Note: Descriptions are shown in the official language in which they were submitted.
HMV-053.02 ~ 02329858 2000-12-28
Expression Cloning of Protein Targets for Phospholipids
Related A,Dplications
This application is contiunuation-in-part of USSN 09/---,--- filed 11 December
2000, which in turn claims priority to US Provisional application 60/170,009
~xled 9
December 2000; the specifications of vsrhich are incorporated by reference
herein.
Government Su ~ >, ort
This invention was partially funded by NIIi Grant No. CA27951 and CA78773
from the National Cancer Institute; the goven~iment has certain rights to the
invention.
Field of the Invention
In an embodiment, this invention relates to the field of expression cloning
of protein targets for phospholipids and to methods and uses therefor. In
another
embodiment, the invention relates to methods and reagents for identifying
proteins or other cellular components which bind to a lipid moiety, to drug
screening assays and to a method of conducting a drug discovery business.
Background of the Invention
It is now well recognized that dynamic changes in the phosphorylatiott state
of
intracellular phosphatidylinositol (PtdIns) play critical roles i» mediating
many cellular
events. Phosphatidylinositol 3'-kinases (PI 3'-Ks) are a subfamily of PtdIns
ltinases that
phosphorylate the 3'-OH (D3) position of Ptdlns to create four different
Ptdlns
derivatives: PtdIas-3-P, PtdIns-3,4-P2, PtdIns-3,5-P2, and Ptdlns-3,4,5-P3.
Nine different
isoforms of PI 3'-K have been identified in mammalian cells and they have been
grouped
into three classes by Domin and Waterfield based on the specific form of PI
that is used
as a substrate.
Singly phosphorylated PtdIns-3-P is constitutively expressed in cells and is
involved in a variety of events associated with membrane protein trafficking.
While all
classes of Pl 3'-Ks can phosphorylate PtdIns to generate this lipid, the
majority of PtdIns-
3-P is probably produced by Class III PI 3'-Ks which is specific for Ptdlz~ts.
PtdIns-3,4-PZ
and PtdIns-3,4,5-P3 arc generated following stimulation by a wide variety of
extracellular
stimuli through many diverse classes of receptozs. Class I PI 3'I~s
phosphorylate PtdIns-
4,5-Pz to generate PtdIns-3,4,5-P3, which can be dephosphorylated to Ptdlns-
3,4-P2 by the
5' lipid phosphatase SHIP. Alternate pathways to Ptdlns-3,4-PZ have also been
described
involving phosphorylation of the 4-position of Ptdlns-3-P by Class II PI 3'K
or an
unidentified Ptdins-3-P 4-kinase, but the extent to which these enzymes
contribute to PI -
1
CA 02329858 2000-12-28
HMV-os~.o~
3,4-Pz synthesis in not clear.
Class I PI 3'-Ks play critical roles in many essential cellular processes.
Perhaps
most importantly, these kinases regulate cell survival. Inhibition of class I
PI 3'-Ks leads
to an induction of programmed cell death ox apoptosis arid constitutive
unregulated
activation of these enzymes or downstream targets of PtdIns-3,4-P2 and PtdIns-
3,4,5-P3
can rescue cells from cell death induced by serum deprivation, loss of matrix
attachment,
myc expression, a»d other apoptotic stimuli. These kiz~ases also control the
activation of
marry iuntz~acellular signaling pathways that regulate cell proliferation
including
Erk/MAPKs, protein translation factors (e.g. eIF-4E), and cyclins /cyclin-
dependent
kinases. Also, membrane trafficking events regulated by 3'PPIs control
receptor
internalization. In additiozt, PI 3'-Ks are necessary for glucose transporter
recruitment to
the plasma membrane and regulation of glycogen synthase kinase 3 and
phosphofructokinase, indicating that 3'PPIs are critically involved in insulin-
mediated
events associated with glucose metabolism. Integrin affinity modulation is
also blocked
by pharmacological inhibitors of PI 3'-K implicating these kinases in critical
events
associated with leukocyte traf~ckiua~g and inflammatory responses. PI 3'-K
also plays an
important role in regulating cell movement and cytoskeletal rearrangements.
For
example, 3'PPIs are necessary for controllieg receptor-induced changes in
actin assembly,
the formation of lamellipodial protrusions, and cell migration through the
small GTP
binding protein Rae.
Because of the central importance o~ PI 3'-Ks in controlling cell
proliferation,
survival, and motility it is likely that class I PI 3'-Ks and 3'PPI binding
proteins play an
importazat role in the pathogenesis of cancer. Overexpression of PI 3'-K in
chicken cells
is sufficient to induce cellular transformation both in vitro and in vivo. PI
3'-K has also
been implicated in the induction of Chronic Myelogenous Leukemia (CML) and
Acute
Lympltocytic Leukemia (ALL) by the $CR-ABL oncogene. ,As might be expected
from
its importance in cellular motility, PI 3'-K has been shown to play a role in
tumor
invasion and metastasis in several model systezaas_ ItecentIy, a role for PI
3'-K in human
carcinogenesis was demonstz~ated by the evidence that the tumor suppressor
PTEN, is a
lipid phosphatase which is specific for 3'phosphate of the inositol head-group
of and that
elimination of the lipid phosphatase activity correlates with the oncogez~ic
potential of
PTEN mutants found in human cancez~s 3'PPIs.
There are several identilaed protein motifs that bind to 3'PPIs: PH domains,
F'YVE domains, SH2 domains, and C2 domains. The pzamary function of these
domains,
2
CA 02329858 2000-12-28
HMV-053.02
each of which is approximately 90 to 120 amiuno acids in size, is believed to
be
localization of the protein to high local concentrations of 3'PPIs found near
active
signaling complexes at the cell membrane. However, there is evidence
indicating that
these domains can regulate protein function as well.
The most diverse and best-cb~aracterized 3'PPI binding dozr~ains are PH
domains
which compzase a largo family of binding modules that are known to bind
proteins as well
as a wide range of lipids. A subset of PH domains binds with a high affinity
to PtdIns-
3,4-P2 and PtdIns-3,4,5-P3. These PH domains are critical for the function of
several
signaling proteins including the serine/threonine kiz~ases Akt and PDK1, the
tyrosine
kinase $tk, and the ARF-GAF Grpl.
L~Y~VE domains are recently characterized domains that contain a zinc finger,
associate exclusively with PtdI,r~s-3-P, and are irnportant for vesicle
sorting. SH2
domains bind primarily to phosphorylated tyrosines; however, the SH2 domains
of PLCy,
the Src tyrosine kinase, and the p85 subunit of PI 3'-K can also bind to
Ptdlns-3,4,5-P3
with micromolar affinity. C2 domains bind to PtdTns-4,5-P2, Ptdlns-3,4-PZ, and
Ptdlns-
3,4,5-P3 and the specificity of lipid binding depends upon the local
concentration of
calcium. C2 domains are found in PKCs, PLA, and in vesicle sorting proteins
such as
synaptotagmin.
Sun, wa of th Invention
The phosphatidylinositol 3-kinase (PI 3'-I~) family of lipid kinases play a
critical
role in cell proliferation, survival, vesicle trafficking, motility,
cytoskeletal
rearrangements, and oncogenesis. To identify downstreara effectors of PI 3'-K,
we
developed a novel scxeen to isolate proteins which bind to the major products
of PZ 3'-K r~
phosphatidylinositol-3,4-bisphosphate (Ptdlns-3,4-PZ) and Ptdlns-3,4,5-P~.
This screen
uses synthetic analogs of these lipids in conjunction with libraries of
proteins that are
produced by coupled in vitro transcription/translation reactions_ The
feasibility of the
screen was initially deianonstrated using avidin-coated beads pre-bound to
biotizzylated
PtdIns-3,4-T~2 and PtdIns-3,4,S,P3 to specifically isolate the PH domain of
the
serinelthreonine kinase Akt. We then demonstrated the utility of this
technique in
isolating novel 3'phosphorylated phosphatidylinositol (3'PPI) binding proteins
through
the preliminary screening of in vitro transcribed/translated cDNAs from a
small pool
expression library derived from mouse spleen. Three proteins were isolated
that bound
3
CA 02329858 2000-12-28
>~INIV-os3.o2
speei~cally to 3'PPIs. Two of these proteins have been previously
characterized as
PIP3$P/p42~P° and the PtdTns-3,4,s-P3-dependent serine/threonine kinase
PDK1. The
third protein is a novel protean that contains an SH2 domain azzd a PH domain
which has
a higher specificity for bout PtdIns-3,4,5-P3 and Ptdlns-3,4-P2 than for
Ptdlns-4,s-Pz.
Transcripts of this novel gene (called PHISI~ for 3' Phosphoiunosztide
Interacting SH2-
Containing protein) are present in every tissue analyzed but are most
prominently
expressed in spleen.
This invention demonstrates the utility of this technique for isolating and
characterizing 3'PPI binding protezns and specifically contemplates broad
applicability
for the isolation of bindiztg domains for other lipid products.
One aspect of the invention pz~ovides a method for identifying a cellular
component which binds to a lipid moiety comprisixtg:
a. providing a lipid bait moiety being derivatized to a solid support;
b. contacting the lipid bait moiety with a library of cellular coraponents;
c. ider~txfying those mezobers of the cellular component library which
specifically bind to the lipid bait moiety.
1n preferred embodiments, the lipid bait uaoiety is a phospholipid, e.g.
selected
from the group consisting of of phosphatidylethanolamines,
phosphatidylcholines,
phosphatidylserines, phosphatidylglycerols, phosphatidylinositols,
polyphosphatidylanositols, and diphosphatidylglycerols. In certain preferred
embodiments, the phospholipid is derivatized to the solid support tlu4ugh a
cross-linking
moiety which is covalently attached to a phosphate head group of the
phospholipid.
In other preferred embodiments, the lipid bait moiety a plasrnalogen or a
sphingolipid.
In certain preferred embodiments, the library of cellular components is a
polypeptide library, e.g., including at least 10 different polypeptides, more
preferably at
least 100, 1000, or even 10,000 di#fezent proteins. For instance, the
polypeptide library
can be an expression library, such as derived from replicable genetic display
packages. Izx
other embodiments, the polypeptide library is a cell lysate or partially
puzif~ed protein
preparation.
The ide~atity of those members of the cellular component library which
specifically bind to the lipid bait uaoiety can eb determined by mass
spectroscopy.
Another aspect of the present invention provides a screening assay comprising:
a. providing a reaction mixture including a cellular component identiFed as
4
CA 02329858 2000-12-28
l3MV-os3.o2
described above fox its ability to specifically bind to the lipid bait moiety;
b, contacting the cellular component with a test compound;
c. determining if the test cuzz~poun~d bxr~ds to the cellular component.
In preferred embodiments, the the assay is repeated for a variegated library
of at
least 100 different test compounds, even mote preferably at least 100, 1000 or
even
10,000 different test compouztds. Exemplary compounds wluicl~ can be screened
for
activity in the subject assays include peptides, nucleic acids, carbohydrates,
small organic
molecules, and natural product extract libraries, such as isolated from
animals, plants,
fungus aad/or microbes
In certain preferred embodizaaents, the reaction mixture is a whole cell. In
other
embodiments, the reaction mixture is a cell lysate or purified protein
composition.
In certain embodiments, a test compound which is identified as able to bind to
the
cellular component is further tested for the ability to inhibit or mimic the
activity of a
lipid moiety.
Still another aspect of the present invention provides a method of conducting
a
drug discovery business comprising:
a. providing a lipid bait moiety being derivatized to a solid support;
b. contacting the lipid bait moiety with a library of cellular components;
e. identifying those members of the cellular component library which
specifically bind to the lipid bait moiety.
d. providing a reaction mixture including a cellular component identified in
step (c) as able to specifically bind to the lipid bait moiety;
e, contacting the cellular component with a test compound;
f. determining if the test compound binds to the cellular component;
g. further tesring those test compound identified in step (f) as able to bind
to
the cellular component for the ability to inhibit or mimic the activity of a
lipid moiety; and
h. formulating a pharmaceutical preparation includixag one or more
compounds identified in step (g) as able to inhibit or mimic the activity of
a lipid moiety.
Xet another aspect of tlae invention pz~ovides a method of conducting a target
discovery business comprising:
a. providizxg a lipid bait moiety being derivatized to a solid support;
s
CA 02329858 2000-12-28
HMV-053.02
b, contacting the Iipid bait moiety with a library of cellular components;
c. identifying those members of the cellular component library which
specifically bind to the lipid bait moiety.
d. licensing, to a third party, the rights for drug development for a cellulax
component identified in step (c) as able to specifically bind to the lipid
bait
moiety.
Brief Description of the Drawings
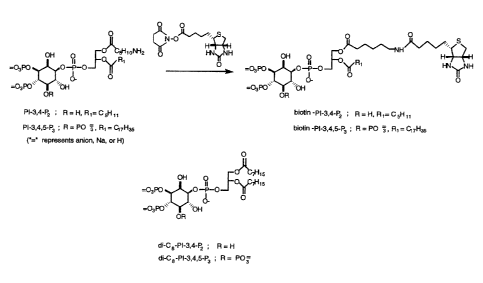
FIG. 1. Synthesis of biotinylated PIPn probes and structure of dioctanoyl
derivatives
FIG. 2. The PH domain. of Akt binds specifically to avidin beads pre-bound
with
biotinylated 3'PPIs. A. 3sS- labeled maltose binding protein (N>BP) or MBP
fused to the
PH domain of Akt (M.BP-PH) (lanes 1 and 5) were incubated with avidin beads
alone
(lanes 2 and G), avidin beads pre-bound with PtdIns-3,4-P2-biotin (lanes 3 and
7), avidin
beads pre-bound with PtdIns-3,4,5-P3 -biotin (lanes 4 and 8). Proteins were
labeled with
3sS-methionine by in vitro transcriptionltxanslation of 0.5 pg of the
respective genes in
pCS2(+) and the binding reactions were done as described in "Experimental
Procedures".
Truncated species of the M)3P and MBP-PH (eMBP, ~MBP-PH) result from
initiation of
translation at start sites after the initial AUG codon. $. Avidin beads pre-
bound with
Ptdlns-3,4-Pz-biotin can speciEcally isolate M13P-PH from a pool of other
proteins. 10
ng of MBP-PH DNA and/or ~ pg of DNA from a random pool from the cDNA library
were transcribedltranslated in the presence 3sS-methionine and binding
reactions were
performed as described in "Experimental Procedwres". Total labeled proteins
(lanes l, 4,
and 7), labeled proteins bound to avidin beads (lanes 2, 5, and 8), labeled
proteins bound
to avidin beads pre-bound with Ptdlns-3,4-P2-biotin (lanes 3, 6, and 9). C.
