Note: Descriptions are shown in the official language in which they were submitted.
CA 02317395 2000-07-10
~rp g9~~Og PCT/US99/03556
Hydrogen Production via the Direct Cracking of Hydrocarbons
Field of the Invention
This invention relates generally to the production of hydrogen, and more
,specifically to hydrogen production by the direct cracking of hydrocarbons
such as
methane and natural gas.
Background of the Invention
Significant progress made in fuel cell technologies during the past decade
has prompted exploration of replacing traditional central large power plants
with so-
called distributed power generators, consisting of a hydrogen generator and a
membrane fuel cell power plant. This latter technology generates electricity
at
locations where it is aimed to be used, and therefore, eliminates the loss of
electricity
during its transmission. In addition, a fuel cell process does not emit any
environmental pollutants such as NOx and SO, which are combustion by-products.
Such a process becomes attractive for the automobile industry as well, since
vehicles
can be propelled by electricity produced from an on-board fuel cell power
plant
rather than by an internal combustion engine.
The current proton-exchange membrane (PElVn fuel cells utilize hydrogen as
the energy source and require essential elimination (ideally below 20 ppmv) of
carbon monoxide from the hydrogen stream to prevent poisoning of the
electrocatalyst. Hydrogen is typically produced through steam reforming,
partial
oxidation or autothermal reforming of natural gas. In all these cases,
however,
carbon monoxide is a co product, which has to be converted into carbon dioxide
in
subsequent steps which adds to the cost of the produced hydrogen.
An alternative route is to directly crack the hydrocarbon fuel into hydrogen
and carbon. In this case, the formation of carbon oxides is avoided and the
need for
downstream reactions such as water-gas shift and selective oxidation for the
conversion of carbon monoxide to carbon dioxide 'is eliminated. Surprisingly,
this
approach has not been extensively studied. While commercial processes exist
that
utilize thermal cracking of methane at extremely high temperatures for the
CA 02317395 2000-07-10
WO 99/43608 PCTNS99/03556
-2-
production of acetylene and carbon black, hydrogen production via the
catalytic
cracking of methane has been only briefly considered in the past.
In U.S. Patent 3,361,535 high temperature catalytic cracking of methane is
taught. The process taught by the 3,361,535 patent, however, results in the
production of undesirable carbon monoxide co-product which requires elaborate
additional processing for its conversion to carbon dioxide and results in
additional
cost.
Recently, Muradov Int. .J. Hydrogen Energy 18,211 (1993), studied the use of
iron and nickel oxides supported on alumina as catalysts for the cracking of
methane
and reported that equilibrium conversions were achieved at temperatures above
800°C. The iron oxide, also appeared to maintain some part of its
activity for
several hours, in contrast to a Pt/A1203 catalyst which deactivated within
minutes
under similar conditions. Muradov Energy & Fuels 12,41 (1998) has also
reported
the use of carbon-based catalysts for the same reaction. Although more stable,
these
catalysts exhibit a lower activity. Furthermore, Ishihara et al. Shokubai
35,324(1993); and Chern. Lett., 93(1995); reported that methane cracking takes
place at low temperatures over a 10% Ni/Si02 catalyst, which does not
deactivate
even after approximately 200 carbon per nickel atoms have been deposited on
it.
The results reported by Ishihara, et al., however, did not demonstrate a level
of
efficiency of hydrogen production which would result in potential commercial
use.
Summary of the Invention
It can therefore be seen from the above review of the prior art that an
effcient method of directly cracking hydrocarbons to produce hydrogen without
the
presence of undesirable co-products, such as carbon monoxide, has been an
objective in the art.
It is therefore an object of the present invention to provide a method of
producing hydrogen by the direct cracking of hydrocarbons.
CA 02317395 2000-07-10
WO 99/43608 PCTNS99/03556
-3-
It is another object of the present invention to provide a method of producing
pure hydrogen without carbon monoxide contamination by the direct cracking of
hydrocarbons.
It is yet another object of the present invention to produce high purity
hydrogen and carbon by the catalytic cracking of hydrocarbons.
It is yet a further object of the present invention to provide a method of
producing hydrogen by direct cracking of methane through the use of a highly
efficient catalyst.
It is another object of the present invention to provide a method of producing
hydrogen by direct cracking of methane or natural gas at low temperature using
a
nickel containing catalyst.
It is yet a further object of the present invention to provide a method of
producing high purity hydrogen by direct cracking of methane at low
temperature
using a silica supported nickel-copper catalyst.
