Note: Descriptions are shown in the official language in which they were submitted.
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127 -
I~.\1'RI:SSION U1~ l~ UNC'1'IUNA1. I?UKrIR~'OTIC PROTEINS
" _
The Government has cetlain rights to this invention pursuant to Grant Nos.
N0014-
96-1-U34U and N00014-98-1-0657, awarded by the United States Navy.
'I~his application claims priority from U.S. application No. 60/094,403 filed
on July
i ?5, I~)~)S and No. ~U/1U6,84U filed November 3, 1998.
I3ACKGRUUND UI~ TI-iC INVENTION
Field of the Invention
I 0 'this invention relates to methods for the selection and production of
polynucleotides
that encode functional poiypeptidcs or proteins, especially eukaryotic
proteins, and
particularly in facile host cell expression systems. Facile expression systems
include robust
prokaryotic cells (e.g. bacteria) and eukaryotic systems (e.g. yeast). In
particular, the
invention concerns the recombinant production of expression-resistant
functional eukaryotic
15 proteins by host cells, in high yield, and without deactivation,
denaturation, inclusion bodies,
or other loss of structure or function. In preferred embodiments, the
expressed proteins are
secreted by the host cells. Preferred proteins of the invention include
peroxidases and heme-
containing proteins, such as horseradish peroxidase (HRP) and cytochrome c
peroxidase
(CCP). Polynucleotides which encode and express these proteins in recombinant
host cell
20 expression systems are also encompassed by the invention.
Description of Related Art
The publications and reference materials noted herein and listed in the
appended
Bibliography are each incorporated by reference in their entirety. They are
referenced
25 numerically in the text and the Bibliography below.
Many proteins of interest are produced by organisms having "eukaryotic" cells.
These are cells having a nucleus surrounded by its own membrane and containing
DNA on
structures called chromosomes. All multicellular organisms, such as humans and
animals,
and many single-cell animals, have eukaryotic cells. Other single-cell
organisms, such as
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127 -
bacteria have "prokaryotic" cells. These cells have a primitive nucleus with
DNA in a
defined structure, but without chromosomes and a nuclear membrane that is
characteristic, _
of eukaryotes. Prokaryotic organisms are generally mucl7 easier and less
costly to grow,
maintain and manipulate than eukaryotic cells.
Genetic engineering and recombinant DNA and RNA technologies have made it
possible to produce proteins, hormones and enzymes that arc native to one
organism, by
using the cells of a different organism as "factories" or host cell expression
systems. In
particular, it is often desirable to express a protein ofeukaryotic origin in
a prokaryotic host
cell, because the prokaryotes can be grown in large quantities of identical
cells, to product
large amounts of the desired foreign protein. For example, certain human
proteins may be
useful as drugs if they can be supplied in sufficient quantity to patients who
have a protein
deficiency. Such proteins may not easily or ethically be obtained by isolating
them from
human cells, nor can they easily be made by direct chemical synthesis or by
growing them
in isolated tissue cultures. Other proteins and enzymes are useful in
industry. For example,
certain enzymes can break down food products, and are useful in laundry
detergent.
However, commercial applications require large amounts of protein and a high
degree of
quality control.
To solve some of these problems, recombinant genetic engineering techniques
have
been developed to use genetic machinery of other cells, such as bacteria and
yeast, to
produce human or other proteins. Selected genetic material, such as a
polynucleotide that
encodes a desired protein, is "recombined" with genetic material in a host
cell, so that the
host cell expresses the introduced foreign genetic material and produces the
desired
polypeptide or protein. Bacteria and yeast can be suitable host cells because
they are easy
and economical to grow and maintain in large quantities, and can be used to
reliably and
repeatably produce foreign proteins.
However, many proteins can not easily be expressed in foreign host cells,
including
bacteria and yeast. Such expression-resistant polypeptides or proteins may not
be expressed
at all, or are expressed inefficiently, e.g. in low yield. The protein may be
expressed, but can
lose some or all of its or function. In some cases the expressed protein may
lose some or all
of its active folded structure, and may even become denatured or completely
inactive.
Expressed proteins may also be encapsulated inside inclusion bodies within a
host cell.
These are discrete particles or globules inside and separate from the rest of
the cell, and
_2_
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00106718 PCT/US99/17127
which contain expressed protein, perhaps in agglomerated or inactive form.
This makes it
difficult to harvest the produced protein from the host cells, as the
isolation and purification'
techniques can be difficult, inefficient, time-consuming and costly. Efforts
to, produce
expression-resistant polypeptides in active or functional form and at
relatively high yields
have spanned many years and have been markedly unsuccessful. In particular,
expression-
resistant enzymes that arc commercially important, such as peroxidase enzymes
like
horseradish peroxidase, have not been functionally expressed in reasonably
high yield or in
convenient, economical or facile host cells. These enzymes arc instead
produced in non-
funetional or inactive form, for example as inclusion bodies, and are
laboriously
manipulated and reconstituted to obtain active enzymes at relatively poor
yields.
Some proteins that are made by cells can be secreted or delivered outside the
cell,
which can improve the yield and the efficiency of subsequent isolation and
purification
steps. However, many proteins are not naturally secreted, and are difficult to
secrete
artificially, for example because they contain chemical groups that do not
easily cross the
cell membrane. in particular, it is difficult to engineer a compatible protein
and host cell
system to secrete a protein that has a tendency to form inclusion bodies.
Therefore,
improved techniques for expressing foreign proteins are needed, particularly
proteins of
eukaryotic origin, and particularly recombinant proteins which can be secreted
by host cells
in high yield, and without loss of activity or function.
As discussed, a particular challenge when producing foreign proteins in a host
cell
expression system is the inability of many foreign proteins to fold properly
into functional
proteins when using common recombinant hosts such as E. coli and yeast ( 1-4).
As a result,
the polypeptide chains that are produced in a recombinant host cell system are
often
degraded upon synthesis or accumulate in inclusion bodies. This is
particularly true for
eukaryotic proteins that contain disulfide bonds or are glycosylated in the
native form. The
underlying reasons, which are not clearly understood and are probably
multifactorial, may
include the "unnatural" recombinant environments in which the proteins
accumulate (35)
and the lack of proper folding cofactors such as molecular chaperones in the
E. coli host (3).
Additionally, glycosylation has been implicated in protein folding in
eukaryotic organisms
(36), which function is absent in bacteria.
The folding problem presents a challenging roadblock to the large-scale
production
of proteins for pharmaceutical or industrial applications. The lack of high-
efficiency
-3-
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127
functional expression systems has also become one of the bottlenecks in
applying directed
evolution techniques Cor optimizing proteins and reaction conditions for
desired uses. ~ _
Employing random mutagcnesis and Lenc recombination follow~cd by screening or
selection,
directed evolution has been successfully applied to improve a variety of
enzyme properties,
_S such as substrate specificity, activity in organic solvents, and stability
at high temperatures,
which arc often critical for industrial applications {5). Eukaryotic enzymes
have a myriad
of existing and potential applications, but improvement of these proteins by
directed
evolution had been limited by tl~c inability to functionally express them in a
facile
recombinant host.
For example, the difficulty of expressing peroxidase enzymes in a facile
expression
host has posed at least two technical challenges for realizing the potential
of peroxidases
as biocatalysts. First, efforts to modi fy these enzymes for industrial
applications by protein
engineering methods have been impeded. Directed evolution, for example,
exploits
expression in a host such as E. colt or S. cerevisiae, organisms in which
large libraries of
mutants or variants can be made. Second, the lack of efficient expression in
an appropriate
foreign (heterologous) host prevents the mass production of some of these
proteins on an
economical scale.
One way to obtain the active form of recombinantly expressed proteins is by
refolding them in vitro from inclusion bodies, but these processes are often
laborious and
inefficient (1-3). Additionally, this is not a viable option for directed
evolution in which
screening of tens of thousands of mutants is required. A more advantageous
means to
resolve the problem may be to identify mutations in a target gene that can
facilitate folding
in host environments. Evidence from a number of studies increasingly suggests
that certain
residues of an amino acid sequence have a profound influence on the folding
per se of the
protein. Thus, it would be highly advantageous if scientists could identify
mutations in a
target gene that facilitate folding in the host environment. This may avoid
the inclusion
body obstacle, but such techniques require the discovery, identification, and
use ofparticular
beneficial mutants.
For example, a series of studies by King and coworkers have shown that several
single amino acid substitutions interfered with the productive folding of the
phage p22
tailspike protein at restrictive temperature iu vivo, and that second-site
suppresser mutations
were able to rescue the defective folding mutants (6). In another study, the
replacement of
-4-
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17IZ7
tyrosine 35 with leucine in bovine pancreatic trypsin inhibitor (BPTI)
eliminated kinetic
traps in the folding pathway i~t vitro (7). Furthem~ore, it was reported that
several mutantb _
of human intcrleukin 1 (3, created by cassette mutagenesis of a tew selected
residues, were
expressed in E. coli in soluble form, while the wild type was largely
insoluble and formed
inclusion bodies (8). In a separate study, a single site-directed mutation was
found to
improve the folding yield of a recombinant antibody (9).
It is difficult to predict which residues arc critical for protein function or
stability,
let alone folding. Thus, it would be advantageous if there was a method for
systematically
searching for beneficial mutations that affect the folding and expression of
proteins, without
l0 compromising biological activity. Directed evolution techniques may prove
useful in the
accomplishment of this goal. This evolutionary approach uses DNA shuffling,
for
simultaneous random mutagenesis and recombination, to generate a variant
having an
improved desirable property over the existing wild type protein. Point
mutations arc
generated due to the intrinsic infidelity of Taq-based polymerase chain
reactions (PCR)
associated with reassembly of nucleic acid sequences. In one example, Stemmer
and
coworkers applied this technique to the gene encoding for green fluorescence
protein (GFP),
which resulted in a protein that folded better than the wild type in E. coli
(10).
One group of proteins of particular interest are heme proteins, that is, they
have iron
containing heme groups. These proteins have many biological and biochemical
uses, and
include certain enzymes called peroxidases, which are enzymes that facilitate
oxidation or
reduction reactions in which a peroxide (e.g. hydrogen peroxide) is one of the
reactants.
Peroxides are compounds, other than molecular Oz, in which oxygen atoms are
joined to
each other. For example, the heme enzyme horseradish peroxidase (HRP) is
widely used
as a reporter in diagnostic assays. HRP catalyzes a reaction in which starting
materials or
substrates are chemically combined in the presence of a peroxide, such as
hydrogen peroxide
(HzOz), with water (Hz0) as a byproduct. This reaction can be exploited to
indicate whether
another reaction of interest has occurred, or whether certain materials, such
as HRP starting
materials, are present in a mixture or sample. It would be beneficial to
provide a means of
producing large quantities of HR.P, and other heme or peroxidase enzymes,
using efficient
and cost-effective systems such as prokaryotic expression systems. However,
native HRP
contains four disulfides and is highly giycosylated (--21%), although the
carbohydrate
moiety has no apparent effect on the activity or stability (11). As a
consequence, previous
-5-
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127
~ ~vi vv ~ v vv ~ ~Vdv
attempts to express HRP in bacteria have yielded inclusion bodies, with no
functional
expression (12-14). Successful expression in yeast has also not been achieved
prior to this»
i nvenuon.
Accordingly, there is a need to develop new and improved methods for
expressing
S groteins which ordinarily have difficulty being expressed in order to
obviate the need for
laborious iu vitro folding protocols. In particular, there is a need for
protein expression
methods which arc well-suited for use in connection with directed evolution
techniques.
In particular, this invention describes methods torscrecning libraries of HRP
mutants
produced by error-prone PCR and DNA shuffling to identify mutations that
facilitate
functional expression in bacteria (E. coli, 13. siibtilis) and yeast (S.
cc~reusine). In one
exemplary embodiment, the variant of the invention is a functional and active
horseradish
peroxidase (HRP) that is expressed in E. coli without inclusion bodies at
levels of about 110
p.g/L. This is comparable to amounts previously obtained from much more
costly, time-
consuming and laborious in vitro refolding techniques used to recover other
HRP enzymes
from inclusion bodies.
SUMMARY OF THE INVENTION
The observed constraints on the use of native proteins are thought to be a
consequence of evolution. Proteins have evolved in the, context and
environment of a living
organism, to carry out specific biological functions under conditions
conducive to life - not
in the laboratory or under industrial conditions. In some cases, evolution may
favor or even
require less than optimally efficient enzymes. The output, efficiency, working
conditions,
stability and other properties of known expression systems are not thought to
be unalterable,
nor are they limitations which should be seen as intrinsic to the nature of
cellular expression
systems. It is possible that the proteins used in these systems can be evolved
in vitro, or that
analogous proteins can be otherwise developed, to alter or enhance the
protein's properties,
for example, to obtain much more efficient expression, folding, and secretion,
while
maintaining activity of the protein. Improved proteins can also be obtained by
screening
cultures of native organisms or expressed gene libraries (3).
Many proteins, when expressed using facile expression systems (e.g., E. coli)
result
in inclusion bodies or are inactive due to an inability to properly fold. The
invention takes
advantage of directed evolution techniques to create novel polynucleotides
encoding for
-6-
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127
mutated functional proteins which have an increased ability to be produced in
an expression
system, without inactivation or inclusion bodies. In preferred embodiments the
protein is -
secreted outside of the cell.
