Note: Descriptions are shown in the official language in which they were submitted.
CA 02330558 2000-10-30
WO 99158514 PCT/US99/10212
-1-
~-ACYLAMINOTHIAZQLE-4-YL1ACETIC ACID DERIVATIVES
BACKGROUND OF THE INVENTION
The prevalence of insulin resistance in glucose intolerant subjects has long
been
recognized. Reaven et al (American Journal of Medicine 1976, 60, 80) used a
continuous infusion of glucose and insulin (insulin/glucose clamp technique)
and oral
glucose tolerance tests to demonstrate that insulin resistance existed in a
diverse group
of nonobese, nonketotic subjects. These subjects ranged from borderline
glucose
tolerant to overt, fasting hyperglycemia. The diabetic groups in these studies
included
both insulin dependent (IDDM) and noninsulin dependent (N>I3DM) subjects.
Coincident with sustained insulin resistance is the more easily determined
hyperinsulinemia, which can be measured by accurate determination of
circulating
plasma insulin concentration in the plasma of subjects. Hyperinsulinemia can
be
present as a result of insulin resistance, such as is in obese and/or diabetic
(NIDDM)
subjects and/or glucose intolerant subjects, or in >DDM subjects, as a
consequence of
over injection of insulin compared with normal physiological release of the
hormone by
the endocrine pancreas.
The association of hyperinsulinemia with obesity and with ischemic diseases of
the large blood vessels (e.g. atherosclerosis) has been well established by
numerous
experimental, clinical and epidemiological studies (summarized by Stout,
Metabolism
1985, 34, 7, and in more detail by Pyorala et al, DiabeteslMetabolism Reviews
1987,
3, 463). Statistically significant plasma insulin elevations at 1 and 2 hours
after oral
glucose load correlates with an increased risk of coronary heart disease.
Since most of these studies actually excluded diabetic subjects, data relating
the
risk of atherosclerotic diseases to the diabetic condition are not as
numerous, but point
in the same direction as for nondiabetic subjects (Pyorala et al). However,
the incidence
of atherosclerotic diseases in morbidity and mortality statistics in the
diabetic population
exceeds that of the nondiabetic population (Pyorala et al; Jarrett
DiabeteslMetabolism
Reviews 1989,5, 547; Harris et al, Mortality from diabetes, in Diabetes in
America
1985).
The independent risk factors obesity and hypertension for atherosclerodc
diseases are also associated with insulin resistance. Using a combination of
insulin/glucose clamps, tracer glucose infusion and indirect calorimetry, it
has been
demonstrated that the insulin resistance of essential hypertension is located
in peripheral
tissues (principally muscle) and correlates directly with the severity of
hypertension
CA 02330558 2000-10-30
WO 99/58514 PCT/US99/10212
-2-
(DeFronzo and Ferrannini, Diabetes Care 1991, 14, 173). In hypertension of the
obese,
insulin resistance generates hyperinsulinemia, which is recruited as a
mechanism to
limit further weight gain via thermogenesis, but insulin also increases renal
sodium
reabsorption and stimulates the sympathetic nervous system in kidneys, heart,
and
vasculature, creating hypertension.
It is now appreciated that insulin resistance is usually the result of a
defect in the
insulin receptor signaling system, at a site post binding of insulin to the
receptor.
Accumulated scientific evidence demonstrating insulin resistance in the major
tissues
which respond to insulin {muscle, liver, adipose), strongly suggests that a
defect in
insulin signal transduction resides at an early step in this cascade,
specifically at the
insulin receptor kinase activity, which appears to be diminished (reviewed by
Haring,
Diabetalogia 1991., 34, 848).
Protein-tyrosine phosphatases (PTPases) play an important role in the
regulation
of phosphorylation of proteins. The interaction of insulin with its receptor
leads to
phosphorylation of certain tyrosine molecules within the receptor protein,
thus
activating the receptor kinase. PTPases dephosphorylate the activated insulin
receptor,
attenuating the tyrosine kinase activity. PTPases can also modulate post-
receptor
signaling by catalyzing the dephosphorylation of cellular substrates of the
insulin
receptor kinase. The enzymes that appear most likely to closely associate with
the
insulin receptor and therefore, most likely to regulate the insulin receptor
kinase
activity, include PTP1B, LAR, PTPa and SH-PTP2 (B. J. Goldstein, J. Cellular
Biochemistry 1992, 48, 33; B. J. Goldstein, Receptor 1993, 3, 1-15,; F. Ahmad
and
B. J. Goldstein Biochim. Biophys Acta 1995,1248, 57-69).
McGuire et al. (Diabetes 1991, 40, 939), demonstrated that nondiabetic glucose
intolerant subjects possessed significantly elevated levels of PTPase activity
in muscle
tissue vs. normal subjects, and that insulin infusion failed to suppress
PTPase activity
as it did in insulin sensitive subjects.