PtdIns-3,4,5-P3
az~d PtdIns-3,4~P2 preferentially displace MBP-PH ~rom Ptdlns-3,4,5-P3-biotin.
3sS-
labeled MBP-PH was bound to avidin beads coated with Ptdlns-3,4,5-P3-biotin in
the
presence of the indicated concentrations of PtdTns-4,S-Pi (squares), Ptd~s-3,4-
PZ
(circles), Ptdlns-3,4,5-P3 (triangles) and processed as described in
ilxperimental
Prncedures?. Points represent the mean of two independent experiments.
FIG 3. Isolation of marine isoforms of PDK1 and Is~P3BP/p42~4 from expression
library via avidin beads pre-bound with biotinylated 3'PPIs. A. PDK1. Top
panel.
SpeeiSc binding of PDK1 in total pool and as single clone. Total 3sS-labeled
proteins
6
CA 02329858 2000-12-28
1"IMV-053.02
(lanes 1 and 5), labeled proteins bound to avidin beads (lanes 2 and 6),
labeled proteins
bound to avidin beads pre-bound with Ptdlns-3,4-P2-biotin (lanes 3 and 7),
labeled
proteins bound to avidin beads pyre-bound with PtdZns-3,4,5-P3 -biotin (lanes
4 and 8).
Bottom panel. Lower diagrams show the protein domain structure of PD1C1 and
corresponding cDNA fragment isolated from the expression library. Nucleotide
positions
of putative stop and start codons (AUG) are indicated. B. PIP38PIp4z~4. ~'op
panel.
Specific binding of Pl~'3BPIp42~4 in total pool and as single clone. Total j5S-
labeled
proteins {lazes 1 and 5), labeled proteins bound to avidin beads (lanes 2 and
6), labeled
proteins bound to avidiz~ beads pre-bound with Ptdlns-3,4-Pz-biotin (lanes 3
and 7),
labeled proteins bound to avidin beads pre-bound with Ptd,Ins-3,4,5-P3 -biotin
{lanes 4
and 8). Bottom panel. Lower diagrams show the protein domain structure of
PIP3BPIp42~p4 and corresponding cDNA fragment isolated from the expression
library.
Nucleotide positions of putative stop and start codons (AUG) are indicated.
FIG. 4. Isolation of PHISH from expressxrn~ library via avidin beads pre-bound
with biotinylated 3'PPIs. A. Specific binding of PHISH in total pool azrd as
sxz~gle clone.
Total 35S-labeled proteins (lanes x and 5), labeled proteins bound to avidin
beads (lanes 2
and 6), labeled proteins bound to avidin beads pre-bound with PtdIns-3,4-Pz-
biotin (lanes
3 and '~), labeled proteins bound to avidin beads pre-bound with PtdIns-3,4,5-
P3 -biotin
(lanes 4 and 8). B. Ptdlns-3,4,5-P3 and Ptdlns-3,4-Pz preferentially displace
PHISH from
Ptdlns-3,4,5-P3 -biotin bound to avidity beads. 35S-labeled PHZSH was bound to
avidity
beads coated with PtdIns-3,4,5-P3-biotin in the presence of the indicated
concentrations
of Ptdins-4,5-P2 (squares), PtdTns-3,4-Pi {circles), Ptdlns-3,4,5-P3
(firiangles) and
processed as described in iExperimental I'roceduresi. Points represent the
mean of two
independent experiments.
FIG. 5. Nucleotide and amino acid sequence of marine PHISH. Sequence of
PHISH isolated via expression elotaing. Putative SH2 domain is outlined in
red, predicted
tyrosine phosphorylation site is outlined in green, putative PH domain is
outlined izt blue.
FIG. 6_ Northern blot of mRNA from various naurine tissues hybridized to
prabes
derived from PHISH and G,A,PDH. Marine tissue RNA (l0ug per lane) was
subjected to
agarose gel electrophoresis, transferred to nylon membrane, and hybridized to
3ZP-labeled
probes derived from the coding regions of PHISH or GAPDH as described in
iExperimental Proceduresl. Lazes: 1-brain, 2-heart, 3-lung, 4-lymph node, 5-
spleen, 6-
thymus. Arrows represent the mobility of the nnajor (large arrow) and minor
(small
arrow) products of an in vitro transcription reaction of the PHISH gene
isolated from the
7
CA 02329858 2000-12-28
HMV-053.02
expression library.
Detailed ~escrintion of the lfnveatioa
A) Overview
One aspect of the present invention relates to methods and reagents fax
identifying
proteins or other cellular components (collectively "LBP" or "lipid binding
partner"),
which bind to lipids such as phospholipids, triacylglycerides, plasmalogens or
sphingolipids. In preferred embodiments, the subject method is useful for
identifying
LBPs that bind to phaspholipids such as phosphatidylserines,
phosphatidylcholines (also
called lecithins), phosphatidylethanolamines, phosphatidylglycerols,
phosphatidylinositols, or sphingomyelins. The Ll~~'s can be naturally
occurring, such as
proteins or fragments of proteins cloned or otherwise dewed from cells, ox can
be
artificial, e.g., polypeptides which are selected from random ox secni~random
polypeptide
libraries.
In general, the method of the present invention comprises providing a lipid
which
includes an "sequestration tag", and contacting the lipid with a structurally
diverse
(variegated) library of polypeptides or other molecules under conditions
wherein binding
of lipids to library molecules can occur such the resulting complexes are
enriched for
library molecules which specifically (as opposed to non-specifically) bind the
lipids.
Library molecules which specifically bind to the lipids are isolated from the
library, or
their identity is otherwise determined, e,g., by the presence of a tag
associated with the
LBP which is a unique identifier of the LBP. The polypepiide library can be
provided as
part of a replicable genetic display package, an expression library
(especially an
intracellular expression library), a synthetic polypeptide library or other
form.
In other embodiments, the system can be reversed and a polypeptide can be used
to screen a library of structurally diverse lipids to identify lipids which
selectively bind to
the polypeptide.
Another aspect of the present invention relates to the LBPs which are
identified by
the subject method. Such zzaolecules can be used as drug screening targets,
e.g., for drugs
which alter the activity of the LBP {such as its ability to bind a lipid) or
which alter the
level of the LBP in the cell. lV,loz~eaver, the level of an LBP in a cell can
be determined
for diagnostic or prognostic purposes.
8
CA 02329858 2000-12-28
z~l~v-os3.o2
Where the LBP is a protein, the invention also relates to nucleic acids which
encode the protein or a fragment thereof ~'he invention also contemplates
nucleic acids
which hybridize to the coding sequence fax an LBP, e.g., which may be useful
as
amplim,ers, probes, primers or antisense.
Another aspect of the present invention relates to antibodies, e.g.,
monoeolonal,
purified and/or recombinant, which are a~unoseleetive for an LBP.
Still another aspect al: the present invention relates to drug screening
assays for
identifying compoutxds, e.g., such as small organic molecules (1V1W~1000amu)
which
inhibit or potentiate the activity of an LBP. For instance, the assay can be
used to identify
compounds which inhibit or potentiate an intrinsic enzymatic activity of an
LBP, or the
ability of the LBP to bind to otb,er molecules, e.g., to lipids, to proteins,
to nucleic acids.
Yet another aspect of the present invention relates to the use the LBPs, or
compounds which agozuize or antagonize, as the case may be, the activity of an
LBP, far
the treatment or prevention of a disorder or unwanted effect mediated by a
lipid.
B) De nitions
Before further description of the inventia~a, certain terms employed in the
specification, examples and appended claims are, for convenience, collected
here.
"Fatty acids" are long-chain hydnxarbon molecules containing a carboxylic acid
moiety at one end. The numbering o,f carbons in fatty acids begins with the
carbon of the
carboxytate group. Fatty acids that contain no carbon-carbazt double bonds are
termed
saturated fatty acids; those that contain double bonds are unsaturated fatty
acids. The
numeric designations used for fatty acids come from the number of carbon
atoms,
followed by the number of sites of unsaturation (eg, pahnitie acid is a 1 b-
carbon fatty acid
with no unsaturation az~d is designated by 16:0). The site of unsaturadan in a
fatty acid is
iundicated by the symbol D and the number of the first carbon of the double
band {e.g.
palmitoleic acid is a I6-carbon fatty acid with on,e site of unsatwration
between carbons 9
and 10, and is designated by 16:109).
"Triacylglycerides" are composed of a glycerol backbone to which 3 fatty acids
are esterified.
'fhe basic structure of '~hospoIipids" is very similar to that of the
triacylglycerides except that C-3 (sn3~f the glycerol bacL~bone is esterihed
to phosphoric
9
CA 02329858 2000-12-28
l3lvw-os3.az
acid. The building block of the phospholipids is phosphacidic acid which
results when the
X substitution in the basic structure shown in the Figure below is a hydrogen
atom.
Substitutions include ethanolamine (phosphatidylethanolarnine), choline
(phosphatidylcholine, also called Iecithins), serine (pbosphatidylserine),
glycerol
(phosphatidylglycerol), myo-inositol (plxosphatidylinositol, these compounds
can have a
variety in the nutnbezs of inositol alcohols that are phosphvrylated
generating
polyphosphatidylinositols), and phosphatidyIglyeerol (diphosphatidylglycerol
more
commonly known as cardiolipins),
"Plasmalogens" are complex membrazxe lipids that reserrxble phospholipids,
principally phosphatidyleholine. The major difference is that the fatty acid
at C-1 (snl) of
glycerol contains either an O-alkyl ax O-alkenyl ether species. A basic O-
alkenyl ether
species is shown in the Figure below. Gne of the most potent biological
molecules is
platelet activating factor (PAF) which is a choline pIasmalogen in which the C-
2 (sn2)
position of glycerol is esterified with an acetyl group insted of a lvng chain
fatty acid.
"Sphingolipids" are corttposed of a backbone of sphingosine which is derived
itself from glycerol. Sphingosizze is N-acetylated by a variety of fatty acids
generating a
family of molecules referred to as ceramides. Sphingolipids predominate in the
myelin
sheath of nerve fibers. Sphingomyelin is an abundant sphingolipid generated by
transfer
of the phasphocholine moiety of phosphatidyIcholine to a ceramide, tluus
sphingomyelin
is a unique form of a phospholipid. The other major class of sphingolipids
(besides the
sphingorayelins) are the glycosphingolipids generated by substitution of
carbohydrates to
the snl carbon of the glycerol backbone of a ceramide. There are 4 major
classes of
glycosphingolipids:
Cerebrosides: contain a single moiety, principally galactose.
Sulfatides: sulfuric acid esters of galactocerebrosides.
Globosides: contain 2 or more sugars.
Gangliosides: similar to globosides except also contain sialic acid.
The term "simultaneously expressing" refers to the expression of a
representative
population of a polypeptide library, e.g., at least 50 percent, more
preferably 7s, 80, 85,
90, 95 or 98 percent of all the different pvlypeptide sequences of a library.
The team "random polypeptide library" refers to a set of random or semi-random
polypeptides.
CA 02329858 2000-12-28
-~S ~ .~~
The language "replicable genetic display package" or "display package"
describes
a biological particle which has genetic information providing the particle
with the ability
to replicate. The package can display a fusion protein iz~cludiz~g a
polypeptide derived
from the variegated pvlypeptide library. The test polypeptide portion of the
fusion
protein is presented by the display package in a context which permits the
polypeptide to
bind to a lipid that is contacted with the display package. The display
package will
generally be derived froz~ra a system that allows the sampling of very large
variegated
polypeptide libraries. The display package can be, for example, derived from
vegetative
bacterial cells, bacterial spores, and bacterial viruses.
The language "differential binding means", as well as "affinity selection" and
"afflnxty enrichment", refer to the separation of members of the polypeptide
display
library based on the differing abilities of polypeptides on the surface of
each of the
display packages of the library to bind to the lipid lipid. The differential
binding of a
lipid by test polypeptides of the display can be used in the affinity
separation of those
polypeptides which specifically bind the lipid from those which do not. For
example, the
affinity selection protocol can also include a pre- or post-exurichznent step
wherein display
packages capable of binding "background lipids", e.g., as a negative
selection, are
removed from the library. Examples of affinity selection means include
aflaz~ity
chromatography, immunoprecipitation, fluorescence activated cell sorting,
agglutination,
and plaque lifts. As described below, the affinity chromatography includes bio-
panning
techniques using either purified, inamabilized lipid proteins or the li~l~e,
as well as whole
cells.
The phrases "individually selective manner" and "individually selective
binding",
with respect to binding of a test polypeptide with a lipid, refers to the
binding of a
polypeptide to a certaiza. protein lipid which binding is specific for, and
dependent oz~, the
molecular identity of the protein lipid.
The term "solid suppork" refers to a material having a rigid or semi-rigid
surface.
Such materials will preferably take the fvz~zn of small beads, pellets, disks,
chips, dishes,
mufti-well plates, wafers ox the like, although other forms may be used. In
some
embodiments, at least one su~'ace of the substrate will be substantially flat.
The term
"surface" refers to any generally two-dimensional structure on a solid
substrate and may
have steps, ridges, kinks, tezxaces, and the like without ceasing to be a
surface.
In an exemplary embodimexxt of the present invention, the display package is a
phage parCicle which comprises a polypeptide fusion coat protein That includes
the amino
11
CA 02329858 2000-12-28
~tvlv-os3.o2
acid sequence of a test polypepcide. Thus, a library of replieable phage
vectors, especially
phagemids (as defined herein), encoding a library of palypeptide fusion coat
proteins is
generated and used to transform suztable host cells. Phage particles formed
from the
chimeric protein can be separated by affnity selection based on the ability of
the
polypeptide associated with a particular phage particle to specifically bind a
lipid. In a
preferred embodiment, each individual phage particle of the library includes a
copy of the
corresponding phagemid encodizzg the polypeptide fizsion coat protein
displayed on the
surface of that package. Exezxtplary phage for generating the present
variegated
polypeptide libraries include M13, fl, fd, Ifl, Ike, Xf, Pfl, Pf3, ~., T4, T7,
P2, P4, ~X-
174, MS2 sari f2.
