Note: Descriptions are shown in the official language in which they were submitted.
CA 02337496 2001-O1-26
WO 00/06759 PCT/tJS99/16388
DESCRIPTION
Anti-Angiogenesis Plasmids And Delivery Systems, And Methods
Of Making And Using The Same
Field Of The Invention
The present invention relates to gene delivery and gene
therapy, and provides novel nucleic acid constructs for
expression of anti-angiogenic gene products in a mammal,
formulations for delivery that incorporate a nucleic acid
construct for expression, and methods for preparing and using
such constructs and formulations.
Background Of The Invention
The following discussion of the background of the
invention is merely provided to aid the reader in
understanding the invention and is not admitted to describe or
constitute prior art to the present invention.
In tumor-bearing hosts, the lack of an effective immune
response is due to weak tumor antigenecity or a tumor-
immunosupressive environment. Immunotherapy is to strengthen
the tumor-host interaction by introduction of tumor-specific
antigens or to boost immune function by local cytokine
expression. Several cytokine gene medicines such as IL-2,
IFN-a and IL-12 have been developed. By direct intratumoral
injection of nucleic acid encoding for IFN-a, IL-2 or IL-12
formulated in a polymeric delivery system, tumor-bearing mice
develop an immune response, which leads to inhibition of tumor
growth in murine syngeneic tumor model. An alternative
approach, anti-angiogenic gene medicine, has been recognized
as therapeutic strategy for both immunogenic and non
immunogenic tumors.
Tumors get oxygen and nutrients through the blood
vessels. The formation of blood vessels, or angiogenesis, is
required for tumor growth and metastases. Moreover,
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
2
microvessel density in the tumor show a positive relationship
with tumor growth, the risk of metastases, tumor recurrence or
death (Folkman, J., J. Natl. Cancer Inst., 82:4-6 (1990);
Folkman, J. Nat. Med., 1:27-31 (1995)). The onset of tumor
angiogensis could be triggered by an upregulation of
angiogenic factors such as VEGF and bFGF or by a
downregulation of anti-angiogenic factors such as endostatin,
angiostatin and thromospondin-1 (Hanahan, D. & Folkman, J.,
Cell, 86:353-64 (1996)). Thus, reconstitution of angiogenic
inhibitors would provide, a plausible strategy for cancer
therapy (Hori, A., Cancer Res., 51:6180-4 (1991): Kim, K.J.,
Nature, 362:841-4 (1993); O'Reilly, M.S. et al., Cell, 79:315-
28 (1994); O'Reilly, M. S. et al., Cell, 88:277-85 (1997);
Clapp, C. et al., Endocrinology, 133:1292-9 (1993); Gupta,
S.K. et al., Proc. Natl. Acad. USA, 92:7799-7803 (1995) ) .
Angiostatin and Endostatin are two members of an
expanding family of proteins that are angiogenesis inhibitors.
Angiostatin is an internal proteolytic fragment of mature
plasminogen. It contains 4 triple loop disulfide-linked
structures, known as kringle domains (Residues 98-440). It
has been shown that a form of 3 kringle domain (residues 98-
333) is more potent in vitro (Coo et al., The Journal of
Biological Chemistry, 271:29461-29467 (1996)) and in vivo
(Griscelli et al., Proc. Natl. Acad. Sci. USA, 95:6367-6372
(1998)). Endostatin is the C-terminal proteolytic fragment of
collagen 18a. Its structure resembles that of E-selectin, an
adhesion molecule on endothelial cell (EC) surface
(Hohenester, E. et al., EMBO J., 17:1656-64 (1998)).
Both angiostatin and endostatin can inhibit endothelial
cell (EC) proliferation in vitro and angiogenesis in vivo.
Moreover, recombinant proteins have been shown to inhibit
tumor growth and metastases in mouse models when injected at
high doses. Recently, it has been shown that combination of
angiostatin and endostatin has synergistic effect in
inhibition of tumor growth and metastases (Bachelot, T. et
al., Abstract from AARC, vol 39, March 1998). It has also
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
3
shown that anti-angiogenic therapy can synergize radiation
therapy in a murine tumor model (Mauceri, et al., Nature,
394:287 (1998)).
Anti-angiogenic therapy targets to EC, but not tumor
cells. EC is more accessible to systemically deliveried drugs,
thus anti-angiogenic therapy is particularly useful in
treatment of disseminated cancer. Moreover, EC is not
transformed and anti-angiogenic therapy of an experimental
model of cancer does not induce acquired drug resistance.
Because angiostatic therapy will require a prolonged
maintenance of therapeutic levels in vivo, the continuous
delivery of a recombinant protein will be expensive and
cumbersome.
Summary of the Invention
The present invention relates to gene delivery and gene
therapy, and provides novel nucleic acid constructs for
expression of anti-angiogenic coding sequences in an organism,
preferably a mammal, formulations for delivery that
incorporate a nucleic acid construct for expression, and
methods for preparing and using such constructs and
formulations. In particular, this invention relates to
plasmid constructs for delivery of therapeutic genes to cells
in order to modulate tumor activity. The invention also
provides methods of using those constructs (including
combination therapy with other treatment methods, such as
radiation therapy, or agents, such as cytokines, preferably
IL-12), as well as methods for preparing such constructs. The
pharmaceutically acceptable, cost effective and highly
efficient delivery system presented herein represents an
unanticipated improvement over the art.
A gene therapy approach utilizing an interactive
polymeric gene delivery system that increases protein
expression by protecting plasmid DNA (pDNA) from nucleases and
controlling the dispersion and retention of pDNA in injected
tissues has been employed. These polymeric interactive non-
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
4
condensing (PINC) systems routinely result in a greater amount
of gene expression from tissues as compared to delivery of
unformulated plasmid in saline. A plasmid expression system
encoding murine IFNa4 or IL-12 formulated as a complex with
PVP could induce an anti-tumor immune response following
direct injection into subcutaneous murine tumors. By using a
plasmid that encodes human insulin-like growth factor-I (hIGF-
I) and formulated as a PINC complex, a long duration of
production of biologically active human IGF-I has shown in
vivo following intra-muscular injection. Since
intramuscularly delivered gene can achieve a high and
persistent expression of therapeutic product in circulating
system, it is conceivable that i.m. delivered cancer gene
medicines could be used in treatment of disseminated disease,
a major limitation of gene therapy.
Alternatively, the administration of a therapeutic gene
into selected organs bearing tumor metastases (e. g. lung),
would be more suitable for a poorly met clinical need. Gene
delivery to the lung for the treatment of genetic defects has
been explored in experimental models as well as in clinical
trials. By using N- [1- (2-3-dioleyloxy) propyl] -N,N,N
trimethylammonium chloride (DOTMA)/cholesterol/ plasmid 3:1
positively charged complexes, enhanced pulmonary expression of
transgenes was shown following intra-tracheal administration
or intravenous injection.
By tail-vein injection of lipid/DNA, detectable levels of
human growth hormone (hGH) in serum, human factor IX (hFIX) in
plasma and chloramphenicol acetyltransferase (CAT) in the lung
and liver were observed with positively charged lipid/plasmid
complexes prepared from 400 nm extruded liposomes with a
cationic lipid to co-lipid ratio of 4:1 mol/mol. Although the
administration of positively charged plasmid/cationic lipid
complexes to the lung airways induces a cytokine pattern
resembling a Thl cell phenotype, even in the absence of
transgene expression, IL-12 transgene expression in murine
lungs following administration of IL-12 plasmid/lipid
CA 02337496 2001-O1-26
WO 00/06759 PCTNS99/16388
complexes inhibits the growth of pulmonary metastases of Renca
tumors in syngeneic BALB/c mice.
Use of tumor EC-specific promoters to express anti
angiogenic factors is described herein. At least two
5 strategies were used to select proliferating EC-specific
promoters. One is to clone promoters of genes that
specifically expressed in tumor EC such as flk-1 and avb3
integrins. Another is to generate chimeric promoters of EC-
specific enhancers such as endothelin-1 enhancer and cell-
cycle-specific promoters such as cyclin A.
The present invention, by using prototype anti-angiogenic
genes, endostatin and angiostatin, demonstrated that anti-
angiogenic gene medicine inhibited growth of solid tumor by
either intratumoral or intramuscular injection. Anti-
angiogenic gene medicines also inhibit lung metastatic tumors
after intramuscular or intravenous delivery of formulated
angiostatic genes.
Thus, in a first aspect, the invention features a plasmid
that contains a tissue specific element and an anti-angiogenic
coding sequence. Preferably the tissue specific element is
specific for endothelial cells and is transcriptionally linked
to the anti-angiogenic coding sequence. As explained in
detail below, the plasmid may optionally include
transcriptional control sequences such as one or more
cytomegalovirus promoter sequences.
The "tissue specific element" may include a promoter,
preferably selected from the group consisting of ET-1, flk-1,
Alpha-V, Beta-3, ICAM-2, cyc A, E2F1, and cdc6, or may include
an enhancer, preferably selected from the group consisting of
CMV, four copies of ET-1, and seven copies of ET-1. Examples
of EC tissue specific promoters are described in detail below.
The tissue specific element preferably is associated or
linked with the anti-angiogenic coding sequence in such a
manner that the anti-angiogenic coding sequence expression is
enhanced, preferably to a predominant (over 50~) nearly
exclusive (about 900 or more) or exclusive level (about 1000 ,
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
6
in the tissue or cells of interest. For example, expression
may be enhanced by about two-fold.
As used herein, the term "plasmid" refers to a construct
made up of genetic material (i.e., nucleic acids). It
includes genetic elements arranged such that an inserted
coding sequence can be transcribed in eukaryotic cells. Also,
while the plasmid may include a sequence from a viral nucleic
acid, such viral sequence does not cause the incorporation of
the plasmid into a viral particle, and the plasmid is
therefore a non-viral vector. Preferably a plasmid is a closed
circular DNA molecule.
"Cytomegalovirus promoter" refers to one or more
sequences from a cytomegalovirus which are functional in
eukaryotic cells as a transcriptional promoter and an upstream
enhancer sequence. The enhancer sequence allows transcription
to occur at a higher frequency from the associated promoter.
In this context, "transcriptionally linked" means that in
a system suitable for transcription, transcription will
initiate under the direction of the control sequences) and
proceed through sequences which are transcriptionally linked
with that control sequence(s). Preferably no mutation is
created in the resulting transcript, which would alter the
resulting translation product.
The term "coding region" or "coding sequence" refers to a
nucleic acid sequence which encodes a particular gene product
for which expression is desired, according to the normal base
pairing and codon usage relationships. Thus, the coding
sequence must be placed in such relationship to
transcriptional control sequences (possibly including control
elements and translational initiation and termination codons)
that a proper length transcript will be produced and will
result in translation in the appropriate reading frame to
produce a functional desired product.
In a preferred embodiment the "anti-angiogenic coding
sequence" encodes a product selected from the group consisting
of endostatin, angiostatin, thrombspondin-1, p53, IL-12, IFN
CA 02337496 2001-O1-26
WO 00/06759 PCTNS99/16388
7
alpha, truncated tissue factor, an integrin av(33 blocking
agent, a VHL gene product, a cell cycle-dependant kinase
inhibitor, a VEGFr, bFGFr, and a bFGF binding protein and
preferably is a synthetic sequence having optimal codon usage
(for the organism receiving the plasmid, preferably a human),
or semi-optimal codon usage (for the organism receiving the
plasmid, preferably a human), or has the nucleotide sequence
of any of the plasmids described herein, more preferably
plasmid pES1281, pIP1316, pAS1095 or pAS 1096.
An anti-angiogenic coding sequence encodes a product that
reduces angiogenesis in the organism of interest, preferably
to a significant extent, (for example to an extent that it
creates a therapeutic effect) or reduces angiogenesis in an in
vitro assay.
A particular example of coding regions suitable for use
in the plasmids of this invention are the natural sequences
coding for anti-angiogenic agents. Thus, in a preferred
embodiment coding region has a nucleotide sequence which is
the same as the natural nucleotide sequence encoding the anti-
angiogenic agent. However, it may be preferable to have an
anti-angiogenic coding sequence which is a synthetic coding
sequence. In a preferred embodiment, the anti-angiogenic
coding sequence is a synthetic sequence utilizing optimal or
semi-optimal codon usage.
Thus, a "sequence coding for a human anti-angiogenic
agent" or "a human anti-angiogenic coding sequence" is a
nucleic acid sequence which encodes the amino acid sequence of
a human anti-angiogenic agent, based on the normal base
pairing and translational codon usage relationships. It is
preferable that the coding sequence encode the exact, full
amino acid sequence of the natural human anti-angiogenic
agent, but this is not essential. The encoded polypeptide may
differ from the natural human anti-angiogenic agent so long as
the polypeptide retains anti-angiogenic activity, preferably
the polypeptide is at least 50%, 75%, 90%, or 97% as active as
natural human anti-angiogenic agent, and more preferably fully
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
8
as active as the natural human anti-angiogenic agent. Thus,
the polypeptide encoded by the anti-angiogenic coding sequence
may differ from a natural human anti-angiogenic agent by a
small amount, preferably less than a 15~, 10$, 5~, or 1~
change. Such a change may be of one of more different types,
such as deletion, addition, or substitution of one or more
amino acids.
The term "transcriptional control sequence" refers to
sequences which control the rate of transcription of a
transcriptionally linked coding region. Thus, the term can
include elements such as promoters, operators, and enhancers.
For a particular transcription unit, the transcriptional
control sequences will include at least a promoter sequence.
The plasmid, in preferred embodiments, may also contain a
growth hormone 3' untranslated region, preferably from a human
growth hormone gene.
A "growth hormone 3' untranslated region" is a sequence
located downstream (i.e., 3') of the region encoding material
polypeptide and including at least part of the sequence of the
natural 3' UTR/poly(a) signal from a growth hormone gene,
preferably the human growth hormone gene. This region is
generally transcribed but not translated. For expression in
eukaryotic cells it is generally preferable to include
sequence which signals the addition of a poly-A tail. As with
other synthetic genetic elements a synthetic 3' UTR/poly(A)
signal has a sequence which differs from naturally-occurring
UTR elements. The sequence may be modified, for example by the
deletion of AZU repeat sequences. Deletion of such ALU repeat
sequences acts to reduce the possibility of homologous
recombination between the expression cassette and genomic
material in a transfected cell.
The plasmid preferably includes a promoter, a TATA box, a
Cap site and a first intron and intron/exon boundary in
appropriate relationship for expression of the coding
sequence. The plasmid may also include a 5' mRNA leader
sequence inserted between the promoter and the coding sequence
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
9
and/or an intron/5' UTR from a chicken skeletal a-actin gene.
Also, the plasmid may have a nucleotide sequence which is the
same as the nucleotide sequence of plasmid any of the plasmids
described herein.
The plasmid may also include: (a) a first transcription
unit containing a first transcriptional control sequence
transcriptionally linked with a first 5'-untranslated region;
a first intron, a first coding sequence, and a first 3'-
untranslated region/poly(A) signal, wherein the first intron
is between the control sequence and the first coding sequence;
and (b) a second transcription unit containing a second
transcriptional control sequence transcriptionally linked with
a second 5'-untranslated region, a second intron, a second
coding sequence, and a second 3'-untranslated region/poly(A)
signal, wherein the second intron is between the control
sequence and the second coding sequence; wherein the first and
second coding sequences contain a sequence coding for any two
different anti-angiogenic agents, preferably angiostatin and
endostatin, although other combinations such as IP-10 and
endostatin or IP-10 and TSPf are also possible.
The present invention also provides plasmids and related
products and methods with a tissue specific element and an
anti-angiogenic coding sequence, wherein the anti-angiogenic
coding sequence encodes a fusion or hybrid peptide or protein.
The fusion or hybrid agent, for example, may be a fusion
product of angiostatin and endostatin. Each portion of the
fusion product is preferably anti-angiogenic on its own, and
has elevated anti-angiogenic effects when presented as part of
a fusion or hybrid product.
The term "transcription unit" or "expression cassette"
refers to a nucleotide sequence which contains at least one
coding sequence along with sequence elements which direct the
initiation and termination of transcription. A transcription
unit may however include additional sequences, which may
include sequences involved in post-transcriptional or post-
translational processes. In preferred embodiments, the first
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
transcriptional control sequence or the second transcriptional
control sequence contain one or more cytomegalovirus promoter
sequences. The first and second transcriptional control
sequences can be the same or different.
5 A "5' untranslated region" or "5' UTR" refers to a
sequence located 3' to promoter region and 5' of the
downstream coding region. Thus, such a sequence, while
transcribed, is upstream of the translation initiation codon
and therefore is not translated into a portion of the
10 polypeptide product.
