Note: Descriptions are shown in the official language in which they were submitted.
CA 02340300 2001-04-02
TITLE OF THE INVENTION
NUCLEOTIDE SEQUENCES THAT CODE 1FOR THE rpiK GENE
AND METHODS OF USE THEREOF
The object of the invention resides in nucleotide sequences from coryneform
bacteria
that code for the rplK gene and a method for enzymatic preparation of amino
acids, especially
L-lysine, while attenuating the rplK gene.
AlI references cited herein are incorporated by referE:nce into this
specification. In
addition, throughout the following disclosure, the term "incorporated by
reference" is
indicated by the notation "I. B. R. "
BACKGROUND OF THE INVENTION
L-Amino acids, especially L-lysine, are used in animal nutrition, in human
medicine
and in the pharmaceutical industry. It is known that these amino acids are
prepared by
fermentation of strains of coryneform bacteria, especially Cwrynebacterium
,glutamicum.
Because of their great importance, work to improve the production methods is
continuously
being carried out. Methods of mutagenesis, selection and mutant selection are
used to impmve
the performance properties of these microorganisms. In this way one obtains
strains that are
resistant to antimetabolites such as the lysine analog S-(2-
anunoethyl)cysteine or auxotrophic
for regulatorily important metabolites and produce L-amino acids.
Page 1
CA 02340300 2001-04-02
For a number of years methods of recombinant DNA, technology have also been
used
for strain improvement of strains of Corynebacterium that produce L-amino
acid, by
amplifying individual amino acid biosynthesis genes and testing the effect on
L-amino acid
production. Review articles on this subject can be found, among other places,
in Kinoshita
("Glutamic Acid Bacteria," in: Biology of Industrial Microorganisms, Demain
and Solomon
(Fds.), Benjamin Cummings, London, UK, 1985, 115-142) LB.R., Hilliger (BioTec
2, 40-4.4
( 1991) 1. B. R. ), Eggeling (Amino Acids 6:261-272 ( 1994) I. B. R. ), Jetten
and Sinskey (Critical
Reviews in Biotechnology 15, 73-103 (1995) I.B.R.) and Sa~hm et al. (Annuals
of the New
York Academy of Science 782, 25-39 (1996) LB.R.).
The rplK protein (ribosomal large subunit protein I~ is a component of the
bacterial
ribosome that was first described for Escherichia coli, the tt~anslation
apparatus of the cell, on
which protein synthesis takes place.
Ribosomes are cellular particles that are composed of three ribonucleic acid
(RNA)
molecules and a specific number of proteins. Ribosomes are; for the most part
obtained from
cell extracts by ultracentrifugation. The further purification of the
remaining cell components
usually takes place by sedimentation in sucrose gradients. This preparation
technique ied to the
usual designations for the components of ribosomes, which refer directly to
the sedimentation
properties. Thus, one functional bacterial ribosome is frequently designated
70S ribosome,
which consists of the small (small subunit) 30S subunit and the large (large
subunit) 50S
subunit. The small 30S subunit of E. coli consists of 21 different
polypeptides and an RNA
molecule with a length of 1542 nucleotides, which is known to the specialist
as 16S-rRNA;
Page 2
CA 02340300 2001-04-02
besides the rplK protein, the large SOS subunit contains an additional 31
different polypeptides,
together with two RNA molecules with lengths of 120 and :2904 nucleotides,
respectively, the
so called SS or 23S rRNA molecules. Meanwhile, an alternative nomenclature has
been
established for the ribosomal proteins. Thus, the polypeptidles of the small
30S subunit are
designated S1 to S21 by the specialist, while those of the large SOS subunit
are designated Ll
to L32. The rplK protein here corresponds to the ribosomal Ll I protein
(Holler and Nomura,
In: Neidhardt et al., Escherichia coli and Salmonella typhir~nurium: Cellular
and molecular
biology. American Society for Microbiology, Washington l~.C., 167-186, 1996)
LB.R.. In
recent years Ll l-Iike proteins have also been identified in other organisms
such as Borrelia
burgdorferi (Eraser et al., Nature, 390, 580-586, 1997) LB'.R.,
Helicobacterpylori (Tomb et
al., Nature, 388, 539-547, 1997) ZB.R., Serratia marcesce~ns (Sor and Nomura,
Molecular
and General Genetics, 210, 52-59, 1987) 1. B. R. , Haemoph;ilus infuenzae
(Fleischmann et al. ,
Science, 269, 496-512, 1995) LB.R. and in the gram positive bacterium Bacillus
subtilis
(Jeong et al., Molecular Microbiology, 10, 133-142, 1993) LB.R..
The process of translation that occurs at the ribosome, thus the messenger RNA-
controlled biosynthesis of polypeptides, is complex. In addition to the
ribosome, other proteins
called protein synthesis factors by the specialist (Holler, Annual Review of
Biochemistry, 60,
191-227, 1991) LB.R., are essential for the translation process. The Lll
protein mediates the
interaction between the n'bosome and some protein synthesis factors; one may
mention as an
example here the elongation factor G (EF-G) and the tenniz~ation factor 1 (RF-
1). The absence
of the Ll l protein in the ribosomes of E. coli Ll l mutants thus leads to a
reduction of the
Page 3
CA 02340300 2001-04-02
translation rate (Xing and Draper, Biochemistry, 35, 1581-1588, 1996) LB.R..
The Ll l protein is likewise essential for the bonding; and activity of the
ReIA protein to
the ribosome. ReIA catalyzes, under conditions where there is a deficiency of
amino acid, the
synthesis of guanosine tetraphosphate (ppGpp) by the transfer of one
pyrophosphate group
from ATP to GDP. In E. coli ppGpp affects the expression of many genes, either
negatively or
positively. In general, the expression of gene products that are effective in
biosynthesis
pathways is stimulated. Gene products that are catabolically effective are as
a rule
correspondingly negatively regulated. A large number of genes and operons that
play a central
role in amino acid biosynthesis are regulated by ppGpp in ls. coli. Among the
genes known up
to now to be positively affected in E. coli are argF, argI, a~rgECBH (arginine
biosynthesis),
gltB, glnA, gdh (glutaminelglutamate biosynthesis), ilvB, IIvA (isoleucine
biosynthesis),
metC, metF, metK (methionine biosynthesis), thrA, lhrB, thrC (threonine
biosynthesis), lysA,
lysC, dapB, asd (lysi~ biosynthesis) (Cashel et al.., In: Ne:idhardt et al.,
Fscherichia call and
Salmonella typhimurium: Cellular and molecular biology, American Society for
Microbiology,
Washington D.C., 1458-1496, 1996) LB.R.. Meanwhile, the function of ppGpp as a
positive
regulator of amino acid biosynthesis was also demonstrated in other bacteria
such as
Salmonella typhimurium tRudd et al. , Journal of Bacteriology, 163, 534-542,
1985) L B. R. ,
Vibrio sp. (Flardh et al., Journal of Bacteriology, 176, 59f~9-5957, 1994)
LB.R. and B.
subtilis (Wendrich and Marahiel, Molecular Microbiology, 26, 65-79, 1997)
LB.R..
Page 4
CA 02340300 2001-04-02
oB.>ECTIVE of THE IrrvENTION
From the prior art it is clear that there is interest in minding out if
knowledge of the
nucleotide sequence of the rplK gene of coryneform bacteria will contribute to
an improvement
of the amino acid production of these bacteria. Thus, an object of the
invention is to make
available new measures for improved enzymatic preparation, of amino acids,
especially L-
lysine.
SUMMARY OF THE INVENTION
L-Amino acids, especially lysine, are used in human medicine and in the
pharmaceutical industry, in the food industry and particularly in animal
nutrition. For this
reason there is general interest in making available new approaches for
improved methods for
preparation of amino acids, especially L-lysine.
A feature of the invention is an isolated polynucleotide containing a
polynucleotide
sequence chosen from the group
a) a polymicleotide that is at least 70 % identical to a polynucleodde that
codes for a
polypeptide that contains the amino acid sequence of SEQ IT> No. 2,
b) a polyrnicleotide that codes for a poIypeptide that contains an amino acid
sequence
that is at least 70% identical to the amino acid sequence of S:EQ ID No. 2.