The language "fusion protein" and "chimeric protein" are art-recognized terms
which are used interchangeably heo~ein, and include contiguous polypeptides
comprising a
first polypeptide covalently linked via an amide band to one or more amino
acid
sequences which define polypeptide domains that are foreign to and not
substantially
homologous with any domain of the first polypeptide. One portion of the fusion
pmtein
comprises a test polypeptide, e.g., which can be random or semi-random. A
second
polypeptide portion of the fusion protein is typically derived from an outer
surface
protein or display anchor protein which directs the "display package" (as
hereafter
defined) to associate the test polypeptide with its outer surface. As
described below,
where the display package is a phage, this anchor protein can be derived from
a surface
protein native to the generic package, such as a viral coat proteiua. Where
the fusion
protein comprises a viral coat protein and a test polypeptide, it will be
referred to as a
"polypeptide fusion coat protein". The fusion protein further comprises a
signal
sequence, which is a short length of amino acid sequence at the amino terminal
end of the
fusion protein, that directs at least the portion of the fusion protein
including the test
polypeptide to be secreted from the cytosol of a cell and localized on the
extracellular
side of the cell membrane.
Gene constructs encoding fusion proteins are likewise referred to a "chimeric
genes" or "fusion genes".
The temp "vector" refers to a DNA, molecule, capable of replication in a host
cell,
into which a gene can be inserted to construct a recombinant DNA xnalecule.
The terms "phage vector" and "phagemid" are art-recognuized and generally
refer
to a vector derived by modification of a phage genome, containing an origin of
replication
for a bacteriophage, and preferably, though optional, an origin (orl~ for a
bacterial
12
CA 02329858 2000-12-28
HMV-05 3 .02
plasmid. The use of phage vectors rather than the phage genome itself provides
greater
flexibility to vary the ratio of ehiznezic polypeptideleoat protein to wild-
type coat protein,
as well as supplement the phage genes with additional genes encoding other
heterologous
polypeptides, such as "auxiliary polypeptides" which znay be useful in the
"dual"
polypeptide display constructs described below.
The language "helper phage" describes a phage which is used to infect cells
containing a defective phage genome or phage vector and which functions to
complement
the defect. The defect can be one which results from removal ox inactivation
of phage
genomic sequence required for pzoduction of phage particles. examples of
helper phage
are M13K07.
As used herein, a "reporter gene construct" is a nucleic acid that includes a
"reporter gene" operatively linked to at least one transcriptional regulatory
seduence.
Transcription of the reporter gene is controlled by these sequences to which
they are
linked.
The term "sequester", as used herein, means to separate, segregate, remove, or
bind a lipid complex, e.g., on a solid support. rn preferred embodiuzzez~ts, a
lipid complex
is sequestered by a solid support such that other non-sequestered LBPs can be
removed,
e.g., by washing or ocher purification techniques. A lipid complex is
"reversibly
sequestered" if the process of sequestering the complex on a solid support can
be reversed
to yield a free complex or free LBP, e.g,, in solution in a reaction mixture.
In preferred
embodiments, the process of sequestering a complex, or of reversing the
sequestration, or
both, occurs under mild cortditivns and in high yield, e.g., greater than at
least about 40%
yield.
The term "polymeric support", as used herein, refers to a soluble or insoluble
polymer to which a lipid can be covalently bonded (e.g., by through an ester
functionality)
by reaction with a functio»al group of the polymeric support. Marry suitable
polymeric
supports arc known, and include soluble polymers such as polyethylene glycols
or
polyvinyl alcohols, as well as insoluble polymers such as polystyrene resins.
A suitable
polyme~c support includes functional groups such as those described below. A
polymeric support is termed "soluble" if a polymer, or a polymer-supported
compound, is
soluble under the conditions employed- However, in genezal, a soluble polymer
can be
rendered insoluble under defined conditions. Accordingly, a polymeric support
can be
soluble under certain conditions and i~x~soluble under other conditions- A
polymeric
support is termed "insoluble" if reaction of a lipid with the polymeric
support results in an
13
CA 02329858 2000-12-28
HMV-os3.oz
iztsoluble polymer-supported lipid under tl~e conditions employed.
Abbreviations used herein include: AID - ADP ribosylation factor; Btk -
Bretons
tyrosine kinase; DTT - dxtlxxothreitol; ErkIMAPK - extracellular regulated
kinase/mitogez~
activated protein kiz~ase; EST - expressed sequence tag; GAP = GTPase
activating
protean; GAPDI~ = glyceraldehyde 3-phosphate dehydragenase; GTP - guanosine
triphosphate; I-iEPES - (N-[z-hydroxyethyl]piperazine-N'-[2-ethanesulfonic
acid); kb
kilobase; SAS - sodium dodecyl sulfate; MBP = maltose binding protein; MBP-PI-
i =
maltose binding protein Akt P~ domain fusion protein; NP-40 -
nonylphenylpolyethylene
glycol; 3'PPI - 3' phosphorylated phosphatidylinositols; PAGE - polyacrylamide
gel
electrophoresis; PCR- polymerise chain reaction; PDK1- phosphoiz~ositide
dependent
kiztase 1; PH = pleckstrin homology; PI3'-K = phosphatidylinositol 3'-kinase;
PKC
protein kinase C; PLA - phopholipase A; PLCy - phospholipase CY; Ptdlns -
phosphatidylinositol; PtdIns-3-P - phosphatidylinositol-3-monophasphate;
PtdIns-3,4-P2
phosphatidylinositol-3,4-bisphosphate; Ptdlns-4,5-Pz - phosphatidyliz~ositol-
4,5-
bisphosphate; PtdIns-3,4,5-P~ - phosphatidylinositol-3,4,5-trisphosphate; SDS -
sodium
dodecyl sulfate; SH2 = Src homology 2.
G) F.xemplarv Embodiments o~'Phos~pleolipid Baits
As set forth above, in certain embodiments, the subject method can be
practiced
by utilizing immobilized lipid moieties, such as phospholipids, as the bait
for identifying
polypeptides and other molecules capable of interacting with, and fonming
complexes
with the lipid moiety. In certain embodiments, the subject lipid moiety is a
phospolipids,
such as selected from the group consisting of phosphatidylethanolamines,
phosphatidylcholines, phosphatidylserines, phosphatidylglycerals,
phosphatidylinositols,
polyphosphatxdylinositols, and diphosphatidylglycerols. E~cemplary
polyphosphaddylinositols include:
di C16, L-a-D-myo-Phosphatidylxriositol 3-monophosphate
di C8, L-a-D-myo-Phosphatidylinositol 3-monophosphate
di C 16, L-a-D-myo-Phosphatidyiinositol 3,4-diphosphate
di C8, L-a-D-myo-Phosphatidylinositol 3,4-diphosphate
di C 15, L-a-D-myo-Phosphatidylinositol 3,4,5-diphosphate
di C8, L-a-D-myo-Phosphatidylinositol 3,4,5-diphosphate
di C16, L-a-D-myo-Phosphatidylinositol 3,5-diphosphate
di C8, L-a-D-xnyo-Phosphataclyliilositol 3,5-ditphosphate
di C16, L-a-D-myo-Phosphatidylinositol 4-monophosphate
14
CA 02329858 2000-12-28
I-1MV-053.02
dl C8, L-a-D-myo-Phosphatidylinositol 4-monophosphate
dl C16, L-a-D-myo-Phosphatidylinositol 4,5-diphosphate
dl C$, L-a-D-myo-Phosphatidylinositol 4,5-dxphosphate
dl C16, L-a-D-myo-Phosphatidylinositol 5-monophosphate
dl C8, L-a-D-myo-Phosphatidylinositol 5-moz~ophosphate
~n other embodiments, the subject lipid moiety is a plasmalogen. In still
other
embodiments, the subject lipid moiety is a sphingolipid, such as may be
selected from the
goup consisting of cerebrosides, sulfatides, globosides, and gar~gliosides.
In certain preferred embodiments, the subject lipid can be immobilized Qr
incorporated into a polymer or other inspluble matrix by, for example,
derivativatior~ with
one oz~ more of subject lipid moieties derivatized to a solid support, such as
glass, silicon,
or a polymeric support. The support can be, inter ills, a bead, a chip, a
hydrogel, etc.
In certain preferred embodiments, the subject lipid moieties are derivatized
by
covalent or non-covalent coupling through one or more of its fatty acid side
chains, e.g.,
in order to present at least a portion of its head group. For example, the
present invention
specx~cally contemplates phosphatidyliz~ositol derivatives represented by the
general
formula:
Hz
P
C7H
H -X-R'
G-X-R'
M2
W~lCreln
X, independently for each occurrence, represents O or S;
R, independently for each occurrence, represents hydrogen or -P03;
R' represents, for each occurrence, -CH~HR3-L or -CORD-L;
R3 and R4, independently for each occurrence, represent a C6-C2a alkyl group,
e.g., which may be saturated or unsaturated, branched or lxz~ear, substituted
or
unsubstituted; and
L represents a linker, or a linker covalently or non-covalently attached to a
solid
support.
CA 02329858 2000-12-28
HMV-053.02
In certain preferred embodiments, X represents O; and L is a Iiz~.ker of 150-
1500amu, such
a biotin.
In certain embodiments, particularly where zztore than one type of lipid-
moiety is
used as a bait (e.g., a library of different Iipid moieties), a spatial array
o~ lipid baits can
be generated, e.g., for libxary versus library scz~eening. For example,
libraries of at least
different lipid moieties can be tested as baits, and zxtaxe preferably
libraries of at least
100 or even 1000 different lipid moieties.
The lipid moiety can be derivatived to the support by any of a number of
means.
In the case of phospholipids, the derivatization is preferably through a
phosphate head
group. As described in the appended examples, biotinylation of the phosphate
head group
can be used to derivatize the lipid moiety to an avidin-displaying support.
There are a large number of other chemical crass-linking agents which could be
used in the present invention are known in the art. For the prese~at
invention, the
preferred cross-linking agents are heterobifunctionaI cross-linkers, which can
be used to
link the lipid bait and solid support in a stepwise mawner. Heterobifunctional
cross-
linkers provide the ability to design more specific coupling methods for
conjugatiung the
subject moieties, thereby reducing the occurrences of unwanted side reactions
such as
homo-lipid polymers. A wide variety of heterobifunetional cross-linkers are
known in the
art. These include: succinimidyl 4-(N-maleimidomethyi) cyclohexane-1-
carboxylate
(SMCC), m-Malezmidobenzoyk-N- hydroxysuccinimide ester (kl~S); N-succinimidyl
(4-
iodoacetyl) aminobenzoate (SI:AB), succinimidyl 4-(p-maleimidophenyl) butyrate
(SMPB), 1-ethyl-3-(3-dimethylamxnopropyl~arbodxirnide hydrochloride (EDC); 4-
succinimidyloxycarbonyl-a-methyl-a-(2-pyridyldithio)-tolune (SMPT), N-
succinimidyl 3-
(2-pyridyldithio)propionate (SPDP), succinimidyl 6->3-(2-
pyridyldithio)propionate!hexanoate (LC-SPDP). Those cross-linking agents
having N-
hydroxysueeinxmide moieties can be obtaiuaed as the N.-hydroxysulfosuccinimide
analogs,
which generally have greater water solubility. In addition, those cross-
linking agents
having disultade bridges within the linking chain can be synthesized instead
as the alkyl
derivatives so as to reduce the amount of linker cleavage in vivo.
I» addition to the heternbifunctional cross-linkers, there exists a number of
other
useful cross-linking agents including homobifunetional a~ad photoreactive
cross-linkers.
Disucciniznidyl suberate (D55), bismaleimidohexane (BMH) and
dimethylpimekiznidate-2
ICI (DMP) are examples of useful ho~naobifunctional cx-oss-linking agents, and
bis-~3-(4-
azidosalicylamido~thyldisulfide (BASED) and N-succinimidyl-6(4'-azido-2'-
16
CA 02329858 2000-12-28
I IMV-053.02
nitrophenylamino)hexaxtvate (SANfAIT) are exanraples of useful photoreactive
cross-
linkers foz use izt this invention. For a review of coupling techztiques which
may be
applied to the subject lipid moieties, see Means et al. (1990) Bioconjugate
Chemistry 1:2-
12.
The third component of the heterobifunctional crass-linker is the spacer arm
or
bridge. The bridge is the structure that connects the two reactive ends. The
nrtost apparent
attribute of the bridge is its effect on steric hindrance. In some instances,
a longer bridge
can more easily span the distance necessary to link two complex biomolecules.
For
xz~stance, SMPB has a span of 1a.5 angstroms.
D) ExemDlarv Embodiments ofPolvpe~ptide Libraries
One goal of the present method is to identify proteins which are bound by the
lipid
bait. Accordingly, the present invention contemplates that any of a number of
methods
for trapping protein complexes using non-protein baits can be used. For
instance, the
proteins which are bound to the lipid bait can be identified by sequencing
using mass
spectroscopy. This technique can be advazztageous when the source of test
proteins is a
cell lysate. In other embodiments, the polypeptides ~e associated with a tags)
which
identi~tes the sequence of the protein, or with the gene which encodes the
protein. In still
other instance, t'he proteins are provided as part of a spatial array for
which the
coordinates on the array provides the identity of the protein.
In certain preferred e~mbvdiznents, the polypeptide library is provided as an
expression. library. For instance, a library of test pvlypeptides is expressed
by a
population of display packages to farm a peptide display library. With respect
to the
display package on which the variegated peptide library is manifest, it will
be appreciated
from the discussion provided herein that the display package will preferably
be able to be
(i) genetically altered to encode heterologous peptide, (ii) rraaintained and
amplified in
culture, (iii) manipulated to display the peptide-containing gene product in a
manner
pezrrxitting the peptide to intezact with a lipid during an affinity
separation step, and (iv)
affinity separated while retaining the nucleotide sequence encoding the test
pvlypeptide
(herein "peptide gene's such that the sequence of the peptide gene can be
obtained. In
preferned embodizxteztts, the display remains viable after affinity
separation.
Ideally, the display package comprises a system that allows the sampling of
very
large variegated peptide display libraries, rapid sorting after each affinity
separation
17
CA 02329858 2000-12-28
HMV-0s3.02
round, and easy xsolatian of the peptide gene from puni~ed display packages yr
further
manipulation of that seduenee in the secretion zr~ode. The most attractive
candidates for
tlxis type of screening are prokaryotic organisms and viruses, as they can be
amplified
quickly, they are relatively easy to xzxanipulate, and large number of clones
can be created.