For the plasmids described herein, one or more of a
promoter, 5' untranslated region (5' UTR), the 3' UTR/poly(A)
signal, and introns may be a synthetic sequence. In this
context the term "synthetic" means that the sequence is not
provided directly by the sequence of a naturally occurring
genetic element of that type but rather is an artificially
created sequence (i.e., created by a person by molecular
biological methods). While one or more portions of such a
synthetic sequence may be the same as portions of naturally
occurring sequences, the full sequence over the specified
genetic element is different from a naturally occurring
genetic element of that type. The use of such synthetic
genetic elements allows the functional characteristics of that
element to be appropriately designed for the desired function.
Thus, a "synthetic intron" refers to a sequence which is
not a naturally occurring intron sequence but which will be
removed from an RNA transcript during normal post
transcriptional processing. Such introns can be designed to
have a variety of different characteristics, in particular
such introns can be designed to have a desired strength of
splice site.
In a preferred embodiment the first and second coding
regions are coding regions for angiostatin then endostatin in
the 5' to 3' direction.
A "sequence coding for angiostatin" is a nucleic acid
sequence which encodes the human angiostatin as described
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
11
above, based on the normal base pairing and translational
codon usage relationships. The sequence coding for endostatin
of similarly defined.
In a preferred embodiment the sequence coding for
angiostatin is 5' to the sequence coding for endostatin.
Those skilled in the art will appreciate that when more than
two anti-angiogenic coding sequences are utilized, the anti
angiogenic coding sequences may all be on a single
transcription unit, that all may be on separate transcription
units, or that any two coding sequences may be on one
transcription unit and the other coding sequence on another
transcription unit (in the case of three coding sequences).
The plasmid may also contain an intron having variable
splicing, a first coding sequence, and a second coding
sequence, wherein the first and second coding sequences
include a sequence coding for any two different anti-
angiogenic agents, preferably angiostatin and endostatin.
In a preferred embodiment, the plasmid also has: (a) a
transcriptional control sequence transcriptionally linked with
a first coding sequence and a second coding sequence; (b) a
5'-untranslated region; (c) an intron 5' to the first coding
sequence: (d) an alternative splice site 3' to the first
coding sequence and 5' to the second coding sequenced and (e)
a 3'-untranslated region/poly(A) signal. The transcriptional
control sequence preferably includes a cytomegalovirus
promoter sequence.
In a preferred embodiment, the plasmid also has: (a) a
transcriptional control sequence transcriptionally linked with
a first coding sequence, an IRES sequence, a second coding
sequence, and a 3'-untranslated region/poly(A) signal, wherein
the IRES sequence is between the first coding sequence and the
second coding sequence; and (b) an intron between the promoter
and the first coding sequence; wherein the first and second
coding sequences include a sequence coding fox any two
different anti-angiogenic agents, preferably angiostatin and
endostatin. The transcriptional control sequence preferably
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
12
includes a cytomegalovirus promoter sequence and the IRES
sequence preferably is from an encephalomyocarditis virus.
For delivery of coding sequences for gene expression, it
is generally useful to provide a delivery composition or
delivery system which includes one or more other components in
addition to the nucleic acid sequences. Such a composition
can, for example, aid in maintaining the integrity of the DNA
and/or in enhancing cellular uptake of the DNA and/or by
acting as an immunogenic enhancer, such as by the non-DNA
components having an immuno-stimulatory effect of their own.
Thus, in another aspect, the invention features a
composition containing a plasmid as described above and a
protective, interactive non-condensing compound (PINC).
The PINC enhances the delivery of the nucleic acid
molecule to mammalian cells in vivo, and preferably the
nucleic acid molecule includes a coding sequence for a gene
product to be expressed in the cell. In many cases, the
relevant gene product is a polypeptide or protein. Preferably
the PINC is used under conditions so that the PINC does not
form a gel, or so that no gel form is present at the time of
administration at about 30-90°C. Thus, in these compositions,
the PINC is present at a concentration of 30% (w/v) or less.
In certain preferred embodiments, the PINC concentration is
still less, for example, 20% or less, 10% or less, 5% or less,
or 1% or less. Thus, these compositions differ in compound
concentration and functional effect from uses of these or
similar compounds in which the compounds are used at higher
concentrations, for example in the ethylene glycol mediated
transfection of plant protoplasts, or the formation of gels
for drug or nucleic acid delivery. In general, the PINCs are
not in gel form in the conditions in which they are used as
PINCs, though certain of the compounds may form gels under
some conditions.
In connection with the compounds and compositions of this
invention, the term "protects" or "protective" refers to an
effect of the interaction between such a compound and a
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
13
nucleic acid such that the rate of degradation of the nucleic
acid is decreased in a particular environment. Such
degradation may be due to a variety of different factors,
which specifically include the enzymatic action of a nuclease.
The protective action may be provided in different ways, for
example, by exclusion of the nuclease molecules or by
exclusion of water.
Some compounds which protect a nucleic acid and/or
prolong the bioavailability of a nucleic acid may also
' 10 interact or associate with the nucleic acid by intermolecular
forces and/or valence bonds such as: Van der Waals forces,
ion-dipole interactions, ion-induced dipole interactions,
hydrogen bonds, or ionic bonds. These interactions may serve
the following functions: (1) Stereoselectively protect
nucleic acids from nucleases by shielding; (2) facilitate the
cellular uptake of nucleic acid by "piggyback endocytosis".
Piggyback endocytosis is the cellular uptake of a drug or
other molecule complexed to a carrier that may be taken up by
endocytosis. CV Uglea and C Dumitriu-Medvichi, Medical
Applications of Synthetic Oligomers, In: Polymeric
Biomaterials, Severian Dumitriu ed., Marcel Dekker, Inc.,
1993, incorporated herein by reference.
To achieve the desired effects set forth it is desirable,
but not necessary, that the compounds which protect the
nucleic acid and/or prolong the bioavailability of a nucleic
acid have amphiphilic properties; that is, have both
hydrophilic and hydrophobic regions. The hydrophilic region
of the compounds may associate with the largely ionic and
hydrophilic regions of the nucleic acid, while the hydrophobic
region of the compounds may act to retard diffusion of nucleic
acid and to protect nucleic acid from nucleases.
Additionally, the hydrophobic region may specifically
interact with cell membranes, possibly facilitating
endocytosis of the compound and thereby also of nucleic acid
associated with the compound. This process may increase the
pericellular concentration of nucleic acid.
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
14
Agents which may have amphiphilic properties and are
generally regarded as being pharmaceutically acceptable are
the following: polyvinylpyrrolidones; polyvinylalcohols;
polyvinylacetates; propylene glycol; polyethylene glycols;
poloxamers (Pluronics); poloxamines (Tetronics); ethylene
vinyl acetates; methylcelluloses, hydroxypropylcelluloses,
hydroxypropylmethylcelluloses; heteropolysaccharides
(pectins); chitosans; phosphatidylcholines (lecithins);
miglyols; polylactic acid; polyhydroxybutyric acid; xanthan
gum. Also, copolymer systems such as polyethylene glycol-
polylactic acid (PEG-PLA), polyethylene glycol-
polyhydroxybutyric acid (PEG-PHB), polyvinylpyrrolidone-
polyvinylalcohol (PVP-PVA), and derivatized copolymers such as
copolymers of N-vinyl purine (or pyrimidine) derivatives and
N-vinylpyrrolidone. However, not all of the above compounds
are protective, interactive, non-condensing compounds as
described below.
In connection with the protective, interactive, non
condensing compounds for these compositions, the term "non
condensing" means that an associated nucleic acid is not
condensed or collapsed by the interaction with the PINC at the
concentrations used in the compositions. Thus, the PINCs
differ in type and/or use concentration from such condensing
polymers. Examples of commonly used condensing polymers
include polylysine, and cascade polymers (spherical
polycations).
Also in connection with such compounds and an associated
nucleic acid molecule, the term "enhances the delivery" means
that at least in conditions such that the amounts of PINC and
nucleic acid is optimized, a greater biological effect is
obtained than with the delivery of nucleic acid in saline.
Thus, in cases where the expression of a gene product encoded
by the nucleic acid is desired, the level of expression
obtained with the PINC:nucleic acid composition is greater
than the expression obtained with the same quantity of nucleic
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
acid in saline for delivery by a method appropriate for the
particular PINC/coding sequence combination.
In preferred embodiments of the above compositions, the
PINC is polyvinyl pyrrolidone (PVP), polyvinyl alcohol (PVA),
5 a PVP-PVA co-polymer, N-methyl-2-pyrrolidone (NM2P), ethylene
glycol, or propylene glycol. In compositions in which a
Poloxamer (Pluronics) is used, the nucleic acid is preferably
not a viral vector, i.e., the nucleic acid is a non-viral
vector.
10 In other preferred embodiments, the PINC is bound with a
targeting ligand. Such targeting ligands can be of a variety
of different types, including but not limited to galactosyl,
residues, fucosal residues, mannosyl residues, carntitine
derivatives, monoclonal antibodies, polyclonal antibodies,
15 peptide ligands, and DNA-binding proteins. The targeting
ligands may bind with receptors on cells such as antigen-
presenting cells, hepatocytes, myocytes, epithelial cells,
endothelial cells, and cancer cells.
In connection with the association of a targeting ligand
and a PING, the term "bound with" means that the parts have an
interaction with each other such that the physical association
is thermodynamically favored, representing at least a local
minimum in the free energy function for that association.
Such interaction may involve covalent binding, or non-covalent
interactions such as ionic, hydrogen bonding, van der Waals
interactions, hydrophobic interactions, and combinations of
such interactions.
While the targeting ligand may be of various types, in
one embodiment the ligand is an antibody. Both monoclonal
antibodies and polyclonal antibodies may be utilized.
The nucleic acid may also be present in various forms.
Preferably the nucleic acid is not associated with a
compounds(s) which alter the physical form, however, in other
embodiments the nucleic acid is condensed (such as with a
condensing polymer), formulated with cationic lipids,
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
16
formulated with peptides, or formulated with cationic
polymers.
In preferred embodiments, the protective, interactive
non-condensing compound is polyvinyl pyrrolidone, and/or the
plasmid is in a solution having between 0.5g and 50~ PVP, more
preferably about 5$ PVP. The DNA preferably is at least about
80~ supercoiled, more preferably at least about 90~
supercoiled, and most preferably at least about 95~
supercoiled.
In another aspect the invention features a composition
containing a protective, interactive non-condensing compound
and a plasmid containing an anti-angiogenic coding sequence.
In yet another aspect, the invention provides a
composition containing a plasmid of the invention (or a
plasmid containing an anti-angiogenic coding sequence) and a
cationic lipid, preferably with a neutral co-lipid.
Preferably the cationic lipid is DOTMA and the neutral
co-lipid is cholesterol (chol). DOTMA is 1,2-di-O-
octadecenyl-3-trimethylammonium propane, which is described
and discussed in Eppstein et al., U.S. Patent 4,897,355,
issued January 30, 1990, which is incorporated herein by
reference. However, other lipids and lipid combinations may
be used in other embodiments. A variety of such lipids are
described in Gao & Huang, 1995, Gene Therapy 2:710-722, which
is hereby incorporated by reference. Other cationic lipid
delivery technology is described in Brigham, U.S. Patent
5,676,954, issued October 14, 1997, incorporated herein by
reference in its entirety, including any drawings.
As the charge ratio of the cationic lipid and the DNA is
also a significant factor, in preferred embodiments the DNA
and the cationic lipid are present is such amounts that the
negative to positive charge ratio is between 1:0.1 and 1:10,
preferably between 1:0.3 and 1:6, more preferably about 1:3.
While preferable, it is not necessary that the ratio be 1:3.
Thus, preferably the charge ratio for the compositions is
CA 02337496 2001-O1-26
WO 00/06759
17
PCT/US99/16388
between about 1:0.1 and 1:10, more preferably between about
1:0.3 and 1:6.
The term "cationic lipid" refers to a lipid which has a
net positive charge at physiological pH, and preferably
carries no negative charges at such pH. An example of such a
lipid is DOTMA. Similarly, "neutral co-lipid" refers to a
lipid which has is usually uncharged at physiological pH. An
example of such a lipid is cholesterol.
Thus, "negative to positive charge ratio" for the DNA and
cationic lipid refers to the ratio between the net negative
charges on the DNA compared to the net positive charges on the
cationic lipid.
As the form of the DNA affects the expression efficiency,
the DNA preferably is at least about 80$ supercoiled, more
preferably at least about 90a supercoiled, and most preferably
at least about 95o supercoiled. The composition preferably
includes an isotonic carbohydrate solution, such as an
isotonic carbohydrate solution that consists essentially of
about 10~ lactose. In preferred embodiments, the composition
the cationic lipid and the neutral co-lipid are prepared as a
liposome having an extrusion size of between 100 and 1,000
nanometers, preferably between 200 and 900 nanometers, more
preferably about 800 nanometers. Preferably the liposomes are
prepared to have an average diameter of between about 20 and
800 nm, more preferably between about 50 and 400 nm, still
more preferably between about 75 and 200 nm, and most
preferably about 100 nm. Microfluidization is the preferred
method of preparation of the liposomes.
In another aspect the invention features a composition
containing: (a) a first component having a plasmid including
an anti-angiogenic coding sequence and a cationic lipid,
preferably with a neutral co-lipid, wherein the cationic lipid
is DOTMA and the neutral co-lipid is cholesterol, wherein the
DNA in the plasmid and the cationic lipid are present in
amounts such that the negative to positive charge ratio is
between 1:0.1 and 1:10, preferably between 1:0.3 and 1:6, more
CA 02337496 2001-O1-26
WO 00/06759 PC'T/US99/16388
18 _ _ _.
preferably about 1:3; and (b) a second component including a
protective, interactive non-condensing compound, wherein the
first component is present within the second component.
In another aspect, the invention provides a composition
having a protective, interactive non-condensing compound, a
first plasmid including a first anti-angiogenic coding
sequence, preferably an angiostatin coding sequence and one or
more other plasmids independently having a second anti
angiogenic subunit coding sequence, preferably an endostatin
coding sequence.
In other aspects, the invention features a composition
comprising a protective, interactive non-condensing compound,
a first plasmid having an angiostatin coding sequence, and one
or more other plasmids having an endostatin coding sequence.
Also provided by the present invention is a composition
containing a plasmid with an anti-angiogenic coding sequence
and a cationic lipid, preferably with a neutral co-lipid.
In another aspect, the invention features a method for
making any of the plasmids described above by inserting a
tissue specific element and an anti-angiogenic coding sequence
into a plasmid.
The invention also provides methods of making the
compositions described above. The method may involve: (a)
preparing a DNA molecule having a transcriptional unit,
wherein the transcriptional unit contains an anti-angiogenic
coding sequence; (b) preparing a protective, interactive non-
condensing compound; and (c) combining the protective,
interactive non-condensing compound with the DNA in conditions
such that a composition capable of delivering a
therapeutically effective amount of an anti-angiogenic coding
sequence to a mammal is formed.
Preferably, the DNA molecule is a plasmid, wherein the
plasmid includes an anti-angiogenic coding sequence, and more
preferably also includes a human growth hormone 3'-
untranslated region/poly(A) signal.
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
19 __
The method may involve the steps of: (a) preparing a DNA
having an anti-angiogenic coding sequence; (b) preparing a
cationic lipid, preferably in a mixture with a neutral co-
lipid, wherein the cationic lipid is DOTMA and the neutral co-
y lipid is cholesterol; and (c) combining the cationic lipid
with the DNA in amounts such that the cationic lipid and the
DNA are present in a negative to positive charge ratio between
1:0.1 and 1:10, preferably between 1:0.3 and 1:6, more
preferably about 1:3.
In another embodiment, the method involves the steps of:
(a) preparing a first component having a plasmid containing an
anti-angiogenic coding sequence and a cationic lipid,
preferably with a neutral co-lipid, wherein the cationic lipid
is DOTMA and the neutral co-lipid is cholesterol, wherein the
DNA in the plasmid and the cationic lipid are present in
amounts such that the negative to positive charge ratio is
between 1:0.1 and 1:10, preferably between 1:0.3 and 1:6, more
preferably about 1:3; (b) preparing a second component having
a protective, interactive non-condensing compound; and (c)
combining the first and second components such that the
resulting composition includes the first component within the
second component.
In another embodiment, the method involves the steps of:
(a) preparing a protective, interactive non-condensing
compound, (b) preparing a first plasmid having a first anti
angiogenic coding sequence, preferably an angiostatin coding
sequence (c) preparing one or more other plasmids
independently having other anti-angiogenic coding sequence,
preferably an endostatin coding sequence and (d) combining the
protective, interactive non-condensing compound, the plasmid
having the first anti-angiogenic coding sequence and the other
plasmids.
The method of making a composition of the invention may
also involve combining a plasmid with an anti-angiogenic
coding sequence and a cationic lipid, and preferably also with
a neutral co-lipid.
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
In another aspect, the invention provides a method for
treatment of a mammalian condition or disease, by
administering to a mammal suffering from the condition or
disease a therapeutically effective amount of a plasmid as
5 described herein.
