmol) was added
over about 10 minutes to a stirred mixture of 2,5-dibromotoluene (2.35 g, 9.4
mmol) in anhydrous ether (50 ml), under nitrogen, at about -75°C. After
a
25 further 1 hour, a solution of t-butyl 4-oxopiperidine-1-carboxylate (1.7 g,
8.5
mmol) in anhydrous tetrahydrofuran (5 ml) was added at such a rate that the
reaction temperature was maintained below -60°C.
The reaction mixture was stirred at about -75°C for 1 hour,
allowed to
warm to about 0°C and quenched with aqueous ammonium chloride solution.
CA 02312935 2000-06-02
wo ~n~~ rcr~c~sioss~o
-70-
The organic phase was separated, washed with water, dried (MgSO,,) and
evaporated under reduced pressure. The residue was purified by flash
chromatography, using ether:hexane (50:50) as eluant, to provide two isomeric
products.
The less polar isomer (0.6 g, 20%) was isolated as a colourless foam
and identified as t-butyl 4-(4-bromo-2-methy!phenyl)-4-hydroxypiperidine-1-
carboxylate. S(CDCl3): 1.46 (s,9H), 1.55 (s,1H), 1.82-2.03 (m,4H), 2.58
(s,3H),
3.23 (m,2H), 4.01 (m,2H), 7.20-7.33 (m,3H).
o LRMS ('fhermospray): 369/371 (M+H)+.
The more polar isomer, was collected as a 4:1 mixture of the title
compounds-butyl 4-oxopiperidine-1-carboxylate (2.15 g), a portion of which was
crystallised from diisopropyl ether to furnish the pure title compound (570
mg)
as a colourless solid, m.p. 102-103°C. Found: C,55.14; H,6.58; N,3.76.
C~~H24BrN03 requires C,55.14; H,6.53; N,3.78%. 8(CDCI3): 1.48 (s,9H), 1.51
(s,1 H), 1.70 (d,2H), 1.96 (m,2H), 2.40 (s,3H), 3.22 (t,2H), 4.02 (m,2H), 7.15
(dd,1 H), 7.36 (d,1 H), 7.50 (d,1 H).
LRMS (Thermospray): 369/371 (M+H)''.
~. A 2.5M solution of n-butyllithium in hexane (38 ml, 94 mmol) was added
over about 10 minutes to a stirred mixture of 2-brorno-5-iodo-toluene (28 g,
94
mmol) in anhydrous ether (500 ml), under nitrogen, at about -75°C.
After a
further 15 minutes, a solution of t-butyl 4-oxopiperidine-1-carboxylate (17 g,
85
mmol) in anhydrous tetrahydrofuran (50 ml) was added at such a rate that the
reaction temperature was maintained below -60°C.
The reaction mixture was stirred at about -75°C for 1 hour,
allowed to
warm to 0°C and quenched with aqueous ammonium chloride solution. The
organic phase was separated, washed with water, dried (MgS04) and
CA 02312935 2000-06-02
WO 99/Z9667 PCT/EP98/a6640
-71-
evaporated under reduced pressure. The residue was dissolved in pentane and
the resulting solution cooled to 0°C, when the title compound
crystallised. It
was collected and dried to afford a colourless solid (20.1 g, 64%), identical
with
that obtained in Preparation 33A.
Obtained as an amorphous solid (74%) from 1,4-dibromobenzene and t-
o butyl 4-oxopiperidine-1-carboxylate, using the procedure of Preparation 33.
8{CDCI3): 1.50 (s,9H), 1.69 (m,2H), 1.95 (m,2H), 3.22 (t,2H), 4.02 (m,2H),
7.34
(d,2H), 7.47 (d,2H).
LRMS (Thefmospray): 357 (M+H)''.
PREPARATION 35
Trifluoroacetic acid (100 ml) was added to a stirred solution of the title
compound of Preparation 33 {20 g) in dichloromethane (100 ml) at room
temperature. After a further 18 hours, the reaction mixture was evaporated
2o under reduced pressure and the residue basified with 2M aqueous sodium
hydroxide solution to pH>12. The resulting mixture was extracted with ether,
then the combined extracts washed with water, dried (MgS04) and evaporated
under reduced pressure to give the title compound (13.6 g) as a low melting
solid. 8(CDCI3): 1.60 (brs,1H), 2.40 (m,SH), 3.10 (t,2H), 3.52 (m,2H), 6.10
(brs,1 H), 7:05 (dd,1 H), 7.22 (d,1 H), 7.46 (d,1 H).
LRMS (Thermospray): 2511253 (M+H)+.
CA 02312935 2000-06-02
wo ~nx~~ rcr~r~so
-72
PREPARATION 36
~4-Bromo h~)~-1.2,3,6-tetra dropa nr 'dine
Obtained as a solid (87%), m.p. 76-78°C, from the title compound
of
Preparation 34 and trifluoroacetic acid, using the procedure of Preparation
35.
8(CDCI3): 2.43 (m,2H), 3.12 (t,2H), 3.53 (m,2H), 6.13 (s,lH), 7.25 (d,2H),
7.44
(d,2H).
LRMS (Thermospray): 239 (M+H}+.
o PREPARATION 37
Methvl2-(4-(4-bromo-3-methyl henyrl)-1,2.3.6-tetrahyrdrop~yrridin-1-
N,O-Bis(trimethylsilyl)acetamide (0.9 ml, 4.0 mmol) was added to a
stirred solution of the title compound of Preparation 35 (2 g, 7.9 mmol) in
~5 anhydrous tetrahydrofuran (40 ml), under nitrogen, at room temperature. A
solution of methyl chlorosulphonylacetate (1.64 g, 9.5 mmol) in anhydrous
tetrahydrofuran (15 ml) was then added and the reaction mixture stirred at
room
temperature for 18 hours. The resulting mixture was evaporated under reduced
pressure, the residue partitioned between ethyl acetate and aqueous sodium
20 bicarbonate solution, then the organic phase separated, washed with water,
dried (MgS04) and evaporated under reduced pressure. The residue was
purified by flash chromatography, using dichloromethane as eluant, followed by
crystallisation from diisopropyl ether, to yield the title compound (1.65 g,
55%)
as a colourless solid, m.p. 110-112°C. Found: C,46.32; H,4.62; N,3.55.
25 C~SH~gBfNO4S requires C,46.40; H,4.67; N,3.61 %. 8(CDCI3): 2.40 (s,3H),
2.60
(m,2H), 3.60 (t,2H), 3.80 (s,3H), 4.01 (s,2H), 4.07 (m,2H), 6.02 (brs,lH),
7.02
(dd,1 H), 7.21 (d,1 H); 7.50 (d,1 H).
LRMS (Thermospray): 404/406 (M+NH4)+.
CA 02312935 2000-06-02
WO 99/Z9667 PCT/EP98~06640
-73
PREPARATION, 38
Methyrl 2-[4-(4-bror~o_~yrl)-1,2.3.6-tetrahyrdronyrridin-1-
yrlsulohony~)acetate
Obtained as a colourless solid (32%), m.p. 100-102°C, from the
title
s compound of Preparation 36 and methyl chlorosulphonylacetate, using the
procedure of Preparation 37. Found: C,44.95; H,4.26; N,3.65. C~4H~sBrN04S
requires C,44.93; H,4.31; N,3.74%. 8{DMSOds): 2.47 (m,2H), 3.46 (t,2H), 3.70
{s,3H), 3.94 (m,2H), 4.37 {s,2H), 6.03 {s,1H), 7.40 (d,2H), 7.55 (d,2H).
LRMS (Thermospray): 393 (M+NH4)''.
~o
Bis-2-iodoethyl ether (3.9 g, 12 mmol) was added to a stirred mixture of
15 the title compound of Preparation 37 (3.6 g, 9.3 mmol), anhydrous potassium
carbonate (3.8 g, 27.8 mmol) and anhydrous dimethylsulfoxide (50 ml), under
nitrogen, at room temperature. After 18 hours, the reaction mixture was
partitioned between ether and water, then the organic phase washed with
water, dried (MgS04) and evaporated under reduced pressure. The residue
2o was purified by flash chromatography, using dichloromethane : methanol
(99:1)
as eluant, followed by crystallisation from diisopropyl ether, to provide the
title
compound (3.43 g, 80%) as a colourless solid, m.p. 128-130°C. Found:
C,49.92; H,5.40; N,2.90. C~9H24BrN05S requires C,49.78; H,5.28; N,3.06%.
8(CDCI3): 2.23 (m,2H), 2.40 (s,3H), 2.42 (m,2H), 2.58 {m,2H), 3.30 (m,2H),
2s 3.58 {m,2H), 3.87 (s,3H), 4.00-4.10 (m,4H), 6.00 (brs,1 H), 7.02 (dd,1 H),
7.21
(d,1 H), 7.49 (d,1 H).
LRMS (Therrnospray): 477 (M+NH4)+.
CA 02312935 2000-06-02
wo ~n9ss~ pc~r~o
-74
PREPARATION 40
s lodomethane (2 ml, 32.1 mmol) was added to a stirred mixture of the title
compound of Preparation 37 (5 g, 12.9 mmol), anhydrous potassium carbonate
(5.4 g, 39.1 mmol) and anhydrous dimethylsulfoxide (50 ml), under nitrogen, at
room temperature. After 24 hours, the reaction mixture was partitioned between
ether and water, then the organic phase washed with water, dried (MgS04) and
o evaporated under reduced pressure. The residue was purified b~,~ flash
chromatography, using an elution gradient of ether:pentane (40:60 to 100:0),
followed by crystallisation from diisopropyl ether, to furnish the title
compound
(4.7 g, 87%) as a colourless solid, m.p. 100-101°C. Found: C,49.00;
H,5.33;
N,3.28. C~~H22BfNO4S requires C,49.04; H,5.33; N,3.36%. 8(CDC13): 1.67
~5 (s,6H), 2.40 (s,3H); 2.58 (m,2H), 3.60 (t,2H), 3.80 (s,3H), 4.08 (m,2H),
6.00
(brs,1 H), 7.03 (dd,1 H), 7.21 (d,1 H), 7.49 (d,1 H).
To a solution of the title compound of Preparation 37 (776 mg, 2 mmol)
in degassed 1,2-dimethoxyethane (20 ml) was added 3-ethoxyphenylboronic
acid (430 mg, 2.6 mmol), cesium fluoride (790 mg, 5.2 mmol), tri-o-
tolylphosphine (61 mg, 0.2 mmol) and tris(dibenzylideneacetone)dipalladium(0)
2s (91 mg, 0.1 mmol), then the reac#ion mixture heated under reflex for about
3
hours under nitrogen. The resulting mixture was allowed to cool to room
temperature, then diluted with dichloromethane and washed with water. The
organic phase was dried (Mg~04) and evaporated under reduced pressure,
then the residue purified by flash chromatography, using dichloromethane as
CA 02312935 2000-06-02
WO 99/29667 PCT/EP98/06640
-75-
eluant, followed by crystallisation from diisopropyl ether, to afford the
title
compound (665 mg, 78%) as a colourless solid, m.p. 79-81°C. Found:
C,64.40; H,6.37; N,3.17. C23H27N05S requires C,64.31; H,6.34; N,3.26%.
s 8(CDC13): 1.43 (t,3H), 2.31 (s,3H), 2.70 (m,2H), 3.63 (t,2H), 3.82 (s,3H),
4.03
(s,2H), 4.10 (m,4H), 6.08 (brs,1H), 6.84-6.93 (m,3H), 7.20-7.37 (m,4H).
LRMS (Thermospray): 447 (M+NH4)+.
p 4-~somo-2-meths,~yrbi hp envy
n-Amyl nitrite (8.1 ml, 60 mmoi) was slowly added to a stirred mixture of
4-bromo-2-methoxyaniline (J.Med.Chem., 1989, 32, 1936; 8.1 g, 60 mmol) and
benzene (175 ml), under nitrogen, at about 50°C. When the addition was
complete, the reaction mixture was heated under reflux for about 3 hours, then
t5 allowed to cool to room temperature and evaporated under reduced pressure.
The residue was azeotroped with tetrahydrofuran, then with ethyl acetate, and
purified by flash chromatography, using an elution gradient of hexane:ethyl
acetate (100:0 to 95:5) to give the title compound (1.66 g) as a colourless
solid,
m.p. 50-52°C. S(CDCI3): 3.77 (s,3H); 7.08 (s,1 H), 7.14 (s,2H), 7.30
(m,1 H),
20 7.36-7.41 (m,2H), 7.41-7.49 (m,2H).
A 2.5M solution of n-butyAithium in hexane (4.4 ml, 11 mmol) was added
2s over about 10 minutes to a stirred mixture of the title compound of
Preparation
42 (2.6 g, 10 mmol) in anhydrous tetrahydrofuran (30 ml), under nitrogen, at
about -75°C. After a further 1 hour, a solution of t-butyl 4-
oxopiperidine-1-
carboxylate (2.2 g, 11 mmol) in anhydrous tetrahydrofuran (10 ml) was added
CA 02312935 2000-06-02
wo ~n~s~ Pc~r~r9s~o~o
-76-
at such a rate that the reaction temperature was maintained below -
60°C. The
reaction mixture was stirred at about -75°C for 1 hour, then slowly
warmed to
room temperature and quenched with aqueous sodium chloride solution. The
organic phase was separated, washed with water, dried (MgS04) and
evaporated under reduced pressure. Purification of the residue by flash
chromatography, using hexane:ethyl acetate (3:1 ) as eluant, yielded the title
compound (3.4 g) as a colourless semi-solid. 8(CDCI3): 1.50 (s,9H), 1.78
(m,2H), 2.04 (m,2H), 3.27 (m,2H), 3.83 (s,3H), 4.08 (m,2H), 7.09 (d,1H), 7.16
o (s,1 H), 7.2 T-7.37 (m,2H), 7.40 (m,2H), 7.52 (d, 2H).
Trifluoroacetic acid (20 ml) was added to a stirred solution of the title
~5 compound of Preparation 43 (3.4 g, 11.9 mmol) in dichloromethane (20 ml) at
room temperature. After a further 72 hours, the reaction mixture was
evaporated under reduced pressure and the residue basified with 1 M aqueous
sodium hydroxide solution. The resulting mixture was extracted with
dichloromethane, then the combined extracts washed with water, dried
20 (MgS04) and evaporated under reduced pressure to provide the title compound
(2.79 g) as a pale yellow viscous oil. 8(CDCI3): 1.73 (s,1 H), 2.51 (m,2H),
3.14
(t,2H), 3.57 (m,2H), 3.83 (s,3H), 6.19 (s,lH), 7.01 (s,1H), 7.05 (d,2H), 7.27-
7.37
(m,2H), 7.40 (t,2H), 7.52 (d, 2H).
LRMS (Thermospray): 266 (M+H)+.
N,O-Bis(trimethylsilyl)acetamide (1.0 ml, 4.4 mmol) was added dropwise
CA 02312935 2000-06-02
WO 99/Z9667 PCT/EP98/06640
-n-
to a stirred solution of the title compound of Preparation 44 (1.95 g, 7.3
mmol)
in anhydrous tetrahydrofuran (40 ml) at room temperature. The reaction mixture
was stirred at room temperature for 1 hour, then a solution of methyl
chlorosulphonylacetate (1.5 g, 8.8 mmol) in tetrahydrofuran (10 ml) was added.
The resulting mixture was stirred at room temperature for about 1.5 hours and
then saturated aqueous sodium bicarbonate solution (50 ml) added. The
mixture was extracted with dichloromethane (3 x 100 ml), then the combined
extracts dried (MgS04) and evaporated under reduced pressure. The residue
o was purified by flash chromatography, using dichloromethane as Eluant, to
furnish the title compound (1.0 g) as a pale yellow solid solid, m.p. 92-
95°C.
Found: C,62.24; H,5.70; N,3.42. C2~H23NO5S requires C,62.82; H,5.77;
N,3.49%. 8(CDCI3): 2.73 (m,2H), 3.67 (t,2H), 3.84 (s,3H), 3.86 (s,3H), 4.06
(s,2H), 4.08 (m,2H), 6.12 (s,1 H), 6.98 (s,1 H), 7.04 (d,2H), 7.27-7.37
(m,2H),
7.44 (t,2H}, 7.56 (d,2H).
