Note: Descriptions are shown in the official language in which they were submitted.
CA 02327097 2000-10-02
WO 99/50203 PCT/US99/06I37
CHEMICAL CONTROL OVER CERAMIC POROSITY USING
CARBOXYLATE-ALUMOXANES
CROSS-REFERENCE TO RELATED APPLICATIONS
S This application claims the benefit of U.S. provisional patent application
Serial Number
60/079,926, filed March 30, 1998, which is incorporated herein by reference in
its entirety.
STATEMENT REGARDING FEDERALLY SPONSORED
RESEARCH ORDEVELOPMENT
Not Applicable.
BACKGROUND OF THE INVENTION
Field of the Invention
The present invention relates generally to compositions of matter and methods
for
1 S synthesizing a composition of matter including controlling the pore size,
pore size distribution
and porosity of aluminum-oxide based ceramics through the choice of
substituents on
carboxylate-alumoxanes and aluminum-oxide nanoparticles. The invention
includes aluminum
and aluminum oxide ceramic bodies with infra-granular pores in the nanometer
range and
methods for forming infra-granular pores in the nanometer range in alumina and
aluminum oxide
ceramic bodies. The invention provides for the control over pore size and pore
size distribution
by the use of chemical substituents on the carboxylate-alumoxanes and aluminum-
oxide
nanoparticles. The invention also includes the use of controlled-porosity
ceramics for ceramic
membrane filters and coatings and interphase layers for fibers and fiber
reinforced composites.
Descritition of the Related Art
The oxides and hydroxides of aluminum are undoubtedly among the most
industrially
important chemicals. Their uses include: precursors for the production of
aluminum metal,
catalysts and absorbents; structural ceramic materials; reinforcing agents for
plastics and rubbers,
antacids and binders for the pharmaceutical industry; and as low dielectric
loss insulators in the
electronics industry. Traditional ceramic processing involves three basic
steps generally referred
to as powder-processing, shape-forming, and densification, often with a final
mechanical
finishing step (Kingery et al. 1976 and Richerson 1992). Whereas traditional
sintering process
CA 02327097 2000-10-02
WO 99/50203 PCT/US99/06137
are primarily for the manufacture of dense parts, solution-gelation processes
have been applied
industrially used for the production of porous materials and coatings.
Solution-gelation involves
a four stage process: dispersion; gelation; drying; and firing. A stable
liquid dispersion or sol of
the colloidal ceramic precursor is initially formed in a solvent with
appropriate additives. By
S change in the concentration (aging) or the pH, the dispersion is polymerized
to form a solid
dispersion or gel. The excess liquid is removed from this gel by drying, and
the final ceramic is
formed by firing the gel at higher temperatures. The common solution-gelation
route to
aluminum oxides employs aluminum hydroxide (or hydroxide-based material) as
the solid
colloid, with the second phase being water and/or an organic solvent. Aluminum
hydroxide gels
have traditionally been prepared by the neutralization of a concentrated
aluminum salt solution
(Serna et al. 1977), however, the strong interactions of the freshly
precipitated alumina gels with
ions from the precursor solutions makes it difficult to prepare these gels in
pure form (Green and
Hem 1974). To avoid this complication alumina. gels may be prepared from the
hydrolysis of
aluminum alkoxides, Al(OR)3 (Eq. 1).
H20 0
Al(OR)3 ----~ AI-gel -~ A1203 (1)
Although this method was originally reported by Adkins in 1922, it was not
until
Teichmer et al. (1976) reported the preparation of alumina aerogels, and
Yoldas (1975) showed
that transparent ceramic bodies can be obtained by the pyrolysis of suitable
alumina gels, that
interest increased significantly. Other pertinent references include: Nogami
(1994), Low et al.
( 1997), Nikolic and Radonj is ( 1997), Rezgui and Gates ( 1997), Rezgui et
al. ( 1994). The exact
composition of the gel in commercial systems is ordinarily proprietary,
however, a typical
composition will include an aluminum compound, a mineral acid and a complexing
agent to
inhibit premature precipitation of the gel. The aluminum compound has
traditionally been the
direct precursor to pseudo-boehmite.
2
CA 02327097 2000-10-02
WO 99/50203 PCT/US99/06137
The aluminum based sol-gels formed during the hydrolysis of aluminum compounds
belong to a general class of compounds, namely alumoxanes. These materials
were first reported
in 1958 (Andrianov and Zhadanov, 1958) with siloxide substituents, however,
they have since
been prepared with a wide variety of substituents on aluminum. Recent work has
shown that the
structure of alumoxanes is as three dimensional cage compounds (Apblett et al.
1992 and Landry
et al. 1993). For example, siloxy-alumoxanes, [Al(O)(OH)X(OSiR3)1_x]n~ consist
of an
aluminum-oxygen core structure (Figure 1 ) analogous to that found in the
mineral boehmite,
[Al(O)(OH)]n, with a siloxide substituents. In the siloxy-alumoxanes, the
"organic" is typically
like that shown in Figure 2. However, the carboxylate anion, [RC02]-, is an
isoelectronic and
structural analog of the organic portion found in the siloxy-alumoxanes
(Figure 3). Based upon
this approach the reaction of boehmite, [Al(O)(OH)]n, with carboxylic acids,
has been developed
(Landry et al. 1995) or Eq. 2.
H02CR
[Al(O)OH)]n --~ [Al(O)x(OH)y(02CR)z]n
Carboxylate-substituted alumoxanes have been well characterized (Landry et al.
1995 and
Callender et al. 1997). Solution particle-size measurements shows that
carboxylate-alumoxanes
are nano-particles with sizes ordinarily ranging from 1 - 1000 nm (Figure 10,
11 and 12). Nano-
particles are ordinarily defined as materials with sizes ranging from 1 nm to
1 ~cn. The
carboxylate ligand is bound to the aluminum surface, and is only removed under
extreme
conditions. The carboxylate-alumoxane materials prepared from the reaction of
boehmite and
carboxylic acids are air and water stable materials and are easily processable
(Figure 7). The
soluble carboxylate-alumoxanes can be dip-coated, spin coated, and spray-
coated onto various
substrates. The physical properties of these alumoxanes are highly dependent
on the identity of
the alkyl substituents, R, and range from those associated with insoluble
crystalline powders to
powders that readily form solutions or gels in hydrocarbon solvents and/or
water. These
alumoxanes are indefinitely stable under ambient conditions, and are adaptable
to a wide range of
processing techniques. The alumoxanes can be easily converted to aluminum
oxide upon mild
thermolysis, while they also react with metal complexes to form doped or mixed
aluminum
oxides (Kareiva et al. 1996).
The control of porosity (pore size, pore size distribution and pore density)
is an important
aspect of ceramics. Lower porosity improves strength, load-bearing capacity,
and corrosion
resistance, but can also lead to catastrophic failure from thermal shock,
because the pores present
3
CA 02327097 2000-10-02
WO 99/50203 PCT/US99/06137
act as crack stoppers in more porous ceramics. Cracks propagate
intergranularly (between grains)
and therefore the grain boundary toughness plays a large role in determining
the fracture mode.
Porosity between grains can promote crack propagation and lower the strength
of a ceramic body.
In both traditional and sol-gel processes, the porosity of the resulting
ceramic is controlled
through physical processing variables (Wilson and Stacey, 1981), such as the
time or temperature
of firing and the addition of pre-fired additives to seed crystal growth
(Shelleman et al. 1986).
Direct chemical control has not been observed. Furthermore, the pore size,
pore size distribution
and porosity are functions of the ceramic particles used to make the ceramic
body, because the
porosity is detenmined by the gaps between the individual particles (Figure 4)
and is therefore
inter-granular, that is between the crystal grains. For example, pores below
0.1 Eun in diameter
require that submicron powders be used (in traditional ceramic processing)"
while smaller pores
require sol-gel processing.
A particularly important area where the strength and porosity of ceramic
materials is
affected by the formation of inter-phase materials in fiber reinforced ceramic
matrix and metal
matrix composites. Fiber reinforced ceramic matrix composites (FRCMCs) are
potential
candidates for use in high temperature structural applications (Courtright,
1991 ). For example,
aerospace applications include high thrust-to-weight ratio gas turbine engines
and high-specific-
impulse rocket motors. Ground based applications include high efficiency
turbine and diesel
engines. In each of these applications there is a need for high performance
ceramic materials that
can be readily fabricated into complex shapes. Compared to current materials
(e.g., nickel based
superalloys) and proposed metallic and intermetallic matrix composites, FRCMCs
have higher
strengths at lower densities, higher maximum use temperatures, and better
oxidation resistance.
Ceramic materials are well known for their stability at high temperatures,
adequate strength and
resistance to corrosion, and can meet most of the requirements for gas turbine
applications.
