Note: Descriptions are shown in the official language in which they were submitted.
CA 02321712 2000-08-30
WO 99/57159 1 PCT/US99/09306
TRIDENTATE LIGAND-CONTAINING METAL CA TALYST
COMPLEXES FOR OLEFIN POLYMERIZATION
FIELD OF THE INVENTION
The present invention relates to transition metal catalyst systems for olefin
polymerization, and more particularly to a reduced oxidation state transition
metal
catalyst system incorporating a tridentate ligand which is not based on
cyclopentadienyl
ligands.
BACKGROUND OF THE INVENTION
The use of discrete biscyclopentadienyl-based and monocyclopentadienyl-based
metal complexes for the polymerization of olefins is well known in the art. In
a few
cases, olefin polymerization has been demonstrated starting from discrete
catalyst
precursor complexes with cyclopentadienyl-based ancillary ligand systems and
reduced
oxidation state metal centers such as in, for example, U.S. Patents 5,374,696
and
5,494,874, both to Rosen, et. al.; WO 96/13529; and Theopold, Acc. Chem. Res.,
vol. 23,
pp. 263-270 (1990). However, these catalyst precursor complexes do not exhibit
CZ or
pseudo-CZ symmetries, useful symmetries with many metallocene catalysts.
Recently, there has been an increased interest in identifying catalytic
systems that
incorporate non-cyclopentadienyl ancillary ligands. For example, Canich and
Turner,
U.S. Patent 5,318,935 discloses bisamido Group 4 transition metal compounds
and
McConville, et. al., Macromolecules, vol. 29, p. 5241 (1996), discloses
bridged,
dianionic, diamide ligands. These catalysts incorporate d metals in their
highest
oxidation states. Both of WO 96/23010 and Gibson, et. al., Chem. Comm., pp.
849-850
(1998), disclose diimine-based ligands for metals in Groups 8-10. These
diimine-based
ancillary ligands are neutral donor ligands. Other demonstrated examples of
catalyst
precursor complexes incorporating non-cyclopentadienyl ancillary ligands and
reduced
oxidation state metals show these compounds to have very low activity, see WO
97/17379.
CA 02321712 2000-08-30
WO 99/57159 PCT/US99/09306
2
Organorrietallic compounds with anionic, non-cyclopentadienyl ligands,
including those with reduced oxidation state metal centers and those
structurally
characterized as having CZ or pseudo-C2 symmetry, are known in the scientific
literature.
See, for example, Fryzuk, Can. J. Chem., vol. 70, p. 2839 (1992); Edwards, et.
al., J.
Chem. Soc., Dalton Trans., p. 1253 (1989); and van Koten, et. al., J. Am.
Chem. Soc.,
vol. 104, p. 5490 (1982). However, the teachings of these documents do not
suggest
potential utility of the compounds as polymerization catalysts or catalyst
precursor
compounds.
SUMMARY OF THE INVENTION
The present invention is directed to a catalyst complex based on a Group 4-9
first
or second row transition metal in a reduced oxidation state and having a
tridentate,
monoanionic, non-cyclopentadienyl ligand containing Group 15 and/or 16
elements, the
complex having been activated for olefin polymerization. The monoanionic
tridentate
ancillary ligand system consists of two Group 15 or 16 atoms bound to the
transition
metal through dative bonds and a Group 15 atom or aromatic ring carbon atom
covalently
bound to the transition metal. The covalently bound atom is linked to the
datively bound
Group 15 or 16 atoms by bridging groups each containing one or more Group 13-
16
elements.
In a preferred embodiment, the present invention is directed to a tridentate
catalyst system for the polymerization of a-olefins comprising the reaction
product of:
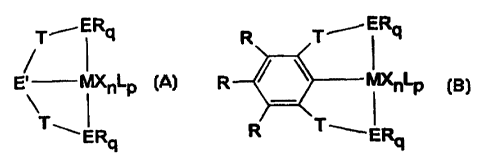
(a) an organometallic complex of one of the formulae:
T ERq R T_-- ERq
~ I
E' MXnLp R O MXnLp
T T-I
ERq R ' ERq
(A) (B)
wherein M is a transition metal from Groups 4-9 in a reduced oxidation state;
each X is
independently halogen, alkoxide, aryloxide, amide, phosphide, hydride,
hydrocarbyl,
CA 02321712 2000-08-30
WO 99/57159 PCT/US99/09306
3
substituted hydrocarbyl, halocarbyl, substituted halocarbyl, hydrocarbyl- or
halocarbyl-
substituted organometalloid, or two X groups are joined and bound to the
transition metal
or an L group to form a ring structure, or one or more of X can contain an L
group; L is a
neutral donor group which stabilizes the complex; each E is independently a
neutral
donor group selected from Groups 15 and 16; E' is a monoanionic donor group
selected
from Group 15; R has the same definition as X but may be the same or
different; T is a
bridging group containing an element or combination of elements from Groups 13-
16; n
is a number from I to 3 which is determined by counterbalancing the charge on
the
transition metal such that the transition metal remains in a reduced oxidation
state and the
overall charge on the complex is neutral; p is a number from 0 to 3 as needed
to stabilize
the complex; q is 1 or 2 such that E remains a neutral donor group; and (b) a
catalyst
activator compound. E is preferably selected from N, P, S and 0 and E' is
preferably N
or P. M is preferably Ti, V, Cr, Mn, Fe or Co. The catalyst activator compound
can be
alkylalumoxane, an alkyl aluminum cocatalyst activator, or an ionizing
noncoordinating
anion precursor compound.
In another aspect, the present invention is directed to a polymerization
process
characterized by contacting one or more monomers polymerizable by coordination
polymerization under suitable coordination polymerization conditions with the
catalyst
system described above. The monomers can be selected from the group consisting
of
ethylene, a-olefins, cyclic olefins, non-conjugated diolefins, acetylenically
unsaturated
monomers, olefinically unsaturated aromatic monomers and CZO CZ.
macromonomers.
The monomers are preferably at least one member selected from the group
consisting of
ethylene and C3-CZO a-olefins. The catalyst system can also include a solid
particulate
support.
DETAILED DESCRIPTION OF THE INVENTION
The catalyst precursor transition metal compounds of the present invention can
be
generically represented by the following chemical formulae:
CA 02321712 2000-08-30
WO 99/57159 PCT/US99/09306
4
T---- ERq R T~ERq
~ I
E' MXnLp R MXnLP
\ T \ I - T
ERq R ERq
(A) (B)
wherein each of the labeled substituents are as defined above.
