Note: Descriptions are shown in the official language in which they were submitted.
CA 02330191 2000-12-04
WO 99!63090 PCT/US99/12276
PROTEASE INHIBITOR PEPTIDES
Bac~our~ of the Invention
The plasma, or serine, proteases of the blood contact system are known to be
activated by interaction with negatively charged surfaces. For example, tissue
injury
during surgery exposes the vascular basement membrane, causing interaction of
the
blood with collagen, which is negatively charged at physiological pH. This
induces a
cascade of proteolytic events, leading to production of plasmin, a
fibrinolytic protease,
and consequent blood loss.
Perioperative blood loss of this type can be particularly severe during
cardiopulinonary bypass (CPB} surgery, in which the patient's blood flow is
diverted to
an artificial heart-lung machine. CPB is an essential component of a number of
lifo
saving surgical procedures. For example, in the United States, it is estimated
that
300,000 patients every year undergo coronary artery bypass grafts involving
the use of
I S CPB.
Although necessary and generally safe, CPB is associated with a significant
rate
of morbidity, some of which may be attributed to a "whole body inflammatory
response"
caused by activation of plasma protease systems and blood cells through
interactions
with the artificial surfaces of the heart-lung machine (Butler et al., Ann.
Thorac. Sung.
55:552 (1993); Edmunds et al., J. Card. Sung. 8:404 (1993)). For example,
during
extracorporeal circulation, exposure of blood to negatively charged surfaces
of the
artificial bypass circuit, e.g., plastic surfaces in the heart-lung machine,
results in direct
activation of plasma factor 7dI.
Factor ?QI is a single-chain 80 kDa protein that circulates in plasma as as
inactive zymogen. Contact with negatively chargod nonendothelial surfaces,
like those
of the bypass circuit, causes surface-bound factor 7~ to be autoactivated to
the active
serine protease factor 3CQa. See Colman, Agents Actions Suppl. 42:125 (1993).
Surfaco-
activated factor XIIa then processes prckallila~ein (PK) to active kallikrein,
which in tum
cleaves more XIIa from 7Qi in a reciprocal activation reaction that results in
a rapid
amplification of the contact pathway. Factor 3~Ia can also activate the first
component
of complement C1, leading to production of the anaphylatoxin CSa through the
classical
complement pathway.
The CPB-induced inflammatory response includes changes in capillary
permeability and interstitial fluid accumulation. Cleavage of high molecular
weight
1
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
kininogen (I-BC) by activated ka11i1Qein generates the potent vasodilator
bradykinin,
which is thought to be responsible for increasing vascular permeability,
resulting in
edema, especially in the lung. The lung is particularly susceptible to damage
associated
with CPB, with some patients exhibiting what has been called "pump lung
syndrome"
S following bypass, a condition indistinguishable from adult respiratory
distress. See
Johnson et al., J. Thorac. Cardiovasc. Surg. 107:1193 ( 1994).
Post-CPB pulmonary injury includes tissue damage thought to be mediated by
neutrophil sequestration and activation in the microvasculature of the lung.
Butler et al.,
supra; Johnson, et al., supra. Activated factor XII can itself stimulate
neutrophil
aggregation. Factor XIIa-generated ka11i1Qein, and complement protein CSa
generated
by Factor XIIa activation of the complement cascade, both induce neutrophil
chemotaxis, aggregation and degranulation. See Edmunds et al., supra.
Activated
neutrophils may damage tissue through release of oxygen-derived free radicals,
proteolytic enzymes such as elastase, and metabolites of arachidonic acid.
Release of
neutrophil products in the lung can cause changes in vascular tone,
endothelial injury
and loss of vascular integrity.
Intrinsic inhibition of the contact system occurs through inhibition of
activated
3CIIa by C1-inhibitor (Cl-INI~. See Colrnan, supra. During CPB, massive
activation of
plasma proteases and consumption of inhibitors overwhelm this natural
inhibitory
mechanism. A potential therapeutic strategy for reducing post-bypass
pulinonary injury
mediated by neutrophil activation would, therefore, be to block the formation
and
activity of the neutiophil agonists kallikrein, factor XIIa, and CSa by
inhibition of
proteolytic activation of the contact system.
Protease inhibitor therapy, which partially attenuates the contact system, is
currently employed clinically in CPB. Aprotinin, also known as basic
pancreaxic
protease inhibitor (BPPI), is a small, basic, 58 amino acid polypeptide
isolated from
bovine Lung. It is a broad-spectrum serine protease inhibitor of the Kunitz
type, and was
first used during bypass in an attempt to reduce the inflammatory response to
CPB. See
Butler et a1, supra. Aprotinin treatment results in a significant reduction in
blood loss
following bypass, but does not appear to significantly reduce neutrophil
activation.
Additionally, since aprotinin is of bovine origin, there is concern that
repeated
administration to patients could lead to the development of an immune response
to
aprotinin in the patients, precluding its further use.
2
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
The proteases inhibited by aprotinin during CPB appear to include plasma
kallikrein and plasmin. See, e.g., Scott, et al., Blood 69:1431 (1987).
Aprotinin is an
inhibitor of plasmin (K; of 0.23nM), and the observed reduction in blood loss
may be
due to inhibition of fibrinolysis through the blocking of plasmin action.
Although
aprotinin inhibits plasma kallikrein (K; of 20nM), it does not inhibit
activated factor 3CB,
and consequently only partially blocks the contact system during CPB.
Another attractive protease target for use of protease inhibitors, such as
those of
the present invention, is factor XIIa, situated at the very first step of
contact activation.
By inhibiting the proteolytic activity of factor 3CIa, kallikrein production
would be
prevented, blocking amplification of the contact system, neutrophil activation
and
bradykinin release. Inhibition of 3QIa would also prevent complement
activation and
production of CSa. More complete inhibition of the contact system during CPB
could,
therefore, be achieved through the use of a better XIIa inhibitor.
Protein inhibitors of factor XIIa are known. For example, active site mutants
of
a,-antitrypsin that inhibit factor 7~QIa have been shown to inhibit contact
activation in
human plasma. See Patston et al., J. Biol. Chem. 265:10786 (1990). The large
siu and
complexity (greater than 400 amino acid residues) of these proteins present a
significant
challenge for recombinant protein production, since large doses will almost
certainly be
required during CPB. For example, although it is a potent inhibitor of both
kallilaein
and plasmin, nearly 1 gram of aprotinin must be infused into a patient to
inhibit the
massive activation of the kallikrein-kinin and fibrinolytic systems during
CPB.
The use of smaller, more potent 7QIa inhibitors such as the com and pumpkin
trypsin inhibitors (Wen, et al., Protein Exp. & Purif. 4:215 (1993); Pedersen,
et al., J.
Mol. Biol. 236:385 (1994)) could be more cost-effective than the large a,-
antitrypsins,
but the infusion of high doses of these non-mammalian inhibitors could result
in
immunologic reactions in patients undergoing repeat bypass operations. The
ideal
protein 7CIIa inhibitor is, therefore, preferably small, potent, and of human
sequence
origin.
One candidate for an inhibitor of human origin is found in circulating
isoforms of
the human amyloid (I-protein precursor (APPn, also known as protease nexin-2.
APPI
contains a Kunitz serine protease inhibitor domain known as KPI (Kunitz
Protease
Inhibitor). See Ponte et al., Nature, 331:525 (1988); Tanzi et al., Nature
331:528
(1988); Johnstone et al., Biochem. Biophys. Res. Commun. 163:1248 (1989);
Oltersdorf
3
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12Z76
et al., Nature 341:144 (1989). Human KPI shares about 45% amino acid sequence
identity with aprotinin. The isolated IS;PI domain has been prepared by
recombinant
expression in a variety of systems, and has been shown to be an active serine_
protease
inhibitor. See, for example, Sinha, et al., J. Biol. Chem. 265:8983 ( 1990).
The measurod
in vitro K; of KPI against plasma kallikrein is 45nM, compared to 20nM for
aprotinin.
Aprotinin, KPI, and other Kunitz-type serine protease inhibitors have been
engineered by site-directed mutagenesis to improve inhibitory activity or
specificity.
Thus, substitution of Lysls of aprotinin with arginine resulted in an
inhibitor with a K; of
0.32nM toward plasma kallikrein, a 100-fold improvement over natural
aprotinin. See
PCT application No. 89/10374. See also Norris et al., Biol. Chem. Hoppe Seyler
371:3742 (1990). Alternatively, substitution of position 15 of aprotinin with
va.line or
substitution of position 13 of KPI with valine resulted in elastase inhibitors
with K;s in
the 100 pM range, although neither native aprotinin nor native KPI
significantly inhibits
eIastase. See Wenzel et al., in: Chemistry of Peptides and Proteins, Yol. 3,
(Walter de
Gruyter, Berlin, New York, 1986); Sinha et al., supra. Methods for
substituting residues
13, 15, 37, and 50 of KPI are shown in general terms in European Patent
Application
No. 0 393 431, but no specific sequences are disclosed, and no protease
inhibition data
are given.
Phage display methods have been recently used for preparing and screening
derivatives of Kunitz-type protease inhibitors. See PCT Application No.
92115605,
which describes specific sequences for 34 derivatives of aprotinin, some of
which were
reportedly active as elastase and cathepsin inhibitors. The amino acid
substitutions in
the derivatives were distributed throughout almost all positions of the
aprotinin
molecule.
Phage display methods have also been used to generate KPI variants that
inhibit
factor VITa and kallila~ein. See Dennis et al., J. Biol. Chem. 269:22129 and
269:22137
(1994). The residues that could be varied in the phage display selection
process were
Limited to positions 9-11, 13-17, 32, 36 and 37, and several of those residues
were also
held constant for each selection experiment. One of those variants was said to
have a IC;
of l.2nM for kallila~ein, and had substitutions at positions 9 ('ThrPro), 13
(ArgLys), 15
(MetLeu), and 37 (GlyTyr). None of the inhibitors was tested for the ability
to inhibit
factor XIIa. PCT application WO 96139515 used phage display methods to vary
the
residues at positions 11-19 and 34. Certain of those variants were tested for
inhibition of
kalIikrein; factors Xia, Xa, and VIZa; thrombin; plasmin; and activated
protein C. PGT
4
CA 02330191 2000-12-04
WO 99/63090 PC'T/US99/12276
application WO 96/35788 used phage display methods to vary the residues at
positions
9, 11, 13-18, 32, and 37-40. Certain of those variants were tested for
inhibition of
kaliikrein, plasmin, and factors Xa, Xia, and XIIa.
It is apparent, therefore, that new protease inhibitors that can bind to and
inhibit
the activity of serine professes are greatly desirable. In particular it is
highly desirable to
prepare peptides, based on human peptide sequences, that can inhibit selected
seriae
professes such as kallikrein; chymotrypsins A and B; trypsin; elastase;
subtilisin;
coagulants and procoagulants, particularly those in active form, including
coagulation
factors such as factors VIIa, IXa, Xa, XIa, and XIIa; plasmin; thrombin;
proteinase-3;
enterokinase; acrosin; cathepsin; urokinase; and tissue plasminogen activator.
It is also
highly desirable to prepare novel protease inhibitors that can ameliorate one
or more of
the undesirable clinical manifestations associated with enhanced serine
protease activity,
for example by reducing pulmonary damage or blood loss during CPB. In
addition, it is
highly desirable to prepare such novel protease inhibitors with high
expression levels, as
well as with high yields.
Summary of the Invention
The present invention relates to peptides that can bind to and preferably
exhibit
inhibition of the activity of serine professes. Those peptides can also
provide a means of
ameliorating, treating or preventing clinical conditions associated with
increased activity
of serine proteases. Particularly, the novel peptides of the present invention
preferably
exhibit a more potent and specific (i.e., greater) inhibitory effect toward
serine professes
of interest in comparison to known serine protease inhibitors. Examples of
such
professes include: kallikrein; chymotrypsins A and B; trypsin; elastase;
subtilisin;
coagulants and procoagulants, particularly those in active form, including
coagulation
factors such as factors V>Za, lXa, Xa, XIa, and XIIa; plasmin; thrombin;
proteinaso-3;
enterokinase; acrosin; cathepsin; urokinase; and tissue plasminogen activator.
In
particular, the peptides of the present invention preferably exhibit a greater
potency and
specificity for inhibiting one or more serine professes of interest (eg.,
kalIila~ein, plasmin
30' and factors VIZa, IXa, Xa, XIa, and XIIa) than the potency and specificity
exhibited by
native KPI or other known setirte protease inhibitors.
The novel peptides of the present invention preferably comprise substituting
the
tyrosine residue at position 48. Such substituted peptides may exhibit an
increased level
of recombinant expression in comparison to the expression levels of serine
professes that
5
CA 02330191 2000-12-04
WO 99/63090 PGT/US99/12276
do not have that substitution. The effect of this substitution may be
manifested not only
on the substituted ICPI peptides of the present invention, but on wild-type
ICPI as well.
Also, the peptides of the present invention that comprise the N-terminal
sequence Glu-
Val-Val-Arg (residues -4 to -I ) may also preferably exhibit increased yields
via a
substitution of that N-terminal sequence to Asp-Val-Val-Arg.
In achieving the inhibition of serine protease activity, the invention
provides
protease inhibitors that can ameliorate one or more of the undesirable
clinical
manifestations associated with enhanced serine protease activity, for example,
by
reducing pulmonary damage or blood loss during CPB.
The present invention relates to protease inhibitors comprising the following
sequences: X'-Val-Cys-Ser-Glu-Gln-Ala-Glu-Xz-Gly-X3-Cys-Arg-Aia-X°-XS-
3C6-X~-
Trp-Tyr-Phe-Asp-Val-Thr-Glu-Gly-Lys-Cys-Ala-Pro-Phe-Xa-Tyr-Gly-Gly-Cys-X9-
X~°-
X~ ~-X~~-Asn-Asn-Phe-Asp-Thr-GIu-Glu-X~3-Cys-Met-Ala-Val-Cys-Gly-Ser-Ala-Ile,
wherein X~ is selected from Glu-Va1-Val-Arg-Glu-, Asp-Val-Val-Arg-GIu-, Asp,
and
Glu; XZ is selected from Thr, Val, Ile and Ser; X' is selected from Pro and
Ala; X'~ is
selected from Arg, Ala, Leu, Gly, and Met; Xs is selected from Ile, His, Leu,
Lys, Ala,
and Phe; X6 is selected from Ser, IIe, Pro, Phe, Tyr, Tip, Asn, Leu, His, Lys,
and Glu; X'
is selected from Arg, His, and Ala; X$ is selected from Phe, Val, Leu, and
Gly; X9 is
selected from Gly, Ala, Lys, Pro, Arg, Leu, Met, and Tyr; X'° is
selected from Ala, Arg,
and Gly; Xl1 is selected from Lys, Ala, and Asn; X~z is selected from Ser,
Ala, and Arg,
X13 is selected from His, Gln, Ala, and Asp.
A further aspect of the present invention provides protease inhibitors wherein
X'
is Asp-Val-Val-Arg-Glu-, XZ is Thr, Val, or Ser, X3 is Pro, X° is Ala
or Met, Xs is Ile, X6
is Ser or Tyr, X7 is His, X$ is Phe, X9 is Gly, X'° is Gly, X~~ is Asn,
and X~~ is Arg.
Another aspect of the present invention provides protease inhibitors wherein
Xt is Asp-
Val-Val-Arg-Glu-, Xi is Pro, X'' is Ala, Xs is Ile, X6 is Phe, X7 is Arg, X$
is Phe, Xs is
Giy, X~° is Gly, X~i is Asn, and X1Z is Arg. Yet another aspect of the
present invention
provides protease inhibitors wherein XZ is Thr or Val. Another aspect of the
present
invention provides protease inhibitors wherein Xi is Thr. A further aspect of
the present
invention provides protease inhibitors wherein XZ is Val. Another aspect of
this
invention provides protease inhibitors wherein X2 is Thr or Val, and X°
is Ala. A further
aspect of the present invention provides protease inhibitors wherein XZ is Thr
or Val, and
X4 is Met. Yet another aspect of the present invention provides protease
inhibitors
wherein XZ is Thr, J~C4 is Ala, ?~ is Tyr, and X~3 is His. A further aspect of
the present
6
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
invention provides protease inhibitors wherein Xz is Thr, X'' is Ala, X6 is
Tyr, and X13 is
Gln. Another aspect of the present invention provides protease inhibitors
wherein XZ is
Thr, X' is Ala, X6 is Tyr, and X'3 is Ala. Another aspect of the present
invention
provides protease inhibitors wherein XZ is Thr, X° is Ala, X~ is Tvr,
and X13 is Asp.
Another aspect of the present invention provides protease inhibitors wherein
Xi is Thr,
X4 is Met, 3C6 is Ser, and X13 is selected from His, Ala, or Gln. Another
aspect of the
present invention provides protease inhibitors wherein XZ is Val, X; is Ala,
X6 is Tyr,
and X13 is selected from His, Ala, or Gln. Another aspect of the present
invention
provides protease inhibitors wherein XZ is Thr, X4 is Ala, X6 is Tyr, and X'3
is selectal
from His, Ala, or Gln. Another aspect of the present invention provides
protease
inhibitors wherein X'3 is selected from His or Ala. Another aspect of the
present
invention provides protease inhibitors wherein X'3 is selected from His or
Ala. Anoiher
aspect of the present invention provides protease inhibitors wherein X13 is
His. A further
aspect of the present invention provides protease inhibitors wherein X13 is
Ala.
A further aspect of the present invention provides an isolated DNA molecule
comprising a DNA sequence encoding a protease inhibitor of the invention.
