Note: Descriptions are shown in the official language in which they were submitted.
CA 02337261 2001-O1-05
Use of prenyltransferase inhibitors for ~re~aring a medicament
intended to treat pathologies resulting from membrane fixing of the
heterotrimeric G protein
The present invention relates. to the use of prenyltransferase inhibitors for
preparing a
medicament intended to trea.t pathologies resulting from the membrane fixing
of the
heterotrime,ric G protein. These diseases include in particular diseases
linked to the
following biological functi.ons or disorders: smell, taste, perception of
light,
neurotransmission, neurode;generation, endocrine and exocrine gland fonctions,
autocrine and paracrine regulation, arterial tension, embryogenesis, viral
infection,
immunological fonctions, diabetes and obesity.
The heterotrimeric G protein coupled with receptors with 7 transmembrane
domains is a
mediator of the extracellular information into the cell. Several intracellular
effectors of
the G protein are identified as adenylate cyclase, phospholipase C or also
ionic channels
(cf. Gilman, A.G. Biorci. Rein. 15, 65-97 (1995)).
The activity of the adenylate cyclase is modulated by the different G proteins
(Gs, Gi,
Gq, Go) which thus regulate; the biosynthesis of the cyclic AMP (cAMP). It is
thus
known that, in order to modu~late the adenylate cyclase, it is necessary for
the G protein
transitionally to be in a heterotrimeric form, in which form the monomer
constituted by
a sub-unit a is associated with the dimer constituted by the sub-units(3 and
y. It is also
known that for the G protein to be functional, it must be fixed to the
membrane by its
sub-unit y, this fixing being possible when the sub-unit Y is prenylated. Unly
in this
situation can the ligand activate the sub-unit a of the G protein, outside the
cell, via the
rece~ptors with seven transme:mbrane domains. Alter disassociation, the a sub-
unit can
modulate the adenylate cyclase and modulate the production of cAMP.
The harmful effects of an abnormal cAMP level are also known and occur in
particular
at the level of the following biological fonctions: smell, taste, perception
of light,
neurotransmission, neurodegeneration, endocrine and exocrine gland fonctions,
CA 02337261 2001-O1-05
-2-
autocrine and paracr~ine regulation, arterial tension, embryogenesis, benign
cell
proliferation, oncogenesis, viral infection, immunological fonctions, diabetes
and
obesity.
Prenyltransferase inhibitors are already used in the field of cancer treatment
(cf. Sebti et
al., Pharmacol. Ther. 74, 103-114 (1997); Sepp-Lorenzino et al., Cancer.Res.
55, 5302-
5309 (1997)). The useful,ness of prenyltransferase inhibitors in this type of
treatment
would appear to derive from their action in preventing prenylation at the
level of the
Ras substrate. However, the prenylation of certain forms of Ras is not
modified by
prenylation inhibitors (Lerner et al., Oncogene, 15, 1283-1288, 1997).
The Applicant has now discovered that prenyltransferase inhibitors can also be
used to
modulate the action of the; heterotrimeric G protein, and thus treat all sorts
of diseases
linked with it.
The invention firstly relates to the use of prenyltransferase inhibitors to
prepare a
medicament intended to treat pathologies resulting from membrane fixing of the
heterotrimeric G protein. In particular, it relates to the use of said
inhibitors to prepare
medicaments intended to treat diseases linked to the following biological
fonctions or
disorders: smell, taste, perception of light, neurotransmission,
neurodegeneration,
endocrine and exocrine ~;land fonctions, autocrine and paracrine regulation,
arterial
tension, embryogenesis, viral infection, immunological fonctions, diabetes and
obesity.
More particularly, the invention relates to the use of certain
prenyltransferase inhibitors
for the preparation of a medicament intended to treat cholera, Acquired Immune
Deficiency Syndrome (~.IDS), travel diarrhea and familial masculine precocious
puberty.
Among the prenyltransferase inhibitors which can be used for the invention,
there can in
particular be mentioned those chosen from a group constituted by the following
compounds: derivatives of general formula (GPI) defined below (in particular
those
descr~ibed in Patent Application WO 97/21701, such as the compound of formula
(I~
defined hereafter, and those described in Patent Application WO 97/16443, such
as the
compound of formula (~~I) defined hereafter), thiazole derivatives such as
those
described in Patent Application WO 98/00409 (for example the compound of
formula
(I) defined below), peptïdomimetic derivatives of 4-aminobenzoic acid (for
example the
compounds of formulae (lI) and (II)a defined hereafter) or peptidomimetic
analogues
such as those described in Patent Applicatïon WO 96/21456 (for e:xample the
compound of formula (III) defined hereafter), tricyclic amides such as those
described
in Patent Application WO 97/24378 (for example the compound of formula (~
CA 02337261 2001-O1-05
-3-
defined hereafter), polyacids such as those described in Patent Application
WO 97/17321 (for exmr~ple the compound of formula (VII) defined hereafter),
piperidine peptidomimetic derivatives such as those described in Patent
Application
WO 97/18813 (for example the compound of formula (VIII) defined hereafter),
benzodiazepine peptidomimetic derivatives such as those described in US Patent
5,532,359 (for example th.e compounds of formulae (IX) and (IX)a defined
hereafter),
tripeptidic derivatives of phosphonic or phosphinic acids such as those
described in US
Patents Nos. 5,523,430 and 5,510,510 (for example the compounds of formulae
(X) and
(X)a defined hereafter), pseudodipeptides based on a N-carbonylpyrazine
structure such
as those described in Patent Application WO 95/00497 (for example the compound
of
formula (XI) defined hereafter), and tetrapeptidic analogues of the type C:ys-
A1-A2-A3
wherein Cys is Cystein, A1 and A2 are aliphatic aminoacids and A3 represents
any
aminoacid (such analogues being described for example in US patent 5,627,202).
Preferably, the inhibitors of prenyltransferases will be chosen from the group
constituted by the compounds corresponding to general formula (GPI) shown
below
R,i /
H3C~N
Ri
(~,)\ ~ I ~ I
N R3
I
R2
(GPI)
in which:
R~ represents OH, SH, NH2 or CH20H;
RZ represents a lower alkyl radical or CORS;
R3 and R4 represent independently a halogen atour or an OH radical;
RS represents a lower alkyl radical, a substituted or non-substituted
carbocyclic
or heterocyclic arylalkyl or aryl radical or a saturated heterocyclic radical
comprising _5 to 6 members, laid members being chosen from CH2, O, NH
and S; and
Y represents CO or CS or is absent;
CA 02337261 2001-O1-05
-4-
and compounds corresponding to one of the following formulae:
SH ~S S ~
N
NH~
HZN~ O NH O~
O
(I)
CH3
HS
OR
H
H2N O
(II) : R = CH 3
(II) a . R = H
HS
H2N ,,,~H
NH
COOCH3
H
(III)
CA 02337261 2001-O1-05
-5-
I
CH3
(IV)
BI'
C1
r CI
N
NH 2
N N' ''
O
v v
--N
HO /~/N
Cl
C1~ v u ~N-
I
CH3
(VI)
CA 02337261 2001-O1-05
-6-
--° \
O ~ O
/
oH
0
OH
/ ~~ N O
O
N~ CH3 O
OH
(VII)
NC
\.
N
H O
N N N
N O O
(VIII)
N
H3C~ ,;
N
~ O O
H2N~~ HN
_ O
~"~CH2R
SH HOO ~C
(IX) . R = CH 2 SCH
(IX) a . R = iPr
CA 02337261 2001-O1-05
_ 'j _
H H
O~ N N~COOH
N
H O ~~SCH3
X
I
HO iP' \ \ \
O
(X) . X = CH 2
(X) a . X = O
SH
O
H2N.
O
(XI)
or
S
S~ O /
H.,N \
(XII)
More particularly, the prenyltransferase inhibitors may be chosen frorr~ the
group
constituted by the followüng compounds : the compounds of general formula
(GPI),
thiazole derivatives such as those described in Patent Application WO 98/00409
(for
example the compound of formula (I)), peptidomimetic derivatives of 4-
aminobenzoic
acid (for example the compounds of formulae (II) and (II)a) or peptidomimetic
analogues such as those de;scribed in Patent Application WO 96/21456 (for
example the
compound of formula (III)), 1-[(2R)-amino-3-mercaptopropyl]-(2S)-(2-
mercaptoethyl)-
4-(1-naphtoyl)-piperazine-1,2-cyclodisulfide (XII), or pseudodipeptides such
as those
described in Patent Application WO 95/00497.