The transition metal centers can be any metal from Groups 4-9, preferably
titanium, vanadium, chromium, manganese, iron or cobalt, in a reduced
oxidation state.
As used in the specification and the appended claims, "a reduced oxidation
state" means
an oxidation number which is less than the highest attainable oxidation number
of the
metal. For example, preferred reduced oxidation state metals include Ti(II),
Ti(III),
V(II), V(III), V(IV), Cr(II), Cr(III), Cr(IV), Cr(V), Mn(II), Mn(HI), Mn(IV),
Fe(II),
Fe(III), Co(H), Co(III), and the like. More preferred reduced oxidation state
metals are
those in the +3 oxidation state, e.g., V(III), Cr(HI), etc.
Source compounds for the neutral donor group(s) L include any neutral Lewis
base compound(s) capable of donating an electron pair to the metal center. Non-
limiting
examples include diethyl ether, trimethylamine, tetrahydrofuran,
dimethylaniline, aniline,
trimethylphosphine, n-butylamine, and the like.
The bridging group T contains any element or group of elements from Groups 13-
16 such as, for example, B, C, N, 0, Al, Si, P, S, Ge, Se or the like. T may
be saturated
or unsaturated. Preferred bridging groups include dialkyl, alkylaryl or diaryl
silicon
radical; a dialkyl, alkylaryl or diaryl germanium radical; alkyl or aryl
phosphine; alkyl or
aryl amine radical; an oxygen or sulfur radical; or a dihydrocarbyl radical
having I or
more carbon atoms such as methylene, ethylene and the like. Specific,
nonlimiting
examples of the T group which are suitable are dimethylsilyl, diethylsiiyl, di-
n-
propylsilyl, diisopropylsilyl, di-n-butylsilyl, di-t-butylsilyl, di-n-
hexylsilyl,
methylphenylsilyl, ethylmethylsilyl, diphenylsilyl, di(p-t-
butylphenethylsilyl), n-
hexylmethylsilyl, cyclopentamethylenesilyl, cyclotetramethylenesilyl,
cyclotrimethylenesilyl, dimethylgermanyl, diethylgermanyl, methylene,
dimethylmethylene, diethylmethylene, ethylene, dimethylethylene,
diethylethylene,
CA 02321712 2000-08-30
WO 99/57159 PCT/US99/09306
dipropylethylene, propylene, dimethylpropylene, diethylpropylene, 1,1-dimethyl-
3,3-
dimethylpropylene, tetramethyldisiloxane, 1,1,4,4-tetramethyl-disilylethylene,
oxygen
and sulfur.
Exemplary hydrocarbyl radicals for X are methyl, ethyl, propyl, isopropyl,
butyl,
5 amyl, isoamyl, hexyl, isobutyl, heptyl, octyl, nonyl, decyl, cetyl, 2-
ethylhexyl, phenyl
and the like, with methyl being preferred. Exemplary halogen atoms for X
include
chlorine, bromine, fluorine and iodine, with chlorine being preferred.
Exemplary
alkoxides and aryloxides for X are methoxide, ethoxide, propoxide, butoxide,
phenoxide
and substituted phenoxides such as 4-methylphenoxide. Exemplary amides of X
are
dimethylamide, diethylamide, methylethylamide, di-t-butylamide,
diisopropylamide and
the like. Exemplary aryl amides are diphenylamide and other substituted phenyl
amides.
Exemplary silyl amides are di-trimethylsilylarnide, di-triethylsilylamide and
triethyl-
trimethyl silylamide, with di-trimethylsilylamide being preferred. Exemplary
phosphides
of X are diphenylphosphide, dicyclohexylphosphide, diethylphosphide,
dimethylphosphide and the like. Exemplary alkylidene radicals for both X's
together are
methylidene, ethylidene, propylidene, or the dianion of ethyleneglycol and the
like.
Suitable hydrocarbyl and substituted hydrocarbyl radicals for the R groups
will
contain from I to about 30 carbon atoms and include singly and multiply
branched alkyl
radicals, cyclic hydrocarbon radicals, alkyl-substituted cyclic hydrocarbon
radicals, aryl-
substituted cyclic hydrocarbon radicals, aromatic radicals and alkyl-
substituted aromatic
radicals, amido-substituted hydrocarbon radicals, phosphido-substituted
hydrocarbon
radicals, alkoxy-substituted hydrocarbon radicals, and halo-substituted
hydrocarbon
radicals or hydrocarbon radicals containing substitutions by any Lewis basic
or acidic
functional group. Suitable organometallic radicals for the R group include
trimethylsilyl,
triphenylsilyl, triphenylgennyl, trimethylgermyl and the like. Other suitable
radicals for
the R group include amido radicals, phosphido radicals, alkoxy radicals, alkyl
boride
radicals and the like. Of the suitable organometallic R groups the radicals of
silicon such
as trimethylsilyl, triethylsilyl, ethyldimethylsilyl and methyldiethylsilyl
are preferred; the
most preferred being trimethylsilyl.
CA 02321712 2000-08-30
WO 99/57159 PCT/US99/09306
6
Illustrative catalyst precursors of the invention include:
[(Me2PCH2SiMeZ)ZN]TiC12 [(EtzPCH2CHZCH2)ZN]FeEt2
[(Me2PCHZSiEt2)ZN]VMeZ [(EtOCHzCH2)ZN]CoI2
[(MeOCH2SiMe2)2N]CrBrz [(EtN=CHCH2)2N]TiC12
[(Et2PCHZSiMeZ)2P]MnC12 [(Ph2NCH2CH2)2N]VH2
[(Et2PCH2SiEt2)2P]FeMe2 [(Ph2PCHZCHZCHZ)2N]CrMe2
[(MeN=CHSiMe2)2N]Co(CH2CHCH)2 [(PhOCHZCH2)2N]MnEt2
[(Ph2PCH2SiMe2)2N]TiCl2 [(PhN=CHCH2)2N]FeC12
[(Ph2PCH2SiEt2)2N]VBrz [(Me2PSiMeZ)2C6H3]TiCIZ
[(PhN=CHSiMez)ZN]CrMe2 [(Me2NCH)ZC6H3]VBr2
[(MeZNCH2CHZ)2N]MnEt2 [(MeSCH2)2C6H3]CrMe2
[((Me3Si)2NCH2CH2)2N]FeBr2 [(MeN=CH)ZC6H3]MnEt2
[(MeZPCH2CH2CH2)2N]CoPh2 [(Et2NCH)ZC6H3]FeC1.