There are several advantages to secreting proteins from bacteria into the
culture
media. since in many cases desired substrates cannot readily pass through the
membranes
of E. coli. Secretion can facilitate screening in directed evolution studies,
because, by
allowing the secreted enzyme to catalyze a reaction in the culture medium,
substrates that
cannot enter the cells can be used. It can also significantly simplify the
production of
recombinant proteins, as the culture supernatant is largely free of
contaminating substances,
lU if the secretion level is high enough. Nonetheless, secretion of proteins
from bacteria into
culture media remains a di fficult task, particularly for enzymes that contain
bulky prosthetic
groups such as heme.
This problem can be solved by using a suitable signal peptide, such as the
signal
from the pectate lyase B (PeIB) of Em~inia carorovora (27), to efficiently
direct the
secretion of a peroxidase such as I-iRP or CCP into the culture medium. This
signal peptide
is also generally applicable to outer proteins containing heme prosthetic
groups, such as
cytochrome P450 enzymes and other peroxidases.
According to one embodiment of the invention, directed evolution or random
mutagenesis is used to produce in vitro proteins which readily fold after
expression, even
in yeast and in prokaryotic expression systems such as E. call, and are easily
secreted
outside the host cell in quantities expected for proteins produced by such
expression
systems. Furthermore, activity of these proteins is not compromised by the
mutagenic step
after appropriate selection is made.
Thus, the invention provides a method for improving the expression of a
~ polynucleotide encoding peroxidase enzymes by using directed evolution, and
polynucleotides encoding for variant horseradish peroxidase which have
improved
expression in conventional expression systems.
The above features and many other attendant advantages of the invention will
become better understood by reference to the following detailed description
when taken in
conjunction with the accompanying drawings.
_7_
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127
BR1EF DESCRIPTION OF THE DRAWTNGS
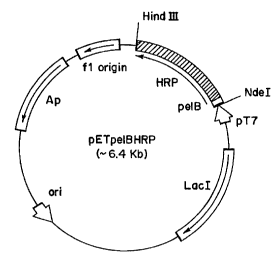
FIG. 1 A is a schematic map of an E. coli HRP expression vector pETHRP, the' -
plasmid pETpeIBI~RP. The 1-iRP gene (with an extra methioninc residue at the N-
terminus)
was inserted into pET-22b(+), immediately downstream of the signal sequence
from the
pectate lyase B (PeIB) of Crwiuia caro~oaora for periplasmic localization.
Expression is
under the control of the T7 promoter.
f IG. 1 Q is a schematic map of a I'. pasioris expression vector pPICZaB-HRP.
The
1-1 RP gene was inscricd immediately downstream of the plasmid's a-factor
signal.
Expression is under the control of the methanol-inducible P~"~, promoter.
FIG. 2 shows the nucleic acid and x1111110 aCld sequences of the pelB signal
peptide
1SEQ. ID. NO. 1 and SEQ. 1D. NO. 2).
F1G. 3 shows a nucleotide and amino acid sequence encoding an HRP enzyme
variant designated HRP1A6 (~SEQ. ID NO. 3 and SEQ. ID. NO. 4]).
FIC. 4 is a map of the expression vector pETpeIBHRP 1 AG.
FIG. SA shows the relative activities ofwild-type and an HRP mutant (I AG)
evolved
in E. coli.
FIG. 5B shows a representative landscape of first generation HRP mutants
sorted
by activity in descending order. Activities are normalized to that of wild-
type.
FIG. 6 shows activity levels of the mutant HRP 1 AG at various ITPG
concentrations.
FIG. 7 is a representation of the structure of HRP, showing the location of
the
Asn255 to Asp mutation in a surface loop of HRP mutant I AG. This figure was
generated
from published HRP coordinates (34), using Insight II software (Molecular
Biosystems).
FIG. 8 is a map of the expression vector pYEXS 1-HRP containing a coding
sequence for HRP cloned into the secretion plasmid pYEX-S I .
FIG. 9 shows the activity levels of HRPIA6 and three other mutants obtained by
directed evolution in S. cerevisiae: HRP 1-77E2, HRP I -11764, and HRP2-28DG.
In this
example HRP 1 A6 was the parent of HRP I -77E2 and HRP 1- I I 764. while HRP I
-11764
was the parent of HRP2-28D6.
FIG. 10 shows the residual activity of several HRP mutants as a function of
temperature, in a thermal inactivation curve that indicates the relative
thermostability of the
mutants.
_g_
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99117127
FIG. I1 shows the residual activity of several HRP mutants as a function of
hvdroeen peroxide concentration, in a titration curve that indicates the
relative ability of the, _
mutants to resist degradation in the presence of hydrogen peroxide.
FIG. 12 shows a nucleotide and amino acid sequence encoding an HRP enzyme
variant dcsi~:natcd HRP1-77E2 ([SEQ. 1D NO. 5 and SEQ. 1D. NO. 6]).
hIG. 13 shows a nucleotide and amino acid sequence encoding an I-1RP enzyme
variant designated HRP1-4B6 ((SEQ. ID NO. 7 and SEQ. ID. NO. 8J).
f IG. 14 shows a nucleotide and amino acid sequence encodine an HRP C117y111C
variant c9csi~.:natcd I-iRPI-2881 1 ([SEQ. ID NO. 9 and SEQ. ID. NO. 10]).
hIG. 15 shows a nucleotide and amino acid sequence encoding an HRP enzyme
variant dcsi~~nated HRPi-24D11 ((SEQ. 1D NO. 11 and SEQ. ID. NO. 12J).
FIG. 16 shows a nucleotide and amino acid sequence encoding an HRP enzyme
variant designated HRP1-1 1764 ((SEQ. ID NO. 12 and SEQ. ID. NO. 13]).
FIG. 17 shows a nucleotide and amino acid sequence encoding an HRP enzyme
variant designated HRP1-80C12 ([SEQ. ID NO. 17 and SEQ. ID. NO. 18]).
FIG. 18 shows a nucleotide and amino acid sequence encoding an HRP enzyme
variant designated HRP2-28D6 ([SEQ. ID NO. 19 and SEQ. ID. NO. 20]).
FIG. 19 shows a nucleotide and amino acid sequence encoding an HRP enzyme
variant designated HRP2-13A10 ([SEQ. ID NO. 21 and SEQ. ID. NO. 22]).
FIG. 20 shows a nucleotide and amino acid sequence encoding an HRP enzyme
variant designated HRP3-17E12 ([SEQ. ID NO. 23 and SEQ. ID. NO. 24J).
FIG. 21 shows the activities of wild-type, parent (HRP1A6) and evolved HRP
mutants in S. cerevisiae strain BJ5465. The values were obtained with the ABTS
assay .
Cells were grown in shaking flasks at 30°C for 64h.
FIG. 22 shows A) The correlation between reactivity and stability (A,~s;~/A;)
ofHRP
mutants.
FIG. 23 shows reactivity of HRP mutants in organic solvent / water systems.
F1G. 24 shows the lineage of the mutants. Nucleotide substitutions are shown
in
parentheses following the corresponding amino acid substitutions, and
synonymous
mutations in Italics. For each generation new mutations are donated with "*".
FIG. 25 shows the accumulation of secreted HRP activity from Pichia for the
variant
HRP3-17E2.
-9-
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127
FIG. 26 is a schematic map of the yeast cytochrome c peroxidase expression
vector
pETCCP
DETAILED DESCRIPTION OF THE INVENTION
_5 This in~~ention concerns methods for improving the expression of proteins
using
conventional expression systems, which proteins would ordinarily result in
inclusion bodies
or arc degraded upon synthesis due to an inability to fold properly in the
environment of the
expression system.
Definilion.s.
As used herein, "about" or "approximately" shall mean within 20 percent,
preferably
within 10 percent, and more preferably within 5 percent of a given value or
range.
The term "substrate" means any substance or compound that is convened or meant
to be converted into another compound by the action of an enzyme catalyst. The
tern
includes aromatic and aliphatic compounds, and includes not only a single
compound, but
also combinations of compounds, such as solutions, mixtures and other
materials which
contain at least one substrate.
An "oxidation reaction" or "oxygenation reaction", as used herein, is a
chemical or
biochemical reaction involving the addition of oxygen to a substrate, to form
an oxygenated
or oxidized substrate or product. An oxidation reaction is typically
accompanied by a
reduction reaction (hence the term "redox" reaction, for oxidation and
reduction). A
compound is "oxidized" when it receives oxygen or loses electrons. A compound
is
"reduced" (it loses oxygen or gains electrons).
The term "enzyme" means any substance composed wholly or largely of protein or
polypeptides that catalyzes or promotes, more or less speci fically, one or
more chemical or
biochemical reactions.
A "polypeptide" (one or more peptides) is a chain of chemical building blocks
called
amino acids that are linked together by chemical bonds called peptide bonds. A
protein or
polypeptide, including an enzyme, may be "native" or "wild-type", meaning that
it occurs
in nature; or it may be a "mutant", "variant" or "modified", meaning that it
has been made,
altered, derived, or is in some way different or changed from a native
protein, or from
another mutant. A "parent" polypeptide or enzyme is any polypeptide or enzyme
from
which any other polypeptide or enzyme is derived or made, using any methods,
tools or
-10-
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127
techniques, and whether or not the parent is itself a native or mutant
polypeptide or enzyme.
A parent polynucleotide is one that encodes a parent polypeptide. A "test
enzyme" is a~ _
protein-containing substance that is tested to dctcm~ine whether it has
properties of an
enzyme. The term "enzyme" can also refer to a catalytic polynucleotide (e.g.
RNA or DNA).
The "activity" of an enzyme is a measure of its ability to catalyze a
reaction, and may
be expressed as the rate at which the product of the reaction is produced. For
example,
enzyme activity can be represented as the amount of product produced per unit
of time, per
unit (e.g. concentration or weight) ofcnzyme. The "stability" ofan enzyme
means its ability
to function, over time, in a particular environment or under particular
conditions. One way
l0 to evaluate stability is to assess its ability to resist a loss of activity
over time, under given
conditions. Enzyme stability can also be evaluated in other ways, for example,
by
determining the relative degree to which the enzyme is in a folded or unfolded
state. Thus,
one enzyme is more stable than another, or has improved stability, when it is
more resistant
than the other enzyme to a loss of activity under the same conditions, is more
resistant to
unfolding, or is more durable by any suitable measure. For example, a more
"thermally
stable" or "thermostable" enzyme is one that is more resistant to loss of
structure (unfolding)
or function (enzyme activity) when exposed to heat or an elevated temperature.
One way
to evaluate this is fo determine the "melting temperature" or Tm for the
protein. The melting
temperature, also called a midpoint, is the temperature at which half of the
protein is
unfolded from its fully folded state. This midpoint is typically determined by
calculating
the midpoint of a titration curve that plots protein unfolding as a function
of temperature.
Thus, a protein with a higher Tm requires more heat to cause unfolding and is
more stable
or more thermostable. Stated another way, a protein with a higher Tm indicates
that fewer
molecules of that protein are unfolded at the same temperature as a protein
with a lower Tm,
again meaning that the protein which is more resistant to unfolding is more
stable (it has less
unfolding at the same temperature). Another measure of stability is T"~, which
is the
transition midpoint of the inactivation curve of the protein as a function of
temperature. T"~
is the temperature at which the protein loses half of its activity. Thus, a
protein with a
higher T"~ requires more heat to deactivate it, and is more stable or more
thermostable.
Stated another way, a protein with a higher T"~ indicates that fewer molecules
of that protein
are inactive at the same temperature as a protein with a lower T,n, again
meaning that the
protein which is more resistant to deactivation is more stable (it has more
activity at the
-11-
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127
same temperature). These assays are also called "thermal shift" assays,
because the
inactivation or unfolding curve, plotted against temperature, is "shifted" to
higher or lower w _
temperatures w~hcn stability increases or decreases. Thcnnostability can also
be measured
in other ways: For example, a longer half-life (t,;,} for the enzyme's
activity at elevated
temperature is an indication of thcmlostability.
An "oxidation enzyme" is an enzyme that catalyzes one or more oxidation
reactions,
typically by adding, inserting, contributing or transferring oxygen from a
source or donor
to a substrate. Such enzymes arc also called oxidorcductases or rcdox enzymes,
and
encompasses oxygcnascs, hydrogcnascs or reductases, oxidascs and pcroxidascs.
The terms "oxygen donor", "oxidizing agent" and "oxidant" mean a substance,
molecule or compound which donates oxygen to a substrate in an oxidation
reaction.
Typically, the oxygen donor is reduced (accepts electrons). Exemplary oxygen
donors,
which are not limiting, include molecular oxygen ordioxygen (02) and
peroxides, including
alkyl peroxides such as t-butyl peroxide, and most preferably hydrogen
peroxide (Hz02).
A peroxide is any compound having two oxygen atoms bound to each other.
A "luminescent" substance means any substance which produces detectable
electromagnetic radiation, or a change in electromagnetic radiation, most
notably visible
light, by any mechanism, including color change, UV absorbance, fluorescence
and
phosphorescence. Preferably, a luminescent substance according to the
invention produces
a detectable color, fluorescence or W absorbance.
The term "chemiluminescent agent" means any substance which enhances the
delectability of a luminescent (e.g., fluorescent) signal, for example by
increasing the
strength or lifetime of the signal. One exemplary and preferred
chemiluminescent agent is
5-amino-2,3-dihydro-1,4-phthalazinedione (luminol) and analogs.
