Note: Descriptions are shown in the official language in which they were submitted.
CA 02324995 2000-10-OS
WO 99/53953 PCTNS99/06874
This invention relates to methods of regulating cell growth and division to
control disease
processes by manipulating mitochondria) metabolism and the expression of cell
surface immune
proteins. The invention also relates to compositions and screening assays.
1o Normal tissue develops and is maintained by normal processes of cell
division and cell
death. in many diseases, such as cancer, diabetes mellitus Type I, and
autoimmune disease,
the normal balance between cell division and cell death is disrupted causing
either a rapid
growth of unwanted and potentially dangerous cells or a loss of cells which
are essential to
maintaining the functions of tissue.
Cell division occurs by a process known as mitosis. During mitosis dividing
cells use
glucose cytolytically at an increased rate as the primary source for energy
(ATP) production
in a process referred to as glycolysis (Brand, K.A., and U. Hermfisse. 1997.
Aerobic
glycolysis by proliferating cells: a protective strategy against reactive
oxygen species. Faseb
J 11, no. 5:388-95). Glycolysis occurs in the cytosol and is required for
mitochondria) energy
2o production. An increased rate of glycolysis occurs when cells divide,
providing more of the
ATP from cytosolic glycolysis. Mitochondria) synthesis of ATP proceeds through
coupling
of electron transport-dependent oxido-reductive reactions to ATP synthetase
(oxidative
phosphorylation) (Harper, M.E. 1997. Obesity research continues to spring
leaks. Clinical
Investigations in Medicine 20, no. 4:239-244). During this process, a proton
gradient is
generated by the pumping of protons out of the mitochondria (Himms-Hagen, J.
1992. Brown
Adipose Tissue. Obesity, eds. P. Bjorntorp and B.N. Brodoff. 1 vols. J.B.
Lippincott,
Philadelphia. 1 pp), increasing mitochondria) membrane potential. Uncoupling
proteins
(UCPs) reversibly uncouple oxidative phosphorylation from electron transport
and thereby can
decrease mitochondria) membrane potential (Harper, M.E. 1997. Obesity research
continues
3o to spring leaks. Clinical Investigations in Medicine 20, no. 4:239-244).
Elevating glucose
concentrations can increase mitochondria) membrane potential (Harper, M.E.
1997. Obesity
research continues to spring leaks. Clinical Investigations in Medicine 20,
no. 4:239-244).
CA 02324995 2000-10-OS
WO 99/53953 PC'T/US99/06874
-2-
Cell death is a physiologic process that ensures homeostasis is maintained
between cell
production and cell turnover in self renewing tissues and is essential to the
proper functioning of
the immune system. Physiological cell death occurs through the processes of
apoptosis and
necrosis. The boundaries between these processes, once thought to be distinct,
have blurred
with the explosion of information on the role of cell death in development,
tissue modeling,
regenerative processes, and in the immune system. However, it is widely
accepted that
necrotic cell death (sometimes called oncosis) typically results in the
osmotic rupture of a cell,
followed by an inflammatory response, while apoptotic death involves cell
shrinkage,
fragmentation of the cell, and phagocytosis of the fragments often without
inflammation. Most
1 o cells die in a form of suicide characteristically apoptotic and tightly
regulated by complex
signals (Zakeri, Z., W. Bursch, M. Tenniswood, and R.A. Lockshin. 1995. Cell
Death:
Programmed, apoptosis, necrosis, or other. Cell Death and Differentiation 2:87-
96). Apoptotic
cell death is particularly important in the reticulo-endothelial system where
the balance between
mitosis and cell death may determine the effectiveness and the nature of an
immune response
(Zakeri, Z., W. Bursch, M. Tenniswood, and R.A. Lockshin. 1995. Cell Death:
Programmed, apoptosis, necrosis, or other. Cell Death and Differentiation 2:87-
96). Failure
results in autoimmune disease or in a lack of immune surveillance.
Inappropriate cell division or cell death results in serious life-threatening
diseases.
Diseases associated with increased cell division include cancer and
atherosclerosis. Disease
2o resulting from increased cell death include AIDS, neurodegenerative
diseases (e.g., Alzheimer's
disease, Parkinson's disease, amyotrophic lateral sclerosis, retinitis
pigmentosa), aplastic anemia,
atherosclerosis (e.g., myocardial infarction, stroke, reperfusion injury), and
toxin induced liver
disease. Many methods for treating these disorders have been proposed Although
these diseases
share the common physiological trait of either excess cell division or
premature cell death,
strategies for identifying potential therapeutic treatments have been
individualized rather than
searching for a common mechanism. It would be desirable to identify a common
mechanism by
which cell division could be interrupted or cell death could be promoted to
treat all of these
diseases.
PC 12 cells, a cell line derived from rat pheochromocytoma (Greene and
Tischler, 1976)
3o have been extensively used as a model for the study of nerve growth factor
(NGF)-induced
neuronal differentiation and dependency (Mills et al., 1997), and of neuronal
cell apoptosis
resulting from serum and/or trophic factor withdrawal (Mesner et al., 1995,
Fulle et al., 1997),
CA 02324995 2000-10-OS
WO 99/53953 PCTNS99/06874
-3-
oxidative stress (Vinard et al., 1996) and, the addition of calcium ionophores
(Fulle et al, 1997).
NGF promotes differentiation, neurite outgrowth and the acquisition of a
mature sympathetic
neuronal morphology on PC 12 cells. Withdrawal of NGF, however, results in
apoptosis of the
PC12 cells which is characterized by prototypic changes, i.e., chromatin
degradation, nuclear
fragmentation, acidification, alterations of surface lipids, cell
fragmentation, blebbing and
nucleosome formation (Gottlieb et al, 1997).
PC 12 transfected variants such as TrkA have been developed to elucidate the
role of NGF
and signal transduction in neuronal function. Nerve growth factor (NGF) binds
to two synergistic
receptors, tyrosine kinase A (TrkA) and p75NGRF (Canossa et al., 1996). The PC
12 TrkA cell
line overexpresses TrkA, a 140 kDa protein with intrinsic tyrosine kinase
activity (Kaplan et al.,
1991 ) and responds more vigorously than native PC 12 cells to NGF
stimulation. It is believed
that the NGF-TrkA complex acts as a messenger that delivers the growth signal
from axon
terminals to sympathetic neuronal cell bodies (Riccio et al., 1997).
Epidermal growth factor (EGF) has different effects on PC12 cells than NGF.
When
tenative PC 12 cells are treated with EGF they are induced to undergo
proliferation rather than
differentiation. In contrast, EGF stimulation of the v-Crk and TrkA cell lines
induce neuronal
differentiation (Teng et al., 1995).
Fas, a member of the tumor necrosis receptor family that includes the nerve
growth factor
receptor, mediates apoptotic cell death in several instances, including TCR (T
cell receptor)/CD3-
2o induced T cell activation (Nagata et. Al., Science). When the Fas molecule
interacts with Fas
ligand or an appropriate anti-Fas antibody, cellular death can ensue (Gottlieb
et al., 1997). Fas
was originally described on the membrane surface of hematopoietic lineage
cells (Itoh et al.,
1991 ), but its presence has been documented on endothelial cells (Richardson
et al., 1994),
hepatocytes (Tanaka et al., 1998) and oligodendrocytes in multiple sclerosis
lesions (Bonetti and
Raine, 1997).
The B7 molecules, B7.1 (CD80) and B7.2 (CD86) are known for their ability to
co-
stimulate T cell proliferation (Linsley et al., 1991 ), the production of
interleukin-2 (Freeman et
al., 1992) and the expression of interleukin-2 receptors (Razi-Wolf et al.,
1996). Expression of
these co-stimulatory molecules on immune cells also may play an important role
in the
3o pathogenesis and response to several bacterial, parasitic and viral
infections as well as
autoimmune disease (Reiser and Stadecker, 1996) such as systemic lupus
erythematosus
(Folzenlogen et al., 1997), experimental allergic encephalomyelitis (Perrin et
al., 1996) and in the
CA 02324995 2000-10- -OSt~~c) '?4~1 f-~ +'1-~ ~~
'C\'. \'ON:EPrI~-~11:F_'VCIiE'V ~1~'k :'~'=- ~- « : -=".=~ . _ < <1
'7:7«~:.1.AF;~:is c-:
U S 009906874
22-05-2000
rejection phase of alloimtnune responses (A~alia et aL, 199.
B7.1 and B7.2 are members of the immunoglobulin gene superfatnily and include
a
V-like and a C2-like extraeellitlar domain. Although originally described on B
cells. B7.1
and B7.2 have also been described on monocytes, dendritic cells and activated
T cells (.tune
et al., 1994). B7.1 (CD80) and particularly Ii7.2 (CD86) are upregulated on
the B Iymphocytc
surface of patients with systemic lupus erythematosus (SLEj (Folzenlogen at
al., 1997).
Street et al, [Grynecological Oncology, 65, 265-272 (199'n~ and the references
cited
therein describe the use of IFN-g to induce expression of MHC class I and I1
and thus
cnhanee lysis of tumor cells by CTLs. Addition of monoclonal antibodies to
class 1 but not
class II blocked cytolysis.
Summary of the Invention
The invention involves the finding that mitochondrial metabolism plays an
essential
role in regulating cellular division and toll death occurriilg in various
diseases. It was found
according to the invent;on th2tt the status of the cellular pmton motor force
which can be
asstsscd by the coupling relationship between electron transport tmd oxidative
phosphorylation plays as important role in the signal which determines Whether
a cell will
undergo cellular division, cellular differentiation ar cellular death. This
finding has important
implications for treating diseases associated with excessive cellular
division, aberrant
differentiation, and premature cellular death, e.g., for the treatment of
cancers, autoimmune
disease, neumdegenerative diseases, etc.
It was also found according to the invention that the expression of immune
recognition molecules on the surface of cells is important in regulating the
processes of cell
division, differentiation and apoptosis occurring in various diseases. It was
discovered, for
instance, according to t.'re invention that the expression of immune
recognition molecules on
the surface of a cell comlates with the ability of the cell to undergo
diffe~rentiation_ For
instance, upon removal of NGF from a nerve cell, the surface expression of B7
molecules is
down regulated and tho nerve cell undergoes apoptosis. ?he induction kinetics
and
expression of Fns, B7.1 and B7.2 molecules on the membrane surface of
differentiated PC12
cells and its mutants, end TrkA ccIls have been examined and are described in
the Examples
below.
The invention includes the d,isc6very that neural differentiation and
apoptosis are
regulated through interaction of the immune recognition molecules on the nerve
cell surface
AMENDED SHEET
:C~'. VU\:t-:YA-hl;~E\CHt~ U4 :Z'_~- >- t~ CA 02324995 2000-10-OS'~'-'() ?44.1-
' +4W R'-~ '_';-'7944U::~ i
- - U S 009906874
22-05-2000
-4A-
with an NC3F producing cell that expresses the counterpart surface immune
recognition molecule, likely CD28 or CTLA4. T'he inteca;etion between the
nerve cell and the
NGF producing cell causes the NGF producing cell to release NGF into the lxal
environment. This NGF then stimulates the nerve cell to undergo nerve cell
dif~'eremiation
and innervation.
AMENDED SHEET
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-5-
Several cell surface proteins have previously been identified as cell death
proteins. These
proteins are believed to be involved in initiating a signal which instructs
the cell to die. Cell
death proteins include for example Fas/CD95 (Trauth et al., Science 245:301,
1989), tumor
necrosis factor receptors, immune cell receptors such as CD40, OX40, CD27 and
4-1BB (Smith
s et al., Cell 76:959, 1994), and RIP (US Patent No., 5,674,734). These
proteins are believed to
be important mediators of cell death. These mediators, however, do not always
instruct a cell to
die. In some cases these mediators actually instruct a cell to undergo cell
division. Prior to the
instant invention the mechanism causing the dual functionality of these cell
death proteins was
not understood. It was discovered according to the invention, that the
intracellular environment
and particularly the status of the proton motor force and source of fuel for
mitochondria)
metabolism determines whether stimulation of the cell death protein will lead
to a signal for death
or cell division.
It was also discovered according to the invention that the regulation of cell
surface
expression of major histocompatibility complex (MHC) class II and co-
stimulatory molecules B7
t s 1 and B7-2 can be manipulated by regulating the intracellular dissipation
of proton motor force
which can be assessed in terms of mitochondria) membrane potential. Under
conditions of low
mitochondria) membrane potential (electron transport and oxidative
phosphorylation are
uncoupled), cells use non-glucose carbon sources for mitochondria) oxygen
consumption (e.g.,
fatty acids or amino acids) and the surface expression of MHC class II and co-
stimulatory
2o molecules B7-1 and B7-2 is increased. Under conditions of high
mitochondria) membrane
potential (electron transport and oxidative phosphorylation are relatively
more coupled and
glucose is being used as a mitochondria) carbon source) the surface expression
of MHC class II
and co-stimulatory molecules B7-1 and B7-2 is decreased.
In one aspect the invention is a method for decreasing mitochondria) membrane
potential
2s in a mammalian cell. The method involves the step of administering an MHC
class II HLA-DR
ligand to the mammalian cell to selectively engage MHC class II HLA-DR on the
surface of the
cell in an amount effective to decrease mitochondria) membrane potential in
the mammalian cell,
wherein the mammalian cell is not an antigen presenting cell. In one
embodiment MHC class II
HLA-DR is expressed on the surface of the mammalian cell. In another
embodiment the method
3o involves the step of contacting the mammalian cell with an amount of an MHC
class II HLA-DR
inducing agent effective to induce the expression of MHC class II HLA-DR on
the surface of the
mammalian cell.
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-6-
The mammalian cell may be any type of cell other than an antigen presenting
cell. In one
embodiment the mammalian cell is a tumor cell. Preferable the MHC class II HLA-
DR ligand
is administered to the tumor cell in vivo in an amount effective for causing
cell Iysis of the tumor
cell. When the mammalian cell is a tumor cell, however, in some embodiments
the MHC class
II HLA-DR inducing agent does not include adriamycin and gamma interferon. In
other
embodiments when the mammalian cell is a tumor cell the MHC class II HLA-DR
inducing agent
does not include adriamycin and gamma interferon.
According to another aspect of the invention a method for decreasing
mitochondria)
membrane potential in a mammalian cell is provided. The method involves the
step of contacting
1o the mammalian cell with an amount of an MHC class II HLA-DR inducing agent
effective to
induce the expression of MHC class II HLA-DR on the surface of the mammalian
cell, wherein
the mammalian cell is not an antigen presenting cell.
The invention in another aspect is a method for increasing mitochondria)
membrane
potential in a mammalian cell. The method involves the step of administering
an MHC class II
HLA-DP/DQ ligand to the mammalian cell to selectively engage MHC class II HLA-
DP/DQ on
the surface of the cell in an amount effective to increase mitochondria)
membrane potential in the
mammalian cell. In this aspect of the invention the mammalian cell is not an
antigen presenting
cell.
In one embodiment MHC class II HLA-DP/DQ is expressed on the surface of the
mammalian cell. In another embodiment the invention includes the step of
contacting the
mammalian cell with an amount of an MHC class II HLA-DP/DQ inducing agent
effective to
induce the expression of MHC class II HLA-DP/DQ on the surface of the
mammalian cell.
According to another embodiment the mammalian cell is a pancreatic ~i cell of
a type I
diabetic and wherein the MHC class II HLA-DP/DQ ligand is administered to the
pancreatic (3
cell in vivo.
The methods of the invention are useful for inducing cell division, cell
lysis, cell
differentiation and cell apoptosis, depending on the metabolic condition of
the cell. In one aspect
the invention is a method for inducing lysis of a mammalian cell. The method
includes the steps
of contacting the mammalian cell with an amount of an MHC class II HLA-DR
inducing agent
3o effective to induce the expression of MHC class II HLA-DR on the surface of
the mammalian
cell, and contacting the MHC class II HLA-DR on the surface of the mammalian
cell with an
amount of an MHC class II HLA-DR ligand effective for causing lysis of the
mammalian cell.
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
_ 'j
In one embodiment the MHC class II HLA-DR ligand is an endogenous MHC class II
HLA-DR ligand and the step of contacting the mammalian cell with the MHC class
II HLA-DR
ligand is a passive step. In another embodiment the step of contacting the
mammalian cell with
the MHC class Ii HLA-DR ligand is an active step.
The mammalian cell may be any type of cell other than an antigen presenting
cell. The
mammalian cell is a tumor cell in another embodiment. Preferably the MHC class
II HLA-DR
ligand is administered to the tumor cell in vivo in an amount effective for
causing cell lysis of the
tumor cell. When the mammalian cell is a tumor cell, however, the MHC class II
HLA-DR
inducing agent does not include adriamycin and gamma interferon.
1 o In one aspect the invention is a method for inducing cell lysis in a tumor
cell. The method
involves the steps of contacting a tumor cell with an amount of an MHC class
II HLA-DR
inducing agent effective to induce the expression of MHC class II HLA-DR on
the surface of the
tumor cell, and contacting the MHC class II HLA-DR on the surface of the tumor
cell with an
amount of an MHC class II HLA-DR ligand effective for causing cell lysis of
the tumor cell.
15 The MHC class II HLA-DR inducing agent is any agent which induces
expression of
MHC class II HLA-DR on a cell surface. Preferably the inducing agent is
selected from the group
consisting of adriamycin, gamma interferon, bacterial byproducts such as
lipopolysaccharides,
mycobacterial antigens such as BCG, a UCP expression vector, a TCRa(3
engagement molecule
and a fatty acid. Once the MHC class II HLA-DR is expressed on the surface of
the cell an MHC
2o class II HLA-DR ligand can interact with the MHC class II HLA-DR and
initiate cell lysis.
Preferably the MHC class II HLA-DR ligand is selected from the group
consisting of an anti-
MHC class Ii HLA-DR antibody, CD4 molecules, a~3 T cell receptor molecules, y8
T cell
receptor molecules and a MHC class II HLA-DR binding peptide.
In one embodiment the MHC class II HLA-DR inducing agent and the MHC class II
25 HLA-DR ligand are administered simultaneously. In another embodiment the
MHC class II
HLA-DR inducing agent and the MHC class II HLA-DR ligand are administered
orally. In yet
another embodiment the MHC class II HLA-DR inducing agent and the MHC class II
HLA-DR
ligand are administered locally.
In another aspect the invention is a method for inducing cell lysis in a tumor
cell by
3o contacting a tumor cell with an amount of an MHC class II HLA-DR inducing
agent effective to
induce the expression of MHC class II HLA-DR on the surface of the tumor cell
in the presence
of an MHC class II HLA-DR ligand. Preferably the MHC class II HLA-DR ligand is
an MHC
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
_g_
class II HLA-DR expressing cell. In one embodiment the inducing agent is
selected from the
group consisting of adriamycin, gamma interferon, bacterial byproducts such as
lipopolysaccharides, mycobacterial antigens such as BCG, a UCP expression
vector, a TCRa(3
engagement molecule and a fatty acid.
S In another embodiment the MHC class II HLA-DR inducing agent and the MHC
class II
HLA-DR ligand are administered orally. In yet another embodiment the MHC class
II HLA-DR
inducing agent and the MHC class II HLA-DR ligand are administered locally.
According to another aspect of the invention a method for inducing apoptosis
in a tumor
cell is provided. The method involves the steps of contacting a tumor cell
with an amount of a
1 o metabolic modifying agent, which when exposed to a cell causes coupling of
electron transport
and oxidative phosphorylation, effective to increase the mitochondria)
membrane potential in the
tumor cell, and contacting the tumor cell with an amount of an apoptotic
chemotherapeutic agent
effective for inducing apoptosis in the tumor cell.
The metabolic modifying agent is added to the tumor cell to induce coupling of
electron
15 transport and oxidative phosphorylation. Preferably the metabolic modifying
agent is selected
from the group consisting of glucose, phorbol myristate acetate in combination
with ionomycin,
MHC class II HLA-DP/DQ ligand, GDP, CD40 binding peptide, UCP antisense,
dominant negative UCP, sodium acetate, and staurosporine. Once electron
transport is coupled
to oxidative phosphorylation, Fas expression is induced on the cell surface
and a apoptotic
2o chemotherapeutic agent can be added to induce apoptosis of the tumor cell.
In one embodiment
the apoptotic chemotherapeutic agent is selected from the group consisting of
adriamycin,
cytarabine, doxorubicin, and methotrexate.
In one embodiment the metabolic modifying agent and the apoptotic
chemotherapeutic
agent are administered simultaneously. In another embodiment the metabolic
modifying agent
25 and the apoptotic chemotherapeutic agent are administered orally. In yet
another embodiment
the metabolic modifying agent and the apoptotic chemotherapeutic agent are
administered locally.
In one embodiment the tumor cell is resistant to the apoptotic
chemotherapeutic agent.
In another embodiment the tumor cell is sensitive to the apoptotic
chemotherapeutic agent, and
wherein the amount of metabolic modifying agent is effective to increase
mitochondria)
3o membrane potential and the amount of apoptotic chemotherapeutic agent is
effective to inhibit
the proliferation of the tumor cell when the mitochondria) membrane potential
is increased.
According to yet another aspect of the invention a method for decreasing
mitochondria)
CA 02324995 2000-10-OS
WO 99/53953 PCTNS99/06874
-9-
membrane potential in a cell of a subject is provided. The method includes the
step of
administering an MHC class II HLA-DR ligand to the subject to selectively
engage MHC class
II HLA-DR on the surface of the cell in an amount effective to decrease
mitochondria) membrane
potential in the mammalian cell. In one embodiment the method is performed in
vivo. In another
embodiment the method is performed ex vivo. In this aspect of the invention
mammalian cells
include but are not limited to antigen presenting cells, T cells, and tumor
cells.
In yet another aspect the invention is a method for increasing mitochondria)
membrane
potential in a mammalian cell expressing MHC class II HLA-DP/DQ. The method
includes the
steps of administering an MHC class II HLA-DP/DQ ligand to the mammalian cell
to selectively
1 o engage MHC class II HLA-DP/DQ on the surface of the cell in an amount
effective to increase
mitochondria) membrane potential in the mammalian cell.
In one embodiment the mammalian cell is a pancreatic ~i cell of a type II
diabetic and
wherein the MHC class II HLA-DP/DQ ligand is administered to the pancreatic ~i
cell in vivo.
According to another aspect the invention is a method for decreasing
mitochondria)
1 s membrane potential in a mammalian cell expressing MHC class II HLA-DR. The
method
involves the steps of administering an MHC class II HLA-DR ligand to the
mammalian cell to
selectively engage MHC class II HLA-DR on the surface of the cell in an amount
effective to
decrease mitochondria) membrane potential in the mammalian cell. Preferably
the mammalian
cell is a pancreatic (3 cell of a type I diabetic and wherein the MHC class II
HLA-DR ligand is
2o administered to the pancreatic ~3 cell in vivo. In one embodiment the
mammalian cell is a tumor
cell and wherein the MHC class II HLA-DR ligand is administered to the tumor
cell in vivo.
The invention in another aspect is a method for treating a subject having a
tumor sensitive
to treatment with a combination of an apoptotic chemotherapeutic agent and a
metabolic
modifying agent. The method includes the steps of administering to a subject
in need of such
25 treatment an apoptotic chemotherapeutic agent and a metabolic modifying
agent in a combined
amount effective to inhibit growth of the tumor, said combined amount being an
amount of
apoptotic chemotherapeutic agent and an amount of metabolic modifying agent,
wherein the
amount of metabolic modifying agent is effective to increase mitochondria)
membrane potential
and the amount of apoptotic chemotherapeutic agent is effective to inhibit the
proliferation of the
30 tumor cell when the mitochondria) membrane potential is increased.
According to another aspect the invention is a method for treating a subject
having a
tumor that is resistant to chemotherapy. The method includes the steps of
administering to the
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-10-
subject an amount of an apoptotic chemotherapeutic agent, and administering
substantially
simultaneously therewith an amount of a metabolic modifying agent, wherein
said amounts when
administered are effective for inhibiting growth of the tumor.
According to another aspect the invention is a method for inducing the
expression of
immune recognition molecules on a cell surface. The method involves the step
of contacting a
cell with an amount of a metabolic inhibition agent effective to decrease
mitochondria) membrane
potential, wherein a decrease in mitochondria) membrane potential causes
induction of the
expression of immune recognition molecules on the cell surface. Preferably the
immune
recognition molecule is selected from the group consisting of MHC class II, B7-
1, B7-2, and CD-
l0 40. Preferably the metabolic inhibition agent is selected from the group
consisting of apoptotic
chemotherapeutic agents, bacterial byproducts, mycobacterial antigens, UCP
expression vectors,
and fatty acids.
The invention in another aspect is a method for inhibiting pancreatic ~3 cell
death in a Type
I diabetic. The progression of pancreatic (3 cell death in type I diabetes
involves two steps. The
~ 5 first phase of type I diabetes is the insulitis phase which results when
membrane potential is
increased, the (3 cells become cell surface Fas positive, but Fas-death
insensitive, . During this
stage it is desirable to decrease the membrane potential and cause the cells
to use fatty acids for
fuel and become cell surface Fas negative. If the diabetes is not treated
during the first phase then
it progresses to a second phase. During the second phase the membrane
potential is decreased and
2o the (3 cell is induced to die if it remains cell surface Fas positive. Thus
the invention contemplates
a two phase approach to the treatment of type I diabetes. In the first phase a
subject is treated to
decrease the membrane potential of the pancreatic (3 cells to prevent or
reduce the chance that the
disease will progress from the insulitis phase to the cell death phase. In the
case when the disease
has already progressed to the cell death phase a subject is treated to
increase the membrane
25 potential of their pancreatic (3 cells. This method involves the steps of
contacting a pancreatic ~i
cell of a Type I diabetic with an amount of a metabolic modifying agent
effective to increase
mitochondria) membrane potential in the pancreatic (3 cell. Preferably the
metabolic modifying
agent is selected from the group consisting of glucose, phorbol myristate
acetate in combination
with ionomycin, MHC class II HLA-DP/DQ ligand, GDP, CD40 binding peptide,
sodium acetate,
30 UCP antisense, dominant negative UCP" and staurosporine. The method is also
useful for
promoting wound healing in a diabetic. In one embodiment the metabolic
modifying agent is
infused with an antagonist of glucose, 2 deoxyglucose.
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-11-
According to another aspect of the invention a method for inhibiting
pancreatic (3 cell
death in a Type I diabetic is provided. The method involves the step of
contacting a pancreatic
(3 cell of a Type I diabetic with an amount of a Fas binding agent effective
to inhibit selective
engagement of Fas on the surface of the pancreatic (3 cell.
According to yet another aspect of the invention a method for inducing
pancreatic (3 cell
death in a Type II diabetic is provided. The method includes the steps of
contacting a pancreatic
(3 cell of a Type II diabetic with an amount of an MHC class II HLA-DR
inducing agent effective
to induce the expression of the MHC class II HLA-DR on the surface of the
pancreatic (3 cell, and
selectively engaging the MHC class II HLA-DR by contacting the cell with an
MHC class II
1 o HLA-DR ligand effective to induce pancreatic (3 cell death. The MHC class
II HLA-DR inducing
agent is selected from the group consisting of adriamycin, gamma interferon,
bacterial byproducts
such as lipopolysaccharides, mycobacterial antigens such as BCG, a UCP
expression vector, a
TCRa~i engagement molecule and a fatty acid in one embodiment.
In another aspect the invention is a method for treating a subject having
autoimmune
~ 5 disease to reduce associated cell death. The method includes the step of
administering an amount
of a y8 binding peptide effective to specifically bind to and inactivate y8
cells in the subject,
wherein the inactivation of the 'y8 cells inhibits cell death associated with
autoimmune disease.
Preferably the y8 binding peptide is an anti-y8 antibody.
According to another aspect of the invention a method for treating a subject
having
2o autoimmune disease to reduce associated cell death is provided. The method
includes the steps
of providing an extracellular environment having a high concentration of
glucose to stimulate
induction of MHC class II HLA-DP/DQ and a low concentration of fatty acids to
inhibit
induction of MHC class II HLA-DR, wherein surface expression of MHC class II
HLA-DP/DQ
is indicative of reduced cell death associated with autoimmune disease.
25 A method for screening a subject for susceptibility to atherosclerosis is
provide according
to another aspect of the invention. The method includes the steps of isolating
a cell selected from
the group consisting of peripheral blood lymphocyte and skin from a subject
and detecting the
presence of an MHC marker selected from the group consisting of an MHC class
II HLA-DP/DQ
and MHC class II HLA-DR on the surface of the cell selected from the group
consisting of
3o peripheral blood lymphocyte and skin, wherein the presence of MHC class II
HLA-DP/DQ is
indicative of susceptibility to atherosclerosis and the presence of MHC class
II HLA-DR is
indicative of resistance to atherosclerosis.
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-12-
The invention in another aspect is a method for selectively killing a Fas
ligand bearing
tumor cell. The method includes the step of contacting the a Fas ligand
bearing tumor cell with
acetate in an amount effective to induce Fas associated cell death. In one
embodiment the a Fas
ligand bearing tumor cell is contacted with the acetate in an amount effective
to sensitize the cell
to a chemotherapeutic agent and further comprising the step of contacting the
cell with a
chemotherapeutic agent. A preferred chemotherapeutic agent is methotrexate.
The method may
also involve the step of administering a Fas ligand to the a Fas ligand
bearing tumor cell. In a
preferred embodiment the Fas ligand bearing tumor cell is selected from the
group consisting of
a melanoma cell and a colon carcinoma cell.
In another aspect the invention is a method for promoting a Thl immune
response. The
method involves the step of administering to a subject who has been exposed to
an antigen an
effective amount for inducing a Th 1 immune response of a MHC class II HLA-DR
inducing agent
to induce DR on a T cell. In one embodiment the MHC class II HLA-DR inducing
agent is fatty
acid.
The invention also includes screening assays. A method for screening a tumor
cell of a
subject for susceptibility to treatment with a chemotherapeutic agent, is one
aspect of the
invention. The assay includes at least the following steps: isolating a tumor
cell from a subject;
exposing the tumor cell to a chemotherapeutic agent; and, detecting the
presence of a cell death
marker selected from the group consisting of a Fas molecule on the surface of
the tumor cell, a
2o B7 molecule on the surface of the tumor cell, an MHC class II HLA-DR on the
surface of the
tumor cell, and a mitochondria) membrane potential indicative of cellular
coupling wherein the
presence of the cell death marker indicates that the cell is susceptible to
treatment with a
chemotherapeutic agent.
In one embodiment the cell death marker is a Fas molecule on the surface of
the tumor
cell and wherein the method comprises the step of contacting the Fas molecule
with a detection
reagent that selectively binds to the Fas molecule to detect the presence of
the Fas molecule. In
another embodiment the cell death marker is a MHC class II HLA-DR molecule on
the surface
of the tumor cell and wherein the method comprises the step of contacting the
MHC class II
HLA-DR molecule with a detection reagent that selectively binds to the MHC
class II HLA-DR
3o molecule to detect the presence of the MHC class II HLA-DR molecule.
Another screening assay of the invention is a method for identifying an anti-
tumor drug
for killing a tumor cell of a subject and includes the steps of isolating a
tumor cell from a subject;
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-13-
detecting the presence of a cell death marker selected from the group
consisting of a Fas molecule
on the surface of the tumor cell, a B7 molecule on the surface of the tumor
cell, an MHC class
II HLA-DR on the surface of the tumor cell, and a mitochondrial membrane
potential indicative
of cellular coupling; exposing the tumor cell to a putative drug; and,
detecting any change in the
presence of the cell death marker to determine whether the putative drug is an
anti-tumor drug
capable of killing the tumor cell of the subject.
A plurality of tumor cells is isolated from the subject and the plurality of
tumor cells is
screened with a panel of putative drugs in one embodiment of the assay. In
another embodiment
the change in the presence of the cell death marker is detected by contacting
the tumor cell with
1o a cell death ligand attached to a solid support. Preferably the cell death
ligand is a Fas ligand.
Yet another assay of the invention is a method for screening a subject for
susceptibility
to disease. This method involves the steps of isolating a cell selected from
the group consisting
of peripheral blood lymphocyte and skin from a subject; and, detecting the
presence of an MHC
marker selected from the group consisting of an MHC class II HLA-DP/DQ, B7-2,
B7-l and
MHC class II HLA-DR on the surface of the cell, wherein the presence of MHC
class II HLA-
DP/DQ is indicative of susceptibility to atherosclerosis and resistance to
autoimmune disease and
the presence of MHC class II HLA-DR, B7-2, or B7-1 is indicative of resistance
to
atherosclerosis and susceptibility to autoimmune disease.
The invention also encompasses kits. One kit of the invention is a kit for
screening a
2o subject for susceptibility to disease. The kit includes a container housing
a first binding
compound that selectively binds to a protein selected from the group
consisting of B7-2, B7-1 and
MHC class II HLA-DR; a container housing a second binding compound that
selectively binds
to a MHC class II HLA-DP/DQ protein; and instructions for determining whether
an isolated cell
of a subject selectively interacts with the first or second binding compound,
wherein the presence
of MHC class II HLA-DP/DQ on the cell surface which interacts with the second
compound is
indicative of susceptibility to atherosclerosis and resistance to autoimmune
disease and the
presence of MHC class II HLA-DR on the cell surface which interacts with the
first compound
is indicative of resistance to atherosclerosis and susceptibility to
autoimmune disease.
