Note: Descriptions are shown in the official language in which they were submitted.
CA 02339553 2001-02-05
WO 00/08454 PCT/CA99/00714
-1-
METHOD FOR SEPARATION OF ISOMERS AND DIFFERENT CONFORMATIONS OF ]ONS IN
GASEOUS PHASE
FIELD OF THE INVENTION
The present invention relates to a method for separating isomers and
different conformations of ions in gaseous phase, based on the principle of
high field
asymmetric waveform ion mobility spectrometry.
BACKGROUND OF THE INVENTION
High sensitivity and amenability to miniaturization for field-portable
applications have helped to make ion mobility spectrometry an important
technique
for the detection of many compounds, including narcotics, explosives, and
chemical
warfare agents (see, for example, G. Eiceman and Z. Karpas, Ion Mobility
Spectrometry (CRC. Boca Raton, FL. 1994); and Plasma Chromatography, edited by
T.W. Carr (Plenum, New York, 1984)). In ion mobility spectrometry, gas-phase
ion
mobilities are determined using a drift tube with a constant electric field.
Ions are
gated into the drift tube and are subsequently separated based upon
differences in
their drift velocity. The ion drift velocity is proportional to the electric
field strength
at low electric fields (e.g., 200 V/cm) and the mobility, K, which is
determined from
experimentation, is independent of the applied field. At high electric fields
(e.g.
5000 or 10000 V/cm), the ion drift velocity may no longer be directly
proportional to
the applied field, and K becomes dependent upon the applied electric field
(see G.
Eiceman and Z. Karpas, Ion Mobility Spectrometry (CRC. Boca Raton, FL. 1994);
and
E.A. Mason and E.W. McDaniel, Transport Properties of Ions in Gases (Wiley,
New
York, 1988)). At high electric fields, K is better represented by Kh, a non-
constant
high field mobility term. The dependence of Kh on the applied electric field
has been
the basis for the development of high field asymmetric waveform ion mobility
spectrometry (FAIMS), a term used by the inventors throughout this disclosure,
and
also referred to as transverse field compensation ion mobility spectrometry,
or field
ion spectrometry (see I. Buryakov, E. Krylov, E. Nazarov, and U. Rasulev, Int.
J.
Mass Spectrom. Ion Proc. 128. 143 (1993); D. Riegner, C. Harden, B. Carnahan,
and
S. Day, Proceedings of the 45th ASMS Conference on Mass Spectrometry and
Allied
Topics, Palm Springs, California, 1-5 June 1997, p. 473; B. Carnahan, S. Day,
V.
CA 02339553 2001-02-05
09-11-2000 CA 009900714
-2-
(see 1. Buryakov, E. Krylov, E. Nazarov, and U. Rasulev, Int. j'. Mass
Spectrom. Ion Proc. 128. 143 (1993); D. Riegner, C. Harden, B. Carnahan,
and S. Dav, Proceedings of the 45th ASMS Conference on Mass
Spectrometry and Allied Topics, Palm Springs, California, 1-5 June 1997, p.
473; B. Carnahan, S. Day, V. Kouznetsov, M. Matyjaszczyk, and A.
Tarassov, Proceedings of the 41st ISA Analysis Division Symposium,
Framingham, MA, 21-224 April 1996, p. 85; and B. Carnahan and A.
Tarassov, U.S. Patent Number 5,420,424). Ions are separated in FAIMS on
the basis of the difference in the mobility of an ion at high field Kh
relative
to its mobility at low field K. That is, the ions are separated because of the
compound dependent behaviour of Kh as a function of the electric field.
This offers a new tool for atmospheric pressure gas-phase ion studies since
it is the change in ion mobility and not the absolute ion mobility that is
being monitored.
An instrument based on the FAIMS concept has been
desigxted and built by Mine Safety Appliances Company of Pittsburgh, P.
("MSA") for use in trace gas analysis. The MSA instrument is described in
U.S. Patent No. 5,420,424 and is available under the trade mark FIS (for
Field Xon Spectrometer). While the use of t,he MSA instrument (and
similar instruments based on the FAIMS concept) for trace gas analysis is
known, the inventors believe that they have identnfied certain, heretofore
unrealized properties of these instruments which, make them more
versatile. Based on this realization, the inventors have developed what is
believed to be a previously tutl:nown method for separation of isomers
and different conformations of ions. A summary and detailed description
of the present invention is provided below.
SUMMARY QF TH1E INVENTYON
The present invention provides a method for identifying an
isomer, comprising the steps ot:
a) providing at least one ionization source for producing ions of
AMENDED SHEET
CA 02339553 2001-02-05
09-11-2000 CA 009900714
-3-
at least one isomer;
b) providing an analyzer region defined by a space between at
least first and second spaced apart electrodes, said analyzer
region being in communication with at least one of each of a
gas inlet, a gas outlet, an ion inlet and an ion outlet, and
introducing said ions into said analyzer region through said
ion inlet;
c) applying an asymmetriC waveform voltage and a direct
current compensation voltage to at least one of said
electrodes;
d) setting said asymutxtetric waveform voltage;
e) varying said direct current compensation voltage and
measuring resulting transmitted ions at said ion outlet, so as
to produce a compensation voltage scan for said transmitted
ions; and
f) identifying at least one peak in said compensation voltage
scan corresponding to at least one of said isomers.
The method may further comprise the step of setting said
direct current compensation voltage to correspond to one of said peaks to
separate a desired ion from other ions with substantially the same mass to
charge ratio.
Ad'~rantageously, the above method is operable at substantially at
atmospheric pressure and substantially at room temperatu.re.
The nnethod may further include detecting said transmitted ions by
mass spectrornetry.
Typically, the method includes providing a gas flow through said
a.naxyzer region, so as to transport said ions along said analyzer region,
although it
will be understood that other ion transport means are possible.
purtharmoxe, in identifying a peak, it will be understood that the term
peak is not l:im3ted to the apex of the peak, and that a peak sarill typically
have a
noticeablewidth, or a compensation voltage range in which the peak appears.
AMENDED SHEET
CA 02339553 2001-02-05
WO 00/08454 PCT/CA99/00714
-4-
with the apparatus of Figures 3A and 3B;
Figures 5A and 5B show schematically the coupling of the FAIMS
apparatus of Figures 3A and 3B together with a mass spectrometer;
Figures 6A and 6B shows schematically a FAIMS apparatus for
measuring the ion distribution in the analyzer region;
Figures 7 illustrates the high voltage, high frequency asymmetric
waveform applied to the FAIMS apparatus shown in Figures 6A and 6B;
Figure 8 illustrates varying ion arrival time profiles at the innermost ion
collector electrode of the FAIMS apparatus in Figures 6A and 6B;
Figure 9 shows an ion selected compensation voltage (IS-CV) spectra at
five different DV values for a leucine/isoleucine mixture;
Figure 10 trace (a) shows the IS-CV spectra, run separately, of a
solution containing leucine and a solution containing isoleucine;
Figure 10 trace (b) shows an IS-CV spectrum of a leucine/isoleucine
mixture;
Figure 11A shows mass spectra for a leucine/isoleucine mixture before
filtering through a FAIMS analyzer;
Figures 11B and 11C show mass spectra for a leucine/isoleucine
mixture after filtering through a FAIMS analyzer at two different CV values;
Figure 12 shows a response curve for leucine plotted as a function of
concentration;
Figure 13 shows an expanded view of an IS-CV spectrum acquired for a
solution containing 0.004 M leucine and 2.496 M isoleucine;
Figure 14A shows an ESI mass spectrum for a solution of bovine
ubiquitin;
Figure 14B shows a total ion current CV (TIC-CV) spectrum of a
solution of bovine ubiquitin;
Figures 14C-14E show mass spectra obtained at several different CV
values;
Figure 15 shows normalized IS-CV spectra for various charge states of
bovine ubiquitin ranging from +5 to +13 using the same solution as that used
for
Figure 14;
RECTIF(ED SHEET (RULE 91)
1SA/EP
_.. w ~.._.._. n.__._
CA 02339553 2001-02-05
WO 00/08454 PCT/CA99/00714
-5-
Figures 16A, 16C, and 16E show mass spectra showing the effect of the
amount of acetic acid on ESI mass spectra of a solution of bovine ubiquitin;
Figures 16B, 16D, and 16F show TIC-CV spectra corresponding to the
ESI-mass spectra obtained in Figures 16A, 16C, and 16E, respectively;
Figures 17A-17I show IS-CV spectra showing the effect of the amount of
acetic acid in a solution of bovine ubiquitin on the charge states +7, +8 and
+9;
Figures 18A-18I show IS-CV spectra showing the effect of the amount of
HC1 in a solution of bovine ubiquitin on the charge states +7, +8 and +9;
Figure 19 shows normalized IS-CV spectra for the charge states +5 to
+13 using a 5 gM solution of bovine ubiquitin (55% water) acidified to pH 2.1
using
HCI;
Figures 20A-20F show the effect of solvent composition on mass
spectra, TIC-CV spectra, and IS-CV spectra of bovine ubiquitin;
Figures 21A and 21B show the effect of adding NaCI to a solution of
bovine ubiquitin on the IS-CV spectrum for charge state +8;
Figures 21C through 21H show mass spectra of different CV values;
Figure 22A shows an IS-CV spectrum showing the dependence of
sodium adduct ion intensity on the conformation for the +6 charge state of
bovine
ubiquitin;
Figures 22B and 22C show mass spectra for the solution used in Figure
21A at two different CV values;
Figure 23A shows an IS-CV spectrum for the +8 charge state of bovine
ubiquitin using a solution containing phosphate;
Figures 23B-23D show mass spectra for the solution used in Figure 23A
at three different CV values; and
Figure 24 is a plot showing the location of the peak maxima for all
conformers of bovine ubiquitin observed in this study.
DETAILED DESCRIPTION OF THE INVENTION
As an important preliminary note, the discussion below generally uses
the term "ion" to mean a charged atomic or molecular entity, the "ion" can be
any
electrically charged particle, solid or liquid, of any size. The discussion
below refers
CA 02339553 2001-02-05
WO 00/08454 PCT/CA99/00714
-6-
to both positively charged and negatively charged ions, and it will be
understood by
a person skilled in the art that, for any individual analysis, only one of
these types of
ions will be used.
