Note: Descriptions are shown in the official language in which they were submitted.
CA 02323422 2000-09-11
WO 99/47514 PCT/EP99/01574
w
CHEMICAL PROCESS FOR THE PRODUCTION OF SULPHINYL DERIVATIVES BY OXIDATION OF
THE CORRESPONDING THIO-DERIUATIYES WITH PERBORATES
The present invention describes an improved process for the preparation of
substituted 2-(2-pyridylmethyl)sulphinyl-1 H benzimidazoles particularly
omeprazole,
lansoprazole and pantoprazole by oxidising the corresponding substituted 2-(2-
pyridylmethylthio)-1 H benzimidazole.
Several proton-pump inhibitors, which are useful in the treatment of duodenal
ulcers, of formula A are known. These include omeprazole (R~=CH3; R2=OCH3;
R3=CH3; R4=OCH3) which is described in EP5129, lansoprazole (R~=CH3;
RZ=OCH2CF3; R3=H; R4=H) which is described in EP174,726 and pantoprazole
(R~=OCH3; R2=OCH3; R3=H and R4=OCHF2) which is described in EP166,287.
Rz
N ~ R~ / R3
/~S NJ A
R ~N
Many methods for preparing such compounds by the oxidation of the
corresponding 2-(2-pyridylmethylthio)-1 H benzimidazole have been described.
Examples of the oxidising agents used are 3-chloroperoxybenzoic acid
(V11091118895, EP533752, US5,386,032, ES43816 and EP484265), magnesium
monoperoxyphthalate (EP533264 and US5,391,752), ammonium molybdate
(EP484,265), iodosobenzene (ES539793), methyliodosobenzene (ES540147),
sodium periodate (ES550070) and vanadium oxide (EP302720).
However, there remains a need for a cheap and efficient process for oxidising
2-(2-pyridylmethylthio)-1 H benzimidazoles which is reliable, produces waste
streams
which are easily disposed of without causing harm to the environment and
produces
a stable final product.
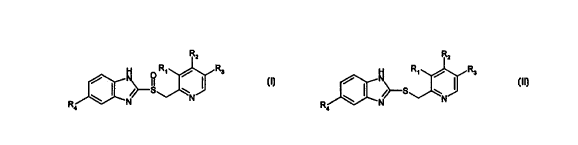
The present invention provides a process for the preparation of a compound
of formula I
CA 02323422 2000-09-11
WO 99/47514 PCT/EP99/01574
2
Rz
O R~ / Rs
J I
\ I /~S N
R N
4
in which R~, R2, R3 and R4 represent
a) (R~=CH3; R2=OCH3; R3=CH3; R4=OCH3) or
b) (R~=CH3; R2=OCH2CF3; R3=H; R4=H) or
c) (R~=OCH3; R2=OCH3; R3=H and R4=OCHFz) respectively
and pharmaceutically acceptable salts thereof
comprising reacting a compound of formula II
R2
N R~ / R3
/ I
\ ( ,~S N J I I
R ~N
4
in which R~, R2, R3 and R4 represent
a) (R~=CH3; R2=OCH3; R3=CH3; R4=OCH3) or
b) (R~=CH3; RZ=OCH2CF3; R3=H; R4=H) or
c) (R~=OCH3; RZ=OCH3; R3=H and R4=OCHF2) respectively
with a perborate salt in a liquid diluent at a pH in the range of 7.5 to 14 at
a
temperature in the range of 0°C to the boiling point of the liquid
diluent employed.
Suitably the perborate salt is a metallic perborate salt or an ammonium
perborate salt. The perborate salt may be anhydrous or hydrated. Preferably
the
perborate salt is potassium or sodium perborate. More preferably the perborate
salt
is sodium perborate. Most preferably the perborate salt is sodium perborate
monohydrate or sodium perborate tetrahydrate.
Suitably the amount of perborate salt employed in the process is in the range
of 0.8 to 3 moles per mole of the compound of formula II employed in the
process.
