Note: Descriptions are shown in the official language in which they were submitted.
CA 02346146 2001-04-04
WO 00/20630 PCT/CA99/00933
TITLE OF THE INVENTION
OLIGONUCLEOTIDE PRIMERS THAT DESTABILIZE NON-
SPECIFIC DUPLEX FORMATION AND USES THEREOF.
5 FIELD OF THE INVENTION
The present invention relates to genetic engineering. More
specifically, a method is presented for reducing mispriming during DNA
synthesis. In particular, the present invention relates to primers containing
modified nucleosides (e.g. universal base) which reduce mispriming during
10 cDNA library construction, thereby increasing the proportion of cDNA clones
having been primed from the bona fide 3' poly A tail. The present invention
further relates to the use of the discriminating oligonucleotides of the
present
invention in other methods such as mRNA purification, PCR-based detection
methods and sequencing.
15
BACKGROUND OF THE INVENTION
The isolation and rapid mapping of complementary DNAs
(cDNAs) is central to characterizing the information that is .of significant
biological relevance in the genome of an organism. A full length cDNA allows
20 one to predict transcription initiation start sites, translation initation
start sites,
deduce certain protein characteristics based on primary amino acid sequence,
predict transcription termination sites, and visually inspect the 5' and 3'
untranslated regions for elements which may be involved in post-
transcriptional
regulation of gene expression. The analysis of several complete cDNAs of a
25 given gene enables one to gather information on alternative splicing,
alternative
promoter usage, and alternative polyadenylation signals - all events known to
be important in gene expression regulation. In addition, the comparison of
genomic and cDNA sequence is essential to determine exon-intron structure and
document the occurrence of RNA editing - a post-transcriptional regulatory
30 mechanism on which there is little information.
CA 02346146 2001-04-04
WO 00/20630
2
PCT/CA99100933
The cloning of mRNA into cDNA for the purposes of
functional studies is a complex, interrelated series of enzyme-catalyzed
reactions involving the in vitro synthesis of a DNA copy of mRNA, its
subsequent
conversion to duplex cDNA, and insertion into an appropriate prokaryotic
vector.
5 The procedure may involve the following series of steps (outlined in Fig.1
):
1) Isolation of high quality mRNA from the tissue or cell line
of interest.
2) Annealing of a DNA oligonucleotide, either a mixture of
oligonucleotides of random sequence or an oligo d(T) primer, to the mRNA.
10 When full-length cDNAs are required, oligo d(T) is utilized, since this is
expected
to anneal to the 3' poly (A) tail of the mRNA.
3) Reverse transcriptase is then utilized to prime from the
DNA primer and copy the RNA template into cDNA.
4) Second strand synthesis is performed utilizing RNAse H,
15 DNA polymerase I, and DNA ligase.
5) The ends of the cDNAs are polished, prepared for cloning,
and the cDNAs are introduced into an appropriate cloning vector.
Although a number of different approaches can be used to
generate cDNA libraries, they suffer from several major problems, often making
20 the isolation of a complete cDNA an arduous task. The cloning of incomplete
cDNAs is widespread, resulting in only partial characterization of mRNA
transcripts and significantly increasing the cost and amount of work required
to
obtain a full-length copy of the cDNA of interest. One major reason why many
clones in current cDNA libraries are not full-length is due to mispriming of
the
25 oligo d(T) primer (de Fatima Bonaldo et al., 1996, Genome Res. 6:791-806).
Many eukaryotic mRNAs contain regions of A-rich stretches within their
sequence. Thus oligo d(T) primers can anneal to these internal A-rich
stretches.
When reverse transcriptase primes from these internal sites, sequence
information from the 3' end of the mRNA is lost during the cDNA cloning
process
30 (Fig. 1 ). Although the genetic code of most organisms is composed of ~ 50%
CA 02346146 2001-04-04
WO 00/20630
3
PCT/CA99/00933
guanosine + cytosine residues and 50% of adenosine + thymidine residues,
there are welt/ known examples of organisms whose genetic code deviates from
this ratio. For example, the genome of the parasite responsible for malaria y
transmission, Plasmodium falciparum, has a genome of >80% adenosine +
5 thymidine residues (Weber, J.L., 1987, Gene 52:103-109). This implies that
cDNA libraries derived from this organism will contain many truncated, less-
than-full-length clones, due to mispriming of the oligo d{T) primer during
first
strand synthesis. Mispriming is thus a serious hindrance to gene discovery and
characterization in general, and more acutely for certain organisms.
10 These technical limitations imply that a set of products of
variable length are often generated during first strand synthesis.
