Note: Descriptions are shown in the official language in which they were submitted.
CA 02343076 2001-03-19
WO 00/15851 I PCT/US99/21525
GEOMETRICAL AND HIERARCHICAL CLASSIFICATION BASED ON
GENE EXPRESSION
FIELD OF THE INVENTION
This invention relates to representations of the extent of relatedness between
cells, cell lines,
tissues, organs, or expressed sequences based on a genomic analysis of gene
expression using software
algorithm based analysis.
RELATED APPLICATIONS
This application claims priority to both United States Application Serial
Number ,
filed September 16, 1999, entitled "GEOMETRICAL AND HIERARCHICAL
CLASSIFICATION BASED ON
GENE EXPRESSION", and United States Provisional Application Serial Number
60/101,009 filed
September 17, 1998, entitled "PHYLOGENOMICS AND PHARMACOGENOMICS", which are
incorporated
herein by reference in their entirety.
BACKGROUND OF THE INVENTION
The rapid development of genomics and proteomics in recent years has led to a
burgeoning of
applications making use of the new information provided. A significant area in
which such information
has been put to use is in the grouping and characterization of pathological
states according to the
differential expression of genes in such states. A corollary application is in
grouping and characterizing
the therapeutic effects of known or candidate pharmaceutical agents used in
treating various pathologies.
Algorithms employing a variety of statistical procedures have been employed to
create heuristic displays
of the information obtained from such analyses. These displays include large
two dimensional, or even
higher dimensional arrays in which the elements are coded, for example by
false color coding, to
represent a particular experimental result. Alternative displays include those
in which the experimental
data is used to generate cladistic or radiating tree structures as a
representation of relatedness.
Furthermore, it is also possible to use similar methods to group expressed
sequences according to
patterns of co-expression over several different biological states.
For example, a system of cluster analysis for genome-wide expression in the
yeast
Saccharomyces cerevisiae and in primary human fibroblasts has been presented
by Eisen et al. (Proc.
Natl. Acad. Sci. USA 95:14863-14868 (1998)). In the yeast work, DNA microchip
arrays carrying
essentially every ORF from this organism were used. Differential expression
was studied by varying the
physiological state, including the diauxic shift, the mitotic cell division
cycle, sporulation, and
temperature and reducing shocks. The human fibroblasts were stimulated with
serum following serum
CA 02343076 2001-03-19
WO 00/15851 _ 2 _ PCT/US99/21525
starvation, and examined using a microarray with 9,800 cDNAs representing
approximately 8,600
distinct human transcripts. Additionally, a further independent variable in
these experiments is the time
at which an assay point was taken. Data reflecting the differential gene
expression in the various studies
were analyzed using pairwise average-linkage cluster analysis (Sokal et al.,
Univ. Kans. Sci. Bull.
38:1409-1438 (1958)), which was used to compute a dendrogram that assembles
all elements into a
single tree.
Colon adenocarcinoma from 40 tumor samples were compared with 22 normal colon
tissue
samples using Affymetrix DNA chips to which sequences from human cDNAs were
bound (Alon et al.,
Proc. Natl. Acad. Sci. USA 96:6745-6750 (June 1999)). 3,200 full-length human
cDNAs and 3,400 ESTs
are represented in sets of 25-by fragments, as well as such sequences
containing a single base mismatch
in the center of the sequence. The gene expression in both the tumor tissue
samples and the normal
colon samples, was assessed by hybridization. The statistical significance of
the correlation between
genes was assessed by calculating pairwise correlation coefficients. The
clustering of the expressed
genes was evaluated using an algorithm based on deterministic-annealing (Rose
et al., Phys. Rev. Lett.
65:945-948 ( 1990); Rose, Proc. IEEE 96: 2210-2239 ( 1998)) to organize the
data in a binary tree. Data
are presented as a large two-dimensional color coded array, with genes
displayed along one dimension
and tissue samples along the other; artificial color values are assigned at
each array point to indicate the
extent of expression in a third dimension. Clustering analysis reveals
patterns in the color distribution
within the array which is disrupted when various randomization procedures are
applied. The clustering
of the genes in the data set reveals groups of genes whose expression is
correlated across tissue types.
The algorithm separated the tissues into distinct clusters.
Pharmacological effects of compounds actually used or being screened for use
in cancer
chemotherapy were analyzed by cluster analysis at the National Cancer
Institute (Weinstein et al.,
Science 275:343-349 ( 1997)). More than 60,000 compounds were screened against
a panel of 60 human
cancer cell lines. A 50% growth-inhibitory concentration of a compound in a
given cell line, when
analyzed across all cell lines, provided detailed information on mechanisms of
drug action and drug
resistance. Patterns of activity were first analyzed by the COMPARE algorithm
(Paull et al., J. Natl.
Cancer Inst. 81:1088 ( 1989); Jayaram, Biochem. Biophys. Res. Commun. 186:1600
( 1992); Paull et al.,
In: CANCER CHEMOTHERAPEUTIC AGENTS, Foye led.), American Chemical Society,
Washington DC,
1993, pp. 1574-1581; Boyd et al., Drug Dev. Res. 34:91 (1995)). The procedures
developed rely on
three databases, an S database characterizing structural information on the
candidate compounds, an A
database related to the 60 cell lines and a T database including information
on molecular targets of
action. In an example of the results of the analysis, a three dimensional
array displaying compounds
CA 02343076 2001-03-19
WO 00/15851 _ 3 _ PCT/US99/21525
versus targets, with a false color code providing a correlation coefficient in
a third dimension for each
position in the array, was developed.
Certain problems arise upon consideration of the procedures currently in use
for the correlation
and clustering of genome-derived attributes. Use of DNA microchips inherently
limits any analysis to
the sampling of the DNA sequence fragments employed as the capture probes
bound to the chips.
Detection of any DNA fragment which does not hybridize with one of the capture
probes is not possible,
so that positive results are potentially lost. Additionally, a mutation or
other allelic polymorphism may
not bind to the capture probe under conditions of moderate or low stringency,
so that again information
relating to a positive result may be lost.
For these reasons there is a need for methods of genomic statistical analysis
based on more
comprehensive accessibility to the genomes of the organisms being studied.
Furthermore there remains a
need for ways of presenting the information obtained in genomic analyses of
relatedness of genes, and in
genomic analysis of response to actual or candidate pharmaceutical agents,
that includes information
gleaned from a comprehensive access to the genomes in question. The present
invention addresses these
needs, for use is made in the invention of partial and full genomic sequences
available from a large
number of sequence databases in clustering analysis of the components
appearing as independent
variables in a particular study.
SUMMARY OF THE INVENTION
The invention provides novel methods of geometric and hierarchical
classification between at
least two classes of data sets. Data sets may represent cells, nucleic acid
sequences, polypeptide
sequences, or the like. The invention is able to utilize both standard DNA
microchip arrays and
non-DNA chip technology to provide input information on nucleic acid moieties
of the specified classes
of cells. The data are then treated in various ways to provide representations
of relatedness that are
readily interpretable by the human eye. The invention additionally provides
novel methods for
generating a representation of the correlation between at least two classes of
cells, the correlation
reflecting any changes in the composition and amount of nucleic acids present
between the classes.
The cell classes may be from different sources for use in comparing
differences between various
cell populations. These differences include, but are not limited to, species
differences, tissue differences,
disease state differences, and drug treatment differences. Computer algorithms
analyze input data
reflecting differences between chosen cell classes and represent them in a
meaningful way.