The relative degree of substitution or mutation in the ;polynucleotide or
amino acid
sequence to produce this percentage of sequence identity can be established or
determined by
well-known methods of sequence analysis. These methods a~~e disclosed and
demonstrated in
Page 5
CA 02340300 2001-04-02
Bishop, et al. "DNA & Protein Sequence Analysis (A Practical Approach"),
Oxford Univ.
Press, Inc. (1997) LB.R. and by Steinberg, Michael "Protein Structure
Prediction" (A
Practical Approach), Oxford Univ. Press, Inc. (1997) LB.R.,
c) a polynucleotide that is complementary to the pcdynucleotides of (a) or
(b), and
d) a polynucleotide containing at least 15 successive bases of the
polynucleotide
sequence of (a), (b) or (c).
Another feature of the invention is the polynucleotide in accordance with (a-
d) above,
where it is preferably a replicable DNA containing:
(i) the nucleotide sequence indicated in SEQ ID No. 1,
or
(ii) at least one sequence that corresponds to the sequence (i) within the
region of
degeneration of the genetic code, or
(iii) at least one sequence that hybridizes with the sequence complementary to
sequence
(i) or (ii) . The degree of stringency required to produce hybridization may
vary from high to
low as described in Sambrook et al. and other documents incorporated by
reference herein and
optionally
(iv) function-neutral sense mutants in (i).
Other features of the invention include:
a polynucleotide which is replicable; preferably recombinant DNA, containing
the
nucleotide sequence as represented in SEQ ID No. 1,
a polynucleotide which is replicable, preferably recombinant DNA, which codes
for a
Page 6
;ii
CA 02340300 2001-04-02
polypeptide that contains the amino acid sequence as represented in SEQ ID No.
2,
a polynucleoride as in (a-d) above, especially item (d), containing the
nucleotide
sequence as represented in SEQ ID No. 3,
a polynucleotide as in (a-d) above, especially item (d;i, which codes for a
polypeptide
that contains the amino acid sequence as represented in SEQ ID No. 4,
a vector containing a mutated polynucleotide as in (a-~d) above, especially
item (d)
represented in SEQ ID No. 3 and Figure 1 (t1 = deHa) and deposited in E. coli
DHSa/p~rplK
as DSM 13158 in accordance with the Budapest Treaty and deposited on November
26, 1999
with the International Depositary Authority of DSMZ-Qeutsche Sammlung von
Mikroorganism
Und Zell Kulturen GmbH, Maschroder Weg lb, D-38124 Braunschweig, Germany,
and coryneform bacteria serving as host cells, which contain an insertion or
deletion in
the rplK gene.
A further feature of the invention is also polynucleotides that essentially
consist of a
polynucleotide sequence that caa be obtained by screening by means of
hybridization, with
varying degrees of stringency as established in Sambrook a=id the other
citations incorporated
by reference herein, of an appropriate gene bank that contains the complete
gene with the
polynucleotide sequence in correspondence with SEQ ID No. 1, with a probe that
contains the
sequence of said polynucleotide in accordance with SEQ ID No. 1 or a fragment
thereof, and
isolation of the said DNA sequence.
Polynucleotide sequences in accordance with the invention are suitable as
hybridization
probes for RNA, cDNA and DNA, in order to isolate cDN.A in its full lengh,
that code for
Page 7
CA 02340300 2001-04-02
the ribosomal protein L1 l and to isolate those cDNA or genes that have high
similarity to the
sequence with the rplK gene.
Polynucleodde sequences in accordance with the invention are also suitable as
primers,
with which the polymerase chain reaction (PCR) of the DN.A of genes that code
for the rplK
gene product or ribosomal protein Ll l can be produced.
Oligonucleotides that serve as probes or primers contain at Ieast 30,
preferably at least
20, really especially preferably at least 15 successive nucleotides. Likewise
suitable are
oligonucleoddes with.a length of at least 40 or 50 base pairs.
"Isolated" means separated from its natural environment.
"Polynucleotide" refers in general to polyribonucleotides and
polydeoxyribornicleotides, where these can be unmodified F;NA or DNA or
modified RNA or
DNA.
"Polypeptides" is understood to mean peptides or proteins that contain two or
more
amino acids bonded via peptide linkages.
The polypeptides in accordance with the invention ir.~clude the polypeptide in
accordance with SEQ ID No. 2, especially ones with the biological activity of
the rplK gene
product and also those that are at least 70 % identical to the :polypeptide in
accordance with
SEQ ID No. 2, preferably at least 80 % and especially at least 90 to 95 %
identical to the
polypeptide in accordance with SEQ ID No. 2 and have the same activity. The
degree of
substitution or mutaxion in the polynucleotide or amino acid sequence to
produce this degree of
identity can be determined by well-known methods of sequence analysis. These
methods are
Page 8
CA 02340300 2001-04-02
disclosed and demonstrated in Bishop, et aI. "DNA & Protein Sequence Analysis
(A Practical
Approach"), Oxford Univ. Press, Inc. (1997) LB.R. and bay Steinberg, Michael
"Protein
Structure Prediction" (A Practical Approach), Oxford Univ. Press, Inc. (1997)
LB.R.
The invention additionally concerns a method for enzymatic preparation of
amino
acids, especially lysine, while using coryneform bacteria that in particular
already produce the
amino acids and in which the nucleotide sequences that co~Ie for the rplK gene
have been
attenuated.
The term "attenuation" in this connection describes the reduction or switching
off of the
intracellular activity or function of one or more enzymes o:r proteins in a
microorganism that
are coded by the corresponding DNA, by using, for example, a weak promoter or
a gene or
allele that codes for a corresponding enzyme or protein with low activity or
that inactivates the
corresponding gene or enzyme or protein and optionally by combining these
measures.
The microorganisms that are objects of this invention can produce amino acids,
especially lysine, from glucose, sucrose, lactose, fructose, maltose,
molasses, starch, cellulose
or from glycerol and ethanol. These can be representatives of coryneform
bacteria, especially
the genus Corynebacterium. In the genus Corynebacterium~ one should especially
mention the
species Corynebacterium glutamicum, which is known among specialists for its
capacity to
produce L-amino acids.
Page 9
CA 02340300 2001-04-02
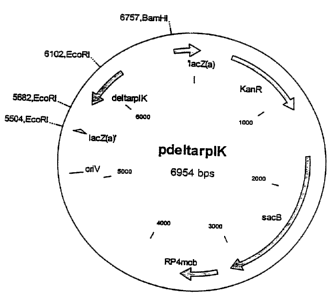
BRIEF DESCRIPTION OF THE (DRAWING
The present invention will be further understood with reference to the drawing
offered
here for illustration only and not in limitation of this inveni:ion.
In the drawing:
FIGURE 1 is a map of the plasmid pOrplK.
The abbreviations and designations that are used have the following meanings:
pdeltarplK parplK
sacB gene sacB gene from Bacillus subti'lis, coded
for the enzyme
levansucrase
lacZ alpha:Part of the S' end of the (3-galactosidase
gene
oriV Replication origin
KmR: Kanamycin resistance
RP4 mob mob region of plasmid RP4
BamHI: Scission site of the restriction. enzyme
BamHI
EcoRI: Scission site of the restriction. enzyme
EcoRI
~rplK rplK allele with a deletion of 12 pb in
the N-terminal region
BRIEF DESCRIPTION OF SEQUIENCE DATA
SEQ ID NO 1 is a new nucleotide sequence that codes for the rplK gene.
SEQ ID NO 2 is the amino acid sequence of the ip:IK gene product (the I. 11
gene).
SEQ ID NO 3 is an allele of the rplK gene.
SEQ ID NO 4 is a variation of protein L-11 coded by the OrplK allele.
SEQ ID NO 5 is a Pl up primer derived on the basis of SEQ ID NO 1.