Consequently,
a number of truncated clones may be present in any given library. Given these
cloning complications, interpretations about gene structure are sometimes
misleading and cDNA cloning is often inefficient, costly, and time-consuming -
15 often requiring the sampling of several different libraries.
The actual procedure for generating cDNA libraries has not
extensively deviated from the original method of Gubler et al., 1983, Gene
_25:263-269. Because of the frequent generation of products of variable length
during first strand synthesis, a number of truncated clones will be present in
20 libraries for any given gene. Priming from the poly (A) tract of mRNAs with
oligo
d(T) is necessary to obtain a copy of the entire 3' untranslated region.
However,
it is the experience of many laboratories screening cDNA libraries, that a
significant proportion of clones do not have a bona fide 3' end, due to
misannealing of the oligo d(T) primer to internal A-rich sites. Indeed, cDNAs
with
25 3' truncations are estimated to occur at frequencies of 10-15% in some
libraires
(de Fatima Bonaldo et al., 1996, supra). Such clones are easily recognized by
the absence of a bona fide polyadenylation signal sequence ~20 nucleotides
upstream of the oligo (dA) tail of the cDNA. If enhanced discrimination could
be
achieved between annealing to the bona fide poly (A) tail versus internal A-
rich
CA 02346146 2001-04-04
WO 00/20630
4
PCT/CA99/00933
sequences by the Reverse Transcriptase primer, then the frequency of this
"mispriming artifact" would be significantly reduced.
Nucleic acid hybridization, in which a DNA or RNA strand
binds to its complement to form a duplex structure is a fundamental process in
5 molecular biology. A critical aspect of this process is the specificity of
molecular
recognition of one strand by the other. Sequence differences as subtle as a
single base change are sufficient to enable discrimination of short (e.g. - 14
mer)
oligomers, and are frequently used to detect point mutations in genes (Conner
et al., 1983, Proc. Natl. Acad. Sci. USA. 80:278-282.). Molecular
discrimination
10 of single point changes using oligonucleotides has been well documented and
the underlying thermodynamics well characterized (Ikuta et al., 1987, Nucl.
Acids Res. 15:797-811; Doktycz et al., 1995, J. Biol. Chem. 270:8439-8445;
Southern et al., 1994, Nucl. Acids Res. 22:1368-1373; Saiki et al., 1989,
Proc.
Natl: Acad. Sci. USA 86:6230-6234). However, in many cases, the stability
15 difference between a perfectly matched complement (e.g. - between a poly
(A)
tail and oligo d(T),5) and a complement mismatched at only one base (e.g.
between A,AAAAAATAAAAA~ and oligo d(T),5) can be quite small,
corresponding to as little as 0.5°C difference in their duplex melting
temperature
(Tms) (Fig. 2). The longer the oligomer of interest (e.g. an oligo d(T)2o
primer
20 versus and oligo d(T),5 primer) the smaller the effect of a single-base
mismatch
on overall duplex stability. This limitation in hybridization is the major
reason why
oligo d(T) primers often hybridize to internal A-rich sequences on mRNA
templates during cDNA library construction, and consequently why a large
number of clones in such libraries do not contain the bona fide 3' end.
25 Guo et al. (1997, Nature Biotech. 15:331-335) have recently
shown that increased discrimination of single nucleotide mismatches by
oligonucleotides can be achieved by introducing artifical mismatches into the
probe oligonucleotide using the base analog 3-nitropyrrole. This base analog
acts as a universal nucleoside that hydrogen bonds minimally with all four
bases
30 without steric disruption of the DNA duplex (Nichols et al., 1994, Nature
CA 02346146 2001-04-04
WO 00/20630
5
PCT/CA99/00933
X9:492-493). Since hydrogen bonding between bases of two complementary
strands of DNA is the major thermodynamic force responsible for maintaining
the integrity of a double stranded DNA duplex, base substitutions with analogs
with lessened hydrogen bond capacity can function as universal nucleosides
5 (Nichols et al., 1994, supra). A number of different nucleoside analogs have
been developed which function in this fashion {Millican et al., 1984, Nucl.
Acids
Res. 12:7435-7453; (none et al., 1985, Nucl. Acids Res. 13:7119-7128; Fukada
et al., 1986, Naturforsch. 8. 41:1571-1579; Seefa et al., 1986, Nucl. Acids
Res.