CA 02343076 2001-03-19
WO 00/15851 _ g _ PCT/US99/21525
Prior to the present invention, input information was obtained only using DNA-
chip technology
to analyze the nucleic acids of the cell classes to be compared. Drawbacks to
these methods are that
identifier sequences need to be already known and isolated, chip technology
has size limitations related
to the number of the nucleic acids immobilized on the chips, and, once the
chips were manufactured, it is
virtually impossible to expand nucleic acid parameters. The invention provides
the use of
GeneCallingT"', a non-DNA chip technology, to assay differences between input
cell classes. An
unexpected result is that GeneCallingT"' is able to provide sensitive
comparisons between disparate
groups above, thereby sidestepping the limitations inherent in the use of DNA
chip technology when
assaying input nucleic acid population.
The invention provides a novel method for generating the extent of relatedness
reflecting
similarities or differences in the presence and quantitation of the fragments
among the classes by
calculating a distance that reflects the amplitude of a difference vector. In
a significant embodiment of
this method for generating the representation of relatedness, the extent of
relatedness is provided by
generating a tree structure reflecting the relatedness between any two
classes. The branches of the tree
1 S structure reflect the difference vectors and are ramified from nodes.
The invention also provides a novel method for generating a representation of
the correlation
between classes of data sets. In a significant embodiment of the method for
generating a representation
of the correlation, the correlation is related to a set of orthonormal
eigenvectors. In another significant
embodiment of the method for generating a representation of the correlation,
the representation is a
cluster diagram or a dendrogram, and includes a tree structure reflecting the
relatedness of the pathways
involved in the biochemical or physiological response to a difference between
cells of the two classes.
The invention additionally relates to providing geometrical representations of
differences
between classes of data sets. The geometrical representations encompass, by
way of nonlimiting
example, principal component analysis and principal factor analysis, as well
as reduced dimensional
representations derived from them. The geometrical representations are based
on differences determined
between classes of cells using any method of analyzing for the presence of
genes, nucleic acids, or
fragments thereof, including nucleic acid microchip arrays and differential
display of expressed genes or
nucleic acid fragments.
The invention also provides display means for displaying the representation of
the extent of
relatedness, the correlation, and the geometrical representations of
differences between classes of data
sets, as well as the representations themselves.
CA 02343076 2001-03-19
WO 00/15851 _ 5 _ PCT/US99/21525
BRIEF DESCRIPTION OF THE DRAWING
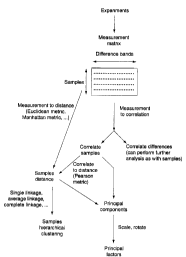
Figure 1 is a schematic flow diagram illustrating the principal steps involved
in generating the
various representations of the invention starting from a set of subsequence-
selected fragments found for
the samples.
Figure 2 is a schematic flow diagram illustrating the primary steps involved
in carrying out a
principal component analysis.
Figure 3 illustrates hierarchical clustering of four drugs with sterile water
as an outgroup.
Figure 4 is a graphical projection of drug treatments and controls onto
principal factors.
DETAILED DESCRIPTION
The present invention relates to methods for preparing representations of the
relatedness between
cells of any two or more different classes of cells. The classes broadly
encompass cells arising in animal
and plant organisms, the cells further being normal cells or cells in a
diseased state, including tumor
cells. They further include cells that have been treated with a putative
pharmaceutical agent. The
representations are obtained using experimental data that provide size and
sequence information on
nucleic acid fragments derived from each of the cellular sources. The
fragments may be prepared from
the nucleic acid content of the cells in each class in any of several ways.
For example, in a particularly
important embodiment, they may be subjected to digestion by particular pairs
of restriction
endonucleases; alternatively, in another important embodiment, cell extracts
may be subjected to
amplification using specially designed primer oligonucleotides. The present
invention also relates to
methods for preparing representations of the relatedness in terms of co-
expression between the nucleic
acid fragments so produced.
The invention further relates to the representations provided by these
methods, and to display
means on which such representations are displayed. The methods for preparing
the fragments, such as
the use of restriction endonucleases or the application of amplification
primers, are chosen to provide
subsequence information relating to the ends of the resulting fragments, while
size determination
provides the length of the fragment. In certain applications of these types of
information, the size and
subsequence results can optionally be scanned against databases providing
known nucleic acid sequences
in order to provide the identity of one or more candidate fragments of known
complete nucleic acid
sequences having the correct length and terminal subsequences (U. S. Patent
No. 5,871,697; Shimkets el
al. 1999 Nature Biotechnology 17:798-803). This database look-up step is not a
required feature of the
current invention. For this reason, the present representations and methods
are more comprehensive and
CA 02343076 2001-03-19
WO 00/15851 _ 6 _ PCT/US99/21525
more informative of genomic variations among the samples than those currently
known. As described in
the Background of the Invention, currently known procedures are restricted in
their comprehensiveness
to those nucleic acid fragments that are applied to DNA microchips as probe
sequences in a given
procedure. Except for a narrowly limited set of model organisms with known
genome sequence, the
number of such probe sequences is considerably fewer than the number of known
nucleic acid sequences
available in sequence databases and employed in the present invention.
Furthermore, even for fully
sequenced genomes, genetic variation is not adequately probed with existing
DNA microchips. This
distinction characterizes an important advantage of the instant invention.
The invention additionally relates to providing geometrical representations of
differences
between classes of cells. The geometrical representations encompass, by way of
nonlimiting example,
principal component analysis and principal factor analysis, as well as reduced
dimensional
representations derived from them. The geometrical representations are based
on differences determined
between classes of cells using any method of analyzing for the presence of
genes, nucleic acids, or
fragments thereof, including nucleic acid microchip arrays and differential
display of expressed genes or
nucleic acid fragments.
As used herein, "sample" relates to a particular experimental state for which
all the variables
being studied in a project are held fixed. By way of nonlimiting example, if a
variable is a class of cell,
the "sample" refers to a particular cell type; if a variable is the
subsequence pairs employed in the
project, a "sample" refers to a particular subsequence pair; or if a variable
is a set of putative
pharmaceutical agents, a "sample" refers to a particular agent from the set.
As used herein,
"representation" relates to any graphical, visual, or equivalent non-verbal
display that provides an image
of the results obtained according to the methods of the present invention.
More specifically, a
"representation" of the invention is obtained by transforming the quantitative
results gathered by
experiments underlying the invention. Examples of such data include, by way of
non-limiting example,
differential gene expression across classes of cell, and/or across a set of
putative therapeutic agents,
and/or equivalent types of experimental parameter.
In important embodiments, a representation of the invention is generated by
algorithms executed
m a computer and is suitable for display on a display means, such as a display
screen or monitor,
employed in the operation of the computer. The representation is also suitable
for storing in a storage
module or data archive of such a computer. It is still further suitable for
printing from the computer onto
a medium such as paper or equivalent physical medium, and for recording it
onto a portable storage
medium, including, for example, magnetic media, CD ROMs and equivalent storage
media. As used
CA 02343076 2001-03-19
WO 00/15851 _ ~ _ PCT/US99/21525
herein, "display means" includes any of the objects and media identified above
in this paragraph, as well
as equivalent apparatuses and objects suitable for displaying the results of
computational processes for
visualinspection.
As used herein, "extent of relatedness" is a characterization according to
methods of the present
invention of a degree of similarity or a degree of non-similarity between any
two members of the same
type of element; in particularly important embodiments, the type of element
may be classes of cells.