A "therapeutically effective amount" of a composition is
an amount which is sufficient to cause at least temporary
relief or improvement in a symptom or indication of a disease
or condition. Thus, the amount is also sufficient to cause a
10 pharmacological effect. The amount of the composition need
not cause permanent improvement or improvement of all symptoms
or indications. A therapeutically effective amount of a
cancer therapeutic would be one that reduces overall tumor
burden in the case of metastatic disease (i.e., the number of
15 metasteses or their size) or one that reduces the mass of the
tumor in localized cancers.
The condition or disease preferably is a cancer, such as
epithelial glandular cancer, including adenoma and
adenocarcinoma; squamous and transitional cancer, including
20 polyp, papilloma, squamous cell and transitional cell
carcinoma; connective tissue cancer, including tissue type
positive, sarcoma and other Coma's); hematopoietic and
lymphoreticular cancer, including lymphoma, leukemia and
Hodgkin's disease; neural tissue cancer, including neuroma,
sarcoma, neurofibroma and blastoma: mixed tissues of origin
cancer, including teratoma and teratocarcinoma. Other
cancerous conditions that are applicable to treatment include
cancer of any of the following: adrenal gland, anus, bile
duct, bladder, brain tumors: adult, breast, cancer of an
unknown primary site, carcinoids of the gastrointestinal
tract, cervix, childhood cancers, colon and rectum, esophagus,
gall bladder, head and neck, islet cell and other pancreatic
carcinomas, Kaposi's sarcoma, kidney, leukemia, liver, lung:
non-small cell, lung: small cell, lymphoma: AIDS-associated,
lymphoma: Hodgkin's disease, Lymphomas: non-Hodgkin's disease,
melanoma, mesothelioma, metastatic cancer, multiple myeloma,
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
21
ovary, ovarian germ cell tumors, pancreas, parathyroid, penis,
pituitary, prostate, sarcomas of bone and soft tissue, skin,
small intestine, stomach, testis, thymus, thyroid,
trophoblastic disease, uterus: endometrial carcinoma, uterus:
uterine sarcomas, vagina, or vulva. The composition
preferably is administered by injection, although the method
may also be performed ex vivo.
In another aspect, the invention provides a method for
transfection (i.e., the delivery and expression of a gene to
cells) of a cell in situ, by contacting the cell with a
plasmid described herein for sufficient time to transfect the
cell. Transfection of the cell preferably is performed in
vivo and the contacting preferably is performed in the
presence of about 5~ PVP solution.
In another aspect, the invention features a method for
delivery and expression of an anti-angiogenic gene in a
plurality of cells, by: (a) transfecting the plurality of
cells with a plasmid or composition of the invention: and (b)
incubating the plurality of cells under conditions allowing
expression of a nucleic acid sequence in the vector, wherein
the nucleic acid sequence encodes an anti-angiogenic agent.
In preferred embodiments, the anti-angiogenic agent is a
human anti-angiogenic agent and the cells are human cells
and/or the contacting is performed in the presence of an about
5~ PVP solution.
In another aspect, the invention features a method for
treating a disease or condition, by transfecting a cell in
situ with a plasmid or composition of the invention. By "in
situ" is meant at the cell's naturally occurring location,
which may be in vivo or in vitro depending upon the cell.
Thus, as sued herein, in situ transfection, for example, is a
term used primarily to distinguish from ex vivo transfection.
The disease or condition can be a localized disease or
condition (e.g., a solid tumor) or a systemic disease or
condition. (e. g., a metastatic cancer).
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
22
In another aspect, the invention features a cell
transfected with a plasmid or composition of the invention.
In yet another aspect, the invention features a method
for treatment of a mammalian condition or disease, by
administering to a mammal suffering from the condition or
disease a therapeutically effective amount of a composition
described herein.
A method for treatment of a mammalian condition or
disease is also featured and involves administering to a
mammal suffering from the condition or disease a
therapeutically effective amount of a composition of a first
plasmid with an angiostatin coding sequence and a second
plasmid with an endostatin coding sequence.
As the compositions are useful for delivery of a nucleic
acid molecule to cells in vivo, in a related aspect the
invention provides a composition at an in vivo site of
administration. In particular this includes at an in vivo
site in a mammal.
In preferred embodiments the nucleic acid molecule
includes a sequence encoding a gene product. Also in
preferred embodiments, the site of administration is in an
interstitial space or a tissue of an animal, particularly of a
mamma 1.
The invention also provides methods for using the above
compositions. Therefore, in further related aspects, methods
of administering the compositions are provided in which the
composition is introduced into a mammal, preferably into a
tissue or an interstitial space.
Various methods of delivery may be utilized, such as are
known in the art, but in preferred embodiments, the
composition is introduced into the tissue or interstitial
space by injection. The compositions may also be delivered to
a variety of different tissues, but in preferred embodiments
the tissue is muscle or tumor.
In another related aspect, the invention provides methods
for treating a mammalian condition or disease by administering
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
23
a therapeutically effective amount of a composition as
described above. In preferred embodiments, the disease or
condition is a cancer.
The summary of the invention described above is non
limiting and other features and advantages of the invention
will be apparent from the following detailed description of
the preferred embodiments, as well as from the claims.
Brief Description Of The Drawings
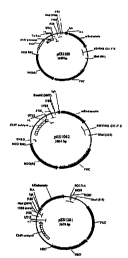
Figure 1 shows plasmid maps for pES1100, pES1062, and
pES1281.
Figure 2 shows plasmid maps for pAS1095 and pAS1096.
Figure 3 shows plasmid information for various
endothelial cell-specific constructs.
Figure 4 shows a plasmid map for pLC1264.
Figure 5 shows luciferase activity and endothelial cell
specificity of plasmids of the invention.
Figure 6 shows a procedure for multimerization of an
endothelial enhancer.
Figure 7 shows plasmid maps for pAS1359 and pES1358.
Figure 8 shows in vitro expression of bioactive
endostatin.
Figure 9 shows that endostatin/PVP inhibits Renca tumor.
Figure 10 shows that endostatin/PVP induces apoptosis of
EC.
Figure 11 shows endostatin a,nd angiostatin expresion in
serum after intramuscular delivery.
Figure 12 shows that endostatin/PVP inhibits sc Renca
tumor after im delivery.
Figure 13 shows that endostatin/PVP inhibits sc Renca
tumor after im delivery.
Figure 14 shows endostatin transgene mRNA in lung.
Figure 15 shows a mouse cornea angiogenesis assay.
Figure 16 shows a preferred codon usage table.
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
24
Detailed Descri tion Of The Preferred Embodiments
The plasmids, related products and methods of the
invention are described in detail below.
I. General
This invention concerns expression systems for the
delivery and expression of anti-angiogenic coding sequences in
mammalian cells, and formulations and methods for delivering
such expression systems or other expression systems to a
mamma 1.
Therefore, particular genetic constructs are described
which include nucleotide sequences coding for anti-angiogenic
agents, preferably human endostatin or angiostatin. Such a
construct can beneficially be formulated and administered' as
described herein, utilizing the expression systems of this
invention.
To allow convenient production of such plasmids, it is
generally preferable that the plasmid be capable of
replication in a cell to high copy number. Generally such
production is carried out in prokaryotic cells, particularly
including Esherichia coli (E.coli) cells. Thus, the plasmid
preferably contains a replication origin functional in a
prokaryotic cell, and preferably the replication origin is one
which will direct replication to a high copy number.
It is also possible to utilize synthetic genetic elements
in the plasmid constructs.
As described below, these elements affect post-
transcriptional processing in eukaryotic systems. Thus, the
use of synthetic sequences allows the design of processing
characteristics as desired for the particular application.
Commonly, the elements will be designed to provide rapid and
accurate processing.
For delivery of DNA encoding a desired expression product
to a mammalian system, it is usually preferable to utilize a
delivery system. Such a system can provide multiple benefits,
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
notably providing stabilization to protect the integrity of
the DNA, as well as assisting in cellular uptake.
In addition, the non-DNA components of the formulation
may contribute to an immune system enhancement or activation.
5 As a result, components of a delivery system can be selected
in conjunction with a particular gene product to enhance or
minimize the immuno-stimulatory effect.
The plasmids may also include elements for expression of
an anti-cancer or anti-tumor agent, such as cytokine, for
10 example IL-12 or one or more subunits thereof.
A "subunit" of a therapeutic molecule is a polypeptide or
RNA molecule which combines with one or more other molecules
to form a complex having the relevant pharmacologic activity.
Examples of such complexes include homodimers and heterodimers
15 as well as complexes having greater numbers of subunits. A
specific example of a heterodimer is IL-12, having the p40 and
p35 subunits.
Similarly, the treatment may involve administration of an
anti-angiogenic coding sequence and one or more cytokine or
20 other anti-cancer or anti-tumor coding sequences whether on a
single plasmid or on separate plasmids. Such plasmids may be
incorporated into compositions for delivery with a protective,
interactive non-condensing compound, a cationic lipid and
neutral co-lipid, or both.
25 While these are specific effective examples, other
components may be utilized in formulations containing the
anti-angiogenic expression vectors of the present invention to
provide effective delivery and expression of anti-angiogenic
agents or with other coding sequences for which manipulation
of the activation of immune system components is desirable.
The selection of delivery system components and
preparation methods in conjunction with the selection of
coding sequences provides the ability to balance the specific
effects of the encoded gene products and the immune system
effects of the overall delivery system composition. This
capacity to control the biological effects of delivery system
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
26
formulation administration represents an aspect of the
invention in addition to the anti-angiogenic agent encoding
constructs and specific formulations of delivery systems. Co-
selection of the encoded gene product and the delivery system
components and parameters provides enhanced desired effects
rather than merely providing high level gene expression. In
particular, such enhancement is described below for the
antitumor effects of the exemplary PVP containing
compositions.
For systems in which IL-12 is also administered, the
antitumor effect may be greater than merely additive (i.e.,
greater than merely the sum of the antitumor effects of the
anti-angiogenic agent alone and IL-12 alone). Enhancement of
immuno-stimulatory effects is also desirable in other
contexts, for example, for vaccine applications.
In contrast, for certain applications, it is preferable
to select a delivery systems which minimizes the immune system
effects. For example, it is often preferred that the immune
system activation be minimized for compositions to be
delivered to the lung in order to minimize lung tissue
swelling.
A useful approach for selecting the delivery system
components and preparation techniques for a particular coding
sequence can proceed as follows, but is not limited to these
steps or step order.
1. Select a particular genetic construct which provides
appropriate expression in vitro.
2. Select delivery system components based on desired
immunostimulatory effects and/or on in vivo physiological
effect. Such effects can be tested or verified in in vivo
model systems.
3. Select the other delivery system parameters
consistent with the desired immuno-stimulatory effects and/or
consistent with enhancing the desired in v.ivo physiological
effect. Such parameters can, for example, include the amount
and ratio of DNA to one or more other composition components,
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
27
the relative amounts of non-DNA composition components, the
size of delivery system formulation particles, the percent
supercoiled DNA for circular dsDNA vectors, and the specific
method of preparation of delivery system formulation
particles. The particular parameters relevant for specific
types of formulations will be apparent or readily determined
by testing.
The description below illustrates the selection of
components and parameters in the context of anti-angiogenic
agent encoding constructs. However, it should be recognized
that the approach is applicable to constructs containing a
variety of other coding sequences.
II. Plasmid Construct Expression Systems
A. Plasmid Design and Construction
For the methods and constructs of this invention, a
number of different plasmids were constructed which are useful
for delivery and expression of sequences encoding anti-
angiogenic agents. Thus, these plasmids contain coding
regions for anti-angiogenic agents, along with genetic
elements necessary or useful for expression of those coding
regions.
While these embodiments utilized cDNA clones or partial
genomic sequences from a particular source, those skilled in
the art could readily obtain anti-angiogenic coding sequences
from other sources, or obtain a coding sequence by identifying
a cDNA clone in a library using a probes) based on the
published anti-angiogenic coding and/or. flanking sequences.
Coding sequences for anti-angiogenic agents were
incorporated into an expression plasmid that contains
eukaryotic and bacterial genetic elements. Eukaryotic genetic
elements include the CMV immediate early promoter and 5' UTR,
and a human growth hormone 3' UTR/poly(a) signal, which
influence gene expression by controlling the accuracy and
efficiency of RNA processing, mRNA stability, and translation.
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
28
The human growth hormone 3' UTR is from a human growth
hormone gene, and preferably includes a poly(a) signal. This
sequence can be linked immediately following the natural
translation termination codon for a cDNA sequence, genomic
sequence, modified genomic sequence, or synthetic sequence
coding for anti-angiogenic agent.
An example of a human growth hormone 3' UTR/poly(a)
signal is shown by the human growth hormone 3' UTR
(nucleotides 1 - 100) and 3' flanking sequence (nucleotides
101 - 191; GenBank accession #J03071, HUMGHCSA) below.
1 GGGTGGCATCCCTGTGACCCCTCCCCAGTGCCTCTCCTGGCCCTGGAAGT
Poly (a)signal
51 TGCCACTCCAGTGCCCACCAGCCTTGTCCTAATAAAATTAAGTTGCATCA
101 TTTTGTCTGACTAGGTGTCCTTCTATAATATTATGGGGTGGAGGGGGGTG
151 GTATGGAGCAAGGGGCAAGTTGGGAAGACAACCTGTAGGGC
The 5' and 3' UTR and flanking regions can be further and
more precisely defined by routine methodology, e.g., deletion
or mutation analysis or their equivalents., and can be
modified to provide other sequences having appropriate
transcriptional and translational functions. Construction of
plasmid, plasmid backbone, human anti-angiogenic cDNA, and
final construct is described below in the examples.
Several EC-specific promoters, discussed below, have been
described in the literature.
The human Von Willebrand factor (vWF) gene flanking
region and the first exon, shown to support high-level and EC-
specific expression in vitro, expressed lacZ in only a sub-
population of EC in vivo and not in the vascular beds of
various organs examined (Aird et al., PNAS 92:4567-4571,
(1995)). It is composed of a non-specific core promoter (-90
to +22), a negative element that inhibits its activity in all
cell types (-478 to -300), and a positive element that
relieves repression only in EC and results in EC-specific
expression (+150 to +247). It has been shown that this
positive element is not present on the bovine vWF promoter.
CA 02337496 2001-O1-26
WO 00/06759 PCT/US9911b388
29
The preproendothelin (ET-1) gene promoter is a 119 base-
pair ("bp") fragment of human Endothelia gene promoter (-240
to -86) which directs EC-specific expression of CAT when fused
to minimal SV40 promoter. Murine ET-1 promoter directs
expression of either LUC or lipid-peroxidating enzyme in
transgenic mice (Harats et al., JCI 95:1335-44, 1995).
However, expression of the transgenes was not confined to
vascular EC, but also present in arteries smooth muscle, and
selected epithium. Moreover, the level of expression ranged
from high in arteries to low in veins and capillaries, and
there was significant variation in expression both between and
withing organs.
The StyI (-336 to +23) fragment of intracellular adhesion
molecule-2 (ICAM-2) gene promoter has been shown to direct
heterologous gene (CD59) expression to kidney and lung
vasculature in trangenic mice. It is TATA-less promoter and
contains Spl, GATA and ETS binding sites.
Alpha v beta 3 integrin is preferentially expressed in
tumor endothelium. In contrast to alpha v beta 3 integrin
(fibronectin receptor), the alpha v beta 3 integrin
(vitronectin receptor) cooperates with certain growth factors.
Inhibition of its expression blocks new vessel formation
during human would healing.
A 15.5 kb DNA fragment that contains the 5' flanking
region, the first exon, and part of the first intron of human
alpha v gene, was determined and named the Human Alpha v gene
promoter. The transcription initiation site was mapped 169 by
upstream of ATG site . The 5' flanking region does not contain
a TATA box or initiation element, but does contain four Spl,
two Ets and one GATA binding site. The 222 by region of alpha
v gene promoter has been shown to exert a strong positive
effect on alpha v promoter activity.
A 6 kb human genomic DNA fragment containing 2.0 kb of
the sequence 5' to the start codon is defined as the human
beta 3 gene promoter. The 584-by fragment 5' to the start
codon promotes expression of the CAT reporter gene by 5-fold
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
over promoter-less control CAT construct. This beta 3
promoter lacks TATA and CART cis-acting elements, but there
are two Spl sites flanking the transcription start site. It
has been shown that beta 3 promoter can be upregulated by PMA
5 and retinoic acid, but not by proinflammatory cytokine such as
TNF/ I FN-gamma .
Vascular endothelial growth factor receptor (VEGFR) and
its two EC-specific receptor tyrosine kinases, Flk-1/KDR and
Flt-1, play key roles in physiological and pathological
10 angiogenesis. The -3118 to +209 fragment of the mouse Flt gene
promoter and the -1829 to +148 fragment of mouse Flk-1 gene
promoter have been cloned. Hypoxia has been shown to be a
major mechanism for up-regulation of VEGF and its receptors in
vivo. In transient transfection assays, hypoxia led to strong
15 transcriptional activation of the Flt-1 promoter, whereas Flk-
1/KDR transcription was essentially unchanged. A 430-by
region of the Flt-1 promoter is required for transcription in
response to hypoxia and this region includes a hypoxia-
inducible factor (HIF) consensus sequence.