_14:1825-1844; Eritja et al.; 1986, Nucl. Acids Res. 14:8135-8153; Habener et
1p -al., 1988, Proc. Natl. Acad. Sci. USA 85:1735-1739; Lin et al., 1989,
Nuci. Acids
Res. 17:10373-10383; Francois et al., 1990, Tetrahedron Lett. 31:6347-6350;
Brown et al., 1991, Carbohydrate Res. 216:129-139.). Guo et al. (1997, supra)
have shown that the introduction of universal analogues into heteropolymeric
oligonucleotides during their synthesis, increases the thermal stability (eTm)
of
15 hybrids formed between an oligonucleotide with the universal nucleoside and
normal and single-nucleotide variant DNA targets by as much as 200%, as
compared to hybrids formed between a wild-type oligonucleotide and normal or
single-nucleotide variant DNA targets.
U.S. patent 5,438,131 of Bergstrom et al. teaches
20 oligonucleotides of at least 10 nucleosides, composed of at least two
different
bases, and containing at least one universal nucleoside and the use thereof to
reduce the element of risk and enhance success in screening DNA libraries.
The universal base is defined in U.S. 5,438,131 as being a modified nucleic
acid
base that can base-pair with its ally, one of the common bases A, T, C and G
25 (as well as U). The aim of the universal base is to reduce degeneracy while
still
preserving the uniqueness of the probe. A variety of compounds have been
investigated as universal bases and a number of them are described in U.S.
5,438,131. In a preferred embodiment, U.S. 5,438,131 relates to
oligonucleotides containing universal nucleosides at degenerate positions,
such
30 that the oligomer allows bonding to unknown bases, enabling the formation
of
CA 02346146 2001-04-04
WO 00/20630
6
PCT/CA99/00933
duplexes with ambiguous or unknown nucleic acid sequences. In a particularly
preferred embodiment, U.S. 5,438,131 relates to 3-nitropyrroie nucleoside as
the universal nucleoside. U.S. 5,438,131 thus relates to the use of universal-
nucleosides in order to stabilize duplex formation between heteropolymers of
5 oligonucleotides and a target nucleic acid.
In view of the technical limitations of current methods of
cDNA synthesis, there remains a need to destabilize artefactual duplex
formation to increase the discrimination between specific and non-specific
duplexes. There also remains a need to provide the means to reduce
10 mismatches in general, and more particularly to reduce mispriming during
DNA
synthesis, cDNA library construction, and PCR applications. The present
invention seeks to meet these and other needs.
The present description refers to a number of documents, the
content of which is herein incorporated by reference, in their entirety.
15
SUMMARY OF THE INVENTION
The invention concerns the identification of primer
modifications that can destabilize artifactual duplex formation and decrease
the
number of mismatches between the primer and its target sequence.
20 In one embodiment, the invention further concerns the
identification of primer modifications that improve the discrimination between
the
binding thereof to a homopolymeric target sequence {the bona fide target
sequence) as compared to a non-homopolymeric target sequence. The
invention therefore provides oligonucleotides which are better at
discriminating
25 between their homopolymeric complementary sequence and a related target
sequence. In addition, the present invention provides assays which can be used
(and adapted) to identify oligonucleotide modifications that destabilize
mismatches.
The invention also concerns the development of primers
30 which decrease mispriming events encountered during DNA synthesis. More
CA 02346146 2001-04-04
WO 00/20630 PCT/CA99100933
7
specifically, the invention concerns the development of primers containing at
least one modified nucleoside, which decrease the number of internal
mispriming events during cDNA generation, thereby improving the efficiency of
correct priming from the bona fide 3' poly (A) tail.
5 The present invention further relates to universal primers
which reduce the proportion of mismatches during genetic engineering methods
such as, for example, mRNA purification, 3' RACE, 5' RACE, PCR, sequencing
and the like. In a particularly preferred embodiment, the present invention
relates to the incorporation of at least one universal base in an
oligonucleotide
10 comprising a homopolymeric stretch in order to reduce mismatches to its
homopolymeric target sequence, and thereby generating a modified
oligonucleotide, The invention concerns more particularly modified
oligonucleotides, wherein a homopolymeric-stretch of the oligos contains a
modification which improves their binding to their target sequence. More
15 specifically, the present invention relates to primers or oligos
incorporating at
feast one 3-nitropyrrole modification in the homopolymeric stretch.
The invention also concerns assays to identify modifications
in oligonucleotides which reduce the proportion of mismatches and mispriming
events, comprising a random or rational design of modifications of a chosen
20 primer, a hybridization thereof with its target sequence to form a duplex,
a
synthesis of DNA priming from this duplex and an analysis of the synthesized
DNA to assess for the presence of mispriming events, wherein the number of
mispriming events produces cDNAs of truncated sizes compared to cDNAs
produced by initiation from the bona fide priming site (i.e. the homopolymeric
25 priming site).