SEQ ID NO 6 is a P2 down primer derived on the basis of SEQ ID NO 1:
SEQ ID NO 7 is a Pl up primer derived on the basis of SEQ ID NO 1.
SEQ ID NO 8 is a P2 down primer derived on the basis of SEQ ID NO 1.
Page 10
CA 02340300 2001-04-02
DETAILED DESCRIPTION OF '.CITE INVENTION
Suitable strains of genus Corynebacterium, espe~~ially the species
Corynebacterium
glutamicum, are in particular the known wild strains
Corynebacterium glutamicum ATCC 13032
Corynebacterium acetoglutamicum ATCC15806
Corynebacterium acetoacidophilum ATCC13870
Corynebacterium melassecola ATCC 17965
Corynebacterium thermoaminogenes FERM BP-1539
Brevibacterium,~lavum ATCC14067
Brevibacterium lactofermentum ATCC 13869 an<i
Brevibaclerium divaricatum ATCC14020
and mutants or strains that produce L-amino acids and are prepared from them,
such as, for
example, the L-lysine producing strains
Corynebacterium glutamicum FERM-P 1709
Brevibacterium,~lavum FERM-P 1708
Brewibacterium lactofermentum FERM-P 1712
Corynebacterium glutamicum FERM-P 6463
Corynebacterium glutamicum FERM-P 6464
Corynebacterium glutamicum DSM 5715
Corynebacterium glutamicum DSM 12866 and
Corynebacterium gluxamicum DM58-1.
The inventors were successful in isolating the nE;w rplK gene from
Corynebacterium
glutamicum.
For isolation of the rplK gene of C. glutamicum a gene bank of Corynebacterium
Page 11
CA 02340300 2001-04-02
glutamicum is first constructed. The formation of gene banks is described in
generally known
textbooks and manuals. Examples that may be mentioned include the textbook by
Winnacker:
Genes and Clones an Introduction to Gene Techriolo~v (Verlag Chemie, Weinheim,
Germany, 1990) LB.R. or the manual by Sambrook et al.: Molecular Cloning. A
Laboratory
Manual (Cold Spring Harbor Laboratory Press, 1989) LB.R.. A very well known
gene bank is
that of E. coli K-12 strain W3110, which was designed by Kohara et al. (Cell
50, 495-508
(1987) LB.R.) in ~.-vectors. Bathe et al. (Molecular and General Genetics,
252:255-265, 199
LB.R. describe a gene bank of C. glutamicum ATCC13032, which was constructed
with the
aid of the cosmid vector SuperCos I (Wahl et al., 1987, Procfxdings of the
National Academy
of Sciences USA, 84:2160-2164) LB.R. in the E. coli K-12 strain NM554 (Raleigh
et al.,
1988, Nucleic Acids Research 16:1563-1575) LB.R.. Borma~m et al. (Molecular
Microbiology 6(3), 317-326) LB.R. again describe a gene bank of C. glutamicum
ATCC13032
using the cosmid pHC79 (Hohn and Collies, Gene 11, 291-298 (1980) LB.R..
Plasmids like pBR322 (Bolivar, Life Sciences, 25, 807-818 (1979) LB.R.) or
pUC9
(Vieira et al., 1982, Gene, 19:259-268) LB.R. can also be used to prepare a
gene bank of C.
gluta~nicum in E. coli. Strains of E. coli that are restriction and
recombination defective are
especially suitable as hosts. An example of this is the strain D~HSamcr, which
was described
by Grant et al. (Proceedings of the National Academy of Scier.~ces USA, 87
(1990) 4645-4649)
LB.R. .
The long DNA fragments cloned with the aid of cosmids can finally main be
subcloned
into common vectors that are suitable for sequencing.
Page 12
CA 02340300 2001-04-02
Methods for DNA sequencing are described, among; other places, in Sanger et
al.
(Proceedings of the National Academy of Sciences of the LJnited States of
America USA,
74:5463-5467, 1977) I.B.R. .
The new DNA sequence of C. glutamicum that codes for the rplK gene, which as
SEQ
ID NO 1 is the object of this invention, is obtained in this way. In addition,
the amino acid
sequence of the corresponding protein was derived from this DNA sequence with
the above
described methods. The resulting amino acid sequence of the rplK gene product
or the L-11
gene is represented in SEQ ID NO 2.
The coding DNA sequences that result from SEQ TD NO 1 through the degeneracy
of
the genetic code are likewise objects of the invention. In the same way, DNA
sequences that
hybridize with SEQ ID NO 1 or parts of SEQ 117 NO 1 an: objects of the
invention.
Hybridization can occur at varying degrees of stringency. Amino acid sequences
that are
obtained in the corresponding way from SEQ ID NO 1 are: likewise objects of
the invention.
The invention has found that coryneform bacteria, .after attenuation of the
rplK gene,
produce L-amino acids, especially L-lysine, in an improved way.
To achieve an attenuation, either the expression of the rplK gene or
preferably the
fetncrional properties of the protein can be reduced. Optionally the two
measures can be
combined.
Reduction of gene expression can take place by suitable culturing ar through
genetic
alteration (mutation) of the signal structures of gene expression. Signal
strictures of gene
expression are, for example, repressor genes, activator genes, operators,
promoters,
Page 13
CA 02340300 2001-04-02
attenuators, ribosome binding sites, the start codons and terminators. The
specialist will find
information in this regard, for example, in Patent Application WO 96/15246
LB.R., in Boyd
and Murphy (Journal of Bacteriology 170: 5949 (1988) LB.R.), in Voskuil and
Chambliss
(Nucleic Acids Research 26: 3548 (1998) LB.R.), in Jensen and Hammer
(Biotechnology and
Bioengineering 58: 191 (1998) LB.R.), in Patek et al. (Microbiology 142: 1297
(1996) LB.R.)
and in well known textbooks on genetics and microbiology such as, for example,
the textbook
by Knippers (Molecular Genetics, 6'~ edition, Georg Thieme Yerlag, Stuttgart,
Germany,
1995) LB.R. or the one by Winnacker (Genes and Clones, 'JCH
Verlagsgesellschaft,
Weinheim, Germany, 1990) 1. B. R. .
Mutations that lead to an alteration or reduction of the functional properties
of proteins
are known from the prior art. One may mention as examples the works by Qiu and
Goodman
(Journal of Biological Chemistry 272: 8611-8617 (1997) LB.R.), Sugimoto et al.
(Bioscience
Biotechnology and Biochemistry 61: 1760-1762 (1997) LB..R.) and Mockel
("Threonine
dehydratase from Corynebacterium glutamicum: elimination. of allosteric
regulation and the
structure of the enzyme," Reports of the Jnlich Research Center, Jiil-2906,
ISSN09442952,
Jiilich, Germany, 1994) LB.R.. Summarizations can be taken from well known
textbooks on
genetics and molecular biology such as the one by Hagemann (General Genetics,
Gustav
Fischer Verlag, Stuttgart, 1986) LB.R..
Transitions, transversions, insertions and deletions am possibilities as
mutations. One
speaks of missense mutations or nonsense mutations; depending on the effect Qf
the amino acid
exchange on activity. Insertions or deletions of at least one base pair in a
gene lead to frame
Page 14
CA 02340300 2001-04-02
shift mutations, as a consequence of which wrong amino acids are incorporated
or the
translation is prematurely cut short. Instructions for producing such
mutations belong to the
prior art and can be taken from well known textbooks on genetics and molecular
biology such
as the textbook by Knippers (Molecular Genetics, 6'" edition, Georg Thieme
Verlag, Stuttgart,
Germany; 1995) LB.R., the one by Winnacker (Genes and Clones, VCH
Verlagsgesellschaft,
Weinheim, Germany, 1990) LB.R. or by Hagemann (Gene:ral Genetics, Gustav
Fischer
Verlag, Stuttgart, 1986) LB.R.. Methods for producing mutations with the aid
of the
polymerase chain reaction (PCR) are described in C. R. Newton and A. Graham,
PCR, 2"°
edition, Spektrum Akademischer Verlag, Heidelberg, 1997, LB.R..