However, the brittle nature of ceramic materials and their tendency to undergo
catastrophic
failure has limited their usefulness. By reinforcing ceramic materials with
fibers, catastrophic
failures can be reduced or eliminated. A major drawback in existing fiber
reinforced ceramic
matrix composites (FRCMCs) is the absence of a fiber-matrix interface (or
interphase) that is
weak and stable over the entire range of expected use. Limitations of such
prior art FRCMCs are
the instability of known interfaces and the chemical reactivity of many weak
interphases with the
fiber and/or matrix. The chemical design of interfaces to optimize the
adhesion or transfer of load
between reinforcing phase and the matrix, to enhance crack deflection through
debonding or to
control interfacial reactivity/stability are an important development. For
both the fiber and
ceramic matrix, material requirements include: high melting points, high
modulus, low density,
freedom from destructive phase transformations, low volatility, oxidative
stability, and creep
4
CA 02327097 2000-10-02
WO 99/50203 PCTNS99/06137
resistance. For structural applications at high temperatures, environmental
stability and creep
resistance are the dominant factors in determining the usefiilness of ceramic
materials. In general,
monolithic polycrystalline oxide ceramics lose strength above 1200 °C.
Therefore, monolithic
ceramics must be strengthened with high modulus fibers. The only materials
that retain strength
at these high temperatures, and under severe oxidative environments, are oxide
fibers (e.g.,
sapphire) or silicon carbide (SiC) fiber. An additional concern is that the
matrix and fiber
materials must be chemically compatible {i.e., not react with each other). In
fiber reinforced
ceramic, the reinforcement is to enhance the fracture toughness. The fiber
reinforcement prevents
catastrophic brittle failure by providing mechanisms to dissipate energy
during fracture. The
operation of various toughening mechanisms, such as crack deflection, fiber
pull out, and fiber
bridging, depend to a large extent on the degree of chemical and/or mechanical
bonding at the
fiber-matrix interface. This chemical bonding is affected by the fiber surface
chemistry and
chemical reactivity between the fiber and matrix. The mechanical bonding is
primarily controlled
by the fiber surface morphology and the fiber/matrix thermal expansion match.
In general,
composites with strong interfacial bonding exhibit brittle behavior,
characterized by high
strength and low fi~acture toughness. If the interfacial interaction is weak
then a composite will
fail by catastrophic manner, and show high fracture toughness but low
strength. It is therefore
highly desirable to control the interfacial bond in order to optimize the
overall mechanical
behavior of the composite. The fiber-matrix interface must be sufficiently
weak to allow
debonding and sliding when a crack impinges upon it from the matrix; otherwise
the crack passes
through the fiber (or the fiber fails near the crack tip) and there is minimal
or no toughening
(Michalke and Hellmann, 1988). To control the strength of fiber coatings and
the interaction
between the coating and both the fiber and matrix, is extremely important to
control the porosity
of the coating materials.
In contrast, control of pore size, pore size distribution and porosity in
ceramics is
important for their applications in ceramic membranes and catalyst supports.
Membrane-based
technologies play a unique and increasingly important role in pollution
prevention, resource
recovery and waste treatment activities {Baker, i 99I ). Due in large part to
cost considerations,
polymeric membranes have dominated these environmental separations
applications. However,
the use of polymeric membranes in separations involving aggressive materials
such as many
solvents, acids, bases, and oxidants may be limited by the tolerance of these
membranes to
extreme conditions (Hsieh, 1988). Ceramic membranes are noted for their
excellent mechanical
strength and tolerance to solvents, pH, oxidant, and temperature extremes. In
addition, the
amphoteric properties of ceramic membrane surfaces result in a uniquely
versatile membranes for
3 S water and waste water treatment. Membrane charge, selectivity, and
permeation rate vary as a
5
CA 02327097 2000-10-02
WO 99/50203 PCT/US99/06137
function of pH, ionic strength and other characteristics of the feed water
solution chemistry,
Baltus (1997) and Kim and Stevens (1997). Membrane characteristics as well as
the properties of
the contaminants can be manipulated through adjustments in the solution
chemistry of the feed
stream in one or more pretreatment steps (Anderson et al. 1988). Ceramic
membranes are
typically produced by slip casting a colloidal suspension on a porous ceramic
support: Okubo, et
al. (1990), Elaloui et al. (1997), Lin et al. {1991), Lao et al. (1994),
Zaspalis et al. (1992). A
schematic view of a typical membrane design is shown in Figure 5. The
individual membranes
are mounted into a membrane module (see Figure 6). Control of the colloidal
suspension in the
sol-gel process and limitations on the size of colloids that can be produced
have constrained the
range of membrane types that can be produced. In addition, a key obstacle to
overcome in
advancing the use of ceramic membranes for pollution prevention applications
is cost. The sol-
gel processes currently used to produce commercially available ceramic
membranes is energy
intensive and difficult to control. Considerable time and expense is invested
in verifying
membrane integrity and re-casting membranes to repair imperfections.
Alternative approaches for
manufacture of ceramic membranes include the anodic oxidization of aluminum
metal
membranes (Furneaux et al. 1989), pore size being controlled by the applied
voltage used in the
anodic oxidation. However, strong dielectric solutions of various acids must
be employed, and
ion beam or chemical etching is performed to produce a working filter. An
ideal ceramic
membrane must be highly selective, highly permeable, and highly durable. The
membrane
selectivity is primarily dependent upon the pore-size distribution: a narrow
distribution
contributes to a highly selective membrane. Membrane permeability is a
function of global
porosity, membrane thickness, connectivity, and pore-size distribution.
Membrane durability is
obtained by high homogeneity and high density; the latter entails a clear
compromise with
permeability. Mechanical integrity is enhanced in such application by slip-
casting a relatively
thin selective membrane onto a larger, durable membrane of poor selectivity
but high
permeability.
SUMMARY OF THE INVENTION
The present invention provides alumina and aluminum-oxide ceramic membranes
filters
of controlled pore size, pore size distribution and porosity, a method to
produce such filters, and
the use of these materials as ceramic membrane filters.
The inventive method is based on the use of carboxylate-alumoxanes that can be
described by the general formula:
6
CA 02327097 2000-10-02
WO 99150203 PC'f/US99/06137
[Al(O)x(OH)y(02CR)z]n and/or [Al(O)x(OHh,(02CR)z(02CR')z~]n and/or
[~(O)x(OH~(02CR)z(02CR~)z'(02CR")z"]n etc.
where RC02- (and R'C02- and R"C02 ) are mono-carboxylates and R (and R' and
R") are the
same or different and are from the group of a hydrogen and/or an organic
group. The organic
group is preferably an alkyl, alkenyl, aromatic, haloalkyl, haloalkenyl,
haloaromatic groups or
alkyl, alkenyl, aromatic ether groups or an organic group containing a hetero-
atom including,
oxygen, nitrogen, sulfur, phosphorous. These components may be prepared by the
methods
described in Landry et al. (1995), Apblett et al. (1992), Kareiva et al.
(1996), and the preferred
method of Callender et al. (1997). The composition of the carboxylate-
alumoxane varies
depending on the starting materials employed and the details of the synthetic
method employed
by Callender et al. (1997). Thermolysis of the carboxylate-alumoxanes results
in alumina being
formed. In accordance with the present invention, the size and distribution of
pores within the
alumina-oxide ceramic is dependent on the identity of the carboxylate
substituents. In particular,
I S the formation of infra- versus inter-granular porosity is dependent on the
identity of the
carboxylate substituents. Similarly, size and distribution of the pores is
controlled by the choice
of the organic substituents.
The invention also provides methods for the manufacture of ceramic coatings on
ceramic
and carbon fibers for composite applications and ceramic membranes with
nanometer sized
pores. Dipping a ceramic or carbon fiber into a solution of the carboxylate-
alumoxane in
accordance with the invention, drying and firing provides a uniform coating of
the aluminum-
oxide based ceramic on the surface of the fiber. The pore size, pore size
distribution and porosity,
and hence the strength, permeability and surface adhesion of the ceramic
coating is controlled by
the choice of the substituent on the carboxylate-alumoxane. Thermolysis of
self supporting spun
layers of the carboxylate-alumoxanes results in disks of alumina with
controlled pore size, pore
size distribution and porosity. Also, a porous substrate may be dipped or
coated with a solution
of the carboxylate-alumoxane, followed by thermolysis to produce a composite
membrane.
Accordingly, in a preferred embodiment, the present invention includes a
ceramic body of
controlled pore size and distribution comprising the thermolysis product of a
carboxylate
alumoxane represented by the formula [Al(O)x(OH)y(02CR)z]n, wherein x is from
0 to 1.5, y is
from 0 to 3, z is from 0 to 3, n is greater than 6, and R is hydrogen or an
organic group.
In another preferred embodiment the invention includes a ce~ic body of
controlled pore
size and distribution comprising the thermolysis products of a carboxylate-
alumoxane
7
CA 02327097 2000-10-02
WO 99/50203 PCT/US99/06137
represented by the formula [Al(O)x(OHh,(02CR)z(02CR')z~jn, wherein x is from 0
to 1.5, y is
from 0 to 3, z is from 0 to 3, z' is from 0 to 3, n is greater than 6, herein
each R, which may be
the same or different, is hy~ogen or an organic group, and wherein each R',
which may be the
same or different, is hydrogen or an organic group.