Otherchemiluminescent
agents include 1,2-dioxetanes such as tetramethyl-1,2-dioxetane (TMD),1,2-
dioxetanones,
and 1,2-dioxetanediones.
The term "polymer" means any substance or compound that is composed of two or
more building blocks ('mgrs') that are repetitively linked to each other. For
example, a
"dimer" is a compound in which two building blocks have been joined together.
The term "cofactor" means any non-protein substance that is necessary or
beneficial
to the activity ofan enzyme. A "coenzyme" means a cofactor that interacts
directly with and
serves to promote a reaction catalyzed by an enzyme. Many coenzymes serve as
carriers.
-12-
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT1US99/17127
For example, NAD' and NADP~ cant' hydrogen atoms from one enzyme to another.
An
"ancillary protein" means any protein substance that is necessary
or.beneficial to the activity, _
of an enzyme.
The term "host cell" means any cell of any organism that is selected,
modified,
transformed, grown, or used or manipulated in any way, for the production of a
substance
by the cell, for example the expression by the cell of a gent, a DNA or RNA
sequence, a
protein or an enzyme.
"DNA" (deoxyribonucleic acid) means any chain or sequence of the chemical
building blocks adenine (A), guanine (G), cytosine (C) and thymine (T), called
nucleotide
bases, that arc linked together on a deoxyribose sugar backbone. DNA can have
one strand
ofnucleotide bases, or two complimentary strands which may form a double helix
structure.
"RNA" (ribonucleic acid) means any chain or sequence of the chemical building
blocks
adenine (A), guanine (G), cytosine (C) and uracil (U), called nucleotide
bases, that arc
linked together on a ribose sugar backbone. RNA typically has one strand of
nucleotide
bases.
A "polynucleotide" or "nucleotide sequence" is a series of nucleotide bases
(also
called "nucleotides") in DNA and RNA, and means any chain of two or more
nucleotides.
A nucleotide sequence typically carries genetic information, including the
information used
by cellular machinery to make proteins and enzymes. These terms include double
or single
stranded genomic and cDNA, RNA, any synthetic and genetically manipulated
polynucleotide, and both sense and anti-sense polynucleotide (although only
sense stands
are being represented herein). This includes single- and double-stranded
molecules, i.e.,
DNA-DNA, DNA-RNA and RNA-RNA hybrids, as well as "protein nucleic acids" (PNA)
formed by conjugating bases to an amino acid backbone. This also includes
nucleic acids
containing modified bases, for example thio-uracil, thio-guanine and fluoro-
uracil.
The polynucleotides herein may be flanked by natural regulatory sequences, or
may
be associated with heterologous sequences, including promoters, enhancers,
response
elements, signal sequences, poiyadenylation sequences, introns, 5'- and 3'-
non-coding
regions, and the like. The nucleic acids may also be modified by many means
known in the
ari. Non-limiting examples of such modifications include methylation, "caps",
substitution
of one or more of the naturally occurring nucleotides with an analog, and
internucleotide
modifications such as, for example, those with uncharged linkages (e.g.,
methyl
-13-
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127
phosphonates, phosphotriesters, phosphoroamidates, carbamates, etc.) and with
charged
linkages (e.g., phosphorothioates, phosphorodithioates, ctc.}. Polynucieotides
may contain» -
one or more additional covalently linked moieties, such as, for example,
proteins (e.g.,
nucleases, toxins, antibodies, signal peptides, poly-L-lysine, etc.),
intercalators (e.g.,
$ acridine, psoralen, etc.), chelators (e.g., metals, radioactive metals,
iron, oxidative metals,
ctc.}, and alkylators. The polynuclcotides may be dcrivatized by formation of
a methyl or
ethyl phosphotricster or an alkyl phosphoramidatc linkage. Furthermore, the
polynucleotides herein may also be modified with a label capable of providing
a detectable
signal, eithec directly or indirectly. Lxemplary labels include radioisotopes,
fluorescent
molecules, biotin, and the like.
Proteins and enzymes are made in the host cell using instructions in DNA and
RNA,
according to the genetic code. Generally, a DNA sequence having instructions
for a
particular protein or enzyme is "transcribed" into a corresponding sequence of
RNA. The
RNA sequence in tum is "translated" into the sequence of amino acids which
forn~ the
protein or enzyme. An "amino acid sequence" is any chain of two or more amino
acids.
Each amino acid is represented in DNA or RNA by one or more triplets of
nucleotides.
Each triplet forms a codon, corresponding to an amino acid. For example, the
amino acid
lysine (Lys) can be coded by the nucleotide triplet or codon AAA or by the
codon AAG.
(The genetic code has some redundancy, also called degeneracy, meaning that
most amino
acids have more than one corresponding codon.) Because the nucleotides in DNA
and RNA
sequences are read in groups of three for protein production, it is important
to begin reading
the sequence at the correct amino acid, so that the correct triplets are read.
The way that a
nucleotide sequence is grouped into codons is called the "reading frame."
The term "gene", also called a "structural gene" means a DNA sequence that
codes
for or corresponds to a particular sequence of amino acids which comprise all
or pan of one
or more proteins or enzymes, and may or may not include regulatory DNA
sequences, such
as promoter sequences, which determine for example the conditions under which
the gene
is expressed. Some genes, which are not structural genes, may be transcribed
from DNA
to RNA, but are not translated into an amino acid sequence. Other genes may
function as
regulators of structural genes or as regulators of DNA transcription.
A "coding sequence" or a sequence "encoding" a polypeptide, protein or enzyme
is
a nucleotide sequence that, when expressed, results in the production of that
polypeptide,
-14-
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127
protein or enzyme, i.e.. the nucleotide sequence encodes an amino acid
sequence for that
polypeptide, protein or enzyme- A coding sequence is "under the control" of
transcriptional~ -
and translational control sequences in a cell when RNA polymerise transcribes
the coding
sequence into mRNA, which is then trans-RNA spliced and translated into the
protein
_S encoded by the coding sequence. Preferably,.the coding sequence is a double-
stranded DNA
sequence which is transcribed and translated into a polypeptide in a cell iu
vitro or irr vivo
when placed under the control of appropriate regulatory sequences. The
boundaries of the
coding sequence arc determined by a start codon at the 5' (amino) terminus and
a translation
S(Ol) COdOn 1l the 3' (carboxyl) terminus. A coding sequence can include, but
is not limited
to, prokaryotic sequences, cDNA from eukaryotic mRNA, genomic DNA sequences
from
eukarvotic (e.g., mammalian) DNA, and even synthetic DNA sequences. If the
coding
sequence is intended for expression in a eukaryotic cell, a polyadenylation
signal and
transcription termination sequence will usually be located 3' to the coding
sequence.
Transcriptional and translational control sequences are DNA regulatory
sequences,
I5 such as promoters, enhancers, terminators, and the like, that provide for
the expression of
a coding sequence in a host cell. In eukaryotic cells, polyadenylation signals
are control
sequences.
A "promoter sequence" is a DNA regulatory region capable of binding RNA
polymerise in a cell arid initiating transcription of a downstream (3'
direction) coding
sequence. For purposes of defining this invention, the promoter sequence is
bounded at its
3' terminus by the transcription initiation site and extends upstream (5'
direction) to include
the minimum number of bases or elements necessary to initiate transcription at
levels
detectable above background. Within the promoter sequence will be found a
transcription
initiation site (conveniently defined for example, by mapping with nuclease
S1), as well as
protein binding domains (consensus sequences) responsible for the binding of
RNA
polymerise. As described above, promoter DNA is a DNA sequence which
initiates,
regulates, or otherwise mediates or controls the expression of the coding DNA.
A promoter
may be "inducible", meaning that it is influenced by the presence or amount of
another
compound (an "inducer"). For example, an inducible promoter includes those
which initiate
or increase the expression of a downstream coding sequence in the presence of
a particular
inducer compound. A "leaky" inducible promoter is a promoter that provides a
high
expression level in the presence of an inducer compound and a comparatively
very low
-15-
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99I17127
expression level, and at minimum a detectable expression level, in the absence
of the
_ " _
inducer.
A "signal sequence" is included at the beeinnint of the Iodine sequence of a
protein
to be expressed in the periplasmic space, or outside the cell. This sequence
encodes a signal
peptide, N-terminal to the mature polypeptide, that directs the host cell to
translocate the
polypeptidc. The tcmt "translocation signal sequence" is also used to refer to
a signal
sequence. Translocation signal sequences can be found associated with a
variety ofproteins
native to eukaryotes and prokaryotes, and arc often functional in both types
of organisms.
Proteins of llte invention may be further modified and improved by adding a
sequence which
1 p directs the secretion of the protein outside the host cell. The addition
of the signal sequence
does not interfere with the folding of the secreted protein, and evidence
thereof is easily
tested for using techniques known in the aru and depending on the protein
(e.g., tests for
activity of a given protein after modification).
Preferred signal sequences ofthe invention include the pelB signal sequence,
which
normally directs a protein to the periplasmic space between the inner and
outer membranes
ofbacteria. Other signal sequences include, for example ompA and ompT (52).
The signal
sequence is iigated upstream of the nucleotide sequence encoding the protein,
such that the
sequence is present at the N-tem~inus of the protein after expression.
Conventional cloning
techniques can be used as described. Some routine experimentation within the
scope of one
skilled in the art may be necessary to optimize addition of the signal
sequence to any given
protein.
The terms "express" and "expression" mean allowing or causing the information
in
a gene or DNA sequence to became manifest, for example producing a protein by
activating
the cellular functions involved in transcription and translation of a
corresponding gene or
DNA sequence. A DNA sequence is expressed in or by a cell to form an
"expression
product" such as a protein. The expression product itself, e.g. the resulting
protein, may also
be said to be "expressed" by the cell. A polynucleotide or polypeptide is
expressed
recombinantly, for example, when it is expressed or produced in a foreign host
cell under
the control of a foreign or native promoter, or in a native host cell under
the control of a
foreign promoter.
A polynucleotide or polypeptide is "over-expressed" when it is expressed or
produced in an amount or yield that is substantially higher than a given base-
line yield, e.g.
-16-
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127
a yield that occurs in nature. For example, a polypeptide is over-expressed
when the yield
is substantially greater than the normal, average or base-line yield of the
native -
polypol~~peptide in native host cells under given conditions, for example
conditions suitable
to the life cycle of the native host cells. Over-expression ofa polypeptide
can be obtained,
for example, by altering any one or more of: (a) the growth or living
conditions of the host
cells; (b) the polynucleotide encoding the polypeptide to be over-expressed;
(c) the promoter
used to control expression of the polynucleotide; and (d) the host cells
themselves. This is
a relative, and thus "over-expression" can also be used to compare or
distinguish the
expression level of one polypcptide to another, without regard for whether
either
polypcptidc is a native polypcptide or is encoded by a native polynucleotide.
Typically,
over-expression means a yield that is at least about two times a normal,
average or given
base-line yield. Thus, a polypeptide is over-expressed when it is produced in
an amount or
yield that is substantially higher than the amount or yield of a parent
polypeptide or under
parent conditions. Likewise, a poiypeptide is "under-expressed" when it is
produced in an
amount or yield that is substantially lower than the amount or yield of a
parent polypeptide
or under parent conditions, e.g. at least half the base-line yield. In this
context, the
expression level or yield refers to the amount or concentration of
polynucleotide that is
expressed, or polypeptide that is produced (i.e. expression product), whether
or not in an
active or functional form. As one example, a polynucleotide or polypeptide may
be said to
he under-expressed when it is expressed in detectable amounts under the
control of an
inducible promoter, but without induction, i.e. in the absence of an inducer
compound.
An expression product can be characterized as intracellular, extracellular or
secreted.
The term "intracellular" means something that is inside a cell. The term
"extracellular"
means something that is outside a cell. A substance is "secreted" by a cell if
it delivered to
the periplasm or outside the cell, from somewhere on or inside the cell.
As used herein, the terms "expression-resistant polypeptide" and "resistant to
functional expression" are synonymous and refer to a polypeptide that is
difficult to
functionally express in selected host cells. For example, an expression-
resistant polypeptide
is not produced, or is produced in very low yield or in non-functional form,
when a
polynucleotide encoding that polypeptide is transformed or introduced into
host cells, e.g.
into a facile host cell expression system.
-17-
SUBSTITUTE SHEET (RULE 2C)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127
These polypeptides include, for example, those which have disulfide bridges,
which
arc composed of mutiplc subunits. or which require glycosylation. Expression-
resistant,
polypeptides also include those which arc sensitive to folding and unfolding
conditions,
particularly intracellular conditions (inside the cell), such as temperature,
pH, protein
_5 concentration, and the presence or absence of certain cofactors, coenzymes,
ancillary
proteins, ctc. Expression-resistant polypcptidcs also include polypeptides
that are encoded
by polynucleotides which arc sensitive to particular promoters or signal
sequences in
particular expression systems. In addition, expression-resistant polypeptides
include those
which tend to agglomerate, form inclusion bodies, or which arc produced in a
non-active or
unfolded fom~.
Particularly suitable for use as expression-resistant parent polypeptides in
the
invention are polypeptides that are inactive (e.g. they agglomerate, etc.)
when produced at
a high yield (e.g. when they are over-expressed), but which are active (e.g.
they do nor
agglomerate, etc.) when produced at a very low yield (e.g. when they are under-
expressed).