20 The Endothelia-1 enhancer, an endothelial cell-specific
regulatory region located between 320 and 364 by upstream of
the transcription initiation site of the mouse endothelia-1
gene, was identified by Bu and Quartermous (J. Biol. Chem.
272:32613-32622, (1997)). Three copies of this enhancer
25 sequence have been shown to activate both the ET-1 promoter
and heterologous promoters.
Gene expression driven by the cell-cycle-specific
promoters cyclin A, E2F1, or cdc6 is regulated in a cell-
cycle-dependent fashion and this regulation is primarily at
30 the transcriptional level. The promoters of these genes
contain common E2F sites which are responsible for repression
in the resting GO (zero) phase, and in some cases for
activation in cycling cells. (Henglein, B., X. Chenivesse, J.
Wang, D. Eick, and C. Brechot. 1994. The structure and cell
cycle-regulated transcription of the human cyclin A gene is
described in Proc. Natl. Acad. Sci. USA, 91:5490-5494.
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
31
Autoregulatory control of E2F1 expression in response to
positive and negative regulators of cell cycle progression is
described in Genes & Dev. 8:1514-1525; Williams, R. S., R. V.
Shohet, and B. Stillman 1997. A human protein related to
yeast Cdc6p expression is described in Proc. Natl. Acad. Sci.
USA 94:142-147; Yan, Z., J. DeGregori, R. Shohet, G. Leone, B.
Stillman, J. R. Nevins, and R. S. Williams, 1998. Cdc6 is
regulated by E2F and is essential for DNA replication in
mammalian cells .
B. Synthetic Genetic Elements
In some embodiments, some or all of the genetic elements
can be synthetic, derived from synthetic oligonucleotides, and
thus are not obtained directly from natural genetic sequences.
These synthetic elements are appropriate for use in many
different expression vectors.
A synthetic intron is designed with splice sites that
ensure that RNA splicing is accurate and efficient. A
synthetic 3' UTR/poly(A) signal is designed to facilitate mRNA
3' end formation and mRNA stability. A synthetic 5' UTR is
designed to facilitate the initiation of translation. The
design of exemplary synthetic elements is described in more
detail below.
Summary of Synthetic Element Features
Exemplary synthetic 5'UTR, intron, and 3'UTR/poly(A)
signal have the general features shown below:
5' UTR Short.
Lack of secondary structure.
Kozak sequence.
Site for intron insertion.
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/1b388
32
Intron 5' splice site sequence matches consensus.
5' splice site sequence is exactly
complementary to 5' end of U1 snRNA.
Branch point sequence matches consensus.
Branch point sequence is complementary to U2
snRNA.
3' splice site matches consensus.
Polypyrimidine tract is 16 bases in length and
contains 7 consecutive T's. (The tract
preferably contains at least 5 consecutive
T's. )
Contains internal restriction enzyme sites.
BbsI cleaves at the 5'ss, Earl cleaves at the
3'ss.
3' UTR/Poly(A) Based on rabbit j3-globin 3' UTR/poly(A) signal.
' Consists of two poly(A) signals in tandem.
Features of the Synthetic 5'UTR {UT6):
The 5' untranslated region (5'UTR) influences the
translational efficiency of messenger RNA, and is therefore an
important determinant of eukaryotic gene expression. The
synthetic 5'UTR sequence (UT6) has been designed to maximize
the.translational efficiency of mRNAs encoded by vectors that
express genes of therapeutic interest.
The sequence of the synthetic 5' UTR (UT6) is shown
below. The Kozak sequence is in boldface and the initiation
codon is double underlined. The location of the intron
(between residues 48 and 49) is indicated by the triangle and
the sequences that form the exonic portion of consensus splice
sites are single underlined. The restriction sites for
HindIII and NcoI are overlined.
HindIII O NcoI
AAGCTTACTCAACACAATAACAAACTTACTTACAATCTTAATTAACAGGCCACCATGG
The 5' untranslated region (5' UTR), located between the
cap site and initiation codon, is known to influence the
efficiency of mRNA translation. Any features that influence
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
33
the accessibility of the 5' cap structure to initiation
factors, the binding and subsequent migration of the 43S
preinitiation complex, or the recognition of the initiation
codon, will influence mRNA translatability. An efficient 5'
UTR is expected to be one that is moderate in length, devoid
of secondary structure, devoid of upstream initiation codons,
and has an AUG within an optimal local context (Kozak, 1994,
Biochimie 76:815-821; Jansen et al., 1994). A 5' UTR with
these characteristics should allow efficient recognition of
the 5' cap structure, followed by rapid and unimpeded ribosome
scanning by the ribosome, thereby facilitating the translation
initiation process.
The sequence of the synthetic 5'UTR was designed to be
moderate in length (54 nucleotides (nts}), to be deficient in
G but rich in C and A residues, to lack an upstream ATG, to
place the intended ATG within the context of a optimal Kozak
sequence (CCACCATGG), and to lack potential secondary
structure. The synthetic 5' UTR sequence was also designed to
lack AU-rich sequences that target mRNAs to be rapidly
degraded in the cytoplasm.
Experiments have demonstrated that introns increase gene
expression from cDNA vectors, and that introns located in the
5' UTR are more effective than ones located in the 3' UTR
(Huang and Gorman, 1990, Mol. Cell. Biol. 10:1805-1810; Evans
and Scarpulla, 1989, Gene 84:135-142; Brinster et al., 1988,
Proc. Natl. Acad. Sci. USA 85:836-840 Palmiter et al., 1991,
Proc. Natl. Acad. Sci. USA 88:978-482; Choi et al., 1991, Mol.
Cell. Biol. 11:3070-3074}. Accordingly, the synthetic 5' UTR
sequence was designed to accommodate an intron with consensus
splice site sequences. The intron may, for example, be
located between residues 48 and 49 (See intron sequence
structure below). The CAG at position 46-48 is the exonic
portion of a consensus 5' splice site. The G at position 49
is the exonic portion of a consensus 3' splice site.
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
34
To facilitate cloning manipulations, the synthetic 5' UTR
sequence was designed to begin with a HindIII site and
terminate with a NcoI site.
Features of the Synthetic Intron
RNA splicing is required for the expression of most
eukaryotic genes. For optimal gene expression, RNA splicing
must be highly efficient and accurate. A synthetic intron,
termed OPTIVSBB, was designed to be maximally efficient and
accurate.
The structure of the exemplary synthetic intron, OPTIVS8
is shown below. Sequences for the 5' splice site (5'ss),
branch point (bp), and 3' splice site (3'ss) are double
underlined. The recognition sequences for the restriction
enzymes BbsI and Earl are overlined. The cleavage site for
BbsI corresponds to the 5'ss, and the cleavage site for Earl
corresponds to the 3'ss.
5'ss by 3'ss
I BbsI I Earl i
5'CAG GTAAGTGTCTTC---(77)---TACTAACGGTTCTTTTTTTCTCTTCACAG G 3'
The 5' splice site (5'ss) sequence matches the
established consensus sequence, MAG ~. GTRAGT, where M = C or
A, and R = G or A. Since the mechanism of splicing involves
an interaction between the 5'ss of the pre-mRNA and U1 snRNA,
the 5'ss sequence of OPTIVS8B was chosen to be exactly
complementary to the 5' end of U1 snRNA.
5'ss 5' CAGGUAAGU 3'
IIIIIIIII
U1 RNA 3' GUCCAUUCA 5'
In mammals, the consensus sequence for branch points
(YNYTRAY, where Y = C or T, R = A or G, N = any base, and the
underlined A residue is the actual branch point) is very
ambiguous. Since the mechanism of splicing involves an
interaction between the branch point (bp) of the pre-mRNA and
U2 snRNA, the branch point sequence of OPTIVSBB was chosen to
maximize this interaction. (Note that the branch point itself
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
is bulged out). The chosen sequence also matches the branch
point sequence that is known to be obligatory for pre-mRNA
splicing in yeast. The branch point is typically located 18-
38 nucleotides (nts) upstream of the 3' splice site. In
5 OPTIVSBB, the branch point is located 24 nts upstream from the
3' splice site.
BP 5' UACUAAC 3'
IIII) I
U2 RNA 3' AUGAU G 5'
10 The sequence of the 3' splice site (3'ss) matches the
established consensus sequence, Y11NYAG .~ G, where Y = C or T,
and N - any base. In 3' splice sites, the polypyrimidine
tract (Y11) is. the major determinant of splice site strength.
For optimal splice site function in OPTIVSBB, the length of
15 the polypyrimidine tract was extended to 16 bases, and its
sequence was adjusted to contain 7 consecutive T residues.
This feature was included because optimal splicing requires
the presence of at least 5 consecutive T residues in the
polypyrimidine tract.
20 Splicing in vitro is generally optimal when introns are
>80 nts in length (Wieringa, et al., 1984; Ulfendahl et al.,
1985, Nucl. Acids Res. 13:6299-6315). Although many introns
rnay be thousands of bases in length, most naturally occurring
introns are 90-200 nt in length (Hawkins, 1988, Nuc.I. Acids
25 Res. 16:9893-9908). The length of the synthetic intron (118
nts) falls within this latter range.
OPTIVSBB was designed with three internal restriction
enzyme sites, BbsI, NheI, and Earl. These restriction sites
facilitate the screening and identification of genes that
30 contain the synthetic intron sequence. In addition, the BbsI
and Earl sites were placed so that their cleavage sites
exactly correspond to the 5'ss (BbsI) or 3'ss (Earl). The
sequence of the polypyrimidine tract was specifically designed
to accommodate the Earl restriction site. Inclusion of the
35 BbsI and Earl sites at these locations is useful because they
permit the intron to be precisely deleted from a gene. They
CA 02337496 2001-O1-26
WO 00/06759 PCT1US99/16388
36
also permit the generation of an "intron cassette" that can be
inserted at other locations within a gene.
The 77 bases between the BbsI site and the branch point
sequence are random in sequence, except for the inclusion of
the NheI restriction site.
Features of the Synthetic 3' UTR/poly(A) Signal:
The 3' ends of eukaryotic mRNAs are formed by the process
of polyadenylation. This process involves site specific site
RNA cleavage, followed by addition of a poly(A) tail. RNAs
that lack a poly(A) tail are highly unstable. Thus, the
efficiency of cleavage/polyadenylation is a major determinant
of mRNA levels, and thereby, of gene expression levels. 2XPA1
is a synthetic sequence, containing two efficient poly(A)
signals, that is designed to be maximally effective in
polyadenylation.
A poly(A) signal is required for the formation of the 3'
end of most eukaryotic mRNA. The signal directs two RNA
processing reactions: site-specific endonucleolytic cleavage
of the RNA transcript, and stepwise addition of adenylates
(approximately 250) to the newly generated 3' end to form the
poly(A) tail. A poly(A) signal has three parts:
hexanucleotide, cleavage site, and downstream element. The
hexanucleotide is typically AAUAAA and cleavage sites are most
frequently 3' to the dinucleotide CA (Sheets et al., 1987).
Downstream elements are required for optimal poly(A) signal
function and are located downstream of. the cleavage site. The
sequence requirement for downstream elements is not yet fully
established, but is generally viewed as UG- or U-rich
sequences (Wickens, 1990; Proudfoot, 1991, Cell 64:671-674;
Wahle, 1992, Bioessays 14:113-118; Chen and Nordstrom, 1992,
Nucl. Acids Res. 20:2565-2572).
Naturally occurring poly(A) signals are highly variable
in their effectiveness (Peterson, 1992). The effectiveness of
a particular poly(A) signal is mostly determined by the
quality of the downstream element. (Wahle, 1992). In
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
37
expression vectors designed to express genes of therapeutic
interest, it is important to have a poly(A) signal that is as
efficient as possible.
Poly(A) efficiency is important for gene expression,
because transcripts that fail to be cleaved and polyadenylated
are rapidly degraded in the nuclear compartment. In fact, the
efficiency of polyadenylation in living cells is difficult to
measure, since nonpolyadenylated RNAs are so unstable. In
addition to being required for mRNA stability, poly(A) tails
contribute to the translatability of mRNA, and may influence
other RNA processing reactions such as splicing or RNA
transport (Jackson and Standart,1990, Cell 62:15-24; Wahle,
1992).
Some eukaryotic genes have more than one poly(A) site,
implying that if the cleavage/polyadenylation reaction fails
to occur at the first site, it will occur at one of the later
sites. In COS cell transfection experiments, a gene with two
strong poly(A) sites yielded approximately two-fold more mRNA
than one with a single strong poly(A) site (Bordonaro, 1995).
These data suggest that a significant fraction of transcripts
remain unprocessed even with a single "efficient" poly(A)
signal. Thus, it may be preferable to include more than one
poly(A) site.
The sequence of the exemplary synthetic poly(A) signal is
shown below. The sequence is named 2XPA. The hexanucleotide
sequences and downstream element sequences are double
underlined, and the two poly(A) sites are labeled as pA#1 and
pA#2. Convenient restriction sites are overlined. The entire
2XPA unit may be transferred in cloning experiments as a XbaI
KpnI fragment. Deletion of the internal BspHI fragment
results in the formation of a 1XPA unit.
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
38 _ _ _
XbaI BspHI
TCTAGAGCATTTTTCCCTCTGCCAAAAATTATGGGGACATCATGAAGCCCCTTGAGCATCTGAC
G
pA#1
Hex - ( Downstream element
TCTGGCTAATAAAGGAAATTTATTTTCATTGCAATAGTGTGTTGGAATTTTTTGTGTCTCTCAC
T
BspHI
CGGTACTAGAGCATTTTTCCCTCTGCCAAAAATTATGGGGACATCATGAAGCCCCTTGAGCATC
T
pA#2
Hex I Downstream element
GACGTCTGGCTAATAAAGGAAATTTATTTTCATTGCAATAGTGTGTTGGAATTTTTTGTGTCTC
T
KpnI
CACTCGGTACC
The sequence of the synthetic poly(A) site shown above is
based on the sequence of the rabbit (3-globin poly(A) signal, a
signal that has been characterized in the literature as strong
(Gil and Proudfoot, 1987, Cell 49:399-406; Gil and Proudfoot,
1984, Nature 312:473-474). One of its key features is the
structure of its downstream element, which contains both UG-
and U-rich domains.
A double-stranded DNA sequence corresponding to the 1XPA
sequence was constructed from synthetic oligonucleotides. Two
copies of the 1XPA sequence were then joined to form the 2XPA
sequence. The sequences were joined in such as way as to have
a unique XbaI site at the 5' end of the first poly (A) signal
containing fragment, and a unique KpnI site at the 3' end of
the second poly(A) signal containing fragment.
C. Coding Sequences
The nucleotide sequences of several natural human anti
angiogenic coding sequences are known, and are provided below,
along with a synthetic sequence which also codes for an anti
angiogenic agent.
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
39 _ _ .
In some cases, instead of the natural sequence coding for
the anti-angiogenic agent of interest, it is advantageous to
utilize synthetic sequences which encode the anti-angiogenic
agent. Such synthetic sequences have alternate codon usage
from the natural sequence, and thus have dramatically
different nucleotide sequences from the natural sequence. In
particular, synthetic sequences can be used which have codon
usage at least partially optimized for expression in a human.
The natural sequences do not have such optimal codon usage.
Preferably, substantially all the codons are optimized.
Optimal codon usage in humans is indicated by codon usage
frequencies for highly expressed human genes, as shown in
Fig. 16. The codon usage chart is from the program
"Human High. cod" from the Wisconsin Sequence Analysis Package,
Version 8.1, Genetics Computer Group, Madison, WI. The codons
which are most frequently used in highly expressed human genes
are presumptively the optimal codons for expression in human
host cells, and thus form the basis for constructing a
synthetic coding sequence. An example of a synthetic anti-
angiogenic coding sequence is shown as the bottom sequence in
the table below.
However, rather than a sequence having fully optimized
codon usage, it may be desirable to utilize a sequence which
has optimized codon usage except in areas where the same amino
acid is too close together or abundant to make uniform codon
usage optimal.
In addition, other synthetic sequences can be used which
have substantial portions of the codon usage optimized, for
example, with at least 500, 70~, 80~ or 90~ optimized codons
as compared to a natural coding sequence. Other particular
synthetic sequences can be selected by reference to the codon
usage chart in Fig. 16. A sequence is selected by choosing a
codon for each of the amino acids of the polypeptide
sequences. DNA molecules corresponding to each of the
polypeptides can then by constructed by routine chemical
synthesis methods. For example, shorter oligonucleotides can
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
be synthesized, and then ligated in the appropriate
relationships to construct the full-length coding sequences.