As used herein, a "putative pharmaceutical agent" relates to a chemical
compound or a
composition comprising at least one chemical compound which is a candidate for
being a therapeutic
agent. Any such therapeutic agent may be used. in treating a mammal suffering
from a disease or a
pathology. In treating the mammal with the therapeutic agent it is intended to
attenuate the symptoms
and/or the underlying causes of the disease or the pathology, to ameliorate
the symptoms and/or the
underlying causes, and/or to contribute to a cure of the disease or the
pathology. Non-limiting examples
of a putative pharmaceutical agent include an agent drawn from a chemical
compound library; an isolate
from a natural source; a compound synthesized specifically as a putative
agent; or a substance derived or
obtained using the practices of genetic engineering and recombinant nucleic
acid technology such as a
recombinant protein, a fragment of a recombinant protein, a recombinant
polypeptide, a fragment of a
recombinant polypeptide, a recombinant peptide, or a nucleic acid including,
for example an
oligonucleotide intended as an antisense agent, and a recombinant gene
intended for administration as a
gene therapeutic agent.
As used herein, a "fragment" of a nucleic acid relates to a contiguous portion
originating from
the genomic or cDNA-derived nucleic acid from a class of cells. The contiguous
portion includes at or
near each end a target subsequence defined according to the operational
procedures disclosed herein, and
includes al l nucleotides in the sequence of the fragment bounded by the two
target subsequences. The
nucleotides between the two target subsequences, together with the
subsequences themselves, define a
"length" of the fragment, as used herein. The target subsequences are
identified, for example, by
contacting the nucleic acid from the cells with a specific pair of restriction
endonucleases, or with a
specific pair of oligonucleotide primers, and in equivalent ways.
The information used in the present invention is obtained from experiments
providing the results
of differential gene expression wherein the difference relates to an
experimental state and a reference
state. Commonly a reference state refers to a normal, or an unperturbed, or a
non-pathological class of
cells. An experimental state may relate to a certain set of conditions applied
to one class of cells, and the
corresponding reference state then relates to the same set of conditions
applied to a second class of cells.
CA 02343076 2001-03-19
WO 00/15851 _ g _ PCT/US99/21525
An experimental state may also relate to a class of cells in the presence of
one or more putative
therapeutic agents, in which case the reference state relates to the same
class of cells in the absence of
any putative therapeutic agent. An experimental state may furthermore be
obtained from a class of cells
that is of interest in a particular set of circumstances. This includes cells
of a given cell type, cells from
a given tissue, and cells from a given organ, and further includes cells that
may be noncancerous or
cancerous. Types of cell encompassed within the present invention include, by
way of non-limiting
example, endothelial cells, rnesothelial cells, and epithelial cells. Tissues
and organs included within the
present invention may be, by way of non-limiting example, lung, heart,
skeletal muscle, smooth muscle,
brain, central nervous system, peripheral nervous system, stomach, liver,
kidney, reproductive tissues
and organs, skin, and bone. Cancerous cells include, by way of non-limiting
example, cells from prostate
cancer, breast cancer, colon cancer, lung cancer, lymphatic or hematopoietic
cancers, and also include
cells obtained from tissue biopsies or from cell lines in the National Cancer
Institute human tumor cell
line panel. The cells subjected to analysis in the present invention may also
originate from plants, yeast,
fungi, and other taxonomic groupings.
1 S The methods of evaluating the extent of relatedness between classes of
cells, for example,
between a first class of cells and a second class of cells, are founded on
evaluating the extent of
relatedness of the expression of particular genes between the cells of the two
classes. In a preferred
embodiment of the invention, similarities and differences in the
susceptibility of the nucleic acid present
in the cells to digestion by specifrc pairs of restriction endonucleases are
determined, according to the
methods of the present invention, by procedures that are disclosed in detail
in co-owned U. S. Patent No.
5,871,697 to Rothberg et al., and in Shimkets et al. 1999 (Nature
Biotechnology 17:798-803), both of
which are incorporated herein by reference in their entirety.
Briefly, for any experimental state of a class of cells, the nucleic acid
content of the cells,
preferably in the form of a preparation of cDNA from the cells, is subjected
to restriction endonuclease
("RE") digestion by specific pairs of endonucleases. Each member of the RE
pair is chosen to optimize
the likelihood that a restriction fragment resulting from the nuclease
digestion will be a unique fragment.
In an important implementation of this method, the restriction nuclease
digestion is carried out on cDNA
prepared from the cells of the class in the given experimental state. This
implementation leads to
emphasis on genes that are expressed in the experimental state, many of which
may be characteristic of
the given experimental state and be more poorly expressed, or not expressed at
all significantly, in a
different experimental state. A large number of specific pairs of nucleases
may be employed.
Alternatively, expression of a gene may be repressed in a characteristic way
in a given experimental state
and be expressed at a higher level, such as at a constitutive level, in a
different experimental state. By
CA 02343076 2001-03-19
WO 00/15851 _ 9 _ PCT/US99/21525
way of non-limiting example, several pairs of restriction nucleases that may
be employed in
implementing the present invention are disclosed in U. S. Patent No.
5,871,697.
In an alternative embodiment, the extent of relatedness may be obtained by
amplification
fragment length polymorphism analysis ("AFLP"). Briefly, amplification ofthe
nucleic acid content of
the class of cells being examined is subjected to a primer-dependent
amplification procedure in which
any of a set of primer pairs is used to initiate amplification. Amplification
procedures are described in
considerable detail in, for example, lnnis et al., PCR PROTOCOLS, A GUIDE TO
METHODS AND
APPLICATIONS, Academic Press, New York (1989), and Innis et al., PCR
STRATEGIES, Academic Press,
New York (1995). The primers of each primer pair are different from each
other, and reflect different
subsequences that are the object of the amplification process. Amplification
may proceed by any
procedure, including polymerase chain reaction, known in the field of
molecular biology. In AFLP, the
length of an amplicon found in a given experimental state differs from the
length found in a different
experimental state. This may arise, for example, if the given experimental
state arises from a mutation
that occurs in a subsequence recognized by a primer used in the amplification
reaction. It may also arise
I 5 from a deletion from, or an insertion into, the nucleic acid of the cells
in that state.
The experimental and computational procedures that may be employed to generate
the
representations of the present invention are described generally below.
Measurements
At the outset, the gene expression levels are determined experimentally. This
can be done, in a
preferred embodiment, by following the general protocols of differential
expression using restriction
endonucleases (U.S. Patent No. 5,871,697). For each pair of restriction
enzymes and each biological
sample, a pool of fluorescently-labeled DNA fragments is generated.
Electrophoresis is then performed
to separate these fragments based on size, and an intensity, designated as
Isrt(x), where s labels the
sample, i.e., the cell class; r labels the restriction enzyme pair, i.e., the
gene fragment; t labels the trial,
and x is the length of the fragment as determined by electrophoresis, is
detected. The length x may be
either a continuous index or a convenient discretization. As an example, the
resolution of the
electropherogram may be set to a discretization of 0.1 nucleotide ("nt").
Commonly three independent
trials are performed. A mean signal I5,(x) is then obtained by averaging over
the n, trials,
ISOx) _ ( 1 /n,) E, I5n(x) y
(1)
CA 02343076 2001-03-19
WO 00/15851 _ ~ p _ PCT/US99/21525
Next, lengths x for each restriction enzyme pair r where some of the samples
have a significant
difference in measured intensity are identified. Such a difference is
determined with respect to cell
types, or with respect to the presence vs. the absence of a putative
pharmaceutical agent. Labeling the d'"
such difference d, the values Isa= Isyx ) are then collected. Any of several
methods for identifying
significant differences may be employed, some of which are outlined herein.
For example, an important
method involves the following computational steps:
1. The mean I,(x) = ES I5,(x) is evaluated.
2. All positions, i. e. lengths, where, for at least one sample, Isf(x) -1~(x)
is larger than some
threshold value, are marked.
3. The largest value of IS~(x) - I,(x), determined as a difference between a
sample state and
the mean for restriction enzyme pair r, is found and the length x, indexing
the difference,
is marked.
