Note: Descriptions are shown in the official language in which they were submitted.
CA 02320828 2000-08-17
WO 99/44056 PCTNS99/04200
METHOD FOR THE DETERMINATION OF HEXAVALElI'T
CHROMTUM USL~TG ULTRAS011'ICATION AND
STROr'G AI\'IOl1' EXCHANGE SOLID PHASE EXTRACTIOl~
Field of the Invention
S This invention relates to a method for the determination of hexavalent
chromium (Cr"). More specifically, the present invention relates to a simple,
fast,
sensitive, and economical method for the determination of Cr'~ which is
especially
adapted for environmental and work place samples (including solid and air
samples).
The present method can be used in both laboratory and field analysis.
Backsround of the Invention
Chromium exists primarily in two valence states, trivalent (Crm) and
hexavalent
(Cr"). 'The trivalent state is relatively non-toxic, and is an essential
nutrient in the
human diet. On the other hand, Cr''' has been shown to be a human respiratory
carcinogen in epidemiological studies of workplace exposures, and has been
classified
1 S by the L'.S. Environmental Protection Agency (EPA) as a Group A inhalation
carcinogen. Hence. analv~tical methods are desired which can be used to easily
speciate chromium so that human exposures to Cr" can be monitored and, thus.
better
controlled.
Workplace exposure to Cr'~ has been associated with a number of industrial
sources, such as metal plating, spray painting, welding, tanning, and abrasive
blasting
operations. Environmental sources of Cr" include, for example, deteriorated or
disturbed chromate-containing paint, combustion sources such as automobiles
and
incinerators. and fugitive dusts from contaminated soils. Because of the
desire to
accurately measure Cr''' at low levels, the development of analytical methods
for the
determination of Cr" has been a subject of significant interest in
occupational and
environmental health.
CA 02320828 2000-08-17
WO 99144056 PCT/US99104200
Chromium has been detected using atomic absorption spectrometry (Mehra et
al., Talanta, 1989, 36(9), 889; Fong et al., Spectrosc. Lett., 1991, 24, 931),
atomic
absorption spectrometry (Jawis et al., Analyst, 1987, 122, 19; Arar et al.,
Environ.
Sci. Technol., 1992, 29, 1944), atomic emission spectrometry (Boumans, Line
Coincidence Tables for Inductively Coupled Plasma Atomic Emission
Spectrometry,
Oxford University Press, Oxford, 2nd ed., 1984;. Ciglio et al., Anal. Chim.
Acta, 1991,
254, 109; Roychowdhury et al., Anal. Chem., 1990, 62, 484), X-ray fluorescence
(Artier et al., Analyst, 1988, I 13, 779), charged-particle X-ray emission
spectrometry
and neutron activation analysis. National Institute for Occupational Safety
and Health
(N10SH) Methods 7024 and 7300 (IVIOSH Manual of Analytical Methods, Eller &
Cassinelli (gds.), National Institute for Occupational Safety and Health,
Cincinnati,
Ohio, 4'~ ed, 1994) use atomic absorption spectrometry and atomic absorption
spectrometry, respectively, for the determination of chromium in workplace air
samples. These methods, however, generally determine only total. Moreover,
these
methods generally involve expensive and complex instrumentation and are not,
therefore, generally suitable for monitoring directly in the field.
Spectrophotometric and colorirrietric methods have been developed for the
determination of Cr'~'. See. for example, Alvarez et al., ?alanta, 1989,
36(9), 919;
Haukka, analyst, 1991, 116, 1055; Abel et al., Am. Ind. Hyg. Assoc. J., 1974,
35, 229.
~- 20 The most~prevalent colorimetric method uses the selective reaction of
Cr''' with 1,5-
diphenylcarbzide (DPC) under acidic conditions to yield a red-violet Cr'''-
diphenylcarbazone complex. A variation of this colorimetric method is used in
NIOSH
method 7600 (1\T10SH Manual of Analytical Methods, Eller & Cassinelli (gds.),
l~'ational Institute for Occupational Safety and Health, Cincinnati, Ohio, 4'"
ed, 1994)
where alkaline extraction is used to help stabilize the Cr''' species.
Stripping
voltammetry (Vfang et al., Analyst, 1992, 117, 1913; Elleouet et al., Anal.
Chim. Acta,
1992, 257, 301 ) and ion chromatographic assays (Powell et al., Anal. Chem.,
1995,
67, 2474: Vercoutere et al., Mikrochim. Acta., 1996, I23, I09; Molina et al.,
Am. Ind.
Hyg. Associ. J., 1987, 48, 830; ASTM D 5281-92, "Standard Test Method for
Collection and Anal~~sis of Hexavalent Chromium," in Annual Book of ASTM
-2-
CA 02320828 2000-08-17
WO 99/44056 PCT/US99104200
Standards, American Society for Testing and Materials, vol. 11.01,
Philadelphia. PA,
1992; U.S. Environmental Agency, Method 218.6. Determination ofDissolved
Hexavalent Chromium in Drinking Water, Groundwater and industrial Waste Vfater
Effects by Ion Chromatography, EPA Office of Research and Development,
Cincinnati, OH, 1990; U.S. Environmental Agency, Method 306UA, "Alkaline
Digestion for Hexavalent Chromium" in Test Methods for Evaluating Solid
Wastes,
EPA, Washington, DC, 1995) have also been used to determine Cr"' in various
samples. Many of these techniques are limited to laboratory-based analysis and
cannot, therefore, be used in field monitoring and/or real-time evaluations.
Over the past decade, solid phase extraction (SPE) has been established in the
analytical chemistry laboratory and has become increasingly popular. The use
of SPE
for the separation and preconcentration of trace polar or non-polar target
anaiytes has
been widely investigated, and the advantages of such a technique over a
conventional
liquid-liquid extraction, coprecipitation, electrochemical deposition and
evaporation
have been well documented. See, for example, Masque et al:, Analyst, 1997,
132,
425-428: Corcla et al., Environ. Sci. Technol., 1994, 28, 850-858; Shahteri et
al., J.
Chromatogr., 1995, 697, 131-136. Some ofthe advantages of SPE over classical
analytical methods include: (l) efficiency and simplicity; (2) solvent
minimization and
enhanced safety with respect to hazardous samples; (3) high preconcentration
factors;
(4) good recoveries; (5) flexibility; and (6) low cost.
Both off line and on-line SPE methodologies have been employed for the
preseparation and preconcentration of a variety of analytes. Off line SPE
methodologies involve the use of packing materials that may contain functional
groups
of different polarity such as C8 or C" bonded silica phases (Falco et al.,
Analyst, I 997,
122, 673-677). 1~'ith on-line SPE, followed by high pressure liquid
chromatography, a
critical parameter is the selection of an adequate precolumn in order to avoid
band
broadening of the first eluded peaks, and to allow for the percolation of
large sample
volumes (Kiss et al., J. Chromatogr., 1996, 725, 261-272). Numerous solid-
phase
extractants, such as pure or modified silica, alumina. magnesia, activated
carbon,
-3-
CA 02320828 2000-08-17
WO 99144056 PGTNS99/04200
polyurethane, and cellulose and its derivatives, have been used in SPD
techniques.