In yet another preferred embodiment the invention includes a porous ceramic
body
comprising the thermolysis product of the reaction product of a carboxylic
acid with boehmite,
represented by the formula [Al(O~(OH)y(02CR)z]n, wherein the porosity and pore
size
distribution of the ceramic body is controlled by the selection of the number,
z, of carboxylate
groups.
In yet another preferred embodiment the invention includes a porous ceramic
composite
comprising a nano-particle comprising the thermolysis product of the reaction
product of a
substituted carboxylate-alumoxane with an aluminum oxide wherein the pore size
and pore
distribution of the ceramic composite are controlled by the substituent on the
carboxylate-
alumoxane.
In yet another preferred embodiment the invention includes a porous ceramic
filter of
controlled pore size and pore size distribution comprising a nano-particle
comprising the
thermolysis product of the reaction product of a substituted carboxylate-
alumoxaae with an
aluminum oxide wherein the pore size and pore distribution of the ceramic
composite are
controlled by the substituent on the carboxylate-alumoxane.
In yet another preferred embodiment the invention includes a fiber reinforced
material
comprising a fiber, and a fiber coating comprising a porous ceramic composite
of a nano-particle
comprising the thermolysis product of the reaction product of a substituted
carboxylate-
alumoxane with an aluminum oxide wherein the pore size and pore distribution
of the ceramic
composite are controlled by the substituent on the carboxylate-alumoxane.
In yet another preferred embodiment the invention includes a method of
controlling the
porosity and pore size distribution of ceramic bodies comprising: reacting
boehmite with a
carboxylic acid to produce carboxylate-alumoxane nanoparticles; drying the
carboxylate-
alumoxane nano-particles; re-dissolving the carboxylate-alumoxane nano-
particles in a solvent;
drying the nano-particles; andfiring the dried nano-particles at a temperature
greater than 300 °C.
BRIEF DESCRIPTION OF THE DRAWINGS
For a more detailed description of the preferred embodiments of the present
invention,
reference will not be made to the accompanying drawings, wherein:
Figure 1 is a schematic representation of the core of an alumoxane sol-gei
material;
Figure 2 is a schematic representation of the periphery of a typical siloxide-
alumoxane;
8
CA 02327097 2000-10-02
WO 99/50203 PCT/US99/06137
Figure 3 is a schematic representation of the periphery of a carboxylate-
alumoxane;
Figure 4 is a schematic representation of a typical spacer ligand;
Figure 5 is schematic representation of intergranular porosity;
Figure 6 is a another schematic representation of intergranular porosity;
Figure 7 is a pictorial representation of the reaction of boehmite with
carboxylic acids;
Figure 8 illustrates thermal processing of alumoxanes by a controlled heating
series;
Figure 9 illustrates a model for inter-granular versus infra-granular
porosity;
Figure 10 illustrates particle size determination by Photon Correlation
Spectroscopy
(PCS);
Figure 11 is a graphical representation of particle size determination of
carboxylate-
alumoxanes in water by PCS;
Figure 12 is a graphical representation of particle size determination of
various aliquots
removed from the reaction of MEA-H with boehmite by PCS;
Figure 13 shows Transmission Electron Microscopy (TEM) images of a-A1203 from
carboxylate-alumoxanes;
Figure 14 is a TEM image of A1203 ceramic material from fired
acetate=alumoxane;
Figure 15 shows TEM negative images of fired acetate-alumoxane illustrating
intra-
granular pores;
Figure 16 shows images of fired acetate-alumoxane illustrating intragranular
porosity;
Figure 17 is a Selected Area Diffraction (SAD) image of fired acetate-
alumoxane ceramic
material;
Figure 18 shows surface images of mixed carboxylate-alumoxanes;
Figure 19 is a schematic representation of the method of formation of a
membrane;
Figure 20 is a schematic representation of the structure of a filter-supported
membrane;
Figure 21 is a SEM image of a coated frit;
Figure 22 shows micrographs of coated carbon fibers;
Figure 23 shows a micrograph of a hibonite coated silicon carbide fiber;
Figure 24 shows micrographs of coated and uncoated sapphire fibers;
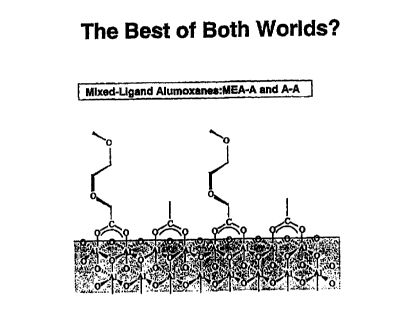
Figure 25 is a schematic representation of a mixed-ligand alumoxane;
Figure 26 is a bar chart comparing the pore size distributions of two
carboxylate
alumoxanes and a physical mixture of two carboxylate alumoxanes; and
Figure 27 is a bar chart comparing the pore size distributions of two
carboxylate
alumoxanes and a chemical mixture of two carboxylate alumoxanes.
9
CA 02327097 2000-10-02
WO 99/50203 PCTNS99/06137
DETAILED DESCRIPTION OF THE INVENTION
This invention discloses the use of carboxylate-alumoxanes
([Al(O~(OHh,(02CR)~],~
and/or aluminum-oxide nano-particles to prepare alumina and aluminum oxide-
based ceramic
bodies, coatings and membranes with chemically controlled pore sizes, pore
size distributions
and porosities. Such ceramics with chemically controlled porosities may be
used as membrane
materials with controlled pore size distributions or as coatings on fibers.
The carboxylato-alumoxanes are precursors to alumina and aluminum oxides
(Table I )
and are prepared by the reaction of boehmite or pseudoboehmite with carboxylic
acids in a
suitable solvent (Tables 2, 3, 4, and 5). The boehmite (or pseudoboehmite)
source can be a
commercial boehmite product such as Catapal (A, B, C, D, or FI, Vista Chemical
Company) or
boehmite prepared by the precipitation of aluminum nitrate with ammonium
hydroxide and then
hydrothermally treated at 200 °C for 24 hours or boehmite prepared by
the hydrolysis of
aluminum trialkoxides followed by hydrothermal treatment at 200 °C. The
carboxylic acid can be
any monocarboxylic acid. The carboxylic acid can be aromatic, aliphatic, and
can contain hetero-
1 S atom functional groups such as hydroxyls, amines, mercaptans, phosphines,
etc. Unlike sol-gel
synthesis the carboxylate alumoxanes are stable both in solution and the solid
state. In addition,
whereas the choice of solvents in sol-gel synthesis is limited, the solubility
of the carboxylate
alumoxanes is dependent only on the identity of the carboxylic acid residue,
which is almost
unrestricted according to the present invention. The solubility of the
alumoxanes is therefore
readily controlled so as to make them compatible with any co-reactants. While
these advantages
are significant, the alumoxanes have yet further benefits with respect to
large scale production of
ternary and quaternary ceramics. The most dramatic of these is the simplicity
of the alumoxane
methodology. The alumoxane route is simple, and can be halted and/or modified
at any stage
without significant effects on the products. A careful control of pH, the use
of additives to inhibit
precipitation, and slow concentration steps are not required, thus making the
alumoxane route
easier and quicker than prior art techniques. Another benefit with respect to
large scale
processing is the relatively low cost of the alumoxane precursors.
Thermogravimetric/differential thermal analysis (TG/DTA) of the carboxylate-
alumoxanes generally indicates two major decomposition regions. The relative
mass loss and
temperatures at which these regions occur is dependent on the identity of the
carboxylic acid. The
volatiles are predominantly the carboxylic acid and water, with traces of the
ketone, i.e., acetone
is liberated from the acetate-alumoxane (A-alumoxane or A-A). As may be
expected, the ceramic
yield is conditional on the identity of the carboxylic acid: greatest for A-A
(ca. 75 %), lowest for
methoxy(ethoxyethoxy)acetate-alumoxane (MEEA-A) (ca. 20 %). All of the
carboxylate-
alumoxanes decompose above 180 °C to give amorphous alumina. Firing
above 900 °C (>_ 3 h.)
CA 02327097 2000-10-02
WO 99/50203 PCT/US99/06137
results in the formation of better ordered mixed phase y-A1203 (JCPDS # 29-63)
and a-A1203
(Corundum, JCPDS # 42-1468), as would be expected based on the known
transformation
sequence of alumina. All of the carboxylate alumoxanes are converted to a-
alumina above 1000
°C with firing times >_ 4 hours. It is interesting to note that the A-
alumoxane is highly reactive
and forms crystalline a-A1203 at temperatures below 850 °C (Table 1 ).
The lower temperature of
this phase formation and failure to observe y-A1203 from A-alumoxane is
consistent with the
very small initial pore size (large surface area) and rapid sintering rates.