Another
aspect of the present invention provides an isolated DNA molecule operably
linked to a
regulatory sequence that controls expression of the coding sequence of the
protease
inhibitor in a host cell. Another aspect of the present invention provides an
isolated
DNA molecule operably linked to a regulatory sequence that controls expression
of the
coding sequence of the protease inhibitor in a host cell fiuther comprising a
DNA
sequence encoding a secretory signal peptide. That secretory signal peptide
may
preferably comprise the signal sequence of yeast x-mating factor. Another
aspect of the
present invention provides a host cell transformed with a DNA molecule.
Another
aspect of the present invention provides a host cell transformed with any of
the DNA
molecules defined above. Such a host cell may preferably comprise E. coli or a
yeast
cell. When said host cell is a yeast cell, the yeast cell may preferably be
Saccharonry~aes
cerevisiae. When said host cell is a yeast cell, the yeast cell may preferably
be Piehia
pastoris.
Another aspect of this invention provides a method for producing a protease
inhibitor, comprising the steps of culturing a host cell as defined above and
isolating and
purifying said protease inhibitor.
7
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
A further aspect of this invention provides a pharma;,eutical composition,
comprising a protease inhibitor together with a pharmaceutically acceptable
sterile
vehicle.
An additional aspect of the present invention provides a method of treatment
of a
clinical condition associated with increased activity of one or more serine
proteases,
comprising administering to a patient suffering from said clinical condition
an effective
amount of a pharmaceutical composition comprising a protease inhibitor of the
present
invention together with a pharmaceutically acceptable sterile vehicle. That
method of
treatment may preferably be used to treat the clinical condition of blood loss
during
surgery.
Yet another aspect of the present invention provides a method for inhibiting
the
activity of serine proteases of interest in a mammal comprising administering
a
therapeutically effective dose of a pharmaceutical composition comprising a
protease
inhibitor of the present invention together with a pharmaceutically acceptable
sterile
vehicle, wherein said serine proteases are selected from the group consisting
of
kallikrein; chymotrypsins A and B; trypsin; elastase; subtilisin; coagulants
aad
procoagulants, particularly those in active form, including coagulation
factors such as
factors VIIa, iXa, Xa, XIa, and XIIa; plasmin; thrombin; proteinase-3;
enterokinase;
acrosin; cathepsin; urokinase; and tissue piasminogen activator.
Another aspect of the present invention provides protease inhibitors
comprising
the sequence: X'-Val-Cys-Ser-Glu-Gln-Ala-Glu-Xz-Gly-Pm-Cys-Arg-Ala-Ala-Ile-Tyr-
His-Trp-Tyr-Phe-Asp-Val-Thr-Glu-Gly-Lys-Cys-Ala-Pro-Phe-Phe-Tyr-Gly-Gly-Cys-
Gfy-Gly-Asn-Arg-Asn-Asn-Phe-Asp-Thr-Glu-Glu-X;-Cys-Met-Ala-Val-Cys-Gly-Set
Ales-Ile, wherein X~ is selected from Glu-Val-Val-Arg-Glu-, Asp-Val-Val-Arg-
Glu-,
Asp, or Glu; XZ is selected from Thr and Val; X3 is selected from His, Gln,
Ala, or Asp.
A further aspect of the present invention relates to protease inhibitors
wherein X'
is Glu-Val-Val-Arg-Glu. Yet another aspect of the present invention provides
for
protease inhibitors wherein XZ is Thr. An additional aspect of the present
invention
provides protease inhibitors wherein Xi is Val. Yet another aspect of the
present
invention provides protease inhibitors wherein X3 is His. Another aspect of
the present
invention provides protease inhibitors wherein X3 is Gln. Another aspect of
the present
invention provides protease inhibitors wherein X' is AIa. Another aspect of
the present
invention provides protease inhibitors wherein X3 is Asp. Another aspect of
the present
invention provides protease inhibitors wherein X~ is Asp-Val-Val-Arg-Glu.
Another
8
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/I2276
aspect of the present invention provides proteaseXz is Another
inhibitors wherein Thr.
aspect of the present invention provides proteaseX~ is Another
inhibitors wherein Val.
aspect of the present invention provides proteaseX3 is Another
inhibitors wherein His.
aspect of the present invention provides proteaseX3 is Another
inhibitors wherein Gln.
aspect of the present invention provides X3 is Another
protease inhibitors wherein Ala.
aspect of the present invention provides proteaseX3 is Another
inhibitors wherein Asp.
aspect of the present invention provides protease A
inhibitors wherein X~ is Glu. further
aspect of the present invention provides proteaseXz is Another
inhibitors wherein Thr.
aspect of the present invention provides proteaseXz is Another
inhibitors wherein Val.
aspect of the present invention provides X3 is Another
protease inhibitors wherein His.
aspect of the present invention provides protease inhibitors wherein X3 is
Gln.
Another aspect of the present invention providesX3
protease inhibitors wherein
is Ala. Another aspect of the present invention providesX3
protease inhibitors wherein
is Asp. Another aspect of the present invention providesXt
protease inhibitors wherein
is Another aspect of the present invention providesX=
Asp. protease inhibitors wherein
is Thr. Another aspect of the present invention providesXi
protease i_~hibitors wherein
is Val. Another aspect of the present invention providesXl
protease inhibitors wherein
is His. Another aspect of the present invention providesX3
protease inhibitors wherein
is Gln. Another aspect of the present invention providesX3
protease inhibitors wherein
is Another aspect of the present invention providesX3
Ala. protease inhibitors wherein
is Asp.
Another aspect of the present invention provides protease inhibitors wherein
Xt
is Glu-Val-Val-Arg-Glu-, Xi is Thr, Val, or Ser, X3 is Pro, X~ is Ala or Met,
XS is Ile, X6
is Ser or Tyr, X' is His, X$ is Phe, X9 is Gly, Xi° is Gly, Xt ~ is
Asn, and Xt= is Arg.
Another aspect of the present invention provides protease inhibitors wherein
Xi is Thr or
Val. Another aspect of the present invention provides protease inhibitors
wherein XZ is
Thr. Another aspect of the present invention provides protease inhibitors
wherein X= is
Val. Another aspect of the present invention provides protease inhibitors
wherein X2 is
Thr or Vat, and X° is Ala Another aspect of the present invention
provides protease
inhibitors wherein XZ is Thr or Val, and X° is Met. Another aspect of
the present
invention provides protease inhibitors wherein XZ is Thr, X4 is AIa, X6 is
Tyr, and Xt3 is
His. Another aspect of the present invention provides protease inhibitors
wherein X= is
Thr, X° is Ala, X6 is Tyr, and X~; is Gln. Another aspect of the
present invention
provides protease inhibitors wherein XZ is Thr, X; is Ala, X6 is Tyr, and Xt3
is Ala.
9
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
Another aspect of the present invention provides protease inhibitors wherein
XZ is Thr,
X4 is Ala, 3.'° is Tyr, and X~~ is Asp. Another aspect of the present
invention provides
protease inhibitors wherein XZ is Thr, X~ is Met, ?C6 is Ser, and X~3 is
selected from His,
Ales, or Gln. Another aspect of the present invention provides a protease
inhibitors
wherein Xz is teal, X4 is Ala, X6 is Tyr, and X'3 is selected from His, Ales,
or Gln.
Another aspect of the present invention provides protease inhibitors wherein
XZ is Thr,
X° is Ales, X6 is Tyr, and X~3 is selected from His, Ala, or Gln.
Another aspect of the
present invention provides protease inhibitors wherein X'3 is selected from
His or Ales.
Another aspect of the present invention provides protease inhibitors wherein
X'3 is
selected fi-om His or Ala. Another aspect of the present invention provides
protease
inhibitors wherein X~3 is His. Another aspect of the present invention
provides protease
inhibitors wherein X'3 is Ala.
Another aspect of the present invention provides an isolated DNA molecule
comprising a DNA sequence encoding a protease inhibitor. Another aspect of the
present invention provides an isolated DNA molecule operably linked to a
regulatory
sequence that controls expression of the coding sequence in a host cell.
Another aspect
of the present invention provides an isolated DNA molecule further comprising
a DNA
sequence encoding a secretory signal peptide. Another aspect of the present
invention
provides an isolated DNA molecule wherein said secretory signal peptide
comprises the
signat sequence of yeast x-mating factor. Another aspect of the present
invention
provides a host cell transformed with any of the DNA molecules defined above.
Such a
host cell may preferably comprise E. coli or a yeast cell. When said host cell
is a yeast
cell, the yeast cell may preferably be Saccharomyces cereviriae. When said
host cell is a
yeast cell, the yeast cell may preferably be Pichia pastoris.
Another aspect of the present invention provides for a method for producing a
protease inhibitor, comprising the steps of culturing a host cell as defined
above and
isolating and purifying said protease inhibitor.
A further aspect of this invention provides a pharmaceutical composition,
comprising a protease inhibitor together with a pharmaceutically acceptable
sterile
vehicle.
An additional aspect of the present invention provides a method of treatment
of a
clinical condition associated with increased activity of one or more serine
proteases,
comprising administering to a patient suffering from said clinical condition
an effective
CA 02330191 2000-12-04
WO 99/63090 PCTNS99/12276
amount of a pharmaceutical composition comprising a protease inhibitor of the
present
invention together with a pharmaceutically acceptable sterile vehicle. That
method of
treatment may preferably be used to treat the clinical condition of blood loss
during
surgery.
S Another aspect of the present invention provides a method for inhibiting the
activity of serine proteases of interest in a mammal comprising administering
a
therapeutically effective dose of a pharmaceutical composition comprising a
protease
inhibitor of the present invention together with a pharmaceutically acceptable
sterile
vehicle, wherein said seiine proteases are selected from the group consisting
of
ka11i1Qein; chymotrypsins A and B; trypsin; elastase; subtilisin; coagulants
and
procoaguiants, particularly those in active form, including coagulation
factors such as
factors VBa, IXa, Xa, XIa, and XIIa; plasmin; thrombin; prnteinase-3;
enterokinase;
acrosin; cathepsin; urokinase; and tissue plasminogen activator.
Yet another aspect of the present invention provides a method for increasing
the.
expression levels of recombinant protease inhibitors comprising the step of
culturing a
host cell transformed with an isolated DNA molecule comprising a DNA sequence
encoding a protease inhibitor. Such a host cell is E. coli or a yeast cell.
When such a
host cell is a yeast cell, the yeast cell may preferably be Saccharomyces
cerevisiae.
When such a host cell is a yeast ccll, the yeast cell may preferably be Pichia
pastoris.
Another aspect of the present invention provides a method for increasing the
yield of recombinant protease inhibitors comprising the step of culturing a
host cell
transformed with an isolated DNA molecule comprising a DNA sequence encoding a
protease inhibitor according to claim 1, wherein X~ is Asp-Val-Val-Arg-Glu-,
and
isolating and purifying said protease inhibitor. When said host cell is a
yeast cell, the
yeast cell may preferably be Saccharomyces cerevisiae. When said host cell is
a yeast
cell, the yeast cull may preferably be Pichia pastoris.
Another aspect of the present invention provides protease inhibitors
comprising
the sequence: X'-Val-Cys-Ser-Glu-Gln-Ala-Glu-Xi-Gly-Pro-Cys-Arg-Ala-X3-DaX'~-
Xs-Trp-Tyr-Phe-Asp-Val-Thr-Glu-Gly-Lys-Cys-Ala-Pro-Phe-Phc-Tyr-Gly-Gly-Cys-
Gly-Gly-Asn-Arg-Asn-Asn-Phe-Asp-Thr-Glu-Glu-7C6-Cys-Met-Ala-Val-Cys-Gly Ser-
Ala-Ile, wherein: X~ is selected from Glu-Val-Val-Arg-Glu-, Asp-Val-Val-Arg-
Glu-,
Asp, or Glu; X~ is selected from Thr or Val; X3 is selected from Arg and Met;
X4 is
selected from Ser and Tyr; Xs is selected from Arg, His, or Ala; and X6 is
selected from
His, Gln, Ala or Asp.
11
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
A further aspect of the present invention provides protease inhibitors
comprising
the sequence: X~-Val-Cys-Ser-Glu-Gln-Ala-Glu-Thr-Gly-Pro-Cys-Arg-Ala-Leu-Phe-
Lys-Arg-Trp-Tyr-Phe-Asp-Val-Thr-Glu-Giy-Lys-Cys-Ala-Pro-Phe-Phe-Tyr-Gly-Gly-
Cys-Leu-Gly-Asp-Arg-Asn-Asn-Phe-Asp-Thr-Glu-GIu-XZ-Cys-Met-Ala-Val-Cys-Gly-
Ser-Ala-Ile, wherein: X' is selected from Glu-Val-Val-Arg-Glu-, Asp-Val-Val-
Arg-
Glu-, Asp, and Glu; XZ is selected from His, Gln, Ala, and Asp.
Yet a further aspect of the present invention provides protease inhibitors
wherein X~ is Asp-Val-Val-Arg-Glu. Another aspect of the present invention
provides protease inhibitors wherein Xi is His. Another aspect of the present
invention provides protease inhibitors wherein XZ is Gln. Another aspect of
the
present invention provides protease inhibitors wherein XZ is Ala. Another
aspect of
the prrsent invention provides protease inhibitors wherein Xi is Asp. Another
aspect
of the present invention provides protease inhibitors wherein X~ is Glu-Val-
Val-Arg-
Glu. Yet another aspect of the present invention provides protease inhibitors
wherein X2
is His. A further aspect of the present invention provides protease inhibitors
wherein XZ
is Gln. Another aspect of the present invention provides protease inhibitors
wherein X~
is Ala. Another aspect of the present invention provides protease inhibitors
wherein X~
is Asp.
Yet another aspect of the present invention provides protease inhibitors
wherein
X' is Asp. A further aspect of the present invention provides protease
inhibitors wherein
Xz is His. Another aspect of the present invention provides protease
inhibitors whettin
XZ is Gln. Another aspect of the present invention provides protease
inhibitors wherein
Xz is Ala. t~nother aspect of the present invention provides protease
inhibitors wherein
X= is Asp. Yet another aspect of the present invention provides protease
inhibitors
wherein X' is Glu. Another aspect of the present invention provides protease
inhibitors
wherein XZ is His. A further aspect of the present invention provides protease
inhibitors
wherein X~ is Gln. Another aspect of the present invention provides protease
inhibitors
wherein X~ is Ala. Another aspect of the present invention provides protease
inhibitors
wherein XZ is Asp. Another aspect of the present invention provides protease
inhibitors
comprising the sequence: Asp-Val-Val-Arg-Glu-Val-Cys-Ser-Glu-Gln-Ala-Glu-Thr-
Gly-Pro-Cys-Arg-Ala-Leu-Phe-Lys-Arg-Trp-Tyr-Phe-Asp-Val-Thr-Glu-Gly-Lys-G~s-
Ala-Pro-Phe-Phe-Tyr-Gly-Gly-Cys-Leu-Gly-Asp-Arg-Asn-Asn-Phe-Asp-Thr-Glu-Glu-
Tyr-Cys-Met-Ala-Val-Cys-Gly-Ser-Ala-Ile.
12
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
Other objects, features and advantages of the present invention will become
apparent from the following detailed description. It should be understood,
however, that
the detailed description and the specific examples, while indicating preferred
embodiments of the invention, are given by way of illustration only, since
various
changes and modifications within the spirit and scope of the invention will
become
apparent io those skilled in the art from this detailed description.
Brief Description of the Drawing
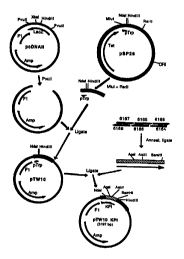
Figure 1 shows the strategy for the construction of plasmid pTW lO:KPI.
Figure 2 shows the sequence of the synthetic gene for KPI (1-r57) fused to the
bacterial phoA secretory signal sequence.
Figure 3 shows the strategy for construction of plasmid pKPI-61.
Figure 4 shows the 192 by XbaI-Hindl>I synthetic gene fragment encoding KPI
(1-X57) and four amino acids from yeast «-mating factor.
Figure 5 shows the synthetic 201 by XbaI-HindIII fragment encoding KPI
(-4-i57) in PKPI-61.
Figure 6 shows the strategy for the construction ofplasnud pTW113.
Figure 7 shows plasmid pTW113, encoding the 445 by synthetic gene for yeast
«-factor-KPI(-4-~57) fusion.
Figure 8 shows the amino acid sequence for KPI (-4-~57).
Figure 9 shows the strategy for constructing plasmid pTW6165.
Figure 10 shows piasmid, pTW6165, encoding the 445 by synthetic gene for
yeast «-factor-KPI(-4-~57; M15A, S17V~ fusion.
Figure I 1 shows the sequences of the annealed oligonucleotide pairs used to
construct plasmids pTW6165, pTW6166, pTW6175, pBG028, pTW6183, pTW6184,
pTW6185, pTW6I73, and pTW6174.
Figure 12 shows the sequence of plasmid pTW6166 encoding the fusion of yeast
«-factor and KPI(-4-~57; M15A, S171~.
Figure 13 shows~the sequence of plasmid pTW6175 encoding the fusion of yeast
«-factor and KPI(-4-~57; MISL, S17F).
Figure 14 shows the sequence of plasmid pBG028 encoding the fusion of yeast
«-factor and KPI(-4157; M15L, S17I~.
13
CA 02330191 2000-12-04
WO 99/63090 PCT/CfS99/12276
Figure 15 shows the sequence of plasmid pTW6183 encoding the fusion of yeast
x-factor and IkPI(-4-~ 57; I 16H, S 17F).
Figure 16 shows the sequence of plasmid pTW6184 encoding the fusion of yeast
x-factor and KPI(-4-~57; I1GH, S17Y).
Figure 17 shows the sequence of plasmid pTW6185 encoding the fusion of yeast
x-factor and KPI(-4-~57; I16H, S 17W).
Figure 18 shows the sequence of plasmid pTW6173 encoding the fusion of yeast
x-factor and KPI(-4-~57; M15A, I16H).