Consequently. solid phase extraction has largely replaced classical Liquid-
liquid
extraction in the analytical laboratory.
More recently, innovative new cartridges for SPE, such as reversed-phase and
ion-exchange of target analytes in a single resin, are being developed and
many are
commercially available. These resins cartridges allow for sorption of the
analyte of
interest while removing non-sorbed interferents, with fast, quantitative
adsorption and
high elution capacities. These cartridges can improve the overall specificity
and
sensiti~7tS~ of trace analysis. Furthermore, the use of commercially
available, low cost
vacuum manifolds for SPE allows for up to, and even greater than, 24 samples
to be
processed simultaneous)}~. Complete automation of procedures based on SPE are
also
available v~~ith commercial instrumentation. Despite these advantages. there
have been
relatively feva~ applications of SPE to inorganic materials, including heavy
metals.
Ultrasonic extraction {UE) for the purpose of dissolving target heavy metal
1 ~ anal~~tes in environmental samples is also a technique that has not been
used
extensively. although it of~'ers great promise {Lugue de Castro et al., Trends
Anal.
Chem.. 1997. 16. 16-24). LrE has been demonstrated to perform well for the
quantitative dissolution of several heavy metals in a variety of environmental
matrices
(Narper et al.. Anal. Chem., 1983, 55, 1553-1557; Sanchez et al., Analusis,
1994. 22.
222-225: Ashley. Electroanalysis, 1995, 7, 1189-1192z), including hexavalent
chromium (James et al., Environ. Sci. Technol., 1995, 29, 2377-2381).
Nonetheless. there remains a need for a simple, reliable, fast, inexpensive.
and
field-based method for the detection of Cr'' in environmental and workplace
samples.
This invention. combining the use of ultrasonic extraction and strong anion
exchange
2> solid phase extraction, provides such a method. This method provides a
novel and
effective approach for the on-site determination of Cr" in environmental and
workplace samples.
-4-
CA 02320828 2000-08-17
WO 99/44056 PGTNS99/04200
Summary of the Invention
7"his invention relates to a method for the determination of hexavalent
chromium (Cr"). Based on the chemical properties of chromium species in
aqueous
solutions, a simple, fast, sensitive. and economical field method has been
developed
and evaluated for the determination of hexavalent chromium (Cr''') in
environmental
and workplace air samples. By means of ultrasonic extraction in combination
with a
strong anion exchange solid phase extraction (SAE-SPE) technique, the
filtration,
preconcentration, and isolation of Cr" in the presence of other chromium
species and
interferents was achieved. ?'he method generally involves: (1) ultrasonication
in basic
buffer solution to extract Cr" from environmental matrices; (2) strong anion
exchange
solid phase extraction to separate Cr" from other chromium species and
potential
interferents; (3) acidification of the eluate containing the Cr" ions: (4)
complexation of
Cr" with a complexin~ agent to form a soluble, colored Cr"-complex: and (5}
spectrophotometric determination of the colored Cr"-complex. Preferably. the
1 ~ ultrasonication step is carried out in the presence of a slightly basic
ammonium buffer
and the complexing agent is I,5-diphenylcarbazide. This present method can
effect the
extraction of both soluble (K.CrO,) and insoluble (KzCrO,) forms of Cr"
without
inducing Crm (Cr:03) oxidation or Cr" reduction. ?he method allows for the
dissolution and purification of Cr''' from environmental and workplace air
sample
?0 ... mat.rices for up to 24 samples (or even higher numbers) simultaneously
in less than
about 20 minutes (excluding the ultrasonic extraction time). the present
method is
simple, fast, quantitative, and sufficiently sensitive for the determination
of
occupational exposures of Cr". The method is especially applicable for on-site
monitoring of Cr" in em~ironmental and industrial hygiene samples, including
both
2~ solid samples {e.g., soil. paint chips, dust, solid residues, and the like)
and air samples.
One objective of the present invention is to provide a method for the
detection
of Cr" in a sample, said method comprising:
-5-
CA 02320828 2000-08-17
WO 99/44056 PGTNS99/04200
( 1 ) ultrasonic extraction of Cr" from the sample utilizing a first buffer
solution
(i. e.. the ultrasonic buffer) having a slightly basic pH, whereby the pN of
the first
buffer solution is such that neither significant Crm oxidation nor Cr"
reduction occurs;
(2) separation of the Cr" in the ultrasonic extractant from step ( 1 ) from
any
Cri° or other interferents that might be present in the sample by
passage of the
ultrasonic ea~tractant through a strong anion exchange solid phase extraction
media;
(3) elution of the Cr'~' from the media with a second buffer solution (i.e.,
the
elution buffer) having a slightly basic pH;
(4) acidification of the eluate containing the Cr''; and
(5) addition of a complexing agent to the acidified eluate to form a colored
Cr"-complex if Cr" is present in the sample. The ultrasonication step employed
to
liberate Cr'~' from the sample matrix is preferably carried out using a
slightly basic
ammonium buffer and the complexing agent is preferably 1,5-diphenylcarbazide.
Preferably. the amount of Cr" present in the sample is determined using any
l 5 appropriate technique. A9ore preferably, the amount of Cr" present in the
sample is
determined using a simple and direct spectrophotometric procedure which can be
used
in the field or on-site.
Another objective of the present invention is to provide a method for the
quantitative detection of Cr'~' in a sample suspected of containing Cr", said
method
comprising:
( 1 ) ultrasonic extraction of Cr" from the sample utilizing a first ammonium
buffer solution (i.e., the ultrasonic buffer) having a slightly basic pH,
whereby the pH
of the ammonium buffer solution is such that neither significant Crm oxidation
nor Cr"
reduction occurs:
(2) separation ofthe Cr" in the ultrasonic extractant from step (1) from any
Cra' or other interferents that might be present in the sample by passage of
the
ultrasonic extractant through a strong anion exchange solid phase extraction
media;
(3) elution ofthe Cr''' from the media with a second ammonium buffer
solution (i.e.. the eultion buffer);
s0 (4) acidification of the eluate containing Cr'~';
-6-
CA 02320828 2000-08-17
WO 99144056 PCT/US99/04200
(5) addition of 1,5-diphenvlcarbazide to the acidified eluate to form a
colored
Cr"-1.5-diphenylcarbazone complex if Cr" is present in the sample; and
(6) subjecting the Cr"-1,5-diphenylcarbazone complex, ifpresent, from step
(5) to a spectrophotometric analysis in order to determine the amount of Cr'~'
present
in the sample.
?here and other objectives and advantages of the present invention will be
apparem to those of ordinary skill in the art upon consideration of the
present
specif canon.
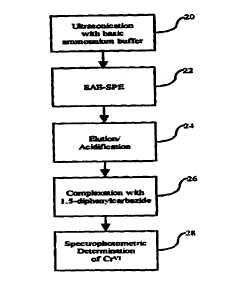
Brief Description of the Drawings
1 U Figure 1 is a flowchart illustrating the general procedures utilized in
the present
method.
Figure ? is a flowchart illustr ating one preferred embodiment of t he present
invention.
