Note: Descriptions are shown in the official language in which they were submitted.
CA 02335347 2000-12-15
WO 99/67345 PCT/KR99/00338
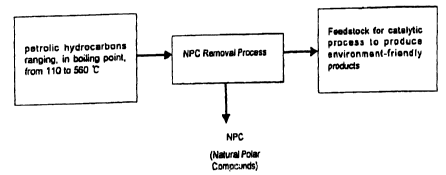
METHOD FOR MANUFACTURING CLEANER FUELS
FIELD OF THE INVENTION
The present invention relates, in general, to a method for manufacturing a
cleaner fuel and, more particularly, to the removal of NPC (Natural Polar
Compounds) from petroleum hydrocarbon feedstocks ranging, in boiling point,
from
110 to 560°C, in advance of a catalytic hydroprocessing process. The
removal of
NPC improves the efficiency of the catalytic process and produces
environmentally
favorable petroleum products, especially diesel fuel with a sulfur content of
below
50 ppm (wt) by deep hydrodesulfurization. Also, the present invention suggests
the
usage of such NPC to improve fuel lubricity.
DESCRIPTION OF THE PRIOR ART
The ever-worsening environmental pollution problem, especially, air
quality degradation, has brought s:ringent environment regulatory policies
throughout the world, and developed countries are imposing tight quality
regulations upon transportation fuels. Of such fuels, diesel fuel is
considered to be a
major contributor of such harmful pollutants as SOx, NOx and PM (particulate
matters). The most severe regulatory standards are being applied to diesel
fuels.
While such diesel quality specifications as sulfur content, aromatics content,
polyaromatics content, cetane number, T95 (95% distillation temperature),
density
and viscosity are known to affect generation of;he aforementioned pollutants,
sulfur
content has become the most critical issue because it forms sulfur dioxide
when
combusted. Further, a portion of sulfur dioxide is readily converted to sulfur
trioxide,
which, with moisture, forms PM. Besides contributing to the formation of PM,
1
CA 02335347 2000-12-15
WO 99/67345 PCT/KR99/00338
sulfur-containing compounds such as sulfur dioxide and sulfate harm automobile
emission after-treatment devices by poisoning the noble metal catalysts
therein.
Recently, automobile manufacturers have claimed that the sulfur content of
diesel fuel should be reduced to below 30 ppm (wt) for their new diesel
engines to
S meet the future tail-pipe emission regulations. Consequently, a ULSD (ultra
low
sulfur diesel) market is now emerging, especially in Western Europe.
Eventually,
such fiuels are expected to replace the conventional 500 ppm sulfur diesel
fuel
market.
In keeping up with the tightening regulations, oil companies have been
making large investments to produce environment-friendly petroleum products,
for
example, by revamping existing facilities or installing new processes. From an
economic standpoint, however, neither existing nor newly developed processes
thus
far appear to be economically feasible under the current price structure of
petroleum
products. Therefore, the United States and many countries in Western Europe
have
implemented refiner-inducing policies such as tax incentives, which reimburse
additional costs incurred in producing cleaner fuels.
An HDS (Hydrodesulfurization) process is most commonly used to reduce
sulfur content from diesel fuel by converting sulfur compounds into hydrogen
sulfide. In the late 1950's, the HDS process was first introduced as a
pretreatment of
naphtha reforming process since catalysts were prone to poisoning by sulfur
compounds. since then, various HDS processes have been developed and an HDS
process for LGO (light gas oil) appeared in the 1960's. Nowadays, most
refineries
are equipped with HDS processes, and statistics shows that, in 1994, the unit
capacity of kerosene and LGO HDS processes in the world amounted to 21 % of
that of the crude distillation units.
Many of the HDS processes currently being used by refiners are non-
licensed processes, and most of related patents pertain to catalyst
preparation and
2
CA 02335347 2000-12-15
WO 99/67345 PCT1KR99/00338
modification. Generally when the process variables are properly modified and
suitable catalysts are selected, diesel fuel with 0.1 weight percent of sulfur
can be
produced. In order to reduce the sulfur content below 50 ppm, however,
innovative
improvement in terms of the following operating parameters is required:
catalyst
activity, reaction temperature, bed volume and hydrogen partial pressure.
Catalyst activity has been doubled since the first generation LGO HDS
catalyst was' introduced in the late 1960s. However, the activity has to be
further
improved to attain deep HDS to desired levels. Deep HDS is understood herein
to
refer to hydrodesulfurization rates greateyw than 95%. An improved activity,
by a
factor of 3.2, compared to that of the first generation catalyst; is required
to reduce
the sulfur content from 2,000 ppm to 500 ppm, and an improvement in activity
by a
factor of 17.6 is needed to reach the 50 ppm level. This means that unless the
catalyst activity is dramatically improved, the number of reactors must be
increased
or the charge rate must be decreased to achieve deep HDS. To make the matters
worse, the catalysts are getting more and more expensive because of the
increase in
the amount of impregnated metals employed in the catalysts and the
sophisticated
modification of support structure, while catalyst lifetime is reduced to 1/2-
1/5 of
conventional catalysts, as reaction conditions get severe.
Reaction temperatures may be increased to reduce the sulfur content.
However, since most HDS processes were designed for a 0.2% sulfur level, the
furnace and the reactor cannot be operated exceeding the design limits. In
addition,
increase in temperature results in product color degradation and/or reduction
in
catalyst life.
In the past, many refiners opted to install additional reactors to meet the
regulatory standards because it seemed to be a simple and straightforward
approach.
However, only a finite number of reactors can be added because there exist
space
limitations, pressure drop considerations across reactors, and huge capital
costs for
3
CA 02335347 2000-12-15
WO 99/67345 PCT/ICR99/00338
additional reactors and compressors.
Increasing the pressure of reactors, as mentioned previously, could be
another alternative. Yet, the revamp costs for high pressure reactors,
compressors,
pumps and heat exchangers are significantly high, not to mention the hydrogen
S consumption increase.
Besides sulfur, it has long been disputed whether the aromatics content
should be a part of the quality standards of diesel fuel. Nevertheless,
automotive
diesel fuels with low aromatic content are already manufactured and sold
regionally
in the United States and Northern Europe. To saturate aromatic compounds,
however, a large amount of hydrogen is necessary with noble metal catalysts,
and
energy consumption also increases noticeably. In addition, the use of noble
metal
catalysts requires an additional HDS process preceding the catalytic
hydrotreating
process in order to prevent sulfur and nitrogen compounds from deactivating
the
catalysts.
Of the catalytic hydroprocessing processes that are designed to produce
cleaner diesel fuels from LGO by removing sulfur and aromatic compounds, only
a
few of them are commercially available, and they can be categorized into the
following three groups.
First, there is a process in which HDS (hydro-desulfuriaation) and HDA
(hydro-dearomatization) are accomplished simultaneously under a high
temperature
and high pressure with a nickel-molybdenum-based catalyst' of high activity in
a
single reactor. The process is, however, not widely used because high
temperature
and high pressure facilities, together with a low processing rate,
significantly
increases the investment cost and still cannot achieve a desirable aromatics
conversion rate.
A second process utilizes t~v~, xea~y~ors placed in series. Deep I-IDS is
achieved in the front reactor while the rear reactor charged with a noble
metal
4
CA 02335347 2000-12-15
WO 99/67345 PCT/KR99/00338
catalyst, reduces aromatic compounds. The process is usually constructed by
adding
a new HDA urilt tri the rear of the existing HDS unit. HDA conversion rate is
significantly improved compared to the stand-alone HDA unit. However,
investment
cost and operation cost also increase significantly.
S Third, there is the Syn-Sat process in which HDS and HDA are conducted
at a high effciency by utilizing countercurrent flow in a single reactor. The
Syn-Sat
process enables higher conversion rate than any other processes, and the
process
economics are superior to two-stage reaction processes. Yet, the Syn-Sat
process
still requires significant amount of investment cost as well as operation cost
compared to deep HDS processes. In additi.m; close attention regarding HDA
catalyst poisoning is required so that the HDS exit stream contains no more
than 10
ppm (wt) of sulfur compounds.
As noted above, conventional processes treating LGO have technical
limitations while breakthroughs in catalyst activity have not been realized.
1 S Therefore, methods using different feedstocks instead of LGO, or using
innovative
reaction pathways, are being studied and practiced in manufacturing cleaner
diesel
fuel.
Hydrocracking processes, using VGO (Vacuum Gas Oil) instead of LGO,
exemplify such methods. Since VGO has very high sulfur content and nitrogen
compound content, HDS and hydrocracking reactions are carried out in two-stage
reactors at high temperature and high pressure. Kerosene and diesel
distillates
obtained from hydrocracking are nearly sulfur-free and contain SO% less
aromatic
compounds compared to that of the products frnm LGO HDS process. However, due
to the high viscosity of the feed, the reaction efficiency is relatively low
and the
investment cost is almost three times higher than that of conventional deep
HDS
processes.