Figure i 9 shows the sequence of plasmid pTW6174 encoding the fusion of yeast
x-factor and KPI(-4-~57; M15L, I16H).
Figure 20 shows the sequence of plasmid pBG022 encoding the fusion of
yeast x-factor and KPI (-4-~57; M15A, S17Y, R18H, Y48H).
Figure 21 shows the sequence of plasmid pBG033 encoding the fusion of yeast
x-factor and KPI (-4-~57; T9V, hilSA, R18H, Y48H).
Figure 22 shows the sequence of plasmid pBG048 encoding the fusion of
yeast x-factor and KPI {-4-~57; Y48H).
Figure 23 shows the sequence of plasmid pBG049 encoding the fusion of
yeast x-factor and KPI (-4--~57; MISA, S17Y, R18H).
Figure 24 shows the sequence of piasmid pBG050 encoding the fusion of
yeast x-factor and KPI (-4-~57; T9V, M15A, S17Y, R18H).
Figure 25 shows the sequence of the coding region for phoA signal: KPI-
BG022: gIII protein contained within the phage display vector pDW 1-L6-16.
Figure 26 shows the sequence of the coding region for yeast x-factor and KPI-
P48 library contained within the P4.8 library.
Figure 27 shows the amino acid sequence of KPI (-4--X57; M15A, S17W).
Figure 28 shows the amino acid sequence of KPI (-4-X57; M15A, S17Y).
Figure 29 shows the amino acid sequence of KPI (-4--X57; M15L, S17F).
Figure 30 shows the amino acid sequence of KPI (-4--~57; M15L, S17Y).
Figure 31 shows the amino acid sequence of KPI (-4--~57; I16H, S17F).
Figure 32 shows the amino acid sequence of KPI (-4-X57; I16H, S17Y).
Figure 33 shows the amino acid sequence ofKPI (-4-X57; I16H, S17W).
Figure 34 shows the amino acid sequence of KPI (-4-~57; M15A, SI7F).
Figure 35 shows the amino acid sequence of KPI (-4-a57; M15A, I16H).
14
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
Figure 36 shows the amino acid sequence ofICPI (-4--~57; MISL, I16H).
Figure 37 shows the amino acid sequence of ICPI (-4-i57; M15A, S17Y,
RIBH, Y48H).
Figure 38 shows the amino acid sequence of ICPI (-4-X57; T9V, M15A, RIBH,
Y48H).
Figure 39 shows the amino acid sequence of KPI (-4-X57; Y48H).
Figure 40 shows the amino acid sequence of KPI (-4--57; MISA, S17Y,
R18H).
Figure 41 shows the amino acid sequence ofICPI (-4-X57; T9V, M15A, S17Y,
R18H).
Figure 42 shows the amino acid sequence of KPI-P48 library (-4-X57; M15A,
S 17Y, Rt 8H, Y28X) encoded by the P48 library.
Figure 43 shows the construction of plasmid pSP26:Amp:Fl.
Figure 44 shows the construction of plasmid pgllI.
Figure 45 shows the construction of plasmid pPhoA:KPI:gIlI.
Figure 46 shows the construction of plasmid pLGI.
Figure 47 shows the construction of plasmid pAL51.
Figure 48 shows the construction of plasmid pAL53.
Figure 49 shows the construction of plasmid pSP26:Amp:FI :PhoA:KPI:gIII.
Figure 50 shows the construction ofplasmid pDWI #14.
Figure 51 shows the construction of plasmid pBG022.
Figure 52 shows the construction of plasmid pBG048.
Figure 53 shows the construction of plasmid pBG049.
Figure 54 shows the construction of plasmid pBG050.
Figure 55 shows the construction of the P48 library.
Figure 56 shows the coding region for the fusion ofphoA-KPI (155)-geneIll.
Figure 57 shows the construction of plasmid pDW 1 14-2.
Figure 58 shows the construction of KPI Library ICrl9.
Figure 59 shows the expression unit encoded by the members of KPI Library 16-
19.
Figure 60 shows the phoA-ICPI(155)-geneItl region encoded by the most
frequently occurring randomized KPI region.
Figure 61 shows the construction ofpDD185 KPI (-4-X57; M15A, S17F).
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
Figure 62 shows the sequence of yeast x-factor fused to ICPI (-4-~57; M15A,
S 17F).
Figure 63 shows the inhibition constants (K;s) determined for purified KPI
variants against the selected serine proteases kallikrein, factor Xa, and
factor XIIa.
S Figure 64 shows the inhibition constants (K;s) determined for KPI variants
against kallikrein, plasmin, and factors Xa, XIa, and XIIa.
Figure 65 shows the post-surgical blood loss in pigs in the presence (KPI) and
absence (NS) of KPI 185-1 (M15A, S17F).
Figure 66 shows the post-surgical hemoglobin loss in pigs in the presence
(KPI)
and absence (NS) of KPI 185-1 (M15A, S17F).
Figure 67 shows the oxygen tension in the presence and absence of KPI, before
CPB, immediately after CPB, and at 60 and 180 minutes after the end of CPB.
Figure 68 summarizes the results shown in Figures 65-67.
Figure 69 shows the inhibitor constants (Kis) determined for KPI variants
against kallikrein in nM and expression levels (mg/ml) of those variants.
Figure 70 shows a comparison of the survival time of rat xenografts in the
presence and absence of KPI-BG022.
Figure 71 shows a comparison of damage in a rat model of TNBS
(trinitrobenzene sulfonic acid) induced colitis in the presence and absence of
KPI-
BG022.
Figure 72 shows a comparison of the HPLC traces, after lyophiiization, of KPI
having the N-tem~inus sequence Glu-Val-Val-Arg (E-KPI) and KPI having the N-
tecminus sequence Asp-Val-Val-Arg (D-KPI).
Detailed Description
The present invention provides peptides that can bind to and preferably
inhibit
the activity of serine proteases. These inhibitory peptides can also provide a
means of
ameliorating, treating or preventing clinical conditions associated with
increased activity
of serine proteases. The novel peptides of the present invention preferably
exhibit a
more potent and specific (i.e., greater) inhibitory effect toward serine
proteases of
interest than known serine protease inhibitors. Examples of such proteases
include:
kallikrein; chymotrypsins A and B; trypsin; elastase; subtilisin; coagulants
and
procoagulants, particularly those in active form, including coagulation
factors such as
16
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
factors VIIa, IXa, Xa, XIa, and. XIIa; plasmin; thrombin; proteinase-3;
enterokinase;
acrosin; cathepsin; urokinase; and tissue plasminogen acrivator. In
particular, the
peptides of the present invention preferably exhibit a greater potency and
specificity for
inhibiting one or more serine proteases of interest (e.g., ka11i1Qein, plasmin
and factors
VIIa, IXa, Xa, XIa, and XIIa) than the potency and specificity exhibited by
native KPI or
other known serine protease inhibitors. In addition, the peptides of the
present invention
preferably comprise a substitution at position 48. Such position 48
substituted peptides
may exhibit an increased level of expression in comparison to the expression
levels of
serine proteases that do not have that substitution. The effect of this
substitution may be
manifested not only on the substituted KPI peptides of the present invention,
but on
wild-type KPI as well. Also, the peptides of the present invention that
comprise the N-
temunal sequence Glu-Val-Val-Arg (residues -4 to -1 ) may also preferably
exhibit
increased yields via a substitution to Asp-Val-Val-Arg.
Peptides of the present invention may be used to reduce the tissue damage
caused by activation of the proteases of the contact pathway of the blood
doting surgical
procedures such as cardiopulmonary bypass (CPB). Inhibition of contact pathway
proteases reduces the 'whole body inflammatory response" that can accompany
contact
pathway activation, and that can lead to tissue damage, and possibly death.
The peptides
of the present invention may also be used in conjunction with surgical
procedures to
ZO roduce activated seiine protease-associated perioperative and postoperative
blood loss.
For instance, perioperative blood loss of this type may be particularly severe
during CPB
surgery. Pharmaceutical compositions comprising the peptides of the present
invention
may be used in conjunction with surgery such as CPB; administration of such
compositions may occur preoperatively, perioperatively or postoperatively.
Examples of
other clinical conditions associated with increased serine protease activity
for which the
peptides of the present invention may be used include: CPB-induced
inflammatory
response; post-CPB pulmonary injury; pancreatitis; allergy-induced protease
release;
deep vein thrombosis; thrombocytoptnia; rheumatoid arthritis; adult
respiratory distress
syndrome; chronic inflammatory bowel disease; psoriasis; hyperfibrinolytic
hemorrhage;
organ preservation; wound healing; and myocardial infarction. Other examples
of
preferable uses of the peptides of the present invention are described in U.S.
Patent No.
5,187,153.
The invention is based upon the novel substitution of amino acid residues in
the
peptide corresponding to the naturally occurring KPI protease inhibitor domain
of
17
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
human arnyloid p-amyloid precursor protein (APPn. These substitutions produce
peptides that can bind to serine proteases and preferably exhibit an
inhibition of the
activity of serine proteases. The peptides also preferably exhibit a more
potent aad
specific serine protease inhibition than known serine protease inhibitors. In
accordance
with the invention, peptides are provided that may exhibit a more potent and
specific
inhibition of one or more serine proteases of interest, e.g., kallila~ein,
plasmin and factors
Xa, XIa, XIIa, and XIIa.
The present invention also includes pharmaceutical compositions comprising an
effective amount of at least one of the peptides of the invention, in
combination with a
pharmaceutically acceptable sterile vehicle, as described in REMINGTONS
PHARMACEUTICAL SCIENCES: DRUG RECEPTORS AND RECEPTOR
THEORY, (18th ed.), Mack Publishing Co., Easton, PA (1990).
A. Selection of sequences of KPI variants
The sequence of KPI is shown in Table 1. Table 2 shows a comparison of this
sequence with that of aprotinin, with which it shares about 45% sequence
identity. The
numbering convention for KPI shown in Table 1 and used hereinafter designates
the first
glutamic acid residue of KPI as residue 1. This corresponds to residue number
3 using
the standard numbering convention for aprotinin.
The crystal structure for KPI complexed with trypsin has been determined. See
Perona et al., J. Mol. Biol. 230:919 (1993). The three-dimensional structure
reveals two
binding loops within KPI that contact the protease. The first loop extends
from residue
Thr9 to Ile~6, and the second loop extends from residue Phe32 to Gly3~. The
two protease
binding loops are joined through the disulfide bridge extending from Cys~Z t0
Cys36.
KPI contains two other disulfide bridges, between Cys3 and Cys33, and between
Cys~ to
Cys°9. '
This structure was used as a guide to inform our strategy for making the amino
acid residue substitutions that will be most likely to affect the protease
inhibitory
properties of KPI. Our examination of the structure indicated that certain
amino acid
residues, including residues 9, 11, 13-18, 32, and 37-40 appear to be of
particular
significance in determining the protease binding properties of the KPI
peptides of the
present invention. It was also found that certain position 48 substitutions
positively
affected the expression levels of the peptide by the transformed host. In a
preferred
18
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
embodiment of the invention one or more of those KPI peptide residues are
substituted,
such substitutions preferably occurring among residues 9, 11, I3-18, 32, 37-
40, and 48.
In particular, those substituted peptides, including peptides comprising
substitutions at
position 9, substitutions of at least two of the four residues at positions IS-
18 and
substitutions at position 48 may exhibit more potent and specific serine
protease
inhibition toward selected serine proteases of interest than exhibited by the
natural KPI
peptide domain.
Specifically, replacement of arginine at position 18 of the native KPI peptide
with histidine (R18H) in combination with one or more additional substitutions
at
residues 9, 15 and 17 were found to exhibit more potent and specific setine
protease
inhibition toward selected serine proteases of interest than the native KPI
peptide. In
particular, the specific substitutions T9V, M15A, S17Y and M15A ,S17Y in the
context
of the R18H substitution exhibited such potent serine protease inhibition. See
Figures
63, 64 and 69D.
In addition, the peptides of the present invention preferably comprise a
substitution at position 48. Such position 48 substituted peptides may exhibit
an
increased level of expression in comparison to the expression levels of serine
proteases
that do not have that substitution. The effect of this substitution may be
manifested not
only on the substituted KPI peptides of the present invention, but on wild-
type KPI as
well. Also, the peptides of the present invention that comprise the N-terminal
soquenee
Glu-Val-Val-Arg (residues -4 to -1) may also preferably exhibit increased
yields via a
substitution to Asp-Val-Val-Arg.
Specifically, substitutions at position 48 may exhibit an increased level of
expression of KPI peptides in comparison to the expression levels of such
peptides not
having such a substitution. These substituted peptides exhibiting an increased
level of
expression also may preferably comprise one or more additional substitutions
at residues
9, 11, 13-18, 32 and 37-40; in particular, such peptides may preferably
comprise a
substitution at positions 9 or 37 and/or substitution of at least two of the
four residua
at positions 15-18. Those additionally substituted peptides may exhibit more
potent and
specific setine protease inhibition toward selected serine proteases of
interest than
exhibited by the natural KPI peptide domain as well as increased expression
levels.
One specific embodiment of the invention is based upon a finding that as
expression vector prepared to express the KPI variant M15A, S17Y, R18H
underwent
a spontaneous mutation at position 48 which changed the native tyrosine to
histidine
19
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
(Y48H) and that this mutation conferred beneficial properties. To assess the
effect of
this mutation, the KPI variant M1 SA, S 17Y, Rl 8H (pBG049) was constructed
using
methods known to those skilled in the art and its expression levels compared
with the
KPI variant M15A, S17Y, R18H, Y48H (pBG022). As detailed infra, the expression
level of KPI variant M15A, S17Y, R18H was increased over five-fold by
replacing
the native tyrosine at position 48 with histidine. See Figures 69A aad B.
Moreover, it
has been determined that this Y48H substitution confers improvements in
expression
levels upon KPI variants as well as upon native sequence KPI.
As an additional example of the position 48 substitution effect on expression
of the recombinant peptides of the present invention, and as delineated in
detail infra,
the expression level of wild-type KPI (pTW 113) was increased on the average
approximately five to six-fold by replacing the native tyrosine at position 48
with
histidine (pBG048;Y48H), glutamine (pBG072; Y48Q) or alanine (pBG073; Y48A).
See Figures 69B and F.
In an additional preferred embodiment of the invention, it was found that
replacement of arginine at position 18 of the native KPI peptide with
histidine
(R18H), in combination with one or more additional substitutions at residues
9, 15 aad
17, exhibited more potent and specific serine protease inhibition toward
selected serine
proteases of interest than the native KPI peptide. In particular, the specific
substitutions
T9V, M15A, S17Y and M15A; S17Y exhibited particularly potent serine protease
inhibition in the context of the R18H substitution. See Figures 63 and 64.
Additionally,
the R18H substitution conferred an increased level of expression in comparison
to tire
expression levels of the corresponding peptides lacking the position 48
substitution. To
assess the effect of the position 48 substitution on these R18H substituted
peptides,
Library P48 was constructed for expression of KPI (M15A, S17Y, R18H) in which
the
amino acids exhibiting at position 48 are randomized. See Figure 55. The amino
acid
sequences of the KPI-P48 Library contained within the P48 Library are shown in
Figure
26. Those substituted peptides included substituting the native tyrosine at
position 48
with histidine (pBG022; SOD4, SOB6,Y48H), glutamine (SOB6, SOL1, SOM1; Y48Q),
alanine (SOPS, SOC4; Y48A) and aspartic acid (SON1; Y48D). See Figures 69B, E
and F.
In yet another preferred embodiment, the KPI peptides of the present invention
may also comprise a substitution at its N-terminus. Specifically, such a
substitution was
found to alleviate the problems associated with the purification and
subsequent isolation
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
of the expressed peptides of the present invention having a giutamic acid
residue at its N-
terminus. This specific substitution changes the additional N-terminal amino
acids from
the ICPI protein sequence (Glu-Val-Val-Arg, designated residues -4 to -1)
immediately
proceeding the KPI domain in APPI to Asp-Val-Val-Atg. Specifically, this
substitution
is thought to prevent cyclization of the N-terminus glutamic acid during
purification of
. the expressed peptides of the present invention. In a preferred embodiment
of the
invention, and as described supra, one or more additional KPI peptide residues
are
substituted, such substitutions preferably occurring among residues 9, 11, 13-
18, 32, 37
40, and 48. in particular, those substituted peptides, including peptides
comprising
substitutions at position 9, substitutions of at least two of the four
residues at positions
15-18 and substitutions at position 48 preferably exhibit the desired potency
and
specificity as well as an increased level of expression in comparison to the
expression
levels of other serine pretenses W thout those specific substitutions.
In particular, the peptides of the present invention preferably exhibit a
greater
potency and specificity for inhibiting one or more serine pretenses of
interest (eg.,
kallila~ein, plasmin and factors VIIa, IXa, Xa, XIa, and XIIa) than the
potency and
specificity exhibited by native ICPI or other known serine protease inhibitors
as well as
an increased level of expression in comparison to the~expression levels of
other setine
pmteases without those specific substitutions. That greater potency and
specificity may
be manifested by the peptides of the present invention by exhibiting binding
constants
for serine pretenses of interest that are less than the binding constants
exhibited by native
KPI, or other known serine protease inhibitors, for such proteases.
By way of example, and as set forth in greater detail below, the serine
protease
inhibitory properties of peptides of the present invention were measured for
the serine
proteases of interest kallikrein, piasmin and factors Xa, XIa, and XIIa.
Methodologies
for measuring the inhibitory properties of the KPI variants of the present
invention are
known to those skilled in the art, e.g., by determining the inhibition
constants of the
variants toward serine pretenses of interest, as described in Example 4,
infra. Such
studies measure the ability of the novel peptides of the present invention to
bind to one
or more serine pretenses of interest and to preferably exhibit a greater
potency and
specificity for inhibiting one or more serirte protease of interest than known
setine
protease inhibitors such as native KPI.