It has been discovered that the catalytic cracking of methane or natural gas
as
a potential route for efficient hydrogen production can be accomplished over
silica-
supported nickel containing catalysts. In one embodiment, activity
measurements
for the methane cracking reaction were conducted with a 16.4 wt.% Ni/Si02
catalyst
in a 20% CH4 in He stream at 550°C and a gas hourly space velocity
(GHSV) of
30000 h-'. Under these conditions the catalyst exhibited a high initial
activity for the
cracking of methane (approximately 35% CH,, conversion). Hydrogen was the only
gaseous product detected. In addition, the rates of methane conversion and
hydrogen formation were found to be in ratio of 1:2, thus, verifying the
reaction
stoichiometry for methane cracking. The amounts of carbon deposited on the
spent
catalyst and methane reacted indicated a good closure of the carbon balance
(10015%). Upon deactivation of the catalyst due to carbon deposition; catalyst
activity may be fully restored by regenerating the catalyst through oxidation
in air or
steam gasification. The process of the invention may be applicable to any
other
suitable hydrocarbon such as ethane, ethylene, propane, propylene, butane,
pentane,
hexane and mixtures thereof, and hydrocarbons with molecular weights in the
gasoline and diesel range. Nevertheless, it is anticipated that the preferred
CA 02317395 2000-07-10
WO 99/43608 PCT/US99/03556
-4-
hydrocarbons will be methane and natural gas. During the catalytic cracking of
higher molecular weight hydrocarbons, it is expected that several other
undesirable
products will be formed in addition to the hydrogen.
In a second embodiment, activity measurements for the methane cracking
reaction were conducted over a set of 9 Ni-Cu/SIOZ catalysts in which the
total metal
amount (on a molar basis) was maintained constant at 2.6 mmole of metal/g of
support while the ratio of Ni:Cu was varied from approximately 8:1 to
approximately 1:8. The reaction was carried out in a pure methane stream, at
650
and 800 °C and at a gas hourly space velocity of 6000 hr'~ The results
indicate that
the presence of small amounts of Cu enhanced significantly the Ni activity at
800 °C.
The initial conversion over the 2.3 mmole Ni/0.3 mmvle Cu/Si02 composition for
example, was measured at 63%, as compared to 14.4% for the 2.3 mmole Ni/Si02
composition. This is a surprising result given that Cu alone is not active for
the
cracking of methane under these conditions (0.3% initial methane conversion
for the
0.3 mmole Cu/Si02 composition). The promoting effect is also more pronounced
when small amounts of Cu are added (i.e., Ni:Cu ratios greater than 1). The
highest
initial methane conversion observed with these set of catalysts is at the 8:1
Ni:Cu
ratio. Even higher initial methane conversions are expected with higher Ni:Cu
ratios
up to about 20:1.
Brief Description of the Drawings
For a fuller understanding of the nature and objects of the invention,
reference should be made to the following detailed description of a preferred
mode
of practicing the invention, read in connection with the accompanying
drawings, in
which:
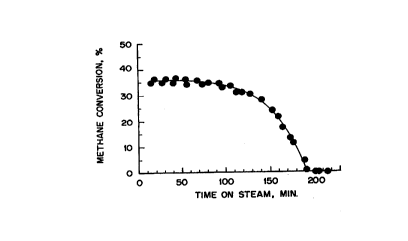
FIG. 1 represents a plot of the deactivation of a Ni/SiOZ catalyst at
550°C and
GHSV = 30,000 h'' in a stream containing 20% CH,,, in He;
FIG. 2 represents a plot of methane conversions obtained over fresh (~,~)
and regenerated (O in air, O in steam) Ni/Si02 catalyst at 550°C at two
different
space velocities;
CA 02317395 2000-07-10
WO 99/43608 PCTNS99/03556
-5-
FIG. 3 represents a plot of selectivities of hydrogen (~ at GHSV =15000 h-',
~ at GHSV =37500 h'') and carbon dioxide (O at GHSV = 15000 h'', O at GHSV =
37500 h-') during steam regeneration of the deactivated Ni/Si02 catalyst at
550°C;
and
Fig. 4 represents a plot of initial methane conversion as a function of
catalyst
composition at two different temperatures over a series of Ni-Cu/Si02
catalysts (O
at 650 °C and D at 800°C).
Detailed Description of the Invention
The catalyst used in the first embodiment of this invention was prepared by
incipient wetness impregnation of an aqueous solution of nickel nitrate onto
the
silica support, followed by calcination in air and in-situ reduction in
flowing
hydrogen. This is a standard method of preparation of supported metal
catalysts and
several different nickel salts can be used instead of nickel nitrate as the
nickel
precursor. Furthermore, other standard methods for the preparation of
supported
metal catalysts could be used without having a detrimental effect on the
properties of
the catalyst. In addition to silica, we investigated other inorganic supports
such as
alumina and titanic. Although nickel supported on these supports was also
found to
be effective for the catalytic cracking of methane, the performance of nickel
supported on silica was superior to those of the other catalysts and
therefore, this
system was chosen to demonstrate the invention in this application. In
addition, we
examined the performance of other transition metals such as Co and Fe,
supported
on silica for this reaction. Although these catalysts were also found to be
effective
for the reaction, at 550°C the performance of nickel was again superior
to the other
catalysts. Finally, by examining several Ni/Si02 catalysts of variable Ni
content, it
was determined that optimum performance for the catalytic cracking of methane
can
be obtained with a nickel content in excess of 5 wt.%, and, in particular a
content of
approximately 16 wt.%. As a result, a 16 wt.% Ni/Si02 catalyst was chosen to
demonstrate the invention in this application.