Another process suggested is to polymerize natural gas to produce a diesel
S
CA 02335347 2000-12-15
WO 99167345 PCTlKR99/00338
distillate such as Shell's middle distillate synthesis (SMDS) process. In the
SMDS
process, natural gas is converted into syn-gas through the Fischer-Tropsch
reaction,
then it undergoes polymerization to produce diesel distillates free of sulfur
and
aromatic compounds. However, since the feed is fairly expensive, and since the
reaction is carried out in three steps, a high investment cost is needed.
Consequently,
it is difficult for most of refiners to attain an economical benefit unless
they have
their own natural gas field and gas-to-liquid conversion process near the
natural gas
field.
Recently, a new technology using a bio-catalyst, referred to as
biodesulfurization process, is under development. Regarded as supplementary
for
HDS processes, biodesulfurization selectively removes the refractory sulfur
compounds, which are difficult to remove by conventional HDS. However, it is
reported that the biodesulfurization process does not yet have the sufficient
reaction
efficiency (space velocity is about 0.1 hr-1 ) applicable for oil refineries
where
large-scale treatments are required. Biodesulfurization also generates
byproducts
such as phenols.
U. S. Pat. No. 5,454,933 discloses an adsorption process to produce sulfur-
free diesel fuel by removing sulfur compounds from an HDS-treated LGO stream.
Despite using the similar adsorption principles, the present invention differs
from
that patent's disclosed invention in chat NPC is removed, instead of sulfur
compounds, upstream of an HDS unit to improve Sulfur conversion rate of the
HDS
unit.
Adsorption, in general, is known to be ineffective in removing the sulfur
compounds from a petroleum hydrocarbon stream. Sulfur compounds have
relatively low polarities compared to nitrogen or oxygen compounds, and an
adsorbent which can adsorb as much sulfur compounds as 0.05% of feedstock is
difficult to come by. Activated carbon usually tends to gradually lose its
adsorption
CA 02335347 2000-12-15
WO 99/67345 PCT/KR99/00338
effectiveness as desorption is repeatedly performed. Therefore, to maintain
sulfur
removal rate, the adsorbent must be regenerated more frequently. This will,
however,
result in yield loss and increased operation cost with less amount of
feedstock
treated and more amount of solvent spent in an operation cycle.
Since the disclosed invention of U. S. Pat. No. 5,454,933 does not indicate
whether sulfur removal rate is maintained, a series of experiments was
performed by
using activated carbon, having similar physical properties to that used in U.
S. Pat.
No. 5,454,933: it was revealed that sulfur removal rate was not satisfactory,
the
sulfur removal rate decreased as desorption was repeated, and the generation
of
desorption extract, the byproduct, was excessive. Results of the experiments
are
tabulated in Comparative Example 18 below.
U. S. Pat. No. 5,730,860 discloses a technique in which the limit in
producing gasoline products 30 ppm (wt) or less in sulfur content can be
overcome
through a conventional hydroprocessing process. According to this technique,
hydrocarbons with high concentrations of sulfur, nitrogen and oxygen compounds
(for example, mercaptan, amine, nitrile and peroxide, exemplified by fluidized
catalytic cracking (FCC) gasoline, a half finished gasoline product) are
treated with
a counter current-type fluidiaing adsorption process and the adsorbent used is
regenerated by use of hot hydrogen, after which the adsorbate concentrated
with
hetero-compounds is subjected to HDS. But this technique has the limitation
that it
can't be applied to a hydrocarbon stream having a boiling range of 260'C or
higher.
In addition, since the by-products produced in the above process must be
treated in
the diesel HDS process, the desulfurization performance under deep I-117S
conditions
may be negatively affected. Therefi~r~, appi~ing this technique is problematic
to the
current situation wherein ult~ a low sulfur diesel fuel has to be produced
concurrently
with gasoline.
Besides, bio-diesel products which are prepared by formulating existing oil
7
CA 02335347 2000-12-15
WO 99/67345 PCT/KR99/00338
products with the oils extracted from plants in an amount of about 20 %, were
found
to produce pollutants at a significantly reduced amount. These bio-diesel
products,
which are developed as an alternative fuel in some countries rich in
agricultural
products, cause a significant problem, so they are suggested to be formulated
at the
amount of about 20 % with conventional diesel fuels. In this case, however,
there is
also caused a significant problem in storage stability.
As explained above, various attempts have been made to produce cleaner
oils, but they are either economically unfavorable because of large-scale
investments or technical limitations.
The intensive and thorough research on the manufacture of cleaner fuels,
carried out by the present inventors, resulted in the finding that the
pretreatment of
LGO with such well known techniques as adsorption or solvent extraction,
permits a
great improvement over the HDS performance of the catalysts used in a deep HDS
zone. Oil fractions removed during the pretreatment step of the present
invention are
composed of various kinds of compounds having such functional groups as -COOH
(naphthenic acids), -OH (phenols), -N(pyridines) and -NH (pyrroles), and
sulfur-
containing compounds having higher polarity other than that of
dibenzothiophene,
as exemplified in Example 4, below. Nitrogen~~containing compounds are mainly
heterocyclic compounds such as carbazoles, benzocarbazoles, indoles,
pyridines,
quinolines, acridines, .and tetrahydroquirmlines. Even though saturated and
aromatic
compounds are also contained in these fractions, the fractions are
characterized by
relatively high polarities due to the high concentration of polar organic
compounds
as described above. Such polar compounds exist in trace amounts, overall, in
petroleum hydrocarbon. Therefore, these polar organic compounds are defined
herein as NPC (Natural Polar Compounds) so as not to be confused with
synthetic
polar compounds, such as process additives or chemicals, and the like.
Depending on the crude ail sources, viscosity and pretreatment of the
8
CA 02335347 2000-12-15
WO 99/67345 PCT/KR99/00338
distillates, NPC have different physical properties and composition. Being
almost
electtiCally neutral, the NPC separated from LGO can be grouped into acidic,
basic
and neutral compounds.
Although NPC content becomes higher in petroleum products with higher
boiling points, NPG exists in relatively small quantities, so that the removal
of the
compounds has little influence on the physical and chemical properties of the
remaining fraction, such as, for example, viscosity range and the content of
sulfur
and aromatic compounds. Therefore, NPC do not harm catalysts or catalytic
processes unlike byproducts or impurities. NPC have not burdened the
achievement
of the sulfur conversion target of the HDS process even though NPC have
relatively
high polarities and densities compared to the distillates that NPC derive
from.
However, it was found by the present invention that even small quantities
of NPC have a significantly negative effect upon the HDS process in the deep
desulfurization zone, which can be achieved only if such compounds as
dibenzothiophene (DBT) and 4,6-dimethyl dibed~zothiophene (4,6-D1V)DBT) are
converted. Consequently, the present inventors conducted extensive research on
removal of NPC, and it is found that such well-known technologies as
adsorption
and solvent extraction can be used as an effective pre-treatment, upstream of
an
HDS unit, to produce cleaner fuels.
To remove impurities or polar compounds from hydrocarbons, adsorption
or solvent extraction has been widely used for a long time. For example, U. S.
Pat.
No. 5,300,218 discloses the use of an optimal adsorbent such as a carbon
molecule
complex in removing diesel smoke-causing materials. U. S. Pat. No. 4,912,873
also
discloses an adsorption process that treats diesel fuel and jet fuel with a
polymer
resin to minimize coloration and filter clogging problems. However, the carbon
molecule complex or the polymer resin are not effective in achieving a
beneficial
NPC removal ratio and are too expensive to be used as an adsorbent for the
present
9
CA 02335347 2000-12-15
WO 99/67345 PCT/KR99/00338
invention. Moreover, the application ranges of the carbon molecule complex or
polymer resin are different from that of the present invention, which is
related to the
improvement of the catalytic activity of hydroprocessing.
Of petroleum and petrochemical manufacturing processes, catalytic
reaction processes take significant portions, and protecting the catalysts
from
permanent performance loss is an important issue. To prevent permanent
performance loss owing to by-products and/or impurities originated from former
stages or feedstock, various pretreatment processes are being utilized. Among
such
pretreatment processes, principle of adsorption or solvent extraction are
commonly
applied thereto. Typical examples include mechanical filters preventing
accumulation of micro impurities; a caustic washing column where naphthenic
acids
in raw materials are neutralized and extracted to protect basic catalysts in
the Merox
process; and an activated clay column that adsorbs sulfur and olefins prior to
naphtha reforming process.
1 S Particularly, the isomerization and etherification process are vulnerable
to
impurities damaging the catalysts, and extensive research has been done on
pretreatment techniques for removing such impurities, as representatively
disclosed
in U. S. Pat. No. 5,516,963, 5,336,834, 5,264,187, 5,271,834, 5,120,881,
5,082,987,
4,795,545 and 4,409,421. However, the application ranges of these, the
feedstock or
processes of these references, are different from that of the present
invention.
U. S. Pat. No. x,344,84!, 4,343,693 and 4,269,694 pertain to adsorption
techniques for preventing water, sediments and additives from causing deposit
formation and equipment fouling in catalytic processes, e.g., subsequent
hydrotreating processes.