Meyerovitch et al (J. Clinical Invest. 1989, 84, 976) observed significantly
increased PTPase activity in the livers of two rodent models of IDDM, the
genetically
diabetic BB rat, and the STZ-induced diabetic rat. Sredy et al (Metabolism,
44, 1074,
1995) observed similar increased PTPase activity in the livers of obese,
diabetic ob/ob
mice, a genetic rodent model of NIDDM.
The compounds of this invention have been shown to inhibit rat-derived and
human-derived recombinant PTPase-1B (rPTP-1B) in vitro . They arewseful in the
CA 02330558 2000-10-30
WO 99/58514 PCT/US99/10212
-3-
treatment of insulin resistance associated with obesity, glucose intolerance,
diabetes
mellitus, hypertension and ischemic diseases of the large and small blood
vessels.
2-Aminothiazoleacetic acid derivatives have been used extensively in chemical
and patent literature as intermediates for penam and cepham classes of
antibiotics, but
the long (C 16, C 1 g ) unsaturated carboxamide chains at C2 of the
thiazoleacetic acid
moiety makes these compounds novel. WO 9616650 and JP 07149745 generically
claim "lower alkyl" amides of 2-aminothiazoleacetic acid are antibacterial and
antiinflammatory (elastase inhibitors) agents, respectively. U.S. patent
5,688,821
( 1997, to AHP) teaches that some of the same compounds that are in this
invention are
inhibitors of the enzymes phospholipase Az derived from human sources (anti-
inflammatory agents), but others are not and vice-versa. Certain long
acylhydrocarbon
chain derivatives of 2-aminothiazoleacetic acid have been prepared (Toth,
Liebigs Ann.
Chem. EN, 7, 685, 1994).
DESCRIPTION OF THE INVENTION
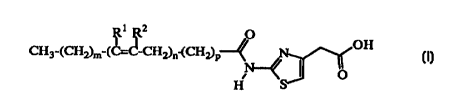
This invention provides a method of using a compound of formula I having the
structure
1 2
CH3-(CH2)n; (~ ~CH2)n-(CH2)~..~ N OH
P \
'O
H S
I
wherein
R1, R2 are both hydrogen or form a bond, or are each, independently, alkyl of
1-6 carbon atoms or aryl of 6-12 carbon atoms;
m=0-10;
n = 1-3; and
p = 0 -10;
with the proviso that m + p is less than or equal to 15;
or a pharmaceutically acceptable salt thereof in the treatment of metabolic
disorders
related to insulin resistance or hyperglycemia, primary hypertension, or
atherosclerosis.
Pharmaceutically acceptable salts can be formed from organic and inorganic
bases, such as alkali metals (sodium, potassium, or lithium), alkaline earth
metals
(calcium or magnesium), ammonium, primary, secondary alkyl amines, or tertiary
alkyl
CA 02330558 2000-10-30
WO 99/58514 PCT/US99l10212
-4-
amines. The use of tromethamine salts of the compounds of this invention
showed
improved water solubility and bioavailability.
It is understood that the compounds of this invention can exhibit E (trans) or
Z
(cis) stereoisomerism about the double bond, and that this invention covers
both the E
and Z isomers, at each double bond, and in particular when Rl and R2 are both
hydrogen, alkyl, or aryl. When R1 and R2 are not a bond, it is preferred that
they both
are hydrogen.
Preferred compounds of this invention are those in which:
m= 1 andn=3 andp=6;
m=4andn=3 andp=3;
m=Sandn=landp=6or8;
m = 7 and n = 1 and p = 6; and
m= l0andn= 1 andp=3.
