Note: Descriptions are shown in the official language in which they were submitted.
CA 02321303 2009-05-05
METHOD FOR DETERMINING A SHAPE SPACE FOR A SET OF MOLECULES
USING MINIMAL METRIC DISTANCES
Field of the Invention
This invention relates to a method and apparatus for
comparing molecules. It is especially useful in comparing
individual molecules against a large library of molecules and
has particular application in the pharmaceutical industry in
drug development.
Background of the Invention
A molecule is normally thought of as a set of atoms of
varying atomic type, with a certain bonding pattern. Indeed
this "chemical" description can uniquely describe the
molecule. It is the language that chemists use to compare
and contrast different molecules. Efficient database models
have been constructed to store such information for fast
retrieval and storage. However, this form of description
does not actually describe the three dimensional structure of
the molecule, e.g. the positions of each atom, and since the
interaction of molecules is a spatial event, the "chemical"
description is incomplete for physical phenomena. One such
phenomenon of commercial importance is the binding of drug
molecules to sites of biological importance, such as the
active areas or "sites" on protein surfaces, which is the
mode of action of nearly all pharmaceuticals.
Drug molecules are often small, on the order of 20 atoms
(excluding hydrogens). They interact with large
- 1 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
macromolecules such as proteins by binding to them. Through
binding, the drug may activate or inhibit the normal action
of the macromolecule. The binding occurs at specific sites
on the macromolecule, and the basis of tight and specific
binding is complementarity in shape and other properties,
such as electrostatic, between the two molecules.
Pharmaceutical companies maintain computer databases of
all molecules they have synthesized, plus other compounds
available on the market. The use of these databases and the
techniques of computer-aided drug design are beginning to
replace trial and error lab testing in new drug development.
Important components of this process are finding new small
molecules similar in shape to ones known to bind a target,
and designing new molecules to fit into known or hypothesized
binding sites.
There have been many attempts to describe or "encode"
the three dimensional information of molecules beyond a
simple list of coordinates. Many involve the distances
between pairs of atoms in a molecule, i.e. an atomistic
approach akin to the chemical description but with extra,
spatial degrees of freedom.
A more radical departure is to adopt an alternate
representation of a molecule: the field representation. A
field is essentially just a number assigned to every point in
space. For instance, the air temperature in a room at every
point in that room forms a field quantity. Molecules have
one fundamental field associated with them, namely the
quantum mechanical field that describes the probability of
- 2 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
electrons and nuclei existing at each point in space.
However, this field can be thought of as giving rise to
other, simpler fields of more use in understanding a
molecule's properties. Chief amongst these are the steric
and electrostatic fields, although others are used, such as
the hydrophobic and the hydrogen bond potential field. An
illustration of a Gaussian representation of a steric field
is shown in Fig. 1. As is customary, on each contour line in
the field, each point has equal value.
Steric fields describe the mass or shape of the
molecule, and at the simplest level such a field might have a
value of one inside the molecule and zero outside.
Electrostatic fields represent the energy it would take to
place an electron at a particular place in space, by
convention positive if the energy is unfavorable and negative
if favorable. These two fields are the most relevant for
molecular interactions because of basic physical laws, i.e.
that two molecules cannot overlap (steric repulsion) and that
positive atoms like to be near negative atoms and vice versa.
These are the basic components of molecular interactions.
If a molecule is known to bind and have effect on some
biological target, it is of great commercial interest to find
other molecules of similar shape and electrostatic properties
(i.e, similar fields) since this enhances the likelihood of
such molecules having similar biological activity. Since
shape and electrostatic character are consequences of the
underlying atoms, which can be efficiently encoded by a
chemical description, such searches have traditionally been
- 3 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
performed at this level, by looking for molecules which are
"chemically" similar. One disadvantage of this approach is
that the relationship between chemical similarity and
structural similarity is not precise; and chemically similar
molecules may be structurally quite different. Another
disadvantage is that a chemically similar compound may well
be covered by the same patents as the original molecule.
Finally, searching only chemically similar compounds
inevitably means one will not find active molecules that are
not chemically similar.
This latter point is key. David Weininger of Daylight
Chemical Information Systems has reported an analysis that
suggests there are 10200 different molecules synthesisable by
known means. (Only 10107 molecules of typical drug size would
fit in the known universe!). As such, any molecule, of any
shape or electrostatic profile, has a potentially
astronomical number of similarly shaped and charged
"doppelgangers". Only a fraction of them are necessarily
chemically similar. Hence by restricting the search to
chemical similarity a vast number of potential drug leads are
never discovered.
Although 10200 molecules is too large a number to ever
enumerate, I believe that it is possible to determine bounds
to the possible variations of molecular fields of this
hypothetical set. Furthermore, I plan to compute for
database storage a very large number of molecular structures
(e.g. of the order of billions) that sample this range such
that I am able to find a match, or "mimic", from this
- 4 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
collection to any novel structure presented. Such a database
would be many thousands of times larger than any currently in
existence and hence crucial to this plan is the efficient
organization of such for fast search and retrieval of such
mimics and the assessment of whether I have indeed "covered"
chemical space. It is these problems that the present
invention addresses.
Prior Art
Much has been done in the use of molecular fields to
compare and contrast molecules and to predict activity from
such operations. Some of these approaches are described
below. I believe that the crucial aspect of my approach
which differs from all prior work is in the application of a
particular property of field comparison, namely the "metric"
property, and in a novel way to decompose fields into
separable domains, wherein each is quantifiably similar to a
geometrically simpler field.
The most widely known "field analysis" approach is that
known as Comparative Molecular Field Activity (COMFA). See
U.S. Patents 5,025,388 and 5,307,287 assigned to Tripos Inc.
of St. Louis, Missouri. The idea behind COMFA is to take a
series of molecules of known activity and to find which parts
of these molecules are responsible for activity. The
procedure is to first overlay the set of molecules onto each
other such that the combined difference of the steric and
electrostatic fields between all pairs of molecules is at a
minimum. (The concept of overlaying, i.e. finding an
- 5 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
orientation between a pair of molecules that minimizes a
field difference is fundamental to all methods that utilize
molecular fields for molecular comparison.)
Given this ensemble average, one then finds values of
properties such as the electrostatic field at a number of
grid points surrounding the set of molecules. These then
become data points in a statistical analysis known as Partial
Least Squares (PLS), which seeks to identify which points
correlate with some measure of activity. For instance, if
all active molecules, once overlaid, had a similar region of
positive potential, while less active or inactive molecules
did not, the procedure would identify this as an important
common motif in activity.
Problems inherent in COMFA are the multiple alignment of
a set of molecules, the placement of grid points near the
molecules, and the interpretation of the PLS output.
Another approach which uses molecular overlay is that
set forth in U.S. Patents 5,526,281 and 5,703,792 of Chapman
et al. of ARRIS Inc. They are interested in selecting a
subset of compounds from a much larger set that retains much
of the diversity of the larger set. The basic concept is to
start with as few as one molecule as the representative set,
then to overlay a candidate molecule to minimize steric
and/or electrostatic field differences to all in the set, and
then to calculate differences between the molecules based
upon this alignment. This is repeated for each of the
candidate molecules. The candidate which is "most different"
from those already in the representative set is added and the
- 6 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
procedure then repeated until the number of compounds chosen
reaches a desired threshold.
In both COMFA and the Chapman approach, field similarity
is used as a tool to solve the alignment issue, and
similarities or differences are then calculated. The value
of the field similarity or difference is of secondary
importance, it merely solves what is called the "assignment"
problem, i.e. which atoms, or areas of a molecule's field are
"equivalent".
In contrast, in Mestres et al., "a Molecular Field-Based
Approach to Pharmacophoric Pattern Recognition," J. Molecular
Graphics and Modelling, Vol. 15, pp. 114-121 (April 1997),
molecules are aligned based upon the overlap of their steric
or electrostatic fields, or by a weighted sum of the two. A
similarity measure is defined that equals one when the fields
are the same, and minus one when they are maximally
different. The Mestres et al. work is embodied in a program
called MIMIC, which performs global and local optimization of
the field overlap. They note that there are several possible
overlays that have the appearance of being the best overlap.
These are so called "local minima", because while small
displacements lead to a decrease in their similarity
function, they may not be the best "global" solution. This
is as expected since field overlay belongs to the class of
problems known to have "multiple minima". Mathematically
this is usually an intractable problem, solvable only by much
computation, e.g., as is evident in the descriptions by
Mestres et al. In fact, the multiple overlay solutions are
- 7 -
CA 02321303 2002-03-22
WO 99/44055 PCT/US99/04343
one of the key aspects of their work, in that one cannot be
sure which is the most "biologically" .relevant overlay, and
what might be the correct weighting of steric to
electrostatic fields.
An additional aspect considered by Mestres et al. is the
issue of molecules existing in multiple structural
conformations, i.e. energetically there may be more than one
possible structure for a given molecule. Mestres et al.
calculate the similarity indexes of all pairs of
conformations of a molecule and perform what is known as a
principal component analysis (PCA). They do this to find
representatives of all possible conformations that are most
distinct. Although this procedure is really akin to finding
the dimensionality of the space in which these conformers
exist, Mestres et al. do not use PCA for this purpose, but
merely to cluster the conformers. They do not apply PCA to
sets of different molecules, only to conformers of the same
molecule, and they do not use any other "metric" property of
their similarity measure. In fact they seem unaware of such.
There is an important distinction to be made between a
"measure" of similarity and a "metric" of similarity,
although these words are often used interchangeably. A
2.5
measure can be any quantity which has a correspondence with
molecular similarity, i.e. the idea that the more similar the
measure the more similar the compounds. A metric has a
precise mathematical interpretation, namely that if the
metric, or more commonly the metric distance, between A and B
is zero then the two items are the same item, that the
- 8 -
CA 02321303 2002-03-22
WO 99/44055 PCT/US99/04343
distance from A to B is the same as the distance from B to A.
and that the distance from A to B plus the distance from B to
a third compound C must be greater than the distance from A
to C. This latter is called the "Triangle Inequality"
because the same conditions can be said of the sides of a
triangle ABC. The Triangle Inequality, or metric upper
bound, also leads to a lower bound, namely that in the case
above, C can be no closer to A than the difference of these
distances A to B and B to C.