Preferred display packages include, for example, vegetative bacterial cells,
bacterial
spores, and most preferably, bacterial viruses (especially DNA viruses).
However, the
present invention also contemplates the use of eukaryotic cells, including
yeast and their
spores, as potential display packages.
In addition to coznnaercially available kits for generating phage display
libraries
(e.g. the Pharmacia Recombinant Phage Antibody System, catalog no. 27-9400-O1;
and
the Stratagene Surfhr4PTM phage display kit, catalog no. 240612), examples of
methods
and reagents particularly amenable for use in gez~erati~ng the variegated
peptide display
library of the present invention can be found in, for example, the Ladner et
al. U.S. Patent
No. 5,223,409; the King et al. International Publication No. WO 92/18619; the
Dower et
al. International Publication No. WO 91/17271; the Winter et al. International
Publication
WO 92120791.; the Marklan,d et al. international Publication No. WO 92/15679;
the
Breitling et al. International Publication WO 93/01288; the MeCat~'eriy et al.
International
Publication No. WO 92101047; the Garrard et al. International Publication No.
WO
92109690;, the L,adner et al. International Publication No. WO 90102$09; Fuchs
et a1.
(1991) BiolTechnology 9:1370-1372; Hay et al. (1992) Hum Antibod Hybridornas
3:81-
85; Huse et al. (1989) Science 246:1275-1281; GTif~hs et al. (/993),E,I~BOJ
12:725-734;
Hawkins et al. (1992) .T ll~lol Biol 226:889-896; Clackson et al. (1991)
Nature 352:624-
628; Gram et al. {1992} PNAS 89:3576-3580; Garrad et al. (1991) NiolTechnology
9:1373-1377; Hoogenboom et al. (/991) Nuc Acid Res 19:4133-4137; and Barbas et
al.
(1991 ) PNAS 88:'797$-7982. These systems can, with. zraadx~cations described
herein, be
adapted for use in the subject method.
When the display is based on a bacterial cell, or a phage which is assembled
periplasmically, the display means of the package will comprise at least two
components.
The first component as a secretion signal which directs the recombinant
peptide to be
localized on the extracellular side a~ the cell membrane (of the host cell
when the display
package is a phage). This secretion signal can be selected so as to be cleaved
off by a
signal peptidase to yield a prneessed, "mature" peptide. The second component
is a
display anchor protein which directs the display package to associate the test
polypeptide
18
CA 02329858 2000-12-28
HMV-053.02
with its outer surface. As described below, this anchor proteizz can be
derived from a
surface or coat protein native to the genetic package.
Whets the display package is a bacterial spore, or a phage whose protein
coating is
assembled intracellularly, a secretion signal directing the peptide to the
inner membrane
of the host cell is unnecessary. In these cases, the means four arraying the
variegated
peptide library comprises a dezavative of a spore or phage coat protein
amenable for use
as a fusion protein.
Irt some instances it may be necessary to introduce an unstructured
polypeptide
linker region between portions of the chimeric protein, e.g., between the test
polypeptide
and display polypeptide. 'this linker cart facilitate enhanced flexibility df
the chimeric
protein allowing the test polypeptide to freely interact with a lipid by
reducing steric
hindrance between the two fragments, as well as allowing appropriate folding
of each
portion to occur. The linker can be of natural origin, such as a sequence
determined to
exist izt random coil between two domains of a protein. Alternatively, the
linker can be of
synthetic origin. For instance, the sequence (Gly4Ser)3 can be used as a
synthetic
unstructured linker. Linkers of this type are described in Hustozt et al.
(1988) PNAS
85:4879; and U.S. Patent Nos. 5,091,513 and S,Z58,498. Naturally occurring
unstructured linkers of human origin are preferred as they reduce the risk of
immunogetticxty.
In the instance wherein the display package is a phage, the cloning site for
the test
polypeptide gene sequences in the phagemid should be placed so that it does
not
substantially interfere with normal phage function. One such locus is the
intergenic
region as described by Zinder and Boske, (1982) Gene 19:1-10.
The number of possible combinations in a peptide library can get large as the
length is increased and selection criteria for degenerating at each position
is relaxed. To
sample as many combinations as possible depends, in part, on the ability to
recover large
numbers of transforrxtattts. For phage with plasznid-like forms (as ~lamentous
phage),
eleetrotransformation provides azx efficiency comparable to that of phage-
transfeetion
with in vitro packaging, in addition to a very high capacity for DNA iun~put.
This allows
large amounts of vector DNA to be used to obtain very large numbers of
transformants.
The method described by Dower et al. (1988) Nucleic Acids Res., 16:6127-6145,
for
example, may be used to transform fd-tet derived recombinants at the rate of
about 107
transformants/ug of ligated vector into E. coli (such as strain MC1.061.), and
libraries may
be constructed in fd-tet Bl of up to about 3 x 108 members or more. Increasing
DNA
19
CA 02329858 2000-12-28
HMV-053.02
input and making modifications to the cloning protocol withizt the ability of
the skilled
artisan may produce increases of Beater than about 10- fold izt the recovery
of
transfoz~zz~ants, providiztg libraries of up to 101 or more recombinants_
,As will be apparent to those skilled in the art, in embodiments wherein high
affinity peptides are sought, an important criteria for the present selection
method can be
that it is able to discriminate between peptides of different affinity far a
particular lipid,
and preferentially enrich for the peptides of highest affinity. Applying the
well k»own
principles of peptide affinity and valence (i.e. avidity), it is understood
that manipulating
the display package to be rendered effectively znonovalent can allow affinity
enrichment
to be carried out for generally higher binding afi~zaities (i.e. binding
constants in the range
of 106 to IUI~ M-~) as compared to the broader range of affinities isalable
using a
multivalent display package. To generate the monovalent display, the natural
(i.e. wild-
type) form of the surface or coat protein used to anchor the peptide to the
display can be
added at a high enough level that it almost entirely eliminates inclusion of
the peptide
fusiazt protein in the display package. Thus, a vast majority of the display
packages can
be generated to include no more than one copy of the peptide fusion protein
(see, for
example, Garrad et al. (1991) BiolT'echnology 9:1373-13??). In a preferred
ezrtbodiment
of a monovalent display library, the library of display packages will comprise
no more
than 5 to 10% polyvalent displays, and more preferably no more than 2% of the
display
will be polyvalent , and most preferably, no more than 1% polyvalent display
packages in
the population. The source of the wild-type anchor protein can be, for
example, provided
by a copy of the wild-type gene present on the same construct as the peptide
fusion
protein, or provided by a separate construct altogether. 1"iowever, it will be
equally clear
that by siz~nilar manipulation, polyvalent displays can be generated to
isolate a broader
range of binding affinities. Such peptides can be useful, for example, in
purification
protocols where avidity can be desirable.
l) ,Phages As Display Packages
Bacteriophage are attractive prokaryotic-related organisms for use in the
subject
method. Bacteriophage are exceIleztt candidates for providing a display system
of the
variegated polypeptide library as there is little or no enzymatic activity
associated with
intact mature pltage, and because their genes are inactive outside a bacterial
host,
rendering the zoatuz~ phage particles metabolically inert. In general, the
phage surface is
a relatively simple structure. Phage can be grown easily in large numbers,
they are
CA 02329858 2000-12-28
HMV-OS 3.02
aztaenable to the practical handling involved in many potential mass screening
programs,
and they carry genetic information for their own synthesis within a small,
simple paclCage_
As the polypeptide gene is inserted into the phage genozne, choosing the
appropriate
phage to be employed in the subject method will generally depend most on
whether (i)
the genome of the phage allows introduction of tlae polypeptide gene either by
tolerating
additional genetic material or by having replaceable genetic material; (ix)
the virion is
capable of packaging the genome after accepting the insertion or substitution
of genetic
material; and (iii) the display of the polypeptide on the phage surface does
not disrupt
virion structure sufficiently to interfere with phage propagation.
One concern presented with the use o~ phage is that the morphogenetic pathway
of the phage determines the environment in which the polypeptide will have
opportunity
to fold. Periplasmically assembled phage are preferred as the displayed
polypeptides may
contain essential disulfides, and such polypeptides may not fold correctly
within a cell.
However, in certain embodiments in which the display package forms
izttraeellularly
(e.g., where ~, phage are used), it has been demonstrated in other instances
that disulfide-
containing polypeptides can assume proper folding after the phage is released
from the
cell.
Another concern related to the use of phage, but also pertinent to the use o~
bacterial cells and spores as well, is that multiple infections could generate
hybrid
displays that carry the gene for one particular test polypeptide yet have two
or more
different test polypeptides on their surfaces. Therefore, it can be
preferable, though
optional, to minimize this possibility by infecting cells with phage under
conditions
resulting in a low multiple-infection.
For a given bacteriophage, the preferred display means is a protein that is
present
on the phage surface (e.g. a coat protein). Filamentous phage can be described
by a
helical lattice; isometric phage, by an icosahedral lattice. Each monomer of
each major
coat protein sits on a lattice point and makes defined interactions with each
of its
neighbors. Proteins that fit into the lattice by making some, but not all, of
the normal
lattice contacts are likely to destabilize the virion by aborting formation of
the virion as
well as by leaving gaps in the virion so that the nucleic acid is not
protected. Thus in
bacteriophage, unlike the cases of bacteria and spores, it is generally
important to retain
in the polypeptide fusion proteins those residues of the coat protein that
interact with
other proteins in the virion. For example, when using the M 13 cpVrII protein,
the entire
mature protein will generally be retained with the polypeptide fragment being
added to
21
CA 02329858 2000-12-28
HMV-os3.o2
tlae N-tezz~ninus of epVl<Il, while on the other hand it can sufl'zce to
xetain only the last 100
carboxy terminal residues (or even fewer) of the M 13 epIII coat protein in
the polypeptide
fusion protein.
Under the appropriate induction, the test polypeptide library is expressed and
exported, as part of the fusion protein, to the bacterial cytoplasm, such as
when the ~.
phage is employed. The induction of the fusion proteins) rnay be delayed until
some
replication of the phage genome, synthesis of some of the phage stzuctural-
proteins, and
assembly of some phage particles 'has occurred. The assembled protein chains
then
interact with the phage particles via the binding of the anchor protein on the
outer surface
of the phage particle. The cells are lysed and the phage bearing the library-
encoded test
polypeptides {that corresponds to the specific library sequences carried in
the DNA of that
phage) axe released and isolated ~~ronrt the bacterial debris.
To enrich for and isolate phage which encodes a selected test polypeptide, and
thus to ultimately isolate the nucleic acid sequences (the polypeptide gene)
themselves,
phage harvested from the bacterial debris are af~'xnity purified. As described
below, when
a test polypeptide which specifically binds a particular lipid is desired, the
lipid can be
used to retrieve phage displaying the desired test polypeptide. The phage so
obtained may
then be amplified by infecting into host cells. Additional rounds of affinity
enrichment
followed by amplification may be employed until the desired level of
enrichment is
reached.
The enriched polypeptide-phage can also be screened with additional detection-
techniques such as expression plaque (or colony) lift (see, e.g., Young and
Davis, Science
(183) 222:778-782) whereby a Labeled lipid is used as a pmbe.
a) Filameritou.r Phage
Filamentous bacteriophages, which include M13, fl, fd, )Cfl, Ilce, Xf, Pfl,
and Pf3,
are a group of related viruses that infect bacteria. They are termed
filamentous because
they are long, thin particles comprised of an elongated capsule that envelopes
the
deoxyribonucleic acid (DNA) that forms the baeteriophage genome. The F pili
filamentous bacteriophage (Ff phage) infect only gram-negative bacteria by
specifically
adsorbing to the tip of F pili, axed include fd, fl and M13.
Compared to othez bacteriophage, filaznentous phage in general are attractive
and
M13 in particular is especially attractive because: {x) the 3-l~ structure of
the virion is
known; (ii) the processing of the coat protein is well understood; (iii) the
genome is
22
CA 02329858 2000-12-28
HMV-053.02
expandable; (iv) the genome is small; (v) the sequence of the genome is
kn.vwn; (vi) the
virior~ is physically resistant to shear, heat, cold, urea, guanidinium
chloride, low p~, and
high salt; {vii) the phage is a sequez~cxng vector so that sequencing is
especially easy;
(viii) antibiotic-resistance genes have been cloned into the genome with
predictable
results (Hines et al. (1980) Gene 11:207-21$); (ix) it is easily cultured and
stored, with no
unusual or expensive media requirements for the infected cells, (x) it has a
higkt burst
size, each iuafected cell yxeldiz~g 100 to 1000 M13 progeny after infection;
and (xi) it is
easily harvested and concentrated (Salivar et al. (1964) Virology 24: 359-371
). The entire
life cycle of the filamentous phage M13, a common clozai~ag az~d sequencing
vector, is
well understood. The genetic structure of M13 is well knvwz~, including the
complete
sequence {Schaller et al. in The Single-Stranded DNA Phages eds. Denhardt et
al. (NY:
CSHL Press, 1978)), the identity and function of the ten genes, and the order
of
transcription and location of the promoters, as well as the physical structure
of the virion
(Smith et al. {1985) Science 228:1315-1317; Raschad et al. (1986) Microbiol
Dev
50:401-427; Kuhn et al. (1987) Science 23$:1413-1415; Zimmerman et al. {1982)
JBiol
Chem 257:6529-6536; and Banner et al. (1981) Nature 289;814-816). Because the
geno~ane is small (6423 bp), cassette mutagenesis is practical on RF M 13
(Current
Protocols in Molecular Biology, eds. ,A,usubel et al. (NX: John Wiley & Sons,
1991 )), as
is single-stranded oligonucleotide dixected mutagenesis (Fritz et al. in DNA
Cloning, ed
by Glover {Oxford, UK: IRC Press, 1985)). M13 is a plasmid and transformation
system
in itself, and an ideal sequez~ciz~g vector. M13 cau. be gown vzt Rec-
stz~ains o~ E. coll.
The M13 genome is expandable (Messing et al. in 2"he Single-Stranded DNA
Phages, eds
Denhardt et al. (NY: CS)EiI. Press, 1978) pages 449-453; and Fritz et al.,
supra) and M13
does not lyre cells. Extra genes can be inserted into M13 and will be
maintained in the
viral genome in a stable manner.
