Note: Descriptions are shown in the official language in which they were submitted.
CA 02338678 2001-O1-26
WO 00/73793 PCT/CA00100605
Identification of Compounds for Modulating Dimeric Receptors
Field of the Invention
The invention relates to methods of using the three dimensional structure of
an
intrinsically covalent dimeric receptor, preferably the insulin receptor, to
identify test
compounds that will interact with the dimeric receptor and modulate its
activity. The
invention also includes compounds identified using the methods of the
invention.
Background of the Invention
Covalent dimeric receptors are found on almost all cells in mammals. These
receptors include IR (insulin receptor), IGF-I R {insulin-like growth factor
I) and IRR
io (the insulin receptor-related receptor). In the case of IR, insulin binding
to IR is
essential for its manifold' effects such as glucose homeostasis, increased
protein
synthesis, growth, and development in mammals. IR belongs to the superfamily
of
transmembrane receptor TKs that include the monomeric epidermal growth factor
receptor (EGFR) and platelet-derived growth factor receptor (PDGFR). In
contrast, iR
and its homologues IGF-I R and IRR are sub-types of this family that are
intrinsic
disulfide-linked dimers of two heterodimers of the form (aJ3)~ (1,2).
Monomeric
receptor TKs are inactive; but are activated by ligand-induced dimerizadon
that results
in autophosphorylation. Dimeric IR-like TKs are also inactive, and are
activated by
ligand binding without further dimerization. Insulin blinding to the
extracellular domain
of IR results in autophosphorylation of specific tyrosines in the cytoplasmic
domain to
initiate an intracellular signal transduction cascade {3). However, the
structural basis for
the mechanism of IR activation by extracellular insulin binding has not been
elucidated
because the quaternary structure of IR was unknown. t?nly some of the smaller
domains
have yielded high resolution structural information.
Diabetes may be caused by mutant IR (eg. acanthosis nigrican or
leprechaunism. Insulin resistance leading to diabetes or similar symptoms may
also
occur.). Diseases are also caused by insu~cient amounts of IR ligand. For
example,
in diabetes, the pancreas produces insufficient amounts of insulin. Insulin
activates IR
and allows cells to absorb and store glucose. In the absence of adequate
insulin,
3o glucose accumulates in excessive amounts in the blood (hyperglycemia). The
CA 02338678 2001-O1-26
WO 00173793 PCT/CA00/00605
symptoms of diabetes may -include poor blood circulation, blindness and organ
damage. These symptoms often lead to premature death.
Diabetes is presently treated by insulin replacement therapy. This treatment
has been very successful, but it still has problems such as glycemic control.
Poor
glycemic control can cause retinopathy, poor blood circulation and the other
problems
associated with diabetes. It is also difficult to formulate insulin for slow
release:
Modified insulins have been created in an attempt to address problems with
insulin
therapy. In some cases, "super-insulins" have been created to increase the
activation
of insulin receptor by. its ligand. In other cases, binding to insulin
receptor is not
1o substantially increased, but the Iigand has more favourable formulation
properties.
For example, in HumalogTM, a lysine and a proline in insulin are switched to
provide
more favourable solubility characteristics.
These drug design strategies have been based on limited information, such as
the chemical properties of the insulin molecule. In some cases, insulin has
been
randomly modified and then assayed to determine the effects on insulin
activity.
While there has been success in producing insulin variants, both of these
approaches
are time consuming because variants are made without a clear understanding of
the
effect of the variation on binding to insulin receptor. There is a need to
obtain
additional information about the insulin receptor in order to provide a
rational basis
for drug design.
For example, it would be helpful if the quaternary structure, including the
Iigand binding site, of IR was available and characterr~zed to the detail of
amino acids.
However, it is very difficult to obtain information about the quaternary
structure of
dimeric receptors. For example, Large transmembra~ne proteins such as cell
surface
hormone receptors have been difficult to crystallizE; as intact molecules for
high-
resolution structural study. They are also too large for NMR spectroscopy. The
480-
kDa insulin receptor (IR) has thus not been crystallized as an intact
molecule, and its
quaternary structure remains unknown to date.
Summary of the Invention
3o We have obtained the quaternary structure of IR. We used low-dose Iow-
temperature dark field scanning transmission electron microscopy (STEM). Using
2
CA 02338678 2001-O1-26
WO 00173793 PCT/CAOOI00605
electron micrographs of the insulin-IR complex we have reconstructed the three-
dimensional quaternary structure of the intact receptor cornplexed with gold-
labeled
insulin ligand. Although IR has been purified and studied for over 1 S years,
this is the
first 3D reconstruction of its entire dimeric structure. Contiguous high
densities
s within the 3D structure indicate a two-fold symmetry for this dimeric
membrane
receptor, as well as a logical sequence for its biochemical subdomains from
the
observed binding of a single insulin on the ectodomain to the juxtaposition of
the pair
of intrinsic tyrosine kinases (TKs) of the intracellular domain.
We determined structural relationships of the IR subdomains in the 3D
1o reconstruction of IR and a structural basis for IR activation by insulin.
In the absence of
ATP which is required to complete the activation of tlhe IR tyrosine kinase,
the structure
of this insulin-bound iR can be considered to be in a transitional state, with
its kinase
domains intermediate between the inactive and activated structures observed by
x-ray
crystallography (4).
15 The quaternary structure of IR, fitted with the atomic co-ordinates of
highly
analogous domains of IR has resulted in a detailed description of the insulin
binding site
on the insulin receptor. Moreover, the combination of structural detail from
20 ~ to
atomic resolution yielded a self consistent model for the mechanism of the
initial phase
of insulin action on binding to effect intracellular receptor tyrosine kinase
activation.
2o The complete IR model provides a simple mechanical paradigm for the
reversible transmembrane signalling response. It explains the need for the
complexity
of structural components to control both inhibition and accommodation of
tyrosine
kinase activation. It gives ready structural explanations for many normal
effects, for
various mutations and for mild chemical reduction of the insulin receptoro It
thus
25 provides a comprehensive structural basis for the mechanics of
transmembrane signal
transduction for the intrinsically dimeric insulin-like membrane receptors.
The details of the insulin binding site provide an explanation of binding of
normal human insulin (including recombinantly produced insulin such as
NovolinTM) as
well as of the lesser or greater binding of insulin from other animals to the
human IR
3o and explains the binding of modified insulins such as "super-insulins",
HumalogT'~ and
other insulin analogs.
3
CA 02338678 2001-O1-26
WO 00/73793 PCT/CA00/00605
One aspect of the invention includes a method of identifying a compound that
modulates insulin receptor activity, including producing a compound that
interacts
with all or part of the fitted quaternary structure of insulin receptor or a
fragment or
derivative thereof and which thereby modulates insulin receptor activity. In
one
s embodiment, the method further includes synthesizing the compounds. The
method
preferably involves producing the compound based on its interaction with the
fitted
quaternary structure of insulin receptor or a fragment or derivative thereof.
For
example, one may produce the compound based on mimicking all or part of the
IR:insulin amino acid interactions.
1o Another aspect of the invention includes a method of identifying a compound
that modulates insulin receptor activity, including comparing the structure of
a
compound for modulating insulin receptor activity to all or part of the f tted
quaternary structure of insulin receptor or a fragment or derivative thereof
to
determine whether the compound is likely to modulate insulin receptor
activity.
I s The method may further include determining whether the compound
modulates the activity of the insulin receptor or a fragment or a derivative
thereof
having IR activity in an in vivo or in vitro assay. The compound identified by
the
method is an IR agonist or an IR antagonist. In one variation, the fitted
quaternary
structure of IR comprises substantially the entire fiti:ed quaternary
structure of IR.
2o The method may further include:
a) introducing into a computer program information defining a ligand binding
site conformation including at least one residue from monomer A in Table I
and at least one residue from monomer B in Table I, the ligand binding site
defined by the approximate amino acid distances listed in Table I, wherein the
2s program displays the quaternary structure thereof, fitted with the atomic
coordinates of the subdomains;
b) comparing the structural coordinates of the compound to the structural
coordinates of the ligand binding site and determining whether the compound
fits spatially into the ligand binding site and is capable of changing IR from
an
4
CA 02338678 2001-O1-26
WO 00/73793 PCT/CA00/00605
inactive conformation to an active conformation or biasing IR toward an active
conformation;
wherein the ability to change IR from an inactive conformation to an active
conformation or bias IR toward an active conformation is predictive of the
ability of the compound to agonize IR activity.
The method may further include preparing the compound that fits spatially into
the
ligand binding site and determining whether the compound agonizes IR activity
in an
IR activity assay. The invention also includes a method of identifying a
compound
which agonizes IR or a fragment or derivative thereof having IR activity, the
IR,
to fragment or derivative including a ligand binding site with at least one of
the residues
and approximate structural coordinates of each of monomer A and monomer B
listed
in Table l, the method including the steps of
a) providing the coordinates of the ligand binding site of the IR to a
computerized modeling system;
b) identifying compounds which interact with the ligand binding site and
change
IR from an inactive conformation to an active conformation or bias IR toward
an active conformation.
The invention also includes a method of drug design including using at least
one of the amino acids of each of monomer A andl monomer B of IR in Table 1 to
2o determine whether a compound interacts with the Iigand binding site of IR
or a
fragment or derivative thereof having IR activity and is capable of changing
IR from
an inactive conformation to an active conformation or biasing IR toward an
active
conformation.
Another aspect of the invention includes a method of agonizing IR including
administering to a mammal a compound that fits spatially into the ligand
binding site
of IR, the compound interacting with at least
a) one IR amino acid in monomer A listed in Table 1; and
b) one IR amino acid in monomer B listed in Table I;
5
CA 02338678 2001-O1-26
WO 00173793 PCTACA00/00605
wherein the compound is capable of changing iR from an inactive
conformation to an active conformation or biasing IR toward an active
conformation.
The method may further include:
a) introducing into a computer program information defining a ligand binding
site conformation including at least one residue from monomer A in Table I
and at least one residue from monomer B in Table I, the ligand binding site
defined by the approximate amino acid coordinates listed in Table I, wherein
the program displays the quaternary structure thereof;
l0 b) comparing the structural coordinates of the . compound to the structural
coordinates of the ligand binding site and determining whether the compound
fits spatially into the ligand binding site and is capable of changing IR from
an
active conformation to an inactive conformation or biasing IR toward an
inactive conformation;
wherein the ability to change IR from an active conformation to an inactive
conformation or bias IR toward an inactive conformation is predictive of the
ability of the compaund to antagonize IR activity.
The method may include preparing the compound that fits spatially into the
ligand
binding site and determining whether the test compound antagonizes IR activity
in an
IR activity assay.
Another aspect of the invention includes a method of identifying a compound
which antagonizes IR or a fragment or derivative thereof having IR activity,
the IR,
fragment or derivative including a ligand binding site with at least one of
the residues
and approximate distances of each of monomer A and monomer B listed in Table
I,
the method including the steps of
a) providing the coordinates of the ligand binding site of the IR to a
computerized modeling system;
6
CA 02338678 2001-O1-26
WD 00/73793 PCTICA00/00605
b) identifying compounds which interact with the ligand binding site and
change
IR from an active conformation to an inactive conformation or bias IR toward
an inactive conformation.
A variation of the invention includes a method of drug design including using
at least one of the structural coordinates from each of monomer A and monomer
B of
IR in Table 1 to determine whether a compound interacts with the ligand
binding site
of IR or a fragment or derivative thereof having IR activity and is capable of
changing
IR from an active conformation to an inactive conformation or biasing IR
toward an
inactive conformation.
The invention also includes a method of antagonizing IR by administering to a
mammal a compound that fits spatially into the ligand binding site of IR, the
compound interacting with at least:
a) one IR amino acid in monomer A listed in Table I ; and
b) one IR amino acid in monomer B listed in Table 1;
wherein the compound is capable of changing IR from an active conformation to
an
inactive conformation or biasing IR toward an active conformation. In a
variation of
the method, the ability of the compound to fit spatially into the ligand
binding site is
determined by comparing the structural _ coordinates of the compound with the
structural coordinates of IR. The ability of the compound to change the
conformation
of IR can be determined by comparing the structural coordinates of the
compound
with the structural coordinates of IR.
Another variation of the invention includes:
a) introducing into a computer program information defining a cam including at
least one residue from the Cam-loop segment in Table 2 and at least one
residue from the L I surface in Table 2, wherein the program displays the
structure thereof and its relation to other IR domains;
b) comparing the structural coordinates of the compound to the structural
coordinates of the cam and determining whether the compound interacts with
7
CA 02338678 2001-O1-26
WO 00/73793 PCT/CA00/00605
the cam and is capable of changing IR from an inactive conformation to an
active conformation or biasing IR toward an active conformation;
wherein the ability to change IR from an inactive conformation to an active
conformation is predictive of the ability of the compound to agonize IR
activity. The
method can further include preparing the compownd that interacts with the cam
and
determining whether the test compound agonizes 1:R activity in an IR activity
assay.
The invention includes a method of identifying a compound which agonizes IR or
a
fragment or derivative thereof having IR activity, the IR, fragment or
derivative
including a cam with at Least one of the residues and approximate structural
coordinates of the cam-loop segment and the L 1 swrface listed in Table 2, the
method
including the steps of
a) providing the coordinates of the cam to a computerized modeling system;
b} determining compounds which interact with the cam and change IR from an
inactive conformation to an active conformation or bias IR toward an active
conformation.
The invention includes a method of drug design including using at Ieast one of
the structural coordinates from each of cam-loop segment and the L 1 surface
listed in
Table 2 to determine whether a compound interacts with the cam of IR or a
fragment
or derivative thereof having IR activity and is capable of changing IR from an
inactive
2o conformation to an active conformation or biasing IR toward an active
conformation.
A variation of the method of agonizing IR includes administering to a mammal a
compound that fits spatially into the cam of IR; the compound interacting with
at least
one of the residues and approximate structural coordinates of the cam-loop
segment
and the L 1 surface listed in Table 2; wherein the compound is capable of
changing IR
from an inactive conformation to an active conformation or biasing IR toward
an
active conformation.
The method can further include:
a) introducing into a computer program information defining a cam conformation
including at least one residue from the Cam-loop segment in Table 2 and at
8
CA 02338678 2001-O1-26
W0 00/73793 PCT/CA00/00605
least one residue from the L 1 surface in Table 2, wherein the program
displays
the structure thereof and its relation to other IR domains;
b) comparing the structural coordinates of the compound to the structural
coordinates of the cam and determining whether the compound interacts with
the cam and is capable of changing IR from an active conformation to an
inactive conformation;
wherein the ability to change IR from an active conformation to an inactive
conformation is predictive of the ability of the compound to antagonize IR
activity.
The method can additionally include preparing the compound that interacts with
the
IO cam and determining whether the test compound antagonizes IR activity in an
IR
activity assay.
The invention also includes a method of identifying a compound which
antagonizes iR or a fragment or derivative thereof having IR activity, the IR,
fragment
or derivative including a cam with at least one of the residues and
approximate
15 structural coordinates of the cam-loop segment and the L 1 surface listed
in Table 2,
the method including the steps of
a) providing the coordinates of the cam to a computerized modeling system;
b) identifying compounds which interact with the cam and change IR from an
active conformation to an inactive conformation or bias IR toward an active
20 conformation.
Another variation of the invention includes a method of producing an IR
modulator
including using at least one of the structural coordinates from each of cam-
loop
segment and the L 1 surface listed in Table 2 to determine whether a compound
interacts with the cam of IR or a fragment of IR or derivative thereof having
IR
25 activity and is capable of changing IR from an active conformation to an
inactive
conformation or biasing IR toward an active conformation.
The method of antagonizing IR can include administering to a mammal a
compound that interacts with the cam of IR, the compound interacting with at
least
one of the residues and approximate structural coordinates of the cam-loop
segment
9
CA 02338678 2001-O1-26
WO 00/73793 PCT/CA00/00605
and the L1 surface listed in Table 2; wherein the compound is capable of
changing IR
from an active conformation to an inactive conformation or biasing IR toward
an
active conformation. The ability of the compound to interact with the cam can
be
determined by comparing the structural coordinates of the compound with the
structural coordinates of IR. In the method of the invention, wherein the
ability of the
compound to change the conformation of IR can be determined by comparing the
structural coordinates of the compound with the structural coordinates of IR.
The methods of the invention may use free IR or IR bound to insulin in an
IR:insulin complex.
