Note: Descriptions are shown in the official language in which they were submitted.
CA 02315296 2000-06-15
WO 99/34214 PCT/US98/Z7850
1
SOLID-PHASE TIPS AND USES RELATING THERETO
TECHNICAL FIELD
The present invention relates generally to the application of solid-phase
techniques to synthesize and to analyze nucleic acid molecules. In particular,
the
present invention relates to improved solid-phase supports that can be used to
perform a
variety of bimolecular procedures.
BACKGROUND OF THE INVENTION
Nucleic acid hybridization provides specificity for the recognition of
biomolecules, such as DNA and RNA sequences, and has become a powerful
technique
in medical diagnosis. For example, nucleic acid hybridization methods have
been
applied to analysis of genetic polymorphisms, diagnosis of genetic disease,
cancer
diagnosis, detection of viral and microbial pathogens, screening of clones,
and ordering
of genomic fragments (for a review, see Chetverin and Kramer, BiolTechnology
12:1093, 1994). The development of automated synthesis of oligonucleotide
probes
has also promoted the development of rapid, simple and inexpensive diagnostic
assays
based on nucleic acid hybridization. The use of DNA probes in analytical
techniques
has been reviewed by Matthews and Kricka, Anal. Biochem. 169:1, 1988 (also
see,
Keller and Mank (eds.), DNA Probes, 2'~ Edition (Stockton Press 1993), Persing
et al.,
Diagnostic Molecular Microbiology (American Society for Microbiology 1993)).
A general approach to nucleic acid hybridization requires
immobilization of target nucleic acid on solid supports, such as
nitrocellulose filters and
nylon membranes, followed by hybridization with a detectable nucleic acid
probe. The
disadvantage of such methods is that the immobilized nucleic acid typically is
not
tightly bound, resulting in loss of target material from the support.
Moreover, only a
small amount of nucleic acid molecules are available for hybridization.
These problems can be overcome by a "sandwich-type" hybridization
assay in which target nucleic acid is hybridized to a "capture"
oligonucleotide that has
been covalently immobilized to a solid support. A detectably-labeled probe is
then
CA 02315296 2000-06-15
WO 99/34214 PCT/US98/27850
2
hybridized with a different region of the captured target nucleic acid, and
the presence
of the probe is measured.
The discovery of new therapeutic targets and diagnostic markers has
been enhanced by techniques for analyzing gene expression patterns derived
from large
expressed sequence tag databases (Fannon, Trends Biotechnvl., I-x:294, 1996).
Such
sequence data, derived from a wide variety of cDNA libraries, offer a wealth
of
information for identifying genes for pharmaceutical product development.
Comparison of expression patterns from normal and disease tissues also
provides
inferences about gene function, and identifies medically relevant genes as
candidates
for therapeutics research and development programs.
A significant barrier to a more widespread use of solid-phase cDNA
synthesis and the use of DNA probes in simple assays has been the lack of
solid
supports and immobilization methods that are fully compatible with the
hybridization
process. The use of solid supports in DNA probe-based hybridization has been
reviewed by Meinkoth and Wahl, Anal. Biochem. 138:267, 1984.
Poly(ethyleneimine) ("PEI") coatings have been extensively used in the
art for binding biomolecules. PEI is very effective in this capacity for a
variety of
reasons. For instance, PEI is very hydrophilic and thus readily wets aqueous
solutions
containing biomolecules. In addition, PEI contains many amino groups, which
can
form salts with acidic groups in a biomolecule. However, the readiness with
which PEI
accepts aqueous solutions of biomolecules is precisely why it has not, to
date, seen use
in the preparation of biomolecular arrays. When aqueous biomolecular solutions
are
placed on a layer of PEI, the solution rapidly wicks throughout the PEI
coating rather
than staying in a discrete location.
Spring probes have become generally well known since they were
introduced early in the development of the printed circuit board industry.
They are
mechanical devices designed to meet the need for precision and reliability in
the
construction and testing of a variety of electronic components and their
connections
when being assembled into functioning circuit boards. Spring probes are
essentially
electro-mechanical devices, typically consisting of a tubular housing encasing
a
CA 02315296 2000-06-15
WO 99/34214 PCTlUS98/2'I850
3
compression spring, ball and plunger. Some probes are specifically designed to
carry
electrical current flow, while others are used to drill, crimp, and secure
components to a
circuit board, and yet others are designed to perform soldering. There is
nothing in the
design or marketing of spring probes that suggests their potential utility as
a mechanical
devise for the transferring and arraying of solutions onto solid support for
use in the
fields of microbiology, biochemistry, or molecular biology.
Accordingly, a need exists for highly-efficient, cost-effective means for
arraying biomolecules on a solid support. The present invention provides these
and
related advantages as disclosed in more detail herein.
SUN)NJfARY OF THE INVENTION
The present invention provides a solid-phase sample-retaining assembly
that overcomes the drawbacks experienced by the prior art and provides further
related
advantages.
In one embodiment of this invention, a solid-phase sample-retaining tip
is provided that is usable in a procedure in synthesizing or detecting a
biomolecule.
The sample-retaining tip includes a solid support tip structure that is
connectable to a
support pin, and a chemical layer coats at least a portion of the tip
structure. The
chemical layer is bindable to the biomolecule to form a solid-phase sample of
the
biomolecule on the tip structure. In one embodiment, the tip structure is
removably
connected to the support pin. The tip structure has a partially conical shape
with a
plurality of flutes formed therein. These flutes define a plurality of heat
exchange fins
that enable the tip structure to quickly heat up and cool down during selected
thermocycling procedures in the synthesizing or detecting of biomolecules.
In another embodiment of the invention, a sample-retaining assembly is
provided for use in a solid-phase procedure for synthesizing or detecting a
biomolecule.
The sample-retaining assembly includes a support pin, a tip structure
connected to the
support pin, and a chemical layer coating at least a portion of the tip
structure. The
chemical layer is bindable to the biomolecule so as to form a solid-phase
sample of the
biomolecule on the tip structure. In one embodiment, the support pin is a
spring probe
CA 02315296 2000-06-15
WO 99/34214 PCT/US98/27850
4
or other spring pin, and the tip structure is a nylon 6/6 member that is
removably
connected to the spring probe.
In another embodiment of the invention, an array of solid-phase sample-
retaining assemblies are provided, wherein a plurality of support pins are
connected to a
S base in a selected array. Each support pin has an end portion that is spaced
apart from
the base and a plurality of tip structures are connected to the end portions.
The
chemical layer coats at least a portion of each tip structure. The chemical
layer is
bindable to a biomolecule to form a solid-phase sample of the biomolecule.
In another embodiment of the invention, a solid-phase sample-retaining
assembly is combined with a microtiter plate. The micotiter plate has a well
that is
shaped to contain a volume of a sample having a biomolecule therein. The solid-
phase
sample-retaining assembly is sized to extend at least partially into the well.
The solid-
phase retaining assembly includes a support pin, a tip structure connected to
the support
pin with the tip structure being removably positionable in the well, and a
chemical layer
1 S coating at least a portion of the tip structure. The chemical layer is
bindable to the
biomolecule and the solution to form a solid-phase sample of the biomolecule.
In one embodiment of the invention, the microtiter plate has a plurality
of wells therein and the solid-phase sample-retaining assembly includes a
plurality of
support pins arranged in a selected array, and a plurality of the tip
structures are
connected to the support pins to form an array of solid-phase sample-retaining
tips that
are positionable into the plurality of wells.
In another aspect of the invention, solid-phase sample-retaining tips are
combined with a microtiter plate having a plurality of wells therein. The
solid-phase
sample-retaining tips are removably positioned within the microtiter plate's
wells. The
microtiter plate and the sample-retaining tips are storable as a unit such
that a solid-
phase sample on the sample-retaining tip can be easily stored until needed for
a
synthesizing or analyzing procedure.
Another aspect of the invention provides a method of manufacturing the
solid-phase sample-retaining tip for use in a solid-phase molecular biology
process.
The method includes the steps of forming a substrate material as a tip
structure that is
CA 02315296 2000-06-15
WO 99/34214 PCTNS98/2'I850
S
attachable to a support pin, coating at least a portion of the substrate
material with a
chemical layer that is bondable to selected biomolecules to form a solid-phase
sample
of the biomolecule, and attaching the chemical layer to the substrate. In one
embodiment, the chemical layer is a poly(ethyleneimine) and the substrate
material is a
nylon 6/6 material, and the step of attaching the chemical layer to the
substrate material
includes covalently attaching the poly(ethyleneimine) to the nylon 6/6
material.
Another embodiment of the invention is directed toward a method of
forming a solid-phase sample of a biomolecule. The method includes the steps
of
immersing a portion of a tip assembly into a solution having a biomolecule
therein.
The tip assembly has a substrate portion and a chemical layer on the substrate
portion,
with the chemical layer being bindable to the biomolecule. The biomolecule is
allowed
to bind to the chemical layer to form a solid-phase sample of the biomolecule
on the tip
assembly, and the tip assembly is removed from the solution after the
biomolecule has
bonded to the chemical layer.
In another aspect of the invention, the method includes the step of
storing the solid-phase tip assembly in a retaining member after a biomolecule
has
bonded to the chemical layer. The retaining member in one embodiment of the
invention is a microtiter plate with a well therein. The step of storing
includes placing
the tip assembly in the well after the biomolecule has bonded to the chemical
layer, and
placing the microtiter plate and tip assembly as a unit in a storage location.
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1 is an isometric view of an array of solid-phase sample-retaining
assemblies in accordance with the exemplary embodiment of the present
invention.
Figure 2A is an enlarged cross-sectional view of a solid-phase sample-
retaining assembly taken substantially along line 2-2 of Figure 1.
Figure 2B is a cross-sectional view of a solid-phase sample-retaining
assembly of an alternate embodiment.
Figure 3 is an enlarged partially cut away view of a tip structure of a
sample-retaining assembly of Figure 1.
CA 02315296 2000-06-15
WO 99/34214 PCT/US98/27850
6
Figure 4 is an enlarged cross-sectional view of the tip structure taken
substantially along line 4-4 of Figure 3.
Figure 5 is a side elevational view of the array of Figure 1 shown in
solid lines positioned above a microtiter plate with a plurality of wells with
liquid
biomolecule samples therein, and shown in phantom lines in lowered position
with the
tip structures positioned within the wells.
Figure 6 is an enlarged side elevation view of the array of Figure 1
shown with a plurality of the tip structure positioned in the wells of a
microtiter plate.
These and other aspects of the present invention will become evident
upon reference to the following detailed description and attached drawings. In
addition, various references are identified below and are incorporated by
reference in
their entirety.
DETAILED DESCRIPTION OF~THE INVENTION
1. Definitions
In the description that follows, a number of terms are used extensively.
The following definitions are provided to facilitate understanding of the
invention.
A "structural gene" is a nucleotide sequence that is transcribed into
messenger RNA (mRNA), which is then translated into a sequence of amino acids
characteristic of a specific polypeptide.
As used herein, "nucleic acid" or "nucleic acid molecule" refers to any
of deoxyribonucleic acid (DNA), ribonucleic acid (RNA), oligonucleotides,
fragments
generated by the polymerase chain reaction (PCR), and fragments generated by
any of
ligation, scission, endonuclease action, and exonuclease action. Nucleic acids
can be
composed of monomers that are naturally-occurring nucleotides (such as
deoxyribonucleotides and ribonucleotides), or analogs of naturally-occurring
nucleotides (e.g., a-enantiomeric forms of naturally-occurring nucleotides),
or a
combination of both. Modified nucleotides can have modifications in sugar
moieties
and/or in pyrimidine or purine base moieties. Sugar modifications include, for
example, replacement of one or more hydroxyl groups with halogens, alkyl
groups,
CA 02315296 2000-06-15
WO 99/34214 PCT1US98/27850
7
amines, and azido groups, or sugars can be functionalized as ethers or esters.
Moreover, the entire sugar moiety can be replaced with sterically and
electronically
similar structures, such as aza-sugars and carbocyclic sugar analogs. Examples
of
modifications in a base moiety include alkylated purines and pyrimidines,
acylated
purines or pyrimidines, or other well-known heterocyclic substitutes. Nucleic
acid
monomers can be linked by phosphodiester bonds or analogs of such linkages.
Analogs
of phosphodiester linkages include phosphorothioate, phosphorodithioate,
phosphoroselenoate, phosphorodiselenoate, phosphoroanilothioate,
phosphoranilidate,
phosphoramidate, and the like. The term "nucleic acid" also includes so-called
"peptide nucleic acids," which comprise naturally-occurring or modified
nucleic acid
bases attached to a polyamide backbone. Nucleic acids can be either single
stranded or
double stranded.
An "isolated nucleic acid molecule" is a nucleic acid molecule that is not
integrated in the genomic DNA of an organism. For example, a DNA molecule that
encodes interleukin-2 that has been separated from the genomic DNA of a
mammalian cell
is an isolated DNA molecule. Another example of an isolated nucleic acid
molecule is a
chemically-synthesized nucleic acid molecule that is not integrated in the
genome of an
organism.