The conversion of
acetate-alumoxane (A-A) to a-aiumina at lower than the expected temperatures
(Figure 17), and
the apparent lack of the y-alumina phase being formed, is useful since the
conversion of y-
alumina to a-alumina is associated with a change in density and a decrease in
volume. This
decrease in volume is detrimental to the formation of stable ceramic
composites.
All the un-doped carboxylate-alumoxanes in the examples reported below
produced
uniform, translucent, fired bodies with differences in microscopic pore size,
pore size distribution
and porosity, but with similarities in macroscopic density. Both MEEA- and MEA-
(methoxy(ethoxyethoxy)acetate)- -alumoxane produce either high porosity
translucent solid
"foam" or slightly translucent bodies consistent with a smaller
porosity/higher microscopic
density.
The a-A1203 formed from MEEA-, MEA-, and MA- (methoxyacetate) alumoxanes
exists
as a nanocrystalline matrix with a very high volume of large interconnecting
pores, as determined
by TEM studies (Figure 13). In contrast, analysis of the a-A1203 formed from A-
alumoxane
revealed very fine uniform infra-granular porosity (Figure 14), in which the
crystallite size is
relatively large (ca. 2 pm). The difference in pore size and structure ~is
more consistent with the
chemical identity of the substituents than the physical processing conditions,
i.e., a higher
organic volume outgassed produces larger pores. Using the alumoxane series, it
is possible to
engineer pore size continuously between these extremes by using mixed ligand
solutions (Figure
25).
Further study of the acetate-alumoxane (A-alumoxane) indicates that instead of
the usual
inter-granular porosity (Figures 15 and 16) the pores are infra-granular, that
is, they are within the
individual crystal grains (Figure 6). This novelty of chemical control over
the formation of intra-
granular (rather than inter-granular) porosity has the aforementioned benefit
of increased fracture
toughness. Intra-granular pores instead of inter-granular therefore allow
increased fracture
toughness and less opportunity for pore/boundary/crack interactions to occur.
The formation of
infra-granular pores for the A-alumoxane is thought to be due to the nano-
particulate nature of
11
CA 02327097 2000-10-02
WO 99/50203 PCT/US99/06137
the acetate-alumoxane and the small length of the organic substituent (Figure
9) Table 2. Other
carboxylate-alumoxanes can produce infra-granular porosity if the nano-
particle size is less than
50 nm.
Control of pore size, pore size distribution and porosity, and hence density,
through
chemical means is an important departure from traditional ceramic processing
in which physical
methods only are applied. The porosity of the resulting alumina is dependent
on the length of the
carboxylate side chain. That is, the pore sizes for carboxylate-alumoxanes
with CH3 substituents
is different from those with CSH~ 1 substituents. Another approach to
controlling pore size, pore
size distribution and porosity described herein is the use of spacer ligands.
The alumoxane can be
cross-linked after fabrication of the membrane with di-acids (Figure 4). Upon
pyrolysis, it then
inhibits the collapse of the ceramic.
Instead of using a single carboxylate-alumoxane, a physical mixture of more
than one
carboxylate-alumoxane may be produced and feed to alumina (Tables 4, 5, 6 and
7). The
porosity (average pore size and pore size distribution) is dependent on the
relative amounts of
each carboxylate-alumoxane (Table 8). In general, the porosity is a mixture of
the values of each
individual carboxylate-alumoxane (Figures 26 and 27). Mixed carboxylate-
alumoxanes may be
synthesized in which more than one type of carboxylate group is bonded to each
of the
alumoxane nano-particles. The resulting porosity is different than the
individual materials, and is
dependent on the relative concentration of each carboxylate used (Table 8).
The relative infra- to
inter-granular porosity can be controlled by the choice of carboxylate group
and/or mixtures or
carboxylate groups.
Solutions of the carboxylate-alumoxanes may be evaporated to leave a thin
membrane
which is glass-like (Figure 19). The resulting glassy membrane can then be
fired to produce a
ceramic membrane in which the porosity is controlled by the choice of the
carboxylate group
and/or ratio of physically and/or chemical mixtures of two or more
carboxylates (Figure 18). As
an alternative to a self supporting ceramic membrane, a porous substrate such
as a glass or
ceramic filter frit may be spun coated, painted, or dip-coated with the
carboxylate-alumoxane
solution, Figure 21 (Tables 9 and 10). After drying and firing the composite
consists of a
membrane supported on a coarse filter (Figure 20). The support for the
carboxylate-alumoxane
derived ceramic membrane does not have to be flat but may be a ceramic tube or
column. If
doped carboxylate-alumoxanes are employed, then the resulting membrane will
have the
composition of the doped carboxylate-alumoxane. In order to ensure that
uniform membranes are
produced, physical mixtures of different carboxylate-alumoxanes can be used.
The lowering of
phase formation/crystal growth temperatures observed for the carboxylate-
alumoxane in
12
CA 02327097 2000-10-02
WO 99/50203 PCT/I1S99/06137
comparison to sol-gel methods, allows for smaller pores to be generated
without being sintered
out during thermal treatment.
Carbon or ceramic fibers can also be dipped or coated with a solution of the
carboxylate-
alumoxanes (Figures 22, 23, and 24). After drying either in air, in an oven or
with a heat gun, the
carboxylate-alumoxane can be thermolyzed to give the appropriate ceramic
coating with a
chemically controlled porosity. Suitable ceramic fibers include (but are not
limited to) silicon
carbide (Figure 23) and sapphire (Figure 24). The conditions of thermolysis of
the alumoxane
coating are dependent on the type of the fiber and the identity of the
carboxylate-alumoxane. The
ceramic coatings produced using the carboxylate-alumoxanes show superior
coverage, better
uniformity, and lower defects than found for sol-gel type coatings, due to the
nano-particle nature
of the carboxylate-alumoxane. Furthermore, the lowering of phase
formation/crystal growth
temperatures observed for the carboxylate-alumoxane allow for less damage to
the fiber substrate
during formation of the ceramic coating.
EXAMPLES
Surface area and pore size analysis were conducted on all samples utilizing a
Coulter SA
3100 Plus. Sample tubes used are all Coulter Rapi-tubes. Samples were
outgassed at 350 °C for 3
hours under nitrogen gas on the SA 3100. All sample masses were in the 0.100 g
to 0.190 g
range. For actual analysis, nitrogen gas was also used as the absorbate and
helium gas was used
to measure the free-space in the sample tube. BET surface area was determined
using 5 data
points. The t-plot method was determined utilizing the Harkins-Jura equation
at normal
resolution. BJH parameters were determined using medium (45 data points)
resolution and the
equation used was Harkins-Jura. Pore size distributions (and weighted
averages) are reported as a
function of the BJH adsorption. AFM images of samples were obtained using a
Nanoscope IIIa
Scanning Probe Microscope, (Digital Instruments, Santa Barbara, CA) in tapping
mode AFM.
FESP tips were used with a pyramidal shape and end radius of 5 - 10 nm (also
from Digital
Instruments). Images were taken at scan sizes of 10 Eun, 1 pm, and 200 nm, and
the scan angle
was changed from 0 to 45° to check the integrity of the images. Images
were later. processed to
obtain roughness, grain size, and section analysis with the accompanying
Nanoscope software.
Permeability was derived from Flux experiments using dead end filtration cells
from Spectrum
and Sartorious. The cells were 400 mL and 200 mL (respectively) and were
connected to a tank
of zero-air for positive pressure. A pressure regulator was used to set
constant pressure for each
flux experiment at 10, 20, or 30 psi, and filtrate was collected in beakers
and measured
volumetrically. Ultrapure deionized water was used, obtained from a Milli-Q
water filter.
Membrane samples were epoxied to precut aluminum foil disks with precut holes
in the center,
of known area, matching each membrane piece. The membrane pieces had an area
between 0.5
13
CA 02327097 2000-10-02
WO 99/50203 PCT/US99/06137
and 2 cm2 and a thickness of 100 to 250 pm. Prewetted glass fiber supports
were placed
underneath the membranes to prevent cracking. Contact angle was measured with
a goniometer.
Samples from flux experiments were used, since they were already mounted on a
pliable foil.
Samples were placed upside-down on top of a glass container full of deionized
water, with the
sample submerged. An air bubble was placed on the sample surface and ten
readings of the
contact angle were read for each side of a bubble. Air bubbles occurring
naturally on the
membrane surface were measured using the same procedure. Surface charge was
determined by
measuring the electrophoretic mobility with a Zeta Meter. Membranes were
crushed with a
mortar and pestle and combined with sodium chloride as an electrolyte to form
a 500 mg.L-1
alumoxane and 500 mg.L-1 NaCI solution. The solutions were set at various pHs
using HCl or
NaOH, and electrophoretic mobility and zeta potential were measured at several
different
voltages.
The following examples are presented to illustrate the ease and versatility of
the approach
and are not to be construed as the only examples of the proposed approach or
as limiting the
scope of the present invention.