Aryl is defined as an organic radical derived from an aromatic hydrocarbon by
the removal of a hydrogen {i.e., phenyl from benzene). It is preferred that
the aryl
moiety is a phenyl or naphthyl group; with phenyl being most preferred. The
aryl
moiety may be optionally mono-, di-, or tri- substituted with a substituent
selected from
the group consisting of alkyl of 1-6 carbon atoms, alkoxy of 1-6 carbon atoms,
trifluoromethyl, halogen, alkoxycarbonyl of 2-7 carbon atoms, alkylamino of 1-
6
carbon atoms, and dialkylamino in which each of the alkyl groups is of 1-6
carbon
atoms, vitro, cyano, -C02H, alkylcarbonyloxy of 2-7 carbon atoms, and
alkylcarbonyl
of 2-7 carbon atoms.
The compounds of this invention can be synthesized by saponification of the
corresponding 2-fatty acylaminothiazole-4-acetic acid ethyl esters, followed
by
acidification of the reaction mixture. Basic salts of these acids are prepared
also in a
conventional manner as is known in the art. In particular the tromethamine
salts of this
invention provide water soluble derivatives for improved bioavailability. The
fatty
acylaminothiazoleacetic acid esters are prepared in one of two ways:
condensation of
ethyl-2-aminothiazoleacetate~with a fatty acid chloride in the presence of a
tertiary amine
base such as triethylamine or diisopropylethylamine in an aprotic solvent
(e.g.
dichloromethane ar tetrahydrofuran) at ice temperature (Procedure A) or,
preferably,
directly from the fatty acid with the aid of an organodiimide coupling agent
as is well
CA 02330558 2000-10-30
WO 99/5$514 PCT/US99/10212
-5-
known in the synthesis of peptides (Procedure B). The fatty aryl starting
materials are
either prepared according to standard chemical methodology (such as Wittig or
Peterson
olefination reactions and or modifications thereof, or by reductive dicarbonyl
coupling
reactions, such as those procedures of McMurry, Corey and Mukiyama), or from
commercially available starting materials General procedures to prepare
representative
compounds of this invention are disclosed in U.S. Patent 5,688,821, which is
hereby
incorporated by reference, and the preparation of specific representative
compounds of
this invention are provided in Examples which follow.
The compounds of this invention are useful in treating metabolic disorders
related to insulin resistance or hyperglycemia, typically associated with
obesity or
glucose intolerance. The compounds of this invention are therefore,
particularly useful
in the treatment or inhibition of type II diabetes. The compounds of this
invention are
also useful in modulating glucose levels in disorders such as type I diabetes.
Additionally, because an association exists between insulin resistance and
hypertension
and between insulin resistance, hypertension and coronary artery disease, the
compounds of this invention are also useful for the treatment of primary
(essential)
hypertension and atheroscierosis.
The ability of compounds of this invention to treat or inhibit disorders
related to
insulin resistance or hyperglycemia was established with representative
compounds of
this invention in the following pharmacological test procedures which measure
the
inhibition of PTPase.
Inhibition of recombinant rat protein rosine nhosphatase 1 B (a°PTP 1
B~ activi , using
p-nitrophenylphospha~te as a substrate
measurement of Np PPase activ'~t3r. The assay is conducted as described by
Moss
(Acid Phosphatases. In: Methods of Enzymatic Analysis, Enzymes 2: Esterases,
Glycosidases, Lyases, Ligases. Bergmeyer, H.U., ed. Weinheim: Verlag Chemie
GmBH, 1984: 92) and Tonks, et al., J. Biol. Chem . 263, 6731, 1988), with
minor
modifications. The incubation mixture contains in a final volume of 0.24 ml:
50 mM
HEPES (pH 7.4), 6.33 mM p-nitrophenyl phosphate and 5/25/100 pNi compound
suspended in 1.25% DMSO. rPTPIB (obtained from the laboratory of Dr. Barry
Goldstein of Thomas Jefferson University. The enzyme (see Goldstein et al.
Mol.
Cell. Bioclzem. 109, 107, 1992), in microvials containing S00-700 pg/ml
protein in
CA 02330558 2000-10-30
WO 99/58514 PCT/US99/10212
-6-
33 mM TRIS-HC1, 2 mM EDTA, 10% glycerol and 10 mM 2-mercaptoethanol) is
preincubated with drug in HEPES buffer for ten minutes at 37°C. The
reaction is
started by the addition of p-nitrophenyl phosphate and after 30 minutes at
37°C, the
reaction is terminated by adding 1 ml of 0.1 N NaOH. The assay is performed in
triplicate and the reaction mixture contains approximately 3.3 lrg/ml protein,
the above
components, plus: 5.50 mM TRIS-HCI, 8.33 mM 2-mercaptoethanol, 0.33 mM EDTA
and 1.67% glycerol. The samples are read at 410 nm in a spectrophotometer and
are
evaluated based on a calibration curve of p-nitrophenol standard solution.