Another aspect of the invention includes a computer medium having recorded
thereon data of an IR receptor, said data sufficient to model all or part of
the
quaternary structure of the receptor. The data can comprise structural
coordinates of
. an IR receptor, the coordinates sufficient to model all or part of the
quaternary
structure of the receptor. The quaternary structure of the receptor can
include
1 s substantially all of the quaternary structure of the receptor.
The invention also includes an insulin analog or other analog or mimetic
identified by the methods of the invention.
The invention also includes a method of identifying agonists of IR by rational
drug design including: producing an agonist for IR that will interact with
amino acids
2o in the IR ligand binding site or IR cam based upon the structure
coordinates of the
IR:insulin complex. The method of may further include synthesizing the agonist
and
determining whether the agonist agonizes the activity of IR in an in vivo or
an in vitro
assay. In a method of the invention, the quaternary structure of the
IR:insulin
complex can be obtained from an IR: insulin complex prepared for EM. The co-
25 ordinates of the IR:insulin complex may be obtained by means of fitting
atomically
known subdomains into the quaternary complex.
The agonist can be designed to interact with at least one amino acid in
monomer A in Table 1 and at least one amino acid in monomer B in Table l and
cause IR to change from an inactive conformation to an active conformation or
bias
3o IR toward an active conformation.
CA 02338678 2001-O1-26
WO OOI73793 PCTICA00/00605
The method of identifying a compound that modulates insulin receptor and
insulin interactions or activity, can include:
a) designing a compound for modulating insulin receptor activity based upon
fitted quaternary structure (eg fitting atomically known subdomains into
quaternary structure) of insulin receptor bound to insulin.
The method can further synthesizing the compound and determining whether the
compound modulates the interactions or activity of the insulin receptor and
insulin.
Another aspect of the invention includes a method of identifying a compound
that modulates insulin receptor and insulin interactions or activity,
including:
Io a) comparing a compound for modulating insulin receptor activity to the
quaternary structure of insulin receptor boiuid to insulin to determine
whether
the compound is likely to modulate insulin receptor and insulin interactions
or
activity;
b) determining whether the potential compound modulates the interactions or
activity of the insulin receptor and insulin.
The compound may agonize or antagonize insulin receptor and insulin
interactions or
activity The method of identifying how a compound interacts W ith IR activity
may
include comparing the compound to all or part of the :fitted quaternary
structures of IR
Another aspect of the invention includes a computer readable medium including
all or
2o part of the fitted quaternary structure of IR as shown in a figure or
described in the
application.
Another aspect relates to an insulin analog identified by a method of the
invention. The invention includes a method of agonizing insulin receptor
inlcuding
administering a an effective amount of the analog. The invention also includes
a
method of medical treatment of diabetes or hyperglycemia including
administering to
a mammal' having diabetes or hyperglycemia a pharmaceutical composition
including
an effective amount of the analog. Mimetics or other insulin variants may also
be
used.
11
CA 02338678 2001-O1-26
WO OOI73793 PCT/CA00/00605
Brief Description of the Drawings
Preferred embodiments are described in relation to the drawings, in which:
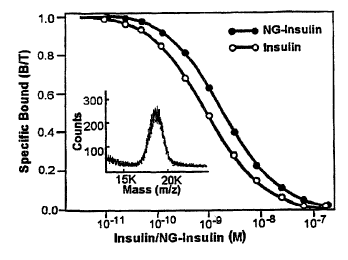
Figure 1. Receptor-binding assay of Nanogold-insulin. Receptor-binding
activity of
purified Nanogold-insulin was compared to that of bovine insulin in a receptor-
binding
assay using human insulin receptor as described (9). Inset shows the mass
spectrum
obtained from the MOLDI-TOF analysis of purified Nanogold-insulin (7).
Figure 2. STEM dark f eld images of human insulin receptor /Nanogold-insulin
(HIRING-BI) complex. A) Raw images showing several complexes. Arrowheads point
to intense signals from Nanogold marker. Scale bar' = 20 nm. B) HIR/NG-BI
images
1 o extracted from image fields, after low pass filtering to 1.0 nm and
boundary
determination (left column). High density threshold representation of
extracted images
showing one (top five images) or two (bottom two images) sites of Nanogold
location
(right column).
Figure 3. Three-dimensional reconstruction of the HIR/NG-BI complex from 704
STEM dark field images. A) Density threshold representing the total expected
volume
far the complex [ I ]; intermediate density threshold, unsymmetrized, showing
higher
contiguous densities [2]; high density threshold of [2] showing only the
NanogoId label
[3]. Circles in the panels indicate location of tl~e gold marker within the
reconstructions.
The resolution was 20 A as measured by Fourier phase residual analysis of two
reconstructions with 352 images each (13). B) Reconstruction with two-fold
symmetry
at intermediate density thresholds in different orientations, indicating the
relationship
and connectivity of the structural domains. Labels, for only one a[i monomer
of the
dimeric HIR, refer to biochemical domains. Arrowhead indicates the proposed
plane of
the cell membrane lipid bilayer. L1, C-R, L2 = L1-Cysteine-rich-L2 domains; CD
=
connecting domain; Fnl, Fn2 = fibronectin III repeats 1 and 2; TK = tyrosine
kinase;
TM = transmembrane domain.
Figure 4. Fitting of biochemical domains and their known x-ray structures to
the 3D
reconstruction. A) Schematic domain structure for one a[i monomer, derived
from i)
connectivity of the 3D reconstruction at intermediate density threshold (Fig.
3), ii) from
3o the primary domain sequence, iii) from the requirement for two disulfides
on the two-
fold symmetry axis between the two oc subunits {4), iv) the fit of the known
domain
12
CA 02338678 2001-O1-26
WO OOI73793 PCTICA00/00605
structures; and v) the principle of keeping domains of unknown structure as
compact as
possible. Distances measured in the 3D reconstruction between locations of
subdomains
CD, Fnl and the ~ symmetrical disulfides were commensurate with numbers of
intervening amino acid residues (structures not shown to scale; , unknown
structures are
spheres or lines): A = TK activation loop; 1 = Cys524; 2 = Cys682, 683, 685; 3
= alpha-
beta disulfide between Cys647 and Cys872; arrowhead = proreceptor cleavage
site;
other labels as described in Fig. 3B. B) Representative fitting of L1-Cys-rich-
L2
domains as approximate cylinders to ectodomain structure of 3D reconstruction
(cf. Fig.
3B, side view, 0; for ribbon structure see Fig 7A}. One insulin molecule
(ribbon, PDB:
i o 1 BEN) inserted with its receptor-binding domain contacting the L 1-Cys-
rich domains of
one subunit and the L2 domain of the other. The Nanogold marker on Phel of
insulin
B chain positioned to coincide with the high-density site of reconstruction.
C) Right
angle side view of (B) (cf. Fig. 3B, side view 90 ) with L1-Cys-rich-L2
domains
(insulin partly hidden), fitted TK structure in symmetric bottom domains
(ribbon, PDB:
IIRK} and two dimeric FnIII structures as symmetric outer structures at mid
height
(ribbons, PDB: lmFn). Activation loop (ribbon) of left TK domain is shown in
its
crystallographic position. A-loop of symmetry-related right TK domain extended
to
overlap peptide substrate position of opposite TK in peptide-bound state (4).
See also
(D}. D) Right angle top view of (B) (c~ Fig. 3B, top view) showing the
positions of the
2o FnIII domains (top and bottom) and the TK domains across centre.
Crystallographic
position of activation loop is uppermost within one TK domain, while extended
activation Ioop of the other TK domain is below centre. One square in the wire
mesh is
6.5 ~.
I3
CA 02338678 2001-O1-26
WO 00/'T3793 PCT/CA00100605
Figure 5
a Three-dimensional structure of the human insulin receptor reconstructed
images of the purified dimeric insulin receptor compiexed with insulin
obtained via low
dose scanning transmission cryomicroscopy [I]. Iyensity threshold at 85% of
total
volume to show contiguity of structure. Maximum diameter is 150 ~. Various
regions
of one a[i monomer of the dimeric structure labelled as determined from
insulin
location, connectivity, mass distribution and fitting of known subdomain
structures.
(i), View as seen from the exterior of the cell, down the two-fold symmetry
axis of the
(a(3)z heterodimer. Partially transparent gray disc represents cell membrane
with fainter
to regions of structure on distal side of membrane. (ii), View at right angles
to A with
extracellular components above gray translucent symbolic cell membrane. {iii),
View
from interior of cell with fainter structures on distal (exterior) side of
modelled
membrane. Arrow head points to cam-Iike feature (see text). For domain
abbreviations
see Fig. 6.
b Simplified, stylized model of insulin-IR in the same orientations as Fig.
Sa.
(i), View from exterior of cell. (ii), Side view (cell membrane edge-on).
(iii), View from
interior of cell. Corresponding subdomains for one a[i monomer are indicated.
The
other a[3 monomer is symmetrically related. Stylized catalytic regions and
activation
loops (spheres and hairpins) are indicated on TK domains. The two a-a
disulphide
bonds (1, 2) modelled on two-fold axis in strained configuration. Cams (arrow
head,
discs) in position permissive for transactivation. Insulin ligand represented
as disc. For
domain abbreviations see Fig. 6.
c Stylized model of IR in the absence of insulin. Same orientations as Fig.
Sb.
(i), View from exterior of cell, with separated LI-Cys-rich domains. (ii),
Side view
{cell membrane edge-on). {iii), View from interior of cell, with separated TK
domains.
Activation loops (arrow) do not reach catalytic loops (spheres on TKs). Cams
(arrow
head, discs) in position to block mutual approach of Fn2/TMfTK assemblies.
Pair of
3o Cys-Cys bonds (1, 2, yellow) in relaxed equilibrium positions. Insulin
(disc) in position
to bind to one a~3 monomer. For domain abbreviations see Fig. 6.
14
CA 02338678 2001-O1-26
WO 00/73793 PCT/CA00100605
Figure 6
Sequential spatial arrangement of the subdomains of one a[i monomer of the
insulin receptor deduced from the 3D structure [1]. The N-terminal of the a
subunit is
at the top, the C-terminal of the (3 subunit near the bottom. The domains and
their
delimiting amino acid sequences [5] are: aN-terminal - 1 - L 1 - 1 S 8I i 59 -
cysteine-rich
(CR) - 310/311 - L2 - 470/471 - connecting-domain/aFibronectin0 {CD/Fn0) -
572/573 -
aFibronectinl (aFnl) - 661/662 - a-insert-domain (ID)- 719 - aC-terminal; ~iN-
terminal - 724 - [i-ID - 779/780 - [iFnl - 816/8 / 7 - ~3Fn2 -913/914 -
juxtamembrane -
929/930 - transmembrane (TM) - 952/953 - juxta~riembrane - 977/978 - tyrosine-
kinase (TK) - 1283/1284 - C-terminal region - 1388 - [iC-terminal. Other
important
residues are Cys524 (denoted by "1 "), which forms an a-a bond on the two-fold
symmetry axis, as does one of Cys682, Cys683 or Cys685 (shown as "2") . An a-
(3 bond
is formed by Cys647 in Fnl of the a subunit and Cys872 in Fn2 of the [i
subunit
(shown as "3 "). "x" marks the cleavage site between the a and [3 subunits in
the pro-
receptor. The catalytic loop and the activation Ioop (shown as "A-C"; residues
1130-37
and 1149-70, respectively) are approximately in the central region of the
tyrosine kinase
structure [ 10,11 ].
Figure 7
a Side view of IR dimer structure at volume corresponding to total receptor
mass, in wire mesh representation rotated 90° with respect to Sa(ii);
fitted centrally with
two L1-CR-L2 regions of IR as adapted from the co-ordinates of the
correspoiading IGF-
1R structure. Aminoacid backbone representation. 7."he diamond-shaped opening
is the
modelled insulin binding site with one Nanagold-insulin fitted into the site
(see Fig. 8).
b End view of full-mass representation of IR dimer. Left half surface
rendering; right half wire mesh representation. Fitted structure of two IR-
adapted L1-
CR-L2 regions. Arrow: cam-like region on CR domain.
c Higher density solid surface representation slightly rotated of view in Fig.
7b
showing location of CR cam regions of atomic stnicture against Fn2 domains of
3D
reconstruction.
CA 02338678 2001-O1-26
WO 00/73793 PCT/CA00/00605
Figure 8
a View in parallel stereo representation of IR insulin-binding region of
docked
L1-CR-L2 regions (cf. Fig. ?a) fitted with insulin. Backbone representation
except for
aminoacid sidechains tabulated in Table 1. See text far details.
b Insulin contacts with one L1-CR-L2 monomer. Slight rotation from Fig. 8a.
The gold sphere represents the Nanogold label on insulin used in the 3D
reconstruction.
See text.
c Insulin contacts with second L1-CR-L2 monomer.
Figure 9
to Simplified schematic of structural changes during activation of insulin
receptor.
a. Inhibitory state. Ectodomain of dimeric a subunits each with two differing
insulin
binding sites and blocking cam. Unbound bivalent insulin. ~i subunits resting
against
cams, crossing membrane, with tyrosine kinase {7;''K) domains separated.
Arrows
indicate thermally induced motion. b, Insulin bound state. Blocking cams
rotated, (i
subunits resting against centre of ectodomain. TK domains juxtaposed for
transphosphorylation:
Figure 10
A. Views (parallel stereo) of fibronectin domains docked into ectodomain
quaternery structure of IR. FnO/CD and aID regions are modelled as
2o extending around L2 to the central 2-fold symmetry axis to form a-a
disulphide bonds. The a-[3 disulphide is shown between aFnl and Fn2.
The domains of one a(3 monomer only are labelled for identification. For
clarity, LCL is shown only with part of the CR domain and all of the L2
domain (amino acids 250 to 470).
B. Complete fit of known IR and IR-Iike domains as docked into 3D EM
reconstruction of quaternary structure of IR dimer. The TM and
juxtamembrane domains, of unknown structure, have been modelled as
helix and loop structures and arbitrarily placed to connect the Fn and TK
domains. The unknown structures of the ~iID region at the N-tenninat of
3o the ~iFnl domain and the C-terminal ~i-domain joined to the TK domains
have not been modelled.
16
CA 02338678 2001-O1-26
WO OOI73793 PCT/CA00/00605
Figure 11
Sequence of (a) human insulin (b) cow insulin (c) pig insulin.
Figure 12
Sequence of human insulin receptor.
Figure 13
System for molecular modeling.
Detailed Description of the Invention
The invention includes new 3D structures for dimeric two state-receptors that
are
activated or inhibited by ligand binding. It also includes aspects such as the
ligand
l0 binding site, binding domains, other functional or structural domains and
the mechanism
of action of the receptors. The invention also includes methods of using these
aspects to
identify compounds capable of modulating (agonizing or antagonizing) the
receptors.
In one embodiment, the receptor is the insulin receptor (amino acid sequence
is
shown in figure 12). In a preferred embodiment the structure is the fitted
quaternary
structure of IR. The "fitted quaternary structure" of IR includes the
structure of the IR
domains fitted together to arrive at a three-dimensional arrangement that fits
into the
corresponding portion of the quaternary structure of IR. Parts of the fitted
quaternary
structure are also useful in the methods of the invention. Prior to this
invention, the
3D structure of the receptor and its mechanism of activity were unknown. The
relative positions of amino acids which bound insulin and provided receptor
activity
were also poorly understood. The invention details the atomic interactions of
insulin
with the dimeric insulin receptor (IR) in the extracellular insulin binding
site of the
receptor. Furthermore, a mechanism is detailed which shows how this binding of
insulin results in transmembrane signalling to activate the intracellular
intrinsic tyrosine
kinase of the insulin receptor dimer. The structure and mechanism explain the
normal
function of the insulin receptor as well as the effect of mutations and of
altered
physiological conditions. The invention provides the first comprehensive
description
of insulin binding to insulin receptor and the mechanical mechanism of insulin
receptor activity. The structure of IR has been determined while complexed to
insulin
and has been modeled in the insulin-free state.
17
CA 02338678 2001-O1-26
WO 00/73793 PCT/CA00/00605
The invention includes the structure of insulin receptor fitted with the
atomic
coordinates of the amino acids comprising the receptor, the use of that
structure to
solve the structure of insulin receptor isoforms, homologues and other forms
of
insulin receptor, mutants and co-complexes of insulin receptor, and the use of
the
insulin receptor structure and that of its isoforms, homologues, mutants, and
co-
complexes to design modulators. The structure is particularly useful for
development
of ingestible (preferably oral) insulin mimicking agents (analogs, mimetics)
that can
be used in place of insulin (which has to be administered by injection) to
treat insulin-
dependent diabetes.
to In one aspect the present invention is directed to the three-dimensional
structure of an isolated and purified IR polypeptide and its structure
coordinates.