CA 02337261 2001-O1-05
_g_
More preferably, the prenyltransferase inhibitors may be chosen from the group
constituted by the compounds corresponding to general formula (GPI) shown
below
R~;
R3
I
R2
(GPI)
in which:
R1 represents OH, SH, NH2 or CHZOH;
R2 represents a louver alkyl radical or CORS;
R3 and R4 represent independently a halogen atour or an OH radical;
RS represents a lover alkyl radical, a substituted or non-substituted
carbocyclic
or heterocyclic arylalkyl or aryl radical or a saturated heterocyclic radical
comprisïng 5 to 6 members, said members being chosen from CH2, O, NH
and S; and
Y represents CO or CS or is absent;
and compounds corresponding to one of the following formulae:
SH ~S S ~
N
NH..~
H2N~ O NH O~
O
(I)
CA 02337261 2001-O1-05
-9-
CH3
HS
OR
H2N H
O
(II) . R = CH 3
(II) a . R = H
HS
HZN ,,~~H
NH
COOCH3
H
(III)
SH
H2N. %~ O
O
(XI)
or
CA 02337261 2001-O1-05
- 10-
s
S~
N N
H.,N
(XII)
According to a preferred variant of the invention, the prenyltransferase
inhibitors used
for the invention correspond to general formula (GPI) and can in particular be
chosen
from the following compounds:
- 1-acetyl-3-(3-chlorophenyl)-5-[(4-chlorophenyl)hydroxy-(1-methyl-1H-
imidazolyl)methyl]indole (GPI~);
- 1-(4-morpholinecarbamoyl)-3-(3-chlorophenyl)-5-[(4-chlorophenyl)hydroxy-(1-
methyl-1H-imidazolyl)methyl]indole (GPI2);
- 4-(3-chlorophenyl)-6-[(4-chlorophenyl)hydroxy]-(1-methyl-1H-imidazol-5-
yl)methyl-
2(1H)-quinolinone (GPI3);
- 4-(3-chlorophenyl)-6-[(4-chlorophenyl)hydroxy]-(1-methyl-1H-imidazol-5-
yl)methyl-
2( 1 H)-quinolinone (GPI4).
According to another pref~rred variant of the invention, the prenyltransferase
inhibitors
will be one of the compounds of formula (I), (II), (II)a, (III) or (XII):
SH ~S S ~
N
NH.~
H2N~ O NH O~
O
(I)
CA 02337261 2001-O1-05
-Il-
CH3
HS
H~I~f H OR
O
(II) . R = CH 3
(II) a : R = H
HS
H2N ,...H
NH
COOCH3
H
(III)
S
O
~' ~i ~ I
H2N w
(XII)
CA 02337261 2001-O1-05
-12-
Among the compounds previously mentioned, one of the following
farnesyltransferase
inhibitors of formula (I), (II) or (II)a, or (XII) will preferably be used as
the
prenyltransferase inhibitor:
SH ~ S ~
N
NH
H2N~ O NH O\
O
(I)
SCH3
H
HS N
N
OR
H 2N H
O
(II) . R = CH
(II) a . R = H
S
S~ O /
U
H.,N \
(XII)
A particularly preferred compound for use according to the invention is the
compound
of formula (XII):
O /
H-, \
(XII)
CA 02337261 2001-O1-05
-13-
The compounds of general formula (GPI) can be prepared according to the
methods
described in Patent Application PCT WO 97/21701 when Y represents CO or CS, or
according to the methods described hereafter when Y is absent. The 1-[(2R)-
amino-3-
mercaptopropyl)-(2S)-( 2-m ercaptoethyl)-4-( 1-naphthyl)-piperazine-1,2-
cyclodisulphide
(XII) can be prepared as described in the experimental part.
The compounds of gen.eral formula (GPI) are described in Patent Application
US 09/098141 dated l6th June 1998 as well as in a very recently filed
subsequent
continuation-in-part of this Application, said Application having been the
subject of a
PCT Patent Application No. US 99/xxxxx not yet published at the date of this
Application. Part of the content of this PCT Application is repeated in the
experimental
part.
The pharmaceutical compositions containing a compound according to the
invention
can be in solid form, for example powders, granules, tablets, gelatin
capsules, liposomes
or suppositories. The appropriate solid supports can be, for example, calcium
phosphate, magnesium stearate, talc, sugars, lactose, dextrin, starch,
gelatin, cellulose,
methyl cellulose, sodium carboxymethyl cellulose, polyvinylpyrrolidine and
wax.
The pharmaceutical compositions containing a compound of the invention. can
also be
presented in liquid form, j~or example, solutions, emulsions, suspensions or
syrups. The
appropriate liquid supports can be, for example, water, organic solvents such
as glycerol
or glycols, as well as their mixtures, in varying proportions, in water.
The administration of a rnedicament according to the invention can be carried
out by
topical, oral or parenteral route, by injection (intramuscular, sub-cutaneous,
intravenous,
etc.), etc. The administration route will of course depend on the type of
illness to be
treated.
The administration dose envisaged for a medicament according to the invention
is
comprised between 0.1 rn;g and 10 g according to the type of pathology to be
treated.
Unless defined differently, all the technical and scientific ternis used here
have the sanie
meaning as that normally understood by an ordinary specialist in the field to
which this
invention belongs. Similarly, all publications, patent applications, patents
and all other
references mentioned here: are incorporated by way of reference.
The following examples are presented in order to illustrate the above
procedures and
should in no event be considered to limit the scope of the invention.
CA 02337261 2001-O1-05
-14-
EXPERIMENTAL PART
Preparation of certain compounds ased in the inventïon
Preparation A:
Compounds of general formula (GPI) in which Y does not exist:
Said compounds are prepared according to the methods described in Patent
Application
PCT US 99/xxxxx, under priority of a Patent Application US 09/098141 dated
l6th
June 1998 as well as a subsequent continuation-in-part of this Application.
Part of the
preparation protocols for the products of this PCT Application is repeated
hereafter.
"" The invention (the subject of PCT Application US 99/xxxxx) relates to the
compound of general fornmla (A)
R'
2 9 N
R R1 R Ra
N
R3 1 R6 (' )p R10
(T ~m
( I )n (-r4 ~ R 11
,,
Z____
R5 R13 R12
Ra
(A)
or a pharmaceutically acceptable sait of said compound,
said general formula (A) being such that:
CA 02337261 2001-O1-05
-15-
------- represents an optional bond, it being understood that only one of the
optional
bonds is presented in a compound of general formula (A);
m, n, p, and q are all independently 0 or 1;
each tune T1, T2, T3 and 'T4 are involved, they are chosen from the group
composed of
CR26R27, S, O, C(O), S(O)2 and NR2g;
X is N-Y, O or S, Y being chosen from the group composed of H, CR14R1sR16,
s(~)R17~ s(~)2R18, C(C~W19~ C(~)~20R21, C(s)~22R23~ C(~)~R24~ C(S)~R2s~
S(O)NR29R3o and S(O)2NR31 R~2;
Z is chosen from the group composed of H, hydroxy, alkoxy, aryloxy, cyano,
halo,
CR14R1sR16, s(O)R17~ s(O)2R18~ C(~)R19~ C(~)~20R21~ C(~)~R24~ C(s)~22R23~
S(O)NR29R3o, C(S)OR2s and S(O)2NR31R32~ it being understood that when the
optional
bond to Z is present then ~'. is an oxygen or sulphur atour;
each time Rl, R2, R3, 1Z4, R5, Rb, R11, R12, R13~ R14~ Rts~ R16~ R26 Wd R27,
are
involved, they are each independently chosen from the group composed of H,
halo,
hydroxy, thio and cyano, or an optionally substituted radical chosen from the
group
composed of the alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkyl-alkyl, aryl,
arylalkyl,
alkyloxy, aryloxy, alkylthio, arylthio, alkylamino, arylamino and alkyl
caibonyl amino
radicals;
or R1 and R2 when they aire in adjacent positions, or R4 and Rs, or R11 and
R12 when
they are in adjacent positions, form together a divalent radical chosen from
the group
composed of -O-CH2-O-, -O-CH2-CH2-O-, -O-CH=CH-, -O-CH2-CH2-,
-O-CH2-CH2-CH2- and -CK33=CR34-CR3s=CR36-;
R7, R8 and R9 are each ündependently chosen from the group composed of H,
halo,
amino, cyano, hydroxycarbonyl, or an optionally substituted radical chosen
from the
group composed of the aryl, alkyl, alkyloxy, alkylthio, aryloxy, arylthio,
mono- or di-
alkylamino, alkoxycarbonyl, alkyl-S(O)-alkyl, alkyl-S(O)2-alkyl,
cyanoarylalkyl and
arylalkyl radicals;
Rlo is chosen from the group composed of H, amino, azido, hydroxy, halo,
alkyl,
substituted alkyl, cyano, hydroxycarbonyl, mono- or di-alkylaminoalkyl, rnono-
or di
alkylamino, alkyloxy, alkylcarbonylalky~, alkyloxy-carbonylalkyl,
carboxyalkyl,
cycloalkyl, cycloalkylamino, cycloalkyloxy, imidazolyl, substituted
imidazolyl,
aminocarbonylalkyl, aryloxy, thio, alkylthio, arylthio, OS(O)2R1H, OC(O)R19,
OC(O)NR2oR21, OC(S)NR22R23, OS(O)NR29R30 and OS(O)2NR31R32~
each tune R17, Ri g, R19, R2o, R21~ R22, R23, R24, R2s, R28, R29, R3o~ R31~
R32 and R37
are involved, they are eaclz independently chosen from the group composed of H
and of
an optionally substituted radical chosen from the group composed of the alkyl,
alkenyl,
cycloalkyl, aryl and arylalkyl radicals;
CA 02337261 2001-O1-05
- 16-
or R2p and RZ1, or Rz2 and R23, or R2ç and R3p, or R31 and R32 form together a
divalent
radical chosen from the group composed of the -(CHZ)r NR3~-(CHZ)S-,
-(CHZ)r O-(CH2)S- and -(CR38R39)t- radicals in which r and s represent
independently
integers from 1 to 3 and t represents an integer from 2 to 6;
and R33, 1234, R3s~ R3f» R3s and R39 are each independently chosen from the
group
composed of H, amine, halo, cyano, alkyl, substituted alkyl, aryl, substituted
aryl,
alkyloxy, aryloxy, alkylth.io, arylthio, mono- or di-alkylamino, arylamine,
hydroxy and
thio.