The ability of the peptides of the present invention to bind one or more
serine
pretenses of interest, particularly the ability of the peptides to exhibit
such greater
21
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
potency and specificity toward serine proteases of interest, manifest the
clinical and
therapeutic applications of such peptides. The clinical and therapeutic
efficacy of the
peptides of the present invention can be assayed by in vitro and in vivo
methodologies
lrnown to those skilled in the art, e.g., as described in Examples 5-8, infra.
Tabte 1: SEQUENCE OF KPI:
1 10 20 30
VREVCSEQAETGPCRAMISRWYFDVTEGKCAP
40 50
FFYGGCGGNRNNFDTEEYCMAVCGSAI
Table 2: COMPARISON OF KPI AND APROTININ SEQUENCES:
1 10 20 30 40 50
KPI: VREVCSEQAETGPCRAMISRWYFDVTEGKCAPF~,NRNNFDTEEYCMAVCGSAI
BP'TI: RPDFCLEPPYTGPCKARIIRYFYNAKA GL CQTFV~'Q~CRNNFfCSAEDCI~TCGGA
1 10 20 30 40 50
B. Methods of producing KPI variants
The peptides of the present invention can be created by synthetic techniques
or
recombinant techniques which employ genomic or cDNA cloning methods.
1. Production by chemical synthesis
Peptides of the present invention can be routinely synthesized using solid
phase
or solution phase peptide synthesis. Methods of preparing relatively short
peptides such
as KPI by chemical synthesis are well known in the art. ICPI variants could,
for example
be produced by solid-phase peptide synthesis techniques using commercially
available
equipment and reagents such as those available from Milligen (Bedford, MA) or
Applied
Biosystems-Perkin Elmer (Foster City, CA). Alternatively, segments of KPI
variants
could be prepared by solid-phase synthesis and linked together using segment
condensation methods such as those described by Dawson et al., Science 266:776
22
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
(1994). During chemical synthesis of the KPI variants, substitution of any
amino acid
can be achieved simply by replacement of the residue that is to be substituted
with a
different amino acid monomer.
2. Production by recombinant DNA technology
(a) Preparation ojgenes encoding KPI variants
In a preferred embodiment of the invention, KPI variants are produced by
recombinant DNA technology. See PCT application WO 96/35788, hereby
incorporated
in its entirety. This requires the preparation of genes encoding each ICPI
variant that is to
be made. Suitable genes can be constructed by oligonucleotide synthesis using
commercially available equipment, such as that provided by Milligen and
Applied
Biosystems, supra. The genes can be prepared by synthesizing the entire coding
and
non-coding strands, followed by annealing the two strands. Alternatively, the
genes can
be prepared by ligation of smaller synthetic oligonucleotides by methods well
known in
the art. Genes encoding ICPI variants are produced by varying the nucleotides
introduced at any step of the synthesis to change the amino acid sequence
encoded by
the gene.
Preferably, however, KPI variants are made by site-directed mutagenesis of a
gene encoding KPI. Methods of site-directed mutagenesis are well known in the
art.
See, for example, Ausubel et al., (eds.) CURRENT PROTOCOLS IN MOLECULAR
BIOLOGY (Wiley Interscience, 1987); PROTEIN ENGINEERING (Oxender & Fox
eds., A. Liss, Inc. 1987). These methods require the availability of a gene
encoding KPI
or a variant thereof, which can then be mutagenized by known methods to
produce the
desired ICPI vaaants. In addition, linker-scanning and polymerase chain
reaction
("PCR'~ mediated techniques can be used for purposes of mutagenesis. See PCR
TECHNOLOGY (Erlich cd., Stockton Press 1989); CURRENT PROTOCOLS IN
MOLECLTL,AR BIOLOGY, vols. 1 & 2, loc. cit.
A gene encoding KPI can be obtained by cloning the naturally occurring gene,
as
described for example in U.S. Patents Nos. 5,223,482 and 5,187,153, which are
hereby
incorporated by reference in their entireties. In particular, see columns 6-9
of U.S.
Patent No. 5,187,153. See aLro PCT Application No. 93/09233. In a nreferned
embodiment of the invention a synthetic gene encoding KPI is produced by
chemical
synthesis, as described above. The gene may encode the 57-amino acid KPI
domain
shown in Table 1, or it may also encode additional N-terminal amino acids from
the
23
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
APPI protein sequence, such as the four amino acid sequence (Glu-Val-Val-Arg
or Asp-
Val-Val-Arg, designated residues -4 to -I) immediately preceding the ICPI
domain in
APPI.
Production of the gene by synthesis allows the codon usage of the KPI gene to
be
altered to introduce convenient restriction endonuciease recognition sites,
without
altering the sequence of the encoded peptide. In a preferred embodiment of the
invention, the synthetic ICPI gene contains restriction endonuclease
recognition sites that
facilitate excision of DNA cassettes from the KPI gene. These cassettes can be
replaced
with small synthetic oligonucleotides encoding the desired changes in the KPI
peptide
sequence. See Ausubel, supra.
This method also allows the production of genes encoding KPI as a fusion
peptide with one or more additional peptide or protein sequences. The DNA
encoding
these additional sequences is arranged in-frame with the sequence encoding KPI
such
that, upon translation of the gene, a fusion protein of KPI and the additional
peptide or
protein sequence is produced. Methods of making such fusion proteins are well
known
in the art. Examples of additional peptide sequences that can be encoded in
the genes
are secretory signal peptide sequences, such as bacterial leader sequences,
for example
ompA and phoA, that direct sxretion of proteins to the bacterial periplasmic
space. Ia a
preferred embodiment of the invention, the additional peptide sequence is a
yeast
secretory signal sequence, such as a-mating factor, that directs secretion of
the peptide
when produced in yeast.
Additional genetic regulatory sequences can also be introduced into the
synthetic
gene that are operably linked to the coding soquence of the gene, thereby
allowing
synthesis of the protein encoded by the gene when the gene is introduced into
a host cell.
Examples of regulatory genetic sequences that can be introduced are: promoter
attd
eahancer sequences and transcriptional and translational control sequences.
Other
regulatory sequences are well known in the art. See Ausubel et al., supra, and
Sambrook
et al., supra.
Sequences encoding other fusion proteins and genetic elements are well known
to those of skill in the art. In a preferred embodiment of the invention, the
ICPI sequence
is prepared by ligating together synthetic oligonucleotides to produce a gene
encoding an
in-frame fusion protein of yeast a-mating factor with either KPI (1-i57) or
KPI
(-4-i ST).
24
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
The gene constructs prepared as described above are conveniently manipulated
in host cells using methods of manipulating recombinant DNA techniques that
are well
known in the art. See, for example Sambrook et al., MOLECULAR CLONING: A
LABORATORY MANUAL, Second Edition, (Cold Spring Harbor Laboratory Press,
Cold Spring Harbor, NY 1989), and Ausubel, supra. In a preferred embodiment of
the
invention the host cell used for manipulating the KPI constructs is E. toll.
For example,
the construct can be ligated into a cloning vector and propagated in E. toll
by methods
that are well known in the art. Suitable cloning vectors are described in
Sambrook,
supra, or are commercially available from suppliers such as Promega (Madison,
WI),
Stratagene (San Diego, CA) and Life Technologies (Gaithersburg, MD).
Once a gene construct encoding KPI has been obtained, genes encoding KPI
variants are obtained by manipulating the coding sequence of the construct by
standard
methods of site-directed mutagenesis, such as excision and replacement of
small DNA
cassettes, as described supra. See Ausubel, supra, and Sinha et al., supra.
See also U.S.
I S Patent 5,373,090, which is herein incorporated by reference in its
entirety. See
particularly, columns 4-12 of U.S. Patent 5,272,090. These genes are then used
to
produce the KPI variant peptides as described below.
Alternatively, KPI variants can be produced using phage display methods. See,
for example, Dennis et al., supra, which is hereby incorporated by reference
in its
entirety. See also U.S. Patent Nos. 5,223,409 and 5,403,484, which are hereby
also
incorporated by reference in their entireties. In these methods, libraries of
genes
encoding variants of KPI are fused in-frame to genes encoding surface proteins
of
Slamentous phage, and the resulting peptides are expressed (displayed) on the
surface of
the phage. The phage are then screened for the ability to bind, under
appropriate
conditions, to serine proteases of interest immobilized on a solid support.
Large libraries
of phage can be used, allowing simultaneous screening of the binding
properties of a
large number of KPI variants. Phage that have desirable binding properties are
isolated
and the sequences of the genes encoding the corresponding KPI variants is
determined.
These genes are then used to pmduce the KPI variant peptides as described
below.
(b) Expression ofKPl variant peptides
Once genes encoding KPI variants have bcen prepared, they are inserted into an
expression vector and used to produce the recombinant peptide. Suitable
expression
vectors and corresponding methods of expressing recombinant proteins and
peptides are
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/1ZZ76
well known in the art. Methods of expressing KPI peptides are described in
U.S. Patent
5,187,153, columns 9-11, U.S. Patent 5,223,482, columns 9-11, PCT application
93/09233, pp. 49-67, and PCT application 96/35788, pp. 31-33. See also Ausubel
et al.,
supra, and Sambrook et al., supra. The gene can be expressed in any number of
different recombinant DNA expression systems to generate large amounts of the
KPI
variant, which can then be purified and tested for its ability to bind to and
inhibit serine
proteascs of interest. Within the context of the present invention,
substitutions at position
48 may exhibit an increased level of expression of KPI peptides, both wild-
type and
substituted, in comparison to the expression levels of such peptides not
having such a
substitution. Such peptides having a substitution at position 48 also may
preferably
comprise one or more additional substitutions at residues 9, 11, 13-18, 32 and
37-40; in
particular, such peptides may preferably comprise a substitution at positions
9. or 37
and/or substitution of at least two of the four residues at positions 15-18.
Those
additionally substituted peptides may exhibit more potent and specific serine
protease
inhibition toward selected serine proteases of interest than exhibited by the
natural KPI
peptide domain as well as increased expression levels.
In particular, replacement of arginine at position 18 of the native KPI
peptide
with histidine (R18H) in combination with one or more additional substitutions
at
residues 9, 15 and 17 was found to exhibit more potent and specific serine
protease
inhibition toward selected serine proteases of interest than the native KPI
peptide. In
particular, the specific substitutions T9V, M15A, S17Y and M15A, S17Y in the
context
of the R18H substitution exhibited such potent serine protease inhibition.
Placing this
position 48 substitution in such substituted peptides resulted in increased
expression
levels of these peptides in comparison to the expression levels of the
peptides without
the position 48 substitution.
Examples of expression systems lrnown to the skilled practitioner in the art
include bacteria such as E. colt, yeast such as Saccharamyces cerevisiae and
Pichia
pastoris, baculovirus, and mammalian expression systems such as in Cos or CHO
cells.
In a preferred embodiment, KPI variants are expressed in S cerevisiae. In
another
preferred embodiment the KPI variants are cloned into expression vectors to
produce a
chimeric gene encoding a fusion protein of the KPI variant with yeast a-mating
factor.
The mating factor acts as a signal sequence to direct secretion of the fusion
pmtein from
the yeast cell, and is then cleaved from the fusion protein by a membrane-
bound protease
26
CA 02330191 2000-12-04
WO 99/63090 PIrT/US99/12276
during the secretion process. The expression vector is transformed into S.
cereviriae, the
transformed yeast cells are cultured by standard methods, and the KPI variant
is purified
from the yeast growth medium.
Recombinant bacterial cells expressing the peptides of the present invention,
for
example, E. colt, are grown in any of a number of suitable media, for example
LB, and
the expression of the recombinant antigen induced by adding IPTG to the media
or
switching incubation to a higher temperature. After culturing the bacteria for
a ftuther
period of between 2 and 24 hours, the cells are collected by centrifugation
and washed to
remove residual media. The bacterial calls are then lysed, for example, by
disruption in
a cell homogenizer and centrifuged to separate dense inclusion bodies and cell
membranes from the soluble cell components. This centrifugation can be
performed
under conditions whereby dense inclusion bodies are selectively enriched by
incorporation of sugars such as sucrose into the buffer and centrifugation at
a selective
speed. If the recombinant peptide is expressed in inclusion bodies, as is the
case in many
instances, these can be washed in any of several solutions to assist in the
removal of any
contaminating host proteins, then solubilized in solutions containing high
concentrations
of urea (e.g., SM) or chaotropic agents such as guanidine hydrochloride in the
presence
of reducing agents such as (i-mercaptoethanol or DTT (dithiothreitol).
At this stage it may be advantageous to incubate the peptides of the present
invention for several hours under conditions suitable for the peptides to
undergo a
refolding process into a conformation which more closely resembles that of
native KPI.
Such conditions generally include low protein concentrations less than 500
uglml, low
levels of reducing agent, concentrations of urea less than 2M and often the
presence of
reagents such as a mixture of reduced and oxidized glutathione which
facilitate the
interchange of disulphide bonds within the protein molecule. The refolding
process can
be monitored, for example, by SDS-PAGE or with antibodies, which are specific
for the
native molecule (which can be obtained from animals vaccinated with the native
molecule isolated from parasites). Following refolding, the peptide can then
be purif ed
ftuther and separated from the refolding mixture by chromatography on any of
several
supports including ion exchange resins, gel permeation resins or on a variety
of affinity
columns.
Purification of KPI variants can be achieved by standard methods of protein
purification, e.g., using various chromatographic methods including high
performance
27
CA 02330191 2000-12-04
WO 99/63090 PCT/US99112276
liquid chromatography and adsorption chromatography. The purity and the
quality of
the peptides can be confirmed by ami.-~o acid analyses, molecular weight
determination,
sequence determination and mass spectrometry. See, for example, PROTEIN
PURIFICATION METHODS: A PRACTICAL APPROACH, Hams et a1, eds. (IRL
Press, Oxford, 1989). In a preferred embodiment, the yeast cells are removed
from the
growth medium by filtration or centrifugation, and the ICPI variant is
purified by amity
chromatography on a column of trypsin-agamse, followed by reversed-phase HPLC.
In yet another embodiment, the ICPI peptides of the present invention may also
comprise a substitution at its N-terminus. Placing the amino acid sequence Asp-
Val
Val-Arg (designated residues -4 to -I ) immediately before the ICPI domain was
found
to alleviate the problems associated with the purification and subsequent
isolation of
the expressed peptides of the present invention having a glutamic acid residue
at its
N-terminus. In a preferred embodiment, this substitution changes the
additional N-
terminaI amino acids from the KPI protein sequence (Glu-Val-Val-Arg,
designated
residues -It to -I) immediately proceeding the ICPI domain to Asp-Val-Val-Arg.
Specifically, this substitution is thought to prevent cyclization of the N-
terminus
glutamic acid in the unsubstituted variant during purification of the
expressed
peptides of the present invention and is thought to be applicable to the
substituted KPI
variants of the present invention, as well as wild-type KPI. By way of
example,
Figure 72 provides a comparison of the HPLC traces, aRer lyophilization, of
KPI having
the N-terminal sequence Glu-Val-Val-Arg (E-KPI) and ICPI having the N-terminus
sequence Asp-Val-Val-Arg (D-ICPI). Those ICPI samples were injected onto a YMC-
Phenyl HPLC column (Cat. No.:PH12S030504WTA, 4x50 mm, 3p particle size, 50
angstrom pore size). Mobile Phase A was 40 nM ammonium phosphate (pH 6.5),
10% acetonitrile, and 90% water. Mobile Phase B was 40 nM ammonium phosphate
(pH 6.5), 60% acetonitrile, and 40% water. The KPI-185 elution point was at
approximately 21% acetonitrile. The HPLC of E-KPI exhibits an additional peak
after 10 minutes, which is the product of the cyclization of the N-temunus
glutamic
acid of E-ICPI. The HPLC of D-KPI exhibits no such peak and thus no such
cyclization pmduct.
C. Measurement of protease inhibitory properties of KPI variants
Once ICPI variants have been purified, they are tested for their ability to
bind to
and inhibit serine proteases of interest in vitro. The peptides of the present
invention
28
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
preferably exhibit a more potent and specific inhibition of serine proteases
of interest
than known serine protease inhibitors, such as the natural ICPI peptide
dcmain. Such
binding and inhibition can be assayed for by determining the inhibition
constants for the
peptides of the present invention toward serine proteases of interest and
comparing those
constants with constants determined for known serine protease inhibitors,
e.g., the native
ICPI domain, toward those proteases. Methods for determining inhibition
constants of
protease inhibitors are well known in the art. See Fersht, ENZYME STRUCT(JRE
AND MECHANISM, 2nd ed., W.H. Freeman and Co., New York, ( i 985).
In a preferred embodiment the inhibition experiments are carried out using a
chromogcnic synthetic protease substrate, as described, for example, in Render
et al., J.
Amer. Chem. Soc. 88:5890 (1966). Measurements taken by this method can be used
to
calculate inhibition constants (K; values) of the peptides of the present
invention toward
serine proteases of interest. See Bieth in BAYER-SYMPOSIUM V "PROTEINASE
INHIBITORS", Fritz et al., eds., pp. 463-69, Springer-Verlag, Berlin,
Heidelberg, New
York, (1974). ICPI variants that exhibit potent and specific inhibition of one
or more
. serine proteases of interest may subsequently be tested in vivo. In vitro
testing, however,
is not a prerequisite for in vivo studies of the peptides of the present
invention.
D. Testing of KPI variants in vivo
The peptides of the present invention may be tested, alone or in combination,
for
their therapeutic efficacy by various in vivo methodologies known to those
skilled in the
art, e.g., the ability of ICPI variants to reduce postoperative bleeding can
be tested in
standard animal models. For example, cardiopulmonary bypass surgery can be
carried
out on animals such as pigs in the presence of ICPI variants, or in control
animals where
the ICPI variant is not used. The use of pigs as a model for studying the
clinical effects
associated with CPB has previously been described. See Redmond et al., Ann.