In the present context, the term "biomolecule" refers either to a nucleic
acid molecule, or to a polymer of amino acids or amino acid analogs.
As used herein, a "detectable tag" or "detectable label" is a molecule or
atom which is conjugated to a nucleic acid molecule to produce a probe that is
useful
for detection methods. Examples of such tags or labels include photoactive
agents or
dyes, radioisotopes, fluorescent agents, mass spectrometer tags, or other
molecules and
marker moieties. Suitable fluorescent labeling compounds include fluorescein
isothiocyanate, rhodamine, phycoerytherin, phycocyanin, allophycocyanin, o-
phthal-
dehyde and fluorescamine. Examples of chemiluminescent labeling compounds
include luminol, isoluminol, an aromatic acridinium ester, an imidazole, an
acridinium
salt and an oxalate ester. Bioluminescent compounds that are useful for such
tags
include luciferin, luciferase and aequorin.
CA 02315296 2000-06-15
WO 99/34214 PCTNS98/27850
8
"Complementary DNA (cDNA)" is a single-stranded DNA molecule that is
formed from an mRNA template by the enzyme reverse transcriptase. Typically, a
primer
complementary to portions of mRNA is employed for the initiation of reverse
transcription.
Those skilled in the art also use the term "cDNA" to refer to a double-
stranded DNA
molecule consisting of such a single-stranded DNA molecule and its
complementary DNA
strand.
The term "expression" refers to the biosynthesis of a gene product. For
example, in the case of a stnactural gene, expression involves transcription
of the structural
gene into mRNA and the translation of mRNA into one or more polypeptides.
A "cloning vector" is a nucleic acid molecule, such as a plasmid, cosmid, or
bacteriophage, that has the capability of replicating autonomously in a host
cell. Cloning
vectors typically contain one or a small number of restriction endonuclease
recognition sites
at which foreign nucleotide sequences can be inserted in a determinable
fashion without loss
of an essential biological function of the vector, as well as nucleotide
sequences encoding a
marker gene that is suitable for use in the identification and selection of
cells transformed
with the cloning vector. Marker genes typically include genes that provide
tetracycline
resistance or ampicillin resistance.
An "expression vector" is a nucleic acid molecule encoding a gene that is
expressed in a host cell. Typically, gene expression is placed under the
control of a
promoter, and optionally, under the control of at least one regulatory
element. Such a gene
is said to be "operably linked to" the promoter. Similarly, a regulatory
element and a
promoter are operably linked if the regulatory element modulates the activity
of the
promoter.
A "recombinant host" may be any prokaryotic or eukaryotic cell that
contains either a cloning vector or expression vector. This term also includes
those
prokaryotic or eukaryotic cells that have been genetically engineered to
contain the cloned
genes) in the chromosome or genome of the host cell.
As used herein, "hybotrope" refers to any chemical or any mixture of a
chemical in an aqueous or organic environment with buffers, chelators, salts
and/or
detergents that changes the enthalpy of a nucleic acid duplex by at least 20%
when
CA 02315296 2000-06-15
WO 99/34214 PCT/US98/27850
9
referenced to a standard salt solution (0.165 M NaCI, 0.01 M Tris pH 7.2, 5 mM
EDTA
and 0.1 % SDS). That is, the energy content of the nucleic acid duplex is
decreased.
The reference oligonucleotide is 5'-GTCATACTCCTGCTTGCTGATCCACATCTG-
3' [SEQ TD N0:9] as the immobilized oligonucleotide and S'-
TGTGGATCAGCAAGCAGGAGTATG-3' [SEQ ID NO:IOJ as the solution
nucleotide which is typically labeled at the 5'-end with a fluorochrome such
as Texas
Red. The oligonucleotide duplex (24 nucleotides in length) has a helical to
coil
transition (HCT) of 25°C or less. The HCT is the difference between the
temperatures
at which 80% and 20% of the duplex is single stranded. The average minimum
slope
for a solution to be defined as a hybotrope is the first derivative of the HCT
and is
equal to 2.4 in units of 1/temperature in degrees C ((80% single strand - 20%
single-
strand)/25°C).
As used herein, "Tm" is the temperature at which half the molecules of a
nucleic acid duplex are single stranded. Tm is measured in solution, while Td
is
measured for the duplex affixed to a solid support, both terms indicate the
temperature
at which half of a duplex are single stranded.
As used herein, "stringency" is the percentage of mismatched base pairs
that are tolerated for hybridization under a given condition.
As used herein, "discrimination" is the difference in Td between a
perfectly base-paired duplex and a duplex containing a mismatch.
As used herein, a "discrimination temperature" is a temperature at which
a hybridization reaction is performed that allows detectable discrimination
between a
mismatched duplex and a perfectly matched duplex.
2. Solid Support
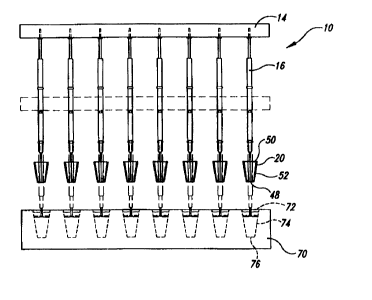
An array 10 of solid-phase sample-retaining assemblies 12 in accordance
with an exemplary embodiment of the present invention is shown in the figures
for
illustrative purposes. As best seen in Figure 1, the array 10 includes a
plurality of
sample-retaining assemblies 12 attached to a base structure 14. Each sample-
retaining
assembly 12 includes a support pin 16 securely fixed at one end 18 to the base
14, and a
CA 02315296 2000-06-15
WO 99/34214 PCT/US98/27850
sample-retaining tip structure 20 is attached to the other end 22 of the
support pin 16.
Each tip structure 20 in the exemplary embodiment is a Nylon 6/6 solid support
structure, and the Nylon 6/6 is coated with a poly(ethyleneinine) (PEI) layer
24 or other
selected chemical layer. The PEI layer 24 or other selected chemical layer is
adapted to
5 bind to a selected biomolecule to form a solid phase sample that is used in
a procedure
for synthesizing or detecting one or more nucleic acids.
The array 10 of the illustrated embodiment includes eight substantially
parallel rows of twelve sample-retaining assemblies 12 to define an array with
ninety-
six sample-retaining assemblies equally spaced along the base structure 14.
Each
10 sample-retaining assembly 12 has approximately the same length so the tip
structures
are equally spaced from the base, thereby defining a substantially coplanar
array of
solid-phase sample-retaining tip structures. The tip structures 20 are spaced
apart to
mate with a conventional 96-well Cetus plate or microtiter plate that is
adapted to
receive and retain selected liquid samples of biomolecules or nucleic acids.
While the
15 exemplary embodiment has an 8 x 12 array of sample-retaining assemblies 12,
alternate
embodiments have other configurations, including a 1 x 8 array, a 1 x 12
array, and a 4
x 12 array, as welt as larger arrays such as a 16 x 24 array.
In the exemplary embodiment, the ninety-six tip structures 20 are
adapted to be dipped into the wells of the Cetus plate with the biomolecules
therein
20 such that the biomolecules chemically bind to the PEI layer 24. When the
tip structures
20 are removed from the sample, the biomolecules are adhered to the PEI layer,
thereby
forming the solid-phase sample of the biomolecule. The tip structures 20 with
the solid
phase sample thereon can then be used in synthesizing or analyzing procedures,
such as
a solid-phase nucleic acid assay and detection process as described in greater
detail
below.
In one embodiment, the array 10 is installed in a robotic or automatic
actuator so the base 14 is clamped into the actuator and the sample-retaining
assemblies
12 project away from the base. The actuator quickly and accurately moves the
array 10
during automated testing to selected controlled positions or stations in
accordance with
a predetermined testing, synthesizing, or analyzing process. Such automated
testing
CA 02315296 2000-06-15
WO 99/34214 PCTNS98/27850
with the array 10 and the ninety-six solid phase samples allows for
substantially faster
testing, synthesizing, or analyzing procedures.
The array 10 is well suited for such automated processing, in part,
because of the support pins 16 of the sample-retaining assemblies 12. As best
seen in
Figure 2A, each support pin 16 is a spring probe that is typically used for
construction
and testing of electronic components, but has been adapted for use in the
present
invention. The spring probe generally includes a housing 28 encasing a biasing
member 30. A plunger 32 extends into the housing 28 so a first end 34 of the
plunger
is within the housing 28 adjacent to the biasing member and a second end 36 is
exterior
of the housing. The biasing member 30 in the exemplary embodiment is a
compression
spring that pushes axially against the plunger 32 toward the base 14. The
plunger's
first end 34 has a shoulder 38 that engages a stop 40 projecting radially
inwardly from
the housing 28 to limit the maximum extension of the plunger 32 with respect
to the
housing. The plunger's second end 36 is fixedly attached to the base 14, and
the
plunger 32 projects substantially perpendicularly away from the base.
In the exemplary embodiment, the housing 28 includes concentric inner
and outer tubular barrels 42 and 44, wherein the biasing member 30 and the
plunger's
first end 34 are contained within the inner barrel. The outer barrel 44
removably
receives the inner barrel 42 therein and frictionally engages the inner barrel
such that
the inner and outer barrels are removably attached to each other. The outer
barrel 44
terminates at a distal end portion 46 that is spaced away from the base 14 and
that
connects to the tip structure 20. Accordingly, the housing's outer barrel 44
and the tip
structure 20 are removable as a unit from the inner barrel 42 and plunger 32,
which
remain fixed to the base 14. Thus, an outer barrel 44 and tip structure 20 can
be easily
and quickly replaced as a unit without having to replace the entire spring
probe.
Suitable spring probes are available from Everett Charles (Pomona,
California),
Interconnect Devices, Inc. (Kansas City, Kansas), Test Connections, Inc.
(Upland,
California), and other manufacturers. While the exemplary embodiment utilizes
spring
probes as the support pins lb, other support pins, including biased or
unbiased support
pins, are used in alternate embodiments of the invention.
CA 02315296 2000-06-15
WO 99/34214 PCT/US98/27850
12
As best seen in Figure 2B, an alternate embodiment of the invention
includes the spring probe as the support pin 16, but the spring probe is
oriented 180°
from the embodiment described above and illustrated in Figure 2A. For example,
the
distal end portion 46 of the outer barrel 44 is fixedly attached to the base
14 and the
second end 36 of the plunger 32 is spaced away from the base and connected to
the tip
structure 20.
The spring probes provide a safety feature that protects the array 10
from being damaged during operation. During a sampling or analyzing process,
for
example, wherein the array 10 is moved to selected positions and the tip
structures 20
are dipped into Cetus plate wells or the like, and if the support pins 16 or
tip structures
inadvertently impacts a surface or other object, the spring probe will
compress axially
to absorb the impact and then return to the uncompressed position.
As best seen in Figure 3, the tip structure 20 of the exemplary
embodiment has a truncated-conical shape with a plurality of channels or
flutes 50
1 S formed therein. The tip structure is connectable to a support pin, or may
be unitary
with the support pin. The flutes 50 are V-shaped flutes that extend axially
between a
flat distal face 48 and a flat proximal face 54. The flutes 50 have veins or
ridges 52 that
converge from the proximal face 54 toward the distal face 48 at a selected
angle. The
truncated conical shape of the tip structure 20 is selected so it virtually
identically
matches the lower cross-sectional shape of a Cetus plate well. Accordingly,
the tip
structure 20 is shaped and sized to fit in a very precise position within the
Cetus plate
well.
The tip structure 20 includes a pin-receiving aperture 56 with an open
proximal end 58 in the proximal face 54 and a closed distal end 60 at a mid-
portion
between the tip structure's proximal and distal faces 54 and 48, respectively.
The pin-
receiving aperture 56 is shaped and sized to removably receive the support
pin's distal
end portion 46. Accordingly, the tip structure 20 is removably connected to
the support
pin 16. However, the tip structure could alternatively be permanently
connected to the
pin, and in fact the pin and tip structure could be a single unitary
structure.
CA 02315296 2000-06-15
WO 99/34214 PCT/US98/27850
13
In the illustrated embodiment, the pin-receiving aperture 56 is coaxially
aligned with the tip structure's longitudinal axis. The aperture's proximal
portion 59 is
generally funnel-shaped such that the aperture's open proximal end 58 has a
larger
diameter than the closed distal end 60. The funnel-shaped proximal portion 59
is
adapted to receive the support pin's distal end portion 46 therein. In the
event the
support pin is slightly misaligned relative to the aperture 56 during an
installation
procedure, the funnel-shaped proximal portion 59 will receive and direct the
support
pin 16 into a position such that the spring probe is coaxially aligned with
the tip
structure 20.