The discussion below also uses the term "isomers" to mean compounds
having identical molecular formulas but which differ in the ways in which the
atoms
are bonded to each other. Generally speaking, isomers may be constitutional
isomers
or stereoisomers. Constitutional isomers differ in the order and the way in
which
atoms are bonded together in their molecules. Stereoisomers differ only in the
arrangement of their atoms in space. Stereoisomers that are nonsuperimposable
mirror images of each other are called enantiomers. Stereoisomers that are not
enantiomers are called diastereomers. Also, the lack of free rotation around
carbon-
carbon bonds may form cis-trans isomers where two substituents may be on
opposite sides of a plane (trans) or on the same side of a place (cis).
Finally,
positional isomers (e.g. ortho, meta, and para positions within a carbon ring)
and
geometrical isomers may form in various types of compounds.
The disclosure also uses the term "ion selected compensation voltage"
(IS-CV) spectra which refers to scanning the compensation voltage applied to a
FAIMS analyzer, as discussed below, typically while monitoring a single mass-
to-
charge (m/z) value. The term "total ion current compensation voltage" (TIC-CV)
spectra is also used to refer to a compensation voltage scan which shows the
sum of
a signal for all detected ions in a given m/z range.
Principles of FAIMS
The principles of operation of FAIMS have been described in Buryakov
et. al. (see I. Buryakov, E. Krylov, E. Nazarov, and U. Rasulev, Int. J. Mass
Spectrom. Ion Proc. 128. 143 (1993)) and are summarized here briefly. The
mobility
of a given ion under the influence of an electric field can be expressed by:
Kh(E) =
K(1+f(E)), where Kh is the mobility of an ion at high field, K is the
coefficient of ion
mobility at low electric field and "f(E)" describes the functional dependence
of the
ion mobility on the electric field (see E.A. Mason and E.W. McDaniel,
Transport
Properties of Ions in Gases (Wiley, New York, 1988); and I. Buryakov, E.
Krylov, E.
Nazarov, and U. Rasulev, Int. J. Mass Spectrom. Ion Proc. 128. 143 (1993)).
CA 02339553 2001-02-05
WO 00/08454 PCT/CA99/00714
-7-
Referring to Figure 1, three examples of changes in ion mobility as a
function of the strength of an electric field are shown: the mobility of type
A ions
increases with increasing electric field strength; the mobility of type C ions
decreases; and the mobility of type B ions increases initially before
decreasing at yet
higher fields. The separation of ions in FAIMS is based upon these changes in
mobility at high electric fields. Consider an ion 1, for example a type A ion
shown in
/
Figure 1, that is being carried by a gas stream 6 between two spaced apart
parallel
plate electrodes 2, 4 as shown in Figure 2. The space between the plates 2, 4
defines
an analyzer region 5 in which the separation of ions may take place. The net
motion
of the ion 1 between the plates 2, 4 is the sum of a horizontal x-axis
component due
to a flowing stream of gas 6 and a transverse y-axis component due to the
electric
field between the plates 2, 4. (The term "net" motion refers to the overall
translation
that the ion 1 experiences, even when this translational motion has a more
rapid
oscillation superimposed upon it.) One of the plates is maintained at ground
potential (here, the lower plate 4) while the other (here, the upper plate 2)
has an
asymmetric waveform, V(t), applied to it. The asymmetric waveform V(t) is
composed of a high voltage component, Vl, lasting for a short period of time
t2 and a
lower voltage component, V2, of opposite polarity, lasting a longer period of
time tl.
The waveform is synthesized such that the integrated voltage-time product
(thus the
field-time product) applied to the plate during a complete cycle of the
waveform is
zero ( i.e., Vl t2 + V2 tl = 0 ); for example +2000 V for 10 gs followed by -
1000 V for
20 gs. Figure 2 illustrates the ion trajectory 8 (as a dashed line) for a
portion of the
waveform shown as V(t). The peak voltage during the shorter, high voltage
portion
of the waveform will be called the "dispersion voltage" or DV in this
disclosure.
During the high voltage portion of the waveform, the electric field will cause
the ion 1
to move with a transverse velocity component vi = KhEhigh, where Ehigh is the
applied field, and Kh is the high field mobility under ambient electric field,
pressure
and temperature conditions. The distance travelled will be dl = v1t2 =
KhEhi$htz,
where t2 is the time period of the applied high voltage. During the longer
duration,
opposite polarity, low voltage portion of the waveform, the velocity component
of
the ion will be v 2= KEIo, where K is the low field ion mobility under ambient
pressure and temperature conditions. The distance travelled is d2 = v2t1 =
KEIoN,tl.
.~..~_.,_... .. .
____-----w-----...
CA 02339553 2001-02-05
WO 00/08454 PCT/CA99/00714
-8-
Since the asymmetric waveform ensures that (Vl t2) +(V2 ti) = 0, the field-
time
products Ehight2 and Elo,~,tl are equal in magnitude. Thus, if Kh and K are
identical,
dl and d2 are equal, and the ion 1 will be returned to its original position
along the
y-axis during the negative cycle of the waveform (as would be expected if both
portions of the waveform were low voltage). If at Ehigh the mobility Kh > K,
the ion 1
will experience a net displacement from its original position relative to the
y-axis.
For example, positive ions of the type A shown in Figure 1 will travel further
during
the positive portion of the waveform (i.e., dl > d2) and the type A ion 1 will
migrate
away from the upper plate 2 (as illustrated by the dashed line 8 in Figure 2).
Similarly, ions of type C will migrate towards the upper plate 2.
If an ion of type A is migrating away from the upper plate 2, a constant
negative dc voltage can be applied to this plate 2 to reverse, or "compensate"
for
this transverse drift. This dc voltage, called the "compensation voltage" or
CV in
this disclosure, prevents the ion 1 from migrating towards either plate 2, 4.
If ions
derived from two compounds respond differently to the applied high electric
fields,
the ratio of Kh to K may be different for each compound. Consequently, the
magnitude of the compensation voltage CV necessary to prevent the drift of the
ion
toward either plate 2, 4 may also be different for each compound. Under
conditions
in which the compensation voltage CV is appropriate for transmission of one
compound, the other will drift towards one of the plates 2, 4 and subsequently
be
lost. The speed at which the compound will move to the wall of the plates 2, 4
depends on the degree to which its high field mobility properties differ from
those of
the compound that will be allowed to pass under the selected condition. A
FAIMS
instrument or apparatus is an ion filter capable of selective transmission of
only
those ions with the appropriate ratio of Kh to K.
The term FAIMS, as used in this disclosure, refers to any device which
can separate ions via the above described mechanism, whether or not the device
has
focussing or trapping behaviour.
Improvements to FAIMS
The FAIMS concept was first shown by Buryakov et. al. using flat plates
as described above. Later, Carnahan et. al. improved the sensor design by
replacing
_..,.._,_..._a.~_
_..._ _-..-......e,..-=-_
CA 02339553 2001-02-05
WO 00/08454 PCT/CA99/00714
-9-
the flat plates used to separate the ions with concentric cylinders (see B.
Carnahan,
S. Day, V. Kouznetsov, M. Matyjaszczyk, and A. Tarassov, Proceedings of the
41st
ISA Analysis Division Symposium, Framingham, MA, 21-24 April 1996, p. 85; U.S.
Patent No. 5,420,424 issued to Carnahan et al.). The concentric cylinder
design has
several advantages including higher sensitivity than the flat plate
configuration (see
R.W. Purves, R. Guevremont, S. Day, C.W. Pipich, and M.S. Matyjaszczyk, Rev.
Sci.
Instrum., 69, 4094 (1998)).
As mentioned earlier, an instrument based on the FAIMS concept has
been built by Mine Safety Appliances Company (MSA). The MSA instrument uses
the concentric cylinder design and is described further below. (For the
purposes of
this disclosure, the MSA instrument is referred to as FAIMS-E, where E refers
to an
electrometer or electric current detection device.)
One previous limitation of the cylindrical FAIMS technology (see D.
Riegner, C. Harden, B. Carnahan, and S. Day, Proceedings of the 45th ASMS
Conference on Mass Spectrometry and Allied Topics, Palm Springs, California, 1-
5
June 1997, p. 473; and B. Carnahan, S. Day, V. Kouznetsov, M. Matyjaszczyk,
and
A. Tarassov, Proceedings of the 41st ISA Analysis Division Symposium,
Framingham, MA, 21-24 April 1996, p. 85) was that the identity of the peaks
appearing in the FAIMS-E CV spectra could not be unambiguously confirmed due
to
the unpredictable changes in Kh at high electric fields.
Thus, one way to extend the capability of instruments based on the
FAIMS concept, such as the FAIMS-E instrument, is to provide a way to
determine
the make-up of the FAIMS-E CV spectra more accurately, for example, by
introducing ions from the FAIMS-E device into a mass spectrometer for mass-to-
charge (m/z) analysis.
In addition, it has been found that a modified FAIMS instrument, or any
similar instrument, can be used in a new method of separating isomers and
different
conformations of gaseous phase ions. The present invention is directed to a
new
method of separating isomers and different conformations of ions and
illustrates the
method by several examples. Details of the method of the present invention are
described below.
. __~- _ _ _ _._~...._..-...-...-..-_ _
CA 02339553 2001-02-05
WO 00/08454 PCT/CA99/00714
-10-
Electrospray Ionization
ESI is one of several related techniques that involves the transfer of ions
(which can be either positively or negatively charged) from liquid phase into
the gas-
phase. Kebarle has described four major processes that occur in electrospray
ionization (intended for use in mass spectrometry): (1) production of charged
droplets, (2) shrinkage of charged droplets by evaporation, (3) droplet
disintegration
(fission), and (4) formation of gas-phase ions (Kebarle, P. and Tang, L.
Analytical
Chemistry, 65 (1993) pp. 972A-986A). In ESI, a liquid solution (e.g. 50/50 w/w
water/methanol) is passed through a metal capillary (e.g., 200 m outer
diameter
and 100 gm ID) which is maintained at a high voltage to generate the charged
droplets, say +2000 V (50 nA) for example. The liquid samples can be pumped
through at, say, l L/rnin. The high voltage creates a very strong, non-
constant
electric field at the exit end of the capillary, which nebulizes the liquid
exiting from
the capillary into small charged droplets and electrically charged ions by
mechanisms
described by Kebarle and many others. Several related methods also exist for
creating gas-phase ions from solution phase. Some examples of these methods
include ionspray, which uses mechanical energy from a high velocity gas to
assist in
nebulization; thermospray, which applies heat instead of a voltage to the
capillary;
and nanospray, which uses small ID capillaries. In this disclosure, the term
ESI is
used to encompass any technique that creates gas-phase ions from solution.
Modified FAIMS-E
As a first step, the FAIMS-E device designed and built by Mine Safety
Appliances Company was modified to permit the introduction of ions using ESI.