Another kit of the invention is a kit for screening a tumor cell of a subject
for
3o susceptibility to treatment with a chemotherapeutic agent. The kit includes
a container housing
a cell death marker detection reagent; and instructions for using the cell
death marker detection
reagent for detecting the presence of a cell death marker selected from the
group consisting of a
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-14-
Fas molecule on the surface of the tumor cell, an MHC class II HLA-DR on the
surface of the
tumor cell, and a mitochondria) membrane potential indicative of cellular
coupling wherein the
presence of the cell death marker indicates that the cell is susceptible to
treatment with a
chemotherapeutic agent.
In some embodiments the kit also includes a container housing a
chemotherapeutic agent
or a panel of chemotherapeutic agents, housed in separate compartments. In
other embodiments
the kit also includes a cell death ligand. Preferably the cell death ligand is
coated on a solid
surface. In another preferred embodiment the cell death ligand is a Fas
ligand.
The invention in another aspect is a method for selectively killing a cell.
The method
1 o involves the step of contacting the cell with a nucleic acid selected form
the group consisting of
a UCP anti-sense nucleic acid and a UCP dominant-negative nucleic acid in an
amount effect to
inhibit UCP function.
In one embodiment the cell death marker is a Fas molecule on the surface of
the tumor
cell and wherein the method comprises the step of contacting the Fas molecule
with a detection
reagent that selectively binds to the Fas molecule to detect the presence of
the Fas molecule. In
another embodiment the cell death marker is a MHC class II HLA-DR molecule on
the surface
of the tumor cell and wherein the method comprises the step of contacting the
MHC class II
HLA-DR molecule with a detection reagent that selectively binds to the MHC
class II HLA-DR
molecule to detect the presence of the MHC class II HLA-DR molecule.
2o In another aspect the invention is a composition of a metabolic modifying
agent and an
apoptotic chemotherapeutic agent. Preferably the metabolic modifying agent is
selected from the
group consisting of glucose, phorbol myristate acetate in combination with
ionomycin, MHC
class II HLA-DP/DQ ligand, GDP, CD40 binding peptide, sodium acetate, UCP
antisense,
dominant negative UCP" and staurosporine. In a preferred embodiment the
apoptatic
chemotherapeutic agent is selected from the group consisting of adriamycin,
cytarabine,
doxorubicin, and methotrexate.
In one embodiment the metabolic modifying agent and the apoptotic
chemotherapeutic
agent are present in an amount effective to inhibit the proliferation of a
tumor cell. In another
embodiment the composition includes a pharmaceutically acceptable carrier.
3o The invention according to another aspect is a composition of an MHC class
II HLA-DR
inducing agent and an MHC class II HLA-DR ligand. In one embodiment the MHC
class II
HLA-DR inducing agent is selected from the group consisting of adriamycin,
gamma interferon,
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-15-
bacterial byproducts such as lipopolysaccharides, mycobacterial antigens such
as BCG, a UCP
expression vector, a TCRa(3 engagement molecule and a fatty acid. In another
embodiment the
MHC class II HLA-DR ligand is selected from the group consisting of an anti-
MHC class II
HLA-DR antibody, CD4 molecules, a~i T cell receptor molecules, y8 T cell
receptor molecules
and a MHC class II HLA-DR binding peptide. According to yet another embodiment
the MHC
class II HLA-DR inducing agent and the MHC class II HLA-DR ligand are present
in an amount
effective to lyse a tumor cell. The composition may be formulated in a
pharmaceutically
acceptable carrier.
The invention also includes the discovery that neural differentiation and
apoptosis are
l0 regulated through interaction of the immune recognition molecules on the
nerve cell surface with
an NGF producing cell that expresses the counterpart surface immune
recognition molecule,
likely CD28 or CTLA4. The interaction between the nerve cell and the NGF
producing cell
causes the NGF producing cell to release NGF into the local environment. This
NGF then
stimulates the nerve cell to undergo nerve cell differentiation and
innervation.
~ 5 The invention in other aspects relates to methods and products for
regulating nerve cell
growth, differentiation, and apoptosis. In one aspect the invention is a
method for inducing nerve
cell differentiation. The method includes the steps of contacting a nerve cell
with an amount of
a B7 inducing agent effective to induce the expression of B7 on the surface of
the nerve cell, and
exposing the nerve cell to a neural activating cell to cause differentiation
of the nerve cell.
20 In another aspect the invention is a method for inducing nerve cell
differentiation. The
method involves the step of contacting a nerve cell with an amount of a B7
inducing agent
effective to induce the expression of B7 on the surface of the nerve cell in
the presence of an
endogenous neural activating cell.
In some embodiments the B7 inducing agent is adriamycin, gamma interferon, a
fatty
2s acid, a lipoprotein, an anti-MHC class II HLA-DR antibody, a MHC class II
HLA-DR binding
peptide, a B7 expression vector, or a UCP expression vector.
In another embodiment the method also includes the step of contacting the
nerve cell with
an amount of a metabolic modifying agent, which when exposed to a cell causes
increased
coupling of electron transport and oxidative phosphorylation, effective to
prevent dissipation of
3o proton motor force in the nerve cell prior to contacting the nerve cell
with the B7 inducing agent.
In some embodiments the metabolic modifying agent is glucose, phorbol
myristate acetate in
combination with ionomycin, MHC class II HLA-DP/DQ ligand, GDP, CD40 binding
peptide,
CA 02324995 2000-10-OS
WO 99/53953 PCTNS99/06874
- 16-
sodium acetate, UCP antisense, dominant negative UCP" and staurosporine. In
other
embodiments the neural activating cell is a T cell, a macrophage, or a
dendritic cell.
In yet another embodiment the method includes the step of administering a
fatty acid to
the nerve cell to stop cell division.
In yet another embodiment the method includes the step of inducing the
expression of a
receptor for nerve growth factor.
According to another aspect the invention is a method for inducing apoptosis
in a nerve
cell. The method includes the steps of contacting a nerve cell with an amount
of a metabolic
modifying agent, which when exposed to a nerve cell causes an increase in
coupling of electron
transport and oxidative phosphorylation, effective to prevent dissipation of
proton motor force
in the nerve cell, and contacting a neural activating cell with an amount of a
B7 receptor blocking
agent effective for inducing apoptosis in the nerve cell.
The metabolic modifying agent, in various embodiments, is glucose, phorbol
myristate
acetate in combination with ionomycin, MHC class II HLA-DP/DQ ligand, GDP,
CD40 binding
peptide, sodium acetate, UCP antisense, dominant negative UCP" or
staurosporine. In various
other embodiments the B7 receptor blocking agent is an anti-CD28 antibody,
CD28 binding
peptide, CTLA4 analog, anti-CTLA4 antibody, or CTLA4 binding peptide.
The invention also includes compositions related to the above methods. In one
aspect the
invention is a composition of a metabolic modifying agent and a B7 receptor
blocking agent.
2o The metabolic modifying agent, in various embodiments, is glucose, phorbol
myristate
acetate in combination with ionomycin, MHC class II HLA-DP/DQ ligand, GDP,
CD40 binding
peptide, sodium acetate, UCP antisense, dominant negative UCP" or
staurosporine. In various
other embodiments the B7 receptor blocking agent is an anti-CD28 antibody,
CD28 binding
peptide, CTLA4 analog, anti-CTLA4 antibody, or CTLA4 binding peptide.
In another embodiment the metabolic modifying agent and the B7 receptor
blocking agent
are present in an amount effective to induce apoptosis of a nerve cell. In yet
another embodiment
the composition also includes a pharmaceutically acceptable carrier.
A composition of a B7 inducing agent and a CD28 inducing agent is provided in
another
aspect of the invention.
3o In some embodiments the B7 inducing agent is adriamycin, gamma interferon,
bacterial
byproducts such as lipopolysaccharides and lipoproteins, mycobacterial
antigens such as BCG,
and fatty acids, an anti-MHC class II HLA-DR antibody, a MHC class II HLA-DR
binding
CA 02324995 2000-10-OS
WO 99/53953 PC'T/US99/06874
-17-
peptide, a B7 expression vector, or a UCP expression vector. In other
embodiments the CD28
inducing agent is a T cell receptor engagement molecule, CD3 engagement
molecule, IL4, or a
CD28 expression vector. In yet another embodiment the composition also
includes a
pharmaceutically acceptable carrier.
According to another aspect of the invention a method for re-innervating an
injured tissue
is provided. The method includes the step of implanting a B7 expressing nerve
cell in the injured
tissue, wherein the implanted B7 expressing nerve cell will undergo neuronal
differentiation in
the presence of a neural activating cell in the injured tissue to re-innervate
the injured tissue.
In one embodiment the B7 expressing nerve cell constitutively expresses B7. In
another
embodiment the B7 expressing nerve cell is a nerve cell which constitutively
expresses a UCP
gene. In yet another embodiment the B7 expressing nerve cell is a nerve cell
which constitutively
expresses a B7 gene.
The method in another embodiment includes the step of administering a B7
inducing
agent effective to induce endogenous B7 expression on the surface of the nerve
cell.
The injured tissue may be any tissue in which a nerve is damaged. In one
embodiment
the injured tissue is a spinal chord. In another embodiment the injured tissue
is a severed limb.
A method for treating a neurodegenerative disorder is provided according to
another
aspect of the invention. The method includes the step of administering an
amount of a B7
inducing agent effective to induce the expression of B7 on the surface of a
nerve cell.
In some embodiments the B7 inducing agent is adriamycin, gamma interferon,
bacterial
byproducts such as lipopolysaccharides and lipoproteins, mycobacterial
antigens such as BCG,
and fatty acids, an anti-MHC class II HLA-DR antibody, a MHC class II HLA-DR
binding
peptide, a B7 expression vector, or a UCP expression vector.
The method may also include the step of inducing expression of CD28 on the
surface of
a neural activating cell. Preferably the neural activating cell is a T cell.
In other preferred
embodiments the neural activating cell is a macrophage, a B cell or a
dendritic cell.
In yet another embodiment the neurodegenerative disorder is selected from the
group
consisting of paralysis, Parkinson's disease, Alzheimer's disease, amyotrophic
lateral sclerosis,
and multiple sclerosis.
3o According to another aspect the invention is a method for selectively
killing a cell. The
method includes the step of contacting the cell with a nucleic acid selected
form the group
consisting of a UCP anti-sense nucleic acid and a UCP dominant-negative
nucleic acid in an
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/Ob874
-18-
amount effect to inhibit UCP function.
In other aspects the invention is a method for selectively killing a tumor
cell. The method
includes the steps of contacting the tumor cell with acetate in an amount
effective to induce cell
surface Fas expression, and administering a Fas ligand to the tumor cell in an
amount effective
to induce Fas associated cell death. In one embodiment the tumor cell is
contacted with the
acetate in an amount effective to sensitize the cell to a chemotherapeutic
agent and further
comprising the step of contacting the cell with an apoptopic chemotherapeutic
agent.
A method for selectively killing a tumor cell is provided according to another
aspect of
the invention. The method includes the step of contacting the tumor cell with
a compound
selected from the group consisting of acetate, GDP and an apoptopic
chemotherapeutic agent in
an amount effective to kill the tumor cell.
Each of the limitations of the invention can encompass various embodiments of
the
invention. It is, therefore, anticipated that each of the limitations of the
invention involving any
one element or combinations of elements can be included in each method and
product.
These and other aspects of the invention are described in greater detail
below.
Brief Description of the Figures
The present invention may be more easily and completely understood when taken
in
conjunction with the accompanying figure.
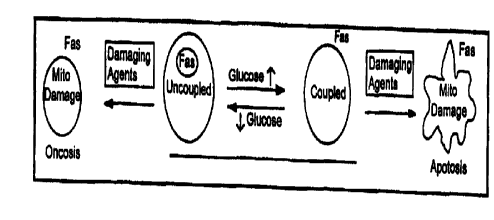
Figure 1 is a schematic diagram showing that increasing environmental glucose
results
in increased cell surface Fas expression and functionally coupled
mitochondria) ATP synthesis,
suggesting a link between mitochondria) glucose metabolism and susceptibility
to Fas-induced
cell death. As glucose levels decrease, levels of cell surface Fas decrease,
newly synthesized Fas
is stored intraceilularly and mitochondria) ATP synthesis is uncoupled from
respiration and less
mitochondria) ATP is produced.
Brief Description of the Sequences
SEQ ID NO: l is the nucleotide sequence of the human B7 (B7.1 ) cDNA with
GenBank
Acc. no.:M27533.
3o SEQ ID N0:2 is the predicted amino acid sequence of the translation product
of human
B7 (B7.1 ) cDNA (SEQ ID NO:1 ).
SEQ ID N0:3 is the nucleotide sequence of the human B7.2 cDNA with GenBank
Acc.
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
- 19-
no.U04343.
SEQ iD N0:4 is the predicted amino acid sequence of the translation product of
human
B7.2 cDNA (SEQ ID N0:3).
SEQ ID NO:S is the nucleotide sequence of the human uncoupling (UCP-1 ) cDNA
with
GenBank Acc. no.U28480.
SEQ ID N0:6 is the predicted amino acid sequence of the translation product of
human
uncoupling cDNA (UCP-1) (SEQ ID NO:S).
SEQ ID N0:7 is the nucleotide sequence of the human uncoupling (UCP-2) cDNA
with
GenBank Acc. no.U82819.
SEQ ID N0:8 is the predicted amino acid sequence of the translation product of
human
uncoupling cDNA (UCP-2) (SEQ ID N0:7).
SEQ ID N0:9 is the nucleotide sequence of the human uncoupling (UCP-3S) cDNA
with
GenBank Acc. no.U82818.
SEQ ID NO:10 is the predicted amino acid sequence of the translation product
of human
~5 uncoupling cDNA (UCP-3S) (SEQ ID N0:9).
SEQ ID NO:11 its the nucleotide sequence of the human CD28 cDNA with GenBank
Acc.
no.J02988.
SEQ ID N0:12 is the predicted amino acid sequence of the translation product
of the
human CD28 cDNA (SEQ ID NO:l 1).
2o SEQ ID N0:13 is the amino acid sequence of a peptide.
The invention relates to methods and products involving the control of cell
division,
differentiation, death, and apoptosis by the regulation of cell surface immune
recognition
25 molecules. It was discovered according to one aspect of the invention that
proton motor force
(assessed as mitochondria) metabolism) is integrally related to the regulation
of cellular division
and cellular apoptosis. The ability to manipulate mitochondria) metabolic
processes has led to
the development of methods for treating diseases associated with excessive
cellular proliferation
or premature cellular death. Additionally, the ability to manipulate the
expression of cell surface
3o immune recognition molecules such that a nerve cell can stimulate local NGF
production from
an NGF producing cell has led to the development of methods for treating
neurodegenerative
diseases associated with premature cellular death. It was also discovered
according to the
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-20-
invention that the regulation of proton motor force (mitochondria) metabolism)
is directly related
to the expression of these cell surface immune recognition molecules involved
in the signaling
process of cell death and in immune response signaling, and thus can be
manipulated as one
method for regulating the expression of the immune recognition molecules. The
ability to control
the expression of these cell surface molecules is a useful and powerful
technique for
therapeutically manipulating the processes of cellular death, apoptosis,
differentiation and
proliferation. Monitoring the expression of these proteins is also useful for
screening assays to
assess disease states as well as the mitochondria) metabolic status of cells.
Based on all these discoveries the invention includes in some aspects methods
for
increasing or decreasing the mitochondria) membrane potential in a mammalian
cell. The ability
to manipulate the mitochondria) membrane potential of a cell provides the
ability to control the
fate of the cell. When the membrane potential of a cell is decreased and the
cell is caused to use
fatty acids for fuel the cell can interpret a signal as a signal for cell
death. If the membrane
potential of a cell is increased, however, and the cell is using glucose for
fuel, the same signal can
~ 5 be interpreted as a signal to divide rather than for cell death. The
invention encompasses
mechanisms for controlling these complex interactions to regulate the
processes of cellular death
and division.
One method for causing a decrease in mitochondria) membrane potential and a
switch to
the use of fatty acids as fuel is by inducing the expression of MHC class II
HLA-DR on the
2o surface of the cell. If low amounts of MHC class II HLA-DR are already
expressed on the
surface the cell can be contacted with an MHC class II HLA-DR ligand to cause
a further
decrease in the mitochondria) membrane potential. When a cell has been induced
to express
MHC class II HLA-DR on the cell surface such that the electron transport is
uncoupled and the
cell is using fatty acids for fuel and the cell is contacted with a MHC class
II HLA-DR ligand,
25 then the cell generally will interpret that signal as a cell death signal,
and cause cell lysis.
The invention also encompasses methods for causing an increase in
mitochondria)
membrane potential. This increase, accompanied by the use of glucose as fuel
is accomplished
in some aspects by inducing the expression of MHC class II HLA-DPDQ on the
surface of the
cell. If low amounts of MHC class II HLA-DPDQ are already expressed on the
surface the cell
3o can be contacted with an MHC class II HLA-DPDQ ligand to cause a further
increase in the
mitochondria) membrane potential and an increase in coupling of electron
transport and oxidative
phosphorylation. When a cell has been induced to express MHC class II HLA-DPDQ
on the cell
CA 02324995 2000-10-OS
WO 99153953 PCTNS99/06874
-21 -
surface such that the electron transport is relatively coupled and the cell is
using glucose for fuel
and the cell is contacted with a MHC class II HLA-DPDQ ligand, then the cell
generally will
interpret that signal as a cell division signal, and cause cellular division.
The methods of the invention have broad utility in regulating mammalian cell
growth and
death in vitro, in vivo and ex vivo. Because mammalian cells utilize the basic
process of
mitochondria) metabolism in regulating their own growth and differentiation,
any type of
mammalian cell can be manipulated according to the methods of the invention.
When the
methods for increasing or decreasing mitochondria) metabolism are performed in
vitro by
contacting an MHC class II HLA-DPDQ or -DR expressing cell with an MHC class
II HLA-
t o DPDQ or -D ligand, respectively, the methods are not performed on antigen
presenting cells.
When the same methods are performed ex vivo or in vivo they may however, be
performed on
antigen presenting cells as well as any other type of mammalian cell. An
"antigen presenting
cell" is used herein consistently with its well known meaning in the art and
includes, for instance,
dendritic cells, macrophage, etc. The in vitro methods of the invention are
useful for a variety
t 5 of purposes. For instance, the methods of the invention may be useful for
identifying drugs which
have an effect, such as a preventative effect, on cellular division or death
by contacting cells
which are caused by the manipulations of the invention to undergo cellular
division or death.
In addition to the in vitro methods, the methods of the invention may be
performed in vivo
or ex vivo in a subject to manipulate one or more cell types within the
subject. An "ex vivo"
2o method as used herein is a method which involves isolation of a cell from a
subject, manipulation
of the cell outside of the body, and reimplantation of the manipulated cell
into the subject. The
ex vivo procedure may be used on autologous or heterologous cells, but is
preferably used on
autologous cells. In preferred embodiments, the ex vivo method is performed on
cells that are
isolated from bodily fluids such as peripheral blood or bone marrow, but may
be isolated from
2s any source of cells. When returned to the subject, the manipulated cell
will be programed for cell
death or division, depending on the treatment to which it was exposed. Ex vivo
manipulation of
cells has been described in several references in the art, including Engleman,
E.G., 1997,
Cytotechnology, 25:1; Van Schooten, W., et al., 1997, Molecular Medicine
Today, June, 255;
Steinman, R.M., 1996, Experimental Hematology, 24, 849; and Gluckman, J.C.,
1997, Cytokines,
30 Cellular and Molecular Therapy, 3:187. The ex vivo activation of cells of
the invention may be
performed by routine ex vivo manipulation steps known in the art. In vivo
methods are also well
known in the art. A subject as used herein means humans, primates, horses,
cows, pigs, sheep,
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-22-
goats, dogs, cats and rodents. The invention thus is useful for therapeutic
purposes and also is
useful for research purposes such as testing in animal or in vitro models of
medical, physiological
or metabolic pathways or conditions.
In preferred embodiments of the invention the mammalian cell is a tumor cell
The
method is useful for inducing cell lysis in many types of mammalian cells but
is particularly
useful for inducing cell lysis in a tumor cell. A "tumor cell" as used herein
is a cell which is
undergoing unwanted mitotic proliferation. A tumor cell when used in the in
vitro aspects of the
invention can be isolated from a tumor within a subject or may be part of an
established cell line.
A tumor cell in a subject may be part of any type of cancer. Cancers include
but are not limited
1 o to biliary tract cancer; brain cancer, including glioblastomas and
medulloblastomas; breast cancer;
cervical cancer; choriocarcinoma; colon cancer; endometrial cancer; esophageal
cancer; gastric
cancer; hematological neoplasms, including acute lymphocytic and myelogenous
leukemia;
multiple myeloma; AIDS associated leukemias and adult T-cell leukemia
lymphoma;
intraepithelial neoplasms, including Bowen's disease and Paget's disease;
liver cancer; lung
cancer; lymphomas, including Hodgkin's disease and lymphocytic lymphomas;
neuroblastomas;
oral cancer, including squamous cell carcinoma; ovarian cancer, including
those arising from
epithelial cells, stromal cells, germ cells and mesenchymal cells; pancreas
cancer; prostate cancer;
rectal cancer; sarcomas, including leiomyosarcoma, rhabdomyosarcoma,
liposarcoma,
fibrosarcoma and osteosarcoma; skin cancer, including melanoma, Kaposi's
sarcoma, basocellular
2o cancer and squamous cell cancer; testicular cancer, including germinal
tumors (seminoma, non-
seminoma[teratomas, choriocarcinomas]), stromal tumors and germ cell tumors;
thyroid cancer,
including thyroid adenocarcinoma and medullar carcinoma; and renal cancer
including
adenocarcinorna and Wilms tumor.
In the aspects of the invention that the mammalian cell is a tumor cell and
the cell is only
treated with an MHC class II HLA-DR inducing agent but not an MHC class II HLA-
DR ligand
the MHC class II HLA-DR inducing agent does not include adriamycin and gamma
interferon.
When the MHC class II HLA-DR inducing agent is adriamycin or gamma interferon
the method
of lysing the tumor cell requires the additional step of contacting the tumor
cell with an MHC
class II HLA-DR ligand to cause cell lysis.
3o Cell lysis is the necrotic death of a cell which occurs by osmotic rupture.
As used herein an "MHC class II HLA-DR inducing agent" is an agent which
causes
MHC class II HLA-DR to be expressed on the cell surface. Preferably the MHC
class II HLA-
CA 02324995 2000-10-OS
WO 99/53953 PCTN599/06874
-23-
DR inducing agent is a pharmacological agent that causes uncoupling of
electron transport and
oxidative phosphorylation, resulting in reduced mitochondrial membrane
potential within the cell.
MHC class II HLA-DR inducing agents include but are not limited to adriamycin,
gamma
interferon, bacterial byproducts such as lipopolysaccharides, mycobacterial
antigens such as
s BCG, a UCP expression vector, a TCRa~i engagement molecule and a fatty acid.
Although
gamma interferon induces expression of both MHC class II HLA-DR and MHC class
II HLA-
DP/DQ it can still be used in combination with an MHC class II HLA-DR Iigand
which
selectively binds to MHC class II HLA-DR and not MHC class II HLA-DP/DQ. The
MHC class
II HLA-DR inducing agent is an isolated molecule. An isolated molecule is one
which has been
removed from its natural surroundings and formulated for administration to an
organism.
Adriamycin, gamma interferon, bacterial byproducts such as
lipopolysaccharides, mycobacterial
antigens such as BCG are all well known compounds which can be purchased from
a variety of
commercial sources. UCP expression vector can be prepared by methods well
known in the art,
such methods are described in detail below. Fatty acids are also well known
compounds that can
~ s be purchased commercially from many sources. Preferred fatty acids include
but are not limited
to oleic acid, palmitate, and myristic acid. A "TCRa~3 engagement molecule" as
used herein
refers to any compound that can bind to and cause cell surface crosslinking of
CD4 and the a(3T
cell receptor (a(3TCR). Such compounds are known in the art. For instance
heterobifunctional
antibodies are capable of crosslinking CD4 and a(3TCR by interacting with both
molecules on
2o the surface of the cell. Other CD4/a~iTCR binding molecules can be
identified with routine
experimentation and are also encompassed by the term TCRa(3 engagement
molecule. Routine
screening methods for identifying such binding molecules are set forth below.
MHC class II HLA-DR refers to a subregion of the human major
histocompatibility class
II genetic locus. As used herein the "MHC class II HLA-DR" is the protein
expressed on the
2s surface of a cell which corresponds to the MHC class II HLA-DR genetic
locus. Although the
term HLA-DR refers to the human subclass of MHC, the invention is intended to
encompass the
corresponding subclass of MHC in other species, which have different
nomenclature, such as the
IE region in the corresponding subclass in the mouse.
As used herein an "MHC class II HLA-DR ligand" is a molecule which binds to
MHC
3o class II HLA-DR and stimulates an MHC class II HLA-DR specific
intracellular signal
stimulating cell lysis. MHC class II HLA-DR ligands are MHC class II HLA-DR
binding
peptides which cause cell surface crosslinking of MHC class II HLA-DR
molecules. Such
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-24-
ligands are well known in the art and include but are not limited to anti-MHC
class II HLA-DR
antibodies such as those commercially available from Becton Dickinson and many
other sources,
CD4 peptides, y8 T cell receptor (TCR) peptides , oc(3 TCR peptides, and other
binding peptides,
optionally bound to a delivery vehicle such as a liposome. CD4 peptides, y8TCR
peptides, and
oc(3 TCR peptides are well known cell surface molecules. These peptides can be
used as a ligand
in a soluble form or may be attached or conjugated to a Garner such as a
liposome or particle
(other chemical/physical vectors useful for this purpose are discussed below).
In addition to these
known binding peptides other MHC class II HLA-DR binding peptides can be
identified with
routine experimentation and are also encompassed by the term MHC class II HLA-
DR ligand.
t o Routine screening methods for identifying such binding molecules are set
forth below.
Cell lysis can be assessed by any method known in the art for making such
measurements.
For example cell lysis can be determined by direct histological analysis,
comparison of intact cell
numbers using a coulter counter, and flow cytometry. These methods are well
known in the art
and some are described in more detail in the examples section below.
The "MHC class II HLA-DR ligand" as used herein is an isolated molecule. An
isolated
molecule is one which has been removed from its natural surroundings and
formulated for
administration to an organism.
The methods of the invention in some aspects may also be performed using
endogenous
MHC class II HLA-DR ligand. An "endogenous MHC class II HLA-DR ligand" is
different than
2o an "MHC class II HLA-DR ligand" used above which is an isolated
composition. For instance
the endogenous MHC class II HLA-DR ligand may be a cell having a cell surface
MHC class II
HLA-DR binding peptide. In this case the method would only include the step of
contacting a
tumor cell with an amount of an MHC class II HLA-DR inducing agent effective
to induce the
expression of MHC class II HLA-DR on the surface of the tumor cell in the
presence of an
endogenous MHC class II HLA-DR ligand.
When the endogenous MHC class II HLA-DR ligand is a cell having a cell surface
MHC
class II HLA-DR binding peptide which is already present in interactive
proximity to the MHC
class II HLA-DR, the cell does not have to be manually brought into contact
with the MHC class
II HLA-DR.
Another aspect of the invention involves the induction of apoptosis in a tumor
cell rather
than cell lysis. In both apoptosis and cell lysis the cell dies but the
processes occur through
different mechanisms and when the cell is in a different metabolic state. As
described above,
CA 02324995 2000-10-OS
WO 99/53953 PCTNS99/06874
-25-
when the methods of the invention are performed to induce cell lysis in a
tumor cell the cell is in
an uncoupled state. When the methods of the invention are performed to induce
apoptosis the cell
is caused to assume a coupled state. The method for inducing apoptosis in a
tumor cell involves
the steps of contacting a tumor cell with an amount of a metabolic modifying
agent, which when
s exposed to a cell causes coupling of electron transport and oxidative
phosphorylation, effective
to increase the mitochondria) membrane potential in the tumor cell, and
contacting the tumor cell
with an amount of a chemotherapeutic agent effective for inducing apoptosis in
the tumor cell.
Apoptosis is a process of cell death in which the cell undergoes shrinkage and
fragmentation, followed by phagocytosis of the cell fragments. Apoptosis is
well known in the
1o art and can be assessed by any art recognized method. For example apoptosis
is easily
determined using flow cytometry, which distinguishes between live and dead
cells. Flow
cytometry is described in more detail in the Examples below.
As used herein a "metabolic modifying agent" is an agent which when exposed to
a cell
causes coupling of electron transport and oxidative phosphorylation, resulting
in increased
mitochondria) membrane potential within the cell. Metabolic modifying agents
include but are
not limited to glucose, sodium acetate, phorbol myristate acetate in
combination with ionomycin,
MHC class II HLA-DP/DQ ligand, guanosine diphosphate (GDP), CD40 binding
peptide, sodium
acetate, UCP antisense, dominant negative UCP" and staurosporine. Glucose,
phorbol myristate
acetate, ionomycin, GDP, and staurosporine are all well known commercially
available
2o compounds which can be obtained form many sources. CD40 binding peptides
are any peptide
molecules which interact with CD40, causing CD40 crosslinking on a cell
surface. These
molecules include, for example, CD40 ligand, which is a well known molecule.
CD40 binding
peptides are not limited to CD40 ligand, however, but include other molecules
which can be
identified with routine experimentation. Routine screening methods for
identifying such binding
25 molecules are set forth below. UCP antisense molecules and dominant
negative UCP molecules
are also known in the art and are described in more detail below.
MHC class II HLA-DP/DQ refers to another subregion of the human major
histocompatibility class II genetic locus. As used herein the "MHC class II
HLA-DP/DQ" is the
protein expressed on the surface of a cell which corresponds to the MHC class
II HLA-DP/DQ
3o genetic locus. Although the term HLA-DP/DQ refers to the human subclass of
MHC, the
invention is intended to encompass the corresponding subclass of MHC in other
species, which
have different nomenclature, such as the IA region in the subclass in the
mouse.
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-26-
As used herein an "MHC class II HLA-DP/DQ ligand" is a molecule which binds to
MHC
class II HLA-DP/DQ and stimulates an MHC class II HLA-DP/DQ specific
intracellular signal
stimulating coupling of electron transport and oxidative phosphorylation
resulting in increased
mitochondria) membrane potential. MHC class II HLA-DP/DQ ligands include but
are not
limited to anti-MHC class II HLA-DP/DQ ligand antibodies, other binding
peptides, and cells
having a cell surface MHC class II HLA-DP/DQ binding antigen. When the MHC
class II HLA-
DP/DQ ligand is a cell having a cell surface MHC class II HLA-DP/DQ binding
antigen which
is already present in interactive proximity to the MHC class II HLA-DP/DQ, the
cell does not
have to be manually brought into contact with the MHC class II HLA-DP/DQ.
t 0 As used herein, the term "dissipation of proton motor force" refers to the
relative amount
of protons in the mitochondria. It can be assessed by measuring mitochondria)
membrane
potential. As used herein "mitochondria) membrane potential" is the pressure
on the inside of the
mitochondria) cell membrane measured relative to the extracellular fluid which
is created by the
generation and dissipation of charge within the mitochondria. The
mitochondria) membrane
potential is maintained by the energy generating system of the mitochondria.
In most tissues
electron transport is coupled to oxidative phosphorylation resulting in the
production of ATP
from glucose. Uncoupling proteins (UCPs) can cause the reversible uncoupling
of electron
transport and oxidative phosphorylation, which leads to a decrease in the
mitochondria)
membrane potential. Other tissue, often referred to as the immuno-privileged
tissue such as the
brain, testis, ovary, eye, and pancreatic ~i cells, express UCPs which cause
electron transport to
be uncoupled to oxidative phosphorylation under normal conditions. In these
tissues glucose
cannot be converted to ATP while the UCP is active because of the uncoupling
and the energy
produced is converted into other energy forms such as heat and released. If
the metabolic
processing systems in these tissues are caused to undergo coupling the
membrane potential would
increase.
The absolute levels of the mitochondria) membrane potential vary depending on
the cell
or tissue type. As used herein an "increase in mitochondria) membrane
potential" is an increase
relative to the normal status of the cell being examined and results from the
prevention of
dissipation of proton motor force. "Prevention" as used herein refers to a
decrease or reduction
3o in the amount of dissipation that would ordinarily occur in the absence of
the stimulus applied
according to the methods of the invention to cause coupling. If electron
transport and oxidative
phosphorylation are normally uncoupled within the cell then the baseline
potential will be
CA 02324995 2000-10-OS
WO 99/53953 PCTNS99/06874
-27-
relatively low and when the ATP generating systems are coupled an increase in
mitochondria)
membrane potential from that baseline level is observed. Likewise, a "decrease
in mitochondria)
membrane potential" is a decrease relative to the normal status of the~cell
being examined and
results from the dissipation of proton motor force. If electron transport and
oxidative
phosphorylation are normally coupled within the cell then the baseline
potential will be relatively
high and when the ATP generating systems are uncoupled a decrease in
mitochondria) membrane
potential from that baseline level is observed.