An example of a plasmid with which a deletion mutagenesis of the rplK gene can
be
carried out is the plasmid pW p1K represented in Figure 1.
Plasmid p~rplK consists of the plasmid pKlBmobsa,cB described by Jager et al.
(Journal of Bacteriology, 1992, 174: 5462-65) LB.R., in which an allele of the
rplK gene,
represented in SEQ ID No. 3, was inserted. The allele designated OrplK carries
a 12 bp-long
deletion in the 5' region of the gene. The variation of protean Ll I coded by
the OrplK allele is
represented as SEQ ID No. 4. The variation of protein L-I1 as represented
differs from the
wild type of protein L 11 by the error in the tetrapeptide proline-alanine-
leucine-glycine
corresponding to the amino acids of position 30 to 33 of SI:Q ID No. 2.
Plasmid p~rplK leads to exchange of the chromosomal rplK gene for the ~rplK
allele
after homologous recombination. Instructions and illustrations for insertion
aiutagenesis can
be found, for example, in Schwarzer and Piihler (BioITechnology 9, 84-87
(I991) LB.R.) or
Page 15
CA 02340300 2001-04-02
Fitzpatrick et al. (Applied Microbiology and Biotechnology 42, 575-580 (1994)
LB.R.).
In addition, it can be advantageous for the production of L-amino acids,
especially
L-lysine, to enhance one or more enzymes of the relevant biosynthesis path,
especially to
overexpress them in addition to attenuation of the rplK gene.
For example, especially for the preparation of L-lysine, one or more of the
genes
chosen from the group
~ the dapA gene coding for dihydrodipicolinate synthase (EP-B 0 197 335)
I.B.R.,
~ an lysC gene coding for a feedback resistant aspartate kinase (Kalinowski et
al.
(1990) LB.R., Molecular and General Genetics 224: 317-324) LB.R. ,
~ the pyc gene coding for pyruvate carboxylase (Eilkmanns (1992), Journal of
Bacteriology 174:6076-6086) 1. B. R. ,
~ the mqo gene coding for malate quinone oxidoreductase (Molenaar et al.,
European
Journal of Biochemistry 254, 395-403 (1998) LB.R.),
~ the lysE gene coding for lysine export (DE-A-195 48 222) LB.R.,
~ the zwal gene (DE 199 59 328.0; DSM 13115) I. B. R. ,
for example, can be simultaneously enhanced, especially overexpressed.
In addition, it can be advantageous for the production of L-amino acids, in
addition to
attenuation of the rplK gene, to attenuate at the same time one or more of the
genes chosen
from the group:
~ the pck gene coding for phosphoenol pyruvate caiiboxykinase (DE 199 50
409.1;
DSM 13047) LB.R.,
Page 16
CA 02340300 2001-04-02
~ the pgi gene coding for glucose 6-phosphate isonierase (US 09/396,478, DSM
12969) LB.R. ,
~ the poxB gene coding for pyruvate oxidase (DE 1199 51 975.7; DSM 13114) 1.
B. R. ,
~ the zwa2 gene (DE: 199 59 327.2; DSM 13113) LB.R.,
~ the relA gene coding for PPGPP synthetase I (EC; 2.7.6.5) LB.R..
In addition, it can be advantageous for the production of L-amino acids, in
addition to
the overexpression of 6-phosphogluconate dehydrogenase, to switch off
undesired side
reactions (Nakayama: "Breeding of Amino Acid Producing Micro-organisms, " in:
Overoroduction of Microbial Products, Krumphanzl, SikyW , Vanek (eds.),
Academic Press,
London, UK, 1982) LB.R..
The microorganisms containing the mutated polynuc:leotide in accordance with
(a-d)
above, particularly item (d), are likewise objects of the invention and can be
cultured
continuously or batchwise in a batch process or in a fed batch process or a
repeated fed batch
process for purposes of producing L-amino acids, especially L-lysine. A
summary of the
known cultivation methods is described in the textbook by C:hmiel (Bioprocess
Ensin~~ 1
Introduction to Bioprocess Techniques (Gustav Fischer Verlag, Stuttgart, 1991)
I B.R.) or in
the textbook by Storhas (Bioreactors and Peripheral Equipment (Vieweg Verlag,
Braunschweig/Wiesbaden, 1994) LB.R:):
The culture medium that ~is to be used must satisfy the requirements of the
relevant
strain in a suitable way. Descriptions of culture media for various
microorga~sms can be
found in the manual "Manual of Methods for General BactenoloQV," of the
American Society
Page 17
CA 02340300 2001-04-02
for Bacteriology (Washington, D.C., USA, 1981) LB.R.. Sugar and carbohydrates
such as
glucose, sucrose, lactose, fructose, maltose, molasses, starch and cellulose,
oils and fats like
soy oil, sunflower seed oil, peanut oil and coconut oil; fatty acids such as
palinitic acid, stearic
acid and linolic acid, alcohols such as glycerol and ethanol! and organic
acids Like acetic acid
can be used as carbon sources. These substances can be used individually or as
a mixture.
Compounds like peptone, yeast extract, meat extract, malt extract, corn steep
liquor, soy flour
and urea or inorganic compounds like ammonium sulfate, ammonium chloride,
ammonium
phosphate, ammonium carbonate and ammonium nitrate can be used as sources of
nitrogen.
The nitrogen sources can be used individually or as a mixbure. Phosphoric
acid, potassium
dihydrogen phosphate or dipotassium hydrogen phosphate'or the corresponding
sodium-
containing salts can be used as phosphorus sources. The culture medium must
additionally
contain salts of metals such as magnesium sulfate or iron sulfate, which are
necessary for
growth. Finally, essential growth-promoter substances like amino acids and
vitamins can be
used in addition to the substances mentioned above. Suitable precursors can
also be added to
the culture medium. Said substances can be added to the culture in the form of
a single batch
or can be dispensed during cultivation in a suitable way.
Basic compounds Iike sodium hydroxide, potassium hydroxide, ammonia or ammonia
water or acid compounds like phosphoric acid or sulfuric acid are used as
appropriate to
control the pH of the culture. Antifoam agents Like fatty a,~id polyglycol
esters can be used to
control the formation of foam. To maintain the stability of plasmids, suitable
selectively
active substances such as antibiotics can be added to the medium. In order to
maintain aerobic
Page 18
CA 02340300 2001-04-02
conditions, oxygen or oxygen-containing gas mixtures such as air can be
introduced into the
culture. The temperature of the culture is normally 20 ° C to 45
° C and preferably 25 ° C to
40°C. The culture is continued until a maximum of the de;>ired product
has formed. This
goal is normally achieved in a period of 10 h to 160 h.
Methods for determining L-amino acids are known i:ram the prior art. The
analysis can
be carried out as described in Spackman et al. (Analytical Chemistry, 30,
(1958), 1190) LB.R.
by anion exchange chromatography followed by ninhydrin <ierivatization, or it
can be carried
out by reversed phase HPLC, as described in Lindroth et al. (Analytical
Chemistry (1979) 51:
1167-1174) LB.R..
The following microorganism was added to the German Collection of
Microorganisms
and Cell Cultures (DSMZ, Braunschweig, Germany) in accordance with the
Budapest Treaty:
~ Escherichia coli strain DHSaIp~rplK, as DSM 13158.
EXAMPLES
Examvle 1
Preparation of a genomic cosmid gene bank from Coryneba~yterium gluramicum
ATCC 13032
Chromsomal DNA was isolated from Corynebacteri~cm glutamicum ATCC 13032 as
described in Tauch et al. (1995, Plasmid 33:168-179) ZB.R., and partially
cleaved with the
restriction enzyme Sau3AI (Amersham Pharmacia, Freiburg, Germany, product
description
Sau3AI, Code No. 27-0913-02) LB.R.. The DNA fragmenits were
dephophosphorylated with
shrimp alkaline phosphatase (Roche Molecular Biochemicals, Mannheim, Germany,
product
Page 19
CA 02340300 2001-04-02
description SAP, Code No. 1758250) I. B. R. , The DNA of the cosmid vector
SupeiCosl
(Wahl et al. (1987) LB.R. Proceedings of the National Academy of Sciences USA
84:2160-
2164) 1. B. R. , purchased from the Stratagene Company {La Jolla, USA, product
description
SuperCosl cosmid Vector Kit, Code No. 251301) LB.R., vvas cleaved with the
restriction
enzyme XbaI (Amersham Pharmacia, Freiburg, Germany, product description Xbal,
Code No.