In accordance with the present invention, there is therefore
provided a method for destabilizing non-specific duplex formation between an
oligonucleotide and a target nucleic acid, wherein at least one of the
oligonucleotide and target nucleic acid comprises a homopolymeric sequence,
30 the method comprising an incubation of the target nucleic acid with a
modified
CA 02346146 2001-04-04
WO 00120630 PCT/CA99100933
8
oligonucleotide, wherein the modified oligonucleotide includes a modification
which decreases or abrogates hydrogen bonding between same and non-
specific target sequences and thus enables a discrimination between a bona
fide
duplex formation and an artifactual one, under the conditions of hybridization
5 used. In accordance with a preferred embodiment of the present invention,
the
target nucleic acid is a homopolymeric sequence.
In accordance with the present invention, there is also
provided a method for increasing the proportion of full length cDNA clones in
a
library, comprising a use of a modified oligo d(T) primer during first strand
10 synthesis, wherein the modification decreases or abrogates hydrogen bonding
between the modified ofigo d(T) primer and a non-specific target sequence,
thereby increasing the proportion of full length cDNA clones.
In accordance with another aspect of the present invention,
there is provided a method for reducing mispriming events during DNA
15 synthesis, comprising a use of a modified oligonucleotide to prime the DNA
synthesis, wherein the modification decreases or abrogates hydrogen bonding
between the modified primer and a non-specific target sequence, thereby
reducing mispriming events.
In accordance with yet another aspect of the present
20 invention, there is provided modified oligonucleotide primers that
destabilize
non-specific duplex formation and reduce mispriming during DNA synthesis.
While the method of the instant invention is demonstrated
during first strand cDNA synthesis to improve the quality of the cDNA
population
by reducing the number of clones containing aberrant 3' ends due to oligo d(T)
25 mispairing, and more specifically using the eIF-4611 mRNA template, the
present
invention, which has broad utility, is not so limited. Although 3' mispairing
is a
general problem encountered when generating cDNA libraries from a number
of organisms, this problem can be particularly exacerbated when generating
cDNA libraries from organisms that have A rich genomes, since the number of
30 internal A-rich stretches will be higher in genes from these organisms.
This type
CA 02346146 2001-04-04
PCT/CA99/00933
WO 00/20630
9
of incomplete A-tract is expected to misanneal to oligo d(T) and produce
truncated cDNAs during library construction. An example of such an organism
is Plasmodium falciparum, the parasite responsible for malaria transmission by
mosquitos. Thus, the present invention provides the means to destabilize
5 mispairing of an oligonucleotide or primer to a non-targeted or non-specific
nucleic acid sequence from any organism or nucleic acid sequence-containing
entity, thereby increasing the proportion of duplexes formed between the
oligonucleotide or primer and its proper targeted sequence. In one preferred
embodiment of the present invention, the modified primer comprises an
10 essentially homopolymeric stretch of nucleotides (including a modification)
which
targets its complementary homopolymeric sequence.
While the instant invention is demonstrated using an oligo
d(T)~Z primer (an oligo d(T) primer in which two of the thymine bases are
substituted by 3-nitropyrrole), the instant invention is not so limited. For
15 example, the position of the modified bases within the exemplified
oligonucleotide, oligo d(T) primer, can be altered (Fig. 2C) without changing
the
discrimination between primer and either complementary template or partially
complementary template. Indeed, Guo et al. (1997, supra) have changed the
position of 2 universal nucleosides within a given heteropolymeric
20 oligonucleotide and shown that in many cases increased discrimination
between
perfect matched template and mismatched template is maintained. Thus, the
instant invention extends to any homopolymeric-stretch-containing
oligonucleotide (or any oligonucleotide designed to bind to a homopolymeric
target sequence) such as an oligo d(T) primer containing modified nucleosides
25 at any position, provided that such modification maintains the
discriminating
ability of the oligonucleotide under suitable assay conditions. It should be
clear
to the person of ordinary skill that the present invention further provides
the
means to assess whether the modifications alter this discriminating activity
of the
oligonucleotide. It should also be clear that any type of homopolymeric-
30 complementary sequence duplex formation could be improved by the instant
CA 02346146 2001-04-04
WO 00/20630 PCT/CA99/00933
10
invention. In a broad sense therefore, the present invention provides the
means
and methods to generate oligos or primers with improved discrimination to
their
complementary homopolymeric sequence compared to non-complementary
sequence.