Figure 3 illustrates the breakthrough of Cr" using a strong anion exchange
solid phase extraction column. Curve 1 uses an elution solution of 0.02 M
ammonia
sulfate and fl.02 M ammonia hydroxide buffer; Curve 2 uses an elution solution
of 0.05
1~9 ammonia sulfate and 0.05 M ammonia hydroxide buffer; Curve 3 uses an
elution
solution of 0.5 M ammonia sulfate and 0.5 M ammonia hydroxide buffer; Curve 4
uses
an elution solution of l .0 M ammonia sulfate and 1.0 M ammonia hydroxide
buffer.
Figure 4 illustrates the elution of varying concentration of Cr" using a
strong
anion exchange solid phase extraction column with varying. Curve 1 uses an
elution
solution of 0.5 M ammonia sulfate and 0.5 M ammonia hydroxide buffer; Curve ?
uses
an elution solution of 1.0 h~s ammonia sulfate and 1.0 M ammonia hydroxide
buffer:
Cuwe ~ uses an elution solution of 2.0 M ammonia sulfate and '.0 M ammonia
2~ hydroxide buffer.
-7-
CA 02320828 2000-08-17
WO 99/44056 PCT/US99/04200
Description of the Preferred Embodiments
This invention provides a simple. fast. sensitive, and economical field method
for the determination of hexavalent chromium (Cr"), especially in
environmental
samples and workplace air samples. The present method combines ultrasonic
extraction and a strong anion exchange solid phase extraction (SAE-SPE)
technique to
allov~~ the filtration, preconcentration, and isolation of Cry' in the
presence of other
chromium species and interferents. The present method is generally illustrated
in
Figure 1. In the first step. ultrasonication 10 is used to extract Cr" in the
sample from
its environmental matrix. After ultrasonic extraction. a strong anion exchange
solid
1 C~ phase extraction (SAE-SPE) step 12. preferably using a column or
cartridge system. is
used to separate Cr" from other chromium species and potential interferents.
The Cr"
species retained on the column or cartridge containing the solid absorbing
system to
allo' separation from Cr'n species and other potential interferents in the
sample. The
Cr°' and other potential interferents pass through the column to
ef~'ect the separation.
1 ~ The Cr" species are then eluted from the column or cartridge and
collected. The
eluant containing the Cr'~' species is then acidified in step 14. The
acidified eluant is
then treated with a complexing agem in step 16 to form a colored Cr"-complex
which
can be detected in step 1 s using any appropriate means (e.g., visual,
spectroscopic, or
similar methods).
20 An especially preferred embodiment of the present invention is illustrated
in
Figure 2 This preferred embodiment involves a first ultrasonication step 20
using a
slightly basic ammonium buffer solution to extract Cr" from the environmental
matrices at a p1-1 to avoid significant Ctl° oxidation and/or Cr'z
reduction. A strong
anion exchange solid phase extraction (SAE-SPE) step 22 is then used to
separate Cr'~'
'_'~ from other chromium species and potential interferents. Step 22 is
preferably carried
out using a column or cartridge containing pre-packed strong anion exchange
material.
Cr" is then removed from the column or cartridge preferably using. for
example. a
slightly basic ammonium solution. The collected eluant containing Cr''' is
then
_g_
CA 02320828 2000-08-17
WO 99/44056 PCTNS99/04200
acidified in step 24. An effective amount of 1.5-diphenylcarbazide is added to
the
eluant containing Cr" in step 26 to form a soluble. colored Crv'-1,5-
diphenylcarbazone
complex. In the last step 28, the amount of Cr" is determined using a suitable
spectrophotometric technique.
The ultrasonication step employed to liberate Cr" from the sample matrix
should be carried out using a slightly basic aqueous buffer solution so as to
reduce the
levels of Crm oxidation and Cr" reduction to low (i.e., insignificant) levels.
Generally,
the pH of buffered solution used in the ultrasonic extraction step is in the
range of
about 7.2 to 9.0, preferably in the range of about 7.5 to 8.5, and most
preferably about
8Ø Preferably the ultrasonic solution is a buffered aqeuous ammonium
solution
containing between about 0.02 A9 to 0.2 M (1VH,),SO, and about 0.02 M to 0.2 M
NH,OH. even more preferably about 0.025 M to 0.1 M {NH4),SO, and about 0.025 M
to 0.1 M NH,OH, and most preferably about 0.05 M (NH,)2SOa and about 0.05 M
1'TI-i40H at a p' T of about 8Ø Other alkaline buffers (e.g.,
tris(hydroxymerhvl)aminomethane hydrochloride (Tris-HCl) where ammonium or
ammonium hydroxide is used to obtain the desired basic pH; ammonium carbonate:
generall~~ basic buffers containing NH,- cations) can also be used. The
ultrasonic
extraction step can be carried out using any conventional ultrasonic bath.
LTltrasonification for about 15 to 60 minutes at about room temperature or
slightly
elevated temperatures should be sufficient to release the Cr'~' contained in
the sample.
Preferably, the ultrasonification is carried out a temperatures less than
about 40°C in
order to minimise oxidation and reduction reactions. Routine experimentation
can
easily be carried out to determine the optimum uitrasonification conditions
for any
particular sample. After the ultrasonication step, the sample can be filtered
if desired
to remove solid material before application to the strong anion exchange
resin.
Once the Cr" species has been liberated by ultrasonification, the sample is
treated using a strong anion exchange resin to separate the Cr'' species from
other
chromium species as well as other potential interferents. The Cr" which is
retained on
the strong anion exch4nge resin can be eluted using a slightly basic aqueous
buffer
_9_
CA 02320828 2000-08-17
WO 99/44056 PCT/US99l04200
system. Suitable strong anion exchange resins include, for example, styrene-
divinylbenzene polvrner resins to which tertiary ammonium groups have been
hound
(e.g.. Dowex 1-X8 from Fluka Chemical. Ronkonkoma, NY) and quaternary amine
bonded silica with Cl' as the counter ion (Supelco. Inc., Bellefontaine, PA).
Other
strong anion exchange resins can be used so long as they allow separation of
Cr'~ from
Crm and other potential interferents. Generally, cartridges pre-packaged with
the
strong anion exchange resins are much easier to use and are, therefore,
preferred.
Strong anion exchange resins with a capacity of about 0.1 to about 2.0 meq/g
are
generally preferred. The strong anion exchange resins are conditioned or
activated
according to known procedures; such procedures are normally supplied by the
resin
manufacturer.
The general procedure for the preferred SPE-SAE procedure will now be
detailed using a quaternary amine bonded silica resin (Cl- counter ion) as the
strong
anion exchange media. . suitable solid-phase extractor (from, for example.
Supelco,
Inc., Bellefontaine, PA) can be attached to an appropriate small vacuum pump
via a
metering valve in order to assist passage of the sample and other reagents
through the
resin. A cartridge (3 ml) containing about 500 mg quaternary amine bonded
silica
sorbent with Cl' as the counter ion (capacity about 0.2 meg/g) is placed in
the solid-
phase extractor and then conditioned with about 3 m1 of deionized water to
activate
the functional groups in order to allow the Cr" to be absorbed on the resin.