27-0948-02) LB.R. and likewise dephosphorylated with shrimp alkaline
phosphatase. Then the
cosmid DNA was cleaved with the restriction enzyme Baml~ (Amersham Pharmacia,
Freiburg, Germany, product description BamHI, Code No. 27-0868-04) L B. R. .
The cosmid
DNA treated in this way was mixed with the treated ATCC 13032 DNA and the
batch was
treated with T4-DNA ligase (Amersham Pharmacia, Freiburg, Germany, product
description
T4-DNA-Iigase, Code No. 27-0870-04) LB.R.. The ligation mixture was then
packaged in
phages using the Gigapack II XL Packing Extracts (Stratagene, La Jolla, USA,
product
description Gigapack II XL. Packing Extract, Code No. 200217) LB.R. . To
infect the E. coli
strain NM554 (Raleigh et al., 1988, Nucleic Acid Research 16:1563-1575) LB.R.,
the cells
were taken up in lOmM MgS04 and mixed with an aliquot portion of the phage
suspension.
Infection and titration of the cosmid bank were carried out a.s described in
Sambrook et al.
(1989, Molecular Cloning: A Laboratory Manual, Cold Spring Harbor) LB.R., with
the cells
being plated out onto LB agar (Leanox, 1955, Virology, 1:1.90) LB.R. with 100
mg/L
ampicillin. After incubation overnight at 37°C recombinant single
clones were selected.
Page 20
CA 02340300 2001-04-02
Example 2
Isolation and sequencing of the rplK gene
The cosmid DNA of a single colony was isolated wiith the Qiaprep Spin Miniprep
Kit
(Product No. 27106, Qiagen, Hilden, Germany) 1. B. R. aa~ording to the
manufacturer's
instructions and partially cleaved with the restriction enzyme Sau3AI
(Amersham Pharmacia,
Freiburg, Germany, product description Sau3AI, Product No. 27-0913-02) LB.R..
The DNA
fragme~s were dephosphorylated with shrimp alkaline phosphatase (Roche
Molecular
Biochemicals, Mannheim, Germany, product description SAP, Product No. 1758250)
LB.R..
After gel electrophoretic separation the cosmid fragments irt the size range
of 1500 to 2000 by
were isolated with the QiaExII gel extraction kit (Product Nlo. 20021, Qiagen,
Hilden,
Germany) LB.R.. The DNA of the sequencing vector pZero-1, purchased from the
Invitrogen
Company (Groningen, Netherlands, product description Zero Background Cloning
Kit,
Product No. K2500-01) LB.R. was cleaved with the restriction enzyme BamHI
(Amersham
Pharmacia, Freiburg, Germany, product description BamHI, Product No. 27-08b8-
04.) LB.R..
Ligation of the cosmid fragments in the sequencing vector p~Zero-1 was carried
out as
described by Sambrook et al. (1989, Molecular Cloning: A Laboratory Manual,
Cold Spring
Harbor) I.B.R., with the DNA mixture having been incubated overnight with T4
ligase
(Pharmacia Biotech, Freiburg, Germany). This ligation mixture was then
electroporated
(Tauch et a1. , 1994, FEMS Microbiol Letters, 123:343-7) 1. B.R. in the E.
coli strain
DHSaMCR (Grant, 1990, Proceedings of the National Acadlemy of Sciences U.S.A.,
87:4645-4649) LB.R. and plated out on LB agar (Lennox, 1955, Virology, 1:190)
LB.R. with
Page 21
CA 02340300 2001-04-02
50 mg/L zeocin. Plasmid preparation of the recombinant clones took place with
the Biorobot
9600 (Product No. 900200, Qiagen, Hilden, Germany) LB.R.. Sequencing followed
the
dideoxy chain termination method of Sanger et al. (1977, Proceedings of the
National
Academies of Sciences U.S.A., 74:5463-5467) LB.R. as mEOdified by Zimmermann
et al.
(1990, Nucleic Acids Research, 18:1067) LB.R.. The "RF; dRhodamin Terminator
Cycle
Sequencing Kit" of PE Applied Biosystems (Product No. 403044, Weiterstadt,
Germany)
ZB.R, was used. The gel electrophoretic separation and analysis of the
sequencing reaction
took place in a Rotiphorese NF acrylamide/bisacrylamide g;el (29:1) (Product
No. A124.1;
Roth, Karlsruhe, Germany) LB:R. with the ABI Prism 377 sequencing device of PE
Applied
Biosystems (Weiterstadt, Germany).
The raw sequencing data that were obtained were then processed using the
Staden
program package (1986, Nucleic Acids Research, 14:217-2:31) LB.R. version 97-
0. The
single sequences of the pZerol derivatives were assembled to a connected
contig. The
computer aided coding region analysis was produced with the program XhTIP
(Staden, 1986,
Nucleic Acids Research, 14:217-231) LB.R.. Further analyses were carried out
with the
BLAST search programs (Altschul et al., 1997, Nucleic Acids Research, 25:3389-
3402) LB.R.
against the nonredundant data bank of the National Center :for Biotechnology
Information
(NCBI, Bethesda, MD, USA) LB.R. in its entirety as of M:ay 5, 2000 (including
all analytical
tools for sequence analysis available as of that date at that web-site).
The resulting nucleotide sequence is represented in ,'SEQ ID NO 1. Analysis of
the
nucleotide sequence gave an open reading frame of 438 ba.~e pairs, which was
characterized as
Page 22
CA 02340300 2001-04-02
the rplK gene. The rplK gene codes for a poIypeptide of 145 amino acids, which
is represented
in SEQ ID No. 2.
Example 3
Preparation of a vector with a copy of the rplK gene
A chromosomal 1200 by DNA fragment, which contained the rplK gene from C.
glutamicum, was cloned by means of PCR.
For this chromosomal DNA was isolated from Corynebacterium glutamicum ATCC
13032 as described in Tauch et al. (1995, Plasmid 33:168-1179) LB.R.. A 1200
by DNA
fragment that contained the rplK gene was amplified by means of the polymerase
chain
reaction. In addition, the following primers were derived on the basis of SEQ
ID No. 1.
Pl up (see also SEQ ID No. 5):
5'- AGG AGC AGG CTG TTG TCA CC -3'
P2 down: (see also SEQ ID No. 6):
5'- GCG GAT AGC TAC TGC GAT GG -3'
The represented oligonucleotides were synthesized by the ARK Scientific
Company
(ARK Scientific GmbH Biosystems, Darmstadt, Germany) and the PCR reaction was
carried
out using the Pfu-DNA polymerase (Stratagene, La Jolla, tfSA) and PTC 100
thermocycler
(MJ Research Inc., Waltharn, USA).
A cycle consisting of thermal denaturing (94°C, 90 aec), annealing
(58°C, 90 sec) and
the polymerise reaction (72 ° C, 90 sec) was carried out 35 times in
the PCR. The resulting
Page 23
CA 02340300 2001-04-02
1200 by DNA fragment was then purified by means of the Qiagen PCR purification
spin kit "'
(Qiiagen, Hilden, Germany).