These include, for example, polypeptides that: (a) tend to agglomerate, form
inclusion
bodies, or are inactive or unfolded, when expressed in the presence of an
inducer, by a
polynucleotide that is under the control of an inducible promoter; and (b)
tend not to
agglomerate, etc., and are active, when expressed without inducer, by a
polynucleotide that
is under the control of the inducible promoter. Such promoters are known and
can be called
"leaky" promoters.
Polypeptides that include, incorporate or are associated with heme groups are
also
examples of expression-resistant polypeptides. Particular expression-resistant
polypeptides
of the invention are prexidase enzymes, such as horseradish peroxidase
enzymes. An
"expression-resistant polynucleotide" is a polynucleotide that encodes an
expression
resistant polypeptide.
A gene encoding a protein of the invention for use in an expression system,
whether
genomic DNA or cDNA, can be isolated from any source, particularly from a
human eDNA
or genomic library. Methods for obtaining genes are well known in the art,
e.g., Sambrook
et al. ( 19).
Accordingly, any animal cell potentially can serve as the nucleic acid source
for the
molecular cloning of the gene of interest. The DNA may be obtained by standard
procedures known in the art, such as from cloned DNA (e.g., a DNA "library"),
from cDNA
_18_
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127
library prepared from tissues with high level expression of the protein, by
chemical
synthesis, by cDNA cloning, or by the cloning of genomic DNA, or fragments
thereof,, _
puri<<ed from the desired cell (19,51). Clones derived from genomic DNA may
contain
regulatory and intros DNA regions in addition to coding regions; clones
derived from
$ cDNA will not contain intros sequences.
In the molecular cloning of the gene from genomic DNA, DNA fragments arc
Lcncrated, sonic of which will encode the desired gene. The DNA may be cleaved
at
specific sifts using various restriction enzymes. Alternatively, one may use
DNAse in the
presence of manganese to fragment the DNA, or the DNA can be physically
sheared, as for
1 p example, by sonication. The linear DNA fragments can then be separated
according to size
by standard techniques, including but not limited to, agarose and
polyacrylamide gel
electrophoresis and column chromatography.
The tem "transformation" means the introduction of a "foreign" (i.e. extrinsic
or
extracellular) gene, DNA or RNA sequence to a host cell, so that the host cell
will express
I S the introduced gene or sequence to produce a desired substance, typically
a protein or
enzyme coded by the introduced gene or sequence. The introduced gene or
sequence may
also be called a "cloned" or "foreign" gene or sequence, may include
regulatory or control
sequences, such as start, stop, promoter, signal, secretion, or other
sequences used by a cell's
genetic machinery. The gene or sequence may include nonfunctional sequences or
20 sequences with no known function. A host cell that receives and expresses
introduced DNA
or RNA has been "transformed" and is a "transformant" or a "clone." The DNA or
RNA
introduced to a host cell can come from any source, including cells of the
same genus or
species as the host cell, or cells of a different genus or species.
The terms "vector", "cloning vector" and "expression vector" mean the vehicle
by
25 which a DNA or RNA sequence (e.g. a foreign gene) can be introduced into a
host cell, so
as to transform the host and promote expression (e.g. transcription and
translation) of the
introduced sequence.
Vectors typically comprise the DNA of a transmissible agent, into which
foreign
DNA is inserted. A common way to insert one segment of DNA into another
segment of
30 DNA involves the use of enzymes called restriction enzymes that cleave DNA
at specif c
sites (specific groups of nucleotides) called restriction sites. Generally,
foreign DNA is
inserted at one or more restriction sites of the vector DNA, and then is
carried by the vector
-19-
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127
into a host cell along with the transmissible vector DNA. A segment or
sequence of DNA
having inserted or added DNA, such as an expression vector, can also be called
a "DNA ~ -
construct."
A common type of vector is a "plasmid", which generally is a self contained
_S molecule of double-stranded DNA, that can readily accept additional
(foreign) DNA and
which can readily introduced into a suitable host cell. A plasmid vector often
contains
coding DNA and promoter DNA and has one or more restriction sites suitable for
inserting
foreign DNA. Promoter DNA and coding DNA may be from the same gene or from
different genes, and may be from the same or different organisms. A large
number of
i 0 vectors, including plasmid alld fungal vectors, have been described for
replication and/or
expression in a variety of eukaryotic and prokaryotic hosts. Non-limiting
examples include
pKK plasmids (Clonetech), pUC plasmids, pET plasmids (Novagen, lnc., Madison,
WI),
pRSET or pREP plasmids (lnvitrogen, San Diego, CA), or pMAL plasmids (New
England
Biolabs, Beverly, MA), and many appropriate host cells, using methods
disclosed or cited
15 herein or otherwise known to those skilled in the relevant art. Recombinant
cloning vectors
will often include one or more replication systems for cloning or expression,
one or more
markers for selection in the host, e.g. antibiotic resistance, and one or more
expression
cassettes. Preferred vectors are described in the Examples, and include
without limitations
pcWori, pET-26b(+), pXTD 14, pYEX-S 1, pMAL, and pET22-b(+). Other vectors may
be
20 employed as desired by one skilled in the art. Routine experimentation in
biotechnology can
be used to determine which vectors are best suited for used with the
invention, if different
than as described in the Examples. In general, the choice of vector depends on
the size of
the polynucleotide sequence and the host cell to be employed in the methods of
this
invention.
25 A "cassette" refers to a segment of DNA that can be inserted into a vector
at specific
restriction sites. The segment of DNA encodes a polypeptide of interest, and
the cassette
and restriction sites are designed to ensure insertion of the cassette in the
proper reading
frame for transcription and translation.
The term "expression system" means a host cell and compatible vector under
suitable
30 conditions, e.g. for the expression of a protein coded for by foreign DNA
carried by the
vector and introduced to the host cell. Common expression systems include
bacteria (e.g.
E. coli and B. subtilis) or yeast (e.g. S. cerevisiae) host cells and plasmid
vectors, and insect
-20-
SUBSTITUTE SHEET (RULE 26~
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127
host cells and Baculovims vectors. As used herein, a "facile expression
system" means any
expression system that is foreign or heterologous to a selected polynucleotide
or, -
polypeptide, and which employs host cells that can be grown or maintained more
advantageously than cells that are native or heterologous to the selected
polynucleotide or
polypeptide, or which can produce the polypeptide more efficiently or in
higher yield. For
example, the use of robust prokaryotic cells to express a protein of
eukaryotic origin would
be a facile expression system. Preferred facile expression systems include !:.
colt, I3. snhlilis
alld S. cereoisiae host cells and any suitable vector.
The terns "mutant" and "mutation" mean any detectable chanLC in
gcnetic.material,
l0 e.g. DNA, or any process, mechanism, or result of such a change. l~his
includes gear
mutations, in which the structure (e.g. DNA sequence) of a gene is altered,
any gene or
DNA arising from any mutation process, and any expression product (e.g.
protein or
enzyme) expressed by a modified gene or DNA sequence. The term "variant" may
also be
used to indicate a modified or altered gene, DNA sequence, enzyme, cell, etc.,
i.e., any kind
of mutant.
"Sequence-conservative variants" of a polynucleotide sequence are those in
which
a change of one or more nucleotides in a given codon position results in no
alteration in the
amino acid encoded at that position.
"Function-conservative variants" are those in which a given amino acid residue
in
a protein or enzyme has been changed without altering the overall conformation
and
function of the polypeptide, including, but not limited to, replacement of an
amino acid with
one having similar properties (such as, for example, acidic, basic,
hydrophobic, and the
like). Amino acids with similar properties are well known in the art. For
example, arginine,
histidine and lysine are hydrophilic-basic amino acids and may be
interchangeable.
Similarly, isoleucine, a hydrophobic amino acid, may be replaced with leucine,
methionine
or valine. Amino acids other than those indicated as conserved may differ in a
protein or
enzyme so that the percent protein or amino acid sequence similarity between
any two
proteins of similar function may vary and may be, for example, from 70% to 99%
as
determined according to an alignment scheme such as by the Cluster Method,
wherein
similarity is based on the MEGALIGN algorithm. A "function-conservative
variant" also
includes a polypeptide or enzyme which has at least 60 % amino acid identity
as determined
by BLAST or FASTA algorithms, preferably at least 75 %, most preferably at
least 85%, and
-21-
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127
even more preferably at least 90%, and which has the same or substantially
similar
properties or functions as the native or parent protein or enzyme to which it
is compared. , _
The teen "DNA reassembly" is used when recombination occurs between identical
sequences. The term "DNA shuffling" indicates recombination between
substantially
homologous but non-identical sequences.
"isolation" or "purification" of a polypcptidc or enzyme refers to the
derivation of
the polypcptide by removing it from its original environment (for example,
from its natural
environment if it is naturally occurring, or form the host cell if it is
produced by
fCC0111b111a17t DNA I11C1110(15). Methods for polypcptidc purification arc
wcll-known 111 the
art, including, ~~~ithout limitation, preparative disc-gel electrophoresis,
isoelectric focusing,
1-IPLC, reversed-phase HPLC, gel filtration, ion exchange and partition
chromatography,
and countercurrent distribution. For some puposes, it is preferable to produce
the
polypeptide in a recombinant system in which the protein contains an
additional sequence
tag that facilitates purification, such as, but not limited to, a
polyhistidine sequence. The
polypeptide can then be purified from a crude lysate of the host cell by
chromatography on
an appropriate solid-phase matrix. Alternatively, antibodies produced against
the protein
or against peptides derived therefrom can be used as purification reagents.
Other
purification methods are possible. A purified polynucleotide or polypeptide
may contain
less than about 50%, preferably less than about 75%, and most preferably less
than about
90%, of the cellular components with which it was originally associated. A
"substantially
pure" enzyme indicates the highest degree of purity which can be achieved
using
conventional purification techniques known in the art.
Polynucleotides are "hybridizable" to each other when at least one strand of
one
polynucleotide can anneal to another polynucleotide under defined stringency
conditions.
Stringency of hybridization is detetzrtined, e.g., by a) the temperature at
which hybridization
and/or washing is performed, and b) the ionic strength and polarity (e.g.,
fotlrtamide) of the
hybridization and washing solutions, as well as other parameters.
Hybridization requires
that the two polynucleotides contain substantially complementary sequences;
depending on
the stringency of hybridization, however, mismatches may be tolerated.
Typically,
hybridization of two sequences at high stringency (such as, for example, in an
aqueous
solution of O.SX SSC at 65 °C) requires that the sequences exhibit some
high degree of
complementarily over their entire sequence. Conditions of intermediate
stringency (such
-22-
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127
as, for example, an aqueous solution of 2X SSC at 65 °C) and low
stringency (such as, for
example, an aqueous solution of 2X SSC at 55°C), require
correspondingly less overall ~ -
complemcntarity between the hybridizing sequences. ( 1 X SSC is 0. I 5 M NaCI,
0.015 M
Na citrate.) Poiynucleotidcs that "hybridize" to the polynucleotides herein
may be of any
length. In one embodiment, such polynucleotides are at least i 0, preferably
at least 15 and
most preferably at least 20 nucleotides long. In another embodiment,
polynucleotides that
hybridizes are of about the same length. In another embodiment,
polynuclcotides that
hybridize include those which anneal under suitable stringency conditions and
which encode
l7plyl)cptldCS Uf Crl'Ly111CS havine the same function, such as the ability to
catalyze an
I 0 oxidation, oxygenase, or coupling reaction of the invention.
The general genetic engineering tools and techniques discussed here, including
transforn~ation and expression, the use of host cells, vectors, expression
systems, etc., arc
well known in the art.
Mutagenesis and Direc~ed Evolution oJProtei~ts.
To improve the expression of proteins using conventional expression systems,
the
invention makes the unexpected discovery that directed evolution can be used
to generate
mutant libraries of polynucleotides which, when expressed using conventional
or facile
expression systems, result in functional proteins having normal or even higher
activity than
the native protein. Inclusion bodies, which commonly form when expressing
proteins
having disulfide bonds, and laborious in vitro refolding procedures can also
be avoided by
directed evolution.
According to the invention, proteins that are more easily expressed in facile
gene
expression systems can be obtained by using directed evolution to generate
mutant
polynucleotides in a library format for selection. General methods for
generating libraries
and isolating and identifying improved proteins (also described as "variants")
according to
the invention using directed evolution are described briefly below and more
extensively, for
example, in U.S. Patent Nos. 5,741,691 and 5,811,238. It should be understood
that any
method for generating mutations in polynucleotide sequences to provide an
evolved
poiynucleotide for use in expression systems can be employed. Proteins
produced by
directed evolution methods can then be screened for improved expression,
folding,
secretion, and function according to conventional methods.
-23-
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127
Anv source of nucleic acid, in purified form can be utilized as the starting
nucleic
acid. Thusrthe process may employ DNA or RNA including messenger RNA, which
DNA _
or RNA may be single or double stranded. In addition, a DNA-RNA hybrid which
contains
one strand of each may be utilized. The nucleic acid sequence may be of
various lengths
depending on the size of the nucleic acid sequence to be mutated. Preferably
the specific
nucleic acid sequence is from 50 to 50,000 base pairs. It is contemplated that
entire vectors
containins~ the nucleic acid encoding the protein of interest may be used in
the methods of
this invention.
Any specific nucleic acid sequenee can be used to produce the population of
mutants
by the present process. An initial population of the specific nucleic acid
sequences having
mutations may be created by a number of di fferent known methods, some of
which are set
forth below.