As best seen in Figure 4, the aperture 56 in the exemplary embodiment
is defined by an axial interior wall 61 of the tip structure 20 and has a
substantially
circular cross-sectional shape. The spring probe's distal end portion 46,
however, has a
substantially square cross-sectional shape with four corners 63. The spring
probe's
distal end portion 46 is sized such that the corners 63 frictionally engage
the tip
structure's interior wall 61 so as to frictionally retain the tip structure 20
on the support
pin 16.
In an alternate embodiment of the invention, the end portion has a
polygonal-shaped cross-sectional area with a plurality of corners that engage
the tip
structure's interior wall 61. As an example, an octagonal-shaped cross-
sectional area
having the eight corners that frictionally engage the interior wall 61. In
another
alternate embodiment, the support pin's distal end portion has a circular
cross-sectional
shape that substantially corresponds to the circular cross-sectional area of
the pin-
receiving aperture 56 such that the tip structure 20 is press-fit onto the
spring probe's
distal end portion 46 and is frictionally retained thereon. In another
embodiment, the
tip structure 20 is adhered to the distal end portion 46 with a conventional
adhesive
such that the tip structure is permanently affixed to the support pin 16.
As best seen in Figure 4, the flutes 50 and ridges 52 define the truncated
conical-shaped tip structure 20 with a generally star-shaped cross-sectional
area. As a
result, the tip structure 20 has an enlarged exterior surface 62 so a greater
amount of
biomolecules can attach to the PEI layer 24 during formation of the solid-
phase sample.
CA 02315296 2000-06-15
WO 99/34214 PCTNS98/27850
14
In the exemplary embodiment, the flutes 50, ridges, distal face 48 and
proximal face 54
of the tip structure 20 define a high-surface area, Nylon 6/6 solid support
that is
covalently bonded to the PEI layer 24. In alternate embodiments, the tip
structure 20 is
made of a solid substrate, such as glass or silicon and the PEI layer 24 is
covalently
bound to the solid substrate using sililating chemistry, as discussed in
greater detail
below.
In an alternate embodiment, the exterior surface 62 of the tip structure
20 along the flutes 50 and ridges 52 is dimpled so as to provide a further
increased
surface area along which the PEI layer 24 will bind. In one embodiment, the
dimples
are generally microscopic, and in an alternate embodiment, the dimples are
macroscopic. Accordingly, the dimpled tip structure 20 provides a larger
reaction
surface for greater efficiency in the synthesizing or analyzing procedures.
During selected synthesizing or analyzing procedures, the tip structure
is thermocycled, wherein the tip structure 20 is cycled between high and low
15 temperatures. The ridges 52 of the tip structure 20 form a plurality of
heat exchange
fins 64 that allow for faster temperature change of the tip structure during
the
thermocycling. As a result, the thermocycIing can be done faster and more
efficiently.
As best seen in Figure 5, the array 10 is adapted to be combined and
used with a Cetus or microtiter plate 70 having a plurality of wells 72
therein. As
20 discussed above, the shape of a portion of the well 72 substantially
matches the
truncated conical shape of the tip structure 20. Accordingly, the ridges 52
substantially
engage sidewalls 74 of the well 72 and the tip structure's flat distal face 48
is positioned
against the bottom 76 of the well. In the preferred embodiment, the microtiter
plate 70
has an array of wells formed by eight substantially parallel rows of twelve
wells 72 to
form the ninety-six well configuration that mates with the tip structures of
the array 10.
In other embodiments, the microtiter plates 70 have arrays of 1 x 8 wells, 1 x
12 wells,
and 4 x 12 wells, as well as larger arrays such as a 16 x 24 well format.
During use of the array 10, the array can be automatically or manually
moved from a raised position, shown in solid lines in Figure 5 with the tip
structures 20
being out of the wells 72, to a lowered position, shown in phantom lines with
the tip
CA 02315296 2000-06-15
WO 99/34214 PCTNS98/27850
l5
structures being positioned within the wells 72. The wells 72, in one example,
contain
a liquid sample with the selected biomolecules therein. When the array 10 is
in the
lowered position and the tip structures 20 are in the liquid sample, the
chemical
reaction occurs between the PEI layer 24 and the biomolecule, so as to form
the
selected solid-phase sample of the biomolecule. In the exemplary embodiment,
the
well 72 has a depth that is approximately 33% larger than that of the tip
structure 20, so
when the tip structure is dunked into the well, the liquid sample flows over
the entire
tip structure to bind as much of the biomolecule as possible.
As best seen in Figure 6, the array 10 of sample-retaining assemblies 12
is also usable by positioning the tip structures 20 within the wells 72 and
separating the
tip structures from the support pins 16, as shown in solid lines, so the tip
structures
remain in the wells. The base 14 and support pins 16 are then moved as a unit
away
from the microtiter plate 70. As a result, the microtiter plate 70 with the
ninety-six tip
structures 20 retained or stored within the wells 72 can be moved as a unit
and, as an
example, placed in cold storage or other suitable storage locations until the
solid-phase
samples are needed for a selected synthesizing or analyzing procedure.
In the exemplary embodiment, the wells 72 retain the tip structures 20 in
a very precise location relative to the microtiter plate 70 so the tip
structures can be
easily and substantially simultaneously installed onto the support pins 16. As
an
example, the microtiter plate 70 is held in a known and fixed location, and
the base 14
and support pins 16 are moved as a unit, either automatically or manually to a
selected
position above the wells 72 such that the support pins substantially coaxially
align with
the pin-receiving aperture 56 in the tip structures. The base 14 and support
pins 16 are
then moved toward the microtiter plate 70 such that the support pins 16 are
pressed into
the apertures in the tip structures, thereby releasably connecting the tip
structures to the
support pins. The base 14, support pins 16, and tip structures 20 are then
moved as a
unit away from the microtiter plate 70, thereby removing the tip structures 20
from the
wells 72. The sample-retaining tip assemblies 12 with solid phase samples
thereon can
be moved to a predetermined location and subjected to selected solid-phase
procedures
for analyzing or synthesizing a nucleic acid.
CA 02315296 2000-06-15
WO 99/34214 PCT/US98/27850
16
The solid supports of the present invention can be used in parallel and
are preferentially configured in a 96-well or 384-well format. The solid
supports can
be attached to pegs, stems, or rods in a 96-well or 384-well configuration,
the solid
supports either being detachable or alternatively integral to the particular
configuration.
The particular configuration of the sold supports is not of critical
importance to the
functioning of the assay, but rather, affects the ease of adapting the assays
to automated
systems.
3. Methods for Binding Nucleic Acid Molecules to a Solid Support
The tips described herein are useful in a variety of methods requiring
attachment of a nucleic acid molecule, peptide, polypeptide, or protein to a
solid
support. Examples of solid-phase assays and detection methods that can be
performed
with such tips are described below.
Standard methods can be used to attach a nucleic acid molecule to the
tips. For example, nucleic acid molecules, modified at their 5'-ends with an
aldehyde
or carboxylic acid, can be attached to a solid support having hydrazide
residues (see,
for example, Kremsky et al., Nucleic Acids Res. 1:2891, 1987). Alternatively,
5'-
aminohexyl phosphoramidate derivatives of oligonucleotides can be coupled with
a
solid support bearing carboxyl groups in a carbodiimide-mediated coupling
reaction
(see, for example, Ghosh et al., Nucleic Acids Res. 1:5353, 1987).
The solid supports are preferentially coated with an amine-polymer such
as polyethylene(imine), acrylamide, amine-dendrimers, etc. The amines on the
polymers are used to covalently immobilize nucleic acids. Preferably, nucleic
acids are
bound to the solid supports described herein using poly{ethyleneimine) (PEI)
coatings.
The chemistry used to adhere a layer of PEI to the substrate depends, in
substantial
part, upon the chemical identity of the substrate. The prior art provides
numerous
examples of suitable chemistries that may adhere PEI to a solid support. For
example,
when the substrate is nylon-6/6, the PEI coating may be applied by the methods
disclosed in Van Ness, et al. Nucleic Acids Res. 19:3345, 1991, and
International
Publication No. WO 94/00600. Suitable methods of applying a layer of PEI to
solid
CA 02315296 2000-06-15
WO 99/34214 PCT/US98/27850
17
supports of glass or silicon are described, for example, by Wasserman,
Biotechnology
and Bioengineering XXl1:271, 1980, and by D'Souza, Biotechnology Letters
8:643,
1986.
Preferably, the PEI coating is covalently attached to the solid substrate.
S When the solid substrate is glass or silicon, the PEI coating may be
covalently bound to
the substrate using silylating chemistry. For example, PEI having reactive
siloxy
endgroups is commercially available from Gelest, Inc. (Tullytown, PA). Such
reactive
PEI may be contacted with a glass or silicon tip, and after gentle agitation,
the PEI will
adhere to the substrate. Alternatively, a bi-functional silylating reagent may
be
employed. According to this process, the glass or silicon substrate is treated
with the
bi-functional silylating reagent to provide the substrate with a reactive
surface. PEI is
then contacted with the reactive surface, and covalently binds to the surface
through the
bi-functional reagent.
PEI coatings are preferably used to immobilize nucleic acids to the
nylon tips, described herein. One suitable method of coating nylon-6/6 with
PET has
been described by Van Ness et al., Nucleic Acids Res. !9:3345, 1991. Briefly,
the
nylon substrate is ethylated using triethyloxonium tetrafluoroborate to form
amine-
reactive imidate esters on the nylon surface. The activated nylon is then
reacted with
PEI to form a polymer coating that provides an extended amine surface.
Following
activation of 5'-aminohexyl-tailed oligonucleotides with 2,4,6-trichloro-1,3,5-
triazine
(cyanuric chloride), the modified oligonucleotides are covalently attached to
the nylon
surface via the triazine moiety.
Accordingly, preferred nucleic acid polymers are "amine-modified" in
that they have been modified to contain a primary amine at the 5'-end of the
nucleic
acid polymer, preferably with one or more methylene groups disposed between
the
primary amine and the nucleic acid portion of the nucleic acid polymer. Six is
a
preferred number of methylene groups.
Nucleic acid molecules can be modified by addition of amine moieties
using standard techniques. Products of a polymerise chain reaction, for
example, can
be arrayed using 5'-hexylamine-modified primers. Nucleic acid duplexes can be
CA 02315296 2000-06-15
WO 99/34214 PCT/US98/27$50
18
arrayed after the introduction of amines by nick translation using amine allyl-
dUTP
(Sigma, St. Louis, MO). Amines can also be introduced into nucleic acids by
polymerases such as terminal transferase with amino allyl-dUTP or by ligation
of short
amine-containing nucleic acid polymers onto nucleic acids by ligases.
Preferably, the nucleic acid polymer is activated prior to be contacted
with the PEI coating. This can be conveniently accomplished by combining amine-
functionalized nucleic acid polymer with a mufti-functional amine-reactive
chemical
such as cyanuric chloride. For example, an excess of cyanuric chloride can be
added to
a solution containing the nucleic acid polymer solution. Preferably, the
solution would
contain a 10- to 1000-fold molar excess of cyanuric chloride over the number
of amines
in the nucleic acid polymer in the arraying solution. In this way, the
majority of amine-
terminated nucleic acid polymers have reacted with one molecule of cyanuric
chloride,
so that the nucleic acid polymer becomes terminated with dichlorotriazine.
An advantageous feature of the present invention is that the
biomolecule-containing arraying solutions may be deposited onto a PEI coating
even
though that arraying solution contains a significant amount of the mufti-
functional
amine-reactive chemical. This provides a significant advantage over methods
wherein
coupling agent needs to be removed from an arraying solution prior to an
arraying
process.
When the nucleic acid polymer is double-stranded, both strands or one
of the strands contains a terminal amino group. The double-stranded nucleic
acid
polymer may be bonded through one terminal amino group to the PEI coating to
immobilize the double-stranded polymer. Since only one of the two strands is
covalently bonded to the PEI coating, the other strand may be removed under
denaturing and washing conditions. This approach provides one convenient
method
according to the present invention of achieving an array of single-stranded
nucleic acid
polymers. The double-stranded nucleic acid polymer may be obtained, for
example, as
a reaction product from PCR.
Preferably, the arraying solution is buffered using a common buffer such
as sodium phosphate, sodium borate, sodium carbonate, or Tris-HCI. A preferred
pH
CA 02315296 2000-06-15
WO 99/34214 PCT/US98/27850
19
range for the arraying solution is 7 to 9, with a preferred buffer being
freshly prepared
sodium borate at pH 8.3 to pH 8.5.
Various methods described below require the use of oligonucleotides
bound to the solid supports of the present invention. Preferably,
oligonucleotides are
synthesized with a 5'-amine (generally a hexylamine which is a six carbon
spacer-arm
and a distal amine). Typically, oligonucleotides are 15 to SO nucleotides in
length, and
are activated with homo-bifunctional or hetero-bifunctional cross-linking
reagents such
as cyanuric chloride. The activated oligonucleotides can be optionally
purified from
excess cross-linking reagent (e.g., cyanuric chloride) by exclusion
chromatography.