In M. Petitjean, "Geometric Molecular Similarity from
Volume-Based Distance Minimization-Application to Saxitoxin
and Tetrodotoxin," J. Computational Chemistry, Vol. 16, No.
1, pp. 80-95 (1995), it is recognized that the quantity that
measures the overlay of fields forms a metric quantity, and
that the measure of the optimum overlay of two fields also
forms a metric which is intrinsic to the molecule, i.e.
independent of orientation or position.
A metric distance may also be used in a technique called
"embedding". The number of links between the elements of a
set of N elements can be shown to be N*(N-1)/2 and each link
can be shown to be a metric distance. While a set of N
elements has N*(N-1)/2 distances, the set can always be
represented by an ordered set of (N-i) numbers, i.e. I can
"embed" from a set of distances to a set of N positions in
(N-i) dimensional space. This is identical to Principal
Component Analysis mentioned previously, except that with PCA
one finds the most "important" dimensions, i.e. the
"principal" directions, which carry most of the variation in
- 9 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
position. Typically with PCA one truncates the
dimensionality at 2 or 3 for graphical display purposes. In
general, the number of dimensions which reproduces the set of
N*(N-1)/2 distances within an acceptable tolerance may be
much smaller than (N-i), yet still be greater than 2 or 3.
Hence one talks of "embedding into a hyper-dimensional
subspace", where hyper-dimensional means more than 3
dimensions, and subspace means less than (N-1). Techniques
for such an embedding are standard linear algebra. When
applied to molecular fields, the result of embedding is a
shape-space of M s N-1 dimensions.
Summary of the Invention
I have invented several techniques for characterizing
molecules based on the shapes of their fields. The minimal
distance between two molecular fields is used as a shape-
based metric, independent of the underlying chemical
structure, and a high-dimensional shape space description of
the molecules is generated. These attributes can be used in
creating, characterizing, and searching databases of
molecules based on field similarity. In particular, they
allow searches of a database in sublinear time. The utility
of this approach can be extended to automatically break
molecules into a series of fragments by using an ellipsoidal
Gaussian decomposition. Not only can these fragments then be
analyzed by the shape metric technique described above, but
the parameters of the decomposition themselves can also be
- 10 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
used to further organize and search databases. The
ellipsoidal method can also be used to describe binding or
active sites on macromolecules, providing a template for
searching for complementary molecules in a database such as I
describe. The most immediate application of these techniques
is to pharmaceutical drug discovery and design.
In a preferred embodiment, I obtain the minimal distance
between a first molecular field and a multiplicity N of other
fields by: selecting a small number M of the fields, for each
of the M fields determining its metric distance to all the
other N fields, for each of the M fields, making an ordered
list of the metric distances between that field and all the
other N fields, determining the metric distances between the
first field and each of the M fields, determining the metric
distances between the first field and the fields on the
ordered list associated with the M field that has the
shortest metric distance between it and the first field.
These metric distances are determined beginning with the
field on the list that has the shortest metric distance
between it and the M field and continuing such determination
with fields having increasingly greater metric distances from
the M field until a field is reached that has a metric
distance from the M field that is more than twice the metric
distance from the first field to the M field.
Advantageously, the invention is practiced on a
computer, the determination of minimal distances and
ellipsoidal Gaussian decomposition are implemented by
computer programs and the molecular field information is
- 11 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
stored in a computer database. Illustrative apparatus for
practicing the invention is a personal computer such as an
IBM-compatible PC or a work station such as a Silicon
Graphics Iris Indigo Elan 4000.
Brief Description of the Drawings
These and other objects, features and advantages of my
invention will be more readily apparent from the following
detailed description of the invention in which:
Fig. 1 is an illustration of a Gaussian representation
of a steric field;
Figs. 2A and 2B illustrate the results of overlaying two
molecules using a prior art technique;
Fig. 3 is an illustration of a molecular field
representation produced in accordance with the invention;
Figs. 4A, 4B and 4C are examples of three molecular
field representations formed using increasing numbers of
ellipsoidal Gaussian functions in accordance with the
invention;
Fig. 5 is a flow chart illustrating one aspect of the
invention;
Fig. 6 is a flow chart illustrating a second aspect of
the invention; and
Fig. 7 is a flow chart illustrating a third aspect of
the invention.
- 12 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
Detailed Description of the Invention
Definitions
The following defined terms are used in the detailed
description:
A structure: A description of the three dimensional
coordinates of each of the atoms that comprise a molecule.
These coordinates may be found from experiment, e.g. X-ray
crystallography, or by computer computation by one of many
methods known in the field of molecular modeling.
A conformer: If a molecule has more than one structure, then
each structure is referred to as a conformer of that
molecule.
A molecular field, or field: A set of numbers that represent
the value of some property in and around a molecule. Such
numbers may be explicitly stated, or listed, for instance as
values associated with each point on a regular lattice, or
grid, which contains the molecule. Or they may be
functionally implied. For instance, if a functional form for
the field property is assigned to each atom then a mechanism
exists to calculate the field value at any point in space.
One example is the electrostatic potential around a molecule,
one form of which may be calculated from the charge
associated at each atom and the functional form for
electrostatic potential from a single charge, i.e. Coulomb's
Law.
13 -
CA 02321303 2000-08-24
A Gaussian molecular field: A particular instance of a
functionally implied molecular field with which I am much
concerned is that produced by assigning a Gaussian function
to each atom to represent the steric field of that molecule.
A Gaussian function is one that has the form:
-wi((x-ai)2+(y-bi)2+(Z-Ci)2) (1)
G. (r) = pi- e
where the position vector is
r= (X, Yi Z)
and where for any atom i, pi is a prefactor, wi is a width
factor, ai, bi, and ci are the Cartesian coordinates of the
atom center, and xi, yi, and zi are the Cartesian coordinates
of any point in space.
Sum and Product forms of Gaussian fields: Upon assignment of
prefactors and widths to the Gaussian associated with each
atom there are many ways of generating a steric field, but
the two that will be referred to in this description are the
sum and product forms.
Sum Form:
14 - NY2 - 673169.1
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
N
FM (r) =~ Gi (r) (2)
i=1
Exclusion Product Form:
N
FM(r)=1-fl (1-Gi(.r)) (3)
i=1
where FM is the steric field of a molecule with structure and
orientation M made up of N atoms represented by Gaussian
functions G1 centered about each atom i. Each form has the
property of having zero values away from the molecule and
usually positive values "inside" the molecule. The exclusion
product form expands to a sum of Gaussians, because the
product of two Gaussians is itself a Gaussian function.
Field Arithmetic: Two fields are added together by adding
their values at corresponding grid points, or by adding
together the functional forms that define the field. A field
can be "scaled" by multiplying either the value at each grid
point, or the functional form, by a number. Two fields can
by multiplied together by multiplying the values at
corresponding grid points by each other or multiplying their
functional forms together.
- 15 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
Field Volume:
For any field function FM the field volume is given by its
integral over all space
V= J FM (z") dV (4)
or if the field is represented for example on a three
dimensional grid with sides of I, J, and K points,
I J K
V =E L r: E FM -vi k (5)
i=1 j=1 k=1 1,j,k ~j1
where FM i,k,j is the value associated with grid point i,j,k and
vi,;,k is the volume associated with that grid point. On a
regular grid vi,j,k would be a constant.
The overlap Q of two fields FM and Fp is given by
Q=IFM(r) Fp(.r) dr(6)
for the continuous case, or
rI rJ rK
QJ: L: 1: FMi, 7, k FPi, J. kV V. (7)
j=1 k=1
- 16 -
CA 02321303 2000-08-24
if the molecule is represented on a grid as described above
in Field Volume.
The norm of a field FM, is defined as the square root of the
overlap of that field with itself, i.e.,
FMI = FM (r) FM (r) dr (8)
The norm of the field difference, also referred to as the
field difference D, is given by
IFM-F pI= (FM(F)- p(r))2dr (9)
The greater the field overlap, the less the field difference,
as can be shown by rewriting the above equation as
D2=f FM(r)dr+fFP(r)dr-2 f FM(F) FP(Fr) dr
(10)
Only the third integral varies if FM and F. are moved relative
to each other. But this third integral is equal to the
17 - NY2 - 673169.1
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
overlap Q, . Thus the larger the overlap, the smaller is D2,
and hence the smaller the field difference D.
The metric triangle inequality:
If the distance dAB from A to B is known, and the distance dBC
from B to C is known, the triangle inequality states that for
the distance dAC:
dAC<_ dAB+dBC (11)
and from this it also follows that
IdAB-C (12)
These are the upper and lower bounds, respectively, for the
value of dAC.
Ellipsoidal Gaussian Function (EGF):
The overlay of molecular fields is a method of finding
global similarities between molecules, i.e., whether molecule
A has the same distribution of properties as molecule B.
While this is extremely useful, it is also the case that
local similarities are of interest, i.e., when a part of
- 18 -
CA 02321303 2000-08-24
molecule A is similar to a part or all of molecule B. The
methods described below are an approach to solve this problem
by defining representations of parts of a molecular field
which have an approximately ellipsoidal character. These
representations conform to visual and chemical intuition as
to possible fragmentations of a molecular structure, and can
be compared between molecules either singly or in combination
with ease.
An EGF for a molecule M is defined as:
Ui(d i-Ai)2-Vi(di-Bi)2-Wi(diCi)2
EGFM, i (r) =pi=e
(13)
for the ith of K ellipsoidal Gaussian functions fit to some
molecular field FM,(r), where K may be as small as 1, and where
the displacement vector is
di= (x-ai, -y-b,, z-Ci)
and the position vector is
r= (x, y, Z)
19 - NY2 - 673169.1
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
and where pi is a prefactor, ui, vi and w, are width factors,
ai, bi and ci are the coordinates of the center of the EGF,
and xi, yi, and zi are the coordinates of any point in space.
Ai, Bi, and Ci are three mutually orthogonal unit vectors that
define the directions of the ellipsoidal axes.