Example 1: Synthesis of methozy(ethozyethogy)acetate-alumoaane (MEEA-A).
Pseudoboehmite (20.0 g) and methoxy(ethoxyethoxy)acetic acid (102 mL) were
refluxed
in water (400 mL) resulting in a clear solution after 72 h. The solution was
centrifuged at 6000
rpm for 1 hour and decanted. Removal of the volatiles in vacuo (10-2 Torr) at
90 °C yielded a gel
which was then dissolved in ethanol (100 mL) while stirring (10 min.) then
triturated with diethyl
ether (200 mL). The white solid powder thus obtained was redissolved in water
( 100 mL) and
dried at 50 °C for 24 h resulting in a clear glassy material. The MEEA-
alumoxane is soluble in
water, methanol, chloroform, and methylene chloride. The alumoxane was heated
from 25 °C to
225 °C at the rate of 1°C/min., soaked for 30 rains. at 225
°C, followed by a temperature ramp up
to 300 °C at the rate of 2 °C/min., and soaked for 80 rains.,
with a final ramp to the maximum
temperature of l I00°C (over 360 minutes} which was then maintained for
400 minutes (Figure
8).
Example 2: Synthesis of methoxy(ethogyethoxy)acetate-alumoxane.
Methoxy(ethoxyethoxy)acetic acid (60 mL) was dissolved in 300 mL of water and
Vista
Captal B boehmite (12 g) was slowly added and allowed to reflux for 96 hours.
The clear/yellow
solution was filtered and the filtrate was evaporated under reduced pressure
to a yellow gel. The
gel was dissolved in ethanol and the white/yellow powder product was obtained
upon addition of
diethyl ether. Yield: 13.6 g. The TGA of the methoxy(ethoxy)acetate-alumoxane
showed 22.3%
ceramic yield (weight loss of 77.7% }. The alumoxane was heated from 25
°C to 200 °C at the
I4
CA 02327097 2000-10-02
WO 99/50203 PCT/US99/06I37
rate of 1.5 °C.min-1, soaked for 2 h. at 200 °C, followed by a
temperature ramp up to 1000 °C at
the rate of 5 °C.min-1, soaked for 2 h.
Example 3: Synthesis of Methoay(ethoxy)acetate-alumogane (MEA-A).
Pseudoboehmite (10.0 g) and methoxy(ethoxy)acetic acid (38.0 mL) were refluxed
in
S water (100 mL) for 24 h, resulting in a clear solution. The solution was
centrifuged at 6000 rpm
for 1 h and decanted. The water was removed in vacuo (10-2 Torr) at SO
°C, resulting in a gel.
The gel was washed with Et20 (3 x 75 mL) then dissolved in EtOH (50 mL) while
stirring (10
minutes). The MEA-alumoxane was precipitated via the addition of Et20 (100 mL)
as a white
powder. After drying overnight at 50 °C the solid yield was
approximately 25 g. The alumoxane
was heated from 25 °C to 225 °C at the rate of 1 °C/min.,
soaked for 30 mins. at 225 °C, followed
by a temperature ramp up to 300 °C at the rate of 2 °C/min., and
soaked for 80 rains., with a final
ramp to the maximum temperature of 1100°C (over 360 minutes) which was
then maintained for
400 minutes.
Ezample 4: Synthesis of Methoxy(ethoxy)acetate-alumogane.
Methoxy(ethoxy)acetic acid (152 mL) was dissolved in 400 mL of water and Vista
Captal
B boehmite (40 g) was slowly added and allowed to reflux for 24 hours. The
clear/yellow
solution was filtered and the filtrate was evaporated under reduced pressure
to a yellow gel. The
gel was dissolved in ethanol and the white/yellow powder product was obtained
upon addition of
diethyl ether. Yield: 82.1 g. The TGA of the methoxy(ethoxy)acetate-alumoxane
showed 27.0%
ceramic yield (weight loss of 73.0 % ). The alumoxane was heated from 25
°C to 200 °C at the
rate of 1.5 °C.min-1, soaked for 2 h. at 200 °C, followed by a
temperature ramp up to 1000 °C at
the rate of 5 °C.min-1, soaked for 2 h.
Example 5: Synthesis of Methoxyacetate-alumoaane (MA-A).
Pseudoboehmite (10.0 g) and methoxyacetic acid (25.6 mL) were refluxed in
water (150
mL) for 24 h. which resulted in a white cloudy solution with a trace of
insoluble particles. The
water was removed in vacuo (10-2 Torr) at 50°C resulting in a white
powder which was washed
with diethyl ether (4 x 150 mL) then dissolved in ethanol (100 mL} while
stirring (50 minutes).
The alumoxane was precipitated via the addition of ether (300 mL). After
drying overnight at
50°C the solid yield was approximately 20 g. The powder was dissolved
in water (100 mL),
isolated by filtration, concentrated under vacuum and dried at 50°C
resulting in a white solid
material. The alumoxane was heated from 25 °C to 225 °C at the
rate of 1 °C/min., soaked for 30
rains. at 225 °C, followed by a temperature ramp up to 300 °C at
the rate of 2 °C/min., and
soaked for 80 rains., with a f nal ramp to the maximum temperature of
1100°C (over 360
minutes) which was then maintained for 400 minutes.
CA 02327097 2000-10-02
WO 99/50203 PCT/US99/06137
Example 6: Synthesis of Acetate-alumoxane.
Pseudoboehmite (20.0 g) was slowly added to a vigorously stirring mixture of
acetic acid
(51.0 mL) in water (200 mL). The resulting slurry was decanted after 10
minutes and then
centrifuged at 6000 rpm for 1 hour to yield a clear viscous solution. Removal
of the volatiles in
vacuo (10-2 Torr) at 90°C results in clear, white granules. The
granules were dissolved in water
and dried for 24 hours at 80 °C to yield a clear glassy material. The
alumoxane was heated from
25 °C to 225 °C at the rate of 1 °C/min., soaked for 30
mins. at 225 °C, followed by a temperature
ramp up to 300 °C at the rate of 2 °C/min., and soaked for 80
rains., with a final ramp to the
maximum temperature of 1100°C (over 360 minutes) which was then
maintained for 400
minutes.
Example 7: Synthesis of Acetate-alumoxane.
Acetic acid (80 mL) was dissolved in water (800 mL) to which Vista Captal B
boehmite
( 100 g) was slowly added, and the reaction was stirred for at room
temperature for 15 minutes.
The clear solution was then decanted into centrifuge bottles and centrifuged
at 4100 rpm for 1
hour. The solution was decanted away from the white powder and evaporated
under reduced
pressure at 80 °C, resulting in a white powder. Yield 81.3 g. The TGA
of the acetate-alumoxane
showed 71.9% ceramic yield (weight loss of 28.1 % ). The alumoxane was heated
from 25 °C to
200 °C at the rate of 1.5 °C.min-I, soaked for 2 h. at 200
°C, followed by a temperature ramp up
to 1000 °C at the rate of 5 °C.min-1, soaked for 2 h.
Example 8: Synthesis of Acetate-alumoxane.
Prepared in an analogous manner to that in Example 7 with the amounts and
conditions
shown in Table 2.
Example 9: Synthesis of Acetate-alumoxane.
Prepared in an analogous manner to that in Example 7 with the amounts and
conditions
shown in Table 2.
Example 10: Synthesis of Malonate-aiumoxane.
Malonic acid (5 g) was dissolved in water (50 mL) to which Vista Captal B
boehmite (5
g) was slowly added and the reaction was stirred for 30 minutes until a thick
gel formed. More
water was added to the solution and the reaction was stirred for another 10
min. The resulting
solution was filtered and the filtrate was evaporated under reduced pressure
resulting in a white
powder. Yield 6.75 g. The TGA of the malonate-alumoxane showed 37.1 % ceramic
yield
(weight loss of 62.9%). The alumoxane was heated from 25 °C to 200
°C at the rate of 1.5
°C.min-1, soaked for 2 h. at 200 °C, followed by a temperature
ramp up to 1000 °C at the rate of
5 °C.min-1, soaked for 2 h.
16
CA 02327097 2000-10-02
WO 99/50203 PCT/US99/06137
Example 11: Synthesis of Malonate-alumoxane.
Prepared in an analogous manner to that in Example i 0 with the amounts and
conditions
shown in Table 3.
Example 12: Synthesis of Malonate-alumozane.
Prepared in an analogous manner to that in Example 10 with the amounts and
conditions
shown in Table 3.
Example 13: Synthesis of Malonate-alumozane.
Prepared in an analogous manner to that in Example 10 with the amounts and
conditions
shown in Table 3.
Example 14: Synthesis of mixed ligand methoxy(ethogy)acetate-acetate-
alumoxane.
Acetic acid (19.0 mL) and methoxy(ethoxy) acetic acid (152.0 mL) was dissolved
in 500
mL of water and Vista Captal B boehmite (20 g) was slowly added and refluxed
for 72 hours.