The activated oligonucleotides are then mixed with the solid supports to
effect covalent
attachment. After covalent attachment of the oligonucleotides, the unreacted
amines of
the solid support are capped (e.g., with succinic anhydride) to eliminate the
positive
charge of the solid support.
Certain methods require the use of biotinylated oligonucleotides that
IS bind to streptavidin, which in turn, is bound to a solid support. Methods
for producing
biotinylated nucleic acid molecules and support-bound streptavidin are well
known to
those of skill in the art. For example, Van Ness et al., Nucleic Acids Res.
19:3345
(1991), describe a method for biotinylation of oligonucleotides, in which
oligonucleotides are treated with activated biotin. Alternatively,
biotinylated
oligonucleotides can be prepared by synthesizing oligonucleotides with biotin-
labeled
dNTPs (see, for example, AusubeI et al. (eds.), Short Protocols in Molecular
Biology,
3rd Edition, pages 12-23 to 12-25 (John Wiley & Sons, Inc. 1995)). Methods for
biotinylating nucleic acids are well known in the art and are described, for
example, in
Avidin-Bioti~t Chemistry. A Handbook (Pierce Chemical Company 1992). Standard
methods that can be used to bind streptavidin to the tips of the present
invention are
provided, for example, by Das and Fox, Ann. Rev. Biophys. Bioeng. 8:165, 1979,
and
by Wilchek and Bayer, Anal. Biochem. 171:1, 1983.
The tips described herein can also be used to assay peptides. General
guidelines for the conjugation of peptides, polypeptides, and proteins to
solid supports
are proved, for example, by Wong, Chemistry of Protein Conj:rgation and Cross
CA 02315296 2000-06-15
WO 99/34214 PCTNS98/27850
linking (CRC Press, Inc. 1991 ), and by Partis et al., J. Protein Chemistry
2:263 ( 1983).
The use of peptides and antibodies in solid phase procedures is known from,
for
example, Vaughn et al. Nature Biotechnology 14:309, 1996 and Huse et al.
Science
246:1275, 1989.
5
4. Use of Solid Supports in cDNA Synthesis
As discussed above, there is an increasing need to synthesize cDNA on
solid supports. These cDNA molecules can be used to create cDNA libraries and
as
probes for gene expression analyses and diagnostic assays. The design of the
disclosed
10 solid support addresses the following problems in current cDNA-dependent
technologies: high input of RNA required, low number of sample throughput,
many
user manipulations, organic extractions and precipitations, vector bias for
insert size
and poor adaptability. One advantage of the solid-phase approach is that cDNA
synthesis in solution requires a large number of steps with intermediate
precipitation
15 steps.
There are various approaches for producing a cDNA molecule on a
specialized solid support as described in this disclosure. The following
general scheme
provides one illustration. First, RNA is prepared using standard techniques.
In the
studies described in Example I, total RNA was prepared by acid-guanidinium-
phenol
20 extraction, using well known methods (see, for example, Ausubel et al.
(eds.), Short
Protocols in Molecular Biology, 3'd Edition, pages 4-4 to 4-6 (John Wiley &
Sons, Inc.
1995); Wu et al., Methods in Gene Biotechnology, pages 33-34, (CRC Press
1997)).
Messenger RNA is then captured on a solid support, for example, that contains
oligonucleotides having oligo(dT) tails.
Alternatively, it is possible to use a simultaneous lysis-mRNA capture
protocol using a chaotrope, such as guanidinium thiocyanate or guanidine
hydrochloride, for both cell lysis and hybridization. This approach permits
the lysis of
a small number of cells. According to this method, DNA is removed by passing
the
lysate through a glass fiber filter, mRNA is captured on the solid support,
and unbound
contaminants and material are washed away. This avoids losses associated with
organic
CA 02315296 2000-06-15
WO 99/34214 PCT/US98/27850
21
phase extractions and serial ethanol precipitations inherent in standard RNA
preparation
procedures.
Support-bound RNA is used as a template to produce the first strand of
cDNA, using standard methods. Then, the second strand of cDNA is synthesized
or an
adapter is placed at the distal end of the first strand cDNA. As an
illustration, it is
possible to place three dNGs at the 3' end of the first strand using, for
example,
terminal transferase. Alternatively, an adaptor can be ligated onto the 3' end
of the
cDNA molecule. A complementary primer is then hybridized to the adaptor. The
second strand cDNA strand is then synthesized using the first strand of cDNA
as a
template.
Alternatively, either random or specific sequences of bound RNA can be
amplified using a polymerase chain reaction (PCR). In brief, PCR is a process
based
on a specialized polymerase, which can synthesize a complementary strand to a
given
DNA strand in a mixture containing deoxyribonucleotides and two DNA primers,
each
about 20 bases long, which flank the target sequence. The mixture is heated to
separate
the strands of double- stranded DNA containing the target sequence and then
cooled to
allow the primers to bind to their complementary sequences on the separated
strands.
The polymerase then extends the primers into new complementary strands.
Repeated
heating and cooling cycles multiply the target DNA exponentially, since each
new
double strand separates to become two templates for further synthesis. In
about one
hour, 20 PCR cycles can amplify the target by a million-fold. Standard methods
for
performing PCR are well-known to those of skill in the art (see, for example,
Delidow
et al., "Polymerase Chain Reaction: Basic Protocols," in PCR Protocols:
Current
Methods and Applications, White (ed.), pages 1-29 (Humana Press, Inc. 1993);
Ausubel et al. (eds.), Short Protocols in Molecular Biology, 3'd Edition,
pages I 5-1 to
15-40 (John Wiley & Sons, Inc. 1995)).
Accordingly, one can add a primer that is complementary to known part
of bound mRNA. It is then possible to amplify using a specific primer and a
primer
complementary to adaptor. Following amplification, typically 5-1 S rounds of
thermocycling, the resulting DNA fragments can then be cloned and the 5' end
of the
CA 02315296 2000-06-15
WO 99/34214 PCT/US98/27850
22
sequence can be determined. It is then possible to synthesize a new hybrid
primer that
contains sequences that are complementary to the adaptor and to 5' end of
gene. The
full length cDNA is then amplified from the solid support.
A modification of PCR, called "anchor PCR," allows amplification of
full-length mRNA even though only a small amount of sequence information is
available (Ausubel et al. (eds.), Short Protocols itt Molecular Biology, 3'd
Edition,
pages 15-27 to 15-32 (John Wiley & Sons, Inc. 1995)). This procedure requires
an
oligo(dT) primer that is either complementary to the poly(A) tail of mature
mRNA,
when amplifying downstream to the known sequence, or complementary to a
synthesized homopolymer tail added to the cDNA following first-strand
synthesis,
when amplifying upstream to the known sequence.
There are at least two branching points in these general cDNA synthesis
methods which provide starting junctures for additional techniques that take
advantage
of solid support methodology. One branch point follows first strand cDNA
synthesis.
At this point, the remaining RNA template can be digested with RNaseH,
hydrolyzed in
sodium hydroxide or removed by heat denaturation, leaving a single-stranded
DNA
template which can be used for oligonucleotide-directed second strand
synthesis, PCR,
random-primed probe production, or gene-expression studies using labeled
oiigonucleotides. The bound cDNA can also be used for preparing a subtracted
library
or differential probes for various applications.
Second strand cDNA synthesis represents a second branch-point in the
solid support cDNA technology. Here, the choice can be made to ligate adapters
to the
cDNA that can support processes such as full-length single-stranded cDNA
probes,
library production, in vitro transcription, and 5' RACE.
An important advantage to solid support cDNA synthesis is the ability to
automate the process. For high through-put cDNA library production or gene
expression studies, it is useful to adapt the solid support described herein
to a 96-well
format. A robotic arm can be used to deliver 96 supports onto 96 gold-plated
pins and
direct cDNA synthesis in a standard 96-well Cetus plate.
CA 02315296 2000-06-15
WO 99/34214 PCT/US98/2'I850
23
5. Analysis of Gene Expression
The solid supports described herein can be used in high through-put
methods for examining the expression of numerous genes (1-2000) in a single
measurement. Such methods can be performed in parallel with greater than one
hundred samples per process. The method is applicable to drug screening,
developmental biology, molecular medicine studies and the like. Thus, within
one
aspect of the invention methods are provided for analyzing the pattern of gene
expression from a selected biological sample, comprising the steps of (a)
exposing
nucleic acids from a biological sample, (b) combining the exposed nucleic
acids with
one or more selected detectably-labeled nucleic acid probes, under conditions
and for a
time sufficient for the probes to hybridize to the nucleic acids, wherein the
detectable
label is correlative with a particular nucleic acid probe and detectable by
spectrometry,
or potentiometry, (c) separating hybridized probes from unhybridized probes,
(d)
detecting the label by spectrometry or potentiometry, and (e) determining
therefrom the
pattern of gene expression of the biological sample.
Within a particularly preferred embodiment of the invention, assays or
methods are provided which are performed as follows. RNA from a target source
is
bound to a solid support through a specific hybridization step (e.g., capture
of poly(A)
mRNA by a tethered oligo(dT) capture probe). The solid support is then washed
and
cDNA is synthesized on the solid support using standard methods (i.e., reverse
transcriptase). The RNA strand is then removed via hydrolysis. The result is
the
generation of a DNA population, covalently immobilized to the solid support,
which
reflects the diversity, abundance, and complexity of the RNA from which the
cDNA
was synthesized. The solid support is then hybridized with one to several
thousand
probes which are complementary to a gene sequence of interest. Each probe type
is
labeled with a tag detectable by spectrometric method, such as mass
spectrometry.
After the interrogation step, excess or unhybridized probe is washed away, the
solid
support is placed, for example, in the well of a microtiter plate and the
detectable tag is
cleaved from the solid support. The solid support is then removed from the
well of
sample container, and the contents of the well are measured with a
spectrometer. The
CA 02315296 2000-06-15
WO 99/34214 PCTNS98/27850
24
appearance of specific tags indicate the presence of RNA in the sample and
evidence
that a specific gene is expressed in a given biological sample. The method can
also be
quantifiable.
The compositions and methods for the rapid measurement of gene
expression using cleavable tags can be described in detail as follows.
Briefly, tissue,
primary or transformed cell lines, isolated or purified cell types or any
other source of
biological material in which determining genetic expression is useful can be
used as a
source of RNA. In the preferred method, the biological source material is
lysed in the
presence of a chaotrope to suppress nucleases and proteases, and to support
stringent
hybridization of target nucleic acid to the solid support. Tissues, cells and
biological
sources can be effectively lysed in one to six molar chaotropic salts
(guanidine
hydrochloride, guanidine thiocyanate, sodium perchlorate, ete. ).
After the source biological sample is lysed, the solution is mixed for i S
minutes to several hours with the solid support to effect immobilization of
the target
nucleic acid. In general, the capture of the target nucleic acid is achieved
through
complementary base pairing of target RNA and the capture probe immobilized on
the
solid support. One permutation utilizes the 3' poly(A) stretch found on most
eukaryotic
messenger RNAs to hybridize to a tethered oligo(dT) on the solid support.
Another
permutation is to utilize a specific oligonucleotide or long probes (greater
than 50
bases) to capture an RNA containing a defined sequence.
Another possibility is to employ degenerate primers (oligonucleotides)
that would effect the capture of numerous related sequences in the target RNA
population. For example, RNA samples can be reversed-transcribed with each of
four
sets of degenerate anchored oligo(dT) primers, having the formula 5'-T,ZMN-3',
where
M can be G, A or C, and N is G, A, T, and C (Ausubel et al. (eds.), Short
Protocols in
Molecular Biology, 3'd Edition, pages 15-35 to 1 S-40 {John Wiley & Sons, Inc.
1995)).
Each primer set is dictated by the 3'-base, with degeneracy in the M position.
Hybridization times are guided by the sequence complexity of the RNA
population and the type of capture probe employed. Hybridization temperatures
are
dictated by the type of chaotrope employed and the final concentration of
chaotrope.
CA 02315296 2000-06-15
WO 99/34214 PCTNS98/27850
General guidelines are provided, for example, by Van Ness and Chen, Nucleic
Acids
Res. 19:5143, 1991. The lysate is preferentially agitated with the solid
support
continually to effect diffusion of the target RNA. After capturing the target
nucleic
acid, the lysate is washed from the solid support and all chaotrope or
hybridization
5 solution is removed. The solid support is preferentially washed with
solutions
containing ionic or non-ionic detergents, buffers and salts.