[(MeN=CHCH2)2N]Ti(OMe)Z [(EtzPSiMe2)2C6H3]Co(CHZPh)2
[(Et2NCH2CH2)2N]VC12 [(Ph2NCH2)2C6H3]Ti(NMe)2
[(EtzNCHZCH2)ZP]CrBrZ [(PhOCH2)2C6H3]VPh2
[(Et2NCH2CH2CH2)2N]MnMe2 [(Ph2PSiEt2)2C6H3]CrMeZ
[(PhN=CH)2C6H3]FeC12 ,
and those illustrated in the examples that follow, where Ph = phenyl, Et =
ethyl and Me =
methyl.
The metal compounds according to the invention may be activated for
coordination or insertion polymerization catalysis by known methods for either
of
Ziegler-Natta or metallocene transition metal compounds suitable for olefin
polymerization. This activation is achieved for coordination polymerization by
the
inclusion of at least one ligand having a reactive metal-ligand sigma bond and
at least one
single vacant orbital adjacent (cis) to the sigma bound ligand, as is achieved
by
activation. The traditional activators of coordination polymerization art are
suitable,
those typically include Lewis acids such as Ziegler organometallic cocatalysts
and
alumoxane compounds, and ionizing, anion precursor compounds that abstract one
ligand
CA 02321712 2000-08-30
WO 99/57159 PCT/US99/09306
7
so as to ionize the metal center into a cationic complex and provide a counter-
balancing
weakly or noncoordinating anion.
The Ziegler cocatalyst will typically be an organometallic compound of a metal
of
Groups 1, 2, 12 or 13 of the Periodic Table of Elements. Preferred are
organoaluminum
compounds selected from the group consisting of aluminum alkyl and aluminum
alkyl
halide. These can be represented by the formulae:
A1(R2)sX'3-=,
wherein R2 is independently a hydride or C, to C,o hydrocarbyl radicals
including
aliphatic, alicyclic or aromatic hydrocarbon radicals, X' is a halogen and s
is an integer
from 1 to 3; and,
A12R23X' 3,
which are hydrocarbylaluminum sesqui-halides. Examples include
triethylaluminum,
triisobutyl-aluminum, diethyl aluminumchloride, A12Et3C13 and A12(i-Bu)3C13.
Alkylalumoxanes and modified alkylalumoxanes are suitable as catalyst
activators, particularly for the invention metal compounds comprising halide
ligands.
The alumoxane component useful as catalyst activator typically is an
oligomeric
aluminum compound represented by the general formula (R."-Al-O),,, which is a
cyclic
compound, or R"(R"-Al-O)nA1R"2i which is a linear compound. In the general
alumoxane formula R" is independently a C, to C,o alkyl radical, for example,
methyl,
ethyl, propyl, butyl or pentyl and n is an integer from 1 to about 50. Most
preferably, R"
is methyl and n is at least 4. Alumoxanes can be prepared by various
procedures known
in the art. For example, an aluminum alkyl may be treated with water dissolved
in an
inert organic solvent, or it may be contacted with a hydrated salt, such as
hydrated copper
sulfate suspended in an inert organic solvent, to yield an alumoxane.
Generally, however
prepared, the reaction of an aluminum alkyl with a limited amount of water
yields a
mixture of the linear and cyclic species of the alumoxane. Methylalumoxane and
modified methylalumoxanes are preferred. For further descriptions, see U.S.
Patents
4,665,208, 4,952,540, 5,041,584, 5,091,352, 5,206,199, 5,204,419, 4,874,734,
4,924,018,
CA 02321712 2006-12-06
8
4,908,463, 4,968,827, 5,329,032, 5,248,801, 5,235,081, 5,157,137, 5,103,031
and EP 0
561476A1,EP0279586B1,EP0516476A,EP0594218A1 and WO 94/10180.
When the activator is an alumoxane, the preferred transition metal compound to
activator molar ratio is from 1:2000 to 10:1, more preferably from about 1:500
to 10:1,
even more preferably from about 1:250 to 1:1.
The term "noncoordinating anion" is recognized to mean an anion which either
does not coordinate to the metal cation or which is only weakly coordinated to
it thereby
remaining sufficiently labile to be displaced by a neutral Lewis base, such as
an
olefinically or acetylenically unsaturated monomer.
Descriptions of ionic catalysts, those comprising a transition metal cationic
complex and a noncoordinating anion, suitable for coordination polymerization
appear in
the early work in U.S. Patents 5,064,802, 5,132,380, 5,198,401, 5,278,119,
5,321,106,
5,347,024, 5,408,017, 5,599,671, and WO 92/00333 and WO 93/14132. These teach
a
preferred method of preparation wherein metallocenes are protonated by
noncoordinating
anion precursors such that an alkyl/hydride group is abstracted by protonation
from a
transition metal to make it both cationic and charge-balanced by the
noncoordinating
anion. Since the abstraction and insertion ligands of such metallocenes also
may be
ligands of the metal compounds of the invention, similar methods of
preparation as active
polymerization catalyst components may be followed.
The use of ionizing ionic compounds not containing an active proton but
capable
of producing both an active metal cationic complex and a noncoordinating anion
is also
possible. See, EP-A-0 426 637, EP-A-0 573 403 and U.S. Patent 5,387,568 for
instructive ionic compounds. Reactive cations of the ionizing ionic compounds,
other
than the Bronsted acids, include ferrocenium, silver, tropylium,
triphenylcarbenium and
triethylsilylium, or alkali metal or alkaline earth metal cations such as
sodium,
magnesium or lithium cations. A further class of noncoordinating anion
precursors
suitable in accordance with this invention are hydrated salts comprising the
alkali metal
or alkaline earth metal cations and a non-coordinating anion as described
above. The
hydrated salts can be prepared by reaction of the metal cation-noncoordinating
anion salt
with water, for example, by hydrolysis of the commercially available or
readily
CA 02321712 2006-12-06
9
synthesized LiB(pfp)4 which yields [LixHzO] [B(pfp),], where (pfp) is
pentafluorophenyl
or perfluorophenyl.
Any metal or metalloid capable of forming a coordination complex which is
resistant to degradation by water (or other Bronsted or Lewis Acids) may be
used or
contained in the noncoordinating anion. Suitable metals include, but are not
limited to,
aluminum, gold, platinum and the like. Suitable metalloids include, but are
not limited
to, boron, phosphorus, silicon and the like.