5 It should be clear to a person of ordinary skill that the present
invention has broad implications since it demonstrates that a modification
which
results in destabilization of a duplex (examplified with oligo d(T), having 2
substitutions, and its poly A target sequence), significantly decreases the
proportion of mismatches and of mispriming events. Hence, it is expected that
10 other types of destabilization of the hydrogen bonds between an
oligonucleotide
and its target sequence would have the same effect. Non-limiting examples of
modifications of the oligonucleotide which would result in such a
destabilization
of the duplex formation, include modifications which reduce or abrogate
hydrogen bonding. Non-limiting more specific examples include known base
15 modifications, base analogs {e.g. inosine, as exemplified hereinbelow),
universal
bases, and partial mismatches. Of course, it will be understood that such
modifications should not favor duplex formation with a non-desired target
sequence.
It should also be understood that the different modifications
20 of the oligonucleotides encompassed by the present invention can be adapted
by the person of ordinary skill to suit particular utilities (e.g. mRNA
purification,
sequencing).
The present invention should not be limited to the
modifications of oligonucleotides with 3-nitropyrrole, since other universal
bases
25 are well known in the art. Indeed, in addition to 3-nitropyrrole, a number
of
universal nucleosides have been synthesized and characterized (Millican et al.
1984, supra; Inone et al., 1985, supra; Fukada et al., 1986, supra; Seela et
a1.,1986, supra; Eritja et al., 1986, supra; Habener et al., 1988, supra; Lin
et al.,
1989, supra; Francois et al., 1990, supra; Brown et al., 1991, supra). Other
30 examples of universal bases can be found at
CA 02346146 2001-04-04
WO 00/20630 PCT/CA99/00933
11
www.Synthegen.com/productslbases.html. Thus, the present invention covers
any homopolymeric-stretch-containing oligo (e.g. oligo d(T) primer),
containing
at least one universal nucleoside which allows for enhanced discrimination
when
hybridizing to perfect versus mismatched templates. In addition, the
5 demonstration that a base analog, such as inosine, inserted into the
homopolymeric stretch also enhances discrimination between the target
sequence and a mismatched sequence, shows that the present invention covers
any homopolymeric-stretch-comprising oligo containing at least one modified
base which reduces or abrogates hydrogen bonding in the sequence which is
10 complementary to the targeted sequence..
A non-limiting example of an alternative use of this
technology is in mRNA purification, by replacing oligo d(T) affinity matrixes
currently employed with modified oligo d(T) according to the instant
invention.
An oligo d(T)~Z affinity matrix would perform the same task, except that
binding
15 to internal A-rich stretches would be minimized and could result in a
purification
method with a higher stringency than currently employed. This matrix, for
example, could provide a better selection between eukaryotic mRNA and
contaminating mycoplasmic RNA (which is A-T rich). Since mycoplasms often
contaminate tissue culture cell lines, co-purification of mycoplasma RNA with
20 eukaryotic mRNA on oligo d(T) column can produce cDNA libraires
contaminated with mycoplasma clones.
Often, the sequence of a particular RNA must be interogated.
Reverse transcriptase (RT), in combination with PCR, can be used to amplify a
given region on an RNA template. The use of oligo d{T)~Z as primer in the RT
25 reaction would ensure that the 3' end of the mRNA is represented on the
cDNA
template. Thus, the present invention can also be incorporated into current 3'
RACE (Rapid Amplification of cDNA Ends) protocols, designed to obtain the 3'
end of a given clone.
In some cloning protocols, first strand synthesis is followed
30 by homopolymeric tailing of the products utilizing terminal
deoxynucleotidyl
CA 02346146 2001-04-04
WO 00/20630 PCT/CA99/00933
12
transferase. For example, dGTP can be utilized to add a homopolymeric stretch
of G's at the 5' end of the cDNA. Thus the DNA polymerase utilized in second
strand synthesis can take advantage of this G-stretch by priming from an oligo
d(C) primer annealed to the G-stretch positioned at the 5' end. This procedure
5 has the advantage of maintaining the sequence at the 5' terminal end of the
cDNA, and is also used in 5' RACE strategies to identify the 5' end of mRNAs
(Frohman -et al., 1988, Proc. Natl. Acad. Sci. USA 85:8998-9002; Loh et al.,
1989, Science 243:217-220) (Fig. 5). One drawback of this approach however
is that, since 5' untranslated regions of mRNAs are usually GC rich, the oiigo
10 d(C) can misprime from internal G-rich regions, producing less than full-
length
cDNAs. It is expected that the incorporation of universal nucleosides into
such
homopolymeric-stretch-containing primers to generate the modified oligos or
primers of the present invention will increase the specificity of binding and
generate cDNAs which terminate at the bona fide 5' end. Thus, the present
15 invention further relates to cloning procedures or RACE protocols involving
priming of second strand synthesis from a homopolymeric tail.