LRMS (Them~ospray): 402 (M)+.
2o Obtained as a colourless oil (67%), from 4-bromo-3-fluorobiphenyl and t-
butyl 4-oxopiperidine-1-carboxylate, using the procedure of Preparation 43.
8(CDCI3): ): 1.50 (s,9H), 1.78 (m,2H), 2.03 (m,2H), 3.26 (t,2H), 4.05 (m,2H),
7.27-7.51 {m,6H}, 7.57 {d, 2H).
PREP~9RAT10N 47
4-{3-Flcoro-4-p_henvlnheny~'~,)- .1 2.3_i6-tetrahyrdroo~ '
Obtained as a colourless solid (90%), m.p. 79-82°C, from the title
compound of Preparation 46 and trifluoroacetic acid, using the procedure of
CA 02312935 2000-06-02
wo ~n~~ pcr~~sross~o
-78-
Preparation 44. 8(CDCI3): 1.85 (s,1 H}, 2.49 (m,2H), 3.13 {t,2H), 3.58 (m,2H),
6.24 (brs, 1 H), 7.12-7.27 (m,2H), 7.35-7.52 (m,4H), 7.59 {d,2H).
LRMS (Thermospray): 253 (M)+.
Obtained as a colourless solid (38%), from the title compound of
Preparation 47 and methyl chlorosulph~nylacetate, using the procedure of
Preparation 45. 8(DMSOds): 2.60 (m,2H), 3.47 (t,2H), 3.68 (s,3H), 3.96 (s,2H},
4.37 (s,2H), 6.33 (brs,1 H), 7.34-7.57 (m,BH).
LRMS (Thermospray): 407 {M+NH4)+.
t5 PRE~~,ARATION 49
lodomethane {0.2 ml, 3.4 mmol) was added to a stirred mixture of the
title compound of Preparation 45 (0.54 g, 1.4 mmol), anhydrous potassium
2o carbonate (0.56 g, 4.1 mmol) and anhydrous dimethylsufphoxide {5 ml), then
the reaction mixture stirred at room temperature for about 16 hours. The
resulting mixture was partitioned between ethyl acetate and water, then the
organic phase washed with water, dried (MgS04) and evaporated under
reduced pressure to afford the title compound (540 mg) as a pale yellow oil.
25 8(CDCI3): 1.69 (s,6H), 2.67 {m,2H), 3.64 (t,2H), 3.82 (s,3H), 3.84 (s,3H},
4.14
(m,2H}, 6.09 (s,1H), 6.98 (s,1H), 7.03 {d,2H), 7.27-7.37 (m,2H), 7.42 (t,2H),
7.54 (d,2H).
LRMS (Thermospray): 430 (M+H)+.
CA 02312935 2000-06-02
WO 99/29667 PCT/EP98/06640
-79-
1 M Aqueous sodium hydroxide solution (1.2 ml, 1.2 mmol) was added to
a stirred solution of the title compound of Preparation 49 (250 mg, 0.58 mmol)
in methanol (5 ml). The resulting solution was heated at 50°C for about
2
hours, then allowed to cool to room temperature and poured into ethyl acetate.
This mixture was washed with 2M hydrochloric acid, then the organic phase
~o dried (MgSG4) and evaporated under reduced pressure to give the title
compound (210 mg) as a pale yellow , semi- solid. 8(CDCI3): 1.69 (s,6H), 2.67
(m,2H), 3.64 (t,2H), 3.83 (s,3H), 4.17 (m,2H), 6.08 (s,1H), 6.97 (s,1H), 7.03
(d,2H), 7.27-7.36 (m,2H), 7.40 (t,2H), 7.53 (d,2H).
LRMS (Thermospray): 433 (M+NH4)+.
A solution of 4-bromo-2-methoxyaniline (J. Med.Chem., 1989, 32, 1936;
17.9 g, 88.6 mmol) in anhydrous ether (350 ml) was added over about 1 hour to
2o boron trifluoride etherate (27 ml, 212 mmol) at -15°C. The resulting
solution
was stirred at -15°C for about 5 minutes and then a solution of t-butyl
nitrite
(11.4 ml, 106 mmol) in anhydrous ether (100 ml) was added slowly, keeping the
internal temperature at around -15°C. The reaction mixture was stirred
at -15°C
for a further 15 minutes and then at 4°C for about 4 hours. Pentane was
added
and the resulting precipitate collected, washed with pentane and dried under
reduced pressure to yield the title compound (20.1 g) as a purple solid.
8(CD3CN): 4.20 (s,3H), 7.58 (d,1H), 7.80 (s,1H), 8.16 (d,1H).
LRMS (Thermospray): 301 (M)+.
CA 02312935 2000-06-02
wo ~n~~ Pc~r~r9sro~eo
-80-
Anhydrous 1,4-dioxan (80 ml) was added to a mixture of the title
compound Preparation 51 (8.0 g, 26.6 mmol), 3-ethoxyphenylboronic acid (5.3
g, 31.9 mmol) and palladium(II) acetate (0.35 g, 1.3 mmol) and the reaction
mixture stirred at room temperature for about 16 hours. The resulting mixture
was diluted with water (100 ml) and ether (100 ml), filtered and the filtrate
extracted with ether (2 x 100 ml). The combined extracts were dried (MgS04)
o and then evaporated under reduced pressure. Purification of the residue by
flash chromatography, using pentane:ether (20:1 ) as eluant, provided the
title
compound (6.9 g) as a colourless oil. S{CDCI3): 1.45 (t,3H), 3.83 (s,3H), 4.10
(q,2H), 6.89 (d,1H), 7.06 {d,2H), 7.10-7.24 (m,3H), 7.27-7.38 (m,1H).
LRMS (APCI): 308 (M+H)+.
4-
Obtained as a colourless oil (60%), from the title compound of
Preparation 52 and t-butyl4-oxopiperidine-1-carboxyiate, using the procedure
of Preparation 43. 8(CDC13): 1.44 (t,3H), 1.50 (s,9H), 1.79 (m,2H), 2.03
(m,2H), 3.27 (t,2H), 3.83 (s,3H), 4.06 (m,4H), 6.86 (d,1H), 7.08 (m,3H), 7.15
(s,
1 H), 7.31 (m,2H).
_P1~,PARATI~N 54
Obtained as a pale yellow viscous oil (91 %), from the title compound of
Preparation 53 and trifluoroacetic acid, using the procedure of Preparation
44.
CA 02312935 2000-06-02
WO 99/29667 PCT/EP98/06640
-81-
S(CDC13): 1.44 (t,3H), 2.53 (m,2H), 3.16 (t,2H), 3.58 {s,2H), 3.83 (s,3H),
4.07
(q,2H), 6.19 (brs,1 H), 6.86 (d,1 H), 7.00 (s,1 H), 7.07 (m,3H), 7.31 (m,2H).
LRMS (Thermospray): 310 (M+H)''.
Obtained as a yellow semi-solid (22%), from the title compound of
1o Preparation 54 and methyl chlorosuphonylacetate, using the procedure of
Preparation 45. S(CDC13): 1.43 (t,3H), 2.69 (m,2H), 3.65 (t,2H), 3.81 {s,3H),
3.84 (s,3H), 4.04 (s,2H), 4.08 (m,4H), 6.10 (brs,1 H), 6.87 (d,1 H), 6.96 (s,1
H),
7.02 (d,1 H), 7.09 (m,2H), 7.31 (m,2H).
LRMS (Thermospray): 446 (M+H)+.
To a solution of the title compound of Preparation 38 (250 mg, 0.7 mmol) in
1,2-
dimethoxyethane (8 ml) was added 3-ethoxyphenylboronic acid (168 mg, 1.0
mmol), cesium fluoride (226 mg, 1.5 mmol), tri-o-tolylphosphine (21 mg, 0.07
mmol) and tris(dibenzyf~deneacetone)dipalladium(0) (31 mg, 0.035 mmol), then
the reaction mixture heated under reflux for about 4 hours under nitrogen. The
resulting mixture was allowed to cool to room temperature, then diluted with
ethyl acetate and washed with saturated aqueous sodium bicarbonate solution:
The organic phase was dried (MgS04) and evaporated under reduced
pressure, then the residue triturated with hexane-ether to furnish the title
CA 02312935 2000-06-02
wo ~n~ss~ Pc~r~r9sro~o
-82-
compound (222 mg) as a pale yellow solid, m.p. 120-122°C. Found:
C,64.25;
H,6.01; N,2.99. C~H25N05S requires C,63.60; H,6.06; N,3.37%. 8(DMSO~):
1.32 (t,3H), 2.62 (m,2H), 3:50 (t,2H), 3.71 {s,3H), 3.98 (d,2H), 4.10 (q,2H),
4.37
{s,2H), 6.27 (brs,1 H), 6.91 (d,1 H), 7.18 (s,1 H), 7.22 (d,1 H), 7.36 (t,1
H), 7.52
(d,2H), 7.66 (d,2H).
LRMS (Thermospray): 433 (M+NH4)~.
4-Bromo-2-meth henyldiazonium tetra~,uoro~~borate
Obtained as a yellow solid (93%), from 4-bromo-2-methylaniline and t-
butyl nitrite, using the procedure of Preparation 51. 8(CD3CN): 2.70 {s,3H),
7.93 {d,1 H), 8.04 (s,1 H), 8.50 (d,1 H).
eJ3EPARATION 58
Obtained as a pale yellow oil (25%), from the title compound of
Preparation 5? and 3-methoxyphenylboronic acid, using the procedure of
Preparation 52. 8(CDCI3): 2.15 (s,3H), 3.84 (s,3H), 6.79-6.94 (m,3H), 7.09
(s,1H), 7.33 (d,1H), 7.37 {d,1H), 7.43 (s,1H).
Obtained as a colourless oil (fi3%), from the title compound of
Preparation 58 and t-butyl 4-oxopiperidine-1-carboxylate, using the procedure
of Preparation 43. 8(CDCI3): 1.50 (s,9H), 1.79 (m,2H), 2.04 (m,2H), 2.19
(s,3H), 3.27 (t,2H), 3.83 (s,3H), 4.06 (m,2H), 6.85 (s,1H), 6.90 (d,2H), 7.2fi
(m,
1 H), 7.34 (t,2H), 7.38 (s,1 H).
3o LRMS (Thermospray): 399 (M+H)+.
CA 02312935 2000-06-02
wo 9gnx~s~ pc~r~~sro~so
-83
PREPARATION 60
4-[4-~(3-Methoxyr~henyrl}-3-methyrl henyrlj-1.2.3x6-tetra yrdrop~ nr 'dine
Obtained as a pale yellow semi-solid (93%), from the title compound of
Preparation 59 and trifluoroacetic acid, using the procedure of Preparation
44.
8(CDCl3), 2.30 (s,3H), 2.42 (s,2H), 3.15 (t,2H), 3.57 (s,2H), 3.83 (s,3H),
6.18
(brs,1H), 6.90 (m,3H}, 7.16-7.36 (m, 4H).
LRMS (Thermospray): 280 (M+H)'.
~o PREPARATION 61
A solution of methyl chlorosulphonylacetate (0.65 g, 3.7 mmol) in
dichloromethane (10 ml) was added dropwise to a stirred solution of the title
compound of Preparation 60 (0.92 g, 3.3 mmol) and 1,8-diazabicyclo
[5.4.0]undec-7-ene (0.76 g, 4.9 mmol) in dichloromethane (20 ml) at about
0°C,
the cooling bath removed and the reaction mixture stirred at room temperature
for 4 hours, then diluted with dichloromethane. The resulting mixture was
washed with 0.1 M hydrochloric acid, dried (MgS04) and evaporated under
2o reduced pressure, then the residue purified by flash chromatography, using
dichloromethane as eluant, to afford the title compound (250 mg) as a
colourless solid, m.p. 83-85°C. 8(CDCI3): 2.30 (s,3H}, 2.69 (m,2H),
3.66 (t,2H),
3.85 (s,3H), 3.86 (s,3H), 4.03 (s,2H}, 4.11 (m,2H), 6.09 (brs,1H), 6.83-
6.97(m,3H), 7.17-7.35 (m, 4H).
LRMS (Thermospray): 416 (M+H)+.
CA 02312935 2000-06-02
WO 99/29667 PCT/EP98/06640
-84-
n-Butyllithium (11 ml of a 2.5M solution in hexane, 28 mmol) was added
to a stirred solution of 3-ethylbromobenzene CChem. Pharm. Bull., 1968, ~,
2456; 4.6 g, 25 mmol) in anhydrous tetrahydrofuran (50 ml), whilst keeping the
internal temperature below -60°C. The mixture was stirred at about -
70°C for 1
hour, then trimethylborate (4.4 ml, 38 mmol) added dropwise, again whilst
keeping the internal temperature below -60°C. The reaction mixture was
stirred
o at -70°C for 30 minutes, then slowly allowad to warm to room
temperature. 2M
Hydrochloric acid was added, the mixture was extracted with dichloromethane
(3 x 50 ml) and the combined extracts concentrated under reduced pressure
The residue was dissolved in ether (50 ml), the solution extracted with 1 M
aqueous sodium hydroxide solution (2 x 30 ml) and the aqueous phase
~5 acidified with 2M hydrochloric acid, then extracted with ether {3 x 50 ml).
The
combined extracts were dried (MgS04) and evaporated under reduced
pressure to give the title compound as a white solid (0.9 g, 24%). 8(DMSO~):
1.17 (t,3H), 2.57 {q,2H), 7.22 (t,2H), 7.57 (t,1 H), 7.61 (s,1 H), 7.93
(s,2H).
20 PREPARATION 63
Obtained as a yellow solid (75%), m.p. 58-60°C, from the title
compounds of Preparation 62 and Preparation 37, using the procedure of
25 Preparation 41. S(CDCI3): 1.27 (t,3H), 2.50 {s,3H), 2.68 (m,4H), 3.64
(t,2H),
3.82 (s,3H), 4.03 (s,2H), 4.10 {s,2H), 6.08 {brs,1H), 7.14-7.37 {m,7H).
LRMS (Thermospray): 431 (M+NH4)+.
CA 02312935 2000-06-02
WO 99/Z9667 PCTIEP98/06640
-85-
yrl~ul onyyt~tra~lyrdroRyrran-4-carboxvlate
s Obtained as a glassy solid (20%), from the title compound of Preparation
61 and bis-2-iodoethyl ether, using the procedure of Preparation 39. 8(CDCI3):
2.20-2.34 (m,SH), 2.45 (m,2H), 2.67 (m,2H), 3.33 (t,2H), 3.62 (m,2H), 3.83
(s,3H), 3.89 (s,3H), 4.01 (m,2H), 4.10 (m,2H), 6.05 (brs,1H), 6.91 (m,3H),
7.23-
7.36 (m,4H).
1o LRMS (APCI): 486 (M+H)+.
15 Obtained as a pale yellow solid (93%), m.p. 180-190°C, from the tile
compound of Preparation 64, using the procedure of Preparation 50. 8(CDCI3):
2.20-2.33 {m,SH), 2.43 {m,2H), 2.65 (m,2H), 3.43 (t,2H), 3.67 (m,2H), 3.82
(s,3H), 4.04 (m,2H), 4.14 (m,2H), 6.04 (brs,1H), 6.88 (m,3H), 7.21-7.36
(m,4H).
