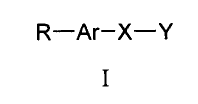Note: Descriptions are shown in the official language in which they were submitted.
CA 02319694 2000-09-15
I
Compound
The present invention relates to a pharmaceutical compound and composition for
use
in medicine.
It is well known in the art that phosphonate compounds, for example
bisphosphonates, have a high affinity for hydroxyapatite crystals and thus
tend to
localise in vivo in regions of bone metabolism. Moreover, it has also been
established that phosphonate compounds are generally low in toxicity.
US 4,880,007 (Amersham International PLC) discloses complexes formed between
(a) an amino di- or polyphosphonate; and (b) a paramagnetic metal ion, such as
gadolinium (III). Such complexes exhibit calcified tissue seeking properties
which
I S render them useful as contrast agents for investigating bone metabolism by
NMR
scanning.
Similarly, US 5,236,695 (Concat Ltd.) discloses polyphosphonate ligands
containing
three or more phosphonate groups, combined with paramagnetic metal cations
which
are administered in the form of pharmacologically acceptable salts. Such
compounds
are useful as MRI contrast agents which tend to localise in bone tissue
without being
conjugated to bone-specific biomolecules.
Bisphosphonates have also been used in combination with technetium-99 (Tc-
99m).
Indeed, Tc-99m is routinely used with carriers such as methylene
bisphosphonate,
for imaging in hospitals. Furthermore, US 4,830,847 discloses diphosphonate-
derivatised macromolecules, such as proteins, suitable for use as technecium-
99m
based scanning agents and anticalcification agents. Typically, the scanning
agents
are prepared by combining Tc-99m in a 3+, 4+ and/or 5+ oxidation state with
the
disphosphonate-derivatised macromolecule. Also disclosed are pharmaceutical
compositions containing such diphosphonate-derivatised macromolecules.
CA 02319694 2000-09-15
2
The therapeutic applications of bisphosphonate compounds are also well
documented in the art. For example, WO 96/39150 (Merck & Co., Inc.) discloses
the
use of bisphosphonates, such as alendronate, in the prevention or treatment of
bone
loss associated with rheumatoid arthritis. Similarly, GB 2,331,459 (SPA)
discloses
an injectable composition for treating skeletal and bone disorders which
comprises a
bisphosphonate in combination with a benzyl alcohol. Eisenhut et al.. (Appln.
Radiat. Isot., Vol 38, No.7, p535-540) disclose the use of 13~I-labelled
benzylidinediphosphonates for the palliative treatment of bone metastases.
Finally,
WO 95/11029 (Merck & Co., Inc.) discloses compositions comprising
bisphosphonate and growth hormone secretagogues, which are useful for reducing
the deleterious effects of osteoporosis in elderly patients.
The present invention seeks to provide improved phosphonate compounds for use
in
medicine. In particular, the invention seeks to provide pharmaceutical
compounds
which exhibit improved activity in the palliative and curative treatment of
bone
disorders, and/or which may also be suitable for use in medical imaging
techniques.
Statement of Invention
Aspects of the invention are presented in the accompanying claims and in the
following description.
In the broadest aspect, the present invention relates to a pharmaceutical
compound
for use in medicine. The pharmaceutical may be for a therapeutic use and/or a
diagnostic use.
More specifically, the present invention provides a pharmaceutical compound,
or
pharmaceutically acceptable salt thereof, for use in medicine, wherein said
compound is of formula I
R-Ar-X-Y
I
CA 02319694 2000-09-15
3
wherein
R is a pharmaceutically active moiety;
Ar is an aromatic moiety;
X is a linker group; and
Y is a moiety comprising two phosphonate groups.
In a preferred aspect of the invention, Y comprises a geminal bisphosphonate
group.
In a further preferred aspect, the invention provides a pharmaceutical
compound, or
pharmaceutically acceptable salt thereof, of formula II
P03H2
R-Ar-X~ p03Hz
~z
II
wherein Z is H, NH2 or an oxy substituent.
Preferably, Z is H or OH.
The pharmaceutical compound of the present invention comprises a linker group,
X.
In a preferred aspect, the linker group of the invention is a substituted or
unsubstituted C ~ ~ alkylene group.
In an alternative preferred aspect of the invention, X is a C, _4 amine group,
C 1 ~ ether
group or a C,_4 thioether group, each of which may be substituted or
unsubstituted.
In another preferred aspect of the invention, X is S=O or S02.
CA 02319694 2000-09-15
4
Where X is substituted, suitable substituents include one or more groups which
do
not interfere with the pharmaceutical activity of the compound in question.
Exemplary non-interfering substituents include hydroxy, amino, halo, alkoxy,
and
alkyl.
The pharmaceutical compound of the present invention also comprises an
aromatic
moiety, Ar.
Preferably, the aromatic moiety of the compound is electron deficient.
In a more preferred aspect, the aromatic moiety of the invention is a single
aromatic
ring. However, other aromatic moieties are also suitable for use in the
invention,
for example, aromatic moieties comprising more than one aromatic ring, where
the
aromatic rings may be fused together or joined via one or more suitable spacer
groups
Examples of aromatic moieties suitable for the present invention include
substituted
or unsubstituted phenyl, naphthyl, thiophenyl, furyl, pyridyl and pyrrole
groups.
Where the aromatic moiety is substituted, suitable substituents include one or
more
groups which do not interfere with the pharmaceutical activity of the compound
in
question. Exemplary non-interfering substituents include hydroxy, amino, halo,
alkoxy, and alkyl.
The pharmaceutical compound of the present invention also comprises a
pharmaceutically active moiety, R.
In a preferred aspect, R comprises a radiolabel. Examples of radiolabels
suitable for
use in the present invention include 124h ~25I, ~3iI, 2uAt (an a-emitter),
~g6Re, Tc
99m, and (3-emitting bromine nuclei.