Another aspect of the invention is to use the structure coordinates of the
insulin
receptor to reveal the atomic details of the ligand binding site and one or
more of the
accessory binding sites of insulin receptor such as a cam. The entire receptor
may be
used or particular regions of interest may be used. Structural and
conformational
changes induced in the receptor may also be studied. Another aspect of the
invention
is to use the structure coordinates of an insulin receptor to solve the
structure of a
different insulin receptor or a mutant, homologue or co-complex of insulin
receptor.
A further aspect of the invention is to provide insulin receptor mutants
characterized
by one or more different properties compared to wild-type insulin receptor.
Another
aspect of this invention is to use the structure coordinates and atomic
details of insulin
receptors or mutants or homologues or co-complexes thereof to design, evaluate
(preferably computationally), synthesize and use modulators of insulin
receptor that
prevent or treat the undesirable pathologies of'inadequately or improperly
functioning
insulin receptor.
The IR structure of the present invention includes the three dimensional
structure of the receptor including the fitted quaternary structure. The IR
structure
includes the ligand binding site that includes the amino acid residues listed
in Table 1
and the cam structures including the amino acid residues in Table 2.
This invention also provides the first rational drug design strategy for
modulating IR activity. It includes methods for identifying compounds that can
18
CA 02338678 2001-O1-26
WO 00/73793 PCT/CA00100605
interact with insulin receptor. The method for identifying insulin mimetics
and insulin
antagonists preferably include fitting the crystal structures, NMR structures
and other
structures of insulin receptor domains into the quaternary structure of the
complete
insulin-bound dimeric insulin receptor determined from electron microscopic
image
reconstruction. These interactions can be easily identified by comparing the
structural,
chemical and spatial characteristics of a test compound to the three
dimensional
structure of the insulin receptor. Since the amino acids that are responsible
for
receptor activity and binding were identified by this invention, drug design
may be
done on a rational basis. Structures such as a cam or a ligand binding site
may be
studied together or separately. Fragments of a cam or a ligand binding site
may also
be studied (e.g. at least one or at least 2 of the amino acids in table 1 or
2, optionally
also including one or more proximate amino acids).
The structure serves as a detailed basis for the design and testing of insulin
analogs, mimetics and insulin antagonists, initially in the computer, but also
in vitro in
1 s cell culture and in viva, providing a method for identifying modulators
(antagonists and
agonists) having specif c contacts with the insulin receptor or an isoform,
homologue,
mutant or co-complex. The effect of a modification to insulin may be readily
viewed
on a computer, without the need to synthesize the compound and assay it in
vitro. As
well, non-protein organic molecules may also be compared to the insulin
receptor on a
computer. One can readily determine if the molecules have suitable structural
and
chemical characteristics to interact with, and activate or inhibit, receptor
activity. The
invention includes the IR modulators discovered using all or part of an IR
structure of
the invention (preferably the fitted quaternary stnacture) and the methods of
the
invention.
Drug design
The determination of the quaternary structure of IR, and in particular its
fitted
quaternary structure, provides a basis for the design of new and specific
compounds
for the diagnosis and/or treatment of IR-related pathologies ("pathology"
includes a
disease, a disorder and/or an abnormal physical state preferably characterized
by
either (i) inadequate or excessive insulin in a mammal (preferably a human) or
inadequate or excessive IR activity. IR related pathologies include those
involving IR
19
CA 02338678 2001-O1-26
WO 00/73793 PCT/CA00/00605
as in fig. 12 or IR variants described in this application.}. This structure
is useful in
the design of modulators (agonists or antagonists), which may be used as
therapeutic
or prophylactic compounds for treating pathologies in which upregulation or
downregulation of receptor activity is beneficial. It will be apparent that
methods
using IR described below may be readily adapted for use with a fragment of IR
or an
IR variant.
The characterization of the novel IR ligarid binding site and cams permit the
design of potent, highly selective IR modulators. Several approaches can be
taken for
the use of the IR structure in the rational design of Iigands of IR. A
computer-
i0 assisted, manual examination of a ligand binding site or cam structure may
be done.
This invention includes the methods for identifying modulators of IR that act
on the IR quaternary structure (preferably the fitted quaternary structure),
ligand
binding site and/or cam, as well as the modulators themselves. The agonist
modulators upregulate IR activity by biasing IR towards its active, closed
1s conformation. The antagonist modulators downregulate IR activity by biasing
IR
towards its inactive, open conformation. Such modulators may bind to all or a
portion
of the ligand binding site of IR. They may also modulate IR activity by
interacting
with other portions of IR, such as the cam structures. One may also select an
IR
amino acid (for example from the IR binding site) to which one could make a
mating
2o amino acid on insulin. Such a new amino acid on insulin would not
necessarily have
to be in the same category as the native amino acid, but could switch
categories to be
more attractive to the mating amino acid on the receptor surface. Amino acids
are
usefully changed in kind (eg. hydrophobic to hydrophilic, non-polar to polar,
non-
polar to charged, etc.) to create a new interaction between amino acids that
are not
25 already used in insulin:IR interactions, or to change the character of an
existing
insulin:IR interaction. Fox example, changes in interactions may increase or
decrease
the strength of the total binding, or make the insulin:IR complex less
sensitive to ionic
conditions around the receptor.
One example is B23 Gly on insulin that is near Ser85 (5.4 Angstr. C alpha to
3o C alpha) and near Argl 14 {9.1 Angstr. C-alpha to C-alpha) on the receptor.
If B23
CA 02338678 2001-O1-26
WO 00/73793 PCT/CA00/08605
GIy on insulin is changed to Thr or Tyr it hydrogen-bonds to Ser85. If it is
changed to
Glu or Asp, it forms a salt bridge with Arg 1 I4.
A change in an amino acid that is already used may also be made, e.g. B22
Arg on insulin is near G1u285 (and others in our Table I) to form a salt
bridge
(electrostatic interaction). It is also near Thr325 and Ser326 on the
receptor. Thus if it
were changed to an amino acid such as Thr, Ser, Tyr, His etc (a hydrogen bond
donor
or acceptor} then this new amino acid forms a hydrogen bond with Thr325 or
Ser326
to change the character of the interaction.
The methods preferably include (a) introducing into a computer program
1o information defining all or part of IR and insulin, for example portions
including the
IR ligand binding site (other regions of IR described in this application,
such as the
cam-loop segment and L1 surface, may also be~used), so that the program
displays the
quaternary structure thereof; b) comparing the structural coordinates of the
compound
to the structural coordinates of the ligand binding site and determining
whether the
compound fits spatially into the ligand binding site and is capable of
changing insulin
receptor from an active conformation to an inactive conformation or biasing
insulin
receptor toward an inactive conformation. The ability to change insulin
receptor from
an active conformation to an inactive conformation or bias insulin receptor
toward an
inactive conformation is predictive of the ability of the compound to
antagonize
2o insulin receptor activity.
One may also adapt the above method to dei:ermine whether the compound is
capable of changing insulin receptor from an inactive conformation to an
active
conformation or biasing insulin receptor toward an active conformation. The
ability
to change insulin receptor from an inactive conformation to an active
conformation or
bias insulin receptor toward an active conformation is predictive of the
ability of the
compound to agonize insulin receptor activity.
The methods preferably further include preparing the compound and
determining whether the test compound agonizes or antagonizes insulin receptor
activity in an insulin receptor activity assay. Other methods described in
this
application may also be readily adapted and used.
2I
CA 02338678 2001-O1-26
WO 00/73993 PCT/CA00/00605
The modulators may be competitive or non-competitive modulators. Once
identified and screened for biological activity, these modulators may be used
therapeutically or prophylactically to affect IR activity.
The invention also includes methods of agonizing or antagonizing insulin
receptor by administering compounds with structural and chemical properties
that
allow the compounds to interact with insulin receptor residues in order to
modulate
receptor activity.
Interaction of modulators of IR ligand binding site
A test compound that is a modulator interacts with at least one insulin
receptor
1 o residue listed in Table 1 on monomer A and at least one residue in Table 1
on
monomer B in order to activate or inhibit insulin receptor. "Interact" refers
to binding
to the receptor which is capable of modulating its activity. Receptor
fragments may
be used in the methods of the invention to predict how the full receptor will
react to a
modulator. Since the IR is a 2-fold symmetric dimer structure, either one of
the IR
monomers can represent monomer A, the other representing monomer B. A
modulator
that is an agonist is capable of changing the IR from an inactive conformation
to an
active conformation. A modulator that is an antagonist is capable of changing
the IR
from an active conformation to an inactive conformation (or may keep or
maintain IR
in its inactive conformation). A modulator may bias the receptor towards a
particular
2o conformation instead of (or in addition to) changing the conformation.
The compound may also interact with at Least: two, three, or four or five of
the
residues on each of monomer A or monomer B that are listed in Table 1. The
test
compound may interact with at Least about: five, sip, seven or eight, nine,
ten, eleven
or twelve amino acid residues on monomer B. The intersidechain distances
between
the modulator and the IR are preferably about those distances (or at least one
of the
distances) listed in Table 1. The distances may be varied by plus or minus
about:
O.lA, 0.2A, 0.25A, 0.3A, 0.4A, O.SA, O.6A, 0.7A, 0.75A, 0.8A, 0:9A, lA or >lA,
>1.SA or 2A as long as the test compound is still able to interact with IR and
modulate its activity. It is apparent that the test compound must be able to
make
appropriate interactions with the IR Iigand binding site if it is to activate
the IR.
22
i,ii
CA 02338678 2001-O1-26
WO 00/73793 PCTlCA00/00605
Table 1
Modeled Approaches between Insulin Side Chains and Insulin Receptor Side
Chains
Insulin Residue Insulin Receptor Residue (Reeion) Intersidechain Interaction
S MonomerA Distance (A)
GluA4$ Arg86 (L1) 2.5 ~ electrostatic
ThrA8 2.6 polar
GluAl7 Arg331 (L2) 2.5 electrostatic
AsnA21 Ser323 (L2) 5.3* H-bond ladder
LysB29 Aspl2 (LI) 2.6 electrostatic
G1n34 2.5 polar
Monomer
B
SerB9 GIn34 (Ll) 2.8 Hbond
HisBlO Argl4 5.0* electrostatic (H20
bridge)
GIuB I3 Arg86 2.5 electrostatic
ValBl2 Phe89 (L1) 2.5 hydrophobic patch
LeuB 17 2.5 hydrophobic patch
TyrB I 6 Leu87 2.5 hydrophobic patch
PheB24 Phe88 2.5 hydrophobic patch
PheB25 hydrophobic patch
TyrB26 Tyr91 hydrophobic patch
G1uB21 His247 (CR) :Z.S electrostatic
G1n249 2.5 polar
ArgB22 G1u250 4.0* electrostatic
2.5 electrostatic
GIu287 (I,2) 2.5 electrostatic
His247 2.5 electrostatic/polar
GlnAS Arg331 (L2) 2.5 polar
GInA 15 2.5 polar
$ Potential vicinal interactions are grouped
Minimum distance of approach modelled at 2.5 ~
* Closest approach; interaction would require a water molecule, hydrogen bond
chain
or a rotation of the entire L2 region
23
CA 02338678 2001-O1-26
WO 00/73793 PCT/CA00/00605
Individual amino acids in insulin that are important in binding 'to the
receptor
include: A 1, A4, A5, A 19, A2 i , B I 2; B 16, B 17, B24, B25 .and B26. On
the insulin
receptor amino acids that are involved in insulin binding include: i2, 14, 15,
34, 36, 39,
64, 86 89, 90, 91, 243-251, 323 and 707-716. Only amino acids 707-716 are not
in the
L1-CR-L2 domains. All others are either in the walls lining the ligand binding
site
tunnel or are at the entrance of the Iigand binding site.
Some examples of insulin derivatives and Humalog derivatives are provided
below.
is
Table I A
Table with Insulin Derivative Products
Insulin ResidueSubstitutions for Insulin Amino Acid
Residue
A chain
GluA4$ acidic amino acids (X,): Asp
GlnAS hyrophilic amino acids (X~): Thr,
Gln, Ser, Thr, Tyr
ThrA8 hyrophilic amino acids (X3): Asn,
Gln, Ser, Thr, Tyr
GhiAlS hyrophilic amino acids (X4): Thr,
Gln, Ser, Thr, Tyr
G1uA17 acidic amino acids (Xs): Asp
AsnA2l hyrophilic amino acids (X6): Thr,
Gln, Ser; Thr, Tyr
B chain
Ser B9 hydrophilic amino acids (Z,): Asn,
Gln, Thr, Tyr
HisB 10 basic amino acids (Z~): Lys, Arg
VaIB 12 hydrophobic (Z3): Ala, Leu, Ile, Pro,
Phe, Trp, Met,
Cys, Gly
GIuB 13 acidic amino acids (Z,): Asp
TyrB 16 hydrophilic amino acids acids (ZS):
Thr, Gln, Ser,
Thr, Asn
LeuBl7 hydrophobic amino acids (Z~): Ala,
- VaI, Ile, Pro,
Phe, Trp, Met, Cys, Gly
GIuB21 acidic amino acids (Z,): Asp
ArgB22 basic amino acids (Z8): Lys, His
PheB24 hydrophobic amino acids (Zg): Ala,
Val, IIe, Pro,
Leu, Trp, Met, Cys, Gly
PheB25 hydrophobic amino acids (Z,m): Ala,
Val, Ile, Pro,
Leu, Trp, Met, Cys, Gly
TyrB26 hydrophilic amino acids acids (Z"):
Thr, Gln, Ser,
Thr, Asn
LysB29 basic amino acids (Z12): His, Arg
24
i!.ii
CA 02338678 2001-O1-26
WO 00/73793 PCT/CAOO/OOb05
Human insulin
B-chain FVNQH LCGZiZ2 LZ3Z4AL ZSZ6VCG Z,Z8GZ9Z~a
ZmTPZI2T
A-chain GIVXIXz CCX3SI CSLYX4 LXSNYC X6 ,
Humalog
B-chain FVNQH LCGZIZz LZ,Z4AL ZSZ6VCG Z~ZgGZ9Zla
ZliTZisPT
Z13 may be substituted with basic amino acids: His,
Arg ,
l0 A-chain CJIVX,XZ CCX3SI CSLYX4 LXSNYC X6
Similar insulin derivatives may be made based on other insulin sequences,
such as bovine insulin and pig insulin in Figure 11.
Bovine Insulin
B-chain FVNQH LCGZ1Z2 LZ3Z4AL ZSZ6VCG Z.,ZgGZ9Z10
Z11TPZ12A
A-chain GIVX1X2 CCX,SV CSLYX4 LXSNYC X6
X7 may be substituted with a hydrophobic amino acid: VaI, Phe,
Ile, Pro, Leu, Trp, Met, Cys, Gly
Pig Insulin
B-chain FVNQH LCGZ~Zz LZ3Z4AL Z5Z6VCG Z.,ZBGZ9Z10
Z11TPZ~ZA
A-chain GIVX1X2 CC X3SI CSLYXQ LXSNYC X6
The invention includes a nucleic acid molecule encoding a polypeptide of the -
invention as well as a host cell including the nucleic acid molecule.
Interaction of modulatorx of IR cam
The invention also provides alternative and new methods to modulate IR
activity. For example, the 3D structure shows that Illt has two "cams" that
change the
conformation of the IR from an inactive conformation to an active
conformation. The
CA 02338678 2001-O1-26
WO OOI73793 PCT/CA00100605
existence of these cams was unknown prior to this invention. Modulators such
as
organic molecules (protein or non-protein) may black or activate cam movements
in
order to modulate the IR toward an inactive state or to an active state.
A modulator interacts with at least one insulin receptor residue listed in
Table
2 on the Carn-loop segment of the Cys-rich region and at least one residue in
Table 2
on the L 1 surface proximate the corn-loop segment in order to activate or
inhibit
insulin receptor. The modulator is capable of changing the IR from an inactive
conformation to an active conformation andlor biasing IR towards an active or
inactive conformation.
io The compound may also interact with at least: two, three, four, five or six
(or
seven, eight, nine, ten, eleven or twelve) of the residues listed in Table 2
on each of
the Cam-loop segment of the Cys-rich region and the L 1 surface proximate the
cam-
loop segment. The intersidechain distances between the test compound and the
IR
may be varied by plus or minus about: O.lA, 0.2A, 0.25A, 0.3A, 0.4A, 0.5A,
0.6A,
0.7A, 0.75A, 0.8A, 0.9A, lA or >lA, >1.5A or 2A as long as the test compound
is
still able to interact with IR and modulate its activity. It is apparent that
the modulator
must be able to make appropriate interactions with the IR cam if it is to
activate or
inactivate the IR.