Compounds were screened robotically at a single concentration of ~25 pNi. The
results are expressed as percent of control, in that the amount of p-
nitrophenol formed
in the compound treated samples (nmol/minute/mg protein) is compared to the
amount
(nrnol/minute/mg protein) formed in the untreated samples. p-
Nitrophenylphosphatase
activity is also determined in each experiment and is expressed per minute per
mg
protein. Representative results are given in Table 1.
Inhibition of recombinant human protein tytosine ~hosDhatase 1B
A representative compound (Example 8) of this invention was also evaluated for
inhibition of recombinant human PTP1B.
Human recombinant PTP1B was prepared as described by Goldstein (see
Goldstein et al. Mol. Cell. Biochem. 109, 107, 1992). The enzyme preparation
used
was in microtubes containing 500-700 pg/ml protein in 33 mM Tris-HCI, 2 mM
EDTA,
10% glycerol and 10 mM 2-mercaptoethanol.
~Vleasurement of PTPase activity The malachite green-ammonium molybdate
method, as described (Lanzetta et al. Anal. Biochem. 100, 95, 1979) and
adapted
for a platereader, is used for the nanomolar detection of liberated phosphate
by
recombinant PTP1B. The assay uses, as substrate, a dodecaphosphopeptide custom
synthesized by AnaSpec, Inc. (San Jose, CA). the peptide, TRDIYETDYYRK,
corresponding to the 1142-1153 catalytic domain of the insulin receptor, is
tyrosine
phosphorylated on the 1146, 1150, and 1151 tyrosine residues. The recombinant
rPTPIB is diluted with buffer (pH 7.4, containing 33 mM Tris-HCI, 2 mM EDTA
and
50 mM b-mercaptoethanol) to obtain an approximate activity of 1000-2000
nmoles/min/mg protein. The diluted enzyme (83.25 mL) is preincubated for 10
min at
37oC with or witlhout test compound (6.25 mL) and 305.5 rnL of the 81.83 mM
HEPES reaction buffer, pH 7.4 peptide substrate, 10.5 ml at a final
concentration of 50
mM, and is equilibrated to 37°C. in a LABLINE Mufti-Blok heater
equipped with a
CA 02330558 2000-10-30
WO 99/58514 PGT/US99/10212
_7_
titerplate adapter. The preincubated recombinant enzyme preparation (39.5 ml)
with or
without drug is added to initiate the dephosphorylation reaction, which
proceeds at
37°C for 30 min. The reaction is terminated by the addition of 200 mL
of the
malachite green-ammonium molybdate-Tween 20 stopping reagent (MG/AM/Tw). The
stopping reagent consists of 3 parts 0.45% malachite green hydrochloride, 1
part 4.2%
ammonium molybdate tetrahydrate in 4 N HCI and 0.5% Tween 20. Sample blanks
are
prepared by the addition of 200 mL MG/AMlTw to substrate and followed by 39.5
ml
of the preincubated recombinant enzyme with or without drug. The color is
allowed to
develop at room temperature for 30 min. and the sample absorbances are
determined at
650 nm using a platereader (Molecular Devices). Sample and blanks are prepared
in
quadruplicates.
PTPase activities, based on a potassium phosphate standard curve, are
expressed as nmoles of phosphate released/min/mg protein. Inhibition of
recombinant
PTP1B by test campounds is calculated as percent of phosphatase control. A
four
parameter non-linear logistic regression of PTPase activities using SAS
release 6.08,
PROC NLIN, is used for determining IC50 values of test compounds.
Representative
results are given in Table 2 .
TABLE 1. Inhibition of recombinant phosphotyrosine phosphatase 1B by compounds
of this invention.