An Ellipsoidal Gaussian Representation (EGR) of a
molecular structure is constructed by fitting one or more
Ellipsoidal Gaussian Functions (EGF) to a field function of a
molecule, for instance a steric or electrostatic field.
The EGR field is defined as the sum or exclusion product of
the fields generated by the EGF's. For the EGR of a
molecular field decomposed into N EGF fragments:
Sum Form:
N
EGRM(r) EGFM' i (r) (14)
~=1
Exclusion Product Form:
N
EGRM(r) =1-fl (l-EGFM,i (r) ) (15)
20 -
CA 02321303 2000-08-24
The Sum Form of the EGR is currently preferred.
The procedure for fitting one or more EGF's to a molecular
field involves ascertaining the parameters of each EGF, i.e.,
center (a,b,c), widths (u,v,w), and axes directions (A,B,C)
that minimize the integral over all space of the square of
the field difference between the molecular field and the EGF
field. This integral is referred to as the EGR Fitness
Function (EFF) .
EFF(EGRM, FM) = f (EGRM(r) -FM(r)) 2dr
(16)
Overview
To compare two molecules A and B, I determine the
difference between their fields A and B using the norm of the
field difference shown in equation (9). The norm of the
field difference constitutes a metric distance, not just a
similarity measure. Furthermore, the minimal value of d, dm,
which occurs at the point of maximal overlap of the two, also
forms a metric.
Various techniques exist to attempt to find the best
overlap of two fields, typically involving repeated searchs
from different starting orientations of the two molecules.
This is necessary because no direct solution for the minimal
distance orientation is available, and most methods tend to
2 1 NY2 - 673169.1
CA 02321303 2002-03-22
WO 99/44055 PCT/US99/04343
get caught in nearby local minima, missing the global
minimum. One such technique is a Gaussian technique
described in J.A. Grant et al., "A Fast Method of Molecular
Shape Comparison: A Simple Application of a Gaussian
Description of Molecular Shape," J. Computational Chemistry,
Vol. 17, No. 14, pp. 1653-66 (1996) Using this technique, I
overlaid the two molecules shown in Fig. 2A to produce the
result shown in Fig. 2B.
This Gaussian technique is less susceptible than other
techniques to being caught in local minima. Because dm, once
found, is an invariant, fundamental distance between the two
molecular fields, it can be used in creating a hyper-
dimensional embedding of a set of molecules. This hyper-
dimensional space is called a "shape space", (but where
"shape" can mean electrostatics or other field properties,
not just steric fields). I believe that for small molecules
the number of dimensions will be on the order of a few dozen.
In the following, I may refer interchangeably to maximal
overlap (or overlay), minimal field difference, and minimal
distance, as they all refer to and measure the same optimal
orientation of two molecules with respect to each other.
Rather than merely use the overlay optimization as a
method of aligning molecules for further similarity
comparison, I use the overlay measure as a metric distance
for the organization of large numbers of structures such that
searches within this dataset can be made "efficient".
Efficiency here can be precisely defined in the language of
database algorithms: the search time can be made "sublinear",
- 22 -
CA 02321303 2002-03-22
WO 99/44055 PCTIUS99/04343
i.e. one does not have to check every entry (a "linear"
search). A large body of literature deals with algorithms
for sublinear searching when a metric exists to pre-organize
a database of knowledge. I have not decided upon an optimum
approach as yet because there are still many aspects not
explored which can impact efficiency, i.e. the nature of the
shape space molecular fields occupy. Basic "vanilla"
database approaches ignore such and merely use the distances
between data entries to organize the data, whereas more
sophisticated approaches utilize shape space characteristics.
In all of my methods for using the field metric, the
steric field for each molecule is constructed either from a
sum of Gaussians centered at each atom, or as one minus the
product of one minus each such Gaussian. These are referred
to as the "sum form" and "product form" respectively. The
product form has the advantage that it removes excess
internal overlap and hence is smoother inside. The sum form
has the advantage that it is numerically simpler. Each
Gaussian is such that its volume is the same as that of the
atom it represents, and the volume, as of any field, is
calculated from the integral of the function over all space.
A simple use of the metric property is to speed the
search for the best global overlay (minimum field difference
or distance) between two molecules. Consider the situation
where the difference measure is calculated for molecule A and
molecule B in one orientation, and I want to know the
difference measure for molecule A and B where B has been
placed in a different orientation. I am free to think of
- 23 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
this second instance of molecule B as a different molecule C.
If I know the difference between B and second B (C) then I
have a measure, via the triangle inequality, of the maximum
and minimum values of the difference between A and this other
B. See equations (11) and (12).
Both upper and lower metric bounds are very useful in
database searching when large numbers of data items must be
compared. For instance, suppose one is looking for the
closest item in the database to a given query example (A) and
the metric distance between this example and one particular
member (B) of the database is small, say D. If one has
precalculated and stored the distances between all pairs of
data entries in the database, then there is no need to search
any entry (C) which has a distance of more than 2D from the
particular member (B) because the metric lower bound
specifies any such entry must be more dissimilar.
If the lower bound to d(AC) is still too large to be
considered for further study I can eliminate entry (C) from
further consideration as a global minimum. Hence, if I know
d(BC) I can "prune" my search space for the best overlay.
But since d(BC) merely involves the overlap of B with itself
in a different orientation, these values can be pre-
calculated, stored along with other quantities associated
with B, and used to speed the search for the best overlap of
B with A or any other molecule.
Finding the shape space position of a molecule not part
of the original shape space characterization is done by a
high-dimensional equivalent of triangulation, i.e. given a
- 24 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
dimensionality of N I need the distance of the molecule to at
most N+1 other molecules which have already had their shape
space vectors calculated. This is again standard linear
algebra.
The positions in shape space can also be used as
characteristics in other procedures, for instance in a
Partial Least Squares analysis to find the components that
correlate with activity. Shape space positions can also be
used to assess the diversity of a set of compounds, i.e. are
they spread evenly or clustered within the shape space
defined by their mutual distance. Various statistical
measures can be applied to this set of positions, e.g.
finding the degree of clustering, the largest "gap" or void
in the shape space so defined etc. Another use is to compare
two sets of compounds which have both had shape space
characterization, i.e. are there parts of shape space in one
set not covered in the second and vice versa; what molecules
can be omitted in a merged set based upon shape similarity;
which compounds fill the largest void in the other set's
shape space, etc. Finally, given a large database whose
dimensionality has been determined, one could find a smaller
representative set of molecules that retain the same
dimensionality, i.e. the same diversity.
These positions can also be used to guide the layout of
the data within a database for efficient search and retrieval
by standard methods. For instance, a method known as N-
dimensional trees orders data by each dimension in turn and
can,reduce the search time for finding all entries within a
- 25 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
certain distance of a query to a constant multiplied by the
logarithm of the number of entries in the database. For
instance, if there are 1,000,000 entries one only has to
check of order 6*constant entries, and for 10,000,000 only
7*constant entries. Another method is known as "Vantage
Trees", wherein the data is organized about certain "vantage"
points, for instance the center of clusters of molecules in
shape space. A third method chooses a set of key compounds,
and for each of these, lists all the molecules in order of
distance from it.
For example, if I have 1000 molecules in my database I
might organize this information thus: select 10 "key"
molecules which are quite different in shape. For each of
these 10 key molecules I then find the distance from each of
these molecules to every other molecule in the database, and
make 10 lists where each list has a different key molecule at
the top and the rest of the 999 molecules are listed in order
of shortest distance from it. To find the closest match
between a test molecule and the 1000 molecules of the
database I begin by determining the metric distances between
the test molecule and each of the key molecules. Suppose the
shortest distance is to key molecule 6 and that distance is
X. I now begin to calculate the distances to the rest of the
molecules, but in the order specified by that key molecule's
list. Since the list has molecules close to key molecule 6
first, it is likely these are also close to my test molecule.
Furthermore, by the triangle inequality, since molecules
which are a distance greater than 2X from key molecule 6 must
- 26 -
CA 02321303 2002-03-22
WO 99/44055 PCTAJS99/04343
be greater than X from my test molecule, I only have to go
down the list until this condition is satisfied, i.e. I may
not have to test all 1000 molecules. Furthermore, if I find
a molecule closer than key molecule 6 early in the list, say
distance X-d, then I. only have to go down the list until the
distance from the key molecule is greater than 2 (X-d), i.e. I
can refine the cutoff distance as I progress down the list.
Thus I can search the database, by shape, in a time sublinear
with the number of molecules in the database. These methods
are not possible without evaluating a shape space description
of the set of molecules that comprise the database.
Ultimately I hope to characterize the shape space of all.
synthesizable molecules, i.e. the 10200 referred to earlier.
But the shape space of smaller sets of molecules can be
determined as well. The "position" of a molecule in such a
high dimensional shape space then allows one to calculate the
best possible overlay between two molecules without any
computer intensive minimization of the overlap of two fields,
or searching through multiple minima. (The distance between
these two points is just the square root of the sum of the
squares of the values assigned to each coordinate in shape
space, i.e. a higher order generalization of finding the
distance between any two points in three-dimensional space.)
I anticipate this will provide nearly a thousand-fold speedup
in the calculation of the best overlap between two molecules,
if their shape space positions are known, relative to my
current best methods (which are already hundreds of times
faster than any reported in the literature).
27 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
The field similarity methods I have described apply to
the total field that represents a molecule, and as such is a
measure of "global" similarity. Yet, very often in searching
for similar molecules one is satisfied with a match to parts
of a molecule, i.e. either all of molecule A matching part of
molecule B, or a part of A matching a part or all of B. The
reason this "subdomain" or "local" match problem is important
is that not all of a molecule's mass may be involved in its
biological function.
The intrinsic problem in subdomain matching is deciding
how to break up a molecule into smaller fragments. Very
quickly the problem can generate combinatorially large
numbers of sub-molecules. When there are good methods
available to avoid this explosion in the number of fragments
(for instance, when a molecule is synthesized from a
collection of smaller subfragments) then each fragment may be
analyzed by the same "global" methods described in previous
sections. In general though, this "submolecule" matching is
a graph theoretic problem upon which many lifetimes of
scholarly energy have been expended.