The inventors believe that the coupling of an ESI source together with a FAIMS-
E
device is not obvious as it is known that ions produced by ESI have a high
degree of
solvation, and that a FAIMS-E device may not function properly when exposed to
high levels of solvent vapour. The inventors have developed various practical
embodiments of an apparatus that combines an ESI source together with a FAIMS
device to show that such coupling is possible.
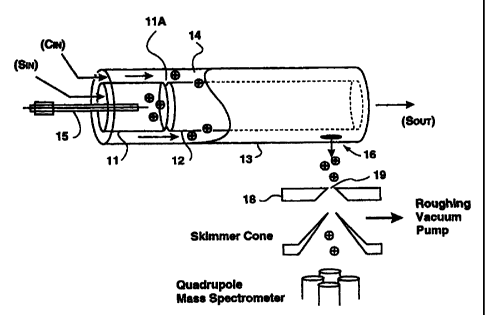
One example is the modified FAIMS-E device 10 shown schematically
~_.
in 3-dimensional view in Figure 3A and in cross section in Figure 3B. The
FAIMS-E
__.-...,.~__~. _...
_.,....a...,..~...-,~._. __
CA 02339553 2001-02-05
WO 00/08454 PCT/CA99/00714
-11-
apparatus 10 is composed of two short inner cylinders or tubes 11, 12 which
are
axially aligned and positioned about 5 mm apart, and a long outer cylinder 13
which
surrounds the two inner cylinders 11, 12. The inner cylinders 11, 12 (12 mm
inner
diameter, 14 mm outer diameter), are about 30 mm and 90 mm long, respectively,
while the outer cylinder 13 (18 mm inner diameter, 20 mm outer diameter) is
about
125 mm long. Ion separation takes place in the 2 mm annular space of FAIMS
analyzer region 14 between the long inner cylinder 12 and the outer cylinder
13. To
produce ions using electrospray ionization (ESI), for introduction into the
FAIMS
analyzer region 14 of the FAIMS device, the metal capillary of the ESI needle
15 was
placed along the central axis of the shorter inner cylinder 11, terminating
about 5 mm
short of the gap or ion inlet between the two inner cylinders 11, 12. The
positioning
of the ESI needle 15 shown in Figures 3(A) and 3(B) differs from the
positioning of
the ionization source found in the MSA FAIMS-E device in that the ESI needle
15
does not extend through the long inner cylinder 12 to which the asymmetric
waveform V(t) is typically applied. By introducing the ESI needle 15 from the
opposite end of the FAIMS-E, i.e. through the short inner cylinder 11, and not
positioning the tip of the ESI needle 15 too dose to the long inner cylinder
12, the
performance of the ESI needle 15 is not compromised by the asymmetric waveform
V(t), which would be the case if the ESI needle 15 was positioned within the
long
inner cylinder 12 (as disdosed in U.S. Patent No. 5,420,424).
As explained above, the FAIMS-E device 10 can be considered as an ion
"filter", with the capability of selectively transmitting one type of ion out
of a
mixture. If a mixture of ions is presented continuously to the entrance of the
FAIMS
analyzer region 14, for example by an ESI needle 15, and the ions are carried
along
the length of the analyzer 14 by a flowing gas under conditions in which no
voltages
are applied to either the inner cylinder 12 or outer cylinder 13 (i.e. the
electrodes are
grounded), some finite level of transmission for every ion is expected, albeit
without
any separation.
It might be expected that the detected current of any selected ion in this
mixture should never exceed the current for that ion when it is transmitted
through
the device 10 in the no-voltages condition. It might also be expected that
application
of high voltages (i.e. application of transverse fields, perpendicular to the
gas flows)
CA 02339553 2001-02-05
WO 00/08454 PCT/CA99/00714
-12-
designed to yield ion separation should not increase the ion transmission, but
should
decrease transmission through collisions with the walls of the cylinders
12,13. That
is, the asymmetric waveform might effectively narrow the "width" of the FAIMS
analyzer region 14, and therefore should decrease the ion transmission.
However,
contrary to this prediction, experiments conducted by the inventors and
described in
this disclosure have shown that the sensitivity of ion detection in the
cylindrical
geometry FAIMS-E 10 increases as the voltage amplitude of the asymmetric
waveform V(t) is increased. As will be explained below, these unusual
observations
suggest that atmospheric pressure ion focussing is occurring in the FAIMS
analyzer
region 14.
Still referring to Figures 3A and 3B, four gas connections to the FAIMS-E
apparatus 10 are shown. Compressed gas (e.g. air or nitrogen) is passed
through a
charcoal/molecular sieve gas purification cylinder (not shown) into the FAIMS-
E 10
through carrier in (Cin) and/or sample in (Sin) ports. The gas exits the FAIMS-
E 10
via the carrier out (Coõt) and/or sample out (Sout) ports. All four gas flow
rates can
be adjusted. Non-volatile analytes are typically introduced into the FAIMS-E
10
using an ESI needle 15. Alternatively, volatile analytes may be introduced
into the
FAIMS-E 10 through the Sin line, and a portion may be ionized as the
compound(s)
pass by a corona discharge needle.
Still referring to Figures 3A and 3B, the outer cylinder 13 of the FAIMS-
E apparatus 10, and the shorter inner cylinder 11, are typically held at an
adjustable
electrical potential (VFAIMs). VFAIMS is usually ground potential in FAIMS-E.
During
operation, a high frequency high voltage asymmetric waveform is applied to the
long
inner cylinder 12 to establish the electric fields between the inner and outer
cylinders
12, 13. In addition to this high frequency (e.g., 210 kHz) high voltage
waveform a dc
offset voltage (i.e. the compensation voltage CV added to FAIMS) is applied to
the
long inner cylinder 12. This leads to the separation of ions in the FAIMS
analyzer
region 14 in the manner discussed earlier.
Still referring to Figures 3A and 3B, some of the ions produced by the
ionization source are carried by the gas stream along the length of the
annular space
between the outer cylinder 13 and the long inner cylinder 12, also referred to
as the
FAIMS analyzer region 14. If the combination of DV and CV are appropriate, and
. _.......~.._,_._._...__- _
_,.m_..._.........._,.-,--_ _
CA 02339553 2001-02-05
WO 00/08454 PCT/CA99/00714
-13-
the ion is not lost to the tube walls, a series of openings or ion outlets 16
near the
downstream end of the outer cylinder 13 allow the ions to be extracted to an
electrical current detector 17 which is biased to about -100 V. (Note that
here the
carrier gas also exits from the ion outlet 16.)
In practice, the simplified square wave version of V(t) shown in Figure
2 cannot be used because of the electrical power demands that such a wave
would
place on the waveform generator. The actual waveforms V(t) appear in Figure 4.
These waveforms are produced by the electronic addition of a sine wave and its
harmonic of twice the frequency. As shown in Figure 4, the FAIMS-E apparatus
10
operates using one of the two waveform modes (with the waveform applied to the
inner cylinder). These reversed polarity waveform modes do not yield "reversed
polarity" CV spectra as might be expected. This is because the reversal of
polarity
in this manner also creates a mirror image effect of the ion focussing
behaviour of
FAIMS. The result of such polarity reversal is that the ions are not focussed,
but
rather collide with the walls of the cylinders 12, 13. The mirror image of a
focussing
valley is a hill-shaped potential surface. (This characteristic, and the
various
"modes" of operation of FAIMS, is discussed further below.)
FAIMS-MS
As discussed earlier, one way to extend the functionality of FAIMS
devices is to couple them together with a mass spectrometer. The use of a mass
spectrometer together with a FAIMS device is advantageous because the mass
spectrometer facilitates a mass-to-charge (m/z) analysis to determine the make-
up
of CV spectra more accurately. One possible FAIMS-MS embodiment is described
here.
Referring to Figures 5A and 5B, the coupling of FAIMS and a mass
spectrometer (FAIMS-MS 20) is shown schematically. The FAIMS-MS 20 of Figures
5A and 5B, and the FAIMS-E 10 shown in Figures 3A and 3B, differ significantly
only at the detection end of the instrument. In accordance with the invention,
the
electrometer 17 has been replaced by a sampler cone 18, placed at the end of
the
FAIMS cylinders 12, 13 as is shown in a simplified form in Figure 5B. The
diameter
of the orifice 19 in the sampler cone 18 is approximately 250 gm. The gas
flows in
___-__..~-=.._.
CA 02339553 2001-02-05
WO 00/08454 PCT/CA99/00714
-14-
the FAIMS-MS 20 are analogous to those in the FAIMS-E 10 except that the Cout
is
divided into two components, namely the original C out and the flow through
the
orifice 19 into the mass spectrometer. The electrical waveforms applied to the
long
inner cylinder 12 are identical to those used in the FAIMS-E apparatus 10. The
sampler cone 18 may be electrically insulated from the other components so a
separate voltage OR can be applied to it. Furthermore, a voltage can be
applied to
the cylinders of the entire FAIMS unit (VFAIMS) for the purpose of enhancing
the
sensitivity of the FAIMS-MS.
Figure 5B shows the FAIMS cylinders 12, 13 at a 45 degree angle in
relation to the sampler cone 18 of the mass spectrometer. Figure 5A showed the
FAIMS cylinders 12, 13 at a 90 degree angle in relation to the sampler cone
18. The
way (i.e., the angle between the two tubes of the FAIMS and the sampler cone
18) in
which the ions are extracted from the cylinders 12, 13 of the FAIMS-MS 20 into
the
mass spectrometer is not limited to these angles. Furthermore, the location in
which
the ions are extracted from the two tubes can also be changed. That is, the
ions can
be extracted anywhere along the separation region of the FAIMS.
Ion Focussing
Referring now to Figures 6A and 6B, to demonstrate the focussing effect
referred to above, a special FAIMS instrument was designed by the inventors
and
constructed to measure the ion distribution between the two cylinders (outer
and
inner cylinders) of a FAIMS device. This instrument will be referred to in
this
disclosure as the FAIMS-Rl-prototype 30 and is illustrated schematically in
Figures
6A and 6B. Ions were generated inside of an electrically grounded cylinder 31
approximately 35 mm long and 20 mm i.d.. The tip of an ionization needle 15
was
typically located near the center of this tube, and at least 15 mm from the
end of the
FAIMS analyzer region 34. The FAIMS analyzer region 34 in this embodiment is
composed of an outer tube 32 which is 70 mm long and 6 mm i.d., and which
surrounds a 2 mm o.d. inner shield electrode 33. The inner shield electrode 33
is an
electrically grounded stainless steel tube which is closed at the end that
faces the
ionization needle 15. This inner electrode 33 surrounds, and shields, an
electrically
isolated conductor 35 passing into its center. This innermost conductor 35
(i.e the
CA 02339553 2001-02-05
WO 00/08454 PCT/CA99/00714
-15-
ion collector electrode) is a collector for ions, and is connected to a fast
current
amplifier or electrometer 36 (e.g. Keithly model 428) and a digital storage
oscilloscope 37 (e.g. LeCroy model 9450).
