Note: Descriptions are shown in the official language in which they were submitted.
CA 02320676 2000-09-20
30041-5D
1
Process and intermediates for the preparation of aminotriazine
derivatives
This application is a divisional of copending
Canadian Application 2,029,838 filed on November 13, 1990.
The present invention relates to a novel process for
the preparation of 4-amino-3-oxo-2,3,4,5-tetrahydro-1,2,4-
triazines.
The invention relates to a process for the
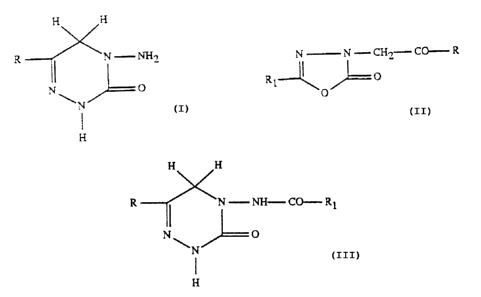
preparation of a compound of formula I
H H
5 4N-NHS ~I)
N 1 2 3~0
\ ,~ /'-
H
wherein R is hydrogen, C1-C6alkyl, C3-C6cycloalkyl, C1-C4a:Lkyl
substituted by from 1 to 10 halogen atoms or by from 1 to 3
radicals from the group C1-C3alkoxy, C1-C3alkylthio and phenyl,
phenyl or phenyl substituted by from 1 to 3 radicals from the
group halogen, methyl, ethyl, methoxy, methylthio and nitro,
which process comprises reacting with hydrazine hydrate a
compound of formula II
N N - CH~_- CO- R
III)
--~'~ O ~
wherein R1 is hydrogen, C1-C4alkyl, C3-C6cycloalkyl, C1-C4alkyl
substituted by from 1 to 9 chlorine atoms, C1-C3alkoxy,
C1-C3alkylthio, C1-C3alkylsulfinyl, C1-C3alkylsulfonyl, phenyl,
phenyl substituted by .from 1 to 3 radicals from the group
CA 02320676 2000-09-20
30041-5D
la
halogen, methyl, ethyl, methoxy, methylthio and nitro, or
pyridyl, and
R is as defined above; and subjecting the resulting
compound of formula III
CA 02320676 2000-09-20
-2-
H H
R ~ -N-NH-CO-R1
N~ O (
N
H
to hydrolysis, preferably acid hydrolysis.
The present process is preferably used for the preparation of compounds of
formula I
wherein R is methyl, ethyl, isopropyl, ten.-butyl or cyclopropyl. The process
is preferably
carried out using compounds of formula II wherein R1 is C1-C4alkyl as starting
materials.
The aminotriazine derivatives of formula I prepared according to the invention
can be
used as intermediates for the preparation of 4-[(pyrid-3-yl)-methyleneamino]-
or
4-[(pyrid-3-yl)-methylamino]-3-oxo-2,3,4,~-tetrahydro-1,2,4-triazines, which
are
distinguished by pronounced insecticidal and acaricidal activity. Such
pesticidal
compounds are, for example, 4-[(pvrid-3-yl)-methyleneamino]-3-oxo-6-methyl-
2,3,4,~-
tetrahydro-1,2,4-triazine, 4-[(pyrid-3-yl)-methyleneamino]-3-oxo-6-cyclopropyl-
2,3,4,5-
tetrahydro-i,2,4-triazine, 4-[(pvrid-3-yl)-methylamino]-3-oxo-6-isopropyl-
2,3,4,x-tetra-
hydro-1,2,4-triazine and 4-[(pvrid-3-yl)-methylamino]-3-oxo-6-tert.-butyl-
2,3,4,x-tetra-
hydro-1,2,4-triazine. Such pesticidal compounds, their preparation and use are
described
in EP Patent Application 314,615.
The process according to the invention can be illustrated by the following
reaction
scheme, the radicals R and R1 being as defined above:
N N-CH2-CO-R
~~ ring
R1~ ~O + HEN-NH2 ~ HBO ---1
~ O expansion
CA 02320676 2000-09-20
-3-
H H acid H H
hydrolysis
R ( ~N-NH-CO-R1 --1 R ~ ~N-NH2 + R1-COOH
N O N~ O
~N N
H H
(I)
The first step (ring expansion) of the process according to the invention for
the preparation
of the compounds of formula i is usually carried out under normal pressure and
preferably
in a solvent. The temperature is from +15 to 120°C, preferably from +20
to 100°C.