An additional method of making the active polymerization catalysts of this
invention uses ionizing anion pre-cursors which are initially neutral Lewis
acids but form
a metal cationic complex and the noncoordinating anion upon ionizing reaction
with the
invention compounds, for example tris(pentafluorophenyl) boron acts to
abstract a
hydrocarbyl, hydride or silyl ligand to yield an invention metal cationic
complex and
stabilizing noncoordinating anion, see EP-A-O 427 697 and EP-A-0 520 732 for
illustration utilizing Group 4 metallocene compounds. See also the methods and
compounds of EP-A-0 495 375.
When the cation portion of an ionic noncoordinating anion precursor is a
Bronsted
acid such as protons or protonated Lewis bases (excluding water), or a
reducible Lewis
acid such as ferrocenium or silver cations, or alkaline metal or alkaline
earth metal
cations such as those of sodium, magnesium or lithium cations, the transition
metal to
activator molar ratio may be any ratio, but preferably from about 10:1 to
1:10, more
preferably from about 5:1 to 1:5, even more preferably from about 2:1 to 1:2
and most
preferably from about 1.2:1 to 1:1.2 with the ratio of about 1:1 being the
most preferred.
The catalyst complexes of the invention are useful in polymerization of
unsaturated monomers conventionally known to be polymerizable under
coo;dination
polymerization conditions using metallocenes. Such conditions are well known
and
include solution polymerization, slurry polymerization, gas-phase
polymerization, and
high pressure polymerization. The catalyst of the invention may be supported
and as
CA 02321712 2000-08-30
WO 99/57159 PCT/US99/09306
such will be particularly useful in the known operating modes employing fixed-
bed,
moving-bed, fluid-bed, slurry or solution processes conducted in single,
series or parallel
reactors.
When using the catalysts of the invention, particularly when immobilized on a
5 support, the total catalyst system will generally additionally comprise one
or more
scavenging compounds. The term "scavenging compounds" as used in this
application
and its claims is meant to include those compounds effective for removing
polar
impurities from the reaction environment. Impurities can be inadvertently
introduced
with any of the polymerization reaction components, particularly with solvent,
monomer
10 and catalyst feed, and adversely affect catalyst activity and stability. It
can result in
decreasing or even elimination of catalytic activity, particularly when
ionizing anion pre-
cursors activate the catalyst system. The polar impurities, or catalyst
poisons include
water, oxygen, metal impurities, etc. Preferably steps are taken before
provision of such
into the reaction vessel, for example by chemical treatment or careful
separation
techniques after or during the synthesis or preparation of the various
components, but
some minor amounts of scavenging compound will still normally be used in the
polymerization process itself.
Typically the scavenging compound will be an organometallic compound such as
the Group 13 organometallic compounds of U.S. Patents 5,153,157, 5,241,025 and
WO-
A-91/09882, WO-A-94/03506, WO-A-93/14132, and that of WO 95/07941. Exemplary
compounds include triethyl aluminum, triethyl borane, triisobutyl aluminum,
methylalumoxane, isobutyl aluminumoxane, and n-octyl aluminum. Those
scavenging
compounds having bulky or C6-CZo linear hydrocarbyl substituents covalently
bound to
the metal or metalloid center are preferred to minimize adverse interaction
with the active
catalyst. Examples include triethylaluminum, but more preferably, bulky
compounds
such as triisobutylaluminum, triisoprenylaluminum, and long-chain linear alkyl-
substituted aluminum compounds, such as tri-n-hexylaluminum, tri-n-
octylaluminum, or
tri-n-dodecylaluminum. When alumoxane is used as activator, any excess over
the
amount needed to activate the catalysts present will act as scavenger
compounds and
additional scavenging compounds may not be necessary. Alumoxanes also may be
used
in scavenging amounts with other means of activation, e.g., methylalumoxane
and
CA 02321712 2006-12-06
11
triisobutyl-aluminoxane. The amount of scavenging agent to be used with Group
4-9
catalyst compounds of the invention is minimized during polymerization
reactions to that
amount effective to enhance activity and avoided altogether if the feeds can
be
sufficiently free of adventitious impurities.
The catalyst according to the invention may be supported for use in gas phase,
bulk, slurry polymerization processes, or otherwise as needed. Numerous
methods of
support are known in the art for copolymerization processes for olefins,
particularly for
catalysts activated by alumoxanes_ Any is suitable for the invention process
in its
broadest scope. See, for example, U.S. Patents 5,057,475 and 5,227,440. An
example of
supported ionic catalysts appears in WO 94/03056. A particularly effective
method is
that described in U.S. Patent 5,643,847, and WO 96/04319. A bulk, or slurry,
process
utilizing supported, invention Group 4-9 metal compounds activated with
alumoxane co-
catalysts can be utilized as described for ethylene-propylene rubber in U.S.
Patents
5,001,205 and 5,229,478, these processes will additionally be suitable with
the catalyst
systems of this application. Thus both inorganic oxide and polymeric
particulate supports
may be utilized in accordance with the knowledge in the field. See
additionally, U.S.
Patents 5,422,325, 5,427,991, 5,498,582 and 5,466,649, and international
publications
WO 93/11172 and WO 94/07928.
In preferred embodiments of the process for this invention, the catalyst
system is
employed in liquid phase (solution, slurry, suspension, bulk phase or
combinations
thereof), in high pressure liquid or supercritical fluid phase, or in gas
phase. Each of
these processes may be employed in singular, parallel or series reactors. The
liquid
processes comprise contacting olefin monomers with the above described
catalyst system
in a suitable diluent or solvent and allowing said monomers to react for a
sufficient time
to produce the invention copolymers. Hydrocarbyl solvents are suitable, both
aliphatic
and aromatic, hexane and toluene are preferred. Halocarbon solvents, e.g.,
methylene
chloride will additionally be suitable. Bulk and slurry processes are
typically done by
contacting the catalysts with a slurry of liquid monomer, the catalyst system
being
supported. Gas phase processes typically use a supported catalyst and are
conducted in
any manner known to be suitable for ethylene homopolymers or copolymers
prepared by
CA 02321712 2006-12-06
12
coordination polymerization. Illustrative examples may be found in U.S.
Patents
4,543,399, 4,588,790, 5,028,670, 5,382,638, 5352,749, 5,436,304, 5,453,471,
and
5,463,999, and WO 95/07942.