CA 02323422 2000-09-11
_ WO 99/47514 PCT/EP99/01574
3
Preferably the amount of perborate employed is in the range 0.95-2.0 moles per
mole of the compound of formula II employed in the process. More preferably
the
amount of perborate employed is in the range 1.0-1.9 moles per mole of the
compound of formula II employed in the process for example 1.1-1.5 moles per
mole
of the compound of formula II. Most preferably the amount of perborate
employed is
in the range 1.4-1.8 moles per mole of the compound of formula 11 employed in
the
process.
The purpose of the liquid diluent is to allow contact between the compound of
formula II and the perborate salt at the required temperature. Any liquid
diluent,
which is inert to the reactants, in which this purpose is achieved may be
used.
Preferably the liquid diluent is selected from water, a C» alcohol, toluene,
tetrahydrofuran, acetone, a CZ_s diol, a C~ triol, ethyl acetate or mixtures
thereof.
More preferably the liquid diluent is a water/alcohol mixture, for example a
water/methanol or a water/ethanol mixture. Most preferably the diluent is a
water/methanol mixture optionally containing toluene.
Preferably the process is carried out at a pH in the range of 8.5 to 12. More
preferably 10 to 12. Most preferably the process is carried out at a pH in the
range
of 10 to 11.
Suitably the pH of the process is controlled by the addition of a base for
example an alkali metal hydroxide an alkali metal carbonate, an alkali metal
bicarbonate or an amine e.g. ammonia or an organic amine or mixtures thereof.
Preferably the base is sodium hydroxide.
It will be appreciated by those skilled in the art that when the reaction is
carried out at high pH a salt of the desired product may be obtained. Lowering
the
pH of the reaction mixture, for example by addition of an acid or preferably
of a less
basic base, allows the isolation of the compound of formula ! as the free
heterocycle.
Preferably the process is carried out at a temperature in the range of 0 to
150°C and more preferably in the range of 15 to 115°C. Most
preferably the process
CA 02323422 2000-09-11
WO 99/47514 PCT/EP99/01574
4
is carried out at a temperature in the range of 40 to 55°C,
particularly at a
temperature in the range of 45 to 50°C.
The process of the present invention has severa! advantages over previously
described oxidation processes. The reagents employed are cheap, non-hazardous
and environmentally friendly, for example sodium perborate is used in domestic
washing powder, in mouth washes and in cleaning fluids for contact lenses.
Sodium
perborate has exceptional storage stability and is not shock sensitive. The
process
gives good yields reproducibly and provides a product of high purity which is
chemically more stable than the products of other oxidation processes
especially
those carried out in acidic conditions. In addition environmentally friendly
liquid
diluents may be used.
The process of the present invention has two further advantages over the
prior art processes. Firstly, this process step may be combined with the
previous
process step and thus avoid isolation of the compound of formula 1i. This
leads to
cost reduction in the process through improved processing times. Secondly, in
comparative experiments sodium perborate appears to give fewer impurities
arising
from over-oxidation, for example formation of a sulphone, or an N oxide, or a
sulphone N oxide, than previously known oxidants, for example 3-chloroperoxy-
benzoic acid.
The desired product can be isolated from the reaction mixture and purified by
conventional means e.g. extraction and recrystallisation or filtration
followed
optionally by recrystallisation.
In a preferred process of the present invention a compound of formula Ila is
reacted with sodium perborate in a mixture of water and methanol at a pH in
the
range of 8.5 to 10 at a temperature in the range of 15 -115°C to give a
compound of
formula la (omeprazole).
In a more preferred process of the present invention the compound of formula
II is prepared by reacting a compound of formula III
CA 02323422 2000-09-11
WO 99/47514 PCT/EP99/Oi574
4
a
I /~SH
R ~ N
4
111
or a salt thereof in which R4 is as previously defined with a compound of
formula IV
Rz
R~ / Rs
cl NJ
IV
5 or a salt thereof in which R~, R2, and R3 are as previously defined, in a
second liquid
diluent at a pH in the range of 7.5 to 14 at a temperature in the range of
0°C to the
boiling point of the second liquid diluent employed and is then reacted with a
perborate salt without isolation.
The purpose of the second liquid diluent is to allow contact between the
compound of formula III and the compound of formula IV at the required
temperature.