CA 02319694 2000-09-15
In an alternative preferred aspect, the pharmaceutically active moiety of the
invention may comprise a functional group (or ligand) to which a metal ion can
be
chelated, or is chelated thereto. Species of the former type, i.e. those
comprising a
functional group to which a metal ion can be chelated, could be potentially
useful for
complexing any excess radiolabel close to the bone, thereby preventing
radiolabel
polsomng.
By way of definition, the term "chelate" refers to a complex in which a ligand
is
coordinated to a metal ion at two or more points, so that there is a ring of
atoms
including the metal, and where the term "ligand" refers to an ion or molecule
that
can donate a pair of electrons to said metal ion.
Suitable functional groups or ligands to which a metal ion may be chelated
include
amine, hydroxy, or carboxylic acid moieties.
In a particularly preferred aspect, the metal ion chelated to the functional
group is
paramagnetic. In the present context, the term "paramagnetic" refers to metal
ions
having net orbital or spin magnetic moments that are capable of being aligned
in the
direction of an applied magnetic field. Such atoms have a positive (but small)
susceptibility and a relative permeability slightly in excess of one.
Paramagnetism
occurs in all atoms with unpaired electrons, e.g. transition metal ions with
unpaired
electron shells.
Examples of suitable paramagnetic metals include those of the lanthanide
elements
with atomic numbers 58 to 70, and those of the transition metals with atomic
numbers 21 to 29, 42 and 44. Typical examples include chromium (III),
manganese
(II), iron (II), iron (III), cobalt (II), nickel (II), copper (II),
praesodymium (III),
neodymium (III), samarium (III), gadolinium (III), terbium (III), dysprosium
(III),
holmium (III), erbium (III) and ytterbium (III).
CA 02319694 2000-09-15
6
The pharmaceutically active moiety, R, may also comprise a paramagnetic
component other than a paramagnetic ion, for example, a moiety comprising the
group NO'.
Compounds of the invention containing paramagnetic moieties or radiolabels may
have applications in the field of medical imaging, especially calcified tissue
imaging. In particular, such compounds may be administered to a patient in
order to
preferentially enhance the NMR image contrast in tissue. By way of definition,
the
term "NMR" also encompasses magnetic resonance imaging (MRI) in which images
of tissue are produced by magnetic resonance techniques.
Thus, in one preferred aspect, the invention provides a pharmaceutical carrier
molecule for a radiolabel. In a particularly preferred aspect, the radiolabel
is Tc-
99m.
The term "calcified tissue" refers to bone, regions of bone metabolism,
regions of
calcified tumours and other diseased tissues.
In a particularly preferred aspect of the invention, the pharmaceutically
active
moiety R is attached directly to the aromatic moiety, Ar.
By way of definition, the term "pharmaceutically acceptable salt" includes any
salt
that has the same general pharmacological properties as the parent species
from
which it is derived, and which is acceptable from a toxicity view-point.
Typical
pharmaceutically acceptable salts include acid addition salts, base salts or
solvates or
hydrates thereof. A review of suitable salts may be found in Berge et al., J.
Pharm.
Sci., 1977, 66, 1-19.
Suitable acid addition salts are formed from acids which form non-toxic salts.
Examples include hydrochloride, hydrobromide, hydroiodide, sulphate,
bisulphate,
nitrate, phosphate, hydrogen phosphate, acetate, maleate, fumarate, lactate,
tartrate,
CA 02319694 2000-09-15
citrate, gluconate, succinate, saccharate, benzoate, methanesulphonate, ethane-
sulphonate, benzenesulphonate, p-toluenesulphonate and pamoate salts.
Suitable base salts are formed from bases which form non-toxic salts and
examples
include alkali metal (sodium and potassium), alkaline earth metal (calcium and
magnesium), aluminium, non-toxic heavy metal (zinc, stannous and indium),
ammonium and low molecular weight substituted ammonium (mono-, di- and
triethanolamine) salts.
Methods for preparing pharmaceutically acceptable salts of compounds of the
invention will be familiar to those skilled in the art. Typically,
pharmaceutically
acceptable salts may be prepared by mixing together a solution of the agent
and the
desired acid or base, as appropriate. The salt may be recovered by evaporation
of
the solvent, or by precipitation from solution followed by filtration.
The compound of the present invention may exist in polymorphic form.
The compound of the present invention may contain one or more asymmetric
carbon
atoms and thus may exist in two or more stereoisomeric forms. The present
invention includes the individual stereoisomers of the compound and, where
appropriate, the individual tautomeric forms thereof, together with mixtures
thereof.
Diastereoisomers of compounds of the invention may be separated by
conventional
techniques such as fractional crystallisation, chromatography or H.P.L.C. of a
stereoisomeric mixture of the agent or a suitable salt or derivative thereof.
Individual
enantiomers of the agent may also be prepared from the corresponding optically
pure intermediate or by resolution, such as by H.P.L.C. of the corresponding
racemate using a suitable chiral support. Alternatively, individual
enantiomers may
be prepared by fractional crystallisation of the diastereoisomeric salts
formed by
reaction of the corresponding racemate with a suitable optically active acid
or base,
as appropriate.
CA 02319694 2000-09-15
8
The present invention also includes all suitable isotopic variations of the
compound,
or pharmaceutically acceptable salts thereof. The term "isotopic variation" as
used
herein refers to a compound in which at least one atom is replaced by an atom
having the same atomic number but an atomic mass different from the atomic
mass
usually found in nature. Examples of isotopes that can be incorporated into
the
compounds of the invention, or pharmaceutically acceptable salts thereof,
include
isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorus, sulphur, fluorine
and
chlorine such as 2H, 3H, ~3C, 14C, ESN, 1~0, ~gp, 31p~ 32P~ 355 ~sF ~d 3601,
respectively. Isotopic variants in which a radioactive isotope is incorporated
may
have applications in drug and/or substrate tissue distribution studies.