U. S. Pat. IVo. 4,176,047 discloses an adsorption pre-treatment process
using waste alumina catalysts in the Delayed Coker process that prevents
silicon-
based antifoaming agents from having a negative influence vn subsequent HDS
CA 02335347 2000-12-15
WO 99/67345 PCT/KR99/00338
processes and processes that improve octane number.
U. S. Pat. No. 4,033,861 discloses a method for reducing nitrogen contents
in hydrocarbon by polymerizing nitrogen compounds that are difficult to be
removed by hydrodenitrification, and separating them with their increased
boiling
points.
U. S. Pat. No. 3,954,603 discloses a method of removing catalyst-poisoning
contaminants, such as arsenic or selenium, from a hydrocarbon stock, such as
Shale
oil, Syncrude and bitumen, in a two-step pretreatment process using iron,
cobalt,
nickel, oxides or sulfides of these metals, or mixtures thereof.
Scrutinizinb the prior arts, as explained above, adsorption and/or solvent
extraction are used only for product quality improvements and for cases where
a
catalytic reaction process cannot be physically operable due to additives,
impurities
or byproducts originating from a former stage and/or from feedstock. Thus far,
there
has been no pretreatment that is developed upon the basis of the fact that the
NPC
1 S removal, the kernel of the invention, has a great influence on the
catalyst activities
in the deep HDS zone.
SUMMARY OF THE INVENTION
The present invention aims to achieve an improvement in catalytic
processes by removing NPC that naturally exist in crude oil. The constituents
of
NPC do not cause a fatal influence on the activity of catalysts used in
general
processes, and are normally converted according to their own reaction pathways
in
catalytic processes. However, where certain sulfur compounds, which require
high
activation energies for their removal, need to be desulfurized in order to
approach a
desulfurization rate of 97% or higher, NPC is found to have a significant
influence
on the reaction pathways and reaction effectiveness of the sulfur compounds.
CA 02335347 2000-12-15
WO 99/67345 PCT/KR99100338
According to the present invention, the influencing factors, in the form of
NPC, can be easily removed through adsorption/desorption or solvent extraction
techniques, and the NPC-removed feedstock enhances HDS rate by 1-2%. This
fraction of improvement may seem to be marginal. However, this additional 1-2%
is
significant in the deep HDS zone, making it possible to produce diesel fuel
with a
sulfur content of 50 ppm (wt) or less in more economical way than any other
processes known to date.
Although various technologies for the desulfurization and dearomatization
of diesel distillates have been developed, oil companies do not regard them as
economically feasible.
With the aim of economically producing petroleum products containing
less sulfur, nitrogen and aromatic compounds for reducing harmful tailpipe
emission
from diesel vehicles, the present invention includes the removal of NPC, which
was
nowhere mentioned in the prior art, improves the efficiency of existing
catalysts,
and has advantages over prior art processes which require excessive
investments
and operation costs. As a consequence of the intensive and thorough
experiments
that the present inventors repeated, in an effort to apply the principle of
the
invention to cornmercialization, it was revealed that some adsorbents can be
continually regenerated in such adsorption/desorption applications, and such
NPC
removal improves the performance of subsequent catalytic reaction processes
for
various feedstocks.
In addition, regarding the lubricity degradation resulting from deep
desulfurization, it has been found that concentrated NPC, obtained by
adsorption, is
effective as a natural lubricity improver.
Although fixed bed adsorption technology was adopted to prove the
invention in most cases, the application ro other types of pretreatment, which
can be
selected depending upon feedstocks, in~Yudog iluidizing bed adsorption and
Solvent
12
CA 02335347 2000-12-15
w0 99!67345 PCT/KR99/00338
extraction, is also included in the scope of the invention.
BRIEF DESCRIPTION'OF THE FIGURE
Figure 1 is a flow diagram illustrating a basic concept of the present
invention.
Figure 2 is a simplified flow scheme of an adsorption process according to
the present invention.
Figure 3 is a graph of product sulfur concentration versus reaction
temperature for two kinds of NPC-removed feedstocks, and base feedstock, in
accordance with Example 13.
Figure 4 is a graph of the nitrogen removal rate versus the number of
regeneration, in accordance with Examplr;, 10.
DETAILED DESCRIPTION OF PREFERRED EMBODIMENTS
The present invention pertains to the substantial removal of NPC from
petroleum hydrocarbon fuel stocks, which improves activity of catalysts in
subsequent hydroprocessing processes, thus facilitating economical production
of
cleaner fuels that emit lower level of pollutants, especially PM, NOx and SOx,
upon
combustion in engines. The overall Concept of thr present invention is
illustrated in
Fig. 1. The petroleum hydrocarbon fuel stocks used in the present invention
range in
boiling point, from 110 to 560°C and preferably from 200 to 400 C. NPC,
naturally
existing in these petroleum hydrocarbon fractions, can be removed by
adsorption or
solvent extraction. Removal of NPC through adsorption utilizing one or more
adsorbents is found to be the most effective method according to a series of
experimental studies carried out for this invention.
13
CA 02335347 2000-12-15
WO 99/67345 PCT/KR99/00338
Hydrocarbon fuel produced in according with the present invention
preferably has a boiling point in the range of 110°C to 400'C and
preferably has a
sulfur content less than 500 ppm (wt), and most preferably less than 50 ppm
(wt).
Adsorption is, therefore, extensively used in the following examples, and
experiments are carried out with a single column to simplify the illustration
of the
present invention. The aciual process can perform adsorption and desorption in
a
continuous manner by alternately switching two or more fixed beds.
In accordance with the invention, NpC are removed from a petroleum
feedstock fraction to substantially decrease the concentration of NPC in the
l0 petroleum feedstock fraction. The substantial decrease in NPC concentration
is at
least 50%. That is, at least 50% of NPC are removed from the petroleum
feedstock
fraction. Preferably, between about 60% and about 90% of the NPC are removed
from the petroleum feedstock fraction.
As shown in the examples below, NPC are removed easily from
hydrocarbon feedstock by alternating adsorption and desorption in a single
adsorption column. The NPC removed or extracted from a petroleum feedstock
fraction preferably comprise between 5.0 and 50wt% oxygen-containing
compounds,
between 5.0 and 50wt% nitrogen-containing heterocyclic compounds and sulfur
content in the range of 0.1 to S.Owt%. The 1VI1'C removed or extracted
preferably
constitutes between 0.1 and 5.0wt% of trie petroleum feedstock fraction.
Generally, as the boiling poiyyt of the feedstock increases, the viscosity
increases and the amount of NPC extracted land the nitrogen content in the
NPC)
tends to become larger. So, depending on the feedstock, the operation
parameters of
the adsorption pretreatment process such as RPA (Ratio of Product to
Adsorbent),
temperature and LHSV (liquid hourly space velocity, h'') may vary. Among these
parameters, RPA is the most important operation parameter of the pretreatment
process. RPA is further defined as z ratio of the amount of the treated
product to that
14
CA 02335347 2000-12-15
WO 99/673d5 PCT/KR99/00338
of the adsorbent within one operating cycle, which consists of adsorption, co-
purging and regeneration step in series. As RPA is lowered, the severity of
the
adsorption process and the adsorption performance increases.
Examples of available adsorbents include active alumina, acid white clay,
Fuller's earth, active carbon, zeolite, hydrated alurnina, silica gel, and ion
exchange
resins. Hydrated alumina and silica gel have ao strong adsorption sites and
such
adsorption mechanism as hydrogen bonding is uniquely desirable for
regenerability.
The aforementioned adsorbents may be used in combinations of two or more, and
a
proper combination may enhance adsorption effectiveness; silica gel and ion
exchange resin, which are charged in an adsorption column in series, are found
to be
more effective in NPC removal than using silica gel or ion exchange resin
alone.
A preferred adsorbent is silica gel, having a pore size ranging from 40 to
200 ~, a specific surface area ranging from 100 to 1000 m~/g, and a pore
volume
ranging from 0.5 to 1.5 cc/g.
With reference to Fig. 2, there is shown an operation scheme of an
adsorption process according to the present imrention. First, liquid
hydrocarbon
stream is fed for a predetermined period of time into one of two or more
adsorption
columns, alternately, wherein NPC is adsorbed. While the NPC-removed
hydrocarbon liquid is fed to a subsequent catalytic process, the NPC adsorbed
in the
adsorption columns are desorbed by the use of a desorption solvent so as to
regenerate the adsorption column. The desorption solvent is usually selected
from
among alcohols, ethers and ketones containing 6 or less carbon atoms, which
are
exemplified by methanol, methyl-tertiary-butyl ether and acetone. Generally,
the
aforementioned solvents have low boiling points, so that they are easily
distilled and
recovered from feedstock or NPC. Instead of the aforementioned scheme where
the
two fixed beds are utilized, either a fluidizing bed or a moving bed may be
applied
to deliver the same results.
CA 02335347 2000-12-15
W O 99/67345 PCT/KR99/00338
The catalytic reaction processes, which follow the adsorption pretreatment
step, can be an HDS, an HAD, a mild hydrocracking, a hydrocracking process, or
combinations thereof. The catalysts used in these processes have acidic active
sites
on the catalyst surfaces and hetero-atom containing polar compounds decreases
the
catalysts' activities due to the tendency of these compounds to be adsorbed
onto the
active sites, while they do riot deactivate the catalysts permanently.