Generally speaking the polymerization reaction temperature can vary from about
-50 C to about 250 C. Preferably the reaction temperature conditions will be
from -20 C
to 220 , more preferably below 200 C. The pressure can vary from about 1 mm Hg
to
2500 bar, preferably from 0.1 bar to 1600 bar, most preferably from 1.0 to 500
bar.
Where lower molecular weight copolymers, e.g., Mn < 10,000, are spught it will
be
suitable to conduct the reaction processes at temperatures above about 0 C and
pressures
under 500 bar. The multiboron activators of U.S. Patent 5,278,119 can
additionally be
employed to facilitate the preparation of the low molecular weight copolymers
of the
invention.
Linear polyethylene, including high and ultra-high molecular weight
polyethylenes, including both homo- and copolymers with other alpha-olefin
monomers,
alpha-olefinic and/or non-conjugated diolefins, for example, C3-C20 olefins,
diolefins or
cyclic olefins, are produced by adding ethylene, and optionally one or more of
the other
monomers, to a reaction vessel under low pressure (typically < 50 bar), at a
typical
temperature of 20-250 C with the invention catalyst that has been slurried
with a solvent,
such as hexane or toluene. Heat of polymerization is typically removed by
cooling. Gas
phase polymerization can be conducted, for example, in a continuous fluid'bed
gas-phase
reactor operated at 2000-3000 kPa and 60-160 C, using hydrogen as a reaction
modifier
(100-200 ppm), C4 CS comonomer feedstream (0.5-1.2 mol%), and C2 feedstream
(25-35
mol%). See, U.S. Patents 4,543,399, 4,588,790, 5,028,670 and 5,405,922 and
5,462,999.
Ethylene-a-olefin (including ethylene-cyclic olefin and ethylene-a-olefin-
diolefin) elastomers of high molecular weight and low crystallinity can be
prepared
utilizing the catalysts of the invention under traditional solution
polymerization processes
or by introducing ethylene gas into a sluiry utilizing the a-olefin or cyclic
olefin or
mixture thereof with other monomers, polymerizable and not, as a
polymerization diluent
in which the invention catalyst is suspended. Typical ethylene pressures will
be between
CA 02321712 2006-12-06
13
and 1000 psig (69-6895 kPa) and the polymerization diluent temperature will
typically
be between -10-160 C. The process can be carried out in a stirred tank
reactor, or more
than one operated in series or parallel. See the general disclosure of U.S.
Patent
5,001,205, WO 96/33,227 and WO 97/22,639.
5 Pre-polymerization of the supported catalyst of the invention may also be
used for
further control of polymer particle morphology in typical slurry or gas phase
reaction
processes in accordance with conventional teachings. For example, such can be
accomplished by pre-polymerizing a Cz C6 aipha-olefin for a limited time, for
example,
ethylene is contacted with the supported catalyst at a temperature of -15 to
30 C and
10 ethylene pressure of up to about 250 psig (1724 kPa) for 75 min to obtain a
polymeric
coating on the support of polyethylene of 30,000-150,000 molecular weight. The
pre-
polymerized catalyst is then available for use in the polymerization processes
referred to
above. The use of polymeric resins as a support coating may additionally be
utilized,
typically by suspending a solid support in dissolved resin of such material as
polystyrene
with subsequent separation and drying.
Other olefinically unsaturated monomers besides those specifically described
above may be polymerized using the catalysts according to the invention, for
example,
styrene, alkyl-substituted styrene, ethylidene norbornene, norbornadiene,
dicyclopentadiene, and other olefinically-unsaturated monomers, including
other cyclic
olefins, such as cyclopentene, norbomene, and alkyl-substituted norbornenes.
Additionally, alpha-olefinic macromonomers of up to 100 mer units, or more,
may also
be incorporated by copolymerization.
The catalyst compositions of the invention can be used as described above
individually for coordination polymerization or can be mixed to prepare
polymer blends
with other known olefin polymerization catalyst compounds. By use of mixtures,
of
coordination catalyst compounds, polymer blends can be prepared under
polymerization
conditions analogous to those using individual catalyst compositions. Polymers
having
increased molecular weight distribution ("MWD") for inihroved processing and
other
CA 02321712 2006-12-06
14
traditional benefits available from polymers made with mixed catalyst systems
can thus
be achieved.
EXAMPLES
The following examples are presented to illustrate the foregoing discussion.
All
parts, proportions, and percentages are by weight unless otherwise indicated.
All
reactions and manipulations have been conducted using dry, oxygen-free
solvents under
an inert nitrogen atmosphere. Although the examples may be directed toward
certain
embodiments of the present invention, they are not to be viewed as limiting
the invention
in any specific respect. In these examples, certain abbreviations are used to
facilitate the
description. These include standard chemical abbreviations for the elements
and certain
commonly accepted abbreviations, such as: Me = methyl, Et = ethyl, Bu = butyl,
Ph =
phenyl, and thf = tetrahydrofuran. Abbreviations used in the accompanying
tables
include Cat = catalyst, T = temperature, P = pressure, t = time, TM =
transition metal and
Br = branches per 1000 C atoms.
All molecular weights are weight average molecular weight unless otherwise
noted. Molecular weights (weight average molecular weight (Mw) and number
average
molecular weight (Mn) were measured by Gel Permeation Chromatography, unless
otherwise noted, using a Waters 150 Gel Permeation Chromatograph equipped with
a
differential refractive index detector and calibrated using polystyrene
standards. Samples
were run in either TfT (45 C) or in 1,2,4-trichlorobenzene (145 C) depending
upon the
sample's solubility using three Shodex GPC AT-80 M/S columns in series. This
general
technique is discussed in "Liquid Chromatography of Polymers and Related
Materials
III"' J. Cazes Ed., Marcel Decker, 1981, page 207. No corrections for column
spreading
were employed; however, data on generally accepted standards, e.g. National
Bureau of
Standards Polyethylene 1475, demonstrated a precision with 0.1 units for M,/Mõ
which
was calculated from elution times. The numerical analyses were performed using
Expert East software available from Waters Corporation.