Typical
examples preferred for their ease of preparation and detectability include
tritium
(3H), and carbon-14 (14C) isotopes. Substitution with other isotopes such as
deuterium (2H) may afford certain therapeutic advantages resulting from
greater
metabolic stability, i.e., increased in vivo half life or reduced dosage
requirements.
Isotopic variations of the agent of the present invention and pharmaceutically
acceptable salts thereof of this invention can generally be prepared by
conventional
procedures familiar to those skilled in the relevant art using the appropriate
isotopic
variations of suitable reagents.
It will be appreciated by those skilled in the art that the compound of the
present
invention may be derived from a prodrug. By way of example, a prodrug includes
any entity having one or more protected groups) and which may not possess
pharmacological activity per se, but may, in certain instances, be
administered (for
example orally or parenterally) and thereafter metabolised in the body to form
the
pharmaceutically active agent of the present invention.
The skilled person in the art will further appreciate that certain moieties
known as
"pro-moieties", for example as described in "Design of Prodrugs" by H.
Bundgaard,
Elsevier, 1985 (the disclosured of which is hereby incorporated by reference),
may
be placed on appropriate functionalities of the compounds. Such prodrugs are
also
intended to fall within the scope of the present invention.
CA 02319694 2000-09-15
9
The present invention also provides a pharmaceutical composition comprising
the
compound provided by the present invention admixed with a pharmaceutically
acceptable carrier, diluent, or excipient (including combinations thereof).
The pharmaceutical compositions may be for human or animal usage in human and
veterinary medicine and will typically comprise any one or more of a
pharmaceutically acceptable diluent, carrier, or excipient. Acceptable
carriers or
diluents for therapeutic use are well known in the pharmaceutical art, and are
described, for example, in Remington's Pharmaceutical Sciences, Mack
Publishing
Co. (A. R. Gennaro edit. 1985).
Examples of suitable carriers include lactose, starch, glucose, methyl
cellulose,
magnesium stearate, mannitol, sorbitol and the like. Examples of suitable
diluents
include ethanol, glycerol and water.
The choice of pharmaceutical carrier, excipient or diluent can be selected
with
regard to the intended route of administration and standard pharmaceutical
practice.
The pharmaceutical compositions may comprise as, or in addition to, the
carrier,
excipient or diluent any suitable binder(s), lubricant(s), suspending
agent(s), coating
agent(s), solubilising agent(s).
Examples of suitable binders include starch, gelatin, natural sugars such as
glucose,
anhydrous lactose, free-flow lactose, beta-lactose, corn sweeteners, natural
and
synthetic gums, such as acacia, tragacanth or sodium alginate, carboxymethyl
cellulose and polyethylene glycol.
Examples of suitable lubricants include sodium oleate, sodium stearate,
magnesium
stearate, sodium benzoate, sodium acetate, sodium chloride and the like.
Preservatives, stabilizers, dyes and even flavoring agents may be provided in
the
pharmaceutical composition. Examples of preservatives include sodium benzoate,
CA 02319694 2000-09-15
sorbic acid and esters of p-hydroxybenzoic acid. Antioxidants and suspending
agents may be also used.
5 The composition/formulation requirements may vary depending on the delivery
systems. By way of example, the pharmaceutical composition of the present
invention may be formulated to be delivered using a mini-pump or by a mucosal
route, for example, as a nasal spray or aerosol for inhalation or ingestable
solution.
Alternatively, the composition may be formulated in an injectable form, for
10 parenteral delivery, for example, by an intravenous, intramuscular or
subcutaneous
route. The formulation may also be formulated so as to be suitable for
delivery by
both routes.
Where the agent is to be delivered mucosally through the gastrointestinal
mucosa, it
should be able to remain stable during transit though the gastrointestinal
tract; for
example, it should be stable at acid pH, resistant to proteolytic degradation
and to
the detergent effects of bile.
The pharmaceutical compositions of the invention can be administered topically
in
the form of a lotion, solution, cream, ointment or dusting powder, or by the
use of a
skin patch. Alternatively, the compositions can be administered orally in the
form of
tablets containing excipients such as starch or lactose, or in capsules or
ovules either
alone or in admixture with excipients, or in the form of elixirs, solutions or
suspensions containing flavouring or colouring agents, or in the form of a
suppository or pessary. Further modes of administration include inhalation, or
parenteral injection, for example intravenously, intramuscularly or
subcutaneously.
For parenteral administration, the compositions may be used in the form of a
sterile
aqueous solution which may contain other substances, for example appropriate
levels of salts and/or monosaccharides to make the solution isotonic with
blood. For
buccal or sublingual administration the compositions may be administered in
the
form of tablets or lozenges which can be formulated in a conventional manner.
CA 02319694 2000-09-15
11
Typically, a physician will determine the actual dosage which will be most
suitable
for an individual patient and it will vary with the age, weight and response
of the
particular patient.
In a preferred aspect, the present invention relates to the use of the
compound or
composition of the invention in the palliative or curative treatment of bone
disorders.
Preferably, the compounds and/or compositions of the invention are used to
treat
bone disorders that are cancer-related skeletal diseases, for example,
skeletal
metastases, osteoblastic osteosarcoma or multiple myelomas. However, the
compounds and/or compositions of the invention may also be used to treat non-
cancerous bone disorders, such as age-related bone loss, rheumatoid-related
bone
loss, or bone loss related to osteoporosis, disuse or steroid therapy.
The treatment of skeletal metastases is one of the main problems encountered
in
practical clinical oncology. It is estimated that up to 85 % of all patients
with
advanced carcinomas to the breast, prostate or lung develop such metastatic
conditions (Bijvoet, O.L.M., Fleisch, H.A., Bisphosphonate on Bones, Elsevier
Science B.V., Amsterdam, 1995; pp. 349). Up to now, the prognosis for these
patients has been poor.