Those skilled in the art will realize that various
nucleic acid sequences with optimized codon usage can be
5 constructed.
The preferred codon usage for human IP-10, endostatin and
angiostatin are set forth below.
Preferred codon sequence for human IP-10
1 aagcttacca tgaaccagac cgccatcctg atctgctgcc tgatcttcct gaccctgagc
10 61 ggcatccagg gcgtgcccct gagccgcacc gtgcgctgca cctgcatcag catcagcaac
121 cagcccgtga acccccgcag cctggagaag ctggagatca tccccgccag ccagttctgc
181 ccccgcgtgg agatcatcgc caccatgaag aagaagggcg agaagcgctg cctgaacccc
291 gagagcaagg ccatcaagaa cctgctgaag gccgtgagca aggagatgag caagcgcagc
301 ccgggcggag gtggcagcgg cggaggtggc agcggcggag gtggcagcgg atcctctaga
15 Preferred codon sequence for human Endostatin
1 CACAGCCACC GCGACTTCCA GCCCGTGCTG CACCTGGTGG CCCTGAACAG
51 CCCCCTGAGC GGCGGCATGC GCGGCATCCG CGGCGCCGAC TTCCAGTGCT
101 TCCAGCAGGC CCGCGCCGTG GGCCTGGCCG GCACCTTCCG CGCCTTCCTG
151 AGCAGCCGCC TGCAGGACCT GTACAGCATC GTGCGCCGCG CCGACCGCGC
20 201 CGCCGTGCCC ATCGTGAACC TGAAGGACGA GCTGCTGTTC CCCAGCTGGG
251 AGGCCCTGTT CAGCGGCAGC GAGGGCCCCC TGAAGCCCGG CGCCCGCATC
301 TTCAGCTTCG ACGGCAAGGA CGTGCTGCGC CACCCCACCT GGCCCCAGAA
351 GAGCGTGTGG CACGGCAGCG ACCCCAACGG CCGCCGCCTG ACCGAGAGCT
401 ACTGCGAGAC CTGGCGCACC GAGGCCCCCA GCGCCACCGG CCAGGCCAGC
25 451 AGCCTGCTGG GCGGCCGCCT GCTGGGCCAG AGCGCCGCCA GCTGCCACCA
501 CGCCTACATC GTGCTGTGCA TCGAGAACAG CTTCATGACC GCCAGCAAGT
551 GA
Preferred codon sequence for human Angiostatin
1 ATGGAGCACA AGGAGGTGGT GCTGCTGCTG CTGCTGTTCC TGAAGAGCGG
30 51 CCAGGGCGAG CCCCTGGACG ACTACGTGAA CACCCAGGGC GCCAGCCTGT
101 TCAGCGTGAC CAAGAAGCAG CTGGGCGCCG GCAGCATCGA GGAGTGCGCC
151 GCCAAGTGCG AGGAGGACGA GGAGTTCACC TGCCGCGCCT TCCAGTACCA
201 CAGCAAGGAG CAGCAGTGCG TGATCATGGC CGAGAACCGC AAGAGCAGCA
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
41 _ _.
251 TCATCATCCG CATGCGCGACGTGGTGCTGTTCGAGAAGAAGGTGTACCTG
301 AGCGAGTGCA AGACCGGCAACGGCAAGAACTACCGCGGCACCATGAGCAA
351 GACCAAGAAC GGCATCACCTGCCAGAAGTGGAGCAGCACCAGCCCCCACC
401 GCCCCCGCTT CAGCCCCGCCACCCACCCCAGCGAGGGCCTGGAGGAGAAC
451 TACTGCCGCA ACCCCGACAACGACCCCCAGGGCCCCTGGTGCTACACCAC
501 CGACCCCGAG AAGCGCTACGACTACTGCGACATCCTGGAGTGCGAGGAGG
551 AGTGCATGCA CTGCAGCGGCGAGAACTACGACGGCAAGATCAGCAAGACC
601 ATGAGCGGCC TGGAGTGCCAGGCCTGGGACAGCCAGAGCCCCCACGCCCA
651 CGGCTACATC CCCAGCAAGTTCCCCAACAAGAACCTGAAGAAGAACTACT
701 GCCGCAACCC CGACCGCGAGCTGCGCCCCTGGTGCTTCACCACCGACCCC
751 AACAAGCGCT GGGAGCTGTGCGACATCCCCCGCTGCACCACCCCCCCCCC
801 CAGCAGCGGC CCCACCTACCAGTGCCTGAAGGGCACCGGCGAGAACTACC
851 GCGGCAACGT GGCCGTGACCGTGAGCGGCCACACCTGCCAGCACTGGAGC
901 GCCCAGACCC CCCACACCCACAACCGCACCCCCGAGAACTTCCCCTGCAA
951 GAACCTGGAC GAGAACTACTGCCGCAACCCCGACGGCAAGCGCGCCCCCT
1001 GGTGCCACAC CACCAACAGCCAGGTGCGCTGGGAGTACTGCAAGATCCCC
1051 AGCTGCGACA GCAGCCCCGTGAGCACCGAGCAGCTGGCCCCCACCGCCCC
1101 CCCCGAGCTG ACCCCCGTGGTGCAGGACTGCTACCACGGCGACGGCCAGA
1151 GCTACCGCGG CACCAGCAGCACCACCACCACCGGCAAGAAGTGCCAGAGC
1201 TGGAGCAGCA TGACCCCCCACCGCCACCAGAAGACCCCCGAGAACTACCC
1251 CAACGCCGGC CTGACCATGAACTACTGCCGCAACCCCGACGCCGACAAGG
1301 GCCCCTGGTG CTTCACCACCGACCCCAGCGTGCGCTGGGAGTACTGCAAC
1351 CTGAAGAAGT GC
Examples anti-angiogenic
of various factors
which
can be
enco ded by the plasmids ention described below.
of the are
inv
Addi tional examples described in Mixson,European Patent
are
Publ ication 0819 75 8 A2, published nuary 21, 1998,
EP Ja
inco rporated in its
herein entirety,
by reference including
any drawings.
1. Endostatin,and
Angiostatin:
Endostatin is a me mber of he expanding
t angiogenensis
inhi bitor family teins. t is a
of pro I 20 KDa
C-terminal
frag ment (189 .a.) of ectively inhibits
a collagen
XVIII
and sel
endo thelial ration vitro and angiogenesis in
cell prolife in
vivo (0'Reilly, Cell 88, 277-285, 1997).
M.S. et
al.,
Angiostatin is an internal proteolytic fragment of mature
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
42
plasminogen (38 Kda and 362 a.a) (0'Reilly, M.S. et al., Cell
79, 315-328, 1994). It contains four triple loop disulfide-
linked structures, known as kringle domains. It has been
shown that three kringle domain form is more potent (Cao, Y.
et al., J. Biol. Chem., 271, 29461-7, 1996). E, coli
expressed rEndostatin or Angiostatin was injected at high
doses (10 mg/kg/d for 15-16 days) to achieve 97~ inhibition of
tumor growth efficacy in preclinical studies. It has been
claimed that Angiostatin has a half-life of 2 days in blood.
2. Tumor suppressor genes:
p53 and its induced anti-angiogenic protein
throbospondin-1 (TSP-1): p53 is mutated in half of human
tumors. Wild-type p53 regulates cell cycle by through p21 (a
inhibitor of cyclin-depedent) and pRB (another tumor
suppressor). Also, p53 induces synthesis of the anti-
angiogenic factor TSP-1, a large trimeric glycoprotein
composed of three identical 180 kd subunits linked by
disulfide bonds. TSPf, a fragment of TSP-1 encoded by 1013-
1650 of TSP-1 gene, has more potent anti-angiogenic activity.
Systemic intravenous administration of a-actin driven p53 gene
complexed to cationic liposomes has been found reduce growth
and metastases of a malignant human breast cancer in nude mice
(Lesoon-Wood et al., PNAS, 36:421, 1995). The primary target
is thought to be the vasculature system of the tumor. It has
been shown that p53 in combination with TSPf reduce tumors
more effectively than p53 alone by gene therapy in mice tumor
model. pRB, p21, p16 and other inhibitors of cell cycle-
dependent kinases are also shown to inhibit endothelial cell
proloiferation and thus block angiogenesis in vivo. Von
Hippel Lindau gene (VHL) gene mutation is the most
characterized in renal carcinoma. Mution of VHL leads to high
expression of VEGF and highly angiogenic. Delivery of wild-
type VHL gene would reduce expression of VEGF and block
angiogenesis.
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
43
3. Integrin av[33 blockage:
Integrin av~i3 are functionally associated on the surface
of angiogenic blood vessels. The RGD-containing peptide
ligand for integrin av~i3 can home to tumors when injected
intravenously into tumor-bearing mice. This ligand has been
recently used to target chemotherapeutic drug to tumor
vasculature for cancer treament in mice model (Arap, W et al.,
Science, 279:377, 1998). Disruption of interaction between
integrin av(33 and matrix metalloproteinase 2 (MMP-2) by a
truncated MMP-2 (PEX) was shown to block angiogenesis
4. Angiogenic factor receptor blockage:
Receptors for VEGF (flt and flk) and for bFGF (bFGF
receptors and bFGF binding proteins) regulate angiogenic
signaling of growth factors. Neutrolizing Antibody against the
25 angiogenic factors could block angiogenesis and tumor growth.
Alternatively, soluble receptor can compete with wild type
receptor in growth factor binding thus block angiogenesis.
5. Cytokines and chemokines:
Cytokine such as IL-12 and IFN-a and chemokine such as
IP-10 have been shown to be potent inhibitors of angiogenesis
in addition to their immunoregulatory effects.
6. Thrombosis factor (stimulator of blood
coagulation:
Tissue factor (TF) is the major initiating receptor for
the thrombogenic cascades. A truncated TF (tTF) has been
targeted to tumor vasculature by a bispecific antibody and
cause tumor infarction in mice (Huang, X. et al., Science,
275:547, 1997)
D. Formulations
Delivery and expression of nucleic acids in many
formulations is limited due to degradation of the nucleic
acids by components of organisms, such as nucleases. Thus,
protection of the nucleic acids when delivered in vivo can
greatly enhance the resulting expression, thereby enhancing a
desired pharmacological or therapeutic effect. It was found
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
44
that certain types of compounds which interact with a nucleic
acid (e. g., DNA) in solution but do not condense the nucleic
acid provide in vivo protection to the nucleic acid, and
correspondingly enhance the expression of an encoded gene
product.
The use of delivery systems designed to interact with
plasmids and protect plasmids from rapid extracellular
nuclease degradation has been described [Mumper, R.J., et al.,
1996, Pharm. Res. 13:701-709; Mumper, R.J., et al., 1997.
Submitted to Gene Therapy]. A characteristic of the PINC
systems is that they are non-condensing systems that allow the
plasmid to maintain flexibility and diffuse freely throughout
the muscle while being protected from nuclease degradation.
While the PINC systems are primarily discussed below, it will
be understood that cationic lipid based systems and systems
utilizing both PINCS and cationic lipids are also within the
scope of the present invention.
A common structural component of the PINC systems is that
they are amphiphilic molecules, having both a hydrophilic and
a hydrophobic portion. The hydrophilic portion of the PINC is
meant to interact with plasmids by hydrogen bonding (via
hydrogen bond acceptor or donor groups), Van der Waals
interactions, or/and by ionic interactions. For example, PVP
and N-methyl-2-pyrrolidone (NM2P) are hydrogen bond acceptors
while PVA and PG are hydrogen bond donors.
All four molecules have been reported to form complexes
with various (poly)anionic molecules [Buhler V., BASF
Aktiengescellschaft Feinchemie, Ludwigshafen, pp 39-42: Galaev
Y, et al., J. Chrom. A. 684:45-54 (1994); Tarantino R, et al.
J. Pharm. Sci. 83:1213-1216 (1994); Zia, H., et al., Pharm.
Res. 8:502-504 (1991);]. The hydrophobic portion of the PINC
systems is designed to result in a coating on the plasmid
rendering its surface more hydrophobic. Kabanov et al. have
described previously the use of cationic polyvinyl derivatives
for plasmid condensation designed to increase plasmid
hydrophobicity, protect plasmid from nuclease degradation,
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
and increase its affinity for biological membranes [Kabanov,
A.V., and Kabanov, V.A., 1995, Bioconj. Chem. 6:7-20; Kabanov,
A.V., et al., 1991, Biopolymers 31:1437-1443 Yaroslavov,
A.A., et al., 1996, FEBS Letters 384:177-180].
5 Substantial protective effect is observed; up to at least
a one log enhancement of gene expression in rat muscle over
plasmid formulated in saline has been demonstrated with these
exemplary PINC systems. The expression of reporter genes in
muscle using plasmids complexed with the PINC systems was more
10 reproducible than when the plasmid was formulated in saline.
For example, the coefficient of variation for reporter gene
expression in muscle using plasmid formulated in saline was 96
+ 35°s (n - 20 studies; 8-12 muscles/study) whereas with
coefficient of variation with plasmids complexed with PINC
15 systems was 40 + 19~ (n - 30 studies; 8-12 muscles/study).
The high coefficient of variation for reporter gene expression
with plasmid formulated in saline has been described
previously [Davis, H.L., et al., 1993, Hum. Gene Ther. 4:151-
9]. In addition, in contrast with the results for DNA: saline,
20 there was no significant difference in gene expression in
muscle when plasmid with different topologies were complexed
with polyvinyl pyrrolidone (PVP). This suggests that PVP is
able to protect all forms of the plasmid from rapid nuclease
degradation.
25 1. Summary of interactions between a PINC polymer
(PVP) and plasmid
Using molecular modeling, it has been demonstrated that
an exemplary PINC polymer, PVP, forms hydrogen bonds with the
base pairs of a plasmid within its major groove and results in
30 a hydrophobic surface on the plasmid due to the vinyl backbone
of PVP. These interactions are supported by the modulation of
plasmid zeta potential by PVP as well as by the inhibition of
ethidium bromide intercalation into complexed plasmid.
Apparent binding between PVP and plasmid has been correlated
35 to pH and salt concentration and have demonstrated the effect
of these parameters on (3-gal expression after intramuscular
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
46
injection of plasmid/PVP complexes [Mumper, R.J., et al.,
1997. Submitted to Gene Therapy] . A summary of the physico-
chemical properties of plasmid/PVP complexes is listed in
Table I below.
Table I: Summary of the Physico-Chemical Properties of
Plasmid/PVP Complexes
Method Result
Molecular modeling Hydrogen bonding and hydrophobic plasmid surface
observed
1 0 Fourier-transformed Infra-red Hydrogen bonding demonstrated
DNase I challenge Decreased rate of plasmid degradation in the
presence of PVP
Microtitration Calorimetry Positive heats of reaction indicative of an
endothermic process
1 5 Potentiometric titration One unit pH drop when plasmid and PVP are
complexed
Dynamic Dialysis Rate of diffusion of PVP reduced in the presence
of plasmid
Zeta potential modulation Surface charge of plasmid decreased by PVP
2 0 Ethidium bromide Intercalation Ethidium bromide intercalation reduced by
plasmid/PVP complexation
Osmotic pressure Hyper-osmotic formulation (i.e., 340 mOsm/kg
H20)
Luminescence Spectroscopy Plasmid/PVP binding decreased in salt and/or at
2 5 pH 7
2. Histology of expression in muscle
Immunohistochemistry for (3-gal using a slide scanning
technology has revealed the uniform distribution of (3-gal
expression sites across the whole cross-sections of rat
30 tibialis muscles. Very localized areas were stained positive
for (3-gal when CMV-~3-gal plasmid was formulated in saline.
gal positive cells were observed exclusively around the needle
tract when plasmid was injected in saline. This is in
agreement with previously published results [Wolff, J.A., et
35 al., 1990, Science 247:1465-68; Davis, H.L., et al., 1993,
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
47
Hum. Gene Ther. 4:151-9~ Davis, H.L., et al., 1993, Hum.
Gene Ther. 4:733-40].
In comparison, immunoreactivity for ~i-gal was observed in
a wide area of muscle tissue after intramuscular injection of
CMV-(3-gal plasmid/PVP complex (1:17 w/w) in 150 mM NaCl. It
appeared that the majority of positive muscle fibers were
located at the edge of muscle bundles. Thus, staining for (3-
gal in rat muscle demonstrated that, using a plasmid/PVP
complex, the number of muscle fibers stained positive for (3-
gal was approximately 8-fold greater than found using a saline
formulation. Positively stained muscle fibers were also
observed over a much larger area in the muscle tissue using
the plasmid/PVP complex providing evidence that the injected
plasmid was widely dispersed after intramuscular injection.