Table 2
Charged and polar amino acids in the region of the cam-loop can bind a
modulator to the receptor, to allow specificity of binding, and to move or
block the
Cam-loop segment.
All specific interactions with the amino acids below would be electrostatic
(ionic) except with Gln (glutamine) and Asn (asparagine) which are polar.
Cam-loop segment L 1 surface
of Cys-rich near cam-loop
region segment
Lys265 electrostatic Glul TTH3+ electrostatic
Lys267 electrostatic AsnlS _____. -poly ___
Asn268 polar Asn l 6 polar
Arg270 electrostatic Arg 19 electrostatic
Arg272 electrostatic G1u22 electrostatic
26
CA 02338678 2001-O1-26
WO OOI73793 PCT/CA00/00605
G1u273 electrostatic G1u24 electrostatic
Asn25 polar
G1u44 polar
Asp45 electrostatic
Arg47 electrostatic
Asp48 electrostatic
Lys53 electrostatic
The invention includes a method of agonizing or antagonizing IR activity by
administering a modulator identified according to the methods of the
invention.
IR modulating compounds
A diagnostic or therapeutic modulating compound of the present invention can
be, but is not limited to, at least one selected from a nucleic acid, a
compound, a
protein, a lipid, a saccharide, an isotope, a carbohydrate, an imaging agent,
a
lipoprotein, a glycoprotein, an enzyme, a detectable probe, and antibody or
fragment
thereof, or any combination thereof. Diagnostic compounds (useful in diagnosis
as a
to research tool in an assay) can be detectably labeled as for labeling
antibodies. Such
labels include, but are not limited to, enzymatic labels, radioisotope or
radioactive
compounds ar elements, fluorescent compounds or metals, chemiluminescent
compounds and bioluminescent compounds. Other types of compounds may also be
useful.
The compound may include an amino acid sequence (including a peptide, a
polypeptide or a protein) or an amino acid sequence derivative (i.e. an
analog,
prepared for example by substituting, deleting, modifying (eg: glycosylating)
ane or
more amino acids - 'see, for example, US Patent Nos. 5,952,297, 5,922,6?S,
5,700,662, 5,693,609, 5,646,242, 5,149,777; 5,00,8241, 4,946,828 and
S,i64,366.
The analog may also be part of a human insulin analog complex, such as that in
US
5,474,978.).
The analog may be an insulin derivative, ata insulin precursor derivative or a
derivative of an already known insulin analog (See for example US Patent Nos.
27
CA 02338678 2001-O1-26
WO 00/73793 PCTICA00100605
5,952,297, 5,922,675, 5,747,642, 5,716,927). One skilled in the art may
analyze
insulin, its precursors, and other known analogs to determine how they
interact with
IR and then prepare improved compounds.
Those of skill in the art recognize that a variety of techniques are available
for
constructing derivatives with the same or similar desired biological activity
insulin but
with more favorable activity than the polypeptide with respect to route of
administration, .solubility, stability, and/or susceptibility to hydrolysis
and proteolysis.
See, for example, Morgan and Gainor, Ann. Rep. Med. Chem., 24:243-252 (1989).
Examples of polypeptide derivatives are described in U.S. Patent Nas.
5,643;873.
I o , Other patents describing how to make and use derivatives include, for
example,
5,786,322, 5,767,075, 5,763,571, 5,753,226, 5,683,983, 5,677,280, 5;672,584,
5,668,110, 5,654,276, 5,643,873. Derivatives may be designed ~ on computer by
comparing compounds to the 3D structures disclosed in this application.
Derivatives
of insulin may also be made according to other techniques known in the art.
For
example, by treating a polypeptide of the invention with an agent that
chemically
alters a side group by converting a hydrogen group to another group such as a
hydroxy or amino group. Derivatives can include sequences that are either
entirely
made of amino acids or sequences that are hybrids including amino acids and
modified amino acids or other organic molecules.
2o The compound may also be a nonprotein organic molecule, such as a mimetic
(i.e. a non-protein molecule which functionally mimics a peptide, polypeptide
or a
protein). For example, a mimetic may functionally mimic insulin by binding to
IR
and activating it. Such a mimetic rnay activate IR to a greater or lesser
extent than
that caused by insulin as long as the mimetic produces the end result of IR
activation.
Examples of mimetics are pyrrolidine compounds such as {2R,3R,4R)-3,4-
dihydroxy-
2-hydroxymethylpyrrolidine and other substituted 2-methylpyrrolidines {e.g. US
No.
5,854,272) or hydroxy alkyl piperidine (e.g. US No. 5,863,903). Small organic
molecules may also be used to antagonize or agonize IR by interacting with a
cam.
A compound can have a therapeutic effect on the target cells, the effect one
of
3o those known to be caused by modulation of IR. The therapeutic effects that
28
CA 02338678 2001-O1-26
WO 00/73793 PCT/CA00/00605
modulates at least one IR in a cell can be provided by therapeutic agent
delivered to a
target cell via pharmaceutical administration (discussed below).
Determining suitable types of modulators from IR structure
One skilled in the art would recognize, in view of the fitted quaternary
structure
of IR, that the type of modulator used may be varied or customized according
to the
portion, of IR targeted. For example, modulators may be simple peptides which
take
advantage of specific hydrophilic, hydrophobic, or charge interactions, or
variously
branched peptides with each branch differentially contributing to a particular
interaction
{such as the loligomer structures of Gariepy and co-workers: PNAS USA 92, 2056-
50,
1995; Bioconjugate Chern. 10, 745-54, 1999). Modulators may be simpler
chemicals
with corresponding interaction sites, in or near the insulin binding contact
sites of IR.
Such agents may also be molecules that act external to' the insulin binding
site to effect
activation or inhibition by interacting with specific sites identified as
important in the
mechanism of transmembrane signal transduction. These include specific
chemicals,
peptides or monoclonal and polyclonal antibodies or subantibody fragments such
as the
Fab, or Fv fragments. They include molecules that specifically remove or
enhance the
natural blockage on the insulin receptor to activation of its intrinsic
tyrosine kinase.
Such agents may also be molecules that enhance or inhibit transphosphorylation
of the
juxtaposing intrinsic pair of tyrosine kinase domains of the dimeric insulin
receptor.
2o Determining structure oJIR, IR variants acrd other receptors
Complete IR Structure
Techniques described in this application {such as those in references 4 and 5
or
US 5,834,228) were used to identify and characterize regions of an insulin
receptor
such as the LI-Cys-rich-L2 domain. We characterize the entire insulin receptor
and its
ligand binding site using these techniques. The fitted quaternary structure of
IR
needed for drug design is disclosed in this application.
IR Variants and Other Receptors
The IR data of the invention may be also used to solve the structure of IR
variants (eg. mutants, homologs) or other dimeric receptors, or of any other
protein
with significant amino acid sequence homology to any functional or structural
domain
29
CA 02338678 2001-O1-26
WO 00/73793 PCT/CA00/00605
of IR. We determine the structure of IR as well as mutants. IR has two
isoforms, A
and B. Isoform A is shorter than isoform B by 12 amino acids which are coded
by
exon 11 of the IR gene (the twelve amino acids are from Lys71$ to Arg 729 as
follows: Lys-Thr-Ser-Ser-Gly-Thr-Gly-Ala-Glu-Asp-Pro-Arg). Isofonn A interacts
with insulin and produces the same effect as isoform B, which is a metabolic
effect.
The insulin receptor described in this application was extracted from human
placenta. Insulin receptor from other sources, such as other tissues, cells or
cDNA
may also be modeled and used in the methods of the invention. The techniques
described in this application to image the receptor may be used with insulin
receptor
from any human, mammalian or other tissue. Insulin receptor homologues and
other
forms of insulin receptor, mutants and co-complexes of insulin receptor may
also be
used. A fragment of the receptor may also be used. A fragment may be from
about
25-50, about 50-I00, about 100-250 or about 250-500, 500-1000 or at least
about
1000 amino acids.
i5 The IR is similar to other dimeric receptors, such as IGFR and IRR. The 3D
structure of IR may be used to determine the 3D structure of these receptors
by
identifying regions of homology (similarity between amino acid, secondary,
tertiary or
quaternary structure) between the receptors and determining the structure of
the
dimeric receptor.
20 One useful method for this purpose is molecular replacement in
crystallography. In this method, the unknown structure in a crystal, whether
it is
another form of IR, an IR mutant, or the structure of some other dimeric
receptor with
significant.-ammo acid sequence homology to any functional domain of IR, may
be
determined using the IR structure coordinates of the IR dimer structure
coordinates of
25 this invention. This method will provide an accurate structural form for
the unknown
structure more quickly and efficiently.
Computer based design
The invention allows computational screening of molecule data bases for
compounds that can bind in whole, or in part, to IR. The IR structure of the
invention
30 permits the design and identif cation of synthetic compounds and/or other
molecules
CA 02338678 2001-O1-26
WO 00173993 PCT/CA00/00605
which have a shape complimentary of the conformation of the IR ligand binding
site
of the invention. Using known computer systems, the coordinates of the IR
structure
of the invention may be provided in machine readable form, the test compounds
designed and/or screened and their conformations superimposed on the
complementary surface structures and surface characteristics of the receptor
or of its
binding site. Subsequently, suitable candidates identified as above may be
screened
for the desired activity, stability, and other characteristics.
In this screening, the quality of fit of such entities or compounds to the
binding
site may be judged either by shape complementary (R.L DesJarlais et al. J.
Med.
to Chern 31:72-729 (1988) or by estimated interaction energy (E.C. Meng et al,
J. Comp.
Chem. 13: 505 - 524 (1992)].
Thus, the IR structure permits the screening of known molecules and/or the
designing of new molecules which bind to the IR structure, particularly at the
iigand
binding site or cams, via the use of computerized evaluation systems. For
example,
computer modeling systems are available in which the sequence of the IR, and
the IR
structure (i.e., atomic coordinates of IR and/or the atomic coordinate of the
Iigand
binding site cavity, bond angles, dihedral angles, distances between amino
acids in the
ligand binding site region, etc. as provided by the fitted quaternary
structure may be
input. A machine readable medium may be encoded with data representing the
2o coordinates of the entire IR structure. The computer then generates
structural details
of the site into which a test compound should bind, thereby enabling the
determination of the complementary structural details of said test compound.
- The production of compounds that bind to or modulate IR generally two
factors. First, the compound must be capable of physically and structurally
associating with IR. Non-covalent molecular interactions important in the
association
of IR with its substrate include hydrogen bonding, ionic interactions van der
Waals
interactions and hydrophobic interactions.
The invention permits the design of agents that bind to the three dirnentional
surfaces of IR by using the pattern on those surfaces of positive charges,
negative
charges, hydrophobic grouping of atoms, dipolar groups and hydrodren bonds
that are
31
CA 02338678 2001-O1-26
WO OOI73793 PCT/CA00/00605
revealed in the structure of the surfaces and in the relative positioning of
these
surfaces with respect to each other in the quaternary structure.
Those skilled in the art can create an agent that places the positions of
chemical groups on the agent near matching atoms or groups of atoms on IR
using
well-known interactions such those as in Table3.
Table 3
Characteristics of atoms or Matching characteristics of atoms or
~eroups of atoms on IR ~rouns of atoms on the aeent
- positive charge - negative charge
- negative charge - positive charge
- hydrophobic group - hydrophobic group
- polar group - polar group
- hydrogen donor - hydrogen acceptor
- hydrogen acceptor - hydrogen donor
~ Second, the compound must be able to assume a conformation that allows it to
associate with IR. The compound will preferably interact with the Iigand
binding site
or a cam and bias or change IR towards either an active conformation or
inactive
conformation. Although certain portions of the compound will not directly
participate
in this association with IR those portions may still influence the overall
conformation
of the molecule. This, in turn, may have a significant impact on potency. Such
conformational requirements include the overall three-dimensional structure
and
orientation of the chemical entity or compound in relation to all or a portion
of the
binding site, e.g., ligand binding site, accessory binding site, or cam of IR
or the
spacing between functional groups of a compound comprising several chemical
entities that directly interact with IR.
The potential modulating effect of a chemical compound with IR may be
estimated prior to its actual synthesis and testing by the use of computer
modeling
techniques. If the structure of the compound shows insufficient interaction
and
association between it and IR the compound is riot synthesized and tested. If
3o computer modeling indicates a suitable interaction, the molecule may then
be
32
CA 02338678 2001-O1-26
w0 00/73793 PCT/CA00/00605
synthesized and tested for its ability to bind to IR in an assay: Synthesis of
ineffective
and inoperative compounds can be avoided.
Computer modeling may be combined with assay techniques. For example,
one could probe the IR (or fragments thereof} with a variety of different
molecules to
determine optimal sites for interaction between candidate modulators and IR.
Small
molecules that bind tightly to IR sites can be designed and synthesized and
tested for
their IR modulatory activity. This information can be combined with computer
modeling information. A modulating compound may be computationally evaluated.
A modulating compound may be further designed by a series of steps in which
to compounds or fragments are screened and selected for their ability to
associate with
the individual binding amino acids, secondary, tertiary or quaternary
structure or other
areas of IR.
One skilled in the art may use one of several methods to screen chemical
entities or fragments for their ability to interact with IR. This process may
begin
generating the ligand binding site on the computer screen based on the IR
amino acids
and distances from the co-ordinates of the IR complex. Selected fragments or
chemical entities are then be positioned against IR. Docking may be
accomplished
using software such as Insight, Quanta, and Sybyl, followed by energy
minimization
and molecular dynamics with standard molecular mechanics forcefields, such as
CHARMM and AMBER.
Specialized computer programs may also assist in the process of selecting
fragmented or chemical entities. These include:
MCSS (Molecular Simulations, Burlington, MA) [A: Miranker and M.
Karpius. "Functionality Maps of Binding Sites: A Multiple Copy Simultaneous
Search Method". Proteins: Structure, Function and Genetics, 11:29-34 (1991)].
GRID (Oxford University, Oxford, UK) [P.J. Goodford, "A Computational
Procedure for Determining Energetically Favorable Binding Sites on
BioIogicaliy
important Macromolecules". J. Med. Chem. 28:849-857 (1985)].
33
CA 02338678 2001-O1-26
WO 00/73793 PCTICA00/00605
DOCK (University of California, San Francisco, CA) [LD. Kuntz et al, "A
_ Geometric Approach to Macromolecule-Ligand Interactions", 3. Mol. Biol. 161:
269-
288 (1982)].
AUTODOCK (Scripps Research Institute, La Jolla, CA) [D.S. Goodsell and
A.J. Olsen, "Automated Docking of Substrates to Proteins by Simulated
Annealing".
Proteins: Structure, Function, and Genetics, 8: i 92-202 ( 1990)].
Additional commercially available computer databases for small molecular
compounds include Cambridge Structural Database and Fine Chemical Database.
For
a review see Rusinko, A., Chem. Des., Auto. News 8.44-47 (1993).
For example, software such as GRID (a program that determines probable
interaction sites between probes with various functional group characteristics
and the
enzyme surface) analyzes the ligand binding site to determine structures of
modulating compounds. The program calculates, with suitable activating or
inhibiting
groups on molecules (e.g. protonated primary amines as the probe) suitable
conformations. The program also identifies potential hot spots around
accessible
positions at suitable energy contour levels. Suitable ligands, such as
inhibiting or
activating compounds or compositions, are then tested for modulating IR.
Once suitable chemical entities or fragments have been selected, they can be
assembled into a single compound. Assembly may be proceeded by visual
inspection
of the relationship of the fragments to each other on the three-dimensional
image
displayed on a computer screen in relation to the structure coordinates of IR.
This
would typically be followed by manual model building using software such as
Quanta
or Sybyl.
Useful programs to aid one of skill in the art in connecting the individual
2s chemical entities or fragments include:
3D Database systems such as, MACCS-3D (MDL Information Systems, San
Leandro, CA). See Y.C. Martin, "3D Database Searching in Drug Design", J.Med.
Chem., 35:2145-2154 (1991).
34
CA 02338678 2001-O1-26
WO 00/73793 PCTlCA00100605
CAVEAT (University of California, Berkeley, CA) [P.A. Barlett et al.
"CAVEAT: A program to Facilitate the Structure Derived design of Biologically
Active Molecules," in Molecular Recognition in Chemical and Biological
Problems."