Error-prone polymerase chain reaction (20,45,4G) and cassette mutagenesis (38-
44),
in which the specific region optimized is replaced with a synthetically
mutagenized
oligonucleotide can be employed in the invention. Error-prone PCR can be used
to
mutagenize a mixture of fragments of unknown sequences. These techniques can
also be
employed under low-fidelity polymerization conditions to introduce a low level
of point
mutations randomly over a long sequence, or to mutagenize a mixture of
fragments of
unknown sequence.
Oligonucleotide-directed mutagenesis, which replaces a short sequence with a
synthetically mutagenized oligonucleotide may also be employed to generate
evolved
polynucleotides having improved expression.
Alternatively, nucleic acid or DNA shuffling, which uses a method of in vitro
or in
vivo homologous recombination of pools of nucleic acid fragments or
polynucleotides, can
be employed to generate polynucleotide molecules having variant sequences of
the
invention.
Parallel PCR is another method that can be used to evolve polynucleotides for
improved expression in conventional expression systems, which uses a large
number of
different PCR reactions that occur in parallel in the same vessel, such that
the product of one
reaction primes the product of another reaction. Sequences can be randomly
mutagenized
at various levels by random fragmentation and reassembly of the fragments by
mutual
priming. Site-specific mutations can be introduced into long sequences by
random
-24-
SUBSTITUTE SHEET (RULE 2fi)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127
fragmentation of the template followed by reassembly of the fragmems in the
presence of
mutagenic oligonucleotides.
A particularly useful application of parallel PCR, which can be used in the
invention,
is called sexual PCR. In sexual PCR, also known as DNA shuttling, parallel PCR
is used
to perform irr vi~ro recombination on a pool of DNA sequences. Sexual PCR can
also be
used to construct libraries of chimacras of genes from different species.
The polynucleotide sequences for use in the invention can also be altered by
chemical mutagenesis. Chemical mutagens include, for example, sodium
bisulfate, nitrous
acid, hydroxylarninc, hydrazine or formic acid. Other agents which arc
analogues of
l0 nucleotide precursors include nitrosoguanidine, 5-bromouracil, 2-
aminopurine, oracridine.
Generally, these agents are added to the PCR reaction in place of the
nucleotide precursor
thereby mutating the sequence. Intercalating agents such as profiavine,
acriflavine,
quinacrine and the like can also be used. Random mutagenesis of the
polynucleotide
sequence can also be achieved by irradiation with X-rays or ultraviolet light,
or by
subjecting the polynucieotide to propagation in a host (such as E. colt) that
is deficient in
thenormal DNA damage repair function. Generally, plasmid DNA or DNA fragments
so
mutagenized are introduced into E. colt and propagated as a pool or library of
mutant
piasmids.
Alternatively a mixed population of specific nucleic acids may be found in
nature
in that they may consist of different alleles of the same gene or the same
gene from different
related species (i.e., cognate genes). Alternatively, they may be related DNA
sequences
found within one species, for example, the peroxidase class of genes. Once the
mixed
population of the specific nucleic acid sequences is generated, the
poiynucleotides can be
used directly or inserted into an appropriate cloning vector, using techniques
well-known
in the art.
Once the evolved polynucleotide molecules are generated they can be cloned
into
a suitable vector selected by the skilled artisan according to methods well
known in the art.
If a mixed population of the specific nucleic acid sequence is cloned into a
vector it can be
clonally amplified by inserting each vector into a host cell and allowing the
host cell to
amplify the vector. The mixed population may be tested to identify the desired
recombinant
nucleic acid fragment. The method of selection will depend on the DNA fragment
desired.
For example, in this invention a DNA fragment which encodes for a protein with
improved
-25-
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/1712?
folding properties can be detern~ined by tests for functional activity of the
protein and
absence of inclusion body fom~ation. Such tests are well known in the art.
Using the methods of directed evolution, the invention provides a novel means
for
producing properly folded, functional, and soluble proteins in conventional or
facile
expression systems such as E. toll or yeast. Conventional tests can be used to
detern~ine
whether a protein of interest produced from an expression system has improved
expression,
folding and/or functional properties. For example, to dctennine whether a
polynuclcotide
subjected to directed evolution and expressed in a foreign host cell products
a protein with
improved folding, one skilled in lhC art Call perform experiments designed to
test the
functional activity of the protein. f3ric(ly, the evolved protein can be
rapidly screened, and
is readily isolated and purified from the expression system or media if
secreted. It can then
be subjected to assays designed to test functional activity of the particular
protein in native
forn~. Such experiments for various proteins are well known in the art, and
are discussed
in the Examples below.
In one embodiment, the invention contemplates the use polynucleotides encoding
for variants of heme-containing proteins. Thus, the invention employs directed
evolution
to generate novel peroxidase enzymes, such as HR.P, which fold properly in the
host cells
(e.g. ~, toll) used in the expression system, retain functional activity, and
avoid the
problems associated with inclusion body formation.
The invention can also be applied to select or optimize an expression system,
including selection of host cells, promoters, and signal sequences. Expression
conditions
can also be optimized according to the invention.
The Examples below further describe the methods of the invention and, in
particular,
teach the use ofdirected evolution to generate variants ofHRP which when
expressed using
conventional expression systems do not form inclusion bodies and retain
functional activity.
Ordinarily, the corresponding native proteins form inclusion bodies and show
little retained
functional activity after expression in conventional expression systems.
Examples of practicing the invention are provided, and are understood to be
exemplary only, and do not limit the scope of the invention or the appended
claims. A
person of ordinary skill in the art will appreciate that the invention can be
practiced in many
forms according to the claims and disclosures here.
-26-
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127
EXAMPLE 1
Functional Expression of Horseradish Peroxidase in E..coli and Yeast
There is growing interest in exploiting eukaryotic peroxidases for use as
industrial
biocatalysts. Protein engineering and directed evolution to improve specific
properties,
however, arc complicated by the lack of facile recombinant expression systems.
In an effort
to develop a functional bacterial expression system suitable for large-volume
screening of
mutants of horseradish peroxidasc (I~RP), the present Example describes the
development
of a bacterial expression system for heme-associated proteins, such as
horseradish
l 0 peroxidase (1-lRP), by inserting a corresponding gene as a fusion to the
signal peptide Pell3.
In addition, by subjecting these genes to directed evolution heme-associated
proteins fold
more ef ~ ciently in E. colt and arc rendered more resistant to heat
(thennostable) and more
resistant to inactivation by H20:. This Example provides an approach for
greatly facilitating
efforts to "fine-tune" many enzymes that are promising industrial
biocatalysts, but for which
suitable bacterial or yeast expression systems are currently lacking because
the proteins
form inclusion bodies or are inefficiently secreted by the cell.
('IoninQ of HRP
The HRP gene (with an extra methionine residue at the N-terminus) was cloned
from
the plasmid pBBGlO (British Biotechnologies, Ltd., Oxford, UK) by PCR
techniques to
introduce an Aat II site at the start codon and a Hind III site immediately
downstream from
the stop codon. This plasmid contains the synthetic horseradish peroxidase
(HRP) gene
described in Smith et al. (13), whose DNA sequence is based on a published
amino acid
sequence for the HRP protein (49). pBBGlO was made by inserting the HRP
sequence
between the HindIII and EcoRl sites of the polylinker in the well-known
plasmid pUCl9.
The PCR product obtained from this plasmid was digested with Aat II first,
blunt-ended
with t4 DNA polymerase, and then further restricted with Hind III. The
digested product
was and ligated into pET-22b(+) (purchased frori~ Novagen) treated with McsI
and Hind III,
to yield the vector pETpeIBHRP. A map of this expression vector shown in FIG
1. In this
construct, the HRP gene was placed under the control of the T7 promoter and is
fused in-
frame to the pelB signal sequence (See ~SEQ. ID NO. 1 and SEQ. ID NO. 2J and
FIG. 2),
which theoretically directs transport of proteins into the periplasmic space,
that is, for
delivery outside the cell cytoplasm (27). The ligation product was transformed
into E. colt
-27-
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCTNS99/17127
strain BL21 (DE3) for expression of the protein in cells both with and without
induction by
1 mM isopropyl-b-D-thiogalactopyranoside (1PTG).
In the cells that were induced with IPTG, no peroxidase activity about
background
was detected, for BL21 (DE3) eelis or pET-22b(+)-harboring BL21 (DE3) cells,
even though
S the level of 1-1RP polypeptides accounted for over 20% of total cellular
proteins. This was
consistent with previous observations ( 12-14).
In the cells thU were trot induced with IPTG, clones were discovered that
showed
weak but mcasurahlc activity atainst azino-di-(cthylbcnzthiazoline sulfonate
(ABTS).
l'he T7 pre~moter in tl~c pCT-22b(+) vector is known to be Icaky (31 ), and in
theory
it is therefore possible that sonic of the HRP polypeptide chains produced at
this basal level
were able to fold into the native form. Conversely, addition of 1PTG leads to
high-level
HRP synthesis, «~hich instead favors aggregation of chains and prevents their
proper folding.
Subsequently, random mutagenesis and screening were used to identify mutations
that lead
to higher expression of HRP activity. ,
Thus, one aspect of the invention includes the use of a promoter that can
regulate
production of small amounts of polypeptide under some conditions, and larger
amounts
under other conditions. For example, a "leaky" inducible promoters can be
used. This type
of promoter produces high levels of a particular protein or proteins in the
presence of an
inducer compound, and much lower levels in the absence of inducer. In some
embodiments,
a polypeptide can be over-expressed under certain conditions (e.g. in the
presence of
inducer) and under-expressed in other conditions (e.g. without inducer).
Polypeptides that
are inactive when expressed at normal levels or when over-expressed, but are
active when
under-expressed, are particularly suitable for use as parent polypeptides of
the invention.
Such expression-resistant polypeptides can be improved, using the methods
ofthe invention,
to provide functional, active expression at suitably high yields and activity
levels.
Random library Qerreration and screening
One of the HRP clones that showed detectable peroxidase activity was used in
the
first generation of error-prone PCR mutagenesis. The random libraries were
generated by
a modification of the error-prone PCR protocol described above (20,21,22), in
which 0.15
mM of MnCl2 was used instead of 0.5 mM MnCIZ. This protocol incorporates both
-28-
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127
manganese ions and unbalanced nucleotides, and has been shown to generate both
transitions and transversions and therefore a broader spectrum of amino acid
changes (50).
" _
Briefly, the PCR reaction solution contained 20 fmolcs template, 30 pmoles of
each
of two primers, 7 mM MgCli, 50 mM KCI, 10 mM Tris-HC1 (pH 8.3), 0.01 %
gelatin, 0.2
mM dGTP, 0.2 mM dATP, 1 mM dCTP, I mM dTTP, 0.15 mM MnCli, and 5 unit of Taq
polymerase in a 100 pi volume. PCR reactions were perfom~ed in a MJ PTC-200
cycler
(MJ Research, MA) for 30 cycles with the following parameters: 94°C for
1 min, 50°C for
1 min, and 72°C for 1 min. The primers used were:
S'-TTATTGCTCAGCGGTGGCAGCAGC [SEQ. ID NO. 15(, and
5'-AAGCGCTCATGAGCCCGAAGTGGC (SEQ. 1D. NO. 16[.
The PCR products were purified with a Promega Wizard PCR kit, and digested
with
Nde 1 and Hind III. The digestion products were subjected to gel-purifica:ion
with a QIAEX
I1 gel extraction kit, and the HRP fragments were ligated back into the
similarly digested
and gel-puri feed pET-22b(+) vector. Ligation mixtures were transformed in the
BL21 (DE3)
I S cells by electroporation with a Gene Pulser II (Bio-Rad). Cell growth and
expression was
carried out in either 96-well or 384-well microplates in LB medium at
30° C. Peroxidase
activity tests were performed with HiOi and ABTS (26).
For each generation, typically 12,000-15,000 colonies were picked and screened
in
96-well plates. This number represents an exhaustive search of all accessible
single
mutants, with a probability of 95% for any mutant to be sampled at least once
(25).
Colonies were either picked manually, or using an automated colony picker at
Caltech, Q-
bot (Genetix, LJK). Of the 12,000 colonies that were screened (no IPTG added),
a mutant
designated HRP1A6 showed 10-14 fold higher peroxidase activity than the parent
clone.
FIG. SA and SB. This mutant clone also showed markedly decreased activity when
as little
as 5 pM of IPTG was added. FIG. 6. Sigma reports that 1 mg of highly purified
HRP from
horseradish has a total activity of 1,000 units, as determined by the ABTS
assay. Other
workers reported similar results (13). Based on this data, the concentration
of active HRP
was estimated to be 100 ug/L. HRP 1 A6 shows a total activity of greater than
100 unitslL.
This compares favorably with the yield obtained from refolding of aggregated
HRP chains
in vitro ( 13). This level of expression for the HRP mutant is also similar to
that for bovine
pancreatic trypsin inhibitor (BPTI) in E. coh (32), an unglyeosylated protein
with three
-29-
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127
disulfide bonds. Greater than 95% of the HRP activity was found in the LB
culture medium
as judged by the ABTS activity.
The mutant HRP remained stable for up to a week at 4°C. IPTG was
omitted in all
HRP expression experiments, unless otherwise specified. Peroxidase activity
tests for HRP
were performed with a classical peroxidase assay, ABTS and hydrogen peroxide
(2G).
Fifteen pl of cell suspension was mixed with 140 pl of ABTS/HiOz (2.9 mM ABTS,
0.5
mM 1-IiO, , pH 4.5) in microplates, and the activity was detcnnined with a
SpectraMax plate
reader (Molecular Devices, Sunnyvale, CA) at 25°C. A unit of HRP is
defined as the
amount of enzyme that oxidizes 1 ymole of ABTS per min at the assay
conditions.