Changes in mitochondria) membrane potential can be assessed by any method
known in
the art for making such measurements. For example the mitochondria) membrane
potential may
1o be measured cytometrically by incubating cells for 20 minutes at room
temperature with 5
mg/ml JC-139 a fluorescent probe able to bind mitochondria. The aggregation
state and
consequently the fluorescence emission of JC-1 changes as the mitochondria)
membrane
potential is altered. Valinomycin, which collapses the mitochondria) membrane
potential can
be used as a positive control treatment. Flow cytometry permits the
examination of up to four
~ 5 fluorescent markers concurrently. This method is described in more detail
in the Examples
section below In addition to examining the mitochondria) membrane potential,
studies can be
performed to determine the rate of glucose utilization and oxidation and
measurements of
proton leak can be assessed by a top-down elasticity analysis, each of which
is described in
more detail in the Examples below.
2o The relationship between mitochondria) metabolism and cell surface Fas
expression is
important to the methods of the invention. When a cell is coupled Fas is
expressed on the cell
surface and when a cell is uncoupled Fas generally is transported to
intracellular stores. When
a cell is coupled and Fas is on the surface engagement of Fas sends a signal
to the cell
instructing the cell to undergo cellular division. If a chemotherapeutic agent
is added then the
2s signal is changed to a signal which instructs the cell to undergo
apoptosis. When a cell is
uncoupled and ordinarily Fas is not expressed on the cell surface. Under
certain disease
conditions such as Type I diabetes (discussed in more detail below), or when
the cell has been
irradiated Fas can be expressed on the surface of uncoupled cells. When this
occurs
engagement of Fas sends a signal to the cell to die.
3o An "apoptotic chemotherapeutic agent" as used herein is a group of
molecules which
function by a variety of mechanisms to induce apoptosis in rapidly dividing
cells. Apoptotic
chemotherapeutic agents are a class of chemotherapeutic agents which are well
known to those
~C1''.VON:I:I':'1-iII.~FNCHLN U4 .~~~~_ 5_ pCA 02324_995 2000-10-OS' 720 l1-1-
l-~ +4~3 8~~ ?n~1~~4.rr:te a
US 009906874
22-05-2000 .
- 28 -
of skill in the art. Clicmothcrapeutic agents include those agents disclosed
in Chapter 52,
Antincoplastic Agents (Paul Calabresi and Bruce A. Chabncr), and the
introduction thereto, 1202-
1263, of Goodman and Gilman's "The Pharmacological Basis of 'Therapeutics",
Eighth Edition,
1990, McGraw-HiII, Inc (Health Professions Division~.iaae~per
s Suitable chcmotherapeutic agents may have various atechanismx of action. The
classes of
suitable chemotherapeutic agents include (a) AlkylaZing Agents such as
nitrogen mustard (e.g.
mechloreihamine, cylophosphanude, ifosfamIde, meiphalan, chlorambucil),
ethylenimines ttnd
metftylmelamines (e.g. hexamethylmelamine, thiotepa), 'alkyl sulfonates (e.g,
busulfan),
nitrosoureas (e.g. carrnustine which is also known as BCNU, lomustine which is
also known as
io CCNU semustinc which is also known as methyl-CCNU, ehlorozoticin,
streptoaocin), and
triarines (e.g, dicarbazine which is also known as DTIC); (b) Antimetabolites
such as folic acid
analogs (e.g. ~thotrexate), pyrimidine analogs (t.g. S-lluorouraeil
floxt;ridinc, cytarabine, and
azauridine and its prodrug form azaribine), and purinc analogs and related
materials (e.g. 6-
mercaptopurine, 6-thiogusnine, perrtostatin); (c) Natural Products such as the
vinca alkaloids (e.g.
~s .vinbIastine, Vincristine), epipodophylotoxins (e.g. etoposide,
teniposide), antibiotics (.c.g
d~ctinomycin which is also known as actir:omycin-D, daunorubicin, doxorubicin,
bieornycin,
plicamycin, mito:nycin, epirubicin, which is 4-epidoxonthicin, idarubicin
which is 4-
dimcthaxydaunorubicin, and raitoxanthrone), enzymes (.e.g L-asparaginase), and
biological
response modifiers (e.g. Interferon nlfa~); (d) Miscellaneous Agents such as
the platinutti
2o coordination complexes (e.g. cisplatin, carboplatia), substituted areas
(e.g. hydroxycuea),
methylhydiazinc derivatives (e.g. procarbazine), sdreocortical suppressants
(e.g. nutotane,
aminoglutethimide) txucol; (c) Hormones and Antagonists such as
adrenocorticostcroids (e.g.
prednisone or the like), progestins (c.g. hydroxyprogesterone caproatc,
modroxyprogesterone
acetate, megestrol acetate), estxogens (c.g, diethyestilbestrol, ethinyl
estradiol, and the like),
2s aatiestrogens (e.g. tamoxifen), androgens (e.g. testosterone propionate,
fluoxymesterone, and the
like), antiandrogens {e.g. flutamide), and gonadotropin-releasing hormone
analogs (e.g.
leuprolide) and (F) DNA damaging compounds such as udriamycin.
In addition to the methods of manipulating cells, the invention is also useful
for screening
cells such as tumor cells to determine if those cells are susceptible to
cellular division or cellular
3o death, alone or in conjunction with treatment with a chemotherapeutic agent
or other cell signal
and kits for performing these screening assays. The screening method can be
accomplished by
isolating a tumor cell from a subject and exposing the tumor cell to a
chen:otherap~utic agent
AMENDED SHEET
CA 02324995 2000-10-OS
WO 99/53953 PCTNS99/06874
-29-
(preferably several different doses of several different chemotherapeutic
agents can be screened
at a time). Then the presence of a cell death marker can be detected. The
level of the cell death
marker indicates that the cell is susceptible to treatment with a
chemotherapeutic agent.
As used herein a "cell death marker" is a cell surface molecule which
indicates that the
cell is susceptible to cell death. A variety of cell death markers exist but
the preferred cell death
markers useful according to the invention include a Fas molecule on the
surface of the tumor cell,
an MHC class II HLA-DR on the surface of the tumor cell, and a mitochondria)
membrane
potential indicative of cellular coupling. The Fas and MHC molecules can be
detected by using
a detection reagent that bind to the protein, such as an antibody.
to The screening methods are particularly useful for determining if a tumor is
sensitive to
a chemotherapeutic agent. A tumor, however, may initially be sensitive to a
particular
chemotherapeutic agent and then as the therapy progresses the tumor may become
resistant to that
chemotherapeutic agent. The methods of the invention can be used to prevent
the tumor from
becoming sensitive to a chemotherapeutic agent during therapy. The method
involves the steps
of administering to a subject in need of such treatment a chemotherapeutic
agent and a metabolic
modifying agent in a combined amount effective to inhibit growth of the tumor.
The metabolic
modifying agent causes the electron transport and oxidative phosphorylation
processes to be
coupled and therefore effects an increased mitochondria) membrane potential in
the cell. As the
cell is held in this coupled state Fas is expressed on the surface and the
chemotherapeutic agent
2o can stimulate Fas mediated apoptosis. The cells will be prevented from
becoming resistant.
The combined amount of metabolic modifying agent and apoptotic
chemotherapeutic
agent effective to inhibit growth of the tumor cell is that amount is
effective to inhibit the
proliferation of the tumor cell when the mitochondria) membrane potential is
increased. An
effective amount means that amount necessary to delay the onset of, inhibit
the progression of,
halt altogether the onset or progression of or diagnose the particular
condition being treated. In
general, an effective amount for treating a tumor cell is that amount
necessary to halt the
proliferation of the cell. In one embodiment, the effective amount is that
amount necessary to kill
the cell. In general, an effective amount for treating cancer will be that
amount necessary to
favorably affect mammalian cancer cell proliferation in-situ. When
administered to a subject,
3o effective amounts will depend, of course, on the particular condition being
treated; the severity
of the condition; individual patient parameters including age, physical
condition, size and weight;
concurrent treatment; frequency of treatment; and the mode of administration.
These factors are
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-30-
well known to those of ordinary skill in the art and can be addressed with no
more than routine
experimentation. It is preferred generally that a maximum dose be used, that
is, the highest safe
dose according to sound medical judgment.
In some cases the screening assay may indicate that the tumor is mostly
resistant to a
chemotherapeutic agent. Resistant tumors may also be treated by the methods of
the invention.
One aspect of the invention involves the discovery that resistant tumors cells
have a
mitochondria) metabolic state in which electron transport is uncoupled from
oxidative
phosphorylation. It was discovered according to the invention that by altering
the metabolic state
of the tumor cell and thereby causing electron transport to be coupled to
oxidative
phosphorylation it is possible to cause the resistant cell to revert such that
it becomes sensitive
to chemotherapy. The method is performed by administering to the subject an
amount of a
chemotherapeutic agent, and substantially simultaneously therewith an amount
of a metabolic
modifying agent which together are effective for inhibiting growth of the
tumor. The metabolic
modifying agent causes electron transport in the cell to be coupled to
oxidative phosphorylation.
As discussed above once these processes are coupled Fas is expressed on the
surface and the cell
becomes susceptible to apoptosis induced by the chemotherapeutic agent.
Other screening assays can be performed according to the invention to identify
an anti-
tumor drug for killing a tumor cell of a subject. These assays are
accomplished by isolating a
tumor cell from a subject; detecting the presence of a cell death marker
selected from the group
2o consisting of a Fas molecule on the surface of the tumor cell, a B7
molecule on the surface of the
tumor cell, an MHC class II HLA-DR on the surface of the tumor cell, and a
mitochondria)
membrane potential indicative of cellular coupling; exposing the tumor cell to
a putative drug;
and, detecting any change in the presence of the cell death marker to
determine whether the
putative drug is an anti-tumor drug capable of killing the tumor cell of the
subject. This assay
may be performed on one or a plurality of tumor cells and with a single drug
or with a panel of
drugs.
The assay can be performed using routine equipment known in the art. For
instance the
change in the presence of the cell death marker can be detected by contacting
the tumor cell with
a cell death ligand attached to a solid support.
The invention also encompasses kits for screening a subject for susceptibility
to disease.
This kit includes at least a container housing a first binding compound that
selectively binds to
a protein selected from the group consisting of B7-2, B7-1 and MHC class II
HLA-DR; a
CA 02324995 2000-10-OS
WO 99/53953 PCTNS99/068'74
-31 -
container housing a second binding compound that selectively binds to a MHC
class II HLA-
DP/DQ protein; and instructions for determining whether an isolated cell of a
subject selectively
interacts with the first or second binding compound, wherein the presence of
MHC class II HLA-
DP/DQ on the cell surface which interacts with the second compound is
indicative of
susceptibility to atherosclerosis and resistance to autoimmune disease and the
presence of MHC
class II HLA-DR on the cell surface which interacts with the first compound is
indicative of
resistance to atherosclerosis and susceptibility to autoimmune disease.
Other kits include kits for screening a tumor cell of a subject for
susceptibility to treatment
with a chemotherapeutic agent. These kits include a container housing a cell
death marker
1 o detection reagent; and instructions for using the cell death marker
detection reagent for detecting
the presence of a cell death marker selected from the group consisting of a
Fas molecule on the
surface of the tumor cell, an MHC class II HLA-DR on the surface of the tumor
cell, and a
mitochondriai membrane potential indicative of cellular coupling wherein the
presence of the cell
death marker indicates that the cell is susceptible to treatment with a
chemotherapeutic agent.
1 s The kit may also include a container housing a chemotherapeutic agent.
Optionally, the kit may
include a panel of chemotherapeutic agents, housed in separate compartments.
The invention also involves the discovery that mitochondria) metabolic
regulation is
directly related to the expression of immune recognition molecules on a cell
surface. As used
herein "immune recognition molecules" are cell surface proteins which mark a
cell for
2o identification by immune cells. Immune recognition molecules include but
are not limited to
MHC, and in particular MHC class II HLA-DR, B7-1, B7-2 and CD-40. When the
mitochondria)
metabolic status of the cell is such that the electron transport is uncoupled
to oxidative
phosphorylation the cell surface expression of the immune recognition
molecules is increased.
When the mitochondria) metabolic status of the cell is such that the electron
transport is coupled
25 to oxidative phosphorylation the cell surface expression of the immune
recognition molecules is
decreased. Under these conditions, however, the expression of MHC class II HLA-
DP/DQ is
actually increased. For purposes of this patent application MHC class II HLA-
DP/DQ is not
defined as an immune recognition molecule.
Based on these findings the invention encompasses a method for inducing the
expression
30 of immune recognition molecules on a cell surface. The method involves
contacting the cell with
an amount of a metabolic inhibition agent effective to decrease mitochondria)
membrane
potential, wherein a decrease in mitochondria) membrane potential causes
induction of the
CA 02324995 2000-10-OS
WO 99/53953 PCTNS99/06874
-32-
expression of immune recognition molecules on the cell surface.
A "metabolic inhibition agent" as used herein is an agent that causes electron
transport
to become uncoupled from oxidative phosphorylation, and includes for example
apoptotic
chemotherapeutic agents, bacterial byproducts, mycobacterial antigens, UCP
expression vectors,
s and fatty acids.
Diabetes mellitus, which encompasses both Type I (i.e., Insulin Dependent
Diabetes
Mellitus (IDDM)) and Type II (i.e., Non-Insulin Dependent Diabetes Mellitus
(NIDDM)), is
known to affect more than one hundred million individuals worldwide. Although
the exact cause
of diabetes is unclear it is believed that diabetes may arise from any of a
variety of physiological
1 o conditions such as genetic syndromes, viral infections, age related
deterioration of structures
responsible for maintaining the glycemic response, pancreatic disease,
hormonal abnormalities,
certain drugs or chemicals, insulin receptor abnormalities, etc. A "type I
diabetic" is a subject
who has diabetes mellitus caused by a destruction of beta cells in the
pancreas. Type I diabetics
require daily insulin administration which may be reduced but not altogether
eliminated by
~ 5 careful restriction of diet.
Neither the genetic/environmental influences nor the inherent (3 cell
characteristics that
trigger immune-mediated destruction are completely understood. However, two
features that
are pivotal in susceptibility to (3 cell destruction are the expression of the
cell surface molecule
Fas and the metabolic state of the (3 cells. Fas can induce mitosis or
apoptosis depending on
2o the cell and the experimental circumstances. During the prediabetic stage
of Type 1 diabetes,
a (3 cell compensatory hypersecretion of insulin occurs and this process is
accompanied by cell
surface expression of the molecule Fas. When NOD mice, an animal model for
Type 1
diabetes, are crossed with mice having the lpr mutation (Fas deficient), the
animals are resistant
to disease. In addition, destruction of ~i cells in the NOD accelerates when
Fas Ligand is
25 placed on the insulin promotor.
It has been discovered according to the invention that changes in
mitochondria)
metabolic processes that alter mitochondria) membrane potential and Fas
expression contribute
to Fas-induced (3 cell destruction. (3 cell glucose-induced insulin secretion
depends on
increased intracellular ATP. The mitochondria) synthesis of ATP occurs through
coupling of
3o electron transport-dependent oxido-reductive reactions to ATP synthetase
(oxidative
phosphorylation). During this process, a proton gradient is generated by the
pumping of
protons out of the mitochondria increasing mitochondria) membrane potential.
Uncoupling
~C\'. VO~J:~1'.1-~llif:MCHE~\ n4 :'?'?- ;~_ f> CA 02324995 2000-10-O5~'?l)
'?441 t4:) Li) '?~t9:)44E;;:~I S1
U S 009906874
22-05-2000
- 33 -
proteins (UCPs) reversibly uncouple oxidative phnsphorylation from electron
transport
decreasiltg mitochondrial membrane poteniiai. Nomnal pancreatic ~i ceh are in
an uncoupled
state and do not express Fas on their cell surface. As diabetes progresses to
a first stage in which
the patient 1s sick but before the pancreatic j3 aeh art destmyod, the
patients pancreatic ~ cells
become coupled and express Fas on the call stu~acc. The disease then pmgresses
to the stage
when pancreatic (~ cell begin to be'lled. Before_thc cells are killed the
metabolic state changes
agaixl to uncoupled and Fns is still expressed on the surface. When d:e cell
is in an uncoupled
state and Fns is expressed on the cell s,uface the cell is killed as soon as
Fns is engaged without
the need for any ether agents.
o The methods of the invention include a rncthod for inhibiting pancreatic (i
cell death in
a Type I diabcdc by altering the mitochondrial metabolic state. The method is
performed by
contacting a pancreatic p cell of a Type I diabetic with an amount of a
metabolic modifying agent
effective to increase mitochondrial mombrane potential in the pancreatic (i
cell. The rnetabolie
modifying agent causes the panctratic ~ cell to avert to or remain in a
coupled state. Although
is . these cells are not in the normal state of a pancreatic ~ cell, they are
not killed and the patients
organ is not destroyed.
Another method for inhibiting the death of a pancreatic ~ cell in a Type I
diabetic can ba
accomplished by contaetirtb a pancreatic p cull of a Type I diabetic with an
amount of a Pas
binding agent effective to inhibit selective engagement of Fns on the surface
of the pancreatic a
2o cell. By inhibiting the selective engagement of Fns on the cell surface and
allowing the cell to
remain in the uncoupled state the cell will remain healthy and have the
phenotype of a normal
pancreatic ~3 cell.
?he Fns binding agents which are useful according to the invention are those
molecules
which bind to Fns t do not activate it. Fns binding agents can be identified
by scn~ning
tttlv~4~c~
2s libraries using the ex~sllt~ regions of Fas, such as the screening methods
described below.
Fas binding agents then can easily be tested without undue experimentation in
vitro for their
ability to bind Fuss but not induce cell death in uncoupled cells. Uncoupled
cells can be prc;pared
according to the methods described above. Fas can be induced to ba expressed
oa the surface of
cells using; irradiulion ~ has previously been identified in tl~c; prior art.
Uncc uncoupled ccEls
3o expressing Fas have been developed potential Fns binding afients can be
incubated with these
cells and cell lysis can be assayed by the methods described herein or by
other methods known
in the art.
AMENDED SHEET
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-34-
The invention is also useful for treating type II diabetics. A "type II
diabetic" is a subject
who has diabetes mellitus caused by abnormal insulin secretion and/or
resistance to insulin action
in target tissues. The physiological problem which occurs in a Type II
diabetic is very different
than that which occurs in a type I diabetic. In type II diabetes the
pancreatic ~3 cells undergo
excessive proliferation. It is desirable to inhibit proliferation of these
cells.
One method for inducing pancreatic j3 cell death in a Type II diabetic
involves the step
of contacting a pancreatic ~i cell of a Type II diabetic with an amount of an
MHC class II HLA-
DR inducing agent effective to induce the expression of the MHC class II HLA-
DR on the surface
of the pancreatic ~i cell, and selectively engaging the MHC class II HLA-DR by
contacting the
l0 cell with an MHC class II HLA-DR ligand effective to induce pancreatic ~3
cell death.
Another finding according to the invention was that mitochondria) metabolism
and the
related expression of MHC class II on the surface of a cell is indicative of
the susceptibility of the
host of that cell to developing atherosclerosis, autoimmune disease or
multiple sclerosis. When
electron transport and oxidative phosphorylation are in a coupled state in a
cell the cell expresses
MHC class II HLA-DP/DQ on the surface. When electron transport and oxidative
phosphorylation are in an uncoupled state in a cell the cell expresses MHC
class II HLA-DR on
the surface. A cell in a coupled state that has MHC class II HLA-DP/DQ on the
surface will be
stimulated to divide when the MHC class II HLA-DP/DQ is engaged. A cell in an
uncoupled
state that has MHC class II HLA-DR on the surface will be stimulated to lyse
when the MHC
2o class II HLA-DR is engaged.
These different metabolic states of the cell have been found according to the
invention to
be predictive of an individuals susceptibility to developing disease. When the
cells of a subject
are coupled and express MHC class II HLA-DP/DQ on the surface the subject is
susceptible to
developing atherosclerosis. When the cells of a subject are uncoupled and
express MHC class
II HLA-DR on the surface the subject is susceptible to developing autoimmune
disease.
The invention encompasses methods for screening a subject for susceptibility
to
atherosclerosis. These methods involve the steps of isolating a cell which is
useful for screening
such as a peripheral blood lymphocyte or a skin cell from a subject and
detecting the presence of
an MHC marker selected from the group consisting of an MHC class II HLA-DP/DQ,
B7-2, B7-1
3o and MHC class II HLA-DR on the surface of peripheral blood lymphocyte,
wherein the presence
of MHC class II HLA-DP/DQ is indicative of susceptibility to atherosclerosis
and the presence
of MHC class II HLA-DR is indicative of resistance to atherosclerosis.
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-35-
Atherosclerosis is a group of diseases affecting the cardiovascular system and
includes
myocardial infarction, stroke, angina pectoris and peripheral cardiovascular
disease. Despite
significant advices in therapy, cardiovascular disease remains the single most
common cause of
morbidity and mortality in the developed world. Many individuals are
susceptible to developing
future cardiovascular disorders, and this susceptibility has usually been
defined in terms of risk
factors such as family history of premature ischemic heart disease,
hyperlipidemia, cigarette
smoking, hypertension, low HDL cholesterol, diabetes mellitus,
hyperinsulinemia, abdominal
obesity, and high lipoprotein. The invention includes a new method for
determining an
individuals susceptibility to developing atherosclerosis. As used herein
susceptibility to
1o atherosclerosis indicates a likelihood of 10% greater than the average of
developing
atherosclerosis.
The invention also encompasses methods for screening a subject for
susceptibility to
autoimmune disease. These methods involve the steps of isolating a peripheral
blood lymphocyte
from a subject and detecting the presence of an MHC marker selected from the
group consisting
of an MHC class II HLA-DP/DQ, B7-2, B7-l and MHC class II HLA-DR on the
surface of
peripheral blood lymphocyte, wherein the presence of MHC class II HLA-DR is
indicative of
susceptibility to autoimmune disease and the presence of MHC class II HLA-
DP/DQ is indicative
of resistance to autoimmune disease.
Autoimmune disease is a class of diseases in which an individuals own
antibodies react
2o with host tissue or in which immune effector T cells are autoreactive to
endogenous self peptides
and cause destruction of tissue. It is well established that MHC class II
alleles act as major
genetic elements in susceptibility to a variety of autoimmune diseases. These
include
rheumatoid arthritis, celiac disease, pemphigus vulgaris, and the prototype
for autoimmune
disease, systemic lupus erythematosus (SLE). The invention includes a new
method for
determining an individuals susceptibility to developing autoimmune disease. As
used herein
susceptibility to Autoimmune disease indicates a likelihood of 10% greater
than the average of
developing autoimmune disease.
The methods of the invention also include methods for treating a subject
having
autoimmune disease to reduce associated cell death. One method is based on the
interaction
between cells expressing MHC class II HLA-DR and y8 T cells. y8 T cells
specifically recognize
MHC class II HLA-DR on the surface of the cell and stimulate cell death. When
the 'y8 T cells
recognize a tissue having significant amounts of MHC class II HLA-DR these T
cells become
:c:V. Vov:FPA-MtiL?NCfiF.:v U4 ;~_r,_ 5_ (~ ;CA 02324995 2000-10-OSt2() '-'-
44!-. +4.~ 8:i '?39~~~t4R~:#m
U S 009906874
22-05-2000
-36-
activated and proliferate in order to kill more of the recognized cells. The
methods of treatment
arc based on the concept of eliminating the activated Y& T cells frem the
body. These cells eau
be removed by isolating a sample of peripheral blood and identifying the
activated r8 T cells by
assessing activation markers using flow eytometry. Antibodies can then be
generated to the
specific activated y~ T cells and the antibodies can be used to selectively
bind to and inactivate
78 cells in the subject. This inactivation of the Y& cells inhibits cell death
associated with
autoimmune disease.
Similarly cells expressing cell surface MHC class II HLA-DR that arc
ordinarily
recognized and killed by y8 T cells can be used for the treatment of diseases
involving excessive
t o cell proliferation such as glioma. The cells can be induced to undergo
cell death by stimulating
excess activated y8 T tills in the subject. 'Ibis can be accomplished using
bacterial byproducts.
It has been found according to the invention that a link exists between Fns
expression,
mitochondriai metabolism, and susceptibility to Fns-degendeut cell death. Thus
by regulating
rnitachondrial metabolism it is possible to control susceptibility to Fas
dependent cell death. This
is .phenomenon is described below with respect to pancreatic ~i cells, but is
applicable to all
biological systems described herein.
Type I diabetes mellitus (DM) a a pancreatic ~i cell-selective autoirn:nune
disease which
results in insulin deficiency. Neither the genetirJcnvironmental influences
nor the inherent ~i cell
cl~racteristics that truer immune-mediated destruction are completely
understood. Apoptosis
2o has been suggested as the mechanism of (3 cell death in mouse models of
Type I diabetes. Two
features that correlate with susceptibility to ~i cell destruction arc the
metabolic state of the ~3 cells
and expression of the cell surface molecule Fns (CZ795), a member of the TNF
f8mily of "death
inducing" roceptorltigand pairs. During the prediabetic stage of Type I DM, a
~i cell glucose-
depeadent ~f Seu'~~a f insulin occurs iu response to high glucose
concentrations and this
25 process is coincident with the cell surface expression of Fns. When NOD
mice arc crossed with
mice having the fpr mutation (Fns deficient), the rutimals arc resistant to
disease. In addition,
destruction of ~i cells in the NOD accelerates when Fns ligand is placed on
the insulin promoter.
In the N4D model, apoptotic a cells have been observed in the islets at 15
weeks of age which
coincides with the earliest onset of diabetes as determined by blood glucose,
urine glucose, and
3o pancreatic inununoresctive insulin measurements. The incidence of apoptosis
decreases by week
18 at which time 50% of the animals have overt diabetes. Virtually all of the
apoptotic cells have
been determined immunohistochemic~lty to be positive for insulin production.
Interestingly,
AMENDED SHEET
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-37-
apoptosis of ~i cells precedes the appearance of T cells in islets. The
ability to upregulate Fas
expression on ~3 cells is also acquired during the early stages of Type I DM.
It is believed according to the invention that the metabolic state of the ~i
cell determines
the susceptibility of ~i cells to Fas mediated death. (3 cell glucose-induced
insulin secretion
depends upon increased intracellular ATP. The mitochondria) synthesis of ATP
results from the
coupling of electron transport-dependent oxido-reductive reactions to ATP
synthetase (oxidative
phosphorylation). During this process, a proton gradient is generated by the
pumping of protons
across the mitochondria) membrane resulting in an increase in mitochondria)
membrane potential.
Uncoupling proteins (UCP) can reversibly dissipate the proton gradient
resulting in decreased
membrane potential. Mitochondria) damage, resulting from viruses,
inflammation, age, or
oxidative stress, can also dissipate the proton gradient and decrease the
mitochondria) membrane
potential. However, in the latter case, the change in mitochondria) metabolism
is irreversible.
For example, increased intracellular NO production in ~i cells is known to
alter (3 cell
mitochondria) membrane potential and sensitize ~i cells to Fas-induced death.
Our data (provided
in the Examples below) demonstrate that ~3 cells express intracellular UCP.
Furthermore, we have
shown that ~i cell surface Fas expression and mitochondria) membrane potential
increase as a
function of environmental glucose concentration. Taken together, these results
are consistent
with the notion that mitochondria) glucose metabolism and consequent
mitochondria) membrane
potential play a critical regulatory role in susceptibility to Fas-induced ~3
cell death.
2o Increasing environmental glucose results in increased cell surface Fas
expression and
functionally coupled mitochondria) ATP synthesis, suggesting a link between
mitochondria)
glucose metabolism and susceptibility to Fas-induced cell death. ATP is
required for insulin
secretion. As glucose levels decrease, levels of cell surface Fas decrease,
newly synthesized Fas
is stored intracellularly and mitochondria) ATP synthesis is uncoupled from
respiration and less
mitochondria) ATP is produced. This is demonstrated schematically in Figure 1.
The
reversibility of this process may account for the pulsatility of insulin
secretion in response to
nutrients. In either state, coupled or uncoupled, damaging agents such as
diabetogenic viruses,
inflammation, ischemia, age, or oxidative stress, may damage mitochondria)
metabolism, increase
cell surface Fas expression, and render the cells susceptible to Fas-induced
apoptosis or oncosis,
3o respectively. One possibility is that during the insulitis phase of Type I
DM, apoptosis (on the
right of the panel), which is thought to occur "silently" without additional
inflammation, occurs
to some of the ~i cells and that oncosis occurs in later stages of disease
resulting from T cell
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-38-
mediated (Fast dependent) ~i cell destruction.
The invention in other aspects relates to methods for selectively killing a
Fas ligand
bearing tumor cell. The method involves the steps of contacting the Fas ligand
bearing tumor cell
with acetate in an amount effective to induce Fas associated cell death. A Fas
ligand bearing
tumor cell is any tumor cell which inducibly or constitutively expressed a Fas
ligand on the cell
surface. Such cells can easily be identified by those of skill in the art
since the Fas ligand is a
well known molecule. These cells include but are not limited to melanoma cells
and colon
carcinoma cells.
Although acetate alone is sufficient to kill a Fas ligand bearing tumor cell,
the cell can
1 o also be treated with a chemotherapeutic agent and/or a Fas ligand to
promotes killing. The use
of these secondary compounds allows the use of less of the acetate to be used
to accomplish the
cell killing. The combination of acetate and chemotherapeutic agents and or
Fas ligands, allows
less of all three reagents to be used than would otherwise be required to kill
the cell.
Additionally, tumor cells that do not express cell surface Fas ligand can also
be killed by
~ 5 the methods of the invention. This killing can be accomplished by
contacting the tumor cell with
acetate in an amount effective to induce cell surface Fas expression, and
administering a Fas
ligand to the tumor cell in an amount effective to induce Fas associated cell
death. Fas ligands
are expressed on the surface of NK yS T cells, CD4 T cells, CD8 T cells, etc.
Other methods for selectively killing a cell include contacting the cell with
a nucleic acid
20 selected form the group consisting of a UCP anti-sense nucleic acid and a
UCP dominant-
negative nucleic acid in an amount effect to inhibit UCP function. A cell can
also be killed
according to the invention by contacting the cell with a compound selected
from the group
consisting of acetate and GDP and an apoptopic chemotherapeutic agent in an
amount effective
to kill the cell.
25 The invention also encompasses methods for promoting a Thl immune response.
The
method is performed by administering to a subject who has been exposed to an
antigen an
effective amount for inducing a Th 1 immune response of a MHC class II HLA-DR
inducing agent
to induce DR on a T cell. MHC class II HLA-DR inducing agents are discussed in
detail above,
and include, for instance, fatty acids.
30 The invention in another aspect is a method for inducing nerve cell
differentiation by
contacting a nerve cell with an amount of a B7 inducing agent effective to
induce the expression
of B7 on the surface of the nerve cell and exposing the nerve cell to a neural
activating cell to
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-39-
cause differentiation of the nerve cell.
The complex process of immune cell activation and proliferation is based on
diverse
interactions such as antigen presentation, cell-cell contact and soluble
immune mediators e.g.,
cytokines or lymphokines. Many of these interactions are mediated in T- and
other immune cells
through surface receptors. T helper cells, for example, require for activation
both the presentation
of an antigen by an antigen presenting cell (APC) in association with major
histocompatibility
complex (MHC) and a secondary signal. The secondary signal may be a soluble
factor or may
involve an interaction with another set of receptors on the surface of T- and
other immune cells.
Antigen presentation in the absence of the secondary signal, however, is not
sufficient to activate
T helper cells.
The CTLA-4/CD28B7 system is a group of proteins involved in regulating T-cell
proliferation through this secondary signaling pathway. The T-cell
proliferative response is
controlled by the interaction of the B7 family of proteins, which are
expressed on the surface of
APCs, with CTLA-4 (cytotoxic T lymphocyte antigen #4) and CD28.
15 The B7 family of proteins is composed of structurally related glycoproteins
including B7-
1, B7-2, and B7-3 (Galea-Lauri et al., Cancer Gene Therapy, v. 3, p. 202-213
(1996); Boussiotis,
et al., Proc. Nat. Acad. Sci. USA, v. 90, p.l 1059-11063 (1993)). The
different B7 proteins appear
to have different expression patterns on the surface of antigen presenting
cells. For example B7-2
is constitutively expressed on the surface of monocytes, whereas B7-1 is not,
although B7-1
2o expression is induced in these cells when the cells are stimulated with
interferon gamma (IFN-y).
The different expression patterns may indicate a different role for each of
the B7 family members.
The B7 proteins are believed to be involved in the events relating to
stimulation of an immune
response by its ability to interact with various immune cell surface
receptors. It is believed, for
example, that B7 plays a role in augmenting T-cell proliferation and cytokine
production through
25 its interaction with the CD28 receptor.
CD28, a homodimeric glycoprotein having two disulfide linked 44-kd subunits,
is found
on 95% of CD4+ and 50% of CD8+ cells. Studies using monoclonal antibodies
reactive with
CD28 have demonstrated that CD28 is involved in a secondary signal pathway in
the activation
of T-cell proliferation. Antibodies which block the interaction of CD28 with
its ligand have been
3o found to inhibit T-cell proliferation in vitro resulting in antigen
specific T cell anergy. (Handing
et al., Nature, v. 356, p. 607 (1991)).
Recently a T-cell surface receptor protein, CTLA-4, having approximately 20%
sequence
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-40-
homology to CD28 was identified. Although CTLA-4 is not endogenously expressed
on T-cell
surfaces, its expression is induced when CD28 interacts with B7 on the surface
of an APC. Once
CTLA-4 is expressed on the surface of the T-cell it is capable of interacting
with B7.