To date, treatment methods in current clinical practice include external
radiotherapy,
hormone therapy and chemotherapy, although the number of patients achieving
complete curation is negligible (Bijvoet, O.L.M., Fleisch, H.A.,
Bisphosphonate on
Bones, Elsevier Science B.V., Amsterdam, 1995; pp. 350). In particular, severe
side-
effects often limit the applicability of these methods. Consequently, there is
a strong
need for improved therapeutic methods for slowing down tumour progression and
for pain palliation.
Normally, the main method for radiation therapy is external beam irradiation.
However, if there are multiple skeletal metastases present, a more efficient
treatment
CA 02319694 2000-09-15
12
may be the targeted radiotherapy of timorous osseous lesions by means of
radioactive compounds with bone affinity. Such regions are characteristic for
several
types of bone-related diseases, malignant as well as benign. Examples of
malignant
lesions are skeletal metastases and osteosarcoma, examples of benign lesions
are
osteoporosis and Paget's disease (Bijvoet, O.L.M., Fleisch, H.A.,
Bisphosphonate on
Bones, Elsevier Science B.V., Amsterdam, 1995; pp. 293). Pathological bone
synthesis in skeletal metastases is observed in very large groups of cancer
patients
who develop metastatic foci originating from different types of soft-tissue
tumours.
Furthermore, there is a small but important, clinically difficult niche of
patients
suffering from metastasised osteoblastic osteosarcoma. In this case, a
primitive
bone-like substance, osteoid, is produced by the tumour cells themselves.
Although
clinically very different from skeletal metastases, the chemistry of these
lesions is
similar, and they may often be targeted by the same type of bone affinity
compounds.
Usually, myelotoxicity sets the limits for the radiation dose that can be
administered
to the tumour by means of radioactive bone-affinity compounds. Any development
which decreases the radiation dose to the bone marrow would therefore
substantially
improve targeted radiotherapy techniques.
In a preferred aspect, the present invention thus seeks to provide improved
bone
affinity compounds which expose the bone marrow to substantially lower levels
of
radiation.
There are two classes of radioactive bone-affinity compounds currently in
clinical
use. The first and most important class includes ions of the radioisotopes of
alkaline
earth elements, such as g9Sr2+, which is the most common bone-seeking agent in
clinical use (Lewington, V.J., Cancer therapy using bone-seeking isotopes,
Physics
in Medicine and Biology, 1996; 41: 2031-2032).
CA 02319694 2000-09-15
13
The other class of bone-seeking compounds include radioactive bis- or
polyphosphonic acids carrying a (3-active radionuclide. The species most
commonly
used in the clinic are ~g6Re-HEDP (Lewington, V.J., Cancer therapy using bone-
s seeking isotopes, Physics in Medicine and Biology, 1996; 41: 2030) and lssSm-
EDTMP (Lewington, V.J., Cancer therapy using bone-seeking isotopes, Physics in
Medicine and Biology, 1996; 41:2029). The latter has also been applied with
success
in the palliative treatment of osteoblastic osteosarcoma (Franzuis, C. et al.,
High
Activity Samarium-153-EDTMP Therapy in Unresectable Osteosarcoma,
Nuklearmedizin,1999; 38:337-340). However, in contrast to the compounds
disclosed in the present invention, none of these nuclides reduce the tumour
progression of small tumours, and hence they are not suitable for curative
treatment.
(3-emitters are usually classified into three groups according to (3-particle
energy and
hence range. With regard to physical properties, ~-particle energy and range
are
parameters which have to be matched to the size of the tumour. The half life
and
chemical properties, on the other hand, are related to the pharmacokinetics
and
metabolism of the carrier molecule. Radionuclides with (3 -energy in the range
Eavg =
0.08-0.18 MeV (mean range 0.4-0.9 mm) are best suited for treatment of small
tumours (Zweit, J., Radionuclides and carrier molecules for therapy, Physics
in
Medicine and Biology, 1996; 41: 1908-1910). ~31I is the most familiar and the
only
radionuclide in this group that has been used clinically. Radionuclides with
medium
~-energy such as ~s3Sm and ~86Re are less suitable for curative treatment,
since the
dose distribution will spare small tumours and the tumour on bone marrow ratio
is
too low. High energy (3 -emitters such as g9Sr are only suited for the
palliative and
curative treatment of large tumours. There is speculation as to whether the
use of a-
emitters in combination with (3-emitters would be superior for curative
treatment.
However, the short range of a-radiation (40-80 ~,m) would most likely require
bonding of the a-emitting carrier molecule to most cancer cells within the
tumour.
CA 02319694 2000-09-15
14
10
In an alternative aspect, the invention also relates to the use of the
compound of the
invention in the preparation of a medicament for the palliative or curative
treatment
of bone disorders.
As used herein the phrase "preparation of a medicament" includes the use of a
compound of the invention directly as the medicament in addition to its use in
a
screening programme for the identification of further active agents or in any
stage of
the manufacture of such a medicament.
Such a screening programme may for example include an assay for determining
whether a candidate substance is capable of mimicking the activity of a
pharmaceutical compound of the present invention.
Another aspect of the invention provides a method of treating a subject in
need of
the palliative or curative treatment of bone disorders, the method comprising
administering to said subject a therapeutically effective amount of the
compound or
composition of the present invention.
The present invention also provides a process for preparing a compound of the
invention, wherein R comprises a radiolabel, said process comprising the
following
steps:
(i) preparing a phosphonate precursor comprising Ar, X and Y;
(ii) radiolabelling said bisphosphonate precursor.
In a preferred aspect, step (ii) of the above-mentioned process is a
deiodosilylation
reaction.
For practical purposes, it is desirable to produce targeting radionuclide
agents at the
site of use, e.g. hospitals.