Special Pub., Royal Chem. Soc. 78, pp 182-196 (1989).
HOOK (Molecular Simulations, Burlington, MA). Instead of proceeding to
build IR modulator in a step-wise fashion one fragernent or chemical entity at
a time
as described above, inhibitory or other type of binding compounds may be
designed as
whole or "de novo" using either an empty Iigand binding site or optionally
including
some portions) of a known compound(s). These methods include:
to LUDI (Biosym Technologies, San Diego.CA) [H.-J. Bohm, "The Computer
Program LUDI: A New method for the De Novo Design of Enzyme Inhibitors", J.
Comp, Aid Molec, Design, 6:61-78 (1992)].
LeapFrog (Tripos Associates, St. Louis, MO). Other molecular modeling
techniques may also be used. For example,., N:C. Cohen et al. "Molecular
Modeling
Software and Methods for Medicinal Chemistry". ~J.Med.Chem., 33:883-894
(1999).
M.A. Navia and M. A. Murcko, "The Use of Structural Information in Drug
Design",
Current Opinions in Structural Bioloev, 2:202-210 (1992). For example, where
the
structures of test compounds are known, a model of the test compound may be
superimposed over the model of the structure of the invention. Numerous
methods
2o and techniques are known in the art for performing this step, any of which
may be
used. See, e.g:, P.S. Farmer, Drug Design, Ariens, E.J., ed" Vol. 10, pp II9-
143
(Academic Press, New York, I980)U.S. Patent No. 5,331,573; U.S. Patent No.
5,500,807; C. Verlinde, Structure. 2:577-587 (1994); and LD. Kuntz, Science,
257:1078-1082 (1992). The model building techniques and computer evaluation
systems described herein are not a limitation on the present invention.
LEGEND (Molecular Simulations, Burlington, MA) [Y. Nishibata and A. Itai,
TetrahedronL47:8985 (1991)].
Using these computer evaluation systems, a large number of compounds may
be quickly and easily examined and expensive and lengthy biochemical testing
CA 02338678 2001-O1-26
WO 00/73793 PCT/CA00/00605.
avoided. Moreover, the need for actual synthesis of many compounds is
effectively
eliminated.
Apparatus including the IR fitted quaternary structure or other IR
structural information
Storage media for the IR fitted quaternary structure or other IR structural
information include, but are not limited to: magnetic storage media, such as
floppy
discs; hard disc storage medium, and magnetic tape; optical storage media such
as
optical discs or CD-ROM; electrical storage media such as RAM and ROM; and
hybrids of these categories such as magnetic/optical storage media. Any
suitable
tp computer readable mediums can be used to create a manufacture comprising a
computer readable medium having recorded on it an amino acid sequence andlor
data
of the present invention.
"Recorded" refers to a process for storing information on computer readable
medium. A skilled artisan can readily adopt any of the presently know methods
for
recording information on computer readable medium to store an amino acid
sequence,
nucleotide sequence and/or EM data information of the present invention.
A variety of data storage structures are available to a skilled artisan for
creating a computer readable medium having recorded thereon an amino acid
sequence and/or data of the present invention. The choice of the data storage
structure
2o will generally be based on the means chosen to access the stored
information. In
addition, a variety of data processor programs and formats can be used to
store the
sequence and data information of the present invention on computer readable
medium.
The sequence information can be represented in a word processing text file,
forinatted
in commercially-available software such as WordPerfect and Microsoft Word, or
represented in the form of an ASCII f le, stored in a database application,
such as
DB2, Sybase, Oracle, or the like. A skilled artisan can readily adapt any
number of
data processor structuring formats (e.g. text file or database) in order to
obtain
computer readable medium having recorded thereon the information of the
present
invention.
36
CA 02338678 2001-O1-26
WO OOI73793 PCT/CA00100605
By providing the sequence andlor data on computer readable medium and the
structural information in this application, a skilled artisan can routinely
access the
sequence and data to model a receptor a subdomain thereof, or a ligand
thereof. As
described above, computer algorithms are publicly and commercially available
which
allow a skilled artisan to access this data provided in a computer readable
medium and
analyze it for molecular modeling or other uses.
The present invention further provides systems, particularly computer-based
systems, which contain the sequence and/or data described herein. Such systems
are
designed to do molecular modeling for an IR or at least one subdomain or
fragment
l0 thereof.
In one embodiment; the system includes a 'means for producing a fitted
quaternary structure of insulin receptor (or a fragment or derivative thereof)
and
means for displaying the fitted quaternary structure of insulin receptor. The
system is
capable of carrying out the methods described in this application. The system
preferably further includes a means for comparing the structural coordinates
of a test
compound to the structural coordinates of the insulin receptor (or ~a fragment
or
derivative thereof, such as a cam-loop, L 1 region, ligand binding site or
other region
described in this application). and means for determining if the test compound
is
capable of modulating insulin receptor between an active conformation and an
2o inactive conformation or biasing insulin receptor toward an active or
inactive
conformation, as described in the methods of the invention.
As used herein, "a computer-based system." refers to the hardware means,
software means; and data storage means used to analyze the sequence and/or
data of
the present invention. The minimum hardware means of the computer-based
systems
of the present invention comprises a central processing unit (CPU), input
means,
output means, and data storage means. A skilled artisan can readily appreciate
which
of the currently available computer-based system are suitable for use in the
present
invention.
As stated above, the computer-based systems of the present invention
comprise a data storage means having stored therein our IR or fragment
sequence
37
CA 02338678 2001-O1-26
WO 00/73793 PCT/CA00/00605
and/or data of the present invention and the necessary hardware means and
software
means for supporting and implementing an analysis means. As used herein, "data
storage means" refers to memory which can store sequence or data (coordinates,
distances, quaternary structure etc.) of the present invention, or a memory
access
means which can access manufactures having recorded thereon the sequence or
data
of the present invention.
As used herein, "search means" or "analysis means" refers to one or more
programs which are implemented on the computer-based system to compare a
target
sequence or target structural motif with the sequence or data stored within
the data
1o storage means. Search means are used to identify fragments or regions of an
IR which
match a particular target sequence or target motif. A variety of known
algorithms are
disclosed- publicly and a variety of commercially available software for
conducting
search means are and can be used in the computer-based systems of the present
invention. A skilled artisan can readily recognize that any one of the
available
is algorithms or implementing software packages for conducting computer
analyses that
can be adapted for use in the present computer-based systems.
As used herein, "a target structural motif," or "target motif," refers to any
rationally selected sequence or combination of sequences in which the
sequences(s)
are chosen based on a three-dimensional configuration or electron density map
which ,
20 is formed upon the folding of the target motif. There are a variety of
target motifs
known in the art. Protein targets include, but are not limited to, ligand
binding sites,
structural subdomains, epitopes, and functional domains. A variety of
structural.
formats for the input and output means can be used to input and output the
information in the computer-based systems of the present invention.
25 One application of this embodiment is provided in Figure 13. This figure
provides a block diagram of a computer system 5 that can be used to implement
the
present invention. The computer system 5 includes a processor 10 connected to
a bus
15. Also connected to the bus 15 are a main memory 20 (preferably implemented
as
random access memory, RAM) and a variety of secondary storage memory 25 such
as
3o a hard drive 30 and a removable storage medium 35. The removable medium
storage
38
CA 02338678 2001-O1-26
WO 00/73793 PCT/CA00l00605
device 35 may represent, for example, a floppy disk drive, A CD-ROM drive, a
magnetic tape drive, etc. A removable storage unit 40 (such as a floppy disk,
a
compact disk, a magnetic tape, etc.) containing control logic and/or data
recorded
therein may be inserted into the removable medium storage medium 35. The
computer system 5 include appropriate software for reading the control Logic
and/or
the data from the removable medium storage device 35 once inserted in the
removable
medium storage device 35. A monitor 45 can be used as connected to the bus 15
to
visualize the structure determination data.
Amino acid, encoding nucleotide or other sequence and/or data of the present
1o invention may be stored in a well known manner in the main,memory 20, any
of the
secondary storage devices 25, andlor a removable storage device 40. Software
for
accessing and processing the amino acid sequence andlor data (such as search
tools,
comparing tools, etc.) reside in main memory 20 during execution.
One or more computer modeling steps and/or computer algorithms are used as
1s described above to provide a molecular 3-D model, preferably showing the
fitted
quaternary structure, of a cleaved dimeric receptor, using amino acid sequence
data
and atomic coordinates for the receptor. The structure of other dimeric
receptors such
as IGFR and IRR may be readily determined using methods of the invention and
the
present knowledge of these receptors.
20 Assays of modulators ident fed from IR structure
Once identified, the modulator may then be tested for bioactivity using
standard techniques (e.g. in vitro or in vivo assays). For example, the
compound
identified by drug design may be used in binding assays using conventional
formats to
screen agonists (e.g by measuring in vivo or in vitro binding of receptor to
insulin
25 after addition of a compound). One assay is the fat cell assay for glucose
uptake and
oxidation which is known in the art. Experiments may also be done with whole
diabetic animals. Suitable assays include, but are not limited to, the enzyme-
linked
immunosorbent assay (ELISA), or a fluorescence quench assay. In evaluating IR
modulators for biological activity in animal models {e.g. rat, mouse, rabbit),
various
30 oral and parenteral routes of administration are evaluated: Using this
approach, it is
39
CA 02338678 2001-O1-26
WO 00/73793 PCT/CA00/00605
expected that modulation of an IR occurs in suitable animal models, using the
ligands
discovered by molecular modeling.
Once identified and screened for biological activity, these inhibitors may be
used therapeutically or prophylactically to modulate IR activity as described
below.
PharmaceuticaUdiagnostic formulations of modulators ident~ed from
quaternary structure, methods of medical treatment and uses
Modulating IR in a Cell
- The present invention also provides a method for modulating the activity of
the IR in a cell using IR modulating compounds or compositions of the
invention. In
~o general; compounds (antagonists or agonists) which have been identified to
inhibit or
enhance the activity of IR can be formulated so that the agent can be
contacted with a
cell expressing a IR protein in vivo. The contacting of such a cell with such
an agent
results in the in vivo modulation of the activity of the IR proteins. So long
as a
formulation barrier or toxicity barrier does not exist, agents identified in
the assays
described above will be effective for in vivo and in vitro use. These
modulators may
be used in therapies that are beneficial in the treatment of diabetes and
other diseases,
disorders and abnormal physical states characterized by improper or inadequate
insulin receptor activity. Even if receptor activity is normal, there may be
therapeutic
benefit in upregulating or downregulating its activity in some circumstances.
Medical Treatments and Uses
Diseases, disorders and abnormal physical states that may be treated by IR
agonists include diabetes and hyperlgycemia. Diseases, disorders and abnormal
physical states that may be treated by IR antagonists include hypoglycemia.
Isoform A of IR is shorter than isoform B by 12 amino acids which are coded
by exon 1 I of the IR gene. Isoform A interacts with insulin and produces the
same
effect as isoform B, which is a metabolic effect. Isoform A acts as an IGF-2
receptor
which may be important in the growth of cancer cells (Frasca, F, Pandini, G,
Scalia, P,
Sciacca, L, Mineo, R, Costantino, A, Goldfine, ID, Delfiore, A, Vigneri, R,
1999,
Insulin receptor isoform A: A newly recognized high affinity insulin like
growth
3o factor II receptor in situ and cancer cells. Molecular and Cellular Biology
19:5 pg.
CA 02338678 2001-O1-26
WO 00/73793 PCT/CA00/00605
3278-3288.). IGF-2 acts on isoform A to produce a growth effect via IR rather
than
just a metabolic effect. The quaternary structure of isoform A is very similar
to
isoform B and can be readily determined according to the information in this
application. IGF I binds to both isoforms with low affinity (1/10) and also
produces a
growth effect (less significant because of the low affinity binding). One may
design
an antagonist of isoform A that does not interact with isoform B (or at least
has Iawer
affinity binding to isoform B) to inhibit cancer cell growth in response to
IGF-2.
Pharmaceutical Compositions
Modulators may be combined in pharmaceutical compositions according to
to known techniques. The compounds of this invention are preferably
incorporated into
pharmaceutical dosage forms suitable for the desired administration route such
as
tablets, dragees, capsules, granules, suppositories, solutions, suspensions
and
lyophilized compositions to be diluted to obtain injectable liquids. The
dosage forms
are prepared by conventional techniques and in addition to the compounds of
this
invention could contain solid or liquid inert diluents and carriers and
pharmaceutically
useful additives such as lipid vesicles liposomes, aggregants, disaggregants,
salts for
regulating the osmotic pressure, buffers, sweeteners and colouring agents.
Slow
release pharmaceutical forms for oral use may be prepared according to
conventional
techniques. Other pharmaceutical formulations are described for example in US
5,192,746.
Pharmaceutical compositions used to treat patients having diseases, disorders
or abnormal physical states could include a compound of the invention and an
~ceptable vehicle or excipient (Remington's Pharmaceutical Sciences 18''' ed,
(1990,
Mack Publishing Company) and subsequent editions). Vehicles include saline and
DSW (5% dextrose. and water). Excipients include additives such as a buffer,
solubilizer, suspending agent, emulsifying agent, viscosity controlling agent,
flavor,
lactose filler, antioxidant, preservative or dye. The compound may be
formulated in
solid or semisolid form, for example pills, tablets, creams, ointments,
powders,
emulsions, gelatin capsules, capsules, suppositories; gels or membranes.
Routes of
3o administration include oral, topical, rectal, parenteral (injectable),
local, inhalant and
41
CA 02338678 2001-O1-26
WO 00173793 PCTICA00100605
epidural administration. The compositions of the invention may also be
conjugated to
transport molecules to facilitate transport of the molecules. The methods for
the
preparation of pharmaceutically acceptable compositions which can be
administered
to patients are known in the art.
The pharmaceutical compositions can be administered to humans or animals.
Dosages to be administered depend on individual patient condition, indication
of the
drug, physical and chemical stability of the drug, toxicity, the desired
effect and on
the chosen route of administration (Robert Rakel, ed., Conn's Current Therapy
(1995,
W.B. Sounders Company, USA))
l0 Polypeptides, such as the insulin derivatives described above, may be
produced for use in pharmaceutical compositions using known techniques. For
example, NovolinTM, a recombinant human insulin, is produced in Saccharmyces
cerevisiae. Other host cells include any cell capable of producing the
polypeptide,
such as a cell selected from the group consisting of a plant, a bacterial,
fungus (eg.
yeast), protozoa, algal or animal cell.
One may prepare a nucleic acid molecule encoding a polypeptide designed by
a method of the invention (including the insulin derivatives described above).
Recombinant nucleic acid molecules include the nucleic acid molecule and a
promoter
sequence, operatively linked so that the promoter enhances transcription of
the nucleic
2o acid molecule in the host cell. The nucleic acid molecules can be cloned
into a variety
of vectors by means that are well known in the art. A number of suitable
vectors may
be used, - including cosmids, plasmids, bacteriophage, baculoviruses and
viruses.
Preferable vectors are capable of reproducing themselves and transforming a
host cell
(Sambrook, J, Fritsch, E.E. & Maniatis, T. (1989}. Molecular Cloning: A
laboratory manual.
Cold Spring Harbor Laboratory Press. New York; AusubeI et al. (1989) Current
Protocols in
Molecular Biology, John Wiley & Sons, Inc.). The metlhods of the invention
further include
preparing nucleic acid molecules, recombinant nucleic acid molecules, vectors
and host
cells (the invention also includes the aforementioned products themselves).
The
nucleic acid molecules, recombinant nucleic acid molecules and vectors are
also
useful for gene therapy, for example, by transforming pancreatic cells that
produce
insulin. Gene therapy methods and compositions are taught, for example, in
U.S.
42
CA 02338678 2001-O1-26
WO 00173793 PCT/CA00/00605
Patent Nos. 5,672,344, 5,645,829, 5,741,486, 5,656,465; 5,547,932, 5,529,774,
5,436,146, 5,399,346 and 5,670,488, 5,240,846. The method can preferably
involve a
method of delivering a nucleic acid molecule encoding a polypeptide of the
invention
to the cells of an individual having diabetes, comprising administering to the
individual a vector comprising DNA encoding a polypeptide of the invention.
The
invention includes methods and compositions for providing a nucleic acid
molecule
encoding the polypeptide to the cells of a subject (preferably a human) such
that
expression of the nucleic acid molecule in the cells provides the biological
activity or
phenotype of the polypeptide to those cells. Sufficient, amounts of the
nucleic acid
molecule are administered and expressed at sufficient levels to provide the
biological
activity or phenotype of the polypeptide to the cells.