Sequencing of the mutant gene found a mutation at position 255, in which the
codon
AAC for the amino acid asparagine (Asn or N) was changed to the codon GAC for
the
amino acid aspartic acid (Asp or D). This residue is a putative glycosylation
site, and is
located at the surface of the protein. The sequence of this mutant (HRP 1 AG)
is shown in
FIG. 3 (SEQ. ID NO. 3]. A map of a plasmid pETpeIBHRP 1 AG containing this
mutant is
shown in F1G. 4.
A representation of the structure of this HRP mutant, showing the Asn255Asp
mutation ~s
shown in FIG. 7.
Functional Expression of HRP in Yeast
The native HRP protein contains four disulfide bonds, and E. coli has only a
limited
capability to support disulfide formation. In theory, these well-conserved
disulfides in HRP
(and other plant peroxidases) are likely to be important for the structural
integrity of the
protein, and may not be replaceable by mutations elsewhere. Yeast has a much
greater
ability to support the formation of disulfide bonds. Thus, yeast can be used
as suitable
expression host, in place of E. coli, particularly if it is desired to relieve
the apparent
limitation on the folding of HRP imposed by any constraints on disulfide
formation in E.
coli. For example, S. cerevisiae can be used as a host for the expression of
mutant HRP
genes and proteins.
The HRP mutant (HRP 1 AG) was cloned into the secretion vector pYEX-S 1
obtained
from Clontech (Palo Alto, CA} (35), yielding pYEXS 1-HRP (FIG. 8). This vector
utilizes
the constitutive phosphoglycerate kinase promoter and a secretion signal
peptide from
Kluveronryces lactic . The plasmid was first propagated in E. coli, and then
transformed into
-30-
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127 -
S. cerevisiae srrai~r BJ54G5, obtained from the Feast Genetic Stock Center
(YGSC),
University of California, Derkeley using the LiAc method as described (3G).
BJ5465 is~ -
protcasc deficient, and has been found to be generally suitable for secretion.
A first generation of error-prone PCR of HRP in yeast was perfom~ed. Anyone
the
$ first 7,400 mutants screened, four variants showed 400% higher activity than
HRP l AG in
yeast. Additional details and results arc given in Example 2.
EaA>\1P1.E 2
IU
Functional Expression of 1-1RP in 1'cast through Directed Evolution
This example describes the use of directed evolution to further improve the
functional expression of HRP. As explained m txampte i, a vanam m ~wm~a~m~
peroxidase (HRP lAG) was isolated. Since HRP contains four well-conserved
disulfides,
and E. coli has only limited ability to support disulfide bond forniation, the
further
15 improvement in bacterial expression of HRP in E. coli maybe constrained by
correct pairing
of disulfide-containing cysteines. Yeast cells, for example S. cerevisiae,
have much greater
ability to support the formation of disulfide bonds, and may be better able to
accommodate
disulfide bonds in peroxidase enzymes. In theory, these well-conserved
disulfides in HRP
(and other plant peroxidases) are likely to be important for the structural
integrity of the
20 protein, and may not be replaceable by mutations elsewhere. Thus, yeast can
be used as
suitable expression host, in place ofE. coli, particularly if it is desired to
relieve the apparent
limitation on the folding of HRP imposed by any constraints on disulfide
formation in E.
col i.
Accordingly, S. cerevisiae was chosen as an alternative host for the
expression of
25 HRP. S. cerevisiae is both a micro-organism and a eukaryote, and possesses
much of the
eukaryotic protein post-translational and secretory machinery, such as ER and
Golgi that
catalyze the formation of disulfide bonds and glycosylate polypeptides.
Genetic
manipulation techniques (in particular gene transformation) are also readily
available. A
drawback is that yeast naturally secrete few proteins. Moreover, yeast
glycosylation differs
30 significantly from that in higher eukaryotic organisms, which might present
problems for
secretion of glycoproteins (4). Nonetheless, several protein~have been
efficiently secreted
from yeast (4). Stategically, the experiments of this example take advantage
of the capacity
-31-
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127
of yeast to catalyze the formation of disulfide bonds while fine-tuning the
glycosylation
factor through the process of directed evolution. " -
Cons~ruc~ion o,~,i~ensl expression systen: for NRP.
The HRP mutant i~RPIAG from Example i was cloned into the yeast secretion
vector pl'EX-S 1 obtained from Clontech (Palo Alto, CA} (35), yielding pYEXS 1-
HRP
(FIC. 8). This vector utilizes the constitutive phosphoglycerate kinase
promoter and a
secretion signal peptide from K. lacri.s. pYEX-S 1 was digested with Scrcl,
and then blunt-
cnded with T4 DNA polymcrasc. TI1C Illallll'C HRP I AG gene was cloned from
pETpeIBHRPI AG by PCR techniques using the proofreading polymcrase pfic
(Stratagenc,
CA) that generate blunt-end products. The forward and reverse primers used
were 5'-
CAGTTAACCCCTACATTC-3' (SEQ ID No. 25J and 5'-
TCATTAAGAGTTGCTGTTGAC-3' (SEQ ID No. 26), respectively. The PCR fragments
were then ligated into the restricted and blunt-ended pYEX-S 1, and
transformed iMo E. coli
DHSa cells. A number of colonies were picked and screened for the presence of
the HRP
gene by colony PCR reactions 18 with these two primers: 5'-
CGTAGTTTTTCAAGTTCTTAG-3 (SEQIDNo.27[ and 5'-
TCCTTACCTTCCAATAATTC-3 [SEQ ID No. 28]. The correct orientation of the HRP
gene was further confirmed by sequencing. This yeast expression vector is
generally
referred to hereinafter as pYEXSl-HRP (FIG. 8). In this construct, the HRP
gene was
placed directly downstream of the secretion signal peptide from K. IacriS, and
the expression
is under the control of the constitutive phosphoglycerate kinase promoter. The
vector also
carries the E. coli Amp resistance gene as well as the yeast selectable
markers leu2-d and
tJRA3 (47).
For expression experiments, the plasmid was first propagated in E. coli strain
DHSa,
and then transformed into S. cerevisiae strain BJ5465, obtained from the Yeast
Genetic
Stock Center (YGSC; University ofCalifomia, Berkeley), using a LiAc method
that utilizes
single strand DNA as described by Gietz et al. (48). BJ5465 is protease
deficient and
generally suitable for secretion (4). Following transformation, cells were
plated on YNB
selective medium supplemented with 20 Pg/ml leucine, 20 ~tg/ml histidine, 20
llg/ml
adenine and 20 ug/ml tryptophan. Colonies were picked, and grown in 96-well
microplates
in YEPD medium at 30°C in an air-circulating incubator for 2 days and
16 hours. HRP
-32
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99I17127
activity tests were performed with a classical peroxidase assay, ABTS and
hydrogen
peroxide (26). The activity obtained from yeast for HRPlA6 was only about 1/10
of tha,( _
from G. coli, and actually slightly lower than obtained for the wild-type iv
this construct.
Gerrerario» n» d Scr-ee»irrQ of HRP A~mants
Libraries of HRP mutants were constructed by error-prone PCR (20) as described
(53) except that the following two primers flanking the HRP gene were used in
the
mutagenic PCR reactions: 5'-CAGTTAACCCCTACATTC-3' [SEQ ID No. 25] and 5'-
'fGA'fGCTGTCGCCGAAGAAG-3' [SfQ tD No. 29[. Also, the thcnnal cycling
lU parameters were: 95 °C for 2 min, (~)4°C for 1 111111, 50
°C for 1 min, and 72 °C for 1 min,
30 cycles).
The PCR products were purified with a Promega Wizard PCR kit (Madison, WI),
digested with Sac l and Bam HI (the first 27 amino acid residues of HRP were
left
unmodified). The digestion products were then subjected to gel-purification
with a QIAEX
II gel extraction kit (QIAGEN, Valencia, CA), and the HRP fragments were
ligated back
into the similarly digested and gel-purified pYEXSI-HRP1A6. Ligation mixtures
were
transformed in E. coli HB 1 O1 cells by electroporation with a Gene Pluser II
(Bio-Rad), and
selected on LB medium supplemented with 100 mg/ml ampiciilin. Colonies were
directly
harvested from LB plates. This plasmid DNA was subsequently used for
transformation into
yeast BJ5465 as described above.
Single colonies were picked from yeast nitrogen base (YNB) plates, and grown
at
°C for 64 h in 96-well microplates containing YEPD medium (I% yeast
extract, I%
peptone, 2% glucose) in an incubator. Microplates were then centrifuged at
1,500 g for 10
min, and l Oml of the supernatant in each well was transferred to a new
microplate with a
25 Beckman 96-channel pipetting station (Multimek, Beckman, Fulerton, CA), and
assayed for
total HRP activity. Overall standard deviations of this measurement {including
pipetting
errors, which was about 2%) did not exceed 10%. Improved mutants (showing the
highest
total HRP activity) were directly retrieved from the microplates, washed three
times with
sterile HZOz, and re-grown in YNB selective medium. Plasmids containing the
HRP mutants
30 were first extracted from the yeast cells with a Zymo yeast plasmid
miniprep kit (Zymo
Research, Orange, CA), and then returned to E. coli X 10-Gold for further
propagation and
preparative isolation.
-33-
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WOliO/06718 PCT/US99/17127 -
Where indicated, pre-screening of HRP-expressing yeast clones were carried out
as
follows. Colonies on YNB plates were replicated onto MSI supported pure
nitrocellulose" -
membranes ( Micron Separations Inc., Wcstboro, MA), which were grown on fresh
YEPD
agar at 30 °C for 34 hr. Membranes were then immersed in 100 ml of TMB
membrane
substrate (O.S mM TMB, 2.9 mM HzOr, and 0.12% (W/V) dextran sulfate as
cnhancer) for
5 min to allow colored product to develop. Those yeast clones that exhibited
bright green
color were traced back to the master YNB selective plates, and picked and
grown in YEPD
for further screening as described above.
Firsr ~eu~rmion HXP r»«rnQC»esis in a~ensl jon it»nrovi«Q exnressio».
A first generation of error-prone PCR of HRP1A6 in yeast was aimed at
improving the expression level. An error-prone PCR protocol incorporating both
unbalanced nucleotide concentrations and manganese ions as described
previously (20, 21 )
was used. This protocol was shown to generate roughly random mutations,
allowing for
sampling of a broader spectrum of amino acid residue changes. The manganese
ion
concentration used was 100 ~tM, which generated an error rate of approximately
1-2
mutations per gene on average (22). The PCR products were purified with a
Promega
Wizard PCR kit, digested with Sac I and Bam H1 (thus the first 27 amino acid
residues of
HRP were left unmodified). The digestion products were then subjected to gel-
purification
with a QIAEX II gel extraction kit, and the HRP fragments were ligated back
into the
similarly digested and gel-purified PEXSI-HRP1A6. Ligation mixtures were
transformed
in HB 1 O 1 cells by electroporation with a Gene Pluser II (Bio-Rad). Colonies
were scratched
from the E. coli plates and resuspended in LB medium, from which plasmids were
prepared.
Then the plasmids were transformed into yeast and yeast colonies were obtained
and grown
as described above.
A total of about 14,000 colonies were picked and screened for this generation,
which
represented an exhaustive search of all accessible single mutants, and a
probability of 95%
for any mutant to be sampled at least once (25). Of these colonies, a number
of mutants
showed significantly higher activity than the parent (HRP1A6) in yeast. Two
exemplary
improved mutants arc designated HRP1-11764 ~SEQ. ID NO. 12 and SEQ. ID NO. 13]
and HRP1-7 7E2 (SEQ. ID NO. 5 and SEQ. ID NO. 6~. HRP1-11764 gave a 16-fold
higher activity than the parent, or a total activity of about 220 units/L
(FIG. 9). HRP 1-77E2
-34
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127
showed a total activity of about 147 units/L. Both of these were higher than
the highest
w level obtained from E. coli. Sce also FIC 12 (HRP1-77E2) and FIG. 16 (HRPI-1
1764). ~ -
Second Qeneraiion ojNRP rnrnaQenesis in yeast for ironrovirrQ ernression.
The second generation of error-prone PCR used HRP 1-1 1764 as the parent. For
this
generation, a higherconccntration ofmanganesc ion was used to increase the
mutation rate.
This change was made based on the following considerations. Since screening
can only
handle a library of about 10, to 10' mutants at the present time, the rate of
mutagencsis has
been conservatively limited to creating predominately single mutants in the
last (1S). In
this example, the fraction of clones more active than the parent for a given
generation
remains relativeiy constant with the error-rate up to G mutations per gene.
The advantage
of using higher error rates is that it would allow neutral mutations to exist
along with
beneficial mutations isolated through screening. These accrued neutral
mutations may
become useful in subsequent generations by either providing a bridge for
generating new
1 S types of mutations, or by synergetic interactions with newly created
mutations. The
manganese ion concentration used in this generation was 3SO lM, which
generated an error
rate of approximately 4-5 mutations per gene on average (22).