Example % inhibidona
1 93.4
2 93.9
3 58.16
4 84.8
94.97
6 93.77
7 83.8
8 96.6
9 92.4
(NH4)Mo7024 4 H20 92.3
Na3V04 86.3
a All compounds administered at 25 p.M
CA 02330558 2000-10-30
WO 99/58514 PCT/US99/10212
_g_
TABLE 2. Dose-response inhibition data and ICSp for Example 8 vs rl?TP1B
(human)
% inhibition Dose (E.~M)
100 10
96.8 2.5
94.94 1
73.21 0.5
43.25 0.25
25.53 0.1
IC50: 0.267
Based on the results obtained in the standard pharmacological test procedures,
representative compounds of this invention have been shown to inhibit PTPase
activity,
and are therefore useful in treating metabolic disorders related to insulin
resistance or
hyperglycemia, typically associated with obesity or glucose intolerance. More
particularly, the compounds of this invention are useful in the treatment or
inhibition of
type II diabetes, and in modulating glucose levels in disorders such as type I
diabetes.
The compounds of this invention are also useful in the treatment of primary
(essential)
hypertension and atherosclerosis. As used herein, the term modulating means
maintaining glucose levels within clinically normal ranges.
Effective administration of these compounds may be given at a daily dosage of
from about 1 mg/kg to about 250 mg/kg, and may given in a single dose or in
two or
more divided doses. Such doses may be administered in any manner useful in
directing
the active compounds herein to the recipient's bloodstream, including orally,
via
implants, parenterally (including intravenous, intraperitoneal and
subcutaneous
injections), rectally, vaginally, and transdermally. For the purposes of this
disclosure,
transdermal administrations are understood to include all administrations
across the
surface of the body and the inner linings of bodily passages including
epithelial and
mucosal tissues. Such administrations may be carried out using the present
compounds, or pharmaceutically acceptable salts thereof, in lotions, creams,
foams,
patches, suspensions, solutions, and suppositories (rectal and vaginal).
Compounds of this invention may be administered neat or with a pharmaceutical
carrier to a patient in need thereof. The pharmaceutical Garner may be solid
or liquid.
CA 02330558 2000-10-30
WO 99/58514 PCT/US99/10212
-9-
Oral formulations containing the active compounds of this invention may
comprise any conventionally used oral forms, including tablets, capsules,
buccal forms,
troches, lozenges and oral liquids, suspensions or solutions. Capsules may
contain
mixtures of the active compounds) with inert fullers and/or diluents such as
the
pharmaceutically acceptable starches (e.g. corn, potato or tapioca starch),
sugars,
artificial sweetening agents, powdered celluloses, such as crystalline and
microcrystalline celluloses, flours, gelatins, gums, etc. Useful tablet
formulations may
be made by conventional compression, wet granulation or dry granulation
methods and
utilize pharmaceutically acceptable diluents, binding agents, lubricants,
disintegrants,
suspending or stabilizing agents, including, but not limited to, magnesium
stearate,
stearic acid, talc, sodium lauryl sulfate, microcrystalline cellulose,
carboxymethyl-
cellulose calcium, polyvinylpyrrolidone, gelatin, alginic acid, acacia gum,
xanthan
gum, sodium citrate, complex silicates, calcium carbonate, glycine, dextrin,
sucrose,
sorbitol, dicalcium phosphate, calcium sulfate, lactose, kaolin, mannitol,
sodium
chloride, talc, dry starches and powdered sugar. Oral formulations herein may
utilize
standard delay or time release formulations to alter the absorption of the
active
compound(s). Suppository formulations may be made from traditional materials,
including cocoa butter, with or without the addition of waxes to alter the
suppository's
melting point, and glycerin. Water soluble suppository bases, such as
polyethylene
glycols of various molecular weights, may also be used.
It is understood that the dosage, regimen and mode of administration of these
compounds will vary according to the malady and the individual being treated
and will
be subject to the judgment of the medical practitioner involved. It is
preferred that the
administration of one or more of the compounds herein begin at a low dose and
be
increased until the desired effects are achieved.
The following procedures describe the preparation of representative examples
of
this invention.