In the system shown in Figures 6A and 6B, the ions which surround the
inner electrode 33 are forced inwards by a pulsed voltage. These ions travel
from the
FAIMS analyzer region 34 to the innermost conductor 35 through a series of 50
m
holes 38 drilled through the inner shield electrode 33. The holes drilled in
the inner
shield electrode 33 are positioned about 2 cm from the end facing the
ionization
needle 15, and are spaced about 0.5 mm apart for a distance of 10 mm on one
side
of the inner shield electrode 33. The holes 38 drilled in the inner shield
electrode 33
are located in this manner to minimize the variability in distance between the
inner
shield electrode 33 and the outer cylinder 32 in the vicinity of these holes
38. It was
the inventors' objective to measure the ion abundance radial profiles of the
ions
located in the annular space (i.e. the FAIMS analyzer region 34) between the
inner
shield electrode 33 and the outer electrode 32 by pulsing the ions toward the
inner
shield electrode 33 and through the holes 38 and against the innermost ion
collector
electrode 35. The time-dependent distribution of ions arriving at the
innermost
conductor 35 is related to the physical radial distribution of ions around the
inner
electrode 33. Excessive variation in the distance between the two cylinders
32, 33
would have increased the uncertainty of the ion arrival times at the innermost
conductor 35, thus decreasing the spatial resolution of the measurements made
with
this device.
Now referring to Figure 7, the high voltage, high frequency asymmetric
waveform V(t), applied to the FAIMS-Rl-prototype of Figures 6A and 6B, is
shown.
The waveform is divided into two parts, the focussing period and the
extraction
period. The waveform was synthesized by an arbitrary waveform generator (e.g.
Stanford Research Systems model DS340, not shown) and amplified by a pulse
generator (e.g. Directed Energy Inc., model GRX-3.OK-H, not shown). The
frequency
of the waveform, and the relative duration of the high and low voltage
portions of
the waveform could easily be modified. Because of the high voltages, and steep
rise-times of the square waves applied to this FAIMS-Rl-prototype 30, the
power
consumption limits were severe, and waveforms in excess of about 1330 pulses
(16
_W._ _....~_._.._..
___...,_....._.,-.W...-.-=.__ _
CA 02339553 2001-02-05
WO 00/08454 PCT/CA99/00714
-16-
ms at 83,000 Hz) could not be delivered by this system without overheating
electronic components of the high voltage pulse generator.
Note that, in the case of the FAIMS-Rl-prototype 30, the high voltage,
high frequency asymmetric waveform was applied to the outer cylinder 32 of the
FAIMS-R1-prototype 30 shown in Figures 6A and 6B. Since all other forms of
FAIMS discussed in this disclosure have the waveform applied to the inner tube
or
electrode, confusion may arise from the "polarity" of the waveform and the
polarity
of CV. In the FAIMS-R1-prototype 30 shown in Figures 6A and 6B, ions of type A
(shown in Figure 1) are focussed during application of the opposite polarity
waveform and CV than that shown for the devices in Figures 3A, 3B, 5A and 5B.
Nevertheless, for simplification, the polarity will be written to be the same
as if the
device was constructed in the same way as those of the more conventional
configuration. In other words the ions transmitted during application of
waveform
#1 will appear with DV positive and with CV negative. (Please note, however,
that
the actual voltages used on the device in Figures 6A and 6B are DV negative
and CV
positive).
As was observed in the conventional parallel plate FAIMS apparatus
described earlier (Figure 2), the application of a high voltage asymmetric
waveform
V(t) will cause ions to migrate towards one of the FAIMS electrodes 2, 4
because of
the changes in ion mobility at high electric fields (shown in Figures 1 and
2). This
migration can be stopped by applying an electric field or compensation voltage
CV in
a direction to oppose the migration. For the FAIMS-R1-prototype 30 of Figures
6A
and 6B, this CV was applied to the same electrode as the high voltage
asymmetric
waveform (i.e. the outer electrode 32), and was added to the waveform as a
small dc
bias (up to 50 V). At an appropriate combination of DV, and compensation
voltage CV, a given ion will pass through the FAIMS device 30. The unit
therefore
acts like an ion filter. It is possible to fix conditions such that a single
type of ion is
isolated in the FAIMS analyzer 34 although a mixture flows uniformly out of
the exit
of the FAIMS device 30 although a mixture of ions are presented to the inlet
of the
FAIMS analyzer region 34.
The second part of the waveform shown in Figure 7 (i.e. the extraction
period) was used to pulse the ions out of the FAIMS analyzer region 34 between
the
_.~ ,._..._~.-_-...
_ __...~....~,-.m... ._
CA 02339553 2001-02-05
WO 00/08454 PCT/CA99/00714
-17-
outer electrode 32, and the inner shield electrode 33 (shown in Figures 6A and
6B).
At the end of the focussing period, i.e. after 16 ms of waveform, the
asymmetric
waveform was replaced by a constant dc bias of approximately +30 V. This
caused
the ions from the annular space 34 between the outer electrode 32 and the
inner
shield electrode 33 to move in the direction of the inner shield electrode 33.
A
detector bias of -5 V, applied to innermost ion collector electrode 35, helped
to carry
the ions from the vicinity of the holes 38 in the inner shield electrode 33,
through the
holes 38 and into contact with the innermost ion collector electrode 35. The
+30 V
bias created an electric field of approximately 150 V/cm across the FAIMS
analyzer
region 34 and most ions located within this region 34 travelled across the 2
mm
space in about 1 ms. The ion current due to the arrival of ions at the center
inner
shield electrode 33 can be predicted. For example, if only one type of ion,
with
mobility of 2.3 cm2/V-s, e.g., (H2O)r,H+ at ambient temperature and pressure
conditions, was located in the FAIMS analyzer region 34, and if this ion was
distributed evenly in the space, an approximately square-topped signal lasting
approximately 0.6 ms should be observed. Deviation from this expected ion
arrival
profile would suggest that the ions were distributed in non-uniform profile
across the
FAIMS analyzer region 34 between the outer and inner cylinders of the FAIMS
device 30.
Still referring to Figures 6A, 6B, and 7, the FAIMS-Rl-prototype 30 was
operated as follows. A 2L/min flow of purified air, Carrier Gas In (Cin), was
passed into the cylinder 31 housing the ionization needle 15. Approximately
2000
V was applied to the needle 15, and the voltage was adjusted to produce a
stable
ionization current. The high voltage asymmetric waveform V(t) was applied to
the
outer FAIMS cylinder 32 for approximately 16 ms; this was followed by a 2 ms
extraction pulse (Figure 7). The ion current striking the innermost ion
collecting
electrode 35 was detected and displayed on a digital oscilloscope 37. A
measurement would typically consist of 100 averaged spectra, collected at a
rate of
approximately 5 Hz. Many experimental parameters were varied, including gas
flow
rates, the voltages of the asymmetric waveform V(t), the dc voltage applied to
the
outer electrode CV, and the extraction voltage.
Figure 8 illustrates the ion arrival times at the innermost ion collector
-----, ._ _
--~ ...~----------
_ .....~.._,-._----._...._
CA 02339553 2001-02-05
WO 00/08454 PCT/CA99/00714
-18-
electrode 35 observed by conducting these experiments. Each trace was recorded
with 2500 V applied DV, but with variable CV voltages. As can be seen, during
application of DV and CV, the radial distribution of ions is not uniform
across the
annular space of the FAIMS analyzer region 34. For example, at CV near -11 V,
the
ions are focussed into a narrow band near the inner electrode 33, and
therefore are
detected as a high intensity pulse occurring very early after the extraction
voltage has
been applied. At low CV, for example at -5.6 V, the ions are much more
uniformly
distributed between the walls of the concentric cylinders 32 33 making up the
FAIMS
analyzer region 34. When no electrical voltages are applied to the cylinders
32, 33,
the radial distribution of ions should be approximately uniform across the
FAIMS
analyzer region 34 (data for this no-voltage experimental condition is not
shown in
this document). The experimental data shown in Figure 8 is evidence that the
ion
focussing is indeed occurring in FAIMS instruments. This focussing results in
the ions
being focussed in a uniform "sheet" or band around the inner cylinder 33
within the
FAIMS analyzer region 34. As mentioned previously, to the inventors'
knowledge,
this focussing effect has never been observed or explained previously.
Modes of Operation of FAIMS
The focussing and trapping of ions by the use of asymmetric waveforms
has been discussed above. For completeness, the behaviour of those ions which
are
not focussed within the FAIMS analyzer region will be described here. As
explained
earlier, the ions which do not have the high field ion mobility properties
suitable for
focussing under a given set of DV, CV and geometric conditions will drift
toward one
or another wall of the device, as shown in Figure 2. The rapidity with which
they
move to the wall depends on the degree to which their Kh/K ratio differs from
that
of the ion that might be focussed under the selected condition. At the very
extreme,
ions of completely the wrong property i.e. type A ion versus type C ion shown
in
Figure 1, will be lost to the walls very quickly.
The loss of ions should be considered one more way. If an ion of type
A (Figure 1) is focussed at DV 2500 volts, CV -11 volts in a given geometry
(for
example, the FAIMS-E device of Figures 3A-3B), is it reasonable to expect that
the
ion will also be focussed if the polarity of DV and CV are reversed, i.e. DV
of -2500
..~.. ~..~..,__..___W ._ ..
_......~-_ ___
CA 02339553 2001-02-05
WO 00/08454 PCT/CA99/00714
-19-
volts and CV of +11 volts (both applied to the inner electrode). It would seem
that
the reversal of polarity is a trivial exercise and the ion should be focussed,
however,
this is not observed. Instead, the reversal of polarity in this manner creates
the
mirror image effect of the ion focussing behaviour of FAIMS. The result of
such
polarity reversal is that the ions are not focussed, but rather are extremely
rapidly
rejected from the device, and collide with the walls of the cylinders 12, 13.
The
mirror image of a trapping valley, is a hill-shaped potential surface. The
ions will
slide to the center of the bottom of a trapping potential valley (2 or 3-
dimensions),
but will slide off of the top of a hill-shaped surface, and hit the wall of an
electrode.
This apparently anomalous behaviour is a consequence of the cylindrical
geometry of
the FAIMS-E.