It was discovered according to one aspect of the invention that nerve cells
can be induced
to express B7 and can interact with T- and other immune cells through
B7/CD28/CTLA4
molecules. The B7 on the nerve cell surface can engage the CD28/CTLA4 on the
immune cell
surface to co-stimulate the immune cell, leading to activation of the immune
cell. The activated
immune cell then releases nerve growth factor which stimulates the nerve cell.
As used herein "B7 inducing agent" is an agent which causes B7 ( and other
related family
members retaining sequence homology with B7) to be expressed on a nerve cell
surface. In one
preferred embodiment the B7 inducing agent is a pharmacological agent that
causes dissipation
of proton motor force such as by uncoupling of electron transport and
oxidative phosphorylation,
resulting in reduced mitochondria) membrane potential within the cell. B7
inducing agents which
cause dissipation of the proton motor force include but are not limited to
adriamycin, gamma
interferon, bacterial byproducts such as lipopolysaccharides, lipoproteins
BCG, fatty acids, cAMP
inducing agents and a UCP expression vector. A "CAMP inducing agent" as used
herein is any
compound which elevates intracellular levels of cAMP. Such compounds include
but are not
limited to isoproterenol, epinephrine, norepinephrine, phosphodiester
inhibitors, theophylline, and
caffeine. In another preferred embodiment the B7 inducing agent is a B7
expression vector.
2o Such a vector can be stably expressed in the nerve cell to produce B7 which
can be expressed on
the cell surface. The B7 inducing agent is an isolated molecule. An isolated
molecule is one
which has been removed from its natural surroundings and formulated for
administration to an
organism.
An "amount of a B7 inducing agent effective to induce the expression of B7 on
the surface
of the nerve cell" as used herein, refers to an amount which is effective to
cause dissipation of a
proton motor force and thus to decrease the mitochondria) membrane potential
in the nerve cell.
Preferably the amount is that amount which is necessary to induce the
expression of at least a
single B7 molecule on the cell surface.
The nerve cell is contacted with the B7 inducing agent to cause expression of
B7 on the
3o surface. As used herein, the step of contacting the cell with B7 inducing
agent can be performed
by any means known in the art. For instance, if the B7 inducing agent is
applied in vitro, it may
simply be added as part of the cellular medium to a tissue culture dish of
nerve cells. If the
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-41 -
method is performed in vivo, then the step of contacting may be performed by
administering the
B7 inducing agent by commonly used therapeutic techniques, such as parenteral
administration,
oral administration, or local administration. Other methods are well known to
those of ordinary
skill in the art.
s According to a method of the invention the B7 expressing nerve cell is
exposed to a neural
activating cell. A "neural activating cell" as used herein, is a cell which is
capable of producing
nerve growth factor when activated and which includes a cell surface B7
receptor. As mentioned
above, B7 receptors include CD28 and CTLA-4. Many cells which are the neural
activating cells
of the invention have been described in the prior art. These cells include,
for example, T cells
(including both gamma, delta and alpha-beta T cells), macrophage, dendritic
cells, CTLA-4 or
CD-28 expressing B cells.
A "B7 receptor" as used herein is a cell surface immune molecule which
interacts with
B7 on a partner cell and cases activation of the cell on which it is
expressed. Preferably the B7
receptor is a CD28 molecule or a CTLA4 molecule.
The nerve cell is exposed to the neural activating cell to cause
differentiation of the nerve
cell. The step of exposing can be performed in vitro, by simply mixing the two
populations of
cells, the nerve cell and the neural activating cell. It can be accomplished
in vivo by causing the
accumulation of the neural activating cells in the local environment of the
nerve cell. For
instance, the neural activating cells may be implanted, or the local
environment may be
2o manipulated to cause accumulation of the neural activating cell. For
instance, stimulating an
immune response in the local environment would cause the accumulation of T
cells, B cells,
dendritic cells and macrophage. The neural activating cell may also be a cell
which produces
nerve growth factor upon activation and which is engineered to express a B7
receptor on its
surface, e.g. by transfection with an inducible or constitutively expressed B7
receptor gene, such
as by the methods described above.
The methods of the invention in some aspects may also be performed using an
endogenous neural activating cell. For instance the endogenous neural
activating cell may be a
cell having a cell surface B7 receptor, such as CD28 and CTLA-4. In this case
the method would
only include the step of contacting a nerve cell with an amount of a B7
inducing agent effective
to induce the expression of B7 on the surface of the nerve cell in the
presence of a neural
activating cell.
When the neural activating cell is a cell having a cell surface B7 receptor
which is already
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
- 42 -
present in interactive proximity to the B7, the cell does not have to be
manually brought into
contact with the B7 on the nerve cell.
When the nerve cell is exposed to a neural activating cell the cell surface B7
can interact
with the B7 receptor to activate the neural activating cell. Once activated,
the neural activating
s cell produces and releases nerve growth factor into the local environment.
This locally produced
nerve growth factor is capable of causing the nerve cell to become
differentiated. Although the
invention is not limited to a specific mechanism of action, applicants believe
that the mechanism
through which neuro-differentiation occurs is that the nerve growth factor
interacts with the nerve
cell surface nerve growth factor receptor such as Trk. It is also believed
that engagement of the
B7 on the cell surface or the induction thereof causes the expression of nerve
growth factor
receptors on the surface of the nerve.
In one embodiment of the invention, the receptors for nerve growth factor may
be induced
to be expressed on the surface of the nerve cell. Two known nerve growth
factors are tyrosine,
kinase A (TrkA) and p75NGRF. When these receptors interact with nerve growth
factor on the
15 surface of a nerve cell, it stimulates the cell to undergo neuronal
differentiation. Expression of
these receptors on the surface of the nerve cell may be performed by any
method known in the
art. For instance, the nerve cell may be recombinantly engineered to
constitutively or inducibly
express the DNA for these receptors, such as by the methods described above.
Nerve growth factor {NGF), originally described by Levi-Montalcini and
Hamburger in
20 1953 (Levi-Montalcini and Hamburger, 1953), contains two copies of three
types of polypeptides
designated a, ~3 and y and exhibits approximately 50% of homology with other
neurotrophins i.e.,
brain-derived neurotrophic factor (BDNF), NT-3, NT-4 and NT-5 (Siegel et al.,
1994). It binds
to tyrosine kinase A (TrkA) and p75NGF receptors in a synergistic manner
(Canossa et al., 1996).
Tyrosine kinase B (TrkB) and tyrosine kinase C {TrkC) receptors preferentially
bind BDNf and
2s NT-3 respectively (Siegel et al., 1994). Intracellular signal proteins via
Src homology 2 (SH20
domain interactions such as phospholipase C-y and the p85 sub-unit of
phosphatidyl-inositol 3-
kinase bind to the tyrosine-phosphorylated receptors and allow multimeric
protein complexes to
form and lead to the activation of specific signal transduction pathways
(Hempstead et al., 1994).
As shown in the Examples below, nerve cells express molecules which are
requisite for
3o T cell activation, indicating that there is a neuro-immunological
intercellular interactive
component that occurs during neuronal differentiation. NGF and EGF have
profound effects on
the differentiation process in utero and early life and on the regeneration
process after pathologic
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-43-
damage. The data provided in the examples is relevant since it not only
demonstrates the
existence of inducible surface molecules on post-mitotic neurons, but their
ability to be kinetically
modified by the presence or absence of specific trophic factors is also
highlighted. The presence
of Fas on the neuronal cell surface suggests that PC 12 cells and their
variants are vulnerable to
apoptosis or that the molecule is capable of transmitting a mitotic signal if
required.
Another aspect of the invention involves a method for inducing apoptosis in a
nerve cell.
The method involves the step of contacting a nerve cell with an amount of a
metabolic modifying
agent which when exposed to a nerve cell causes coupling of electron transport
and oxidative
phosphorylation effective to increase the mitochondria) membrane potential in
the nerve cell and
1 o contacting a neural activating cell with an amount of a B7 receptor
blocking agent effective for
inducing apoptosis in the nerve cell. "Metabolic modifying agents" and "Fas
binding agents"
are discussed above.
A "B7 receptor blocking agent" as used herein is any agent which interacts
with a B7
receptor but does not cause activation of the cell and prevents that receptor
from binding to B7.
These agents include, for example, but are not limited to anti-CD28
antibodies, CD28 binding
peptides, anti-CTLA-4 antibodies, CTLA-4 analogs and CTLA-4 binding peptides
which do not
cause activation of the receptor. Other B7 receptor blocking agents can be
identified by those of
skill in the art by routine experimentation using immune cell activation
assays such as a T cell
activation assay.
2o This method is useful whenever it is desirable to induce apoptosis of a
nerve cell. For
instance, it may be useful to induce apoptosis of a nerve cell in vitro in
order to screen molecules
for their ability to prevent apoptosis of nerve cells. Other uses will be
apparent to those of
ordinary skill in the art.
As discussed above, when a cell is coupled, Fas is expressed on the cell
surface and when
2s a cell is uncoupled Fas generally is transported to intracellular stores.
When a cell is coupled and
Fas is on the surface engagement of Fas sends a signal to the cell instructing
the cell to undergo
cellular division. When a cell is uncoupled ordinarily Fas is not expressed on
the cell surface.
In the presence of NGF, however, Fas is down regulated and is no longer
expressed on the cell
surface. In a damaged tissue if a nerve cell is in an uncoupled state, and
expresses both Fas and
3o B7 on the surface, then the presence or absence of NGF will determine the
fate of the cell. If an
NGF producing cell according to the invention is present in the local
environment, the B7 of the
nerve cell will stimulate production of NGF by interacting with that cell. The
local NGF
CA 02324995 2000-10-OS
WO 99/53953 PCTNS99/06874
-44-
produced will cause the down regulation of Fas and the cell will undergo
differentiation. If an
NGF producing cell is not available or if B7 is not expressed on the surface
of the nerve cell, then
environmental factors can stimulate Fas to cause apoptosis.
Another aspect of the invention is a method for reinnervating an injured
tissue. The
method involves the step of implanting a B7 expressing nerve cell in the
injured tissue, wherein
the implanted B7 expressing nerve cell will undergo neuronal differentiation
in the presence of
a neural activating cell in the injured tissue to reinnervate the injured
tissue. Methods are known
in the art implanting nerve cells into living tissue. For example, nerves can
be implanted directly
into exposed tissue or may be implanted in biodegradable tubes which will
guide the extension
1 o of the nerve into surrounding tissue where it can be differentiated.
A B7 expressing nerve cell can be prepared by an means known in the art. For
instance,
a B7 expressing nerve cell may be genetically engineered to constitutively or
inducibly express
B7. The gene encoding a B7 protein can be constitutively expressed in a nerve
cell by
transfection procedures known in the art, such as by the methods described
above. B71 gene is
provided herein as SEQ ID No. 1 and listed under Accession No. M27533 in
Genebank and the
nucleic acid sequence for B72 is provided herein as SEQ ID No. 2 and listed
under Accession No.
U04343 in Genebank. Alternatively the nerve cell may be engineered to
inducibly or
constitutively express UCP which will induce expression of endogenous B7. In
another
embodiment, the implanted B7 expressing nerve cell may also constitutively or
inducibly express
2o at least one of the nerve growth factor receptors, which would induce
expression of endogenous
B7.
An injured tissue is a tissue in which nerve damage has been sustained. An
injured tissue
may include for example, a spinal chord injury, a severed or severely damaged
limb or any other
tissue which can be innervated and in which the nerve has been damaged. Neural
activating cells
are generally found in skin and muscle surrounding the nerves of an injured
tissue. These neural
activating cells can stimulate the differentiation of the nerve cell once they
are activated by
interaction with the B7 on the surface of the nerve cell.
The invention also includes a method for treating a neurodegenerative disorder
by
administering an amount of a B7 inducing agent effective to induce the
expression of B7 on the
3o surface of a nerve cell. An amount that is effective to induce the
expression on the surface is an
amount which is effective to cause dissipation of a proton motor force and
thus to decrease the
mitochondria) membrane potential in the nerve cell.
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
- 45 -
A "neurodegenerative disorder" as used herein, is a disorder associated with
the death or
injury of neuronal cells. For example, the loss of dopaminergic neurons in the
substantia nigra
ultimately leads to Parkinson's Disease. The deposition of ~3-amyloid protein
in the brain
generally causes neural damage leading to Alzheimer's Disease. Conditions
involving injuries
such as brain ischemia, spinal chord damage, and severance of limbs often
causes extensive
neuronal cell death. When a nerve is severed, the regions of the nerve cells
which are distal to
the severance become separated from the nerve cell body and degenerate. After
such a severance,
it is possible for the nerve cell body to regenerate by re-extension of the
severed axons. This
process of nerve regeneration does not occur naturally in the absence of
certain environmental
conditions. In some cases in the prior art, various factors such as nerve
growth factor have been
added to the nerve to attempt to stimulate the regeneration. The methods of
the invention
describe a different system in which the nerve cell is manipulated to express
an immune
recognition molecule on its surface which can then cause the local expression
of nerve growth
factor leading to differentiation. This method more closely simulates the
natural processes of
neuronal regeneration. Other neurodegenerative diseases include for example
but are not limited
to epileptic seizures and amyotrophic lateral sclerosis.
The invention also includes compositions of the above described agents. One
composition
of the invention includes a metabolic modifying agent and an apoptotic
chemotherapeutic agent.
The pharmaceutical preparations of the invention are administered to subjects
in effective
2o amounts. An effective amount means that amount necessary to delay the onset
of, inhibit the
progression of, halt altogether the onset or progression of or diagnose the
particular condition
being treated. In one embodiment the ",Prahr,~;~ f".,.~;~,;".. ".o.,. ,...a .,-
.. ..____.__
chemotherapeutic agent are present in an effective dose for treating a tumor.
In another
embodiment the metabolic modifying agent and the apoptotic chemotherapeutic
agent are present
in an effective dose for treating type II diabetes. In general, an effective
amount for treating
cancer and type I diabetes will be that amount necessary to favorably affect
mammalian cell
proliferation in-situ. When administered to a subject, effective amounts will
depend, of course,
on the particular condition being treated; the severity of the condition;
individual patient
parameters including age, physical condition, size and weight; concurrent
treatment; frequency
of treatment; and the mode of administration. These factors are well known to
those of ordinary
skill in the art and can be addressed with no more than routine
experimentation. It is preferred
generally that a maximum dose be used, that is, the highest safe dose
according to sound medical
«v. vw:t:eA-vll~E:VCHI=a ~!4 :''2- p- a CA 02324995 2000-10-O5~'-'0 ~-'4~'1-
+'t't t3~d 2:~~'l~~t>;:~+t t
- US 009906874
22-05-2000
-46-
judgment.
Anotteer composition according to the invention is an MHC class II HLA-DR
inducing
agent and an MHC class II HLA-DR Iigaad. In one embodiment the NfHC class II
HLA-DR
inducing agent and MHC class II HI,A-DR tigand are present in an cflective
dose for treating type
II diabetes. In general, an effective amount for treating type iI diabetes
will be that amount
necessary to favorably affect trtarnmaiian cell proliferation in-situ. When
admiriistcred to a
subject, ei~cctive amounts will depend, of course, on the particular condition
being treated; the
severity of the condition; individual patient paratnetcrs including age,
physical condition, size and
weight; concurrent treatment; frequency of treatment; and the mode of
administration. These
t o factors are well known to those of ordinary skill in the art and can be
addc~ess~d with no more than
routine experimentation. It is preferred generally that a maximum dose be
used, that is, the
highest safe dose according to sound medical judgment.
Ono composition of the invention is a H7 inducing agent and a B7 receptor
inducing
agent. In one embodiment the B7 inducing agent and H7 receptor inducing agent
present in an
t s effective dose for treating neurodcgenerative disease. In general, an
effective amount for
neurodegencrative disease will be that amount necessary to favorably affect
nerve cell
differentiation in-situ. When administered to a subject, effective.amounts
will depend, of cotusc,
on the particular condition being treated; the severity of the condition;
individual patient
parameters including age, physical condition, size and weight; concurrent
treatment; frequency
2p of treatment; and the mode of administration. These fFtCtOrS are well known
to those of ordinary
skill in the art and can be addressed with no more than mutinc
experimentation. It is preferred
generally that a maximum dose be used, that is, the highest safe dose
according to sound medical
judgment.
Generally, doses of active compounds will be fmon about Q.Olmg/kg per day to
1000
3s tng/ks per day. It is expected that doses rmge of 50-S00 mg/kg will be
suitable, in one or several
administrations per day. In the event that a response in a subj act is
insufFcient at the initial doses
applied, hi6her doses (or effectively higher doses by a different, more
localized delivery route)
may be employed to zhc extent that patient tolerance permits. Multiple doses
per day arc
contemplated to achieve appropriate levels of compounds. ~o~ p ~~l es
so . The invention involves the use of several different types of binding
peptides or-rnofecaefs,
MHC class II HLA-DR binding peptides, CD4la(iTCR binding molecules, CD40
binding
pOEpides, MI3C class II I~LA-DP/DQ binding peptides, CD28/CTLA4 binding
peptides, and Fns
pQp~;~es
AMENDED SHEET
CA 02324995 2000-10-OS
WO 99/53953 PCTNS99/06874
-47-
biding peptides. The binding peptides of the invention can be identified using
routine assays,
such as the binding and activation assays described in the Examples and
elsewhere throughout
this patent application.
The binding peptides of the invention are isolated peptides. As used herein,
with respect
s to peptides, the term "isolated peptides" means that the peptides are
substantially pure and are
essentially free of other substances with which they may be found in nature or
in vivo systems to
an extent practical and appropriate for their intended use. In particular, the
peptides are
sufficiently pure and are su~ciently free from other biological constituents
of their hosts cells
so as to be useful in, for example, producing pharmaceutical preparations or
sequencing. Because
an isolated peptide of the invention may be admixed with a pharmaceutically
acceptable carrier
in a pharmaceutical preparation, the peptide may comprise only a small
percentage by weight of
the preparation. The peptide is nonetheless substantially pure in that it has
been substantially
separated from the substances with which it may be associated in living
systems.
The binding peptides also may easily be synthesized or produced by recombinant
means
15 by those of skill in the art. Methods for preparing or identifying peptides
which bind to a
particular target are well known in the art. Molecular imprinting, for
instance, may be used for
the de novo construction of macromolecular structures such as peptides which
bind to a particular
molecule. See for example Kenneth J. Shea, Molecular Imprinting of Synthetic
Network
Polymers: The De Novo synthesis of Macromolecular Binding and Catalytic Sites,
TRIP Vol.
2o 2, No. 5, May 1994; Klaus Mosbach, Molecular Imprinting, Trends in Biochem.
Sci., 19(9)
January 1994; and Wulff, G., in Polymeric Reagents and Catalysts (Ford, W. T.,
Ed.) ACS
Symposium Series No. 308, pp 186-230, American Chemical Society (1986). One
method for
preparing mimics of the known binding peptides involves the steps of (i)
polymerization of
functional monomers around a known binding peptide or the binding region of an
antibody
2s which also binds to the targets (the template) that exhibits a desired
activity; (ii) removal of the
template molecule; and then (iii) polymerization of a second class of monomers
in the void left
by the template, to provide a new molecule which exhibits one or more desired
properties which
are similar to that of the template. In addition to preparing peptides in this
manner other binding
molecules which have the same function as the binding peptides useful
according to the invention
3o such as polysaccharides, nucleosides, drugs, nucleoproteins, lipoproteins,
carbohydrates,
glycoproteins, steroids, lipids, and other biologically active materials can
also be prepared. This
method is useful for designing a wide variety of biological mimics that are
more stable than their
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-48-
natural counterparts, because they are typically prepared by the free radical
polymerization of
functional monomers, resulting in a compound with a nonbiodegradable backbone.
Other
methods for designing such molecules include for example drug design based on
structure activity
relationships which require the synthesis and evaluation of a number of
compounds and molecular
modeling.
The binding peptides may also be identified by conventional screening methods
such as
phage display procedures (e.g., methods described in Hart, et al., J. Biol.
Chem. 269:12468
(1994)). Hart et al. report a filamentous phage display library for
identifying novel peptide
ligands for mammalian cell receptors. In general, phage display libraries
using, e.g., M13 or fd
1 o phage, are prepared using conventional procedures such as those described
in the foregoing
reference. The libraries display inserts containing from 4 to 80 amino acid
residues. The inserts
optionally represent a completely degenerate or a biased array of peptides.
Ligands having the
appropriate binding properties are obtained by selecting those phages which
express on their
surface a ligand that binds to the target molecule. These phages then are
subjected to several
~ 5 cycles of reselection to identify the peptide ligand-expressing phages
that have the most useful
binding characteristics. Typically, phages that exhibit the best binding
characteristics (e.g.,
highest affinity) are further characterized by nucleic acid analysis to
identify the particular amino
acid sequences of the peptides expressed on the phage surface and the optimum
length of the
expressed peptide to achieve optimum binding. Alternatively, such peptide
ligands can be
2o selected from combinatorial libraries of peptides containing one or more
amino acids. Such
libraries can further be synthesized which contain non-peptide synthetic
moieties which are less
subject to enzymatic degradation compared to their naturally-occurring
counterparts.
To determine whether a peptide binds to the appropriate target any known
binding assay
may be employed. For example, in the case of a peptide that binds to the MHC
class II HLA-DR
25 the peptide may be immobilized on a surface and then contacted with a
labeled MHC class II
HLA-DR (or vice versa). The amount of MHC class II HLA-DR which interacts with
the peptide
or the amount which does not bind to the peptide may then be quantitated to
determine whether
the peptide binds to MHC class II HLA-DR. A surface having a known peptide
that binds to
MHC class II HLA-DR such as a commercially available monoclonal antibody
immobilized
3o thereto may serve as a positive control.
Screening of peptides of the invention, also can be carried out utilizing a
competition
assay. If the peptide being tested competes with the known monoclonal
antibody, as shown by
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/068~4
-49-
a decrease in binding of the known monoclonal antibody, then it is likely that
the peptide and the
known monoclonal antibody bind to the same, or a closely related, epitope.
Still another way to
determine whether a peptide has the specificity of the known monoclonal
antibody is to pre-
incubate the known monoclonal antibody with the target with which it is
normally reactive, and
then add the peptide being tested to determine if the peptide being tested is
inhibited in its ability
to bind the target. If the peptide being tested is inhibited then, in all
likelihood, it has the same,
or a functionally equivalent, epitope and specificity as the known monoclonal
antibody.
By using the known MHC class II HLA-DR (and other target) monoclonal
antibodies of
the invention, it is also possible to produce anti-idiotypic antibodies which
can be used to screen
other antibodies to identify whether the antibody has the same binding
specificity as the known
monoclonal antibody. Such anti-idiotypic antibodies can be produced using well-
known
hybridoma techniques (Kohler and Milstein, Nature, 256:495, 1975). An anti-
idiotypic antibody
is an antibody which recognizes unique determinants present on the known
monoclonal
antibodies. These determinants are located in the hypervariable region of the
antibody. It is this
region which binds to a given epitope and, thus, is responsible for the
specificity of the antibody.
An anti-idiotypic antibody can be prepared by immunizing an animal with the
known monoclonal
antibodies. The immunized animal will recognize and respond to the idiotypic
determinants of
the immunizing known monoclonal antibodies and produce an antibody to these
idiotypic
determinants. By using the anti-idiotypic antibodies of the immunized animal,
which are specific
2o for the known monoclonal antibodies of the invention, it is possible to
identify other clones with
the same idiotype as the known monoclonal antibody used for immunization.
Idiotypic identity
between monoclonal antibodies of two cell lines demonstrates that the two
monoclonal antibodies
are the same with respect to their recognition of the same epitopic
determinant. Thus, by using
anti-idiotypic antibodies, it is possible to identify other hybridomas
expressing monoclonal
antibodies having the same epitopic specificity.
It is also possible to use the anti-idiotype technology to produce monoclonal
antibodies
which mimic an epitope. For example, an anti-idiotypic monoclonal antibody
made to a first
monoclonal antibody will have a binding domain in the hypervariable region
which is the image
of the epitope bound by the first monoclonal antibody.
3o In one embodiment the binding peptides useful according to the invention
are antibodies
or functionally active antibody fragments. Antibodies are well known to those
of ordinary skill
in the science of immunology. Many of the binding peptides described herein
are available from
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-50-
commercial sources as intact functional antibodies. As used herein, the term
"antibody" means
not only intact antibody molecules but also fragments of antibody molecules
retaining specific
binding ability. Such fragments are also well known in the art and are
regularly employed both
in vitro and in vivo. In particular, as used herein, the term "antibody" means
not only intact
immunoglobulin molecules but also the well-known active fragments F(ab')2, and
Fab. F(ab')2,
and Fab fragments which lack the Fc fragment of intact antibody, clear more
rapidly from the
circulation, and may have less non-specific tissue binding of an intact
antibody (Wahl et al., J.
Nucl. Med. 24:316-325 (1983)).
As is well-known in the art, the complementarity determining regions (CDRs) of
an
l0 antibody are the portions of the antibody which are largely responsible for
antibody specificity.
The CDR's directly interact with the epitope of the antigen (see, in general,
Clark, 1986; Roitt,
1991 ). In both the heavy chain and the light chain variable regions of IgG
immunoglobulins,
there are four framework regions (FRl through FR4) separated respectively by
three
complementarity determining regions (CDRI through CDR3). The framework regions
{FRs)
maintain the tertiary structure of the paratope, which is the portion of the
antibody which is
involved in the interaction with the antigen. The CDRs, and in particular the
CDR3 regions, and
more particularly the heavy chain CDR3 contribute to antibody specificity.
Because these CDR
regions and in particular the CDR3 region confer antigen specificity on the
antibody these regions
may be incorporated into other antibodies or peptides to confer the identical
specificity onto that
antibody or peptide.
According to one embodiment, the peptide of the invention is an intact soluble
monoclonal antibody in an isolated form or in a pharmaceutical preparation. An
intact soluble
monoclonal antibody, as is well known in the art, is an assembly of
polypeptide chains linked by
disulfide bridges. Two principle polypeptide chains, referred to as the light
chain and heavy
chain, make up all major structural classes (isotypes) of antibody. Both heavy
chains and light
chains are further divided into subregions referred to as variable regions and
constant regions.
As used herein the term "monoclonal antibody" refers to a homogenous
population of
irnmunoglobulins which specifically bind to an epitope (i.e. antigenic
determinant) , e.g., of MHC
class II HLA-DR.
The peptide useful according to the methods of the present invention may be an
intact
humanized a monoclonal antibody. A "humanized monoclonal antibody" as used
herein is a
human monoclonal antibody or functionally active fragment thereof having human
constant
rm.v. vum:r.t'A-~~ut''Lt~t'n' u4 ""~- ~.- U :CA 02324995 2000-10 OSlt) ~'441~
+49 89 '?3999-At~~~:1t72
= US 009906874
22-05-2000
-51 -
regions and a binding CDR3 region from a matnma,l of a species other than a
human. I3urnanized
monoclonal antibodies may be made by any mcthad known in the art I3umaniud
monoclonal
antibodies, for example, may be constructed by replacing the non-CDR regions
of a rton-human
mammalian antibody wftii similar regions of human antibodies while retaining
the epitopic
s specificity of the original antibody. For exaruplc, non-human CDRs and
optionally some of the
framework regions may be covalently joined to human FR and/or FclpFc' regions
to produce a
functional antibody. There are entities is the United States which will
synthesize humanized
antibodies from specific marine antibody regions commercially, such as Protein
Design Labs
(Mountain Vicw Califorctia). For instance, a humanized form of the Pltarmingen
anti-Fns
~ o antibody used in t5c attached F,xatnples could be easily prepared arid
used according to the
methods of the invention.
European Patent Application (?239400, red
1~.~, provides an exemplary teaching of the production and use of humanized
monoclonal
antibodies in which at least the CDR portion of a marine (or other non-human
mammal) antibody
i s . is included in the humanized antibody. Briefly, the following methods
arc useful for consin~cting
ti itumanizcd CDIZ monoclonal antibody including at !cast a portion of a
ntousc CDR. A Gcst
replicabie expression vector including a suitable pmmotcr operably linked to a
DNA sequence
encoding at least a variable domain of an Ig heavy or tiglit chain and the
variable domain
comprising framework regions from a human antibody and a CDR region of a
marine antibody
Zo is prepared. Optionally a second replicablc expression vector is prepared
wrich includes a
suitable promoter opcrabiy linked to a DNA sequence eneodirtg at least the
variable domain of
a corrtplemeatary human Ig light or heavy chain respectively. A cell line is
then transformed with
the vectors. Preferably the ceh line is an irnmortaliud mammalian cell line of
lymphoid origin,
such as a mycloma, hybridoma, trioma, or quadroma cell line, or is a normal
lymphoid cell which
2s ..has been immortalized by t<aasfOnmttion with a virus' 'Ihe transformed
cell line is then cultured
under conditions known to those of skill in the art to produce the humanized
antibody.
As set forth in European Patent Application 0239400 several techniques are
well known
in the an for creating the particular antibody.domains to be inserted into the
replicable vector.
(Preferred vectors and recombinant techniq~,ies are discussed in greater
detail below.) For
3 o example, the DNA sequence encoding the domain may be prepared by
oligoaucleotide synthesis.
Alternatively a synthetic gent lacking the CDR regions in which four framework
regions arc
fused together with suitable restriction sites at the junctions, such that
double stranded synthetic
AMENDED SHEET
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-52-
or restricted subcloned CDR cassettes with sticky ends could be ligated at the
junctions of the
framework regions. Another method involves the preparation of the DNA sequence
encoding the
variable CDR containing domain by oligonucleotide site-directed mutagenesis.
Each of these
methods is well known in the art. Therefore, those skilled in the art may
construct humanized
antibodies containing a marine CDR region without destroying the specificity
of the antibody for
its epitope.
Human monoclonal antibodies may be made by any of the methods known in the
art, such
as those disclosed in US Patent No. 5,567,610, issued to Borrebaeck et al., US
Patent No.
565,354, issued to Ostberg, US Patent No. 5,571,893, issued to Baker et al,
Kozber, J. Immunol.
to 133: 3001 (1984), Brodeur, et al., MonoclonalAntibodyProduction Techniques
andApplications,
p. 51-63 (Marcel Dekker, Inc, new York, 1987), and Boerner el al., J.
Immunol., 147: 86-95
(1991). In addition to the conventional methods for preparing human monoclonal
antibodies,
such antibodies may also be prepared by immunizing transgenic animals that are
capable of
producing human antibodies (e.g., Jakobovits et al., PNAS USA, 90: 2551
(1993), Jakobovits et
al., Nature, 362: 255-258 (1993), Bruggermann et al., Year in Immuno., 7:33
(1993) and US
Patent No. 5,569,825 issued to Lonberg).
The binding peptides may also be functionally active antibody fragments.
Significantly,
as is well-known in the art, only a small portion of an antibody molecule, the
paratope, is
involved in the binding of the antibody to its epitope (see, in general,
Clark, W.R. (1986) The
2o Experimental Foundations of Modern Immunology Wiley & Sons, Inc., New York;
Roitt, I.
(1991) Essentiallmmunology, 7th Ed., Blackwell Scientific Publications,
Oxford). The pFc' and
Fc regions of the antibody, for example, are effectors of the complement
cascade but are not
involved in antigen binding. An antibody from which the pFc' region has been
enzymatically
cleaved, or which has been produced without the pFc' region, designated an
F(ab')2 fragment,
retains both of the antigen binding sites of an intact antibody. An isolated
F(ab')2 fragment is
referred to as a bivalent monoclonal fragment because of its two antigen
binding sites. Similarly,
an antibody from which the Fc region has been enzymatically cleaved, or which
has been
produced without the Fc region, designated an Fab fragment, retains one of the
antigen binding
sites of an intact antibody molecule. Proceeding further, Fab fragments
consist of a covalently
3o bound antibody light chain and a portion of the antibody heavy chain
denoted Fd (heavy chain
variable region). The Fd fragments are the major determinant of antibody
specificity (a single
Fd fragment may be associated with up to ten different light chains without
altering antibody
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-53-
specificity) and Fd fragments retain epitope-binding ability in isolation.
The terms Fab, Fc, pFc', F(ab')z and Fv are used consistently with their
standard
immunological meanings [Klein, Immunology (John Wiley, New York, NY, 1982);
Clark, W.R.
(1986) The Experimental Foundations ofModern Immunology (Wiley & Sons, Inc.,
New York);
Roitt, I. (1991) Essential Immunology, 7th Ed., (Blackwell Scientific
Publications, Oxford)].