Example 1 (Procedure A): 2-[((Z)-1-Oxo-9-octadecenyl)amino]-4-thiazole-
acetic acid
A mixture of ethyl 2-aminothiazoleacetic acid (4:6 g, 24.7 mmol) and
triethylamine (4.2 mL, 20.3 mmol) in dichloromethane (125 mL) was cooled in an
ice
bath under N2 atmosphere. Oleoyl chloride (neat 75%, 11 mL, 25 mmol) was added
CA 02330558 2000-10-30
WO 99/58514 PC'T/US99/10212
- 10-
dropwise and the reaction was then allowed to warm to room temperature. After
15 h at
room temperature 10% aqueous HCI solution was added to the reaction mixture,
stirred
for 2 h and then poured onto saturated aqueous brine solution in a separatory
funnel.
The organic layer was separated, dried over anhydrous MgS04, filtered and
concentrated on a rotary evaporator to a yellow oil. The crude ester was
purified by
HPLC (70% hexane, 30% EtOAc) to give 4 g of ethyl-2-(((Z)-1-oxo-9-
octadecenyl)amino)-4-thiazoleacetate, as a yellow oil. IR (film) a (crri 1 ):
1745, 1700.
MS (EI) m/z 450 (M+).
Ethyl-2-[((Z)-1-oxo-9-octadecenyl)amino]-4-thiazoleacetate (4 g, 8.89 mmol),
sodium hydroxide (0.79 g, 19.8 mmol) and tetrahydrofuran (50 mL) were combined
and cooled in an ice bath under N2 atmosphere. Enough distilled water was
added to
the mixture to dissolve the hydroxide and provide a homogeneous solution. The
reaction was kept at 0°C for 2 h, then allowed to warm to room
temperature and stirred
overnight. Aqueous 10% HCl solution was added to the mixture, and stirring
continued for 1 h. Ethyl acetate and saturated aqueous brine solution were
added to the
mixture, and the organic phase was separated and dried over MgS04. After
filtration
and concentration the crude dark amber oil was treated with ether, stirred for
0.5 h and
the product was collected on a Buchner funnel and air dried to give 1.5 g of
the title
compound as a white solid, mp 106-108.5 °C.
Analysis for: C23H38N2O3S
Calc'd: C, 65.36; H, 9.06; N, 6.63
Found: C, 64.99; H, 9.19; N, 6.57
Example 2 (Procedure B): 2-[((E)-1-Oxo-9-octadecenyl)amino]-4-thiazole-
acetic acid
A mixture of elaidic acid (5.1 g, 17.7 mmol; 98%), ethyl-2-
aminothiazoleacetate
(3.3 g, 17.7 mmol), triethylamine (2.8 g, 27.7 mmol), 1-(3-
dimethylaminopropyl)-3-
ethyl carbodiimide hydrochloride (5.3 g, 26.9 mmol) and 4-
dimethylaminopyridine
(0.4 g, 3.3 mmol) was combined in dichloromethane (225 mL) at ice temperature
under
N2 atmosphere. The mixture was stirred at 0 °C for 1 h, then allowed to
warm to room
temperature and stirred for I S h. The reaction mixture was diluted with water
and the
organic phase was separated, washed with water and saturated brine solution,
dried
over MgS04, filtered and concentrated to give 7.1 g of ethyl-2-[((E)-1-oxo-9-
octadecenyl)-amino]-4-thiazoleacetate, which was used directly without
purification.
CA 02330558 2000-10-30
WO 99/58514 PCT/US99/10212
-11-
A mixture of the above ester {7.4 g, 16.5 mmol) and sodium hydroxide (1.44 g,
36.1 mmol) was combined in THF ( 150 mL) and cooled in ice under N2
atmosphere.
Enough distilled water was added portionwise until the hydroxide was dissolved
(- 15
mL) and the mixture was maintained at 0 °C for 2 h, then allowed to
warm to ambient
temperatures with stirring for 15 h. The reaction mixture was evaporated in
vacuo, the
residue cooled in ice and treated with 2M aqueous HCl solution with stirring.
The
white precipitate was collected by vacuum filtration, washed with water and
air dried.
This material was crystallized from boiling heptane, filtered hot, and dried
overnight on
an abderhalden apparatus (refluxing acetone) to give 4.6 g of the title
compound as a
white powder, mp 116-118 °C.