The following definitions are used for formula (A) above:
- In the part of the compound of formula (A) in which two optional bonds are
indicated,
only one of these optional bonds can be present in a compound at a tune. When
the
optional bond directly attached to the variable Z is present, then Z is an
oxygen or
sulphur atour.
- The terni "alkyl" refers to non-substituted linear or branched hydrocarbon
drains
contairllng 1 to 20 carbon atours, and preferably 1 to 7 carbon atours.
The terni "substituted alkyl" refers to an alkyl radical group substituted,
for example,
by one to four substituents, such as halo, hydroxy, alkoxy, oxo, alkanoyl,
aryloxy,
alkanoyloxy, amine, alkylamino, arylamine, aralkylamino, disubstituted amines
in
which the 2 substituents of the amine fonction are chosen from alkyl, aryl or
aralkyl;
alkanoylamino, aroylamino, aralkanoylamino, substituted alkanoylamino,
substituted
arylamine, substituted aralkanoylamino, thiol, alkylthio, arylthio,
aralkylthio,
alkylthiono, arylthiono, aralkylthiono, alkylsulphonyl, arylsulphonyl,
aralkylsulphonyl,
sulphonamido, e.g. SOZNH2, substituted sulphonamido, nitre, cyano, carboxy,
carbamyl, for example CONH2, substituted carbamyl for example CONf-I alkyl,
CONH
aryl, CONH aralkyl or in the case in which there are two substituents on the
nitrogen
they are chosen from al:kyl, aryl and aralkyl; alkoxycarbonyl, aryl,
substituted aryl,
guanidino and heterocycles, such as indolyl, imidazolyl, furyl, thienyl,
thiazolyl,
pyrrolidyl, pyridyl, pyrimidyl and analogues. When it is indicated that the
substituent is
itself substituted, it is substituted by an alkyl, alkoxy, aryl or aralkyl
radical.
- The terni "halogen" or "halo" refers to fluorine, chlorine, bromine and
iodine.
- The terni "aryl" refers to monocyclic or bicyclic aromatic groups having 3
to 12
carbon atours and 0 to 2 nitrogen atours and 0 to 1 sulphur atour in their
cyclic part such
as phenyl, naphthyl, biphenyl, imidazoyl, pyridyl and diphenyl, each being
optionally
substituted.
- The terni "arylalkyl" refers to an aryl group directly linked by an alkyl
group, such as
benzyl.
CA 02337261 2001-O1-05
_ 17_
- The tenu "substituted ar~l" refers to an aryl group substituted by, for
example, one to
five substituenis such as alkyl; substituted alkyl, halo, trifluoromethoxy,
trifluoromethyl, hydroxy, alkoxy, alkanoyl, alkanoyloxy, amino, alkylamino,
arylalkylamino, arylalkylamino, dialkylamino, alkanoylamino, thiol, alkylthio,
ureido,
nitro, cyano, carboxy, carboxyalkyl, carbamyl, alkoxycarbonyl, alkylthiono,
arylthiono,
alkylsulphonyl, sulphonamido, aryloxy and analogues. The substituent cran
itself be
substituted by hydroxy, alkyl, alkoxy, aryl, substituted aryl, substituted
alkyl, or
arylalkyl.
- The terni " alkenyl" refc;rs to non substituted linear or branched
hydracarbon drains
containing 2 to 20 carbon atours, preferably 2 to 15 caubon atours, and more
preferentially 2 to 8 carbon atours, having one to four double bonds.
The terni "substituted allcenyl" refers to an alkenyl group substituted by,
for example,
one to four substituents, such as halo, hydroxy, alkoxy, alkanoyl,
alkanoyloxy, amino,
alkylamino, dialkylamino, alkanoylamino, thiol, alkylthio, alkylthiono,
alkylsulphonyl,
sulphonamido, nitro, cyamo, carboxy, carbamyl, substituted carbamyl,
guanidino,
indolyl, imidazolyl, furyl, thienyl, thiazolyl, pyrrolidyl, pyridyl, pyr-
imidyl and
analogues.
- The terni "alkynyl" refers to non substituted linear or branched hydrocarbon
drains
containing 2 to 20 carb~on atours, preferably 2 to 15 carbon atours, and more
preferentially 2 to 8 carbon atours, having one to four triple bonds.
The terni "substituted alkynyl" refers to an alkenyl group substituted by one
to four
substituents, for examplc: a substituent such as halo, hydroxy, alkoxy,
alkanoyl,
alkanoyloxy, amino, alkylamino, dialkylamino, alkanoylamino, thiol, alkylthio,
alkylthiono, alkylsulphonyl, sulphonamido, nitro, cyano, carboxy, carbamyl,
substituted
carbamyl, guanidïno and heterocyclo, for example imidazolyl, furyl, thienyl,
thiazolyl,
pyrrolidyl, pyridyl, pyrimidyl and analogues.
- The terni "cycloalkyl" refers to optionally substituted saturated
hydrocarbon rings or
systems of saturated hydrocarbon rings, preferably comprising 1 to 3 rings and
3 to 7
carbon atours per ring, which can themselves be joined with an unsaturated C3-
C7
carbocyclic ring. Sach groups include for example cyclopropyl, cyclobutyl,
cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclodecyl, cyclododecyl and
adamantyl. The substituents include for example one or more alkyl groups as
described
above, or one or more groups described above as a substituent of an alkyl
group.
- The ternis "heterocycle", "heterocyclic" and "heterocyclo" refer to an
optionally
substituted, saturated or unsaturated, aromatic or non-aromatic cyclic group,
which is,
for example, a monocyclic system containing 4 to 7 atours, a bicyclic system
containing
7 to 11 atours, or a tricyclic system containing 10 to 15 atours, which
contains at least
one heteroatom in a ring comprising at least one carbon atour. Each ring of
the
CA 02337261 2001-O1-05
-18-
heterocyclic group comprising a heteroatom can contain l, 2 or 3 heteroatoms
chosen
from nitrogen, oxygen and sulphur, the oxygen and sulphur atours being
optionally
oxidized and the nitrogen atours being optionally in the form of quaternary
cations. The
heterocyclic group can be attached to any carbon atour or heteroatom.
- Monocyclic heterocyclic groups include for example pyrrolidinyl, pyrrolyl,
indolyl,
pyrazolyl, oxetanyl, pyra~zolinyl, imidazolyl, imidazolinyl, imidazolidinyl,
oxazolyl,
oxazolidinyl, isoxazolinyl, isoxazolyl, thiazolyl, thiadiazolyl,
thiazolidinyl, isothiazolyl,
isothiazolidinyl, furyl, tetrahydrofuryl, thienyl, oxadiazolyl, piperidinyl,
piperazinyl, 2-
oxazepinyl, azepinyl, 4-piperidonyl, pyridyl, N-oxo-pyridyl, pyrazinyl,
pyrimidinyl,
pyridazinyl, tetrahydropyranyl, morpholinyl, thiamorpholinyl, thiamorpholinyl
sulphoxide, thiamorpholinyl sulphone, 1,3-dioxolane and tetrahydro-1,1-
dioxothienyl,
dioxanyl, isothiazolidinyl, thietanyl, thüranyl, triazinyl, and triazolyl, and
analogues.