Also, the present invention pertains to the use of NPC as a natural lubricity
improver against the lubricity degradation resulting from the deep
desulfurization.
In such an application, the NPC is concentrated such that the content of
nitrogen in the NPC becomes substantially higher than the feedstock by a
factor of
10 or higher (preferably 50 times greater) and the content of oxygen-
containing
organic acids or phenols is in the region of 10 % or greater and preferably I
S % or
higher. NPC is preferably concentrated by adsorption processes, preferably
utilizing
adsorbents selected from the group consisting of activated carbon, zeolite,
hydrated
alumina, silica gel, ion exchange resin, and combinations thereof.
As more NPC is extracted by adsorption, nitrogen content, sulfur content
and total acid number decrease. Nitrogen content, in particular, turns out to
be
closely related with NPC removal ratio, as illustrated in Example 4. While it
is also
expected that content of oxygen-containing compounds should vary with NPC
removal, tracking down the changes in oxygen content of treated hydrocarbons
is
extremely di~fcult since the change occurs within the error margin of oxygen
content analysis. The NPC removal ratio would be exactly quantified if changes
in
NPC weight are measured and compared. however, the measurement takes
impractically long time. Consequently, NPC removal ratio is represented by
changes
in nitrogen content in the following examples, since nitrogen content is easy
to
analyze with reasonably small error margins as shown in Example 4.
16
CA 02335347 2000-12-15
WO 99/67345 PCT/KR99/00338
EXAMPLE 1
LGO and light cycle oil (LCO), used as a feedstock in the present invention,
vary in their properties according to the crude oil type. In Table l, the
properties and
the compositions of various LGOs and an LCO are given. These oils are
exclusively
used in connection with the present invention. As mentioned previously, the
composition and properties of NPC may vary with the feedstock used, but such
variation does not limit the present invention. In Table 1, "A", "B" and "C"
are
LGOs with dif~'erent boiling points, sulfur contents and nitrogen contents,
while "D"
is an LCO produced from an atmospheric residue (AR) fluid catalytic conversion
(FCC) process.
TABLE 1
Feed CharacteristicsA B C . D
Sulfur, ppm(wt) 12,286 15,420 14,056 8,738
Nitro en, m wt 226 173 156 2,503
Distillation,
C
IBP 228 220 227 285
10% 270 261 274 323
50% 311 308 306 343
90% 367 375 353 355.
EP 388 382 368 n/a
ttsr= mt~a~ boiling point
EP= end point (final boiling point;
EXANiILE 2
Silica gel, alumina and ion exchange resins, which are commonly used in
column chromatography, were selected as adsorbents in the present invention.
Physical properties of the adsorbents used are given in Table 2.
17
CA 02335347 2000-12-15
WO 99/67345 PCT/KR99/00338
TABLE 2
I.D, Adsorbent Pore Avg. Pore BET
Vol. Size, A Surface Area
cc/ mI~
a Silica gel 0.3 20.19 733.2
8
b _ 25.9 700
0.45 27
c 0.74 48.55 .
607
95
d 1.05 68.98 .
608
05
1.07 104.39 .
410
94
f ' 1.16 164.34 .
283
47
' 1.16 234.4 .
198.24
h Alumina 0.79 _ 100200
50-70
i ~ Ion Exchange Resin0.55 450-S00 >400
T
EXAMPLE 3
To compare the NPC rers~o-val e~'ectiver~ess of different adsorbents, a series
of experiments were conducted using the silica gels in Table I, identified as
"a"
through "g", having diameters ranging from 0.3 to 0.5 mm. The adsorption
desorption procedure was as follows:
1) 40 cc of the adsorbent "a" v.~as oadeo in the inner tube ofthe concentric
glass column.
2) The temperature of the adsorbent bed was maintained constant by
circulating water through the outer jacket of the concentric column at 50
°C .
3) 400 ce of the LGO "A" was fed at a flow rate of 200 cc/hr into the inner
tube where adsorbent was charged.
4) Upon completion of step, 3), 80 cc of a non-polar solvent, hexane, was
pumped into the inner tube at 200 cc/hr.
5) The inner tube was purged with nitrogen.
6) The products obtained from steps 3), 4), end 5) were mixed together.
7) The products of step 6) were separated from the solvent by a rotary
evaporator, keeping the remnant as "NPC-removed LGO".
l8
CA 02335347 2000-12-15
WO 99/67345 PCT/KR99I00338
8) Upon completion of step 5), 80 cc of a highly polar solvent, rnethyl-
tertiary-butyl-ether, was introduced at 200 cc/hr to the inner tube.
9) The inner tube was purged again with nitrogen.
10) Product obtained from steps 8) and 9) was mixed together.
11) The product of step 10) was separated from the solvent by the use of
rotary evaporator, keeping the remnant us "NPC".
12) The procedure from the steps 3) to 11) was repeated two more times.
13) For the adsorbent "b" through "g", the procedure from steps 1 ) to 12)
was repeated, respectively.
TABLE 3
Adsorbents _N_itro Removal
en Ratio
1st 2nd 3rd
~
a b 5 7
b 22 21 21
c S3 52 52
d 62 61 60
a 51 54 53
f 46 47 47
49 50 50
Nitrogen removal ratio is determined as [(feed N - product N) x 100]/(feed
N content), wherein product N is the nitrogen content of adsorption-treated
hydrocarbon.
The comparison between the performances and physical properties of
adsorbents showed that their adsorption performance was closely related with
their
pore volume, pore size and specific surface area: the larger the pore volume
was, the
better the adsorption performance was. As the pore volume increased, the pore
size
increased while the specific surface area decreased.
From the results, the adsorbent, silica gel, with pore volume from 0.5 to 1.5
cc/g, pore size from 40 to 200 A and specific surface area from 10 to 1,000
m~/g is
19
CA 02335347 2000-12-15
WO 99/67345 PCT/KIt99100338
desirable for treating LGO. For example, if the pore volume is less than 0.5
cc/g or
if the pore size is less than 40 A, adsorption would not be effective. On the
other
hand, if an adsorbent has too large a pore volume, physical strength of the
adsorbent
is significantly weakened and the surface area is drastically reduced.
EXAMPLE 4
The NPC obtained in Example 3 were analyzed for chemical species as
follows:
1) 103.47 g (200 ml) of silica gel (P~Ierck Silica gel 60, 70-230 mesh
ASTM) were charged in a glass column ( I m x 2. Scm) for medium pressure
chromatography.
2) 10.00 g of the NPC obtained in Example 3 were dissolved in n-pentane
and this solution was poured onto the gtass column, followed by flowing 500 ml
of
n-pentane, 500 ml of a mixed solvent of 1:1 n-pentaneaoluene, 500 ml of
toluene
and 500 ml of methanol, in sequence, through the column.
3) Six effluent fractions FI to F6 were obtained such that the aliquot
amount was 250 ml each far the first four fractions (F1-F4), 500 ml for the
fraction
FS and 300 ml for the last effluent, F6.
4) Each fraction was introduced to a rotary evaporator for solvent removal
and the residue was weighed.
5) Qualitative analyses were conducted using an Antek Analyzer for
nitrogen and sulfur content, a FT-IR analyzer, and a GC-MSD and a GC-AED for N
and S species.
6) Using a field desorption (i~D)-mass spectrometer, a semi-quantitative
analysis was done for certain chemical species in each of the fractions
through
molecular weight comparison. A series of mass peaks were selected at a mass
CA 02335347 2000-12-15
WO 99/67345 PCT/KR99I00338
interval of 14 if coincided within the allowed limit of mass difference of
0.05 with
comparison t0 the calculated molecular weight, which are regarded as a
chemical
group with alkyl substitutes. In order to confirm the accuracy of the mass
measurement, a mass correcting standard (polyethylene glycol: PEG) was
analyzed
aRer the mass measurement of the samples. The PEG mass measured was consistent
with the calculated mass within an error range of .03.
The results are given below in 'Table 4.
'fI~BLE 4
F1 F2 F~ F4 F5 F6 ~I
Yield 18.0 2.8 11.8 5.0 19.5 42.9
%
S, m wt 1746 44496 42514 31077 16365 17569
N, m(wt 0 0 1688 30450 20434 27798
FT-IR Typical - Aromatic Pyrrole Pyrrole COON
NH
n-alkane NH Aromatic
s ectra AromaticCOON
GC-AED- Non- DBTs DBTs DBTs: DBTs: n.d.DBTs:n.d
MSD DBTs CBZs: n.d. CBZs CBZs:n.d
n.d. CBZs Amines
ridine
FD-Mass Acids,
Phenols
: 29%
Pyridines,
Quinolines,
Carbazoles,
Henzocarbazoles,
Indole,
Acridines:
32%
Parafftns:5%
Naphthenes:
+"/
Aromatics,
Sulfur
cnm
ounds
& Unknown
:30%
* DBTs (Dibenzothiophenes); CBZs (Carbazoles) ; n.d (not detected)
As is apparent from Table 4, NPC was found to be a polar mixture of polar
compounds, in which such nitrogen-containing compounds as pyridines,
quinolines,
acridines, carbazoles benzocarbazoles, indoles. and such oxveen-containing
compounds as organic acids, and phenols, comprise over half of the total
weight. In
fact, the change in the properties and compositions of LGO before and after
the
adsorption resulted mainly from changes in its nitrogen and oxygen contents.