20 P$EPARATION 66
A stirred mixture of the title compound of Preparation 64 (315 mg, 0.65
mmol), ammonium formate (200 mg, 3.2 mmol), 10% palladium on carbon (50
2s mg) and methanol (5 ml) was heated under reflux for 1.5 hours, then allowed
to
cool and filtered. The filtrate was evaporated under reduced pressure and the
residue partitioned between ether and water. The organic phase was dried
(MgS04) and evaporated under reduced pressure, then the residue crystallised
CA 02312935 2000-06-02
wo ~n9~s~ Pcr~r~sro~o
-86-
from methanol to yield the title compound (215 mg) as a white solid, m.p. 137-
139°C. 8(CDCI3): 1.75-1.94 (m,4H), 2.19 (d,1 H), 2.22 (d,1 H), 2.27
(s,3H), 2.43
(m,2H), 2.66 (m,1H), 3.07 (t,2H), 3.32 (t,2H), 3.83 (s,3H), 3.90 (s,3H), 3.96
(m,2H), 4.00 (d,1 H), 4.02 (d,1 H), 6.87 (m,3H), 7.08 (m,2H), 7.19 (d,1 H),
7.32
(m,1 H).
LRMS (APCI): 486 (M+H)~.
0 4-~~ -~(,,~-Metho~nhenyl)-3-meth~pheny~)gir~eridin-1-
Obtained as a white solid (84%), m.p. 225-228°C, from the title
compound of Preparation 66, using the procedure of Preparation 50, but with a
mixture of methanol (5 ml) and tetrahydrofuran (10 ml) as solvent: Found:
~5 C,63.04; H, 6.59; N, 2.91. C25H3~NO6S requires C, 63.40; H, 6.60; N, 2.96%.
8(DMSO~): 1.60 (m,2H), 1.80 (m,2H), 1.93 (dt,2H), 2.20 (s,3H), 2.24 (m,2H),
2.68 (m,1 H), 3.05 (t,2H), 3.20 (t,2H), 3.75 (s,3H), 3.77 (m,2H), 3.88 (d,1
H), 3.92
(d,1 H), 6.86 (m,3H), 7.11 (m,3H), 7.31 (m,1 H), 13.80 (brs,1 H).
LRMS (APCI): 474 (M)+.
Obtained as a pale yellow oil (78%), from the title compound of
Preparation 51 and 3-methoxyphenylboronic acid, using the procedure of
Preparation 52. Found: C, 57.77; H, 4.51. C~4H~3Br02 requires C, 57.36; H,
4.47. S(CDCI3): 3.82 (s,3H), 3.86 (s,3H), 6.91 (d,1H), 7.03-7.39 (m,6H).
LRMS (Thermospray): 311 (M+NH4)'~.
CA 02312935 2000-06-02
wo ~n~s~ pcr~r9a~o~o
_87_
Obtained as a colourless oil (67%), from the title compound of
Preparation 68 and t-butyl 4-oxopiperidine-1-carboxyfate, using the procedure
of Preparation 43. 8(CDCI3): 1.50 (s,9H), 1.79 {m,2H), 2.06 (m,2H), 3.28
(t,2H),
3.83 {s,6H), 4.06 (m,2H), 6.88 {d,1 H), 7.08 (m,3H), 7.14 (s, 1 H), 7.32
{m,2H).
LRMS (Thermospray): 436 {M+Na)+.
0
A stirred solution of the title compound of Preparation 69 (6.7 g, 16.2
mmol) and p-toluenesulphonic acid (6.17 g, 32.5 mmol) in toluene (70 ml) was
t5 heated under reflux in a Dean-Stark apparatus until water removal was
complete (~. 4 hours), then allowed to cool and diluted with ethyl acetate
(100
ml). The resulting mixture was washed with 1 M aqueous sodium hydroxide
solution (3 x 50 ml), then the organic phase dried (MgS04) and evaporated
under reduced pressure to provide the title compound (3.3 g) as a yellow oil.
2o b{CDC13): 1.80 {brs,1H), 2.50 {m,2H), 3.14 (t,2H), 3.57 (m,2H), 3.84
(s,6H), 6.20
(brs,1H), 6.88 (d,1H), 6.98-7.36 (m, 6H).
LRMS (Thermospray): 296 (M+H)+.
Obtained as a yellow semi-solid (40%), from the title compound of
Preparation 70 and methyl chlorosulphonylacetate, using the procedure of
CA 02312935 2000-06-02
wo ~n~s~ rcr~r9sios~o
-88-
Preparation 45, Found: C,58.87; H, 5.65; N, 3.11. C22H~NO6S; 1.00 H20
requires C, 58.78; H, 6.05; N, 3.12%. 8(CDCI3): 2.71 (m,2H), 3.64 (t,2H), 3.81
(s,3H), 3.84 (s,6H), 4.02 (s,2H), 4.11 (m,2H), 6.10 (brs,1 H), 6.88 (d,1 H),
6.9?
(s,1 H), 7.02 (d,1 H), 7.12 (m,2H), 7.32 (m,2H).
LRMS (Thermospray): 449 (M+H)''.
to 1-yrlsulphonyrl}-2-met ,yrl~ ap noate
Obtained as a colourless semi-solid (40%), from the title compound of
Preparation 71 and iodomethane, using the procedure of Preparation 49.
Found: C,62.30; H, 6.29; N, 3.00. C24H2sNOsS requires C, 62.73; H, 6.36; N,
3.05%. 8(CDCI3): 1.68 (s,6H), 2.67 (m,2H), 3.64 (t,2H), 3.82 (s,3H), 3.84
~5 (s,3H), 3.85 (s,3H), 4.13 (m,2H), 6.07 (brs,1H), 6.88 (d,1H), 6.97 (s,1H),
7.02
{d,1 H), 7.13 (m,2H), 7.31 (m,2H).
LRMS (Thermospray): 460 (M+H)+.
2o Methyrl~[3-methox~~-~(3-methoxvnhenyrl~phen~~j~ir~eridin-1-yrlsulphonvll-2-
methyrj~,rop~p,~~
Obtained as a colourless solid (80%), m.p. 140-142°C, from the
title
compound of Preparation 72, using the procedure of Preparation 66. Found:
C,62.31; H, 6.87; N, 2.91. C24Hg~NOgS requires C, 62.45; H, 6.77; N, 3.03%.
25 8(CDCI3): 1.66 (s,6H), 1.87 (m,4H), 2.49 (m,1H), 3.09 (t,2H), 3.82 (s,6H),
3.84
(s,3H), 3.93 (m,2H), 6.81 {s,1H), 6.86 (d,2H), 7.08 (m,2H), 7.29 (m,2H).
LRMS (Thermospray): 462 (M+H)+.
CA 02312935 2000-06-02
WO 99/29667 PCT/EP98/06640
-89
PREP RATION 74
Obtained as a colourless solid (80%), m.p. 164-165°C, from the
title
compound of Preparation 73, using the procedure of Preparation 50. Found:
C,61.64; H, 6.53; N, 3.06. CZ3H29NO6S requires C, 61.73; H, 6.53; N, 3.13%.
8(CDCI3): 1.69 (s,6H), 1.87 (m,4H), 2.69 (m,1H), 3.10 (t,2H), 3.81 (s,3H),
3.82
(s,3H), 4.02 (m,2H), 6.80 (s,lH), 6.87 (m,2H), 7.07 (m,2H), 7.27 (m,2H).
o LRMS (Thermospray): 465 (M+NH4)+.
r
~5 Obtained as a pale yellow oil (29%), from the title compound of
Preparation 55 and iodomethane; using the procedure of Preparation 49.
Found: C,62.99; H, 6.64; N, 2.88. C25H3~NOsS requires C, 63.40; H, 6.60; N,
2.96%. s(CDCI3): 1.43 (t,3H), 1.67 (s,6H), 2.66 (m,2H), 3.63 (t,2H), 3.81
(s,3H),
3.83 (s,3H), 4.07 (q,2H), 4.13 (m,2H), 6.07 (brs,1 H), 6.87 (d,1 H), 6.96 (s,1
H),
20 7.02 (d,1H), 7.10 (m,2H), 7.31 (m,2H).
LRMS (Thermospray): 474 (M+H)+.
Obtained as a colourless semi-solid (83%) from the title compound of
Preparation 75, using the procedure of Preparation 66. Found: C,62.86; H,
7.12; N, 2.68. C25HssNOsS requires C, 63.14; H, 6.99; N, 2.95%. 8(CDCl3):
1.43 (t,3H), 1.67 (s,6H), 1.86 (m,4H), 2.70 (m,1H), 3.09 (t,2H), 3.82 (s,6H),
3.97
ao (m,2H), 4.06 (q,2H), 6.80 (s,1H), 6.86 (m,2H), 7.08 (m,2H), 7.27 (m,2H).
LRMS (Thermospray): 476 (M+H)+.
CA 02312935 2000-06-02
wo ~nx~~ pc r~r9o
-90
PREPARATION 77
Obtained as a colourless solid (95%), from the title compound of
Preparation 76, using the procedure of Preparation 50. Found: C,61.92; H,
7.00; N, 2.72. C24H3~NO6S requires C, 62.45; H, 6.77; N, 3.03%. 8(CDCI3):
1.42 (t,3H), 1.70 (s,6H), 1.87 (m,4H), 2.70 (m,1 H), 3.11 (t,2H), 3.80 (s,3H),
4.04
(m,4H), 6.80 (s,1H), 6.85 (d,2H), 7.08 (m,2H), 7.27 (m,2H).
o LRMS (Thermospray): 479 (M+NH,~)+.
~5 Obtained as a colourless foam (86%), from the title compound of
Preparation 39 and 3-ethoxyphenylboronic acid, using the procedure of
Preparation 41, but with methanol:dichloromethane (1:99) as eluant. 8(CDCl3):
1.42 (t,3H), 2.22 (m,2H), 2.28 (s,3H), 2.44 (d,2H), 2.65 (m,2H), 3.34 (dd,2H),
3.60 (m,2H), 3.90 (s,3H), 4.00-4.15 (m,6H), 6.03 (brs,1H), 6.83-6.92 (m,3H),
20 7.20-7.36 (m,4H).
LRMS (Them~ospray): 500 (M+H)+.
4..-(~(4~~E~klQXy~l~.y!1)~thylQb~D.1!I)-1.2.3.6-tetrahydroRy 'd~ in-1-
25 ytsulnhony~}tetrahydroRyrran-4-carbo~,r(~~c acid
1 M Aqueous sodium hydroxide solution (2.3 ml, 2.3 mmol) was added to
a stirred solution of the title compound of Preparation 78 (290 mg, 0.58 mmol)
in a mixture of methanol (10 ml) and 1,4-dioxan (2 ml). The resulting solution
was heated at 80°C for about 5 hours, then allowed to cool to room
CA 02312935 2000-06-02
wo ~n~~ Pc~r~~s~o
-91-
temperature and evaporated under reduced pressure. The residue was
partitioned between 1 M hydrochloric acid and ethyl acetate, then the organic
phase dried (MgS04) and evaporated under reduced pressure. The residue
was crystallised from diisopropyl ether to furnish the title compound (220 mg)
as a colourless solid, m.p. 203-205°C. Found: C,64.14; H, 6.47; N,
2.87.
C26H3~NO6S requires C, 64.31; H, 6.44; N, 2.89%. 8(CDCI3): 1.43 (t,3H), 2.27
(m,2H), 2.29 (s,3H), 2.42 (d,2H), 2.68 (m,2H), 3.42 (dd,2H), 3.67 (m,2H), 4.00-
4.18 (m,6H), 6.04 (brs,1H), 6.82-6.93 (m,3H), 7.20-7.35 (m,4H).
o LRMS {Thermospray): 486 (M+H)+.
~s Obtained as a colourless solid (80%), m.p. 75-76°C, from the title
compound of Preparation 41 and iodomethane, using the procedure of
Preparation 40. Found: C,65.55; H, 6.82; N, 2.98. C25H3~ NO5S requires C,
65.62; H, 6.83; N, 3.06%. 8(CDCI3): 1.43 (t,3H), 1.68 (s,6H), 2.28 (s,3H},
2.65
(m,2H), 3.62 (m,2H), 3.81 (s,3H), 4.06 (q,2H), 4.12 (m,2H), 6.06 (brs,1H),
6.83-
20 6.92 (m,3H), 7.20-7.35 (m,4H).
ARMS (Thermospray): 458 (M+H)+.
?-(~-j~~-~ Ftho~q~y1)_~_mPt yl~,y~~-1,2.3.6-tetrahydro~~ rid-r, in-1-
25 ylsul~yrjj)--2-methyl~panoic acid
Obtained as a colourless amorphous solid (50%), from the title
compound of Preparation 80, using the procedure of Preparation 79, but with
purification by flash chromatography using methanol:dichloromethane (2:98).
CA 02312935 2000-06-02
wo ~n~~ rcr~~sros~ao
-92-
8(CDCI3): 1.43 (t,3H), 1.68 (s,6H), 2.25 (s,3H), 2.60 (m,2H), 3.62 (m,2H),
4.05
(q,2H), 4.15 (m,2H), 6.03 (brs,1 H), 6.78-6.90 (m,3H), 7.20-7.35 (m,4H).
LRMS (APCI): 444 (M+H)+.
Obtained as a colourless amorphous solid (99%), from the title
compound of Preparation 41, using the procedure of Preparation 66. 8(CDCI3):
0 1.43 (t,3H), 1.85 (m,2H), 1.97 (m,2H), 2.28 (s,3H), 2.67 (m,1H), 3.01
(m,~H),
3.84 (s,3H), 3.98 (s,2H), 4.01 (m,2H), 4.05 (q,2H), 6.80-6.90 (m,3H), 7.05-
7.34
(m,4H).
LRMS (Thermospray): 432 (M+H)''.
pREPARATI~ON 83
Obtained as a colourless gum (91 %), from the title compound of
Preparation 82 and iodomethane, using the procedure of Preparation 40.
8(CDCI3): 1.42 (t,3H), 1.67 (s,6H), 1.80-1.95 (m,4H), 2.29 (s,3H), 2.67
{m,1H),
3.10 (m,2H), 3.82 (s,3H), 3.97 (m,2H), 4.06 (q,2H), 6.82-6.90 (m,3H), 7.06-
7.35
(m,4H).
LRMS (Thennospray): 460 (M+H)+.
p~REPARATlOI~ 84
Obtained as a colourless solid (72%), m.p. 125-128°C, from the
title
CA 02312935 2000-06-02
wo ~n9~~ rcr~~o~
-93-
compound of Preparation 83, using the procedure of Preparation 79, except
that the crude product was flash chromatographed using methanol
dichloromethane (3:97), before crystallisation from diisopropyl ether. Found:
C,64.14; H, 7.01; N, 3.06. C2,,H3~NO5S requires C, 64.69; H, 7.01; N, 3.14%.
8(CDCI3): 1.41 (t,3H), 1.68 (s,6H), 1.77-1.97 (m,4H), 2.26 (s,3H), 2.66
(m,1H),
3.10 (m,2H), 4.00-4.10 (m,4H), 6.80-6.90 (m,3H), 7.03-7.35 (m,4H).
LRMS (APCI): 446 (M+H)+.
1o PREPARATION $5
Methyrl 2-(4-[3-methyl-4-(pyrid i n-2-yrl)i phenyrl]-1.2.3.6-tetra h yd
roRyrid i n-1-
A stirred solution of the title compound of Preparation37 (206 mg, 0.53
mmol), 2-(tri-n-butylstannyl)pyridine (Tetrahedron, 1997 ~, 859; 295 mg, 0.80
~5 mmol), tri-o-tolylphosphine (50 mg, 0.16 mmol), palladium(II) acetate (12
mg,
0.05 mmol) and triethylamine (0.2 ml, 1.44 mmol) in anhydrous acetonitrile (6
ml), under nitrogen, was heated under reflux for 6 hours. Additional portions
of
tri-o-tolylphosphine (50 mg, 0.16 mmol) and palladium(II) acetate (12 mg, 0.05
mmol) were added, then reflux continued for a further 24 hours. The resulting,
2o cool mixture was partitioned between ethyl acetate and aqueous sodium
bicarbonate solution, then the separated organic phase washed with water,
dried (MgS04) and evaporated under reduced pressure. The residue was
purified by flash chromatography, using an elution gradient of hexane:ethyl
acetate (100:0 to 55:45), to afford the title compound (20 mg, 10%) as a
25 colourless amorphous solid. 8(CDCI3): 2.39 (s,3H), 2.68 (m,2H), 3.65
(t,2H),
3.81 (s,3H), 4.02 (s,2H), 4.10 (m,2H), 6.10 (brs,1H), 7.23-7.30 (m,3H), 7.40
(m,2H), 7.75 (dd,1 H), 8.70 (d,1 H).