ThoraG
Surg. 56:474 (1993).
The ICPI variant is supplied to the animals in a pharmaceutical sterile
vehicle by
methods known in the art, for example by continuous intravenous infusion. Chat
tubes
can be used to collect shed blood for a defined period of time. The shed
blood, together
with the residual intrathoracic blood found after sacrifice of the animal can
be usod to
calculate hemoglobin (Hgb) loss. The postoperative blood and Hgb loss is then
compared between the test and control animals to detemnirte the effect of the
ICPI
vanants.
29
CA 02330191 2000-12-04
WO 99/63090 PCTNS99/12276
E. Therapeutic use of KPI variants
ICPI variants of the present invention found to exhibit therapeutic efficacy
(eg.,
reduction of blood loss following surgery in animal models) may preferably be
used anti
administered, alone or in combination or as a fusion protein, in a manner
analogous to
that currently used for aprotinin or other known serine protease inhibitors.
See Butler et
aL, supra. Peptides of the present invention generally may be administered in
the
manner that natural peptides are administered. A therapeutically effective
dose of the
peptides of the present invention preferably affects the activity of the
serine proteases of
interest such that the clinical condition may be treated, ameliorated or
prevented
Therapeutically effective dosages of the peptides of the present invention can
be
determined by those skilled in the art, e.g., through in vivo or in vitro
models. Generally,
the peptides of the present invention may be administered in total amounts of
approximately 0.01 to approximately 500, specifically 0.1 to 100 mg/kg body
weight, if
desired in the form of one or more administrations, to achieve therapeutic
effect. It may,
however, be necessary to deviate from such administration amounts, in
particular
depending on the nature and body weight of the individual to be treated, the
nature of the
medical condition to be treated, the type of preparation and the
administration of the
peptide, and the time interval over which such administration occurs. Thus, it
may in
some cases be sufficient to use less than the above amount of the peptides of
the present
invention, while in other cases the above amount is preferably exceeded. The
optimal
dose required in each case and the type of administration of the peptides of
the present
invention can be determined by one skilled in the art in view of the
circumstances
surrounding such administration. Such peptides can be administered by
intravenous
injections, in situ injections, local applications, inhalation, oral
administration using
coated polymers, dermal patches or other appropriate means. Compositions
comprising
peptides of the present invention are advantageously administered in the form
of
injectable compositions. Such peptides may be preferably administered to
patients via
continuous intiavtnous infusion, but can also be administered by single or
multiple
injections. A typical composition for such purpose comprises a
pharmaceutically
acceptable carrier. Pharmaceutically acceptable carriers include aqueous
solutions, non-
toxic excipients, including salts, preservatives, buffers and the like, as
described in
REMIT1GTONS PF-IARMACEUTICAL SCIENCES, pp. 1405-12 and 1461-87 (1975)
and THE NATIONAL FORMULARY XIV., 14th Ed. Washington: American
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
Pharmaceutical Association ( 1975). Aqueous carriers include water,
alcoholic/aqueous
solutions, saline solutions, parenteral vehicles such as sodium chloride,
Ringer's
dextrose, etc. Intravenous vehicles include fluid and nutrient replenishers.
Preservatives
include antimicrobials, anti-oxidants, chelating agents and inert gases. The
pH and exact
concentration of the various components of the composition are adjusted
according to
routine skills in the art. See GOODMAN AND GILMANS 'ITS
PHARMACOLOGICAL BASIS FOR THERAPEUTICS (7th ed.). The peptides of the
present invention may be present in such pharmaceutical preparations in a
concentration
of approximately 0.1 to 99.5% by weight, specifically 0.5 to 95% by weight,
relative to
the total mixture. Such pharmaceutical preparations may also comprise other
pharmaceutically active substances in addition to the peptides of the present
invention.
Other methods of delivering the peptides to patients will be readily apparent
to the
skilled artisan.
Examples of mammalian serine proteases that may exhibit inhibition by the
peptides of the present invention include: kallikrein; chymotrypsins A and B;
trypsin;
elastase; subtilisin; coagulants and procoagulants, particularly those in
active form,
including coagulation factors such as thrombin and factors V>Za, IXa, Xa, XIa,
and XBa;
plasmin; proteinase-3; enterokinase; acrosin; cathepsin; urokinase; and tissue
plasminogen activator. Examples of conditions associated with increased serine
protease
activity include: CPB-induced inflammatory response; post-CPB pulmonary
injury;
pancreatitis; allergy-induced protease release; deep vein thrombosis;
thrombocytopenia;
rheumatoid arthritis; adult respiratory distress syndrome; chronic
inflammatory bowel
disease; psoriasis; hyperfibtinolytic hemorrhage; organ preservation; wound
healing; and
myocardial infarction. Other examples of the use of the peptides of the
present invention
are described in U.S. Patent No. 5,1$7,153.
The inhibitors of the present invention may also be used for inhibition of
serine
protease activity in vitro, for example during the preparation of cellular
extracts to
prevent degradation of cellular proteins. For this purpose the inhibitors of
the preseat
invention may preferably be used in a manner analogous to the way that
aprotinin, or
other known serine protease inhibitors, are used. The use of aprotinin as a
protease
inhibitor for preparation of cellular extracts is well known in the art, and
aprotinin is sold
commercially for this purpose.
31
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
The present invention, thus generally described, will be understood more
readily
by reference to the following examples, which are provided by way of
illustration and
are nut intended to be limiting of the present invention.
EXAMPLES
Example I. Expression of wild-type KPI (-4-~5'n
A. Construction of pTWIO:KPl
Plasmid pTWIO:KPI is a bacterial expression vector encoding the 57 amino acid
form of ICPI fused to the bacterial phoA signal sequence. The strategy for the
construction of pTW l O:ICPI is shown in Figure 1.
Plasmid pcDNAII (Invitrogen, San Diego, CA) was digested with PvuII and the
larger of the two resulting PvuII fragments (3013 bp) was isolated. Bacterial
expression
plasmid pSP26 was digested with MIuI and RrrII, and the 409 by MIuI-RsrII
fragment
containing the pTrp promoter element and transcription termination signals was
isolated
by electrophoresis in a 3% NuSieve Agarose geI (FMC Corp., Rockland, ME).
Plasmid
pSP26, containing a heparin-binding EGF-tike growth factor (I~-EGF) insert
between
the NdeI and HindllI sites, is described as pNA28 in Thompson et aL, J. Biol.
Chem.
269:2541 (1994). Plasmid pSP26 was deposited in host E. coli W3110, pSP26 with
the
American Type Culture Collection (ATCC), 10801 University Boulevard,
Mantissas,
Virginia 20110-2209, USA under the conditions specified by the Budapest Treaty
on the
International Recognition of the Deposit of Microorganisms (Budapest Treaty).
Host E.
coli W3110, pSP26 was deposited on 3 May 1995 and given Accession No. 69$00.
Availability of the deposited plasmid is not to be construed as a license to
practice the
invention in contravention of the rights granted under the authority of any
government in
accordance with its patent laws.
The ends of the MIuI-RsrII fragment were blunted using DNA polymerise
Klenow fragment by standard techniques. The blunted fiagrrtent of pSP26 was
then
ligated into the large PvuII fragment of plasmid pCDNAII, and the ligation
mixture was
used to transform E. coli strain MC1061. Ampicillin-resistant colonies were
selected
and used to isolate plasmid pTW 10 by standard techniques.
A synthetic gene was constructed encoding the bacterial phoA secretory signal
sequence fused to the amino terminus of KPI(1-X57). The synthetic gene
contains
cohesive ends for NdeI and HindBI, and also incorporates restriction
endonuclease
recognition sites for AgeI, RrrII, AatII and BamHI, as shown in Figure 2. The
synthetic
32
CA 02330191 2000-12-04
WO 99!63090 PCT/US99/12276
phoA-KPI gene was constructed from 6 oligonucleotides of the following
sequences
(shown 5'-3'):
S 6167:
TATGAAACAAAGCACTATTGCACTGGCACTCTTACCGTTACTGTTTAC
CCCTGTGACAAAAGCCGAGGTGTGCTCTGAA
6169:
CTCGGCTTTTGTCACAGGGGTAAACAGTAACGGTAAGAGTGCCAGTG
CAATAGTGCTTTGTTTCATA
6165:
CAAGCTGAGACCGGTCCGTGCCGTGCAATGATCTCCCGCTGGTACTTT
GACGTCACTGAAGGT__AAGTGCGCTCCATTCTTT
6166:
GCACTTACCTTCAGTGACGTCAAAGTACCAGCGGGAGATCATTGCAC
GGCACGGACCGGTCTCAGCTTGTTCAGAGCACAC
6168:
TACGGCGGTTGCGGCGGCAACCGTAACAACTTTGACACTGAAGAGTA
CTGCATGGCAGTGTGCGGATCCGCTATTTAAGCT
6164:
AGCTTAAATAGCGGATCCGCACACTGCCATGCAGTACTCTTCAGTGTC
AAAGTTGTTACGGTTGCCGCCGCAACCGCCGTAAAAGAATGGAGC
The oligonucleotides were phosphorylated and annealed in pairs: 6167 + 6169,
6165 + 6166, 6168 + 6164. In 20 ~.1 T4 DNA Ligase Buffer (New England Biolabs,
Beverly, MA), 1 g of each oiigonucleotide pair was incubated with 10 U T4
Polynucleotide Kinase (New England Biolabs) for 1 h at 37°C, then
heated to 95°C for 1
minute, and slow-cooled to room temperature to allow annealing. All three
annealed
33
CA 02330191 2000-12-04
WO 99/63090 PCT/I1S99/12276
oligo pairs were then mixed for ligation to one another in a total volume of
100 1 T4
DNA Ligase Buffer, and incubated with 400 U T4 DNA Ligase (New England
Biolabs)
overnight at I S°C. The ligation mixture was extracted with an equal
volume of
phenol:CHCl3 (1:1), ethanol-precipitated, resuspended in SO Restriction
Endonuclease
Buffer #4 (New England Biolabs) and digested with NdeI and Hindllt. The
annealod,
ligated and digested oligos were then subjected to electrophoresis in a 3%
NuSieve
Agarose gel, and the 240 by NdeI-HindlTI fragment was excised. This gel-
ptuified
synthetic gene was ligated into plasn~.id pTWlO, which had previously been
digested
with NdeI and HindllI, and the ligation mixture was used to transform E. coli
strain
MC1061. Ampicillin-resistant colonies were selected and used to prepare
plasmid
pTWIO:KPI. This plasmid contains the phoA-KPI(1-X57)-fiuion pmtein inserted
between the pTrp promoter element and the transcription termination signals.
B. Constraction of pKPI 61
The strategy for constructing pKPI-61 is shown in Figure 3. Plasmid pTWIO:KPI
was digested with AgeI and HindIll; the resulting 152 by AgeI-HindllI fragment
containing a portion of the KPI synthetic gene was isolated by preparative gel
electrophoresis. An oligonucleotide pair (I29 + 130) encoding the 9 amino-
terminal
residues of KPI(1-~57) and 4 amino acids of yeast a-mating factor was
phosphorylated
and annealed as described above.
129: CTAGATAAAAGAGAGGTGTGCTCTGAACAAGCTGAGA
130: CCGGTCTCAGCTTGTTCAGAGCACACCTCTCTITI'AT
The annealed oligonucieotides were then ligated to the AgeI-HindJII fragarent
of
the KPI (1-X57) synthetic gene. The resulting 192 by XbaI-Hi~tdIII synthetic
gene
(shown in Figure 4) was purified by preparative gei electrophoresis, and
ligated into
plasmid pUC 19 which had previously been digested with Xbal and HindlTI. The
ligation
products were used to transform E. coli strain MC1061. Ampicillin-resistant
colonies
were picked and used to prepare plasmid pKPI-57 by standard methods. To crate
a
synthetic gene encoding KPI(-4-~ 57), pKPI-57 was digested with ~'baI and Agel
and the
smaller fragment replaced with annealed oligos 234 + 235, which encode 4 amino
acid
34
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
residues of yeast a-mating factor fused a 4 amino acid residue amino-terminal
extension
of KPI(1-~57).
234:
CTAGATAAAAGAGAGGTTGTTAGAGAGGTGTGCTCTGAACAAGCTGAGA
235: CCGGTCTCAGCTTGTTCAGAGCAGACCTCTCTAACAACCTCTCI'ITI'AT
The 4 extra amino acids are encoded in the amyloid [3-protein
precursor/protease
nexin-2 (APPn which contains the KPI domain. The synthetic 201 by XbaI-HindIB
fragment encoding KPI(-4-X57) in pKPI-61 is shown in Figure 5.
C. Assembly of pTWI !3
The strategy for the construction of pTW113 is shown in Figure 6. Plasmid
pSP35 was constructed from yeast expression plasmid pYES2 (Invitrogen, San
Diego,
CA) as follows. A 267 by PvuII XbaI fragment was generated by PCR from yeast a
mating factor DNA using oiigos 6274 and 6273:
6274: GGGGGCAGCTGTATAAACGATTAAAA
6273: GGGGGTCTAGAGATACCCCTTCTTCTTTAG
This PCR fragment, encoding an 82 amino acid portion of yeast a-mating factor,
including the secrctory signal peptide and pro-region, was inserted into pYES2
that had
been previously digested with PvuII and XbaI. The resulting plasmid is denoted
pSP34.
Two oligonucleotide pairs, 6294 + 6292 were then ligated to 6290 + 6291, and
the resulting 135 by fragment was purified by gel electrophoresis.
6294: CTAGATAAAAGAGAGGCTGAGGCTCACGCTGAAGGTACTTTCACTTC
6290:
TGACGTCTCTTCTTACTTGGAAGGTCAAGCTGCTAAGGAATTCATCG
CTTGGTTGGTCAAAGGTAGAGGTTAAGCTTA
6291:
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
CTAGTAAGCTTAACCTCTACCTTTGACCAACCAAGCGATGAATTCCT
TAGCA
6292:
GCTTGACCTTCCAAGTAAGAAGAGACGTCAGAAGTGAAAGTACCTT
CAGCGTGAGCCTCAGCCTCTCTTTTAT
The resulting synthetic fragment was ligated into the XbaI site of pSP34,
resulting in plasmid pSP35. pSP35 was digested with XbaI and HindBI to remove
the
insert, and ligated with the 201 by .YbaI-HindBI fragment of pKPI-61, encoding
KPI
(-4-X57). The resulting plasmid pTW 113, encodes the 445 by synthetic gene for
the a-
factor-KPI(-4-X57) fusion. See Figure 7.
D. Transformation of yeast with pTWll3
Saccharomyces cerevisiae strain ABLi 15 was transformed with plasmid pTW113
by electroporation by the method of Becker et al., Methods Enrymol. 194:182
(1991).
An overnight culture of yeast strain ABL115 was used to inoculate 200 ml YPD
medium. The inoculated culture was grown with vigorous shaking at 30°C
to an ODD
of 1.3-1.5, at which time the cells were harvested by centrifugation at 5000
rpm for 5
minutes. The cell pellet was resuspended in 200 ml ice-cold water, respun, and
resuspended in 100 ml ice-cold water, then pelleted again. The washed cell
pellet was
resuspended in 10 ml ice-cold 1M sorbitol, recentrifuged, then resuspended in
a final
volume of 0.2 ml ice-cold IM sorbitol. A 40 ml aliquot of cells was placed
into the
chamber of a cold 0.2 cm electroporation cuvette (Invitrogen), along with 100
ng
plasmid DNA for pTW 113. The cuvette was placed into an Invitrogen
Electroporator II
and pulsed at 1500 V, 25F, 100 s2. Electroporated cells were diluted with 0.5
mI 1M
sorbitol, and 0.25 ml was spread on an SD agar plate containing 1M sorbitol.
After 3
days' growth at 30°C, individual colonies were streaked on SD + CAA
agar plates.
E. Induction ofpTWll3/ABLIIS, purification ofKPl(-4-~57)
Yeast cultures were grown in a rich broth and the galactose promoter of the
KPI
expression vector induced with the addition of galactose as described by
Sherman,
Methods Enzymol. 194:3 (1991). A single well-isolated colony of pTW113/ABL115
36
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/1ZZ76
was used to inoculate a 10 ml overnight culture in Yeast Batch Medium. The
next day,
IL Yeast Batch Medium which had been made 0.2% glucose was inoculated to an
ODD
of 0.1 with the overnight culture. Following 24 hours at 30°C with
vigorous shaking, the
1L culture was induced by the addition of 20 ml Yeast Galactose Feed Medium.
Following induction, the culture was fed every 12 hours with the addition of
20 ml Yeast
Galactose Feed Medium. At 48 hours after induction, the yeast broth was
harvested by
centrifugation, then adjusted to pH 7.0 with 2M Tris, pH 10. The broth was
subjected to
trypsin-Sepharose affinity ~ chromatography, and bound KPI(-4-~57) was eluted
with
20mM Tris pH 2.5. See Schilling et al., Gene 98:225 (I991). Final purification
of ICPI(-
4-~57) was accomplished by HPLC chromatography on a semi-prep Vydac C4 column
in a gradient of 20% to 35% acetonitrile. The sample was dried and resuspended
in PBS
at 1-2 mg/ml. The amino acid sequence of KPI(-4-X57) is shown in Figure 8.
Example 2. Recombinant Expression of site-directed KPI(-4--X57) variants
Expression vectors for the production of specific variants of KPI(-4-~57) were
all constructed using the pTW 113 backbone as a starting point. For aach KPI
variant, an
expression construct was created by replacing the 40 by RsrII AatlI fi-agment
of the
synthetic ICPI gene contained in pTW 113 with a pair of anneaked
oligonucleotides which
encode specific codons mutated from the wild-type KPI(-4-X57) sequence. In the
following examples, the convention used for designating the amino substituents
in the
ICPI variants indicates first the single letter code for the amino acid found
in wild-type
ICPI, followed by the position of the residue, followed by the code for the
replacement
amino acid. Thus, for example, M15R indicates that the methionine residue at
position
15 is replaced by an arginine.