CA 02319694 2000-09-15
1$
To date, the demand for convenient labelling chemistry, high stability and
favourable biological behaviour has proven difficult to meet with
radiohalogenated
bisphosphonates. In recent studies, pre-labelled compounds have been connected
to
bisphosphonic acid functionalities (Fritzberg, A.R. et al., US5202109:
Conjugates
for bone imaging and bone cancer therapy; Murud, k. et al., Synthesis,
Purification,
and in Vitro Stability of 21 ~At- and ~25I-Labeled Amidobisphosphonates;
Nuclear
Medicine and Biology, 1999, 26). Favourable results have been obtained with
such
radioconjugates. However, this strategy requires two reactions involving
radioactivity and three purification steps to obtain the final product. When
working
with therapeutic doses of radioactivity such procedures are inappropriate in
view of
radiation safety standards. Up to now, no therapeutic experiments have been
conducted with such radioconjugated bisphosphonic acids.
The present invention thus provides an improved labelling technique for
bisphosphonic acids. More particularly, the invention focuses on precursors in
which
the label is incorporated in the final stages of the synthesis. The present
process is
therefore advantageous compared to the preparation of many of the
radionucleotide
agents currently in clinical use.
More specifically, the present invention employs trialkylarylsilyl precursors.
These
substances are easy to synthesise and appear to be very stable; moreover
iododesilylation affords the radioiodinated bisphosphonic acids in very high
yield.
Dialkylaryltriazene precursors were also investigated, but labelling yields
were
lower, and the final purification was hampered by the presence of many side
products.
From a chemical perspective, it is widely known that non-radioactive
bisphosphonic
acids may be used as pharmaceuticals for treating bone related disorders.
Moreover,
it is known that other moieties linked to bisphosphonic acids may also affect
bone
affinity. However, it is the bisphosphonic acid functionality that is
primarily
responsible for the bone affinity of such molecules. This was the motivation
for
CA 02319694 2000-09-15
16
synthesising a series of radioiodinated aromatic bisphosphonic acids with
different
alpha functionalities and with various linker groups inserted between the aryl
group
and the bisphosphonic acid moiety (Fig. 1 ).
The synthesis of compound 2g is depicted in Figure 2. The transformation of m-
chlorotoluene (2a) to m-trimethylsilyltoluene (2b) by use of magnesium and
trimethylchlorosilane in HMPTA is described in the literature (Effenberger, F.
and
Habich, D., Liebigs Ann. Chem., 1979, pp. 842-857). However, prolonged heating
is
required and the work-up uses large amounts of this strongly carcinogenic
solvent. A
second way of synthesising m-trimethylsilyltoluene is by reacting m-
bromotoluene
with molten sodium in toluene (Clark, H. A. et al., J. Am. Chem. Soc., 1951;
73:3798). However, this route is troublesome and hazardous. In contrast, by
refluxing m-chlorotoluene with magnesium in THF followed by the addition of
trimethylchlorosilane, m-trimethylsilyltoluene can be obtained in 88-90%
yield.
Trimethylsilyltoluene was then brominated with N-bromosuccinimide in carbon
tetrachloride as described in the literature (Severson, R. G. et al., J. Am.
Chem. Soc.,
1957; 79:6540). The resulting m-trimethylsilylbenzyl bromide (2c) was reacted
with
the lithium ylide of diethyl methylenephosphonate providing diethyl m-
trimethylsilylphenylethylidene-phosphonate (2d) in moderate yield. The
phosphonate was then converted to the corresponding lithium ylide using
butyllithium and the subsequent reaction with diethyl chlorophosphate gave the
tetraethyl bisphosphonate 2e in high yield. Hydrolysis was achieved by trans-
esterification to the corresponding tetramethylsilyl ester 2f, followed by the
addition
of aqueous ethanol. The bisphosphonic acid was isolated as the corresponding
disodium salt 2g.
The synthesis of compound 3g is depicted in Figure 3. m-Trimethylsilylbenzyl
bromide (2c) was converted to trimethylsilylbenzyl cyanide (3a) with potassium
cyanide under phase transfer conditions. Hydrolysis to m-
trimethylsilylphenylacetic
acid (3b) was then achieved by refluxing the cyanide with sodium hydroxide in
CA 02319694 2000-09-15
17
methanol. Overall yields in the range of 59-67% were obtained. The acid was
then
transformed to the acid chloride 3c employing thionyl chloride at ambient
temperature. The ultrasound-assisted reaction of the acid chloride with
trimethylphosphite afforded a cis/trans stereoisomeric mixture of the
enolphosphonate 3d. The second phosphonate moiety was introduced by adding
dimethylphosphite to the enolphosphonate under basic conditions. The
corresponding tetramethyl bisphosphonate 3e was trans-esterified to the
tetratrimethylsilyl ester 3f, which was subsequently hydrolysed by stirring
with
aqueous ethanol. The bisphosphonic acid was isolated as its disodium salt 3g.
Compounds 2g and 3g were labelled using an iododesilylation reaction (Figure
4).
This was achieved by adding n.c.a. (non carrier added) Na~3lI and N-
chlorosuccinimide as an oxidising agent to a solution of the precursor (2g,
3g) in a
mixture of acetic acid and trifluoroacetic acid at room temperature. The
yields
obtained were greater than 95%, as measured by HPLC. The radioiodinated
compounds were purified by HPLC and their structures were confirmed by
coelution
with the corresponding non-radioactive compounds.