LRMS (APCI): 387 (M+H)+.
CA 02312935 2000-06-02
wo ~n~ss~ rcT~o
-94-
Obtained as a colourless foam (78%), from the title compound of
Preparation 37 and 3-(tri-n-butylstannyl}pyridine, using the procedure of
Preparation 85, except that the reaction time was 5 hours at reflux followed
by
72 hours at room temperature. Found: C, 62.07; H, 5.78; N, 7.09.
~20H22N2~4S requires C, 62.16; H, 5.74; N, 7.25%. 8(CDCl3): 2.30 (s,3H}, 2.70
(m,2H), 3.68 (t,2H), 3.83 (s,3H), 4.03 (s,2H), 4.14 {m,2H), 6.10 (brs,1H),
7.20-
7.40 (m,4H), 7.66 (d,lH), 8.60 {m,2H).
LRMS {APCI): 387 (IVI+H)''.
'S M~11YJ..2~(~(~J~t111!!=~Qy~.-.Y1)~t~h.~.D.)dI-1.2.3.6-tetrahydro~yrridin-1-
Obtained as a colourless foam (54%), from the title compound of
Preparation 37 and 4-(tri-n-butylstannyl)pyridine, using the procedure of
Preparation 85. Found: C, 61.98; H, 5.80; N, 7.13. C2oH~N204S requires C,
20 62.16; H, 5.74; N, 7.25%. 8(CDC13}: 2.30 (s,3H), 2.70 (m,2H), 3.64 (t,2H),
3.81
(s,3H), 4.02 (s,2H), 4.10 (m,2H), 6.10 (brs,1H), 7.20-7.30 (m,SH}, 8.66
(d,2H}.
LRMS (APCI): 387 (M+H)+.
25 A 2.5M solution of n-butyllithium in hexane (4.5 ml, 11.3 mmol) was
added to a stirred solution of 2-bromo-6-ethoxypyridine (Rec. Trav. chim.,
1965,
$4, 53; 2.1g, 11.3 mmol) in anhydrous ether (25 ml), under nitrogen, at about -
40°C. After about 20 minutes, tri-n-butyltin chloride (3.1 ml, 11.4
mmol) was
CA 02312935 2000-06-02
wo ~n~~ Pc~r~r9~oss~o
-95-
slowly added and, after a further 15 minutes, the reaction mixhrre was allowed
to warm to room temperature. The resulting mixture was quenched by the
addition of aqueous ammonium chloride solution, then the organic phase
separated, washed with water, dried (MgSO4) and evaporated under reduced
pressure. The residue was purified by flash chromatography, using an elution
gradient of pentane:dichloromethane (100:0 to 80:20), to give the title
compound (1.6 g, 34%) as a colourless oil. 8(CDCI3): 0.90 (t,9H), 1.08 (t,6H),
1.30-1.42 (m,9H), 1:58 (m,6H), 4.40 (q,2H), 6.53 (d,1H), 6.97 (d,1H), 7.39
o (dd,1 H).
~5 A stirred mixture of the title compounds of Preparation 40 (500 mg, 1.2
mmol) and Preparation 88 (745 mg, 1.8 mmol), tri-o-tolylphosphine (109 mg,
0.36 mmol), palladium(II) acetate (30 mg, 0.13 mmol),
tetrakis(triphenyfphosphine)palladium(0) (30 mg, 0.025mmo1), triethylamine
(0.45 ml, 3.2 mmol) and anhydrous acetonitrile (15 ml), under nitrogen, was
2o heated under reflux for 18 hours. The cool mixture was partitioned between
ethyl acetate and aqueous sodium bicarbonate solution, then the organic
phase separated, washed with water, dried (MgS04) and evaporated under
reduced pressure. The residue was purified by flash chromatography, using an
elution gradient of pentane:ether (95:5 to 80:20), to yield the title compound
25 (290 mg, 52%) as a colourless foam. 8(CDCI3): 1.40 (t,3H), 1.68 (s,6H),
2.46
(s,3H), 2.66 (m,2H), 3.63 (m,2H), 3.81 (s,3H), 4.12 (m,2H), 4.40 (q,2H), 6.07
(brs,1 H), 6.68 (d,1 H), 6.98 (d,1 H), 7.26 (m,2H), 7.40 (d,1 H), 7.60 (dd,1
H).
t_RMS (APCI): 459 (M+H)+.
CA 02312935 2000-06-02
wo 99n~u~ prrn~~s~o
_g6_
PREPARA'[I~Q
A stirred solution of the title compound of Preparation 89 (280 mg, 0.6
mmol) in methanol {12 ml) was hydrogenated at 345 kPa (50psi) pressure over
10% palladium on carbon {50 mg) for 18 hours, then the resulting mixture
filtered. The filtrate was evaporated under reduced pressure and the residue
purified by flash chromatography, using an elution gradient of pentane:ether
{90:10 to 70:30), to provide the title compound (70 mg, 25%) as a colourless
foam. s(CDCI3): 1.40 (t,3H), 1.67 (s,6H), 1.82 (m,2H), 1.89 (m,2H), 2.43
(s,3H), 2.67 (m,1H), 3.08 (m,2H), 3.81 (s,3H), 3.95 (brd,2H), 4.38 (q,2H),
6.67
(d,1 H), 6.96 (d,1 H), 7.10 (m,2H), 7.38 {d,1 H), 7.60 {dd,1 H).
LRMS (APCI): 461 (M+H)+.
A solution of the title compound of Preparation 91 (68 mg, 0.15 mmol) in
2o a mixture of 1,4-dioxan (2 ml) and 1M aqueous sodium hydroxide solution
(0.26
ml, 0.26 mmol) was stirred at room temperature for 18 hours. The resulting
solution was diluted with water (20 ml), acidified with glacial acetic acid to
pH
~4 and extracted with ethyl acetate. The extract was dried (MgS04) and
evaporated under reduced pressure to furnish the title compound (60 mg, 87%)
as a colourless solid, m.p. 178-179°C. Found: C, 61.53; H, 6.81; N,
6.09.
C23H~N2O5S requires C, 61.86; H, 6.77; N, 6.27%. S(CDCI3): 1.39 (t,3H), 1.68
(s,6H), 1.82 (m,2H), 1.90 (m,2H), 2.43 (s,3H), 2.67 (m,1H), 3.10 (m,2H), 4.00
(brd,2H), 4.38 (q,2H), 6.65 (d,1 H), 6.96 (d,1 H), 7.10 (m,2H), 7.38 (d,1 H),
7.60
{dd,1 H).
CA 02312935 2000-06-02
wo ~nx~~ rc~r~~srots~o
-97-
s Obtained as a colourless solid (67%), m.p. 203-206°C, from the title
compound of Preparation 8 and bis-2-iodoethyl ether, using the procedure of
Preparation 39. 8(CDCI3): 2.25 (m,2H), 2.44 (d,2H), 2.66 (m,2H), 3.32 (t,2H),
3.61 (m,2H}, 3.90 (s,3H), 4.01 (dd,2H}, 4.10 (m,2H), 6.08 (brs,1 H), 7.30-7.62
(m,9H).
1o LRMS (Thermospray): 442 (M+H)+.
~s Obtained as a colourless solid (66%), m.p. 214°C, from the title
compound of Preparation 92, using the procedure of Preparation 79. 8(CDCI3):
2.27 (m,2H), 2.42 (d,2H}, 2.66 (m,2H), 3.41 (t,2H), 3.62 (m,2H), 4.04 (dd,2H),
4.15 (m,2H}, 6.08 (brs,1H), 7.30-7.48 (m,SH}, 7.58 (m,4H).
LRMS (APCI}: 427 (M+H)+.
Obtained as a colourless solid (66%), 140-141 °C, from the title
compound of Preparation 39 and 4-ethoxyphenyfboronic acid, using the
procedure of Preparation 41, but with ethyl acetate:hexane (30:70) as eluant.
Found: C, 64.59; H, 6.60; N, 2.74. C2~H33NOgS requires C, 64.91; H, 6.66; N,
2.80%. b( DMSO~}: 1.34 (t,3H), 2.00 (m,2H}, 2.22 (s,3H), 2.28 (d,2H), 2.55
CA 02312935 2000-06-02
wo ~n~~ . Pcrn~~s~ot~to
-98-
(brs,2H), 3.19 (t,2H), 3.50 (brs,2H), 3.80 (s,3H), 3.90 (dd,2H), 3.99
(brs,2H),
4.06 (q,2H), 6.17 (brs,1 H), 6.96 (d,2H), 7.14 (d,1 H), 7.22 (d,2H), 7.28 (d,1
H),
7.33 (s,1 H).
LRMS (Thermospray): 500 (M+H)'".
4-{4-(4-(4-Ethoxy~~~,-3-methyrlpheny~]'-1.2.3.6-tetrah~~pyr 'din-1
yisulphonyr~J~tetra ~,rdropvran-4-carbo~rlic acid
~o Obtained as a colourless solid (56%), m.p. 212-214°C, from the title
compound of Preparation 94, using the procedure of Preparation 79. S(CDCI3):
1.34 (t,3H), 1.98 (m,2H), 2.22 (s,3H), 2.24 (d,2H), 2.55 (brs,2H), 3.19
(t,2H),
3.52 (brs,2H), 3.90 (dd,2H), 4.01 (brs,2H), 4.04 (q,2H), 6.17 (brs,1H), 6.96
(d,2H), 7.13 (d,1 H), 7.22 (d,2H), 7.28 (d,1 H), 7.33 (s,1 H).
~5 LRMS (Thermospray): 486 (M+H)+.
2o Obtained as a colourless gum {86%),from the title compound of
Preparation 37 and 3-formylphenylboronic acid, using the procedure of
Preparation 41, but with methanol:dichloromethane (1:99) as eluant. 8(CDCI3):
2.30 (s,3H), 2.70 (m,2H), 3.64 (t,2H), 3.82 (s,3H), 4.02 (s,2H), 4.12 (m,2H),
6.10 (brs,1H), 7.20-7.33 (m,3H), 7.60 (m,2H), 7.85 (m,2H), 10.08 (s,1H).
25 LRMS (APCI): 414 (M+H)''.
j~,~,~l~yrl2~4-(4-(3-h~oxymethyr_I~~henyl)~-3-methyr hen~~]-1.2.3.6
~Q~ra dropyrridin-1-vlsulphonyj}acetate
3o The title compound of Preparation 96 (303 mg, 0.73 mmol) was
CA 02312935 2000-06-02
wo ~n~s~ rc~r~r9sio~o
-99-
dissolved in a mixture of methanol (15 ml) and 1,2-dimethoxyethane (5 ml),
polymer supported borohydride on AmberliteTM IRA-400 (360 mg, 0.91 mmol)
added and the reaction mixture stirred for 3 hours at room temperature. The
resin was removed by filtration and the filtrate evaporated under reduced
pressure to afford the title compound as a colourless foam (270 mg; 90%).
8(CDCJ3): 2.30 (s,3H), 2.70 (m,2H), 3.64 (t,2H), 3.82 (s;3H), 4.02 (s,2H),
4.12
(m,2H), 4.77 (brs,2H), 6.10 (brs,1 H), 7.20-7.50 (m,7H).
LRMS {Thermospray): 416 (M+H)+.
A 2.5M solution of n-butyllithium in hexane (4.4 ml, 11 mmol) was slowly
added to a stirred solution of 3-bromoquinoline (2.08 g, 10 mmol) in anhydrous
ether (20 ml), under nitrogen, at -75°C. After a further 20 minutes at -
75°C,
trimethylborate {1.46 ml, 13 mmol) was added, whereupon the red colour
changed to yellow. The reaction mixture was allowed to warm to room
temperature and quenched with water, followed by 1M aqueous sodium
hydroxide solution (10 ml). The resulting mixture was stirred for 30 minutes
2o and then glacial acetic acid added until a pH ~5-6 was attained, which
generated a gummy precipitate. Diisopropyl ether was added to this mixture,
stirring continued for 1 hour and then the clear aqueous and organic phases
were decanted from the solid and discarded. The solid residue was dissolved in
ethyl acetate and the solution washed with water, dried (MgS04) and
evaporated under reduced pressure to give the title compound as a pale yellow
solid (580 mg, 34%). Found: C, 62.74; H, 4.11; N, 7.92. C9H8BN02 requires
C, 62.49; H, 4.66; N, 8.10%. b( DMSOds): 7.59 (t,1 H), 7.76 (t,1 H), 7.98
(m,2H),
8.42 (brs,2H,exchangeable), 8.70 (s,1 H), 9.18 (s,1 H).
CA 02312935 2000-06-02
wo ~n~s~ rc~r~~srs~o~o
-100-
Obtained as a colourless solid (82%), m.p. 149-151 °C, from the
title
compounds of Preparation 40 and Preparation 98, using the procedure of
Preparation 41, but using 25% 1-methylpyrrolidin-2-one in 1,2-
dimethoxyethane as the reaction solvent and ether:pentane (80:20) as eluant
for flash chromatography. Found: C, 67.02; H, 6.20; N, 5.78. C~H2gN2O4S
o requires C, 67.22; H, E.08; N, 6.03%. S(CDCI3): 1.67 (s,6H), 2.36 (s,3H),
2.67
(m,2H), 3.65 (m,2H), 3.82 (s,3H), 4.13 (m,2H), 6.10 (brs,1 H), 7.32 (m,3H),
7.60
(t,1 H), 7.75 (t,1 H), 7.87 (d,1 H), 8.10 (s,1 H), 8.16 (d,1 H), 8.93 (s,1 H).
LRMS (Thermospray): 465 (M+H)+.
PREPARATION 100
A solution of the title compound of Preparation 99 (500 mg, 1.08 mmol)
in a mixture of 1,4-dioxan (5 ml), methanol (10 ml) and 1 M aqueous sodium
2o hydroxide solution (3.2 ml, 3.2 mmol) was stirred under reflux for 30
minutes.
The resulting solution was allowed to cool, acidified with glacial acetic acid
to
pH ~4, diluted with water (15 ml) and the resulting mixture partially
evaporated
under reduced pressure until crystallisation occun-ed. The solid was collected
and dried to yield the title compound (440 mg, 90%) as a colourless solid,
m.p.
222-224°C. Found: C, 66.17; H, 5.77; N, 6.15. C25H~N2O4S requires C,
66.64; H, 65.82; N, 6.22%. b( DMSO~): 1.51 (s,6H), 2.31 (s,3H), 2.59 (m,2H),
3.58 (m,2H), 4.08 (m,2H), 6.26 (brs,1 H), 7.33-7.50 (m,3H), 7.65 (t,1 H), 7.78
(t,1 H), 8.05 (t,2H), 8.37 (s,1 H), 8.90 (s,1 H), 13.4 (brs,1 H).
LRMS (APCI): 451 (M+H);. ,
CA 02312935 2000-06-02
wo ~n~~ Pc~r~ns~o~o
-101
P~<~EPARATION 101
3-Met ~yrlthio~pyrlboronic acid
A solution of 3-bromothioanisoie (10.3 g, 50.9 mmoi) in anhydrous
tetrahydrofuran (15 ml) was added dropwise to a stirred mixture of magnesium
turnings (1.86 g, 75 mmol) and a crystal of iodine under nitrogen. Once the
reaction was initiated, the remainder of the solution was added at such a rate
as to keep the reaction mixture under reflux. When the addition was complete,
the mixture was stirred under reflux for a further 1 hour, allowed to cool to
room
o temperature and then added to a solution of trimethyl borate (5.8 ml, 51
mmol)
in anhydrous tetrahydrofuran (25 ml), whilst keeping the internal temperature
at
about -10°C. The reaction mixture was allowed to warm to about
0°C, stirred for
30 minutes and then quenched with 2M hydrochloric acid. The resulting mixture
was extracted with ether, then the combined extracts extracted, in turn, with
2M
~5 aqueous sodium hydroxide solution. The combined aqueous extracts were
acidified with concentrated hydrochloric acid and extracted with ether. The
combined ether extracts were dried (MgS04) and evaporated under redacted
pressure to provide the title compound (7.8 g, 100%) as a white solid.