The
sample (generally about I to 5 ml) obtained from the sonification extraction
step is
then loaded on the cartridge and passed through the resin at a flow rate of
about 1
ml!min to about 2 ml/min. The Cr'~ remains on the resin. After washing the
cartridge
with deionized water (generally about I to 5 ml), the Cr" is removed from the
cartridge with a slightly basic buffer solution (generallyabout 4 to 10 ml in
1 to 3
portions) and the elutant containing the Cr'? is collected. Washing the column
w.~ith
water prior to removal of the Cr" helps to remove potentially interfering
materials. Of
course. as one of ordinary skill in the art will realize, larger or smaller
cartridges or
columns will generally required larger or smaller amounts. respectively, of
the relevant
s0 reagents.
- 10-
CA 02320828 2000-08-17
WO 99144056 PGT/US99104200
The buffer system used to elute the Cr" from the strong anion exchange resin
may be the same or different from that used in the ultrasonification step. It
should,
however, be slightly basic to avoid Cr" reduction. It is generally preferred
that a
buffered aqueous ammonium solution containing between about 0.25 M to 1.0 M
(NH,)=SO, and about 0.25 M to I .0 M I~TH40H is used as the eluting buffer.
More
preferably, the eluting buffer solution contains about 0.5 M (NH,)~SO, and
about 0.5
M NH,OH at a pH of about 8Ø Generally, the eluting buffer system is more
concentrated (preferably by a factor of about 10) than the ultrasonic buffer
system.
Other alkaline buffers (e.g., tris(hydroxymethyl)aminomethane hydrochloride
(?ris-
HCl) where ammonium or ammonium hydroxide is used to obtain the desired basic
pH: ammonium carbonate; generally basic buffers containing NH,' canons) can
also be
used.
After elution from the SAE column or cartridge, the Cr"-containing eluant is
acidif ed using suitable acids (e.g., ~Ifuric acid, hydrochloric acid, acetic
acid, and the
l 5 like). Generally the pH is not critical so long as the eluant is acidic,
After
acidification, the complexing agent is added to form a detectable Cr"-complex.
The
complexing agent is added in excess in order to maximize the~amount of Cr"-
complex
formed. In other words, the amount of complexing agent should be suffcient to
complex with essentially all of the Cr" present in the sample. Examples of
suitable
complexine agents include 1,5-diphenylcarbazide, 2-(5-bromo-2-pyridylazo-5-
(diethylamino)phenol. L-penicillamine, 2-mercaptonicotinic acid, and the like.
1,5-
Diphenylcarbazide is the preferred complexing agent. The Cr"-I,5-
dipenylcarbazone
complex is red-violet with an absorbance maxima at about 520-540 nm. Generally
color formation occurs within a few seconds and is stable for several hours.
Nonetheless, it is generally preferred that the detection step be undertaken
shortly after
complex formation. The dynanuc range of the chromium-diphenylcarbazone complex
was generally linear from about 10 pg/1 to about 5 mg/1 (RZ = 0.99997) using
spectrophotometric determination. Selectivity was also excellent since typical
interferences. including Cr'n , Fem. and CuD, are essentially eliminated in
the SAE
procedure.
CA 02320828 2000-08-17
WO 99/44056 Pt:T/US99/04200
Quantitative or qualitative detection of the resulting Cr'''-complex can be by
any suitable means. For example. the development of the colored Cr"-complex
itself
visually indicates the presence of Cr" in the sample. Preferably W/~'IS-based
spectrophotometric detection methods are preferred for quantitative
colorimetric
measurement. A standard cataloging spectrophotometer, especially one adapted
for
field operation, is generally preferred so as to provide a portable, field-
adapted
detection method. For detection of Cr" using the 1,5-diphenylcarbazide
complexing
agent, a spectrophotometer operating at about 540 nm is preferred; this
wavelength
provides maximum sensitivity and generally eliminates interference of excess
(i.e.,
l0 uncomplexed) 1.5-diphenylcarbazide complexing reagent.
The present method may be either manual, semi-automated. or fully automated.
Manual or semi-automated analysis and detection techniques might be used, for
example, for screening or spot-checking a workplace environment to determine
if Cr"
exposure is likely and if further, more extensi~ ~, sampling is necessary.
Generally.
15 automated techniques wherein multiple samples can be evaluated at the same
time are
preferred. For example. a flov~J injection analysis system as described in
Wang et al.
(Analyst, 1997, 12, 1307-13l 2, which is hereby incorporated by reference) can
be
employed. Such a system. employing a pump and an autosampler operating under
low
pressure and computer control can be used to automate the precise
r»anipulation of
20 microliter amounts of samples and deliver the products to a flow-through
colormetric
detector for the detection of Cr". In addition, commercially available, low
cost
vacuum manifolds for SPE can allow multiple samples to be run; generally, 24
samples
(or more with appropriate modifications) at a time can easily be evaluated.
Commercial instrumentation is also available by which the SPE process can be
25 automated. Thus, the present method can provide a low cost, simple, fast,
quantitative, and sensitive method for the determination of occupational
exposure of
Cr". This method is ideally suited for on-site monitoring of Cr''' in
environmental arid
industrial hygiene applications.
- 12-
CA 02320828 2000-08-17
WO 99/44056 PGT/US99/04200
Examples
The following examples are provided to illustrate the invention and not to
limit
the invention. All references cited in the present specification are hereby
incorporated
by reference.
Instrumentation and Reagents. Ultrasonic extraction was performed using a
Sonicor 115V, 60 Hz laboratoy sonicator (Sonicor Instruments, Farningdale, NY,
USA). The solid-phase extractor was obtained from Supelco, Inc.
(Bellefontaine, PA.
USA), and was attached to a small vacuum pump via a pressure metering valve.
The
strong anion exchange cartridge used in the solid phase extraction contained
500 mg
quaternan~ amine bonded silica sorbent with Cf as counter ion for strong anion
exchange (capacity 0.2 meg/g): the tube size was 3 ml. These pre-packaged
cartridges
were obtained from Supelco, Inc., Bellefontaine, PA. A portable cataloging
spectrophotometer (HACH DR'2010, HACH Company '~oveland, CO) was used for
spectrophotometric measurement.
Hexavalent and trivalent chromium standards, ammonium sulfate, ammonium
hydroxide. 1,5-diphenvlcarbazide (DPC). chromium oxide (Cr~03), potassium
chromate (K:CrO,), hydrochloric acid, sulfuric acid, and nitric acid were all
reagent
grade from Aldrich (Milwaukee, V'1). T'ris(hydroxymethyl)aminomethane
hydrochloride (Tris-HCI)) was from Sigma, Inc. (St. Louis, MO). Lead chromate
PbCrOd) was from Fisher Scientific (Elizabeth, NJ). Chromium-containing
reference
materials v~~ere used as supplied: ( 1 ) paint chips -- US EPA Certified
Reference
Material (CRM) Ol3-050, Laramie, VVY; (2) coal fly ash -- National Institute
of
Standards and Technology (NIST) Standard Reference Material (SRM) 1633a,
Gaithersburg, MD; and (3) welding dust loaded on glass fiber filters --
Institute for
Reference AJaterials and Measurements (IRMJ1~I) CRM 545) from the European
Commission (IR~T1T. Geel, Belgium). For air samples, preloaded filter
cassettes
containing mixed cellulose ester membrane filters (0.8 lr pore size, .37 mm
diameter)
were obtained from SKC Inc. (Eighty Four, PA).