Analysis for: C23H38N203S
Calc'd: C, 65.37; H, 9.06; N, 6.63
Found: C, 65.33; H, 9.15; N, 6.59
Example 3: 2-[((E)-1-Oxo-9-octadecenyl)amino)-4-thiazoleacetic acid,
tromethamine (2-amino-2-hydroxymethyl)-1,3-propanediol) salt
A mixture of the title compound of Example 2 (3.0 g 7.1 mmol), tris
(hydroxymethyl)amino methane (0.86 g, 7.1 mmol) and absolute ethanol (75 mL)
were
heated on a hot plate until a homogeneous solution was obtained. The solution
was
allowed to stand at room temperature for several hours and the solvent removed
on a
rotary evaporator. The residue was slurried in toluene and the solvent removed
in-
vacuo. The glassy solid was heated under vacuum at 35°C for 24 h to
give 3.39 g of
the title compound, as a white solid, mp 163-164°C.
Analysis for: C27H49N306S
Calc'd: C, 59.64; H, 9.08; N, 7.73
Found: C, 59.46; H, 9.05; N, ?.89
Example 4: 2-[(1-Oxo-9-octadecenyl)amino)-4-thiazoleacetic acid
The title compound was prepared by Procedure A (example 1). The crude
product was crystallized from hot heptane to give lemon colored crystals, mp
95.5-
96.5°C.
Analysis for: C23H36N2~3S
Calc'd: C, 65.68; H, 8.63; N, 6.66
Found: C, 65.46; H, 8.53; N, 6.74
CA 02330558 2000-10-30
WO 99/5$514 PCT/US99/10212
-12-
Example S: 2-[((Z)-1-Oxo-6-octadecynyl)amino]-4-thiazoleacetic acid
The title compound was prepared by the method of procedure B (example 2).
The crude product was recrystallized from heptane on a steam bath to provide
white
S crystals, mp 104.5-IOS.S°C.
Analysis for: C23H38N2~3s
Calc'd: C, 65.37; H, 9.06; N, 6.63
Found: C, 6S.4S; H, 9.06; N, 6.58
Example 6: 2-[((Z)-1-Oxo-9-hexadecenyl)amino]-4-thiazoleacetic acid
The title compound was prepared by the method of procedure B. The crude
product was treated with hot heptane (steam bath) cooled to room temperature
and
1 S collected on a Buchner funnel. The material was air dried for several
hours, then in an
Abderhalden apparatus (refluxing acetone) overnight to provide the title
compound as a
white waxy solid, mp 108-110°C.
Analysis for: C21H34N203S
Calc'd: C, 63.92; H, 8.68; N, 7.10
Found: C, 64.27; H, 8.86; N, 7.32
Example 7: 2-[((E)-1-Oxo-9-hexadecenyl)amino]-4-thiazoleacetic acid
2S The title compound was prepared by the method of procedure B. The crude
acid was recrystallized from hot heptane to give the product as white
crystals, mp 117-
118oC.
Analysis for: C21H34N2~3S
Calc'd: C, 63.92 ; H, 8.69 ; N, 7.10
Found: C, 63.78 ; H, 8.73 ; N, 6.79
CA 02330558 2000-10-30
WO 99/585r4 PCT/US99/102I2
-13-
Example 8: 2-[((E)-1-Oxo-11-octadecenyl)amino]-4-thiazoleacetic acid
The title compound was prepared according to the method of procedure B. The
crude product was crystallized from hot heptane to give white crystals, mp i
15.8
117.1oC.
Analysis for: C23H38N2O3S
Calc'd: C, 61.99; H, 9.06; N, 6.63
Found: C, 65.74; H, 8.85; N, 6.32
Example 9: 2-[((Z,Z,Z)-1-Oxo-9,12,15-octadecatrienyl)amino]-4-
thiazoleacetic acid
The title compound was prepared by procedure B and precipitated from heptane
as a hygroscopic yellow wax.
Analysis for: C23H34N2~3S' 1.5 H20
Calc'd: C, 61.99; H, 7.92; N, 6.29
Found: C, 62.22; H, 7.68; N, 6.02
Example 10: 2-[((Z,Z,Z)-1-Oxo-6,9,12-octatrienyl)amino]-4-thiazole-
acetic acid
The title compound was prepared according to procedure B. The crude product
was chromatographed on Si02 (flash column, 40 wt. eq., elution with hexane
(70%),
ethyl acetate (30%), acetic acid ( 1 %)) to give the title compound as a pale
yellow wax.
Analysis for: C23H34N2~3S
Calc'd: C, 65.99 ; H, 8.91; N, 6.69
Found: C, 65.76 ; H, 8.33; N, 5.50