8(DMSO~): 2.45 (s,3H), 7.27 (m,2H), 7.54 (m,1 H), 7.67 (s,1 H), 8.05 (brs,2H).
Obtained as a colourless solid (83%), from the title compounds of
Preparation 101 and Preparation 37, using the procedure of Preparation 41, but
using dichloromethane:hexane (3:1) as eluant. 8(CDCl3): 2.28 (s,3H), 2.50
(s,3H), 2.68 (m,2H), 3.64 (t,2H), 3.81 (s,3H), 4.02 (s,2H), 4.10 (m,2H), 6.08
(brs,1 H), 7.07 (d,1 H), 7.20-7.36 (m, 6H).
LRMS (APCI): 432 (M+H)+.
CA 02312935 2000-06-02
wo ~n~6~ pc~r~,p9s~o~eo
-102-
Obtained as a yellow solid (100%), from 1-bromo-3-
s methoxymethylbenzene (J. Amer. Chem. Soc., 1989,11x.., 6311; Tetrahedron
1985, 41, 1435) and trimethyl borate, using the procedure of Preparation 101.
8(DMSO~): 3.27 (s,3H}, 4.38 (s,2H), 7.31 (m,2H), 7.68 (m,2H}, 7.98 (brs,2H).
o J'~"°~h1!L2_~4 (4_!(~1~11t~h~D.YI,-~-~~Athvlohenvll-1.2.3.6-
Obtained as a colourless oil (35%), from the title compounds of
Preparation 103 and Preparation 37, using the procedure of Preparation 41, but
using ether:hexane (60:40) as eluant. S(CDCI3): 2.27 (s,3H), 2.68 (m,2H), 3.42
~5 (s,3H), 3.64 (t,2H), 3.81 (s,3H), 4.02 (s,2H), 4.10 (m,2H}, 4.51 (s,2H),
6.08
(brs,1H), 7.20-7.32 (m, 7H}.
LRMS (Thermospray): 430 (M+H)+.
20 1-Bromo-3-( -m tho i to hoxy)benzene
Anhydrous potassium carbonate (4.2 g, 30.4 mmol) was added to a
stirred solution of 3-bromophenol (5.0 g, 28.9 mmol) in anhydrous
dimethylformamide (100 ml). After 5 minutes, 1-iodo-2-methoxyethane
(Annalen, 1967, ~Q, 59; 5.9 g, 31.8 mmol} was added and the reaction mixture
25 stirred at room temperature for about 16 hours. At this point the mixture
was
heated at about 50°C for approximately 72 hours, before 1-chloro-2-
methoxyethane (1.8 ml, 19.8 mmol) was added and heating continued for a
further 24 hours. The resulting mixture was evaporated ~rnder reduced pressure
and the residue partitioned between ethyl acetate and water. The layers were
CA 02312935 2000-06-02
WO 99/29667 PCT/EP98~6640
-103-
separated and the aqueous layer was further extracted with ethyl acetate. The
combined organic phases were washed with saturated aqueous sodium
chloride solution, dried (MgS04) and concentrated to a red oil, which was
s purified by flash chromatography using hexane:ethyl acetate (3:1 ) as
eluant, to
furnish the title compound as a colourless oil (1.7 g, 25%). 8(CDCI3): 3.43
(s,3H), 3:74 (t,2H), 4.10 (t,2H), 6.87 (d,1H), 7.10 (m,3H)
~2-Methoxy eti hoxy) hoe yjboronic acid
Obtained as a colourless solid (74%), m.p. 101-103°C, from the
title
compound of Preparation 105 and trimethyl borate, using the procedure of
Preparation 101. Found: C, 55.09; H, 6.70. C9H~3B04 requires C, 55.15; H,
6.69%. 8(DMSO~): 3.30 (s,3H), 3.63 (t,2H), 4.06 (t,2H), 6.94 {dd,1H), 7.22
~ 5 (t,1 H), 7.32 (m,2H), 7.98 (brs,2H).
LRMS (Thermospray): 214 (M+NH4)+.
20 tetrah~Lolwridin-1-yrlsul~~~yl}acetate
Obtained as a colourless oil (59%), from the title compounds of
Preparation 106 and Preparation 37, using the procedure of Preparation 41, but
using ether:hexane {1:1) as eluant. 8(CDCI3): 2.28 {s,3H), 2.67 (m,2H), 3.45
{s,3H), 3.63 (t,2H), 3.77 (t,2H), 3.80 (s,3H), 4.02 (s,2H), 4.09 {s,2H), 4.15
2s (s,2H), 6.07 {brs,1H), 6.90 {m,3H), 7.19-7.34 (m, 4H).
Obtained as a colourless solid (38%), m.p. >240°C (decomp.), from
5-
CA 02312935 2000-06-02
WO 99/Z9667 PCT/EP98~6640
-104-
bromo-2,3-dihydrobenzofuran (Synthesis, 1988, 952) and trimethyl borate,
using the procedure of Preparation 101. S(DMSO~): 3.33 {t,2H), 4.48 (t,2H),
6.68 (d,1 H), 7.56 (d,1 H), 7.63 (s,1 H), 7.70 {brs,2H).
5-(4-Bromo-2-methy~~~)2..3-di~ydrobenzofuran
The title compound of Preparation 108 (2.0 g, 12.2 mmol) was added
portionwise over 5 minutes to a stirred mixture of the title compound of
1G Preparation 57 (3.4 g, 12.0 mmol) and palladium(II) acetate (0.15 g, 0.6
mmol)
in anhydrous methanol (30 ml) and the reaction mixture heated under reflux for
1.5 hours. The resulting mixture was allowed to cool to room temperature,
filtered, then the filtrate diluted with water (100 ml) and extracted with
ether (2 x
100 ml). The combined extracts were dried (MgSO,,) and evaporated under
reduced pressure, then the residue purified by flash chromatography, using
hexane as eluant, to afford the title compound (1.7 g) as a pale orange oil.
8(CDCI3): 2.25 (s,3H), 3.25 (t,2H), 4.62 (t,2H), 6.83 (d,1 H), 7.02 (d,1 H),
7.09
(m,2H), 7.34 (d, 1 H), 7.60 (s,1 H).
LRMS (APCI): 289 (M)+.
Obtained as a white solid (66%), from the title compound of Preparation
109 and t-butyl 4-oxopiperidine-1-carboxylate, using the procedure of
Preparation 43. 8(CDCI3): 1.50 (s,9H), 1.78 (m,2H), 2,06 (m,2H), 2.29 (s,3H),
3.26 (m,4H), 4.05 (m,2H), 4.62 (t,2H), 6.82 (d,1 H), 7.05 (d,1 H), 7.15 (s,1
H),
7.21 (d, 1 H), 7.30 (m,1 H), 7.36 (s,1 H).
LRMS (Thermospray): 432 (M+Na)+.
CA 02312935 2000-06-02
WO 99/29667 p~yEp9g/p(~p
-105-
Obtained as a colourless viscous oil (98%), from the title compound of
Preparation 110 and trifluoroacetic acid, using the procedure of Preparation
44.
8(CDCI3): 2.30 (s,3H), 2.49 (m,2H), 3.12 (t,2H), 3.26 (t,2H), 3.56 (s,2H),
4.62
(t,2H), 6.17 (brs,1 H), 6.82 (d,1 H), 7.07 (d,1 H), 7.16(m,2H), 7.26 (m,2H).
LRMS (Thermospray): 292 (M+H)+.
1o PREPARATION 112
Obtained as a pale yellow, foamy solid (45%), m.p. 118-122°C, from
the
title compound of Preparation 111 and methyl chlorosulphonylacetate, using
~ 5 the procedure of Preparation 61. 8(CDCI3): 2.30 (s,3H), 2.67 (m,2H), 3.24
(t,2H), 3.62 (t,2H), 3.79 (s,3H), 4.01 (s,2H), 4.06 (s,2H), 4.62 (t,2H), 6.08
(brs,1 H), 6.82 (d,1 H), 7.05 (d,1 H), 7.26 (m,4H).
LRMS (Thermospray): 428 (M+H)+.
2o PREPARATION 113
Obtained as a colourless oil (66%), from the title compound of
Preparation 57 and 3-tr'~fluoromethylphenylboronic acid, using the procedure
of
Preparation 109. 8(CDCI3): 2.23 (s,3H), 7.08 (d,1H), 7.40-7.64 (m, 6H).
Obtained as a yellow oil (54%), from the title compound of Preparation
CA 02312935 2000-06-02
wo 99n~~ rc~r~o
-10fi-
113 and t-butyl 4-oxopiperidine-1-carboxylate, using the procedure of
Preparation 43, but with hexane:ethyl acetate (85:15) as eluant. 8(CDCI3):
1.50 (s,9H), 1.77 (m,2H), 2.04 (m,2H), 2.27 (s,3H), 3.26 (m,2H), 4.05 (m,2H),
7.04 (d,1 H), 7:15 (s,1 H), 7.37 (m,2H), 7.55 (m,4H}.
LRMS (Thermospray): 436 (M+H)+.
o Obtained as a colourless oil (98%), from the title compound of
Preparation 114 and trifluoroacetic acid, using the procedure of Preparation
44.
Found: C, 70.90; H, 5.84; N, 4.38. C~9H~8F3N; 0.25 H20 requires C, 70.90; H,
5.79; N, 4.35%. 8(CDCl3): 2.26 (s,3H), 2.49 (brs,2H), 3.12 (brs,2H), 3.56
(brs,2H), 6.20 (brs,1 H), 7.18 (d,1 H), 7.26 (m,2H), 7.55 (m,4H).
~5 LRMS (APCI): 318 (M)+.
Methyr~..~~(~-methyrt-4-(3-trifluoromethyrl~hernyrl)yheny ~]-1.2.3.6
etrahxdropyrridin-~-yl~~~~)acetate
2o Obtained as a pale yellow foam (31%), from the title compound of
Preparation 115 and methyl chlorosulphonylacetate, using the procedure of
Preparation 61, but with dichloromethane:hexane (80:20) as eluant. Found: C,
56.38; H, 4.77; N, 2.96. C~H~F3N04S; 0.25 CH2CI2 requires C, 56.30; H, 4.78;
N, 2.95%. 8(CDCI3): 2.27 (s,3H), 2.70 (m,2H), 3.64 (t,2H}, 3.82 (s,3H), 4.03
25 (s,2H), 4.11 (s,2H), 6.10 (brs,1 H), 7.24 (m,3H}, 7.55 (m,4H).
LRMS (APCI): 453 (M)+.
t~yl4-h ro -4-(4- eno rphenyl)~pir~eridine-1-carboxvlate
3o Obtained as a white foam (54%), from 4-phenoxybromobenzene and t-
CA 02312935 2000-06-02
wo ~n9~~ rc~r~r9sross4o
-107-
butyl 4-oxopiperidine-1-carboxylate, using the procedure of Preparation 43,
but
with a mixture of anhydrous ether and anhydrous tetrahydrofuran as solvent
and ether:hexane (60:40) as eluant. S(CDCI3): 1.50 (s,9H), 1.75 (m,2H), 1.99
(m,2H), 3.25 (m,2H), 4.04 (m,2H), 7.00 (m,4H), 7.12 (t,1H), 7.37 (t,2H), 7.44
(d,2H).
Triethylsilane (3.0 ml, 18.9 mmol) was added to a stirred solution of the
title compound of Preparation 117 (775 mg, 2.1 mmol) in anhydrous
dichloromethane (10 ml), the resulting solution was cooled to about 0°C
and
then trifluoroacetic acid (10 ml) was slowly added. The reaction mixture was
allowed to warm to room temperature and then stirred for about 1.5 hours. The
~ 5 resulting mixture was evaporated under reduced pressure, then the residue
dissolved in methanol and this solution basified with 2M aqueous sodium
hydroxide solution. The mixture was extracted with ethyl acetate and the
combined extracts dried (MgS04) and evaporated under reduced pressure.
This residue was dissolved in glacial acetic acid (20 ml) and the solution
2o hydrogenated over palladium on carbon (60 mg) at 345 kPa (50psi) and room
temperature for 2 hours. The mixture was filtered, the filtrate evaporated
under
reduced pressure and the residue dissolved in methanol. This solution was
then basified with 2M aqueous sodium hydroxide solution, extracted with ethyl
acetate and the combined extracts dried (Na2S04) and evaporated under
25 reduced pressure to give the title compound as a yellow oil (550 mg, 100%).
8(CDCl3): 1.63 (m,2H), 1.84 (m,2H), 2.60 (m,1 H), 2.74 (t,2H), 3.20 (m,2H),
6.95
(d,2H), 7.00 (d,2H), 7.07 (t,1 H), 7.18 (d,2H), 7.33 (m,2H).
LRMS (Thermospray): 254 (M+H)''.
CA 02312935 2000-06-02
wo ~rn~~ pcr~~orou~o
-108-
Obtained as a colourless solid (38%), from the title compound of
Preparation 118 and methyl chlorosulphonylacetate, using the procedure of
Preparation 45. 8(CDCI3): 1.80 {m,2H), 1.94 (m,2H), 2.64 (m,1 H), 3.00 (t,2H),
3.83 (s,3H), 3.95 (m,4H), 6.95 (m,4H), 7.10 (t,1 H), 7.16 (d,2H), 7.33 (m,2H).
LRMS (Thermospray): 407 (M+NH4)+.
~o PREPARATION 120
Obtained as a pale yellow oil (97%), from the title compounds of
Preparation 106 and Preparation 40, using the procedure of Preparation 41, but
with ethyl acetate:hexane (1:3) as eluant. 8(CDCI3): 1.67 (s,6H), 2.28 (s,3H),
2.65 (m,2H), 3.45 (s,3H), 3.62 (m,2H), 3.76 (m,2H), 3.80 (s,3H), 4.13 (m,4H),
6.06 (brs,1H), 6.90 {m,3H), 7.19-7.35 (m,4H).
Obtained as a pale yellow oil (83%), from the title compound of
Preparation 120, using the procedure of Preparation 90. 8(CDCI3): 1.66 (s,6H),
1.78-1.88 (m,4H), 2.27 {s,3H), 2.68 (m,1H), 3.09 (m,2H), 3.45 (s,3H), 3.77
(t,2H), 3.81 (s,3H), 3.9fi (d,2H), 4.15 (t,2H), 6.90 (m,3H), 7.10 (m,2H), 7.18
(d,1 H), 7.30 (t,1 H).
LRMS (Thermospray): 490 (M+H)'.
CA 02312935 2000-06-02
wo ~nx~~ Pcr~~s~os~o
-109-
Obtained as a colourless solid (78%), m.p. 140-141 °C, from the
title
compound of Preparation 121, using the procedure of Preparation 79. Found:
C, 62.89; H, 7.06; N, 2.85. C25HssNOgS requires C, 63.14; H, 6.99; N, 2.95%.
8(CDC13): 1.68 (s,6H), 1.78-1.88 (m,4H), 2.27 (s,3H), 2.68 (m,1H), 3.11
(m,2H),
3.45 (s,3H), 3.77 (t,2H), 4.00 (d,2H), 4.15 (t,2H), 6.90 (m,3H), 7.10 (m,2H),
7.18
~o (d,1H), 7.30 (t,1H).
LRMS (Thermospray): 475 (M+H)+.
tetra rdroQyrridin-1-yl~ulphonyl)butanoate
60% Sodium hydride dispersion in mineral oil (23 mg, 0.57 mmol) was
added to a stirred solution of the title compound of Preparation 9 (200 mg,
0.52
mmol) in anhydrous 1-methylpyrrolidin-2-one (3 ml), under nitrogen, at room
temperature. After 30 minutes, 1-iodo-2-methoxyethane (101 mg, 0.57 mmol)
2o was added and stirring continued for a further 16 hours, then the resulting
mixture was partitioned between ethyl acetate and water The organic phase
was separated, washed with brine, dried (MgS04) and evaporated under
reduced pressure. The residue was purled by flash chromatography, using
dichloromethane as eluant, followed by trituration with diisopropyl ether, to
yield
the title compound (148 mg) as a colourless solid, m.p. 95-96°C. S
(CDCI3):
2.28 (s,3H), 2.39 (m,2H), 2.67 (m,2H), 3.30 (s,3H), 3.40 (m,1H), 3.54 (m,2H),
3.67 (m,1 H), 3.80 (s,3H), 4.10 (brs,2H), 4.17 (dd,1 H), 6.07 (brs, 1 H), 7.22
(m,3H), 7.32 (m,3H), 7.41 (m,2H).