-IS-
CA 02320828 2000-08-17
WO 99/44056 PCT/US99J04200
Solid-Phase Extraction Procedures. Solid-phase extraction (SPE) was
performed with the aid of an extractor and a strong anion exchange (SAE)
cartridge.
The SAE cartridge contained a silica sorbeni to which a quaternary amine was
bound;
chloride ion functioned as the counter ion. Cartridges were conditioned with 3
ml of
deionized water. which activated the functional groups on the sorbent,
allowing for
binding of Cr'~I onto the SAE sorbent. Subsequently, a 3 ml aliquot of sample
solution
was loaded on the SAE cartridge. After sample loading, the cartridge was
rinsed with
3 ml of deionized water to remove potential interferents. Then, elution of the
Cr"
concentrated on the SAE cartridge was performed with 9 ml of 0.5 M ammonium
sulfate buffer solution in three 3 ml fractions.
Sample Collection. Workplace air samples were collected from aircraft
painting operations at LT.S. Air Force bases using 0.8 micron, 37-mm cellulose
ester
membrane filters. Samples were obtained from different work practices
including
priminL. sanding. alodining, cutting, and grinding. The samples w~ -e
transported to
the laboraton~ immediately after collection, and stored in a refrigerator at
4°C until
prepared for analysis. Environmental samples were collected using standard
techniques.
General Procedure for Em~ironmental and Workplace Air Samples. Samples
were placed into Z 5 ml plastic centrifuge tubes. Sample preparation consisted
of
?0 adding 10 ml of 0.05 M (NH,)SO, and 0.05 M NH,OH (pH 8) buffer solution to
the
sample followed by sonication in an ultrasonic bath for 30 minutes at ambient
temperature (<40 ° C). After ultrasonication, a 3 ml aliquot of the
supernatant was
loaded onto a strong anion exchange cartridge. The Cr'~ was eluted with 9 ml
0.5 M
(1v'H,)SO, and 0.1 M NH,OH (pH 8) buffer solution in three 3 ml fractions at a
flow
rate of 2 mllmin. After isolation and purification, the eluate was acidif ed
with 100 pl
37°.-o HCl solution. This was followed by mixing with 2 ml of20 mM 1,5-
diphem~lcarbazide complexing reagent. The reaction of 1,5-diphenylcarbazide
va~ith
Cr" is completed in a few seconds and the color of the complex can be stable
for at
- 14-
CA 02320828 2000-08-17
WO 99/44056 PCT/US99/04200
least 8 hours. If analysis is not to be completed shortly after complex
formation, it is
preferred that they be stored at refrieerated temperatures.
Detection of Hexayalent Chromium. Quantification of hexavalent chromium
was done by ea-ternal standard or standard addition methods with the
spectrophotometer set at 540 nm. For the air samples, a standard addition
calibration
was used. Blank filters and quality controUquality assurance samples (standard
solutions of known hexavalent chromium concentration) were analyzed at a
minimum
frequency of one per twenty experimental samples.
Example 1 -- Absorption Capacity Studies. Absorption capacities were
l 0 determined using spiked Cr''' solutions produced by dissolving KZCrO, in
0.02 M, 0.05
M, and 0.5 M ammonium sulfate and ammonium hydroxide buffer solutions (pH 8).
Four different Cr" spiked solutions (1.0 mM, 2.0 mM, 4.0 mM, and 8.0 mM) were
prepared and loaded onto the cartridge. eluted, and analyzed in triplicate to
a ~ablish
the reproducibility of the procedure. Breakthrough of the anaiyte was
determined by
analysis of the solution that passed through the SAE cartridge after loading
of sample
aliquots.
The results are showm in Figure 3 which shows that the initial breakthrough
that was observed at various Cr''' loading volumes with the flow rate
controlled at 2
ml/min. Similar results (not shown) were obtained with matrix spikes of
cellulose ester
filters. The adsorption capacities of SAE cartridges changed with increasing
concentration of the ammonium sulfate and ammonium hydroxide buffer in the
sample
solutions (Figure 3). The adsorption capacity of SAE cartridges for Cr'''
decreased
more than threefold when the concentration of ammonium sulfate and ammonium
hydroxide buffer solution was changed from 0.02 M (Figure 3, Curve 1 ) to 1.0
M
(Figure 3, Curare 4) at the same controlled flow rate of 2 mUmin. The
adsorption
capacities of SAE cartridges were also related to sample loading flow rates.
At a high
flow rate of loading (e.g., 4-5 ml/min). the breakthrough of the Cr''' was
more evident
than was observed at a lower loading flow rate (e.g., 1 ml/min) under the same
other
- IS -
CA 02320828 2000-08-17
WO 99/4405b PCT/US99/04200
experimental parameters. The percentage breakthrough increased with increased
loading speed, and an optimum flow rate of about 2 ml/min was arrived at by
trial and
error. Generally. the throughput flow rate is an important function of SPE.
Loading
flow rates are often more critical to optimizing recoveries than are the
conditioning
and rinse flow rates. In the conditioning step, there is nothing bound to the
cartridge,
and the flow rate can sometimes be increased without a loss of recovery. Flow
rates
may also be increased in the rinse step depending upon the banding mechanism
and the
sorbent/adsorbate chemistry. These trials (Figure 3) have demonstrated that
reducing
the concentrations of ammonium sulfate and ammonium hydroxide buffer in sample
solution can improve the adsorption capacity and minimize cartridge
breakthrough.
V1'hen air samples containing chromium were tested to study SAE cartridge
breakthrough, they were treated in an identical manner as spiked Cr" solutions
in
order to evaluate the effects of ammonium sulfate and ammonium hydroxide
buffer
solution (pH 8) used in the extraction procedure. In these trials, the samples
were
treated with ultrasonication for 30 min at ambient temperature (<40 °C
bath
temperature) in ammonium sulfate buffer solution. The results demonstrated no
significant breakthrough of Cr'~' when. an ammonium sulfate and ammonium
hydroxide
buffer solution (< 0.05 M) was used to extract Cr" from samples via
sonication.
Example 2 - Elution Studies. Ammonium sulfate and ammonium hydroxide
buffer was used as the elution buffer solution. 'To obtain optimum recoveries,
spiked
Cr" solutions of four levels (10.0 pl, 20.0 ul, 40.0 pl, 80.0 pl) were eluted
using
different number of fractions of 0.5 M ammonium sulfate and ammonium hydroxide
buffer solution (pH 8). Eluting power was investigated for five levels of Gr'~
solutions
(50.0 lrg, 100.0 Ng, 200.0 Irg, 400.0 pg, 600.0 ug) at various flow rates. In
each
2~ experiment, only one parameter was changed at a time. When elution flow
rate is
considered. it should not be too fast, since the analyte of interest may not
have enough
time to desorb from the cartridge. I~Tor should the flow rate be too slow, or
sample
preparation may be too time-consuming. As those skilled in the art will
understand.