The mature capsule or Ff phage is comprised of a coat of eve phage-encoded
gene
products: cpVBl, the major coat protein product of gene 'VT>I that forms the
bulk of the
capsule; and four minor coat proteins, cpl~ and cpIV at one end of the capsule
and cpV~I
and cpIX at the other end of the capsule. The length of the capsule is formed
by 2500 to
3000 copies of cpVIII in an ordered helix array that forms the characteristic
filament
structure. The gene iII-encoded protein (cp)~T} is typically present in 4 to 6
copies at one
end of the capsule az~d serves as the receptor for binding of the phage to its
bacterial host
in the initial phase of infection. For detailed reviews of Ff phage structure,
see Ranched et
al., ,~I~TGTOb101. Rev., 50:401-427 (1986); and Model et al.,in The
.8acteriophages. Yolurne
1, R. Calendar, Ed., Plenum Press, pp. 375-456 ( 1988).
23
CA 02329858 2000-12-28
HMV-OS3.02
The phage particle assembly ixlvolves extrusion of the viral genome through
the
host cell's membrane. Prior to extrusion, the major coat protein cpVIII and
the minor coat
protein cpIll are synthesized and transported to the host cell's xnezz~braz~e.
Both cpVnl and
cpflT are anchored in the host cell membrane prior to their iztcorporatiozz
into the zxxature
particle. In addition, tb~e viral genome is produced and coated with cpV
protein. During
the extrusion process, cpV-coated genomic DNA is stripped of the cpV coat and
simultaneously recoated with the mature coat proteins.
Bath cpIII and cpV3ZI proteins include two domains that provide signals fox
assembly of the mature phage particle. The first domain is a Secretion Signal
that directs
the newly synthesized proteixi to the host cell zzae~onbrane. The secretion
signal is located
at the amino terminus of the polypeptide and lipids the polypeptide at least
to the cell
membrane. The second domain is a membrane anchor domain that provides signals
for
association with the host cell n aembrane and for association with the phage
particle
during assembly. This second signal for bath cpVI~ and cplLI comprises at
least a
hydrophobic region for spanning the membrane.
The 50 amino acid mature gene Vl'1I coat protein (cpVll)~ is synthesized as a
73
amino acid precoat (,lto et al. {1979) PNAS 76:1199-1203). cpVIil has been
extensively
studied as a model membrane protein because it can integrate into lipid
bilayers such as
the cell txaembrane in an asymmetric orientation with the acidic amino
terminus toward
the outside and the basic carboxy terminus toward the i;aside of the membrane.
The first
23 amino acids constitute a typical signal-sequence which causes the nascent
polypeptide
to be inserted into the inner cell membrane. An B. coli signal peptidase {SP-
17 recognizes
amino acids 18, 21, and 23, and, to a lesser extent, residue 22, and cuts
between residues
23 and 24 of the precoat (Kahn et al. (1985) .I. Biol. Chem. 260:15914-15918;
and Kuhzt
et al. (1985) d. Biol. Chem. 2b0:15907-15913). After removal of the signal
sequexace, the
amino terminus of the mature coat is located on the periplasmic side of the
inner mem-
braxte; the carboxy terminus is on the cytoplasmic side. About 3000 copies of
the mature
coat protein associate side-by-side in the inner membrane.
The sequence of gene VIII is known, axxd the amino acid sequence can be
encoded
on a synthetic gene. Mature gene VIII protean znalces up the sheath around the
circular
ssDNA. The gene VL'.~ protein can be a suitable anchor protein because its
location and
orientation in the virion are known (Banner et al. (1981) Nature 289:81.4-
816). Prefera-
bly, the polypeptide is attached to the amino terminus of the mature M13 coat
protein to
generate the phage display library. As set out above, manipulation of the
concentration of
24
CA 02329858 2000-12-28
HMV-OS 3.02
both the wild-type cpVIII and AbIcpV~ fusion in an infected cell can be
utilized to
decrease the avidity of the display and thereby enhance the detection of high
affinity
polypeptides directed to the lipid(s).
Anothez vehicle for displaying the polypeptide is by expressing it as a domain
of a
chimeric gene containing part or all of gene III, e.g., encoding cpIII. When
monovalent
displays are required, expressing the polypeptide as a fizsion protein with
cpIII can be a
preferred embodiment, as manipulation of the ratio of wild-type cps to
chimeric cps
during formation of the phage particles can b$ readily controlled. This gene
encodes one
of the minor coat proteins of M13. Genes VI, V1I, and IX also encode minor
coat
proteins. Each of these minor proteins is present in about 5 copies per virion
and is
related to morphogenesis or infection. In contrast, the major coat protein is
present in
more than 2500 copies per vizion. The gene VI, VII, and IX proteins are
present at the
ends of the virion; these three proteins are not posttranslationally processed
(Rasched et
al. (1986) Ann ,Rev. Microbiol. 41:507-541). In particular, the single-
stranded circular
phage DNA associates with about five copies of the gene T1I protein and is
then extruded
through the patch of membrane-associated coat protein in such a way that the
DNA is
encased in a helical sheath o~ protein (Webster et al. in The Single-Stranded
DNA
Phages, eds Dressier et al. (NY:CSHL Press, 1978).
Manipulation of the sequence of cpIII has demonstrated that the C-terminal 23
amino acid residue stretch of hydrophobic amino acids normally responsible for
a
membrane anchor function can be altered in a variety of ways and retain the
capacity to
associate with membranes. Ff phage-based expression vector's were first
described in
which the cps amino acid residue sequence was modified by insertion of
heterologous
polypeptide (Parmely et al., Gene (1988) 73:305-318; and Cwirla et al., PNAS
(1990)
87:637$-6382) or an amino acid residue sequence defining a single chain
polypeptide
domain (McCafferty et al., Science (1990) 348:552-554). It has been
demonstrated that
insertions into gene III can result in the prodution of novel protein domains
on the virion
outer surface. (Smith (19$5) Science 228:1315-1317; and de la Cruz et al.
(1988) J. Biol.
Chem. 263:4318-4322). The polypeptide gene may be fused to gene III at the
site used by
Smith and by de la Cruz et al., at a codon corresponding to another domain
bouztdary or
to a surface loop of the proteuo, or to the amino terminus of the mature
protein.
Generally, the successful cloning strategy utilizing a phage coat protein,
such as
cpDI of filamentous phage fd, will provide expression of a polypeptide chain
fused to the
N-terminus of a coat protein (e.g., cpl~ and transport to the inner membrane
of the host
CA 02329858 2000-12-28
HNIV-os3.02
where the hydrophobic domain iz~ the C-terminal region of the coat pmtein
anchors the
fusion protein in the membrane, with the ~1-tezmxnus containing the
polypeptide chain
protruding into the periplasmic space.
Similar constructions could be made with other filamentous phage. Pf3 is a
well
known filamentous phage that infects Pseudomonos aerugenosa cells that harbor
an
IncP-I plasmid. The entire genome has been sequenced ((Luiten et al. (19$5) J.
Virol.
5G:268-276) and the genetic signals involved in replication and assembly are
known
(Luitezt et al. (19$7) DNA 6:129-137). The major coat protein of PF3 is
unusual in
having no signal peptide to direct its secretion. The sequence has changed
residues ASP-
7, ARG-37, LYS-40, and PHE44 which is consistent with the amino terminus being
exposed. Thus, to cause a polypeptide to appear on the surface of Pf3, a
tripartite gene
can be constructed which comprises a signal sequence lsa~own to cause
secretion in P.
aerugenosa, fused in-frame to a gene fragment encoding the polypeptide
sequence, which
is fused in-fi-ame to DNA encoding the mature Pf3 coat protein. optionally,
DNA
encoding a flexible linker o~ one to 10 amino acids is introduced between the
polypeptide
gene fragment and the Pf3 coat-protein gene. This tripartite gene is
introduced into Pf3
so that it does not interfere with expression o~any P~'3 genes. Qrtce the
signal sequence is
cleaved off, the polypeptide is in the periplasm and the mature coat protein
acts as an
anchor and phage-assembly signal.
b) Bacteriophage ø1~C174
The bacteriophage X174 is a very small icosahedral vizus which has been
thoroughly studied by genetics, biochemistry, and electron microscopy (see The
Single
Stranded DNA ,Phages (eds. Den hardt et al. (NY:CSHL Press, 1978)). Three gene
products of X174 are present on the outside of the nnature virioz~: F
(capsid), G (major
spike protein, 60 copies per virion), and H (minor spike protein, 12 copies
per virion).
The G protein comprises 1.75 amino acids, while H comprises 32$ amino acids.
The F
protein interacts with the single-stranded DNA of the virus. The proteins F,
G, and H are
translated from a single mRNA in the viral infected cells. As the virus is so
tightly
constrained because several of its genes overlap, X174 is not typicahy used as
a cloning
vector due to the fact that it cart accept very little additional DNA.
However, mutations
in the viral G gene (encoding the G protein) can be rescued by a copy of the
wild-type G
gene carried on a plasmid that is expressed in the same host cell (Chambers et
al. (19$2)
Nue Acid Res 10:f465-6473). In one embodiment, one or cnot~e stop eodons are
introduced into the G gene so that no G protein is produced ~ro~oa the viral
genome. The
26
CA 02329858 2000-12-28
HMV-053.02
variegated polypeptide gene library can then be fused with the nucleic acid
sequence of
the H gene. An amount of the viral G gene equal to the size of polypeptide
gene fragment
is eliminated from the X174 genome, such that the size of the genome is
ultimately
unchanged. Thus, in host cells also transformed with a second plasmid
expressing the
wild-type G protein, the praductiott of viral panicles from the mutant virus
is rescued by
the exogenous G protein source. Where it is desirable that only acne test
polypeptide be
displayed per ~X 174 particle, the second plasmid caz~ fitrthex include one or
more copies
of the wild-type H protein gene so that a zttix of H and test polypeptide/H
proteins will be
predominated by the wild-type H upon incorporation into phage particles.
c) Large DNA Phage
Phage such as ~. ax 'f4 have much larger genomes than do M13 or X174, azxd
have more complicated 3-D eapsid structures titan 1VI13 or ~PX174, with more
coat
proteins to choose &om. In embodiments a~ the invention whereby the test
palypeptide
library is processed az~d assembled into a fuxtctional form and associates
with the
bacteriophage particles within the cytoplasm of the host cell, bacteriophage ~
and
derivatives thereof are examples of suitable vectors. The intracellular
morphogenesis of
phage ~. can potentially pz~event protean domains that ordinarily contain
disulfide bonds
from folding correctly. However, variegated libraries expressing a population
of
functional polypeptides, which include such bonds, have been generated in ~,
phage.
(Hose et al. (1989) Science 246:1275-1281; Mullinax et al. {1990) PNAS 87:8095-
8099;
and Pearson et al. (1991) PNAS 88:2432-2436). Such strategies take advantage
of the
rapid construction and eflxcient transformation abilities of ~, phage.
When used for expression of polypeptide sequences (exogenous nucleotide
sequences), may be readily inserted into a ~, vector. Pox instance, variegated
polypeptide
libraries cans be constructed by modification of ~, ZAP TI through use of the
multiple
cloning site of a ~ ZAP II vector (Hose et al. supra).
ii) Bacterial Cells a~ Dirplay Packages
Recombinant polypeptides are able to cross bacterial membranes after the
addition of appropriate secretion signal sequences to the N-terminus of the
protein (Better
et al (1988) Science 240:1041-1043; and Skerra et al. (19$8) Science 240:1038-
1041). In
addition, recombinant polypeptides have been fused to outer membrane proteins
for
surface presentation. p'or example, one strategy for displaying poly~peptides
on bacterial
27
CA 02329858 2000-12-28
HMV-p~ 3 .02
cells comprises generating a fusion protein by inserting tl~e polypeptide into
cell surface
exposed portions of an integral outer membrane protein (Fucks et al. (1991)
BiolZ'echnology 9:1370-1372). 1n selecting a bactez'ial cell to serve as the
display
package, any well-characterized bacterial strain will typically be suitable,
provided the
bacteria may be gown in culture, engineered to display the test polypeptide
library on its
surface, and is compatible with the particular affinity selection process
practiced iz~ the
subject method. Among bacterial cells, the preferred display systems include
Salmonella
typhirnurium, Bacillus subtilis, Pseudomonas aeruginosa, Yibrio cholerae,
Klebsiella
pneumonia, Neisseria gonorrhaeae, Neisseria meningitides, Bacteroides nodosus,
.~l~oraxella bovis, and especially Escherichia coli. Many bacterial cell
surface proteins
useful in the present izxvez~tion have been characterized, and works on the
localization of
these proteins and the methods of determining their structure xz~clude Benz et
al. (1988)
Ann Rev Microbial 42: 359-393; Balduyck et a1. (1985) Biol Chem Hoppe-Seyler
366:9-
14; Ehrmann et al {1990) PNAS 87:7574-7578; Heijne et al. {1990) Protein
Engineering
4:109-112; Ladnex et al. C1.S. Patent No. 5,223,409; Ladner et al. W088/06630;
Fucks et
al. (1991) Biotechnology 9:1370-1372; and Goward et a1. (1992) TIBS 18:136-
140.
To further illustrate, the Lama protein of E coli is a well understood surface
protein that can be used to generate a variegated library of test polypeptides
on the
surl'ace of a bacterial cell (see, for example, Ronco et al. (1990) Biochemfe
72:183-189;
van der Weit et al. (1990) Vaccine 8:269-277; Charabit et al. (1988) Gene
70:181-189;
and Ladner TJ.S. Patent No. 5,222,409). Lama of ~ cvli is a porin for maltose
and
maltodextrin transport, and serves as the receptor for adsorption of
bacteriophages ~, arld
K10. Lama is transpot'ted to the outer membrane if a functional N-terminal
signal
sequence is present (Benson et al. (1984) PNAS 81:3830-3834). As with other
cell
surface proteins, Lama is synthesized with a typical signal-sequence which is
subsequently removed. Thus, the variegated polypeptide gene library can be
cloned into
the Lama gene such that the resulting library of fusion proteins comprise a
portion of
Lama sufficient to anchor the protein to the cell membrane with tlae test
polypeptide
fragnnent ozaented on the extracellular side o;f the nnembrane. Secretion of
the
extracellular portion of the fusion protein can be facilitated by inclusiozz
of the Lama
signal sequence, or othex suitable signal sequence, as the N-terminus of the
protein.