LRMS (APCI): 444 (M+H)+.
CA 02312935 2000-06-02
WO 99/29667 PCT/EP98J06640
-110
PREPARATION 124
s 1 M Aqueous sodium hydroxide solution (1.0 ml, 1.0 mmol) was added to
a stirred solution of the title compound of Preparation 123 (148 mg, 0.33
mmol)
in a mixture of methanol (5 ml) and 1,4-dioxan (2 ml). The resulting solution
was heated at 80°C for about 4 hours, then allowed to cool to room
temperature and evaporated under reduced pressure. The residue was diluted
o with water, then the resulting mixture acidified with glacial acetic acid
and
extracted with ethyl acetate. The combined organic phases were dried
(MgS04) and evaporated under reduced pressure to provide the title compound
(145 mg) as a colourless solid, m.p. 108-109°C. b (CDC13): 2.28 (s,3H),
2.39
(rn,2H), 2.67 (m,2H), 3.36 (s,3H), 3.53-3.73 (m,SH), 4.12 (brs,2H), 4.20
(dd,1H),
~ 5 6.07 (brs, 1 H), 7.19-7.47 (m,BH).
ARMS (APCI): 429 (M+H)+.
60% Sodium hydride dispersion in mineral oil (34 mg, 0.86 mmol) was
added to a stirred solution of the title compound of Preparation 9 (300 mg,
0.78
mmol) in anhydrous 1-methylpyrrolidin-2-one (3 ml), under nitrogen, at room
temperature. After 30 minutes, bis-2-iodoethyl ether (380 rng, 0.78 mmol) was
2s added and stirring continued for a further 4 hours, then more 60% sodium
hydride dispersion in mineral oil (34 mg, 0.86 mmol) was added and the mixture
stirred for a further 16 hours. The resulting mixture was partitioned between
ethyl acetate and water, then the organic phase separated, washed with water,
dried (MgS04) and evaporated under reduced pressure. The residue was
CA 02312935 2000-06-02
wo ~nx~~ p,c.-r~~8
-111-
purled by crystallisation from diisopropyl ether to furnish the title compound
(188 mg) as a colourless solid, m.p. 117-119°C. Found: C,65.70; H,6.44;
N,2.98. C25H2sNOsS requires C,fi5.91; H,6.42; N,3.08%. 8(CDCI3): 2.22
(m,2H), 2.29 (s,3H), 2.47 (m,2H), 2.64 (m,2H), 3.33 (t,2H), 3.60 {m,2H), 3.8?
(s,3H), 4.00 (dd,2H), 4.10 (m,2H}, 6.07 (brs,1H), 7.20-7.43 (m,BH).
LRMS (Thermospray): 456 (M+H)+.
't0 4-[4-{3-Methyrl-4-phenyrlRhenyl~~-1.2,3.6-tetra ~yrdroQyrridin-1-
ylsy honyr[ltetra vd~pyrar~~-4-carbr~~riic acid
1 M Aqueous sodium hydroxide solution (1.4 ml, 1.4 mmol) was added to
a stirred solution of the title compound of Preparation 125 (160 mg, 0.35
mmol)
in a mixture of methanol (5 ml) and 1,4-dioxan (2 ml). The resulting solution
~5 was heated at 80°C for 4 hours, then allowed to cool to room
temperature,
diluted with water and concentrated under reduced pressure. The resulting
mixture was acidified with 1 M hydrochloric acid and the precipitate thus
obtained was collected, washed with water and dried to afford the title
compound (135 mg) as a colourless solid, m.p. 211-213°C. Found:
C,64.89;
2o H,6.14; N,3.07. C24HZ7NOsS requires C,65.28; H,6.14; N,3.17%. 8(DMSO~):
1.96 (m,2H), 2.03 (s,3H), 2.10 (m,2H), 2.53 (m,2H), 3.23 (t,2H), 3.54 (m,2H),
3.92 (dd,2H), 4.03 (m,2H), 6.18 (brs,1H), 7.16 (d,1H), 7.28-7.43 (m,7H), 13.7
(brs,1 H).
LRMS (APCI): 442 (M+H)''. ,
~f4-l3-Methoxvnhenyrl)~-3-methy~,~yrl]~~y eridine
Obtained as a pink oil (74%), from the title compound of Preparation 59,
CA 02312935 2000-06-02
wo ~nx~~ pc~r~o
-112-
using the procedure of Preparation 118. 8(CDCI3): 1.67 {m,2H), 1.88 (m,2H),
2.29 (s,3H), 2.63 (m,1H), 2.57 (t,2H), 3.22 (m,2H), 3.83 (s,3H), 6.89 (m,3H),
7.10 (m,2H), 7.18 (d,1 H), 7.32 (t,1 H).
LRMS (Thermospray): 282 (M+H)''.
Obtained as a colourless solid (47%), m.p. 96-98°C, from the title
1o compound of Preparation 127 and methyl chlorosulphonylacetate, using the
procedure of Preparation 45. 8(CDCI3): 1.83 (m,2H), 1.96 (m,2H), 2.28 (s,3H),
2.63 (m,1H), 3.02 (t,2H), 3.83 (s,6H), 4.01 (m,4H), 6.89 (m,3H), 7.10 (m,2H),
7.20 {d,1 H), 7.34 (t,1 H).
LRMS (Thermospray): 418 (M+H)+.
1,2-Di{bromomethyl)benzene (409 mg, 1.55 mmol) was added to a
2o stirred mixture of the title compound of Preparation 128 (500 mg, 1.2 mmol)
and
anhydrous potassium carbonate (497 mg, 3.6 mmol) in anhydrous 1,2-
dimethoxyethane (5 ml) and the resulting mixture stirred at room temperature
for 17 hours. Little reaction had occurred, so the solvent was evaporated
under
reduced pressure and the residue dissolved in 1-methylpyrrolidin-2-one (5 ml)
and the solution heated at 100°C for 2 hours. This mixture was allowed
to cool
to room temperature, partitioned between ether and water, then the organic
phase washed with water, dried (MgS04) and evaporated under reduced
pressure to give a yellow oil. Purification by flash chromatography, using an
CA 02312935 2000-06-02
wo ~n9ss~ pc~r~o
-113-
elution gradient of pentane:ethyl acetate (10:1 to 3:1 ), yielded the title
compound as a white crystalline solid (160 mg), m.p. 174-176°C. Found:
C,68.95; H,6.48; N,2.56. C~H33N05S requires C,69.34; H,6.40; N,2.70%.
8(CDCI3): 1.74 (m,2H), 1.85 (m,2H), 2.27 (s,3H), 2.57 (m,1H), 2.89 (t,2H),
3.73-
3.86 (m,4H), 3.83 (s,6H), 3.96 (m,2H), 6.87 (m,3H), 7.06 (m,2H), 7.18-7.33
(m,6H).
LRMS (Thermospray): 520 (M+H)+.
1o PREPARATION 130
Obtained as a colourless solid (83%), m.p. 204-206°C, from the
title
compound of Preparation 129, using the procedure of Preparation 79. Found:
~5 C,68.17; H,6.22; N,2.74. C~H31N05S; 0.30 HZO requires C,68.16; H,6.23;
N,2.74%. 8(DMSO~): 1.54 (m,2H), 1.76 (m,2H), 2.21 (s,3H), 2.57 (m,1H),
2.89 (t,2H), 3.55 (d,2H), 3.72 (d,2H), 3.77 (s,3H), 3.81 (m,2H), 6.87 (m,3H),
7.07 (m,3H), 7.19 (m,2H), 7.28 (m,3H), 13.65 (brs,1H).
LRMS (Thermospray): 520 (M+NH4)+.
1,3-Diiodopropane (513 mg, 1.73 mmol) was added to a stirred mixture
of the title compound of Preparation 128 (557 mg, 1.33 mmol), anhydrous
potassium carbonate (553 mg, 4.0 mmol) and anhydrous 1,2-dimethoxyethane
(8 ml), then the mixture was stirred at room temperature for 17 hours and at
reflux for 72 hours. The resulting mixture was allowed to cool to room
CA 02312935 2000-06-02
wo ~n~6~ rcr~r~rsios~eo
-114-
temperature and partitioned between ethyl acetate and water, then the organic
phase dried (MgS04) and evaporated under reduced pressure to give a yellow
oil. Purification by flash chromatography, using pentane:ethyi acetate (3:1)
as
eluant, provided the title compound (472 mg) as a white crystalline solid,
m.p.
97-101°C. 8(CDCl3): 1.75-2.02 (m,SH), 2.13 (m,1H), 2.28 (s,3H), 2.59-
2.75
(m,3H), 2.90 {m,2H), 3.00 {t,2H), 3.82 (s,3H), 3.87 (s,3H), 3.93 {m,2H), 6.87
(m,3H), 7.06 (m,2H), 7.18 (d,1 H), 7.32 (t,1 H).
LRMS (Thermospray}: 458 (M+H)+.
Obtained as a colourless solid (100%), m.p. 155-160°C, from the
title
compound of Preparation 131, using the procedure of Preparation 79.
8(CDCI3}: 1.80 (m,2H), 1.91 (m,2H), 2.12 (m,2H), 2.27 (s,3H), 2.62 {m,1H),
2.74 (m,2H), 2.91 (m,2H), 3.04 (t,2H), 3.82 {s,3H), 3.99 {m,2H), 6.87 (m,3H),
7.06 (m,2H), 7.18 (d,1 H), 7.32 (t,1 H).
LRMS (Thermospray): 444 {M+H)+.
The title compound of Preparation 128 (500 mg, '! .2 mmol) and
anhydrous potassium carbonate (553 mg, 4.0 mmol), followed by anhydrous
1,2-dimethoxyethane (8 ml), were added to N-methyl-bis{2-chloroethyl)amine
hydrochloride {231 mg, 1.2 mmol) and the mixture was heated under reflux for
48 hours. The resulting mixture was allowed to cool to room temperature,
diluted with ethyl acetate, washed with 5% aqueous citric acid solution, dried
CA 02312935 2000-06-02
wo ~n~s~ Pc~r~po
-115-
(MgS04) and evaporated under reduced pressure to give a yellow oil. The
residual oil was dissolved in ethyl acetate and the solution washed
successively
with aqueous sodium bicarbonate solution/aqueous sodium hydroxide solution
(pH 12) and aqueous sodium chloride solution, then dried (MgS04) and
evaporated under reduced pressure to furnish the title compound as a yellow
gum (175 mg). 8(CDCI3): 1.87 (m,6H), 2.20 (m,2H), 2.25 (s,3H), 2.27 (s,3H),
2.53 (m,2H), 2.66 (m,1H), 2.90 (m,2H), 3.07 (t,2H), 3.82 (s,3H), 3.88 (s,3H),
3.93 (m,2H), 6.88 (m,3H), 7.08 (m,2H), 7.20 (d,1 H), 7.32 (t,1 H).
LRMS (Thermospray): 501 (M+H)+.
~-(~~~-(~; AllethoxyRheny~l)-3-methylr~hen)L~ia~heridin-1-yrlsul~yl)~-1_
methylpioeridine-4sarbo~yrh acid hyrdrochloride
1 M Aqueous sodium hydroxide solution (1.4 ml, 1.4 mmol) was added to
a stirred solution of the title compound of Preparation 133 (172 mg, 0.34
mmol)
in a mixture of methanol (5 ml) and 1,4-dioxan (3 ml). The reaction solution
was heated at 80°C for 10 hours, then allowed to cool to room
temperature and
concentrated under reduced pressure. The resulting mixture was acidified with
1 M hydrochloric acid, washed with dichloromethane and evaporated under
reduced pressure, then the residue was washed with water to afford the title
compound (143 mg) as a colourless solid. 8(CDCI3): 1.67 (m,2H), 1.84 (m,2H),
2.20 (s,3H), 2.30 (m,2H), 2.62-2.90 (m,7H), 3.12 (t,2H), 3.48 (m,2H), 3.77
(brs,6H), 6.88 (m,3H), 7.10 (brs,2H), 7.18 (brs,1 H), 7.32 (t,1 H).
LRMS (Thermospray): 523 (M+HCI)+.
60% Sodium hydride dispersion in mineral oil (34 mg, 0.77 mmol) was
CA 02312935 2000-06-02
wo ~n966~ rcr~r9sro6fao
-116-
added to a stirred solution of the title compound of Preparation 8 (288 mg,
0.77
mmol) in anhydrous dimethylformamide (5 ml), under nitrogen, at room
temperature. After 30 minutes, benzyt bromide (0.1 ml, 0.82 mmol) was added
and stirring continued for a further 16 hours, then the resulting mixture was
partitioned between ethyl acetate and water The organic phase was separated
and the aqueous phase washed with ethyl acetate. The combined organic
solutions were washed sequentially with water and aqueous sodium chloride
solution, dried (MgS04) and evaporated under reduced pressure. The resulting
residue was triturated with diisopropyl ether to give the title compound (170
mg)
as a colourless solid, m.p. 137-138°C. b (CDCI3): 2.68 (m,2H), 3.42
(m,2H),
3.59 {m,1 H), 3.67 (s,3H), 3.72 (m,1 H), 4.14 (brs,2H), 4.21 (dd,1 H), 6.10
(brs,1H), 7.18-7.37 {m,6H), 7.44 (m,4H), 7.59 (m,4H).
LRMS (Thermospray): 462 {M+H)+.
yrlsulphony~Jplo~aanoic acid
Obtained as a colourless solid (50%), m.p. 164-165°C, from the
tile
2o compound of Preparation 135, using the procedure of Preparation 79. 8
(CDCI3): 2.68 (m,2H), 3.41 (m,2H), 3.60 (m,1 H), 3.72 (m,1 H), 4.14 (brs,2H),
4.24 (dd,1 H), 6.08 (brs,1 H), 7.20-7.37 (m,6H), 7.43 (m,4H), 7.59 (m,4H).
LRMS {APCI): 447 (M+H);.
PREPARATION 137
60% Sodium hydride dispersion in mineral oil (30 mg, 0.73 mmol) was
added to a stirred solution of the title compound of Preparation 8 (250 mg,
0.67
CA 02312935 2000-06-02
wo ~n~~ ~cr~r9sios~ao
-117-
mmol) in anhydrous dimethylformamide (5 ml), under nitrogen, at room
temperature. After 30 minutes, 1,2-di(bromomethyl)benzene (267 mg, 1.0
mmol) was added and stirring continued for a further 16 hours. Next, an
s additional quantity of sodium hydride dispersion in mineral oil (30 mg, 0.73
mmol) was added and the reaction mixture stirred at room temperature for a
further 2 hours. The resulting mixture was partitioned between ethyl acetate
and water, the organic phase was separated and the aqueous phase was
extracted with ethyl acetate. The combined organic solutions were washed
successively with water and aqueous sodium chloride solution, dried (MgS04)
and evaporated under reduced pressure, then the residue purified by flash
chromatography, using pentane:ether (3:1 ) as eluant, followed by trituration
with diisopropyl ether, to yield the title compound (154 mg) as a colourless
solid, m.p. 186-188°C. b (CDCI3): 2.60 (m,2H), 3.56 (m,2H), 3.75-3.88
(m,4H),
~5 3.82 {s,3H), 4.07 (brs,2H), 6.05 (brs,1H), 7.19-7.28 (rn,4H), 7.34-7.45
(m,SH),
7.59 {m,4H).
LRMS (APCI): 474 (M+H)'.