The next step is the synthesis of DNA complementary to the captured
RNA, in which the tethered capture oligonucleotide serves as the extension
primer for
reverse transcriptase. The reaction is generally performed at 25 to
37°C, and preferably
10 agitated during the polymerization reaction. After the cDNA is synthesized,
it becomes
covalently attached to the solid support since the capture oligonucleotide
serves as the
extension primer. The RNA is then hydrolyzed from the cDNA/RNA duplex. The
step
can be effected by the use of heat which denatures the duplex or the use of
base (i.e.,
0.1 N NaOH) to chemically hydrolyze the RNA. The objective of this step is to
make
15 the cDNA available for subsequent hybridization with defined probes. The
solid
support or set of solid supports are then further washed to remove RNA or RNA
fragments. At this point, the solid support contains an approximate
representative
population of cDNA molecules that represents the RNA population in terms of
sequence abundance, complexity, and diversity.
20 The next step is to hybridize selected probes to the solid support to
identify the presence or absence and the relative abundance specific cDNA
sequences.
Probes are preferentially oligonucleotides in length of 15 to 50 nucleotides.
The
sequence of the probes is dictated by the end-user of the assay. For example,
if the
end-user intends to study gene expression in an inflammatory response in a
tissue,
25 probes would be selected to be complementary to numerous cytokine mRNAs,
RNAs
that encode enzymes that modulate lipids, RNAs that encode factors that
regulate cells
involved in an inflammatory response, etc. Once a set of desired sequences is
defined
for study, each sequence is used to design an oligonucleotide probe, and each
probe is
assigned a specific cleavable tag. The tags) is then attached to the
respective
oligonucleotide(s). The oligonucleotides are then hybridized to the cDNA on
the solid
CA 02315296 2000-06-15
WO 99/34214 PCT/US98/27850
26
support under appropriate hybridization conditions. After completion of the
hybridization step, the solid support is washed to remove any unhybridized
probe. The
solid support or array of supports are then placed in solutions which effect
the cleavage
of the detectable tags. The presence (and abundance) of an expressed mRNA is
S determined by measuring the amount of detectable tags. For example, mass
spectrometer tags are examined using a mass spectrometer.
There are numerous variations of the above-described method for
analyzing differential expression. For example, differential expression can be
examined using a subtracted library. Subtraction of redundant messages to
reveal
patterns of gene expression representative of particular states of activation
or
development is a desirable capability of any gene discovery program. Many
protocols
exist for production of subtracted cDNA libraries, but most require either
large amounts
of RNA or pre-existing cDNA libraries. The use of a solid support to capture
mRNA,
allows the message source representing the background to be subtracted.
Desired
mRNA species are reverse transcribed and RNA templates are destroyed with
alkali.
The resulting "subtraction template" can be re-used indefinitely.
To make a subtracted library, RNA from the source under investigation
is heat-denatured then hybridized to the Frst strand of cDNA on the
subtraction
template. Unbound RNA is then washed away and either captured directly or
hybridized a second time after all the bound, subtracted RNA has been eluted
from the
subtraction template. After capture, cDNA synthesis continues as described
previously.
In a related approach, subtractive cDNA probes are prepared by
hybridizing single-stranded cDNA from one cell type with immobilized mRNA from
a
closely-related cell type, and isolating the small fraction of unhybridized
cDNA. As a
result of this enrichment, DNA fragments of subtractive cDNA can be used to
identify
cDNA clones containing differentially expressed sequences. Subtractive cDNA
can
also be used to prepare subtractive cDNA libraries. PCR can be used to amplify
subtractive cDNA for use as probes or for cloning (see, for example, Kuel and
Battey,
"Generation of a Polymerase Chain Reaction Renewable Source of Subtractive
cDNA,"
in PCR Protocols: Current Methods and Applications, White (ed.), pages 287-304
CA 02315296 2000-06-15
WO 99/34214 PCT/US98/27850
27
(Humans Press, Inc. 1993); Wu et al., Methods in Gene Biotechnology, pages 29-
65,
(CRC Press 1997)).
In yet another variation, a biotinylated oligo(dT)MN molecule, the
degenerate primer described above, is used to bind mRNA to a solid support,
and to
prime first strand synthesis (see, for example, Ra~sok et al., BioTechniques
21:114,
1996). After reverse transcription, the solid-phase cDNA is used as a template
in a
polymerase chain reaction, with the biotinylated oligo(dT)MN and an arbitrary
decamer
as primers. PCR products obtained from two cell populations are then compared
following fractionation by polyacrylamide gel electrophoresis, PCR bands of
interest
are eluted from the gel, and purified PCR products are used as templates for
an
additional polymerase chain reaction, which amplifies the selected bands. The
products
of the second polymerase chain reaction can be used as probes, or can be
further
examined by sequence analysis.
I S 6. Solid-Phase Diagnostic Assays
(A) Detection of Polymorphisms
Restriction endonucleases recognize short DNA sequences and cleave
DNA molecules at those specific sites. Certain restriction enzymes cleave DNA
very
infrequently, generating a small number of very large fragments (several
thousand to a
million base pairs). Most restriction enzymes cleave DNA more frequently, thus
generating a large number of small fragments (less than a hundred to more than
a
thousand base pairs). On average, restriction enzymes with four-base
recognition sites
will yield pieces 256 bases long, six-base recognition sites will yield pieces
4000 bases
long, and eight-base recognition sites will yield pieces 64,000 bases long.
Since
hundreds of different restriction enzymes have been characterized, DNA can be
cleaved
into many different small fragments.
Although a few known human DNA polymorphisms are based upon
insertions, deletions or other rearrangements of non-repeated sequences, the
vast
majority are based either upon single base substitutions or upon variations in
the
number of tandem repeats. Base substitutions are very abundant in the human
genome,
CA 02315296 2000-06-15
WO 99/34214 PCT/US98/27850
28
occurring on average once every 200-500 base pairs. Length variations in
blocks of
tandem repeats are also common in the genome, with at least tens of thousands
of
interspersed polymorphic sites, or "loci." Repeat lengths for tandem repeat
polymorphisms range from one base pair in (dA)~(dT)~ sequences to at least 170
base
pairs in a-satellite DNA. Tandem repeat polymorphisms can be divided into two
major
groups which consist of minisatellites/variable number of tandem repeats
(VNTRs),
with typical repeat lengths of tens of base pairs and with tens to thousands
of total
repeat units, and microsatellites, with repeat lengths of up to six base pairs
and with
maximum total lengths of about 70 base pairs. Most of the microsatellite
polymorphisms identified to date have been based on (dC-dA)n or (dG-dT)"
dinucleotide repeat sequences. Analysis of microsatellite polymorphisms
involves
amplification by the polymerase chain reaction of a small fragment of DNA
containing
a block of repeats followed by electrophoresis of the amplified DNA on
denaturing
polyacrylamide gel. The PCR primers are complementary to unique sequences that
flank the blocks of repeats. Polyacrylamide gels, rather than agarose gels,
are
traditionally used for microsatellites because the alleles often only differ
in size by a
single repeat.
A wide variety of techniques have been developed for the analysis of
DNA polymorphisms. The most widely used method, the restriction fragment
length
polymorphism (RFLP) approach, combines restriction enzyme digestion, gel
electrophoresis, blotting to a membrane and hybridization to a cloned DNA
probe.
Polymorphisms are detected as variations in the lengths of the labeled
fragments on the
blots. The RFLP approach can be used to analyze base substitutions when the
sequence
change falls within a restriction enzyme site, or to analyze
minisatellites/VNTRs by
choosing restriction enzymes that cut outside the repeat units. The agarose
gels do not
usually afford the resolution necessary to distinguish minisatellite/VNTR
alleles
differing by a single repeat unit, but many of the minisatellites/VNTRs are so
variable
that highly informative markers can still be obtained. (Vos et al., Nuc. Acids
Res.
23:4407, 1995).
CA 02315296 2000-06-15
WO 99/34214 PCTNS98/27850
29
Solid-phase techniques have enhanced the ability to detect
polymorphisms. For example, a biotinylated primer is used with allele-specific
PCR
primers to amplify one form of an allele. After amplification, the PCR
products can be
detected by solution hybridization to a fluorophore-labeled probe, and hybrids
are
captured on a solid-phase support bearing streptavidin (see, for example,
Syvanen and
Landegren, Human Mutation 3:172, 1994).
In another approach, termed "solid-phase minisequencing," a
biotinylated amplification product is immobilized by a streptavidin-coated
support.
The amplification product is then used as a template in a sequence-specific
extension
reaction in the presence of a single nucleoside triphosphate complementary to
one of
the sequence variants to be distinguished (see, for example, Syvanen and
Landegren,
Human Martation 3:172, 1994; Syvanen, Clir~. Chem. Acta 226:225, 1994; Jarvela
et al.,
J. Med. Genet. 33:1041, 1996).
In the "oligonucleotide-ligation assay," two differentially-labeled allele-
specific oligonucleotides are compared for their ability to ligate to a
biotinylated
downstream oligonucleotide (see, for example, Nickerson et al., Prvc. Natl.
Acad. Sci.
USA 87:8923, 1990; Nickerson et al., Genomics 12:377,1992). The unlabeled
oligonucleotide is immobilized on an avidin-coated solid support that projects
into a
test well. The presence of the target sequence is determined by measuring the
particular signal generated by the bound labeled oligonucleotide. For example,
the
allele-specific oligonucleotides can be labeled with different fluorophores,
and the
presence of the target is determined by measuring fluorescence.
Nepom et al., J. Rhematvl.23:5 (1996), describe a method for
genotyping analysis in which a selected sequence is amplified using PCR with a
biological sample of nucleic acid and a biotinylated primer. A small amount of
amplified product is transferred to an automated processor instrument that
perfonms
allele-specific hybridization and detection.
DNA fingerprinting represents another aspect of polymorphism
detection. A variety of DNA fingerprinting techniques are presently available,
most of
which use PCR to generate fragments (see, for example, Jeffries et al.,
Nat:~re 314:67,
CA 02315296 2000-06-15
WO 99/34214 PCT/US98/27850
1985; Welsh and McClelland, Nzlcleic Acids Res. 19:861, 1991). The choice of
which
fingerprinting technique to use is dependent on the application (e.g., DNA
typing, DNA
marker mapping) and the organisms under investigation, (e.g., prokaryotes,
plants,
animals, humans).
5 In general, DNA fingerprinting can be performed by synthesizing full-
length cDNA on a tip of the present invention. The cDNA is then digested with
restriction endonucleases, and the unbound material is rinsed from the tip.
Adapters are
then ligated onto the bound cDNA, and the product can be amplified and
analyzed.
A number of fingerprinting methods have been developed over the past
10 few years, including random amplified polymorphic DNA (RAPD), DNA
amplification fingerprinting (DAF), and arbitrarily primed PCR (AP-PCR). These
methods are all based on the amplification of random genomic DNA fragments by
arbitrarily selected PCR primers. DNA fragment patterns may be generated of
any
DNA without prior sequence knowledge. The patterns generated depend on the
15 sequence of the PCR primers and the nature of the template DNA. PCR is
performed at
low annealing temperatures to allow the primers to anneal to multiple loci on
the DNA.
DNA fragments are generated when primer binding sites are within a distance
that
allows amplification. In principle, a single primer is sufficient for
generating band
patterns.
20 A more recent technique for DNA fingerprinting and for detection of
polymorphism is the amplified fragment length polymorphism (AFLP) technique
(see,
for example, Vos et al., Nucleic Acids Res. 23:4407, 1995; Schreiner et al.,
J. Immunol.
Methods 196:93, 1996). Briefly, genomic DNA is digested with restriction
endonuclease and ligated with oligonucleotide adapters, PCR provides selected
25 amplification of sets of restriction fragments, and the amplified fragments
are analyzed
following fractionation by polyacrylamide gel electrophoresis.
This method is readily adapted to a solid phase using the tips of the
present invention. Here, full-length genomic DNA is covalently immobilized on
a tip.
Bound genomic DNA is then digested with restriction endonucleases, and the
unbound
CA 02315296 2000-06-15
WO 99/34214 PCT1US98/27850
31
material is washed from the tip. Adapters are then ligated to bound genomic
DNA
fragments, and the bound DNA molecules are amplified and analyzed.
The AFLP technique has also been applied to mRNA fingerprinting
(Habu et al., Biochem. Biophys. Res. Commura. 23~I:516, 1997). In this
approach,
S double-stranded cDNA is synthesized with anchored oligo(dT) primers, and
digested
with Tayl, which recognizes a four-base sequence. A TadI adapter is then
ligated to the
ends of the cDNA fragments, and PCR amplification is performed with selected
primers, following the general methods of AFLP-based genomic fingerprinting.
Advantageously, mRNA fingerprinting by the AFLP technique is performed with
the
solid supports described herein to anchor the oligo(dT) primers.
Mutations can also be identified via their destabilizing effects on the
hybridization of short oligonucleotide probes to a target sequence (see, for
example,
Wetmur, Crii. Rev. Biochem. Mol. Biol. 26:227, 1991). Generally, this
technique of
allele-specific oligonucleotide hybridization, involves amplification of
target sequences
and subsequent hybridization with short oligonucleotide probes. An amplified
product
can be scanned for many possible sequence variants by determining its
hybridization
pattern to an array of immobilized oligonucleotide probes. Illustrations of
this
approach are provided by Examples 6 and 7.