The E_ coli lama has also been expressed in functional form in S. typhimurium
(Harkki et al. (1987) ,Col Gen Genet 209:607-611), Y. cholerae (Harkki et al.
(1986)
Microb Pathol 1:283-288), and K. pneumonia (Wehmeier et al. (1989) Mol Gen
Genet
z8
CA 02329858 2000-12-28
HMV-053.02
215:529-536), so that one could display a population of test poiypeptides in
any of these
species as a fusion to E. coli Lama. Moreover, K. ,pneumonia e~cpresses a
maltoporin
similar to Lama which could also be used. In .P_ aeruginosa, the Dl protein (a
honnologue
of Lama) can be used (Trios et al. (1988) Biochem Biophys Acta 938:493-496).
Similarly, other bacterial surface proteins, such as PAL, OxnpA, OmpC, OmpF,
PhaB,
pilin, BtuB, FepA, FhuA, IutA, FecA and FhuE, may be used in place of LanaB as
a
portion of the display means in a bacterial cell.
In another exemplary embodiment, the fusion protein can be derived using the
k'liTrxT"s Random Polypeptide Display Library (Invitragexa). That library is a
diverse
population of random dodecapolypeptides inserted within the thioredoxin active-
site loop
inside the dispensable region of the bacterial flagellin gene (fliC). The
resultaztt
recombinant fusion protein (FLITR?~ is exported and assembled into partially
Functional
flagella on the bacterial cell surface, displaying the random palypeptide
library.
Polypeptides are fused in the nn~iddle of thioredoxin, therefore, both their N-
az~d
C-termini are anchored by thioredoxin's tertiary structure. This results in
the display of a
constrained polypeptide. By contrast, phage display proteins are fused to the
N-terminus
of phage coat proteins in an unconstrained manxaer. The unconstrained
molecules possess
many degrees of cozifoxroatiaztal Freedom which may result in the lack of
proper
interaction with the lipid ax~olecule. Without proper interaction, many
potential protein-
pratein interactions may be missed.
Moreover, phage display is limited by the low expression levels of
bacteriophage
coat proteins. FliTrxT"t and similar methods can overcome this limitation by
using a
strong promoter to drive expression of the test polypeptide fusions that arre
displayed as
multiple copies.
According to the present invention, it is contemplated that the FliTtx vector
can
be modified to provide, similar to the illustrated vectors of the attached
figures, a vector
which is differezttially spliced in mammalian cells to yield a secreted,
soluble test
polypeptide.
iii) Bacterial Spores as Display Packages
Bacterial spores also have desirable properties as display package candidates
in
the subject xxaethod. For example, spores are much more resistant than
vegetative bacterial
cells or phage to chemical and physical agents, and hence permit the use of a
great variety
29
CA 02329858 2000-12-28
1~V-053.02
of affinity selection conditions. Also, Bacillus spores neither actively
nnetabolize nor alter
the proteins on their surface. However, spores have the disadvantage that the
molecular
mechanisms that tzagger sporulation are less well worked out than is the
formation of
MX3 or the export of protein to the outer membrane of E. cvli, though such a
limitation is
not a serious detractant from their use in the present invention.
Bacteria of the genus $acillus form endospores that are extremely resistant to
damage by heat, radiation, desiccation, and toxic chemicals (reviewed by
Losick et al.
{1986) Ann Rev Genet 20:625--b69). This phenomenon is attributed to extensive
intermolecular cross-lxnkang of the coat proteins. In certain embodiments of
the subject
method, such as those which include relatively harsh affinity separation
steps, Bacillus
spores can be the preferred display package. Endospores from the genus
Bacillus are
wore stable than are, for example, exospores from Streptornyces. lv,Iareover,
Bacillus
subtilis forms spores in 4 to b hours, whereas Streptomyces species may
require days or
weeks to sporulate. In addition, genetic knowledge and manipulation is much
more
developed for B, subtilis than for other spore-forming bacteria.
Viable spores that dif>'er only slightly from wild-type are produced in B.
subtilis
even if any one of four coat proteins is missing (Donovan et al. (1987) J,tKol
Biol 196:1-
10). ~areover, plasmid DNA is commonly included in spores, and plasmid encoded
proteins have been observed on the surface of Bacillus spores (Debro et al.
(1986) J
Bacteriol 165:258-268). Thus, it can be possible during spozulatio~a to
express a gene
encoding a chimaric cast protein comprising a polypeptide of the variegated
gene library,
without interfering materially with spore formation.
To illustrate, several galypeptide components of B. subtilis spore coat
(Danavan
et al. {x9$7) J Mol Biol 196:1-10) have been characterized. The sequences of
two
complete coat proteins and amino-terminal fragments of two others have been
determined. Fusion of the test polypeptide sequence to cotC or cotD fragments
is likely
to cause the polypeptide to appear on the spore surface. The genes of each of
these spore
coat proteins are prefez~red as neither eotC or eotD are post-translationally
modified (see
Ladner et al. U.S, Patent No. 5,223,409).
iv) Selecting Peptides from the .Dfsplay Mode
Upon expression, the variegated polypeptide display is subjected to affinity
enrichment in order to select for test poIypeptides which bind preselected
lipids. The
CA 02329858 2000-12-28
HMV-053.02
term "affinity separation" or "affrnity enrichment" includes, but is not
limited to: ( 1 )
affinity chromatography utilizing immobilized lipids, and (2) precipitation
using soluble
lipids. In each embodiment, the libxa;ty of display packages are ultimately
separated
based oz~ the ability of the associated test polypeptide to bind the lipid of
interest. See, for
example, the Ladner et al. U.S. Patent No. 5,223,409; the l~.ang et al.
International
Publication No. WO 92118619; the Dower et al. International Publication Nv. WO
91117271; the Winter et al. lntenn~atxonal Publication WO 92120791; the
Markland et al.
International Publication No. WO 92/15679; the Bz~eitling et al. International
Publication
WO 93/01288; the MeCafferty et al. International Publication No. W4 92101047;
the
Garrard et al. International Publication No. WO 92/09b90; and the Ladner et
al.
International Publication No. WO 90102809. In most preferred embodiments, the
display
library will be pre-enriched for peptides specific for the lipid by lxrst
contacting the
display library with any negative controls or other lipids for which
differential binding by
the test polypeptide is desired. Subsequently, the non-binding fraction from
that pre-
treatment step is contacted with the lipid and peptides from the display which
are able to
specifically bind the lipid are isolated.
With respect to affinity chromatography, it will be generally understood by
those
skilled in the art that a great number of chromatography techniques can be
adapted for use
in the present invention, ranging from column chromatography to batch elution,
and
including ELISA and biopanning techniques. Typically, whexe lipid is or can be
immobilized on an insoluble carrier, such as sepharvse or polyacrylamide
beads, or,
alternatively, the wells of a micrvtitre plate.
The population of display packages is applied to the affinity matrix under
conditions compatible with the binding of the test polypeptide to the lipid.
The population
is then fractionated by washing with a solute that does not greatly effect
specilac binding
of polypeptides to the lipid, but which substantially disrupts any non-
specific binding of
the display package to the lipid or matrix. A certain degree of control can be
exerted over
the biunding characteristics of the polypeptides recovered from the display
library by
adjusting the conditions of the binding incubation and subsequent washing.
'fhe
temperature, pH, ionic strength, divalent cation concentration, and the volume
and
duration of the washing can select for polypeptides within a particular range
of affnity
aztd specificity. Selection based on slow dissociation rate, which is usually
predictive of
bagh a~fanity, is a very practical route. This may be done either by continued
incubation in
the presence of a saturating amount of free lipid (if available), or by
increasing the
31
CA 02329858 2000-12-28
HlvlV-053.02
volume, number, and length of the washes. Zn each case, the rebinding of
dissociated
polypeptide-display package is prevented, axzd with increasing time, display
packages of
higher and higher affinity are recovered. Moreover, additional modifications
of the
binding and washing procedures may be applied to find polypeptides with
special
characteristics. The affinities of some peptides are dependent on ionic
strength or cation
concentration. 'This is a useful characteristic for peptides to be used in
affinity purification
of various proteins when gentle conditions fox removing the protein from the
peptide are
required. Specific examples are polypeptides which depend on Cap for lipid
binding
activity and which lose or gain binding affinity in the presence of EGTA or
other metal
ehelating agent. Such peptides may be identified in the recozzibinant
polypeptide library
by a double screening technique isolating first those that bind the lipid in
tlae presence of
Cap, and by subsequently identifying those in this group that fail to bind in
the presence
of EGTA, or vice-versa.
After "washing" to remove nozi-specifically bound display packages, when
desired, specifically bound display packages can be eluted by either specific
desorption
(using excess lipid) ox non-specific desorption (using pH, polazity reducing
agents, or
chaotropic agents). In preferred embodiments, the elution protocol does not
kill the
oxganistxi used as the display package such that the enriched population of
display
packages can be further amplified by reproduction. The list of potential
eluants includes
salts (such as those in which one of the counter ions is Na+, NH4+, Rb+, S04W,
H~E04
citrate, K+, Li+, Cs+, HS04 , CO~2', Ca2+, Sr~'~, Cl-, POa2', HC03-, Mg2~',
Ba2+,
Br, HP0~2', or acetate), acid, heat, and, when available, soluble forms of the
lipid.
Because bacteria continue to metabolize durizig the affinity separation step
and are
generally more susceptible to damage by harsh conditions, the choice of buffer
compoziexats (especially eluates) can be more restricted when the display
package is a
bacteria rather than for phage or spores. Neutral solutes, such as ethanol,
acetone, ether,
or urea, are examples of other agents useful for eluting the bound display
packages.
In preferred embodiments, afl"inity enriched display packages are iteratively
amplified and subjected to further rounds of affinity separation until
enrichment of the
desired binding activity is detected. In certain embodiments, the specifically
bound
display packages, especially bacterial cells, need not be eluted per se, but
rather, the
rz~atrxx bound display packages can be used directly to inoculate a suitable
growth media
for amplification.
32
CA 02329858 2000-12-28
1~V-053.02
Where the display package is a phage particle, the fusion protein generated
with
tlae coat protein can interfere substantially with the subsequent
amplilieation of eluted
phage particles, particularly in embodiments wherein the cpIII protein is used
as the
display anchor. Even though present in only one of the 5-6 tail fibers, some
peptide
constructs because of their size and/or sequence, may cause severe defects in
the
infectivity of their carrier phage_ "his causes a loss of phage from the
population during
reinfection and amplification following each cycle of panning. In one
embodiment, the
peptide can be derived on the surface of the display package so as to be
susceptible to
proteolytic cleavage which severs the covalent linkage of at least the target
binding sites
of the displayed peptide from the remaining package. Eor instance, where the
cpI>I coat
protein of M13 is employed, such a strategy can be used to obtain infectious
phage by
treatment with an enzyme which cleaves between the test polypeptide portion
and cp)TI
portion of a tail ibex fusion protein (e.g. such as the use of an enterokinase
cleavage
recognition sequence).
To further minimize problems associated with defective infectivity, DNA
prepared from the eluted phage can be transformed into host cells by
electroporation or
well known chemical means. The cells are cultivated for a period of tune
sufficient for
marker expression, and selection is applied as typically done for 1~NA,
transformation.
The colonies are arnplihed, and phage harvested for a subsequent rounds) of
panning.
ARer isolation of display packages which encode polypeptides having a desired
binding specificity for the lipid, the test polypeptides for each of the
purred display
packages can be tested for biological activity in the secretion mode of the
subject
method.
(v) C'renerations of Polypeptide Libraries
The variegated polypepdde libraries of the subject method can be generated by
any of a number of methods, and, though not limited by, preferably exploit
recent trends
in the preparation of chemical libraries. For instance, chemical synthesis of
a degenerate
gene sequence can be carried out in an automatic DNA synthesizer, and the
synthetic
genes then ligated into an appropriate expression vector. The purpose of a
degenerate set
of genes is to provide, in one mixture, all of the sequences encoding the
desired set of
potential test sequences. The synthesis of degenerate oligonucleotides is well
la~own ire
the art (see for example, Narang, SA ( 1983) Tetrahedron 39:3; Itakura et al.
( 1981 )
33
CA 02329858 2000-12-28
HMV-053.02
Recombinant DNA, Proc 3rd Cleveland Sympos. Macromolecules, ed. AG Walton,
Amstexdatn: Elsevier pp273-289; ltakura et al. (1984) Annu. Rev. Biochem.
53:323;
Itakura et al. (1984) Science 198:1056; Ike et al. (1983) Nucleic Acid Res. 1
X:477. Such
techniques have been employed in the directed evolution of other proteins
(see, fox
example, Scott et al. (1990) Science 249:386-390; Roberts et al. (1992) PNAS
89:2429-
2433; Devlin et al. (1990) Science 249: 404-406; Cwirla et al. (1990) PNAS 87:
6378-
6382; as well as U.S. Patents Nos. 5,223,409, 5,198,346, and 5,096,815).
As used hezein, "variegated" refers to the fact that a population of peptides
is
chat-actezized by having a peptide sequence which differ from one member of
the library
to the next. For example, in a given peptide library of n amino acids in
length, the total
number of different peptide sequences in the library is given by the product
of
y1 x y2 X~~~~ vn-1 x vn~ where each on represents the number different amino
acid
residues occurring at position n of the peptide. In a preferred embodiment of
the present
invention, the peptide display collectively produces a peptide library
including at least 96
to 10~ different peptides, so that diverse peptides may be simultaneously
assayed for the
ability to interact with the lipid.
In one embodiment, the test polypeptide library is derived to express a
combinatorial library of peptides which are not based on any known sequeztce,
nor
derived from cDNA. That is, the sequences of the library are largely, if not
entirely,
random. It will be evident that the peptides of the library may range in size
from
dipeptides to large proteins.
In another embodiment, tlae peptide library is derived to express a
combinatorial
library o~ peptides which are based at least in part on a known polypeptide
sequence or a
portion thereof (though preferably not a cDNA library). That is, the sequences
of the
library is semi-random, being derived by combinatorial mutagenesis of a known
sequence{s). See, for example, Ladner et al. PCT publication WO 90/42909;
Garrard et
al., PCT publication WO 92/09690; lVlarks et al. (1992) J. Biol. Chem.