20 ?~j4-y4-Phen henyl)-1.2.3,6-tetrahyrdro~yridin-1 yrIsuIRhon~~[]' ane-2-
Obtained as a colourless solid (67%), from the title compound of
Preparation 137, using the procedure of Preparation 50, except that the
residue
was triturated with diisopropyl ether. 8 (CDCI3): 2.60 (m,2H), 3.56-3.84
(m,6H),
25 4.07 (brs,2H), 6.05 (brs,1H), 7.19-7.60(m,l3H).
4-(3-Chloro-4-fluorol,~~~)-~ imethylbromobenzene
Obtained as a colourless oil (20%), from the title compound of
CA 02312935 2000-06-02
wo ~n~s~s~ Pcrn~w~s~s~o
-118-
Preparation 57 and 3-chloro-4-fluorophenylboronic acid, using the procedure of
Preparation 109. S(CDCI3): 2.22 (s,3H), 7.04 (d,1 H), 7.10-7.20 (m,2H), 7.28-
7.39 (m,2H), 7.42 (s,1 H).
Obtained as a colourless gum (39%), from the title compound of
Preparation 139 and t-butyl 4-oxopiperidine-1-carboxylate, using the procedure
of Preparation 43. Found: C, 65.96; H, 6.64; N, 3.36. C23HZ~CIFNOa requires
C, 65.79; H, 6.48; N, 3.34%. 8(CDCI3): 1.50 (s,9H), 1.76 (m,2H), 2.04 (m,2H),
2.28 (s,3H). 3.2$ (t,2H), 4.07 (m,2H), 7.16-7.20 (m,3H), 7.30-7.40 {m,3H).
LRMS (APCI): 420 (M+H)+.
Obtained as a pale yellow oil {99%), from the title compound of
2o Preparation 140 and trifluoroacetic acid, using the procedure of
Preparation 35.
8(CDCI3): 2.26 (s,3H), 2.50 (m,2H), 3.12 (t,2H), 3.5fi (m,2H), 6.16 (brs,1 H),
7.15 (m,3H), 7.27 (m,2H), 7.35 (d,1 H).
LRMS (APCI): 302 (M+H)+.
PREPARATION 142
~arahvd ro Ryrrid i n-1-ylsul r~honyrl~ acetate
Obtained as a colourless solid (3?%), m.p. 125-126°C, from the
title
CA 02312935 2000-06-02
WO 99129667 PCT/EP98/Ob640
-119-
compound of Preparation 141 and methyl chlorosulphonylacetate, using the
procedure of. Preparation 61. Found: C, 57.46; H, 4.83; N, 3.14.
C2~H2~CIFN04S requires C, 57.60; H, 4.83; N, 3.20%. 8(CDCI3): 2.28 (s,3H),
2.67 {m,2H), 3.62 (t,2H), 3.80 {s,3H), 4.01 (s,2H), 4.06 (s,2H), 6.08
(brs,1H),
7.17 (m,3H), 7.23 (m,2H), 7.36 (d,1H).
LRMS (Thermospray): 438 (M+H)+.
~ l1.,~Benz~dioxol-5-yl)-3-methy~lbromobenzene
Obtained as a colourless oil (34%), from the title compound of
Preparation 57 and 1,3-benzodioxol-5-ylboronic acid, using the procedure of
Preparation 109. 8(CDCI3): 2.22 (s,3H), 6.00 {s,2H), 6.70 (d,lH), 6.75 (s,1H),
6.85 (d,1 H), 7.06 (d,1 H), 7.33 (d,1 H), 7.40 (s,1 H).
t-Buhr~ 4-[4-~(1 3-benzodioxol-5-vll-3-methylpy~~-4-hyrdroxy~ir,~eridine-1-
Obtained as a colourless solid (39%), m.p. 135-138°C, from the
title
2o compound of Preparation 143 and t-butyl 4-oxopiperidine-1-carboxylate,
using
the procedure of Preparation 43. Found: C, 69.82; H, 7.15; N, 3.44.
C24H29N05 requires C, 70.05; H, 7.10; N, 3.40%. 8(CDCI3): 1.50 (s,9H), 1.76
(m,2H), 2.04 (m,2H), 2.29 (s,3H). 3.28 (t,2H), 4.04 (m,2H), 6.00 (s,2H), 6.76
(d,1 H), 6.79 (s,1 H), 6.87 {d,1 H), 7.20 {d,1 H), 7.30 (d,1 H), 7.37 (s,1 H).
LRMS (APCI): 412 {M+H)''.
Obtained as a pate yellow solid (96%), m.p. 105-108°C, from the
title
3o compound of Preparation 144 and trifluoroacetic acid, using the procedure
of
CA 02312935 2000-06-02
wo ~n~~ Pc~r~w~sro~o
-120-
Preparation 35. S(CDCI3): 2.28 (s,3H), 2.50 (m,2H), 3.12 (t,2H), 3.56 (m,2H),
6.00 (s,2H), 6.17 (brs,1 H), fi.75-6.82 (m,2H), 6.87 {d,1 H), 7.17 (d,1 H),
7.22-
7.30 {m,2H).
LRMS (APCI): 294 (M+H)''.
o Obtained as a colourless solid (57%), m.p. 133-134°C, from the title
. compound of Preparation 145 and methyl chlorosulphonylacetate, using the
procedure of Preparation 61. Found: C, 61.15; H, 5.41; N, 3.15. CZZH23NOsS
requires C, 61.52; H, 5.40; N, 3.26%. 8(CDCI3): 2.28 {s,3H), 2.67 (m,2H), 3.62
(t,2H), 3.80 (s,3H), 4.01 (s,2H), 4.06 (s,2H), 6.00 (s,2H), 6.08 (brs,1 H),
6.78
~ 5 (m,2H), 6.8? (d,1 H), 7.20 (m,2H), 7.26 (m,1 H).
LRMS (APCI): 430 (M+H)+.
2o Obtained as a colourless oil (33%), from the title compound of
Preparation 57 and 3-fluorophenylboronic acid, using the procedure of
Preparation 109. 8(CDCI3): 2.20 (s,3H), 7.06-7.25 (m,4H), 7.30-7.40 (m,2H),
7.43 (s,1 H).
25 PREPARATION 148
t~~rl 4 '[~-~( fluoro~yj~~.-methy!~ heny~]-4-hydroxyRineridine-1-carboxvlate
Obtained as a pale yellow, amorphous solid (53%), from the title
compound of Preparation 147 and t-butyl 4-oxopiperidine-1-carboxylate, using
CA 02312935 2000-06-02
wo ~n9s6~ Pcr~r9s~o
-121-
the procedure of Preparation 43. Found: C, 71.39; H, 7.37; N, 3.69.
CZaH2aFN03 requires C, 71.67; H, 7.32; N, 3.63%. 8(CDCI3): 1.50 (s,9H), 1.78
(d,2H), 2.04 (m,2H), 2.22 {s,3H). 3.28 (t,2H), 4.04 (m,2H), 7.12 (t,1 H), 7.16-
7.26
(m,3H), 7.35 (m,2H), 7.40 (s,1H).
LRMS (APCI): 386 (M+H)+.
4-[~(2-Fly~oro enyrl)i-3-meth~~phenyrl]-1.2.3.6-tetrahydro~yrridine
o Obtained as a pale yellow oil (93%), from the title compound of
Preparation 148 and trifluoroacetic acid, using the procedure of Preparation
35.
8(CDCI3): 1.80 (brs,1 H), 2.21 (s,3H), 2.50 (m,2H), 3.12 (t,2H), 3.57 (m,2H),
6.19 (brs,1H), 7.10-7.38 {m,7H).
LRMS (APCI): 268 (M+H)+.
Obtained as a colourless solid (30%), m.p. 128-130°C, from the
title
2o compound of Preparation 149 and methyl chlorosulphonylacetate, using the
procedure of Preparation 61. Found: C, 62.57; H, 5.71; N, 3.32. CZ~ H22FNO4S
requires C, 62.52; H, 5.50; N, 3.47%. b(CDCI3): 2.21 (s,3H), 2.69 (m,2H), 3.63
(t,2H), 3.81 (s,3H), 4.01 (s,2H), 4.10 (m,2H), fi.09 (brs,1 H), 7.14 (t,1 H),
7.17-
7.30 (m,SH), 7.35 (m,1H).
LRMS (APCI): 404 (M+H)+.
3o Obtained as a colourless gum (76%), from the title compound of
CA 02312935 2000-06-02
wo ~n~~
-122-
Preparation 37 and 3,4-dimethoxyphenylboronic acid, using the procedure of
Preparation 41. Found: C, 61.71; H, 6.10; N, 2.91. C~H2~N06S requires C,
62.01; H, 6.11; N, 3.14%. 8(CDCI3): 2.30 (s,3H), 2.67 (m,2H), 3.62 (t,2H},
3.82
(s,3H), 3.87 (s,3H), 3.92 (s,3H), 4.02 (s,2H), 4.10 (m,2H), 6.08 (brs,1H),
6.83-
6.97 (m,3H}, 7.20-7.30 (m,3H).
LRMS (APCI): 446 (M+H)+.
~~hyrl 2-~4-(~yndan-5-yl)-3-methyrl~enyrl)-1.2.3.6-tetrahydroRyridin-1-
Obtained as a pale yellow solid (75%}, from the title compound of
Preparation 37 and indan-5-ylboronic acid (WO A-97/32853), using the
procedure of Preparation 41. 8(CDCI3): 2.10 (m,2H), 2.30 (s,3H), 2.69 (m,2H),
~5 2.98 (m,4H), 3.62 (t,2H), 3.82 (s,3H), 4.02 (s,2H), 4.10 (m,2H), 6.08
(brs,1H),
7.09 (d,1 H), 7.18-7.35 (m,SH).
LRMS (Thermospray): 443 (M+NH4)+.
2o Me~~,vl ~~4-![3-methyl~3-trifluoromethoxvrohenyrl)phenyrl]-1.2.3.6-
Obtained as an amorphous solid (28%), from the title compound of
Preparation 37 and 3 trifluoromethoxyphenylboronic acid (WO-A-96/13500},
using the procedure of Preparation 41. 8(CDCI3): 2:30 (s,3H), 2.69 (m,2H),
25 3.64 (t,2H), 3.81 (s,3H), 4.02 (s,2H), 4.10 (m,2H}, 6.10 (brs,1H), 7.15-
7.30
(m,6H), 7.43 (t,1H).
LRMS (APCI): 471 (M+H)+.
CA 02312935 2000-06-02
wo ~nx~~
-123-
Obtained as an orange oil (37%), from 4-bromo-2-trifluoromethylaniline,
using the procedure of Preparation 42. Found: C, 51.70; H, 2.61. C~3H88rF3
requires C, 51.86; H, 2.68%. 8(CDCI3): 7.19 (d,1 H), 7.26 (m,2H), 7.38 (m,3H),
7.65 (d,1 H), 7.86 (s,1 H).
Obtained as a colourless solid (53%), m.p. 153-155°C (from
hexane),
from the title compound of Preparation 154 and t-butyl 4-oxopiperidine-1-
carboxylate, using the procedure of Preparation 43. Found: C, 65.34; H, 6.22;
N, 3.26. C23HZSFsNOs requires C, 65.55; H, 6.22; N, 3.32%. 8(CDCI3): 1.50
s (s,9H), 1.78 (d,2H), 2.04 (m,2H), 3.28 (t,2H), 4.10 (m,2H), 7.28-7.42
(m,6H),
7.66 (d,1 H), 7.88 (s,1 H).
LRMS (Thermospray): 422 {M+H)+.
4-(4-Phenxl-,_ 3-triflyoromethyrlphenyrll-1.2.3.6-tetrahyrdropyridine
Obtained as a pale brown oil (90%), from the title compound of
Preparation 155 and p-toluenesulphonic acid, using the procedure of
Preparation 70. 8(CDCI3): 2.50 (m,2H), 3.17 (t,2H), 3.58 (m,2H), 6.27 (brs,1
H),
7.25-7.42 (m,6H), 7.56 (d,1 H), 7.75 (s,1 H).
LRMS (Thermospray): 304 (M+H)''.
CA 02312935 2000-06-02
WO 99/Z9667 PCT/EP98/06640
-124-
Obtained as a pate yellow oil (59%), from the title compound of
Preparation 156 and methyl chlorosulphonylacetate, using the procedure of
Preparation 37. 8(CDCI3): 2.71 (m,2H), 3.66 (t,2H), 3.82 (s,3H), 4.02 (s,2H),
4.12 (m,2H), 6.18 (brs,1 H), 7.28-7.42 (m,6H), 7.55 (d,1 H), 7.72 (s,1 H).
LRMS (APCI): 440 (M+H)+.
Obtained as a green solid (47%), m.p. 174-176°C, from 5-bromo-2,2-
dimethyl-1,3-benzodioxole (GB-A-2187452) and trimethyl borate, using the
procedure of Preparation 101. 8(DMSOds): 1.60 (s,6H), 6.77 (d,1 H), 7.17
(s,1 H), 7.28 (d,1 H), 7.80 (s,2H).
tetra,~ydrooyr~din-1-yrlsul~honyrl~ 2-methyrlp~panoate
Obtained as a colourless, amorphous solid (33%), from the title
compounds of Preparation 158 and Preparation 40, using the procedure of
Preparation 41, but with ether:hexane (1:4) as eluant. 8(CDCI3): 1.67 (s,6H),
1.73 (s,6H), 2.30 (s,3H), 2.65 (m,2H), 3.62 (t,2H), 3.80 (s,3H), 4.13 (m,2H),
6.05 (brs,1 H), 6.70-6.78 (m,3H), 7.19-7.30 (m,3H).
LRMS (Thermospray): 486 (M+H)+.
CA 02312935 2000-06-02
WO 99/Z9669 PGT/EP98/06640
-125
PREPA~?ATION 160
Obtained as a colourless, amorphous solid (96%), from the title
compound of Preparation 159, using the procedure of Preparation 90.
8(CDC13): 1.64 (s,6H), 1.72 (s,6H), 1.78-1.88 (m,4H), 2.27 (s,3H), 2.63 (m,1
H),
3.09 (m,2H), 3.81 (s,3H), 3.98 (d,2H), 6.68-6.77 (m,3H), 7.07 (m,2H), 7.17
(d,1 H).
LRMS (Thermospray): 488 (M+H)+.
Obtained as a colourless, amorphous solid (47%) from the title
compound of Preparation 160, using the procedure of Preparation 79.
8(CDC13): 1.67 (s,6H), 1.72 (s,6H), 1.78-1.88 (m,4H), 2.27 (s,3H), 2.63
(m,1H),
3.10 (m,2H), 4.00 (d,2H), 6.68-6.77 (m,3H), 7.05 (m,2H), 7.16 (d,1H).
LRMS (Thermospray): 474 (M+H)+.
1.2-Dimethyl-5-(,~.4.5.5-tetramethyrl-1,3.2-dioxaborolan-2~yrllbenzimidazole
A mixture of 5-bromo-1,2-dimethylbenzimidazole (J. Chem. Soc., 1931,
1143; 2 g, 8.88 mmol), 4,4,5,5-tetramethyl-1,3,2-dioxaborolane (2.06 ml, 14
mmol), triethylamine (3.9 ml, 28 mmol), [1,1'-
bis(diphenylphosphino)ferrocene)dichloro palladium(II) (224 mg, 0.27 mmol)
and anhydrous 1,4-dioxan (35m1), under nitrogen, was stirred under reflux for
44 hours, then allowed to cool and partitioned between ethyl acetate and
water.
CA 02312935 2000-06-02
wo ~n9~s~ rc~r~co
-126-
This mixture was filtered to remove palladium residues, the layers separated
and the aqueous phase washed with ethyl acetate. The combined organic
solutions were dried (MgS04) and evaporated under reduced pressure, then
the residue was purified by flash chromatography, using
methanol:dichloromethane (1:3) as eluant, followed by trituration with
diisopropyl ether, to provide the title compound (356 mg, 15%) as a
colourless,
amorphous solid. S(CDCI3): 1.37 (s,12H), 2.60 (s,3H), 3.72 (s,3H), 7.27
(d,1H),
7.70 (d,1 H), 8.15 (s,1 H).