- 16-
CA 02320828 2000-08-17
WO 99/44056 PCT/US99I04200
the optimum flowrates can be determined experimentally for a given
experimental
setup.
To evaluate the effect of ion exchange due to the elution power. eluting
solutions of 0.5 M, 1.0 M, or 2.0 M ammonium sulfate and ammonium hydroxide
buffer solutions (pH 8) were tested by eluting spiked Cr" solutions at 5
levels (50 lrg,
100 pg, 200 pg, 400 pg, 600 pg). The results are shown in Figure 4. The Cr'~
started
to initially elute at various volumes of buffer solution, corresponding to
different
contents of Cr''' loaded the cartridges. Increasing the concentrations of the
ammonium sulfate and ammonium hydroxide in elution solution from 0.5 M (Figure
4,
Curve 1) to 2.0 M (Figure 4. Curve 3) did not significantly change the Cr'''
eluting
time. By eluting with 0.5 M ammonium sulfate and ammonium hydroxide buffer
solution, the Cr" at all 5 Levels started to elute at flow rates lower than 1
ml/min,
which is lower than the generally used flow rate of 2 ml/min. Therefore, by
considering the adsorption capacity of SAE cartridges (Figure 4, Curve I ),
0.5 M
ammonium sulfate and ammonium hydroxide buffer solution was determined to
perform best as an elution buffer solution in these experiments with the
particular
experimental setup employed.
Table I shows the recoveries of Cr" obtained from spiked Cr" solutions at 4
levels (10.0 lrg. 20.0 ug 40.0 pg. 80.0 pg) using varying number of eluting
fractions
(each fraction consisted of 3 ml of 0.5 M (''rH,),SO, and 0. I M. NH40H (pH 8)
huffer).
_ 17_
CA 02320828 2000-08-17
WO 99/44056 PCT/US99/04200
TABLE I
Recovery
ik (%) of Cr"
S ~ RSD
d C - -
'~'
p .
e Two Three Four Five
r Fractions Fractions Fractions Fractions
(pg)
10.0 85.3f2.7 95.3f3.3 95.52.7 96.312.4
~ 20.0 83. l f 3.4 94. l f 3.7 94.914.1 95.0 f 2.9
40.0 83.9 ~ 4.8 93.3 f 3.6 94.5 f 3.2 94.8 t 2.8
80.0 82.5 t 4.5 93.0 f 4.2 94.1 t 3.7 94.6 t 3.4
"RSD" in the various Tables indicates the relative standard deviation of the
samples
(normally carried out in triplicate).
This data show that eluting with three fractions (3 ml for each fraction) of
eluting solution allows for high recoveries (93.3 to 95.3%) and that no
significant
improvement in recoveries could be obtained by increasing the number of
eluting
fractions to more than three. Since a small volume of eluate is preferred to
achieve a
high enrichment factor, all the Cr" concentrated on the cartridge should be
eluted in as
small a volume as possible. Each fraction of 3 ml ammonium sulfate buffer
solution
(pH 8) was tested and good results were obtained. Under these optimum
conditions,
recoveries from duplicate spike solutions exhibited reproducible results. It
was also
confirmed from these experiments that the presence of Cr'° did not
significantly affect
Cr'~ recovery. These results indicated that the conditions used stabilized
both Crz° and
Cr" species.
These trials indicated tha: increasing either ammonium sulfate concentration
above 0.5 M or using more than three eluting fractions did not significantly
change
Cr'7 retention abilities. Therefore, an ammonium sulfate buffer of 0.5 W with
3
fractions (3 ml for each fraction) was found to give optimal elution power of
Cr" from
2~ these SAE cartridees.
_l8_
CA 02320828 2000-08-17
WO 99/44056 PCT/US99104200
As shown in Table I, relative standard deviations for the triplicate runs were
between 2.4 and 4.8%. The limit of detection (LOD), estimated as the mass of
analyte
which gives a signal that is 3 times o above the mean blank signal (where o is
the
standard deviation of the blank signal), was about I .0 ng/mI for the
spectrophotometric determination. A calibration curve was obtained by using a
linear
plot of the peak area as a function of standard concentrations of Cr" by least
squares
regression analysis.
Example 3 - Ultraso is Fxtrartinn of Soluble and lncnlnhlP ~"rt~ Although
the soluble fractions of Cr" are useful parameters for estimating levels of
Cr" that mav_
be directh~ absorbed b~~ humans, quantifying insoluble forms of Cr"' is
pertinent to
occupational hazards (such as PbCr04 in chromate ore processing and painting)
associated with airborne respirable dust. Therefore, an effective and reliable
method
for extracting both soluble and insoluble forms of Cr" without inducing Crm
oxidation
or Cr'~ reduction is required. Thus, it is important to evaluate ultrasonic
extraction for
I 5 the dissolution of both soluble Cr" (KZCr04) and insoluble Cr" (PbCrO,)
with basic
buffer solutions. In this portion of the study, both ammonium sulfate and
ammonium
hydroxide buffer (pH 8) and/or Tris-HCI buffer solutions (with adjustment to
pH 8
using I~'HaOH) were used as the ultrasonic ea-traction buffer.
As noted above, a slightly basic ammonium buffer solution may help
stabilization of chromium species in this system. Ammonium sulfate and
ammonium
hydroxide buffer solutions were used to dissolve hexavalent chromium from
environmental matrices. It is expected that conditions of the hot ammonium
buffer
sonication procedure might oxidize CrB' to Cr" under diverse redox conditions
of
various matrices. Hence no heating and relatively low concentration of the
ammonium
sulfate and ammonium hydroxide buffer solution (pH 8) were used to quantify
and
define operationally soluble and insoluble forms of Cr"
To maximize dissolution of all forms of Cr" while minimizing method-induced
oxidation and reduction. an extraction protocol was employed using 0.05 M
- 19-
CA 02320828 2000-08-17
WO 99/44056 PCT/US99I04200
ammonium sulfate and ammonium hydroxide buffer solution with uitrasonication
at
temperatures lower than about 40'C. These conditions should maintain a low
activity
of Crm, thereby minimizing oxidation of this species to Cr'''. Lead chromate
{PbCrO~,
Ksp = 1.8 x 10'") was used to represent insoluble Cr''i because it contained a
greater
percentage of its total Cr''' in an insoluble form. In initial trials, 300~cg
(w/w) of
PbCrO,, K,CrO,, and Cr20j were spiked into triplicate samples of the 10 ml
0.05 M
ammonium sulfate and ammonium hydroxide buffer solution. Since this method
does
not employ a high temperature to dissolve insoluble forms of Cr''', it was
hypothesized
that it would minimize method-induced oxidation and reduction while
effectively
dissolving Cr'~'
Despite the demonstrated efficacy ofthe ammonium sulfate solution to dissolve
insoluble forms of Cr" in solution while minimizing method-induced oxidation
of Crm
or reduction of Cr''', a second procedure employing T'ris-HCl buffer (with
ammonium
hydroxide adjustment to pH 8) ultrasonication dispersion was also tested in an
attempt
to quantiy Cr" in spiked solutions. As T'ris-HCL buffer contains ammonia, this
ion
can complex with Cr'~ and therefore may help stabilization of the trivalent
'chromium
species.