For the cloning of the DNA amplificate containing the rpiK gene to a plasmid
that can
replicate in C. glutamicum, the plasmid pECM3, a deletion derivative of the
plasmid pECM2
described in Tauch et al. (FEMS Microbiological Letters, :123, 343-347 (1994)
LB.R.) was
prepared. For this the plasmid pECM2 was digested with tl~e restriction
enzymes BamHI
(Amersham-Pharmacia, Freiburg, Germany) and BgIII (Amersham-Pharmacia,
Freiburg,
Germany) and treated with T4 ligase (Amersham-Pharmacia, Freiburg, Germany),
as
described in Sambrook et al. (Molecular Cloning: A Laboratory Manual, Cold
Spring Harbor
Laboratory (1989) LB.R.), thus producing the plasmid pEC'M3. Transformation of
the E. coli
strain DHSaMCR described in Grant et a1. (Proceedings of the National Academy
of Science
USA, 87, 4645-4649 (1990) LB.R.) with the plasmid pECrrI3 took place as
described in Tauch
et al. LB.R. {FEMS Microbiological Letters, 123, 343-347 (1994) LB.R.). The
transformants
were selected on LBG agar (10 g trypton, 5 g yeast extract, 5 g NaCI, 2 g
glucose, 15 g agar
per Iiter), to which chloramghenicol (Merck, Darmstadt, GE:rmany) (50 mg/L)
had been
added.
Then the rplK gene-containing 1200 by DNA ampIificate was mixed with the
plasmid
pECM3, which had been linearized previously with the resb-iction enzyme SmaI
(Amersham-
Pharmacia, Freiburg, Germany) and treated with T4 DNA ligase (Amersham-
Pharmacia,
Freiburg, Germany), thus producing the plasmid prplK. Transformation of the E.
codi strain
DHSaMCR with the plasmid prplK took place as described iin Tauch et al. (hFMS
Page 24
CA 02340300 2001-04-02
Microbiological Letters, 123, 34.3-347 (1994) LB.R.), and the transformants
were selected on
LBG agar, to which chloramphenicol (Merck, Darmstadt, Germany) (50 mg/L) had
been
added. The plasmid prplK thus carries the complete rplK gene of C. glutamicum
and can
replicate autonomously both in E: coli and in C. glutamicu~m.
Example 4
Insertion of a deletion into the rplK gene
An allele of the rplK gene that carries a 12 by long deletion and is
characterized as
~rplK was prepared by means of PCR. The resulting 12 bp~ deletion in the rplK
gene leads to
loss of the tetrapeptide proline-alanine-leucine-glycine in the N-terminal
region of the Ll l
protein of C. glutamicum.
As primer for production of the rplK deletion allele, the following
oligonucleotides,
which were prepared by ARK Scientific (ARK Scientific G~mbH Biosystems,
Darmstadt,
Germany), were used in addition to the primers P1 up and IP2 down described in
Example 3.
Pl down (see also SEQ ID No. 7):
5' -Extension-CGC CGT GAG C-5' -side-GCC AA,C TGG AGG AGC AGG GT-3 '
P2 up: (see also SEQ ID No. 8):
5' -Extension-TCC AGT TGG C-5' -side-GCT CAC GGC GTC AAC ATC AG-3'
The primers used for PCR were derived from the known DNA sequence. This has in
each case a 10 by 5' extension, which is exactly complementary to the 5' sides
of the primers
Pl up and P2 down. Chromosomal DNA was extracted fronn C. glutamicum ATCC
13032 and
Page 25
CA 02340300 2001-04-02
two 600 by DNA fragments were first produced in separate PCR reactions with it
as matrix
using the given oligonucleotides P1 up, P1 down, P2 up anal P2 down, the Pfu
DNA
polymerase (Stratagene, La Jolla, USA) and the PTC 100 ihermocycler (MJ
Research Inc. ,
Waltham, USA). A cycle consisting of thermal denaturing (94°C, 90 sec),
annealing (58°C,
90 sec) and the polymerase reaction {72 ° C, 90 sec} was carried out 35
times in each of these
PCR reactions.
The first 600 by DNA amplificate, designated as rplK part I, was obtained with
the
oligonucleotides Pl up and Pl down. It contains the 5' region of the rplK gene
(nucleotide 1-
8~ and additionally the 10 by extension deriving from oligonucleotide P1 down,
which
corresponds to the nucleotides 100-109 of the rplK gene (S1JQ ID No. 1). Thus
this amplified
rplK gene region has a 12 by gap compared to the chromosomal DNA template that
was used.
The second 600 by DNA fragment, rplK part 2, was obtained with the
oligonucleotides P2
down and P2 up and contains the 3' region of the rplK gene: (nucleotide 78-
435) and carries
the identical gap (nucleotide 88-99) within the amplified rplK gene region.
These two 600 by
DNA amplificates accordingly have a 20 by overlapping DNA region. The two 600
by PCR
products rplK part 1 and rplK part 2 were now used in an additional PCR
reaction together as
DNA template, where the leading strand of rplK part 1 could be bonded to the
lagging strand
of rplK part 2 because of the overlapping complementary DIVA region. The
extension of this
overlapping DNA region or the addition of the oligonucleotiides P1 up and P2
down led to the
formation of a 1200 by PCR amplificate, which contains a 1.2 by deletion
derivative of the C.
gkctamicum rplK gene. A cycle consisting of thermal denaW ring (94°C,
90 sec), annealing
Page 26
CA 02340300 2001-04-02
(58°C, 90 sec) and the polymerase reaction (72°C, 90 sec) vvas
carried out 35 times in each of
these PCR reactions.
The nucleotide sequence of the OrplK allele is represented in SEQ ID No. 3 and
the
variation of the Ll l protein coded by this allele is represented in SEQ ID
No. 4. This Ll l
protein variation lacks the tetrapeptide proline-alanine-leucine-glycine
corresponding to the
amino acid positions 30 to 33 of the wild type form of the L1.1 protein
represented in SEQ ID
No. 2.
Example 5
Insertion of the OrpIK allele into the chromosome
The OrplK allele, which contains a 12 by deletion in dhe rplK gene, was
inserted into
the chromosome of C. gluxamicum by means of integration mutagenesis with the
help of the
sacB system described in Schafer et al., Gene, 14, 69-73 (1994) LB.R.. This
system enables
the specialist to make the identification or selection of allele exchanges
that are executed
through homologous recombination.
1. Construction of the exchange vector p~rplK
The 1200 by rplK deletion allele ~rplK obtained in Ex~unple 4 was purified by
means
of the Qiagen PCR purification spin kit (Qiagen, Hilden, Gernnany) and used
for ligation with
the mobilizable cloning vector pKl8mobsacB described in Schafer et al., Gene,
14, 69-73
(1994) LB.R.: This vector was linearized beforehand with the restriction
enzyme SmaI
Page 27
CA 02340300 2001-04-02
(Amersham-Pharmacia, Freiburg, Germany), mixed with ths: rplK deletion allele
and treated
with T4 DNA ligase (Amersham-Pharmacia, Freiburg, Germany).
The result was the plasmid p~rplK.
Transformation of the E. coli strain DHSa with the plasmid pOrpiK took place
as
described in Tauch et al., FEMS Microbiological Letters, 1f.3, 343-347 (1994)
LB.R.. The
transformants were selected on LBG agar, to which kanamyc:in (Merck,
Darmstadt, Germany)
(50 mg/L) had been added. The strain DHSaIp~rplK was obtained in this way.
A clone was selected and characterized as ATCC1303~2~rplK.
2. Conduct of the allele exchange
A chromosomal I2 by deletion in the rplK gene of C. glutamicum was obtained by
means of integration mutagenesis using the sacB system described in Schafer et
al., Gene, 14,
69-73 (1994) LB.R.. This system allows the specialist to make the
identification or selection of
allele exchanges that are executed by homologous recombination.
The mobilizable plasmid p~rplK was then inserted in ahe strain C. glutamicum
ATCC
13032 as recipient starting from the E. coli donor strain S17-',l described in
Simon et aL,
Bio/Technology, 1, 784-794 ( 1993) I. B. R. using the conjugation method
described by Schafer
et al., Journal of Bacteriology, 172, 1663-1666 (1990) LB.R.. Since the
plasmid p~rplK
cannot replicate in C. glutamicum, establishing it is possible only by
integration into the G
glutamicum chromosome via homologous recombination between the plasmid-coded
rplK
deletion fragment and the identical chromosomal rplK gene region. The
transconjugants were
Page 28
CA 02340300 2001-04-02
selected on LBG agar, to which kanamycin (25 mg/L) (Merck, Darmstadt, Germany)
and
nalidixic acid (Merck, Darmstadt, Germany) (50 mg/L) had been added.