My approach is simple and yet quite powerful. I seek to
represent the molecular field by certain simpler shapes,
namely ellipsoidal Gaussian functions (EGF). An EGF is a
mathematical form that specifies a value at each point in
space that is equal to:
p*exponential of( -a*x*x -b*y*y -c*z*z)
where x, y and z are distances in mutually orthogonal
directions from a particular point in space, a, b and c are
- 28 -
CA 02321303 2002-03-22
WO 99/44055 PCTIUS99/04343
positive constants, and p may be positive or negative but is
usually positive. This function falls off rapidly far from
its center and has the symmetry of an ellipsoid.
The procedure is as follows:
a) Choose the number of EGFs that I want to represent the
field.
b) Choose random positions for the center of each EGF and make
each spherical, i.e. a==b=c=1.
c) Let the center, directions and magnitudes of each EGF
change such as to maximize the field. overlap between this set
and the molecular field. Many standard numerical techniques,
e.g., steepest descent, conjugate gradient method, can
perform this minimization. I have encoded the first and
second derivatives of this overlap function, which leads to
particularly efficient minimizations. An example of the EGF
produced by this method is shown in :Fig. 3.
The above is also a multiple minima problem, and once
this procedure has been completed one typically repeats it
with new starting positions, i.e. there is more than one
possible representation of a molecular field by a set of
EGF's. One also tries increasing numbers of EGF's, i.e. one,
2 5
then two, then three, etc., as illustrated by Figs. 4A, 4B
and 4C. Small drug-like molecules typically require at most
3 or 4 ellipsoids to represent them well. I can construct a
measure of how many EGF's are needed by also measuring the
field overlap of each EGF with the atoms that lie within its
domain, where each such atom is represented as a spherical
29 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
Gaussian. This value of fit typically gets worse as the
overall fit gets better (i.e. as the fit gets tighter, atoms
may be split across EGF boundaries) and hence the sum of the
two is a quantity which first increases with number of EGF's
but then decreases, the maximal value indicating the "best"
number of EGF's for that molecule, although I also keep "sub-
optimal" representations since these may also be useful.
One can now use the EGF's in the molecular comparison
problem in numerous ways. For instance, the simplest is to
deconstruct the molecule into separate sets of atoms, each of
which is then operated upon by my shape space methods, i.e.
as an automatic, non-chemistry based method of fragment
construction.
One can also use the EGF decomposition to organize the
storage of molecular shape information. For instance the
volumes and orientations of each EGF can be used as keys in
an index for rapidly finding similarly decomposed molecules.
Because the actual number of EGF's in any representation is
small, e.g. 3 or 4, one can also store all subsets of EGFs.
For instance a four EGF set ABCD could be stored on the basis
of ABCD, ABC, ABD, ACD, BCD, AB, AC, AD, BC, BD, CD, A, B, C
and D. This then provides a very simple method for subdomain
matching, i.e. a molecule might have EGF's very similar to A,
B and C but no fourth domain, or might have a different
fourth domain from D and yet still be rapidly returned as a
potential match.
The relationship between organizing molecular structural
information by the field metric and by EGF's is synergistic.
- 30 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99104343
For example, rather than start with random positions for the
EGF's, the algorithm will run much faster if one starts near
the final solution. If one can use the overlap metric to
pull up a similarly shaped molecule that has already been
EGF-processed, it is very likely that the EGF composition of
this molecule will form a good starting point. Similarly, if
one needs shape space coordinates of a molecule relative to a
set for which shape space has been characterized, then it is
better to start by calculating distances to molecules that
are close in shape. The EGF breakdown of the new molecule,
when compared to that of those within the database, provides
an "initial" embedding, i.e. by finding its rough position
within the shape space.
In addition to representing shapes of molecules one can
also use the EGF's to compare properties of the underlying
atoms, i.e. to solve the "assignment problem", i.e. which
atoms correspond to each other between two molecules. This
is because when one compares two EGF's there are no
ambiguities in their relative overlay (save for a four-fold
degeneracy from rotations about the major and minor axes,
i.e. there are just four ways to overlay two EGF's). As such
one can either directly compare atoms belonging to each EGF
(e.g. find distances from similar types of atoms to each
other), or one can project such properties onto the pseudo-
surface of each ellipsoid. I define a pseudo surface of an
EGF as the surface of an ellipsoid that has axes in the same
direction as the EGF and with the same relative axis (i.e.
a/b/c) as the EGF, and which has the same volume as the EGF.
- 31 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
(The volume of an EGF, or any field function, is the integral
of that function over all space). This also then defines an
object that can be graphically displayed as representing the
EGF, and this is included in the body of my software.
Properties associated with the surface of the EGF could be
the electrostatic potential at each point, the distance from
the nearest hydrogen bond acceptor etc. The difference
between the properties "painted" on the surfaces of two EGF's
is a measure of the similarity of these two EGF's with
respect to these properties and also forms a metric. As
such, an alternate method of storing molecular information is
on the basis of this metric, such that one can perform
sublinear searches to find similarly "painted" EGF's.
Advantageously, the fields, metrics and ellipsoids I
have been discussing can be used in drug design. If the
structure of the "active site" of a target protein is known,
this can be used to guide the design of the drug molecule.
This is because drugs tend to "fit" into the active site,
i.e. there is a "lock and key" or "hand in glove"
relationship between the shape of the drug and the shape of
the active site, which is often a cleft or groove-like. In
addition to representing the field of a molecule, one can
also use EGF's to represent the "absence" of a molecular
field. A long-standing theoretical challenge has been how to
represent the space in these binding pockets, since a
representation of such can then be used as a template to fit
possible tight binding drug molecules. Such an approach,
using spheres of varying size, has been used for over a
- 32 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
decade in the program by Kuntz known as DOCK, (the procedure
of estimating the binding arrangements of a small molecule in
a protein pocket is known as "docking").
Instead of using spheres to represent the vacant space
in active site protein pockets I use EGF's. The functional
form that I use to adjust the centers, axes, and axis
magnitudes of the EGF is such that for a spherical EGF the
correct distance of approach to an atom of similar radius is
reproduced. Such a function has already been devised for
this purpose, although other functions may perform better.
Given a representation of the protein pocket in terms of
EGF's, which may well be painted with the properties of the
protein atoms in proximity to each EGF, the process of
finding a match in a database of structures which have been
decomposed into EGF's becomes a matter of matching EGF's,
painted or not. This can be done by taking combinations of
EGF's from the pocket and treating this as the "target".
Since the number of EGF's for any pocket is expected to be
small, and because my proposed search strategies are
"sublinear", I anticipate that this will be practical for
libraries of compounds in excess of one billion structures.
This will dramatically simplify the process of finding both
potential tight-binders and also initial starting points for
docking algorithms for predicting binding orientations. It
will also help design libraries of compounds biased towards
fitting a particular protein pocket by selecting those
compounds likely to have similar EGF's to those filling the
active site.
- 33 -
CA 02321303 2000-08-24
WO 99/44055 PCTIUS99/04343
IMPLEMENTATION AND EXAMPLES
Specific implementations and examples of the foregoing
include the following:
1: Finding The Maximal Overlap (Minimal Field
Difference) Between Two Fields A And B
2: Refining The Search Position Via Numerical Or
Analytical Derivatives
3: Determining The Shape Space Of A Set Of Molecules
4: Calculating The Position Of A New Structure In A
Preconstructed Shape Space
5: Extending The Shape Space
6: Calculating The Maximal Overlap Between A New
Structure And A Large, Previously Shape-Space
Decomposed Set Of Molecules
7: Using The Shape Space Description To Correlate With
Known Biological Activity
8: Examples Of Using The Minimum Field Difference
Metric To Organize A Database Of Molecules
9: Examples Of Using The Shape Space Positions To
Organize A Database Of Molecules
10: Local Domain Decomposition
11: Constructing An Ellipsoidal Gaussian Representation
(EGR).
12: Construction Of Multiple EGR's Containing The Same
Number Of EGF's.
13: Constructing Molecular Fragments From An EGR.
14: Evaluating An EGR Fit: The Fragment Adjusted EFF.
15: Construction Of Multiple EGR's With Different
Numbers Of EGF's
16: Storage In A Database
17: Comparing Single EGR's To Solve The Atom Assignment
Problem
18: EGF Pseudo-Surfaces And Painted EGF's
19: Using A Database Of EGR's For (Molecular/Molecular
Fragment) Similarity Evaluation
20: Using EGF's In Similarity searches
21: Using EGR's To Find A Place In Shape Space; Using
Shape Space To Find EGR's
22: Defining An EGR For A Negative Space
- 34 -
CA 02321303 2002-03-22
WO 99/44055 PCTIUS99/04343
Finding the maximal overlap (minimal field difference)
between two fields A and B
A) Exhaustive search:
The optimal overlay of two molecules depends upon six
variables, i.e. three translational degrees of freedom and
three rotational degrees of freedom. Since the time taken
for any search technique rises as the power of the
dimensionality, the problem of optimal overlay is
computationally expensive. However, the metric property of
the field difference allows for certain improvements in
performance because of the triangle inequality. Thus
suppose I am trying to overlay molecule B on molecule A and
I already have one relatively "good" overlay, with a field
difference value of d. Suppose I have just calculated the
field difference value for a particular orientation and
central position of B with respect to A, d', which is
greater than d. I am now in a position to determine which
orientations and central positionings relative to this
orientation and positioning might have a field difference
of less than d. To see this note that the triangle
equality lower bound on metric properties states that:
Id(AB)- d(BC) s d(AC)
where d(AB) is the field difference between molecule A and
molecule B at a given orientation and translation relative
to each other, and similarly for d(BC) and d(AC), and the
3 C-
-
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
I ...I symbol of the left hand side indicates the positive
value is taken for the difference.
Now I am free to decide that molecule C is actually
molecule B, but at a different orientation and separation,
call it B'. The inequality then reads:
d(AB)-d(BB') < d(AB')
i.e. I have a limit on how much better the overlap of B'
can be with A than that of B with A, given that I know the
overlap of B with itself, but at a different orientation
and separation. But this quantity, d(BB') is independent
of A, i.e. depends only upon B. Hence it could have been
calculated before the overlap with A was considered, and
indeed can be reused in finding the best overlap with
molecules other than A. The use of d(BB') is that if the
left hand side of the preceding equation is still greater
than the best (lowest) value of the field difference so
far, then there is no need to calculate the overlap
quantity of A with B'.