The enhanced plasmid distribution and expression in rat
skeletal muscle was a result of both protection from
extracellular nuclease degradation due to complexation and
hyper-osmotic effects of the plasmid/PVP complex. However,
Dowty and Wolff et al. have demonstrated that osmolarity, up
to twice physiologic osmolarity, did not significantly effect
gene expression in muscle [Dowty, M.E., and Wolff, J.A. In:
J.A. Wolff (Ed.), 1994, Gene Therapeutics: Methods and
Applications of Direct Gene Transfer. Birkhauser, Boston, pp.
82-98]. This suggests that the enhanced expression of plasmid
due to PVP complexation is most likely due to nuclease
protection and less to osmotic effects. Further, the surface
modification of plasmids by PVP (e. g., increased
hydrophobicity and decreased negative surface charge) may also
facilitate the uptake of plasmids by muscle cells.
3. Structure-activity relationship of PINC
polymers
There is a linear relationship between the structure of a
series of co-polymers of vinyl pyrrolidone and vinyl acetate
and the levels of gene expression in rat muscle. The
substitution of some vinyl pyrrolidone monomers with vinyl
acetate monomers in PVP resulted in a co-polymer with reduced
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
48
ability to form hydrogen bonds with plasmids. The reduced
interaction subsequently led to decreased levels of gene
expression in rat muscle after intramuscular injection. The
expression of ~i-gal decreased linearly (R - 0.97) as the
extent of vinyl pyrrolidone monomer (VPM) content in the co-
polymers decreased.
These data demonstrate that pH and viscosity are not the
most important parameters effecting delivery of plasmid to
muscle cells since these values were equivalent for all
complexes. These data suggest that enhanced binding of the
PINC polymers to plasmid results in increased protection and
bioavailability of plasmid in muscle.
4. Additional PINC systems
The structure-activity relationship described above can
be used to design novel co-polymers that will also have
enhanced interaction with plasmids. It is expected that there
is "an interactive window of opportunity" whereby enhanced
binding affinity of the PINC systems will result in a further
enhancement of gene expression after their intramuscular
injection due to more extensive protection of plasmids from
nuclease degradation. It is expected that there will be an
optimal interaction beyond which either condensation of
plasmids will occur or "triplex" type formation, either of
which can result in decreased bioavailability in muscle and
consequently reduced gene expression.
As indicated above, the PINC compounds are generally
amphiphilic compounds having both a hydrophobic portion and a
hydrophilic portion. In many cases the hydrophilic portion is
provided by a polar group. It is recognized in the art that
such polar groups can be provided by groups such as, but not
limited to, pyrrolidone, alcohol, acetate, amine or
heterocyclic groups such as those shown on pp. 2-73 and 2-74
of CRC Handbook of Chemistry and Physics (72nd Edition), David
R. Lide, editor, including pyrroles, pyrazoles, imidazoles,
triazoles, dithiols, oxazoles, (iso)thiazoles, oxadiazoles,
oxatriazoles, diaoxazoles, oxathioles, pyrones, dioxins,
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
49
pyridines, pyridazines, pyrimidines, pyrazines, piperazines,
(iso)oxazines, indoles, indazoles, carpazoles, and purines and
derivatives of these groups, hereby incorporated by reference.
Compounds also contain hydrophobic groups which, in the
case of a polymer, are typically contained in the backbone of
the molecule, but which may also be part of a non-polymeric
molecule. Examples of such hydrophobic backbone groups
include, but are not limited to, vinyls, ethyls, acrylates,
acrylamides, esters, celluloses, amides, hydrides, ethers,
carbonates, phosphazenes, sulfones, propylenes, and
derivatives of these groups. The polarity characteristics of
various groups are quite well known to those skilled in the
art as illustrated, for example, by discussions of polarity in
any introductory organic chemistry textbook.
The ability of such molecules to interact with nucleic
acids is also understood by those skilled in the art, and can
be predicted by the use of computer programs which model such
intermolecular interactions. Alternatively or in addition to
such modeling, effective compounds can readily be identified
using one or more of such tests as 1) determination of
inhibition of the rate of nuclease digestion, 2) alteration of
the zeta potential of the DNA, which indicates coating of DNA,
3) or inhibition of the ability of intercalating agents, such
as ethidium bromide to intercalate with DNA.
5. Targeting Ligands
In addition to the nucleic acid/PINC complexes described
above for delivery and expression of nucleic acid sequences,
in particular embodiments it is also useful to provide a
targeting ligand in order to preferentially obtain expression
in particular tissues, cells, or cellular regions or
compartments.
Such a targeted PINC complex includes a PINC system
(monomeric or polymeric PINC compound) complexed to plasmid
(or other nucleic acid molecule). The PINC system is
covalently or non-covalently attached to (bound to) a
targeting ligand (TL) which binds to receptors having an
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
50 -- -
affinity for the ligand. Such receptors may be on the surface
or within compartments of a cell. Such targeting provides
enhanced uptake or intracellular trafficking of the nucleic
acid.
The targeting ligand may include, but is not limited to,
galactosyl residues, fucosal residues, mannosyl residues,
carnitine derivatives, monoclonal antibodies, polyclonal
antibodies, peptide ligands, and DNA-binding proteins.
Examples of cells which may usefully be targeted include, but
are not limited to, antigen-presenting cells, hepatocytes,
myocytes, epithelial cells, endothelial cells, and cancer
cells.
Formation of such a targeted complex is illustrated by
the following example of covalently attached targeting ligand~
(TL) to PINC system:
TL-PINC + Plasmid ----------> TL-PINC::...:Plasmid
Formation of such a targeted complex is also illustrated
by the following example of non-covalently attached targeting
ligand (TL) to PINC system
TL::...:PINC + Plasmid --------> TL::...:PINC::...:Plasmid
or alternatively,
PINC + Plasmid ------------> PINC:.....:Plasmid + TL -------
---> TL::...:PINC::....:Plasmid
In these examples ........ is non-covalent interaction
such as ionic, hydrogen-bonding, Van der Waals interaction,
hydrophobic interaction, or combinations of such interactions.
A targeting method for cytotoxic agents is described in
Subramanian et al., International Application No.
PCT/US96/08852, International Publication No. WO 96/39124,
hereby incorporated by reference in its entirety, including
any drawings. This application describes the use of polymer
affinity systems for targeting cytotoxic materials using a
two-step targeting method involving zip polymers, i.e., pairs
of interacting polymers. An antibody attached to one of the
interacting polymers binds to a cellular target. That polymer
then acts as a target for a second polymer attached to a
CA 02337496 2001-O1-26
WO 00/06759 PGT/US99/16388
51
cytotoxic agent. As referenced in Subramanian et al., other
two-step (or multi-step) systems for delivery of toxic agents
are also described.
In another aspect, nucleic acid coding sequences can be
delivered and expressed using a two-step targeting approach
involving a non-natural target for a PINC system or PINC
targeting ligand complex. Thus, for example, a PINC-plasmid
complex can target a binding pair member which is itself
attached to a ligand which binds to a cellular target (e.g., a
MAB). Binding pairs for certain of the compounds identified
herein as PINC compounds as identified in Subramanian et al.
Alternatively, the PINC can be complexed to a tareting ligand,
such as an antibody. That antibody can be targeted to a non-
natural target which binds to, for example, a second antibody.
III. Model Systems for Evaluation of Anti-Angiogenic
Constructs and Formulations
In accord with the concept of using anti-angiogenic
expressing plasmid constructs and formulations in anti-cancer
treatment, murine model systems were utilized based on murine
tumor cell lines. The line primarily used was S.C. VII/SF,
which is a cell line derived from murine squamous cell
carcinoma (S.C.).
Squamous cell carcinoma of the head and neck begins with
the cells lining the oral and pharyngeal cavities. Clinical
disease progresses via infiltration and spreads into the
underlying tissues and lymphatics. The undifferentiated, in
vivo passage tumor line S.C. VII/SF displays this typical
growth pattern. In addition, its rapid growth rate provides a
relatively short test period for individual experiments.
Other murine tumor cell lines include another SCC line KLN-
205, a keratinocyte line I-7, and a colon adenocarcinoma line
MC-38.
An optimal model system preferably satisfies the criteria
based on having tumor growth rate in vivo (i.e., tumors are
ready for treatment in 4-10 days post implant), invasiveness,
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
52
and local spread similar to those observed in clinical
disease, and providing accessibility for experimental
treatment. As indicated, the SCC VII/SF cell line was
utilized as the primary model system cell line. This cell
line typically grows rapidly, resulting in death of untreated
syngeneic mice 14-17 days after tumor cell implantation.
This cell line can be utilized in a variety of ways to
provide model system suitable for a variety of different
tests. Four such possibilities are described below.
First, SCCVII cells can be utilized in cell culture to
provide an in vitro evaluation of anti-angiogenic agent
expression construct and formulation characteristics, such as
expression levels and cellular toxicities.
Second, the cells can be implanted subcutaneously in
mice. This system can be utilized in tests in which
accessibility of the implant site is beneficial. As an
example, the method was utilized in evaluations of expression
efficiencies based on the expression of chloramphenicol
acetyltransferase (CAT).
Third, the cells can be implanted transcutaneously into
the fascia of digastric muscle.
Fourth, the cells can be implanted transcutaneously into
digrastric/mylohyoid muscles. The important features of
models 3 and 4 are shown in the table below.
CA 02337496 2001-O1-26
WO 00/06759 PC'f/US99/16388
53 -- -
TAH?~E II
f'~r"nr,nri ann of submandibular tumor models
_-_r __ -
Feature Mouse Tumor Model 3 Mouse Tumor Model 4
Tumor implant 2-4 x 105 cells 5 x 105 transcutaneously
procedure transcutaneously into into digastric/mylohyoid
fascia
of digastric muscle muscles
Tumor growth Prominent submandibularMore variable, invasion
and of
invasiveness bulge: invasion of digastric/mylohyoid
characteristics digastric/mylohyoid muscles and lymphatics
muscles
and lymphatics
Treatment Transcutaneous, needle Lower jaw skin flap
raised
procedure (primaryinserted and moved withinto expose tumor, needle
treatment) tumor to produce a 4 inserted and moved
within
quadrant distribution tumor to produce a
of 4
gene medicine quadrant distribution
of
gene medicine
Days treated Day 5, day 10 (both Day 5 (tumor exposed),
day
1 5 (post-implant) transcutaneously) 8 (transcutaneously)
Measurement External calipering First caliper when
2-3 x tumor
procedure per week until death exposed for treatment,
second caliper at
sacrifice
Advantages Non-surgical, closed Surgical, open model
model
allows larger experimentsallows direct treatment
of
2 O and more frequent exposed tumor: Local
treatments: Sacrifice inflammation from surgery
unnecessary to caliper may additionally stimulate
(=more time points) immune response: More
like
clinical situation
for
protocol development
Disadvantages Transcutaneous treatmentLabor intensive; Smaller,
is
2 5 potentially less accuratefewer experiments
and intensive; less possible: Tumors deeper
like
expected clinical treatmentsand more difficult
to
than surgical approachestreat transcutaneously
(for secondary
treatments): Fewer
treatments and caliperings
possible
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
54 --
The tumor size treated in the mouse models is generally
20-50 mm3. A 50 mm3 mouse tumor is approximately equivalent
to 150 cc3 human tumor having an average diameter of about 6.6
cm. This tumor size is approximately 10-fold larger than the
size proposed to be treated in the phase I clinical trials.
This indicates that the mouse models are strongly biased
towards over estimating the expected tumor burden in human
patients.
IV. Formulations for In Vivo Delivery
A. General
While expression systems such as those described above
provide the potential for expression when delivered to an
appropriate location, it is beneficial to provide the
expression system constructs) in a delivery system which can
assist both the delivery and the cellular uptake of the
construct. Thus, this invention also provides particular
formulations which include one or more expression system
constructs (e.g., DNA plasmids as described above), and a
protective, interactive non-condensing compound.
An additional significant factor relating to the plasmid
construct is the percentage of plasmids which are in a
supercoiled (SC) form rather than the open circular (OC) form.
B. Delivery and Expression
A variety of delivery methods can be used with the
constructs and formulations described above, in particular,
delivery by injection to the site of a tumor can be used. The
submandibular tumor models utilized injection into four
quadrants of the tumor being treated.
C. Anti Cancer Efficacy of Human Anti-Anqiogenic
Formulations
The effects of the administration of the anti-angiogenic
formulations described above were evaluated using the S.C. VII
mouse tumor models. Plasmid constructs as described above
were incorporated in delivery formulations. The formulations
were delivered by injection. The effects of the expression of
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
55 --
the human anti-angiogenic plasmids in tumor cells on the
progress of the mouse tumors demonstrates that such anti-
angiogenic expression is effective against such tumors.
D. Toxicity Evaluation of Exemplary Formulations
The exemplary formulations do not show high cellular
toxicity at the concentrations tested, suggesting that the
formulations do not significantly kill cells by direct toxic
action in vivo.
V. Administration
Administration as used herein refers to the route of
introduction of a plasmid or carrier of DNA into the body. In
addition to the methods of delivery described above, the
expression systems constructs and the delivery system
formulations can be administered by a variety of different
methods.
Administration can be directly to a target tissue or by
targeted delivery to the target tissue after systemic
administration. In particular, the present invention can be
used for treating disease by administration of the expression
system or formulation to the body in order to establishing
controlled expression of any specific nucleic acid sequence
within tissues at certain levels that are useful for gene
therapy.
The preferred means for administration of vector
(plasmid) and use of formulations for delivery are described
above. The preferred embodiments are by direct injection
using needle injection.
The route of administration of any selected vector
construct will depend on the particular use for the expression
vectors. In general, a specific formulation for each vector
construct used will focus on vector uptake with regard to the
particular targeted tissue, followed by demonstration of
efficacy. Uptake studies will include uptake assays to
evaluate cellular uptake of the vectors and expression of the
DNA of choice. Such assays will also determine the
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
56
localization of the target DNA after uptake, and establishing
the requirements for maintenance of steady-state
concentrations of expressed protein. Efficacy and
cytotoxicity can then be tested. Toxicity will not only
include cell viability but also cell function.
Muscle cells have the unique ability to take up DNA from
the extracellular space after simple injection of DNA
particles as a solution, suspension, or colloid into the
muscle. Expression of DNA by this method can be sustained for
several months.
Delivery of formulated DNA vectors involves incorporating
DNA into macromolecular complexes that undergo endocytosis by
the target cell. Such complexes may include lipids, proteins,
carbohydrates, synthetic organic compounds, or inorganic
compounds. Preferably, the complex includes DNA, a cationic
lipid, and a neutral lipid in particular proportions. The
characteristics of the complex formed with the vector (size,
charge, surface characteristics, composition) determines the
bioavailability of the vector within the body. Other elements
of the formulation function as ligand which interact with
specific receptors on the surface or interior of the cell.
Other elements of the formulation function to enhance entry
into the cell, release from the endosome, and entry into the
nucleus.
Delivery can also be through use of DNA transporters.
DNA transporters refers to molecules which bind to DNA vectors
and are capable of being taken up by epidermal cells. DNA
transporters contain a molecular complex capable of non-
covalently binding to DNA and efficiently transporting the DNA
through the cell membrane. It is preferable that the
transporter also transport the DNA through the nuclear
membrane. See, e.g., the following applications all of which
(including drawings) are hereby incorporated by reference
herein: (1) Woo et al., U.S. Serial No. 07/855,389, entitled
"A DNA Transporter System and Method of Use", filed March 20,
1992, now abandoned: (2) Woo et al., PCT/US93/02725,
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
57 _ _ _
International Publ. W093/18759, entitled "A DNA Transporter
System and Method of Use", (designating the U.S. and other
countries) filed March 19, 1993; (3) continuation-in-part
application by Woo et al., entitled "Nucleic Acid Transporter
Systems and Methods of Use", filed December 14, 1993, U.S.
Serial No. 08/167,641; (4) Szoka et al., U.S. Serial No.
07/913,669, entitled "Self-Assembling Polynucleotide Delivery
System", filed July 14, 1992 and (5) Szoka et al.,
PCT/US93/03406, International Publ. W093/19768 entitled "Self-
Assembling Polynucleotide Delivery System", (designating the
U.S. and other countries) filed April 5, 1993.
A DNA transporter system can consist of particles
containing several elements that are independently and non-
covalently bound to DNA. Each element consists of a ligand
which recognizes specific receptors or other functional groups
such as a protein complexed with a cationic group that binds
to DNA. Examples of cations which may be used are spermine,
spermine derivatives, histone, cationic peptides and/or
polylysine. One element is capable of binding both to the DNA
vector and to a cell surface receptor on the target cell.
Examples of such elements are organic compounds which interact
with the asialoglycoprotein receptor, the folate receptor, the
mannose-6-phosphate receptor, or the carnitine receptor. A
second element is capable of binding both to the DNA vector
and to a receptor on the nuclear membrane. The nuclear ligand
is capable of recognizing and transporting a transporter
system through a nuclear membrane. An example of such ligand
is the nuclear targeting sequence from SV40 large T antigen or
histone. A third element is capable of binding to both the DNA
vector and to elements which induce episomal lysis. Examples
include inactivated virus particles such as adenovirus,
peptides related to influenza virus hemagglutinin, or the GALA
peptide described in the Szoka patent cited above.