- Bicyclic heterocyclic groups include for example benzothiazolyl,
benzoxazolyl,
benzothienyl, quinuclidinyl, quinolinyl, quinolinyl-N-oxide,
tetrahydroisoquinolinyl,
isoquinolinyl, benzimidazolyl, benzopyranyl, indolizinyl, benzofuryl,
chromonyl,
coumarinyl, cinnolinyl, quinoxalinyl, indazolyl, pyrrolopyridyl, furopyridinyl
(such as
furo[2,3-c]pyridinyl, furo[3,1-b]pyridinyl) or furo[2,3-b]pyridinyl),
dihydroisoindolyl,
dihydroquinazolinyl (such as 3,4-dihydro-4-oxo-quinazolinyl),
benzisothiazolyl,
benzisoxaxolyl, benzodiazinyl, benzofurazanyl, benzothiopyranyl,
benzotriazolyl,
benzopyrazolyl, dihydrolbenzofuryl, dihydrobenzothienyl,
dihydrobenzathiopyranyl,
dihydrobenzothiopyranyl sulphone, dihydrobenzopyranyl, indolinyl,
isochromanyl,
isoindolinyl, naphthyriclinyl, phthalazinyl, piperonyl, purinyl,
pyridopyridyl,
quinazolinyl, tetrahydroquinolinyl, thienofuryl, thienopyridyl, thienothienyl,
and
analogues.
- The substituents for the heterocyclic systems include for example one or
more alkyl
groups as described abovc:, or one or more groups described above as
substituent of an
alkyl group. Smaller heterocycles such as epoxides and aziridines are also i
ncluded.
- The terni "heteroatom" includes oxygen, sulphur and nitrogen.
The compounds of formula (A) can be prepared according to the methods
described
hereafter or by analogy to these methods:
EXAMPLE 1: ~ -3- 3~-chloroPhen~ll-5 ~j~,4-chlorophenyl)h dy roxy(1-methyl-1H
imidazol-S~llmethyl] indole
A solution of butyllithium in hexane (1.6 M, 430 pl) at approximately -78
°C is added
dropwise to a solution of' 1-methylimidazole (53 mg) in anhydrous THF (3 ml).
The
mixture is agitated at approximately -78 °C for approximately 15
minutes. A solution
of chlorotriethylsilane in THF (1.0 M; 660 pl) is added dropwise to the
solution. The
mixture is brought progressively back to ambient temperature then agitated at
ambient
CA 02337261 2001-O1-05
- 19-
temperature for one hour. The mixture is cooled down to approximately -78
°C and a
solution of' butyllithium in hexane (1.6 M; 430 ~tl) is added dropwise. The
solution is
agitated at approximately -78 °C for approxirnately I hour and, over
the next 15
minutes, brought back to a temperature of approximately -15 °C. The
solution is cooled
down to approximately -78 °C. A solution of I-methylsulphonyl-3-(3-
chlorophenyl)-5-
(4-chlorobenzoyl)indole (95 mg, see preparation 7) in THF (2 ml) is added
dropwise.
The mixture is brought progressively back to ambient temperature then agitated
for
approximately 2 hours. The solution is cooled down to approximately 0
°C and
methanol and water are ad~ded. The mixture is agitated for approximately 2
hours. The
solution is concentrated by evaporation under reduced pressure. The residue is
dissolved in dichlorometh<me (DCM) and washed once with water. The organic
phase
is dried over anhydrous MgS04, filtered and concentrated by evaporation under
reduced
pressure. The crude product is purified by column chromatography, on silica,
eluting
with a DCMlMeOH mixture 95:5 in order to produce the expected compound (60 mg;
yield 63 %).
R~ 0.20 (silica, DCM/MeOH 9:1). MS (ES) 447.2 (Calc. MM = 447.4).
Alternatively, the compound of Example 1, (t)-3-(3-chlorophenyl)-5-[(4-
chlorophenyl)hydroxy(1-m:~ethyl-1H-imidazol-5-yl)methyl]indole can be
synthesized
according to the procedure: hereafter. A solution of butyllithium in hexane
(1.6 M, 430
pl) is added dropwise to a solution of 1-methylimidazole (53 mg) in anhydrous
THF (3
ml) at approximately -78 °C. The mixture is agitated at approximately -
78 °C for
approximately 15 minutes. A solution of chlorotriethylsilane in THF (1.0 M;
660 p.l) is
added dropwise to the solution. The mixture is brought progressively to
ambient
temperature then agitated pat ambient temperature for one hour. The mixture is
cooled
down to approximately -78 °C and a solution of butyllithium in hexane
(1.6 M; 430 pl)
is added dropwise. The solution is agitated at approximately -78 °C for
approximately
1 hour and, over the next 15 minutes, brought to a temperature of
approximately -15 °C.
The solutïon is cooled down to approximately -78 °C. A solution of
1-
methylsulphonyl-3-(3-chlorophenyl)-5-(4-chlorobenzoyl)indoline (95 mg, see
prEparation 6) in THF (2 nnl) is added dropwise. The mixture is brought
progressively
to ambient temperature the:n agitated for approximately 2 hours. The solutian
is cooled
down to approximately 0 °'C and methanol and water are added. The
mixture is agitated
for approximately 2 hours. The solution is concentrated by evaporation under
reduced
pressure. The residue is dissolved in dichloromethane (DCM) and washed once
with
water. The organic phase is dried over anhydrous MgS04, filtered and
concentrated by
evaporation under reduced pressure. The crude product is purified by column
chromatography, on silica, eluting with a DCM/MeOH mixture 95:5 in order to
produce
the expected compound.
CA 02337261 2001-O1-05
-20-
The enantiomers of the compound of Example 1 can be separated using techniques
known to a person skilled i,n the art, such as preparative HPLC on a chiral
column.
EXAMPLE 2: ~ -1-methylsulphonyl-3-(3-chlorophenyll-5-(j4-
chlorophen~l)hydroxy(1-rr~ethyl-1H imidazol-5-yl)methyl]indole
A solution of butyllithium in hexane (1.6 M; 694 ~1) is added dropwise tu a
solution of
1-methylimidazole (88 ml;) in anhydrous THF (3 ml) at approximately -78
°C. The
mixture is agitated at approximately -78 °C for approximately 15
minutes. A solution
of chlorotriethylsilane in THF (1.0 M; 1.08 ml) is added dropwise to the
solution. The
mixture is brought pro,gressively to ambient temperature then agitated at
ambient
temperature for one hour. 'The mixture is cooled down to approximately -78
°C and a
solution of butyllithium in hexane (1.6 M, 694 pl) is added dropwise. The
solution is
agitated at approximatel:y -78 °C for approximately 1 hour then brought
to a
temperature of approximately -15 °C. It is agitated at approximately -
15 °C for
approximately 15 minutes. The solution is cooled down to approximately -78
°C. A
solution of 1-methylsulphonyl-3-(3-chlorophenyl)-5-(4-chlorobenzoyl)ïndoline
(150
mg, see preparation 6) in THF (2 ml) is added dropwise. The mixture is brought
progressively to ambient temperature then agitated for approximately 2 hours.
The
solution is cooled down to approximately -78 °C. Methanesulphonyl
chlaride ( 116 mg)
is added dropwise. The solution is brought slowly to ambient temperature and
agitated
overnight. The solution is cooled down to approximately 0 °C. Water is
added and the
mixture is agitated for approximately 2 hours. The solution is diluted using
DCM and
the organic phase is separated and concentrated by evaporation under reduced
pressure.
The crude product is purified by column chromatography, on silica, eluting
with a
CHC13/MeOH mixture 95:5 in order to produce the expected compound in the form
of a
solid (30 mg; yield 17 %). MS (ES): 525.1 (Calc. MM= 525.5).
Alternatively, the compound of Example 2, can be prepared as follows. A
solution of
butyllithium in hexane (1.6 M; 694 pl) is added dropwise to a solution of 1-
methylimidozole (88 mg) in anhydrous THF (1.5 ml) at approximately -78
°C . The
mixture is agitated at approximately -78 °C for approximately 30
minutes. A solution
of chlorotriethylsilane in '1CHF (1.0 M; I .11 ml) is added dropwise to the
solution. The
mixture is brought progressively to ambient temperature then agitated at
ambient
temperature for one hour. The mixture is cooled down to approximately -78
°C and a
solution of butyllithium in hexane (1.6 M, 694 Vil) is added dropwise. The
mixture is
agitated at approximately -78 °C for approximately 1 hour then brought
to
approximately -15 °C for the next 30 minutes. The solution is cooled
down to
approximately -78 °C. A solution of 1-methylsulphonyl-3-(3-
chlorophenyl)-5-(4-
chlorobenzoyl)indole (150 mg, see preparation 7) in THF (I ml) is added
dropwise.