'Table Il below shoves the results for Cr'~' recoveries from ammonium sulfate
and ?ris-HCL buffer solutions.
-20-
CA 02320828 2000-08-17
WO 99/44056 PCT/US99/04200
TABLE II
Recovery (%) of
Cr" ~ RSD
Ult
i
rason
c
Buffer Solution Spiked ~KZCrO, Spiked PbCrO,
(300 Ng/IU ml) (300 pg/10 ml)
Ultrasonication
30 min, Temp. <40C
0.02M(IVH,)ZSO,+ 94.513.5 86.513./
0.02 M NH,OH
0.05M(NH,)ZSO,+ 94.32.4 90.52.9
0.05 M NH,OH
0.02 M Tris-HCl + 93.9 ~ 3.8 82.5 t 4.3
NH,OH (pH 8)
0.05 M Tris-HCl + 93.7 t 2.5 85.9 ~ 3.6
NH,OH (pH 8)
Ultrasonication
60 min, 'Temp.
<40C
0.02M(NH,)~SO,+ 94.413.8 90.413.9
0.02 M NH,OH
O.OSM(NH,):SO,+ 95.513.7 92.514.7
0.05 M NH,OH
0.05 M Tris-HC1 + 94.9 ~ 4.9 89.5 t 5.3
NH,OH (pH 8)
One hour sonication in ammonium sulfate and ammonium hydroxide buffer
solution dissolved slightly greater quantities of lead chromate (92.5%) than
Tris-HCl
buffer (89.5%) under the same ultrasonic extraction conditions; however, these
values
are not statistically different (t-test, 95% C.L). Based on the incomplete
dissolution of
lead chromate (Ksp =I .8 x 10'"), comparisons were made between lead chromate
and
potassium chromate both spiked into same buffer solutions. As indicated in
Table II,
Cr" is more difficult to extract from lead chromate (recovery 90.5%) than with
potassium chromate (95.1 %) under the same ultrasonication time and
temperature
conditions. Similar results were observed for recoveries of soluble Cr" spiked
in Tris-
HCl buffer. Close evaluation of the ultrasonication process revealed that lead
-21 -
CA 02320828 2000-08-17
WO 99/44056 PCTNS99/04200
chromate crystals were slowly dissolved, and this is evident in the nearly
quantitative
recoveries obtained from insoluble Cr". In addition, it was noticed that
extension of
the ultrasonication period from 30 min to 60 min can enhance the dissolution
of
insoluble Cr''' and increase the recoveries from 90.5% to 92.6%.
S Samples spiked with both soluble and insolube Crm and Cr" were also
evaluated. Ultrasonication was for 30 minutes at a temperature less than about
40 °C.
The results are shown in Table III below.
TABLE I11
Recovery (%)
of Cr" t RSD
Ul
i
trason
c
l0 Buffer SolutionSpiked K~CrO' Spiked KZCr04 Spiked PbCrO~
(300 pg/10 ml) + Cr'm (both + Cr~03 (both
at 300 at
pg/10 ml) 300 NgIlO ml)
0.02 M (NH,),SO,/94.5 ~ 3.5 94.0 ~ 2.6 85.2 t 3.7
0.02 M NH,OH
0.05 M (NH,).SO,/95.3 t 2.4 95.1 t 3.9 89.2 ~ 4.1
0.05 M NH,OH
15 0.02 M Tris-HCl93.9 t 3.8 93.3 t 2.8 83.0 t 4.8
+
'VH,OH (pH
8)
0.05 M Tris-HCl94.7 ~ 2.5 94.0 ~ 3.6 95.1 t 3.7
+
l~'H,OH (pH
8)
20 Neither method-induced reduction of Cr" nor oxidation of Cri° was
observed under
these experimental conditions. When Cts° was present in solutions
containing Cr",
Cr" was essentially unchanged. Thus, the conditions used appear to stabilize
both Crj°
and Cr" species.
Examote 4 - Analysis of Reference materials. Three certified reference
25 materials (CRMs) were chosen to evaluate the basic isolation and
determination
procedure of the present invention. One certified reference material, US EPA
CRl~9
- z~ -
CA 02320828 2000-08-17
WO 99/44056 PCTIUS99/04200
013-050 (paint chips) contained relatively high levels ofnotal chromium
(reference
value 617.6 pg/g with a 95% confidence interval of 595 to 670 ~tg/g). Another
hulk
standard reference material, I~TIST SRM 1633a (coal fly ash) contained a much
lower
level of total chromium ( I 96 ~ 6 pg/g). No standard or reference data
appears to be
available for the Cr" content of these CRM samples. A third reference
material,
IRIvfM CRM 545, contained approximately 3 mg of total welding dust with about
100
pg of hexavalent chromium per filter sample. This Last reference material has
only just
recently become available and appears to be the only known CRM for particulate
hexavalent chromium.
In operation, 10 ml of 0.05 M anvnonium sulfate and 0.05 M ammonium
hydroxide (pN 8) buffer solution was added to about 1 g EPA CRM 013-50 paint
chips or about 2 g MST SRM 1633a coal fly ash to release the hexavalent
chromium
by sonication. For the mMM CRM 545 sample, the filters were sonicated in the
same
buffer solution. Follov~~ing sonication, aliquots of extraction solution were
subjected
to solid phase extraction, complexation with diphenylcarbazide, and
spectrophotometric detection. The analysis results showed that the hexavalent
chromium content was 54.4 t 2.3 pg/g for EPA CRM 0134-50 paint chips, and 0.19
+
0.01 ug/g for :v'IST SRM 1633a coal fly ash. These values are virtually
identical to
those found in using a flow-injection analysis procedure (see Example 6 below
and
Vfang et aL, Analyst. 1997, 122, 1307-1313). . -
The recovery results for hexava 1 ent chromium from 1RMM CiRM 545 are
shown in Table IV.
- 23 -
CA 02320828 2000-08-17
WO 99/44056 PCTIUS99/04200
TABLE n'
Cenified Experimental
Value Value
for Cr"
in CRM
545
Sample Vt'elding Crv' Cr''' . Cr" Recovery
dust {pg/mg) (ug/filter)(pg/filter)(%)
(~tg/filter)
. 1 2.895 39.5 114.3 112.3 98.1
2 2.953 39.5 1 ]6.6 115.2 98.8
3 3.053 39.5 120.5 119.4 99.1
4 2.994 3 9. 5 I ] 8.2 117.4 99.3
5 2.939 39.5 116.1 115.5 99.5
Average 2.967 ~ 39.5 I 17.1 115.9 99.0 ~
t ~ 0.55
1.98 2.33
The recovery of hexavalent chromium from the iRMM CRM is quantitative (99%).