(B) General Diagnostic Methods
DNA probes can be used to detect the presence of infectious agents or
diseased cells, such as tumor cells expressing tumor-associated antigens.
Typically, a
test biological sample is subjected to a lysis step using ionic detergents or
choatropes to
release nucleic acid targets. Typical nucleic acid targets include mRNA,
genomic
DNA, plasmid DNA or RNA, and rRNA viral DNA or RNA. To effect detection of the
target nucleic acid, the target requires some type of immobilization. For
example,
nucleic acids are immobilized on a solid support or substrate which possesses
some
affinity for nucleic acid. The solid supports are then probed with tagged
oligonucleotides of pre-determined sequence to identify the target nucleic
acid of
interest. Unhybridized probe is removed is a washing step, the tags are
cleaved form
CA 02315296 2000-06-15
WO 99/34214 PCT/US98/27850
32
their respective probes, and then measured (for a review, see Reischl and
Kochanowski,
Molec. Biotech. 3:55, 1995).
In one general type of assay method, oligonucleotides representing a
characteristic part of an amplified sequence are attached to a solid support.
The
S attachment may be covalent or via a biotinatreptavidin, or similar, type of
linkage.
Target nucleic acid is used as a template to produce detectably labeled PCR
products.
These PCR products are hybridized with the capture oligonucleotides, and the
presence
of the PCR products is determined by a label-mediated detection reaction.
In a variation of this approach, biotin-labeled PCR products are attached
to a streptavidin-coated solid support. Immobilized PCR products are
hybridized with
a labeled probe complementary to internal sequences of the amplification
product.
As a further illustration of a diagnostic method, Wilber, Immzinol.
Invest. 26:9 (1997), describes a solid-phase nucleic acid hybridization assay
based on
branched DNA signal amplification methods. In this study, HIV RNA was detected
in
plasma by hybridization of multiple oligonucleotides to the target, 10 of
which captured
the target onto the surface, and 39 of which mediated hybridization of
branched DNA
molecules to the pol region of the RNA. Detectably labeled probes bound to
each arm
of the branched DNA molecules.
Additional detection methods are well-known to those of skill in the art.
For example, solid phase detection can be achieved using amplified colormetric
means
such as an alkaline phosphatase system, a streptavidin system, or a
horseradish
peroxidase system. Radiometric detection is another alternative. Suitable
radiolabels
for radiometric detection include 3H, 'ZSI, '3'I, 355, '4C, 32P, and the like.
Fluorescently-
labeled molecules provide yet another means of detection.
The present invention, thus generally described, will be understood more
readily by reference to the following examples, which are provided by way of
illustration and are not intended to be limiting of the present invention.
CA 02315296 2000-06-15
WO 99/34214 PCT/US98/27850
33
EXAMPLES
EXAMPLE l:
CDNA SYNTHESIS ON THE SOLID SUPPORT
S (A) RNA Capture
In these studies, total RNA was prepared by acid-guanidinium-phenol
extraction, using standard techniques (see, for example, Ausubel et al.
(eds.), Short
Protocols in Molecular Biology, 3'd Edition, pages 4-4 to 4-6 (John Wiley &
Sons, Inc.
1995)). Polyadenylated mRNA was captured on tips by first heating the mixture
of
isolated total RNA to 70°C, adding a high salt hybridization solution
to the RNA,
adding the RNA to the tip solid support having oligo(dT), and then placing the
support
on a moving platform, such as a rotary mixer. Sufficient mixing was
accomplished by
a variety of instruments including continual vortexer, orbital shaker, rotary
rocker, and
a hybridization oven equipped with a rotator.
The time required for RNA capture was found to be dependent on the
quantity of input RNA. At ambient temperature, 10 p.g of total RNA were
sufficient to
achieve 90% saturation of a tip in approximately two hours. In a typical
resting cell,
this amount of total RNA corresponds to approximately 100 ng poly(A)+ mRNA
bound
per tip. Forty micrograms of total RNA from the same source gave the same
level of
saturation in 30 minutes.
In another series of experiments, a stimulated human T-cell line was
used as the RNA source and saturation was reached in one hour under the same
capture
conditions using a 10 pg RNA input. Several additional RNA sources have been
captured including mouse, hamster and human cell lines, and all tend to fall
within this
range of 90% capture in 1-2 hours per 10 ~g total RNA, defining a maximum and
minimum incubation time.
CA 02315296 2000-06-15
WO 99/34214 PCT/US9$/27850
34
(B) First Strand cDNA Synthesis
After capture of poly(A)+ mRNA, the tip was washed in hybridization
buffer three times to remove unbound RNA. Reverse transcription was performed
in a
30 ~1 volume containing MMLV-reverse transcriptase (NflIZLV-RT) and an
optimized
buffer system for 1-2 hours at 42°C on a hybridization oven rotator
rack. As with the
capture step, efficient first stand synthesis requires constant mixing of the
reagents.
Initial experiments yielded 50-100 ng cDNA per solid support. The
products of reverse transcription have been assayed indirectly by
autoradiography of
labelled double-stranded cDNA cleaved from the tip and sized by agarose gel
electrophoresis. In several experiments, the size span of copied mRNA species
was
comparable to conventional methods and was often longer. The size distribution
of the
cDNA ranged from 0.5 kilobases - 20 kilobases, with the average being
approximately
2.0 kilobases.
(C) Second Strand cDNA S, n
After reverse transcription, the tip was washed three times to remove
reactants and enzyme. Second strand synthesis was performed in a 40 ~1 volume
using
one unit of RNaseH per 25 units E. coli DNA polymerase I. The reaction was
incubated at room temperature on a rotary rocker for six hours or overnight.
Unevenly extended ends were "polished" for the subsequent ligation step
by removing the second strand reaction and adding T4 DNA polymerase and
dNTP's.
This incubation proceeded for 30 minutes at 37°C on the rotator in a
hybridization
oven. Products are visualized directly by running the reaction in the presence
of a 32P-
labelled dNTP, and either boiling the second strand products, or by cleavage
from the
support with the restriction enzyme AscI. The radiolabelled products are run
on a gel
and placed on film for visualization. From a typical input of 10 p.g total
RNA, it is
possible to recover 50-120 ng double-stranded cDNA, depending on the
particular
RNA.
The use of a thermostable DNA polymerase after reverse transcription
with an enzyme possessing RNaseH activity, such as MMLV-RT is also a
possibility.
CA 02315296 2000-06-15
WO 99/34214 PCT/US98/27850
In this case, the thermostable polymerise digests the RNA template,
eliminating the
need to remove RNA via a thermal denaturation step.
An experiment comparing RNaseH/DNA pol 1 versus TthI DNA
polymerise in second strand synthesis demonstrated little difference in either
the
5 quantity of the AscI-cleaved product or the content of that product, which
was tested by
PCR amplification of selected genes such as GAPDH and IL-2. Using TthI
decreases
the incubation time from six hours at room temperature to one hour at
70°C. TthI
polymerise also shows reverse transcriptase activity in the presence of
manganese ions,
but incubations need to be kept very short due to the possibility of RNA
hydrolysis at
10 high temperature in the presence of divalent cations. Another advantage of
performing
DNA polymerization at high temperature is that secondary structure is
decreased, which
aids the synthesis of highly structured messages.
(D) Adapter Ligation-LiQation To Vector
Hemi-phosphorylated adapters were ligated to the double-stranded
15 cDNA in a 30 p.l volume with a 5-10:1 molar ratio of adapter:cDNA ends. A
preferred
ligation buffer contained 10% PEG. The adapter-cDNA mixture was incubated with
T4 DNA ligase overnight at room temperature on a rotary rocker. The solid
support
was then washed three times to remove excess linkers. After ligation, the 5'-
hydroxyl
group on the adapter was phosphorylated with T4 polynucleotide kinase and ATP
for
20 one hour at 37°C on a hybridization oven rotating rack. This
reaction was stopped by
washing the tip three times in TE buffer. If the cDNA will be used for another
application, the phosphorylation step may not be necessary.
In some applications, second strand synthesis was specifically primed
from an adapter added immediately after reverse transcription. According to
this
25 approach, after hydrolysis of the mRNA, a partially single-stranded,
heteroduplex
adapter would be ligated using T4 RNA ligase. The partial double-stranded
nature of
this adapter would provide a 3'-hydroxyl group from which second strand
synthesis
could begin, and would prevent concatemerization of the adapter during
ligation since
T4 RNA ligase cannot ligate double-stranded nucleic acids.
CA 02315296 2000-06-15
WO 99/34214 . PCTIUS98/27850
36
(E) Cleavage From the Support/Recircularization
In certain studies, a vector was legated to cDNA that had been
synthesized on a tip. cDNA or vector:cDNA was cleaved from the solid support
in a 40
Itl volume with Ascl at 37°C in a hybridization oven rotator rack for
four hours.
Cleaved DNA was then heated to 70°C for 20 minutes with or without
mixing.
Vector:cDNA was recircularized directly by bringing the volume to 50 ~.1 with
the
addition of legation buffer and T4 DNA ligase. cDNA not previously legated to
vector
was split into several aliquots for test legations to determine which
vector:insert ratio
gives the best result in subsequent transformations. In this case, since the
total amount
of cDNA is small (50-120 ng total in 40 pl volume), the entire legation must
be
transformed, making some type of desalting step necessary.
It is possible to cleave more than 90% of the cDNA from a tip in four
hours. There appears to be no advantage to increasing the incubation time
under these
conditions. Increasing enzyme concentration may cut the time necessary for
cleavage,
but a balance must be struck between incubation time and cleavage volume.
(F) Transformation
cDNA clones were propagated by electroporation transformation of
electrocompetent E. coli with DNA aliquots from a legation. Transformation
frequencies can vary between 109 - 10'° cfu per pg DNA. As those of
skill in the art
know, the major limitation in this procedure is the salt sensitivity of the
electroporation.
Only 1-2 pl of a standard legation (5-10% of the total volume) can be
electroporated per
aliquot of electrocompetent cells before the threshold salt tolerance is
exceeded and the
applied current arcs between the electrodes, vaporizing the cells and wasting
the portion
of legation used. Many separate aliquots of cells can be electroporated to
keep this
from happening, but this is both laborious and wasteful. A better scheme is to
use a
desalting step that can be added to the current cDNA methodology which is
flexible
enough to be adapted to an in-line or scaled-down version of the technology
without
sacrificing yield.
CA 02315296 2000-06-15
WO 99/34214 PCT/US98/27850
37
Transformants were plated on standard growth medium, such as LB agar
containing antibiotics. Only bacteria harboring the gene for antibiotic
resistance form
colonies on media containing that antibiotic. Differentiating between true
recombinants
and colonies containing only vector sequences is often accomplished by blue-
white
color selection which is a result of the expression of the IacZ gene which
runs through
the multiple cloning sites of many common plasmid vectors. Expression of the
IacZ
gene product is interrupted by an insert ligated into the multiple cloning
site resulting in
a white colony phenotype. In this scenario, recombinants can be visually
identified
from non-recombinants, but must either be picked from the non-recombinants or
a
certain level of background non-recombinants must be tolerated in the final
product.
Even a relatively low background of non-recombinants can cause problems when
the
library is amplified since bacteria containing vectors without inserts tend to
be faster
growing and are more stable than those containing inserts, especially those
recombinants with large inserts.
EXAMPLE 2:
SCALING DOWN INPUT RNA FOR CDNA. SYNTHESIS ON THE SOLID SUPPORT
By minimizing loss of material during the many manipulations involved
in standard cDNA library production, it is possible to scale down the input
RNA levels
by 10-100 fold. For example, roughly 100 ng of double-stranded cDNA can be
synthesized on a solid support from 10 ~g total RNA, which is 50-fold less
starting
material than that called for in commercially available kits. It may be
possible to scale
this down as much as 10-fold without changing the protocol described above
significantly, although this will probably require the addition of an
amplification step.
An alternative would be to follow cDNA synthesis through adapter
ligation using a T7 promoter-adapter. This would allow the amplification by in
vitro
transcription rather than by PCR, which may produce a more representative
library by
eliminating length bias inherent in PCR amplifications. In vitro synthesized
transcripts
could then be captured and processed just as any other RNA source. The success
of
CA 02315296 2000-06-15
WO 99/34214 PCT/US98/Z7850
38
this amplification would rely on careful optimization of the irr vitro
transcription
conditions to insure full-length transcripts. Uncaptured transcripts could be
polyadenylated using yeast poly(A) polymerise, then captured by adding them
back to
the same support. Although the re-adenylated transcripts would not be full-
length, not
all their information would be lost from the RNA pool.
In one study, a T7 promoter-adapter was synthesized and ligated to
double-stranded cDNA from 10 ~g total RNA on a tip. Using the manufacturer's
suggested conditions for in vitro transcription, a significant amount of
transcripts were
produced that ranged in size from 300 base pairs to 2,000 base pairs. Although
these
sizes are not optimal for an effective amplification step, the study shows
that producing
in vitro transcripts is possible on a solid support.