CA 02337261 2001-O1-05
-21-
The mixture is brought progressively to ambient temperature then agitated at
ambient
temperature for 19 hours. The solution is cooled down to approximately -78
°C.
Methanesulphonyl chloride (163 mg) is added dropwise, followed by
düsopropylethylamine (8'7 mg). The solution is brought to ambient temperature
over
one hour and agitated at ambient temperature for 3 hours. 4 ml of a 1N aqueous
solution of HCI and 4 ml of THF are added to the solution. The solution is
agitated at
0 °C for 1 hour 30 minul:es. The organic solvent is eliminated by
evaporation under
reduced pressure. The adueous solution is adjusted to pH 8 by adding a 6N
aqueous
solution of KOH at 0 °C. The aqueous solution is extracted twice with
DCM. The
combined organic phases are washed once with sait water, dried over MgS04,
filtered
and reduced by evaporation under reduced pressure. The residue is purified by
column
chromatography, on silïca, eluting with a CH3Cl/MeOH/Et3N mixture, 98:2:0.1.
The
expected compound is obtained in the form of a solid (44 mg; yield 25%).
MS(ES) 525.2 (Calc. MM= 525.3).
The enantiomers of the compound of Example 2 car be separated using techniques
known to a person skilled in the art, such as preparative HPLC on a chiral
column.
Example 3: f -1- rd-dimethylcarbamoyl)-3-(3-chlorophenyll-5-[(4-chlorophenyll
)~droxy,~l -methvl-1 H-irnidazol-5-yl)methyl] indole
This compound is synthesized in a marner analogous to the first method
described for
preparing the compound of Example 2, using dimethylcarbamyl chloride instead
of the
methanesulphonyl chloride. MS (electrospray): 518.2 (Calc. MM: 518.5).
Example 4: ~ -1-methylsulphonyl-3-(3-chlorophenyl)-5-[amino 4-chloro-phenyll(1-
methyl-1H imidazol-5-vl)methyll indole
Methanesulphonyl chloridle (66 mg) at -78 °C is added dropwise to a
solution of (~)-1-
methylsulphonyl-3-(3-chlorophenyl)-5-[(4-chloro-phenyl)hydroxy(1-methyl-1H
imidazol-5-yl)methyl] indole (100 mg) and triethylamine (58 mg) in anhydrous
DCM (2
ml). The solution is brought slowly to ambient temperature then agitated at
ambient
temperature for 19 hours. The solution is diluted by adding DCM and washed
twice
with salt water, dried over MgS04, filtered and reduced by evaporation under
reduced
pressure. The residue is dissolved in DMF. Sodium azide (124 mg) is then
added. The
mixture is heated to approximately 60 °C overnight. The solvent is
eliminated by
evaporation under reduced pressure. The residue is dissolved in water and DCM.
The
organic phase is separated, washed once with salt water, dried over MgS04,
filtered and
reduced by evaporation under reduced pressure. The residue is dissolved in an
EtOAc/HOAc mixture 9:1. 200 mg of palladium on carbon (Pd/C 10%) is added. The
mixture is agitated under HZ (40 psi) for approximately 3 hours. Thc; solvent
is
eliminated by evaporation under reduced pressure. The residue is purified by
column
CA 02337261 2001-O1-05
-22-
chromatography, on silica~, eluting with a CH3C1/MeOH/Et3N mixture, 98:2:0.1,
in
order to produce the expected compound.
Example 21: ~ -1-acet~l-3-(3-chlorophenyll-5-((4-chlorophenyl)hydroxy(1-methyl-
1H-imidazol-5-yl)methyllindole
This compound is synthesüzed in a marner analogous to the first method
described for
preparing the compound of Example 2, using acetic anhydride instead of
methanesulphonyl chloride;. MS (electrospray): 489.2 (Cale. MM: 489.4).
The following compounds car be prepared by procedures analogous to the
procedures
detailed in Examples 1 to 4 using the appropriate starting reagents and
modifications
which are well known to a person skilled in the art. The compounds of Examples
5, 6,
8, 10, 12, 16, 18, 20 and 2:Z car be synthesized in a marner analogous to the
compound
of Example 4. Examples 7, 9, 11, 15, 17 and 19 car be synthesized in a marner
analogous to the compound of Example 2.
PRÉPARATION 1: 2-(3-chlorophenyl)-N-methoxy-N-methyl-acetamide
A solution of 3-chlorophenylacetic acid (5.00 g; 29.3 mmol), l -(3-dimethyl
aminopropyl)-3-ethylcarbodümide hydrochloride (6.18 g; 32.2 mmol), and
1-hydroxybenzotriazole (HOBt; 4.00 g; 29.3 mmol) in dichloromethane (DCM; 40
ml)
is agitated at ambient temp~erature for approximately 10 minutes. The solution
is cooled
down to approximately 0 "C. N,O-dimethylhydroxylamine hydrochloride (2.86 g;
29.0
mmol) and düsopropylethylamine (DIEA; 3.80 g, 29.3 mmol) are added. The
reaction
mixture is brought to ambi.ent temperature and agitated for approximately 5
hours. The
solution is diluted with 100 ml of DCM and washed with a saturated aqueous
solution
of NaHC03 (twice), a 1N aqueous solution of HCl (twice) and salt water
(twice), dried
over anhydrous MgS04, filtered and concentrated by evaporation under reduced
pressure. The liquid obtained is purified by silica column chromatography
eluting with
an EtOAc/hexane mixture 1:1. The expccted compound is obtained in the form of
a
colourless liquid (5.60 g, 89%). Rf = 0.44 (silica, EtOAc/hexane 1:1 ).
CA 02337261 2001-O1-05
-23-
NMR 1H (300 MHz, CDC13): 7.18-7.34 (m, 4H); 3.76 (S, 2H); 3.66 (S, 3H);
3.22 (S, 3H).
PRÉPARATION 2: 2~3-chloro~phenylLacetaldehyde
A suspension of LiAIH~ (1.90 g, 51 mmol) in anhydrous ether (250 ml) is
agitated at
ambient temperature under a nitrogen atmosphere for approximately 1 hour. The
suspension is cooled down to approximately -45 °C. A solution of 2-(3-
chlorophenyl)
N-methoxy-N-methyl-acetarnide (8.19 g; 38.3 mmol; see Preparation 1) in 10 ml
of
anhydrous tetrahydrofuran (THF) is added dropwise. The mixture is brought to
approximately 0 °C and ag;itated for approximately 3 hours. The
solution is then cooled
down to approximately -4-'i °C. A solution of KHS04 (13 g) in water
(approximately 30
ml) is slowly added to this solution. The resulting mixture is filtered on
Cl?LITE. The
filtrate is concentrated by c:vaporation under reduced pressure, and the
resulting solution
is diluted with DCM and washed with a 1 N aqueous solution of HCl (twice), and
salt
water (twice), dried over anhydrous MgS04, filtered and concentrated by
evaporation
under reduced pressure. 'The expected compound is obtained in the form of a
liquid
(5.80 g), which is used. immediately in the additional stage without
additional
purification.
Rf= 0.71 (silica, EtOAc/h~exane 1:3).
PRÉPARATION 3: 3 ~3-~chlorophenyl)indole
A solution of 2-(3-chlorophenyl)-acetaldehyde (5.80 g; 37.5 mmol) and
phenylhydrazine (6.22 g; 57.5 mmol) in glacial acetic acid (150 ml) is
saturated with
nitrogen by passing N2 th~rough the solution. The solution is then taken to
reflux for
approximately 2.5 hours. The solvent is eliminated by evaporation under
reduced
pressure and the residue o~btained is dissolved in DCM and washed with a 1N
aqueous
solution of HCl (twice), a saturated aqueous solution of NaHC03 (twice) and
sait water
(twice), dried over anhydrous MgS04, filtered and concentrated by evaporation
under
reduced pressure. The c;rude product is purified by silica column
chromatography
eluting with an EtOAc/he:xane mixture 1:6. The expected compound is obt.ained
in the
form of a reddish oil (5.30 g; 62%). Rf= 0.26 (silica, EtOAc/hexane 1:4)
PRÉPARATION 4: 3-~3_--chlorophen~l-indoline
3-(3-chlorophenyl)indole (5.30 g; 23.3 mmol) is dissolved in 50 ml of a 1M
solution of
BH3 in THF. The mixture is cooled down to approximately 0 °C.