7;
CA 02335347 2000-12-15
WO 99/67345 PCT/KR99/00338
The data of Table 4 also demonstrate that most of the sulfur compounds in
NPC have a longer retention time than that of DBTs. Also, the sulfur compounds
are
concentrated twice as much as DBTs in terms of the number of molecules. It is
generally known that polycyclic sulfur compounds, e.g. having 3 or more
aromatic
rings, have stronger adsorptivity than DBTs in the gas oil. Hence, it can be
deduced
that polycyclic sulfur compounds are concentrated in NPC. However, the change
in
sulfur content before and after the adsorption was only trace amount. The
reason is
that the content of the polycyclic sulfur compounds in unit volume of
feedstocks
was extremely low. From these analytical data, it can be deduced that oxygen,
nitrogen and polycyclic sulfur~coraaini~~g compounds. which tend to be
adsorbed on
the active sites of the catalysts in desulfurization or denitrification
reactions, were
separated and accumulated in the NPC during the adsorption pretreatment,
resulting
in a significant increase in reaction rate cc:nhared to the case in which NPC
remained in the reactant and played the role as "activity inhibitor",
especially in the
deep hydrodesulfurization.
To further examine the correlation between physical properties of NPC-
removed LGO and the NPC removal ratio, the following experiments were
conducted in a similar manner to Example 3, but adsorbent, "c" was used to
adsorb
feedstock "B". Physical properties of NPC-removed LGOs were analysed and
compared, varying with the RPA.
TABLE 5
~A ~ 10 20 40 80 R~
NPC over Feed 4.86 j:;1 _ 1.11
~~ 1.99
LGO r./Liter
Nitrogen Removal Ratio,%55 39 29 13 0.973
TAN reduction rate, 94 74 66 26 0.849
Sulfur Removal Rate, 3.7 2.3 0.7 1.7 0.721
%
22
CA 02335347 2000-12-15
W O 99/67345 PCT/KR99/00338
EXAMPLE 5
The same procedure as in Example 3 was repeated, except that the
feedstock B was used, along with 40 cc of an adsorbent selected from the
adsorbents
"d", "h" and "i" and the combinations thereof. 200 cc of "feedstock B" was
introduced at a rate of 200 cc/hr through the bed charged with the adsorbents
ranging, in diameter, from 0.3 to 0.5 mm. The procedure was repeated 12 times
to
test the adsorbents for regenerability. Table 6 shows the nitrogen removal
ratio of
the adsorbents from the feedstock B deprived of RfPC.
TABLE 6
Adsorbents _ Nitro
en Removal
Ratio
Char ed _3rd 6th 9th l2th
d 72 73 74 73
i 48 n/a n/a n/a
d:h=l:l 92 90 85 80
d:i=1:1 _7_6 73 74 ~a-
d:i=1:2 77 77 77 n/a
In the above Examples 2, 3 and 4, it was revealed that the NPC removal
could be achieved by various adsorbents, such as ion exchange resins; the
nitrogen
removal ratio, however, may vary with difierenr adsorbents. In addition, it
was also
found that combinations of two or more adsorbents could enhance the nitrogen
removal ratio. For example, in the cast of d:i in Table 6, where an adsorption
column was prepared with ion exchange resin "i", which was charged immediately
after the silica ge! "d", then the nitrogen removal ratio improved as much as
3~ 5%
2o points compared tv the case "d", where silica gel is used alone,
Ear a,n~f,F b
23
CA 02335347 2000-12-15
WO 99167345 PCT/KR99/00338
The same procedure as in Example 3 was repeated, except that the
feedstocks "A", "B", "C" and "D" were used, along with 40 cc of the adsorbent
"d"
having a particle diameter of 0.3 to 0.5 mm. Effluent stream fractions from
the
feedstocks "A", "B", "C" and "D" were designated A-1, B-1, C-1 and D-1,
respectively. The nitrogen removal ratio of the fractions A-1, B-1, C-1 and D-
1 are
given in Table 7.
TABLE 7
_A-1 B-l~~C-1 D-1
Nitrogen Removal Ratio (%) 60 61 61 13
As is apparent from Table 7, no differences in the nitrogen removal ratios
were found among various LGOs, while an extremely low nitrogen removal ratio
was given to the LCO, which contained almost ten times higher nitrogen
compounds
content and was also high in viscosity and aromatics content. Therefore,
adsorption
turned out to be an effective pretreatment technology for LGO that has a
relatively
low level of nitrogen contents, viscosity and aromatics, but may not be a good
one
for LCO.
EXAMPLE 7
The same procedure as in Example 3 was repeated, except that the
feedstock A of 2,000, 3,000 and 4,000 cc was introduced at a rate of 1,000,
2,000
and 4,000 cc/hr through a bed charged with 400 cc of the adsorbent d ranging,
in
diameter size, from 0.85 to I.0 mm. Together with pressure drop across the
adsorption bed and the amounts of the polar solvent used, the nitrogen removal
ratio
for LGO is given in Table 8, below. Also, there is shown the pressure drop
variation with space velocity.
24
CA 02335347 2000-12-15
WO 99/67345 PCT/KR99/00338
TABLE 8
Flaw Treated Nitrogen Solvent Pressure
Rate LGO RPA Removal to LGO Difference
cc/hr cc Ratio Vol. Ratiok cm2
%
2,000 5.0 74 0.40
1,000 3,000 7.5 64 0.27 0.75
4 000 10.0 58 0.20
2,000 5.0 66 0.40
2,000 3,000 7.5 62 0.27 0.95
4,000 10.0 52 0.20
2,000 5.0 59 0.40
4,000 3,000 7.5 50 0.27 1.50
4,000 10.0 46 0.20
As the space velocity increases at the same RPA, the pressure drop
increased while the nitrogen removal ratio decreased. On the other hand, as
the RPA
decreases at the same space velocity, the nitrogen removal ratio increased.
The same
tendency, which constitutes a basic operational rule in the removal of NPC by
adsorption, is expected for other adsorbent types or adsorption techniques as
well.
The particle diameter size of an adsorbent is closely related to the pressure
drop: the pressure drop is inversely proportional to the square of the
particle
diameter. Increasing the particle size may reduce the pressure drop, but also
reduces
the adsorption performance of the adsorbent. As the particle size increased,
the
nitrogen removal ratios were shown to be mare sensitive to the space
velocities. In
addition, the NPC removal tends to change with the adsorption temperatures,
and
the optimal bed temperature is found to be in the range between 40 and 80
°C for
LGO. Such a temperature range happens to be very close to the storage
temperature
of the LGO.
EXAMPLE 8
25
CA 02335347 2000-12-15
w0 99/6345 PCT/KR99/00338
The same procedure as in Example 3 was repeated, except that only one
polar solvent was used along with 40 cc of the adsorbent "d" ranging, in
particle
diameter from 0.3 to 0.5 mm. The nitrogen removal ratio for the feedstock A
was
given in Table 9, below.
An experiment was conducted as follows:
1 ) 40 cc of the adsorbent d was loaded in the inner tube of the concentric
glass column.
2) The temperature of the adsorbent bed was ' maintained constantly by
circulating water through the outer jacket of the concentric tube at
50°C.
3) 400 cc of the LGO "A" was fed at a flow rate of 2C0 cc/hr into the inner
tube where the adsorbent was charged.
4) Upon completion of step 3 ), 40 cc of MTBE vapor was introduced at a
rate of 200 cc/hr through the adsorption bed. To vaporize the solvent, a
preheating
tube was installed and heated to 90 °C and the temperature of the
adsorbent bed was
maintained constant by circulating water through the outer jacket of the
concentric
tube at 80 °C .
5) The product obtained from steps 3) and 4) were mixed together and the
solvent was remaved by the use of a rotary evaporator, keeping the remnant as
"NPC-removed LGO".
6) Upon completion of step 5), 80 cc of liquid MTBE was injected at 200
cc!hr to the inner tutre.
7) The product of step 6) was separated from the solvent by the use of a
rotary evaporator, keeping the remnant as "NPC".
TABLE 9
Nitrogen Removal Ratio (°/a) 6
In contrast to Example 4,, in which two di~'erent solvents were used,
26
CA 02335347 2000-12-15
WO 99/67345 PCT/KR99/00338
Example 8 employed only one polar .solvent, but resulted in a similar nitrogen
removal ratio. This result bears in determining what regeneration techniques
should
be used for the adsorbents. For .instance,. ebullated bed or fluidized bed,
which
cannot be operated with two different solvents, could be applicable for the
adsorption pretreatment step.