267:16007-16010;
Griffths et al. (1993) EMBO J 12:725-734; Clackson et al. (1991) Nature
352:524-628;
and Baxbas et al. (1992) PNAS ~89:a457-4461. Accordingly, polypeptide(s) which
are
known binding partners far a lipid can be n~utagenized by standard techniques
to derive a
variegated library of polypeptide sequences which can fiuther be screened far
agonists
andlor antagonists. The purpose of screening such combinatorial peptide
libraries is to
generatC, for example, hoxnologs o~ known polypcptidcs which can act as either
agonists
34
CA 02329858 2000-12-28
HMV-053.02
or antagonists, or alternatively, possess novel activities all together. To
illustrate, a ligand
can be engiz~eezed by the present method to provide more efflciez~t biz~dxz~g
or specibcity
to a cogitate receptor, yet still retain at least a portion of an activity
associated with wild-
type ligand. Thus, combinatorially-derived homologs can be generated to have
an
increased poteztcy relative to a naturally occurring form of the protein.
Likewise,
homologs can, be generated by the present approach to act as antagonists, in
that they are
able to mimic, for exaaxaple, birtdizxg to the lipid, yet not iztduce aray
biological response,
thereby inhibiting the action of authentic ligand.
Ln pzeferred embodiments, the combinatorial polypeptides are in the range of 3-
100 amino acids in length, more preferably at least 5-50, and even more
preferably at
least 10, 13, 15, 20 or 25 amino acid residues in length. Preferably, the
polypeptides of
the library are of uniform length. It will be understood that the length of
the
combinatorial peptide does not reflect any extraneous sequences which may be
present in
order to facilitate expression, e.g., such as signal sequences yr invazaazxt
portivzts of a
fusion protein.
The harnessizig of biological systems for the generation of polypeptide
diversity is
now a well established technique which can be exploited to ge~aerate the
peptide libraries
of the subject method. The source of diversity is the combinatorial chemical
synthesis of
mixtures of oligonucleotides. 0ligonucleotide synthesis is a well-
characterized chemistry
that allows tight control of the composition of the mixtures created.
Degezterate DNA
sequences produced are subsequently placed into an appropriate genetic context
for
expression as polypeptxdes.
There are two principal ways in which to prepare the required degenerate
mixture.
In one method, the DNAs are synthesized a base at a time. When variation is
desired at a
base position dictated by the genetic code a suitable mixture of nucleotides
is reacted
with the nascent DNA, rather than the pure nucleotide reagent of conventional
polynucleotide syntheses. The second method provides more exact ca~ntral aver
the
amino acid variatioxx. First, trinucleotide reagents are prepared, each
trinucleotide being a
codon of one (and anly one) of the amino acids to be featured in the
polypeptide library.
When a particular variable residue is to be synthesized, a mixture is made of
the
appropriate trinucleotides and reacted with the nascent DNA. Once the
necessary
"degenerate" DN,A is complete, it must be joined with the DNA sequences
necessary to
assure the expression of the polypeptide, as discussed in more detail below,
and the
complete DN,A, construct must be introduced into the cell.
CA 02329858 2000-12-28
H.IvIV-053.02
Whatever the method may be for generating diversity at the codon level,
chemical
synthesis of a degenerate gene sequence can be carried out xzt an automatic
DNA
synthesizer, and the syz~thetac ge~aes can then. be higated into an
appropriate gene for
expression. The purpose of a degenerate set of genes is to provide, iz~ ox~e
mixture, all Qf
the sequences encoding the desired set of potential test polypolypeptide
sequences. The
synthesis of degenerate ohagonucleotides is well known in the art (see for
example,
Narang, SA (1983) Tetrahedron 39:3; Itakura et al. (1981) Recombinant DNA,
.Pros 3rd
Cleveland Sympos. Macromolecules, ea. AG Wahton, Amsterdam: Elsevier pp273-
289;
Itakura et al. (1984) Annu. Rev. Biochem. 53:323; Itakura et aI. (1984)
Science 198:1056;
lke et al. (1983) Nucleic Acid Res. 11:477. Such techniques have been employed
in the
directed errolution of other proteins (see, for example, Scott et al. ( 1990)
Science
249:386-390; Roberts et al. (1992) PNAS 89:2429-2433; Devlin et al. (1990)
Science
249: 404-406; Cwirla et al. (1990} PNAS 87: 6378-6382; as well as U.S. Patents
Nos.
5,223,409, 5,198,346, and 5,096,815).
E) ExemnliRcation
Given, the diversity of cellular responses dependent on the products of PI 3'-
K, it is
probable that many as yet unidentified proteins bind to and are regulated by
PtdLns-3,4,5-
P3 and Ptdins-3,4-Pi. Identification of these proteins and the elucidation of
their role in
cellular signaling will be critical to our understanding of these cellular
functions as well
as to diseases such as cancer. In order to isolate novel 3'PPI binding
proteins we
developed a screen using in vitro coupled transc~ptionltranslation technology,
izz
conjunction with the use of syaathetic biotinylated Ptdins-3,4-Ps and PtdIns-
3,4,5-P3
ligands. We show in this report that this screen can isolate 3'PPI binding
proteins with
high specificity and selectivity ~rom among a vast excess and diversity of
other non-
speci~c proteins. We report an initial demonstration of the effectiveness of
the systenn
using the PH domain of the serinelthreonine kinase Akt, a known 3'PPI binder,
as a
model. Second, we demonstrate the utility of this technique in isolating other
3'PPI
binding proteins by screening a small pool expression library derived from
mouse spleen
mRNA. Three 3'PPI binding proteins were adenti~ed in this initial screen. Two
of these
proteins have been previously characterized, PfP3BPIp42~" az~d PDKI, thereby
establishing positive controls that the system operates as predicted.
Izoportez~tly, the
tlurd protein is a novel protein of unknown fiuactioz~ that contains both a PH
domain and
an SH2 domain.
36
CA 02329858 2000-12-28
HMV~053.02
EXPERIMENTAL PROCEDURES
Cloning and Expression of Akt PH domain. The PH domain of Akt was cloned as
a fusion protein to maltose binding protein (MBP) in the expression vector
pMAL2B
(New England Bivlabs). The N-terminal 130 amino acids of murine Aktl in the
vector
pJ3? (a gift of P. Tsichlis, Fox-Chase Cancer Center) was amplified by PCR
using Pfu
polymerase with the coding strand primer Si-cgatcgggatccatggaacag-3e (upstream
of the
myc tag in p13? and contains a BamHl site) and the z~on coding strand primer
Si-
ccctgaattctcactgggtga-3' (~rozn Akt amino acid 130 and encodes an EcoRl site).
Both
maltose binding protein alone and the MBP - Akt PH domain fusion construct
were
subeloned into the in vitro translation vector pCS2(+) under the control of
tl~e SP6
promoter. The proteins were expressed by transcribing/translating U.Ol-1 ~g of
DNA of
MBP oz MBP-Akt PH with the Promega TnT Coupled Reticulocyte Lysate System
using
SP6 RNA polymerase and 35S-methionine (in vivo labelizxg grade, Arnersham)
according
to the manufacturers protocol.
~nthesis of Biotinylated and Non-~iotinylated PhQSphatidylinositols. All
chemucally synthesized probes were prepared using a tztethyl D-glueopyranoside
as the
chiral starting mate~al fox the inositol head group; different phosphate
substitution
patterns were elaborated using the Ferrier rearrangement of suitably protected
glucose-
derived precursors. Di-Cs PtdIns-3,4-P2 and Ptdlns-3,4,5-P3 were prepared by
modifications of the synthesis o~ the dipalmitoyl analogs. Biotinylated
phosphoinositides
were prepared ~roxa the sn-2-aminohexanoyl derivatives of Ptdlns-3,4-P2 and
Ptdlns-
3,4,5-P3 by condensation of the active ester of biotin with the water-soluble
PIP" analog
in the presence of a mild base (Figure 1 ). The biotinylated probes, bPtdIns-
3,4-Pi and
bPtdTns-3,4,5-P3, were purified by ion exchange chromatography and employed as
aqueous solutions fox attachment to streptavidin-coated surfaces
Binding and Isolation of Radiolabeled In Vitrd Translated Proteins with
.8iotinylated Phosphoinositides. Avidim beads (Ultralink immobilized
Neutravidin~,
Pierce Chenaacal) were washed twice with 5 volumes of wash/binding (WB) buffer
(IOmM H.EPES pH=7.4, 150 mM NaCI, 0.5% NP-40, SmM DTT). The beads were then
reconstituted in 2x volume of W$ buffer and the biotinylated phosphoinositide
was
added. Generally, 0.1 ~1 of 100 ~lvl biotinylated lipid was bound per 1 ~1 of
packed avidin
beads. The biotinylated lipid was incubated with the beads for 1-2 hours at
4°C with
gentle agitation and then washed twice with lOx-bead volume of WB buf~'er to
remove
37
CA 02329858 2000-12-28
HMV-053.02
excess ligand. Control beads without biotinylated lipid were prepared in an
identical
tzaaz~ner but without the addition of the lipid- S u1 of the 35S-labelling
reaction containing
the in vitro trattscribed/translated protein was then added to the tubes and
the protein was
allowed to bind for 2 hours at 4°C with gentle agitation. The tubes
were then centrifuged
briefly, the beads were washed 2x with 0.5 txtl W B buffer, and the bound
proteins were
eluted by boiling the beads in 20 ~1 Laemmli sample buffer containing 5% 2-
mercaptoethanol. The proteins were then separated on a x 3.75% SDS-PAGE gel.
Following electrophoresis the gel was soaked in EztHance (Dupoztt/NEN) For 1
hour and
then izt a 7.5% wlv solution of PEG-3350 for 1 hour. The gel was then dried
and
subjected to autoradiography.
Competition Binding of Isolated Proteins for Non-biotinylated Lipids over
Biotinylated Lipidr. Competition binding experiments were performed exactly as
described above for standard binding of radiolabeled proteins to biotinylated
lipids except
that avidin beads which had been pre-bound with the biotinylated dice Ptdlns-
3,4,5-P3 or
Ptdlns-3,4-P2 were incubated with the in vitro tralaslated proteins in the
presence of 1 pM
to 100~.~M of non-biotinylated lipids (diC8 forms of PtdIns-3,4,5-P3, Ptdlns-
3,4-PZ, PtdIns-
4,5-PZ [Echelon Research Laboratories, Salt Lake City, UtaltJ). Binding was
quantitated
by measuring the intensities of bands from the autoradiogram or by
scintillation counting
of the eluted proteins in Laemrnli sample buffer usiatg Opti-fluor (Packard).
Binding
curves were modeled by the equation:
bound[ICsol(ICso + C)] x x 00
where % bound represents the quantity of protein bauud to the biotinylated
lipid coated
beads in the presence of a concentration C of competing lipid. ICSO is the
molar excess of
competitor C required to reduce the % bound to 50% of its maximal value in the
absence
of competitor.
In Vitro Translation F.~rpression Cloning. A marine spleen cDNA library
containing 2x105 independent clones was divided into 1700 individual small
pools. The
eDNAs in this library had beezt clotted into the in vitro transcription vector
pCS2(+)
under control of the SP6 promoter. Individual cDNAs from positive pools were
isolated
from the other cDNAs of the pool using a 9fi-well format as described
pzeviously. The
individual cDNAs were sequenced (Harvard Biopolymer Facility) and compared
with
known sequences by searchixtg GenBank databases.
Northern Blotting. Northeztt blotting was performed as described in Current
38
CA 02329858 2000-12-28
HMV-053.02
Protocols in Ivlolecular Biology ( 1999). A. 32P-labeled double stranded DNA
probe was
made a PCR fragment &oxn the respective gene using random prirraed
oligonucleotide
synthesis (Prime-a-Gene, Proz~c~ega). Murine tissue RN,A was a gift of Dr. W.
Swat
(Harvard Medical School).
RESULTS AND DISCUSSION
Biotinylated forms of Ptdlns-3,4-P~ and .F'tdlns-3,4,5-P3 specifcally bind to
the
in vitro translated PF~I domain of Akt and can be used for amity isolation. In
order to
test whether cloning of phvsphonnositide binding proteins through coupled in
vitro
transcription/translation expression was feasible, we examined whether
biotinylated
forms of 3'PPIs could be used to affinity isolate a known 3'PPI binding domain
that had
been prnduced by a coupled in vitro transcription/translation system. We first
pre-bound
avidin-coated beads with diC~-analogs of Ptdlns-3,4,5-P3 and Ptdlns-3,4-PZ
that had been
biotinylated on the co-end of the sn-1-aminohexanoyl derivatives (see
Materials and
Methods and Figure 1). The beads were then incubated with 35S-methionine-
labeled
prnteins generated by in vitro transcription/translation of cDNAs encoding
either maltose
binding pmtein (MBP) or MBP fused to the PH domain of Akt. Both genes were
cloned
into the in vitro transcription vector pCS2(+) under the control of the SP6
promoter. The
PH domain of Akt is known to bind 3'PPIs witb~ b~iglZ affinity and served as a
positive
control to determine optimal binding conditions and specificity. As shown in
Figure 2A,
the fusion pzvtein of MBP-Akt PH bound to avidin beads that were pre-bound
with the
biotinylated forms of either PtdIns-3,4,5-P~ ox Ptdlns-3,4-Pz. The MBP-PH
fusion
protein failed to bind to the unmodified avidin beads, and MBP failed to bind
to either the
avidin beads alone or to the beads pre-bound with the biotinylated
phosphoinositides.
These data demonstrate that the biotinylated 3'PPIs can effectively and
specifically
isolate an in vitro translated form of a phosphoinositide-binding domain.
The goal of this study is to isolate novel 3'PPI binding proteins from among
pools
of in vitro transeribedltranslated proteins from a small pool expression
library. In order to
specifically isolate 3'PPI binding proteins fxom among other proteins
expressed in the
library it is necessary that the system used for screening have a high
specificity for 3'PPI
binding proteins and a low background affinity for non-specific binding
proteins. The
system must also be su~ci,ently sensitive to detect small amounts of a 3'PPI
binding
protein in a large background of non-specific binding pxoteiuas. To determine
if our
system conformed to these criteria we iunvestigated whether biotinylated
PtdIns-3,4,5-P3
39
CA 02329858 2000-12-28
HNIV-os3.oz
azxd Ptdlras-3,4-Pz could specifically isolate the ~P-Akt PH domain fusion
protein after
having been diluted into a pool from the in vitro translation library.