A. Corutruction of pTW6165
The strategy for constructing pTW6165 is shown in Figure 9. Plasmid pTW113
was digested with RsrlI and AatII, and the larger of the two resulting
fragments was
isolated. An okigonucleotide pair (812 + 813) was phosphorylated, annealed and
gel-
purified as described above.
812: GTCCGTGCCGTGCAGCTATCTGGCGCTGGTACTTTGACGT
813: CAAAGTACCAGCGCCAGATAGCTGCACGGCACG
37
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
The annealed oligonucleotides were ligated into the RrrII and AatII-digested
pTW113,
and the ligation product was used to transform E. coli strain MC1061.
Transformed
colonies were selected by ampicillin resistance. The resulting plasmid,
pTW616S,
encodes the 44S by synthetic gene for the a-factor-KPI(-4--57; M1SA, S17VV)
fusion.
S See Figure 10.
B. Corutruction of pTW6166, pT'W6175, pBG028, pTW6183, pTW6184,
pTW6185, pT'W6173, pTW6174.
Construction of the following KPI(-4-i57) variants was accomplished exactly as
outlined for pTW6165. The oligonucleotides utilized for each construct are
denoted
below, and the sequences of annealed oligonucleotide pairs are shown in Figure
11.
Figures 12-19 show the synthetic genes for the a-factor fusions with each KPI(-
4-iS7)
variant.
1S pTW6166: KPI(-4-~S7; MISA, S17~ See Figure 12.
814: GTCCGTGCCGTGCAGCTATCTACCGCTGGTACTTTGACGT
815: CAAAGTACCAGCGGTAGATAGCTGCACGGCACG
pTW6175: KPI(-4-iS7; M1SL, S17F) See Figure 13.
2S 867: GTCCGTGCCGTGCATTGATCTTCCGCTGGTACTTTGACGT
868: CAAAGTACCAGCGGAAGATCAATGCACGGCACG
pBG028: KPI(-4-~57; M15L, S17I~ See Figure 14.
1493: GTCCGTGCCGTGCTTTGATCTACCGCTGGTACTTTGACGT
38
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
1494: CAAAGTACCAGCGGTAGATCAAAGCACGGCACG
pTW6183: KPI(-4-X57; I 16H, S 17F j See Figure 15.
925: GTCCGTGCCGTGCAATGCACTTCCGCTGGTACTTTGACGT
926: CAAAGTACCAGCGGAAGTGCATTGCACGGCACG
pTW6184: KPI(-4-~57; I16H, S 17I~ See Figure 16.
927: GTCCGTGCCGTGCAATGCACTACCGCTGGTACTTTGACGT
928: CAAAGTACCAGCGGTAGTGCATTGCACGGCACG
pTW6185: KPI(-4-~57; I16H, S17W) Sec Figure 17.
929: GTCCGTGCCGTGCAATGCACTGGCGCTGGTACTTTGACGT
930: CAAAGTACCAGCGCCAGTGCATTGCACGGCACG
pTW6173: KPI(-4-~57; M15A, I16H) See Figure 18.
863: GTCCGTGCCGTGCAGCTCACTCCCGCTGGTACTTTGACGT
864: CAAAGTACCAGCGGGAGTGAGCTGCACGGCACG
pTW6174: KPI(-4--~57; M15L, I16H) See Figure 19.
865: GTCCGTGCCGTGCATTGCACTCCCGCTGGTACTTTGACGT
39
CA 02330191 2000-12-04
WO 99/63090 PCTlUS99/12276
866: CAAAGTACCAGCGGGAGTGCAATGCACGGCACG
C. Transformation ojyeast with expression vectors
S Yeast strain ABL115 was transformed by electroporation exactly according to
the protocol described for transformation by pTW 113.
D. Induction of transformed yeast strains, puriftcation of KPI(-4-iS7)
variants.
Cultures of yeast strains were grown and induced, and recombinant secreted
ICPI
(-4-X57) variants were purified according to the procedure described for KPI {-
4-X57).
The amino acid sequences of KPI(-4--~57) variants are shown in Figures 27-36.
Example 3. Identification of ICPI (-4-~57; M15A, S17F) DD185 by phage
display.
A. Construction of vector pSPZ6:Amp:F1
The construction of pSP26:Amp:F1 is outlined in Figure 43. Vector
pSP26:Amp:F1 contributes the basic plasmid backbone for the construction of
the phage
display vector for the phoA:KPI fusion, pDWI #14. pSP26Amp:F1 contains a low-
copy number origin of replication, the ampicillin-resistance gene (Amp) and
the Fl
origin for production of single-stranded phagemid DNA.
The ampicillin-resistance gene (Amp) was generated through poiymerase chain
reaction (PCR) amplification from the plasmid genome of PUC19 using
oligonucleotides 176 and 177.
176: GCCATCGATGGTTTCTTAAGCGTCAGGTGGCACTTTTC
177: GCGCCAATTCTTGGTCTACGGGGTCTGACGCTCAGTGGAACGAA
The PCR amplification ofAmp was done according to standard techniques, using
Taq polymerase (Perkin-Elmer Cetus, Norwallc, CT). Amplification from plasmid
pUCl9 with these oligonucleotides yielded a fragment of 1159 bp, containing
P,~INB and
CIaI restriction sites. The PCR product was digested with PJIIvB and CIaI and
purified
by agarose gel electrophoresis in 3% NuSieve Agarose (FMC Corp.). Bacterial
expression vector pSP26 (supra) was digested with PfINl1 and CIaI and the
larger vector
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
fragment was purified. The PJIMI-CIaI PCR fi~agment was ligated into the
previously
digested pSP26 containing the Amp gene. The ligation product was used to
transform E.
call strain MC1061 and colonies were selected by ampicillin resistance. The
resulting
plasmid is denoted pSP26Amp.
The F1 origin of replication firom the mammalian expression vector pcDNAII
(Invitrogen) was isolated in a 692 by Earl figment. Piasmid pcDNAII was
digested
with Earl and the resulting 692 by figment purified by agarose gel
electrophoresis.
Earl-NotI adapters were added to the 692 by Earl fragment by ligation of two
annealed
oligonucleotide pairs, 179 + 180 and 181 + 182. The oligo pairs were annealed
as
described above.
179: GGCCGCTCTTCC
180: AAAGGAAGAGC
181: CTAGAATTGC
182: GGCCGCAATTC
The oligonucleotide-ligated fragment was then ligated into the single NotI
site of
PSP26:Amp to yield the vector pSP26:Amp:Fl.
B. Constraetion of vector pglll
The construction of pgIl1 is outlined in Figure 44. The portion of the phage
gene)TI protein gene contained by the PDW1 #14 phagemid vector was originally
obtained as a PCR amplification product from vector m13mp8. A portion of
m13mp8
geneIll encoding the carboxyl-terminal 158 amino acid rcsiducs of the geneIT1
prnduct
was isolated by PCR amplification of m13mp8 nucleotide residues 2307-2781
using
PCR oligos 6162 and 6160.
6162: GCCGGATCCGCTATTTCCGGTGGTGGCTCTGGTTCC
6160: GCCAAGCTTATTAAGACTCCTTATTACGCAG
The PCR oligos contain BamHI and HindIII restriction recognition sites such
that PCR from ml3mp8 plasmid DNA with the oligo pair yielded a 490 by BamHI-
HindIII figment encoding the appropriate portion of geneIlT. The PCR product
was
41
CA 02330191 2000-12-04
WO 99/63090 PCTNS99/12276
ligated between the BamHI and Hina~I sites within the poiylinker of PUC19 to
yield
plasmid pgIII.
C Cor~straciion ojpPhoA:KPLglll
Construction of pPhoA:KPI:gIII is outlined in Figure 45. A portion of the phoA
signal sequence and KPI firsion encoded by the phage display vector PDWI #14
originates with pPhoA:KPI:gIIl. The 237 by NdeI-HindIII fiagment of pTWIO:KPI
encoding the entire phoA:KPI (I-X57) fission was isolated by preparative
agarose gel
electrophoresis, and inserted between the NdeI and HindIII sites of pUCI9 to
yield
plasmid pPhoA:KPI. The 490 by BamHI-HindIII fi-agment of pgllI encoding the C-
terminal portion of the geneIII product was then isolated and ligated between
the BamHI
and Hina'ITI sites of pPhoA:KPI to yield vector pPhoa:KPI:gITI. The
pPhoA:KPI:gIIi
vector encodes a 236 amino acid residue fission of the phoA signal peptide,
KPI (1-~57)
and the carboxyl-terminal portion of the geneIII product.
D. Constraction oJpLGI
Construction of pLGI is illustrated in Figure 46. The exact geneBI sequences
contained in vector PDWI #14 originate with phage display vector pLGI. A
modified
genellI segment was generated by PCR amplification of the genellI region from
pgIl1
using PCR oligonucleotides 6308 and 6305.
6308: AGCTCCGATCTAGGATCCGGTGGTGGCTCTGGTTCCGGT
6305: Gc:AGCGGCCGTTAAGCTTATTAAGACTCCT
PCR amplification fi-om pgIlI with these oligonucleotides yielded a 481 by
BamHI-HinaHI fiagment encoding a geneIII product shortened by 3 amino acid
residues
at the amino-terminal portion of the segment of the genellI fiagment encoded
by pgIll.
A 161 by NdeI-BamHI fragment was generated by PCR amplification firm bacterial
expression plasmid pTHW05 using oligonucleotides 6306 and 6307.
6306: GATCCTTGTGTCCATATGAAACAAAGC
6307: CACGTCGGTCGAGGATCCCTAACCACGGCCTTTAACCAG
42
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
The 161 by NdeI-BamHI fragment and the 481 by BamHI-HindIll fragment were
gel-purified, and then iigated in a three-way ligation into pTW 10 which had
previously
been digested with NdeI and HindIII. The resulting plasmid pLGl encodes. a
phoA
signal peptide-insert-geneIII fusion for phage display purposes.
E. Construction ojpALS!
Construction of pALS 1 is illustrated in Figure 47.
Vector pAL51 contains the geneIII sequences of pLGI which are to be
incorporated in
vector pDW 1 # 14.
A 1693 by fragment of plasmid pBR322 was isolated, extending from the
BamHI site at nucleotide 375 to the PvuII site at position 2064. Plasmid pLGI
was
digested with Asp718I and BamHI, removing an 87 by fiagment. The overhanging
Asp718I end was blunted by treatment with Klenow fragment, and the PvuII-BamHI
fragment isolated from pBR322 was ligated into this vector, resulting in the
insertion of
a 1693 by "stuffer" region between the Asp718I and BamHI sites. The 78 by NdeI-
Asp718I region of the resulting plasmid was removed and replaced with the
annealed
oligo pair 6512 + 6513.
6512:
TATGAAACAAAGCACTATTGCACTGGCACTCTTACCGTTACTGTITA
CCCCGGTGACCAAAGCCCACGCTGAAG
6513:
GTACCTTCAGCGTGGGCTI"TGGTCACCGGGGTAAACAGTAACGGTAA
GAGTGCCAGTGCAATAGTGCTITGTTTCA
The newly created 74 by NdeI Asp718I fragment encodes the phoA signal
peptide, and contains a BstEII cloning site. The resulting plasmid is denoted
pALS I.
F. Construction ojpAL53
Construction of pAL53 is outlined in Figure 48. Plasmid pAL53 contributes
most of the vector sequence of pDWI #14, including the basic vector backbone
with
Amp gene, F1 origin, low copy number origin of replication, genellI segment,
phoA
promoter and phoA signal sequence.
43
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
Plasmid pAL51 was digested with NdeI and Hindlil and the resulting 2248 by
NdeI-HindIII fragment encoding the phoA signal peptide, stuffer region and
genellI
region was isolated by preparative agarose gel electrophoresis. The NdeI-
HindllI
fragment was ligated into plasmid pSP26:Amp:F1 between the NdeI and HindIII
sites,
resulting in plasmid pALS2.
The phoA promoter region and signal peptide was generated by amplification of
a portion of the E. coli genome by PCR, using oligonucleotide primers 405 and
406.
405: CCGGACGCGTGGAGATTATCGTCACTG
406: GCTTTGGTCACCGGGGTAAACAGTAACGG
The resulting PCR product is a 332 by MIuI-BstEII fragment, which contains the
phoA promoter region and signal peptide sequence. This fragment was used to
replace
the 148 by MIuI-BstEII segment of pAL52, resulring in vector pALS3.
G. Construction ofpSP26:Amp:Fl:PhoA:KPLglll
Construction of pSP26:Amp:Fl:PhoA:ICPI:gIII is illustrated in Figure 49. This
particular vector is the source of the KPI coding sequence found in vector
pDWI #14.
Plasmid pPhoa:ICPI:gIII was digested with NdeI and HindIll, and the resulting
714 by
NdeI-HindIll fragment was purified, and then inserted into vector pSP26:Amp:F1
between the NdeI and HindIII sites. The resulting plasmid is denoted
pSP26:Amp:F 1:PhoA:KPI:gIII.
H. Construction of pDWI #l4
Construction ofpDWl #14 is illustrated in Figure 50. The sequences encoding
KPI were amplified from plasmid pSP26Amp:Fl:PhoA:KPI:gIl1 by PCR, using
oligonucleotide primers 424 and 425.
424: CTGTTTACCCCGGTGACCAAAGCCGAGGTGTGCTCTGAACAA
425: AATAGCGGATCCGCACACTGCCATGCAGTACTCTTC
The resuiting 172 by BstEII-BamHI fragment encodes most of KPI (155). This
fragment was used to replace the stuff'er region in pAL53 between the BstEII
and BamHI
sites. The resulting piasmid, pDWI #14, is the parent ICPI phage display
vector for
44
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
preparation of randomized itPI phage libraries. The coding region for the phoA-
KPI
(155)-geneIII fusion is shown in Figure 55.
I. Corutruction ojpDWl l4-2
Construction of pDWI 14-2 is illustrated in Figure 57. The first step in the
construction of the KPI phage libraries in pDW 1 #14 was the replacement of
the AgeI-
BamHI fragment within the KPI coding sequence with. a stuffer fragment. This
greatly
aids in preparation of randomized KPI libraries, which are substantially fi~ee
of
contamination of phagemid genomes encoding wild-type KPI sequence.
Plasmid pDWI #14 was digested with AgeI and BamHI, and the 135 by AgeI-
BamHI fragment encoding KPI was discarded. A stuffer fiagment was created by
PCR
amplification of a portion of the pBR322 Tet gene, extending from the BamHI
site at
nucleotide 375 to nucleotide 1284, using oligo primers 266 and 252.
266: GCTTTAAACCGGTAGGTGGCCCGGCTCCATGCACC
252: CGAATTCACCGGTGTCATCCTCGGCACCGTCACCCT
The resulting 894 by AgeI-BamHI stuffer fi~agment was then inserted into the
Age1/BamHI-digested pDWI #14 to yield the phagcmid vector pDWI 14-2. This
vector
was the starting point for construction of the randomized KPI libraries.
J. Construction of KPI Library l619
Construction of KPI Library 16-19 is outlined in Figure 58. Library 16-19 was
constructed to display KPI-genelTI fi~sions in which amino acid positions
Ala~4, Met~s,
Ile~b and Sere are randomized. For preparation of the library, plasmid pDWl 14-
2 was
digested with AgeI and BamHI to remove the stuffcr region, and the resulting
vector was
purified by preparative agarose gel elxtrophoresis. Plasmid pDWI #14 was usod
as
template in a PCR amplification of the KPI region extending from the AgeI site
to the
BamHI site. The oligonucleotide primers used were 544 and 551.
544: GGGCTGAGACCGGTCCGTGCCGT(NNS)4CGCTGGTACTTTGACGTC
551: GGAATAGCGGATCCGCACACTGCCATGCAG
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/1Z276
Oligonucleotide primer 544 contains four randomized colons of the sequence
NNS, where N represents equal mixtures of A/G/C/T and S an equal mixture of G
or C.
Each NNS colon thus encodes all 20 amino acids plus a single possible stop
colon, in
32 different DNA sequences. PCR amplification fi-om the wild-type KPI gene
resulted
in the production of a mixture of 135 by AgeI-BamHI fragments all containing
diffet~ent
sequences in the randomized region. The PCR product was purified by
preparative
agarose gel electrophoresis and ligated into the AgeIlBamHI digested pDW 1 14-
2 vector.
The ligation mixture was used to transform E. coli TopIOF~ cells (Invitrogtn)
by
electroporation according to the manufacturer's directions. The resulting
Library 16-19
contained approximately 400,000 independent clones. The potential size of the
library,
based upon the degeneracy of the priming PCR oligo #544 was 1,048,576 members.
The expression unit encoded by the members of Library 16-19 is shown in Figure
59.
K. Selection ofLibrary 16-19 with human plasma kallikrein
KPI phage were prepared and amplified by infecting transformed cells with
M13K07 helper phage as described by Matthews et a1, Science 260:1113 (1993).
Human plasma kallikrein (Enzyme Research Laboratories, South Bend, INJ, was
coupled to Sepharose 6B resin. Prior to phage binding, the immobilized
kallilQein resin
was washed three times with 0.5 ml assay buffer (AB = 100mM Tris-HCI, pH 7.5,
O.SM
NaCI, SmM each of KCI, CaCIZ, MgCIZ, 0.1% gelatin, and 0.05% Triton X-100).
Approximately 5x109 phage particles of the amplified Library 16-19 in PBS, pH
7.5,
containing 300mM NaCI and 0.1% gelatin, were bound to 50 pl ka11i1Qein resin
containing 15 pmoles of active human plasma kallila~ein in a total volume of
250 pl.