This is the reason for the existence, in the FAIMS, of the independent
"modes" called 1 and 2. In this disclosure, the FAIMS instrument is operated
in
four modes: P1, P2, N1, and N2. The "P" and "N" describe the ion polarity,
positive (P) and negative (N). The waveform (Figure 4, wave #1) with positive
DV
(where DV describes the peak voltage of the high voltage portion of the
asymmetric
waveform) yields spectra of type P1 and N2, whereas the reversed polarity
(Figure
4, wave #2, negative DV) waveform yields P2 and N1. The discussion thus far
has
considered positive ions but, in general, the same principles can be applied
to the
negative ions, as explained in the preliminary note to the Detailed
Description.
Separation Experiments
Based on the FAIMS principles discussed above and the experiments
conducted by the inventors to demonstrate the concept of ion focussing, the
inventors have developed what is believed to be a previously unknown method
for
separation of isomers and different conformations of ions at substantially
atmospheric pressure and substantially room temperature. Ion separation
experiments involving several different types of ions are provided by way of
example.
A) Leucine/Isoleucine Separation
In the prior art, there have been several attempts to distinguish the
_ .-,,.~-....~..-____. ._.. .
_.~.._._._.,,.,-_.-_...._.
-....w_.._
..._._..~..,..,....,
CA 02339553 2001-02-05
WO 00/08454 PCT/CA99/00714
-20-
amino acids leucine (Leu) and isoleucine (lie) using sector, ion trap and
quadrupole
mass spectrometry. However, since Leu and Ile are structural isomers (i.e.,
identical
elemental composition and molecular weight), their mass spectrometric
differentiation has been limited to the interpretation of fragment ion mass
spectra.
While differences in relative abundances of certain fragment ions can be used
to
unambiguously identify either of the two isomers in pure or pre-fractionated
samples, mass spectrometric differentiation is incapable of selective
determination
within a mixture. In fact, the identification of the molecular ions of Leu and
Ile, or
those of their derivatives, has been demonstrated only following their
chromatographic separation prior to mass spectrometric analysis. The
separation of
these structural isomers has been achieved using ion exchange chromatography,
high
performance liquid chromatography, gas chromatography, and micellar
electrokinetic
chromatography. In general, these chromatographic methods require 5-15 minutes
for
separation and produce a 5-30 second transient pulse of analyte. The length of
time
required for separation and the relatively short duration of the transient
pulse of
analyte limit this method of separation.
Based on the principles of high field asymmetric waveform ion mobility
spectrometry, and based upon various experiments conducted by the inventors, a
new and significantly improved method of separation of Leu and Ile has been
developed and is described here.
For this experiment, the FAIMS device coupled to a mass spectrometer
as shown in Figure 5A was used. To generate negative ions, the electrospray
needle
was held at approximately -1900 V, giving an electrospray current of about 40
nA.
The actual asymmetric waveform that was applied to the long inner cylinder of
FAIMS is shown in Figure 4 (Waveform #2). The maximum voltage of this
waveform,
referred to as the dispersion voltage (DV) was varied between 0 and -3300 V
(which
was the limit of the instrument). The frequency of the asymmetric waveform was
constant at about 210 kHz. The CV, which was also applied to the long inner
cylinder of the FAIMS analyzer, was scanned over specified voltage ranges.
As explained earlier, if the combination of DV and CV was
appropriate, ions were not lost to the cylinder walls during their passage
through the
FAIMS analyzer and were transferred through an approximately 250 m orifice 19
to
.._~.....~........,..~ _ _ _,. _...._....~... _._ . .
CA 02339553 2001-02-05
WO 00/08454 PCT/CA99/00714
-21-
the vacuum chamber of a mass spectrometer (PE SCIEX API 300 triple
quadrupole).
The MS orifice was electrically insulated from the FAIMS and a separate
orifice
voltage of -45 V was applied to it. Optionally, an offset voltage of -45 V was
also
applied to the entire FAIMS unit (VFAIMS) to enhance the sensitivity of the
FAIMS-MS. The skimmer cone 18A of the MS was held at ground potential and the
small ring electrode normally located behind the orifice of the API 300 was
not
incorporated into the present interface, resulting in some loss of sensitivity
for low
mass ions such as Leu and Ile. Compressed air was introduced into the carrier
gas
inlet (C;n) at a flow rate of 3 L/min. Gas exited through the carrier gas
outlet (Cout)
at 2 L/min and through the sample gas out port (Sout) at 1 L/min. There was no
flow through the "sample gas in" port (Sin) in this study. The pressure inside
the
FAIMS analyzer was kept at approximately 770 torr.
For this experiment, commercially available samples of L-leucine and
L-isoleucine were obtained. All standard solutions were prepared in 9:1
methanol/water (v/v) containing 0.2 mM ammonium hydroxide.
As explained earlier, FAIMS can be operated in any one of four modes,
namely P1, P2, Nl or N2, where P and N describe ion polarity (positive and
negative), and "1" and "2" are indicative of instrumental conditions. In
general, low
mass ions (m/z < 300) such as Leu and Ile are transmitted in mode 1, while
larger
ions are transmitted in mode 2. In the present study, the ESI source was tuned
to
generate negative ions. Hence, all CV and mass spectra were collected using N1
mode. The asymmetric waveform used for N1 operation is shown in Figure 4
(Waveform #2).
The capability of FAIMS to separate ions generated from ESI of a
mixture of Leu and Ile (5 M each) is shown in Figure 9. In each trace, (a) to
(e), the
voltage of the asymmetric waveform was set, and an ion-selected CV spectrum
(IS-CV) was collected by scanning the CV while monitoring m/z -130, the mass
of
the (M - H)- ion of both compounds (where M= C6H13NO2). The dwell time and
number of scans were kept constant for each spectrum. The IS-CV spectrum
acquired without application of V(t) is shown in Figure 9 trace (a). Since the
transmitted ions were not subjected to the electric fields caused by V(t)
within the
FAIMS analyzer they have experienced no change in mobility and appear in the
.._.~__.~
__ ._,._.~.w.....~._.._..__ _
CA 02339553 2001-02-05
WO 00/08454 PCT/CA99/00714
-22-
spectrum near CV = 0 V. An increase to DV = -1700 V, Figure 9 trace (b),
results in
the observation of three distinct peaks located at CV values of -0.1 V, 0.7 V
and 1.3
V. Increasing DV to -2300 V, Figure 9 trace (c), caused most of the peaks in
the
IS-CV spectrum to shift to higher CV values, indicating significant changes in
their
high field mobility terms, Kh. The one exception is the first peak in spectrum
at CV =
-0.3 V. This peak was identified as an amino acid dimer (M2 - H)- and is an N2
type
ion, and as such is transmitted through FAIMS in N1 mode in a defocusing
electric
field. At DV = -2700 V, Figure 9 trace (d), the single peak seen in Figure
4(c) at CV
2.9 V has separated into two partially resolved peaks at CV values of 4.7 and
5.0 V.
The separation of these two peaks may be improved by increasing DV to -3300 V
(the limit of the power supply), as shown in Figure 9 trace (e), giving CV
values of
7.7 and 8.4 V. The position and separation of the group of smaller peaks at CV
values of 2.9, 3.9 and 4.3 V in Figure 9 trace (e) also increased. The
unambiguous
assignment of any of these five peaks to Leu or Ile cannot be determined from
this
experiment. However, the mass spectrometer ensures that all of the peaks
correspond to ions with m/z -130.
Identification of peaks in IS-CV spectra is analogous to using mass
spectrometry as a selective detector for a chromatographic method such as CE,
LC
or GC. As with chromatography, it is necessary to have a set of matching
standard
solutions to identify the peaks in the CV spectrum. Determination of the
identities of
the peaks observed at CV values of 7.7 V and 8.4 V in Figure 9 trace (e) was
accomplished by collecting IS-CV spectra (m/z -130, DV = -3300 V) for 5 M
solutions of either Leu or Ile. The spectra are shown in Figure 10 trace (a),
where the
dashed trace is the IS-CV spectrum of Leu and the solid trace is that of Ile.
Note,
Figure 10 trace (b) is the IS-CV spectrum of the mixture, as shown previously
in
Figure 9 trace (e), plotted over a narrower range of CV values. The peaks at
CV
values of 7.7 V and 8.4 V in the IS-CV spectrum shown in Figure 10 trace (a)
may
therefore be attributed to Leu and Ile, respectively.
The three small peaks seen in Figure 9 trace (e) at CV values of 2.9, 3.9
and 4.3 V were also present in the IS-CV spectra for both standard solutions.
Identification of these peaks involved alternately tuning the FAIMS analyzer
to a
fixed CV value to selectively transmit one of the three ions, and collecting
mass
CA 02339553 2001-02-05
WO 00/08454 PCT/CA99/00714
-23-
spectra using varying FAIMS-MS sampling conditions. Gentle conditions,
attained
by reducing the collisional voltage in the MS interface, showed that these
species
correspond to the following adduct ions: (M(CH3O(CO2))- at CV 2.9 V;
M(CH3COO)- at CV 3.9 V; and M(N03)- at CV 4.3 V where M is the neutral
molecule of leucine or isoleucine. Under the more energetic conditions used to
acquire
the IS-CV spectra, these ion adducts were readily fragmented to yield the
molecular
ions (M -H)' of Leu and Ile at m/z -130.
As realized by the inventors, FAIMS, which continuously transmits one
type of ion from a complex mixture is a significant improvement over
conventional
chromatographic methods of ion separation, especially when interfaced to
relatively
slow scanning mass spectrometers. As mentioned above, commonly used
chromatographic methods for Leu and Ile are time-consuming (5-15 minute
retention
times) and result in narrow, finite impulses of analyte (5-30 seconds). The
transient
nature of these separation methods offers little flexibility in varying
detection
parameters and generally limits the degree to which the capabilities of the
mass
spectrometer may be exploited. With FAIMS, ion separation is independent of
several experimental parameters associated with classical chromatography such
as
the stationary phase. Advantageously, problems encountered with the
compatibility
of LC and CE buffers (e.g. high salt content) and flow rates with the
electrospray
process are also eliminated.
In addition to its impressive separation ability, the FAIMS analyzer also
functions to focus ions. (An experiment conducted by the inventors to show
this
focussing effect was described above.) A comparison of the observed ion
current in
Figure 9 trace (a) and 9 trace (e) shows an increase in signal of more than
two orders
of magnitude when DV is increased from 0 to -3300 V. This increase in ion
current is
attributed to the two-dimensional atmospheric pressure ion focusing discussed
in
detail earlier.