Any liquid diluent, which is inert to the reactants, in which this purpose is
achieved
may be used. Preferably the reaction of III and IV is carried out at a
temperature in
the range of 10-100°C, preferably at a temperature in the range of 20-
80°C and more
preferably at a temperature in the range of 40-60°C.
Preferably the second liquid diluent is selected from water, a C~~ alcohol,
toluene, tetrahydrofuran, acetone, a CZ~ diol, a C3.s triol, ethyl acetate or
mixtures
thereof. More preferably the second liquid diluent is a water/alcohoi mixture,
for
example a wateNmethanol or a waterlethanol mixture. Most preferably the
diluent is
a water/methanol mixture optionally containing toluene. Especially preferably
the
second liquid diluent is the same as the first liquid diluent. This avoids
further
processing for example diluent exchange.
CA 02323422 2000-09-11
WO 99/47514 PCT/EP99/01574
6
In a preferred embodiment the compound of formula III is present as the free
thiol, initially, and the process is carried out in the presence of a base.
Preferably the
base is an alkali metal hydroxide for example sodium hydroxide or potassium
hydroxide. More preferably the base is sodium hydroxide.
In a preferred embodiment the compound of formula IV is present as a salt
and sufficient base is used in the process to neutralise the salt of the
compound of
formula 1V and to form a salt of the compound of formula III. Preferably the
salt of the
compound of formula IV is the hydrochloride salt, the hydrobromide salt, the
acetate
salt, the nitrate salt or a salt of sulphuric acid or the salt of a phosphoric
acid. Most
preferably the salt is the hydrochloride salt.
Preferably the amount of base employed is in the range of 2.0 to 5.0 moles
per mole of the compound of formula III. More preferably the amount of base
employed is in the range of 3 to 4 moles per mole of the compound of formula
II(.
1n a preferred embodiment of the process a purification solvent is added at
the end of the oxidation reaction. The purification solvent has been found to
remove
certain impurities from the crude reaction product by dissolving these
impurities so
that on filtration the product obtained requires fewer recrystallisations than
would
otherwise be necessary. This provides time and energy and therefore cost
savings
in the process. The purification solvent also aids the filtration process by
changing
the physical nature of the product so that it can be more readily filtered.
Preferably
the purification solvent is immiscible with the liquid diluent.
Preferred purification solvents are hydrocarbons, including aliphatic and
aromatic hydrocarbons, and ethers, particularly di(C~_salkyl) ethers in which
the alkyl
groups are the same or different, and esters, for example ethyl acetate and
mixtures
thereof. More preferably the purification solvent is tent butyl methyl ether,
diisopropyl
ether, hexane , heptane or toluene and mixtures thereof. Most preferably the
purification solvent is tent butyl methyl ether, diisopropyl ether or hexane
and
mixtures thereof. Especially preferably the purification solvent is tern butyl
methyl
ether or diisopropyl ether.
CA 02323422 2000-09-11
_ WO 99/47514 PCT/EP99/01574
7
The invention is illustrated by the following Examples which are given by way
of example only. The final product of each of these Examples was characterised
by
one or more of the following procedures: high pertormance liquid
chromatography;
elemental analysis, nuclear magnetic resonance spectroscopy, infrared
spectroscopy
and high resolution mass spectroscopy. The compounds of formula II, III and IV
used in the Examples were either commercially available or were prepared by
the
methods given in EP5129, EP174,726 or EP166,287 which are incorporated herein
by reference.
Example 1
A solution of sodium hydroxide pellets (0.32 g), sodium perborate
tetrahydrate (1.43 g) and water (35 ml) was prepared by stirring and heating
the
mixed components until a solution was obtained, and was then added dropwise
with
stirring over 2.5 hours to a solution of 5-methoxy-2-{((4-methoxy-3,5-dimethyl-
pyridin-2-y!)methyl]thio}-1 H benzimidazole (2.0 g) in methanol (20 ml) and
toluene
(2 ml) which was boiling under reflux. The methanol was removed under reduced
pressure and the residue was cooled to 50°C and then added to saturated
sodium
bicarbonate solution (20 ml). The mixture was extracted with dichloromethane
(2 x 10 ml), the combined extracts were dried, filtered and evaporated to give
5-methoxy-2-{((4-methoxy-3, 5-dimethyl-pyridin-2-yl)methyl]sulphinyl}-1 H-
benzimidazole (1.60 g). Yield 86.5%.