It may be desirable in some PCR protocols to utilize modified
oligonucleotides according to the present invention, wherein the modified
oligo
comprises a homopolymeric stretch containing at least one universal nucleoside
20 (or other non-specific duplex destabilizing modifications), to achieve
increased
discrimination between a target site (or several target sites) of interest
when
generating a specific product or a set of products (for example use of an
oligo
d(T) primer to prime DNA synthesis from the A-rich stretch of Alu repeats in
humans). Since these products can be developed to be used as genetic markers
25 (by identifying polymorphisms residing with the sequence of the product),
changing the specificity of targeting by altering the specificity of the oligo
d(T)
primer, could result in a more consistent representation of the final PCR
products. The present invention thus further relates to the use of universal
oligonucleotides or other modified oligonucleotides, during PCR amplification.
30
CA 02346146 2001-04-04
WO 00/20630 PCT/CA99/00933
13
DEFINITIONS
Nucleotide sequences are presented herein by single strand,
in the 5' to 3' direction, from left to right, using the one letter nucleotide
symbols
as commonly used in the art and in accordance with the recommendations of
5 the IUPAC-IUB Biochemical Nomenclature Commission.
Unless defined otherwise, the scientific and technological
terms and nomenclature used herein have the same meaning as commonly
understood by a person of ordinary skill to which this invention pertains.
Generally, the procedures for cell cultures, infection, molecular biology
methods
10 and the like are common methods used in the art. Such standard techniques
can be found in reference manuals such as for example Sambrook et al. (1989,
Molecular Cloning - A Laboratory Manual, Cold Spring Harbor Laboratories) and
Ausubel et al. (1994, Current Protocols in Molecular Biology, Wiley, New
York).
The present description refers to a number of routinely used
15 recombinant DNA (rDNA) technology terms. Nevertheless, definitions of
selected examples of such rDNA terms are provided for clarity and consistency.
For certainty, it is emphasized that the present invention finds utility with
nucleic
acids in general. Non-limiting examples of nucleic acids which can be used in
accordance with the teachings of the present invention include that from
20 eukaryotic cells such as that of animal cells, plant cells, or
microorganisms as
well as that from prokaryotic cells.
As used herein, the term "homopoiymeric sequences" refers
to a sequence composed of a single type of nucleotide base (adenosine A;
cytosine C; guanine G; thymine T; uracil U) or of a less common base (non-
25 limiting examples including inosine, I; and pseudouridine, ~).
As used herein, "nucleic acid molecule", "nucleic acid
sequence" or "sequence" refer to a polymer of nucleotides. Non-limiting
examples thereof include DNA (e.g. genomic DNA, cDNA) and RNA molecules
(e.g. mRNA). The nucleic acid molecule can be obtained by cloning techniques
CA 02346146 2001-04-04
WO 00/20630 PCT/CA99/00933
14
or synthesized. DNA can be double-stranded or single-stranded (coding strand
or non-coding strand [antisense]).
The term "recombinant DNA" as known in the art refers to a
DNA molecule resulting from the joining of DNA segments. This is often
referred
5 to as genetic engineering.
The terminology "amplification pair" refers herein to a pair of
oligonucleotides (oligos) of the present invention, which are selected to be
used
together in amplifying a selected nucleic acid sequence by one of a number of
types of amplification processes, preferably a polymerase chain reaction.
Other
10 types of amplification processes include ligase chain reaction, strand
displacement amplification, or nucleic acid sequence-based amplification, as
expiained in greater detail below. As commonly known in the art, the
oligonucleotides are designed to bind to a complementary sequence under
selected conditions.
15 The nucleic acid (e.g. DNA or RNA) for practicing the present
invention may be obtained according to well known methods.
"Oligonucleotides" or "oligos" or "primers" define a nucleic
acid molecule composed of nucleotides (ribo or deoxyribonucleotides).
Oligonucleotide probes or primers of the present invention may be of any
20 suitable length, depending on the particular assay format and the
particular
needs and targeted genomes employed. In general, the oligonucleotide probes
or primers are at least 10 nucleotides in length, preferably below 50
nucleotides.