Additionally, a prescreening of the colonies using nitrocellulose membranes
was
performed. This was possible because the higher error-rate significantly
reduce the number
of colonies that showed similar or higher activity than the parent. The
procedures were as
follows. Colonies were first replicated from the master plates onto
nitrocellulose
membranes and grown on YEPD plates at 30°C for one day and 6 hours. The
membranes
were then retrieved from the plates and immersed in a mixture of TMB
(tetramethylbenzidine) and Hz02. The colonies with the brightest color were
identified, and
2S corresponding mother colonies were picked and grown from the master plates.
For this
generation, about 120,000 colonies were screened (about 5,000 were actually
picked and
grown), and the mutant HRP2-28D6 was obtained. It showed an activity 85%
higher than
its parent, HRPI-11764, or a total activity of 410 units/L (FIG. 9).
Third Qerrera~ion ojHRP mutaQenesis in .Least for imnrovirrQ expression.
The third round of random mutagenesis was carried out under similar conditions
with HRP2-28D6 as the parent. For this generation, a total of 90,000 colonies
were pre-
-35-
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99117127
screened, and 3,000 picked and grown. The best mutant, HRP3-17E12, gives an
expression
level of 1080 units/L, an increase of 1 GO% over the parent HRP2-28DG, or 85
fold over the, _
starting mutant, 1-IRPIAG.
I-'irsr ~c~uermion o~HHP mmnQmresis in vens~ (or inrnr'oainQ stnhilitt~
Onc ~,encration of random mutagencsis of HRP for improving thern~ostability
and
resistance towards 1~,0: ~~~as can-led out using: HRP1-77):2 as the parent.
The random
muta~cncsis (with 100 pM manganese) and cell growth was essentially performed
as
described above (with no prescreening). Thennostability tests were performed
with a MJ
I O fTC-200 cycler (MJ Research, MA) at 73 °C with an incubation time
of 1 U min. I~zOZ
resistance tests were separately perforn~ed in 25 mM HzOi at room temperature
and a pre-
incubation tin~c of 30 min., followed by ABTS screening in 25 mM HiOZ. Mutants
that
were more thennostable or chemically stable (H,O; resistant) than the parent
were further
characterized at various temperatures (far thennostability) or H~02
concer;trations (for HzOi
stability).
Out of 3,000 colonies screened, one thermostable mutant (HRP I -4BG) showed a
T"~
of over G°C higher than that of the parent (T"z is the transition
midpoint of the HRP
inactivation curve as a function of temperature) (FIG. 10). Another mutant,
HRP 1-28B 11
also showed some improvement in thermostability. The mutant HRPI-24D11 was not
markedly more thennostable than its parent HRP1-77E2, but was more resistant
to HiOZ
degradation. (A feedback mechanism common to HRP enzymes is that they are
degraded
by HIOZ, which is a reactant in the enzymatic reactions that HRP facilitates.)
The HRP1-
24D 11 mutant retained about 60% of activity after incubation with 25 mM HiOi
for 30 min,
while the parent exhibited a 42% residual activity under the same conditions
(FIG. 11).
1%ctor Constrrrctiorr for HRP expression irr Pichia nastoris.
The improved HRP mutants were further cloned into the Piehia expression vector
pPIZaB (Invitrogen Cotp., Carlsbad, CA) to facilitate production of the
mutants for
biochemical characterization (54). This vector contains the a-factor signal
peptide including
a spacer sequence of four residues Glu-Ala-GLu-Ala at the C-terminus of the
secretion
signal, and the methanol-inducible PAOX 1 promoter. pPIZaB was restricted with
Pst I first,
blunt-ended with T4 DNA polymerase, and then further digested with EcoR I. The
sample
-36-
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127
was purified with a Promega DNA purification kit. The coding sequences for the
HRP
- variants were obtained from the corresponding pYEXS 1-HRP plasmids by PCR
techniques ~ _
using the proofreading polvmcrasc Pfu. The following two primers were used in
the PCR
reactions: 5'-TCAGTTAACCCCTACATTC-3' (forward) ESEQ ID No. 30] and 5'
S CCACCACCAGTAGAGACATGG-3' (reverse) ]SEQ 1 D No. 31 ). The PCR products were
restricted with Eco Rl, and iigated into digested and purified pPIZaB,
yieldingpPlZaB-HRP
(Fig. lb) in which the HRP genes were placed immediately downstream of the a-
factor
signal. The ligation products were first transfonned into E. ~oli strain ~L10-
Gold and
selected on low salt LB IllCdll1111 ( 1 % tryptophan, 0.5% yeast extract,
0.5°/, NaCI, pl-1
adjusted to 7.5) supplemented with 25 mg/ml Zeocin (Cayla, Toulouse codex,
France).
Colonies were screened for the presence of the HRP genes by colony PCR
reactions (55)
with these two primers: 5'- GAGAAAAGAGAGGCTGAAG TC-3' (fonvard) ~SEQ ID
No.3Z) and 5'-TCCTTACCTTCCAATAATTC-3 (reverse) )SEQ 1D No. 33). The forward
primer contained the last three nucleotides of the signal sequence and the
first nucleotide of
the HRP sequence (as underlined), which ensured that the positive colonies
carried the fuli-
length HRP genes in the correct orientation. Plasmids were isolated with a
QIAgen
miniprep kit from liquid cultures of positive transformants, and used for
further
transformation into Pichia for the expression of HRP.
Transformation of Pichia was performed with electroporation according to the
manufacturer's instructions (Invitrogen). Before transformation, plasmids were
linearized
with Pme I, purified with a Promega DNA purification kit, and further treated
with
Princeton Centri-Sep columns equilibrated in d.d. HBO to remove any residue
impurities.
The linearized vectors were integrated into the Pichia genome upon
transformation via
homologous recombination between the transforming DNA and the Pichia genome.
The
transformed cells were plated on YPDS medium (1% yeast extract, 2% peptone, 2%
glucose, 1 M sorbitol) supplemented with 100 mg/m1 Zeocin. For each construct
containing
a distinct HRP mutant, typically 4-6 transformants were picked, and purified
on new YPDS
plates (supplemented with 100 mg/ml Zeocin) to isolate single colonies, which
were then
screened to identify the clones that conferred the highest expression levels.
The Pichia
strain X-33 was used in all expression experiments. It was determined in
initial tests that X-
33 (Mut+) afforded significantly better HR.P expression than KMI7 (MutS).
-37-
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127
NRP ernressio» i» Pichia nasroris.
Pichia cell growth was carried out at 30oC in a shaker. pPIZaB-HRP-harboring
cells ~ -
were first grown overnight in BMGI' ( 1% yeast extract, 2% peptone, 100 111M
polaSSlunl
phosphate, pH G.O, 1.34% YNB, 4X 10-5% biotin, 1 % glycerol) supplemented with
1
casamino acids to an OD G00 of 1.2-1.G. The cells were then pelleted and
resuspended to
an ODD of 1.0 in BMMY medium (identical to BMG>' except 0.5% methanol in lieu
of 1
glycerol) supplemented with 1 % casamino acids. Growth was continued for
another 54-72
h. Sterile methanol was added every 24 h to maintain induction conditions. HRP
Icveis in
the supernatants peaked around 54-GO h post-induction (at whlCh llllle the
OD~,«, reached
about 8.0-10.0). Where applicable, at the point of induction, 1.0 mM vitamin B
I, 1.0 mM
d-ALA, and 0.5 ml/L trace element mix (0.5 g/L MgCli, 30 g/L FeCIz.GHZO, 1 gIL
ZnCIZ.4H:0, 0.2 g/L CoCI,.GHZO, 1 g/L Na,Mo0,.2H20, O.Sg/L CaCl,.2H,0, 1 g/L
CuClz,
and 0.2 g/L HZB03) were added to the growth medium.
I S Peroxidase Activity Assay.
Peroxidase activity tests for HRP were performed with a classical peroxidase
assay, ABTS and hydrogen peroxide (2G). 10 ul (or 15 pl) of cell suspension
were mixed
with 140 ~tl (or 150 ~I1) of ABTS/H,Oz (the concentrations of ABTS and H=O,
are 0.5 mM
and 2.9 mM respectively, pH 4.5) in a microplate, and the increase of
absorbance at 405nm
(e of oxidized ARTS is 34.700 cm''M-~) was determined with a SpectraMax plate
reader
(Molecular Devices, Sunnyvale, CA) at 25°C. A unit of HRP is defined as
the amount of
enzyme that oxidizes 1 ~t mole of ABTS per min under the assay conditions.
Guaiaco! assay.
The assay is perfonrted with 1 mM HZOZ and SmM Guajacol in SOmM
phosphate buffer pH 7.0 and an increase of absorbance at 470nm is followed (t:
of oxidized
product at 470nm is 26.000 cm~'M~') after adding the yeast supernatant.
The stability of mutants was assessed using assays for initial activity (A;)
and
residual activity (A,~s;~, perfotzrted as described above with ABTS as
substrate. A,K;d is
measured after incubation of HRP mutants in NaOAc buffer pH 4.5 containing no
H~Oz or
1 mM HiO~ and incubating at 50°C for 10 min.
-38-
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127
The assay for stability in organic solvent/buffer (NaOAc buffer SOmM pH4.5)
mixture was done with 1 mM H202 and 2mM ABTS using supernatant of HRP mutants -
expressed in yeast ( l0ul) in dioxanc/buffcr (20/SO).
l'ro~locrion o~llXP 111111111115 lI1 Piclrin.
To obtain sufficient quantities of purified enzymes, l'iclliel was used in an
further
effort to increase production of 1-iRP mutants. 1-iRP-C (wild-type) , HRP 2-
13A 10 (FIG. 19,
[Sh.Q ID No. 21 and SEQ ID No. 22[) and HRP 3-17E12 (hlG. 20, ~SEQ ID No. 23
end SEQ ID No. 24[) were cloned into the l'icllicr secretion vector pPICZaB.
In this
construct (pPICZaB-HRP, FIG. 1 b), HRP was fused to the a-factor signal
peptide, and the
expression was induced with methanol. A typical expression curve is shown in
F1G. 25.
For HRP3-17E12, after SS h of cultivation, about 6,500 units/L of HRP activity
was
detected in the supernatant (F1G. 25, open squares), or G.5 fold of that
obtained from yeast.
The work from others as well as from our laboratory found that the addition of
trace metal
elements, heme synthesis intenrediate aminolevulinic acid, and vitamin
supplements to
growth medium (such as thiamine) resulted in substantial improvement in the
yields of
holoenzymes of heme-containing prtoeins in E. coli (59-62). Addition of these
additives to
the Pichia growth medium in our experiements led to a 32% increase in HRP3-
17EI2
activity detected in the supernatant (FIG.25, solid squares).
Se4~g Data
Sequencing revealed that HRPI-77E2, the parent used for thennostability and
HzO~
stability studies carries a reverent D255 to N255 (GAC to AAC), and a second
mutation
L37I (TTA to ATA). This residue is part of the helix 2, and is near the heme
pocket (34).
See, FIG. 12, (SEQ. ID. NO. 5) and [SEQ. ID. NO. 6[.
The mutant HRPI-4B6 carries K232M (AAG to ATG) in addition to L37I. This
residue is part of the helix 14, and is exposed to solvent on the surface.
See, FIG.13, (SEQ.
ID. NO. 7[ and (SEQ. ID. NO. 8].
HRP I -2SB 11, the mutant with thennostability between HRP 1-77E2 and HRP 1-
4B6
has the mutation F221 L (TTT to TTA) in addition to L37I. This residue is in a
structural
-39-
SUBSTITUTE SHEET (RULE 2b)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127
loop and pare of the substrate access channel (34). Sec, FIG. 14, [SEQ. ID.
NO. 9] and
[SEQ. ID. NO. 10]. -
1'he mutant HRP1-24D1 1 contains the mutation L131P (CTA to CCA) in addition
to L371. This residue is at the tip of the helix 7, and is on the surface.
See, F1G. 15, [SEQ.
1D. NO. 11] and [SEQ. 1D. NO. 12[.
The mutant HRPI-11764, a preferred mutant from the first generation in tcm~s
of
total activity, contains five mutations with respect to its parent: ( i ) a
reversion of D255 to
N255 (GAC to AAC) (the wild-type sequence); (2) L131 P (CTA to CCA); (3) L223Q
(C'fG
to CAG); ~~~itli silent mutations (4) at N135 (AAC to AAT) and (5) T257 (ACT
to ACA).
For the mutation L223Q, this amino acid residue is in a loop and is exposed to
solvent. See,
FIG. 1G, [SEQ. ID. NO. 13[ and [SEQ. 1D. NO. 14).
Strikingly, the improved HRP mutants, HRP1-80C i 2 (FIG. 17, [SEQ. ID. NO. 17[
and (SEQ. ID. NO. 18)). and HRP1-77E2 (FIG. 20, [SEQ. 1D. NO. 23[ and [SEQ.
ID.
NO. 24]) also carry the revenant D255 to N255 (GAC to AAC). In addition, HRP 1-
80C12
contains L131P (CTA->CCA), found in HRP1-7764. On the other hand, HRP-77E2 has
a second mutation L37I (TTA --> ATA) which is part of the helix B, and is in
the heme
pocket, presumably accessible to solvent as well.
HRP2-28D6 (FIG. 18, [SEQ. ID. NO. 19] and [SEQ. ID. NO. 20]) contains two
additional mutations with respect to HRP1-11764: T102A (ACT --> GCT) and P226Q
(CCA --> CAA). T102A is pan of the helix D, and is the only mutation found to
be buried
inside the structure. P226Q is located in the same loop as L223Q. HRP2-13A10,
on the
other hand, contains four more mutations with respect to HRP1-I 1764: R93L
(CGA -->
CTA); T102A (ACT --> GCT); K241T (AAA --> ACA); and V303E (GTG --> GAG).