As illustrated in the flow chart of Fig. 5, an example
of a procedure to use the metric property of the field
overlap is then as follows:
(i) Calculate the field difference norms of B with itself
at a series of orientations and separations, B'. Each
new orientation and separation is described by a
rotation and translation matrix, respectively. Store
the field difference norm along with the rotation and
translation matrices, and along with any other
information on molecule B. Note that in practice this
- 36 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
set of orientations and separations will range over
all such that give an overlap greater than some
critical value close to zero.
(ii) Find what might be a good overlay between A and B so
as to establish a reasonable value of d. One does
this by trying a set of translations and orientations,
B'', to coarsely sample all reasonable overlays (as in
(i), this means that the overlap is greater than some
small threshold value), and setting drain equal to the
best such value obtained.
(iii) Sample around each such overlay tried in (ii) using
translations and rotations B '' ' of B'' from these
positions as precalculated in (i), such that the
triangle lower bound can be used to prevent needless
calculation of possible overlays.
(iv) Accept the best overlay from (iii) as the best
possible overlay, or select the best N such as
candidates for further refinement, e.g. via numerical
optimization as described below.
B) Selective Search:
Align the two fields based upon aspects of the
structures or fields that suggest this alignment might be
good. For instance, if the two molecules are long and thin
then it makes sense to align them such that these axes are in
the same direction. Other examples might be to align similar
chemical fragments. In my approach I calculate quantities
- 37 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
such as the center of mass and the moment of inertia and use
these to align the molecules.
(i) Calculate properties, e.g. center of mass and moment
of inertia for A and B.
(ii) Translate and rotate B to match its properties to
those of A, i.e. superimpose the centers of mass, and align
the moments of inertia. Techniques for this are known in the
art.
2: Refining the search position via numerical or analytical
derivatives
Since a field overlap is defined for all relative
orientations and separations, one can calculate derivatives
of this function with respect to such variables. These are
then used to adjust the relative orientations and separations
such as to minimize the field difference (maximize the
overlap) by any number of standard numerical techniques, e.g.
steepest descent, conjugate gradient, Newton-Raphson etc. If
the field is explicitly defined (i.e. as values on a grid)
the gradient quantities are calculated numerically by
standard techniques. If the field is defined functionally,
derivatives may be obtained either analytically or via
numerical approximation.
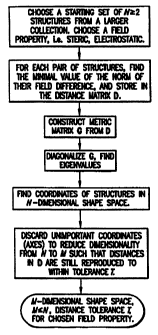
3: Determining the shape space of a set of molecules
As illustrated in the flow chart of Fig. 6, the shape
space of a set of molecules is determined by the following
steps:
(a)Calculate the maximal overlap (minimal field difference)
of all pairs of N structures that constitute my initial
- 38 -
CA 02321303 2000-08-24
WO 99/44055 PCTIUS99/04343
set. Note than N will not be the total set of molecules
and may be as small as two.
(b)Given that the norm of the minimal field difference
between any two molecules is a metric distance,
construct the distance matrix D, where the element ij of
D is the minimal field difference between molecule i and
molecule j.
(c)Construct what is known as the metric matrix G from D, as
described in any description of distance geometry, e.g.
Blaney and Dixon, "Distance Geometry in Molecular
Modeling", Reviews in Computational Chemistry, Volume V,
VCH publishers, 1994.
(d)Diagonalize G using any standard technique to find the
eigenvalues of a matrix.
(e)From this diagonalization procedure find a set of
positions in N-1 dimensional space that reproduce the
distances in the matrix D (see the Blaney/Dixon
reference for details)
(f)Determine which coordinates can be set to zero for each
and every molecule and still enable the distance matrix
to be reproduced to within a given tolerance T. (This is
equivalent to setting all eigenvalues of absolute
magnitude less than some cut-off value to zero). The
procedure for this is to a) set all the coordinates I
want to keep to zero, b) find the geometric center
(average coordinates) of the remaining coordinates (the
ones I might be able to discard), c) find the largest
distance from this center to any one point. d) ascertain
39 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
if this distance is less than 0.5*T, if so I conclude I
can discard these coordinates. (Alternatively, I find
the two most widely separated points in the set and
determine if the distance between them is less than T.
However, this is a more time intensive procedure).
In doing so I determine the M dimensional subspace that the N
molecules occupy, subject to tolerance T, where MSN-1.
This is the shape space for the field property used to
derive the minimal field differences.
A shape space, once determined, allows for various
geometric characterizations of that space and the molecules
whose positions have been determined within that space, a
characterization that would not have been possible otherwise.
These typically involve the degree of uniformity in the
coverage of the shape space so defined, which is a useful
concept since it relates to the extent this set of molecules
represents all possible shapes defined by this shape space.
Examples of such characterizations include:
(i) The volume each molecule occupies within the shape space
that is closer to it than to any other molecule in the
set, i.e. the Voronoi volume. This can then be used to
"cull" those molecules with very small neighborhoods,
i.e. which are most redundant within that shape space.
The distribution of the Voronoi volumes also gives a
measure of the uniformity of coverage of the shape
space.
- 40 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
(ii) The largest void within the space, i.e. the largest
hypersphere of the same dimensionality as the shape
space that can fit between molecules. This can be used
to ascertain which molecules from another set would best
fill that gap and hence make the coverage of shape space
more uniform.
(iii) The volume of the space occupied by the complete
set of molecules (i.e. the volume of what is known
geometrically as the "convex hull" defined by those
points in the shape space). This quantity is
useful in the context of deciding what fraction of
the shape space a subset of molecules covers
compared to the complete set.
(iv) The smallest subset of molecules which reproduces (iii),
i.e. molecules whose shape space positions lie on the
convex hull of that set of molecules. These molecules
define the boundaries of shape space and hence are
useful as the smallest subset of molecules which has the
same shape space volume as the total collection.
(v) The local dimensionality around a particular molecule.
Given a distance cut-off and a particular molecule, I
can calculate the dimensionality of the local shape
space of the set of molecules consisting of this
molecule and all those closer than the cut-off. The use
of this is that certain subsets of molecules may embed
within the global shape space in a space of much lower
dimensionality (imagine a set of points lying on a
curved surface in 3 dimensional space; the global
- 41 -
CA 02321303 2002-03-22
WO 99/44055 PCT/US99/04343
dimension is 3 but the local dimension is 2). This has
import for the efficient storage of the shape
information of molecules.
4: Calculating the position of a new structure in a
preconstructed shape space
Once I have a shape space for N molecules, of dimension
M, the next step is to calculate the position within this
shape space for ,a molecule not used in the construction of
that shape space. This position is found by analogy with
:L 0
triangulation in three dimensions, i.e. if one has a set of
distances from an object to four reference objects the exact
position can be ascertained. In two dimensions one needs
three distances. In M dimensional shape space one needs M+1
:L5 distances. (In each of these cases, the M+1 distances must
be from points which cannot as a set be described at a
dimensionality less than M, e.g. for the case of three
dimensions, the four reference points cannot all lie in a 2
dimensional plane). The actual procedure for going from
distances to a position is simply that a linear equation for
the coordinates can be generated from each distance, such
that the solution of the set of such produces the position.
This set of linear equations can be solved by any standard
method, for instance, Gauss-Jordan elimination (see, for
example Stoer and Bulirsch, "Introduction to Numerical
Analysis", 2nd Ed., Springer-Verlag, chapter 4). An important
note here is that this procedure can fail, i.e. it will
produce a position which will underestimate the M+1 distances
by a constant amount. This is an indication that the
- 42 -
CA 02321303 2000-08-24
WO 99/44055 PCTIUS99/04343
structure under study actually lies in a higher dimensional
space than the shape space previously constructed. As such,
that shape space needs to be extended.
5: Extending the Shape Space
If the position determining procedure fails, then it is
necessary to increase the dimensionality of the shape space
description. This is straightforward to accomplish in that I
merely need to add an additional variable to all the previous
positions, set the additional variable to zero for these
structures, and to then find what the new M+1 coordinate for
the new molecule needs to be such that the new coordinates
now reproduce the distances correctly. But this is simple to
calculate from the shortfall in distances calculated by the
previous method, since there is only the need to add one more
coordinate.
6: Calculating the maximal overlap between a new structure
and a large, previously shape-space decomposed set of
molecules
Suppose I have a set of N structures for which a shape
space decomposition of dimension M is known. If I have a new
structure and wish to find the closest such structure within
this set I could use the minimal field distance method
between this one molecule and all molecules within the set.
Many of the ideas within this document concern how to avoid
doing this since N may be very large. An additional
component is avoiding the performance of more applications of
the minimal field distance method than are necessary.
- 43 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
(i) Determine the shape space position of the new structure
as previously described.
(ii) Once the position in shape space is calculated then the
procedure to calculate the minimal field difference to
any other molecules from within the set (i.e. other than
the M+1 used to find its shape space position) is much
simpler, i.e. it is just a matter of calculating the
distance from the position assigned this new molecule
and those positions already calculated for any of the N
molecules in the set.
(iii) Since this is simply the square root of the sum of
the differences squared for each coordinate, it
only involves on the order of 2*M arithmetic
operations, i.e. is likely to be several orders of
magnitude faster than the minimal field distance
method.
7: Using the shape space description to correlate with
known biological activity
Partial Least Squares (PLS) analysis is a method of
calculating the importance of a set of quantities in
determining some "resultant" property. For instance, one
might have a set of measurements of physical properties for
each member of a set of compounds and wish to know the
correlation of each property with the biological activity in
some assay. PLS returns a set of weights for each input
property such that the activity results can be reproduced
from the input values. These then quantify the correlation
of the input quantities with the activity and can be used to
- 44 -
CA 02321303 2000-08-24
WO 99/44055 PCTIUS99/04343
predict the activity of molecules for which the input
quantities are known, but not the biological activity. The
use with the shape space decomposition is as follows:
(i) Calculate the shape vector for each molecule for which a
biological activity value is known. Note here that the
shape vector can be relative to a shape space defined by
a completely separate set of compounds, or to the space
calculated from that very set of compounds.