4. Step 3 is repeated for succeedingly smaller values of the intensity
difference. If the
length x that marks the current largest difference is within a distance w from
the length
of a previously identified difference, the current difference is skipped and
the next
smaller difference is considered.
5. Step 4 is repeated until there are no more differences to consider.
Another method involves finding differences that meet a statistical criterion.
A particular
example of such a method involves the computational steps of:
1. defining a set of sample classes and assigning each sample to a particular
class c;
2. for each restriction enzyme pair r and length x, evaluating the F-statistic
for the set of
measurements IS~(x) and the classes c to which samples are assigned, thereby
providing
the probability p,(x) that any differences between sample classes may be
explained by
random variation (See, for example, P. Hinton, Statistics Explained, Routledge
1995);
3. ordering the probabilities p,(x) from smallest (most significant) to
largest (least
significant);
4. optionally truncating the list at some threshold value of p~(x) above which
differences are
no longer considered significant (accepted values are p~(x) = 0.01 to 0.05);
5. finding the smallest value of p~(x) and marking the length x as a
difference for restriction
enzyme pair r;
CA 02343076 2001-03-19
WO 00/15851 _ 1 ~ _ PCT/US99/21525
6. repeating step 4 and determining whether the length x that marks the
current difference
is in a region that is within a distance w of a previous difference, in which
case the
current difference is skipped and the next smaller distance is considered; and
7. continuing until there are no more differences to consider.
These exemplary computational procedures provide a set of measures of
intensity Isa for the class
of cells in sample s at difference d.
Distances
For hierarchical clustering, a distance DSS, may be defined as the distance in
vector space
between pairs of samples s and s'. A variety of methods for calculating Dss.
are available. Some
examples, which are intended as being nonlimiting, are provided below.
Dfs. as a scaled correlation function:
1. One calculates Ila= ( 1 /ns) ES I5a and as = [( 1/ns)ES (Isa - pa)zJo.s, If
data is missing, for
example no measurement of Isa exists for some sample s, that sample is
excluded from the
sum and ns is reduced by 1.
2. One calculates Jsa = (Isa - Via) / 6a~ If data is missing for Isa, then Jsa
is defined as Jsa = 0.
3. One calculates us = ( I/na) Ea Jsa and as = [( 1/na)Ea (Jsa - ~5)ZJo.s
4. One calculates Ksa = (J5a - !~S) / 6s
5. One calculates the covariance matrix S55. _ ( 1 /na) Ea Ksa Ks~a
6. One calculates the correlation matrix CS,. = SSS. / [ SSS Ssw]° s .
7. One calculates D55. _ [2 - 2 CSS. J°-j .
D55~ as a Euclidean distance: DSS~ _ [ Ea (ha -15a)z Jo.s
DSS. as a Pearson distance: DSS~ -- [ ~a (I$a - Is~a)- / aa2 ]° 5 Where
as is defined in step 1 of scaled
correlation function above.
CA 02343076 2001-03-19
WO 00/15851 _ IZ _ PCT/US99/21525
DSS. as a pairwise Pearson distance:
I . One calculates the covariance matrix SSS. _ ( 1 /na)[ Ea balsa - (~a ha )
(Ea 15~a ) / na ]-
2. One calculates the correlation matrix CSS. = SSS. / [ S55 SSS~ ]°~5
.
3. One calculates DSS. _ [ 2 - 2 CSS. ]°.s
D55. as a Mahalanobis distance:
1. One calculates the covariance matrix Saa. _ (ES Isa Isa) - (~5 15a) (E5
ISeO/ns
2. One calculates the correlation matrix Caa, = Saa~ / [ Saa Sw~ ]° 5
and its matrix inverse C-'aa. .
3. One calculates DSS. _ [ Eaa (I5a - I5a) C-~aa~ (I5a - Isw) ]° s
It is contemplated that other distance methods known in the art may be used in
the invention,
such as Spearman correlation, and the like. Other methods known in the art can
be found, for example
and not be means of limitation, in K. V. Mardia, J. T. Kent, and J. M. Bibby,
MULTIVARIATE ANALYSIS,
Academic Press, New York, 1979.
Hierarchical Clustering
The distances can be used to perform hierarchical clustering of the samples. A
general algorithm
for clustering is described below.
1. Each sample s is assigned its own initial cluster c.
2. One calculates all the distances between pairs of clusters and finds the
smallest distance.
These two clusters are joined into a single cluster and the number of clusters
is decreased
by 1.
3. Step 2 is repeated until only a single cluster remains.
In order to implement this algorithm, a method to calculate the distance
between pairs of clusters
is also required. Some nonlimiting examples of such calculations, using well-
known methods, are
indicated below.
Nearest neighbor, single linkage: The distance between clusters c and c' is
the smallest distance
DSf. , where s ranges over all samples in cluster c and s' ranges over all
samples in cluster c'.
Unweighted pair group method using arithmetic averages (UPGMA), also known as
average
linkage: The distance between clusters c and c' is (ESS. DSS.) / (n~ n~.)
where s ranges over all samples in
CA 02343076 2001-03-19
WO 00/15851 - 13 - PCT/US99/21525
cluster c, s' ranges over all samples in cluster c', n~ is the number of
samples in cluster c, and n~. is the
number of samples in cluster c'.
Furthest neighbor, complete linkage: The distance between clusters c and c' is
the largest
distance D55. where s ranges over all samples in cluster c and s' ranges over
all samples in cluster c'.
S Other distance-based hierarchical clustering methods are well-known. See,
for example, Wen-
Hsiung Li, MOLECULAR EVOLUTION, Sinauer Assoc, 1997.
Software packages are available to perform the clustering and display the
results. See, for
example, Phylip, Joe Felsenstein, http://evolution.Qenetics washin~aon edu for
clustering, and Treeview,
Rod Page, htto://taxonomy.zoology cla ac uk/rod/treeview html for display. The
source code for the unit
within Phylip employed for the clustering, and the downloaded executable file
of Treeview for Windows
95 and Windows NT, as well as a manual for Treeview, are available from the
owner of the present
application.
Two-Dimensional Clustering
It is also possible to cluster the distances, rather than clustering the
samples. One simply
1 S exchanges the roles of the samples and differences in the equations above.
Furthermore, it is possible to
perform clustering of both samples and differences, and then to display the
measurements Isd in which
both samples and differences are presented in cluster order.
Principal Component Analysis and Principal Factor Analysis
Principal component analysis is described in standard texts.. See, for
example, Mardia, Kent, and
Bibby. To perform principal component analysis, one begins with a correlation
matrix C55. as defined
above, in the section "Distances". (Alternatively, one could use the
covariance matrix SSS.). Eigenvalues
and eigenvectors, defined such that Css.gs.; = a;gs; , where the i'"
eigenvalue is a; and its eigenvector is gs; ,
are calculated. The eigenvalues are ordered from largest to smallest: a, ~ a,
>_ ... > as . To obtain a
reduced dimensional depiction of the samples, a number of desired dimensions k
is chosen. Then, in k-
dimensional space, sample s is represented as the point (gs,, gsz, ..., gSk).
Samples that are close in the k-
dimensional space have similar expression profiles and may be considered to be
related.
As an alternative to using the correlation matrix C55. as the starting point
for principal component
analysis, it is possible to calculate principal components using the inner
product matrix from
multidimensional scaling defined as
B = H C H (2)
CA 02343076 2001-03-19
WO 00/15851 _ ~ 4 - PCT/US99/21525 __
where C is the correlation matrix, H is the centering matrix with diagonal
elements given by 1 - (I/n) and
off diagonal elements - (i/n), where n is the number of items being
correlated. (See, for example,
Mardia, Kent, and Bibby, Multivariate Analysis, and Arkin, Shen, and Ross,
Science 277: 1275 (1997)).