Trifluoroacetic acid
(TFA, 50 ml) is then added slowly. Alter this addition, the solution is
agitated for
approximately 10 minute:.. A 1M solution of BH3 in THF (40 ml) is added slowly
to
the solution. The mixture is agitated for approximately 5 minutes then
concentrated by
evaporation under reduced pressure. The residue is purified by silica column
CA 02337261 2001-O1-05
-24-
chromatography eluting with an EtOAc/hexane mixture 1:6. The expected compound
is
obtained in the form of an oil (3.93 g; 74%).
Rf= 0.20 (Silica, EtOAc/hexane 1:4). MS (ES): 229.1 (Calc. MM = 229.7).
PRÉPARATION 5: 1-me hYlsu~honyl-3-(3-chlorophenyl indoline
Methanesulphonyl chloride (2.13 g; 18.6 mmol) is added dropwise to a solution
of 3-(3-
chlorophenyl)indoline (3.8.8 g; 16.9 mmol) and DIEA (2.40 g; 18.6 mmol) i.n
DCM (40
ml) at approximately 0 °C. The mixture is agitated for approximately
one hour 30
minutes. The solution is; diluted with DCM and washed with a saturated aqueous
solution of NaHC03 (twice), a 1N aqueous solution of HCl (twice) and sait
water
(twice) then dried over anhydrous MgS04, filtered and concentrated by
evaporation
under reduced pressure. The crude product is purified by silica column
chromatography
eluting with an EtOAc/hexane mixture 1:4. The expected compound is obtained in
the
form of an oil (4.40 g; 85°rb). Rf= 0.41 (silica, EtOAc/hexane 1:2).
NMR ~H (300 MHz, CDC',13): 7.52 (d, 1H); 7,24-7.34 (m, 3H); 7.20 (s, 1H); 7,02-
7.14
(m, 3H); 4.59 (t, 1H); 4.38 (t, 1H); 3.87 (dd, 1H); 2.92 (s, 3H).
PRÉPARATION 6: 1-meth~phonyl-3-(3-chlorophenyll-5-(4-chlorobenzovll
indoline
AlCl3 (7.62 g; 57.2 mmol) is added by portions to a solution of 1-
methylsulphonyl-3-
(3-chlorophenyl)indoline (4.40 g; 14.3 mmol, see Preparation 5) and 4-
chlorobenzoyl
chloride (3.25 g; 18.6 nunol) at approximately 0 °C in CS2 (25 ml). A
brown
precipitate immediately fonns. The mixture is agitated for approximately 2
hours. 100
ml of cold water containin.g 3 ml of concentrated HCl are added slowly to this
mixture.
The solution is diluted with DCM and the organic phase is separated and washed
with a
1N aqueous solution of HC',1 (twice), a saturated aqueous solution of NaHC03
(twice)
and sait water (twice), then dried over anhydrous MgS04, filtered and
concentrated by
evaporation under reduced pressure. The crude product is purified by silica
column
chromatography eluting with an EtOAc/hexane mixture 1:2. The expected compound
is
obtained in the form of a solid (3.90 g; 61 %). R f = 0.24 (silica,
EtOAc/hexane 1:2).
MS (ES): 445.2 (Calc. MM=445.4). NMR ~H (300 MHz, CDC13): 7,67-7.76 (m, 3H);
7,53-7.59 (m, 2H); 7.48 (s, 1H); 7.44 (s, 1H); 7,30-7.32 (m, 2H); 7.20 (m,,
1H); 7,09-
7.14 (m, 1H); 4.65 (t, 1H); 4.50 (t, 1H); 3.98 (dd, 1H); 3.02 (s, 3H).
PRÉPARATION 7: 1-methylsul~honyl-3-(3-chlorophenyl)-5-(4-chlorobenzo~)indole
A solution of 1-methylsulphonyl-3-(3-chlorophenyl)-5-(4-chlorobenzoyl)indoline
(350 mg) and 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (356 mg) in dioxane (6
ml) is
taken to reflux under a nitrogen atmosphere for approximately 6 hours then
heated to
approximately 95 °C overnight. The solvent is eliminated and the
residue purified by
CA 02337261 2001-O1-05
-25-
silica column chromatography eluting with an EtOAc/hexane mixture, 1:4. The
expected compound is obtained in the form of a solid (195 mg; 56%). MS(ES):
443.2
(talc. MM=443.4). Rf=0.38 (silica, EtOAc/hexane, 1:2).
,> >,
Preparation B:
1-~(2R)-amino-3-mercaptopropylJ-(2S)-(2-mercaptoethyl)-4-(I-naphthoyl)
~iperazine-
1,2-cyclodisulphide (XII):
a) Synthesis of 1-benzyl-3(S)-benzyloxycarbonylmethyl piperazine-2,5-dione:
A cold solution of dicyclohexylcarbodümide (DCC, 7.1 g) in 20 ml of CH2C12 is
added
to a solution cooled down by an ice bath of the ~3-benzylic ester of BO(:-
aspartic acid
(10 g), hydroxybenzotriaz,ole (HOBT, 4.2 g) and the ethyl ester of N-
benzylglycine
(6.4 g) in 80 ml of CH2C12.. The reaction medium is agitated for approximately
1 hour
at a temperature of 0 to 5 °C, then overnight at ambient temperature.
The precipitate is
eliminated by filtration and the filtrate is evaporated to dryness under
reduced pressure.
The residue is dïvided beoween ethyl acetate and water. The organic phase is
washed
with 100 ml of aqueous NaHC03, then with water, and dried (MgS04). The solvent
is
eliminated by evaporation to dryness under reduced pressure in order to
produce 16 g of
a solid. Thin layer chromatography (TLC) (silica gel: CHC13/acetone = 9:1, Rf=
0.55).
The product obtained is th.en treated with 50% trifluoroacetic acid in CHCI_;
(40 ml) for
approximately 1 hour and the volatile substances are eliminated by evaporation
to
dryness under reduced pressure. The residue is divided between ethyl acetate
and a
saturated aqueous solution of NaHC03. The organic phase is dried (MgS04) and
the
solvent is eliminated by evaporation to dryness under reduced pressure in
order to
produce 10 g. TLC (silica gel: CHCI3/acetone = 9:1, Rf= 0.14).
b) Synthesis of 4-benzyl- L-tert-butoxycarbonyl-2(S)-(2-hydroxyethyl)
piperazine:
A 50% mineral dispersion of lithium aluminium hydride (LAH) (12.5 g) under a
nitrogen atmosphere is ad<ied by portions to a solution of the product of
Stage a) (9.73g)
in 200 ml of tetrahydrofuran (THF) cooled down by an iced water bath. The
reaction
medium is maintained a1: reflux overnight. Alter cooling down in an ice bath,
a
saturated aqueous solution of Na2S04 is added dropwise in order to decompose
the
excess LAH and the white molasse obtained is eliminated by filtration. The
filtrate is
evaporated to dryness under reduced pressure and dissolved in dichlorornethane
(55
CA 02337261 2001-O1-05
-26-
ml), treated with tert-butyll dicarbonate (5.9 g), and agitated for
approximately 1 hour.
A saturated aqueous solution of NaHC03 (25 ml) is added and the mixture is
agitated
for approximately 2 hours. The organic phase is washed [with aJ saturated
aqueous
solution of sodium chlorid~e and dried (MgS04). Alter evaporation of the
solvent, the
residue is chromatographed on silica gel ( 160 g) using a CHC13/MeOH rnixaure
19:1 as
eluant. The appropriate fractions are collected, and the solvents are
elïminated by
evaporation to dryness undler reduced pressure in order to produce 10 g of a
glass. TLC
(silica gel: CHCl3lMe01-I == 9:1, Rf= 0.56).
c) Synthesis of 1-tert-butoxycarbonyl-2-(S)-(2-hydroxyethyl) piperazine:
The product of Stage b (8.7g) is dissolved in ethanol (35 ml) and treated with
Pd(OH)Z
on carbon (0.8 g) and acetic acid (3 ml). Hydrogenation is then carried out
under a
pressure of 30 p.s.i. ove:rnight. The reaction mixture is filtered and the
solvent
eliminated by evaporation to dryness under reduced pressure in order to
produce the
expected product.
d) Synthesis of 1-tert-butoxycarbonyl-2(S)-(2-hydroxyethyl)-4-(1.-naphthoyl)
piperazme:
110 ml of a 1N aqueous solution of NaOH then a solution of 1-naphthoyl
chloride
(5.14 g) in acetonitrile (20 ml) are added to a solution of the product of
Stage c) (8.4 g)
in acetonitrile (40 ml). Aj~er approximately 3 hours under agitation, a large
part of the
acetonitrile is eliminated by evaporation under reduced pressure and the
residual
mixture is extracted with chloroform. The extract is dried (MgS04) and the
solvent
eliminated by evaporation to dryness under reduced pressure in order to
produce 8.12 g
of the expected product. TLC (silica gel: CHC13/MeOH = 9:1, Rf = 0.64).