Example 1 - Determination of the 3D structure of ~R
Preparation of IR
Insulin receptor protein (HIR) was solubilized from human placental membranes
and
purified by affinity chromatography on an insulin column (9) followed by
further FPLC
purification on Sephacryl S-200. The purity of HIR was better than 95% by
sodium
dodecyl sulfate polyacrylamide gel electrophoresis. HIR was incubated with NG-
BI
(final concentration of ~ 0.5 x 10'~ M) at 4° C overnight in 20 mM
HEPES buffer (pH
7.5) at a molar ratio of insulin:HIR of ~ 10:1. Free NG-BI was removed by
2o microfiltration with a cut-off of 300 kDa (Sigma). The mixture was diluted
to 7.5 p,g of
receptor protein/ml with 20 mM HEPES buffer, pH 7.5, prior to loading on the
grid.
Preparation of Specimen for STEM
The specimen (5 p,l) was injected into 5 p,l of the dilution buffer on 300-
mesh
copper grid coated with a holey plastic film overland with a carbon film 23 A
thick,
washed with HEPES buffer and 10 mM ammonium acetate (pH 7.5). The grid was
drained by wicking with f lter paper, leaving a very thin solution layer, then
immediately quick-frozen by plunging into liquid ethane at -150°C. The
frozen
s~cimen was transferred at liquid nitrogen temperature into the STEM (Vacuum
Generators, Model HB601LJ~ and freeze-dried at -140° C in the STEM cold-
stage.
3o Images in a 480 x 480 pixel format were acquired with the specimen at -
150°C using
43
CA 02338678 2001-O1-26
WO 00/73793 PCT/CA00/00605
cold field emission at 100 kV, a dose of 6e1~2 and a pixel size of 6.5 t~. The
beam size
was 3A. Inelastic and annular dark field signals were detected simultaneously.
Nanogold Marking
The quaternary structure of IR bound to insulin was determined by marking with
Nanogold. The 70 atom gold marker localized and delimited the insulin binding
site.
Compared to native bovine insulin, Nanogold-bovine-insulin (NG-BI),
derivatized at the
B-chain Phel(5), a location not directly involved in receptor binding (6),
bound to
human insulin receptor (HIR) with only a slightly reduced affinity (Fig. 1 ).
Purified
solubilized HIR used in this study has been shown to be fully active (7). Such
HIR,
to incubated with NG-BI to form the HIRING-BI complex in the absence of ATP,
was
subjected to low-dose dark field STEM imaging at -150° C. Figure 2A
shows a
representative f eld of individual molecules. On average, each HIRING-BI
complex
measured 15 nm across. Based on its strong scattering, the I .4 nm gold ligand
of NG-BI
was located on the itriage directly as a clear site of highest density, or
could be
demonstrated as such by thresholding. Figure 2B shows examples of molecules
with 1
or 2 sites of highest density, indicative of binding of one or occasionally
twa NG-BI
particles, consistent with the known binding of between one and two insulins
per IR (3).
When two NG-BI particles were detected, they were in close proximity to each
other.
Image Reconstruction
Approximately 700 images were selected far reconstruction on the basis of
having a definite site of high density, the expected mass for the complex,
being
structurally contiguous, and being separated from neighbouring images. The 3D
reconstructions of the HIR/NG-BI complex are shown in Figure 3. The
interpreted
alignment and the fit of the biochemical domains to this structure are
detailed in Fig. 4.
The 3D structure at the full expected volume is compact and globular (Fig. 3A,
top
panel). The NG-BI particle was located on the 3D reconstruction by increasing
the
density threshold without imposing symmetry (Fig. ~A, panel 2 and 3), to
pinpoint the
binding site and to Iimit the fit of insulin to its vicinity within the IR
complex. Since
3o insulin binds to the L1-Cys-rich-L2 regions of the ectodomain of IR, the NG
cluster
identifies this region of IR in the reconstruction.
44
CA 02338678 2001-O1-26
WO 00/73793 PCT/CA00/00605
Paired elastic and inelastic images were combined to increase the signal-to-
noise
ratio two-fold. Single particles were interactively selected in 64x64 pixel
windows
using the program WEB (Wadsworth Laboratories, Albany NY), and low-pass
filtered
to 1.0 nm using a Gaussian filter in the program SPIDER (Wadsworth
Laboratories,
Albany NYJ. The molecular mass was calculated relative to the 23 ~ carbon
support
with a density of 2.0 g/cm3. The particles had a Gaussian mass distribution
with a
modal mass of 570 kDa, which includes the mass of 480 kDa for the HIR and NG-
BI
plus the weight for an estimated 150 Triton X-100 molecules. Particle images
were
"grown" from a central high density in expanding contiguous contour levels to
a global
l0 cut-off corresponding to the average mass. Relative orientations were
computed as
before (N. A. Farrow and F. P. Ottensmeyer, J. Opt. Soc: Am. A9, 1749 (1992);
N. A.
Farrow and F. P. Ottensmeyer, Ultramicroscopy 52, 141 (1993); G: J. Czarnota,
D. W.
Andrews, N. A. Farrow, F. P. Ottensmeyer, J. Structural Biology 113, 35
(1994); G. J.
Czarnota, D. P. Bazett-Jones, E. Mendez, V. G. Allfrey, F. P. Ottensmeyer,
Micron 28,
419 (1997)) and 3D reconstructions were performed by filtered back-projection
using an
angular distribution-dependent filter. Measurements of resolution were
obtained via
Fourier shell phase residual calculations between reconstructions of two
independent
sets of half of the 704 images each (G.J. Czarnota, D.W. Andrews, N.A. Farrow,
F.P.
Ottensmeyer, J. Struct. Biol. 113, 35 (1994)). Calculations were carried out
on an SGI
2o Indigo workstation (Silicon Graphics Inc., Mountain View, CA). The program
IRIS
EXPLORER 2.0 (SGI, Mountain View, CA) displayed the 3D reconstructions. To
show domain relationships and structural links, the reconstructions were
displayed with
intermediate densities between 5% and 10% higher than the average density for
the full
volume. INSIGHT II (Molecular Simulations Inc., San Diego, CA) was used to
dock
known crystal structures and approximate models. Handedness of the construct
was
determined by fitting the x-ray crystallographic structure of tyrosine kinase
domain into
mirror pairs of the 3D reconstruction.
Example 2 - Structural Characteristics of IR
Domain-like features of the structure become evident at intermediate density
3o thresholds (Figure 3A, panel 2), and, except for the NG-BI region, these
indicate a
strong 2-fold vertical rotational symmetry as anticipated from the dimeric
configuration
CA 02338678 2001-O1-26
WO OOI73793 PCT/CA00l00605
of the oligotetrameric (a~i)2 structure of IR. This symmetry was used to
reduce noise in
the reconstructions and render the structures shown in panel l and in Figure
3B, as being
viewed in the plane of the membrane, and in the extracellular (top) and
intracellular
(bottom) perspectives. Views of these structures are reminiscent of the X- and
Y-shaped
electron microscopic images previously observed for IR or its ectodomain.
In the side views, the top part of the structure, where NG is located, is
identified
as the ectodomain of the a subunit. The dog-bone-shaped substructure of the 3D
reconstruction, (Fig. 3B, top view), and equivalently the top-most, bow-tie-
shaped
structure (Fig. 3B, 0°), are designated as the two L1 domains of the
dimeric receptor on
to the basis of the x-ray structure of the Ll-Cys-rich-L2 domains. The side
view at 65°
shows the L1-Cys-rich-L2 domains as contiguous substructures across the upper
central
region of the molecule, with enough additional volume in this region to
account for most
of the remaining mass of the two a subunits, primarily the connecting domains
{CD).
The contiguity of the domain structure (Fig. 3B, top and side view
90°), along
i5 with the primary domain sequence (Fig. 4A), shows that the two (3 subunits
occupy the
Lower half of the structure, distal from L 1, reaching up and out as a
contiguous mass.
The intracellular TK domain of IR would then occupy the bottom portion of this
structure with two IR fibronectin type III (FnIII) repeats in each receptor
half being in
the top portion of the crescent-shaped spiral of the ~i subunit at the same
level as the L2
20 domain in the a subunit. One of the FnIII repeats, composed of residues
from both the
a and ~i subunit, is assigned to the upper left end of the crescent (side
view, 0°) where it
is contiguous with the CD portion of the a subunit (top view). Fig. 4C and 4D
(cf. Fig.
3B, 90°, top view, respectively) show the fitting of the crystal
structure of the TK
domain of the j3 subunit and of the two FnIII repeats modelled as the
canonical
25 fibronectin type III structures (16).
The masses of the kinase domains are connected via a slender horizontal bridge
(Fig. 3B; side view 90°) that was not observed in the x-ray structures
of the TKs, but can
be explained in terms of the reconstruction being in a transition between free
IR and its
ligand-activated form. In the two symmetrically fitted TK (Fig. 4C and 4D)
crystal
3o structures the catalytic loops are separated by 4 nm. This distance is just
sufficient to
permit the tyrosine triplet (Tyr1158, 1162 and 1163) in a fully extended
flexible
46
CA 02338678 2001-O1-26
WD 00173793 PCT/CAOOI00605
activation loop of one TK to reach the catalytic loop of the opposite TK as
modelled
from the x-ray coordinates (PDB l IRp). The extension of the activation loops,
equivalent in cross-section to four extended polypeptide chains, easily
accounts for the
linking density observed between the lower portions of the ~i subunits (Fig.
3B, 90°).
This is an important difference frnrn the x-ray structures of the inactive and
activated
TKs as discussed below.
The spatial relationship between the domains of the a and ~i subunits (e.g:
side
view, 90°) shows the location of the cell membrane lipid bilayer as the
space below the
a subunits and above the bridge linking the two assigned TK domains. Instead
of a flat
open region, this space in the 3D reconstruction forms a thick dome-like slab
above the
bridge with a thickness variation of 2.2 to 2.7 nm. This spacing is a change
in shape
from, and a decrease in the thickness expected for a membrane bilayer that
would
accommodate an alpha-helical transmembrane domain (TM) of 23-26 hydrophobic
amino acids. However, since the purified IR in the absence of its native
membrane was
fully active, the relative positions of the extracellular and intracellular
domains must still
represent a close to native arrangement.
The crossing Ll-Cys-rich-L2 domains of the dimeric a subunits were
presented (Fig. 4B and 4C). We determined the x-ray coordinates with IR from
the
domain structures (5) (See Fig. 7). Using this structure, the localization of
the gold
2o cluster, and the known receptor-binding domain of insulin (8), we have
fitted an NG-
BI molecule into this region. The best fit is obtained with a molecule of
insulin,
partially on the two-fold symmetry axis of the dimer, being in contact with
the L I -
Cys-rich domains of one a subunit and with the L2 domain of the other a
subunit. A
model involving both a subunits in the high-affinity binding of insulin has
previously
been proposed based on studies of insulin analogues binding to IR and IR/IGF-1
R
chimeras (8). Our 3D reconstruction shows this involvement. Although two
molecules of insulin can be fitted to this configuration, two molecules of
Nanogold-
labeled insulin were observed only rarely in the STEM images. The high-
affinity
binding of the first insulin molecule to the IR has induced a conformational
change in
3o the binding domain so that the second insulin molecule would bind only at
law
affinity. Likewise the binding of a second molecule of insulin could effect a
47
CA 02338678 2001-O1-26
WO OOI73793 PCTICA00/00605
conformational change that enhances the dissociation of the bound insulin.
Thus the
curvilinear Scatchard plot and the negative cooperativity of insulin binding
(9} can be
explained on the basis of the 3D reconstruction. The reconstruction also
explains why
only low-affinity binding is obtained with purified a(3 monomer.
Superimposition of known crystal structures of smaller domains of the receptor
on substructures of the 3D reconstruction has made it possible to deduce the
spatial
relationship among the domains in the complex. The structure shows the
division of the
complex into the extracellular and the cytoplasmic segments along a plane, the
cell
membrane, on which the fibronectin type III repeats lie (16-18). These repeats
appear
1o pontoon-like to support the centrally located insulin-binding segment of
the ectodornain.
Monomeric inactive receptor TKs such as EGFR are brought, together by iigand
binding and become activated as dimers resulting in TK autophosphorylation. In
the
intrinsically dimeric IR-family receptors, the distance between the two
cytoplasmic (3-
subunit TKs within the dimer must be too great without ligand binding for the
activation
t 5 of the kinase. Hubbard et al. (4) suggested that insulin binding to IR
decreased this
distance by disengaging Tyrl 162 fromthe catalytic loop to enable traces
phosphorylation
in the presence of ATP. In our reconstruction a good f t to the ligand-
receptor complex
is obtained when the two TK domains are oriented with their catalytic loops
juxtaposed.
In this orientation the extended flexible activation loop of each TK, which
moves 30 ~
2o between the inactive and activated states in the crystal structures (4),
can just reach the
catalytic loop of the opposing TK to be activated. These two loops can easily
form the
linking mass density between the TKs seen in the 3D reconstruction in the
absence of
ATP.
The 3D structure obtained from images of the HIR-complex containing only a
25 single NG-BI, shows that one molecule of insulin is sufficient to bring the
two a(3
monomers to an activating configuration. The dimeric receptor with a Ser323Leu
mutation in the L2 domain of both a subunits showed a severe impairment in
insulin
binding, whereas a hybrid receptor with only one of the two a subunits mutated
was
found to bind insulin with high affinity and was fully active as a tyrosine
kinase. Based
30 on our 3D reconstruction, insulin bound to the LI damain of the mutant a
subunit and
48
CA 02338678 2001-O1-26
WO 00/73793 PCT/CA00/00605
the wild-type L2 domain of the hybrid IR and the binding of only a single
molecule of
insulin is sufficient for TK activation.
Thus we have obtained the 3D quaternary structure of the IR-insulin complex
formed in the absence of ATP. The structure was a~n intermediate between
insulin-free
IR and the fully activated, phosphorylated IR. The reconstruction is readily
interpreted
as such: as a receptor poised for activation by traps-phosphorylation. We
determine the
full extent of conformational changes induced by insulin binding. We
reconstruct the
initial state of free IR and the final activated state for comparison. The 3D
reconstruction presented here provides concrete structural information towards
the full
1 o understanding of transrnembrane signal transmission in insulin action:
Furthermore, the
approach used in this study can be applied to obtain the quaternary structure
of other
membrane proteins or receptors that are refractory to crystallization. The
invention
includes the methods for studying polypeptide structure described in this
application.
Example 3 - Mechanics of Transmembrane Signalling of the Insulin Receptor
The binding of insulin to the extracellular domain of the insulin receptor
(IR)
begins an intracellular signal cascade that ends in numerous insulin-specific
cellular
responses. The binding event activates the intracellular tyrosine kinase {TK)
domain of
the receptor. How the signal is transmitted across the cell membrane has
remained a
mechanistic puzzle, since complete membrane receptors have been refractory to
high
2o resolution structural studies by NMR spectroscopy or by crystallography. In
an
alternative approach we have used low-dose low-temperature dark field scanning
transmission electron microscopy (STEM) to determine the three-dimensional
quaternary structure of the entire isolated 480 kDa human insulin receptor
bound to
insulin'. Recently the atomic co-ordinates of individual N-terminal domains of
the
extracellular region of a highly homologous receptor, the insulin-Iike growth
factor type
1 receptor (IGF-1R} have become available, as have models of the three
individual
fibronectin type III (Fn) domains of IR'°~': We have modified these
domain structures
substituting the IR amino acid sequence and accommodating the covalent dimeric
character of IR. The IR TK domain structures were available previouslyg~9. All
of these
3o domains were fitted into the quaternary structure calculated from STEM
micrographs.
The fit provides a detailed description of the insulin binding site of IR and
of its
49
CA 02338678 2001-O1-26
WO 00!73793 PCTICA00100605
interactions with insulin. Moreover, the entire 3D complex is a molecular
machine with
intrinsic linkages that provides a mechanistic model for transmembrane signal
transduction by IR. Since IR is constitutively dimeric2, the mechanism of IR
signal
transduction is of necessity different from that of many receptors activated
by ligand-
induced dimerization. Instead, the binding of insulin changes the IR dimer
from a
configuration that inhibits TK activation to one that is openly permissive of
TK
transphosphorylation.
The structure and model explain observations on insulin binding, on disulphide
modifications linking the two monomers and linking their constituent domains,
the
io block to TK activation, dominant negative- mutations, insulin-dependent and
insulin-
independent autophosphorylation, and transmembrane modifications. Moreover,
the
model is sufficiently general to serve as an archetype for dimeric two-state
receptors like
IR that are activated or inhibited by ligand binding.