Chemical and radiochemical purity is essential for the in vivo application of
radiopharmaceuticals in man. Chemical purity demands the isolation of the
radiolabelled compounds from all non-radioactive starting compounds and side-
products. The bisphosphonates described in the present invention were all
labelled
with n.c.a. (non carrier added) radioiodine. Purification and confirmation of
their
structures was achieved by HPLC. The demand for a non-toxic mobile phase along
with the complex aqueous chemistry of bisphosphonates made the development of
HPLC systems problematic. In particular, bisphosphonates are apparently
strongly
associated in aqueous solutions and form clusters with a range of sizes all
due to a
single compound (Wiedmer, W. H. et al., Ultrafiltrability and Chromatographic
Properties of Pyrophosphate, 1-Hydroxyethylidene-1,1-Bisphosphonate, Calci~
Tissue Int., 1983; 35:397-400). However, these problems were successfully
solved
CA 02319694 2000-09-15
I8
for analytical and purification purposes, resulting in single peaks for each
compound, whilst avoiding severe pressure build-up in the HPLC system
With regard to biological activity, the success of radionuclide targeting
therapy is
strongly dependent on the following conditions:
i) choice of radionuclide;
ii) the ability of the targeting compound to home in on the target quickly and
in
high amounts;
iii) the retention time at the site;
iv) resistance towards degradation;
v) little accumulation in other organs.
Details of the biological activity of the compounds of the invention are
discussed
further in the Examples section below.
In summary, the biological results clearly indicate that the compounds
disclosed
herein exhibit superior properties to previously reported radiohalogenated
bisphosphonic acids, in terms of bone affinity, selectivity, kinetics and
stability in
vivo. Moreover, the compounds of the invention are readily available and can
be
labelled in high yields by simple means from stable precursors
With regard to cancer treatment, the compounds of the invention that are
labelled
with radioiodine have been shown to seek out the desired target quickly,
selectively,
and in extraordinarily high amounts. In addition, the compounds display
resistance
to enzymatic dehalogenation.
The present invention will now be described only by way of example and with
reference to the accompanying figures, wherein:
Figure 1 shows the structure of bisphosphonate compounds 2h, 3h, 4d and Sx.
CA 02319694 2000-09-15
19
Figure 2 shows the reaction scheme for the preparation of compound 2g.
Figure 3 shows the reaction scheme for the preparation of compound 3g.
Figure 4 shows the reaction scheme for radiolabelling compounds 2g and 3g.
Figure 5 shows the biodistribution of intravenously administered compound 2h
in
balb/c mice (20-25g) after 30 min, 2 hours, 5 hours and 24 hours.
Figure 6 shows a bioscope picture of the spinal cord (balb/c mice) employing
compound 2h.
Figure 7 shows the biodistribution of intravenously administered compound 3h
in
balb/c mice (20-25g) after 30 min, 2 hours, 5 hours and 24 hours.
Figure 8 shows the biodistribution of intravenously administered compound 3h
in
nude/nude rats 60g) for HPLC purified 3h and crude 3h.
Figure 9 shows a scintillation picture of intravenously administered compound
3h in
nude/nude rat (60g) for HPLC purified compound.
Examples
m-Trimethylsilyltoluene
A solution of m-chlorotoluene (25.3 g, 0.20 mol), 1,2-dibromoethane (2.18 g,
11.6
mmol) and THF (40 cm3) was added to magnesium turnings (5.35 g, 0.22 mol). The
mixture was heated under reflux in an atmosphere of argon for 4 h, after which
the
heating bath was removed and trimethylchlorosilane (23.9 g, 0.22 mol) was
added so
that gently reflux was maintained. The mixture was stirred for another 30 min
and
CA 02319694 2000-09-15
then separated between 5% NaHC03 (100 cm3) and dichloromethane (3 x 100 cm3).
The organic extract was washed with brine and dried (MgS04). The solvent was
removed under reduced pressure affording the title compound (29.4 g, 89%) as a
5 pale yellow liquid, which was used without purification in the next
reaction. All
spectroscopic data were identical to those reported in the literature
(Eisenhut et al.,
ibiclJ.
m-Trimethylsilylbenzylbromide
10 The reaction was carried out as described in the literature (Bijvoet,
O.L.M. et al.,
Bisphosphonates on Bones, Elsevier Sciences B.V., Amsterdam, 1995, pp. 131 and
pp. 142) with the following modifications. The reaction mixture was
partitioned
between water and dichloromethane, the organic extract was washed with brine,
dried (MgS04) and the solvent evaporated. The residue was chromatographed on
15 silica gel using hexane as an eluent to give the title compound (96%) as an
orange
liquid. The product was used without further purification in the next
reaction.
m-Trimethvlsilvlbenzylcyanide
A mixture of KCN (5.5 g, 84 mmol), m-trimethylsilylbenzylbromide (5.31 g, 21.8
20 mmol), tributylamine (0.11 g, 0.59 mmol) and water (12.5 cm3) was stirred
overnight. The product was extracted with dichloromethane (3 x 30 cm3), the
extract
was filtered through a silica plug (5 g) and the solvent was removed under
reduced
pressure. The residue was distilled to give the product (2.27 g, 55%) as a
colourless
liquid.
Bp. 134-136°C (2 mm); V",~(film)/cni ~ 2956, 2255, 1412, 1249, 885 and
867; 8H
(200 MHz; CDC13; Me4Si) 0.31 (9H, s, CH3), 3.76 (2H, s, CH2) and 7.27-7.56(4H,
m, Ar); 8~ (50 MHz; CDCl3; Me4Si) 0.3, 24.3, 117.5, 127.7, 127.9, 128.6,
132.0,
132.3 and 141.2; m/z (EI) 189.0972 (M+-C~1H15NSi requires 189.0974).
CA 02319694 2000-09-15
21
m-Trimethylsil~phen~lacetic acid
To a solution of NaOH (4.0 g, 0.1 mol) in MeOH (20 cm3) m-trimethyl-
silylbenzylcyanide (2.84 g, 15.0 mmol) was added and the mixture was heated
under
reflux for 4 h. The solvent was evaporated and the residue was dissolved in
water
(50 cm3). The resulting solution was washed with dichloromethane (2 x 20 cm3),
the
aqueous phase was acidified with phosphoric acid to pH 2 and extracted with
dichloromethane (3 x 30 cm3). The extract washed with brine, dried (MgS04) and
the solvent evaporated to give the title compound (2.93 g, 94%) as a yellow
liquid.