The white solution was filtered and the filtrate was dissolved under reduced
pressure to yield a
brown gel. The gel was dissolved in ethanol ( 100 mL) and the white powder
product was
obtained by the addition of diethyl ether. Yield: 8.9 g. The TGA of the
product showed 32.5
ceramic yield (weight loss of 67.5%). The alumoxane was heated from 25
°C to 200 °C at the
rate of 1.5 °C.min-1, soaked for 2 h. at 200 °C, followed by a
temperature ramp up to 1000 °C at
the rate of 5 °C.min-1, soaked for 2 h.
Ezample 15: Synthesis of mixed ligand methoxy(ethoxy)acetate-acetate-
alumoxane.
Prepared in an analogous manner to that in Example 14 with the amounts and
conditions
shown in Table 4.
Example 16: Synthesis of mixed ligand methoxy(ethoay)acetate-acetate-
alumoxane.
Prepared in an analogous manner to that in Example 14 with the amounts and
conditions
shown in Table 4.
Example 17: Synthesis of mixed ligand methoxy(ethoxy)acetate-acetate-
alumoxane.
Prepared in an analogous manner to that in Example 14 with the amounts and
conditions
shown in Table 4.
Example 18: Synthesis of mined ligand methoxy(ethoxy)acetate-acetate-
alumoxane.
Prepared in an analogous manner to that in Example 14 with the amounts and
conditions
shown in Table 4.
Example 19: Synthesis of mixed ligand methoxy(ethoxy)acetate-acetate-
alumoxane.
Prepared in an analogous manner to that in Example 14 with the amounts and
conditions
shown in Table 4.
17
CA 02327097 2000-10-02
WO 99/50203 PCT/US99106137
Example 20: Synthesis of mixed Ggand methoay(ethoxyethoxy)acetate-acetate-
alumozane.
Acetic acid (28.6 mL) and methoxy(ethoxyethoxy) acetic acid (76.7 mL) was
dissolved in
500 mL of water and Vista Captal B boehmite (20 g) was slowly added and the
solution was
allowed to reflux for 72 hours. The solution was filtered and the filtrate was
evaporated under
reduced pressure resulting in a white%lear gel. The gel was dissolved in
ethanol and the product
was collected as a white powder upon the addition of diethyl ether. Yield:
25.4 g. The TGA of
the product showed a 28.5 % ceramic yield (weight loss of 71.5%). The
alamoxnne was heated
from 25 °C to 200 °C at the rate of 1.5 °C.min-1, soaked
for 2 h. at 200 °C, followed by a
temperature ramp up to 1000 °C at the rate of 5 °C.min-I, soaked
for 2 h.
Example 21: Synthesis of mixed ligand methoay(ethoayethoxy)acetate-acetate-
alamoxnne.
Prepared in an analogous manner to that in Example 20 with the amounts and
conditions
shown in Table 5.
Example 22: Physical Mixing of methoxy(ethogy)acetate-alamoxnne (MEA-A) and
acetate-
alumogane (A-A).
1 S MEA-A ( 1.0 g) and A-A ( 1.0 g) were dissolved into about 20 mL of water.
After stirring
for approximately 0.5 hours the solutions were poured into drying containers.
After
approximately 36 hours, the solutions had evaporated to leave a thin membrane
which is glass-
like. The glassy membrane is then fired as described in example 2.
Example 23: Physical Mixing of methozy(ethogy)acetate-alamoxnne (MEA-A) and
acetate-
alamoxnne (A-A).
Prepared in an analogous manner to that in Example 22 with the amounts and
conditions
shown in Table 6.
Example 24: Physical Mixing of methogy(ethogy)acetate-alamoxnne (MEA-A) and
acetate-
alumozane (A-A).
Prepared in an analogous manner to that in Example 22 with the amounts and
conditions
shown in Table 6.
Example 25: Physical Mixing of methoay(etho~cy)acetate-alamoxnne (MEA-A) and
acetate-
alumoxane (A-A).
Prepared in an analogous manner to that in Example 22 with the amounts and
conditions
shown in Table 6.
Example 26: Physical Mixing of chemically mined methoxy(ethoxyethoxy)acetate-
acetate-
alumozane (MEA/A-A) and acetate-alumoaane (A-A).
MEA/A-A ( 1.0 g) and A-A ( 1.0 g) were dissolved into 20 mL of water. After
stirnng for
approximately 0.5 hours the solutions were poured into drying containers.
After approximately
18
CA 02327097 2000-10-02
WO 99/50203 PCT/US99/06137
36 hours, the solutions had evaporated to leave a thin membrane which is glass-
like. The glassy
membrane is then fired by the conditions listed in Example 2.
Example 27: Physical Mixing of chemically mixed methoxy(ethozyethoxy)acetate-
acetate-
alumoxane (MEA/A-A) and acetate-alumoxane (A-A).
Prepared in an analogous manner to that in Example 26 with the amounts and
conditions
shown in Table 7.
Example 28: Physical Mixing of chemically mixed methory(ethoxyethoxy)acetate-
acetate-
alumoxane (MEA/A-A) and acetate-alumoaane (A-A).
Prepared in an analogous manner to that in Example 26 with the amounts and
conditions
shown in Table 7.
Example 29: Physical Mixing of chemically mixed methoay(ethoxyethoxy)acetate-
acetate-
alumoxane (MEA/A-A) and acetate-alumoaane (A-A).
Prepared in an analogous manner to that in Example 26 with the amounts and
conditions
shown in Table 7.
Example 30: Physical Mixing of chemically mixed methoxy(ethoxyethoxy)acetate-
acetate-
alumoxane (MEA/A-A) and acetate-alumoxane (A-A).
Prepared in an analogous manner to that in Example 26 with the amounts and
conditions
shown in Table 7.
Example 31. Infiltration of alumino-silicate filters.
A filter frit (pore size ca. 25 p,m) was placed in a Schleck flask and
evacuated. A solution
of A-A (10 g) in 100 mL of water was introduced into the Schlenk by canula
under vacuum
which resulted in the ceramic frit "soaking up" the alumoxane solution. The
frit was allowed to
sit for approximately 0.5 hours under reduced pressure with an excess of the
alumoxane solution
covering the frit in the schlenk. The frit was then allowed to dry at room
temperature. The frit
was then either infiltrated again, fired, or fired then infiltrated again.
Example 32. Infiltration of alumino-silicate filters.
Prepared in an analogous manner to that in Example 31 with the number of
infiltrations
(# dips) and firing sequence shown in Table 9.
Example 33. Infiltration of alumino-silicate filters.
Prepared in an analogous manner to that in Example 31 with the number of
infiltrations
(# dips) and firing sequence shown in Table 9.
Example 34. Infiltration of alumino-silicate filters.
Prepared in an analogous manner to that in Example 31 with the number of
infiltrations
(# dips) and firing sequence shown in Table 9.
19
CA 02327097 2000-10-02
WO 99/50203 PCT/US99/06137
Example 35. Infiltration of alumino-silicate filters.
Prepared in an analogous manner to that in Example 31 with the number of
infiltrations
(# dips) and firing sequence shown in Table 9.
Example 36. Infiltration of alumino-silicate filters.
Prepared in an analogous manner to that in Example 31 with the number of
infiltrations
(# dips) and firing sequence shown in Table 9.
Example 37. Infiltration of alumino-silicate filters.
Prepared in an analogous manner to that in Example 31 with the number of
infiltrations
(# dips) and firing sequence shown in Table 9.
Example 38. Infiltration of alumino-silicate filters.
Prepared in an analogous manner to that in Example 31 with the number of
infiltradons
(# dips) and firing sequence shown in Table 9.
Ezample 39. Inf ltration of alumino-silicate filters.
Prepared in an analogous manner to that in Example 31 with the number of
infiltrations
(# dips) and firing sequence shown in Table 9.
Example 40. Infiltration of alumino-silicate filters.
Prepared in an analogous manner to that in Example 31 with the number of
infiltrations
(# dips) and firing sequence shown in Table 9.
Example 41. Infiltration of alumino-silicate filters.
Prepared in an analogous manner to that in Example 31 with the number of
infiltrations
(# dips) and firing sequence shown in Table 9.
Example 42. Infiltration of alumino-silicate filters.
Prepared in an analogous manner to that in Example 31 with the number of
infiltrations
(# dips) and firing sequence shown in Table 9.
Ezample 43. Infiltration of alumino-silicate filters.
Prepared in an analogous manner to that in Example 31 with the number of
infiitrations
(# dips) and firing sequence shown in Table 9.
Example 44. Infiltration of glass filters.
A glass filter frit (pore size D) was placed in a Schleck flask and evacuated.