(ii) Use the numbers that make up this vector as input to a
PLS procedure, with or without other quantities known
for each molecule under consideration.
(iii) Use the resultant "weights" to predict the activity
of other molecules not in the original set, i.e. by
calculating their shape space vector.
Note that more than one shape field can be used as input to a
PLS calculation, i.e. the shape vector for the electrostatic
field as well as that for the steric field.
8: Examples of using the minimum field difference metric to
organize a database of molecules
Required is a set of N molecular structures. (These
will belong to L molecules where L can be less than N if
there is more than one conformer of a molecule in the set.)
These structures may also have unique chemical identifiers
(e.g. chemical names, SMILES strings, catalog numbers etc).
1) Constructing and using a Distance Tree
45 -
CA 02321303 2002-03-22
WO 99/44055 PCT1US99/04343
(i) Choose a structure at random from the N possible
structures.
(ii) Find the field distances from this root structure to all
N-i other structures.
(iii) Calculate the median of the distances found in (ii).
(iv) Use the median value as a threshold distance T to
subdivide the N-1 other structures into two lists, or
"branches", based upon this criterion, with the lower branch
containing all structures with distances below the threshold,
and the upper branch containing all structures with distances
greater than the threshold.
(v) Store the threshold value along with the root structure
in the root node data. structure.
(vi) Repeat this process for each list from i) onwards, but
with N decremented by one until the repeatedly divided trees
are of size one or zero
Now, faced with a problem of finding the closest structure in
the database to a novel example, i.e. one not in the
database, I. proceed as follows.
(i) Find the distance to the root structure.
(ii) If this distance is less than half the threshold
distance (T/2) for this node, then :I need never check any
structure along the upper branch of the tree, which contains
structures whose distance from the root is greater than that
threshold, since by the lower bound of the triangle
- 46 -
CA 02321303 2002-03-22
WO 99/44055 PCT/US99/04343
inequality none of them could be as close to the test
structure as the roct structure is.
(iii) If the distance to the root structure is more than T/2,
I must check the structures on both branches.
(iv) For each branch selected in (ii) and (iii), the
structure in the next node is made a new root structure, and
the process repeats starting at step (i), continuing until
all relevant branches have been checked.
Typical searches based upon this procedure reduce the number
of structures to be actually checked, ideally around the
logarithm of N multiplied by some constant factor.
2) Ordered lists
A. Creating, the lists.
(i) (Optional) Determine the shape space of a set of N
structures.
(ii) From the set of N structures, select K key structures
that are quite different from each other (i.e. are
remote from each other in shape space). For instance,
the structures may simply be different from each other
in total volume, or be chosen by more computationally
intensive methods, e.g. as representatives of clusters
of molecular shapes found by standard clustering
techniques (e.g. Jarvis-Patrick, etc). These more
sophisticated methods may be greatly speeded if the
shape space has been determined.
- 47 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
(iii) For each of the K key structures, find the minimal
distance m from it to every other structure in the
database. If K is large, this step may also be speeded
if the shape space has been determined, allowing simple
distance calculations rather than complete overlay
calculations for every structure.
(iv) For each of the K key structures, create a list
associated with it, and place into the list, in order of
increasing distance, references (name, number etc) to
each database structure along with its distance m from
the key structure.
B. Using the lists to find the structure closest to a test
molecule.
As illustrated in the flow chart of Fig. 7, the closest
structure is found by the following steps:
(i) For a test molecule, find its minimal distance x to
each of the K key molecules.
(ii) Choose the list whose key molecule k is closest to the
test molecule, where this distance is X. Since the list
has molecules close to k first it is likely these are
also close to my test molecule.
(iii) Set as current list position n the top of list k.
Set a variable BEST equal to X.
- 48 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
(iv) Otherwise, if the distance m from key k to list
structure n is greater than X + BEST, then stop, as by the
lower bound of the triangle inequality no structure further
down the list can be closer to the test molecule than STRUCT,
i.e. if m = X + BEST + a for any positive distance a, then
the triangle inequality (m - XI < d can be rewritten as d >
BEST + a.
(v) Find the minimal distance d from the test molecule
to the current structure n on the list.
(vi) If d < BEST, store d in BEST, and n in STRUCT.
(vii) If more structures, increment the list position n
by one and continue at (iv).
(viii) When the procedure terminates, the index n of the
closest structure to the test molecule is found in
STRUCT.
Thus I can search the database, by minimum field difference,
in a time sublinear with the number of molecules in the
database. This is because, by the triangle inequality, I
know the cutoff distance for evaluating structures in the
list is at most equal to 2X (when BEST = X) and is
potentially further refined as I progress down the list and
find better (smaller) values for BEST. As noted above, the
list creation process can be speeded if the shape space of
the structures has already been determined. Whether the time
- 49 -
CA 02321303 2002-03-22
WO 99/44055 PCT/US99/04343
saved will be justified by the time spent constructing the shape
space depends on the number of key structures K and the
number of structures in the database.
9: Examples Of Using The Shape Space Positions To Organize
A Database Of Molecules
Making and Using an M-dimensional tree:
M dimensional data points may be stored in a tree-like
data structure such that an efficient search can be made to
find all points within a distance d of a new point. These
algorithms are standard in art. Although the performance of
this tree lookup is not guaranteed efficient, i.e. there are
pathological cases where it is no better than testing all
points, on the average it allows the number of search steps
to be reduced from N (the number of points in the tree) to
some multiple of the logarithm of N.
1) Constructing and using an M-dimensional tree.
(i) Find the shape space positions for a set of N structures.
(ii) Choose a structure at random from this set and record its
name in the zero level node of a tree structure which is such
that each "node", or "slot", has two child nodes, called
"left" and "right", at what I refer to as a level one greater
than this node.
(iii) Select a second structure at random and store its name
in either the left level one node of the tree if its first
shape space coordinate is less than that of the first
structure, otherwise place it in the right level one node.
(iv) Chose another structure at random. As before, test its
first shape space coordinate. This time, however, if the
- 50 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
node it should be placed in is already occupied, place it
under that node, i.e. level two, in either the left or right
hand node based upon comparison to the second shape space
coordinate of the node it now lies under.
v) Continue in this fashion, where the right-left decision is
based upon the level of the node it is to lie under. If that
level is greater than M, then the test coordinate is equal to
the depth of the tree, modulo M, i.e. on the M+1 level the
coordinate used to test upon returns to the first coordinate,
M+2 to the second etc.
2) Searching an M-dimensional Tree
(i) Find the shape space position of the new structure for
which a similarly shaped molecule is required.
(ii) Perform the following test: is the first coordinate of
the new structure within distance d of that of the zero
level structure? If not, is it (a) greater than or (b)
less than that coordinate of the zero level structure?
(iii) In the first instance calculate the distance of the
zero level structure to the new structure, adding
it to a "hit" list if it is closer than d.
(iv) In the second instance (a) I can now discard all
structures lying under and including the left node from
consideration
(v) In the third instance (b) I can discard all structures
lying under and including the right node from
consideration.
(vi) Test the structures assigned to nodes on level one
remaining (after the test in ii)
- 51 -
CA 02321303 2002-03-22
WO 99/44055 PCT/US99/04343
(vii) Perform the same test as in (ii)-(v) on each of
these structures, except that the test is now
performed using the second coordinate.
(viii) Proceed down the tree, adding the "hit" list where
appropriate, culling portions of the tree were
possible until all structures have either been
tested for addition to the list or culled.
In (1) above, rather than choosing structures at random for
insertion into the tree, they could instead be sorted into a
list, for example in order of increasing volume, and then
taken sequentially from the list for insertion into the tree.
This allows additional criteria to be used to terminate a
search of the branches of the tree.
10: Local Domain Decomposition
The overlay of molecular fields is a method of finding
global similarities between molecules, i.e. whether molecule
A has the same distribution of properties as molecule B.
While this is extremely useful, it is also the case that
local similarities are of interest, i.e. when a part of
molecule A is similar to a part or all of molecule B. The
methods described below are an approach to solve this problem
by defining representations of parts of a molecular field
which have an approximately ellipsoidal character. These
representations conform to visual and chemical intuition as
to possible fragmentations of a molecular structure, and can
- 52 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
be compared between molecules either singly or in combination
with ease.
11: Constructing An Ellipsoidal Gaussian Representation
(EGR)
An Ellipsoidal Gaussian Representation (EGR) of a
molecular structure is constructed by fitting one or more
Ellipsoidal Gaussian Functions (EGF) to a field function of a
molecule, for instance a steric or electrostatic field.
A variety of methods can be used to find the optimal
parameters for one or more EGF's so as to minimize the EFF.
Two such methods for calculating the EFF for a given set of
parameters for each EGF are:
1) Numeric:
(i) Define a grid of regularly spaced points (a lattice)
large enough so that it contains all points for which
the functional value of the molecular field and/or the
EGR field has an absolute value greater than zero by a
given amount. For example, choose the extent of such a
lattice such that all atom centers and all EGF centers
are within the boundaries of the lattice by a given
distance.
(ii) Calculate the square of the difference at each grid
point and sum over all such points.
2) Analytic:
- 53 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
(i) Define an analytic description of the molecular field.
An example would be the steric field generated by
representing each atom by a Gaussian function (defined
above) and forming the sum or exclusion product field
(defined above) from each such Gaussian.
(ii) Given such an analytic description the EFF becomes the
sum of a series of integrals since the square of the
difference field is now a sum of integrable functions.
If the molecular field is defined from Gaussian
functions, then the integrals are of a particularly
simple form.
Given a procedure to calculate the EFF, the EGF parameters
can be optimized to minimize the EFF. Standard methods exist
in the literature for such an optimization, whether the EFF
is calculated numerically or analytically.