The B7 and UCP expression vectors and other relevant expression vectors
described
herein can be prepared and inserted into cells using routine procedures known
in the art. These
procedures are set forth below in more detail. The term "IRM" (immune
recognition molecule)
nucleic acid is used herein to refer to each of the nucleic acids encompassed
by the expression
vectors described herein. Although UCP is not an immune molecule the term IRM
is used to
encompass UCP nucleic acids to simplify the discussion. "IRM nucleic acid", as
used herein,
refers to a nucleic acid molecule which: (1) hybridizes under stringent
conditions to a nucleic
acid having the sequence of SEQ ID NO:I, 3, 5, 7, 9, and 1 l and (2) codes for
a IRM polypeptide
(i.e., the respective immune recognition polypeptide). The preferred IRM
nucleic acid has the
t5 nucleic acid sequence of SEQ ID NO:1, 3, 5, 7, 9, and 11 (the nucleic acids
encoding the human
B7.1, B7.2, UCP-1, UCP-2, UCP-3S, and CD28 polypeptides respectively). The IRM
nucleic
acids may be intact IRM nucleic acids which include the nucleic acid sequence
of Sequence ID
No. 1-S as well as homologs and alleles of a nucleic acid having the sequence
of SEQ ID NO:l,
3, 5, 7, 9, and 11. Intact IRM nucleic acids further embrace nucleic acid
molecules which differ
2o from the sequence of SEQ ID NO:1, 3, 5, 7, 9, and 11 in codon sequence due
to the degeneracy
of the genetic code. The IRM nucleic acids of the invention may also be
functionally equivalent
variants, analogs and fragments of the foregoing nucleic acids. "Functionally
equivalent", in
reference to a IRM nucleic acid variant, analog or fragment, refers to a
nucleic acid that codes for
a IRM polypeptide that is capable of functioning as an immune recognition
molecule or an
25 uncoupling protein. The invention further embraces complements of the
foregoing nucleic acids
or of unique fragments of the foregoing nucleic acids. Such complements can be
used, for
example, as antisense nucleic acids for inhibiting the expression of IRM in a
cell in order to create
an experimental model of a cell in which IRM is not expressed.
The IRM nucleic acid molecules can be identified by conventional techniques,
e.g., by
3o identifying nucleic acid sequences which code for IRM polypeptides and
which hybridize to a
nucleic acid molecule having the sequence of SEQ ID NO:1, 3, 5, 7, 9, and 11
under stringent
conditions. The term "stringent conditions", as used herein, refers to
parameters with which the
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-54-
art is familiar. More specifically, stringent conditions, as used herein,
refer to hybridization at
65°C in hybridization buffer (3.5 x SSC, 0.02% Ficoll, 0.02% polyvinyl
pyrolidone, 0.02%
bovine serum albumin, 2.SmM NaH2P04 (pH 7), 0.5% SDS, 2mM EDTA). SSC is O.15M
sodium chloride/O.15M sodium citrate, pH 7; SDS is sodium dodecyl sulphate;
and EDTA is
ethylenediaminetetraacetic acid. After hybridization, the membrane to which
the DNA is
transferred is washed at 2x SSC at room temperature and then at O.ix SSC/O.lx
SDS at 65°C.
There are other conditions, reagents, and so forth which can be used, which
result in a
similar degree of stringency. The skilled artisan will be familiar with such
conditions and, thus,
they are not given here. It will be understood, however, that the skilled
artisan will be able to
manipulate the conditions in a manner to permit the clear identification of
homologs and alleles
of the IRM nucleic acid of the invention. The skilled artisan also is familiar
with the
methodology for screening cells and libraries for the expression of molecules,
such as IRM,
which can be isolated, followed by purification and sequencing of the
pertinent nucleic acid
molecule. In screening for IRM nucleic acid sequences, a Southern blot may be
performed using
the foregoing conditions, together with a radioactive probe. After washing the
membrane to
which the DNA is finally transferred, the membrane can be placed against x-ray
film to detect the
radioactive signal.
In general, homologs and alleles typically will share at least 40% nucleotide
identity with
SEQ ID NO:1, 3, 5, 7, 9, and 11; in some instances, will share at least 50%
nucleotide identity;
2o and in still other instances, will share at least 60% nucleotide identity.
The preferred homologs
have at least 70% sequence homology to SEQ ID NO:1, 3, 5, 7, 9, and 11. More
preferably the
preferred homologs have at least 80% and, most preferably, at least 90%
sequence homology to
SEQ ID NO:1, 3, 5, 7, 9, and 11 -5.
The invention also includes degenerate nucleic acids which include alternative
codons to
those present in the naturally occurring nucleic acid that codes for the human
IRM polypeptide.
For example, serine residues are encoded by the codons TCA, AGT, TCC, TCG, TCT
and AGC.
Each of the six codons is equivalent for the purposes of encoding a serine
residue. Thus, it will
be apparent to one of ordinary skill in the art that any of the serine-
encoding nucleotide codons
may be employed to direct the protein synthesis apparatus, in vitro or in
vivo, to incorporate a
3o serine residue. Similarly, nucleotide sequence triplets which encode other
amino acid residues
include, but are not limited to, CCA, CCC, CCG and CCT (proline codons); CGA,
CGC, CGG,
CGT, AGA and AGG (arginine codons); ACA, ACC, ACG and ACT (threonine codons);
AAC
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-55-
and AAT (asparagine codons); and ATA, ATC and ATT (isoleucine codons). Other
amino acid
residues may be encoded similarly by multiple nucleotide sequences. Thus, the
invention
embraces degenerate nucleic acids that differ from the naturally occurring
nucleic acids in codon
sequence due to the degeneracy of the genetic code.
The IRM nucleic acid, in one embodiment, is operably linked to a gene
expression
sequence which directs the expression of the IRM nucleic acid within a
eukaryotic cell. The
"gene expression sequence" is any regulatory nucleotide sequence, such as a
promoter sequence
or promoter-enhancer combination, which facilitates the efficient
transcription and translation of
the IRM nucleic acid to which it is operably linked. The gene expression
sequence may, for
to example, be a mammalian or viral promoter, such as a constitutive or
inducible promoter.
Constitutive mammalian promoters include, but are not limited to, the
promoters for the following
genes: hypoxanthine phosphoribosyl transferase (HPTR), adenosine deaminase,
pyruvate kinase,
and (3-actin. Exemplary viral promoters which function constitutively in
eukaryotic cells include,
for example, promoters from the simian virus, papilloma virus, adenovirus,
human
t 5 immunodeficiency virus (HIV), Rous sarcoma virus, cytomegalovirus, the
long terminal repeats
(LTR) of moloney leukemia virus and other retroviruses, and the thymidine
kinase promoter of
herpes simplex virus. Other constitutive promoters are known to those of
ordinary skill in the art.
The promoters useful as gene expression sequences of the invention also
include inducible
promoters. Inducible promoters are expressed in the presence of an inducing
agent. For example,
2o the metallothionein promoter is induced to promote transcription and
translation in the presence
of certain metal ions. Other inducible promoters are known to those of
ordinary skill in the art.
In general, the gene expression sequence shall include, as necessary, 5' non-
transcribing
and 5' non-translating sequences involved with the initiation of transcription
and translation,
respectively, such as a TATA box, capping sequence, CAAT sequence, and the
like. Especially,
25 such 5' non-transcribing sequences will include a promoter region which
includes a promoter
sequence for transcriptional control of the operably joined IRM nucleic acid.
The gene
expression sequences optionally include enhancer sequences or upstream
activator sequences as
desired.
Preferably, the IRM nucleic acid of the invention is linked to a gene
expression sequence
3o which permits expression of the IRM nucleic acid in the local environment
of a cell, e.g. a
damaged nerve cell. In some embodiments the gene expression sequence permits
expression of
the IRM nucleic acid in a human nerve cell or a neural activating cell. A
sequence which permits
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-56-
expression of the IRM nucleic acid in a nerve cell or a neural activating cell
is one which is
selectively active in nerve cell or a neural activating cell and thereby
causes the expression of the
IRM nucleic acid in these cells. Those of ordinary skill in the art will be
able to easily identify
promoters that are capable of expressing a IRM nucleic acid in a nerve cell or
a neural activating
cell, as well as other known cells.
The IRM nucleic acid sequence and the gene expression sequence are said to be
"operably
linked" when they are covalently linked in such a way as to place the
transcription and/or
translation of the IRM coding sequence under the influence or control of the
gene expression
sequence. If it is desired that the IRM sequence be translated into a
functional protein, two DNA
sequences are said to be operably linked if induction of a promoter in the 5'
gene expression
sequence results in the transcription of the IRM sequence and if the nature of
the linkage between
the two DNA sequences does not ( I ) result in the introduction of a frame-
shift mutation, (2)
interfere with the ability of the promoter region to direct the transcription
of the IRM sequence,
or (3) interfere with the ability of the corresponding RNA transcript to be
translated into a protein.
Thus, a gene expression sequence would be operably linked to a IRM nucleic
acid sequence if
the gene expression sequence were capable of effecting transcription of that
IRM nucleic acid
sequence such that the resulting transcript might be translated into the
desired protein or
polypeptide.
The IRM nucleic acid of the invention can be delivered to the cell alone or in
association
2o with a vector. In its broadest sense, a "vector" is any vehicle capable of
facilitating: ( 1 ) delivery
of a IRM molecule to a target cell or (2) uptake of a IRM molecule by a target
cell. Preferably,
the vectors transport the IRM molecule into the target cell with reduced
degradation relative to
the extent of degradation that would result in the absence of the vector.
Optionally, a "targeting
ligand" can be attached to the vector to selectively deliver the vector to a
cell which expresses on
its surface the cognate receptor for the targeting ligand. In this manner, the
vector (containing
a IRM nucleic acid) can be selectively delivered to a cell in, e.g., an
injured nerve tissue. In
general, the vectors useful in the invention are divided into two classes:
biological vectors and
chemical/physical vectors. Biological vectors are useful for delivery/uptake
of IRM nucleic acids
to/by a target cell. Chemical/physical vectors are also useful for
delivery/uptake of IRM nucleic
3o acids to/by a target cell.
Biological vectors include, but are not limited to, plasmids, phagemids,
viruses, other
vehicles derived from viral or bacterial sources that have been manipulated by
the insertion or
CA 02324995 2000-10-OS
WO 99/53953 PCTNS99106874
-57-
incorporation of the nucleic acid sequences of the invention, and free nucleic
acid fragments
which can be attached to the nucleic acid sequences of the invention. Viral
vectors are a preferred
type of biological vector and include, but are not limited to, nucleic acid
sequences from the
following viruses: retroviruses, such as: Moloney marine leukemia virus;
Harvey marine sarcoma
s virus; marine mammary tumor virus; Rous sarcoma virus; adenovirus; adeno-
associated virus;
SV40-type viruses; polyoma viruses; Epstein-Barr viruses; papilloma viruses;
herpes viruses;
vaccinia viruses; polio viruses; and RNA viruses such as any retrovirus. One
can readily employ
other vectors not named but known in the art.
Preferred viral vectors are based on non-cytopathic eukaryotic viruses in
which non-
1o essential genes have been replaced with the gene of interest. Non-
cytopathic viruses include
retroviruses, the life cycle of which involves reverse transcription of
genomic viral RNA into
DNA with subsequent proviral integration into host cellular DNA. Retroviruses
have been
approved for human gene therapy trials. In general, the retroviruses are
replication-deficient (i.e.,
capable of directing synthesis of the desired proteins, but incapable of
manufacturing an
15 infectious particle). Such genetically altered retroviral expression
vectors have general utility for
the high-efficiency transduction of genes in vivo. Standard protocols for
producing repIication-
deficient retroviruses (including the steps of incorporation of exogenous
genetic material into a
plasmid, transfection of a packaging cell lined with plasmid, production of
recombinant
retroviruses by the packaging cell line, collection of viral particles from
tissue culture media, and
2o infection of the target cells with viral particles) are provided in
Kriegler, M., "Gene Transfer and
Expression, A Laboratory Manual," W.H. Freeman Co., New York (1990) and Marry,
E.J. Ed.
"Methods in Molecular Biology," vol. 7, Humana Press, Inc., Cliffton, New
Jersey ( 1991 ).
Another preferred virus for certain applications is the adeno-associated
virus, a double-
stranded DNA virus. The adeno-associated virus can be engineered to be
replication -deficient
25 and is capable of infecting a wide range of cell types and species. It
further has advantages, such
as heat and lipid solvent stability; high transduction frequencies in cells of
diverse lineages; and
lack of superinfection inhibition thus allowing multiple series of
transductions. Reportedly, the
adeno-associated virus can integrate into human cellular DNA in a site-
specific manner, thereby
minimizing the possibility of insertional mutagenesis and variability of
inserted gene expression.
3o In addition, wild-type adeno-associated virus infections have been followed
in tissue culture for
greater than 100 passages in the absence of selective pressure, implying that
the adeno-associated
virus genomic integration is a relatively stable event. The adeno-associated
virus can also
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-58-
function in an extrachromosomal fashion.
In addition to the biological vectors, chemical/physical vectors may be used
to deliver a
IRM molecule to a target cell and facilitate uptake thereby. As used herein, a
"chemical/physical
vector" refers to a natural or synthetic molecule, other than those derived
from bacteriological or
viral sources, capable of delivering the IRM molecule to a cell.
A preferred chemical/physical vector of the invention is a colloidal
dispersion system.
Colloidal dispersion systems include lipid-based systems including oil-in-
water emulsions,
micelles, mixed micelles, and liposomes. A preferred colloidal system of the
invention is a
liposome. Liposomes are artificial membrane vessels which are useful as a
delivery vector in
i o vivo or in vitro. It has been shown that large unilamellar vessels (LI1V),
which range in size from
0.2 - 4.0 ~m can encapsulate large macromolecules. RNA, DNA, and intact
virions can be
encapsulated within the aqueous interior and be delivered to cells in a
biologically active form
(Fraley, et al., Trends Biockem. Sci., (1981) 6:77). In order for a liposome
to be an efficient gene
transfer vector, one or more of the following characteristics should be
present: ( 1 ) encapsulation
t 5 of the gene of interest at high efficiency with retention of biological
activity; (2) preferential and
substantial binding to a target cell in comparison to non-target cells; (3)
delivery of the aqueous
contents of the vesicle to the target cell cytoplasm at high efficiency; and
(4) accurate and
effective expression of genetic information.
Liposomes may be targeted to a particular tissue, such as the site of a tumor,
by coupling
2o the liposome to a specific ligand such as a monoclonal antibody, sugar,
glycolipid, or protein.
Ligands which may be useful for targeting a liposome to a tumor cell include,
but are not limited
to: intact or fragments of IRM which interact with tumor cell specific
receptor and molecules
which interact with the cell surface markers of tumor cells such as
antibodies. Such ligands may
easily be identified by binding assays well known to those of skill in the
art. Additionally, the
25 vector may be coupled to a nuclear targeting peptide, which will direct the
IRM nucleic acid to
the nucleus of the host cell.
Liposomes are commercially available from Gibco BRL, for example, as
LIPOFECTINTM
and LIPOFECTACETM, which are formed of cationic lipids such as N-[1-(2, 3
dioleyloxy)-
propyl]-N, N, N-trimethylammonium chloride (DOTMA) and dimethyl
dioctadecylammonium
3o bromide (DDAB). Methods for making liposomes are well known in the art and
have been
described in many publications. Liposomes also have been reviewed by
Gregoriadis, G. in
Trends in Biotechnology, {1985) 3:235-241.
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-59-
In one particular embodiment, the preferred vehicle is a biocompatible
microparticle or
implant that is suitable for implantation into the mammalian recipient.
Exemplary bioerodible
implants that are useful in accordance with this method are described in PCT
International
application no. PCT/US/03307 (Publication No. WO 95/24929, entitled "Polymeric
Gene
Delivery System", claiming priority to U.S. patent application serial no.
213,668, filed March 15,
1994). PCT/US/0307 describes a biocompatible, preferably biodegradable
polymeric matrix for
containing an exogenous gene under the control of an appropriate promotor. The
polymeric
matrix is used to achieve sustained release of the exogenous gene in the
patient. In accordance
with the instant invention, the IRM nucleic acids described herein are
encapsulated or dispersed
1o within the biocompatible, preferably biodegradable polymeric matrix
disclosed in
PCT/US/03307.
The polymeric matrix preferably is in the form of a microparticle such as a
microsphere
(wherein the IRM molecule is dispersed throughout a solid polymeric matrix) or
a microcapsule
(wherein the IRM molecule is stored in the core of a polymeric shell). Other
forms of the
polymeric matrix for containing the IRM molecule include films, coatings,
gels, implants, and
stents. The size and composition of the polymeric matrix device is selected to
result in favorable
release kinetics in the tissue into which the matrix is introduced. The size
of the polymeric matrix
further is selected according to the method of delivery which is to be used,
typically injection into
a tissue or administration of a suspension by aerosol into the nasal and/or
pulmonary areas.
2o Preferably when an aerosol route is used the polymeric matrix and IRM
molecule are
encompassed in a surfactant vehicle. The polymeric matrix composition can be
selected to have
both favorable degradation rates and also to be formed of a material which is
bioadhesive, to
further increase the effectiveness of transfer when the matrix is administered
to a nasal andlor
pulmonary surface that has sustained an injury. The matrix composition also
can be selected not
to degrade, but rather, to release by diffusion over an extended period of
time.
In another embodiment the chemical/physical vector is a biocompatibIe
microsphere that
is suitable for oral delivery. Such microspheres are disclosed in Chickering
et al., Biotech. And
Bioeng., (1996) 52:96-101 and Mathiowitz et al., Nature, (1997) 386:.410-414.
Both non-biodegradable and biodegradable polymeric matrices can be used to
deliver the
3o IRM nucleic acids of the invention to the subject. Biodegradable matrices
are preferred. Such
polymers may be natural or synthetic polymers. Synthetic polymers are
preferred. The polymer
is selected based on the period of time over which release is desired,
generally in the order of a
CA 02324995 2000-10-OS
WO 99/53953 PCTNS99/06874
-60-
few hours to a year or longer. Typically, release over a period ranging from
between a few hours
and three to twelve months is most desirable. The polymer optionally is in the
form of a hydrogel
that can absorb up to about 90% of its weight in water and further, optionally
is cross-linked with
multi-valent ions or other polymers.
In general, the IRM nucleic acids are delivered using a bioerodible implant by
way of
diffusion, or more preferably, by degradation of the polymeric matrix.
Exemplary synthetic
polymers which can be used to form the biodegradable delivery system include:
polyamides,
polycarbonates, polyalkylenes, polyalkylene glycols, polyalkylene oxides,
polyalkylene
terepthalates, polyvinyl alcohols, polyvinyl ethers, polyvinyl esters, poly-
vinyl halides,
polyvinylpyrrolidone, polyglycolides, polysiloxanes, polyurethanes and co-
polymers thereof,
alkyl cellulose, hydroxyalkyl celluloses, cellulose ethers, cellulose esters,
nitro celluloses,
polymers of acrylic and methacrylic esters, methyl cellulose, ethyl cellulose,
hydroxypropyl
cellulose, hydroxy-propyl methyl cellulose, hydroxybutyl methyl cellulose,
cellulose acetate,
cellulose propionate, cellulose acetate butyrate, cellulose acetate phthalate,
carboxylethyl
cellulose, cellulose triacetate, cellulose sulphate sodium salt, poly(methyl
methacrylate),
poly(ethyl methacrylate), poly(butylmethacrylate), poly(isobutyl
methacrylate),
poly(hexylmethacrylate), poly(isodecyl methacrylate), poly(lauryl
methacrylate), poly(phenyl
methacrylate), poly(methyl acrylate), poly(isopropyl acrylate), poly(isobutyl
acrylate),
poly(octadecyl acrylate), polyethylene, polypropylene, polyethylene glycol),
polyethylene
oxide), polyethylene terephthalate), polyvinyl alcohols), polyvinyl acetate,
poly vinyl chloride,
polystyrene, polyvinylpyrrolidone, and polymers of lactic acid and glycolic
acid, polyanhydrides,
poly(ortho)esters, poly(butic acid), poly(valeric acid), and poly(lactide-
cocaprolactone), and
natural polymers such as alginate and other polysaccharides including dextran
and cellulose,
collagen, chemical derivatives thereof (substitutions, additions of chemical
groups, for example,
alkyl, alkylene, hydroxylations, oxidations, and other modifications routinely
made by those
skilled in the art), albumin and other hydrophilic proteins, zero and other
prolamines and
hydrophobic proteins, copolymers and mixtures thereof. In general, these
materials degrade
either by enzymatic hydrolysis or exposure to water in vivo, by surface or
bulk erosion.
Examples of non-biodegradable polymers include ethylene vinyl acetate,
3o poly(meth)acrylic acid, polyamides, copolymers and mixtures thereof.
Bioadhesive polymers of particular interest include bioerodible hydrogels
described by
H.S. Sawhney, C.P. Pathak and J.A. Hubell in Macromolecules, (1993) 26:581-
587, the teachings
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-61 -
of which are incorporated herein, polyhyaluronic acids, casein, gelatin,
glutin, polyanhydrides,
polyacrylic acid, alginate, chitosan, poly(methyl methacrylates), poly(ethyl
methacrylates),
poly(butylmethacrylate), poly(isobutyl methacrylate), poly(hexylmethacrylate),
poly(isodecyl
methacrylate), poly(lauryl methacrylate), poly(phenyl methacrylate),
poly(methyl acrylate),
poly(isopropyl acrylate), poly(isobutyl acrylate), and poly(octadecyl
acrylate).
Compaction agents also can be used alone, or in combination with, a biological
or
chemical/physical vector of the invention. A "compaction agent", as used
herein, refers to an
agent, such as a histone, that neutralizes the negative charges on the nucleic
acid and thereby
permits compaction of the nucleic acid into a fine granule. Compaction of the
nucleic acid
to facilitates the uptake of the nucleic acid by the target cell. The
compaction agents can be used
alone, i.e., to deliver the IRM molecule in a form that is more efficiently
taken up by the cell or,
more preferably, in combination with one or more of the above-described
vectors.
Other exemplary compositions that can be used to facilitate uptake by a target
cell of the
IRM nucleic acids include calcium phosphate and other chemical mediators of
intracellular
transport, microinjection compositions, electroporation and homologous
recombination
compositions (e.g., for integrating a IRM nucleic acid into a preselected
location within the target
cell chromosome).
In addition to the expression vectors, the invention also encompasses the use
of antisense
oligonucleotides that selectively bind to a IRM nucleic acid molecule, and
dominant negative
2o IRM to reduce the expression of IRM. Antisense oligonucleotides are useful,
for example, for
preparing an animal model of a subject having a neurodegenerative disorder.
Such animal models
can be used in screening assays for identifying therapeutic drugs for treating
neurodegenerative
disorders..
As used herein, the term "antisense oligonucleotide" or "antisense" describes
an
oligonucleotide which hybridizes under physiological conditions to DNA
comprising a particular
gene or to an RNA transcript of that gene and, thereby, inhibits the
transcription of that gene
and/or the translation of the mRNA. The antisense molecules are designed so as
to hybridize with
the target gene or target gene product and thereby, interfere with
transcription or translation of
the target mammalian cell gene. Those skilled in the art will recognize that
the exact length of
3o the antisense oligonucleotide and its degree of complementarity with its
target will depend upon
the specific target selected, including the sequence of the target and the
particular bases which
comprise that sequence. The antisense must be a unique fragment. A unique
fragment is one that
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-62-
is a 'signature' for the larger nucleic acid. It, for example, is long enough
to assure that its precise
sequence is not found in molecules outside of the IRM gene. As will be
recognized by those
skilled in the art, the size of the unique fragment will depend upon its
conservancy in the genetic
code. Thus, some regions of SEQ ID NO:1, 3, 5, 7, 9, and 11, will require
longer segments to be
unique while others will require only short segments, typically between 12 and
32 base pairs (e.g.
12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31
and 32 bases long).
It is preferred that the antisense oligonucleotide be constructed and arranged
so as to bind
selectively with the target under physiological conditions, i.e., to hybridize
substantially more to
the target sequence than to any other sequence in the target cell under
physiological conditions.
to Based upon the known sequence of a gene that is targeted for inhibition by
antisense
hybridization, or upon allelic or homologous genomic and/or cDNA sequences,
one of skill in the
art can easily choose and synthesize any of a number of appropriate antisense
molecules for use
in accordance with the present invention. In order to be sufficiently
selective and potent for
inhibition, such antisense oligonucleotides should comprise at least 7 and,
more preferably, at
~ 5 least 15 consecutive bases which are complementary to the target. Most
preferably, the antisense
oligonucleotides comprise a complementary sequence of 20-30 bases. Although
oligonucleotides
may be chosen which are antisense to any region of the gene or RNA (e.g.,
mRNA) transcripts,
in preferred embodiments the antisense oligonucleotides are complementary to
5' sites, such as
translation initiation, transcription initiation or promoter sites, that are
upstream of the gene that
20 is targeted for inhibition by the antisense oligonucleotides. In addition,
3'-untranslated regions
may be targeted. Furthermore, 5' or 3' enhancers may be targeted. Targeting to
mRNA splice
sites has also been used in the art but may be less preferred if alternative
mRNA splicing occurs.
In at least some embodiments, the antisense is targeted, preferably, to sites
in which mRNA
secondary structure is not expected (see, e.g., Sainio et al., Cell Mol.
Neurobiol., (1994)
25 14(5):439-457) and at which proteins are not expected to bind. The
selective binding of the
antisense oligonucleotide to a mammalian target cell nucleic acid effectively
decreases or
eliminates the transcription or translation of the mammalian target cell
nucleic acid molecule.
Reduction in transcription or translation of the nucleic acid molecule is
desirable in preparing an
animal model for further defining the role played by the mammalian target cell
nucleic acid in
3o modulating an adverse medical condition.
The invention also includes the use of a "dominant negative UCP" polypeptide.
A
dominant negative polypeptide is an inactive variant of a protein, which, by
interacting with the
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
- 63 -
cellular machinery, displaces an active protein from its interaction with the
cellular machinery
or competes with the active protein, thereby reducing the effect of the active
protein. For
example, a dominant negative receptor which binds a ligand but does not
transmit a signal in
response to binding of the ligand can reduce the biological effect of
expression of the ligand.
Likewise, a dominant negative catalytically-inactive kinase which interacts
normally with target
proteins but does not phosphorylate the target proteins can reduce
phosphorylation of the target
proteins in response to a cellular signal. Similarly, a dominant negative
transcription factor which
binds to a promoter site in the control region of a gene but does not increase
gene transcription
can reduce the effect of a normal transcription factor by occupying promoter
binding sites without
increasing transcription.
The end result of the expression of a dominant negative polypeptide as used
herein in a
cell is a reduction in function of active UCP. One of ordinary skill in the
art can assess the
potential for a dominant negative variant of a protein, and using standard
mutagenesis techniques
to create one or more dominant negative variant polypeptides. For example, one
of ordinary skill
in the art can modify the sequence of the UCP by site-specific mutagenesis,
scanning
mutagenesis, partial gene deletion or truncation, and the like. See, e.g.,
U.S. Patent No. 5,580,723
and Sambrook et al., Molecular Cloning: A Laboratory Manual, Second Edition,
Cold Spring
Harbor Laboratory Press, 1989. The skilled artisan then can test the
population of mutagenized
polypeptides for diminution in a selected and/or for retention of such an
activity. Other similar
2o methods for creating and testing dominant negative variants of a protein
will be apparent to one
of ordinary skill in the art.
In other aspects the invention includes transgenic animals and cells
transfected with the
IRM's. Additionally, complements of the IRM nucleic acids described above can
be useful as
anti-sense oligonucleotides, e.g., by delivering the anti-sense
oligonucleotide to an animal to
induce a "knockout" phenotype. The administration of anti-sense RNA probes to
block gene
expression is discussed in Lichtenstein, C., Nature 333:801-802 (1988).
Alternatively, the IRM nucleic acids can be used to prepare a non-human
transgenic
animal. A "transgenic animal" is an animal having cells that contain DNA which
has been
artificially inserted into a cell, which DNA becomes part of the genome of the
animal which
develops from that cell. Preferred transgenic animals are primates, mice,
rats, cows, pigs, horses,
goats, sheep, dogs and cats. Animals suitable for transgenic experiments can
be obtained from
standard commercial sources such as Charles River (Wilmington, MA), Taconic
(Germantown,
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
N~, Harlan Sprague Dawley (Indianapolis, IN), etc. Transgenic animals having a
particular
property associated with a particular disease can be used to study the affects
of a variety of drugs
and treatment methods on the disease, and thus serve as genetic models for the
study of a number
of human diseases. The invention, therefore, contemplates the use of IRM
knockout and
s transgenic animals as models for the study of neurodegenerative disorders.
A variety of methods are available for the production of transgenic animals
associated
with this invention. DNA can be injected into the pronucleus of a fertilized
egg before fusion of
the male and female pronuclei, or injected into the nucleus of an embryonic
cell (e.g., the nucleus
of a two-cell embryo) following the initiation of cell division. See e.g.,
Brinster et ~1., Proc. Nat.
1 o Acad. Sci. USA, 82: 4438 (1985); Brinster et al., cell 27: 223 ( 1981 );
Costantini et al., Nature 294:
982 ( 1981 ); Harpers et al., Nature 293: 540 ( 1981 ); Wagner et al., Proc.
Nat. Acad. Sci. USA
78:5016 (1981); Gordon et al., Proc. Nat. Acad. Sci. USA 73: 1260 (1976). The
fertilized egg is
then implanted into the uterus of the recipient female and allowed to develop
into an animal.
An alternative method for producing transgenic animals involves the
incorporation of the
~ s desired gene sequence into a virus which is capable of affecting the cells
of a host animal. See
e.g., Elbrecht et al., Molec. Cell. Biol. 7: 1276 (1987); Lacey et al., Nature
322: 609 (1986);
Leopol et al., Cell S1: 885 (1987). Embryos can be infected with viruses,
especially retroviruses,
modified to carry the nucleotide sequences which encode IRM proteins or
sequences which
disrupt the native IRM gene to produce a knockout animal.
2o Another method for producing transgenic animals involves the injection of
pluripotent
embryonic stem cells into a blastocyst of a developing embryo. Pluripotent
stem cells derived
from the inner cell mass of the embryo and stabilized in culture can be
manipulated in culture to
incorporate nucleotide sequences of the invention. A transgenic animal can be
produced from
such cells through implantation into a blastocyst that is implanted into a
foster mother and
2s allowed to come to term. See e.g., Robertson et al., Cold Spring Harbor
Conference Cell
Proliferation 10: 647 (1983); Bradley et al., Nature 309: 255 (1984); Wagner
et al., Cold Spring
Harbor Symposium Quantitative Biology 50: 691 (1985).
The procedures for manipulation of the rodent embryo and for microinj ection
of DNA into
the pronucleus of the zygote are well known to those of ordinary skill in the
art (Hogan gI ~1.,
3o supra). Microinjection procedures for fish, amphibian eggs and birds are
detailed in Houdebine
and Chourrout, E~erientia, 47: 897-905 (1991 ). Other procedures for
introduction of DNA into
tissues of animals are described in U.S. Patent No., 4,945,OS0 (Sandford gI
~1., July 30, 1990).
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-65-
By way of example only, to prepare a transgenic mouse, female mice are induced
to
superovulate. Females are placed with males, and the mated females are
sacrificed by COZ
asphyxiation or cervical dislocation and embryos are recovered from excised
oviducts.
Surrounding cumulus cells are removed. Pronuclear embryos are then washed and
stored until
the time of injection. Randomly cycling adult female mice are paired with
vasectomized males.
Recipient females are mated at the same time as donor females. Embryos then
are transferred
surgically. The procedure for generating transgenic rats is similar to that of
mice. See Hammer
~ ~., ~g]~t, 63:1099-1112 (1990).
Methods for the culturing of embryonic stem (ES) cells and the subsequent
production of
to transgenic animals by the introduction of DNA into ES cells using methods
such as
electroporation, calcium phosphate/DNA precipitation and direct injection also
are well known
to those of ordinary skill in the art. See, for example, Teratocarcinomas and
Embryonic Stem
Cells A Practical Approz~ch, E.J. Robertson, ed., IRL Press (1987).
In cases involving random gene integration, a clone containing the sequences)
of the
~ 5 invention is co-transfected with a gene encoding resistance.
Alternatively, the gene encoding
neomycin resistance is physically linked to the sequences) of the invention.
Transfection and
isolation of desired clones are carried out by any one of several methods well
known to those of
ordinary skill in the art (E.J. Robertson, supra).
DNA molecules introduced into ES cells can also be integrated into the
chromosome
2o through the process of homologous recombination. Capecchi, Scie. ,nce, 244:
1288-1292 (1989).
Methods for positive selection of the recombination event (e.g., neo
resistance) and dual
positive-negative selection (e.g., neo resistance and gangcyclovir resistance)
and the subsequent
identification of the desired clones by PCR have been described by Capecchi,
supra and Joyner
et ~1., Nature, 338: 153-156 (1989). The final phase of the procedure is to
inject targeted ES cells
25 into blastocysts and to transfer the blastocysts into pseudopregnant
females. The resulting
chimeric animals are bred and the offspring are analyzed by Southern blotting
to identify
individuals that carry the transgene.
Procedures for the production of non-rodent mammals and other animals have
been
discussed by others. See Houdebine and Chourrout, supra; Pursel ~t ~1., i nc
244: 1281-1288
30 (1989); and Simms ~ ~., Bio/Technolo~v, 6: 179-183 (1988).
Inactivation or replacement of the endogenous IRM genes can be achieved by a
homologous recombination system using embryonic stem cells. The resultant
transgenic non-
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-66-
human mammals having a knockout characteristic may be used as a model for
neurodegenerative
disorders. Nerve cells which do not express IRMs may be predisposed to
apoptosis and unable
to differentiate and thus, produce a neurodegenerative phenotype. A variety of
therapeutic drugs
can be administered to the phenotypically neurodegenerative animals to
determine the affect of
the therapeutic drugs on nerve cell differentiation. In this manner,
therapeutic drugs which are
useful for preventing or reducing neurodegenerative disorders can be
identified. Such agents are
useful for, e.g., treating spinal chord injuries or Parkinson's disease.
Additionally, a normal or mutant version of IRM can be inserted into the mouse
germ line
to produce transgenic animals which constitutively or inducible express the
normal or mutant
l0 form of IRM. These animals are useful in studies to define the role and
function of IRM in cells.