e) Synthesis of 1-tert-butoxycarbonyl-2(S)-(2-triphenylmethylthioethyl)-4-(1-
naphthoyl)-piperazine
A solution of diethylazodicarboxylate (DEAD, 0.25 g) in 2 ml of THF is added
dropwise to a solution of t.riphenylphosphine (0.53 g) in 5 ml of anhydrous
THF cooled
down by an ice bath. At~er agitation for approximately 30 minutes at a
temperature of 0
to 5 °C, a solution of the product of Stage d) (0.4 g) and
triphenylmercaptan (0.55 g) in
10 ml of THF is added dropwise. The mixture ïs agitated for approximately 1
hour at a
temperature of 0 to 5 °C and for approximately 1 hour at ambient
temperature. The
solvent is eliminated by evaporation to dryness under reduced pressure and the
residue
is chromatographed on ~,ilica gel (40 g) using CHC13 as eluant. The
appropriate
fractions are collected and the solvent is eliminated by evaporation to
dryness under
CA 02337261 2001-O1-05
-27-
reduced pressure in order to produce 420 mg of a pale yellow foam. Mass
spectrometry
(MS) (Electrospray) 665.2 (643 + 23(sodium)). TLC (silica gel: CHCl3/acetone =
9:1,
Rf= 0.53)
fJ Synthesis of 2(S)-(2-triphenylmethylthioethyl)-4-(1-naphthoyl) piperazine
10 ml of trifluoroacetic acid (TFA) is added to an agitated solution of the
product of
Stage e) (2.2 g) in 30 ml of CHZC12. The mixture is agitated for approximately
30
minutes. The volatile substances are eliminated by evaporation to dryness
under
reduced pressure. The residue is dissolved in CHCl3 (50 ml) and treated with
triethylamine in excess (4 ml). The mixture is washed with water, then dried
(MgS04)
and the volatile substances are eliminated by evaporation to dryness under
reduced
pressure in order to produce 2.1 g of a pale yellow glass. TLC (silica gel:
CHC13/MeOH = 9:1, Rf = 0.63)
g) Synthesis of 1-[2(F;)-N-tert-butoxycarbonylamino-3-
triphenylmethylthiopropyl]-
2(S)-(2-triphenylmethyl-thioethyl)-4-( 1-naphthoyl)-piperazine
4 g of 4~ molecular sieve then by portions Na(OAc)3BH (1 g) are added over a
period
of 30 minutes to a solution of the product of Stage f) (0.9g) and 2(R)-N-tert-
butoxycarbonylamino-3-triphenylmethylthiopropan-al (1.2 g), prepared according
to the
procedure of O.P. Goel, et al., (Org. Syn. 1988, 67, 69-75), in CH2C12 (20 ml)
containing 1 % of acetic acid. Alter agitation for approximately 2 hours, the
mixture is
filtered and the filtrate is washed with water, with a 5% aqueous solution of
NaHC03,
with water, then dried (M:gS04). The solvent is eliminated by evaporation to
dryness
under reduced pressure, and the residue is chromatographed on silica gel (60
g) using
CHCl3 as eluant. The ap~propriate fractions are collected and the solvent is
eliminated
by evaporation to dryness, under reduced pressure in order to produce 0.6 g of
a white
foam. TLC (silica gel: CHCI~/acetone = 9:1; Rf= 0.55); MS (Electro Spray)
974.3.
h) Synthesis of 1.-[2(R)-amino-3-mercaptopropyl]-2(S)-2-mercaptoethyl)-4-(1-
naphthoyl )-piperazine
The product of Stage g) (450 mg) is treated for approximately 30 minutes with
50%
TFA in CHZCIz (10 ml) c;ontaining 1 ml of triethylsilane. The volatile
substances are
eliminated to dryness by evaporation under reduced pressure. The residue is
triturated
with ether, filtered, then dried in order to produce 280 mg of 1-[2(R)-amino-3-
mercaptopropyl]-2(S)-(2-mercaptoethyl)-4-( 1-naphthoyl)-piperazine. MS
(electrospray) 390.3
CA 02337261 2001-O1-05
-28-
i) Cyclizatïon of 1-[2(R)-amino-3-mercaptopropylJ-2(S)-2-mercaptoethyl)-4-(1-
naphthoyl)-piperazine in order to form 1-[2(R)-amino-3-mercaptopropyl]-2(S)-2-
mercaptoethyl)-4-(1-naphthoyl)-piperazine-1,2-cyclodisulphide (XII)
100 mg of the product of Stage a) is dissolved in 10 ml of an aqueous solution
of
CH3CN (H20/CH3CN - 7:3), and treated with 3 g of EKATHIOXC~ resin (0.34
mmoles/g). The reaction medium is agitated for approximately 6 hours at
ambient
temperature. The mixture is then filtered, the resin is washed with an aqueous
solution
of methanol (water/methanol 1:3), and most of the organic solvent is
eliminated by
evaporation under reduced pressure, retaining only a small volume. The
concentrate is
subjected to preparative HPLC chromatography casing 0.1 % aqueous TFA and
CH3CN
as the mobile phase. The appropriate fractions are collected and most of the
solvents
are eliminated by evaporation under reduced pressure, only retaining a small
volume.
The concentrate is then lyoyphilized in order to produce the expected product.
Alternatively, a solution of 1-[2(R)-amino-3-mercaptopropyl]-2(S)-(2-
mercaptoethyl)-
4-(1-naphthoyl)-piperazine in aqueous CH3CN is agitated with air at a pI-1:
comprised
between 6 and 8. In both cases, the reaction medium comprises a mixture of
cyclized
disulphide and its dimer in a proportion of approximately 4 to 1.
PHARMACOLOGICAL STUDY OF THE PRODUCTS OF THE INVENTION
As an example, the effect of treating an MCF-7 human cell fine with the
compounds of
formulae (I), (II), (XI), (XII), (GPI1) and (GPI2) described previously will
be studied.
Electrophoresis will also be carried out on pancreatic cancer cens treated
with the
compounds of formulae (I) and (III). All these compounds are prepared casing
the
methods described in the patents mentioned or, in the case of (XII), according
to the
particular method described in Preparation 1 above.
Procedures
Cell line
The MCF-7 (human pleural cells, breast cancer) and Mia-PaCa2 (pancreatic
cancer) cell
fines were acquired from the American Tissue Culture Collection (Rockville,
Maryland,
USA).
CA 02337261 2001-O1-05
-29-
Measurement of tlze intracellular guantity of cyclic AMP for MCF 7 tells
MCF-7 tells (2.104) seeded on a 24-well plate are cultured for 5 days in
Dulbecco's
modified Eagle medium ((Jibco-Brl, Cergy-Pontoise, France) completed with 10%
of
foetal calf serum inactivated by heating (Gibco-Brl, Cergy-Pontoise, France ),
50000
units/1 of penicillïn and 50 mg/1 streptomycin (Gibco-Brl, Cergy-Pontoise,
France), and
2 mM of glutamine (Gibco-Brl, Cergy-Pontoise, France). The culture medium is
replaced alter two washes with a medium without serum completed or net
completed
with the specified agents for a tune indicated in the different figures.
Agents activating
the production of cAMP are then added at 37 °C. The reaction is stopped
alter 30
minutes by suppressing the medium and rapidly adding 200 pl of a O. l h1
solution of
HCI. These extracts are frozen at -80 °C until they are used. The
concentration of
cAMP is measured using a commercial measurement kit (reference NEK033 from
NEN, Les Ulis, France) following the manufacturer's instructions. The
radioactivity is
determined using a Gamma counter (Gamma Master-1277, LKB, Turku, Finland).
Cell proliferation tests
3000 MCF-7 tells placed in 80 pl of Dulbecco's modified Eagle medium (Gibco-
Brl,
Cergy-Pontoise, France) completed with 10% foetal calf serum inactivated by
heating
(Gibco-Brl, Cergy-Pontoise, France ), 50000 units/1 of penicillin and 50 mg/1
streptomycin (Gibco-Brl, Cergy-Pontoise, France), and 2 mM of glutamine (Gibco-
Brl,
Cergy-Pontoise, France) vvere seeded on a 96-well plate on day 0. The tells
were
treated on day 1 for 96 hours with increasing concentrations up to 50 pM of
each of the
compounds to be tested. At the end of this period, the quantification of tell
proliferation is evaluated by a colorimetric test based on the cleavage of the
WST1
tetrazolium sait by mitoclhondrial dehydrogenases in the viable tells leading
to the
formation of formazan (Boehringer Mannheim, Meylan, France). These tests are
carned out in duplicata ~with 8 determinations per concentration tested. For
each
compound to be tested, thc: values included in the linear part of the sigmoid
were used
for linear regression analys;is and to estimate the ICSO inhibitory
concentration.