The k'" principal component is then the k'" eigenvector of B normalized to
unit length and ordered by
decreasing eigenvalue ~,k, and the k'" principal factor is obtained by scaling
the eigenvector by ~,k'~-'. The
projection of sample s onto the k'" principal factor is the element of the
factor for row s. The
components or factors are ordered from 1 (corresponding to the most
informative) to n (corresponding to
the least informative). By using some, but not all, of the components or
factors, the samples can be
represented in a small-dimensional geometric space. Furthermore, the amount of
information retained in
the representation can be related to the eigenvalues of the components that
are used (See Mardia, Kent,
and Bibby).
A centered inner product matrix B appropriate for principal component or
prinicpal factor
analysis can also be obtained from any distance matrix Dss, as
B=HAH (3)
where
ASS _ -I/2 (D55.)-''. (
To perform principal factor analysis, factor i is defined as hs; = a;
°~5 gs; where, as before, a; is the
eigenvalue of the i'" eigenvector gs;. An orthonormal rotation matrix G (E~
G;~Gk~ is I if i = k and 0
otherwise, det(G) _ +1 ) is introduced and the factors are rotated to obtain
rotated coordinates for the
samples. Thus, to obtain a k-dimensional representation of the locations of
the samples, the following
operations are performed:
I . One calculates the correlation matrix C,S. or the covariance matrix SSS,
where s and s'
label individual samples.
2. One calculates the eigenvalues a; and eigenvectors gs; for the matrix, with
a, >_ a2 >_...>_ as.
3. Unrotated factor loadings hs; = a°'S gs; are defined.
4. The first k factor loadings and an orthonormal rotation matrix G are
selected. The j'n
coordinate of sample s in the rotated space is E~. h5~. G~.~
The rotation matrix G may be optimized according to standard criteria. See,
for example,
Mardia, Kent, and Bibby, Ch. 9.6 on Varimax rotation, supra. The rotated axes
represent factors that
influence the observed measurements for the samples.
CA 02343076 2001-03-19
WO 00/15851 _ ~ 5 _ PCTNS99/21525
In implementing the methods of the present invention, these operations may be
sequentially
combined in any of several ways according to the intended display, i.e., the
nature of the relatedness that
is intended to be shown.
Also, the information from the principal factors can be used to help filter
the experimental noise
from the correlation functions. For example, it is possible to select a cut-
off principal factor j < n, then
compute distances and correlations between samples based on their
representation in the j-dimensional
principal factor space.
As a nonlimiting example of the computational procedures that may be employed
in the present
invention, a schematic overview of procedures that may be adopted is presented
in Figure I . The
experimental results represent the sample-dependent and selection-dependent
intensities obtained in an
experiment, arrayed in a measurement matrix. In the implementation shown in
Figure l, the difference
bands having various, defined, nucleotide lengths are arrayed as the columns
of the matrix; they are
obtained in various experiments that are selected using different members of
the sets of subsequence
pairs. The samples represent the classes of cells, or cells treated with a set
of putative pharmaceutical
I S agents, or analogous sample sets, and are arrayed as the rows.
The values arrayed in the measurement matrix may then be subjected to
correlation analysis to
provide either direct sample correlations or correlations of differences. The
measurement matrix can
also be subjected to a calculation providing a vectoral distance between
samples; such a sample distance
may also be obtained from the sample correlation result. The distance vector
can further be subjected to
a linkage analysis to provide hierarchical clustering of the samples.
Additionally, the correlated samples
may be subjected to principal component analysis providing the principal
factors contributing to a state
or to a difference.
A nonlimiting example of the way in which a principal component analysis may
be carried out,
using methods described herein, is presented in Figure 2. The correlation
matrix or the centered inner
product matrix described above is subjected to appropriate operations to
provide the principal
components and the principal factors, based on their eigenvalues and
eigenvectors. Advantageously a
reduction in the number of dimensions employed in the number of eigenstates
may provide a filtering
effect, reducing the noise in the vector distances calculated.
The representations provided in the present invention find use in various
applications of
genomics in the biological and medical fields. Extents of relatedness and
correlations provide rapid
overviews of enzymatic reactions, metabolic pathways, and physiological
effects that become
distinguished when comparing states. When a pathological state is compared
with a normal state, for
CA 02343076 2001-03-19
WO 00/15851 _ 16 _ PCT/US99/21525
example in a mammal, and especially in a human, the display of distinguished
pathways is instructive in
the development of therapeutic approaches and/or therapeutic agents for the
treatment of the pathological
state. When a putative pharmaceutical agent is compared to a state that omits
the agent, or when one
such agent is compared with another, important information is provided
relating to the metabolic
reactions induced by or undergone by the agent or agents, leading to optimal
choice of such agents. This
information may also provide leads to the development of novel pharmaceutical
agents. If the genome
being studied is a plant genome, such as the genome of an important crop
plant, analogous principles
apply.
Nucleic acid assays
The present invention provides a method for generating a representation of the
extent of
relatedness between at least two classes of cells. In this method, the cells
in each class are chosen from
among cells of a given cell type, cells from a given tissue, and cells from a
given organ. Generation of
nucleic acids from the cell samples of choice may be as described in the
GeneCallingTM methodology.
See U.S. Patent No. 5,871,697. The method includes the steps of: (a) defining
a plurality of pairs of
nucleotide subsequences, each pair consisting of a first subsequence and a
second subsequence;
(b) isolating the nucleic acid of each class of cells and assaying for the
presence of a nucleic acid
fragment with the first subsequence at one end and the second subsequence at
another end and having a
length separated by the first and second subsequences, and quantitating the
extent to which each
fragment is present; and (c) determining the extent of relatedness reflecting
similarities or differences in
the presence and quantitation of the fragments among the classes using
software algorithm programs
known in the art.
One important embodiment of this method, i.e., determining the presence of the
fragments and
quantitating the amounts present, as described in step (b) above, is carried
out by a process that includes
the steps as follow. First, samples of the nucleic acid from the cells of each
class are digested with a
plurality of specific pairs of restriction endonucleases ("REs"). Each sample
is treated by one RE pair,
where one RE of the pair targets the frrst subsequence described in step (a)
above, and the second RE of
the pair targets the second subsequence, with each digestion providing
specific restriction fragments.
Second, double stranded adapter DNA molecules are hybridized to the fragments.
Each adapter
DNA molecule comprises: (i) a shorter strand, preferably having no 5' terminal
phosphate, consisting of
a first and second portion, the first portion being a region at the S' end
that is complementary to the
overhang produced by one of the REs of the given pair and a second portion
hybridizable to the opposite
longer strand of the adaptor, and (ii) a longer strand, preferably having no
5' terminal phosphate,
CA 02343076 2001-03-19
WO 00/15851 _ 1 ~ _ PCT/US99/21525
consisting of a first portion at its 3' end complementary to the above-
mentioned second portion of the
shorter strand, and an optional second portion at its 5' end comprising a
unique region not hybridizable
to any sequence present in the original sample population. See U.S. Patent No.
5,871,697. The longer
strand is optionally labeled with fluorochrome 208, although any DNA labeling
system that preferably
allows multiple labels to be simultaneously distinguished is usable in this
invention. See, e.g., Ausubel,
et al. CURRENT PROTOCOLS IN MOLECULAR BIOLOGY, John Wiley & Sons, New York,
NY, 1993.