A solution of
A-A (10 g) in 100 mL of water was introduced into the Schlenk by canula under
vacuum which
resulted in the glass frit "soaking up" the alumoxane solution. The frit was
allowed to sit for
approximately 0.5 hours under reduced pressure with an excess of the alumoxane
solution
covering the frit in the schlenk. The frit was then allowed to dry at room
temperature. The
infiltration was repeated twice. The infiltrated glass frit was heated from 25
°C to 350 °C,
analyzed by SEM, heated from 25 °C to 700 °C and analyzed.
CA 02327097 2000-10-02
WO 99/50203 PCT/US99/06137
Example 45. Infiltration of glass filters.
Prepared in an analogous manner to that in Example 44 with the number of
infiltrations
(# dips) and firing sequence shown in Table 10.
Example 46. Infiltration of glass filters.
S Prepared in an analogous manner to that in Example 44 with the number of
infiltrations
(# dips) and firing sequence shown in Table 10.
Example 47. Infiltration of glass filters.
Prepared in an analogous manner to that in Example 44 with the number of
infiltrations
(# dips) and firing sequence shown in Table 10.
Ezample 48. Preparation of alumina coated carbon fibers.
MEEA-alumoxane (0.1 g) was dissolved in CHC13 (5 mL) at room temperature. The
fiber
is dipped in MEEA-alumoxane solution and allowed to fully air dry, at room
temperature. Repeat
dipping/drying until desired coating thickness is obtained. The coated fiber
was heated from 25
°C to 225 °C at the rate of 1 °C.min-1, soaked for 30
mins. at 225 °C, followed by a temperature
ramp up to 300 °C at the rate of 2 °C.min-1, and soaked for 80
mins., with a final ramp to the
maximum temperature of 1100 °C (over 360 minutes) which was then
maintained for 400
minutes.
Eaample 49. Preparation of alumina coated carbon fibers.
Prepared in an analogous manner to that in Example 48 using the amounts and
conditions
shown in Table 11.
Example 50. Preparation of alumina coated carbon fibers.
Prepared in an analogous manner to that in Example 48 using the amounts and
conditions
shown in Table 11.
Ezample 51. Preparation of alumina coated carbon fibers.
Prepared in an analogous manner to that in Example 48 using the amounts and
conditions
shown in Table 11.
Example 52. Preparation of aiumina coated carbon fibers.
Prepared in an analogous manner to that in Example 48 using the amounts and
conditions
shown in Table 11.
Ezample 53. Preparation of alumina coated carbon fibers.
MEEA-alumoxane (0.1 g) was dissolved in H20 (5 mL) with low heat (40
°C) and
stirring. The fiber is dipped in MEEA-Alumoxane solution and allowed to
partially dry at room
temperature then dried in oven (45° C) for 24h. Repeat dipping/drying
until desired coating
thickness is obtained. The coated fiber was heated from 25 °C to 225
°C at the rate of 1 °C.min-i,
21
CA 02327097 2000-10-02
WO 99/50203 PCT/US99/06137
soaked for 30 rains. at 225 °C, followed by a temperature ramp up to
300 °C at the rate of 2
°C.min-1, and soaked for 80 rains., with a final ramp to the maximum
temperature of 1100 °C
(over 360 minutes) which was then maintained for 400 minutes.
Eaample 54. Preparation of alumina coated carbon fibers.
Prepared in an analogous manner to that in Example 53 using the amounts and
conditions
shown in Table 11.
Example 55. Preparation of alumina coated carbon fibers.
Prepared in an analogous manner to that in Example 53 using the amounts and
conditions
shown in Table 11.
Example 56. Preparation of alumina coated carbon fibers.
Prepared in an analogous manner to that in Example 53 using the amounts and
conditions
shown in Table 11.
Example 57. Preparation of alumina coated carbon fibers.
Prepared in an analogous manner to that in Example 53 using the amounts and
conditions
shown in Table 11.
Ezample 58. Preparation of YAG coated carbon fibers.
Yttrium-doped MEEA-alumoxane (0.5 g) was dissolved in H20 (5 mL) with low heat
(40
°C} and stirring. The fiber is dipped in the Y-doped MEEA-alumoxane
solution and allowed to
partially dry at room temperature then dried in oven (45° C) for 24h.
Repeat dipping/drying until
desired coating thickness is obtained. The coated fiber was heated from 25
°C to 225 °C at the
rate of 1 °C.min-I, soaked for 30 rains. at 225 °C, followed by
a temperature ramp up to 300 °C
at the rate of 2 °C.min-1, and soaked for 80 rains., with a final ramp
to the maximum temperature
of 1100 °C (over 360 minutes) which was then maintained for 400
minutes.
Ezample 59. Preparation of YAG coated carbon fibers.
Prepared in an analogous manner to that in Example 58 using the amounts and
conditions
shown in Table 11.
Eaampie 60. Preparation of YAG coated carbon fibers.
Prepared in an analogous manner to that in Example 58 using the amounts and
conditions
shown in Table 11.
Example 61. Preparation of bibonite coated carbon fibers.
Calcium-doped MEEA-alumoxane (0.5 g) was dissolved in H20 (5 mL) with low heat
(40 °C) and stirring. The fiber is dipped in the Ca-doped MEEA-
alumoxane solution and allowed
to partially dry at room temperature then dried in oven (45° C) for
24h. Repeat dipping/drying
until desired coating thickness is obtained. The coated fiber was heated from
25 °C to 225 °C at
22
CA 02327097 2000-10-02
WO 99/50203 PCT/US99/06137
the rate of 1 °C.min-1, soaked for 30 rains. at 225 °C, followed
by a temperature ramp up to 300
°C at the rate of 2 °C.min-1, and soaked for 80 rains., with a
final ramp to the maximum
temperature of 1100 °C (over 360 minutes) which was then maintained for
400 minutes.
Example 62. Preparation of hibonite coated carbon fibers.
Prepared in an analogous manner to that in Example 61 using the amounts and
conditions
shown in Table 11.
Example 63. Preparation of hibonite coated carbon fibers.
Prepared in an analogous manner to that in Example 61 using the amounts and
conditions
shown in Table 11.
Example 64. Preparation of hibonite coated carbon fibers.
Calcium-doped MEEA-alumoxane (0.1 g) was dissolved in CHC13 (5 mL) at room
temperature. The fiber is dipped in Ca-doped MEEA-alumoxane solution and
allowed to fully air
dry, at room temperature. Repeat dipping/drying until desired coating
thickness is obtained. The
coated fiber was heated from 25 °C to 225 °C at the rate of I
°C.min-1, soaked for 30 rains. at
225 °C, followed by a temperature ramp up to 300 °C at the rate
of 2 °C.min-~, and soaked for 80
rains., with a final ramp to the maximum temperature of 1100 °C (over
360 minutes) which was
then maintained for 400 minutes.
Example 65. Preparation of hibonite coated carbon fibers.
Prepared in an analogous manner to that in Example 61 using the amounts and
conditions
shown in Table 11.
Example 66. Preparation of hibonite coated carbon fibers.
Prepared in an analogous manner to that in Example 61 using the amounts and
conditions
shown in Table 11.
Example 67. Preparation of hibonite silicon carbide fibers.
Calcium-doped MEA-alumoxane (0.1 g) was dissolved in CHCl3 (5 mL) at room
temperature. The SiC fiber was cleaned with acetone and dipped in a Ca-doped
MEA-alumoxane
solution and allowed to fully air dry, at room temperature. Repeat
dipping/drying until desired
coating thickness is obtained. The coated fiber was heated from 25 °C
to 225 °C at the rate of 1
°C.min-1, soaked for 30 rains. at 225 °C, followed by a
temperature ramp up to 300 °C at the rate
of 2 °C.min-Z, and soaked for 80 rains., with a final ramp to the
maximum temperature of 1100
°C (over 360 minutes) which was then maintained for 400 minutes.
Example 68. Preparation of hibonite coated silicon carbide fibers.
Prepared in an analogous manner to that in Example 67 using the amounts and
conditions
shown in Table 12.
23
CA 02327097 2000-10-02
WO 99/50203 PCT/US99/06137
Example 69. Preparation of hibonite coated silicon carbide fibers.
Prepared in an analogous manner to that in Example 67 using the amounts and
conditions
shown in Table 12.
Example 70. Preparation of hibonite coated silicon carbide fibers.
S Prepared in an analogous manner to that in Example 67 using the amounts and
conditions
shown in Table 12.
Example 7I. Preparation of hibonite coated silicon carbide fibers.
Prepared in an analogous manner to that in Example 67 using the amounts and
conditions
shown in Table 12.
Example 72. Preparation of hibonite coated silicon carbide fibers.
Prepared in an analogous manner to that in Example 67 using the amounts and
conditions
shown in Table 12.
Example 73. Preparation of hibonite coated silicon carbide fibers.
Prepared in an analogous manner to that in Example 67 using the amounts and
conditions
shown in Table 12.
Example 74. Preparation of hibonite silicon carbide fibers.