Example 2
A solution was prepared by dissolving sodium hydroxide pellets (17.7 g) and
sodium perborate tetrahydrate (68.3 g) in water (1085 ml) with stirring and
heating
and this solution was then added dropwise to a solution of 5-methoxy-2-{[(4-
methoxy-3,5-dimethyl-pyridin-2-yl)methylJthio}-1H-benzimidazole (83.4 g) in
methanol (834 ml) whilst the mixture was boiled under reflux. The methanol was
removed under reduced pressure and the residue was cooled to 50°C and
then
added to saturated sodium bicarbonate solution (830 ml). The mixture was
cooled to
30°C and extracted with dichloromethane (2 x 400 ml). The combined
dichloromethane extracts were dried over magnesium sulphate, filtered and
CA 02323422 2000-09-11
WO 99/47514 PC1'/EP99/01574
8
evaporated to give give 5-methoxy-2-{[(4-methoxy-3,5-dimethyl-pyridin-2-
yl)methyl]sulphinyl}-1H benzimidazole (74.0 g, 84.fi% yield). This material
was
stirred in ethyl acetate {222 ml) for 1 hour then filtered. The residue was
washed with
ethyl acetate (2 x 25 ml) and dried to give a product which was 96.7% pure by
HPLC.
Example 3
A solution of sodium hydroxide (1.0 g) and sodium perborate tetrahydrate
(3.8 g) in water (65.0 ml) was prepared by heating and stirring. This solution
was
then added dropwise to a solution of 2-[3-methyl-4-(2,2,2-
trifluoroethoxy)pyrid-2-
ylmethylthio]-1 H-benzimidazole (5.0 g) in methanol (50.0 ml) which was being
boiled
over 2 hours at reflux with stirring. The mixture was stirred and boiled for a
further 15
minutes, then the methanol and water were removed under reduced pressure to
give
a residue which was cooled to 50°C and added to saturated sodium
bicarbonate
solution (50.0 ml). This mixture was cooled to 30°C and then extracted
with
dichloromethane (2 x 25 ml). The combined extracts were dried, filtered and
evaporated to give 2-[3-methyl-4-(2,2,2-trifluoroethoxy)-2-
pyridinylmethylsulphinyl]-
1 H-benzimidazole (4.8 g, 92.3% yield). The purity of this material was 90.3%
by
HPLC. This solid was stirred with ethyl acetate (14.4 ml) for 1 hour and then
the
solid collected by filtration, washed with ethyl acetate and dried to give
material which
was 91.4% pure by HPLC.
Example 4
A solution of sodium hydroxide (9.8 g) and sodium perborate tetrahydrate
(37.3 g) in water (638.3 g) was prepared by heating and stirring. This
solution was
then added dropwise over 2.5 hours to a solution of 2-[3-methyl-4-(2,2,2-
trifluoroethoxy)pyrid-2-ylmethylthio]-1 H-benzimidazole (49.1 g) in methanol
(491.0 ml) which was being boiled at reflux with stirring. The mixture was
stirred and
boiled for a further 15 minutes, then the methanol and water were removed
under
reduced pressure to give a residue which was cooled to 50°C and added
to
saturated sodium bicarbonate solution (491 ml). This mixture was cooled to
30°C
and then extracted with dichloromethane (2 x 245.5 ml). The combined extracts
CA 02323422 2000-09-11
WO 92/47514 PCT/EP99/01574
were dried, filtered and evaporated to give 2-[3-methyl-4-(2,2,2-
trifluoroethoxy)-2-
pyridinylmethylsulphinylj-1H benzimidazole in quantitative yield.
Example 5
In a similar manner to Example 1, 5-(difluoromethoxy)-2-[(3,4-dimethoxy-2-
pyridinyl)methylthio]-1 H benzimidazole is reacted with sodium perborate to
give 5-
(difluoromethoxy)-2-[(3,4-dimethoxy-2-pyridinyl)methylsulphinylj-1H
benzimidazole.