Another strategy for scale-down is to take advantage of asymmetric
(linear) PCR in the same manner as in vitro transcripts to amplify and
recapture the
product. Since the amplified product is DNA rather than RNA, a DNA polymerise
can
1 S be used to generate double-stranded cDNA.
EXAMPLE 3:
SOLID SUPPORT PROBES
Poly(A)+ mRNA from 10 ~g total RNA was captured, reverse
transcribed, second strand cDNA was synthesized, and a T7 promoter-adapter was
ligated to the cDNA. In one study, the solid support was placed in a standard
Cetus
PCR tube (ABI, Foster City, CA) and cycled for 3 S rounds using a long PCR
polymerise (Ex-Taq,TAKARA) in a format where the 70°C extension steps
were
2S increased in length one minute for every five cycles. Only one primer used
in the PCR
was complementary to the adapter so that amplification would be primed from
the S'-
most end of the bound cDNA's, producing many copies of the (+) strand. After
cycling, PCR products were heat denatured and run on an agarose gel to
visualize the
range of lengths of the single-stranded cDNA. Sizes ranged from roughly S00
base
pairs to over 20 kilobase pairs, which was in good agreement with the sizes of
double-
CA 02315296 2000-06-15
WO 99/34214 PCT/US98/27850
39
stranded cDNAs labelled, cleaved, electrophoresed and visualized by
autoradiography
in parallel control experiments.
To determine whether or not the single-stranded PCR product was
representative of the original mRNA population, PCR primers were designed for
several genes known to be present at a high level, low level, or not at all.
The design of
the primers was such that all products would be approximately the same length
(400-
600 base pairs) and would be situated at or near the 5'-end of the cDNA. This
primer
design provided a good approximation of the quality of the first strand
synthesis. Since
the RNA source was the human Jurkat T-cell line, IL-2, IL-4, GM-CSF, GAPDH,
CTLA4, c-fos, and Werner's helicase sequences were used for the primers. Mouse
guanylate kinase was used as a negative control.
All polymerase chain reactions produced the expected product size with
the exception of CTLA4 and mouse guanylate kinase, as expected. The product
for IL-
2 was confirmed by Northern blot where 50 ng of the putative IL-2 was 3zP-
labelled
1 S and hybridized to a blot containing immobilized RNA from both stimulated
(PMA +
ionomycin) Jurkat and unstimulated Jurkat. After a high stringency wash, the
unstimulated RNA showed almost no signal whereas the stimulated sample showed
an
intense signal consistent with the size expected for IL-2 message.
EXAMPLE 4:
USE OF THE SOLID SUPPORT FOR RAPID AMPLIFICATION OF CDNA ENDS
Rapid amplification of cDNA ends, or RACE, is also a technique that is
adaptable to the solid support cDNA technology, described herein. Either 5'-
or 3'-
RACE can be performed after adapter ligation of double-stranded cDNA using the
back-end of the capture oligonucleotide (3'-RACE) or the proper 5' adapter
oligonucleotide (5'-RACE) as anchors. Solid support RACE offers advantages
over
currently available techniques since little material is used in generating the
cDNA and
the product can be re-used. This application would rely on the ability to run
a PCR
directly on the solid support described above. Initial experiments have shown
that this
CA 02315296 2000-06-15
WO 99/34214 PCT/US98/27850
can be done for a high copy number housekeeping gene, GAPDH, and that the end
product is directly proportional to the. quality of the captured RNA and
subsequently
synthesized cDNA. Standard methods for performing 3'-RACE and S'-RACE are well-
known to those of skill in the art (see, for example, Wu et al., Methods in
Gene
5 Biotechnology, pages 15-28, (CRC Press 1997)).
EXAMPLE 5:
SOLID SUPPORT CDNA SYNTHESIS FOR GENE: EXPRESSION ASSAY
(A) Cell Stimulation and RNA Preparation
10 In one set of studies, Jurkat line JRT 3.5 was stimulated for six hours at
a cell density of 1x106 cells/ml in serum-free RPMI medium (Life Technologies,
Gaithersburg, MD) in the presence of 10 ng/ml phorbol-12-myristate-13 acetate
(Calbiochem, San Diego, CA) and 100 ng/ml ionomycin (Calbiochem). Cells were
pelleted, washed in lx PBS (Life Technologies), re-pelleted and lysed in O.SmI
per 106
15 cells with buffer containing 4 M guanidine isothiocyanate/1% N-lauryl
sarcosine/25
mM sodium citrate (pH 7.1 ) (Fisher Scientific, Pittsburg, PA). One-tenth
volume 2 M
sodium acetate (pH 4.2) (Fisher Scientific) was added followed by one volume
of
water-saturated phenol (Amresco, Solon, OH). After mixing, one-fourth volume
chloroform:isoamyl alcohol (29:1), (Fisher Scientific) was added, the solution
was
20 mixed vigorously, then incubated on ice for 10 minutes. The lysate was then
centrifuged, the aqueous phase removed, and extracted with an equal volume of
chloroform:isoamyl alcohol. The aqueous phase was then pooled and the RNA
precipitated with two volumes of ethanol (Quantum Chemical Corp, Tuscola, IL).
After centrifugation, the ethanol was decanted and the RNA was air-dried
briefly, then
25 resuspended in RNase-free water to a concentration of between 1 and 5
mg/ml.
(B) Capture and First Strand Synthesis
One solid support bearing the covalently linked oligonucleotide, 5'-
ACTACTGATCAGGCGCGCCTTTTTTTTTTTTTTTTTTTT-3' [SEQ ID NO:1
CA 02315296 2000-06-15
WO 99/34214 PCT1US98/27850
41
(Genset, La Jolla, CA), was added to 10 pg total cellular RNA, and diluted in
sufficient
RNase-free water to cover the tip in a sterile 1.5 ml microfuge tube (Fisher
Scientific).
This oligonucleotide contains a spacer and an AscI cleavage site 5' of the
Oligo(dT)
sequence. The RNA and tip were incubated at 65°C for 5 minutes. An
equal volume
S of 2x mRNA hybridization buffer consisting of 50 mM Tris (pH 7.5), 1 M NaCI
(Fisher
Scientific) and 20 ~g/ml acetylated-BSA (New England Biolabs, Beverly, MA) was
added to each tube, and the tubes rocked gently for two hours at room
temperature.
The supernatant was removed and the tip was then washed three times in lx mRNA
hybridization buffer. After the final wash was complete, a reverse
transcription mix
consisting of lx MMLV-reverse transcriptase buffer, 1 mM dNTP mix , 2 mM DTT
(Life Technologies), 20 units RNasin (Promega, Madison, WI) and 10 pg/ml
acetylated-BSA (New England Biolabs) were added to each tube followed by
addition
of 600 units MMLV-reverse transcriptase (Life Technologies). This reaction was
rocked gently at 42°C for two hours. One unit of RNase H (Boehringer-
Mannheim,
Indianapolis, IN) was then added and the reaction was allowed to continue for
another
half hour. The supernatant was again removed and each tip was washed three
times in
10 mM Tris (pH 8) with 1 mM EDTA (pH 8) (Fisher Scientific). Remaining RNA
template was removed by boiling the tips in TE buffer with 0.01% SDS (Fisher
Scientific).
EXAMPLE 6:
HIGH THROUGH-PUT MELTING PROCEDURE USWG THE SOLID SUPPORT
A capture oligonucleotide (36-mer) was covalently linked to a
polyethyleneimine-coated nylon pin assembly via a C6-amine tail. Shorter
oligonucleotides (18-mer) labeled via a C6 amine tail with Texas Red were
hybridized
to the capture oligonucleotide in a 1.5 M guanidinium thiocyanate solution for
15
minutes at ambient temperature. The pin assemblies were then washed to remove
unhybridized signal oligonucleotide twice with TEN buffer (0.01 M Tris (pH
7.5), 1
mM EDTA, 100 mM NaCI) and then once with TENS buffer (0.01 M Tris (pH 7.5), 1
CA 02315296 2000-06-15
WO 99/34214 PCT/US98/27850
42
mM EDTA, 100 mM NaCI, 0.1% SDS) followed by two washes with TEN buffer.
Test solutions were aliquoted into wells of a polycarbonate thermowell plate
(Corning
Costar Corp. Cambridge, MA), and the plate was placed in an MJ thermal cycler
(MJ
Research Company Watertown, MA). The pins were serially transferred between
the
wells of the plate. Every five minutes the temperature was increased by
5°C, starting at
10°C and reaching 85°C at the final point. The liquid was
transferred to a black 96
well microtiter plate and fluorescence was measured.
The level of fluorescence in each well correlates with the amount of
signal oligonucleotide that has melted from the capture oligonucleotide. The
"melting"
or duplex dissociation was conducted over a temperature range of 10°C
to 95°C.
Fluorescence was measured with a commercial fluorescence plate reader. To
calculate
the Td, cumulative counts eluted at each temperature were plotted against
temperature.
The temperature at which 50% of the material had been dissociated from the tip
was
taken as the Td. The helical coil transition is defined as the temperature at
which a
value of alpha equals 0.2 for a given oligonucleotide duplex (or nucleic acid
duplex,
containing or not containing a mismatch at any place in the duplex) to the
temperature
at which a value for alpha equals 0.8 for the same given oligonucleotide
duplex (or
nucleic acid duplex). The data are exported into a spreadsheet and melt curves
are
generated for each solution. From these melt curves, Td, OHCT, and 0 Td are
calculated.
In one study using a 1 x 12 Pin assembly, each test solution was placed
into 16 separate wells of two thermowell plates (corning Costar Cambridge, MA)
containing 100 ul aliquots, one tube for each temperature point. The pin
assembly was
transferred to a new row of the plate before each temperature jump. Just
before
, reaching the 50°C temperature point, the first plate was removed from
the thermal
cycler and replaced with the second thermowell plate. When the thermal cycling
program was complete, the liquid was transferred to wells of two black
microfluor
plates (Dynatech). Fluorescence was measured using excitation wavelength 584
nm
and emission wavelength 612 nm. The data were exported to a spreadsheet
program
for analysis.
CA 02315296 2000-06-15
WO 99/34214 PCT/US98/27850
43
In a study using a 4 x 12 Pin Assembly, eight thermowell plates were cut
in half to make sixteen 4 x 12 well plates. A 100 p,l aliquot of each test
solution was
placed into one well of each half plate until all wells contained test
solution. All
sixteen half plates were identical in solution configuration. The pin assembly
was
transferred to a new half plate before each temperature jump. When the thermal
cycling program was complete, the liquid was transferred to wells of eight
black
microtiter plates (Dynatech). Fluorescence was measured using excitation
wavelength
584 nm and emission wavelength 612 nm. The data were exported to a spreadsheet
program for analysis.
EXAMPLE 7:
DETERMINATION OF THE MELTING TEMPERATURE OF OLIGONUCLEOTIUE DUPLEXES 1N
VARIOUS HYBOTROPE AND NON-HYBOTROPE BASED HYBRIDIZATION SU1.UTIONS.
This example describes the determination of the Td of wild type and
mutant oligonucleotides when hybridized to a target nucleic acid. It is shown
that
hybotrope based hybridization solutions allow the detection of single base
pair
mutations in a nucleic acid target with a probe up to a 30 nucleotides in
length.
(A) Solutions and Reagents
The filter wash (FW) was 0.09 M NaCI, 540 mM Tris (pH 7.6), 25 mM
EDTA. "SDS/FW" is FW with 0.1% sodium dodecyl sulfate (SDS). Hybridization
solutions contained the text specified concentration of hybotrope, 2% N-
lauroylsarcosine (sarcosyl), 50 mM Tris (pH 7.6) and 25 mM EDTA. Formamide
hybridization solution contained 30% formamide, 0.09 M NaCI, 40 mM Tris-HCl
(pH
7.6), 5 mM EDTA, and 0. I % SDS. Guanidinium thiocyanate was purchased from
Kodak (Rochester, NY). GuCI, lithium hydroxide, trichloroacetic acid, NaSCN,
NaC104 and KI, were purchased from Sigma (St. Louis, MO). Rubidium hydroxide
was purchased from CFS Chemicals (Columbus, OH). CsTFA was purchased from
Pharmacia (Piscataway, NJ).
CA 02315296 2000-06-15
WO 99/34214 PCT/US98/27850
44
LiTCA and TMATCA, and TEATCA were prepared by the dropwise
titration of a 3 N solution of LiOH, TEAOH and TMAOH respectively, with
trichloracetic acid (100% w/v, 6.1 N) to pH 7.0 on ice with constant stirring.
The salt
was evaporated to dryness under vacuum, washed once with ether and dried.