To illustrate the improvement in the mass spectra collected with FAIMS,
spectra were acquired for the 5 M Leu/Ile mixture and are shown in Figures
11A-
11C. The spectrum acquired with FAIMS disabled (DV = 0 V) and CV = 0 V is
shown in Figure 11A. With no applied DV, there is no ion filtering effect, and
hence
no discrimination of the ions passing through the FAIMS analyzer. The mass
RECTIFIED SHEET (RULE 91)
ISA / EP
_..- . _...._~.~......
.._.__.~,_.,.~.~...-...,.-- _
CA 02339553 2001-02-05
WO 00/08454 PCT/CA99/00714
- 24 -
spectrum is complex, a commonly observed and often detrimental characteristic
of
electrospray mass spectra in the low-mass region. Peaks attributable to
(C02(CH3O)-; m/z -75), oxalate (m/z -89), Leu/Ile (M-H; m/z -130), ((M2 - H)-;
m/z -261) and ((Na(M - H)2-; m/z -283), among others, are present. The peak
observed at m/z -135 is due to an impurity in the solvent or the ammonium
hydroxide buffer. From this spectrum, the signal intensity for the dimer (M2 -
H) -, is
roughly twice that of the molecular ions of Leu and Ile at a total analyte
concentration of 10 M. At DV = -3300 V, the mass spectra collected for the
same
sample mixture at CV values of 7.7 V and 8.4 V, i.e., the CV values of
transmission
of Leu and Ile, respectively, are simple and show one intense peak at m/z -130
as
shown in Figs. 11B and 11C. The FAIMS analyzer has effectively filtered out
almost
all of the background ions. This filtering action of the FAIMS analyzer was
observed
to improve signal to background ratios (S/B) for these analytes over spectra
observed in conventional ESI-MS by at least a factor of 50.
The difference in the CV values required for the transmission of Leu and
Ile is sufficient to permit selective monitoring of one of the species without
interference from the other. This was illustrated by establishing response
curves for
both analytes present in a mixture. The response curve for Leu is shown in
Figure 12.
The total analyte concentration (i.e., [Leu] + [Ile]) in solution was kept
constant at
2.500 gM, with the individual concentrations of each analyte varying over more
than
two orders of magnitude (i.e., from 0.004 to 2.496 M). The plot is linear,
(y=3.3[Leu)-0.01, R2= 0.9998), indicating that there is no overlap of the Leu
signal
from Ile when it is monitored at CV = 7.7 V, the value corresponding to its
peak
maximum. If an overlap of Leu and Ile was present at the CV values monitored,
the
Leu signal at low concentration would have been most notably affected,
resulting in
non-linearity in the response curve. An expanded view of an IS-CV plot
acquired for
the solution containing 0.004 M Leu and 2.496 M Ile, Figure 13, shows that
the
peaks are still well resolved at these concentrations. Note that the total
concentration of 2.500 pM was sufficiently low that no dimer ions were
observed in
the mass spectra. If the concentration of dimer ions had been significant, a
negative
deviation in the analytical curve at high analyte concentration would have
resulted.
RECTIFIED SHEET (RULE 91)
ISA / EP
_ _....~..........._-_
__...~..._.._._....-.--w_. _
CA 02339553 2001-02-05
WO 00/08454 PCT/CA99/00714
-25-
B) Separation of Different Conformations of Gas-phase Molecular Ions using
FAIMS
Recently, considerable effort has been focused on attempting to
understand the relationships between the amino acid sequence, the structure,
and
properties of proteins. A protein is composed of a series of linked amino
acids,
chemically covalently bonded to each other. Since there are about 20 different
types
of amino acids which can be included in this chain, the first level of the
description
of the structure of a protein is the listing of the names of these amino acids
in the
sequence that they appear in the protein. This is called the amino acid
sequence.
Some of the amino acids have side groups which have the capability of
creating chemical bonds to the side group of another amino acids someplace
else in
the amino acid sequence. This creates cross-linking. This cross linking is a
very
important structural element of proteins, because it forces certain areas of
the protein
sequence to be physically in close proximity to each other, in the final
protein
structure.
The chains of amino acids have the capability of forming small
structures including loops, and hairpin shape structures that involve only a
small
number of amino acids. These structures are formed because some of the side
chains
of the amino acids interact weakly (non-covalently) with one another, and if
the
appropriate amino acids are in close proximity, then these weakly held
structures
will spontaneously form.
Finally, the combination of all of the smaller structures, and cross-links,
give the protein an overall 3 dimensional structure. This structure is called
the
'conformation'. This structure can be disrupted or modified many ways. The
heating
of the protein will 'denature' the protein. This usually means that the
protein loses
its functional capability because the 3-dimensional structure has been
modified.
This can occur because of the breaking of a cross-linking bridge, or the
disruption of
small or large scale structures via addition of thermal energy to the
molecule.
Conformation, therefore, describes the 3-dimensional structure of the
protein. The protein has a conformation whether or not the protein is capable
of
performing it's normal chemical activity, i.e. native, or denatured. Some
terminology
which describes the 3-dimensional structure may be 'extended', 'elongated',
which
CA 02339553 2001-02-05
WO 00/08454 PCT/CA99/00714
-26-
describes in a very non-specific way what we imagine the overall 3-dimensional
structure will look like. Electrospray ionization (ESI), described above, has
enabled
the formation of intact gas-phase pseudo-molecular ions from large molecules,
such
as proteins. By coupling an ESI to a mass spectrometer (MS), ESI-MS has been
used
to provide information about conformations in solution. Since aqueous
solutions at
nearly physiological conditions are used in ESI-MS, this technique has been
used to
provide complementary structural information with other solution based methods
such as Nuclear Magnetic Resonance (NMR).
In the prior art, information on gas-phase protein conformations has
been gathered using several techniques. It has been proposed that these
techniques
can be divided into two general categories: chemical reactivity studies, and
non-reactive studies. Chemical reactive studies, for example hydrogen-
deuterium
(H/D) exchange and proton transfer kinetics, examine the differences of
reactivity of
different conformations. Non-reactive studies use collisions with inert
species to
derive conformational information.
Based on the principles of high field asymmetric waveform ion mobility
spectrometry discussed above, and based on experiments conducted by the
inventors, a new and complementary method of measuring and studying different
conformers of protein ions has been developed by the inventors and is
described
here.
Figure 5A shows a schematic view of a ESI-FAIMS-MS instrument of the
type that was used in this study of different conformations. The electrospray
needle
15 and associated liquid delivery system were constructed by threading a 30 cm
piece of fused silica capillary (50 gm i.d., 180 gm o.d.) through a 5 cm long
stainless
steel capillary (200 .rn i.d., 430 m o.d), with the fused silica capillary
protruding
about 1 mm beyond the end of the stainless steel capillary. This stainless
steel
capillary, in turn, protruded about 5 mm beyond the end of a larger stainless
steel
capillary (500 m i.d., 1.6 mm o.d.) that was used for structural support and
application of the high voltage. Solutions were delivered to the electrospray
needle
by a syringe pump (Harvard Apparatus model 22), at a flow rate of 1 L/min.
For
the generation of positive ions, the needle was held at approximately +2200 V
giving
an electrospray current of about 0.03 gA. The electrospray needle was placed
on the
_,.
_.-..Y.__....._....._.----
CA 02339553 2001-02-05
WO 00/08454 PCT/CA99/00714
- 27 -
center axis of the short inner cylinder, terminating about 5 mm short of the
gap 11A
between the two inner cylinders 11 and 12. The electrospray ions were driven
radially outward by the electric field to the analyzer region through the 5 mm
gap
11A between the two inner cylinders.
A high frequency (210 kHz), high voltage (0 to 4950 V p-p), asymmetric
waveform (Figure 4) was applied to the long inner cylinder 12, thereby
establishing
the electric field between the inner and outer tubes. For the purposes of
demonstrating separation of the conformers of the protein bovine ubiquitin all
spectra were collected using P2 mode with DV =-3300 V. In addition to the high
frequency waveform, a compensation voltage CV was also applied to the long
inner
cylinder 12. Although the CV can be scanned from -50 V to +50 V, the CV
spectra
herein are only shown from -12 V to OV since the ions of bovine ubiquitin were
transmitted through FAIMS within this CV window.
In this study, nitrogen gas was passed through a charcoal/molecular
sieve gas purification cylinder and introduced into the FAIMS device through
the
carrier in (Cin) port at a flow rate of 6 L/min. The gas exited through the
sample out
(Sout) port at 1 L/min and through the carrier out (Cout) port at 5 L/min. The
sample in (Sin) port was plugged in this study. A fraction of C;n was directed
radially inward through the 5 mm gap 11A between the inner cylinders 11, 12,
and
acted to help desolvate the ions. While the ions formed by ESI were driven
radially
outward through the gap by the electric field, the inward flow of curtain gas
prevented neutrals from entering the annular analyzer region. This portion of
Cin,
along with the neutrals, exited the FAIMS device via the Sout port.
The electrospray ions were carried by the gas stream along the length of
the annular space between the outer cylinder and the long inner cylinder. If
the
combination of DV and CV was appropriate, and the ions were not lost to the
tube
walls, ions were transferred to the vacuum chamber of a mass spectrometer
through
the orifice 19 in the "sampler cone" 18 placed at the end of the FAIMS
analyzer.
A custom interface was constructed for a tandem combination of
FAIMS and a PE Sciex API 300 triple quadrupole mass spectrometer. The voltage
of
the sampler cone 18 was set to 44 V, whereas the skimmer cone 18A of the API
300
remained at ground potential for all experiments. The small ring electrode
normally
CA 02339553 2001-02-05
WO 00/08454 PCT/CA99/00714
-28-
located behind the orifice of the conventional API 300 interface was not
incorporated
into the new interface, resulting in some loss of sensitivity. Behind the
skimmer cone,
in a low pressure region (~7 x 10-3 torr), was an rf-only quadrupole (QO) mass
spectrometer 19A which acted to collisionally focus ions prior to their
transmission
into the first analyzer quadrupole (Q1). The voltage drop between the skimmer
cone
18A and QO controlled the energy of the collisions in this region. The higher
the
voltage drop, the greater the extent of fragmentation. Unless otherwise
stated, QO =
-1 V was used.
Ion-selected CV spectra (IS-CV spectra) were obtained by scanning the
compensation voltage applied to the FAIMS, while monitoring a single m/z
value.
"Total ion current" CV spectra (TIC-CV spectra) show the sum of the signal for
all
detected ions in a given m/z range as CV was scanned. The mass spectrum
collected
at fixed values of DV and CV revealed the identity of any ions transmitted
through
the FAIMS under those conditions.