CA 02317395 2000-07-10
WO 99/43608 PCTNS99/03556
-6-
When the catalyst was placed in a conventional fixed bed reactor and
exposed to a stream containing 20% CH,, (by volume) in He, at 550°C and
under a
GHSV of 30,000 h-', a high initial activity was observed for the cracking of
methane
(approximately 35% CH4 conversion). Hydrogen was the only gaseous product
detected and the rates of methane consumption and hydrogen production were
found
to be in a ratio of 1:2, thus, verifying the reaction stoichiometry for
methane
cracking.
The catalyst used in the present invention will eventually deactivate as a
result of carbon deposition. Carbon may deposit on the surface to cover the
active
sites (site-blocking) or accumulate at the entrance of the pores to block
further
access of the reactants to the interior (pore-mouth plugging). It has been
estimated
that in both cases catalyst deactivation would occur within a short period of
time.
Even if 10 carbon atoms are needed to block each surface Ni atom, for example,
11
mg of carbon deposition would be enough to completely deactivate one gram of
the
16.4% Ni/Si02 catalyst. Furthermore, if pore-mouth plugging was the main
deactivation mechanism, approximately 250 mg of carbon would be sufficient to
clog the external 10% of the pores, in one gram of the Ni/Si02 catalyst
sample.
It has been.discovered that a significantly higher amount of carbon
deposition on the Ni/Si02 catalysts occurs before deactivation occurs. At a
temperature of 550°C for example, a very slow deactivation of the
Ni/SiOZ catalyst
was observed for the first 2 hours (Figure 1) followed by a more rapid loss of
activity during the third hour. By the time the catalyst was completely
deactivated
(200 minutes), approximately 0.59 g of carbon had accumulated on the 0.2 g of
the
Ni/SiO~ catalyst sample. This amount is in very good agreement with the amount
of
carbon calculated based on the integration of the methane conversion (0.61 g),
and
corresponds to approximately 2700 carbon atoms accumulated on the catalyst per
surface nickel atom.
It is therefore apparent that the capability of the silica supported nickel
catalyst to accommodate carbon is significantly higher than those predicted by
either
the site-blocking or pore-mouth plugging models. Scanning Electron Microscopy
CA 02317395 2000-07-10
WO 99/43608 PG"f/US99/03556
-
(SEM) and Transmission Electron Microscopy (TEM) analyses of the spent
catalysts
were utilized to further understand the deactivation mechanism. SEM
micrographs
indicate the formation of filamentous carbon on the catalyst surface. These
filaments appear to grow out of the silica support surface, with their length
increasing with time-on-stream. Each filament has a bright tip, identified by
the use
of SEMBDS .(Energy Dispersive X-Ray Spectroscopy) to be a nickel particle.
Spent catalyst samples were further studied by the use of X-Ray Diffraction
(XRD).
The XRD patterns, suggest that graphitic carbon constituents with different
degrees
of defect or distortion are present in the deactivated samples. TEM
micrographs of
the fully deactivated sample show that the growth of the carbon is terminated
as a
result of spatial limitations. The modes of filament termination include the
nickel
particle's restriction by the silica surface, the arm and the tip of another
carbon
filament. Formation of carbon filaments as a result of hydrocarbon cracking
has
been extensively reported in the literature with higher molecular weight
hydrocarbons over supported nickel, iron, cobalt and several alloy catalysts.
The
carbon deposited on the catalyst in carrying out the present invention may be
used in
electrochemical applications such as superconductors, electrodes and fuel
cells.
The reversibility of filament growth and the regeneration of the catalyst has
been previously considered by others, but no results have been reported on
whether
the catalyst activity can be restored. In fact, some workers have suggested
that
catalyst regeneration may be futile in view of the changes caused to the
catalyst
support structure as a result of the filament growth process. Conventional
oxidation
in air (C + OZ = COZ) and steam gasification (C + xH20 = COx + xH2) were
considered as potential routes for the regeneration of the catalyst at 823 K.
Surprisingly, both methods appear to be able to fully restore the activity of
the
catalyst, as shown in Figure 2 where the methane conversion is plotted against
time-
on-stream for the fresh and the regenerated catalysts. The oxidation process
was
faster than the steam gasification, but caused a high temperature front. This
front
gradually moved through the catalyst bed, causing the collapse of the sample
to a
fine powder. XRD analysis suggests that the oxidation process completely
removed
CA 02317395 2000-07-10
WO 99/43608 PCTNS99/03556
_g_
the deposited carbon and converted the metallic nickel into nickel oxide which
had
to be reduced in flowing hydrogen before the next reaction cycle shown in
figure 2.