E.,ampie 5 - Workplace Air Samples. This example illustrates the use of the
present method to monitor Cr" exposure at various aircraft painting and
maintenance
operations at U. S. Air Force bases. Air filters used to collect airborne
particulate
(including Cr") were placed in a 15 ml plastic tube to which 10 ml of 0.05 M
(NH,)~SO, and 0.051-4 I''1-i,OH (pH 8) buffer solution was added. This was
followed
by ultrasonication in an ultrasonic bath for 30 minutes (<40°C). After
ultrasonication,
a portion (about 3 ml) of the supernatant obtained (total of about 10 ml) was
loaded
onto a strong anion exchange cartridge. Washing of the SAE cartridge was
performed
by flushing it with 3 ml deionized water. The Cr" was eluted with 9 ml of 0.5
M
(:v'H,)=SO, and 0.1 M NH,OH (pH 8) buffer solution in three fractions at about
2
ml/min. After isolation and purification by SAE-SPE, the eluate was acidified
with
100 ul 37% HCl solution, followed by reaction with 2 ml of 20 mM 1,5-
phenylcarbazide complexing reagent. The reaction of 1,5-diphenylcarbazide with
Cr'''
is completed in a feu~ seconds.
-24-
CA 02320828 2000-08-17
WO 99/44056 PCTIUS99/04200
Quantification of hexavalent chromium for the air filter samples was preformed
using the standard addition method. A blank filter (non exposure) was used as
a blank
sample and was subjected to the same procedure as all other air samples, and
analyzed
with the same technique to check for any influence of baseline shift, and to
ensure
matrix matching. The linear dynamic range was IO pg/1 to 3.0 mg/1 with a
correlation
coefficient (R') of 0.9991-0.9998 for all quality assurance/quality control
samples
(clean filters spiked with known quantities of hexavalent chromium). The
calibration
equation for the external standard or standard addition was calculated by
linear
regression.
The results obtained with these workplace air samples are shown in Table V
below.
-ZS-
CA 02320828 2000-08-17
WO 99/44056 PCTIUS99I04200
Sample Cr" Content Sample Location
lrg/sampleAir Volumelrg/m'
1 0.664 147.1 4.51 Alodining F-16
2 0.707 72.67 9.73 Alodining spry>'
F016 swipe and
water rinse)
3 1.034 126. I 8.19 Cutting/grinding
steel
4 0.643 117.2 5.48 Sanding C0130 beaver
tail
5 0.52 I 73.01 . 7.14 Sanding F-I6 bare
metal
6 2.952 191.5 I 5.41 Sanding F-16 under
wing ~.
under couch
7 4.175 192.1 21.73 Sanding F-16 to bare
metal
under wing
8 4.523 173.1 26.12 Sanding F- I 6 landing
gear
9 2.678 72.45 36.97 Sanding C-13U door
edges
10 2.224 I 59.03 37.67 Sanding F-16 air
intake
11 3.641 36.04 101.02 Priming F-16 vellovc
spray
12 4.341 39.11 110.99 Priming C-130 door
13 6.922 67.01 103.32 Priming F-115
1 14 - 6.567 39.53 166.12 Priming F-16
S
8.535 48.91 174.51 Priming C-130
16 34.41 136.5 252.08 Priming F-I6
17 20.84 79.01 263.76 Ping C-130 cargo
door
18 6.822 25.28 269.86 Priming water base
I9 14.18 39.02 363.65 Priming F-16 landing
gear
20 8.011 19.86 403.37 Priming small parts
& panels
The Cr" contents detected reasonably match those expected in the various
workplace
locations. The highest contents of Cr" were detected in paint priming
operations
(where the primers are known to contain high levels of Cr'''). and the lowest
hexavalent chromium exposures were observed in alodining and cutting
operations.
-26-
CA 02320828 2000-08-17
WO 99/44056 PCT/US99104200
Sanding cave rise to Cry'' exposures that were intermediate in hexavalent
chromium
content.
Example 6 - Flow Infection Anah~sis. This example illustrates the use of flow
injection analysis (FIA) using UV/VIS detection of the Cr-diphenylcarbazone
complex.
Except as noted, the general procedures and reagents used in the previous
examples
were employed here. (This procedure is described in more detail in Wang et
al.,
Analyst, 1997, 122, 1307-1312, which is hereby incorporated by reference in
its
entirety.) The flow injection system consisted of a Waters 600-MS system
controller
pump, a Waters 717 Plus autosampler (Millipore, Milford, MA), and a Model 783
programmable ultraviolet absorbance detector set at 540 nm (Applied
Biosystems,
Ramsey, NJ). The flow rate of the pump was I .0 mUmin for the mobile phase.
Upon
initial start up, the system was allowed to equilibrate for about 15 minutes.
A sample
volume of 10 pl was used for injections.
For elution, a 10 cm x I .5 cm id anion-exchange column containing 1.0 g resin
~~as used. The strong anion-exchange resin was Dowex 1-X8 (Fluka Chemical,
Ronkonkoma. Nl'), a styrene-divinylbenzene polymer to which tertiary ammonium
groups have been bound. The resin capacity was 1.3 mequiv/ml; the chloride
form
was used. The resin was cleaned prior to use by slurrying it with 3 M HCI,
allowing it
to stand for 10 minutes, and then decanting offthe acid. This procedure was
repeated
three times. After pouring offthe last portion ofacid, the resin was slurried
with 1 M
HCl and dried prior to use.
This FIA-W/VIS method was used on a variety of spiked samples, certified
reference materials, and workplace samples. In general, the samples (spiked
filters
(0.8 u. 37 mm). 1.0 g spiked sand, 0.1 g CRMs, and 1.0 g other fly ash and
paint chip
materials) were treated with 0.05 M (IVH4)~SO; 0.05 M NH3 (pH 8.0) in an
ultrasonic
bath for 30 minutes at about 40°C. The samples were then subjected to
anion
exchange separation (1.0 g Dowex 1-X8). The eluate was acidified and reacted
with 2
_27_
CA 02320828 2000-08-17
WO 99/44056 PCT/US99/04200
ml of 20 rr~'vI 1,5-diphenylcarbazide complexing solution. Finally, the Cr"
content was
determined by flov~~ injection analysis. The following results were obtained.
TABLE ~'I
Sample Cr" Recovery
(p8/8 ~RSD) (%)
~ Spiked filter - 40 Ng/filter 37.9 f 3.2 95.6
Spiked sand - 25 Itg Cr"/g 23.8 t 4.5 95.2
Spiked sand - 25 pe Cr"lg + 23.3 t 7.6 93.8
250 erg Crmlg
EPA 013-050 CRM (paint chip) 54.4 f 3.4 NIA
NIST 1633a CRM (coal fly ash) 0.19 t 3.9 NIA
Power company coal fly ash 0.5313.4 NIA
Gas pipe paint chips 352.6 t 4.4 NIA
Laboraton~ door paint chips bE .w detectionNIA
i imits
University building paint chipsbelo.,~ detectionNIA
limits
"NIA" in the Table indicates that no standard or reference data for Cr" was
available
for those particular samples and that Cr" recovery cannot be determined. The
detection limit was estimated at 0.11 ng Cr'''. The results for the two CRM
samples
are in good agreement with those reported in Example 4 above.
-28-