The metabolic modifying agent, apoptotic chemotherapeutic agent, MHC class II
HLA-
DR inducing agent, MHC class II HLA-DR ligand, B7 receptor blocking agent, B7
inducing
agent, and B7 receptor inducing agent described herein are commercially
available compounds,
are derived from commercially available compounds or are synthesized de novo
using routine
chemical synthetic procedures known to those of ordinary skill in the art.
When administered, the pharmaceutical preparations of the invention are
applied in
pharmaceutically-acceptable amounts and in pharmaceutically-acceptably
compositions. Such
preparations may routinely contain salt, buffering agents, preservatives,
compatible carriers, and
optionally other therapeutic agents. When used in medicine, the salts should
be pharmaceutically
acceptable, but non-pharmaceutically acceptable salts may conveniently be used
to prepare
pharmaceutically-acceptable salts thereof and are not excluded from the scope
of the invention.
Such pharmacologically and pharmaceutically-acceptable salts include, but are
not limited to,
those prepared from the following acids: hydrochloric, hydrobromic, sulfuric,
nitric, phosphoric,
malefic, acetic, salicylic, citric, formic, malonic, succinic, and the like.
Also, pharmaceutically-
acceptable salts can be prepared as alkaline metal or alkaline earth salts,
such as sodium,
potassium or calcium salts. As used herein, a composition of an metabolic
modifying agent and
an apoptotic chemotherapeutic agent means the compounds described above as
well as salts
thereof and a composition of an MHC class II HLA-DR inducing agent and an MHC
class II
HLA-DR ligand means the compounds described above as well as salts thereof.
3o The compositions of the invention may be combined, optionally, with a
pharmaceutically-
acceptable carrier. The term "pharmaceutically-acceptable carrier" as used
herein means one or
more compatible solid or liquid filler, diluents or encapsulating substances
which are suitable for
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-67-
administration into a human or other animal. The term "carrier" denotes an
organic or inorganic
ingredient, natural or synthetic, with which the active ingredient is combined
to facilitate the
application. The components of the pharmaceutical compositions also are
capable of being co-
mingled with the molecules of the present invention, and with each other, in a
manner such that
there is no interaction which would substantially impair the desired
pharmaceutical efficacy.
The pharmaceutical compositions may contain suitable buffering agents,
including: acetic
acid in a salt; citric acid in a salt; boric acid in a salt; and phosphoric
acid in a salt.
The pharmaceutical compositions also may contain, optionally, suitable
preservatives,
such as: benzalkonium chloride; chlorobutanol; parabens and thimerosal.
1 o Compositions suitable for parenteral administration conveniently comprise
a sterile
aqueous preparation of the compositions of the invention, which is preferably
isotonic with the
blood of the recipient. This aqueous preparation may be formulated according
to known methods
using suitable dispersing or wetting agents and suspending agents. The sterile
injectable
preparation also may be a sterile injectable solution or suspension in a non-
toxic parenterally-
~ 5 acceptable diluent or solvent, for example, as a solution in 1,3-butane
diol. Among the acceptable
vehicles and solvents that may be employed are water, Ringer's solution, and
isotonic sodium
chloride solution. In addition, sterile, fixed oils are conventionally
employed as a solvent or
suspending medium. For this purpose any bland fixed oil may be employed
including synthetic
mono- or di-glycerides. In addition, fatty acids such as oleic acid may be
used in the preparation
20 of injectables. Carrier formulation suitable for oral, subcutaneous,
intravenous, intramuscular,
etc. administrations can be found in Remington's Pharmaceutical Sciences, Mack
Publishing Co.,
Easton, PA.
A variety of administration routes are available. The particular mode selected
will depend
of course, upon the particular drug selected, the severity of the condition
being treated and the
25 dosage required for therapeutic efficacy. The methods of the invention,
generally speaking, may
be practiced using any mode of administration that is medically acceptable,
meaning any mode
that produces effective levels of the active compounds without causing
clinically unacceptable
adverse effects. Such modes of administration include oral, rectal, topical,
nasal, interdermal, or
parenteral routes. The term "parenteral" includes subcutaneous, intravenous,
intramuscular, or
3o infusion. Intravenous or intramuscular routes are not particularly suitable
for long-term therapy
and prophylaxis. They could, however, be preferred in emergency situations.
Oral administration
will be preferred for prophylactic treatment because of the convenience to the
patient as well as
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-68-
the dosing schedule.
The pharmaceutical compositions may conveniently be presented in unit dosage
form and
may be prepared by any of the methods well-known in the art of pharmacy. Ail
methods include
the step of bringing the compositions of the invention into association with a
carrier which
constitutes one or more accessory ingredients. In general, the compositions
are prepared by
uniformly and intimately bringing the compositions of the invention into
association with a liquid
carrier, a finely divided solid carrier, or both, and then, if necessary,
shaping the product.
Compositions suitable for oral administration may be presented as discrete
units, such as
capsules, tablets, lozenges, each containing a predetermined amount of the
compositions of the
1o invention. Other compositions include suspensions in aqueous liquids or non-
aqueous liquids
such as a syrup, elixir or an emulsion.
Other delivery systems can include time-release, delayed release or sustained
release
delivery systems. Such systems can avoid repeated administrations of the
compositions of the
invention described above, increasing convenience to the subject and the
physician. Many types
15 of release delivery systems are available and known to those of ordinary
skill in the art. They
include polymer base systems such as poly(lactide-glycolide), copolyoxalates,
polycaprolactones,
polyesteramides, polyorthoesters, polyhydroxybutyric acid, and polyanhydrides.
Microcapsules
of the foregoing polymers containing drugs are described in, for example, U.S.
Patent 5,075,109.
Delivery systems also include non-polymer systems that are: lipids including
sterols such as
20 cholesterol, cholesterol esters and fatty acids or neutral fats such as
mono- di- and tri-glycerides;
hydrogel release systems; sylastic systems; peptide based systems; wax
coatings; compressed
tablets using conventional binders and excipients; partially fused implants;
and the like. Specific
examples include, but are not limited to: (a) erosional systems in which the
compositions of the
invention is contained in a form within a matrix such as those described in
U.S. Patent Nos.
25 4,452,775, 4,667,014, 4,748,034 and 5,239,660 and (b) difusional systems in
which an active
component permeates at a controlled rate from a polymer such as described in
U.S. Patent Nos.
3,832,253, and 3,854,480. In addition, pump-based hardware delivery systems
can be used, some
of which are adapted for implantation.
Use of a long-term sustained release implant may be particularly suitable for
treatment
30 of chronic conditions. Long-term release, are used herein, means that the
implant is constructed
and arranged to delivery therapeutic levels of the active ingredient fox at
least 30 days, and
preferably 60 days. Long-term sustained release implants are well-known to
those of ordinary
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-69-
skill in the art and include some of the release systems described above.
The following examples are provided to illustrate specific instances of the
practice of the
present invention and are not to be construed as limiting the present
invention to these examples.
As will be apparent to one of ordinary skill in the art, the present invention
will find application
in a variety of compositions and methods.
Example 1: Metabolic state of a cell is indicative of cell surface Fas
expression and
sensitivity/resistance to cell death.
1. Re~iat~nce to a~t~sis is characterized by failure to express Fas: The cell
lines
utilized herein include L 12I 0, a leukemic cell line; HL60, a human pro-
myelocytic cell line; and
PC12, a pheochromocytoma cell line which can be induced to differentiate into
a neuronal cell
line in the presence of NGF (Lindenboim, L, et al., Cancer Res, 1995, 55:1242-
7). Each cell line
was examined in parallel with apoptotic resistant sublines: L 1210 DDP, HL60
MDR, and
~5 PCl2Trk. L1210 DDP are resistant to cisplatin and methotrexate; HL60 MDR
are resistant to
adriamycin induced apoptosis; PC 12 TrkA, which have been transfected with
TrkA which results
in constitutively expression the NGF receptors, are not susceptible to alcohol
and NGF
withdrawal as are the PC 12 cells.
The apoptosis sensitive cells from each tissue origin were morphologically
round,
2o non-adherent, rapidly dividing cells, with the exception of the PC 12 cell
line. The apoptosis
resistant cells from all tissue origins were morphologically large, adherent,
and slowly dividing
cells.
The recently characterized molecules, Fas (CD95) and Fas Ligand (CD95L), have
been
strongly implicated in the process of apoptotic death (Muller, M, et al.,. J
Clin Invest,1997,
25 99:403-413). We examined expression of Fas on the above-identified cell
lines. Flow cytometric
analysis of Fas expression was performed using an isotype versus FITC-anti-
Fas(Pharmingen)
on L 1210; PC 12; and HL60 cells and resistant cell lines L 121 ODDP, PC
12Trk; and HL60MDR.
A Coulter Epics Elite flow cytometer with a single excitation wavelength (488
nm) and band
filters for PE (575 nm), FITC (525 nm) and Red613 (613 nm) was used to analyze
the stained
3o cells. Each sample population was classified for cell size (forward
scatter) and complexity (side
scatter), gated on a population of interest and evaluated using 40,000 cells.
Criteria for positive
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-70-
staining were established by comparison with the intensity of the isotype
controls, thick lines.
Independent of tissue origin, all of the apoptosis resistant lines fail to
express cell surface Fas
both constitutively and in the presence of agents that induce apoptosis in the
parental cell lines.
2. 'sch a' f a~
and utilization: We performed experiments to examine the correlation between
cell surface Fas
expression and glucose metabolism. As a prototype for the Fas positive and Fas
negative cells
we used the L 1210 and the L 121 ODDP cell lines, as Fas positive and Fas
negative, respectively.
We directly measured the rates of glucose utilization and oxidation of L1210
and L1210DDP.
Ratios were generated by using nanomolar values.
to Rate of glucose utilization was measured by the method of Ashcroft et al.
Briefly, cells
were incubated 90 min at 37°C in 100 ~l KRB, glucose (5.5 mM), 1.3 pCi
D-[5 3H] glucose
(Amersham, Arlington Heights, IL). The reaction was carried out in a 1 ml cup
contained in a
rubber stoppered 20 ml scintillation vial that had 500 ~I of distilled water
surrounding the cup.
Glucose metabolism was stopped by injecting 100 ~.l 1 M Hcl through the
stopper into the cup.
~5 An overnight incubation at 37°C was carried out to allow
equilibration of the [3H]-Hz0 in the
reaction cup and the distilled water, followed by liquid scintillation
counting of the distilled
water.
Rate of glucose oxidation was measured by incubating cells for 90 min at 37
°C in 100 ml
of reaction buffer, glucose (2.8, 8.3, 27.7 mmol/1), 1.7 mCi (U-14C glucose).
The reaction was
2o carried out in a I ml cup in a 20 ml scintillation vial capped by a rubber
stopper with a center well
that contains filter paper. Metabolism was stopped and COZ liberated with 300
ml 1 mol/1 HCl
injected through the stopper into the cup containing the cells. COZwas trapped
in the filter paper
by injecting 10 ml 1 mol/1 KOH into the center well, followed 2 hours later by
liquid scintillation
counting. Tubes containing NaHC03 and no cells were used to estimate the
recovery of'4COz
25 in the filter paper, routinely close to 100%.
The results are presented in Table I .
Table 1. Glucose Metabolic~n in L1210/0 and L1210/DI~P
L 1210/0 ~ L 1210/DDP
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-71 -
Glucose Utilization 1740920 3470460
(pmol glucose/ 90 min/ 50,000 cells)
Glucose Oxidation 2357 4281124
(pmol glucose/ 90 min/ 50,000 cells)
Glucose UtilizationlOxidation 7.4 8.1
Because the L I 210 and L 121 ODDP cells are tumor cell lines and are likely
to have
increased ratios of glucose oxidation to utilization (Warburg, O, et al., Klin
Woch, 1926,
5:829-832), we measured glucose utilization on normal lymphocytes. We isolated
106 splenic
lymphocytes from C57BL/6 animals, Fas-deficient C57BL/6 (B6.lpr), and Fast
defective
C57BL/6 (B6.gld) animals. The rate of glucose utilization and oxidation of the
Fas deficient and
the Fast deficient lymphocytes are demonstrated in Table 2. The ratio of
glucose utilization to
oxidation is highest in lpr lymphocytes and lowest in wild type normal,
quiescent lymphocytes.
Table 2. Glucose Metabolism in Lymphocytes from Normal, Fas Deficient and Fast
Deficient Mice
b6 Ipr gld
GLUCOSE UTILIZATION
(nmol glucose/90 mins/50,0000.04 0.36 0.22
cells)
GLUCOSE OXIDATION
(pmol glucose/90 mins/50,00073.24 164.51 122.82
cells)
CELL TYPE RATIO
GLUCOSE UTILIZATION / b6 0.55
GLUCOSE OXIDATION
lpr 2.19
gld 1.79
i mc~C ua~a ~ ~ aoie me ~~ aemonstrate mgn rates of glucose utilization and
oxidation of
3o both tumor lines relative to the normal lymphocytes; and higher rates of
glucose utilization and
oxidation of the apoptotic resistant line relative to the wild type. There is
an important difference
CA 02324995 2000-10-OS
WO 99/53953 PCTNS99/06874
-72-
in the ratio of glucose utilization to oxidation between normal and Fas or
Fast deficient animals,
with the ratio being higher for lymphocytes from both mutant strains of
animals. The
consequences of uncoupling are a decrease in mitochondria) membrane potential;
use of fat as a
carbon source increased rate of glycolysis, increased rate of electron
transport, and energy
dissipation, in a form other than ATP. These data suggest that there is an
increase in proton leak
in the cells with high rates of glucose oxidation and utilization relative to
the normal cells,
suggesting some degree of uncoupling may have occurred in these cells.
3. Fas xpression Increase as a Fu_nctinn of C',ln~ncP; We investigated the
effect of
increasing concentrations of glucose on cell surface Fas expression. L1210 and
L1210/DDP cells
1 o were cultured in glucose free RPMI media or in media supplemented with
insulin and glucose for
16 hours. Intra- and extracellular Fas expression was determined by labelling
the cells with
FITC-conjugated anti-Fas antibodies (Pharmingen), or FITC-conjugated isotype
control, then
subtracting the fluorescence intensity of the isotype staining from Fas
staining for each treatment
group.
These data showed that Fas expression increases as a function of glucose
concentration
and that as a result the cell surface Fas negative L 1210/DDP begin to express
cell surface Fas.
4. Treatment of L 1210 DDP cell v~ith stauroshorin restores Fas expression and
susce tp ibiliyto drug-ind ice a o tOSIS; L1210, but not L1210 DDP, undergo
apoptotic cell
death. We treated L1210 or L1210 DDP cells with the staurosporin, which
inhibits protein kinase
2o C and increases mitochondria) membrane potential, or an anti-cancer agent
to which both cells
are sensitive, adriamycin. Fas expression was increased or induced on both L
1210 and L 1210
DDP, respectively, in the presence of staurosporin or adriamycin. The L 1210
DDP changed
morphologically and began to divide rapidly, changes which appeared to
correspond with a
reversion back to the phenotype of the L 1210 cells. These results demonstrate
that Fas expression
results in parallel with altered metabolic activity.
S. Confocal microsconv reveals that resistance to a~ontosis i~ charactPrzed b
intra (but
not extra) cellular Fas expression: L 1210 DDP cells express no cell surface
Fas. To address the
possibility that Fas is expressed, but has been targeted to a subcellular
organelle, we
permeabilized and stained L1210 and L1210DDP cells with fluorochrome
conjugated anti-Fas
3o antibody (J02.2, Pharmingen). The cells were examined by confocal
microscopy. (This
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-73-
experiment was representative of four experiments).
Our data indicate that L 1210 DDP cells express Fas in an intracellular,
cytosolic
compartment. Fluorochrome-conjugated isotype matched antibody was used as
control.
Additionally, these data also demonstrate that the Fas negative, apoptosis
resistant cells, express
intracellular Fas.
6. Fas- a - b not ex r - a Fa I
We isolated lymphocytes from spleens of C57BL/6 mice and from C57BL6
transgenics having
the lpr mutation (loss of Fas sensitivity). Cells were stained with
fluorescein conjugated hamster
anti-Fas and examined by confocal microscopy.
Results demonstrate that unstimulated, non-permeabilized splenocytes from
C57BL/6
animals express Fas at low levels relative to isotype controls. Interestingly,
significant levels
of Fas expression were detected in permeabilized normal lymphocytes. As
expected,
non-permeabilized cells from C57BL6.lpr animals express no detectable cell
surface Fas relative
to isotype control. Interestingly, intracellular Fas staining of permeabilized
splenocytes from
C57B1/6.lpr animals reveals intracellular expression of Fas. These results
demonstrate that
mutations affecting susceptibility to Fas-induced death prevent cell surface,
but not intracellular
expression of the Fas molecule.
7. Anti-cancer agents induce suscentibilitv to Fas induced cell ~iPath: To
determine if the
anti-cancer agent methotrexate sensitizes L 1210 or L 1210/DDP cells to Fas
induced cell death,
we cultured L 1210 cells in the presence or absence of 10-g M methotrexate for
72 hours. Each
group of cells was cultured on uncoated plates or plates coated with l Og/ml
anti-Fas (Jo.2.2,
Pharmingen). We analyzed cell death using flow cytometry. Forward angle and 90
degree light
scatter were used to distinguish between live and dead cells. Dead cells were
gated as forward
angle light scatter low/high ethidium bromide retaining cells. Percent death
was calculated over
2s the total number of cells acquired. In Table 3 below, values indicate %
dead cells over
background of untreated cells.
Table 3. Fas-induced cell death
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-74-
L1210/0 L1210/DDP
Control 4.72 40.88
anti-Fas Coated 79.98 46.60
Plates
s Additionally, L1210 and L1210/DDP cells were treated with 10-8 M
methotrexate for 24
hours. Flow cytometric analysis revealed two populations based on forward side
scatter. The
forward scatter high populations did not take ethidium bromide and were
therefore viable. The
forward scatter low populations took up ethidium bromide differentially. The L
1210 cells took
up a moderate amount. Analysis of DNA fragments reveals that L1210 produced a
ladder of
1o nucleosome sized fragments indicative of apoptosis, whereas L1210/DDP cells
did not. This
latter phenotype - loss in forward scatter and membrane permeability with no
"DNA laddering" -
is the hall mark of oncosis.
8. Fas Deficient Lvm hocytes are al~c~ dig resistant to methotrexatP; We
isolated
splenic lymphocytes from aged-matched wild type C57BL/6 mice and C57BL6.Ipr
and
15 C57BL.gld. Splenocytes from C57BL/6 Ipr or gld animals were isolated, red
cells depleted, and
single cell suspensions prepared. Cells were cultured in the absence or
presence of 5 x 10-g M
methotrexate for 18 or 32 hours. Cells were harvested and viability was
determined by flow
cytometric analysis and confirmed with trypan blue exclusion.
The data demonstrate decreased susceptibility to methotrexate-induced
apoptosis in Fas
2o deficient lymphocytes. These data are consistent with the notion that Fas
is required for drug
susceptibility.
9. Dig resistant ~Pll~ ress intracellular f s T MCP a_ d bcl 2: We determined
if wild
type and/or drug resistant cells express intracellular and surface fas, UCP
and bcl-2. We stained
non-permeabilized L 1210 and L 1210/DDP cells for cell surface or
intracellular Fas. The data
25 show that while there is no cell surface expression of Fas on the
drug/apoptotic resistant cells, the
drug resistant cells express high levels of intracellular Fas. The drug
resistant cells are cell
surface Fas negative and protected from death resulting from changes in
mitochondria) membrane
permeability transitions.
3o Example 2: Pancreatic B cells express UCP and have no cell surface fas
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-75-
1. Loss of antigen in (3-cell tumors: Proliferation with two responder T cell
clones,
BDC-2.5 and BDC-6.9, was tested using NOD peritoneal cells as APC and as
antigen, either
freshly prepared NOD islet cells (control) or ~i tumor cells, or NIT-l, an
established beta tumor
cell line from the NOD-RIPTag mouse. Upon harvesting the islet tumors, the (3-
cells obtained
are fully as antigenic as normal NOD islet cells. The NIT-1 line is also
antigenic for these T cell
clones, but only at low passage numbers; with continued culture, the line
changes its morphology
and growth kinetics and undergoes complete loss of antigen.
2. R~snonse of gancreatirr~i-cells to glucose: The experiments described below
were
designed to test the hypothesis that [3 cell metabolism may be linked to
immune recognition and
to destruction. Glucose utilization was measured as [3H]Hz0 production from 5-
[3H]glucose in
normal rat islets. Glucose oxidation was measured as ['4C]COZ production from
U-['4C]glucose.
The data show increasing glucose utilization and oxidation in [i-cells as a
function of increasing
glucose concentration.
3. ~lormal -cells E ~nre~s Intracellular UCP2 and No Cell Surface Fas: Normal
[i-cells
15 have a specialized glucose response which is based on the cell being
responsive to physiologic
glucose concentrations. The process that mediates the glucose responsiveness
is the process
involving flux through glycolysis. [3-cell glucose usage is mediated through a
relatively unique
system that entails specialized high K", glucose transporter (GLUT2) and
glucose phosphorylation
isoforms (glucokinase). We isolated (3-cells from C3H mice, stained the
isolated cells with
2o anti-Fas, and electronically gated viable cells. In parallel, cells were
permeabilized and stained
with an antibody to UCP2 (kindly provided by Drs. Jean Himms-Hagen and M.E.
Harper).
The results show that normal ~i-cells express intracellular UCP2 and no cell
surface Fas.
4 Fas expression and mitochondrial membrane ~notential are ~ unction of lug
Cose
concentration in mouse (3 cells
25 The central question is whether Fas expression is altered by changes in
physiological
glucose concentrations in normal (3 cells and does the mitochondrial membrane
potential increase,
suggesting that the cell has ATP synthesis resulting from increased rates of
electron transport.
Islets were isolated and dispersed with trypsin and a cell strainer. Debris
and dead cells were
removed and applying the cells to a 1.066 Percoll gradient. Electronic gating
of the cells was
3o used to segregate the populations of islets cells. The region with larger
cells were gated [3 cells
where the region with smaller cells were gated as alpha cells. Other larger
cells were excluded
<CV.vuN:E,t'~-vtUHNCHt~, ~>4 :zy_ S_ (, CA 02324995 2000-10-057'~U 1441-» +~~9
Hy '?3'~5'~4~":~1:;
U S 009906874
22-05-2000
-7s-
because they contained a cells. The cells were trtatod ovettiight with either
physiological I 1.1
mM glucose or high glucose 55.5 mM glucose. Fas expression was determined by
staining with
a EITC conjugated antibody. Mcaa ffuorcscence of staining with isotype control
antibody wes
subtracted. Measurement of mitochondria) membrane pvtertial was measured using
JC-1 as a
s fluorescence probe. The relative membrane potential was read by taking the
red mean
fluorescence (aggregatod JC-1 laboled) divided by mean green fluorescent
(rnonornerie 1C-I)
Labeled fluorescence. Our data suggest that as glucose concentration
increasos, the large ~3 cell
subset of gated cells have increased Fas expression and concomitant increased
mitochondria)
membrane potential, while the smaller (possibly alpha, glucagon producins
cells) dv not.
~ ~etermin~t;',p,~~',~,~ttgrhondri~rl rneny,ratrg.~,'ptential m ~~~ es~Qjat~~
fr3m four
strains ofanimals.
Mitochondria) tnembtxne potential is assessed flow cytometrically using
mitotracker red.
The amount of membrane potential was rneasurcd in the four strains of animals
AB-, AB-Fra,
C59B1/6, BITgEa, described in more detail below. Mitochondria) membrane
potential was
t 5 highest in the AB~ strain, foI'._owed by the AB-EA stain. The CS?B I/6 and
BITgEa sins had
much lower mitochondria) membrane potential.
Example 3: Relationship between the metabolic state of a cell and expression
of I1~C class
Ix molecules
1, i 1
i. Perturbation of class ll on resting B cells results in the generation of
CAMP.
Early studies demonstrated that ligation of iE molecules on resting B cells
resulted in the
,rapid generation of intracellular cAMP in those cells (Cambier, et al.,Nature
(1987). Based on
this observation and on our more recent evidence that elevated levels of CAMP
correlate with
z5 death in resting B cells, we have studied the generation of CAMP in more
detail. The data
provide a comparative analysis of alterations in levels of CAMP as induced by
antibodies to IE
and IA. Cells were stftnulated with antibodies to MHC class I (From ATCC) ,
with anti-Ig
i 5t~ t'~-ere~o l
(laclcson Itnmunoehemicals) and IL-4 (Genzymc) , or with isop~ar~ol (Sigma).
We isolated
B cells from CS7BL (which express 1A, but not IE molex~ules) wild type, lpr,
or gld animals.
3a The datashow that the Ipr, or gld mutation does not alter the signal
delivered by MHC
AMENDED SHEET
~CY.VC)li:Gt'A-MI!fiNCHt~ 04 :Z'?- 5- () CA 023_2_4995 2000-10-OSl'?Q '?441-~
+9-;1 ti.l _~~~y'~~4f;~:er~n
U S 009906874
22-05-2000
-77-
class II engagement at the dose of the anti-IA mAb that we have used. T>zese
data show that
under the conditions we have employed to stiraulate the colts, the anti-IE
antibodies arc more
effective at increasing intracellular cAMP. To investigate the possibility
that the differences in
the ability of IA and IE to alter levels of CAMP may be the consequence of
dose dependent
differences and to determine if anti-IA antibodies ever induce increase in
cAMP, we performed
a dose titration of anti~IA stimulation on B cells from C57BIJ6 animals.
T"nese data demonstrate
an oscillation of CAMP Ievals, atu3 reflect alteration in signaling by
differences in aggregation
state of MHC class II.
An analysis of DNA fragmentation from resting B cells was also performed.
Cells were;
incubated w~th medium alone, with anti class II (IA), anti-class II (IA beta
chain), aaL-clsss II
s~o~ee..no~
IE, ~ape~a~rral, or dibutyryl CAMP and cultured overnight. Cells were
harvested, nuclei
separated, and fragmented (non-clear) DNA precipitated and the samples wore
electrophorcsod
on agarose gels. Bands were visualized using ethidium bromide and ultraviolet
light deicction.
The data is presen~ed in the Table bolow.
t5 MHC class II indnees
apoptotic cell death
C
TREATM~tT NORMALIZED AREA APOPTOTIC IIv'DEX
medium alone 0.07I I 0.0000
I 7r227 0.4168 0.3722
14-4-4S 0.2309 0.1721
zo 10-2.I6 ' 0.6850 0.6b09
H 11632 0.7183 0.6968
Isoproterenol 1.00 t .0000
dbcAMP 1.0304 1.032$
25 ii. Anti-class II rr~Ab irrdreee an increase in opaptotic cell death in
resting B cells.
Our data demonstrate that treatment of resting B lymphocytes with anti-class
II mAb
results in B c811 apoptasis, as measured by inct~eases in nucleosome-sized
DN?~ fra6ments.
Apoptotic indices were generated by comparison to maximum anaptotic death as
stimulated by
isoproterenol. The data shows that ligation of class II molecules on resting,
but not activated
so .B cells, results in apopootic death. Resting B cells, ~n vitro activated B
cells [anti-Ig and
AMENDED SHEET
U ,CA 023_24995 2000-10-057zU 2441-~ i49 8C1 ~?;39~14.'~(":t~lr
acv. vav : f:~A-n~u~.ncrm ~r~
U S 009906874
22-05-2000
_ 78 -
recombinant IL-4], or freshly ex vivo activated cells, were treated with 10
pg/mI anti-I-A~
so~rvker ero t
rnAb (17/227); anti Ig and 16 units of recombinant IL.~, or IO ~tM
iaepcaterertvt. $ cells were
treated with the stimuli indicated for 10 min at 37°C, cul~red
overnight, harvested and
assayed. (The in vitro activated cells were not tttated with additional anti-
Ig plus rIL-4.)
Fragmented DNA was isolated by ccntt'ifugation and run on agarose gels. The
ethidium
bromide stained gel was photographed and scanned. An Apoptotic Index was
calculated by
taking: [(experimental area - area with maiiwn alone) I (area with
isoproterettoI - area with
medium alone)]. Treamaents that produce a score of 0 show background levels of
apoptosls,
whereas treatments that arc protective produce scores < 0. Data from four
ipdepetuiet?t
to experiments are averaged for normal animals; means and standard errors are
shown.
iii. MXC class ll induced death in resting B-cells from normal mouse sttairrs,
but not
mouse strains having the Ipr and gld mtetatiorrs.
To test the hypothesis that the mechanism of IA-mediated death involves the
receptor
ligand pairs CD95/CD95L, we have used mouse strains that have the Ipr
:autation, or the gld
i 5 mutation, which have deflects in CD95 and CD95L, respectively (Watanabe-
Fukunaga, R, et al.>
Nature (London), I992, 35b, 314-3I7; Suds, T et al., Cell, 1993, 75:1 l69-
1178). Total splenic
B-cells were isolated from C3H, AItR, C3H.lpr, and MRL.gId mice. AlI of these
strains are H-2x.
The cells were cultured overnight, harvested, permeabilized in saponin,
stained with propidium
iodide (1'1) which intercalates into DNA, and analyzed by flow cytometry.
After a IS hour
2o culture, a significant percentage of cultured B-oeIls fragment their DNA,
with no stimulation
(Newell, MK, et al., Proc Nat Acad Sri USA, 1993, 90:104.59-10463).
Crosstinking MHC class
II IA (HLA-D>'/D~ in humans) oo B-cells from the wild type animal cause an
increase in
apoptosis. Unlike the normal B-cells, there is no increase in less than 2X DNA
a~~r cmsslinking
~MHC class II on $-cells from Ipr or gld mice.
2s 2. lnr~~ 'on of B-cells wit_tt T callt~: The results of mAb binding to MHC
class II does
not, a priori , reSeet tho result of an interaction with a physiolobiealiy
relevant li6and. To
address the possibility that the physiological ligand for MHC class II is
expressed on a CL74+ T
cell, we examined the effect of class II signaling resulting from T cetl:l3
cell interactions, Table
4. Rcstin~splenic B-cells were isoIa:ed by T depletion and density gradient
centrifugation
F4R CDtL
30 (perea~). The B-cells were then combined with wither an sutoreactive I-Ak-
specific T cell
.hybridoma (Kal-68.4) or with a hen egg lysozyme (HEL) peptide-specific, I-Ak-
restricted T cell
AMENDED SHEET
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-79-
hybridoma (A6.A2) either with or without a tryptic digest of lysozyme as the
source of the
required peptide. Cells were cultured overnight at 37°C and then
examined under a fluorescence
microscope. Apoptotic cells were scored based on their morphology and on their
uptake of
Hoechst Dye 33342 at 5 ~g/ml final concentration (Cohen, J.J, et al., Ann Rev
Immun,1992, 10,
267-293). B and T cells were distinguished by morphology.
Table 4. Induction of apoptosis in resting, but not activated, AKR B cells by
interaction
with T cells
Resting B cells Activated B cells
Apoptotic IL-2 Titer, % Apoptotic IL-2 Titer,
1 o Culture AdditionsB cells U/ml B cells U/ml
"
Medium Alone 14 <20 25 <20
A6.A2 13 <20 18 <20
A6.A2 + tryp-HEL ' 54 1280 25 1280
Kal-68.4 '' 30 160 22 320
Equal numbers (5 x 105) of B-cells and T cells were incubated for 16 hr at
37°C in a 24 well
microtiter plate.
A6.A2 is I-Ak-restricted T cell hybridoma specific for the hen egg lysozyme
peptide
HEL(aa34-45). IL-2 titers were determined using HT-2 cells as previously
described (36).
' Tryptic digest of HEL, containing HEL(aa34-45), was used at 1 mg/ml.
'~ Kal-68.4 is an autoreactive I-Ak-specific T cell hybridoma.
3. Pheno pic characterizanr,n of apontofi~ B-cell: We adapted the technique of
using
terminal deoxynucleotidyl-transferase (TdT) to add fluorochrome-conjugated
2s deoxyribonucleotides to the free ends of DNA to flow cytometric analysis of
apoptosis (Gold,
R, et al., J Histochem Cytochem, 1993, 41:1023-1030). Because the fragmented
DNA of
apoptotic cells has significantly more free ends that DNA of non-apoptotic
cells, the apoptotic
cells stain bright green with dUTP-FITC (deoxyuridine triphosphate) whereas
viable cells remain
dull. After a 16 hr incubation with the Kal-68.4 autoreactive T cell
hybridoma, resting B-cells
from AKR mice showed 46-47% apoptotic cells, confirming the results using the
two other
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-80-
methods. This method allows a counter-stain of cells with antibodies directed
against various
surface receptors expressed by the B-cells.
A two-color flow cytometric analysis of apoptotic resting B-cells was
performed. AKR
resting B-cells and Kal-68.4 T hybridomas cells were incubated overnight.
Cells were stained
with biotin-conjugated mAb directed against B7-1, B7-2, or Fas followed by PE-
streptavidin, and
apoptotic cells were detected using TdT/dUTP-FITC. The contour plots generated
showed
labeling with dUTP-FITC (as a measure of apoptotic death) versus counter-
staining with the
indicated mAb for B-cells harvested from culture with T cells. The percentages
indicate the
relative number of cells in the viable (dUTP-FITC dull) and apoptotic (dUTP-
FITC bright)
1o populations.