Third, output signals from each ligated fragment are detected for each sample
population so
treated. Each ligated fragment generates output signals that characterize (a)
the presence of the given
subsequences corresponding to the RE pair used in a particular run, (b) the
length between the two
subsequences corresponding to the two REs employed in a given run, and (c) the
quantitation of the
relative amounts present of each fragment so generated in a given run.
Optionally, a nucleotide sequence database may be searched for sequences that
are predicted to
produce, or alternatively, not produce, the one or more output signals
generated by the nucleic acid from
the cells of each class, given the parameters described above. The analysis
methods comprise, first,
selecting a database of DNA sequences representative of the DNA sample to be
analyzed, second, using
this database and a description of the experiment to derive the pattern of
simulated signals that would be
generated, contained in a database of simulated signals, that will be produced
by DNA fragments
generated in the experiment, and third, for any particular detected signal,
using the pattern or database of
simulated signals to predict the sequences in the original sample likely to
cause this signal. Further
analysis methods present an easy to use user interface and permit
determination of the sequences actually
causing a signal in cases where the signal may arise from multiple sequences.
and perform statistical
correlations to quickly determine signals of interest in multiple samples. A
sequence from a searched
database is predicted to produce the one or more output signals when that
sequence has both (a) the same
length between occurrences of target nucleotide subsequences as is represented
by the one or more
output signals, and (b) the same target nucleotide sub-sequences that are
represented by said one or more
output signals, or target nucleotide subsequences that are members of the same
sets of target nucleotide
sub-sequences represented by the one or more output signals.
A first analysis method is selecting a database of DNA sequences
representative of the sample to
be analyzed. In the preferred use of this invention, the DNA sequences to be
analyzed will be derived
from a tissue sample, typically a human sample examined for diagnostic or
research purposes. In this
use, database selection begins with one or more publicly available databases
which comprehensively
record all observed DNA sequences. Such databases are GenBank from the
National Center for
CA 02343076 2001-03-19
WO 00/15851 _ I g _ PCT/US99/21525
Biotechnology Information (Bethesda, Md.), the EMBL Data Library at the
European Bioinformatics
Institute (Hinxton Hall, UK) and databases from the National Center for Genome
Research (Santa Fe,
N.Mex.). However, as any sample of a plurality of DNA sequences of any
provenance can be analyzed
by the methods of this invention, any database containing entries for the
sequences likely to be present in
such a sample to be analyzed is usable in the further steps of the computer
methods.
A second analysis method uses the previously selected database of sequences
likely to be present
in a sample and a description of an intended experiment to derive a pattern of
the signals which will be
produced by DNA fragments generated in the experiment. This pattern can be
stored in a computer
implementation in any convenient manner, In the following, without limitation,
it is described as being
stored as a table of information. This table may be stored as individual
records or by using a database
system, such as any conventionally available relational database.
Alternatively, the pattern may simply
be stored as the image of the in-memory structures which represent the
pattern.
A second important embodiment of this method, i.e., determining the presence
of the fragments
and their quantitation, as described in step (b) above, is carried out by a
process that includes the steps as
I 5 follow. First, for each pair of nucleotide subsequences selected, a pair
of oligonucleotide primers are
provides, the pair consisting of a first primer and a second primer, wherein
the first primer is
complementary to the first subsequence and the second primer is complementary
to the second
subsequence. Second, the nucleotide sequence between the first subsequence and
the second
subsequence are amplified using the oligonucieotide primers to prime the
amplification, thereby
providing an amplicon characterized by the subsequence pair, a length between
the two subsequences
corresponding to the two primers employed in each pair and a quantitation of
the extent to which each
amplicon is present. Third, output signals are generated as above for each
arnplicon, each output signal
characterizing (a) the subsequences of the pairs of primers, (b) the length,
and (c) the quantitation.
Optionally, a nucleotide sequence database may be searched for sequences that
are predicted to produce,
or alternatively, not produce, the one or more output signals generated by the
nucleic acid from the cells
of each class, given the parameters described above. Analysis methods are as
described above.
This invention can be applied, for example and not by way of limitation, to in
vitro cell
populations or cell lines, to in vivo animal models of disease or other
processes, to human samples, to
purified cell populations perhaps drawn from actual wild-type occurrences, and
to tissue samples
containing mixed cell populations. The cell or tissue sources can
advantageously be a plant, a single
celled animal, a multicellular animal, a bacterium, a virus, a fungus, or a
yeast, etc. The animal can
CA 02343076 2001-03-19
WO 00/15851 _ I g _ PCT/US99/21525
advantageously be laboratory animals used in research, such as mice engineered
or bred to have certain
genomes or disease conditions or tendencies.
Cells used in the invention may be obtained from a mammal, preferably a human,
having or
suspected of having a diseased condition. In one embodiment, the diseased
condition is a malignancy.
The in vitro cell populations or cell lines can be exposed to various
exogenous factors to determine the
effect of such factors on gene expression. In a preferred embodiment, the
exogenous factor is a putative
pharmaceutical agent. Cells so contacted with a putative pharmaceutical agent
are treated with an
amount of the agent sufficient to effect a change in the state of those cells
or with an amount of the agent
less than or equal to a predetermined upper limit of dosing concentration,
prior to their being assayed.
Measures of relatedness and extent of correlation may be made between cells so
contacted with putative
pharmaceutical agent and, for example, cells not so contacted.
Extent of relatedness methodology
The present invention provides a representation of the extent of relatedness
between a f rst class
of cells and a second class of cells. The cells in each class are chosen from
among cells of a given cell
I S type, cells from a given tissue, and cells from a given organ, as
described above. The extent of
relatedness reflects similarities or differences in the presence of pairs of
nucleotide subsequences, each
pair consisting of a first subsequence and a second subsequence, in a
nucleotide length separating the
first and second subsequences of the pair and in a quantitation of the extent
to which each pair having the
determined length is in the classes of cells. Input information of the
fragments to be analyzed are
obtained by methods of nucleic acid analysis and quantitation as described in
the NUCLEIC ACtD ASSAYS
section above.
The measure of relatedness is provided by calculating a distance that reflects
the amplitude of a
difference vector. A difference vector is defined as a difference between a
first vector and a second
vector. Herein, the first vector reflects information derived from the
quantitation for each subsequence
pair obtained for the first class of cells, and correspondingly, the second
vector reflects the analogous
information derived from the second class. The different elements of each
vector relate to data obtained
using different subsequence pairs.
In an embodiment of the representation, the extent of relatedness is related
to a distance. This
distance reflects the amplitude of a difference vector that is a difference
between a first vector which
reflects information derived from the quantitation for each subsequence pair
obtained for the first class
and a second vector which reflects the corresponding information obtained for
the second class. The
different elements of each vector relate to data obtained using different
subsequence pairs.
CA 02343076 2001-03-19
WO 00/15851 _ 2p _ PCT/US99/21525
In an additional significant embodiment, the representation includes a tree
structure reflecting
the extent of relatedness is provided by generating a tree structure
reflecting the relatedness between any
two classes. The branches of the tree structure reflect the difference vectors
and are ramified from
nodes.
In important embodiments of the representation of the extent of relatedness,
the representation is
obtained employing the methods of the invention, including the methods that
have been summarized in
the paragraphs immediately above.
In additional significant embodiments of the representation of the extent of
relatedness, the cells
in at least one class are obtained as described in the NUCLEIC ACID ANALYSIS
section above.
Correlation analysis methodology
The invention also provides a method for generating a representation of the
correlation between
a first class of cells and a second class of cells. The correlation reflects a
change in the nature and
amount of nucleic acids present in the classes. In this method, the cells in
each class are chosen from
among cells of a given cell type, cells from a given tissue, and cells from a
given organ. The method of
nucleic acid analysis and quantitation are as describe in the NUCLEIC ACID
ASSAYS section above.