EXAMPLE 9
In addition to adsorption, a series of solvent extraction experiments were
conducted using a polar solvent to verify whether solvent extraction removed
NPC,
and whether solvent extraction brought the same degree of improvement as
adsorption did to the HDS process. The following procedure was used.
1) 500 cc of the feedstock "B" was mixed and stirred along with an equal
volume of methanol in a mixer.
2) After completing the mixing and stirring, the mixture was allowed to
settle for S min to give phase separation, followed by draining LGO, which
underwent solvent extraction, from the bottom of the mixer.
3) The extracted LGO was treated with a rotary evaporator to remove
residual methanol, thereby yielding pure LGO deprived of NPC. The nitrogen
removal ratios varying the volume ratios of feedstock "B" to methanol are
given in
Table 10, below.
4) The same procedure as in steps 1 ) through 3) was repeated, except that
the feedstock was "D" instead of "B". The nitrogen removal ratios, varying
with the
volume ratio of feedstock "D" to methanol, are given in Table 10, below.
5) In order to prepare the feedstock for the dcep desulfurization reaction
tests, steps 1) to 3) were repeated to obtain 14 liters of the LGO deprived of
NPC,
which was designated as "B-SX"
27
CA 02335347 2000-12-15
WO 99/67345 PC?/KR99I00338
6) Step 5) was repeated, except that the feedstock was "D" instead of "B",
to obtain 14 liters of LGO deprived of NPC, which was designated as "D-SX".
TABLE 10
Feedstock:MeOH B-SX, nitrogen D-SX, nitrogen
Removal ratio removal ratio
%
250cc :750cc 72 65
333cc:667cc 65 -
SOOcc: SOOcc 55 23
667cc:3 33 cc 40 -
750cc:250cc 31 -
7) In order to determine the nitrogen removal capacity of methanol, steps 1)
to 3) were repeated to prepare the NPC-deprived LGO and designated a "B-SX1".
8) A fresh feedstock "B" was fed agaan into the mixer, mixed, and stirred
for 20 min, together with remaining nn~ethano! of step 2). From this mixture,
an LGO
fraction was extracted and designated as "B-SX2".
9) "B-SX3" was prepared by repeating step 8) in the same manner.
The nitrogen removal ratios for another fresh feed and with the same
methanol solvent are given in Table 11, below.
TABLE 11
~Feedstock ~ ' B-SXI ~~( B-SX2 ~ B-
Nitrogen Removal Ratio(%) ~55 32 22
The nitrogen removal ratio for feedste;.k ' :r?" w~~s ver'; ;:.ar as shown in
Table 7 of Example 6. However, by using he solvent extraction method, the
nitrogen removal ratio of the feedstocks such as fluidized catalytic cracking
(FCC)
cycle oil, eoker gas oil, or vacuum gas oil, which tend to make it rather
difficult to
remove NPC through adsorption, can be improved to almost the same level as
that
of LGO obtained by adsorption, as shown in the data of Table 10. Solvent
2$
CA 02335347 2000-12-15
WO 99/67345 PCT/ICIt99l00338
extraction permits removal of NPC from petroleum feedstock contains heavy gas
oils having a final boiling point over 400°C, FCC cycle oil, and coker
gas oil.
As illustrated in Tables 10 and 11, as the ratio of the amount of oil to that
of
solvent for extraction increased, or as the number of solvent recycle
increased, the
nitrogen removal ratio gradually decreased, which means that the solubility of
NPC
in the solvent phase approached the saturation point. With the proper
selection of a
solvent that promotes high NPC solubility, solvent extraction might be a good
scheme for removing NPC from heavier distillates.
E7~AMPLE ~.0
The silica gel "d" (Example 2) was tested for its regenerability using the
feedstock "B". The LGO that was passed through the adsorption bed was
designated
as "B I". After 40 cc of chromatographic silica gel was charged in the inner
tube of
I S the concentric column through which water of 50'C was circulated, 200 cc
of the
LGO "B" was passed through the bed. Immediately after this, 80 cc of MTBE was
passed through. Fractions of "B 1+MTBE" and "NPC+MTBE" were collected after
repeating the above procedure 10 times and then MTBE was removed from the
fractions by the use of a rotary evaporator. By measuring the nitrogen content
of
"B" and "B1", the nitrogen removal ratio was calculated. The results are shown
in
Fig. 4.
After having undergone 400 operating cycles, the adsorbent did not
produce a degraded nitrogen removal ratio at all, as shown in Fig. 4, Such
regenerability is very important for industrial application and the economics
thereof.
In general, the adsorption mechanism of silica gel is known to be through
hydrogen bonding. Silica gel does riot have strong adsorptive sites, unlike
activated alumina which has many strong acid sites. Such characteristics
explain
29
CA 02335347 2000-12-15
WO 99/67345 PCT/KR99/00338
why silica gel shows superior regenerability. Adsorption to strong acidic or
basic
sites makes the reverse action (desorption) difficult. It can be recognized
from the
data of Table 6 in Example 5, where the adsorbent combination d:h = 1:1 shows
high nitrogen removal ratio in the early stage, but the nitrogen removal ratio
falls
sharply as the number of regeneration iterations increases.
The regeneration of such adsorbents is possible by heating or by the use of
a highly polar solvent, which is also included in the scope of this invention,
but is
considered to have somewhat limited application. Desirable adsorbents,
therefore,
must have such regenerative adsorption characteristics as hydrogen bonding,
exemplified by silica gel and hydrated alumina. The performance of the
adsorbent
also depends on the structural characteristics of the adsorbent and the
feedstock
properties, such as boiling range, NPC content and the feedstock's
composition.
EXAMPLE 11
In order to examine whether and how the NPC-removed LGOs affect the
catalyst performance in the I-lDS process, the feeds for HDS reaction unit
were
prepared as below.
1 ) 400 c;, of the adsorbent d with a particle diameter from 0.88 to 1.0 mm
was loaded at the concentric glass column.
2) The temperature of the adsorbent bed was maintained to 50 C by
circulating water through the outer jacket.
3) 4,000 cc of feedstock "A" was fed at a flow rate of 2,000 cc/hr into the
adsorbent bed.
4) Upon completion of step 3)y 800 cc of hexane was pumped into the
adsorbent bed at a flow rate of 2,000 cc/hr for co-purging.
5) The adsorption bed was purged with nitrogen.
CA 02335347 2000-12-15
WO 99/67345 PCT/KR99/00338
6) Products obtained from step 3), 4) and 5) were mixed together.
7) The solvent was removed from the products of step 6) by a rotary
evaporator, keeping the remnant as "A-2".
8) Upon completion of step 5), 800 cc of methyl-tertiary-butyl-ether was
introduced to the adsorbent bed at a flow rate of 2,000 cc/hr.
9) The inner tube was purged again with nitrogen.
10) The procedure from steps 3) to 9) was repeated until the remnant of
step 7) amounted to 14 liters.
11) For the feedstock B and C, the procedure from steps 3) to 10) was
repeated and the remnants of step 7) were designated as "B-2" and "C-2",
respectively.
12) For the feedstock B, the procedure from steps 3) to 10) was repeated,
except that 2,000 cc of feedstock "B" was fed in step 3) and the remnant of
step 7)
was designated as "B-3".
13) For comparison, 3 liters of o~~ I~,':~C-: emoved LGO fraction "D-SX" was
prepared in a manner similar to that of E.~3mple 9, except that NPC was
removed by
solvent extraction using the feedstock D at methanol ratio of 1:3.
The resulting nitrogen removal ratios are given in Table 12, below.
TABLE 12
A-2~ C-2 B-2 B-3 D-SX
Nitrogen 1~ er:zo°~ aI i2a ~"' ' j j S ~ ] nU ~ SO ~ !l 64
~i;y ~u
EXAMPLE 12
For the purpose of examining thG ibvprovement of the catalyst performance
as in Example 11, deep desulfurization tests were carried out using the feed A
of
Example I and the feed "A-2" of Example 1 t . Tested in this example was a
catalyst
31
CA 02335347 2000-12-15
WO 99I6~345 PCT/1CR99100338
currently being used in a commercial HISS process practiced by the present
applicant. Its physical properties are given, together with its chemical
composition,
in Table 13, below.
TABLE 13
Chemical Com Ph sical Pro
osition erties
ICP,CoO 4.09 wt.% B.E.T surface 214 m2/
~ Area
MoO 16.35 Pore Volume 0.41 cc/
wt.%
Ni0 0.01 wt.% Avg. Pore size76 A
Na O 0.09 wt.% Loadin Densit 836 k /m'
Al O Balance Av . Length 1 /20 inch
400 cc of the catalyst was charged in a HDS pilot-plant facility for deep
desulfurization and was subjected to pry.-sulfiding in which dirnethyl-
disulfide was
mixed at an amount of 1 wt% with an LGO. Then, the raw LGO "A" was introduced
to the reactor and the product samples were collected for sulfur analyses at 3
different reaction temperatures. Before sampling the product, the catalyst bed
was
maintained for 24 hours at the same temperature for stabilisation.
Determination of
catalyst activity with the adsorption-treated LGO "A-2" was done in the same
manner. The results are given in Table 14, below.