There are approximately 100 iundependent clones in each pool of cDNA from vur
pCS2znouse spleen cDNA library and the total DNA cozzcentration was
approximately 1
~glNl for each pool. Therefore 1.0 ng of the plasmid encoding Iv~P-Akt PH
fusion
protein was mixed with 1 ug of DNA of a random pool from the library iz~ order
to
approximate the amount of 3'PPI binding protein that would be found in a
random pool
under the conditions of our scxeezt. As is shown in Figure 2B, biotiz~ylated
PtdIns-3,4-PZ-
beads captured the MBP-Akt PH fusion protein from among the other proteins i»
the
pool. However, some proteins in the library, e.g. the 25 kDa protein, bound to
the avidin
beads in both the presence and absence of lxgand, most likely through a non-
specific
hydrophobic interaction. These results provided the proof of concept that this
approach is
feasible for isolation of 3'PPI binding domains.
Fx~nally we examined whether our binding conditions allowed the PH domain of
Akt to distinguish between phosphoinositides phosphorylated at the 3' position
from
phosphoinositides lacking a phosphate at the 3' position. We performed
competition
binding experiments usi»g unbiotinylated forms of PtdIns-3,4,5-P3, PtdIns-3,4-
Ps, and
PtdIns-4,5-PZ to compete for biotinylated PtdIns-3,4,5-P3 binding to the MBP-
Akt PH
fusion protein (Figure 2C). Ptdlns-3, 4,5-P3 and PtdIns-3,4-PZ displaced the
biotin~ylated
PtdIns-3,4,5-P3 at concentrations of 1000 to10,00o-fold lower than Ptdlns-4,5-
P2 showing
that the specificity of the Alst PH domain for the 3' phosphate is preserved
under our
conditions. The data also shows that PtdIns-3,4,5-P3 binds more tightly to the
Akt PH
domain than does PtdTns-3,4-PZ and this is consistexat with two published
studies on the
a~zaity of the PH domain of Akt for phosphorylated phosphoinositides which
rank the
order of affinities as Ptdlns-3,4,5-P3~Ptdlns-3,4-P2»Ptdlns-4,5-P2. This
result
demonstrates that our system preserves the specificity of protein binding to
different
species of phosphorylated phosphoinositides.
Isolation of the murine isoforms of PDKI and PIP3BPlp42~p' by small pool
expression cloning. After an initial screening of 500 pools of the library
using the
biotinylated phosphoinositides, we isolated three 3'PPT binding proteins. We
obserrred a
protein of approximately 25 kDa in a single pool which. appeared to bind
exclusively to
biotinylated PtdIns-3,4,5-P3 (Figure 3A, top panel left). When the cDNA for
the protein
was isolated from the other cpNAs in the pool, the expressed purified protein
was also
found to bind to PtdIns-3,4-Pz but with an apparent lower affinity than for
PtdIns-3,4,s-P3
CA 02329858 2000-12-28
HMV-053.02
(Figure 3A, top panel right). Sequencing of the cloned eL7NA revealed that it
was
identical to the C-terminal 319 amino acids of murine phosphoinositide
dependent kinase
1 (P1~K1). PD1C1 is a ubiquitously expressed 559 amino acid (65 kDa) protein
which
contains an N-terminal serine/threonine kinase domain and a C-terminal PH
domain. In
agreement with our results, the PH domain of PDKl has recently been found to
bind with
a four- fold higher affinity to Ptdlns-3,4,5-P3 than to 1'tdIns-3,4-PZ but
with much lower
affinity to PtdIns-4,5-PZ. The fragment of PDIC1 that we isolated contains the
entire C-
terminal PH domain and approximately half of the serine/threonine kinase
domain
(Figure 3A, bottom panel). However, the f~Zrst 230 nucleotides of the isolated
rnKNA are
not the sequence of PDKI and the first in-frame AUG codon appears at the end
of the
kinase domain such that axaly the entire PH domain of PDK1 is translated. The
initial 230
non-coding nucleotides could either be due to an alternative splicing of PDKI,
or a~n
artifact from the construction of the library.
Anothex 3'PPl binding protein identified in our screen was a prntein of
approximately 25 kDa that bound tightly to both biotinylated PtdIns-3,4-Pz and
Ptdlns-
3,4,5-P3 but also had same residual biutading to the avidin beads (Figure 3B,
top panel).
Upon isolation and sequencing of the cDNA frarn the pool, the gene was found
to encode
the C-terminal 210 amino acids of a protein which has >95% amino acid homology
to 42
kDa inositol-1,3,4,5-tetrakisphosphate binding proteins isolated from both
porcine brain
called p42~4, and bovine brain called PIP3BP. PiP3BP/p42~4 is a protein of 374
amino
acids which contains an N-terminal zinc-f~ngcr domain which has homology to
GTPase
activating proteins For the ARF family of small G-proteins, and two tandem PH
domains
at the C-terminal end of the protein (Figure 3B, bottom panel). The fragment
of the gene
that we isolated contains the entire C-terminal PH domain and approximately
half of the
N-terminal PH domain but contains none of the putative A'RF-GAP domain. The C-
tcnninal PH domain ca~atanns a consensus sequence fax high affinity binding of
3'PPIs.
The N-terminal PH domain does not contain this sequence but two studies have
shown
that it displays some specificity far 3'PPIs.
The function of PIP3BP/p42~° is currently unknown although it is
highly
expressed in the brain and is thought to play a role in vesicle and membrane
transport
because of the putative ARF'-GAP domain. Interestingly, a closely related
yeast protein
called Gcsl has been shown to be an ARF-GAP and is important in proper
cytoskeletal
organization and actin polymerization in yeast. A recent study has also shown
that
PIP3BP is localized to the nucleus but translocates to the plasma membrane
upon
41
CA 02329858 2000-12-28
H~J-053.02
activation of PI 3'-~; however, the functional significance of both the
nuclear localization
and the PI 3'-K dependent translocation is unknown.
Isolation of 1',HISH, a Novel 3 PPI Binddng Protein Containing both ~H and SH2
Domains. We isolated a novel 3'PPI binding protein frarn the library that
migrated at
approximately 30 kDa and, siznilax to PIP3BP/p42'~4, bound tightly to both
biotinylated
PtdIns-3,4-Pi and I'tdlns-3,4,5-P3 but not to the avidin beads alone (Figure
4A). In
competition binding studies the in vitro transcribedltranslated protein was
found to bind
with equal affinity to both Ptdlns-3,4,5-P3 and Ptdlns-3,4-P2 but with
significantly lower
affinity to PtdIns-4,5-P2, thus demonstrating the specificity of the protein
for 3'PPIs
(Figure 4B).
The genc fragment containing the sequence for the protein was approximately
1.3
kb in length and encoded an open reading fraz~a~e from the beginning of the
fragment to a
step codon at nucleotide position 927 (Figure 5). A putative ICozak initiator
(ATG)
codon for methionine is present at position 87 and initiation of translation
from this
codon is consistent with the observed the size of the translated protein (280
amino acids
and 30 kDa). ~Iowevex, since the sequence upstream of this ATG did not contain
any in-
frame stop codons it was difficult to determine if the isolated gene fragment
encoded the
entire coding sequence of the gene or if more coding sequences existed
upstream.
The amino acid sequence contains coding regions for both an SH2 domain and a
C-terminal PH domain. The SH2 domain, which encompasses nucleotides 200 to
470, is
most similar to the SH2 domain of the neural adaptor pt~otein Nck (35%
identical, 59%
homologous at the amino acid level). The PH domain, which encompasses
nucleotides
590 to 840, is most similar to the PH domain of Akt (39% identical, fi3%
homologous at
the amino acid level) and contains the consensus sequence for high affinity
biz~di~ng of
3'PPIs. There is also a tyrosine (Y139) located between the SH2 and PH domains
which
could be phosphorylated in stimulated cells since the sequence surrounding
this tyrosine
is a putative consensus motif for phosphorylation by tyrosine kinases. 'fhe
existence of a
putative phosphotyrosine bi~odi~ug SH2 domain, a 3'PPI binding PH domain, and
a
sequence for phospharylatxoz~ of tyrosine. strongly suggest that this protein
plays a rate in
cellular signaling. We have nazxaed this protein PHISI~ for 13'
Phosphoinositide
Interacting SH2-Containing proteini.
In order to determine the tissue distribution of PHISH we performed Northern
blots on total RNA from several marine tissues, spleen, brain, heart, lung,
thymus, and
lymph using a probe derived from nucleotides 87 to 927, the putative coding
region of the
42
CA 02329858 2000-12-28
HMV-053.02
protein. TwQ R~tA species that are approximately 1.2 to 1.4 kb were detected.
These
could represent products of alternative spliciztg of the gene. The larger of
the two
transcripts was detected in all tissues, and the smaller of the two
transcripts was highly
expressed in spleen and at lower levels in both heart and lung tissue (Figure
6). In vitro
transcription of the cDNA for PHISH isolated from the expression library also
produced
two transcripts that were approximately 1.2 -1.4 kb izx size. The major
transcript is very
close in size to the larger of the two transcripts present iz~ all marine
tissues tested, while
the minor product is close in size to the smaller transcript present in heart,
lung, card
spleen. This data suggests that the gene fragment isolated from the expression
cloning
library encodes the complete sequence of the gene.
Upon searching the human EST database with the nucleotide sequence of PHISH
we found that PHISH had an 87% rxucleotide sequence identity with that of a
human EST
derived from stem cells (locus AF150266)_ This EST encodes a cDNA, that is 1.4
kb long
and contains entire coding region of PHISH. The sequences of PHISH and the
human
EST differ significantly outside of the protein coding sequence. Moreover,
three colons
upstream from the putative start colon in the human EST are in frame stop
colons. The
high degree of sequence homology between PHISH and the human EST implies that
they
encode species-specific homologues of the same protein and that the gene for
PHISH
isolated from the expression screen encodes the entire protein.
Since PHISH contains only a PH domain and an SH2 domain, it is possible that
PHISH functions as an adaptor protein. The PH doraain could dock PHISH to
3'PPI
generated at membrane receptor complexes and the SH2 domain could recruit
phosphotymsine containing proteins) to such complexes. It is interesting to
note that
Skolnik and coworkers have isolated the PH domain of EST684797 from a homology
search of human ESTs for PH domains that would be predicted to bind tightly to
3'PPIs.
They have also shown that the PH domain of ESTb$4797, which is highly
homologous to
the PH dozraain of PHISH binds tightly and selectively to Ptdlns-3,4-Pz and
Ptdlns-3,4,5-
P3 but not to Ptdlns-4,5-Pi.
These studies demonstrate that coupled in vitro transcriptiom/translation
libraries
can be used in conjunction with a#Enity isolation technology using synthetic
phosphoinositides to isolate 3'PPI binding domains. Recently, several other
methods
have been described to isolate and clone 3'PPX binding proteins_ Qne example
is the use
of resins coupled to phosphorylated inositol phosphates such as IP3, or
inositol
phosphates linked to a glycerol moiety, to extract proteins from tissue
extracts. Using
43
CA 02329858 2000-12-28
HIV-053.02
these techniques PIP3BP and p42~° were extracted from total brain
extracts. However,
proteins expressed iz~ Iow abundance are difficult to isolate by this
procedure az~d
furtHezzz~ore, cDhTA cloz~iUag z~equxres zz~ultiple steps following affinity
isolation.
Pxpression cloning of genes from cDNA Iibraries can circumvent some of the
limitations of protein affinity isolation techniques. Two expression cloning
techni9ues
have been recently developed to identify 3'PPI binding proteins. The first
involved the
screening of a ~.gtl l library with a crude mixture of brain phospholipids
that had been
phosphorylated in vitro by PI 3'-K. Screening of two marine derived cDNA
libraries
(from brain and adipocytes) yielded only oz~e protean that bound tightly to
Ptdlns-3,4,5-P3.
This protein, called Grpl (General Receptor for Phosphoinositides), contained
a PH
domain and an ARF-GEF domain that catalyzes 3'PPI dependent nucleotide
exchange on
mammalian AItFs. However, no other 3'PPI dependent binding protein was
isolated izt
the screen possibly due to improper folding of the proteins.
Another recent expression cloning strategy for isolating 3'PPI binding
proteins is
the use of a modified yeast two-hybrid system. In this system, genes f~Qrn a
mammalian
eDNA libzary were fused to a constitutively active Ras. The fusion proteins
were then
tested for their ability to rescue a temperature sensitive phenotype, due to a
defect
upstream of Ras, when coexpressed with constitutively active PZ 3'-~,. This
system
worked well in experiments where constitutively active Ras was fused to PH
domains
already lrnown to bind to 3'PPIs. However, when tested with an expression
library in
yeast only Akty was isolated.
The screen described here is the first to isolate multiple 3'PPI binding
targets from
an expression library, including a novel protein. The basis for the better
effciency of this
screen may stem from the fact that in vitro translated proteins are more
likely to be
properly folded. bo, addition, many internal initiation sites for translation
allow for the
independent and unoccluded expression of PH and other 3'PPI binding domains.
We also
used analogs of PtdIns-3,4-PZ and PtdIns-3,4,5-P3 that are synthetically pure
and, as a
result of modification in a distal zegiota of the acyl chain, closely resemble
the
phosphorylated phosphatidylinositols that occur naturally. It has beezt shown
that
although the main binding of tlae protein is to the inositoI head grnup, the
contribution of
the glycerol chain and the fatty acid side chains of the phosphatidylinositol
are essential
far specificity and tight bindiztg to the protein. Our screening technique is
not without
drawbacks. For example, there can be non-specific binding of proteins to the
avidin-
coated beads and this non-speciftc binding may obscure the biutding of other
3'PPI
44
CA 02329858 2000-12-28
binding proteins with the same electrophoretic mobility. In addition, we
isolated only C-
terminal PH domains, possibly because our library was made using oligo-dT to
prime
cDbI,A synthesis from the 3' poly-A tail of mRNAs. In addition, our screen
thus far has
isolated only strong binders of 3'PPIs indicating that our bindxx~g conditions
may be too
striz~gez~t to accommodate weaker binding proteins.
In summary, we have described a novel and effective way for isolating and
cloning 3'PPl binding proteins from expression libraries. The technique
described here
has broad applications for the isolation of binding partners for other
phosphoinositide
polyphosphates or other lipid products.