Transfer of genes directly into a tumor has been very
effective. Experiments show that administration by direct
injection of DNA into tumor cells results in expression of the
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
58 _ _
gene in the area of injection. The injected DNA preferably
persists in an unintegrated extrachromosomal state. This
means of transfer is a preferred embodiment.
Administration may also involve lipids as described in
preferred embodiments above. The lipids may form liposomes
which are hollow spherical vesicles composed of lipids
arranged in unilamellar, bilamellar, or multilamellar fashion
and an internal aqueous space for entrapping water soluble
compounds, such as DNA, ranging in size from 0.05 to several
microns in diameter. Lipids may be useful without forming
liposomes. Specific examples include the use of cationic
lipids and complexes containing DOPE which interact with DNA
and with the membrane of the target cell to facilitate entry
of DNA into the cell.
Gene delivery can also be performed by transplanting
genetically engineered cells. For example, immature muscle
cells called myoblasts may be used to carry genes into the
muscle fibers. Myoblast genetically engineered to express
recombinant human growth hormone can secrete the growth
hormone into the animal's blood. Secretion of the incorpor-
ated gene can be sustained over periods up to 3 months.
Myoblasts eventually differentiate and fuse to existing
muscle tissue. Because the cell is incorporated into an
existing structure, it is not just tolerated but nurtured.
Myoblasts can easily be obtained by taking muscle tissue from
an individual who needs gene therapy and the genetically
engineered cells can also be easily put back with out causing
damage to the patient's muscle. Similarly, keratinocytes may
be used to delivery genes to tissues. Large numbers of kera-
tinocytes can be generated by cultivation of a small biopsy.
The cultures can be prepared as stratified sheets and when
grafted to humans, generate epidermis which continues to
improve in histotypic quality over many years. The keratino-
cytes are genetically engineered while in culture by
transfecting the keratinocytes with the appropriate vector.
Although keratinocytes are separated from the circulation by
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
59 _ _ _
the basement membrane dividing the epidermis from the dermis,
human keratinocytes secrete into circulation the protein
produced.
The chosen method of delivery should result in expression
of the gene product encoded within the nucleic acid cassette
at levels which exert an appropriate biological effect. The
rate of expression will depend upon the disease, the
pharmacokinetics of the vector and gene product, and the route
of administration, but should be in the range 0.001-100 mg/kg
of body weight /day, and preferably 0.01-10 mg/kg of body
weight/day. This level is readily determinable by standard
methods. It could be more or less depending on the optimal
dosing. The duration of treatment will extend through the
course of the disease symptoms, possibly continuously. The
number of doses will depend upon the disease, delivery
vehicle, and efficacy data from clinical trials.
Examples
The present invention will be more fully described in
conjunction with the following specific examples which are not
to be construed in any way as limiting the scope of the
invention:
_Example 1:
Cloning EC-specific enhancer and promoters
Promoters: Two sets of reporter vectors - CMV enh+/pro
and CMV enh-/pro- with either CAT (pCT1132 and pCT1133) or LUC
(pLC1137 and pLC1138 as reporter gene) were constructed. SacI
site is an unique site in thses vectors. The promoter
sequence will be amplified from human genomic DNA by PCR with
two primers (5' primer, 3'primer with SacI site) followed by
cloning into TA vector. The construct with right orientation
(3' is away from the SacI site on TA vector so that SacI
digest give PCR insert). The Sacl fragment will be inserted
into Sacl site of a vector above.
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
60 -- -
Multimerized endothelin enhancer: The endothelin
enhancer (ETe) was synthesized with overhangs to create Bgl II
and Bam HI sites as shown below.
gatctGTACTTCATACTTTTCATTCCAATGGGGTGACTTTGCTTCTGGAG
aCATGAAGTATGAAAAGTAAGGTTACCCCACTGAAACGAAGACCTCctag
This DNA fragment was multimerized by ligation at high
concentration and digested with Bgl II and Bam HI to eliminate
inverted and everted repeats. DNA species containing four and
seven tandem copies were gel-purified and inserted into
plasmids containing various endothelial-specific or
proliferation-specific promoters.
Materials and Methods
_Construction of EC-specific promoter-driven reporter
constructs: Plasmids containing endothelial-specific
promoters were constructed as follows. The minimal promoter
sequences of endothelin-1 (ET-1), KDR/flk-1, ICAM-2, (33, and
aV, and cell cycle-dependent genes (cyclin A, E2F1, or cdc6)
were directly amplified by PCR from human genomic DNA. The
amplified promoter sequence was then subcloned into pCR 2.1
(Invitrogen). The promoter sequence was then subcloned as a
SacI-SacI fragment into an expression plasmid pLC1136 which
contains the luciferase reporter gene, a synthetic intron, and
the human growth hormone 3' untranslated region/poly (A)
signal to create the promoter specific expression constructs.
Plasmids were grown under kanamycin selection in E. coli host
strains DH5a and purified using alkaline lysis and
chromatographic methods. Purified plasmid utilized for
injections had the following specifications: < 50 Eu/mg
endotoxin.
Endothelial Cell Culture and transfection
Human umbilical aortic endothelial cells (HUVEC) were
grown in six well plates in EBM-1 media (Clonetics, Inc.)
supplemented with 5~s fetal bovine serum with supplement of
endothelial cell growth factors. HUVEC at passages 2-4 were
used for transfection. HeLa cells were also grown in 6-well
plates in Dulbecco's modified Eagle's medium supplemented with
CA 02337496 2001-O1-26
WO 00/06759 PCTNS99/16388
61 -- -
10~ fetal bovine serum, 1~ glutamine, and 1$
penicillin/streptomycin. Cultured cells were transfected by
DEAE-dextran with 2 ~tg of reporter constructs. To correct for
transfection efficiency, 0.5 ~.g of the plasmid, pBG0965,
expressing the ~i-galactosidase gene, driven by the
cytomeglovirus immediate early promoter, was also included in
each transfection. Cell extracts were prepared 48 hr after
transfection and luciferase and (3-galactosidase assays were
performed. The relative luciferase activity was calculated as
the ratio of light units to (3-galactosidase units. The
corrected light units for HUVECs were divided by the corrected
light units for HeLa cells to obtain fold endothelial-
specificity.
Analysis of proliferating cell-specific promoters: The
resulting reporter plasmids containing EC-enhancer and cell
cylce-specific promter into two different endothelial cell
lines, HUVEC and BAEC, and compared the activity to that in a
non-endothelial cell line, NIH3T3. The reporter activities
was assayed in these samples.
Results
_Construction of plasmids: The resulting reporter
constructs are listed in the figures as are the representative
constructs ET-enhancer/ET-promoter, pLC1264, and pLC1265.
_EC-specific activity analysis: The activity of ETe/ETp
in HUVEC and HeLa cells and the specificity (EC vs HeLa) are
shown in the figures. The data indicated that ETe enhances
ETp expression specifically in EC by 10-fold.
Four or seven copies of the ETe inserted upstream of the
cyclin A, E2F1, or cdc6 promoter increases expression several
fold specifically in endothelial cells. These chimeric
regulatory elements, consisting of a proliferation-specific
promoter and an endothelial-specific enhancer, may provide a
means to achieve robust expression specifically in dividing
endothelial cells in vivo.
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
62 _ . _.
_EC specific expression of anti-angiogenic genes: The
coding sequence for endostatin or angiostatin was inserted
into vector pLC1265 to generate pES1358 and pAS1359. Thus,
the expression of endostatin and angiostatin was driven by
ETe/ETp. The specificity of endostatin and angiostatin
expression will be determinced.
Example 2: Anti-angiogenic gene medicines for cancer thera
Materials and Methods
Plasmid construction: Plasmids containing an expression
cassette for endostatin or angiostatin were constructed as
follows. The coding sequences of endostatin is the 184 as of
C-terminal of collagen 18a1. (human collagen type XVIII alpha
1 Accession # L22548: Oh, S . P. , Warman, M. L. , Seldin, M. F. ,
Cheng, S.D., Knoll, J. H., Timmons, S., and Olsen, B. R.
Cloning of cDNA and genomic DNA encoding human type XVIII
collagen and localization of the alpha 1(XVIII) colagen gene
to mouse chromosome 10 and human chromosome 21. Genomics
19(3), 494-499 (1994). Angiostatin is internal fragment (97-
440aa) of human plasminogen (Accession # M74220; Browne,
M.J., Chapman, C.G., Dodd, I., Carey, J.E., Lawrence, G.M.P.,
Mitchell, D., and Robinson, J. H. Expression of recombinant
human plasminogen and aglycolplasminiogen in HeLa cells.
Fibrinolysis, (1991) Endostatin and angiostatin coding
sequences were directly amplified by PCR from human liver cDNA
(Clontech) using oligonucleotide primers (shown below), which
add a BamHI site at the 5'end and an XbaI site at the 3'end:
human angiostatin 5' primer
ATg gAA CAT AAg gAA gTg gTT CTT
human angiostatin 3' primer (with Xhol site)
gC CTCgAg gCA TTT TTT CAg gTT gCA gTA CTC
human angiostatin 3' primer (internal primer) K3
gC ggATcc AAA gTg TAT CTC TCA gAg TgC AAg
mouse angiostatin 5' primer
ATg gAC CAT AAg gAA gTA ATC CTT
mouse angiostatin 3' primer (with XhoI site added)
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
63 --
gC CTCgAg gCA CCg CTT CAg gTT gCA gTA TTC
mouse angiostatin 3' primer (internal primer) K3?
gC ggATCC gTg TAT CTg TCA gAA TgT AAg ACC
mouse endostatin 5' primer (with BamHI added)
gCggATCC CAT ACT CAT CAg gAC TTT CAg CCA
mouse endostatin 3' primer (with Xho I added)
gCCTCgAg CTA TTT ggA gAA AgA ggT CAT gAA
human endostatin 5' primer
gAATTC CAC AgC CAC CgC gAC TTC CAg CCg
human endostatin 3' primer
CTCgAg CTA CTT ggA ggC AgT CAT gAA gCT
The amplified endostatin or angiostatin sequence was then
subcloned into pCR 2.1 (Invitrogen). The coding sequence for
endostatin or angiostatin was then subcloned as a BamHl-Xbal
fragment into Sfil-XbaI sites of an intermediate vector,
pHookl (Invitrogen). Thus the coding sequence for an Igk
sigal peptide and HA epitope from influenza virus was upstream
of that of endostatin or angiostatin. The coding sequence for
Igk-HA-endostatin or -angiostatin was then subcloned as an
BamHl-XbaI fragment into the expression plasmid containing the
cytomegalovirus immediate early promoter, a synthetic intron,
and the bovine growth hormone 3' untranslated region/poly (A)
signal to create the endostatin or angiostatin expression
systems, pES1100 (murine endostatin, mE), pES1281 (human
endostatin, hE), pAS1095 (human angiostatin kl-k4, hAk4), or
pAS1096 (human angiostatin kl-k3, hAk3). The HA-epitope was
deleted from pES1100 by recombinant PCR to generate expression
plasmid for HA-free mouse endostatin (pES1062, mE-HA-) (Fig.
1). Plasmid pVC0612 (empty plasmid, EP) contains expression
elements including the cytomegalovirus immediate early
promoter and the 3' UTR/poly(A) signal from the bovine growth
gene in the pVC0289 backbone described by Alila et al., Human
Gene Therapy, 8:1785-1795 (1997). Plasmid pVC0612 was used as
a control plasmid in all in vivo experiments. Plasmids for
intra-tumoral injection were grown under kanamycin selection
in E. coli host strains DHSa and purified using alkaline lysis
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
6 4 _ _ _.
and chromatographic methods. Purified plasmid utilized for
intra-tumoral injections had the following specifications: <
50 Eu/mg endotoxin; <1~ protein; and < 20~ chromosomal DNA.
Plasmid formulation:
DNA/PVP com lp ex: Purified expression plasmid and control
plasmids were formulated at a concentration of 3 mg DNA/ml in
a PINC delivery system as described previously (Mumper et al.,
Pharmaceutical Research, Vol. 13, No. 5, (1996) and Mumper et
al., Journal of Controlled Release, 52:191-203 (1998))
Preparation of liposomes and DNA/lipid complexes: Small
unilamellar vesicles (SUVs), composed of the cationic lipid
DOTMA (N-(1-(2-3-dioleyloxy)propyl)-n-n-n-trimethylammonium
chloride):cholesterol at 4:1 mole ratio, were prepared by
extrusion (400 nm). Positively charged plasmid/lipid
complexes were prepared at a 1:3 -/+ charge ratio in l00 (w/v)
lactose by mixing the plasmid with the liposomes under
controlled conditions (Freimark et al., The Journal of
Immunology, 160:4580-4586 (1998)). The mean diameter and zeta
potential of the complexes were characterized using dynamic
light scattering and Doppler electrophoretic light scattering.
The complexation efficiency was determined by agarose gel
electrophoresis.
Cell culture: Cos-1 cells were cultured in DMEM.
Endothelial cells (HUVEC, HLMEC and BAEC) were from Clonetics)
and cultured in specified medium according to the
Manufacturer. TS/A is a tumor cell line established by Dr. P.
Nanni, University of Bologna, Italy, from the first in vivo
transplant of a moderately differentiated mammary
adenocarcinoma that spontaneously arose in a BALB/c mouse
(Nanni et al., Clin. Exp. Metastasis, 1:373-376 (1983)) A
number of pre-immunization-challenge experiments suggested
that TS/A does not elicit long-lasting anti-tumor immunity
Renca, a spontaneously arising murine renal cell carcinoma,
and CT-26, a colon adenocarcinoma, were also used, as was
lewis lung carcinoma (LLC, metastatic variant from ATCC).
Tumor cell cultures were maintained in sterile disposable
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
65 -- --
flasks from Corning (Corning, NY) at 37° C in a humidified 5$
C02 atmosphere, using either RPMI 1640 (Renca, TS/A) or DMEM
LLC) supplemented with 10$ FBS, 100 U/ml penicillin, 100 U/ml
streptomycin and 50 ~.g/ml gentamycin; all from Life
Technologies.
Production of mouse antibody against human endostatin and
angiostatin:
75 ~~g of pES1281 or pAS1096 formulated in saline was
injected intramuscularly and boosted on day 14 and 28.
Antibodies were determined by western blotting.
Western blot analysis of endostatin and angiostatin: Cos
cells, Ecs or tumor cells' were plated in 6 well plates at 2.5
x 105 cells/well, and transfected using 2 ~.g of plasmids
pES1100, pES1062, pES1281, pAS1095, or pAS1096 and 3 ~g of
Lipofectamine (Life Technologies, Inc., Gaithersburg, MD) in
serum-free DMEM. Supernatants were harvested 24 hours later
and endostatin or angiostatin were immunoprecipitated using
mouse monoclonal anti-HA and protein A and G agarose
(Boehringer Mannheim, Indianapolis, IN). Samples were run on
a 12~ Tris-glycine gel and electroblotted to Millipore PVDF
membrane. Monoclonal anti-HA (Boehringer Mannheim), anti-
angiostatin (IgG (Enzyme Research Laboratories, Inc.) or anti-
endostatin was used at 1:1000, followed by anti-mouse (or
rabbit) Ig HRP (Amersham Life Science) at 1:1000. Rainbow
molecular weight markers (Amersham Life Science) were used to
determine protein size. Detection was performed using the
Amersham ECL kit.
Angiostatin/Endostatin ELISA: Supernatants (1 ml/well)
were collected from transfected cells above and levels of
endostatin or angiostatin were assayed by enzyme-linked
immunosorbent assay (ELISA). ELISA plates (Falcon flexible
PVC #3912) were coated with affinity purified goat anti-PG
(plasminogen) IgG (Enzyme Research Laboratories, Inc.) or
anti-endostatin (Folkman lab) in 50 mM carbonate buffer (1/500
dilution) at 4° C overnight and blocked with 150 ~,1/well of 20
BSA in PBS at room temperature for 4 hr. Supernatants (100
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
66
~,1/well) were added and incubated for 1 hr at 37° C. Mouse
monoclonal anti-HA (Boehringer Mannheim) diluted 1/500 in HBS-
Tween-BSA buffer (23.8 M HEPES, 5.84 M NaCl, 1.0$ BSA, 0.1~
Tween~ pH 7.2) was added (100 ~,1/well) and incubated for 1 hr
at 37 ° C. Peroxidase conjugated anti-mouse IgG (Amersham
Life Sciences) diluted 1/500 in HBS-Tween-BSA buffer was added
(100 ~1/well) and incubated for 1 hr at 37° C. 100~1/well of
OPD substrate (o-phenylenediamine (Sigma 5 mg tablets) diluted
in citrate-phosphate buffer (5.2 M citric acid, 13.8 M
Na2HP04; pH 5) with 0.1~ H202 was applied and color was
developed for 2-5 min. The reaction was stopped by the
addition of 50~.1/well of 2.5 M H2S09. Absorbance was
determined at 450 nm in an EL340 Microplate reader (Bio-tek
Instruments). Serial two-fold dilutions of plasminogen
lysine-binding site I (Sigma), HA-angiostatin or HA-endostatin
from transfected cells was used as a standard.