Preferably, the oligos or primers have lengths between 15 and 40 nucleotides,
more preferably between 20 to 30 nucleotides. Of course, the probes or primers
25 of the present invention may be adapted to be especially suited to a chosen
nucleic acid amplification system. As commonly known in the art, the
oligonucleotide probes and primers can be designed by taking into
consideration
the melting point of hydrizidation thereof with its targeted sequence (see
below
and in Sambrook et al., 1989, Molecular Cloning - A Laboratory Manual, 2nd
30 Edition, CSH Laboratories; Ausubel et al., 1989, in Current Protocols in
CA 02346146 2001-04-04
WO 00/20630 PCT/CA99/00933
15
Molecular Biology, John Wiley & Sons Inc., N.Y.). The size of the
oligonucleotide will be dictated by the particular situation and ultimately on
the
particular use thereof and adapted accordingly by the person of ordinary
skill.
An oligonucleotide can be synthesized chemically or derived by cloning
5 according to well known methods. For example, the skilled artisan will be
able
to adapt the length of the essentially homopolymeric stretch-containing oligo
(the
targeting stretch wherein the homopolymeric stretch has been modified in
accordance with the teachings of the present invention), to particular needs,
as
a function of the targeted stretch and other parameters such as the sequence
10 of the duplex, the conditions of the assay (and hence of the Tm) and the
presence of additional sequences, flanking the essentially homopolymeric
stretch (at the 5' and/or 3' end thereof).
The term "oligonucleotide" or "DNA" molecule or sequence
refers to a molecule comprised of the deoxyribonucleotides adenine (A),
guanine
15 (G}, thymine (T) and/or cytosine (C), in a double-stranded form, and
comprises
or includes a "regulatory element" according to the present invention, as the
term is defined herein. The term "oligonucleotide" or "DNA" can be found in
linear DNA molecules or fragments, viruses, plasmids, vectors, chromosomes
or synthetically derived DNA. As used herein, particular double-stranded DNA
20 sequences may be described according to the normal convention of giving
only
the sequence in the 5' to 3' direction.
Probes and oligonucleotides of the invention can be utilized
with naturally occurring sugar-phosphate backbones as well as modified
backbones including phosphorothioates, dithionates, alkyl phosphonates and
25 a-nucleotides and the like. Modified sugar-phosphate backbones are
generally
taught by Miller, 1988, Ann. Reports Med. Chem. 23:295 and Moran et al., 1987,
Nucleic acid molecule. Acids Res., 14:5019. Probes of the invention can be
constructed of either ribonucleic acid (RNA) or deoxyribonucleic acid (DNA),
and
preferably of DNA. General teachings on the synthesis of oligonucleotides and
30 substituents and modifications thereof can be found for example in US
CA 02346146 2001-04-04
WO 00/20630 PCT/CA99/00933
16
5,438,131. The selection of the best suited synthesis pathway of an
oligonucleotide and of the appropriate modifications, and substituents to be
used, may be selected accordingly by the person of ordinary skill to which the
instant invention pertains.
5 The modified oligonucleotides of the present invention can be
synthesized chemically or produced through recombinant DNA technology. All
these methods are well known in the art. According to the present invention,
the
modified oligonucleotides are molecules comprising an essentially
homopolymeric stretch or sequence composed of a single type of nucleotide and
10 at least one type of modification which enables a destabilization of
mismatches.
In a preferred embodiment, these modified oligonucleotides are a molecule
composed of a single type of nucleotide (ribo- or deoxyribonucleotides, A, C,
G,
T or U) and containing at least one universal nucleoside. As mentioned above,
the length is between 10 and 50 nucleotides. Of course, it should be
recognized
15 that in a case where more than one modification or nucleotide which
destabilizes
mismatches is used, it need not be the same type of "modification". In some
embodiments of the present invention, the modified oligonucleotides of the
present invention comprise an essentially homopolymeric stretch and a "3'
lock"
(see below) or a sequence enabling the creation of a restriction site.
20 As used herein, a "primer" defines an oligonucleotide which
is capable of annealing to a target sequence, thereby creating a double
stranded
region or duplex which can serve as an initiation point for DNA synthesis
under
suitable conditions.
"Nucleic acid hybridization" refers generally to the
25 hybridization of two single-stranded nucleic acid molecules having
complementary base sequences, which under appropriate conditions will form
a thermodynamically favored double-stranded structure. Examples of
hybridization conditions can be found in the two laboratory manuals referred
above (Sambrook et al., 1989, supra and Ausubel et al., 1989, supra) and are
30 commonly known in the art. In the case of a hybridization to a
nitrocellulose
CA 02346146 2001-04-04
WO 00/20630 PCT/CA99/00933
17
filter, as for example in the well known Southern blotting procedure, a
nitrocellulose filter can be incubated overnight at 65°C with a labeled
probe in
a solution containing 50% formamide, high salt (5 x SSC or 5 x SSPE), 5 x
Denhardt's solution, 1% SDS, and 100 Ng/ml denatured carrier DNA (e.g.