TABLE 14
H~ Partial 58.8
Pr~;ssure,
kgf/cm~
Reaction ConditionsHz/~rl Ratio 170
,Nm'/KI
LHSV, hi' l,gg
Catal st 400
Volume
cc
Rxn. Results,
Feed A A-2
Product Sulfur
BAT 300 3,943 m 2,547 m
BAT 320 1,960 m 1,298 m
I ~ BAT 340 756 ppm j 325 ppm
,
'~t3Al~ : t3ed Average Temperature
32
CA 02335347 2000-12-15
WO 99/67345 PCT/KR99/00338
As is apparent from the data, the level of sulfur reduction in the product
improved substantially with the adsorption-pretreated feed at the same
operating
temperatures, compared to the feed that was not pretreated.
EXAMPLE 13
To examine and compare improved catalyst activity, deep desulfurization
tests were carried out using 3 diB'erent NPC-removed feedstocks that were
adsorption-treated with different RPAs: "B" of Example 1, and "B-2" and "B-3"
of
Example 11.
100 cc of the same catalyst as in Example 12 was charged in a high-
pressure, continuous type reactor, and was subjected to pre-sulfiding, in
which
dimethyl disulfide was mixed at an amount of 1 wt% with LGO. Deep HDS was
conducted under the same conditions as in Example 12. After being stabilized
at the
same reaction temperature for 24 hours, the product sample was collected for
sulfur
analysis. The results are given in Table 15, below.
TABLE 15
Product Sulfur Content Versus reaction Temp. (wt. ppm)
Feed B _~ B-2 B-3
BAT 324 C 1503 756 47Q
BAT 334 C 671 399 182
BAT 344 ~C 301 99 63
BAT 354C 108 39 18
As illustrated in Table 15, the LGO feedstocks which were denitrified to the
extent of 60 % or higher by adsorptive pretreatment of the present invention
resulted
in LGO products with sulfur cont$nt belo~.v : JO ppm (wt) at the same HDS
operating conditions that would have produced 300 ppm (wt) product sulfur for
the
3:1
CA 02335347 2000-12-15
WO 99167345 PCT/KR99J00338
same LGO feed.
EXAMPLE 14
100 cc of sample was taken from each of the products hydrodesulfurized at
334°C described in Example 13 arid analyzed for Saybolt color using
Minolta
Digital Colorimeter CT-320. The results are given in Table 16, below.
TABLE 16
Feedstock B feed B-2 feed B-3 feed
Product Colorx-12 +20 +18
Product Sulfur671 m 399 m 182 m
While product color degradation is often encountered in deep HDS, the
adsorption pretreated feed significantly improved product color, as shown in
Table
16. Such results suggest that deep I-~S after adsorptive pretreatment of the
present
invention can bring about a substantial improvement in the product color as
well as
to the product sulfur content.
EXAMPLE 15
Deep HDS reaction tescF ~Nr"~e carried out with the feedstocks "A" and "C"
of Example 1 and the NPC-removed LGOs "A-Z" and "C-2" of Example 11.
The same high pressure, continuous type reactor and catalyst described in
Example 12 were used and the resu~ts are ~;~~et~, ;ogetn~r with the operating
conditions, in Table 17, below.
34
CA 02335347 2000-12-15
WO 99/67345 PCT/KR99/00338
TABL,)r 17 ' . .
H~ Partial 40.0
Pressu~~e,
kgf/cm~
Condition Hz~0i1 250
Rxn Ratio
,Nm,/KI
. LHSV, hr 1.35
Catal st 100
Volurne
cc
Feed A A-2 C C-2
BAT 324C 1,348 779 1,355 846
Rxn. Results, gAT 334 684 281 774 426
C
lf
ur,ppm
Product su
BAT 334 296 155 355 180
C
BAT 354C 115 ~ 40 ~ 158 ~
75
Regardless of the difference in boiling points, sulfur content and nitrogen
content of LGO, similar deep HDS improvements were obtained.
EXAMPLE 16
A deep HISS reaction test was carried out for the NPC-removed LGO "B-
SX", prepared by solvent extraction in Example 9, under the same deep IBS
conditions and employing the reactor described in Example 12. The results are
given in Table 18, below.
TABLE 18
Feedstock Nitrogen, ppm 57
(wt)
__ _ .Sulfur~~pm 15 400
wt
B~ 3~4r ~'g0
Product SulfurBAT 344 C 279
'
ppm (~)
BAT 354 118
The purpose of this example was to examine whether identical or similar
effects could be attained by other NPC removal methods such as solvent
extraction.
The LGOs obtained in Example 9 were found to have a similar effect improving
the
HISS catalyst activity compared to the LGOs obtained by adsorption if the
nitrogen
CA 02335347 2000-12-15
WO 99/67345 PCT/1CR99100338
removal ratios are the same. Therefore, the nitrogen removal ratio of the
solvent
extraction had the same effect as adsorption proposed in the previous
examples. The
solvent extraction could well be one of the pretreatment methods for deep HDS.
However, an excessive quantity of solvent was needed to achieve the same
nitrogen
removal ratio as obtained by the adsorption. In fact, solvent extraction would
require
two or more distillation columns for solvent recovery, of which capacities
could be
as large as the subsequent deep HDS process The solvent extraction method is,
therefore, disadvantageous in operation and irmestment costs. If only a small
amount of feedstock is to be treated with solvent extraction, such
disadvantage can
be overcome with suitable solvent. Such. commercial disadvantage, however,
does
not limit the scope of the present invention.
EXA1VIPLE l7
A series of tests were carried out to examine the deep IBS effect of NPC-
removed LCO.
At first, the LCO feedstock "D" was mixed with feedstock "B" at a volume
ratio of 3:7, followed by subjecting the mixtures to deep HDS. The same
catalyst
and HDS conditions as in Example 12 were used. The NPC-removed LCO "D-SX",
prepared by solvent extraction in Example 11, was also mixed with feedstock
"B" at
a volume ratio of 3:7, followed by subjecting the mixtures to deep HDS. The
results
are given in Table 19, below.
3ti
CA 02335347 2000-12-15
WO 99/67345 PCTlKR99/00338
TABLE 19
Product S Contents
_
ReactionBAT Feed B 70 v.% Feed B 70 v.%
+Feed D 30 v.~ +Feed D-Sx 30
v.%
334C 2,449 ppm S 1,859 ppm S
344 ~ 1,649 ppm S 1,201 pprn S
354 C I,OfiO ppm S 749 ppm S
364 C 665 ppm S 393 ppm S
Even in the case that cycle oils were added up to 30% of the feedstock,
significant deep HDS improvements resulted, which could be ascribed to the
removal of NPC. This is believed to have some economic significance to
refinery
operations to desulfurize middle distillates with a high portion of cycle oil
in the
feed.
COMPARATIVE EXAMPLE 18
A series of experiments were conducted to investigate the difference
between U. S. Patent No. 5,454,933 and the present invention in the following
aspects: adsorbent regenerability, RPA and by-product amount. To make a valid
comparison, an adsorbent, of which surface features were closed to that of
adsorbent
I S used in the disclosed invention, Filtrosorb 400, were carefully selected.
BET
properties of the two adsorben!s acre ~h.o~m i.r Tzb!e 20, below.
TABLE 20
BET Property Activated Cw: hon suggr~stedActivated Carbon
by ,
U. S. Pat. 5 454,933 DARCO
Surface Area 800-110U ~~ 627
mZ/
Pore Size 20100 43.5
The adsorption/desorption procedure was as follows.
37
CA 02335347 2000-12-15
WO 99/b7345 PCT/KR99/00338
1) Activated carbon (ACROS organies, DARCO 20-40 mesh) was dried 6
hours at 150'C .
2) 40 cc of dried activated carbon was loaded in the inner tube of the
concentric glass column and the activated carbon bed was maintained at 90~ by
circulating hot water through the outer jacket of the concentric tube.
3) 250 cc of toluene was fed at a flow rate of 8 cc/min into the adsorbent
bed.
4) Upon completion of Step 3), 400 cc of deep hydro-desulfurized LGO,
which had been produced from the LGO HDS process of SK Corporation and had
contained 240 ppm (wt) sulfur , was introduced at a flow rate of 15 cc/min.
5) The first 75 cc of the product mixture of LGO and toluene, which is
equivalent to 35 ee of LGO, was col:ected and separated from the toluene by a
rotary evaporator, keeping the remnant as T1.
6) The rest of the product mixture was collected and separated from the
toluene by rotary evaporator, keeping the remnant as T2.
7) Upon completion of Step 4), 250 cc of toluene was introduced to
regenerate the activated carbon at a flow rate of R ec/min.
8) The sulfur contents of "T1" and "T2" were analyzed by an ANTEK
Sulfur analyzer.
9) The procedure from steps 4) to 8) was repeated once more.
10) The procedure from steps 4) to 8) was repeated three more times except
Step 4), in which 100 cc of deep hydro-desulfurized LGO was introduced instead
of
400 cc.
The results are given in Table 21.