The 3D structure determined at 20 ~ by reconstruction from electron
i5 micrographs of sets of single insulin-bound IR complexes' is shown in Fig.
5, with
views as seen from the exterior of the cell membrane (Fig. Sa{i)), the
interior of the cell
(Fig. Sa(iii)), and at 90° from these in the plane ofthe membrane (Fig.
Sa(ii)). Antibody
labelling has recently confirmed the location of three pairs of the assigned
ectodornain
regions3.
2o Covalent linking of the two monomers of IR occurs between Cys524 of each
monomer, and also between corresponding Cys682 (or 683 or 685)
moieties°''. Each
monomer itself contains a 135 kDa a subunit and a 95 kDa ~i subunit linked by
a single
disulphide bond (aCys647 to (3Cys872)°. The structure of one monomer is
diagrairuned
in Fig. 6. From considerations of symmetry of the {a~3)2 dimer, the two a-a
disulphide
25 bondss~' occur one above the other on the two-fold symmetry axis of the
dimer {labelled
1 and 2, Fig. 6). In the interpretation of the 3D stnacture, two polypeptide
chains link
the (3 subunit from fibronectin domain Fnl to the connecting domain CD/Fn0 and
insert
domain ID of the central a subunit.
Crystal structures were determined only for parts of IR: the intracellular TK
3o domain in the unphosphorylated state as well as phosphorylated and bound to
a peptide
substrate8~9, and the first three extracellular domains, Ll, Cys-rich, and L2,
of the
CA 02338678 2001-O1-26
WO 00/73793 PCTlCA00/00605
homologous type 1 insulin-like growth factor receptor (IGF-1R)'°. From
analysis of
sequence homology each a~i monomer contains three fibronectin type IIi
repeatsl'~'3~'.
The ID of the a subunit, the transmembrane and juxtamernbrane regions and the
ID and
C-terminal domains of the ~i suburut are still of unknown structure.
Example 4 -Docking of Ll-CR-L2
The atomic co-ordinates of the L1-CR-L2 regions of IGF-1R (PDB: IIGR) were
used to substitute and insert corresponding amino acids far IR into the IGF-1
R structure.
Additional loops that do not exist in IGF-1R, e.g. amino acids 272-275, were
inserted
where necessary. This was followed by - several rounds of molecular dynamic
1o calculations using the program InsightIl (Molecular Simulations, San Diego,
CA} to
eliminate atomic clashes and to approach a corresponding energy minimum for
the IR
sequence. No rotations of the L1, CR, or L2 domains relative to each other
were carried
out during any of the procedures. Two IR-based Ll-CR-L2 structures, one for
each IR
monomer, were then docked symmetrically into the central ectodomain of the
quaternary IR dimer structure according to the domain sequence scheme proposed
previously'. Several other symmetric configurations were tested as well, such
as
reversing the positions for Ll and L2 or rotating the L1-CR-L2 structure to
extend L2
into the regions designated for the CDlFnO domains. The final fit maximized
overlap of
the EM-based mass with the atomic structure, while avoiding overlap of the
atoms of the
2o two L1-CR-L2 cross-over regions (Fig. 7a). Moreover, this configuration
resulted in an
additional fit of loops in the L 1 regions to slender masses extending from
the
corresponding regions of the EM structure (Fig. 7b) and provided atomic
confirmation
for the cam-like structures on the CR regions (Fig. 7c). These cam-like
structures are
formed by a loop of amino acids from 250 to 280 that is stabilized by a
disulphide bond
between Cys266 and Cys27432.
Ezample 5 - Insulin Binding Region
The fit of the two L1-CR-L2 regions formed a diamond-shaped central tunnel
(Fig. 7a). Each CR domain and the juxtaposing L2 surface of the opposite
monomer
formed one side of the diamond, proximal to the membrane. The other two sides
were
formed, one each, by the L2-facing surface of Ll'°. This arrangement
lined the tunnel
SI
CA 02338678 2001-O1-26
WO 00/73793 PCT/CA00/00605
with almost all of the amino acids that are linked to the binding of insulin.
The atomic
structure of human insulin (PDB:1BEN) fitted into this tunnel as shown in the
stereo
view in Fig. 8a, involving binding sites on both monomers. Insulin interaction
with one
monomer involved major hydrophobic areas on the insulin B chain (ValBl2,
TyrBl6,
s LeuB 17, and PheB24 to TyrB26) and on L 1 (Leu87 to Phe89, and Tyr91 ), as
well as
interactions between G1uB21 on insulin and His247 and G1n249 of the CR region
(Fig. 8b). Interaction with the other monomer was predominantly electrostatic
with no
obvious hydrophobic components (Fig. 8c). These interactions and others are
given in
Table 1, as are some of the distances between interacting side chains.
t o One overriding constraint on the docking of insulin was the need to
satisfy the
location of the Nanogold label attached to PheB 1 of insulin for electron
microscopy'.
This requirement was easily satisfied by flexing the insulin B chain between
aminoacids
1 to 6, a motion that appears to occur naturally, as judged by the position of
the B chain
in different crystal structures of the molecule'4. The fit indicated that the
gold marker
15 location was closest to L 1 of the monomer interacting electrostatically
with insulin
(Figs. 8a and 8c).
Example 6 - Fibronectin Linkers
The linkage in the ectodomain between the L1-CR-L2 regions and the IR
2o transmembrane domain is via three fibronectin type III (Fn) domains and two
so-called
insert domains, one each on the a and (3 subunits of each monomer. This region
also
provides the two disulphide bonds that covalently link the a~i monomers to
form the
constitutive IR dimer. ~ne disulphide bond occurs between the Fn0 domains of
the a
subunits; the other between corresponding oc insert domains (Fig. 6). Two of
the Fn
25 domains, Fnl and Fn2 are not involved in dirner formation, and have
beenmodelled into
the 3D reconstruction previously as the normal seven-beta-strand fibronectin
type III
structure', even though Fnl is made up of four beta strands from the a
subunits and
three from the [3 subunitb.
In relation to our quaternary IR dimer structure, the a insert domain is
modelled
3o to Iead out of the Fnl domain across to the CD/Fn0 region, and then to lie
against the
near side of the L2 domain until it reaches the diad axis of the dimer. Here
it forms a
52
CA 02338678 2001-O1-26
WO OOI73793 PCT/CA00/00605
disulphide bond with its symmetric partner insert domain. The location of the
remaining 34 amino acids of this domain is unknown, although the final 12
residues
appear to assist in insulin bindingz. This shows that the peptide chain either
remains
near the central region or returns to the centrally located binding site.
The structure of the most N-terminal Fn domain, FnO; designated CD in prior
descriptions"~', is more problematical. The domain sequence of the quaternary
structure shows that Fn0 is located at the extreme ends of the central region
of the IR
ectodornain'. The same conclusion is reached fro~r~ the location and
accessibility of
monoclonal antibodies and Fab fragments against this region3~3. At the same
time, the
lU location of the a-a disulphide bond at Cys524 within this region requires
that this
domain extend to the diad symmetry axis of the IR dimer. To accommodate both
requirements, the Fn0 domains were placed at the ends of the central
ectodomain.
However, a hairpin structure, containing the Cys524 loop and two neighbouring
beta
strands of the seven-stranded Fn configuration, was unfolded from the Fn beta
sandwich
and layed against the contiguous L2 domain on the side opposite the insert
domain loop
placement above. This manoeuver permitted the Cys524 residue to reach the diad
axis
and form the second a-a disulphide bond. In addition, Fn-like configuration of
this
domain still easily accommodated the internal linkage to the C-terminal of L2,
provided
an exposed location of the monoclonal epitope between residues 535 and 5483'3,
and
2o retained the normal location of the Fn0 C-terminal, suitably positioned for
the flexible
linkage leading into Fnl (Fig. 6). Moreover, the additional size of this Fn
region (122
aminoacids versus 106 and 97 for Fnl and Fn2, respectively) provided enough
mass to
accommodate the volume of this region in the EM reconstruction.
Example 7 - Physical model for transmembrane signalling
In contrast to activation of monomer membrane receptors, activation of the IR
tyrosine kinase cannot be caused by ligand-induced dimerization, since IR is
intrinsically dimeric. However, the articulated structural features of the 1R
dimer
indicate obvious mechanical arrangement that permits transmembrane signalling
and
intracellular recognition both of the absence of insulin on the receptor and
of insulin
3o binding to it.
53
CA 02338678 2001-O1-26
WO 00/73793 PCT/CA00/00605
Figure 5a shows that the central, extracellular region of the two sets of
contiguous domains from L1 to Fn0 is flanked on both sides by the .pontoon-
like
Fnl/Fn2 domains, which are tethered asymmetrically only between Fnl and FnO.
The
two Fn2 ends, which terminated at the juxtamembrane and transmembrane (TM)
domains; are held away from the central regions by the bumper-like cam
structures of
the two symmetry-related CR domains. The intracellular TK domains are then
influenced by the TM and juxtamembrane domains to which they are attached.
Nuclear magnetic resonance studies have shown that helical TM domains,
similar to the IR TM, cannot transmit a signal longitudinally along their
lengths3'. At
to most a torsional force can be exerted by them. However, they can shift
laterally within
the membrane. This provides a simple and direct means for transmembrane
signalling
for IR.
The structural basis for the proposed mechanism of IR transmembrane signal
transduction is depicted in Fig. 9, pared to a two-dimensional representation.
In the
inactive state (Fig.9a) the ~i subunit transmembrane regions and the
associated
intracellular TKs are held apart by the cam-like blocks on the central portion
of the
dimeric oc ectodomain. The open extracellular structure of the IR dimer shows
that the
two sets of L1-CR regions are splayed apart. When a single insulin molecule
with its
two different binding regions's attaches to a contralateral pair of the four
binding sites of
2o the two a subunits'6, the bumper-like caQn regions are rotated and lifted
out of the way
of the extracellular domains of the [3 subunits (Fig. 9b). The closed
structure is based on
the 3D reconstruction'.
A more realistic depiction of the contiguous three-dimensional structural
features of the TR dimer (Fig. Sa), that alternately permit and prevent TK
activation, is
the set of connected cylinders in Figs. 5b and Sc. The perspective of Figs.
Sb(ii) and
5c(ii) is similar to Fig. 9. The insulin-binding domains, L1 and Cys-rich
(CR); of each
monomer (one blue, one fuchsia), cross symmetrically near the middle of the
structure.
They are attached to the L2, CDlFnO and ID domains, modelled as contiguous
central
barrel structures joined together on the two-fold symmetry axis via the two
inter-
3o monomer disulphides (labelled 1,2 in Figs. Sb and Sc). The cam-like
protrusions on the
CR domains, represented as discs, abut the Fn2 domains of the ~i subunits.
These
54
CA 02338678 2001-O1-26
WO 00/73793 PCT/CA00100605
protrusions can just be seen in the high-density representation of the 3D
reconstruction
(cam, Fig. Sa). The mass of the cam reaches across from the centre to the Fn2
region in
the full-volume representation (Fig. 7b). Near the CD/Fn0 ends of the barrels,
each a
subunit structure extends sideways to help form the Fnl repeat and to tether
each ~3
subunit by a flexible joint to the central structure.
The N-terminal domain of the (3' subunit starts near the CDlFnO side arm of
the a
subunit (Fig.6), leading into Fnl and Fn2 of the extracellular domain of IR
(Figs. Sb and Sc). At that point the ~3 subunit forms an axle-like
transmembrane (TM)
region4, crossing the membrane before folding into tlxe TK domain. Flexible
activation
loops (A) of both TKsg~9 are modelled as extending towards the catalytic
region of the
opposite TK (Fig. Sc(iii}).
The insulin ligand, depicted as a disk, binds slightly asymmetrically with
respect
to the two-fold axis between the two a(3 monomers', representative of the high
affinity
binding position (Fig. Sb). It is shown attached to only one monomer at the
inception of
binding to the open, insulin-free IR dimer (Fig. Sc).
Example 8 - Mechanism
In the inhibitory, insulin-flee state (Fig. 5c), a minimum separation is
maintained
between the two intracellular TKs, in spite of thermal motion, by the oc-
ectodomain CR
2o cam regions that contact the ~i-ectodomains at the Fn'~'TM domains.
Consequently, the
distance between the intraceIlularly attached TKs prevents the flexible TK
activation
loop of one TK from reaching the catalytic transphosphorylation site of the
other TK8~9
(Figs. Sc(ii and iii), "A" arrow).
High affinity binding of a single insulin molecule joins the two Ll-CR-L2
domains of the ectodomain (Fig. Sb) against a small torsional resistance
offered by the
two on-axis disulphide bonds (cf. Fig. Sb(ii) and Fig. 5c(ii}). This action
rotates and lifts
the cam protrusions, such that thermal motion can bring the pair of Fn2/ i"M-
axle regions
closer to the central barrel of the ectodomain. The reduction in separation
between the
TM axles permits a sufficiently close approach of the associated TK domains to
allow
transphosphorylation of the activation loop at the catalytic locus of the
opposite TK
(Fig. Sb(ii and iii}).
CA 02338678 2001-O1-26
WO 00173793 PCT/CA00/00605
When insulin detaches from the receptor, the two L1-Cys-rich domains spring
apart again, as the two strained Cys-Cys linkages return to their equilibrium
positions
(1 and 2, Fig. 5c(ii)). At the same time the CR-region cams again restrict the
approach
of the TK domains (Fig. 5c(ii and iii)); increasing their separation, possibly
to. facilitate
downstream signalling actions.
Example 9 - Functional Consequences of the Model
The detailed model of insulin binding, the relative positioning of the known
domain structures into the quaternary structure of the IR dimer, and the
proposed
mechanism for transmembrane signal transduction explain many observations on
the
t o behaviour of IR. A few examples are detailed here.
The Insulin Binding Site
The. symmetric juxtaposition of the IR adapted L1-CR-L2 domains in the
stricture concentrated virtually all of the known binding interactions to
insulin into a
tunnel-like space that readily accomodated the insulin ligand. Both
hydrophobic and
ionic interactions are accommodated involving L1, L2 and the CR region. A
number of
insulin interactions change in character as either insulin or IR is modified.
These now
have structural explanations. Experimentally, the interaction of insulin with
the CR loop
from 243 to 251 had indicated a strengthening of binding with the introduction
of
positively charged aminoacids into this region'6. The fitting of insulin into
the model
2o binding site indicates an interaction of GiuB21 of insulin with His247 and
possibly
Asn249 in the CR loop. The presence of the negatively charged Asp250 in this
vicinity
weakens this interaction. Thus the addition of a positive charge in the
243/251 loop
would clearly enhance the binding of insulin by providing a potential salt
bridge to the
GluB21 residue, while the substitution of this liis247Asp permits a new ionic
interaction with ArgB22.
Experimentally, a mutation in Phe89 of the L 1 domain reduces insulin
binding3°.
As indicated in Table 1, Phe89 forms part of a hydrophobic region in the
insulin binding
tunnel, that is juxtaposed to a hydrophobic surface on insulin. Any decrease
in this
hydrophobic region would be expected to decrease the strength of insulin
binding.
56
CA 02338678 2001-O1-26
WO 00/73793 PCT/CA00100605
A mutation of HisB 10 in insulin to AspB 10 creates a superactive insulin35.
In the
fit to the model HisB 10 interacts with Arg 14 of L 1. A stronger ionic
interaction would
be expected to result with the introduction of aspexagine in insulin at
position B 10.
Modification of IR on Insulin finding
High affinity binding of insulin is initially augmented, then diminished, by
reduction of the disulphides of IR with increasing concentrations of
dithiothreotol
(D'T"I~". In the model, normal high affinity insulin binding must overcome an
energy
barrier created by the binding-induced elastic strain ixi the two a-a
disulphide bonds on
the diad axis of the IR dimer, due to rotation of the two Ll-CR-L2 regions to
the closed
position. Reduction of one of the two disulphide bonds eliminates this
torsional strain,
removing the energy barner, and facilitating high amity binding. Further
reduction
separates IR into monomers, abrngating high affinity binding, which involves
two a
subunits in close proximity". A similar effect would be expected for a
deletion that
includes one of the a-a disulphide bonds'$.