Animals: Normal 8-week-old female BALB/c or C57b1 mice
were purchased from Harlan Laboratories, Houston, TX. Mice
were maintained on ad libitum rodent feed and water at 23° C,
40o humidity, and a 12-h/12-h light-dark cycle. Animals were
acclimated for at least 4 days before the start of the study.
_In v.ivo evaluation of tumor growth and treatments:
BALB/c or C57b1 mice were challenged s.c. in the middle of the
left flank with 30 ~~tl of a single-cell suspension contained
the specified number of cells. Seven days later when the
tumor size reached approximately 10 mm3, treatments with
endostatin/PVP or EP/PVP started and were repeated at 1-2 day
intervals for 2 weeks (total of 8 treatments: 4/week). Tumor
volume was measured with electronic caliper in the two
perpendicular diameters and in the depth. Measurements of the
tumor masses (mm3) were performed twice a week for 40-50 days.
All mice bearing tumor masses exceeding 1 cm3 volume were
sacrificed for humane reasons. Data for the effects of
endostatin or angiostatin gene therapy on tumor growth were
analyzed by repeated measures analysis. Individual treatment
means were compared using Duncan's multiple range test when
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
67 _ _ _
the main effect was significant. Data for the effect of
endostatin ar angiostatin gene therapy on tumor rejection were
analyzed by ANOVA. In all cases a p value of less than 0.05
was considered to be statistically significant.
In vivo evaluation of Lung metastases and treatments:
Lung metastases were established in mice by injecting 3 x
105 Renca cells in 100 ~L HBSS (Hank's Balanced Salt Solution,
without Ca++ or Mg++, Life Technologies) into the tail vein.
Animals were warmed using a 150-watt lamp and placed in a
mouse restrainer prior to tail vein injection. 4 or 7 day
after tumor injection, mice were injected intravenously with
endostatin/DC, angiostatin/DC or EP/Dconce a week for 2-3
weeks. The lungs were insufflated with 1-2 mL India ink
solution (150 mL distilled H20, 30 mL India ink, 4 drops
ammonium hydroxide), using a 22 gauge gavage needle, then
fixed with Fekete's solution (90 mL Formaldehyde, 37~
solution, 900 mL 70 o EtOH, 45 mL glacial acetic acid) for at
least 24 hours. Metastases counts were performed under a
dissecting microscope. Survival data were analyzed using the
Kaplan-Meier log-ranked test. All other data were analyzed
using the Neuman-Keul's Test on StatMost for Windows software
(DataMost Corporation, Sandy, UT). Data were considered
statistically significant if p values were < 0.05.
LLC model: Subcutaneous tumor (8-12 mm in diameter) were
resected aseptically. All necrotic zones were removed and the
viable tissue was minced and dissociated with collagen (Type
I, 200 U/ml) and Dnase (270 U/ml) (Sigma Chemical Co., St.
Louis, MO). Cells were susupended in DMED with nsupplement
and plated at 5-10x106 viable cells/T175 flask. After a 3 hr
adherence, the culture were rinsed and given fresh medium. 48
h later, to adherent tumor cells were harvested by brief
trypsinization, wahsed once with medium and resuspend in HBSS.
Aliquotes of 106 cells in 0.1 ml of HBSS were injected
subcutaneously. When tumors were 12-15 in diameter, the mice
were anesthetized with methoxyflurane. The tumors in one
group of mice were surgical excised, and the area closed with
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
68 _ _ ..
metal woud clips. The other group of mice underwent a sham
surgical procedure which left the sc tumors intact. The mice
were monitored daily and killed 10-14 days after surgery. The
lungs were weighed and stained as described above.
Intramuscular injection and electroporation: 200-300 ug
of DNA/PVP (3mg/ml) were injected into tibialis (25 ul) and
gastrocnemia (50 ul) in one mouse leg, 2 min after injection,
electroporation (500V/cm, 96 usec with 4 pulses) was applied
on the injected leg.
Histological analysis: For CD-31, CD3, CD4, MAC-1
Immunostaining: Frozen sections were cut at 5~M and then
subsequently fixed in Acetone for 10 minutes at room
temperature. Immunohistochemistry was accomplished utilizing
an avidin biotin technique. Endogenous peroxidases were
quenched by incubating the sections with a 1~ H202 solution
for 10 minutes at room temperature. Nonspecific binding was
blocked with an incubation of 5 minutes at room temperature
with PowerBlock (Cat# HK085-5K, Biogenex, San Ramon, CA, USA).
The sections were then incubated with the appropriate dilution
of primary antibody for 1 hour at room temperature. Following
washes in PBS, the secondary antibody, biotinylated anti-Rat
(Cat#BA-4001, Vector Laboratories, Burlingame, CA, USA) 1:400,
was added and the sections were incubated for 1 hour at room
temperature. Following washes in PBS, the vectastain reagent
(Cat#pk-6001, Vector Laboratories, Burlingame, CA, USA) was
added at a dilution of 1:80 and the sections were incubated
for 1 hour at room temperature. 3,3-'diaminobenzidine (Stable
DAB, cat#750118, Research Genetics, Huntsville, AL, USA) was
used as the chromagen. The primary antibodies used were all
Rat anti-Mouse Monoclonal Antibodies. The antibodies and
dilutions were: CD3 (Cat#19914-D19. Gibco, Gaithersburg, MD,
USA) 1:250, CD4 (Cat#380220, Seikagaku, America, Inc.,
Ijamsville, MD, USA) 1:10,000, MAC-1 (Cat# CL8941AP, Cedar
Lane Laboratories, LTD, Westbury, NY, USA) 1:100, CD31
(Cat#0951D, Pharmingen, San Diego, CA, USA) 1:800.
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
6g __ ..
Apoptotic cells were detected utilizing the ApopTag
inSitu Apoptosis detection kit (Cat# S7100-KIT, Oncor,
Gathersburg, MD, USA). The residues of digoxigenin-nucleotide
were catalitically added to the DNA by terminal
deoxynucleotidyl transferase.
PCNA immunostaining was accomplished utilizing the Mouse
to Mouse HRP kit (Cat#MTM001, Scytek, Logan, UT, USA) and
following the manufacturer's instructions. The PCNA antibody
(Cat#32251, Pharmingen, San Diego, CA, USA) at a dilution of
1:10,000.
Anti-angiogenesis assay: In vitro inhibition of EC
proliferation assay was performed as follows. 5,000 cells of
human lung microvessel nendothelial cells (HLMEC) were plated
onto gelatinized 96-well culture plates and incubate (37°C, 5~
C02) for 24 h in 100u1 HLMEC medium containing growth factors
(bFGF). The media was replaced with 80u1 of endostatin- or
angiostatin-containing supernatant from transfected cells.
After 20 min of incubation, 20 ul of HLMEC media was added.
After 72 h, cell numbers were analyzed by WST-1 assay
(Boeringer).
In vivo neovascularization was assayed in mouse cornea.
Hydron pellet (Hydron; Interferon Sciences) of <1 ul (Aluminum
sucrose sulfate; Sigma) were formulated containing bFGF at 1
ug/ml and implanted into the cornea of C57b1 mouse 0.3-0.5 mm
from the limbus. Next day, DNA/PVP was injected im or DNA/DC
was injected iv. Angiogenesis was assessed by slit-lamp
microscopy on day 3, 4, and 5 after implantation. Maximal
angiogenesis was achieved on day 5. Inhibition of
angiogenesis by endostatin or angiostatin gene medicine was
examined by comparing to control vector.
Results
Construction of expression plasmids for endostatin and
angiostatin: The coding sequence for endostatin or
angiostatin (kl-k3) was PCR amplified from liver cDNA library
since collagen 18a (endostatin precusor) and plasminogen
(angiostatin precusors are rich in liver). Due to the fact
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
70 _
that Angiostatin and Endostatin are internal fragment devoid
of their natural secretion sequence, and that there are no
commercially available antibodies against angiostatin or
endostatin, HA epitope which is 9aa from influenza
hemaglutinin protein is tagged to the N-terminus of
angiostatin or endostatin so that antibody against this
peptide can be used for detection. Ig-kappa signal peptide is
added to upstream of HA-angiostatin or -endostatin to direct
secretion of the fusion protein. The expression plasmids for
HA-tagged mouse endostatin (mE, pES1100), HA-free mouse
endostatin (mE-HA , pES1062), HA-human endostatin (hE,
pES1281), HA-human angiostatin kl-k4 (hAk4), HA-angiostatin
kl-k3 (hAk3, pAS1096) were constructed and the maps were shown
in Fig. 1.
Expression of bioactive endostatin and angiostatin: To
assess their expression, the expression plasmids are
transfected into cos-1 cells, human endothelial cells (HUVEC),
or Renca tumor cells. The transgene expression was examined
for mRNA in cells by RT-PCR and for protein in media by
western blotting (Fig 2). The results showed Endostatin and
Angiostatin were transcribed as correct sizes as indicated and
no missplicing products were detected. The recombinant
protein was present as a single band with an approximate
molecular weight of 22 KD (HA-mE), 21 KD (mE), 20 KD (HA-hE)
or 30 KD (HA-hAk3). The protein expression was quantitatively
assessed an ELISA with anti-Endosattin or plasminogen antibody
and anti-HA epitope antibody. The result indicated that mE and
hAk3 was about 4 ng/ml in the culture medium. hAk3 was
expressed at higher level than hAk4. mE-HA expression was
determined by western blttoing with anti-endosattin antibody.
The conditioned media containing endostatin and angiostatin
showed strong inhibitory effects on endothelial cell
proliferation.
In vivo expression
Intratumoral injection: 24 ug DNA/PVP was injected
intratumorally and tumor was harvested at 24 h. The protein
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
71 __
expression of endostatin or angiostatin was determined by
ELISA. The tumor culture media was also examined for
bioactivity of endostatin and angiostatin.
Intramuscular delivery: 400-420 ~g DNA/PVP was injected
intramulscularly followed by electroporation (see Materials
and Methods). Serum was collected at day 2, 5 and 10. The
data showed that 3-5 ng/ml of Endostatin or 10-15 ng/ml of
Angiostatin in serum was produced on day 5, and
electroporation increased the level of trangenes by 3-5-fold.
The expression declined on day 10.
Intravascular delivery: DOTMA:chol formulated endostatin
or angiostatin expression plasmid was injected i.v. to normal
mice. Expression of endostatin and angiostatin were determined
by RT-PCR for mRNA in lung and ELISA for protein expression in
serum. 400 nm size particle resulted in higher expression and
may have more secondary cytokine effect than the SUV
formulation (see in vivo efficacy).
In vivo efficacy: Three approaches to assess Endostatin
in v.ivo anti-tumor activity are described below- sc tumor by
intratumoral injection, sc tumor by im injection and lung
tumor model by systemic delivery (iv and im) . The data from
these three models is summarized below.
Renca s c tumor model with intratumoral injection:
Intratumoral administration of 24 beg Endostatin gene/PVP at 4
times/week for two weeks (total 7 treatments) induced complete
regression in 7 out of 14 mice (50~ regression rate, p<0.05).
Survival rate was increased from 21~ (3/14) in vector/PVP
group to 780 (11/14) in Endostatin/PVP group on day 21. Human
endostatin with HA-tag was constructed. Intratumoral
injection of human endostatin/PVP resulted in 51~ tumor growth
inhibition in Renca sc tumor model.
The regressed tumors by Endostatin gene medicine remain
in a microscopic dormant state by day 29, whereas 6 cycles
treatments (400 ~tg/mouse/day for 27 days each cycle) with
recombinant protein is needed to keep tumor dormancy (Boehm et
al., Nature 390, 404-7). The data presented herein clearly
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
72 __ ..
demonstrates that Endostatin gene medicine has strong anti-
tumor activity in mouse tumor model, and may have many
advantages over the recombinant protein. Histological
analysis of the regressing tumors by Endostatin/PVP treatment
showed 3-5-fold reduced vascularization (CD31 staining), 3-
fold increased tumor apoptosis (TdT immunostaining). No
change in tumor proliferation (PCNA staining) and tumor
infiltration lymphocytes (CD3, CD4 and Mac-1 staining).
Renca s.c. tumor with systemic delivery: Im injection of
120 ~.g of Endostatin/PVP followed by electroporation (2x week,
switch legs) induced 40o Renca tumor injection. The
regressing tumors had reduced vascularization. Repeated
experiment showed that 34 $ tumor growth inhibition and
prolonged survival.
However, intravascular delivery of vector or
Endostatin/Dotma:chol (SUV, 30 ~g/2x week) didn't show a gene-
specific effect, although overall 50-60o reduction of tumor
growth was observed. Use of other lipids is expected to yield
improved results.
Lung metastasis model with systemic delivery: It has
been shown that IL-12 at 15 ~,g DNA in 400 nm particle induced
prolonged survival. It is ongoing to see if Endostatin,
Angiostatin can prolong survival by iv or im delivery of the
gene medicine.
LLC can metastize to lung when sc primary tumor is
excised. This lung metastasis model will be established to
assess inhibitory effects of lung metastases by anti-
angiogenic gene medicines.
Mouse_ cornea assay: Mouse cornea angiogenesis was
induced by implantating bFGF pellet. Endostatin gene medicine
was delivered by either iv of DNA/DC or DNA/PVP on next day
(see Materials and Methods). The results showed Endostatin by
iv strongly inhibited bFGF-induced cornea angiogenesis by day
5.
New anti-angiogenic genes: A double-barrel expression
plasmid for endostatin-angiostatin (pAS1254), in which
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
73
endostatin and angiostatin are driven by separate
transcription units, has also been also constructed.
IP-10, interferon-inducing factor 10, is a potent
chemokine and anti-angiogenic factor. Thrombospondin-1 (TSP
1), a p53-induced glycoprotein on EC surface, is a potent
inhibitor of angiogensis. An internal fragment (TSPf) is
essential for its activity. IP-10 cDNA was reverse
transcribed from published protein sequence with optimal
humanized codons. The cDNA was synthesized by Operon and the
expression plasmids for IP-10 (pIP-1316), IP-10/endostatin
fusion protein (pIP1311), and IP-10/TSPf fusion protein have
been constructed. The expression of IP-10, IP-10/hE fusion
protein, and IP-10/TSPf fusion protein were determined by RT-
PCR for mRNA and by western blotting for protein. About 150-
250 ng/ml protein was expressed in culture medium. These
constructs will be exmained in Renca sc model to see if
synergistic anti-tumor efficacy could be achieved.
One skilled in the art would readily appreciate that the
present invention is well adapted to carry out the objects and
obtain the ends and advantages mentioned, as well as those
inherent therein. The molecular complexes and the methods,
procedures, treatments, molecules, specific compounds
described herein are presently representative of preferred
embodiments are exemplary and are not intended as limitations
on the scope of the invention. Changes therein and other uses
will occur to those skilled in the art which are encompassed
within the spirit of the invention are defined by the scope of
the claims.
It will be readily apparent to one skilled in the art
that varying substitutions and modifications may be made to
the invention disclosed herein without departing from the
scope and spirit of the invention.
All patents and publications mentioned in the speci
fication are indicative of the levels of those skilled in the
art to which the invention pertains. All patents and
publications are herein incorporated by reference to the same
CA 02337496 2001-O1-26
WO 00/06759 PCT/US99/16388
74 _ _ _
extent as if each individual publication was specifically and
individually indicated to be incorporated by reference.
The invention illustratively described herein suitably
may be practiced in the absence of any element or elements,
limitation or limitations which is not specifically disclosed
herein. Thus, for example, in each instance herein any of the
terms "comprising", "consisting essentially of" and
"consisting of" may be replaced with either of the other two
terms. The terms and expressions which have been employed are
used as terms of description and not of limitation, and there
is no intention that in the use of such terms and expressions
of excluding any equivalents of the features shown and
described or portions thereof, but it is recognized that
various modifications are possible within the scope of the
invention claimed. Thus, it should be understood that
although the present invention has been specifically disclosed
by preferred embodiments and optional features, modification
and variation of the concepts herein disclosed may be resorted
to by those skilled in the art, and that such modifications
and variations are considered to be within the scope of this
invention as defined by the appended claims.
In addition, where features or aspects of the invention
are described in terms of Markush groups, those skilled in the
art will recognize that the invention is also thereby
described in terms of any individual member or subgroup of
members of the Markush group. For example, if X is described
as selected from the group consisting of bromine, chlorine,
and iodine, claims for X being bromine and claims for X being
bromine and chlorine are fully described.
Other embodiments are within the following claims.