Analysis of the subcellular localization of the,Qycomplex by the western-bloc
method
The Mia-PaCa2 tells (400,000) are seeded in Petri dishes (10 cm diameter) in
Dulbecco's modified Eagl.e medium (Gibco-Brl, Cergy-Pontoise, France)
completed
with 10% foetal calf serurn inactivated by heating (Gibco-Brl, Cergy-Pontoise,
France),
50,000 units/1 of penicillin and 50 mg/1 of streptomycin (Gibco-Brl, Cergy-
Pontoise,
France), and 2 mM of glutamine (Gibco-Brl, Cergy-Pontoise, France). The
compounds
are added on day 1 at a concentration of 30pM. The tells are harvested by
scraping on
day 3 alter washing twice with the phosphate buffer (PBS, Gibco-Brl, Cergy-
Pontoise,
CA 02337261 2001-O1-05
-30-
France) at 4°C. The cells are separated by low speed centrifugation
(800g, 5 minutes).
The cells are resuspended in an A buffer containing SOmM Hepes, pH 7.5, SmM
MgCl2, 1mM EDTA, pro~tease inhibitors (Cocktail tablets, no. 1836-170,
Boehringer
Mannheim, Meylan, France). The cells are lysed by three cycles of " freezing
in liquid
nitrogen - thawing at 4 °C' ". The lysate is centrifuged first at a low
speed (1200g, 5
minutes, 4 °C) in order to eliminate the unlysed cens. The supernatant
is centrifuged at
high speed (200,OOOg, SU minutes, 4 °C) in order to separate the
cytosolic fraction and
the membrane fraction (pellet). This pellet is resuspended in the A buffer.
The
concentration of proteins in each fraction is determined by Bradford
colorimetric assay
(Bio-Rad protein assay, Ivry, France). The proteins (20pg) are separated by
electrophoresis on denatuong polyacrylamide gel (16% Gel-tricine, Novex,
Prolabo,
Fontenay sous Bois) following the manufacturer's recommendations. The proteins
thus
separated are transferred onto a nitrocellulose membrane (C-hybond, RPN 2020C,
Amersham, Les Ulis, France) in a semi-dry transfer. The (3 protein is detected
by the
T20 antibody, sc378, Santa-Cruz, TEBU, Le Perray en Yvelines, France), itself
recognized by the second antibody coupled with the peroxydase enzyme. The
chemoluminescence is obtaïned with the ECL-Plus system (RPN2132, Amersham, Les
Ulis, France). The signal is revealed on Kodak BioMax-light autoradiographie
films,
237358, Sigma, St Quentin, France). The quantity of chemolurninescence is
proportional to the quantity of proteins present.
Eguipment
The vaso-intestinal peptide (VIP) and the mevastatin were acquired from Bachem
(Voisins le Bretonneux, France) and TEBU (Le Perray en Yvelines, France)
respectively. The comp~ounds of formulae (I), (II) and (III) were supplied by
Biomeasure Inc. (Milford, MA, USA). All these compounds were used following
their
manufacturers' recommend~ations.
Results
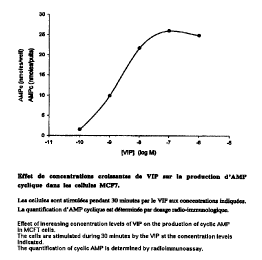
VIP has been presented as an extracellular ligand of a receptor coupled with
protein G
which stimulates the synth.esis of cAMP in human breast cancer cens. Figure 1
shows
that treatment of MCF-7 human breast cancer cens with VIP increases the
intracellular
quantity of cAMP in a concentration-dependent manner. A VIP concentration of
10
nM, which offers quasi-optimum production of cAMP, will be used for the
following
tests. This concentration agrees with data already published concerning the
human
breast cancer cell fine T47D.
CA 02337261 2001-O1-05
-31-
While post-transcriptional prenylation of G proteins is a necessary stage to
allow it to
fonction, the addition of prenyltransferase inhibitors should alter it
significantly. The
compound of formula (1) is known to be a powerful specific inhibitor of
farnesyltransferases, capable of inhibiting the farnesylation of ras at
concentrations in
the region of a nanomole. Figure 2 shows that pre-treatment for 24 hours of
MCF-7
cens taken from in vitro cultures with the compound of formula (I) inhibited,
in a
concentration-dependent marner, the accumulation of cAMP stimulated by the
VIP.
Almost complete inhibition was obtained at a concentration of 30 pM of the
compound
of formula (I). These results clearly show that treatment with a specific
inhibitor of
farnesyltransferases is sufficient to block the transduction of the signal
l:he route of
which uses the heterotrimeric G proteins as medïators.
Figure 3 shows that treatment for 1 hour with the compound of formula (I) is
rot
sufficient to modify the re;sponse to the VIP. Despite a slight but
reproducible effect
alter treatment for 8 hours,, only treatment for 24 hours clearly inhibits the
stimulation
of the VIP. The action kinetics observed agrees with that expected for a
prenyltransferase inhibitor allowing the appearance of non-prenylated forms of
sub-
units of the G protein.
The ïnhibition of the stinnulation by the VIP is rot restricted to compounds
with a
structure analogous to that of the compound of formula (I) but tan be extended
to other
prenyltransferase inhibitors, as shown by the results obtained in particular
for the
compound of formula (II) which is also a powerful inhibitor of
farnesyltransferases
capable of inhibiting the farnesylation of H-ras of human tumours at
concentrations of
the order of a nanomole (Table I).
Moreover, Figure 4 shows that the pretreatment of tells for 24 hours by the
compound
of formula (I) does rot modify the production of cAMP induced by the direct
activator
of the adenylate cyclase, forskolin. This shows that the adenylate cyclase
itself is rot
modified by treatment witlh the compound of formula (I).
Figure 5 shows that the pre-treatment of tells for 24 hours with the compound
of
formula (I) decreases the production of cAMP induced by the direct activator
of the
a sub-unit, the choleric toxin. Now this toxin tan only fix itself to the
heterotrimeric
complex (Warner et al., C'ell Signal, 8, 43-53 (1996)). This therefore
suggests that
pretreatment with the compound of formula (I) prevents the formation of the
heterotrimeric complex.
The ~3y dimeric complex is very stable and tan be identified in a proteinic
extract with
an antibody directed against the (3 sub-unit. Figure 6 shows the results of a
'western-blot
CA 02337261 2001-O1-05
-32-
carried out on untreated cens, cens treated for 48 hours with mevastatin (a
mevalonate
synthesis inhibitor), wïth a geranyl-geranyl transferase inhibitor, the
compound of
formula (III), or with a pure inhibitor of farnesyltransferase, the compound
of formula
(I). The proteins of the cyosolic and membrane fractions of the control or
treated cens
are separated by denaturing electrophoresis on polyacrylamide gel before being
transferred onto a semi-solid membrane allowing identification with an
antibody
(western-blot) directed against all forms of (3 proteins.
The quantity of the (3y c;amplex detected on the cell membrane of untreated
cells
(control) decreases greatly in cens treated for 48 hours with mevastatin, with
a geranyl-
geranyl transferase inhibitor (compound of formula (III)) or also with a pure
inhibitor
of farnesyl-transferase (compound of formula (I)). It can therefore be
concluded that
alter treatment with a prc~nyltransferase inhibitor, the y sub-unit therefbre
no longer
plays its rote in anchoring the ~iy complex to the membrane.
Finally, Table Il shows the in vitro values for the proliferation inhibition
activities of
the compounds of formula.e (I) and (II) with respect to MCF-7 human tumoral
cens not
carrying a Ras oncogen which has been subject to a mutation.
CA 02337261 2001-O1-05
-33-
Compound Dose (pM) Inhibition of n
the G
rotein
(I) 10 52 3
II) 100 69 2
XI) 30 27 5
(XII 10 53 2
(GPI1 ) 10 22 1
(GPI2) 10 44 3
Table I
Effects of compounds incubated for 24 hours on the production of cyclic AMP
stimulated by VIP in MC.'F7 cells.
The cells are incubated for 24 hours in the presence of the tested compounds
at the
doses indicated and then stimulated by 10-g M of VIP. The quantification of
cyclic
AMP is determined by radioimmunoassay.
(~om ounds ICso (NM)
Compound 19.4
(I)
Com ound 35.8
II
Table II
Proliferation inhibition of the compounds of formulae (I) and (II) with
respect to
MCF-7 human tumoral tells not carrying a Ras oncogen having been subjected to
a mutation.