Vm~(fllm)/crri ~ 3600-2600, 1717, 1418, 1254 and 847; 8H (200 MHz; CDC13;
Me4Si) 0.32 (9H, s, CH3), 3.70 (2H, s, CH2) and 7.30-7.53 (4H, m, Ar); 8~ (50
MHz;
CDC13; Me4Si) 0.53, 41.6, 127.8, 129.6, 132.1, 132.3, 134.1, 140.8 and 177.7;
m/z
(EI) 208.0918 (M+-C11Hi602Si requires 208.0920).
Dimethyl m-trimethylsilyl~henylacetylphosphonate
To m-trimethylsilylphenylacetic acid (1.04 g, 5.0 mmol) was added SOCl2 (0.82
g,
6.9 mmol) and the mixture was stirred overnight. Excess SOC12 was removed by
adding toluene (2 cm3) and then evaporating the solvent under reduced
pressure. The
residue was dissolved in THF (5 cm3) and trimethylphosphite (0.69 g, 5.5 mmol)
was added at -20°C. The resulting solution was kept in an ultrasound
bath at 0°C for
min, after which the solvent was evaporated to leave the phosphonate as a
cream
coloured semisolid (1.63 g, 5.0 mmol). The product decomposed upon exposure to
25 the atmosphere. An analytical sample was prepared by washing the crude
product
with hexane and was shown to consist of a mixture of two stereoisomeric enols.
The
spectral data represents the major component.
VmaX(fllm)/crri ~ 3600-2600, 1698, 1249 and 1036; 8H (200 MHz; CDC13; Me4Si)
30 0.19 (9H, s, CH3), 3.86 (6H, d , J 14), 6.11 (1H, d, J 13), 7.20-7.78 (4H,
m, Ar) and
7.92 (1H, d, J 7); 8(50 MHz; CDC13; Me4Si) 0.68, 54.6, 117.5, 118.1, 128.1,
130.4,
CA 02319694 2000-09-15
22
132.9, 133.6, 134.0, 134.9, 138.3, 140.6 and 142.3; 8P(500 MHz; CDC13; H3POa)
16.3; ; m/z (EI) 300.0929 (M+-C,3H21OaPSi requires 300.0947).
Tetramethyl m-trimethylsilylphenyl-1-hydroxy-ethylidenebis(phosphonate)
To a solution of dimethyl m-trimethylsilylphenylacetylphosphonate ( 1.63 g,
5.0
mmol) in Et20 ( 10 cm3) was added a solution of dimethylphosphite (0.83 g, 7.5
mmol), dibutylamine (0.32 g, 2.5 mmol) and hexane (10 cm3) at 0°C. A
white solid
began to form after a few minutes. After 20 min the mixture was filtrated to
give the
title compound (1.34 g, 65%) as a white solid. Recrystallization was achieved
by
adding water (9.5 cm3) to a solution of the title compound (0.50 g) in acetic
acid (0.5
cm3) and then leaving the resulting mixture for several days in the
refrigerator.
Filtration afforded the bisphosphonate (0.33 g, 66%) as white crystals.
VmaX(neat)/crri ~ 3700-2815, 2958, 1645 and 1251; 8H (200 MHz; CDC13; MeaSi)
0.25 (9H, s, CH3), 3.38 (2H, t, J 14), 3.71-382 (12H, m, OCH3) and 7.26-7.51
(4H,
m, Ar); 8~ (50 MHz; CDC13; MeaSi) 0.1, 40.1, 54.9, 73.3, 76.3, 79.3, 127.7,
132.1,
132.4, 133.8, 136.6 and 140.2; bP(500 MHz; CDC13; H3POa; m/z (EI) 410.1080 (M+-
CISH2gO~P2Si requires 410.1080).
Disodium m-trimethylsil~phen~l-1-h dy roxy-ethylidenebis phosphonic acid)
To tetramethyl m-trimethylsilylphenyl-1-hydroxy-ethylidenebis(phosphonate)
(0.82
g, 2.0 mmol) was added trimethylbromosilane (2.45 g, 16 mmol) and the
resulting
solution was stirred for 1 h. Excess reagent was then removed under reduced
pressure and the residue was dissolved in EtOH (75%, 8 cm3) at 0°C and
stirred for
0.5 h. The resultant solution was neutralised to pH 8 with aqueous Na2C03 (1.0
M, 3
cm3) and filtrated to give the disodium bis(phosphonic acid) salt (0.75 g,
94%) as a
white solid. The product was used without purification in the next reaction.
CA 02319694 2000-09-15
23
Tetramethyl m-iodophenyl-1-hydroxy-ethylidenebis(phos~honate)
Tetramethyl m-trimethylsilylphenyl-1-hydroxy-ethylidenebis(phosphonate) (0.41
g,
1.0 mmol) was added to a solution of ICl (0.32 g, 2.0 mmol) in acetic acid (2
cm3)
and the mixture was stirred for 5 h. The solvent was then evaporated and the
residue
was extracted with 1:1 hexane:Et20 (2 x 10 cm3). Remaining solvents were
removed
from the residue under reduced pressure affording the title compound (0.38 g,
81%)
as an orange liquid.