LRMS (Thermospray): 273 (M+H)+.
Methyrl~..{4 j~1.2-dimethyrlbenzimidazol-5-yrl)-3-me~ll~l~~l]-
tetrahyrdronvridin-1-yrlsu hony~(Jw2-met rlTpanoate
~5 To a stirred solution of the title compound of Preparation 40 (400 mg, 0.96
mmol) in degassed 1,2-dimethoxyethane (20 ml) was added the title compound
of Preparation 162 (351 mg, 1.29 mmol), cesium fluoride (380 mg, 2.5 mmol),
tri-o-tolylphosphine (31 mg, 0.1 mmol) and
tris(dibenzylideneacetone)dipalladium(0) (47 mg, 0.05 mmol), then the reaction
2o mixture heated under reflux for about 3 hours, under nitrogen. Because of
limited solubility, a portion of 1-methylpyrrolidin-2-one (4 ml) was added and
the
resulting mixture refluxed for 9 hours, then allowed to cool to room
temperature,
diluted with ethyl acetate, washed with water, dried (MgS04) and evaporated
under reduced pressure. The residue was purified by flash chromatography,
25 using an elution gradient of methanol:dichloromethane (0:100 to 2:98),
followed
by crystallisation from diisopropyl ether, to furnish the title compound (261
mg,
56%) as a colourless solid, m.p. 148-151°C. 8(CDCI3): 1.67 (s,6H), 2:30
(s,3H),
2.63 (s,3H), 2.67 (m,2H), 3.63 (m,2H), 3.77 (s,3H), 3.81 (s,3H), 4.13 (m,2H),
6.07 (brs,1 H), 7.19-7.32 (m,SH), 7.62 (s,1 H).
3o LRMS (Thermospray): 482 (M+H)+.
CA 02312935 2000-06-02
wo ~n~sc~ rc~r~wmo
-127-
Obtained as a pale yellow gum (32%), from the title compound of
Preparation 163, using the procedure of Preparation 90, except that the
hydrogenation was conducted at 414 kPa (60 psi) and 70°C for 24 hours
and
methanol:dichloromethane (3:97) was used as chromatography eluant.
8(CDCI3): 1.65 (s,6H), 1.78-1.88 (m,4H), 2.27 (s,3H), 2.62 (s,3H), 2.65
(m,lH),
3.09 (m,2H), 3.75 (s,3H), 3.81 (s,3H), 3.97 (m,2H), 7.05-7.32 (m,SH), 7.61
(s,1 H).
LRMS (Thermospray): 484 (M+H)+.
Obtained as a colourless solid (88%), m.p. 125-127°C, from the
title
compound of Preparation 164, using the procedure of Preparation 91.
8(DMSO~): 1.50 (s,6H), 1.62 (m,2H), 1.82 (m,2H), 2.20 (s,3H), 2.70 (m,1H),
2.78 (s,3H), 3.08 (t,2H), 3.81 (d,2H), 3.92 (s,3H), 7.10-7.20 (m,3H), 7.46
(d,1H),
7.65 (s,1 H), 7.88 (d,1 H).
LRMS (Thermospray): 471 (M+H)+.
Z:j~~4-Brom-o-3-methyrlphenvll-1.2.3.,6 tetrahyrdro~yrridin-1-yrlsulphonv~]-2-
1 M Aqueous sodium hydroxide solution (4.2 ml, 4.2 mmol) was added to
CA 02312935 2000-06-02
wo ~n9~~ Pcr~r9sio~o
-128-
a stirred solution of the title compound of Preparation 40 (500 mg, 1.2 mmol)
in
a mixture of methanol (3 ml) and 1,4-dioxan (3 ml). The resulting solution was
heated at 50°C for 2 hours, then allowed to cool to room temperature
and
poured into ethyl acetate. The mixture was washed with 2M hydrochloric acid,
then the organic phase dried (MgS04} and evaporated under reduced pressure
to afford the title compound (439 mg, 91 %) as a colourless, amorphous solid.
8(CDC13): 1.67 (s,6H), 2.40 (s,3H), 2.58 (m,2H), 3.64 (t,2H), 4.11 (m,2H),
6.00
(brs,1 H), 7.03 (d,1 H), 7.21 {d,1 H), 7.48 (d,1 H).
o LRMS (Elec~rospray): 425 (M+Na)+.
N-Beer 2;j4-(4-bromo-3-methylphenyy-1.2.3.6-tetrahvdroRyridin-1-
yrlsulnhon-~~]!-2-methyrt~nionamide
1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (274 mg,
1.43 mmol) was added to a stirred mixture of the title compound of Preparation
166 {439 mg, 1.19 mmol), N-hydroxybenzotriazole (176 mg, 1.3 mmol), O-
benzylhydroxylamine hydrochloride (200 mg, 1.25 mmol), N-methylmorpholine
(0.29 ml, 2.62 mmol) and anhydrous dichloromethane (8 ml). The reaction
mixture was stirred at room temperature for 18 hours, diluted with
dichloromethane, washed sequentially with dilute aqueous citric acid, water
and
aqueous sodium bicarbonate solution, dried (MgS04) and evaporated under
reduced pressure. The residue was flash chromatographed, using an elution
gradient of methanol:dichloromethane {1:99 to 2:98), to give the title
compound
(553 mg, 91 %) as a colourless oil. s(CDCI3): 1.60 (s,6H), 2.40 (s,3H), 2.53
(m,2H), 3.58 (t,2H), 4.04 (m,2H), 4.93 (s,2H), 5.95 (brs,1H), 7.00 (d,1H),
7.20
(s,1 H), 7.36-7.50 (m,6H), 9.20 {brs,1 H).
LRMS (Electrospray): 531 (M+Na)+.
CA 02312935 2000-06-02
wo 99n~~ rcr~~sros~eo
-129-
-vlsulpho_nyrj~~-2-me~h~~p~ropionamide
Obtained as an amorphous solid (30%), from the title compound of
Preparation 167 and 3-cyanophenylboronic acid (Arch. Pharm. 1996, ~Q, 73),
using the procedure of Preparation 41, but using an elution gradient of ethyl
acetate:pentane (10:90 to 50:50) for the flash chromatographic purification
step. 8(CDCI3): 1.60 (s,6H), 2.24 (s,3H), 2.61 (m,2H), 3.60 (t,2H), 4.08
(m,2H),
4.95 (s,2H), 6.03 (brs,1 H), 7.15 (d,1 H), 7.25 (m,2H), 7.40 (m,SH), 7.55
(m,2H),
7.62 (m,2H), 9:20 (s,1 H).
t_RMS (Thermospray): 530 (M+H)+.
PREPARATION 169
A stirred mixture of 3-bromo-5-etho~cypyridine (Rec. Trav. chim., 1948,
67, 377; 930 mg, 4.6 mmol), bis(tri-n-butyltin) (3.46 ml, 6.9 mmol), tri-o-
tolylphosphine (420 mg, 1.37 mmol), palladium(II) acetate (78 mg, 0.35 mmol),
2o triethylamine (1.23 ml, 8.84 mmol) and acetonitrile (15 ml), under
nitrogen, was
heated under reflux for 18 hours, then allowed to cool. The solution was
decanted from the black, tang residue and evaporated under reduced pressure,
then the resulting residue flash chromatographed, using an elution gradient of
ethyl acetate:pentane (0:100 to 5:95), to yield the title compound (600 mg,
32%) as a colourless oil. 8(CDCI3): 0.90 (t,9H), 1.08 (t,6H), 1.30-1.42
(m,6H),
1.42 (t,3H), 1.58 (m,6H), 4.08 (q,2H), 7.25 (s,1 H), 8.17 (s,1 H), 8.19 (s,1
H).
CA 02312935 2000-06-02
wo ~n9~~ rc~r~~o
-130
PREepR~,~ O'I N 170
s Obtained as a gum (30%), from the title compounds of Preparation 169
and Preparation 40, using the procedure of Preparation 89. 8(CDCI3): 1.46
(t,3H), 1.68 (s,6H), 2.28 {s,3H), 2.65 (m,2H), 3.63 (m,2H), 3.81 (s,3H), 4.10
(m,4H), 6.08 (brs,1 H), 7.12 {s,1 H), 7.20 (d,1 H), 7.28 (m,2H), 8.18 {s,1 H),
8.28
(s,1 H).
o LRMS (APCI): 459 (M+H}''.
~5 Obtained as a colourless solid (88%), m.p. 110-112°C, from the title
compound of Preparation 170, using the procedure of Preparation 66. Found:
C, 62.24; H, 6.96; N, 5.97. C24H32N2~5s r~luires C, 62.59; H, 7.00; N, 6.08%.
8(CDCI3): 1.44 (t,3H), 1.67 {s,6H), 1.82 (m,2H), 1.89 (m,2H), 2.28 (s,3H),
2.67
(m,1 H), 3.08 (m,2H), 3.81 (s,3H), 3.96 (m,2H), 4.12 (q,2H), 7.10-7.20 (m,4H),
20 8.17 (s,1 H), 8.27 (s,1 H).
LRMS (Electrospray): 461 (M+H)'.
25 ~roR~ oi~ c acid
Obtained as a colourless solid (98%), m.p. 202-203°C, from the
title
compound of Preparation 171, using the procedure of Preparation 91.
8{CDCI3): 1.44 (t,3H), 1.70 (s,6H), 1.80 (m,4H), 2.18 (s,3H), 2.60 (m,1H),
3.08
CA 02312935 2000-06-02
WO 99/29667 PCT/EP98/06640
-131-
(m,2H), 3.96 (m,2H), 4.13 (q,2H), 7.05-7.15 (m,3H), 7.26 (obscured by CHCI3,
1 H), 8.22 (s,1 H), 8.33 (s,1 H).
LRMS (Thermospray): 447 (M+H)'.
Anhydrous potassium carbonate (18.0 g, 130 mmol) was added to a
stirred solution of 3-bromophenol (6.0 ml, 52 mmol) in anhydrous
dimethylformamide (120 ml) and the mixture was heated under reflex for 45
minutes, then allowed to cool to about 50°C. 2-Bromoethanol (3.1 ml, 43
mmol)
was added and the reaction mixture heated under reflex for a further 2 hours,
before being allowed to slowly cool to room temperature. The resulting mixture
was poured into ether, the mixture washed with water and the organic phase
~5 dried (MgS04) and evaporated under reduced pressure. The residue was
purified by flash chromatography, using an elution gradient of pentane:ethyl
acetate (10:1 to 5:1 ), to provide the title compound as a colourless oil (6.4
g,
57%). 8(CDCI3): 1.98 (t,1H), 3.95 (t,2H), 4.07 (m,2H), 6.87 (d,1H), 7.08-7.17
(m,3H).
zo
(2-" t-Bul~,rldi~yr~,~ilyrl~rQ~(Lo,~,rllbromobenzene
Triethylamine (1.7 ml, 9.2 mmol) was added to a stirred solution of the
title compound of Preparation 173 (1.8 g, 8.2 mmol) in anhydrous
25 dimethylformamide (30 ml) and the mixture was cooled to about 0°C. t-
Butyldiphenylsilyl chloride (2.4 ml, 9.2 mmol) was added and the reaction
mixture stirred at 0°C for 1 hour and at room temperature for about 16
hours,
then poured into ether. The resulting mixture was washed with 0.5M
hydrochloric acid, then the aqueous washings back-washed with ether. The
CA 02312935 2000-06-02
wo ~n~s~ pcr~r9sio6soo
-132-
combined organic solutions were washed with water, dried (MgSO,,) and
evaporated under reduced pressure, then the residue purified by flash
chromatography, using an elution gradient of pentane:dichloromethane (3:1 to
2:1 to 1:1 ), to furnish the title compound as a colourless oil (2.2 g, 62%).
8(CDCI3): 1.10 (s,9H), 4.00 (t,2H), 4.08 (t,2H), 6.82 (m,1H), 7.03-7.29
(m,4H),
7.38-7.48 (m,SH), 7.71 (m,4H).
LRMS (Thermospray): 474 (M+NH4)+.
PREPARATION 175
3-y~-t-Bu ildi henylsilyrl~ret_ hoxy)phenyrlboronic acid
n-Butyllithium (2.3m1 of a 2.5M solution in hexane, 5.9 mmol) was added
to a stirred solution of the title compound of Preparation 174 (2.5 g, 5.6
mmol)
in anhydrous tetrahydrofuran (25 ml), keeping the internal temperature below
-60°C. The reaction mixture was stirred at about -70°C for 1
hour, then
trimethylborate (4.4 ml, 38 mmol) was added dropwise, again keeping the
internal temperature below -60°C . The reaction mixture was stirred at -
70°C
for 30 min, allowed to slowly warm to room temperature, quenched with a
mixture of concentrated hydrochloric acid (12.5 ml) and water (30 ml), then
2o ether (30 ml) added. The layers were separated and the aqueous layer was
washed with ether. The combined organic solutions were dried (MgSO~) and
evaporated under reduced pressure, then the residue purified by flash
chromatography, using ether as eluant, to afford the title compound as a
colourless oil (1.14 g, 50%). 8(CDC13): 1.08 (s,9H), 4.06 (t,2H), 4.19 (t,2H),
7.'t2 (m,1 H), 7.36-7.45 (m,BH), 7.74 (m,6H), 7.82 (m,1 H).
LRMS (Thermospray): 438 (M+NH4)~.
CA 02312935 2000-06-02
wo ~nx~s~ Pc~r~rr9sro~4o
-133-
Obtained as an oil (73%), from the title compounds of Preparation 175
and Preparation 40, using the procedure of Preparation 41, except that
purification by flash chromatography involved an elution gradient of
pentane:ethyl acetate {10:1 to 5:1 ). 8(CDCI3): 1.07 (s,9H), 1.67 (s,6H}, 2.28
(s,3H), 2.65 (m,2H), 3.62 (m,2H), 3.81 (s,3H), 4.02 (m,2H), 4.08-4.16 {m,4H),
6.08 (brs,1 H), 6.90 (m,3H), 7.19-7.42 (m,10H), 7.71 (d,4H).
LRMS (Thermospray): 438 (M+NH4}+.
me y~p~,g~,x[]Qjr~eridin-1-ylsulnhonyrj}-2-methyrlpro anoate
Obtained as a colourless oil (88%}, from the title compound of
Preparation 176, using the procedure of Preparation 66. 8(CDCI3): 1.07 (s,9H},
1.67 {s,6H), 1.80-1.95 (m,4H), 2.28 (s,3H), 2.65 (m;1H), 3.10 {t,2H), 3.81
(s,3H), 3.95 (m,2H), 3.97 (t,2H), 4.11 (t,2H}, 6.86 (m,3H}, 7.10 (m,2H), 7.19
(d,1 H), 7.34-7.47 (m,6H), 7.71 (d,4H).
LRMS: (Thermospray}: 438 {M+NH4)+.
yrlsulphonyt~-2-rnethyrlp,~,panoi~c acid
Obtained as a colourless foam (88%), from the title compound of
Preparation 177, using the procedure of Preparation 79, but with the reaction
being carried out at room temperature. S(CDCI3): 1.07 (s,9H), 1.67 (s,6H),
1.78-1.95 (m,4H), 2.27 {s,3H), 2.65 (m,1H), 3.11 (t,2H), 3.97 (t,2H), 4.02
(m,2H), 4.11 (t,2H), 6.86 (m,3H), 7.10 (m,2H), 7.18 (d,1H), 7.30 (d,1H), 7.34-
7.43 (m,6H), 7.71 (d,4H).
CA 02312935 2000-06-02
WO 99/29667 PCTlEP98/06640
-134-
The following Table illustrates the ja y.~CQ activities for a range of the
compounds of the invention as MMP inhibitors.
s
EXAMPLE 1C~{nM)
NO.
MMP-3 MMP-2 MMP-13
2 16 315 28
3 26 38 25
15 20 173 NR
28 24 432 NR
40 18 525 NR
50 23 1907 NR
56 15 387 NR
to NR = no result