Selection on the subsequent excision of the plasmid pArpIK with the aid of the
sacB
system could be carried out only using a wild type allele of :rplK. For this
the plasmid prplK
constructed in Example 3, which carries the complete rplK l;ene, was
transferred into the
integrant strain by electroporation by the method of Liebl et al., FEMS
Microbiology Letters
65, 299-304 (1989) LB.R.. Selection of the strain took place on LBG agar, to
which
kanamycin (25 mglL) (Merck, Darmstadt, Germany) and chloramphenicol ( 10 mg/L)
(Merck,
Darmstadt, Germany) had been added.
A selected transformed colony was transinoculated in 100 mL LBG liquid medium
(in a
250 mL Erlenmeyer flask with baffles) and incubated for 24 h at 30°C
and 300 rpm. Then 2 x
106 cell/mL of this liquid culture was applied to LBG agar that contained 10 %
sucrose (Merck,
Darmstadt, Germany) and incubated for 48 h at 30°C. C. glutamicum cells
that were capable
of growing on this medium had lost the integrated plasmid phrpiK as a
consequence of a
second recombination event between the rplK deletion allele and the natural
rplK region. This
second recombination event leads either to reformation of the natural
chromosomal rplK gene
arrangement or it results in the generation of a C. glutamicum! p~rplK mutant
in which the 12
by N-terminal DNA fragment is missing. The chromosomal IDNA was extracted from
the
selected "sucrose-resistant" and potential pOrpIK-bearing C, glutamicum cells.
This served as
matrix with which the oligonucleotides Pdel up and Pdel down2 were derived
using the rplK
sequence (ARK Scientific GmbH Biosystems, Darmstadt, Germany). The PCR
experiments
Page 29
CA 02340300 2001-04-02
were carried out using the primers, Pfu DNA polymerase (Stratagene, La Jolla,
USA) and the
PTC 100 thermocycler (MJ Research Inc., Waltham, USA). A cycle consisting of
thermal
denaturing (94°C, 90 sec), annealing (58°C, 90 sec) and the
polymerase reaction (72°C, 90
sec) was carried out 35 times in the PCR. Then the resulting DNA amplificates
were purified
by means of the Qiagen PCR purification spin kit (Qiagen, :fiilden, Germany).
Analysis of the
nucleotide sequences of the purified DNA amplificates, which was carried out
as described
above, showed that in 43 % of the cases a DNA amplificate :had been produced
that lacked the
12 by DNA region. Accordingly, in the relevant patplK-be;iring transconjugants
the second
recombination event had led to the formation of the chromosomal 12 by deletion
in the rplK
gene.
Then the plasmid prplK was removed from a selectedl deletion-bearing
transconjugant
by the plasmid curing method described in Schafer et al., Journal of
Bacteriology, 176, 7309-
7319 (1994) LB.R.. The resulting strain of C. glutamicum A.TCC13032 OrplK thus
carries a
chromosomal 12 by deletion within the rplK gene, which leads to the Ioss of
the tetrapeptide
proline-alanine-leucine-glycine of the Lll protein.
E~le 6
Preparation of lysine
The strain C. glutamicum ATCC13032 arplK obtained in Example 5 was cultured in
a
nutrient medium suitable for production of lysine and the lysine content in
the culture
supernatant was determined.
Page 30
CA 02340300 2001-04-02
For this the strain was first incubated on agar plates I;brain-heart agar) for
24 h at
33°C. Starting from these agar plate cultures a preculture was
inoculated (10 mL medium in
100 mL Erlenmeyer flasks). The complete medium CgIII (1.0 g/L bactopeptone, 10
g/L yeast
extract, 2.5 g/L NaCI, 20 gIL glucose, pH 7.4) was used as medium for the
preculture. The
preculture was incubated for 24 h at 130 ° C and 240 rpm on the shaker.
A primary culture
was inoculated from this preculture, so that the starting optical density (660
nm) of the main
culture was 0.1 OD. The medium MM was used for the main culture.
Medium MM
CSL (corn steep liquor) 5 g/L
MOPS (morpholinopropane sulfonic20 g/L
acid)
Glucose (separately autoclaved)S~D gIL
(NH~2(SO~ 2;5 gIL
KHZPO4 0.1 g/L
MgS04 ~ 7 H20 1.0 gIL
CaCl2 ~ 2 H20 1~D mg/L
FeS04 ~ 7 HZO 10 mg/L
.
MnS04 ~ H20 5.0 mg/L
Biotin (sterile filtered) 0.3 mg/L
Thiamine ~ HCl (sterile filtered)0.2 mg/L
CaC03 2.5 gIL
The CSL, MOPS and salt solution are adjusted to pH ? with ammonia water and
autoclaved. Then the sterile substrate and vitamin solutions ~~re added, as
well as the dry
Page 31
CA 02340300 2001-04-02
autoclaved CaC03.
Culturing takes place in 10 mL volume in a 100 mL, Erlenmeyer flask with
baffles.
Culturing took place at 33°C and 80~ air humidity.
After 48 h the OD at a measurement wavelength of 660 nm was determined with
the
Biomek 1000 (Beclanann Instnunents GmbH, Munich). ThE: amount of lysine that
formed was
determined by means of an amino acid analyzer from the Eppendorf BioTmnik
Company
~~bm'g~ Germany) by ion exchange chromatography and subsequent column
derivatization
with ninhydrin detection.
The result of the experiment is shown in Table 1.
Table 1
S~ OD (660)
Lysine~HCl g/L
ATCC13032 ~rplK I3.0 0.98
ATCC13032 13.8 0.0
It is understood that the foregoing detailed description. is given merely by
way of
illustration and that many variations may be made therein without departing
from the spirit of
this invention and are intended to be encompassed by the claims appended
hereto.