CA 02321712 2000-08-30
WO 99/57159 PCT/US99/09306
Example 1
Synthesis of [(Ph2PCH2SiMe2)2NJTiCl2 (1). To a solution of
(Ph2PCHZSiMe2)ZNH (0.500 g, 0.944 mmol) in pentane (15 mL) was added a 1.6 M
solution of BuLi in hexanes (0.59 mL, 0.94 mmol). Upon addition, a white
precipitate
5 formed and the mixture was stirred at room temperature for 30 min. The solid
was
allowed to settle and the solvent was removed by pipette. The solid was washed
with
pentane (10 mL) and then dissolved in thf (10 mL). The thf solution was added
dropwise
to a suspension of TiC13=3thf (0.350 g, 0.944 mmol) in thf (30 mL). Upon
addition, the
color changed from light blue to olive green. After stirring at room
temperature for 16 h,
10 the volatiles were removed under reduced pressure and the residue was
extracted with
toluene (30 mL). The deep green solution was filtered through Celite ,
concentrated to
10 mL, and cooled to -30 C. The product was isolated as lime green crystals
(0.291 g,
0.449 mmol, 48 %). The elemental analysis, IR spectrum, and magnetic moment
were
consistent with the title compound.
15 Example 2
Synthesis of [(Ph2PCH2SiMe2)2N1VC12 (2). This compound was synthesized as
described for the Ti derivative in Example I from VCl3 3thf (0.353 g, 0.944
mmol) and
(Ph2PCH2SiMe2)2NH (0.500 g, 0.944 mmol). The product was isolated as deep red
crystals from toluene/pentane. The yield was 0.415 g (0.638 mmol, 68%). The
elemental analysis, IR spectrum, magnetic moment, and X-ray crystallographic
data were
consistent with the title compound.
Example 3
Synthesis of [(Ph2PCH2SiMe2)2N1CrCl.2(th, f) (3). This compound was
synthesized as described for the Ti derivative in Example I from CrC13 3thf
(0.707 g,
1.89 mmol) and (Ph2PCH2SiMeZ)ZNH (1.00 g, 1.89 mmol). The product was isolated
as
magenta microcrystals from toluene/pentane. One molecule of thf was
coordinated to the
metal. The yield was 0.644 g (0.890 mmol, 47%). The elemental analysis, IR
spectrum,
magnetic moment, and X-ray crystallographic data were consistent with the
title
compound.
CA 02321712 2000-08-30
WO 99/57159 PCT/US99/09306
16
Example 4
Synthesis of [(Ph2PCH2SiMe2)2N1FeC12 (4). This compound was synthesized as
described for the Ti derivative in Example I from FeC13 (0.153 g, 0.944 mmol)
and
(Ph2PCH2SiMeZ)ZNH (0.500 g, 0.944 mmol). The product was isolated as dark
green-
brown crystals from toluene/pentane. The yield was 0.383 g (0.584 mmol, 62%).
The
elemental analysis, IR spectrum, and magnetic moment were consistent with the
title
compound.
Example 5 (Comparative)
Synthesis of [(Et2NCH2CH2)2N1ScC12 (5). A solution of (Et2NCH2CH2)2NLi
(0.731 g, 3.30 mmol) in thf (7 mL) was added dropwise to a suspension of ScC13
(0.500g,
3.30 mmol) in thf (30 mL) at -78 C. After addition, the mixture was slowly
warmed to
room temperature and stirred for 18 h. The volatiles were removed under
reduced
pressure and the residue was extracted with pentane (80 mL). The solution was
filtered
through Celite and cooled to -25 C. The product was isolated as white, blocky
crystals
(0.244 g, 0.739 mmol, 22%). The elemental analysis and 'H NMR spectrum were
consistent with the title compound.
Examples 6-7
Synthesis of [(Et2NCH2CH2)NJTiC12 (6) and [(Et2NCH2CH2)N]VC12 (7). The
title compounds were synthesized as described in Wills et al., J. Chem.Soc.,
Dalton
Trans., p. 1253 (1989).
Example 8
Synthesis of [(Et2NCH2CH2)2N]CrC12 (8). This compound was synthesized as
described for the Sc derivative of Example 5 from CrCl3 3thf (1.69 g, 4.52
mmol) and
(EtZNCHZCHZ)ZNLi (1.00 g, 4.52 mmol) except that the reaction was stirred at
room
temperature for 3 h and the product was crystallized from pentane/toluene. The
product
was isolated as dark, green-brown crystals (0.528 g, 1.57 mmol, 35%). The
elemental
analysis, IR spectrum, and magnetic moment were consistent with the title
compound.
Example 9
Synthesis of [(Me2NCH2)2C6F131VC12 (9). A solution of (Me2NCH2)2C6H3Li
(0.500 g, 2.52 mmol) in benzene (10 mL) was added dropwise to a suspension of
VC13 3thf (0.943 g, 2.52 mmol) in benzene (40 mL). Upon addition, the solids
dissolved
CA 02321712 2006-12-06
17
leaving a deep red solution. After stirring at room temperature for 18 h, the
solution was
filtered through Celite and concentrated to 20 mL. The solution was diluted
with
pentane (20 mL) and filtered. The solution was cooled to -30 C resulting in
the
formation of a mass of red needles and partial crystallization of the benzene.
The
mixture was warmed until the benzene just melted and the crystalline product
was
collected. The yield of product was 0.287 g (0.916 mmol, 36%). The elemental
analysis,
IR spectrum, and magnetic moment were consistent with the title compound.
Example 10
Synthesis of [(Me2NCH2)2C6H3]CrC12 (10). This compound was synthesized as
described for the V derivative of Example 9 from CrCl3=3thf (0.453 g, 1.21
mmol) and
(Me.NCH2)ZC6H.Li (0.240 g, 1.21 mmoi). The product was isolated as green
microcrystals (0.152 g, 0.484 mmol, 40%). The elemental analysis, IR spectrum,
and
magnetic moment were consistent with the title compound.
Examples 11
Synthesis of [(Me2NCH2)2C6H3]FeCl2 (11). The title compound was
synthesized as described in Kanters, et al., Acta. Cryst., vol. C41, p. 893
(1985).
Examples 12-13
Synthesis of [(Et2NCH2CH2)2N1TiMe2 (12) and [E12NCH2CH2)2NJVMe2 (13).
The title compounds were prepared as described in Wills et al., J. Chem. Soc.,
Dalton
Trans., p. 1253 (1989).