Relative to resting B-cells, B-cells cultured overnight with Kal-68.4
upregulated B7-1,
B7-2 and Fas, with the upregulation of B7-1 being the most striking and giving
rise to a bimodal
distribution. Two-color analysis reveals that the B-cells from these cultures
may be divided into
viable (deoxyuridine-FITC tow) and apoptotic (deoxyuridine-FITC high)
populations with
~ 5 apparent differential expression of the counter-stained receptors on the
two populations.
Histograms of fluorescence intensity of the stained receptors show that Fas
and especially B7-1
are upregulated on the apoptotic population whereas B7-2 is expressed at
higher levels on the
viable population.
4. Class II-mediated si~naling~(NZR x N7W~F1 end (NZB x SWR1F1 snicP
2o i. Engagement of class II on resting B-cells from autoimmune strains of
mice does not
result in increases in cAMP over background levels.
Experiments with B-cells from (NZB x NZ~F 1 and (NZB x SWR)F 1 mice suggested
a potential link between class II-mediated B cell signaling and autoimmunity
in these mice.
Following the protocols as described in Newell, MK, et al. (Proc Natl Acad
Sci, ZISA, 1993,
25 90:10459-10463) for resting B lymphocytes from normal mice, we isolated
resting B-cells from
spleens of (NZB x NZVi~F 1 and (NZB x S WR)F 1 mice by Percoll gradient
separation and
treated the cells with antibodies to class II molecules. Experiments utilized
mice younger than
3 months of age. These mice do not have elevated serum levels of IgG
autoantibodies to histone
and DNA and do not demonstrate evidence of an immune-complex
glomerulonephritis.
3o The data demonstrated that in contrast to B-cells from normal mice, resting
B-cells
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-81-
from (NZB x S WR)F 1 and (NZB x NZW)F 1 mice do not elevate intracellular cAMP
in response
to ligation of their class II molecules. In contrast, the response to
isoproterenol is normal.
Resting ( I .079 < r < 1.085) B-cells from normal, ; (NZB x S WR)F 1, ; or
(NZB x S WR)F 1,
animals were treated for 10 min at 37oC with stimuli. It should be noted that
there are no
significant differences between resting cells in normal versus (NZB x S WR)F I
mice, as defined
by surface MHC class II expression and by density (Julius, M and Haughn, L,
Eur J Immun,
1992, 22, 2323-2329).
ii. Class II mediated apoptosis does not occur in resting B lymphocytes from
autoimmune
mice.
We stimulated both resting and activated B-cells from (NZB x NZW)F1 and (NZB x
S WR)F 1 mice with antibodies to class II molecules. Young animals, prior to
the development
of lupus-like autoantibody and renal disease, were used in these studies.
Agarose gel
electrophoresis of DNA fragments from high density resting B-cells (1.079 < r
< 1.085) was
carried out as described above. Normalized areas produced by scanning
densitometry on the gels
for normal; (NZB x S WR)F 1; or (NZB x NZW)F 1 animals. The data were
normalized to
background levels rather than calculating an Apoptotic Index since (NZB x
NZW)F I cells did not
show a significant increase in apoptosis when treated with isoproterenol.
Because experiments
were performed on different days it was difficult to compare background levels
of apoptosis
between strains; however, it should be noted that there is significant
variability in background
levels of apoptosis, with those for the (NZB x NZW)F 1 animals apparently
tending to be higher
than for the other strains.
The data demonstrated that in contrast to normal B-cells, resting B-cells from
(NZB x
NZW) F 1 and (NZB x S WR) F 1 mice are refractory to MHC class II-mediated
apoptosis. The
data also showed that despite normal cAMP generation after isoproterenol, this
treatment induced
minimal evidence of apoptosis in (NZB x NZW)F1 B-cells. An intermediate level
of
isoproterenol-induced apoptosis was apparent in B-cells from (NZB x SWR)F1
mice. Thus, the
collective results demonstrate that, while resting B-cells from both
autoimmune strains are
defective in coupling the ligation of class II molecules to the generation of
cAMP, cells from
(NZB x NZW) F 1 animals also appear to have a second lesion in the apoptotic
pathway that is
downstream from the generation of CAMP. Thus the data shows a ligation of
class II molecules
on resting B-cells from (NZB x NZW)F I and (NZB x S WR)F 1 animals does not
result in
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-82-
apoptotic death.
iii. Phenotypic characterization of apoptotic B-cells from autoimmune prone
mice
Our results show that when using B-cells from (NZB xNZW)F1 animals resting B-
cells
from these animals are refractory to class II-induced apoptosis. This
indicates that a failure of
class II mediated apoptosis provides a mechanism for polyclonal hyper-
gammaglobulinemia,
characteristic of autoimmune disease. Resting B-cells are able to present
sufficient self peptides
to allow their interaction with the "autoreactive" T cells and that the
failure to obtain class
II-mediated apoptosis in resting B-cells may drive polyclonal hyper-
gammaglobulinemia.
5. Stn.~ctural features and cell surface expression of MHC IA and IE molecules
sociated
1 o with cell death
UCP expression in L 1210 and L 1210/DDP cells was measured in response to
staurosporin
and PMA. L 1210 cells expressed the lowest levels of UCP (approximately 110
Geo MFI over
background). UCP levels were increased in staurosporin (approximately 175 Geo
MFI over
background) and PMA (approximately 175 Geo MFI over background) treated cells.
XIA
(approximately 190 Geo MFI over background) and XIE ( approximately 240 Geo
MFI over
background) also had higher levels of UCP. UCP levels in L1210/DDP cells were
also
significantly higher than L1210 cells (approximately 300 Geo MFI over
background).
Seguence of MHC IA and MHC IE molecules
Comparison of the transmembrane (TM) and cytoplasmic (Cy) domain sequences of
the
2o beta and alpha chains of IAA and IEk reveals both conserved and unique
sequences. The
differences between IA and IE and the human equivalents are generally shared.
The beta chains
of IAk and IE'' have 18 of 22 amino acids that are conserved in the TM domain.
These changes
are basically conserved, whereas the Cy domains differ in length and
composition. The Cy
domains of IAk has more two more prolines and an extra two positive chargres
(R, H) at the
proximal end of the Cy domain next to the inner leaflet. The area (RH_RS~P)
(SEQ ID NO.
13) of the Cy domain of IAk that has been mapped by Wade et al (Int Immunol,
1994,
6:1457-1465) as being required for PKC translocation and cAMP, respectively,
is different from
the sequence seen in IE, but the residues QKG are the same. This sequence
similarities may
explain the observation that ligation of IA or IE can signal increases in
intracellular cAMP. The
lack of the RH or PP residues in IE could explain the lack of Fas induction,
due to either the loss
CA 02324995 2000-10-OS
WO 99/53953 PCTNS99/06874
-83-
of a binding site defined by the positive charges of the kinks introduced in
the Cy domain by the
multiple P residues.
6 Co t' w a r ac o c l
Phenotype of MHC Clays II+ Cells. Much of our work is based upon the use of
model cell lines
which have been transfected with wild type or mutated MHC class II molecules
and which exhibit
each of the prototypic signalling phenotypes, M12.C3 and K46J, representing
cAMP and Ca++
generating responses, respectively (Wade, WF et al., Int Immunol, 1994 6:1457-
1465). The .
results of flow cytometric analysis to phenotype the cell surface marker
expression on each of
these lines is combined with a summary of what we know about the way cells die
in Table 5.
to These data were generated either flow cytometrically by assessing changes
in forward and side
scatter with uptake of ethidium bromide, or by morphological assessment and
trypan blue
exclusion.
Table 5. Summary of Cell Surface Expression and Cell Death Phenotype
Cell Surface Phenotype Type of class II induced death (if any)
~ I~ ~Z2 B~- Ano otic Osmotic R~ t ~r
M12.C3 wt/wt ++ ~ __ + _ ++++
M12.C3 411 -12/-18 -- -- -_ + --
2o M12.C3 7D3 -12/wt + -- -- + -- +
K46J wt/wt ++ +++ ++ + ++ +.~.
K46J -12/-18 + + -- +
Example 4: Involvement of IA versus IE in resistance and susceptibility to
immune-mediated cardiovascular disease
Coxsackievirus-mediated myocarditis
To evaluate the role of MHC class II antigens in immune-mediated myocarditis
susceptibility, transgenic mice were graciously provided by Dr. Chella David
of Mayo Clinic.
Dr. David supplied the following strains: 1 ) AB° mice lack MHC class
II IA and IE molecules
(class II knockout (KO) mice); 2) AB° Eab are MHC class II KO mice
which have a functional
transgenic IE chain, so that the animal express IE but not IA; 3) BI.Tg.Eab
mice express the wild
type IA molecules as well as the IE molecules; and 4) wild type C57B16 express
IA only.
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-84-
Male mice, 4-5 weeks of age were injected ip with 100 pg GL3-3A (anti-ya)
monoclonal
antibody in 0.5 ml PBS, or PBS alone on days -2 and +2 relative to virus.
Animals received 104
PFU CVB3 on day 0 and surviving animals were euthanized on day 7. Hearts were
removed
from animals between days 5 and 7 for analysis. Hearts were divided and the
apex was formalin
fixed, sectioned and evaluated by image analysis for percent of the myocardium
affected. The
remaining tissue was titered by plaque forming assay for virus. Groups
consisted of 4 mice each.
The results are summarized in Table 6 below.
Table 6. Effect of Depleting y8+ T Cells on CYB3-Induced Myocarditis
STRAIN ANTIBODY MORTALITY VIRUS TITER MYOCARDITIS
C57BL/6 0 5.1 t 0.7 0.5 10.3
Anti-y8TcR 0 S.St0.9 Ot0
AB 0 6.5 f 1.4 0 t 0
Anti-ydTcR 0 7.1 10.8 1.3+0.8
AB Eak 100 6.2 + 0.9 5.1 t 2.0
Anti - yS 25 6.5 ~ 0.7 1.8 + 1.1
TcR
B1 Tg Eak SO 4.3 10.5 8.31 1.6
Anti-yBTcR 0 5.3t0.4* 1.7t0.5*
* Q:,...,:~:.......a_..t_rr_____.
m_ _
--p------.,.--..> ...~~.....w uacua uvia-aiu1uv14y-LIGaLGCI illl(:.~ C1L I-
SU.UJ.
Mice expressing either no class II MHC antigen or IA only were myocarditis
resistant
having little or no cardiac inflammation and no animal mortality. In contrast,
IE-bearing mice
showed increased mortality accompanied by substantial myocardial necrosis.
AB° Ea mice,
expressing IE only, began dying earlier (day 3 post-infection) and had more
extensive
coaggulative myocardial necrosis with limited cardiac inflammation compared to
BI.Tg.Ea mice
(both IA+ and IE+). Cardiac lesions in BI.Tg.Ea mice were confined to regions
of mononuclear
cell infiltration and were characterized by extensive myocyte dropout. Viral
titers also differed
between mouse strains with the highest titers occurring in AB° and
AB° Ea mice. This suggests
that IA expression is important in virus clearance. Also, the elevated viral
titers in AB° Ea mice
3o must not be directly responsible for the necrotic heart lesions in this
strain since AB° mice also
have elevated virus concentrations but no histological evidence of cardiac
injury. Thus, by either
animal mortality or histology, the presence of IE in C57BL/6 mice aggravated
CVB3-induced
CA 02324995 2000-10-OS
WO 99/53953 PCT1US99/06874
-85-
disease. Treating the BL tg Ea'' strain with antibodies to deplete y8 T cells
conferred resistance
to myocarditis. We measured cytokine profiles from total splenocytes of the
animals before and
after infection. We observed increased y-interferon in all strains and higher
levels of IL4 in the
BI.Tg.Ea'' animals. Depletion of y8 T cells resulted in an increased
percentage of cells producing
IL4 post infection (not shown). This result demonstrates that the cytokine
bias is important in
the development of myocarditic lesions.
Exaraple 5: MHC class II IE molecules confer protection from early
atherosclerotic fatty
lesions
io Several studies suggest that CD4+T lymphocytes contribute to the
pathogenesis of
fat-induced atherosclerotic lesions (Emeson, EE, et al., Am J Path, 1996,
149:675-685). We
addressed the possibility that expression of IA and/or IE impacted the
development of lesions
which result from a high fat diet.
C57BL/6 transgenic mice differing in MHC class II antigen expression were
kindly
supplied by Dr. Chella David (Mayo Clinic). Between 4 and 10 mice of each
strain were placed
on high-fat, high-cholesterol diet (Teklad #96354;20% total fat, 1.5%
cholesterol, 0.5 % sodium
cholate) at three weeks of age and were killed 15 weeks later for evaluation
of the aorta and
splenocytes. An additional group of 7 C57BL/6 mice were placed on high-fat
diet as above, but
were injected ip every two weeks with 100 ~.g monoclonal rat anti-CD4 antibody
(cione GK1.5;
2o American Type Tissue Collection, Bethesda, MD). This protocol has ben used
previously to
maintain CD4+ T cell-deficient mice for extended periods in the experimental
allergic
encephalomyelitis (EAE) model. The heart and ascending aorta including the
aortic arch were
removed and evaluated for atherosclerotic lesions according to the method of
Plump et al. using
oil red-0 stained serial sections. Briefly, hearts were fixed in 10% buffered
formalin, embedded
in 25% gelatin, grossly cut through the ventricles parallel to the atria,
frozen in OCT and
sectioned by cryostat. Ten micron thick sections were placed on 5% gelatin
coated slides, stained
with 0.24% oil Red-0 (neutral lipids) and counterstained with 2.4% hematoxylin
(nuclei and
basophilic tissue) and 0.25% light green (remaining tissue). Lesions were
quantified by area
morphometry using a compound light microscope.
Table 7. MHC class II IE molecules confer protection from early
atherosclerotic
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-86-
fatty lesions
Strain Treatment Fatty Cholesterol
Lesion
Heart Liver
C57BL/b + - No Treatment++ - Not Significant
(++
Lymp
h)
C57BL/6 + - Anti-CD4 + - Not Significant
AB - - No Treatment+/f +++ Not Significant
ABEak - + No Treatment-/f +++ Not Significant
BITgEak + + No Treatment-/f - Not Significant
(++
Lymp
h)
Our data demonstrate that the presence of MHC class II IA correlated with
susceptibility
to fatty lesions in the hearts of the C57BL/6 animals and that the presence of
IE molecules
conferred protection from the fatty lesions. The role of CD4 cells in this
process was confirmed
by the finding that removal of CD4 cells from the susceptible C57BL/6
abrogated the pathology
in the heart. Note the correlation between expression of IE, increased
production of IL-4, and
protection from fatty lesions resulting from a high fat diet. The results in
Table 7 demonstrate
t 5 that CD4+T cells contribute to early fat deposition in the aortic sinus
and that IE molecules
suppress lesions. MHC class II molecules, IA or IE, regulate susceptibility to
development of
early atherosclerotic plaques, and that cytokine profiles are altered (not
shown).
2o Example 6: NGF and EGF- dependent changes in Fas (CD95), B7.1 (CD80) and
B7.2
(CD86) expression on PC12, TrkA, and v-Crk neuronal cell surface.
materials and llilethnrls
Cell Culture: Rat pheochromocytoma cell lines, including PC12, Trk and v-Crk
cells
were a kind gift from Dr. Raymond Birge. They were maintained in complete RPMI
1640
25 (GIBCO) supplemented with 7% heat inactivated fetal calf serum and 3% heat
inactivated horse
serum at 37° C in a humidified incubator with S% C02. The PC12
transfectants were generated
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
_87_
as described previously (Glassman et al., 1997, Hempstead et al., 1994). Cells
were plated in 6
well culture plates at a concentration ranging from 2-8 x 106 cells/well,
according to the cell
type's growth kinetics, and 5 ml complete medium. NGF-7S from mouse
submaxillary glands
(Sigma Chemical Co.) or EGF, kindly provided by Dr. Raymond Birge, was added
at a
s concentration of 50 ng/ml culture medium and the cells were incubated for 24
or 48 hours.
Cytofluorometric Analysis: Cells were harvested after a 10 minute incubation
period on
ice in order to diminish their adherence to the plastic culture flask. They
were spun at 1210 rpm,
for 7 minutes, resuspended in medium and counted after trypan blue staining.
Equal numbers of
cells were placed in 12 x 75 mm flow tubes {range: 0.1-1 x 106 cells/tube),
washed in PBS and
1o S% FBS and stained at 4°C. The following stains were used: a)
fluorescein isothiocyanate
(FITC) hamster IgG isotype standard, b) FITC anti-mouse Fas, c) FITC anti-
mouse CD80 (B7.1 ),
d) phycoerythrin (PE) mouse IgG2b K isotype standard and e) PE anti-mouse CD86
(B7.2). All
antibodies were obtained from Pharmingen.
After incubation on ice for 20 minutes, cells were washed, resuspended in PBS
and 5%
15 FCS and analyzed by flow cytometry (Becton Dickinson). Histogram plots were
derived from
dot plots gated on the live cell population according to the forward versus
side scatter ratio (Cell
Quest Program). The absolute Fas, B7.1 and B7.2 values presented on the graphs
were obtained
by subtracting the geometric mean fluorescence of the specific antibody from
the mean geometric
fluorescence of the corresponding isotype. Values were considered subjectively
positive (+) if
20 the difference was statistically significant (p<0.001) according to the
applied Kilmogorov-
Smirnov statistical analysis.
Fas cell surface expression after NGF stimulation: The data showed that Fas is
25 constitutively expressed on the surface of PC 12 and TrkA cells. NGF
stimulation abrogates Fas
expression on PC 12 cells and paradoxically increases its expression on Trk
cells at 24 hours. Fas
levels tend to return to basal values on PC 12 cells and are maintained
constant on TrkA cells after
48 hours of NGF stimulation. V-Crk constitutive cell surface Fas expression is
minimal but
statistically significant, and it totally abrogated after NGF stimulation at
48 hours.
3o Fas cell surface expression after EGF stimulation: EGF stimulation at 24
and 48 hours
also downregulates Fas expression on PC 12 and Trk cells; EGF stimulation at
48 hours totally
abrogates Fas expression on both cell types. On the other hand, EGF
stimulation at 24 hours
CA 02324995 2000-10-OS
WO 99/53953 PCf/US99/06874
_88_
significantly up-regulates Fas expression on v-Crk cells, but it is again down-
regulated and
abrogated by EGF stimulation at 48 hours.
B7.1 cell surface expression after NGF stimulation: B7.1 is constitutively
highly
expressed on the surface of unstimulated PC12 and Trk cells. Its expression is
minimal on
unstimulated v-Crk cells. NGF stimulation initially downregulates B7.1
expression on PC12 cells
at 24 hours, but it tends to return to basal values at 48 hours. NGF
stimulation has no effect on
B7.1 expression on the surface of Trk cells and v-crk cells.
B7.1 cell surface expression after EGF stimulation: EGF stimulation at 24
hours
significantly lowers the detection of high B7. I levels on the surface of PC
12 cells; EGF
1o stimulation at 48 hours reestablishes the basal values. As in PC12 cells,
B7.1 expression is
lowered after EGF stimulation of Trk cells at 24 hours and reestablished at
the 48 hour time point.
EGF stimulation significantly increases B7.1 expression on v-Crk cells after
24 and 48 hours of
culture.
B7.2 cell surface expression after NGF stimulation: B7.2 cell surface
expression is
minimal on unstimulated PC 12 cells. NGF stimulation at 24 hours has no effect
on its expression
but stimulation at 48 hours completely abrogates its expression. Trk B7.2 cell
surface expression
is also minimal on unstimulated cells, it is slightly down-regulated by NGF
stimulation at 24
hours and abrogated by NGF withdrawal. There is no B7.2 cell surface
expression on v-Crk cells
nor is it induced by NGF stimulation.
2o B7.2 cell surface expression after EGF stimulation: EGF stimulation at 48
hours up-
regulates B7.2 expression on PC12 but this is most significant on the surface
of Trk and v-crk
cells. EGF stimulating at 48 hours is the only instance whereby there is
significant induction of
the constitutively absent B7.2 molecule on v-Crk cells. EGF withdrawal totally
rescinds this
effect and B7.2 levels are again undetectable.
In PC 12 cells and its mutant variants, NGF induces proteins required for the
acquisition
of a sympathetic neuronal phenotype, potentiating cellular differentiation as
reflected by an
increase in the size and flattening of the neuronal soma and particularly by
inducing neurite
outgrowth (Ray Paper, J. Bio Chem 1995). By contrast, EGF stimulation of these
cells induces
their entry into the cell cycle and thus, cellular proliferation, by binding
to another receptor also
3o belonging to the tyrosine kinase receptor family (Hempstead et al., 1994;
Siegel et al., 1994). The
NGF and EGF receptor pathways appear to be very similar since they both
activate the receptor-
type tyrosine kineses, the Erk2/MAPK pathway and involve the Ras proteins
(Chao, 1992; Ray
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
-89-
Id Menendez Iglesias et al., 1997) It has been found, according to the
invention, however, that
the effects of these molecules are quite different on the induction and
abrogation of Fas, B7.1 and
B7.2 expression on the neuronal cell surface suggesting that the induction of
these molecules by
growth factors NGF and EGF is mediated by a different infra-cellular signaling
pathways albeit
dependent on tyrosine kinase activation.
As described above Fas B7.1 and B7.2 are constitutively expressed on PC 12 and
TrkA
cells maintained in culture. Fas expression on PC 12 cells is significantly
decreased by NGF
stimulation (NGFS). Early NGFS of TrkA cells induces an increase in Fas
expression over basal
levels. Microglia constitutively express Fas ligand (Bonetti and Raine, 1997;
Menendez Iglesias
et al., 1997) and perhaps direct cell-cell contact between the neurons and
microglia is required
for the interaction of Fas and Fas ligand and the development of apoptosis or
a co-stimulatory
mitotic signal. In TrkA cells that overexpress the Trk receptor, NGF-induced
Fas expression can
promote cell division as NGF stimulates mitosis at that time period and
synchronously play a role
in the differentiation process and the development of filopodia. On TrkA
cells, the higher
induction of Fas expression correlates with the increased numbers of tyrosine
kinase A surface
receptors and thus, the development of a sustained increased stimulus for the
mRNA translation
of the Fas protein and its secondary expression on the cell surface.
EGF stimulation (EGFS) significantly diminishes and even abrogates Fas
expression on
PC12 and TrkA cells. However, its expression increases three-fold in v-Crk
cells transiently after
EGFS at 24 hours and disappears after EGFS at 48 hours. It has been shown that
EGFS induces
the development of neurite processes on the PC 12 v-crk mutant not on native
PC 12 cells
(Hempstead et al., 1994) and this clearly correlates with the induction of Fas
on the v-Crk
membrane cell surface, but does not explain the down-regulation of Fas
observed in Trk and
PC12 cells. EGF receptors are also expressed on cortical neurons, the
cerebellum and
hippocampus, and appear to act on mitotic cells and postmitotic neurons
(Yamada et al., 1997).
NGFS and NGFW condition a minor upregulation of B7.1 on PC12, TrkA and v-Crk
cells; however, NGFW at 48 hours does diminish B7.1 expression by 60% on v-Crk
cells. In
contrast, we consider that the effect of NGF on B7.2 on all three cell types
is negligible.
However, EGFS at 48 hours, significantly increases B7.1 expression on all cell
variants but its
expression clearly decreases after EGFW. EGFS at 48 hours also significantly
increase B7.2
expression on PC12 and Trk cells and induces its expression on v-Crk cells.
EGFW at 24 and 48
hours correlates with B7.2 down-regulation in PC12 and TrkA cells and
paradoxically increases
KCB'. VU~:L.F'.4-b1t'ENC'fli:~ 0~6 w'2- 5- a CA 0_2324995 2000-10-05720 '_'~1-
41~ +49 (3~~ -~3y~4.1-E ~ u~E;
U S 009906874
22-05-2000
-90-
87.2 cxprfrssion no v-Crk cells at 24 hours; it is immediately down-regulated
after EGF'f~V at 48
hours. Therefore, Fas expression on v-Crk cells and B~.I-B7.2 expression on
all cell types, but
particularly oa v~Crk cells, appear to be EGF-dependetrt, V-Crk cells are
characterized by the
presence of a fusion protein with viral gag sequences fused to the cellular
sequences of the Src
s, homology regions 2 and 3 (SH2 aad SI~i3) (Hempstead at at.,1994). C-arc
also possesses tyrosine
kiaasc activity (Vaingankar sad Mafins-Green, 1998) and perhaps these
modifications in its
sequence allow it to aM similarly to the EGF recxptor per se and increase the
signal intensity for
the expression of these cell surface molecules. Withdrawal of the stimulus
(IrGFW} reverts the
expression of these molecnlcs towards basal levels.
r o Idan~~cation oflmniune Recognition Molecules on treated versus rron-
lreatad Ganglia_
Ganglia, are removed from Po (one-day old mice) brain and plated into
cultures. The
~aparolad asu~~
sensory neurons do not have to be separabedavaay from Schwann cells. Isolated
ganglia arc
eultun~d for at least 72 hours under the following conditions:
f s . 1 } No Treatment
2} In the presence of nerve growth factor (NGF) for 55 hours
3) followed by harvest, wash, and replating in the presence of antibodies to
NGF.
Cells are harvested by cell scraping and dispersed into single cell
suspensions. Cells are sr.,ained
for cell surfac:c $?-1, B7-2, CDRB, and Trk A (NGF receptor) using monoclonal
antibodies to
2o these nlolxules.
The cells are cultured as described above but in the presence of CTLA-4~Hulg
to inhibit
cell interactions (synapses) which will protect Group I from death. This shows
that B7-bearing
cells cause the CD28+ pr CTLA4+ cell to release NGF and promote innovation.
Additionally
histological sections are stained by immv:ctorluoresccna (using the salt 87
and TrkA antibodies)
.,
2s inunediately rx vivo intact mouse brain.
Example 7 UCP i~ present is a panel of Tumor Cells
We extended our analysis of intracellular expression of UCf to other tumor
cells.
~"~4t~,lS ~1~
jatracUCP expression was examined flow cytametricaily on celis which had bc(:n
30 permcabilizcd anf stained as indicated. the histograms represent FITC
isotype control versus
stained with Rabbit anti-UCP (a kind gift of Mtuy Ellen I-Iurpcr) PITC-anti-
Rabbit outerstcp. A
Coulter Epics Elite flaw cytometcr with a single excitation waveiettgth (488
nm) and bid itlte:s
AMENDED SHEET
~tCV.~~(>f~:Et'A-41UEnCHt?~ u4 :2'3- ~- U CA 02324995 2000-10-05720 2441-~ +4~
~a 1:~9H44.E;~:nm
U S 009906874
22-05-2000
-91 -
for PE (575 nm}, FITC (525 nm) and Red613 {6I3 nm) was used to analyze the
stained cells.
Each sample population was classified for cell size (forward scatter) and
complexity (side scatter),
gated on a population of interest and evaluated using 40,000 cells. Crteria
for positive staining
were established by comparison with isotype controls, thin lines to apec~c
stain, thick lines.
AlI of the tumor cells lines examined express UCP intracellularly. These data
are
consistent with the possibility that expression of UCP in tumor cells is
gencralizable to all tunc~or
cells, and likely results from the well documented shift in subccllular
production of ATP from
mitochondria to cytosol as eetls divide. Importantly, these data also
demonstrate that expression
of UCP2 is not specific to lymphoid tumors. The L929 cells are fibroblasts and
the PC12 Trk
t0 cells which are derived from pheochromocytoma cell Lines, respectively. The
EL4 cells arc a
mouse thymoma cell lint and Jurkat arc hutttan T cell tumor cells.
To confirm that tlow cytometrically defeated UCP expression was nutochondrial,
we
isolated mitochondria from L1210 and LI2I0 DDP, and performed Western Blot
analysis
blotting with rabbit anti-UCP antibodies. Mitochondria were isolated using
differential
t s , centrifugation as adapted firom REF, REF (Reinhart, PfI, Taylor, WM and
Bygravc FL (1982)
A procedure far the rapid preparation of mitochondria from rat liver.
Biochern. J. 204: 73 i-735.
and Sims hIR (1990~Rapi~isoladon of metabolically active mitochondria from rat
brain and
~ cou.
subregions using Pdensity gradient centrifugation J. Ncurochem. 55:698-707.)
The
following samples were run: molecular weight markers (BIORAD Biotinylated SDS-
PACE
2o standards; LI210/0 mitochoridtial protein (40 ugh from two distinct
cnitochoadrial preparations;
LI210/DDP mitochondria) protein (40 ~zg) from two distinct mitochondria)
preparations; and
uncoupling protein standard (0.75 ug) ~rom rat brown adipose tissue (which
expresses UCPs 1-
3}. Rabbit anti-hamster UCP was used at a dilation of 16,000. The secondary
antibody: goat
anti-rabbit IgG conjugated to I3RP at 1:10,000. Chemilumineseent detection:
A.mcrsharrt ECL kit.
25 , ~ The blot showed greater levels of mitocliondrial UCP in the drug
resistant L 1210/DDP
than in L1210/0. The detectsd mitochondria protein has an approximate
molecular weight of 30
kDa, close to the predicted molecular weight of UCP2 (33 kDa).
To determine whether increased UCP. corresponded to increased mit~ochondrial
proton
leak and a lower mitochondria) membrane potential (O~Fm) we assessed
characteristics of non-
phosphorylating respiration in intact L 1210 wild type and L 1210 DDP cells.
State 4 aim in DDP
cells, x mV, was siSniFcantly lower than in wild type cells, y mV (p < 0.001
), and state 4 oxygen
consumption in DDP cells is signi$cantly higher than in wild type cells,
indicating increased
AMENDED SHEET
Kw.W wL:YA-btU~~CHI~~ u4 :y?- v- i, CA 0_2324995 2000-10-057''i> '-"~'~1~
+'~'~ 89 '?a99~4E :at9
U S 009906874
22-05-2000
-92-
mitochondria) proton leak.
Example 8: Rates of Glucose Utilization, oxidation and Cell surface and
Intrac>cUularFas
levels in Melanoma Dells.
s H I 6 cells were cultured in the .~.t~re presence of different
concentrations of sodium
acetate and Fas expression was measured. The data shows the level of cell
surface Fns expression
on non-permeabilized and intracellular Fas expression in permeabili~xd H 16
melanoma cells.
With increasing concentrations of sodium acetate, the levels of intracellular
Fas declined and the
levels of eel l surface Fns iracreascd, demonstrating a tzanstoeation of Fns
from intracellular stores
~ o to the surface.
Next vhe rates of glucose utilization and oxidation in B 16 melanoma cells was
determined.
Alain cells were cultured is the presence of varying concentration of sodium
acetate. Both
glucose utilization and glucose oxidation (measured is nmoles) decreased with
increasing
concentrations of sodium acetate, demonstrating a correlation witH expression
of cell surface Fns
t 5 . in the same cells.
Example 9: hIormal mouse T cells express lE
1°revious work with human T cells indicates that activation of the T
cells by antigens
or engagement of CD4 results in expression of I3LA pR on the T cell surface.
Expression of
2o MHC class II on mouse T cells is controversial. Reports indicate both
posftive and negative
results. The studies to date did not distinguish bctwecxi failure to express
IA versus IE. To
address the possibility that normal mouse T cells express IE we isolated Iymph
nodes or spleens
as indicated from strains of animals which express IF., Balblc, CBA,, and AKR
mice, spleens or
nodes were taken from 4 week old mice, minced to single cell suspension, and
red blood cells
,,
2s were removed via Gey's treatrnent. Splenocytes were then passed over
Cellect Columns
(Cytovax, Edmonton, Canada) to purify CD4' T cells. CD4~ T cells were
collected, found to be
98.5% puck and contaminants were identifeed as NK and yd T cells flow
cytometrically. The
CD4 T cells vr~rc treated with antibody to CD4 (C3KI.S) at lOUgIrnUIO' cells.
washed and
treated with rabbit anti-rat antibody for 45 minutes at 37° C, followed
by washing. The coils were
30 cultured overnight and stained with FTTC conijugated anti-IE antibody (14-
4..4 S), or 14-4.45 and
counterstained with anti-T'CR.
ror the PCR experiment purified CD4' T cells, Sx t0°Iml, were incubated
for 8 hrs.
AMENDED SHEET
IZC\'.vU~V:La'A-h1l~rNCtIE:V 04 :po_ ~_ ,~ CA _02324995 2000-10-057'?u 2441-~
+4.~a 8J '':~ys~.l.~~s~:at~~
U S 009906874
22-05-2000
-93-
with biotinylated antibodies for CD4 (GKI.S), CD28, CD3 (145.2C11) alone, CD4
and CD28,
CD3 and CD28, a: no treatment. lxperitneutal setup included wells of purified
T ells and
~EKCou..~
Percoil isolated 8 cells added to control for potential MHC Class II'
contaminants. 8 cells, Sx 1 OS
cells, which is 5% of purified T cells (far greater than the I.5°lo
contaminants seen following
s CelIect Coluam purificetion)werc added to T cell wells. Cells were then
washed, collected and
total RNA was isolated using an RNA isolation kit, RIv'Easy (Qiegen,
Chatsworth CA). Single
SuPH~.SC~~
strand DNA was generated from 2~tg of RNA using-6~spg~s~t II reverse
transcriptase
(GIBCoIBRL Gaithersburg, MD). PCR was done using MHC Class II, (I-E, exon 3 5'-
TAGCTGAGCCCAAGGTGACT and 5' TCACCAGCrGTCTGGTAGGTC) primers. PCR
to protocol was: 1 min at 94°C, 1 min at 60°C, and 2 min. at
72°C for 35 cycles. Followir~ PCR,
samples were loaded onto 1% agarose gals, stained vtrith ethidium bromide and
visualized with
Uv light.