Suitable solvents are, for example, water, nitrites, such as acetonitrile,
alcohols, dioxane or
tetrahydrofuran. The subsequent hydrolysis of the acylamino compounds of
formula III to
form the free amino compounds of formula I is preferably carried out with
inorganic acids,
such as 1N hydrochloric acid to conc. hydrochloric acid or 1N to lON sulfuric
acid, at
temperatures of from 0 to 120°C, especially from +20 to 100°C,
in an aqueous medium or
in organic solvents, such as alcohols, dioxane, tetrahydrofuran> nitrites,
etc..
The 1,3,4-oxadiazolon-3-yl-ketones of formula II used as starting materials
according to
the invention are novel. They can be prepared analogously to known procedures,
for
example as follows (see, for example, EP Patent Application No. 314,615):
N NH ~ ,
R1~ ~O + X-CHZ-C-R ~ II
O
(IV) (V)
In the above formulae IV and V, R and R1 are as defined above and X is a
halogen atom,
preferably chlorine or bromine. The above process for the preparation of the
oxadiazolone
ketones of formula II is generally carried out under normal pressure in the
presence of a
base and in a solvent. The temperature is from 0 to +150°C, preferably
from +20 to
100°C. Suitable bases are organic and inorganic bases, for example
trimethylamine,
alcoholates, sodium hydroxide or sodium hydride. Suitable solvents are, inter
alia,
alcohols, halogenated hydrocarbons, for example chloroform, nitrites, for
example
acetonitrile, tetrahydrofuran, dioxane, dimethyl sulfoxide, dimethylformamide
or water.
CA 02320676 2000-09-20
3 ~ v z 1 - S
4
The oxadiazolones of formula ~IV [see EP Patent
Application No. 321,833; J. Pharm. Soc. Japan 76, 1300-1303
(1956); B. 82, 121-123 (1949)] and their preparation, and also
the haloketones of formula V, are for the most part known.
It is known from Liebigs Ann. Chem. 749, 125 ff.
(1971) that 4-amino-6-phenyl-3-oxo-2,3,4,5-tetrahydro-1.,2,4-
triazines can be obtained starting from 2-amino-5-methyl-3-
phenacyl-1,3,4-oxadiazolium bromide by reaction with hydrazine
hydrate. The main disadvantage of this process is that. it is
limited to the preparation of 1,2,4-triazine rings that. are
phenyl-substituted in the &-position; in addition, this process
comprises several steps and its yield is poor. Furthermore, it
is known from EP Patent Application No. 314,615 to prepare
4-amino-3-oxo-2,3,4,5-tetrahydro-1,2,4-triazines that are
substituted in the 6-positon, by reacting corresponding
5-trifluoromethyl-1,3,4-oxadiazolon-3-yl-ketones with excess
hydrazine in a one-step reaction:
N-CH2-CO-A H H
CF3--~ j O + H N~NH2 ~ A''' N NH2 .._
2 ~~ I
N~ N~ O
I
H
(II) (Ia)
wherein A may be an unsubstituted or substituted alkyl or aryl
substituent. The disadvantages of this process are primarily
the high cost of the trifluoroacetic acid ethyl ester required
for preparing the trifluoroacethydrazide, the instability of
that trifiuoroacethydrazide at room temperature, and the not
very high yield in the reaction of the trifluoroacethydrazide
with phosgene in water (see Helv. Chim. Acta 1986, 333) to
prepare the 5-trifluoromethyl-1,3,4-oxadiazol-2(3H)-one from
CA 02320676 2000-09-20
30,. rl-S
4a
which the 5-trifluoromethyl-1,3,4-oxadiazolon-3-yl-ketones of
formula IIa above (starting compound) are obtained. Moreover,
the reaction according to EP Patent Application No. 314,615
inevitably produces toxic trifluoroacetic acid derivatives as a
by-product, which present ecological problems and require a
considerable outlay for their disposal.
In contrast, within the scope of the present
invention it has now surprisingly been found that the presence
of a 5-CF3 group in the starting compounds of formula II is not
necessary for the preparation of the 4-amino-3-oxo-2,3,4,5-
tetrahydro-1,2,4-triazines of formula I by
CA 02320676 2000-09-20
-5-
ring expansion. The present starting compounds of formula II, which contain
one of the
mentioned radicals R1 in the ~-position instead of the mentioned CF3 group,
react readily
with hydrazine hydrate to form the acylamino compounds of formula III, from
which,
however, the radical -CO-R1 must subsequently be removed by acid hydrolysis to
obtain
the compounds of formula I. With the process according to the invention, the
disadvantages of the procedures hitherto available are eliminated since, in
the process
according to the invention, inexpensive and readily available starting
compounds can be
used, high yields are obtained and, instead of toxic trifluoroacetic acid
derivatives,
ecologically harmless carboxylic acid derivatives, for example acetic acid,
are formed as a
by-product.