Examples 14-54
Ethylene homopolymerization method (MAO activation). Polymerizations were
performed in a hot, nitrogen purged 500 mL Zipperclave reactor (Autoclave
Engineers)
in dry hexane (250 mL) as the polymerization solvent/diluent. The catalyst was
activated
with methylalumoxane (MAO) in a 10 wt % toluene solution (Albemarle) unless
otherwise noted. Usually, 2.5 mL of the MAO solution was diluted with fresh
toluene
prior to injection into the hexane-filled reactor. The hexane in the reactor
was then
saturated with ethylene at the dcsignated pressure and temperature. The
catalyst solution
was prepared in the drybox by mixing from 5 to 50 mg of catalyst precursor
with toluene
(50 mL). The catalyst precursor solution was pumped to the reactor, and
combined with
the previously added cocatalyst solution, until the ethylene make-up flow
became
CA 02321712 2000-08-30
WO 99/57159 PCT/US99/09306
18
constant during the polymerization. The reactor temperature was controlled by
a
steam/water mixture flowing through the reactor jacket. The polymerizations
were run
for 30 min. At the end of the run, the ethylene was vented and the reactor was
cooled
down. The reactor contents were poured into a 1 L beaker and treated with
isopropyl
alcohol or acetone. The polymer solvent mixture was blown down with nitrogen
or
filtered to recover the polymer. The final product was dried under vacuum at
60 to 90 C
for about 12 h. The polymerization conditions and results are shown in Table
1.
CA 02321712 2000-08-30
WO 99/57159 PCT/US99/09306
19
Table 1
Example Cat. T P t TM A1:TM Yield g_E Mõ(k) MWD
( C) (psi) (min) ( mol) (g) mmo1TM=h
14 1 30 76 30 25.6 149 2.2 169 506 2.4
15 1 60 125 30 9.6 400 2.9 612 244 77
16 1 115 275 30 9.6 400 3.6 758 33 22
17 1 140 375 30 24.7 155 4.0 325 16 11
18 2 30 75 30 0.6 6079 0.6 1809 1437 2.6
19 2 60 125 30 1.1 3367 1.2 2145 1136 2.5
20 2 115 276 30 5.9 649 1.1 386 135 159
21 2 140 350 30 21.2 180 0.9 88 75 89
22 3 30 75 30 2.2 1733 2.8 2569 448 16
23 3 60 125 30 8.6 447 4.0 934 134 2.9
24 3 115 275 30 8.4 454 8.2 1936 53 2.2
25 3 140 351 30 12.1 318 0.4 66 42 7.2
26 4 30 75 30 44.9 171 0.5 22 NM NM
27 4 60 125 30 43.9 174 0.6 27 NM NM
28 4 115 275 30 44.9 171 0.7 29 NM NM
29 5 30 75 30 30.2 127 0.4 23 NM NM
30 5 60 125 30 60.5 63 0.5 15 NM NM
31 5 115 275 30 95.2 40 0.3 6 NM NM
32 5 140 350 30 82.0 47 1.2 29 182 54
33 6 30 75 30 87.3 44 0.3 6 NM NM
34 6 60 125 30 88.2 43 1.3 29 504 6.4
35 6 115 275 30 11.3 338 7.6 1345 190 11
36 6 140 350 30 49.8 77 11.0 442 278 6.2
37 7 30 75 30 83.0 46 0.7 17 NM NM
38 7 60 125 30 86.6 44 1.7 39 177 27
39 7 115 275 30 47.3 81 7.9 332 59 4.0
40 7 140 350 30 57.1 67 11.6 406 53 2.9
41 8 30 75 30 85.3 90 0.6 15 NM NM
42 8 60 125 30 87.2 88 0.5 12 NM NM
43 8 115 275 30 85.4 90 0.7 17 NM NM
44 9 30 75 30 20.1 190 4.7 468 NM NM
45 9 60 125 30 21.1 182 3.3 313 NM NM
46 9 115 275 30 10.5 364 0.8 142 NM NM
47 9 140 350 30 84.3 45 0.8 19 NM NM
48 10 30 75 30 61.1 125 0.8 26 NM NM
49 10 60 125 30 91.7 84 0.9 19 NM NM
50 10 115 275 30 91.7 84 0.6 14 NM NM
51 10 140 350 30 91.7 84 0.9 20 NM NM
52 11 30 75 30 86.8 88 0.5 11 NM NM
53 11 60 125 30 90.5 85 0.7 15 NM NM
54 11 115 275 30 90.5 85 0.9 19 NM NM
NM - not measured
Examples 55-59
Propylene homopolymerization method (MAO activation). Polymerizations were
performed in a hot, nitrogen purged 500 mL Zipperclave reactor (Autoclave
Engineers).
CA 02321712 2000-08-30
WO 99/57159 PCT/US99/09306
The catalyst was activated with methylalumoxane (MAO) irt a 10 wt % toluene
solution
(Albemarle) unless otherwise noted. Usually, 2.5 mL of the MAO solution was
diluted
with fresh toluene prior to injection into the reactor. Liquid propylene (300
mL) was
then added to the reactor at room temperature and the contents were heated to
the desired
5 polymerization temperature. The catalyst solution was prepared in the drybox
by mixing
from 5 to 50 mg of catalyst precursor with toluene (50 mL). The catalyst
precursor
solution was pumped to the reactor, and combined with the previously added
cocatalyst
solution, until the reactor temperature and pressure became constant during
the
polymerization. The reactor temperature was controlled by a steam/water
mixture
10 flowing through the reactor jacket. The polymerizations were run for 30
min. At the end
of the run, the propylene was vented and the reactor was cooled down. The
reactor
contents were rinsed into a 1 L beaker with hexane or toluene and treated with
isopropyl
alcohol or acetone. The polymer solvent mixture was blown down with nitrogen
or
filtered to recover the polymer. The final product was dried under vacuum at
60 to 90 C
15 for about 12 h. The polymerization conditions and results are shown in
Table 2.
Table 2
Example Cat. T( C) C3(mL) t(min) mol TM A1:TM Yield (g) gPP/mmo1TM=h
55 3 60 300 30 1.1 3466 0.3 615
56 4 60 300 30 43.9 174 1.0 46
57 5 60 300 30 84.1 46 0.3 7
58 8 60 300 30 85.4 90 1.1 25
59 10 60 300 30 100.9 76 0.9 17
11 20
Examples 60-72
Ethylene/1 hexene copolymerization method (MAO activation). The ethylene/ 1-
hexene copolymerizations were run as previously described for the ethylene
homopolymerizations except that 1-hexene was added to the reactor immediately
prior to
the addition of the MAO. The polymerization conditions and results are shown
in Table
3 and the polymer characterization for selected samples is shown in Table 4.