Example 6
A mixture of 5-methoxy-2-mercapto-1 H-benzimidazole (198.8 g), methanol
(380 ml) and water (760 ml) was stirred while sodium hydroxide solution (215
ml,
46-48% w/w) was added over 5 minutes. The mixture was stirred at 45-
50°C and a
solution of 2-chloromethyl-4-methoxy-3,5-dimethylpyridine hydrochloride (245
g) in
water (1136 ml) was added over 1 hour. The mixture was stirred at 45-
50°C for
2 hours and then sodium perborate tetrahydrate (202.4 g) was added. The
mixture
was stirred at 45-50°C for 18 hours. Further sodium perborate
tetrahydrate (16 g)
was added and the mixture was stirred for a further 4 hours. The mixture was
cooled
to 30-35°C and sodium hydrogen carbonate (221.9 g) was added followed
by water
(763.4 ml) and tert-butyl methyl ether (763.4 ml). The mixture was stirred
vigorously
for 2 hours then filtered to give a product which was washed with ter# butyl
methyl
ether (500 ml) and then dried under vacuum at 45-50°C for 21 hours to
give
5-methoxy-2-{[(4-methoxy-3,5-dimethyl-pyridin-2-yl)methyljsulphinyl}-1 H-
benzimidazole (293.4 g, 77% yield, purity by HPLC 98.3%).
Example 7
A mixture of 5-methoxy-2-mercapto-1 H-benzimidazole (4.3 g), methanol
(8.4 ml) and water (16.7 ml) was stin-ed while sodium hydroxide solution (4.7
ml,
46-48% wlw) was added over 5 minutes. The mixture was stirred at 45-
50°C and a
solution of 2-chloromethyl-4-methoxy-3,5-dimethylpyridine hydrochloride (5.3
g) in
water (25 ml) was added over 35 minutes at 45-50°C. The mixture was
stirred at
45-50°C for 1.75 hours and then sodium perborate tetrahydrate (4.5 g)
was added.
CA 02323422 2000-09-11
WO 99/47514 PCT/EP99/01574
The mixture was stirred at 45-50°C for 20 hours. Further sodium
perborate
tetrahydrate (0.35 g) was added and the mixture was stirred at 45-50°C
for a further
3 hours. A final batch of sodium perborate tetrahydrate (0.35 g) was added and
the
mixture stirred for a further 2 hours at 45-50°C. The mixture was
cooled to 35°C and
5 sodium hydrogen carbonate (4.9 g) was added followed by water (16.7 ml) and
diisopropyl ether (16.2 ml). The mixture was stirred rapidly at 20-25°C
for 1.5 hours.
The mixture was filtered to give a product which was washed with water and
dried
under vacuum at 45-50°C to give 5-methoxy-2-{[(4-methoxy-3,5-dimethyl-
pyridin-2-
yl)methyl]sulphinyl}-1 H benzimidazole (6.5 g, 78.9% yield, purity by HPLC
95.5%).
Example 8
A mixture of 5-methoxy-2-mercapto-1 H-benzimidazole (4.3 g), methanol
(8.4 ml) and water (16.7 ml) was stirred while sodium hydroxide solution (4.7
ml,
46-48% w/w) was added over 5 minutes. The mixture was stirred at 45-
50°C and a
solution of 2-chloromethyl-4-methoxy-3,5-dimethylpyridine hydrochloride (5.3
g) in
water (25 ml) was added over 35 minutes at 45-50°C. The mixture was
stirred at
45-50°C for 1.75 hours and then sodium perborate tetrahydrate (4.5 g)
was added.
The mixture was stirred at 45-50°C for 20 hours. Further sodium
perborate
tetrahydrate (0.35 g) was added and the mixture was stirred at 45-50°C
for a further
3 hours. The mixture was cooled to 35°C and sodium , hydrogen carbonate
(4.9 g)
was added followed by water (16.7 ml) and hexane (16.7 ml). The mixture was
stirred rapidly at 20-25°C for 1.5 hours. The mixture was filtered to
give a product
which was washed with water and dried under vacuum at 45-50°C to give
5-methoxy-2-~[(4-methoxy-3,5-dimethyl-pyridin-2-yi)methyl]sulphinyl}-1H
benzimidazole (6.6 g, 80.4% yield, purity by HPLC 94.45%).