Of significance to the practical implementation of any
method are the initial EGF parameters. My work suggests that
the initial parameters must be such that there is significant
overlap between the each EGF and the molecular field. Thus,
my typical starting configuration is a center for each EGF
within the molecule (defined as within an atoms radius of at
least one atom within the molecule), with axes A, B and C set
to the x, y and z Cartesian axes, and widths u, v and w set
to 1.0, and prefactor p set to 1Ø
12: Construction Of Multiple EGR's Containing The Same
Number Of EGF's.
- 54 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
Most methods of optimizing EGF parameters, such as
minimizing the EFF, suffer from one drawback, namely that
they converge to a set of parameters that do not necessarily
correspond to the lowest possible EFF value, rather such
parameters are "locally stable", i.e. any small change in any
one parameter results in an increase in the EFF value. Such
"multiple minima" are intrinsic in the nature of the problem.
The local minimum with the lowest EFF value is referred to as
the "global minimum". I utilize the multiple minima nature
of the problem in that I do not necessarily require a single
EGF for a particular molecular structure, in fact I welcome
multiple representations for uses described later in this
document.
Procedure:
(i) Choose how many EGF's I want in the EGR.
(ii) Find the EGR by optimizing the EGF parameters to
minimize the EFF.
(iii) Add the characteristics of this EGR (i.e. the EGF
parameters) to a list of possible EGR's.
(iv) Find a new EGR by repeating (ii) but with different
starting positions (which, as described above, are
chosen to have random centers within the molecule).
(v) Compare this EGR's characteristic to all those on the
EGR list.
(vi) If these characteristics are "similar" to any on the
list discard this EGR, otherwise add it to the list.
- 55 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
"Similar" here can be defined as having EGF parameters
that differ by no more than a preset amount. If the EGR
is discarded I increment a parameter "OLD" by one,
otherwise set OLD to zero.
(vii) If OLD is equal to a predetermined number END, I
quit the procedure, otherwise I return to (iv),
i.e. if I have not generated a new structure in the
previous END tries end the procedure, otherwise try
again.
13: Constructing Molecular Fragments From An EGR.
Given an EGR containing more than one EGF, a procedure
of particular utility is to assign each atom in the molecular
structure to a particular EGF. This assignment operator can
have various forms. The form I have adopted is as follows:
(i) For each atom calculate the EFF of the functional form
representing that atom's contribution to the molecular
field with each EGF in turn.
(ii) Based upon these "atomic-EGF" EFF's, assign each atom to
the EGF for which its EFF is lowest.
(iii). The collection of atoms assigned to a particular
EGF forms a set referred to here as a molecular
fragment, or subdomain.
14: Evaluating An EGR Fit: The Fragment Adjusted EFF.
Given that the number of EGF's I include in an EGR is a
variable, there is an infinite number of possible EGR's for a
molecule. When comparing two different EGR's with the same
number of EGF's I can use the EFF as a guide to which
- 56 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
representation is the more appropriate representation, i.e.
lower EFF means a better fit. Comparing EGR's with different
numbers of EGF's is more problematic because the number of
adjustable parameters is proportional to the number of EFF's,
hence the more EGF's the lower the EFF tends to be. Yet, more
EGF's in an EGR are not necessarily a more useful domain
decomposition since this will ultimately tend towards one EGF
per atom. Decomposing a molecule into each of its component
atoms is a trivial exercise of little utility. A more useful
measure of fit can be obtained as follows:
(i) Calculate the molecular fragments induced by the EGR as
described above.
(ii) Calculate the EFF of the field produced by each
molecular fragment with the EGR to which it has been
assigned.
(iii) Sum these domain EFF's together to form a quantity
I will refer to as the sum fragment EPP.
(iv) Calculate the EFF of the whole molecule's field vs. its
EGR. I will term this quantity the molecular EFF.
(v) Add together the sum fragment EFF and the molecular EFF
to form what I refer to as the fragment adjusted EFF.
Although the fragment adjusted EFF typically is smaller for
two EGF's versus one, I have observed that as the number of
EGF's further increases, the fragment-adjusted EFF eventually
starts increasing again. The number of EGF's for which the
- 57 -
CA 02321303 2002-03-22
WO 99/44055 PCT/US99104343
best obtained EGR has the lowest fragment adjusted EFF is
defined as that structure's optimal EGF count.
15: Construction Of Multiple EGR's With Different Numbers of
EG '
In light of the above, the procedure for generating a
set of EGR's that covers possible domain decompositions of a
molecular structure is then as follows:
(i) Set the number of EGF's in the EGR to one.
(ii) Perform the "multiple EGR" procedure as described above.
(iii) Store each EGR so generated as a representation of
the molecular structure.
(iv) Find the best fragment adjusted EFF amongst the EGR so
generated.
(v) If the number of EGF's used in (ii) is equal to one set
the parameter "BEST" equal to this fragment adjusted
EFF.
(vi) If the number of EGF's used in (ii) is greater than one
check. to see if this fragment adjusted EFF is greater
than BEST. If so then quit the procedure, otherwise
increment the number of EGF's to be used in (ii) by one
and return to (ii).
16: Storage In A Database
The utility of having various EGR's for a single
structure is limited. The true utility comes in using the
information contained in the EGR to compare different
structures. Typically this will involve finding aspects in
common between a single structure and a set of structures,
- 58 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
each of which has had an EGR decomposition performed and the
parameters that describe each EGR stored in association with
a unique identifier for that structure (e.g. the structure's
name, number). The way the EGF information is stored and
retrieved can determine the efficiency and utility of its
use, as is typical of all database applications. Below are
described examples of such database applications.
17: Comparing Single EGR's To Solve The Atom Assignment
Problem
A recurrent problem in the comparison of molecules or
parts of molecules is known as the "assignment" problem. Put
simply, it is to associate each atom from one molecule with
an atom in a second molecule. Given an alignment between two
molecules the assignment problem can be solved via such
operators as "closest" or "closest of similar type", where
type might be element type or any number of characterizations
of an atom. However, choosing the alignment that in some way
optimizes some characteristic of the assignment is a hard
problem, since there are an infinite number of possible
alignments. However, the EGR does provide one solution, as
follows:
(i) Calculate an EGR for molecule X and molecule Y.
(ii) Each EGF within an EGR has a center (a,b,c) and an
orientation defined by axes A, B and C.
(iii) Orient an EGF from the EGR of molecule Y such that
the center and axes of Y coincide with that of X.
The alignment of the axes is such that the axis
- 59 -
CA 02321303 2002-03-22
WO 99/44055 PCT/US99/04343
from X with the largest value from its u, v, and w
values is aligned with the axis from Y with the
largest of its u, v and w values and similarly for
the smallest of such coefficients. There are
actually four different ways this can be achieved,
given the symmetry of an ellipsoidal function.
(iv) For each of the four alignments, make the atom to atom
assignments for the atoms which belong to the pair of
EGF's being aligned together based upon "closest" or
"closest of similar type".
(v) Rather than have an infinite number of possible
alignments I now have just four to choose from, and given
any kind of measure for the assignment (e.g. minimize
the sum of the distances of each atom pair) this is
straightforward.
Note that if the number of EGF's for X and Y is one then this
provides a match between all atoms of X with all of those of
Y. Otherwise, the choice of EGF's from X and Y induce a
partial match between X and Y. Given a measure of the
quality of this partial assignment, all possible combinations
of EGF's between X and Y can be chosen to find the best EGF
induced assignment.
18: EGF Pseudo-Surfaces And Painted EGF's
Rather than compare molecular fragments via an atom
assignment procedure one can compare the properties in the
vicinity of each fragment. To do so I introduce the concept
60 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
of an EGF pseudo-surface. The concept is simple enough,
namely that just as a spherical Gaussian function (i.e. an
EGF where the widths u, v and w are all equal) is the field
function for a single atom, which is normally thought of as
having a surface at that atom's radius, so I can associate an
ellipsoidal surface with each EGF. This surface can be
thought of as an isovalue contour of the EGF, and as such has
the same shape implicit in the EGF representation. I have
found that creating this ellipsoidal contour such that it has
the same volume as that of the EGF is effective in enclosing
the atoms belonging to that EGF. The procedure is as
follows:
(i) Calculate the volume of the EGF (the volume of an EGF is
defined as the integral of that function over all
space).
(ii) Find the factor R such that an ellipsoid with axes
(R/square root of (u), R/ square root of (v), R/ square
root of (w)) would have the same volume as the EGF.
(iii) Define the pseudo-surface of the EGF as the surface
of the solid ellipsoid defined in (ii) which has
its center coincident with the EGF and its axes
aligned along A, B and C of that EGF (the axis of
length R/square root of(u) is aligned with axis A,
the axis of length R/square root of(v) is aligned
with axis B, the axis of length R/square root of(w)
is aligned with axis C).
- 61 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
The use of the EGF pseudo-surface lies in the fact that
if properties are assigned to each point on this surface,
then such properties can be compared with those belonging to
a different EGF pseudo-surface. I refer to an EGF with a
pseudo-surface that has properties associated with it as a
"painted EGF". Properties can "paint" an EGF in a variety of
ways, for example:
(i) Be analytically defined on the surface of the ellipsoid
from the underlying analytical form of the property.
(ii) Be calculated at a number of "sample" points distributed
on the surface of the ellipsoid, either from analytic
functional forms of the property, or interpolated from
values of the property at nearby points in space (e.g.
from a grid of values).
(iii) Be assigned values at sample points based upon
proximity to underlying atoms, e.g. the atomic
properties are "projected" to this surface.
The information in the pseudo-surface values at a set of
points (as in (ii) above) may be stored in several ways. (a)
The first is just as a list of values associated with each
point. (b) If the values at points are of binary nature (one
or zero) then the storage may take the form of a bit-pattern.
(c) The pattern of values at points may be transformed into
an approximate functional form, typically spherical
harmonics, i.e. an analytic form as (i) above. (d) Finally,
the patterns may be stored "virtually", i.e. not actually
- 62 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
calculated until required but rather "stored" as a method of
calculation.
The comparison of the properties "painted" on two such
surfaces is as follows:
(i) Align the two EGF's relative to each other in one of the
four possible orientations wherein centers and axes are
aligned as above.