38
CA 02335347 2000-12-15
WO 99/67345 PCTIKR99/00338
Table 21
No. of regeneration 0 1 2 3 4
T1, cc 35 35 35 35 35
RPA 0.88 0.88 0.88 0.88 0.88
Desulfurization % 80 64 66 47 48
T1+T2, cc 400 400 100 100 100
RPA 10 10 2.5 2.5 2.5
Desulfurization,% 26 23 55 43 42
Extract, gram 33.0 32.3 32.1 31.7 32.4
Ratio of Extract over Feed 9.7 9.5 37.8 37.3 38.1
w/w % .
* RPA : Ratio of Product volume to Adsorbent volume per one cycle
Since Filtrosorb 400 was not available, DARCO activated carbon, having
physical properties similar to Filtrosorb 400, was used in the experiment. For
RPA
of 0.88, the adsorbent removes up to 80% of sulfur compounds from a deep HDS-
treated LGO stream. The adsorbent, however, tends to lose its adsorption
effectiveness as desorption is repeatedly conducted. Therefore, it is
questionable
whether the disclosed invention can be continuously operable with 11.75 RPA,
even with Filtrosorb 400. the disclosed invention of Pat. No. 5,454,933 does
not
provide examples describing continuous operation, where adsorption and
desorption
are conducted at more than one cycle.
As given in Table 21, approximately 40% of feed was converted to the
byproduct at RPA of 2.5. Should Filtrosorb 400 be much more selective in
adsorbing sulfur compounds, byproduct generation at RPA of 1--1.75 could be
considerably higher. The byproduct cannot be used other than as a blending
stock
for high sulfur heavy oils, and this could be greatly disadvantageous for the
disclosed invention of the patent from an economic standpoint.
EXA~LE 19
The IVP~: obtained as in Example 4 was added to a diesel fuel "LL" with
39
CA 02335347 2000-12-15
WO 99/67345 PCT/KR99/00338
poor lubricity as much as 100 ppm (wt) and 300 ppm (wt), respectively. The
prepared samples were then subjected'to lubricity tests by the use of an HFRR
(high
frequent reciprocating rig), which is a standard ISO diesel fuel lubricity
measuring
instrument. The results are given in Table 22, be~ow.
TABLE 22
Amount added ppm (wt~ 0(Base) 100 300
Avg. Abraded Diameter, HFRR 588 498 415
The above results indicate that the NPC, which is a by-product of the
adsorption pretreatment process, can be used as an effective diesel lubricity
additive
for ultra-low sulfur diesel fuel, wl-uch tends to have very poor lubricity.
The pretreatment process of the present invention, thus, not only improves
subsequent catalytic processes to produce ultra low sulfur fuels but also
provides
solutions to the lubricity degradation problems of the fuels by using the by-
product
as a lubricity additive.
EXAMPLE 20
Tests were carried out to examine how the NPC removal influenced the
emission characteristics of the produced diesel fuel. NPC-removed diesel and
"regular" diesel with the same sulfur level were subjected to the emission
test and
the test was conducted as follows:
1) The feedstock "A" was subjected to deep HDS at 356°C under the same
conditions as in Example 12, to produce desulfurized LGO, which was designated
as "A-em-1".
2) The feedstock "A" was subjected to the adsorption pretreatment to
remove NPC in the same way as in Example 6, and then, was subjected to deep
CA 02335347 2000-12-15
WO 99/67345 PCT/KR99/00338
~S reaction at 339°C to produce desulfurized LGO, which was designated
as "A-
em-2".
3) Commercially available kerosene with.a sulfur content of 10 ppm (wt)
was blended with the "A-em-1" and "A-em-2" at 30% level to prepare emission
test
fuels which had similar distillation characteristics comparable to that of
commercially available diesel fuels and designated "A-em-1-D" and "A-ern-2-D",
respectively.
4) The above two samples were tested in a diesel engine along with the
reference fuel to stabilize the engine. Commercial diesel fuel (SK Diesel) was
used
as a reference fuel. The characteristics of the samples are shown in Table 23,
below.
5) The emission test was carried out v~ith a bus diesel engine having a
displacement of 11,050 cc, such as sold by Daewoo Motors Co. Ltd., Korea,
identified as Model D2366. The amount of PM emission was measured according to
the D-13 mode, which is an emission test mode for heavy-duty diesel vehicles
in
Korea. In addition, smoke was measured with the 3 samples according to smoke 3
mode. Details and measurements are given in Tables 24 and 25, respectively.
6) To minimize errors due to environmental changes, the test was
conducted continuously. 'The engine was checked for its repeatability using a
reference fuel before and after the test session. In addition, 4.pre-tests
were carried
out with the same reference diesel fuel to evaluate the reproducibility of an
MDT
(Mini Dilution Tunnel) used for PM measurement and an exhaust gas analyzer.
41
CA 02335347 2000-12-15
WO 99167345 PCT/KR99/00338
TABLE 23
Characteristics SK Diesel A-em-1-D A-em-2-D
Gravity 15/4 C 0.8374 0.8218 0.8209
ASTM D86,
jgp 152 159 165
10% 190 200 204
SO% 259 278 282
90% 344 35I 353
95% - 368 370
gp 3 76 3 82 378
Residue, vol. % 0.8 1.6 1. S
Flash Point, C - 63 63
S m wt 33 220 220
0
N m wt _ 23 8
__
10% C residue, wt% 0.10 0.04 0.02
* SK Diesel: commercial SK corporation diesel products
TABLE 24
Operating conditions by Test Modes
D-13 PM Measurement
Mode En % Load ~ Wt. factor
ine
RPM
1 1 _- _ 025
dle ~ ~
~
2 1926 10 0.08
~
3 1920 25 0.08
_
~
4 1920 ~ 50 0.08
5 1920 75 0.08
6 1920 100 0.25
_
7 1 - 0.25
dle
8 3200 100 0.1
9 3200 75 0.02
3200 50 0.02
11 3200 25 0.02
12 3200 10 0.02
13 ~ _ 0.25/3
~ylL
i
Smoke 3 Smoke
measurement
Mode En ine %
RPM Load
1 1000 100
2 1_320 100
~
w. __ 100
2200
~x
CA 02335347 2000-12-15
WO 99/67345 PCT/KR99/00338
*RPM at maximal engine output: 3200 rpm
60% of the RPM at maximal engine output: 1920 rpm
TABLlr 25
Test Items Total PM SOF Sulfate Smoke
r/KW-h W-h ~h
A-em-1-D 0.766 0.051 0.007 50
A-em-2-D 0.596 0.040 0.005 48
Improvement,% 22 21 28 4
SOF (Soluble Organic Fraction)
As shown in Table 25, the NPC-removed-then-deep-hydrodesulfurized
diesel fuel showed 22% lower level of PM emission compared to the other fuel
at
the same sulfur level. Such an improvement in emission characteristics likely
resulted from removal of precursor material for PM; such precursor material
might
well be removed as part of NPC. Such emission characteristics make the
pretreatment process of the present invention even more attractive because it
can
produce cleaner diesel fuels, which are low in sulfur content as well as emit
less
pollutant compared to other diesel fuels with the same sulfur contents.
Although the present invention may be applied to various catalytic
processes far producing hydrocarbon fuels, it is more preferably applied to
the
upstream of deep I~I)S processes man~~faauring Icerosene and diesel fuels to
improve effectiveness of the I-IDS processes and qualities of the products
therefrom.
Due to the ever-tightening stringent environmental regulations, refineries
call for effective and economic deep desulfurization technologies to produce
cleaner
diesel fixels. The present invention suggests a pimple but efficient
pretreatment
process that will enable a conventional I-~S process to economically produce
ULSD from high-sulfur LGO feedstock.
In addition, the present invention provides such advantages as extending
43
CA 02335347 2000-12-15
W O 99167345 PCT/KR99/0033$
the catalyst life, reducing hydrogen consumption and saving operation cost by
making the best use of low-quality feedstocks.
Furthermore, for the same sulfur content level, adsorption-treated diesel
fuel shows better emission characteristics than conventional diesel fuel. When
combusted, adsorption-treated diesel fuel emits lower amounts of PM and NOx,
two
of the most strictly regulated pollutants, compared to conventional diesel
fuel. The
color of diesel fuel is improved because HDS reaction temperature is decreased
and
color body precursor level gets substantially reduced in the pretreatment
process.
Operating conditions of the pretreatment process are close to ambient
temperature and pressure. In addition, the pretreatment process can treat a
hydrocarbon stream at higher space velocities t:ian ;'iDS processes, and
therefore the
size requirement becomes substantially smaller than other conventional
reaction
units. The investment cost of the pretreatment process is estimated to be
approximately 10 % of that of HDS process. Since the pretreatment process uses
common adsorbent and solvent without catalyst and hydrogen, the operating cost
is
also estimated to be around 10-20% of that of HDS process.
The present invention has been described in an illustrative manner, and it is
to be understood the terminology used is intended to be in the nature of
description
rather than of limitation. Many modifications and variations of the present
invention
are possible in light of the above teachings. Therefore, it is to be
understood that
within the scope of the appended claims, the im~ention may be practiced
otherwise
than as specifically described.
44