Phage were allowed to bind for 4 h at room temperature, with rocking. Unbound
phage
were removed by washing the kallilarein resin three times in 0.5 ml AB. Bound
phage
were eluted sequentially by successive 5 minute washes: 0.5 ml SOmM sodium
citrate,
pH 6.0, 150mM NaCI; 0.5 ml SOmM sodium citrate, pH 4.0, 154mM NaCI; and 0.5 ml
SOmM glycine, pH 2.0, i50mM NaCI. Eluted phage were neutralized immediately
and
phagemids from the pH 2.0 elution were titered and amplified for reselection.
After
three rounds of selection on kallikrein-Scpharose, phagemid DNA was isolated
finm 22
individual colonies and subjected to DNA sequence analysis.
The most frequently occurring randomized KPI region encoded: Alai°-Alms-
Ile~6-
Phe~'. The phoA-KPI-geneBI region encoded by this class of selected KPI phage
is
46
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12Z76
shown in Figure 60. The KPI variant encoded by these phagemids is denoted ICPI
(155;
M15A, S17F).
L. Construction ojpDDl85 KPI (-4-X57; MISA, S17F)
Figure 61 outlines the construction ofpDD185 KPI (-4-~57; M15A, S17F). The
sequences encoding ICPI ( 155; M 15A, S 17F) were moved from one phagemid
vector,
pDWI (16-19) 185, to the yeast expression vector so that the KPI variant could
be
purified and tested.
Plasmid pTW113 encoding wild-type ICPI (-4-X57) was digested with AgeI and
BamHI and the 135 by AgeI-BamHI fi~agment was discarded. The 135 by AgeI-BamHI
fragment of pDWI (16-19) 185 was isolated and Iigated into the yeast vector to
yield
plasmid pDD185, encoding a-factor firsed to KPI (-4-X57; M15A, S17F). See
Figure
62.
M. Purification ojKPl ( 4-~57; Ml SA, SI7F) pDDl85
Transformation of yeast strain ABL115 with pDD185, induction of yeast
cultures,
and purification of KPI (-4-X57; M15A, S17~ pDD185 was accomplished as
describai
for the other KPI variants.
N. Construction ojKPl Library 6 MI SA, with residues 14, l6-18 random.
Library 6 was constructed to display KPI-genellI firsions in which amino acid
positions Ala'4, Ile'6, Ser" and Arg'8 are randomized, but position 15 was
held constant
as Ala. For preparation of the library, plasmid pDWI #14 was used as the
template in a
PCR amplification of the KPI region extending from the AgeI site to the BamHI
site.
The oligonucleotide primers used were 551 and 1003.
1003: GCTGAGACCGGTCCGTGCCGTNNSGCA(NNS)3TGGTACTTTGACGTC
551: GGAATAGCGGATCCGCACACTGCCATGCAG
Oligonucleotide primer 1003 contained four randomized codons of the sequence
NNS, where N represents equal mixtures of A/G/CrT and S an equal mixture of G
or C.
Each NNS codon thus encodes all 20 amino acids plus a single possible stop, in
32
47
CA 02330191 2000-12-04
WO 99/63090 PCTIUS99/12276
different DNA sequences. PCR amplification from the wild-type ICPI gene
resulted in
the production of a mixture of 135 by AgeI-BamHI fragments all containing
different
sequences in the randomized region. The PCR product was phenol extracted,
ethanol
precipitated, digested with BamHI and purified by preparative agarose gel
electrophoresis. Plasmid pDWI 14-2 was digested with BamHI, phenol extracted
and
ethanol precipitated. The insert was ligated at high molar ratio to the
vector, which was
then digested with AgeI to remove the stuffer region. The vector containing
the insert
was purified by agarose gel electrophoresis and recircularized. The resulting
library
contains approximately Sx 1 O6 independent clones.
D. Construction ojKPl Library 7 with residues 14-18 random.
Library 7 was constructed to display ICPI-geneIll fusions in which amino acid
positions Alai°, Met~s, Ile~6, Sere and Arg~$ are randomized. For
preparation of the
library, nlasmid pDWI #14 was used as template in a PCR amplification of the
KPI
region extending from the AgeI site to the BamHI site. The oligonucleotide
primers used
were 551 and 1179.
1179: GCTGAGACCGGTCCGTGCCGT(NNS)STGGTACTTTGACGTC
551: GGAATAGCGGATCCGCACACTGCCATGCAG
Oligonucleotide primer 1179 contains five randomized codons of the sequence
NNS, where N represents equal mixtures of A/G/C/T and S an equal mixture of G
or C.
Each NNS codon thus encoded ail 20 amino acids plus a single possible stop, in
32
different DNA sequences. PCR amplification from the wild-type KPI gene
resulted in
the production of a mixture of 135 by AgeI BamHI fiagments all containing
different
sequences in the randomized region. The PCR product was phenol extracted,
ethaaol
precipitated, digested with BamHI and purified by preparative agarose gel
electrophoresis. Plasntid pDWI 14-2 was digested with BamHI, phenol extracted
and
ethanol precipitated. The insert was ligated at high molar ratio to the
vector, which was
then digested with AgeI to remove the stuffer.region. The vector containing
the insert
was purified by agarose gel electrophoresis and recircularized. The resulting
library
contains approximately 1x10' independent clones.
48
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
P. Selection ofLioraries 6 & 7 with human factorXlla
KPI phage were prepared and amplified by infecting transformed cells with
M13K07 helper phage (Matthews and Wells, 1993). Human factor 3~Ia (Enzyme
Research Laboratories, South Bend, IN), was biotinylated as follows. Factor
XIIa (0.5
mg) in SmM sodium acetate pH 8.3 was incubated with Biotin Ester (Zymed) at
room
temperature for 1.5 h, then buffer-exchanged into assay buffer (AB).
Approximately
1x10'° phage particles of each amplified Library 6 or 7 in PBS, pH 7.5,
containing
300mM NaCI and 0.1% gelatin, were incubated with SO pmoles of active
biotinylated
human factor XIIa in a total volume of 200 Icl. Phage were allowed to bind for
2 h at
room temperature, with rocking. Following the binding period, 100 ul
Strepavidin
Magnetic Particles (Boehringer Mannheim) were added to the mixture and
incubated at
room temperature for 30 minutes. Separation of magnetic particles from the
supernatant
and wash/elution buffers was carried out using MPC-E-I Neodymium-iron-boron
permanent magnets (Dynal). Unbound phage were removed by washing the
magnetically bound biotinylated 3QIa-phage complexes three times with 0.5 ml
AB.
Bound phage were eluted sequentially by successive 5 minute washes: 0.5 ml
SOmM
sodium citrate, pH 6.0, 150mM NaCI; 0.5 ml SOmM sodium citrate, pH 4.0, 150mM
NaCI; and 0.5 ml SOmM glycine, pH 2.0, 150mM NaCI. Eluted phage were
neutralized
immediately and phagemids fiom the pH 2.0 elution were titered and amplified
for
reselection. After 3 or 4 rounds of selection with factor 3HIa, phagemid DNA
was
isolated fiom individual colonies and subjected to DNA sequence analysis.
Sequences in the randomized regions were compared with one another to
identify consensus sequences appearing more than once. From Library 6 a
phagemid
was identified which encoded M15L, S17Y, RIBH. Fmm Library 7 a phagemid was
identified which encoded M15A, S17Y, R18H.
Q. Construction of KPI Library P48 with residue 48 random.
Library P48 was constructed to for expression of KPI (M15A, S17Y, R18H) IN
WHICH AMINO ACID n which amino acid position Tyr4g is randomized. Construction
of Library P48 is detailed in Figure 55. For preparation of the library,
plasmid pDWI
L6-16, encoding the pBG022 KPI peptide as a fusion with the m13 gI)1 protein,
was
used as template in a PCR amplification of the KPI region extending from the
RsrII site
to the BamHI site. The oligonucleotide primers used were 1663 and 1945.
49
CA 02330191 2000-12-04
WO 99/63090 PCT/tJS99/12276
1663: GCTTTACTGTTTACCCCGGTGACCAAAGCCGAGGTGTGC
1945: ATTAGCGGATCCGCACACTGCCATGCASNNCTCTTCAGTGTCAAAG
Oligonucleotide primer 1945 contains a single randomized codon of the
sequence SNN, where N represents equal mixtures of AIG/CrI' and S an equal
mixture
of G or C. Following the procedure delineated supra, PCR amplification from
the wild-
type ICPI gene resulted in the production of a mixture of RsrII-BamHI
fiagments all
containing different sequences in the randomized region. The PCR product was
phenol
extracted, ethanol precipitated, digested with RsrII and BamHI and purified by
preparative agarose gel electrophoresis. Plasmid pBG022 was digested with
RsrII and
BamHI, phenol extracted and ethanol precipitated. The insert was ligated at
high molar
ratio to the vector. The vector containing the insert was purified by agarose
gel
electrophoresis and recircularized.
R. Construction ojpBG015 KPI (-4-~57; MISL, S17Y, RIBH), pBG012
(-4--~57; MI SA, SI7Y, RI BH, having spontaneous mutation Y48H)
The sequences encoding KPI (155; MISL, S17Y, Ri8H) and KPI (155; M17A,
S 17Y, R18H) were moved from the pragemid vectors to the yeast expression
vector so
that the ICPI variant could be purified and tested. Plasmid pTWlI3 encoding
wild-type
KPI (-4-~57) was digested with AgeI and BamHI and the 135 by AgeI-BamHI
fi~agment
was discarded. The 135 by AgeI-BamHI fi-agment of the phagemid vectors were
isolated
and Iigated into the yeast vector to yield plasmids pBG015 and pBG022,
encoding yeast
x-factor fused to KPI (-4-~57; M15L, S17Y, R18I~, and KPI (-4-X57; M15A, SI7Y,
RI8H, having spontaneous mutation Y48H), respectively. Figure 20 shows the
synthetic gene for the x-factor fusion with KPI variant (-4-~57; M15A, S17Y,
RIBFi,
having spontaneous mutation Y48H). Figure 37 shows the amino acid sequence of
KPI
variant (-4-X57; M15A, S17Y, RI8H, having spontaner~us mutation Y48H).
S. Construction of pBG033 KPI ( 4--57; T~9V, M15A, S17Y, R18H)
Plasmid pBG022 was digested with XbaI and RsrII, and the larger of the two
resulting fi-agments was isolated. An oligonucleotide pair ( 1593 + 1642) was
phosphorylated, annealed and gel-purified as described previously. The
annealed
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
oligonucieotides were ligated into the.YbaI and RsrII-digested pBG022, and the
Iigation
product was used to transform E. cell strain MC1061 to ampicillin resistance.
The
resulting plasmid pBG033, encodes the 445 by synthetic gene for the yeast x-
factor-KPI
(-4-X57; T9V, M15A, SI7F, R18H) fusion. Figure 21 shows the synthetic gene for
the
x-factor fusion with KPI variant (-4-X57; T9V, M15A, S17Y, R18H, Y48H). ).
Figure
38 shows the amino acid sequence of KPI variant (-4-~57; T9V, M15A, SI7Y,
R18H,
Y48H).
T. Construction of pBG048 KPI ( 4-~57; Y48H)
Figure 52 outlines the construction of pBG048 KPI (-4-X57; Y48H). Plasmid
pTW113 encoding wild-type KPI (-4-i57) was digested with AatII and BamHI and
the
92 by AatII-BamHI fragment was discarded. Plasmid pBG022 encoding KPI (-4-X57;
M15L, S 17Y, R18H, Y48H) was digested with AatII and BamHI. The resulting 92
by
AatII-BamHI fragment was isolated and ligated into the yeast vector to yield
plasmid
pBG048, encoding yeast x-factor fused to KPI (-4-~57; Y48H). Figure 22 shows
the
synthetic gene for the x-factor fusion with KPI variant (-4-~57; Y48I-~.
Figure 39
shows the amino acid sequence of KPI variant (-4--~57; 48H).
U. Constraction ofpBG049KP1 (-4-X57; MISA, SI7Y, RI8H)
Figure 53 outlines the construction of pBG049 KPI (-4-X57; M15A, S17Y,
R18H). Plasmid pBG022 encoding KPI (-4-~57; M15A, S17Y, R18H, Y48H) was
digested with AatII and BamHI and the 92 by AatII-BarnHI fragment was
discarded.
Plasmid pTW 113 encoding wild-type ICPI (-4-X57) was digested with AatII and
BamHI.
The resulting 92 by AatII-BamHI fragment was isolated and ligated into the
yeast vector
to yield piasmid pBG048, encoding yeast x-factor fused to KPI (-4-X57; M15A,
S17Y,
R18H). ). Figure 23 shows the synthetic gene for the x-factor fusion with KPI
variant
(-4-X57; M15A, S17Y, R18H). Figure 40 shows the amino acid sequence ofKPI
variaat
(-4-~57; M15A, S17Y, R18H).
V. Construction oJpBG050 KPI ( 4-~57; ?'9V, Ml SA, SI7Y, R18H)
Figure 54 outlines the construction of pBG050 KPI (-4-i57; T9V, M15A, S17Y,
RIBH). Plasmid pBG033 encoding KPI (-4-~57; T9V, M15A, R18H, Y48H) was
digested with AatII and BamHI and the 92 by AatII-BamHI fragment was discarded
51
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
Plasmid pTW113 encoding wild-type KPI (-4-i57) was digested with.4atII and
BamHL
The resulting 92 by AatII-BamHI 6ragrnent was isolated and ligated into the
yeast vector
to yield plasmid pBG050, encoding yeast x-factor fined to KPI (-4-~57; T9V;
M15A,
S17Y, R18H). ). Figure 24 shows the synthetic gene for the x-factor fusion
with KPI
variant (-4-~57; T9V, MISA, S17Y, RIBH). Figure 41 shows the amino acid
sequence
ofKPI variant (-4-X57; T9V, M15A, S17Y, R18H).
W. Construction of pBG019 KPI (-4-X57, ?'9V, MISL, SI7Y, R18H)
Plasmid pBG015 was digested with XbaI and RsrII, and the larger of the two
resulting fi~agments was isolated. An oligonucleotide pair (1593 + 1642) was
phosphorylated, annealed and gel-purified as described previously.
1593:
CTAGATAAAAGAGAGGTTGTTAGAGAGGTGTGCTCTGAACAAGCTG
AGGTTG
1642:
GACCAACCTCAGCTTGTTCAGAGCACACCTCTCTAACAA
CCTCTCIT)'TAT
The annealed oligonucleotides were ligated into the XbaI and RsrII-digested
pBG015,
and the ligation product was used to transform E. colt strain MC1061 to
ampicillin
resistance. The resulting piasmid pBG029, encodes the 445 by synthetic gene
for the
yeast x-factor-KPI (-4-~57; T9V, M15L, S17F, R18H) fission.
X. Selection ofLibrary l6-19 with human factorXa
KPI phage were prepared and amplified by infecting transformed cells with
M13K07 helper phage (Matthews and Wells, 1993). Human factor Xa (Haematologic
Technologies, Inc., Essex function, VT) was coupled to Sepharose 6B resin.
Prior to
phage binding, the immobilized Xa resin was washed three times with 0.5 ml
assay
buffer (AB = 100mM Tris-HCI, pH 7.5, O.SM NaCI, SmM each of KCI, CaCl2, MgCl2,
0.1% gelatin, and 0.05% Triton X-100). Approximately 4x10'° phage
particles of the
amplified Library 16-19 in PBS, pH 7.5, containing 300mM NaCI and 0.1%
gelatin,
52
CA 02330191 2000-12-04
WO 99/63090 PCTNS99/12276
were bound to 50 pl Xa resin in a total volume of 250 pl. Phage were allowed
to bind
for 4 h at room temperaturz, with rocking. Unbound phage were removed by
washing
the Xa resin three times in 0.5 ml AB. Bound phage were eluted sequentially by
successive 5 minute washes: 0.5 ml SOmM sodium citrate, pH 6.0, 150mM NaCI;
0.5 ml
SOmM sodium citrate, pH 4.0 150mM NaCI; and 0.5 ml 50mM glycine, pH 2.0, 150mM
NaCI. Eluted phage were neutralized immediately and phagemids from the pH 2.0
elution were titered and amplified for reselection. After three rounds of
selection on Xa-
Sepharose, phagemid DNA was isolated and subjected to DNA sequence analysis.
Sequences in the randomized Alai°-Sere region were compared with one
another
to identify consensus sequences appearing more than once. A phagemid was
identified
which encoded ICPI (155; M15L, I16F, S17K).
Y. Construction ofpDDl31 KPI (-4-X57; MISL,116F, S17K)
The sequences encoding KPI (155; M15L, I16F, S17K) were moved from the
phagemid vector to the yeast expression vector so that the ICPI variant could
be purified
and tested.
Plasmid pTW113 encoding wild-type KPI (-4-X57) was digested with AgeI and
BamHI and the 135 by AgeI-BamHI fragment was discarded. The 135 by AgeI-BamHI
fragment of the phagemid vector was isolated and ligated into the yeast vector
to yield
plasmid pDDl3l, encoding yeast x-factor fused to ICPI {-4-~57; M15L, I16F,
S17K).
Z. Constraction of pDDl34 KPI ( 4-~57; MI SL, I16F, S17K, G37~
Plasmid pDD 131 was digested with AatI and BamHI, and the larger of the two
resulting fragments was isolated. An oligonucleotide pair (738 + 739) was
phosphorylated, annealed and gel-purified as described previously.
738:
CACTGAAGGTAAGTGCGCTCCATTCTTTTACGGCGGTTGCTACGGCA
ACCGT
AACAACTT.TGACACTGAAGAGTACTGCATGGCAGTGTGCG
739:
53
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12Z76
GATCCGCACACTGCCATGCAGTACTCTTCAGTGTCAAAGTTGTTACG
GTTGC
CGTAGCAACCGCCGTAAAAGAATGGAGCGCACTTACCTTCAGTGAC
GT
The annealed oligonucleotides were ligated into the AatI and BamHI-digested
pDD131, and the ligation product was used to transform E. coli strain MC1061
to
ampicillin resistance. The resulting plasmid pDD134 encodes the 445 by
synthetic gene
for the yeast x-factor-KPI (-4-~57; M1SL, I16F, S17K, G37I~ fusion.