Page 32
;ii
CA 02340300 2001-04-02
SEQUENZPROTOKOLL
<110> Degussa-Hiils AG
<120> Neue fiir das rplK-Gen kodierende Nukl.eotidsequenzen
<13Q> 990178 HT
<140>
<141>
<160> 8
<170> PatentIn Ver. 2.1
<210> 1
<211> 835
<212> DNA
<213> Corynebacterium glutamic~,m~
<220>
<221> CDS
<222> (201)..(635)
<223> rplK-Gen
<400> 1
ttgcgtgtag ggtagacaat cgcgtgt~=~ ttaagcatgc: tcaaaatcat tcatccccgg 60
3 0 tggcccggtt acgtaaagat cagcaaagat gatcaactaa. agcgatcatc tgaagttgta 120
gcgggaccga~gcatccggac ggttactagt ggggtttcat cgtcccagtt gtqgccggta 180
acaaggaagc aggtttaacg atg get cct aag aag a.ag aag aag gtc act ggc 233
Met Ala Pro Lys Lys L~ys Lys Lys Val Thr Gly
1 5 10
ctc atc aag ctc cag atc cag gca gga cag gca aac cct get cct cca 281
Leu Ile Lys Leu Gln Ile Gln Ala Gly Gln Ala Asn Pro Ala Pro Pro
15 20 25
gtt ggc cca gca ctt ggt get cac ggc gtc aac atc atg gaa ttc tgc 329
Val Gly Pro Ala Leu Gly Ala E~is Gly Val Asn Ile Met Glu Phe Cys
30 35 40
aag get tac aac get gcg act gaa aac cag cgc ggc aac gtt gtt cct 377
Lys Ala Tyr Asn Ala Ala Thr Glu Asn Gln Arg Gly Asn Val Val Pro
45 50 55
gtt qag atc acc gtt tac gaa gac cgt tca ttc gac ttc aag ctg aag 425
Val Glu Ile Thr Val Tyr Glu Aso Arg Ser Phe Asp Phe Lys Leu Lys
65 70 75
act cct cca get gca-aag ctt ctt ctg aag get get ggc ctg cag aag 473
Thr Pro Pro Ala Ala Lys Leu Leu Leu Lys Ala Ala Gly Leu Gln Lys
55 80 85 =90
ggc tcc ggc gtt cct cac acc cag aag gtc ggc aag gtt tcc atg get 521
Gly Ser Gly Val Pro His Thr G?n Lys Val Gly Lys Val Ser Met Ala
95 I00 105
caq gtt cgt gag atc get gag acc aag aag gaa gac ctg aac get cgc 569
Gln Val Arg Glu Ile Ala Glu Thr Lys Lys Glu Asp Leu Asn Ala Arg
lI0 li5 I20
Page 33
CA 02340300 2001-04-02
gat atc gac get get gcg aag atc atc get ggt acc get cgt tcc atg 6I7
Asp Ile Asp Ala Ala Ala Lys Ile Ile Ala Gly Thr Ala Arg Ser Met
125 I30 135
S ggc atc acc gtc gaa ggc taaaagcttt cacaccggta agtggctcat 665
Gly Ile Thr Val Glu Gly
140 145
tcaaaatgaa tggccaccaa ccaattttca ccaaagtttt atgtggcagg gccagctccg 725
gcccgttaaa ccacagaatt ccatgaaagg gaatttctaa tgagcaagaa ctctaaggcg 785
taccgcgagg ccgctgagaa gatcgacgct ggtcgcatct actccccact
835
<210> 2
<211> 145
<212> PRT
<2I3> Corynebacterium
glutamicum
<400> 2
Mei Ala Lys Ly5 Lys LysVal ThrGlyLeu IleLys Leu
Pro Lys Gln
10 15
2 S Ile Gln Gly Gln~AlaAsn ProAla ProProVal GlyPro Ala
Ala Leu
20 25 30
Gly Ala Gly Val Ile MetGlu PheCysLys AlaTyr Asn
His Asn Ala
35 40 45
Ala Thr Asn Gln Gly AsnVal ValProVal GluIle Thr
Glu Arg Val
50 55 60
Tyr Glu Arg Ser Asp PheLys LeuLys'ThrProPro Ala
Asp Phe Ala
6s 70 75 80
Lys Leu Leu L85 Ala GlyLeu GlnLysGly SerGly Val
Leu Ala Pro
90 g5
4 0 His Thr Val Gly Lys ValSer MetAlatslnValArg Glu
Gln i Ile
00 105 I10
Ala Glu Lys Lys Asp LeuAsn AlaArg~~.spIleAsp Ala
Thr Glu Ala
115 I2 0 125
Ala Lys Ile Ala Thr AlaArg SerMet(plyIleThr Val
IIe Gly Glu
130 135 7.40
Gly
145
<210> 3
SS <211> 825
<212> DNA
<213> Corynebacterium glutamicum
<220>
<221> CDS
<222> (200)..(622)
<223> delta rplK
<400> 3
tgcgtgtagg gtagacaatc gcgtgttttt taagcatgct caaaatcatt catccccggt 60
Page 34
CA 02340300 2001-04-02
ggcccggtta cgtaaagatc agcaaagatg atcaactaaa gcgatcatct gaagttgtag I20
cgggaccgag catccggacg gttactagtg gggtttcatc: gtcccagttg tggccggtaa 180
caagqaagca ggtttaacg atg get cc~ aag aag aag aag aag gtc act ggc 232
Met Ala Po Lys Lys Lys Lys Lys Val Thr Gly
1. 5 10
ctc atc aag ctc cag atc cag gca gga cag gca. aac cct gc~ cct cca 280
Leu Ile Lys Leu Gln Ile Gln Ala Gly Gln-Ala. Asn Pro Ala Pro Pro
20 25
gtt ggc get cac ggc gtc aac atc atg gaa ttc tgc aag get tac aac 328
Val Gly Ala His Gly Val Asn Ile Met Glu Phe Cys Lys Ala Tyr Asn
15 30 35 40
get gcg act gaa aac cag cgc ggc aac gtt gtt cct gtt gag atc acc 376
Ala Ala Thr Glu Asn Gln Arg Gly Asn Val Val Pro Val Glu Ile Thr
45 50 55
gtt tac gaa gac cgt tca ttc gac ttc aag ctg aag act cct cca get 424
Val Tyr Glu Asg Arg Ser Phe Asp Phe Lys Leu Lys Thr P=o Pro Ala
60 65 70 75
2 5 gca aag ctt ctt ctg aag.gct gc~ ggc ctg cag aag ggc tcc ggc gtt 472
Ala Lys Leu Leu Leu Lys Ala Ala Gly Leu Gln Lys Gly Ser Gly Val
80 85 90
cct cac acc cag aag gtc ggc aag gtt tcc atg get cag gtt cgt gag 520
Pro His Thr Gln Lys Val Gly Lys Val Ser Met Ala Gln Val Arg Glu
95 100 105
Ile Ala Glu Thr Lys Lys Glu Asp Leu Asn Ala Arg Asp Ile Asp Ala 568
lI0 II5 120
get gcg aag atc atc get ggt acc get cgt tcc atg ggc atc acc gtc 616
Ala Ala Lys Ile Ile Ala Gly Thr Ala Arg Ser Met G1y Ile Thr Val
125 130 135
gaa ggc taaaagcttt cacaccggtt ag ggctcat tc<~aaatgaa tggccaccaa 672
GIu Gly
140
ccaattttca ccaaagtttt atgtggcagg gccagctccg gcccgttaaa ccacagaatt 732
ccatgaaagq gaatttctaa tgagcaagaa ctctaaggcg taccgcgagg ccgctgagaa 792
gatcgacgct ggtcgcatct actccccact cga 825
<210> 4
<211> 141
<212> PRT
SS <2I3> Corynebacterium giutamicum
<400> 4
Met Ala Pro Lys LysLys Lys ValThr Gly Ile Leu
Lys Leu Lys Gln
I 5 10 15
Ile Gln Ala Gly GlnAla Asn AlaPro Pro GLy His
Pro 'Val Aia Gly
20 25 30
Va1 Asn Ile Met GluPhe Cys AlaTyr Asn Ala Glu
Lys ~41a Thr Asn
35 40 45
Page 35
CA 02340300 2001-04-02
Gln Gly AsnVal ValPro ValGluIle ThrVal TyrGlu Arg
4rg Asp
50 55 SO
Ser PheAsp PheLys LeuLys ThrProPro Ala.Ala LysLeuLeu Leu
65 70 75 80
Lys AlaAla GlyLeu GlnLys G1ySerGly ValPro HisThrGln Lys
85 90 95
GiyLys ValSer MetAla GlnValArg GluIle AlaGluThr Lys
Val
100 105 110
Lys GluAsp LeuAsn AlaArg AspIleAsp AlaAla AlaLysIle Ile
115 I20 125
Ala GlyThr AlaArg SerMet GlyIleThr ValGlu Gly
130 135 140
<210> 5
<21I> 20
<2I2> ONA
<213> Kiinstliche Secuenz
<220>
<223> Primer P1-ua
<220>
<223> Beschreibung der kiinstlichen Sequenz: Primer
<400> 5
aggagcaggc tgttgtcacc
35
<210> 6
<211> 20
<212> DNA
<213> Kiinstliche Sequenz
<220>
<223> P2-down
<220>
4 5 <223> Beschreibung der. kiinstlichen Sequenz: Primer
<400> 6
gcqgatagct acqtcgatgg 20
<210> 7
<211> 30
<212> DNA
<213> Kiinstliche Seqiienz
<220>
<223> PI-down
<220>
\60 <223> Beschreibung der kiinstlichen Seguenz: Primer
<a00> 7
cgccgtgagc gccaactgga ggagcagggt 30
<210> 8
Page 36
CA 02340300 2001-04-02
<21I> 30
<212> DNA
<213> Kiinstliche Sequenz
<220>
<223> p2-up
<220>
<223> Beschreibung der kiinstlichen Sequenz: Primer
<400> 8
tccagttggc gctcacgqcg tcaacatcag
I5
Page 37