On the contrary, the catalyst bed maintained a uniform temperature profile
during
the steam regeneration process and the catalyst preserved its metallic nickel
form at
the end of the process.
Another difference between the two regeneration methods is that the steam
gasification leads to the production of additional hydrogen. This is at the
expense of
external thermal energy, since the oxidation of carbon releases a large amount
of
heat. Nevertheless, the additional production of hydrogen may be of
significant
practical importance. Theoretical yields of four and three moles of hydrogen
per
mole of methane can be achieved respectively, with steam reforming and partial
oxidation of methane, provided that all carbon monoxide is converted to carbon
dioxide by the water-gas shift reaction. In comparison, methane cracking
produces
less hydrogen {two moles) from each mole of methane. The hydrogen yield
however, can be dramatically improved if the hydrogen produced during the
regeneration step is also accounted for. In this case, 2 moles of hydrogen
were
obtained from each mole of methane during the cracking step and an additional
1.4
moles were produced during the subsequent steam gasification of the deposited
carbon, leading to an overall hydrogen yield of 3.4 moles for each mole of
methane.
This overall hydrogen yield is slightly less than that from steam reforming,
but
better than that for partial oxidation.
Formation of carbon monoxide and methane were also detected during the
initial stage of the steam regeneration, where, as shown in Figure 3, the
carbon
dioxide and hydrogen selectivities are less than 100%. Increased selectivities
of
carbon dioxide and hydrogen, obtained when the regeneration was conducted at a
higher space velocity, appear to suggest that carbon monoxide and methane may
be
secondary products, formed due to the reverse water-gas shift (HZ + C02 = CO +
HZO) and methanation (CO + 3 HZ = CH,+H20) reactions. These observations
suggest that one may be able to e#ficiently control the selectivity of
hydrogen
production during the regeneration step by adjusting the space velocity.
CA 02317395 2000-07-10
WO 99/43608 PGT/US99/03556
-9-
TEM micrographs of the steam regenerated sample show that some remains
of the large filaments are still present on this catalyst after the
regeneration has been
completed. The presence of these remains however, does not appear to have a
negative effect on catalytic activity which is fully restored following the
steam
regeneration step (Figure 2). Following a few cycles of deactivation and steam
regeneration however, an air oxidation step will be probably required, to
completely
remove the accumulated remains.
The set of Ni-Cu/SiOZ catalysts used in the second embodiment of this
invention had the total metal amount (on a molar basis) maintained constant at
2.6
mmole of metal/g of support while the ratio of Ni:Cu was varied from
approximately
8:1 to approximately 1:8. The catalysts were prepared by incipient wetness
impregnation of nickel and copper nitrates (Ni(N03)2x6HZ0 and Cu(N03)2x2.5Hz0)
obtained from Aldrich {with a purity of 99.999%) onto commercially available
SiOz
(Davison Syloid 74). Prior to impregnation the silica support was dried,
pressed into
pellets under a pressure of 15,000 psig, crushed and sieved to obtain a
granulometric
fraction in the 20-35 mesh size. The impregnated samples were dried in a
vacuum
oven at 120 °C overnight and subsequently calcined in a muffler furnace
at 700 °C
for 6 hours. The Ni and Cu loadings were estimated by the weight difference
between the blank support and the catalyst reduced overnight in a 1:2 HZ/NZ
mixture
(total flow rate of 120 ml/min) at 650 °C .
Following the reduction treatment, the samples were exposed to methane
(GHSV - 6000 hr'') at 650 and 800 °C . Activity measurements were
conducted at
two different temperatures and the results are presented in Fig. 4. The
results
indicate that the presence of small amounts of Cu enhanced significantly the
initial
activity at 800 °C , while the presence of Cu had no significant effect
at 650 °C. The
initial conversion over the 2.3 mmole Ni/0.3 mmole Cu/Si02 composition for
example, was measured at 63%, as compared to 14.4% for the 2.3 mmole Ni/SiO~
composition. This is a surprising result given that Cu alone is not active for
the
cracking of methane under these conditions (0.3% initial methane conversion
for the
CA 02317395 2000-07-10
WO 99/43608 PCTNS99/03556
-10-
0.3 mmole Cu/SiOz composition). The promoting effect is also more pronounced
when small amounts of Cu are added (i.e., Ni:Cu ratios greater than 1).
While the present invention has been particularly shown and described with
reference to the preferred mode as illustrated in the drawing, it will be
understood by
one skilled in the art that various changes in detail may be effected therein
without
departing from the spirit and scope of the invention as defined by the claims.