Example 1: Preparation of the starting compound
2 3-Dihvdro-~-inethvl-2-oxo-1.3.4-oxadiazole-3-acetone
g of 2,3-dihydro-~-methyl-2-oxo-1,3,4-oxadiazole (prepared in customary manner
from
acethydrazide and phosgene) are added to a solution of ?.3 g of sodium in 100
ml of
methanol, the mixture is stirred for a short time and then the solvent is
removed in vacuo
at a bath temperature of 60°C. The sodium salt so formed is introduced
in portions into a
solution of 9.2 g of chloroacetone and 0.2 g of tetrabutylammonium bromide in
~0 ml of
chloroform, and the reaction mixture is stirred for 4 hours at 65°C.
After the salts have
been filtered off, the solvent is removed in vacuo at a bath temperature of
50°C. The
residue that remains is recrystallised from tert.-butyl methyl ether, yielding
the title
compound having a melting point of »-~7°C. ,
The following compounds of formula II are also prepared in a manner
corresponding to
that described above:
CA 02320676 2000-09-20
-6-
R1 R . phys. data
H -CH3 b.p. 0.08 toa/80°C
-CH3 -CH3 m.p. ~5-58°C
-C(CH3)3 -CH3 b.p. 0.07 torr/102°C
oZN ~ ~ -CH3 m.p. 179-181°C
NO~
v -CH3 m.p. 98-102°C
02N
-CH3 m.p. 126-129 °C
02N
-CH3 m. p. 116-118 ° C
02N
N02 '
o2N ~ ~ -CH3 m.p. 164-168°C
-CH3 m.p. 146-14$°C
N
-CH3 -CF3
-CCl3 -CH3
CA 02320676 2000-09-20
Example 2:
a) Preparation of 4 acetylamino 6 methyl-3-oxo-2,3 4,5-tetrahydro-1 2,4-
triazine (ring
ex ansion
3.12 g of the 2,3-dihydro-5-methyl-2-oxo-1,3,4-oxadiazole-3-acetone prepared
according
to Example 1 are stirred in 40 ml of alcohol together with 2 g of hydrazine
hydrate for
16 hours at a bath temperature of 35°C. After the solvent and the
excess hydrazine have
been evaporated off in vacuo, recrystallisation from isopropanol yields the
title compound
having a melting point of 197-199°C.
The following compounds of formula III are also prepared in a manner
corresponding to
that described above:
CA 02320676 2000-09-20
_g_
R1 R . m.p. [°Cl
H -CH3 185-187°
-CH3 -CH3 197-199°
-CH 205-207°
-C(CH3)3 s
O2N ~ ~ -CH3 252-255°
NOZ
-CH3 255-257°
02N
-CH3 228-231 °
02N
-CH3 259-262°
O.,N
-CH3 259-262°
N -
-CH3 H
-CH3 -~3
H H
CA 02320676 2000-09-20
-9-
b) Preparation of 4-amino-6-methyl-3-oxo-2,3,4,5-tetrahydro-1 2 4-triazine
(acid
hydrolysis):
1.7 g of the 4-acetylamino-6-methyl-3-oxo-2,3,4,5-tetrahydro-1,2,4-triazine
prepared
according to a) above are stirred for 5 hours in 10 ml of 2N hydrochloric acid
at 80°C.
After cooling of the solution, 1.7 g of sodium acetate are added and the
solution is
concentrated by evaporation in a rotary evaporator at a bath temperature of
60°C. The
residue formed is stirred with ethanol and freed of salt precipitates by
filtration. The
resulting solution is concentrated to a small volume and caused to
crystallise. The tide
compound is obtained in the form of colourless crystals having a melting point
of
116-119°C.
The following~compvunds of formula I are also prepared in a manner
corresponding to
that described above:
R m.p. [°C]
-CH3 116-119
-C~HS 143-145
-C3H7(i) 79- 81
-C(CH3)3 148-150
94- 95 '
199-202°
208-210°
H
-CF3