Autophosphorylation
Basal insulin-independent autophosphorylation of IR occurs naturally at a low
Level. In the model the low levels of autophosphoryiation reflect the
torsional resistance
of the two on-axis disulphide bonds which control the position of blocking
cams in the
insulin-free equilibrium position (Fig. Sc}. However, random thermally induced
motion
2o is occasionally sufficient to rotate the blocking CR cams momentarily to
the permissive
positions. If random motion simultaneously brings the TM regions with their
associated
TK domains close enough together, then a round of transphosphorylation can
occur even
in the absence of insulin. Experimentally, such autophosphorylation is
stimulated by
mild reduction with DTT, then drops off to zero 'at higher DTT
concentrations". The
breakage of either of the disulphide bonds would remove the resistance to
random
rotation to the permissive position, resulting in a more frequent random
approach of the
TK domains for transphosphorylation. The reduction of both bonds would result
in
monomeric IR, halting transphosphorylation altogether.
Deletional Activation
The IR is activated artificially by removal of amino acids 1 to 578 through
tryptic digestion'9. This cleavage still retains covalent links between the
monomers and
57
CA 02338678 2001-O1-26
WO 00/73793 PCT/CA00/00605
between the alpha and beta subunits. However, the insulin-binding region and
the CR
domains have been removed, along with their physical "cam structures". Thus
the (3
domains and their TKs can move closer together and transphosphorylate,
independent of
the presence of insulin. A more limited deletion which removes part of L2 and
most of
the CD region activates IR and blunts the action of insulin'8. Such a deletion
removes
the physical support for the CR cam region of the partner monomer, thus partly
collapsing the cam to permit rapprochement of the TK regions. At the same time
the
geometry of the insulin binding site in the L2 and CR region would be
affected, as well
as the insulin-induced change in the relative configuration of the entire Ll-
CR-L2
1 o regions.
Point Mutations
More subtle alterations of IR are the mutations Phe383VaI and Asp919G1u, both
of which impair TK actions~°~'. Phe383 is midway in the L2
domain'°, which in the
model is straddled by the Fn0 linkage to the a-a, Cys524 disulphide bond and
by the CR
cam region of the partner monomer that contacts the Fn2lZ'M region. The
Asp919G1u
mutation is at the C-terminal edge of the Fn2 domain of the ~3 subunit, which
in the
model contacts the cam. Size modifications in either of these complementary
extracellular contact sites may prevent proper mating of the intracellular TK
domains.
Other aspects of the function of IR that can be explained by the arrangement
of
2o the domains in the 3D sizucture include the negative or positive
cooperativity of binding
of insulin to native or mutant receptors~'z4, the loss of intracellular TK
activity from the
extracellular Cys647Ser mutation2, the effect on extracellular binding of
insulin by the
intracellular TK mutant Met1153I1ezs, the predominantly passive role of the
transmembrane regionZ~'Z8, and the relative down-stream kinase activity of
monomeric
and dimeric IRS.
As three further tests, the model predicts (a) that an antibody linking the
two TK
domains at their most distal intracellular ends to induce transphosporylation,
would
increase the high affinity binding of insulin; (b) that a helix breaking amino
acid in the
transmembrane region would affect TK activation without modifying insulin
binding
3o characteristics; and (c) that a genetically engineered shift of the cam
bulge via judicious
58
CA 02338678 2001-O1-26
WO 00173793 PCT/CA00/00605
insertion/deletion mutations would invert the response to insulin such that TK
activation
would be constitutive, but abrogated in the presence of the iigand.
Eacampte 10 - Method of Identifying Modulators
The three dimensional atomic structure can be readily used as a template for
selecting potent modulators. Various computer programs and databases are
available
for the purpose. A good modulator should at least have excellent steric and
electrostatic camplementarity to the target, a fair amount of hydrophobic
surface
buried and sufficient conformational rigidity to minimize entropy loss upon
binding.
l0 The approach usually comprises several steps.
One must first def ne a region to target. The ligand binding site of IR or an
IR
cam can be used, but any place that is essential to the IR activity could
become a
potential target. Other protein targets include, but are not limited to,
structural
subdomains, epitopes, and functional domains. Since the fitted quaternary
structure
has been determined, the spatial and chemical properties of the target region
is known.
A compound is then docked onto the target. Many methods can be used to
achieve this. Computer databases of three-dimensional structures are available
for
screening millions of molecular compounds. A negative image of these compounds
can be calculated and used to match the shape of the target cavity. The
profiles of
ionic, hydrophobic, hydrophilic, hydrogen bond donor-acceptor, and lipophilic
points
of these compounds can be calculated and used to match the shape of the
target.
Anyone skilled in the art would be able to identify many small molecules or
fragment
as hits. .
One then utilizes linking and extending recognition fragments. Using the hits
identified by above procedure, one can incorporate different functional groups
or
molecules into a single, large molecule. The resulting molecule is likely to
be more
potent and have higher specificity. It is also possible to try to improve the
modulator
by adding more atoms or fragments that will interact with the target protein.
The
originally defined target region can be readily expanded to allow further
necessary
extension.
59
CA 02338678 2001-O1-26
WO 00/73793 PCT/CA00/00605
A number of promising compounds can be selected through the process. They can
then be synthesized and assayed for their agonizing or antagonizing
properties.
The present invention has been described in detail and with particular
reference to the preferred embodiments; however, it will be understood by one
having
ordinary skill in the art that changes can be made thereto without departing
from the
spirit and scope thereof.
Ali publications, patents and patent applications (including Canadian patent
application nos. 2,273,576, 2,292,258 and US patent application na.
09/461,791) are
to herein incorporated by reference in their entirety to the same extent as if
each
individual publication, patent or patent application was specifically and
individually
indicated to be incorporated by reference in its entirety.
CA 02338678 2001-O1-26
WO 00/73793 pCTICA00100605
REFERENCES AND NOTES
1. M. Bajaj, M. D. Wate~eld,. J. Schlessinger, W. R. Taylor, T. Blundell,
Biochim.
Biophys. Acta 916, 220 { 1987).
2. A. Ullrich and J. Schlessinger, Cell 61, 203 (1990).
3. M. R. White and R. Kahn, J. Biol. Chem. 269, 1 (1994):
4. S. R. Hubbard, L. Wei, L. Ellis, W. A. Hendrickson, Nature 372, 746 (
1994). S. R.
Hubbard, EMBOJ. 16, 5572 {1997).
S. T. P. J. Garrett et al., Nature 394, 395 (1998).
5. Bis-(t-Boc)-bovine-insulin (BBI) was prepared as described [R. Geiger, H.
H.
Schone, W. Pfaff, Physiol. Chem. 352, 1487 (1971)j. 1 mg of BBI and 30 nmole
of
mono-NHS-Nanogold (Nanoprobes Inc., Stoneybrook, NY) were dissolved in 200
pl of N'N-dimethylacetamide containing 2 p.l di-iso-propylethylamine, pH 7.6.
After vigorous mixing at room temperature for 60 minutes the mixture was dried
in
a SpeedVac (Savant Instruments, Inc. Farmingdaie. N.Y.). The pellet obtained
was
dissolved in 20 ~tl of trifluoroacetic acid, kept at room temperature for 5
minutes,
and then dried again. The pellet, resuspended in 120 pl of 1 M acetic acid,
was
chrornatographed twice on a Bio-Gel P-10 column {1.7 x 25 cm) in 1 M acetic
acid.
The purified NG-BI was more than 95% pure and had a molecular mass of 19796
Da by matrix-assisted laser desorptionJionization (MALDI) time-of flight (TOF}
mass spectrometry (Figure 1, inset), consistent with one insulin molecule
having
been derivatized with one Nanogold cluster.
61
SUBSTITUTE SHEET (RULE 25~
CA 02338678 2001-O1-26
WO 00!73793 PCT/CA00/00605
6. J. Murray-Rust, A. N. McLeod, T. L. Blundell, S. P. Wood, BioAssay 14, 325
( 1992).
7. Y. Fujita-Yamaguchi, S. Choi, Y. Sakamoto, K. Itakura, J. Biol. Chem. 258,
5045
(1983).
8. C.C. Yip, J. Cell. Biochem. 48, 19 (1992); L. Schaffer, Exp. Clin.
Endocrinol. I01,
7 (1993}; P. De Meyts et al., Exp. Clin. Endocrinol. I01, 17 (1993).
9. P. De Meyts, A. R. Bianco, J. Roth,.7. Biol. Chem. 2~1, 1877 (1976).
References
1. Luo, R.Z.-T., Beniac D.R., Fernandes; A.B., Yip, C.C. & Ottensmeyer, F.P.
Quaternary Structure of the Insulin-Insulin Receptor Complex. Science 285,
1077-80 ( 1999).
2. White, M.F. & Kahn, R. The insulin signaling system. J. Biol. Chem. 269, 1-
4
( 1994).
3. Tulloch, P.A. et al. Single-molecule imaging of human insulin receptor
ectodomain andits Fab complexes. J. Struct. Biol. 125, 11-18 (1999).
4. Ebina, Y. et al. The human insulin receptor cDNA: the structural basis for
hormone-activated transmembrane signalling. Cell 40, 747-58 (1985).
5. Taylor, S.I. et al. Mutations in the insulin receptor gene. Endocrine Rev.
13,
566-95 ( 1992).
6. Sparrow, L.G. et al. The disulfide bonds in the C-terminal domains of the
human
insulin receptor ectodomain. J. Biol. Chem. 272, 29460-7 (1997).
62
SUBSTITUTE SHEET (RULE 26)
CA 02338678 2001-O1-26
WO 00/73793 PCT/CA00/00605
7. Kristensen, C., Wiberg, F.C., Schaffer, L. & Andersen, A.S. Expression and
characterization of a 70-kDa fragment of the insulin receptor that binds
insulin.
Minimizing ligand binding domain of the insulin receptor. J. Biol. Chem. 273,
I 7780-6 ( 1998).
8. Hubbard, S.R., Wei, L., Ellis, L. & Henderson, W.A. Crystal structure of
the
tyrosine kinase domain of the human insulin receptor. Nature 372, 746-54
(1994).
9. Hubbard, S.R Crystal structure of the activated insulin receptor tyrosine
kinase
in complex with peptide substrate and ATP analog. EMBO J. I6, 5572-81
{ 1997).
10. Garrett, T.P.J. et al. Crystal structure of the first three domains of the
type-1
insulin-Iike growth factor receptor. Nature 394, 395-99 {1998).
11. Mulhern, T.D., Brooker, G.W. & Cosgrove, L.A. Third firbronectin-type-III
domain in the insulin-family receptors. Trends Biochem. Sci. 23, 465-466
( 1998}.
12. Casasnovas, J.M., Springer, T.A., Liu, J.H., Harrison, S.C. & Wang, J.H.
Crystal structure of ICAM-2 reveals a distinctive integrin recognition. Nature
387, 312-5 ( 1997).
13. Copie, V. et al. Solution structure and dynamics of linked cell attachment
modules of mouse fibronectin containing the RGD and synergy regions:
comparison with the human fibronectin crystal structure. J. Mol. Biol. 277,
663-82 ( I 998).
63
SUBSTITUTE SHEET' (RULE 26)
CA 02338678 2001-O1-26
WO OOI73793 PCT/CA00/00605
14. Schaefer, E.M., -Erickson. H.P., Federwisch, M., Wollmer, A. & Ellis, L.
Structural organization of the human insulin receptor ectodomain. .l. Biol.
Chem.
267, 23393-402 (1992) .
15. Schaffer, L. A Model for insulin binding to the insulin receptor. Eur. J.
Biochem.
221, 1127-32 (1994).
16. Yip, C.C. The insuiind-binding domain of insulin receptor is encoded by
exon 2
and exon 3. J. Cell. Biochem. 48, 19-25 (1992).
17. Boni-Schnetzler, M., Scott, W., Waugh, S.M., DiBeIla, E. & Pilch, P.F. The
insulin receptor. Structural basis for high affinity ligand binding. J. Biol.
Chem.
262,8395-401(1987).
18. Sung, C.K., Wong, K.Y., Yip, C.C., Hawley, D.M. & Goldfine, LD. Deletion
of residues 485-599 from the human insulin receptor abolishes antireceptor
antibody binding and influences tyrosine kinase activation. Mol. Endocrinol.
8,
3 I 5-24 ( 1994).
19. Shoelson, S.E., White, M.F. & Kahn, C.R. Tryptic activation of the insulin
receptor. Protealytic truncation of the alpha-subunit releases the beta-
subunit
from inhibitory control. J. Bfol. Chem. 263, 4852-60 ( 1988).
20. Grunberger, G., Zick, Y. & Gorden, P. Defect in phosphorylation of insulin
receptors in cells from an insulin-resistant patient with normal insulin
binding.
Science 223, 932-4 { 1984).
21. Roach, P., Arakaki, R.F., Acciii, D. & Taylor, S.I. Extreme insulin
resistance
associated with decreased tyrosine kinase activity: mutation in the insulin
receptor ~i-subunit. Clin Res. 40, 311A (1992).
64
SUBSTITUTE SHEET (RULE 2S)
CA 02338678 2001-O1-26
WO 00/73793 PCT/CA00100605
22. Meyts, P., de. Roth, J., Neville, D.M. Jr., Gavin, J.R. 3d: & Lesniak,
M.A:
Insulin interactions with its receptors: experimental evidence for negative
cooperativity. Biochem. Biophys. Res. Comm. S5, 154-61 {1973).
23. Yip, C.C. et al. Localization of the insulin-binding site to the cysteine-
rich
region of the insulin receptor alpha-subunit. Biochem. Biophys. Res. Comm.
157,
321-9 {1988).
24. Bilan, P.J. & Yip, C.C. Unusual insulin binding to cells expressing an
insulin
receptor mutated at cysteine 524. Biochem. Biophys. Res. Commun. 205, i 891-8
( 1994).
25. Kahn. R.C. et al. The insulin receptor and its substrate: Molecular
determinants
of early events in insulin action. Recent Prog. Hormone Res. 48, 291-339
(1993).
26. Lee, J. & Pilch, P.F. The insulin receptor: structure, function, and
signaling.
Amer. J. Physiol. 266, C319-34 {1994).
27. Frattali, A.L., Treadway, J.L. & Pessin, J.E. Evidence supporting a
passive role
for the insulin receptor transmembrane domain in insulin-dependent signal
transduction. J. Biol. Chem. 266, 9829-34 (1991).
28. Longo, N., Shuster, R.C., Griffin, L.D., Langley, S.D. & Elsas, L.J.
Activation
of insulin receptor signaling by a single amino acid substitution in the
transmembrane domain. J. Biol. Chem. 267, 12416-9 ( 1992).
29. Boni-Schnetzler, M., Rubin, J.B. & Filch, P.F. Stracturai requirements for
the
transmembrane activation of the insulin receptor kinase. J. Biol. Chem. 261,
15281-7 (1986).
SUBSTITUTE SHEET (RULE 26)
CA 02338678 2001-O1-26
WO 00/73793 . PCT/CA00/00605
30. De Meyts, P. et al. Identification of a ligand-binding region of the human
insulin
receptor encoded by eht second exon of the gene. Molec. Endocrinol. 4, 409-16
( 1990).
31. Marino-Buslje, C., Mizuguchi, K., Siddle, K., & Blundell T.L. A third
fibronectin type III domain in the extracellular region of the insulin
receptor
family. FEBSLetters 441, 33I-36 (1998).
32. Chiacchia, K.B. Quantitation of the class I disulfides of the insulin
receptor.
Biochem. Biophys. Res. Comm. 176, 1178-82, 1991.
33. Zhang B. & Roth RA. A region of the insulin receptor important far ligand
binding (residues 450-601 ) is recognized by patients' autoimmune antibodies
and inhibitory monoclonal antibodies. Proc. Natl. Acad. Sci. 88, 9858-62, 1991
34. Murray-Rust, J., NcLeod, A.N., Blundell, T.L. & Wood, S.P. Structure and
evolution of insulins: Implications for recepto binding. BioEssays 14, 325-31.
1992.
35. Schwartz GP. Burke GT. Katsoyannis PG. A highly potent insulin:
des-(B26-B30)-[AspBlO,TyrB25-NH2jinsulin(human). Proc. Natl. Acad. Sci.
USA 86, 458-61, 1989
3b. Williams, F.P. Mynarcik, D.C., Yu, G.Q. & Whittaker, J. Mapping of an NHZ-
terminal ligand binding site of the insulin receptor by alanine scanning
matagenesis. J. Biol. Chem. 270, 3012-16, 1995
37. Rigby, A.C. , Barber, K.R. Shaw, G.S. & Grant, C. W. Transmembrane region
of the epidermal growth factor recepto: Behaviour and interactions via zH NMR.
Biochemistry 35, 12591-601, 1996.
66
SUBSTTfUTE SHEET (RULE Z6)