The ubiquitin ions described and used in this experiment behave as type
C ions, as shown in Figure 1. Analogous to the description of the behavior of
positive type A ions, provided earlier, and referring back to Figures 1 and 2,
a type C
ion will travel further during the negative portion of the waveform (dl < d2),
and will
migrate toward the upper plate.
As explained earlier, there are a total of four modes of operation for
FAIMS. The waveform with negative DV (Figure 4B) yields spectra of types P2
and
N1, whereas the reversed polarity waveform yields P1 and N2 type spectra. In
general, low mass ions (m/z is usually less than 300) are of type A (Figure 1)
and are
detected in mode 1, while larger ions, including the positively charged
ubiquitin ions
studied here, are type C ions and are detected in mode 2 (i.e. mode P2).
Now referring to Figure 14A, an ESI-FAIMS-MS mass spectrum is
shown for a solution of 5 gM bovine ubiquitin in 50/50/0.05
methanol/water/acetic
acid (v/v/v) collected with FAIMS disabled. The solvent combination was
selected
for this illustration because several charge states are present. This mass
spectrum
essentially represents a conventional ESI-MS spectrum with somewhat lower
sensitivity. Figures 14B-14E are ESI-FAIMS-MS spectra of the same solution
used
for Figure 14A with FAIMS in operation (DV =-3300V): Figure 14B shows a TIC-CV
CA 02339553 2001-02-05
WO 00/08454 PCT/CA99/00714
-29-
spectrum (m/z 30 to 2300), collected by scanning the CV from -12 V to 0 V,
while
Figures 14C-14E are mass spectra taken at specified CV values. The TIC-CV
spectrum in Figure 14B shows two distinct peaks with maxima at CV = -8.0 V and
CV = -5.4 V. Figures 14C-14E illustrate ESI-FAIMS-MS mass spectra of protein
ions,
taken at CV values indicated by the arrows in Figure 14B. The mass spectrum
collected at CV = -8.0 volts (Figure 14C) is dominated by the [M + 7H]7+ ion
of
bovine ubiquitin. Unlike the conventional ESI-MS spectrum, Figure 14A, charge
states higher than +7 are virtually absent in this mass spectrum. A mass
spectrum
(Figure 14D) taken at the CV corresponding to the small shoulder off of the
main
peak, at CV = -7.0 V (Figure 14B), shows a very different charge state
distribution
than Figure 14C. The [M + 6H] 6+ ion is the most abundant ion in this mass
spectrum
and several higher charge states are also observed. Finally, the mass spectrum
at CV
= -5.4 volts (Figure 14E) shows yet another charge state distribution which is
quite
unlike the previous two mass spectra. In this spectrum, the [M + 7H]7+ ion is
significantly reduced and the higher charge states (along with charge state
+5) have
increased in intensity. The increase in sensitivity in the mass spectra
collected with
the FAIMS operating (Figures 14C-14E), compared with the mass spectra with the
FAIMS disabled (Figure 14A), is a result of the atmospheric pressure ion
focusing
mechanism discussed earlier.
The changes in the charge state distribution of the mass spectra
collected at different CV values suggested that ion separation in FAIMS was
sensitive to the structure of the protein ion. Consequently, IS-CV spectra for
the
individual charge states of bovine ubiquitin were collected using mass
spectrometry,
for the same solution as used to collect the data shown on Figure 14A, for
charge
states +5 to +13.
Now referring to Figure 15, some charge states (e.g., +10) show only one
peak while others show multiple peaks (e.g., +8). Since the CV values for
charge
state +7 (m/z 1224.6) are more negative than the CV values for charge state +6
(m/z 1426.6) which are in turn more negative than the CV values for charge
state
+10 (m/z 857.5), the position of the peak in an IS-CV spectrum is clearly not
a
function of m/z. The multiple peaks which appear in several of the IS-CV
spectra in
Figure 15 are attributed to coexisting, and distinct conformations of bovine
CA 02339553 2001-02-05
WO 00/08454 PCT/CA99/00714
-30-
ubiquitin. In the following discussion we will illustrate the behavior of
several of the
conformers of bovine ubiquitin, as a function of solution phase conditions
including
the solvent composition and the pH.
E~'ect o Acid:
Different concentrations of acetic acid were used with 5 gM solutions
(55:45 H20/MeOH v/v) of bovine ubiquitin to observe changes in the mass
spectra
and the CV spectra as a function of pH. The composition of the solvent has
been
selected to yield suitable mixtures of conformations for illustrating the
effects of pH
in the experiments discussed below. This solvent mixture was used for the data
appearing in Figures 16 to 19. Figures 16A, 16C, and 16E, show mass spectra
collected with the FAIMS disabled for 3 acetic acid (HOAc) concentrations from
0.04% to 4%, while Figures 16B, 16D, and 16F show the corresponding TIC-CV
spectra (m/z from 30 to 2300) collected with the FAIMS in operation (DV =-
3300V). At 0.04% acetic acid Figure 16A, the higher charge states are present
at very
low abundances (with 60% H20 by volume these charge states are no longer
observed). This spectrum is very similar to one reported in an earlier study
for bovine
ubiquitin is in its native state.
The TIC-CV spectrum in Figure 16B for the 0.04% acetic acid solution
shows a strong peak at CV = -8.2 V with a small shoulder at CV = -6.0 V. The
mass
spectrum (FAIMS disabled) collected using a solution containing 0.4% HOAc
(Figure
16C) shows a second charge state distribution, centered around [M + 12H]12+,
in
addition to the distribution centered at [M + 7H]7+. This second distribution
is
consistent with spectra collected for bovine ubiquitin in its denatured form
as
reported in an earlier study. The TIC-CV spectrum in Figure 16D for the 0.4%
HOAc
solution has a second peak at CV = -5.8 V, presumably due to the presence of
the
denatured bovine ubiquitin. Finally, at 4% acetic acid, the low sensitivity
ESI-MS
spectrum (Figure 16E) shows almost exclusively the higher charge states. The
TIC-CV spectrum (Figure 16F) shows essentially one peak located at CV = - 6.0
V.
Consequently, it is clear that the changes in the TIC-CV spectra collected
with the
FAIMS in operation (DV = -3300 V) reflect changes seen in the mass spectra of
bovine ubiquitin with the FAIMS disabled.
CA 02339553 2001-02-05
WO 00/08454 PCT/CA99/00714
-31-
Figures 17A-171 show IS-CV spectra for some of the charge states (i.e.,
+7, +8, and +9) of bovine ubiquitin as a function of pH. Traces were obtained
using
the same solutions as traces in Figures 16A-16F. The CV spectra of some of the
charge states, such as +10 to +13, did not show significant changes as a
function of
pH and were excluded from the Figure.
Figures 17A-17C are the IS-CV spectra of the +7, +8, and +9 charge
states, respectively, collected using 0.04% acetic acid. These spectra reflect
conditions in which the bovine ubiquitin is essentially in its native state.
For charge
state +8 in Figure 17B, two main peaks are observed at CV ~ -9 V and - -7 V.
With
0.4% acetic acid, in the same solution, Figure 17E, the IS-CV spectrum for
charge
state +8 changes significantly. The peak that is observed CV - -9V is now
virtually
absent from the spectrum and a new peak at CV - -5 V is visible. This change
suggests that as the bovine ubiquitin begins to unfold the conformer of bovine
ubiquitin, reflected by the peak at CV - -9 V, is no longer favorable. At a
concentration of 4 % acetic acid, Figure 17H, the peak at - -9 V is now
completely
absent from the spectrum. Charge state +7, Figures 17A, 17D, and 17G, also
experiences significant changes with increasing concentrations of acetic acid.
At low
acetic acid levels, Figure 17 A, the IS-CV spectrum is dominated by a peak
located at
CV - -8 V. However, when 4 % acetic acid is used, Figure 17G, the IS-CV
spectrum
shows a shift that now favors the conformer which is present at CV - -7 V.
Figures
17C, 17F, and 171 show that charge state +9 did not show significant changes
as a
function of pH.
To investigate behavior of the conformers at even lower pH values,
hydrochloric acid, HCI, was added to solutions of 5 M bovine ubiquitin
(solutions
were still 55% H20 by volume). IS-CV spectra at pH 2.8, 2.1, and 1.8 are shown
in
Figures 18A-18I. The top traces (Figures 18A, 18B, and 18C) were obtained at
the
same pH as the last experiment in Figures 17G, 17H, and 171 (i.e., pH - 2.8).
This
was done to permit a comparison based on changing the acids only. The IS-CV
traces for charge states +8 and +9 at pH 2.8 (Figures 18B, and 18C) in HCl are
very
similar to that observed in Figures 17H, and 171 using acetic acid at pH 2.8.
The
IS-CV spectra for the other charge states not shown also gave similar results.
However, the IS-CV spectrum for charge state +7 in HCI at pH 2.8 (Figure 18A)
CA 02339553 2001-02-05
WO 00/08454 PCT/CA99/00714
-32-
shows significant differences from the IS-CV spectrum (Figure 17G) for charge
state
+7 at pH 2.8 in acetic acid. The former spectrum more closely resembles IS-CV
spectra for acetic acid at slightly higher pH values,but with an additional
peak at
CV - -5 V that was not observed when using acetic acid.
By decreasing the pH from 2.8 (Figures 18A, 18B, and 18C) to 2.1 in
Figures 18D, 18E, and 18F, there are significant changes that are observed for
charge
states +7, +8, and +9 respectively (and others). In all instances, a peak at a
CV of
between -5 and -6 V becomes favored. This trend is continued as the pH is
lowered
even further to 1.8 (Figures 18G, 18H, and 181). The IS-CV spectra that were
obtained for this solution were markedly more noisy and less intense. However,
the
dominance of the peak at the least negative CV value is still apparent.
The data illustrated in Figures 15 and 19 permit a comparison of the CV
spectra of bovine ubiquitin at several charge states, using solutions of
acetic acid at
pH 3.4 (Figure 15, 50:50 water/MeOH) with CV spectra collected using a
solution
acidified to pH 2.1 with HCl (Figure 19, 55:45 water/MeOH). The traces for
charge
states +10 through +13, in Figure 19, are very similar to those observed in
Figure 15.
Changes in the CV spectra of charge states +5 through +9 were observed.