Calcium-doped MEA-alumoxane (0.5 g) was dissolved in H20 (5 mL) with low heat
(40°C) and stirring. The fiber is cleaned with acetone and dipped in a
metal-doped MEA-
alumoxane solution and allowed to partially dry at room temperature then dried
in oven (45° C)
for 24 h. Repeat dipping/drying until desired coating thickness is obtained.
The coated fiber was
heated from 25 °C to 225 °C at the rate of 1 °C.min-i,
soaked for 30 miss. at 225 °C, followed by
a temperature ramp up to 300 °C at the rate of 2 °C.min-1, and
soaked for 80 mins., with a final
ramp to the maximum temperature of 1100 °C (over 360 minutes) which was
then maintained for
400 minutes.
Example 75. Preparation of hibonite coated silicon carbide fibers.
Prepared in an analogous manner to that in Example 74 using the amounts and
conditions
shown in Table 12.
Example 76. Preparation of hibonite coated silicon carbide fibers.
Prepared in an analogous manner to that in Example 74 using the amounts and
conditions
shown in Table 12.
Example 77. Preparation of hibonite coated silicon carbide fibers.
Prepared in an analogous manner to that in Example 74 using the amounts and
conditions
shown in Table 12.
24
CA 02327097 2000-10-02
WO 99/50203 PCTlUS99/06137
Example 78. Preparation of hibonite coated silicon carbide fibers.
Prepared in an analogous manner to that in Example 74 using the amounts and
conditions
shown in Table 12.
Example 79. Preparation of hibonite sapphire fibers.
Calcium-doped MEA-alumoxane (0.1 g) was dissolved in CHC13 (5 mL) at room
temperature. The sapphire fiber was cleaned with acetone and dipped in a Ca-
doped MEA-
alumoxane solution and allowed to fully air dry, at room temperature. Repeat
dipping/drying
until desired coating thickness is obtained. The coated fiber was heated from
25 °C to 225 °C at
the rate of 1 °C.min-i, soaked for 30 rains. at 225 °C,,followed
by a temperature ramp up to 300
°C at the rate of 2 °C.min-1, and soaked for 80 rains., with a
final ramp to the maximum
temperature of 1100 °C (over 360 minutes) which was then maintained for
400 minutes.
Example 80. Preparation of hibonite coated sapphire fibers.
Prepared in an analogous manner to that in Example 79 using the amounts and
conditions
shown in Table 13.
Example 81. Preparation of hibonite coated sapphire fibers.
Prepared in an analogous manner to that in Example 79 using the amounts and
conditions
shown in Table 13.
Example 82. Preparation of hibonite coated sapphire fibers.
Prepared in an analogous manner to that in Example 79 using the amounts and
conditions
shown in Table 13.
Example 83. Preparation of hibonite coated sapphire fibers.
Prepared in an analogous manner to that in Example 79 using the amounts and
conditions
shown in Table 13.
Example 84. Preparation of hibonite coated sapphire fibers.
Prepared in an analogous manner to that in Example 79 using the amounts and
conditions
shown in Table 13.
Example 85. Preparation of hibonite coated sapphire fibers.
Prepared in an analogous manner to that in Example 79 using the amounts and
conditions
shown in Table 13.
Example 86. Preparation of hibonite sapphire fibers.
Calcium-doped MEA-alumoxane (0.5 g) was dissolved in H20 (5 mL) with low heat
(40°C) and stirnng. The fiber is cleaned with acetone and dipped in a
metal-doped MEA-
alumoxane solution and allowed to dry at room temperature. Repeat
dipping/drying until desired
coating thickness is obtained. The coated fiber was heated from 25 °C
to 225 °C at the rate of 1
CA 02327097 2000-10-02
WO 99/50203 PCT/US99/06137
°C.min-1, soaked for 30 rains. at 225 °C, followed by a
temperature ramp up to 300 °C at the rate
of 2 °C.min-I, and soaked for 80 rains., with a final ramp to the
maximum temperature of 1100
°C (over 360 minutes) which was then maintained for 400 minutes.
Ezample 87. Preparation of hibonite coated sapphire fibers.
Prepared in an analogous manner to that in Example 86 using the amounts and
conditions
shown in Table 13.
Ezample 88. Preparation of hibonite coated sapphire fibers.
Prepared in an analogous manner to that in Example 86 using the amounts and
conditions
shown in Table 13.
Ezample 89. Preparation of hibonite coated sapphire fibers.
Prepared in an analogous manner to that in Example 86 using the amounts and
conditions
shown in Table 13.
Ezample 90. Preparation of hibonite coated sapphire fibers.
Prepared in an analogous manner to that in Example 86 using the amounts and
conditions
shown in Table 13.
REFERENCES
The following are incorporated herein in their entirety for all purposes:
Adkins, A., J. Am. Chem. Soc. , 44, 2175 ( 1922).
Anderson, M. A.; Gieselman, M. L.; Xu, Q., J. Membrane Sci., 39, 243 (1988).
Andrianov K. A.; Zhadanov, A. A., J. Polym. Sci, 30, 513 (1958).
Apblett, A. W.; Warren, A. C.; Baryon, A. R., Chem. Mater., 4, 167 (1992).
Baker, R. W., Membrane Separation Systems: Recent Developments and Future
Directions,
Noyes Data Corp., Park Ridge, NJ ( 1991 ).
Baltus, J. Mater. Sci.,123, 165 (1997).
Callender, R.L., Harlan, C.J., Shapiro, N.M., Jones, C.D., Callahan, D.L.,
Wiesner, M.R.,
MacQueen, D.B., Cook, R., Baryon, A.R., Chem. Mater., 9, 2418 (1997).
Courtright, E. L., Ceramic Engineering and Science Proceedings,12, 1725
(1991).
Elaloui, E.; Pierre, A. C.; Pajonk, G. M., J. Catalysis, 166, 340 (1997).
Furneaux, R. D.; Rigby, W. R.; Davidson, A. P., Nature, 337, 147 (1989).
Green R. H.; Hem, S. L., J. Pharm. Sci , 63, 635 (1974).
Hsieh H. D. in New Membrane Materials and Processes for Separation, Ed. K. K.
Sirkar and D.
R. Lloyd, American Institute of Chemical Engineering, New York, Vol. 84
(1988).
Kareiva, A.; Harlan, C. J.; MacQueen, D. B.; Cook, R.; Baryon, A. R., Chem.
Mater., 8, 2331
(1996).
26
CA 02327097 2000-10-02
WO 99/50203 PCT/US99/06137
Kim, K. J.; Stevens, P. V., J. Membrane Sci.,123, 303 (1997).
Kingery, W. D.; Bowen, H. K.; Uhlmann, D. R. Introduction to Ceramics, 2nd Ed.
Wiley, New
York, Chapter 1 (1976).
Landry, C. C.; Davis, J. A.; Apblett, A. W.; Baryon, A. R., J. Mater. Chem.,
3, 597 (1993).
Landry, C. C.; Pappe, N.; Mason, M. R.; Apblett, A. W.; Tyler, A. N.;
MacInnes, A. N.; Baryon,
A. R., J. Mater. Chem., 5, 331 (I995).
Lao, H.; Detellier, C.; Matsuura, T.; Tremblay, A. Y., J. Mater. Sci.
Letters,13, 895 (1994).
Lin, Y. S.; de Vries, K. J.; Burggraaf, A. J., J. Mater. Sci., 26, 715 ( 1991
).
Low, I. M.; Suherman, P. M.; Rhillips, P. N., J. Mater. Sci. Letters,16, 982 (
1997).
Michalke T. A.; Hellmann, T. R., J. Am. Ceram. Soc., 71, 725 (1988).
Nikolic, L.; Radonjic, L., Thin Solid Films, 295, 101 (1997).
Nogami, M., J. Non-Cryst. Solids.,178, 320 (1994).
Okubo, T.; Watanabe, M.; Kusakabe, K.; Morooka, S., J. Mater. Sci., 25, 4822
(1990).
Rezgui, S; Gates, B. C., J. Non-Cryst. Solids., 210, 287 (1997).
Rezgui, S; Gates, B. C.; Burkett, S. L.; Davis, M. E., Chem. Mater., 6, 2390
(1994).
Richerson, D. W., Modern Ceramic Engineering, Marcel Dekker, New York, p 373
(1992).
Serna, C. J.; White, J. L.; Hem, S. L., Soil. Sci., 41, 1009 (1977).
Shelleman, R A.; Messing, G. L.; Kumagai, M., J. Non-Cryst. Solids, 82, 277
(1986).
Teichner, S. J.; Nicolaon, G. A.; Vicarini, M. A.; Gardes, G. E. E., Adv.
Coll. Interf. Sci., 5, 245
(1976).
Wilson, S. J.; Stacey, M. H., J. Colloid Interface Sci., 82, 507 (198I).
Yoldas, B. E., J. Mat. Sci.,10, 1856 (1975).
Zaspalis, V. T.; van Praag, W.; Keizer, K.; Ross, J. R. H.; Burggraaf, A. J.,
J. Mater. Sci., 27,
1023 (1992).
27