(ii) Change the shape of each pseudo-ellipsoid to that of a
sphere of unit radius via a series of three
compressions, one along each axis of the ellipsoid.
(iii) Compare the properties, point by point, of the now
identically shaped surfaces and superimposed
surfaces.
The comparison step in (iii) can actually be made a
metric distance, i.e. if the integral over the difference in
properties at the surface of the unit spheres is found, then
the square root of this quantity is a metric distance for
exactly the same reason the similar quantity is such for
three dimensional fields. Technically speaking the field
described by a property on the surface of a unit sphere has
the mathematical designation S2, as opposed to R3 for a
molecular field, but is still a field. However, the problem
of finding the minimal distance between two such spherical
fields is much simpler in my case than for general S2 fields
because the overlay of the ellipsoids only allows for four
63 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
possible orientations, rather than an infinite number (mine
is actually an oriented S2 field).
Given that the properties on the surface of two pseudo-
surfaces can be compared via a metric distance, I can apply
all of the techniques discussed previously on using metrics
to organize such information, now using them for fast
similarity searches on pseudo-surface property patterns. I
can also form an M dimensional representation of the minimal
difference between all pairs of a set of painted EGF's. Note
that this is not as useful as in the case where the alignment
problem is paramount, since the ellipsoid's alignment only
allows for four possible such cases.
19: Using A Database Of EGR's For (Molecular/Molecular
Fragment) Similarity Evaluation
A typical use of an EGR database is to extend the
concept of global similarity matches to parts of a molecule,
i.e. the local domain match problem. A typical procedure for
this is:
(i) Construct a database of EGR's for a set of molecular
structures.
(ii) Each EGF in each EGR so generated defines a set of atoms
which "belong" to it, hence each EGR defines various
molecular fragments. For instance, if EGR 1 contains
EGF's A, B and C then there are seven molecular
fragments defined, namely those from A, B, C, AB, BC,
- 64 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
AC, ABC. Note that the last such is simply the whole
molecule.
(iii) Store details of each molecular fragment. Note
that such details might include the shape vector of
this fragment from a predefined shape space, the
nature of the EGF's which "enclose" these
fragments, as well as other physical properties.
(iv) For a new structure not represented in this database of
molecular structures, generate a set of EGR's and a
corresponding set of molecular fragments for each.
(v) For each such fragment search the database generated in
(iii) for similar fragments, where "similar" may mean
similar in steric shape or any of a variety of other
measures, e.g. electrostatic field, hydrogen bond
positioning etc. These fragment searches may or may not
use metric quantities.
(vi) If such a search generates similarities above a certain,
specified level then report these matches either
graphically or via a listing to file or screen.
20: Using EGF's In Similarity Searches.
In addition to using the fragmentation properties of the
EGF's within an EGR, EGF's can also be used to directly aid a
similarity search. The following describes a linear search:
(i) Construct a database of EGR's for a set of molecular
structures.
- 65 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
(ii) Find the optimal number, M, of EGF's needed in an EGR
for the new structure I wish to compare to the
structures in this database.
(iv) Construct a set of M EGR's for the new structure, one of
each containing 1 up to M EGF's (i.e. if M = 3 construct
EGR's with one, two and three EGF's).
(v) Find the metric field difference value of the new
structure with the first molecule in the database, store
this value in the parameter BEST, and store the value
one in the parameter BESTSTRUCTURE.
(vi) Retrieve the best EGR description containing M EGF's of
the next structure in the database, or if this structure
has at most L EGF's in any of its EGR's, the best EGR
with L EGF's.
(vii) Compare this retrieved EGR with the EGR with the
same number of EGF's from the new structure as
follows: Pair the EGF's between the two EGR's by
ranking each set by volume, i.e. the "biggest" EGF
in the test molecule with the "biggest" EGF in the
database structure. Calculate the EFF for each
such pair assuming the EGF's are superimposed
optimally (i.e. the centers and major and minor
axes are aligned). This is easy to do since there
is an analytic formula for the EFF of two EGF's
optimally aligned to each other. This gives a
measure of the best possible EFF that could be
- 66 -
CA 02321303 2002-03-22
WO 99/44055 PCT/US99/04343
expected between the new structure and the current
structure from the database.
(viii) If this estimate of the best EFF between the new
structure and the current database structure is
greater than the BEST parameter go to (v).
(ix) Otherwise actually find the best metric field difference
between the new molecule and the current database
structure. If this value is less than BEST, set BEST
equal to this value, set the value of BESTSTRUCTURE to
indicate this structure. Go to (v) unless this is the
last structure in the database.
In the above procedure I am using the EGF's that make up
1:5
the best EGR of a molecule to save time in actually having to
calculate the best orientations between two molecules, which
is analogous to the use of the triangle inequality of the
metric field difference distance. In fact I am using this
210 metric nature, but in a more convoluted way than before.
Note that I can improve on the above method by
organizing the structures based upon the EGF's that make up
the best EGR for each structure, i.e. such that instead of
starting with the first structure in the database I start
with the structure which has the most similar set of EGF's
("most similar" defined here as having the most similar
volumes). Starting with a structure that is more likely to
have a small resultant BEST parameter means that step (vii)
is more likely to reject the next structure, i.e. the time
intensive step of finding the best overlay is avoided more
- 67 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
often. A simple method of organizing the structures such
that those with similar EGF descriptions to a test structure
can be rapidly found is via an N-dimensional hash table, but
many other methods exist in the literature.
21: Using EGR's To Find A Place In Shape Space; Using Shape
Space To Find EGR's
There is a synergistic relationship between the shape
space concept and the EGR concept in that having one makes
finding the other easier. In the following, I assume that a
database has been constructed which holds both the shape-
space description and the EGR description of each structure.
Given an EGR for a new structure, and requiring a shape space
description:
(i) Use the EGR description to find the structure of most
similar shape in the database of structures.
(ii) Find the minimal metric field difference between this
structure and the new structure.
(iii) Find the next most similarly shaped structure in
the database.
(iv) Find the minimal metric field difference between this
structure and the new structure.
(v) Find the position in shape space assuming that the
structures retrieved so far from the database define the
whole of the shape space. If the distances between the
new molecule and those structures are correctly
68 -
CA 02321303 2000-08-24
WO 99/44055 PCTIUS99/04343
reproduced then finish the procedure, otherwise go to
(iii).
The rationale behind this procedure is that although the
shape space dimensionality of the database of structures may
be M, the local dimensionality of molecules like the new
molecule may be much less than M. As an example, if in three
dimensions points lie on a line which is curving through
space, and if my test point lies on this line, close to two
other points, then the distances to just these two points can
define quite accurately the position on this line, not the
normal four such points that would be required if the points
were randomly distributed in all three dimensions. Hence,
given similar structures I find the position in shape space
with fewer than M+1 comparisons, if the local distribution of
such structures is not smooth in all M directions.
Given a shape space vector of a new molecule but not an
EGR
(i) Find the most similar structure, or set of structures
from within a database of such.
(ii) Use the EGF positions of each of the EGR's associated
with each such structure as a starting point for an EFF
minimization for the new molecule.
(iii) Then perform a "normal" EFF minimization but with
random starting positions.
The rationale here is that since the structures found in
the database are similar then the EGR's will also be similar.
- 69 -
CA 02321303 2002-03-22
WO 99/44055 PCT/US99/04343
If so, much time will be saved in step (iii) because many of
the potential EGR's will have already been found.
22: Defining An EGR For A Negative Space
Of particular interest in the use of EGR descriptions is
finding molecules that might fit in the active site of a
protein, as this is the mode of action of most pharmaceutical
compounds. The procedure is as follows:
Finding active site EGF's:
(i) Define a fitting function f between any two EGF's such
that if both were spherical this function would be a
minimum when the inter-EGF distance is the same as the
1.5 sum of the radii of each EGF (defining the radii of the
EGF as that of a sphere of equivalent volume). Such a
function for two EGF's, EGF1 and EGF2, is:
f = a*V - b* (Q (EGF1, V) - Q (EGF2, V) )
where V = Q (EGF1, EGF2) where Q is defined in equation
(6) above.
(ii) Define a molecular field function from either a sum or
exclusion product of a set of N atomically centered
spherical EGF's, where each such EGF has the same volume
as,-the atom upon which it is placed, and where the N
atom centers belong to all of the atoms in the protein
within a specified distance of the active site (the
active site may be defined in various ways, for instance
- 70 -
CA 02321303 2002-03-22
WO 99/44055 PCT/US99/04343
as consisting of all atoms known to be involved in the
function of the active site). It should be noted that
the molecular field function generated by the exclusion
product method is itself a sum of EGF's, as EGF's
multiplied together result in another EGF.
(iii) Minimize a function F which is the sum of functions
f defined in (i) over the individual EGF components
of the molecular field defined in (ii) each
overlapped with a test EGF, where this test EGF is
placed at a random starting point in the active
site and is initially spherical with a volume set
to that of a single carbon atom and where all
parameters that define this test EGF are allowed to
vary.
F = sum( f(MolecFieldEGFn, TestEGF) ) for n from 1 to
the number of individual EGF's making up the molecular
field.
(iv) Repeat step (iii) with different random starting points
until no significantly new final EGF's are generated
(criteria for "new" being those used in the generation
of a standard EGR).
This procedure produces a series of single EGF descriptions
of the active site. These EGF's may be painted, based upon
properties of the nearest protein atoms, or of any field
- 71 -
CA 02321303 2000-08-24
WO 99/44055 PCT/US99/04343
quantity generated by such atoms, e.g. electrostatic
potential.
Searching an EGR database for a fit to the active site:
(i) Form an EGR from the single EGF descriptions generated
by the above procedure by combining two or more such
EGF's.
(ii) Treat these EGF combinations exactly as one would those
from any molecular EGR in searching for similar
molecules, with the one exception that one may want to
change the sign of some EGF pseudo-surface properties,
e.g. electrostatics so that one finds electrostatically
complementary molecules to the active site.
(iii) Use the alignments with any molecular structures
found to be similar to this combination of active
site EGF's as starting points to procedures to
optimize any fitness function of the molecule with
the active site (e.g. an energy function, or
docking function).
30
72 -