The results demonstrated that IE was expressed in CD4 or GD4 and CD3 treatod
cells
but not in cells receiving no treatment. The cells treated with CD3 or CD28
showed low levels
~ s . o f expression.
lrxample 10: Use of fgtty acids as a mitochondria) carbon source
The use of fatty acid (Oleic Acid) as a mitochondria.L carbon source was
measured.
Rate of oleatc oxidation wits maasurcd by incubating cells for 90 min at
37°C in 1 OOUI of reaction
buffer, glucose {2,8. 8.3, 27.7 mmoUl~ l.7mCi (U-14C olcaic acid). The
reaction was carried out
zo in a 1 ml cup in a 20 ml scintillation vial capped by a rubber stopper with
a center welt that
contains filter paper. Metabolism was stoppod and COZ liberated with 300~e! 1
rnol/1 I-iCl
injected through the stopper into the cup containing the cells. COi was
trapped it: the fitter paper
by injecting 1 Omt I moll) KOH into the center welt, followed 2 hours Inter by
liquid scintillation
.,
counting. Tubca containing NaHCO~ and no cells were used to estimate the
recovery of "COa
zs in the filter paper, routinely close to 100%. Values indicate the rate of
C02 production by
L 1 Z 1 ODDP cells or L I 21 G ctIls. The L 1210 DDP use oleic acid at much
higher rates than ti:e
L.I210 cells.
Example l I: CAMP Iovtls io LI2I0 and L1ZIODDP
3o cAMP levels in L1210 versus L1210DDP were exami:led. Increasing
intracellular
levels of cAMP are necessary for the activity of uncoupling proteins. We have
shown that class
LT engagement results in increased CAMP and we have determined that the
mitochondria)
AMENDED SHEET
rcw. ~U~:t~1'A-11UGVCHi~:~ U4. :y°o_ ~_ ~~ C_A 023_2_4995 2000-10-
057'?0 x441- +;~~ t.:t :"
U S 009906874
22-05-2000
-94-
membrane potential of L1210DDP cells is lower then LI210 cells. Thus, we used
a
radioimmunoassay to detaminc the levels of ctLMP in L 1210, Icft panel versus
L I2I ODDP, right
panel. Cells were treated for I O minutes with nothing, antibodies to IA, IE,
or with a beta
adrenergic agonise', isoprotsrenol (10 microMolar). Cells were harvested and
CAMP was
s extracted from the cells and cAMP levels determined using 125 I labeled cAMP
in competitive
inhibition in the presence of antibodies to cAMP, radioimmunoassay.
eAMP levels were significantly highs in L1210DDP cells than L1210 cells, in
untreated cells as wetI as cells tre~tttd with antibodies to IA, IF, or with a
beta adrGncrgic agonise,
isoproterenol.
Example 12: Sodium Acetate as a mitochondrisl modifying agent.
Sodium Acetate as a ttiitochondrial rtiodifying agent was examined. L1210 or
L121 ODDP cxiIs were culturod in the prtsoncc of graded concentrations of
sodium acetate in the
medium. Cells were stained with Jo2.2, a tluorescein conjugatod anti-Fns
antibody, or an aotype
15 . control. CeII surface staining was measured flow cytometrically. The
Percentage of mean
fluorescence intensity over the isotype control was plotted. The data indicate
that the presence
of acetate increases cell surface Fns expression in both cell lines.
'phe effects of acetate on susceptibility to Fas~iependent cell death were
also
examined. Calls cultured with acetate were loaded with 51 Cr and plated onto
FasLbearing or
2o mock transfected fibrobIast to determine sensitivity to Fns-induced cell
death. Results are
reported as percent chromium release from cells in the presence of Fast
bearing oetls over mock-
traasfcictants. The data indicate that in a dose dependent manner, culture of
both cell types with
acetate results in susceptibility to Fns-dependent cell death.
Cacl~ of the fo oing patents, patent applications and re ~rences is hereby
'.
25 incorporated by reference, include US Provisional Applica ' erial No.
60!082,250 f led
April 17, 1998, US Provisional Applicatt Serial hio 01,580 fried Septemoer 24,
1998 and
US Provisional Application Serial No. 4,519 filed July 24, 1998, from which
this
application claims priority under SC $119(e). ile the invention has boon
described with
respect to certain embo ' ts, it should be appreciate t many modifications and
changes
3o may be made b~ ase of ordinary skill in the art without parting from the
spirit of the
invc;ntion is intended that such modiClcation, changes and rquiva is fall
within the scope of
ClalIllS.
AMENDED SHEET ** TOTC,I. PRGE.20 **
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
SEQUENCE LISTING
<110> University of Vermont and State Agricultural College
<120> METHODS AND PRODUCTS RELATED TO
METABOLIC INTERACTIONS IN DISEASE
<130> V0139/7028W0/HK
<150> U.S. 60/082,250
<151> 1998-04-17
<150> U.S. 60/094,519
<151> 1998-07-29
<150> U.S. 60/101,580
<151> 1998-09-24
<160> 13
<170> FastSEQ for Windows Version 3.0
<210> 1
<211> 1491
<212> DNA
<213> Homo Sapiens
<400>
1
ccaaagaaaaagtgatttgtcattgctttatagactgtaagaagagaacatctcagaagt 60
ggagtcttaccctgaaatcaaaggatttaaagaaaaagtggaatttttcttcagcaagct 120
gtgaaactaaatccacaacctttggagacccaggaacaccctccaatctctgtgtgtttt 180
gtaaacatcactggagggtcttctacgtgagcaattggattgtcatcagccctgcctgtt 240
ttgcacctgggaagtgccctggtcttacttgggtccaaattgttggctttcacttttgac 300
cctaagcatctgaagccatgggccacacacggaggcagggaacatcaccatccaagtgtc 360
catacctcaatttctttcagctcttggtgctggctggtctttctcacttctgttcaggtg 420
ttatccacgtgaccaaggaagtgaaagaagtggcaacgctgtcctgtggtcacaatgttt 480
ctgttgaagagctggcacaaactcgcatctactggcaaaaggagaagaaaatggtgctga 540
ctatgatgtctggggacatgaatatatggcccgagtacaagaaccggaccatctttgata 600
tcactaataacctctccattgtgatcctggctctgcgcccatctgacgagggcacatacg 660
agtgtgttgttctgaagtatgaaaaagacgctttcaagcgggaacacctggctgaagtga 720
cgttatcagtcaaagctgacttccctacacctagtatatctgactttgaaattccaactt 780
ctaatattagaaggataatttgctcaacctctggaggttttccagagcctcacctctcct 840
ggttggaaaatggagaagaattaaatgccatcaacacaacagtttcccaagatcctgaaa 900
ctgagctctatgctgttagcagcaaactggatttcaatatgacaaccaaccacagcttca 960
tgtgtctcatcaagtatggacatttaagagtgaatcagaccttcaactggaatacaacca 1020
agcaagagcattttcctgataacctgctcccatcctgggccattaccttaatctcagtaa 1080
atggaatttttgtgatatgctgcctgacctactgctttgccccaagatgcagagagagaa 1140
ggaggaatgagagattgagaagggaaagtgtacgccctgtataacagtgtccgcagaagc 1200
aaggggctgaaaagatctgaaggtagcctccgtcatctcttctgggatacatggatcgtg 1260
gggatcatgaggcattcttcccttaacaaatttaagctgttttacccactacctcacctt 1320
cttaaaaacctctttcagattaagctgaacagttacaagatggctggcatccctctcctt 1380
tctccccatatgcaatttgcttaatgtaacctcttcttttgccatgtttccattctgcca 1440
tcttgaattgtcttgtcagccaattcattatctattaaacactaatttgag 1491
-1-
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99106874
<210> 2
<211> 288
<212> PRT
<213> Homo Sapiens
<400> 2
Met Gly His Thr Arg Arg Gln Gly Thr Ser Pro Ser Lys Cys Pro Tyr
1 5 10 15
Leu Asn Phe Phe Gln Leu Leu Val Leu Ala Gly Leu Ser His Phe Cys
20 25 30
Ser Gly Val Ile His Val Thr Lys Glu Val Lys Glu Val Ala Thr Leu
35 40 45
Ser Cys Gly His Asn Val Ser Val Glu Glu Leu Ala Gln Thr Arg Ile
50 55 60
Tyr Trp Gln Lys Glu Lys Lys Met Val Leu Thr Met Met Ser Gly Asp
65 70 75 BO
Met Asn Ile Trp Pro Glu Tyr Lys Asn Arg Thr Ile Phe Asp Ile Thr
85 90 95
Asn Asn Leu Ser Ile Val Ile Leu Ala Leu Arg Pro Ser Asp Glu Gly
100 105 110
Thr Tyr Glu Cys Val Val Leu Lys Tyr Glu Lys Asp Ala Phe Lys Arg
115 120 125
Glu His Leu Ala Glu Val Thr Leu Ser Val Lys Ala Asp Phe Pro Thr
130 135 140
Pro Ser Ile Ser Asp Phe Glu Ile Pro Thr Ser Asn Ile Arg Arg Ile
145 150 155 160
Ile Cys Ser Thr Ser Gly Gly Phe Pro Glu Pro His Leu Ser Trp Leu
165 170 175
Glu Asn Gly Glu Glu Leu Asn Ala Ile Asn Thr Thr Val Ser Gln Asp
180 185 190
Pro Glu Thr Glu Leu Tyr Ala Val Ser Ser Lys Leu Asp Phe Asn Met
195 200 205
Thr Thr Asn His Ser Phe Met Cys Leu Ile Lys Tyr Gly His Leu Arg
210 215 220
Val Asn Gln Thr Phe Asn Trp Asn Thr Thr Lys Gln Glu His Phe Pro
225 230 235 240
Asp Asn Leu Leu Pro Ser Trp Ala Ile Thr Leu Ile Ser Val Asn Gly
245 250 255
Ile Phe Val Ile Cys Cys Leu Thr Tyr Cys Phe Ala Pro Arg Cys Arg
260 265 270
Glu Arg Arg Arg Asn Glu Arg Leu Arg Arg Glu Ser Val Arg Pro Val
275 280 285
<210> 3
<211> 1424
<212> DNA
<213> Homo Sapiens
<400> 3
aggagccttaggaggtacggggagctcgcaaatactccttttggtttattcttaccacct 60
tgcttctgtgttccttgggaatgctgctgtgcttatgcatctggtctctttttggagcta 120
cagtggacaggcatttgtgacagcactatgggactgagtaacattctctttgtgatggcc 180
ttcctgctctctggtgctgctcctctgaagattcaagcttatttcaatgagactgcagac 240
ctgccatgccaatttgcaaactctcaaaaccaaagcctgagtgagctagtagtattttgg 300
caggaccaggaaaacttggttctgaatgaggtatacttaggcaaagagaaatttgacagt 360
gttcattccaagtatatgggccgcacaagttttgattcggacagttggaccctgagactt 420
cacaatcttcagatcaaggacaagggcttgtatcaatgtatcatccatcacaaaaagccc 480
-2-
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
acaggaatgattcgcatccaccagatgaattctgaactgtcagtgcttgctaacttcagt 540
caacctgaaatagtaccaatttctaatataacagaaaatgtgtacataaatttgacctgc 600
tcatctatacacggttacccagaacctaagaagatgagtgttttgctaagaaccaagaat 660
tcaactatcgagtatgatggtattatgcagaaatctcaagataatgtcacagaactgtac 720
gacgtttccatcagcttgtctgtttcattccctgatgttacgagcaatatgaccatcttc 780
tgtattctggaaactgacaagacgcggcttttatcttcacctttctctatagagcttgag 840
gaccctcagcctcccccagaccacattccttggattacagctgtacttccaacagttatt 900
atatgtgtgatggttttctgtctaattctatggaaatggaagaagaagaagcggcctcgc 960
aactcttataaatgtggaaccaacacaatggagagggaagagagtgaacagaccaagaaa 1020
agagaaaaaatccatatacctgaaagatctgatgaagcccagcgtgtttttaaaagttcg 1080
aagacatcttcatgcgacaaaagtgatacatgtttttaattaaagagtaaagcccataca 1140
agtattcattttttctaccctttcctttgtaagttcctgggcaacctttttgatttcttc 1200
cagaaggcaaaaagacattaccatgagtaataagggggctccaggactccctctaagtgg 1260
aatagcctccctgtaactccagctctgctccgtatgccaagaggagactttaattctctt 1320
actgcttcttttcacttcagagcacacttatgggccaagcccagcttaatggctcatgac 1380
ctggaaataaaatttaggaccaataaaaaaaaaaaaaaaaaaaa 1424
<210> 4
<211> 323
<212> PRT
<213> Homo Sapiens
<400> 4
Met Gly Leu Ser Asn Ile Leu Phe Val Met Ala Phe Leu Leu Ser Gly
1 5 10 15
Ala Ala Pro Leu Lys Ile Gln Ala Tyr Phe Asn Glu Thr Ala Asp Leu
20 25 30
Pro Cys Gln Phe Ala Asn Ser Gln Asn Gln Ser Leu Ser Glu Leu Val
35 40 45
Val Phe Trp Gln Asp Gln Glu Asn Leu Val Leu Asn Glu Val Tyr Leu
50 55 60
Gly Lys Glu Lys Phe Asp Ser Val His Ser Lys Tyr Met Gly Arg Thr
65 70 75 80
Ser Phe Asp Ser Asp Ser Trp Thr Leu Arg Leu His Asn Leu Gln Ile
85 90 95
Lys Asp Lys Gly Leu Tyr Gln Cys Ile Ile His His Lys Lys Pro Thr
100 105 110
Gly Met Ile Arg Ile His Gln Met Asn Ser Glu Leu Ser Val Leu Ala
115 120 125
Asn Phe Ser Gln Pro Glu Ile Val Pro Ile Ser Asn Ile Thr Glu Asn
130 135 140
Val Tyr Ile Asn Leu Thr Cys Ser Ser Ile His Gly Tyr Pro Glu Pro
145 150 155 160
Lys Lys Met Ser Val Leu Leu Arg Thr Lys Asn Ser Thr Ile Glu Tyr
165 170 175
Asp Gly Ile Met Gln Lys Ser Gln Asp Asn Val Thr Glu Leu Tyr Asp
180 185 190
Val Ser Ile Ser Leu Ser Val Ser Phe Pro Asp Val Thr Ser Asn Met
195 200 205
Thr Ile Phe Cys Ile Leu Glu Thr Asp Lys Thr Arg Leu Leu Ser Ser
210 215 220
Pro Phe Ser Ile Glu Leu Glu Asp Pro Gln Pro Pro Pro Asp His Ile
225 230 235 240
Pro Trp Ile Thr Ala Val Leu Pro Thr Val Ile Ile Cys Val Met Val
245 250 255
Phe Cys Leu Ile Leu Trp Lys Trp Lys Lys Lys Lys Arg Pro Arg Asn
260 265 270
-3-
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
Ser Tyr Lys Cys Gly Thr Asn Thr Met Glu Arg Glu Glu Ser Glu Gln
275 280 285
Thr Lys Lys Arg Glu Lys Ile His Ile Pro Glu Arg Ser Asp Glu Ala
290 295 300
Gln Arg Val Phe Lys Ser Ser Lys Thr Ser Ser Cys Asp Lys Ser Asp
305 310 315 320
Thr Cys Phe
<210> 5
<211> 924
<212> DNA
<213> Homo Sapiens
<400>
atggggggcctgacagcctcggacgtacacccgaccctgggggtccagctcttctcagct 60
ggaatagcggcgtgcttggcggacgtgatcaccttcccgctggacacggccaaagtccgg 120
ctccaggtccaaggtgaatgcccgacgtccagtgttattaggtataaaggtgtcctggga 180
acaatcaccgctgtggtaaaaacagaagggcggatgaaactctacagcgggctgcctgcg 240
gggcttcagcggcaaatcagctccgcctctctcaggatcggcctctacgacacggtccag 300
gagttcctcaccgcagggaaagaaacagcacctagtttaggaagcaagattttagctggt 360
ctaacgactggaggagtggcagtattcattgggcaacccacagaggtcgtgaaagtcaga 420
cttcaagcacagagccatctccacggaatcaaacctcgctacacggggacttataatgcg 480
tacagaataatagcaacaaccgaaggcttgacgggtctttggaaagggactactcccaat 540
ctgatgagaagtgtcatcatcaattgtacagagctagtaacatatgatctaatgaaggag 600
gcctttgtgaaaaacaacatattagcagatgacgtcccctgccacttggtgtcggctctt 660
atcgctggattttgcgcaacagctatgtcctccccggtggatgtagtaaaaaccagattt 720
attaattctccaccaggacagtacaaaagtgtgcccaactgtgcaatgaaagtgttcact 780
aacgaaggaccaacggctttcttcaaggggttggtaccttccttcttgcgacttggatcc 840
tggaacgtcattatgtttgtgtgctttgaacaactgaaacgagaactgtcaaagtcaagg 900
cagactatggactgtgccacataa 924
<210> 6
<211> 307
<212> PRT
<213> Homo Sapiens
<400> 6
Met Gly Gly Leu Thr Ala Ser Asp Val His Pro Thr Leu Gly Val Gln
1 5 10 15
Leu Phe Ser Ala Gly Ile Ala Ala Cys Leu Ala Asp Val Ile Thr Phe
20 25 30
Pro Leu Asp Thr Ala Lys Val Arg Leu Gln Val Gln Gly Glu Cys Pro
35 40 45
Thr Ser Ser Val Ile Arg Tyr Lys Gly Val Leu Gly Thr Ile Thr Ala
50 55 60
Val Val Lys Thr Glu Gly Arg Met Lys Leu Tyr Ser Gly Leu Pro Ala
65 70 75 80
Gly Leu Gln Arg Gln Ile Ser Ser Ala Ser Leu Arg Ile Gly Leu Tyr
85 90 95
Asp Thr Val Gln Glu Phe Leu Thr Ala Gly Lys Glu Thr Ala Pro Ser
100 105 110
Leu Gly Ser Lys Ile Leu Ala Gly Leu Thr Thr Gly Gly Val Ala Val
115 120 125
Phe Ile Gly Gln Pro Thr Glu Val Val Lys Val Arg Leu Gln Ala Gln
130 135 140
Ser His Leu His Gly Ile Lys Pro Arg Tyr Thr Gly Thr Tyr Asn Ala
-4-
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
145 150 155 160
Tyr Arg Ile Ile Ala Thr Thr Glu Gly Leu Thr Gly Leu Trp Lys Gly
165 170 175
Thr Thr Pro Asn Leu Met Arg Ser Val Ile Ile Asn Cys Thr Glu Leu
180 185 190
Val Thr Tyr Asp Leu Met Lys Glu Ala Phe Val Lys Asn Asn Ile Leu
195 200 205
Ala Asp Asp Val Pro Cys His Leu Val Ser Ala Leu Ile Ala Gly Phe
210 215 220
Cys Ala Thr Ala Met Ser Ser Pro Val Asp Val Val Lys Thr Arg Phe
225 230 235 240
Ile Asn Ser Pro Pro Gly Gln Tyr Lys Ser Val Pro Asn Cys Ala Met
245 250 255
Lys Val Phe Thr Asn Glu Gly Pro Thr Ala Phe Phe Lys Gly Leu Val
260 265 270
Pro Ser Phe Leu Arg Leu Gly Ser Trp Asn Val Ile Met Phe Val Cys
275 280 285
Phe Glu Gln Leu Lys Arg Glu Leu Ser Lys Ser Arg Gln Thr Met Asp
290 295 300
Cys Ala Thr
305
<210> 7
<211> 1105
<212> DNA
<213> Homo Sapiens
<400>
7
gttcctctatctcgtcttgttgctgattaaaggtgcccctgtctccagtttttctccatc60
tcctgggacgtagcaggaaatcagcatcatggttgggttcaaggccacagatgtgccccc120
tactgccactgtgaagtttcttggggctggcacagctgcctgcatcgcagatctcatcac180
ctttcctctggatactgctaaagtccggttacagatccaaggagaaagtcaggggccagt240
gcgcgctacagccagcgcccagtaccgcggtgtgatgggcaccattctgaccatggtgcg300
tactgagggcccccgaagcctctacaatgggctggttgccggcctgcagcgccaaatgag360
ctttgcctctgtccgcatcggcctgtatgattctgtcaaacagttctacaccaagggctc420
tgagcatgccagcattgggagccgcctcctagcaggcagcaccacaggtgccctggctgt480
ggctgtggcccagcccacggatgtggtaaaggtccgattccaagctcaggcccgggctgg540
aggtggtcggagataccaaagcaccgtcaatgcctacaagaccattgcccgagaggaagg600
gttccggggcctctggaaagggacctctcccaatgttgctcgtaatgccattgtcaactg660
tgctgagctggtgacctatgacctcatcaaggatgccctcctgaaagccaacctcatgac720
agatgacctcccttgccacttcacttctgcctttggggcaggcttctgcaccactgtcat780
cgcctcccctgtagacgtggtcaagacgagatacatgaactctgccctgggccagtacag840
tagcgctggccactgtgcccttaccatgctccagaaggaggggccccgagccttctacaa900
agggttcatgccctcctttctccgcttgggttcctggaacgtggtgatgttcgtcaccta960
tgagcagctgaaacgagccctcatggctgcctgcacttcccgagaggctcccttctgagc1020
ctctcctgctgctgacctgatcacctctggctttgtctctagccgggccatgctttcctt1080
ttcttccttctttctcttccctccg 1105
<210> 8
<211> 309
<212> PRT
<213> Homo Sapiens
<400> 8
Met Val Gly Phe Lys Ala Thr Asp Val Pro Pro Thr Ala Thr Val Lys
1 5 10 15
Phe Leu Gly Ala Gly Thr Ala Ala Cys Ile Ala Asp Leu Ile Thr Phe
-5-
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
20 25 30
Pro Leu Asp Thr Ala Lys Val Arg Leu Gln Ile Gln Gly Glu Ser Gln
35 40 45
Gly Pro Val Arg Ala Thr Ala Ser Ala Gln Tyr Arg Gly Val Met Gly
50 55 60
Thr Ile Leu Thr Met Val Arg Thr Glu Gly Pro Arg Ser Leu Tyr Asn
65 70 75 80
Gly Leu Val Ala Gly Leu Gln Arg Gln Met Ser Phe Ala Ser Val Arg
85 90 95
Ile Gly Leu Tyr Asp Sex Val Lys Gln Phe Tyr Thr Lys Gly Ser Glu
100 105 110
His Ala Ser Ile Gly Ser Arg Leu Leu Ala Gly Ser Thr Thr Gly Ala
115 120 125
Leu Ala Val Ala Val Ala Gln Pro Thr Asp Val Val Lys Val Arg Phe
130 135 140
Gln Ala Gln Ala Arg Ala Gly Gly Gly Arg Arg Tyr Gln Ser Thr Val
145 150 155 160
Asn Ala Tyr Lys Thr Ile Ala Arg Glu Glu Gly Phe Arg Gly Leu Trp
165 170 175
Lys Gly Thr Ser Pro Asn Val Ala Arg Asn Ala Ile Val Asn Cys Ala
180 185 190
Glu Leu Val Thr Tyr Asp Leu Ile Lys Asp Ala Leu Leu Lys Ala Asn
195 200 205
Leu Met Thr Asp Asp Leu Pro Cys His Phe Thr Ser Ala Phe Gly Ala
210 215 220
Gly Phe Cys Thr Thr Val Ile Ala Ser Pro Val Asp Val Val Lys Thr
225 230 235 240
Arg Tyr Met Asn Ser Ala Leu Gly Gln Tyr Ser Ser Ala Gly His Cys
245 250 255
Ala Leu Thr Met Leu Gln Lys Glu Gly Pro Arg Ala Phe Tyr Lys Gly
260 265 270
Phe Met Pro Ser Phe Leu Arg Leu Gly Ser Trp Asn Val Val Met Phe
275 280 285
Val Thr Tyr Glu Gln Leu Lys Arg Ala Leu Met Ala Ala Cys Thr Ser
290 295 300
Arg Glu Ala Pro Phe
305
<210> 9
<211> 1132
<212> DNA
<213> Homo Sapiens
<400>
9
tcctgggatggagccctagggagcccctgtgctgcccctgccgtggcaggactcacagcc 60
ccaccgctgcactgaagcccagggctgtggagcagcctctctccttggacctcctctcgg 120
ccctaaagggactgggcagagccttccaggactatggttggactgaagccttcagacgtg 180
cctcccaccatggctgtgaagttcctgggggcaggcacagcagcctgttttgctgacctc 240
gttacctttccactggacacagccaaggtccgcctgcagatccagggggagaaccaggcg 300
gtccagacggcccggctcgtgcagtaccgtggcgtgctgggcaccatcctgaccatggtg 360
cggactgagggtccctgcagcccctacaatgggctggtggccggcctgcagcgccagatg 420
agcttcgcctccatccgcatcggcctctatgactccgtcaagcaggtgtacacccccaaa 480
ggcgcggacaactccagcctcactacccggattttggccggctgcaccacaggagccatg 540
gcggtgacctgtgcccagcccacagatgtggtgaaggtccgatttcaggccagcatacac 600
ctcgggccatccaggagcgacagaaaatacagcgggactatggacgcctacagaaccatc 660
gccagggaggaaggagtcaggggcctgtggaaaggaactttgcccaacatcatgaggaat 720
gctatcgtcaactgtgctgaggtggtgacctacgacatcctcaaggagaagctgctggac 780
-6-
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
taccacctgctcactgacaacttcccctgccactttgtctctgcctttggagccggcttc 840
tgtgccacagtggtggcctccccggtggacgtggtgaagacccggtatatgaactcacct 900
ccaggccagtacttcagccccctcgactgtatgataaagatggtggcccaggagggcccc 960
acagccttctacaaggggtgagcctcctcctgcctccagcactccctcccagagaacagg 1020
ggcttctttcttttcgaatgtggctaccgtgggtcaacctgggatgtagcggtgaagagt 1080
acagatgtaaatgccacaaagaagaagtttaaaaaaccatgcaaaaaaaaas 1132
<210> 10
<211> 275
<212> PRT
<213> Homo Sapiens
<400> 10
Met Val Gly Leu Lys Pro Ser Asp Val Pro Pro Thr Met Ala Val Lys
1 5 10 15
Phe Leu Gly Ala Gly Thr Ala Ala Cys Phe Ala Asp Leu Val Thr Phe
20 25 30
Pro Leu Asp Thr Ala Lys Val Arg Leu Gln Ile Gln Gly Glu Asn Gln
35 40 45
Ala Val Gln Thr Ala Arg Leu Val Gln Tyr Arg Gly Val Leu Gly Thr
50 55 60
Ile Leu Thr Met Val Arg Thr Glu Gly Pro Cys Ser Pro Tyr Asn Gly
65 70 75 BO
Leu Val Ala Gly Leu Gln Arg Gln Met Ser Phe Ala Ser Ile Arg Ile
85 90 95
Gly Leu Tyr Asp Ser Val Lys Gln Val Tyr Thr Pro Lys Gly Ala Asp
100 105 110
Asn Ser Ser Leu Thr Thr Arg Ile Leu Ala Gly Cys Thr Thr Gly Ala
115 120 125
Met Ala Val Thr Cys Ala Gln Pro Thr Asp Val Val Lys Val Arg Phe
130 135 140
Gln Ala Ser Ile His Leu Gly Pro Ser Arg Ser Asp Arg Lys Tyr Ser
145 150 155 160
Gly Thr Met Asp Ala Tyr Arg Thr Ile Ala Arg Glu Glu Gly Val Arg
165 170 175
Gly Leu Trp Lys Gly Thr Leu Pro Asn Ile Met Arg Asn Ala Ile Val
180 185 190
Asn Cys Ala Glu Val Val Thr Tyr Asp Ile Leu Lys Glu Lys Leu Leu
195 200 205
Asp Tyr His Leu Leu Thr Asp Asn Phe Pro Cys His Phe Val Ser Ala
210 215 220
Phe Gly Ala Gly Phe Cys Ala Thr Val Val Ala Ser Pro Val Asp Val
225 230 235 240
Val Lys Thr Arg Tyr Met Asn Ser Pro Pro Gly Gln Tyr Phe Ser Pro
245 250 255
Leu Asp Cys Met Ile Lys Met Val Ala Gln Glu Gly Pro Thr Ala Phe
260 265 270
Tyr Lys Gly
275
<210> 11
<211> 1514
<212> DNA
<213> Homo Sapiens
<400> 11
agactctcag gccttggcag gtgcgtcttt cagttcccct cacacttcgg gttcctcggg 60
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
gaggaggggctggaaccctagcccatcgtcaggacaaagatgctcaggctgctcttggct 120
ctcaacttattcccttcaattcaagtaacaggaaacaagattttggtgaagcagtcgccc 180
atgcttgtagcgtacgacaatgcggtcaaccttagctgcaagtattcctacaatctcttc 240
tcaagggagttccgggcatcccttcacaaaggactggatagtgctgtggaagtctgtgtt 300
gtatatgggaattactcccagcagcttcaggtttactcaaaaacggggttcaactgtgat 360
gggaaattgggcaatgaatcagtgacattctacctccagaatttgtatgttaaccaaaca 420
gatatttacttctgcaaaattgaagttatgtatcctcctccttacctagacaatgagaag 480
agcaatggaaccattatccatgtgaaagggaaacacctttgtccaagtcccctatttccc 540
ggaccttctaagcccttttgggtgctggtggtggttggtggagtcctggcttgctatagc 600
ttgctagtaacagtggcctttattattttctgggtgaggagtaagaggagcaggctcctg 660
cacagtgactacatgaacatgactccccgccgccccgggcccacccgcaagcattaccag 720
ccctatgccccaccacgcgacttcgcagcctatcgctcctgacacggacgcctatccaga 780
agccagccggctggcagcccccatctgctcaatatcactgctctggataggaaatgaccg 840
ccatctccagccggccacctcagcccctgttgggccaccaatgccaatttttctcgagtg 900
actagaccaaatatcaagatcattttgagactctgaaatgaagtaaaagagatttcctgt 960
gacaggccaagtcttacagtgccatggcccacattccaacttaccatgtacttagtgact 1020
tgactgagaagttagggtagaaaacaaaaagggagtggattctgggagcctcttcccttt 1080
ctcactcacctgcacatctcagtcaagcaaagtgtggtatccacagacattttagttgca 1140
gaagaaaggctaggaaatcattccttttggttaaatgggtgtttaatcttttggttagtg 1200
ggttaaacggggtaagttagagtagggggagggataggaagacatatttaaaaaccatta 1260
aaacactgtctcccactcatgaaatgagccacgtagttcctatttaatgctgttttcctt 1320
tagtttagaaatacatagacattgtcttttatgaattctgatcatatttagtcattttga 1380
ccaaatgagggatttggtcaaatgagggattccctcaaagcaatatcaggtaaaccaagt 1440
tgctttcctcactccctgtcatgagacttcagtgttaatgttcacaatatactttcgaaa 1500
gaataaaatagttc 1514
<210> 12
<211> 220
<212> PRT
<213> Homo Sapiens
<400> 12
Met Leu Arg Leu Leu Leu Ala Leu Asn Leu Phe Pro Ser Ile Gln Val
1 5 10 15
Thr Gly Asn Lys Ile Leu Val Lys Gln Ser Pro Met Leu Val Ala Tyr
20 25 30
Asp Asn Ala Val Asn Leu Ser Cys Lys Tyr Ser Tyr Asn Leu Phe Ser
35 40 45
Arg Glu Phe Arg Ala Ser Leu His Lys Gly Leu Asp Ser Ala Val Glu
50 55 60
Val Cys Val Val Tyr Gly Asn Tyr Ser Gln Gln Leu Gln Val Tyr Ser
65 70 75 80
Lys Thr Gly Phe Asn Cys Asp Gly Lys Leu Gly Asn Glu Ser Val Thr
85 90 95
Phe Tyr Leu Gln Asn Leu Tyr Val Asn Gln Thr Asp Ile Tyr Phe Cys
100 105 110
Lys Ile Glu Val Met Tyr Pro Pro Pro Tyr Leu Asp Asn Glu Lys Ser
115 120 125
Asn Gly Thr Ile Ile His Val Lys Gly Lys His Leu Cys Pro Ser Pro
130 135 140
Leu Phe Pro Gly Pro Ser Lys Pro Phe Trp Val Leu Val Val Val Gly
145 150 155 160
Gly Val Leu Ala Cys Tyr Ser Leu Leu Val Thr Val Ala Phe Ile Ile
165 170 175
Phe Trp Val Arg Ser Lys Arg Ser Arg Leu Leu His Ser Asp Tyr Met
180 185 190
Asn Met Thr Pro Arg Arg Pro Gly Pro Thr Arg Lys His Tyr Gln Pro
_g_
CA 02324995 2000-10-OS
WO 99/53953 PCT/US99/06874
195 200 205
Tyr Ala Pro Pro Arg Asp Phe Ala Ala Tyr Arg Ser
210 215 220
<210> 13
<211> 8
<212> PRT
<213> Homo Sapiens
<400> 13
Arg His Arg Ser Gln Lys Gly Pro
1 5
-9-