AA. Corrstraction of pDD135 KPI ( 4-~57; MI SL,116F, SI7K, G37L)
Plasmid pDD131 was digested with AatII and BamHI, and the larger of the two
resulting fragments was isolated. An oligonucleotide pair (738 + 739) was
phosphorylated, annealed and gel-purified as described previously.
738:
CACTGAAGGTAAGTGCGCTCCATTCTTTZ'ACGGCGGTTGCTACGGCA
ACCGT
AACAACTTTGACACTGAAGAGTACTGCATGGCAGTGTGCG
739:
GATCCGCACACTGCCATGCAGTACTCTTCAGTGTCAAAGTTGTTACG
GTTGC
CGTAGCAACCGCCGTAAAAGAATGGAGCGCACTTACCTTCAGTGAC
GT
The annealed oligonucleoddes were ligated into the AatII and BamHI-digested
pDD131, and the ligation product was used to transform E. coli strain MC1061
to
ampicillin resistance. The resulting plasmid pDD135 encodes the 445 by
synthetic gene
for the yeast x-factor-KPI (-4-~57; M 15L, I16F, S 17K, G37L) fusion.
Example 4. Kinetic analysis of Kpi(-4-~5'n variants
The concentrations of active human plasma kallikrein, factor 3QIa, and trypsin
were determined by titration with p-nitrophenyl p'-guanidinobenzoate as
described by
54
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
Bender et ai., supra, and Chase et al., Biochem. Biophys. Res. Commun. 29:508
(1967).
Accurate concentrations of active KPI(-4--~S7) inhibitors were determined by
titration of
the activity of a known amount of active-site-titrated trypsin. For testing
against
kallikrein and trypsin, each KPI(-4-rS7) variant (0.5 to 100nM) was incubated
with
S protease in low-binding 96-well microtiter plates at 30°C for 1 S-2S
min, in 100mM Tris-
HCI, pH 7.5, with SOOmM NaCI, SmM KCI, SmM CaCl2, SmM MgCl2, 0.1% Difco
gelatin, and 0.05% Triton X-100. Chromogenic synthetic substrate was then be
added,
and initial rates at 30°C recorded by the SOFTmax kinetics program via
a
T'1-iERMOmax microplate reader (Molecular Devices Corp., Menlo Park, CA). The
substrates used were N-a-benzoyl-L-Arg p-nitroanilide (lrriM) for trypsin
(20nM), and
N-benzoyl-Pro-Phe-Arg p-nitroanilide (0.3rnM) for plasma kallikrein (1nM). The
Enzfitter (Elsevier) program was used both to plot fractional activity (i.e.,
activity with
inhibitor, divided by activity without inhibitor), a, versus total
concentration of inhibitor,
I,, and to calculate the dissociation constant of the inhibitor (K;) by
fitting the curve to
1 S the following equarion:
a=I-!E>~+!I J,+K.- flElr+!I J.+Kil'-4IEJ.lII~
The K;s determined for purified KPI variants are shown in Figures 63 and 69.
The most potent variant, KPI (-4-~57; M1SA, S 17F) DD185 is 11 S-fold more
potent as a
human kallikrein inhibitor than wild-type KPI (-4-~S7). The least potent
variant, KPI
(-4--~S7; I16H, S 17V1~ TW618S is still 3S-fold more potent than wild-type
KPI.
Replacement of arginine at position 18 of the native KPI peptide with
histidine
(R18H) in combination with one or more additional substitutions at residues 9,
15 and
17 of the native KPI peptide also exhibited potent and specific serine
protease inhibition
toward selected serine proteases of interest than the native KPI peptide. In
particular, the
2S specific substitutions T9V, M1SA, S17Y and M1SA ,S17Y in the context of the
R18H
substitution exhibited such potent serine protease inhibition. See Figures 63
and 64.
Substituting the native tyrosine at position 48 in these R18H substituted
peptides with
histidine (pBG022, SOD4, SOB6; Y48H), glutamine (SOB6, SOL1, SOM1; Y48~,
alanine (SOPS, SOC4; Y48A) or aspartic acid (SON1; Y48D) produced a
significant
SS
CA 02330191 2000-12-04
WO 99163090 PCT/US99112276
increase in their level of expression in comparison to the R18H substituted
peptides
without the position 48 substitution. See Figures 69B, E and F.
For testing against factor XIIa, essentially the same reaction conditibns were
used, except that the substrate was N-benzoyl-Ile-GIu-Gly-Arg p-nitroaniline
hydrochloride and its methyl ester (obtained from Pharmacia Hepar, Franklin,
OH), and
corn trypsin inhibitor (Enzyme Research Laboratories, South Bend, IN) was used
as a
control inhibitor. Factor XTIa was also obtained from Enzyme Research
Laboratories.
Various data for inhibition of the serine proteases of interest kallikrein,
plasmin,
and factors Xa, XIa, and XIIa by a series of ICPI variants are given in Figure
64. The
results indicate that KPI variants can be produced that can bind to and
preferably inhibit
the activity ~f serine proteases. The results also indicate that the peptides
of the
invention may exhibit the preferable more potent and specific inhibition of
one or morn
serene proteases of interest.
Example 5. Effect of KPI variant KPI185-1 on postoperative bleeding
A randomized, double-blinded study using an acute porcine cardiopulmonary
bypass (CPB) model was used to investigate the effect of ICPI185-1 on
postoperative
bleeding. Sixteen pigs (55-65 kg) underwent 60 minutes of hypothermic
(28°C) open-
chest CPB with 30 minutes of cardioplegic cardiac arrest. Pigs were randomized
against
a control solution of physiological saline (NS; n=8) or ICPI-I85 (n=8) groups.
Duriag
aortic cross-clamping, the tricuspid valve was inspected through an atriotomy
which was
subsequently repaired. Following reversal of heparin with protamine, dilateral
thoracostomy tubes were placed and shed blood collected for 3 hours. Shed
blood
volume and hemoglobin (Hgb) loss were calculated from total chest tube output
and
residual intrathoracic blood at time of sacrifice.
Total blood loss was significantly reduced in the ICPI185-1 group (245.75 ~
66.24 ml vs. 344.25 ~ 63.97 ml, p~.009). In addition, there was a marked
reduction in
total Hgb loss in the treatment group (13.59 ~ 4.26 gm vs. 23.61 ~ 4.69 gm,
p~.0005).
Thoracostomy drainage Hgb was significantly increased at 30 and 60 minutes in
the
control group [6.89 + 1.44 vs. 4.41 + 1.45 gm/dl (p~.004) and 7.6 ~ 1.03 vs.
5.26 ~
1.04 gm/dl (p=0.0002), respectively]. Preoperative and post-CPB hematocrits
were not
statistically different between the groups. These results are shown in
graphical form in
Figures 65-68.
56
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
Example 6. Effect of KPI Variant hPI-BG022 on Transplant Rejection
ICPI-BG022 was tested for its ability to delay transplant rejection in a tat
model of acute xenograft rejection. Xenotransplantation of vascularized organs
between discordant species results in hyperacute graft rejection within
minutes to
hours after graft reperfusion. Cardiac xenografts from male Hartley guinea
pigs were
hetemtopically grafted into male rats that were complement deficient.
Experimental
animals received 5 mg/kg ICPI-BG022 N prior to reperfusion, and control
animals
received saline placebo. The data in Figure 70 demonstrate that a single ICPI-
BG022
dose significantly prolongs survival of guinea pig hearts grafted into
complement-
deficient rats.
Example 7. Effect of KPI Variant KPI-BG022 on Ulcerative Colitis
KPI-BG022 was tested in a rat model of TNBS (trinitrobenzene sulfonic acid)
induced colitis. Animals were subjected to intracolonic instillation of TNBS
to
induce inflammation and ulceration. Tail-vein injection of ICPI or vehicle was
begun
at the time of TNBS infusion and continued with three different dosing
regimens:
twice daily injections for 7 days; once daily injections for 7 days; and, two
injections
only in the day following injury. In each treatment group, half of the animals
were
sacrificed and scored for colonic injury 8 days following injury, and the
remaining
animals were sacrificed at 14 days. There were no significant differences in
damage
scores between saline or ICPI treated animals sacrificed 8 days following
injury. As
shown in Figure 71, in all three dosing groups there was a significant
reduction in
damage in ICPI-treated animals at 14 days after injury. Even the animals
receiving
only three doses of ICPI in the 24 hours following injury showed significant
reduction
in colonic damage two weeks after the TNBS instillation.
Example $. Effect of KPI Variant KPI-BG022 on Postoperative Bleeding
ICPI-BG022 will be tested in an ovine model of cardiopulmonary bypass-
associated pulmonary pathophysiology and blood loss and conducted as described
in
Friedman, M., Sellke, F.W., Wang, S.Y., Weintraub, R.M., and Johnson, R.G.
(1994)
Circulation 90: II262-II268; Friedman, M., Wang, S.Y., Sellke, F.W., Cohn,
W.E.,
Weintraub, R.M., and Johnson, R.G. ( 1996) J. Thorac. Cardiovasc. Surg. lll:
460-
468) with modifications as follows:
57
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
Surgical procedures:
Dorset-Rambouillet sheep (n=10 in each group) weighing 25-30 kg each will
be anesthetized with intravenous 80 mg/kg alpha-chlorarose and S00 mg/kg
urethane.
Animals will be intubated and mechanically ventilated (Harvard Apparatus).
Arterial
blood gas and pH measurements will be performed during the procedure (pH blood
gas analyzer 1306, Instruments Lab, Lexington, MA) and alpha-stat pH
management
will be used.during CPB. Systemic arterial pressure will be continuously
monitored
afrer direct cannulation of the femoral artery, and a separate femoral artery
cannula
will be used for blood collection. A jugular vein cannula will be used for
drug
administration. Lymph fluid will be collected from the lungs as follows: the
efferent
duct of the caudal mediastinal lymph node will be cannulated through a right
thoracotomy in the fiRh intercostal space using a silicone, heparin-coated
catheter.
CPB preparation:
A midline sternotomy will be performed and the pulmonary artery (PA)
isolated and surrounded with an ultrasonic flowmeter (Transonic System,
Ithaca, N~.
Animals will be heparinized to achieve an activated clotting time (ACT) > 750
seconds as monitored using a Hemochron device. At the end of CPB the heparin
will
be reversed with protamine sulphate to baseline ACT. A catheter will be
inserted into
the left atrium (LA) for blood withdrawl and pressure recording, and the PA
will be
cannulated for continuous pressure monitoring. Venous drainage will be
provided by
a cannula in the right atrium (R.A) and an aortic perfusion catheter will be
placed in
the aorta. The extracorporeal circuit will consist of a roller pump
(Cardiovascular
Instruments, Wakef eld, MA) and bubble oxygenator (Bently Bio-2, Baxter Health
Care). The circuit will be primed with 1 l lactated Ringer's solution.
Myocardial protection will be provided by antegrade cold blood cardioplegia
at 4°C using a 4:1 ratio of autologous blood to crystalloid
cardioplegia (KCI 60 meq,
mannitoi 12.5 g, citrate-phosphate-dextrose solution 50 mL, THAM 10 meq, 5%
dextrose and saline 0.225% QS). Iced slush will be used for topicial cooling
to
augment the cardioplegia. Immediately after application of the aortic cross-
clamp
cardioplegia will be given until arrest of the heart and then reinfused every
20
58
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
minutes. With institution of CPB all animals will be cooled to a core
temperature of
27°C. After a mean time of SC minutes, rewarming will be commenced
approximately 10 minutes before removal of aortic cross-clamp to achieve a
core
temperature of 37°C at the termination of bypass. Flow will be
maintained to keep
aortic mean pressure not less than 40 mm/Iig. Norepinephrine bitartrate
injection will
be given through the CVP line to all animals after termination of CPB with an
incrementaily decreasing infusion rate until the infusion is stopped one hour
post-
CPB.
Physiologic and biochemical determinations:
Hemodyamic measurements will be made before institution of CPB (baseline),
every 30 minutes during bypass and every 15 minutes for the first hour after
termination of CPB. Thereafter measurements will be made every 30 minutes for
3
hours. Cardiac output will be determined as pulmonary artery flow (Qpa in
L/min) or,
during CPB, as pump flow. Cardiac index (CI), systemic vascular index (SVRI),
pulmonary vascular resistance index (PVRI), will be calculated by standard
equations.
Simultaneous with the hemodynamic measurements, 2 ml blood samples will be
collected from left and right artria and placed into ice-cooled EDTA tubes.
Hematocrit, blood gases, and oxygen content will be measwed for each sample.
After
blood is centrifuged, supernatant platelet. counts and white blood cell counts
will be
performed.
Lymph collection and measurements:
Lymph volume will be measured and the protein content detemtined. ~ Lymph
protein clearance will be calculated as milliliters lung lymph flow per 30
minutes x
lymph:plasma protein ratio. Protein clearance is considered reflective of the
degree to
which larger molecules leak into the lymph, as an indication of damage greater
than
that seen with lymph fluid flow alone.
Study protocol:
A double-blind study will be performed. Sheep will be randomized to 3
groups of 10 animals each: Group 1 = saline control; Group 2 = ICPI-BG022 dose
1;
and, Group 3 = ICPI-BG022 dose 2. Vehicle and KPI-BG022 will be formulated and
aliquoted into coded tubes such that after anesthesia each animal wiii receive
a
59
CA 02330191 2000-12-04
WO 99/63090 PCT/US99/12276
loading dose of 100 ml, a 100 ml pump prime and a 25 ml/hr infusion during the
course of CPB. We propose to test two total doses of the KPI-BG022 variant: 5
mg/kg, and 0.5 mg/kg. Therefore, Group 2 will receive a 70 mg loading dose of
ICPI-
BG022, a 70 mg pump prime and 18 mg/hr infusion. Group 3 will receive a 7 mg
loading dose, a 7 mg pump prime and 1.8 mglhr infusion..
Total, non-pulsatile hypothermic CPB will be continued for 90 minutes with a
cross-clamp time of 1 hour. Rewarming will start 10 minutes before removal of
the
cross-clamp and will be continued until a core temperature of 37°C is
attained. CPB
will be terminated when core temperature has stabilized at 37°C. Post-
CPB
monitoring will continue for 3 hours. Protamine will be given in the first 30
minutes
post-CPB, and when ACT has been reduced to baseline levels the chest will be
closed
with a large-bore thoracostomy tube left in place for drainage.
Blood and hemoglobin loss measurements:
The thoracostomy tube will be connected to a drainage system and suction
applied at a force of 10 lcPa. Drain losses will be collected for a total of
two hours
post-CPB, and then the sternotomy wound wilt be reopened and all shed blood
will be
aspirated from the thorax and pericardium. The volume of blood loss and
hemoglobin
will be measured and used to calculate the total hemoglobin loss in grams.
Based on previous experience with this (Friedman et al., 1994; Friedman et
al., 1996) model the control group should demonstrate several parameters of
pulmonary injury, including increases in: pulmonary vascular resistance (PVR)
(170% increase reported), pulmonary lymph flow (233% reported), and lung water
(15% reported). An increase in sequestration of WBCs and platelets in the lung
should be seen in the control group. Arterial oxygenation (PaOZ) should fall
significantly upon cessation of CPB with a gradual recovery in the post-bypass
period.
With respect to blood and hemoglobin loss in the post-bypass period, our
experience with KPI-wt in another sheep model of CPB (Ohri et al, Ann. Thorac.
Surg. 1996 Apr;61(4):1223-1230) leads us to anticipate collection of 200-400
mI
blood in chest drains in the control group, containing 10-20 g hemoglobin. In
that
study, recombinant ICPI was assessed in an ovine model of CPB as a hemostatic
agent
after CPB. Sheep (n = 22) underwent CPB for 90 minutes. Two thoracic drains
were
sited and drain losses collected for a period of 3 hours after CPB. Wounds
were
CA 02330191 2000-12-04
WO 99/63090 PCT/US99I12276
subjectively assessed before closure for "dryness" using a visual analogue
scale.
Sheep were randorr~ized to control (n = 8), aprotinin (n = 8), and rKPI (n =
6) groups.
Control animals had a drain loss of 409.4 +/- 39.4 mL/3 h, compared with 131.3
+/-
20.3 ml,/3 h for the aprotinin group and 163.7 +/_ 34.3 mL3 h for the rICPI
group (p =
0.16). Hemoglobin loss was 11.6 +/- 3.6, 6.02 +/- 2.1, and 4.6 +.~- 1.2 g/3 h
for the
control, rKPI, and aprotinin groups respectively (p = 0.25). The subjective
analysis of
the wounds at the end of CPB found aprotinin (1.25 +/- 0.16; p < 0.05) and
rKPI (I.17
+/- 0.17; p < 0.05) animals to score significantly lower than control animals
(2.63 +/-
0.42).
Indeed, significant reductions in blood and hemoglobin Ioss in the KPI-BG022
treated groups are expected. Measureable reductions in one or more of the
other
parameters of post-bypass pulmonary injury are also expected in the KPI-BG022
treated group. Positive results in this regard would include smaller increases
or no
increases in PVR, pulmonary lymph flow or lung water content in the KPI-
treated
groups, as well as reduced WBCs and platelets sequestered in the lungs. One
significant indication of improved pulmonary function in the KPI groups would
be
improved arterial oxygenation in the immediate post-bypass period.
The invention has been disclosed broadly and illustrated in reference to
representative embodiments described above. Those skilled in the art will
recognize that
various modifications can be made to the present invention without departing
from the
spirit and scope thereof.
61