Upon generation of a signal output, the correlation between the cells of.the
first class and cells of
the second class are correlated, and a representation of the correlation is
prepared. The quantitation of
the fragments in the invention corresponding to the RE pair used in a given
run and the length of each
fragment so generated; thereby providing a quantitative measure of the extent
to which the nucleic acid
present in the cells in each class contains fragments having the specific
subsequence pairs and the
nucleotide length between the pairs.
In a significant embodiment of the method for generating a representation of
the correlation, the
correlation is related to a set of orthonormal eigenvectors, as described in
the DISTANCES section above.
The elements of the basis set upon which the eigenvectors are constructed
reflect particular biochemical
or physiological pathways correlated between the cells of the two classes Each
of these eigenvectors is
associated with an eigenvalue that is an integer greater than zero. After
defining an upper limit of the
eigenvalues to be used, the coefficients of the basis set elements in each
eigenvector whose eigenvalue is
less than or equal to this upper limit reflects the contribution of the
corresponding pathway to the
biochemical or physiological differences correlated between the cells of the
first class and the cells of the
second class.
CA 02343076 2001-03-19
WO 00/15851 _ 21 _ PCT/US99/21525
In another significant embodiment of the method for generating a
representation of the
correlation, the representation is a cluster diagram or a dendrogram, and
includes a tree structure
reflecting the relatedness of the pathways involved in the biochemical or
physiological response to a
difference between cells of the two classes. In obtaining this representation,
a correlation matrix is
calculated that provides a distance determination in which the distance
reflects the amplitude of a
difference vector. This vector is a difference between two vectors each of
which reflects information
obtained for the response of one of the two classes to the difference between
the classes, and wherein the
branches of the tree structure reflect the difference vectors and the branches
are ramified from nodes.
In additional significant embodiments of the representation of the extent of
correlation, the cells
in at least one class obtained as described in the NUCLEIC ACID ANALYSIS
section above.
Display means
The present invention also provides a display means displaying a
representation of the extent of
relatedness between a first class of cells and a second class of cells. The
cells in each class are chosen
from among cells of a given cell type, cells from a given tissue, and cells
from a given organ, as
described above. The extent of relatedness reflects similarities or
differences in the presence of pairs of
nucleotide subsequences, each pair consisting of a first subsequence and a
second subsequence, in a
nucleotide length separating the first and second subsequences of the pair and
in a quantitation of the
extent to which each pair having the determined length is in the classes of
cells.
In a significant embodiment of the display means, the extent of relatedness is
related to a
distance. This distance reflects the amplitude of a difference vector that is
a difference between a first
vector which reflects information derived from the quantitation for each
subsequence pair obtained for
the first class and a second vector which reflects the corresponding
information obtained for the second
class. The different elements of each vector relate to data obtained using
different subsequence pairs.
In an additional significant embodiment of the display means, the
representation includes a tree
structure reflecting the relatedness between any two classes, in which the
branches of the tree structure
reflect the difference vectors and the branches are ramified from nodes.
In important embodiments of the display means displaying a representation of
the extent of
relatedness, the representation is obtained employing the methods of the
invention, including the
methods that have been summarized in the paragraphs immediately above.
CA 02343076 2001-03-19
WO 00/15851 - 22 - PCT/US99/21525
In additional significant embodiments of the display means displaying a
representation of the
extent of relatedness, the cells in at least one class obtained as described
in the NUCLEIC ACID ANALYSIS
section above.
The present invention additionally provides a display means displaying a
representation of the
correlation between a first class of cells and a second class of cells. The
cells in each class are chosen
from among cells of a given cell type, cells from a given tissue, and cells
from a given organ, as
described above. The correlation reflects differences between the first class
and the second class in the
presence of a pair of nucleotide subsequences, each pair consisting of a first
subsequence and a second
subsequence and the nucleotide length separating the first and second
subsequences of the pair, and in a
quantitation of the extent to which each pair having the determined length is
present in the cells.
In an advantageous embodiment of this display means, the correlation is
related to a set of
orthonormal eigenvectors. The elements of the basis set upon which the
eigenvectors are constructed
reflect particular biochemical or physiological pathways correlated between
the cells of the two classes
Each of these eigenvectors is associated with an eigenvalue that is an integer
greater than zero. After
I S defining an upper limit of the eigenvalues to be used, the coefficients of
the basis set elements in each
eigenvector whose eigenvalue is less than or equal to this upper limit reflect
the contribution of the
corresponding pathway to the biochemical or physiological differences
correlated between the cells of
the first class and the cells of the second class.
In an additional advantageous embodiment of the display means displaying a
representation of
the correlation, the representation is a cluster diagram or a dendrogram, and
includes a tree structure
reflecting the relatedness of the pathways involved in the biochemical or
physiological response to a
difference between cells of the two classes. In obtaining this representation,
a correlation matrix is
calculated that provides a distance determination in which the distance
reflects the amplitude of a
difference vector. This vector is a difference between two vectors each of
which reflects information
obtained for the response of one of the two classes to the difference between
the classes. The branches
of the tree structure reflect the difference vectors and the branches are
ramified from nodes.
1n important embodiments of the display means displaying a representation of
the correlation,
the representation is obtained employing the methods of the invention,
including the methods that have
been summarized in the paragraphs immediately above.
In important embodiments of the representation of the correlation, the
representation is obtained
employing the methods of the invention, including the methods that have been
summarized in the
paragraphs immediately above.
CA 02343076 2001-03-19
WO 00/15851 _ z3 _ PCT/US99/21525
In additional significant embodiments of the display means displaying a
representation of the
correlation, the cells in at least one class obtained as described in the
NuCLEtC ACID ANALYSIS section
above.
Other Aspects
In addition to providing representations of cells, the techniques described
here are also useful for
providing representations of nucleic acid fragments or genes. The starting
point for the analysis is the
matrix Isd described previously, where s labels the sample (or group of
samples or distinct types of cells)
and d labels a particular measurement of the expression level of a particular
gene in that class. Rather
than generating representations based on the rows of I, each representing a
different sample or group of
samples, it is possible to generate representations based on the columns of I,
each representing a different
nucleic acid. Hierarchical and geometrical representations of nucleic acids,
based on their relative
abundance across a series of cells, can be used to infer genes that are co-
expressed and are likely to have
related biological function.
Other Embodiments
The data matrix of intensities I can be described more generally as a
representation in which
each row corresponds to a particular biological sample or group of samples,
and each column
corresponds to a particular nucleic acid molecule or class of molecules whose
quantities are measured in
each of the biological states.
In addition to the differential-display methods described to provide
measurements of nucleic acid
quantities, other methods for obtaining measurements of the nucleic acids
present in a cell are available.
These include restriction fragment length polymorphism, amplification fragment
length polymorphism,
EST sequencing, serial analysis of gene expression, hybridization to
oligonucleotide probes, and other
methods known in the art. Other methods, such as quantification by TaqMan or
Northern blots, are also
used. All of these methods generate data sets that can be analyzed according
to the methods described
here. The measurements Isd for each biological state and nucleic acid can
correspond to absolute
concentrations, concentrations relative to a standard (either ratio or numeric
difference), or other
convenient measures.
The methods of the invention includes analysis of populations ranging from 5,
10, 25, 50, 100,
1000, 10,000 or 100,000 or more members.