F~,.fFert of Solvent:
The CV spectra and mass spectra shown in Figure 20 illustrate the
effect of changing the solvent mixture from 50:50 water/MeOH Figures 20A, 20C,
and 20E, to 55:45 water/MeOH v/v Figures 20B, 20D, and 20F, while maintaining
low acid (0.04% HOAc) concentration. The mass spectra (Figures 20A and 20B)
were collected with the FAIMS disabled while the CV spectra (Figures 20C
through
20F) were collected with the FAIMS in operation (DV =-3300V). As is consistent
with previous studies, increasing amounts of organic solvent cause bovine
ubiquitin
to denature. This is shown by the slight increase in the higher charge states
of mass
spectra acquired with the solution containing the higher percentage of organic
solvent
(Figure 20A, 50% MeOH) relative to that shown for lower percent organic
solvent
(Figure 20B). The FAIMS CV spectra also reflect this change in solvent
composition
as indicated in the TIC-CV spectra in Figures 20C and 20D. Furthermore, the IS-
CV
spectrum for charge state +8 (Figures 20E, and 20F) also undergoes a
significant
CA 02339553 2001-02-05
WO 00/08454 PCT/CA99/00714
-33-
change over this solvent range. The peak at -9 V in the CV spectrum of the +8
charge state collected using a solution containing 50% MeOH (Figure 20E) is
much
less intense than that observed at -9 V for a solution which contained 45%
MeOH
(Figure 20F).
Exchange Reactions:
Mass spectra of the three distinct peaks observed in the CV spectrum of
charge state +8 (Figure 21A) were collected to investigate differences among
the
conformers of this charge state. The CV spectrum (Figure 21A) and the mass
spectrum collected for each peak, for a solution containing 5 gM bovine
ubiquitin in
50% H20 and 0.04% acetic acid are shown in Figures 21C, 21E, and 21G. The
expanded views of the mass spectra show that several sodium ions have replaced
protons, (M+nH+mNa)+n+m where n+m=8, in each conformation of the +8 charge
state of bovine ubiquitin. The mass spectrum of the peak at CV = -4.8 V
(Figure
21G) clearly shows up to four sodium replacements, the mass spectrum of the
peak
at CV = -8.9 V (Figure 21C) shows virtually no proton replacement, and the
mass
spectrum of the peak at CV = -6.9 volts (Figure 21E) shows that up to two
sodium
ions have replaced protons in the +8 ion. We speculate that the differences in
the
aqueous phase, three-dimensional structures of the conformers of the +8 charge
state
of the bovine ubiquitin ion have resulted in varying degrees of replacement of
protons
by sodium ions in the gas-phase conformer.
It could be argued that the location of these adducts in the CV spectrum
is not a consequence of the different conformations of bovine ubiquitin, but
instead
that the sodium adduct actually causes the ions to be located in the CV
spectrum as
shown in Figure 21A. Previous results with ESI-FAIMS-MS obtained by the
inventors
have shown that for a smaller species (e.g., leucine enkephalin, MW 555.5),
adduct
ions can alter the CV of a given species in the compensation voltage spectrum.
Consequently, two additional experimental results are described to show that
the
presence of different degrees of sodium replacement is a result of the
different
conformations, and that the location of the ion in these CV spectra is not
altered by
replacement of protons with sodium ions.
The spectra in Figures 21B, 21D, 21F, and 21H were obtained after 1
CA 02339553 2001-02-05
WO 00/08454 PCT/CA99/00714
-34-
mM of sodium chloride was added to the solution that was used to collect the
data
for Figures 21A, 21C, 21E, and 21G. Note that due to the distribution of the
sodium-containing ions over several m/z values, the IS-CV spectrum collected
in
Figure 21B was collected over the m/z range 1071-1080 rather than at m/z
1071.6
exclusively. The IS-CV spectra in Figures 21A and 21B are very similar, and
the CV
values of the three peaks corresponding to +8 of bovine ubiquitin have not
changed
significantly. However, the expanded views of the mass spectra collected for
these
three species have changed. Furthermore, the mass spectrum for the species at
CV =
-4.8 V with no sodium added (Figure 21G) is very similar to the mass spectra
for the
species at CV = -6.9 V with 1 mM sodium added (Figure 21F). The question to be
asked is whether the number of sodium ions bound to the protein ion determine
its
position in the CV spectrum or does the conformation of the protein ion
determine
the degree of sodium substitution. If the replacement of protons with sodium
ions
was causing the CV shift, these two mass spectra should be observed at
virtually
identical locations in the CV spectra. Thus, the number of sodium replacements
reflects structural differences in the conformers represented by the three
peaks
observed for the +8 state of bovine ubiquitin.
An IS-CV spectrum for the +6 charge state (Figure 22A) is dominated
by a peak at CV = -7.5 V, however there is also a smaller peak at CV = -5.7 V.
Figures 22B and 22C show mass spectra that were collected for these peaks at
CV =
-5.7 V and CV = -7.5 V (note Figure 22B represents a sum of 50 mass spectra).
For
this charge state, the ion which is amenable to higher level of sodium
replacement of
protons is observed at the more negative CV of the two conformers. Conversely,
the
data for the +8 charge state of bovine ubiquitin (Figure 21), indicates that
the
conformer which accumulates the higher number of sodium ions was observed at
less
negative CV values.
The effect of other species, beside sodium, on CV spectra for bovine
ubiquitin was also investigated. Figure 23 shows ESI-FAIMS-MS data collected
using
an excess of potassium di-hydrogen phosphate added to a solution of 5 M
bovine
ubiquitin in 50% H20 and 0.02% acetic acid. Figure 23A shows an IS-CV spectrum
for the +8 charge state of bovine ubiquitin that is very similar to that
observed
previously in this study for solutions of 50% H20, Figure 21A. The change in
the
CA 02339553 2001-02-05
WO 00/08454 PCT/CA99/00714
-35-
relative abundances of the conformers at CV - -9 V and - -5 V compared with,
for
example, Figure 21A, can be attributed to the change in pH caused by the
difference
in HOAc concentration, and added KH2PO4. Expanded views of the mass spectra,
obtained at CV values -8.9 V, -6.9 V, and -4.8 V in Figure 23A, are shown in
Figures
23B, 23C, and 23D, respectively, and show significant differences in the
abundances
of phosphate adducts. As was observed with sodium, the degree of adduct
formation is a function of the conformer. However, the results in Figure 23
show the
opposite trend compared with the results shown in Figure 21. That is, in
Figure 23,
the conformer that appears in the CV spectrum at CV - -9 V (Figure 23B) shows
the
most intense phosphate adduct ion whereas the conformer that appears in the CV
spectrum at CV - -5 V shows the least intense phosphate ion adduct (Figure
23D).
Note also that the addition of phosphate did not modify the CV at which each
of
the three conformers appeared (compare to Figure 21A). We also note that none
of
the three conformers of the +8 ion had a tendency to replace protons with
potassium
ions.
These examples illustrate that by examining 'spectator' ions such as
sodium and phosphate, we can gain clues into the differences in the individual
conformations. Experiments have indicated that the replacement/addition of the
'spectator' has not affected the ion conformation. It is possible that the
formation of
some replacements/additions may lead to significant changes in the
conformation of
an ion. Such a change would be expected (in most cases) to be reflected by
peak
shifts in the IS-CV spectra.
Concentration Effects:
n
In an earlier study, the inventors observed more than one peak in an
IS-CV spectrum of a solution of leucine enkephalin (m/z 556.5) with ESI-FAIMS-
MS
(see R. Purves and R. Guevremont, Anal. Chem. 71, 2346 (1999), R. Guevremont
and
R. Purves J. Am. Soc. Mass Spectrom. 10, 492 (1999)), and attributed the
additional
peaks to a series of cluster ions of the type (nM+nH)n+. To ensure that the
different
peaks observed in the IS-CV spectra in this study (e.g. Figure 15) were not a
consequence of the formation of multimers or other cluster ions, different
concentrations of bovine ubiquitin were studied. For the concentration range
from 1
CA 02339553 2001-02-05
WO 00/08454 PCT/CA99/00714
- 36 -
M to 100 M bovine ubiquitin (in 50% water and 0.04% acetic acid), no
significant
changes in the shapes of the IS-CV spectra for any charge state were observed.
If the
multiple peaks in an IS-CV spectrum were caused by the formation of cluster
ions,
the relative amounts of the various peaks in an IS-CV spectrum would change as
a
function of concentration.
Comparison with Drift Tube MobilituSpectrrLmetty
ESI-drift tube mobility spectrometry/MS has been used to examine the
conformations of a number of proteins. The separation of conformers in a drift
tube
is based on the ion cross section, whereas the separation of ions in FAIMS is
based
on heretofore unknown properties of the ions. It is expected therefore, that
there will
be many similarities, and also significant differences in the array of
conformations
detected by these two, independent approaches.
Valentine et. al. have used their ESI-drift tube mobility
spectrometry/MS, and proton transfer reagents, to study the conformers of
bovine
ubiquitin (S.J. Valentine, A.E. Counterman, D.E. Clemrner, J. Am. Soc. Mass
Spectrom. 8, 954 (1997)). In their work, the authors divided their results for
collision
cross section into three types of conformations: elongated, partially folded,
and
compact. Figure 24 summarizes the conformational information obtained for
bovine
ubiquitin by ESI-FAIMS-MS as a plot of CV values for each of the peaks
appearing
in the CV spectra for each of the individual charge states. The "low acid"
solution
contained 55% H20 and 0.04% acetic acid while the "high acid" solution
contained
55% H20 and the pH adjusted to 2.1 with HC1. The peak maxima that were
observed in the IS-CV spectra were classified as being observed either in the
"low
acid" solution, the "high acid" solution, or both.
For some charge states in Figure 24 (i.e., +10 through +13), the CV
remains approximately constant between -5 and -6 V independent of the acid
concentration (pH). For the remaining charge states (i.e., +5 through +9),
there are
several resolved conformers, and the relative abundances of the conformers is
dependent on the solution conditions. The most negative CV values are observed
for
conformers only present in the "low acid" solution. For charge states +5
through +8,
Valentine et. al. reported the co-existence of elongated and partially folded
CA 02339553 2001-02-05
WO 00/08454 PCT/CA99/00714
-37-
conformations. Consequently, both FAIMS, and drift tube mobility spectrometry
techniques identified the same charge states having a multiplicity of
conformations.
Although the invention has been described and illustrated with reference
to specific embodiments, those skilled in the art will recognize that the
invention may
be otherwise embodied within the scope of the following daims. Specifically,
while
the experiments describe the use of a mass spectrometer to measure transmitted
ions
at an ion outlet of a FAIMS device, it will be understood that a mass
spectrometer is
not necessary once the values for DV, CV and other operating conditions for
the
FATMS device have been determined for separating a desired ion. Thus,
collection of
desired transmitted ions may occur at the ion outlet of a FAIMS device, rather
than
entering a mass spectrometer for measurement.