CA 02321712 2000-08-30
WO 99/57159 PCT/US99/09306
21
Table 3
Example Cat. T( C) P C2' C6 t(min) TM AI:TM Yield (g) EgH
(psi) (mL) ( mon mmolTM=h
60 1 60 60 15 30 11.1 345 4.1 736
61 2 60 60 15 30 45.7 84 0.9 40
62 3 60 61 15 30 4.4 866 4.3 1922
63 4 60 60 15 30 43.9 174 0.6 25
64 5 60 60 15 30 91.7 42 0.2 5
65 6 60 60 15 30 85.5 45 0.5 11
66 6 115 115 15 30 14.4 532 8.9 1231
67 7 60 60 15 30 86.6 44 0.3 7
68 7 115 275 15 30 73.2 105 3.0 81
69 8 60 60 15 30 85.4 90 0.7 16
70 9 60 60 15 30 93.9 41 0.9 19
71 10 60 60 15 30 84.0 91 0.5 13
72 11 60 60 15 30 90.5 85 0.7 15
Table 4
Example Cat. M,,. (k) MWD Br
60 1 65 2.0 2
62 3 68 1.8 1
66 6 75 8.3 5
68 7 49 12.0 8
Example 73
Ethylene/norbornene copolymerization with [(Ph2PCH2SiMe2)2N] CrCl2(th, f)
(MAO activation). The ethylene/norbornene copolymerization was run as
previously
described for the ethylene homopolymerizations except that norbornene (22 mL
of an 80
wt% solution in hexane) was added to the reactor immediately prior to the
addition of the
MAO. The polymerization was run at 60 C with 61 psi ethylene using 3.6 mmols
of
[(PhZPCH2SiMeZ)2N]CrCIZ(thf) and an AI:TM ratio of 1075. The yield of
copolymer was
2.6 g giving an activity of 1448 g ENB/mmol TM=h. The copolymer had Mw = 647k
and
MWD = 2.9
Examples 74-81
Ethylene homopolymerization method ([Ph3C][B(C6F5)4J activation).
Polymerizations were performed in a hot, nitrogen purged 500 mL Zipperclave
reactor
CA 02321712 2000-08-30
WO 99/57159 PCT/US99/09306
22
(Autoclave Engineers) in dry hexarie (250 mL) as the polymerization
solvent/diluent. To
the diluent was added 0.2 mL of a 25 wt% solution of triisobutylaluminum in
heptane
(Akzo Nobel). The hexane in the reactor was then saturated with ethylene at
the
designated pressure and temperature. The catalyst solution was prepared in the
drybox
by mixing from 5 to 50 mg of catalyst precursor with an equimolar amount of
[Ph3C][B(C6F5)4] in toluene (50 mL). The catalyst solution was pumped to the
reactor
until the ethylene make-up flow became constant during the polymerization. The
reactor
temperature was controlled by a steam/water mixture flowing through the
reactor jacket.
The polymerizations were run for 30 min. At the end of the run, the ethylene
was vented
and the reactor was cooled down. The reactor contents were poured into a I L
beaker
and treated with isopropyl alcohol or acetone. The polymer solvent mixture was
blown
down with nitrogen or filtered to recover the polymer. The final product was
dried under
vacuum at 60 to 90 C for about 12 h. The polymerization conditions and results
are
shown in Table 5.
Table 5
Example Cat. T P (psi) t TM Yield (g) gPE/mmol Mw(k) MWD
( C) (min) ( mon TM-h
74 12 30 75 30 31.4 1.4 86 772 4.3
75 12 60 125 23 7.8 11.0 3679 NM NM
76 12 115 275 30 7.2 7.4 2059 NM NM
77 12 140 350 30 6.8 5.0 1463 NM NM
78 13 32 75 30 29.2 0.1 4 NM NM
79 13 60 125 30 28.4 0.3 18 NM NM
80 13 115 275 30 65.7 3.3 100 241 3.5
81 13 140 350 30 38.3 0.8 41 NM NM
Examples 82-83
Propylene homopolymerization method ([Ph3CJ[B(C6F5)4] activation).
Polymerizations were perfonmed in a hot, nitrogen purged 500 mL Zipperclave
reactor
(Autoclave Engineers). To the reactor was added 0.2 mL of a 25 wt.% solution
of
triisobutylaluminum in heptane (Akzo Nobel). Liquid propylene (300 mL) was
then
added to the reactor at room temperature and the contents were heated to the
desired
polymerization temperature. The catalyst solution was prepared in the drybox
by mixing
from 5 to 50 mg of catalyst precursor with an equimolar amount of
[Ph3C][B(C6F5)4] in
CA 02321712 2000-08-30
WO 99/57159 PCTIUS99/09306
23
toluene (50 mL). The catalyst precursor solutioii was pumped to the reactor
until the
reactor temperature and pressure became constant during the polymerization.
The reactor
temperature was controlled by a steam/water mixture flowing through the
reactor jacket.
The polymerizations were run for 30 min. At the end of the run, the propylene
was
vented and the reactor was cooled down. The reactor contents were rinsed into
a 1 L
beaker with hexane or toluene and treated with isopropyl alcohol or acetone.
The
polymer solvent mixture was blown down with nitrogen or filtered to recover
the
polymer. The final product was dried under vacuum at 60 to 90 C for about 12
h. The
polymerization conditions and results are shown in Table 6.
Table 6
Example Cat. T C3 (mL) t(min) TM Yield (g) gPP/mmol Mw(k) MWD
( C) ( mol) TM=h
82 12 60 300 30 33.8 6.6 387 394 6.5
83 13 60 300 30 33.5 0.6 35 NM NM
Examples 84-87
Ethylene/I-hexene copolymerization method ([Ph3C][B(C6F5)4J activation).
The ethylene/1-hexene copolymerizations were run as previously described for
the
ethylene homopolymerizations with [Ph3C][B(C6F5)4] activation except that 1-
hexene was
added to the reactor immediately prior to the addition of the
triisobutylaluminum. The
polymerization conditions and results are shown in Table 7.
Table 7
Example Cat. T P Cz' C6' t TM Yiel EH Mw MWD Br
( C) (psi) (mL) (min) ( mol) d (g) mmo1TM=h (k)
84 12 60 60 15 30 12.2 5.1 829 549 2.6 34
85 12 115 150 15 30 11.6 4.6 785 157 4.0 12
86 13 60 60 15 30 59.9 0.1 3 NM NM NM
11
87 13 115 150 15 30 73.1 0.3 7 NM NM NM