CA 02343076 2001-03-19
WO 00/15851 _ 2q _ PCT/US99/21525
EXAMPLE
Male Sprague-Dawley rats (Harlan Sprague Dawley, Inc., Indianapolis, Indiana)
of 10-14 weeks
of age were gavage-fed and dosed once a day for three days with the following
drugs, dissolved in sterile
water, at the following levels:
phenobarbitol 3.81 mg/kg/day
gabapentin 34.29 mg/kg/day
vigabatrin 150 mg/kg/day
paraldehyde 77.08 mg/kg/day.
These dosages correspond to the ED100 (the upper limit of the effective dose
for humans)
adjusted for the difference in metabolic rate between rats and humans. Three
rats were used for each
drug treatment, and an additional three rats to match each drug were treated
with sterile water to serve as
a control.
Rats were sacrificed 24 hours after the final dose and their brains were
harvested. Collection of
mRNA, synthesis of cDNA, and differential display protocols were carried out
according to methods
I S described in U. S. Patent No. 5,871,697 and Shimkets et al. 1999 (Nature
Biotechnology 17:798-803).
The following steps were followed to analyze the differential display pattern:
1. The intensities IS~(x) for each of the three animals treated with the same
drug were combined
into a single average h,(x), where the subscript a labels the drug. The
standard deviation se~(x) was also
computed for the measurements from the individual animals treated with the
drug.
2. The averages Ia~(x) and standard deviations s"(x) for each drug were
compared with the
average I~~(x) and standard deviation s~~(x) for the sterile water control
treatment. A difference at length
x was marked if
ABS(In [I,r(x)/I~~(x)]) ? ln( 1.5) (5)
and if the significance was smaller than 0.15 for a two-tailed t-test with
t = LIe~(x) - I~~(x)l / ~( sa~(x)~ + s~~(x)~ 3/ 2 ~uz (6)
and infinite degrees of freedom. The difference intensities marked according
to this procedure
may then be inspected by eye and visually significant differences may be
retained.
CA 02343076 2001-03-19
WO 00/15851 - 25 - PCT/US99/21525
3. For each of the differences d, defined by a restriction enzyme pair r and a
position x, the
intensity h,(x) =18d was determined for each of the drug treatments, whether
or not that particular
treatment has a difference compared to the control.
In this example, the final data matrix Ied has 8 rows: I row for each of the 4
drugs, and 1 row for
each of 4 replicates of the water control data. The matrix has as many columns
as the number of
differences detected in the differential display pattern.
The Pearson correlation coefficient Cab between the 8 classes of samples (4
drugs, 4 water
controls) was determined using methods provided in the Detailed Description of
the Invention. If a data
element for a particular difference was missing for a particular treatment,
that difference did not
contribute to the correlation coefficient. The correlations are shown in the
Table 1 below, with the
standard deviation within a drug shown as the diagonal elements.
Table 1.
vigabatnn gabapentinparaldehydewater_1water water water
phenobarbitol 2 3 4
vigabatrin261.360t:0.9982Ø99 0.9728 0.6548 _ 0.36740.3786
T3 0.6673
phenobarbitol0.9982'26344690.9973 0.9325 0.5678 0.4724 0.65690.6601
gabapentin0.9913 0.9973556.81140.9922 0.6177 0.6057 0.34000.2704
paraldehyde0.9728 0.93250.9922 423.19160.6485 0.5307 0.59520.5702
water_1 0.6548 0.5678O.6TI7 0.6485 59.45730.9718 0.98960.9735
water_2 0.6673 0.47240.6057 0.530T 0.9718 62.35680.9960:0.9886
waler 0.3874 0 65690.3400 0.5952 0.9896 0.9960 107.26300.9975
3
_ 0.3786 0.66010.2704 0.5702 0.9735,0.9886 0.9978123.0743
water .
4 .
Next the pairwise Pearson distance was calculated as described previously. The
distance matrix
is shown in Table 2 below.
Table 2.
vigabatrin gabapentinparaldehydewater_1water water water
phenobarbitol 2 3 4
vigabatrin0.0000 0.05970.1319 0.2333 0.8309 0.8157 _ _
1.1248 1.1148
phenobarbitol0.0597 0.00000.0741 0.3675 0.9297 1.0272 0.8284 0.8245
gabapentin0.13 0.07410.0000 0.1245 0.8744 0.8880 t1489 1.2080
paraldehydeØ2333 0.36750.1245 0.000 0.8384 0.9688 0.8997 0.9271
.
water_t 0.8309 0.92970.8744 0.8384 0.0000 0.2376 0.1446 0.2302
water_2 0.8157 1.02726.8880 0.9688 0.2376 0.0000 0.0895 0.150
water_3 1.1248 0 1.1489 0.8997 0.1446 0.0895 0.0000 0.0663
, 8284
water,.4.11148 0.8245.:1.208D 0.9271 x.2302 U..f509x.0663 0.0000
..
The distances were then used as input to a nearest-neighbor clustering
algorithm. The resulting
clusters, using sterile H.,O as an outgroup, was shown in Fig. 3. The
horizontal distances in Fig. lA were
proportional to the pairwise Pearson distance between clusters.
The correlation matrix C,b also served as the starting point for principal
factor analysis. First,
principal components were calculated using the inner product matrix from
multidimensional scaling
B= HCH (2)
CA 02343076 2001-03-19
WO 00/15851 - 26 - PCT/US99/21525
where C is the correlation matrix and H is the centering matrix. The k'"
principal component is
then the k'" eigenvector of B normalized to unit length and ordered by
decreasing eigenvalue ~,,;, and the
k'" principal factor was obtained by scaling the eigenvector by ~.A"'.
Projections of the treatments and
controls onto principal factors are shown in Table 3 below.
Table 3.
factor: 1 ~ 3.
eigenvalue:1841 0.347 0.093Ø036 0.00>'0.000 -0.036-0.38&
vigabatrin-0.513-0.163-0.0990.083 -0.0040.000 0.000 0.000
pheno.barbitol-0,.3970.317 -0.132-D.038-0.0190.000 0.000 0.000
gabapentin-0.580-0.157Ø028 -0.094-0.0040.000 0.000 0.000
..
paraldehyde-0.4040.127 019$,.Ø057 , 0.034Ø000 0.000 0.000
,
water_1 0.368'-0.16901D9 0.017 -O.O6i. 0.000 0.000
~ 0.000
water 0.422 -0.300-0 -0.0170.038 0.000 0.000 0.000
2 091
water.3 0.542 0.156 0.04~r'-0.0900.012 O.OOp 0.000 0.000
water.4 0.561 0.190 -0.053Ø082 0.001 0.000 0.000 0.000
,.
The components are ordered from I (most informative) to 8 (least informative).
The negative
eigenvalues arise from the method used to account for missing data. If missing
data had been handled in
an alternate manner, for example if a missing element had been set to the
average value or if the analysis
were restricted to differences for which no data was missing, the eigenvalues
would alt be non-negative.
In Fig. 4, the treatments are displayed by projection onto principal factors.
Factor 1
discriminates between drugs, where it has a negative value, and controls,
where it has a positive value.
Factor 2 discriminates between the drug treatments.
EQUIVALENTS
1 S From the foregoing detailed description of the specific embodiments of the
invention, it should
be apparent that unique methods for representing the extent of relatedness
between cells, cell lines,
tissues, organs, or expressed sequences based on a genomic analysis of gene
expression have been
described. Although particular embodiments have been disclosed herein in
detail, this has been done by
way of example for purposes of illustration only, and is not intended to be
limiting with respect to the
scope of the appended claims which follow. In particular, it is contemplated
by the inventor that various
substitutions, alterations, and modifications may be made to the invention
without departing from the
spirit and scope of the invention as defined by the claims. For instance, the
choice of source material,
subsequences used, or software algorithm used is believed to be a matter of
routine for a person of
ordinary skill in the art with knowledge of the embodiments described herein.