8H (200 MHz; CDC13; Me4Si) 3.18 (2H ,t, J 14) 3.68-3.79 (12H, m, OCH3) and
6.95-7.69 (4H, m, Ar); 8~ (50 MHz; CDC13; Me4Si) 39.7, 55.8, 56.3, 73.3, 76.3,
79.3, 94.2, 129.9, 130.8, 136.3, 136.8 and 140.2; 8P(500 MHz; CDC13; H3P04)
19.6.
m-Iodophenyl-1-hydroxy-ethylidenebisl;phosphonic acid)
A sample of tetramethyl m-iodophenyl-1-hydroxy-ethylidenebis(phosphonic acid)
was prepared by refluxing the parent tetraester in concentrated hydrochloric
acid for
3 h. Evaporation to dryness gave the title compound as an orange powder.
m-ji3il]_Iodophenyl -1-hydroxy-ethylidenebis(phosphonic acid)
To a solution of Na131I in water (1-2 ~l) was added a solution of N-
chlorosuccinimide (0.4 mg, 3 p.mol) in 9:1 TFA:AcOH (10 ~,1) followed by the
addition of disodium m-trimethylsilylphenyl-1-hydroxy-ethylidenebis(phosphonic
acid) (0.1 mg, 0.25 pmol) in TFA (2 pl). The mixture was vortexed and left for
5
min.
CA 02319694 2000-09-15
24
Biolo;~ical Testing
In order to assess the targeting ability of the radioiodinated bisphosphonic
acids
described, tissue distributions were carried out in healthy mice.
24 h after injection, the bone uptake of phenylethylidenebisphosphonic acid
(2h)
was averaging 15 ~ 5 % ID/g (injected dose per gram bone tissue) in femur,
with a
peak value of 21 ~ 4 % ID/g after 2 h, indicating that the entire molecule is
involved
in the bone affinity.
Compound 2h contains a hydrogen atom alpha to the bisphosphonate moiety and it
is known in the art that hydroxybisphosphonic acids possess a superior bone
affinity
in general (Bijvoet, O.L.M. et al., ibiclJ. Consequently, the alpha hydrogen
in the
phenylethylidenebisphosphonic acid was substituted with a hydroxy group. The
resulting hydroxybisphosphonic acid 3h showed a two-fold increase in bone
affinity
compared to the unsubstituted derivative 2h (Fig. 7). 30 min after injection
as much
as 45 ~ 7 % ID/g was observed in femur. The bone uptake remained high 24 h
after
injection (38 ~ 12 % ID/g), with a peak value of 46 ~ 4 % ID/g femur after 2
h. The
compound cleared rapidly from all other organs, except the kidneys (5 ~ 2 %
ID/g
24 h) and liver (2 ~ 0.7 % ID/g 24 h).
A fair resemblance has been found regarding uptake of bone seeking agents in
various specimens (Murud, K. et al., Evaluation of blood clearance, tissue
distribution, and bone microdistribution of 21'At and ~25I labeled
bisphosphonates in
dogs. Eur. J. Nucl. Med., 1999, submitted). However, care must be exercised
when
comparing different studies, since the age of the animals used is known to
greatly
affect bone uptake as well as kidney clearance (Hassfjell, S.P. et al., An
alpha
particle emitting bone seeking agent for targeted radiotherapy, Nucl. Med.
Biol.,
1997, in press). To overcome some of these problems an alternative parameter
for
the targeting ability of each compound was measured, namely % injected dose
per
gram tissue x animal body weight.
CA 02319694 2000-09-15
In this respect, the above-described compounds possess a bone uptake in the
range
900 to 1100 (% injected dose per gram tissue x animal body weight). In
contrast, the
most promising radioiodinated compound reported in the literature to date
possesses
5 a bone uptake of only slightly greater than 400 (Larsen, R.H. et al., 2uAt-
and ~31I-
labeled bisphosphonates with high in vivo stability and bone accumulation. J.
Nucl.
Med., 1999, in press). Since radioiodine is known to accumulate to a high
degree in
the thyroid gland (McNabb, F. M. Anne, Thyroid hormones, Englewood Cliffs,
N.J.:
Prentice Hall, 1992, pp. 23-25), uptake in the neck area is a good measurement
for
10 the stability of radioiodinated compounds. Indeed, about 0.05% of injected
dose
remained in the neck 24 h after injection, while values reported previously in
the
literature for this class of compounds have been in the range of 0.5% and
above.
Biodistribution of compound 3h in nude/nude rats
15 Nude/nude rats with a body weight averaging 60 g were used in the
biodistribution
experiments. The aim of the study was to compare crude 3h with HPLC-purified
3h.
The term "crude compound" refers to the diluted and neutralised labelling
reaction
mixture described hereinbefore. The compounds were administrated by tail vein
injections of 200 ~1 for each animal, using three animals for each sample. 24
h after
20 injection the animals were killed by a lethal injection and the tissue
distribution
determined.
A significant difference was observed between crude 3h and HPLC purified 3h
(Fig.
8). The purified compound possessed a 3-fold higher bone affinity than the
crude
25 product. Both samples showed high stability and selectivity although the
purified
sample was again superior. The bone affinity found for HPLC purified 3h was
consistent with the results obtained with balb/C mice. Femur uptake was found
to be
1020 %g dose/g. In contrast, the most promising radioiodinated compound
reported
in the literature to date possesses a bone uptake of only slightly above 400
%g
dose/g.
CA 02319694 2000-09-15
26
It is interesting to compare the selectivity found in this experiment with the
results
obtained with balb/c mice. In mice, the highest uptake in non target tissues
was
found in kidneys. After 24 h the ratio between femur and kidney was found to
be 7.
In rats the ratio between femur and kidney was found to be above 45,
indicating a
remarkable selectivity. A scintillation picture of the HPLC purified compound
in
nude/nude rat further demonstrated the high selectivity of this compound (Fig.
9).
To date, the most promising radioiodinated compound reported in the literature
possesses ratios between femur uptake and uptake in non target tissues of 3/2
(femurapleen), 7/3 (femur/liver) and 20 (femur/kidneys) after 24 h.
Various modifications and variations of the described methods of the invention
will
be apparent to those skilled in the art without departing from the scope and
spirit of
the invention. Although the invention has been described in connection with
specific preferred embodiments, various modifications of the described modes
for
carrying out the invention which are obvious to those skilled in chemistry or
related
fields are intended to be within the scope of the following claims.