R93L, which is solvent accessible, is in the structure loop connecting helices
C and D.
K241T is in the structural loop connecting helices G and H. This residue is
again exposed
to the solvent. Finally, V303E is pan of the long strand extending from helix
J at the C-
terminus of the protein. These three mutations seem to contribute to the
increased stability
of this mutant compared to the others.
HRP3-I7E12 contains three more mutations with respect to the parent HRP2-28D6:
N47S (AAT --> AGT); K241 T (AAA --> ACA), and one silent mutation at G 121
(GGT -->
GGC). It is noteworthy that K241T was also found in HRP2-13A10. N47S is
located in
a structural loop which connects helix B and a 3-helix, and is also solvent
accessible.
-40-
SUBSTITUTE SHEET (RULE 2b)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127 -
Analysis o~Mutations.
All three improved mutant forms of the first generation of evolution carry
they _
revenant D255N with respect to the parent t~RPIAb. 'Plus appears to suggest
that the
giycosylation sites on HRP are beneficial for folding and expression. The
function of
glycosylation in proteins has been an intriguing matter, but its role in
protein folding,
processing and secretion is being gradually recognized (SG-58).
The three synonymous mutations can not be easily explained by changes in codon
usage (G3). Two of them, N 135 (AAC --> AAT) and ~f257 (ACT --> ACA), resulted
in few
changes in the frequencies of used, while for G 121 (GG'r --> GGC) , a more
frequently used
1 Q codon (GCT, G 1 %) was replaced by a Icss frequent ot~e (GGC,
16°io). However, it is unclear
how this substitution would significantly affect the translation of HRP mRNA.
Char ucteri°atiorr o~rrrrrtants reear-dine reaclivify and
stabilil.
Besides the ABTS assay also as a second independent activity assay for HRP
mutants the guajacol system is used (FIG. 21 ). Both assays show a good
correlation
regarding activity of the mutants.
Figures 22a and 22b show the correlation between reactivity and stability
after incubation
at 50°C without H~O~ (a) and with ImM H20~ (b). In both cases mutant
HRP 2-13A10
shows the highest stability in combination with a good reactivity. As revealed
by sequencing
three amino acid changes seem to be responsible for this stability.
A similiar pattern of stability is observed in organic solvent system (Figure
23) where
mutant HRP 2-13A10 shows the best ratios of activity in dioxane/buffer system
versus those
in buffer only.
EXAMPLE 3
Expression and Secretion of CCP in E. coli
Corrsrrrrction of expression vector for CCP.
The S. cerevisiae cytochrome c peroxidase (CCP) gene from pT7CCP (16,17),
donated by Dr. Dave Goodin, The Scripps Research Institute, La Jolla, CA) was
recloned
by PCR techniques to introduce an Msc I site at the start codon and a Hind III
site
immediately downstream from the stop codon. The PCR product was restricted
with Msc
-41-
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127 -
i and Hind III, and then iigatcd into similarly digested pET-22b(+), yielding
pETCCP (Fig.
26). The pT7CCP carries a gene for CCP in which the N-terminal sequence has
bee ~ -
modified to code for amino acids Met-Lys-Thr, as described in Goodin et al.
(17) and
Fitzgerald et al. ( 1 G). Thus, in this construct, the CCP gene was placed
under the control
of the T7 promoter, and was fused in-frame to the pelB signal sequence for
periplasmic
Vocalization.
Frnrcs.sio~r ojCC'l'.
Eaprcssion experiments of CCP in E roll BL21 (DE3) were carried out in LB
I U medium containing 100 pg/ml ampicillin. Cells were grown at 37°C to
an A~ of 0.7-0.8,
at which time IPTG was added to a final concentration of 1 mM to induce the
synthesis of
CCP from the T7 promoter. Growth was continued at 30°C for an
additional 20 hours, and
cells and supenlatant were harvested by centrifugation.
CCP is known to fold correctly inside E. coli. Surprisingly, greater than 95%
of the
1 S CCP protein was found in the LB culture medium at high levels
(approximately 100
mg/liter, as assessed by SDS-PAGE). The protein was active towards ABTS,
showing that
the secreted CCP is folded and contains the required ferric heme.
Having thus described exemplary embodiments of the invention, it should be
noted
by those skilled in the art that the within disclosures are exemplary only and
that various
20 other alternatives, adaptations, and modifications may be made within the
scope of the
invention. For example, it will be understood by practitioners that the steps
of any method
of the invention can generally be performed in any order, including
simultaneously or
contemporaneously, unless a particular order is expressly required, or is
necessarily inherent
or implicit in order to practice the invention. Accordingly, the invention is
not limited to
25 any specific embodiments or illustrations herein. The invention is defined
according to the
appended claims, and is limited only according to the claims.
BIBL10GRA.PHY
30 1. Cleland, J. L; Wang, D. I. C. BiolTechnology 8, Z 274 ( 1990).
-4Z-
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127
2. Bernarderz-Clark, E. D.; Georgiou, G. Inclusion Bodies and Recovery of
Proteins
from the Aggrcgatcd Statcs. In Protein Rejoldirrg; Bcrnarderz-Clark, E. D., ~ -
Gcorgiou, G., Eds,; ACS: Washington, D. C. p. 1-20 (1990)
3. Thatcher, D. R.; Hitchcock, A. Protein Folding in Biotechnology. In
A~eclrarrisms
ojProtcirr Folding; Pain, R. H., Ed.; 1RL Prcss: Oxford p. 229-2G1 (1994).
4. Parckh, R.; Forrcstcr, K.; Wittrup, D. Protein I:.rlrres. I'rrrij. 6, 537 (
1995).
5. Amold, F. 1J. ACCUrlrrIS C'hem. Rcs. 31, 125 ( 1998).
(,, Mitraki, A.; King, J. FEBS Lett. 307, 20 ( 1992).
7. Zhang, J. X.; Goldenberg, D. P. Biochenristy 32, 14075 (1993).
g, Wetzel, R.; Peny, L. P.; Veilleux, C. BiolTechnology 9, 731 (1991 ).
9_ Knappik, A.; Pluckthun, A. Protein Eng. 8, 81 ( 1995).
10. Crameri, A.; Whiteborn, E. A.; Tate, E.; Stemmer, W. P. C. Nature
Biotechrrol. 14,
315 ( 1996).
11. Tams, J. W.; Welinder, K. G. FEBSLett. 421, 234 (1998).
12. Ortlepp, S. A.; Pollard-Knight, D.; Chiswelt, D. J. J. Bioteclrnol. 11,
353 (1989).
13. Smith, A. T. et al. J. Biol. Cheer. 265, 13335-13343 (1990).
14. Egorov, A. M.; Gazaryan, I. G.; Savelyev, S. V.; Fechina, V. A.;
Veryovkin, A. N.;
Kim, B. B. Ann. N. Y. Acad. Sci. 646, 3 5 ( 1991 ).
15. Moore, J. C.; Arnold, F. H. Nature Biotechnol. 14, 458 (1996).
-43-
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127
16. Fitzgerald; M. 1~1.: Churchill, M. J.; McRee, D. E.; Goodie, D. B.
Biochemisry 33~
3807 ( 1994).
17. Goodie, D. B.; Davidson, M. G.; Roe, J. A.; Mauk, A. G.; Smith. M.
Bioclre»ristry
30, 4953 ( 1991 )
1 S. Dc Suttcr, K.; 1-lostcns, K.; Vandckcrckhovc, J.; Ficrs, W. GEArI: 141,
163 ( 1994).
19. Salllbl'UOk, J.; Fritsch, E. F.; Maniatis, T. AlOlccr(I(rr'C~IOrrlrrb': A
l.crborcrtor_vA4u»rlul;
Cold Spring Harbor Laboratory: New York (1989).
20. Caldwcll, R. C.; Joyce, G. F. PCR Metlrods Applic. 2, 28 (1992).
21. Beckman, R. A.; Mildvan, A. S.; Loeb, L. A. Biochemistry 24, 5810 (1994).
22. Shafikhani, S.; Siegel, R. A.; Ferrari, E.; Schellenberger, V.
Biotech»iques 23, 304
( 1997).
23. Stemmer, W. P. C. Proc. Natl. Acad. Sci. USA 91, 10747 (1994).
24. Zhao, H. M.; Arnold, F. H. Nucleic Acids Res. 25, 1307 (1997).
25. Carbon, J.; Clarke, L.; Ilgen, C.; Ratzkin, B. The Construction and Use of
Hybrid
Plasmid Gene Banks in Escherichia coli. In Recombinant Molecules: In:pact o»
Science and Society; Beers, R. F. J., Bassett, E. G., Eds; Raven Press: New
York, pp
355-378 (1977).
26. Shindler, J. S.; Childs, R. E.; Bardsley, W. G. Eur. J. Biochenr. 65, 325
(1976).
27. Lei, S. P.; Lin, H. C.; Wang, S. S.; Callaway, J.; Wilcox, G. J.
Bacteriol. 169, 4379
( 1987).
-44-
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCTNS99/17127 -
28. Better, M.; Chang, C. P.; Robinson, R. R.; Horwitz, A. H. Science 240,
1041
(1988).
29. Goshorn, S. C.; Svensson, H. R.; Kerr, D. E.; Somerville, J. E.; Senter,
P. D.; Fell,
H. P. Cancer Res. 53, 2123 (1993).
30. Rathore, D.; Nayak, S. K.: Batra, J. K. FEBSLett. 392, 259 (1996).
31. Studier, F. W.; Rosenberg, A. H.; Dunn, J. J.; Dubendorff, J. W. Meth.
Enzymol.
185, 60 ( 1990).
32. Ostermeier, M.; Desutter, K.; Georgiou, G. Eukaryotic J. Biol. Chem.
271,10616
{ 1996).
33. Savenkova, M. L; Kuo, J. M.; Ortiz de Montellano, P. R. Biochemistry
37,10828
(1998).
34. Gajhede, M.; Schuller, D. J.; Henriksen, A.; Smith, A. T.; Poulos, T. L.
Nature
Struct. Bio14, 1032 (1997).
35. Anfmsen, C. B. Science 181, 223 (1973).
36. Schein, C. H. BiolTechnology 8, 308 (1990).
37. Martineau, P.; Jones, P.; Winter, G. J. Mol. Biol. 20, 117 (1998).
38. Stemmer, W. P. C. et al., Biotechniques 14, 256 (1992).
39. Arkin, A. and Youvan, D. C. Proc. Natl. Acaa'. Sci. USA 89, 7811 (1992).
40. Oliphant, A. R. et al., Gene 44, 177 (1986).
-45-
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127
41. Hermes, J. D. et aL, Proc. Natl. Acad. Sci. USA 87, 696 (1990).
42. Delagrave et al. Protein Engineering 6, 327 (1993).
43. Delagrave et al. BiolTechnology 11, 1548 (1993)
44. Goldman, E. R. and Youvan D. C. BiolTechnology 10,1557 (1992).
45. Leung, D. W. et al., Technique 1, (1989).
46. Grarnm, H. et al., Proc. Natl. Acad Sci. USA 89, 3576 (1992).
47. Castelli, M. C. et al., Gene 142, 113 (1994}.
48. Gietz, D., Schiestl, R. H., Willems, A., Woods, R. A., Yeast 11, 355
(1995).
49. Welinder, K. G., Eur J. Biochem. 96, 483-502 (19?9).
25
50. Sirotkin, K. J. Theor. Biol. 123, 261 (1986).
51. Glover, D.M. (ed.), 1985, DNA Cloning: A Practical Approach, MRL Press,
Ltd.,
Oxford, U.K. Vol. I, II.
52. Schatz, P.J. Et al., Annu. Rev. Genet. 24, 215-248 (1990).
53. Lin, Z., Thorsen, T. & Amold, F.H. Biotechnol. Prog. 15, 467-471 (1999).
54. Cregg, J.M., Vedvick, T.S. & Raschke, W.C. BiolTechnology 11, 905-910
(1993).
55. Gussow, D. & Clackson, T. Nucleic Acids Res., 17(10):4000. (1989).
56. Helenius, A. Mol. Biol. Cell 5, 253-265 (1994).
-46-
SUBSTITUTE SHEET (RULE 26)
CA 02331777 2000-12-21
WO 00/06718 PCT/US99/17127
57. Fiedler, K. & Simons, K. Cell 81, 309-312 (1995).
58. Nagayama, Y., Namba, H., Yokoyama, N., Yamashita, S. & Niwa, M. J. Biol.
Chem. 273, 33423-33428 (1998).
59. Gillam, E.M., Guo, Z., Martin, M.V., ]enkins, C.M. & Guengerich, F.P.
Arch.
Biochem. Biophys. 319, 540-550 (1995).
60. Guengerich, F.P., MArtin, M.V., Guo, Z. & Chun, Y.J. Meth Enzymol 272, 35-
44 (1996).
61. Khosla, C., Curtis, J.E., Demodena, J., Itinas, U. & Bailey, J.E.
Bio/Technology
8, 849-853 (1990).
62. Joo, H., Arisawa, A., Lin, Z. & Arnold, F.H. Chem. Biol. submitted(1999).
63. Ausubel, F.M. et al. Current Protocols in Molecular Biology, (Greene
Publishing
Associates and Wiley-Interscience, New York,1987).
-47-
SUBSTITUTE SHEET (RULE 26)