S Oligonucleotides were synthesized on a commercial synthesizer using
standard cyanoethyl-N,N-diisorpropylamino-phosphoramidite (CED-
phosphoramidite)
chemistry. Amine tails were incorporated onto the 5'-end using the
commercially
available N-monomethoxytritylaminohex-6-yloxy-CED-phosphoramidite.
Alternatively, oligonucleotides were commercially purchased (Midland Certified
Reagents, Midland, Tx.).
Table I shows the oligonucleotides that were used to measure the
difference in Td between a wild type oligonucleotide and a mutant
oligonucleotide. The
wild type oligonucleotide represents fully and perfectly base-paired duplex
and a
mutant oligonucleotide represents a single base pair mismatch (generally in
the middle
1 S of the oligonucleotide).
Table I
Identity of Nuceotide Sequence SEQ ID
Oligonucleotide NO.
"capture" oligonucleotide5'-GTCATACTCCTGCTTGCTGATCCACATCTG-3'2
wild type 30-mer 5'-CAGATGGGTATCAGCAAGCAGGAGTATGAC-3'3
mutant 30-mer 5'-CAGATGGGTATCAGGAAGCAGGAGTATGAC-3'4
wild type 24-mer 5'-ATGGGTATCAGCAAGCAGGAGTAT-3' S
mutant 24-mer 5'-ATGGGTATCAGGAAGCAGGAGTAT-3' 6
wild type 18-mer 5'-GGTATCAGCAAGCAGGAG-3'
mutant 18-mer 5'-GGTATCAGGAAGCAGGAG-3' g
Oligonucleotides were bound to the tips described herein. In these
studies, the oligonucleotides were attached to the tips using the approach
described by
CA 02315296 2000-06-15
WO 99/34214 PCT/US98/27850
Van Ness et al., Nucl. Acidr Res. 19:3345, 1991. The oligonucleotide-tips
contained
0.1 to 1.2 pg/tip of covalently immobilized oligonucleotide.
(B) Solid-phase Hybridization
5 To label the probe oligonucleotides, amine oligonucleotides were
reacted with amine-reactive fluorochromes. The derived oligonucleotide
preparation
was divided into three portions and each portion was reacted with either (a)
20-fold
molar excess of Texas Red sulfonyl chloride (Molecular Probes, Eugene, OR),
with
(b) 20-fold molar excess of Lissamine sulfonyl chloride (Molecular Probes,
Eugene,
10 OR), or (c) 20-fold molar excess of fluorescein isothiocyanate. The final
reaction
conditions consisted of 0.15 M sodium borate (pH 8.3) for one hour at room
temperature. The unreacted fluorochromes were removed by size exclusion
chromatography on a G-50 Sephadex column.
A high throughput method for the measurement of the thermodynamic
15 properties of oligonucleotide duplexes has been developed. The method
allows
thousands of solution samples to be scanned for their ability to modulate the
thermodynamic parameters of the helical to coil transition of oligonucleotide
duplexes.
This method employs a solid support which has been designed to fit in a Cetus
plate (or
the well of a plate designed for 96 well PCR format) and requires about 40 p.l
of
20 volume to be completely covered by liquid. The design of the tip is shown
in Figure 1.
This tip is also designed to be compatible with the square end of a spring
probe that can
be used as an attachment site in order to array the nylon tips in a 1x8, 1x12,
4x8, 4x12,
or 8x12 format. A depiction of such a device is shown in Figure 2.
One member of the oligonucleotide duplex is immobilized on the nylon
25 tip as described by Van Ness and Chen, Nucleic Acids Res. 19:5143, 1991. A
hybridization step is then used to form the oligonucleotide duplexes on a tip.
The
hybridization step can be performed en masse in a single container or
individually in
the wells of a plate used for the PCR. It is therefore possible for every tip
of a 96
member array of tips to possess a different oligonucleotide duplex.
CA 02315296 2000-06-15
WO 99/34214 PCT/US98/27850
46
After the hybridization step, the tips are washed and then placed in a
PCR plate mounted on a thermocycler. In the case of the I x8 or 1 x 12 format,
the tips
are then moved through a series of wells each time the temperature is
increased by 5°C.
Typically, the temperature increments are in S°C steps and the period
of the melting at
each temperature is I to 5 minutes. For example, tips in a 1x12 format are
placed in
row H at 10°C. The thermocycler is then programmed to ramp through 16
steps at 2
minute intervals with 5°C increments of temperature. The tip array is
moved from row
to row I 5 seconds prior to the temperature increase. In this format, 12
solutions can be
studied using two plates of solution. In a 96 tip format, entire plates of
solution are
moved off and on the thermocycler at the timed interval.
Fluorescent probes are commonly used in this format and have little
effect on the measured Td values described herein. The use of radiolabeled or
fluorescent probes permit a wide variety of solutions to be measured since
there is no
requirement of optical clarity, in contrast to the case for melt curves
derived by UV
I S spectrometry (hyperchromicity shifts). Fluorescence is measured with a
microtiter
plate fluorescence reader, the data are directly imported into a spreadsheet
program,
such as Excel, which then calculates the stability, enthalpy, helical coil
transition, and
temperature range, and then graphs the results. Typically, a 1x12 format that
measures
12 solutions at once can be completed within one hour, including set up and
data
reduction.
For the determination of oligonucleotide/oligonucleotide Td from the
oligonucleotide-tip, fluorescently-labeled oligonucleotide is incubated in
various
hybridization solutions with a complementary oligonucleotide immobilized on
oligonucleotide-tips. From 5 to 5000 ng of oligonucleotide are hybridized in
300-400
p,l volumes at various temperatures (19-65°C) for 5 to 30 minutes. The
tips are washed
three times with one milliliter of the respective hybridization solution, and
then once
with the respective melting solution at the starting temperature of the
melting process.
The tips in 100 p.l of the respective melting solution are then placed on top
of a
thermocycler. At one to five minute intervals, the temperature is raised
5°C, and the tip
is moved into a new well of the microtiter plate. The melting, or duplex
dissociation, is
CA 02315296 2000-06-15
WO 99/34214 PCT/US98/27850
47
conducted over a temperature range of 10°C to 95°C. Fluorescence
is measured with a
commercial fluorescence plate reader.
To calculate the Td, cumulative relative fluorescent units (RFUs) eluted
at each temperature were platted against temperature. The temperature at which
50%
of the material had been dissociated from the tip is the Td or Tm. The helical
coil
transition is defined as the temperature at which a value of a equals 0.2 for
a given
oligonucleotide duplex (or nucleic acid duplex, containing or not containing a
mismatch at any place in the duplex) to the temperature at which a value for a
equals
0.8 for the same given oligonucleotide duplex (or nucleic acid duplex).
The following Tas were obtained in the hybridizations described below:
CA 02315296 2000-06-15
WO 99/34214 PCT/US98/27850
48
Table II
Ta Ta
Solution TypeLength (Mutant)(Wild Type)~-Td HCT
of Probe(C) (C) (C) (C)
2.5 m LiTCA 30-mer 27 33 6 13/14
2.5 m LiTCA 24-mer 25.5 32 6.5 13/14.5
2.5 m LiTCA 18-mer 24 31 7 9/14
2.0 m LiTCA 30-mer 42 47 5 13.5/16
2.0 m LiTCA 24-mer 38 44 6 14/15
2.0 m LiTCA 18-mer 37 43 6 14.5/16.5
3.0 m GuSCN 30-mer 37 42.5 S.5 13.5/17.5
3.0 m GuSCN 24-mer 34.5 41 6.6 12.5/17
3.0 m GuSCN 18-mer 33.5 40.5 7 14.5/15
3.0 m GuHCI 30-mer 55.5 60 4.5 16/21
3.0 m GuHCI 24-mer 52.5 58 5.5 15/20
3.0 m GuHCI 18-mer 50 57 7 18/20
Ra id H be 30-mer 80 80 0 na*
Ra id H be 24-mer 80 80 0 na
Ra id H be 18-mer 68 70 2 18/23
Sx SSC 30-mer 72.5 72.5 0 18/18
Sx SSC 24-mer 69 70 I 18/18
Sx SSC 18-mer 67 72 5 16/18
Prome a Y 30-mer 80 80 0 na
Prome a Y 24-mer 80 80 0 na
Prome a Y 18-mer 62 65 3 20/23
* na indicates not applicable or too large to accurately determine.
The data indicate that the hybotropic solutions (LiTCA, GuSCN and
GuHCI) permit the detection of a single base-pair mismatch in a 24-mer and 30-
mer
S probe, whereas the detection of a single base-pair mismatch in standard
hybridization
solutions (Rapid Hybe, Promega QY or Sx SSC) was not possible.
CA 02315296 2000-06-15
WO 99/34214 PCTlUS98/2'1850
49
A similar experiment was performed for the 24-mers described above in
a series of hybridization solutions.
Table III
Hybridization SolutionSlope ([..J, HCT ~-Td
Type k)
LiTCA,3M 19 8C 7.SC
GuSCN, 3 M 13 10 6.0
NaSCN, 3 M 8.5 11 5:5
NaClO , 3 M 7 12 4. S
KI,3M 5 15 3.0
NaCI, 0.165 M 4.5 17.5 1.5
GuCI, 3 M 3.5 18 1.2
CsTFA, 2M 2.5 18 1.2
30% formamide ND 20 1.5
Td(wt) is the Td of a perfectly base-paired oligonucleotide duplex and
Tm(mt) is the Td of a oligonucleotide duplex containing a single mismatch. The
values
shown are for a 24-mer duplex of the sequence described above. From the data
presented in Table III, the stringency factor is directly proportional to the
difference
between a perfectly base paired duplex and a duplex containing a mismatch.
That is,
the stringency factor predicts the ability of given hybridization solution to
discriminate
mismatched duplexes.
Although the foregoing refers to particular preferred embodiments, it
will be understood that the present invention is not so limited. It will occur
to those of
ordinary skill in the art that various modifications may be made to the
disclosed
embodiments and that such modifications are intended to be within the scope of
the
present invention, which is defined by the following claims.
CA 02315296 2000-06-15
WO 99/34214 PCTNS98/27850
1
SEQUENCE LISTING
<110> Rapigene, Incorporated
Garrison, Lcri K.
Tabone; John C.
Van Ness, Jeffrey
<120> SOLID-PHASE TIPS AND USES RELATING THERETO
<130> 780068.430PC
<140> PCT
<141> 1998-12-29
<160> 10
<170> PatentIn Ver. 2.0
<210> 1
<211> 39
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
Oligonucleotide for capture and first strand
synthesis used in gene expression assay
<400> 1
actactgatc aggcgcgcct tttttttttt ttttttttt 39
<210> 2
<211> 30
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
Oligonucleotide used to measure the difference in
temperature at which half of the molecules of
nucleic acid duplex are single stranded for wild
type and mutant oligonucleotides
<400> 2
gtcatactcc tgcttgctga tccacatctg 30
<210> 3
<211> 30
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
Oligonucleotide used to measure the difference in
temperature at which half of the molecules of
CA 02315296 2000-06-15
WO 99/34214 PCT/US98/27850
2
nucleic acid duplex are single stranded for wild
type and mutant oligonucleotides
<400> 3
cagatgggta tcagcaagca ggagtatgac 30
<210> 4
<211> 30
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
Oligonucleotide used to measure the difference in
temperature at which half of the molecules of_
nucleic acid duplex are single stranded for wild
type and mutant oligonucleotides
<400> 4
cagatgggta tcaggaagca ggagtatgac 30
<210> 5
<211> 24
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
Oligonucleotide used to measure the difference in
temperature at which half of the molecules of
nucleic acid duplex are single stranded for wild
type and mutant oligonucleotides
<400> S
atgggtatca gcaagcagga gtat 24
<210> 6
<211> 24
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
Oligonucleotide used to measure the difference in
temperature at which half of the molecules of
nucleic acid duplex are single stranded for wild
type and mutant oligonucleotides
<400> 6
atgggtatca ggaagcagga gtat 24
<210> 7
<211> 18
<212> DNA
<213> Artificial Sequence
CA 02315296 2000-06-15
WO 99/34214 PCT/US98/27850
3
<220>
<223> Description of Artificial Sequence:
Oligonucleotide used to measure the difference in
temperature at which half of the molecules of
nucleic acid duplex are single stranded for wild
type and mutant oligonucleotides
<400> 7
ggtatcagca agcaggag 18
<210> 8
<211> 18
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:
Oligonucleotide used to measure the difference in
temperature at which half of the molecules of
nucleic acid duplex are single stranded for wild
type and mutant oligonucleotides
<400> 8
ggtatcagga agcaggag 18
<210> 9
<211> 30
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Immoblized
oligonucleotide of the reference oligonucleotide
representing a nucleic acid duplex
<400> 9
gtcatactcc tgcttgctga tccacatctg 30
<210> 10
<211> 24
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: Solution
oligonucleotide of the reference oligonucleotide
representing a nucleic acid duplex
<400> 10
tgtggatcag caagcaggag tatg
24