5 salmon sperm DNA). The non-specifically binding probe can then be washed
off the filter by several washes in 0.2 x SSC/0.1 % SDS at a temperature which
is selected in view of the desired stringency: room temperature (low
stringency),
42°C (moderate stringency) or 65°C (high stringency). The
selected temperature
is based on the melting temperature (Tm) of the DNA hybrid. Of course, RNA-
10 DNA hybrids can also be formed and detected. In such cases, the conditions
of
hybridization and washing can be adapted according to well known methods by
the person of ordinary skill. Stringent conditions will be preferably used
(Sambrook et al.,1989, supra).
The types of detection methods in which probes can be used
15 include Southern blots (DNA detection), dot or slot blots (DNA, RNA), and
Northern blots (RNA detection).
Probes or oligonucleotides can be labeled according to
numerous well known methods (Sambrook et al., 1989, supra). Non-limiting
examples of labels include 3H, '4C, 32P, and 35S. Non-limiting examples of
20 detectable markers include ligands, fluorophores, chemiluminescent agents,
enzymes, and antibodies. Other detectable markers far use with probes, which
can enable an increase in sensitivity of the method of the invention, include
biotin and radionucleotides. It will become evident to the person of ordinary
skill
that the choice of a particular label dictates the manner in which it is bound
to
25 the probe.
As commonly known, radioactive nucleotides can be
incorporated into probes of the invention by several methods. Non-limiting
examples thereof include kinasing the 5' ends of the probes using gamma 32P
ATP and poiynucleotide kinase, using the Klenow fragment of Pol I of E. coli
or
30 reverse transcriptase in the presence of radioactive dNTP {e.g. uniformly
labeled
CA 02346146 2001-04-04
WO 00/20630 PCT/CA99/00933
18
DNA probe using random oligonucleotide primers in low-melt gels), using the
SP6IT7 system to transcribe a DNA segment in the presence of one or more
radioactive NTP, and the like.
Amplification of a selected, or target, nucleic acid sequence
5 may be carried out by a number of suitable methods. See generally Kwoh et
al.,
1990, Am. Biotechnol. Lab. _8:14-25. Numerous amplification techniques have
been described and can be readily adapted to suit particular needs of a person
of ordinary skill. Non-limiting examples of amplification techniques include
polymerise chain reaction (PCR), ligase chain reaction (LCR), strand
10 displacement amplification (SDA), transcription-based amplification, the
Q~i
replicase system and NASBA (Kwoh et al., 1989, Proc. Natl. Acid. Sci. USA $~,
1173-1177; Lizardi et al., 1988, BioTechnology 6:1197-1202; Malek et al.,
1994,
Methods Mol. Biol., 28:253-260; and Sambrook et al., 1989, supra). Preferably,
amplification will be carried out using PCR.
15 Polymerise chain reaction (PCR) is carried out in accordance
with known techniques. See, e.g., U.S. Pat. Nos. 4,683,195; 4,683,202;
4,800,159; and 4,965,188 (the disclosures of ali three U.S. Patent are
incorporated herein by reference). In. general, PCR involves, a treatment of a
nucleic acid sample (e.g., in the presence of a heat stable DNA polymerise)
20 under hybridizing conditions, with one oligonucleotide primer for each
strand of
the specific sequence to be detected. An extension product of each primer
which
is synthesized is complementary to each of the two nucleic acid strands, with
the
primers sufficiently complementary to each strand of the specific sequence to
hybridize therewith. The extension product synthesized from each primer can
25 also serve as a template for further synthesis of extension products using
the
same primers. Following a sufficient number of rounds of synthesis of
extension
products, the sample is analysed to assess whether the sequence or sequences
to be detected are present. Detection of the amplified sequence may be carried
out by visualization following EtBr staining of the DNA following gel
30 electrophores, or using a detectable label in accordance with known
techniques,
CA 02346146 2001-04-04
WO 00/20630 PCT/CA99/00933
19
and the like. For a review on PCR techniques (see PCR Protocols, A Guide to
Methods and Amplifications, Michael et al. Eds, Acad. Press, 1990).
Ligase chain reaction (LCR) is carried out in accordance with
known techniques (Weiss, 1991, Science 254:1292). Adaptation of the protocol
to meet the desired needs can be carried out by a person of ordinary skill.
Strand displacement amplification (SDA) is also carried out in accordance with
known techniques or adaptations thereof to meet the particular needs (Walker
et al., 1992, Proc. Natl. Acad. Sci. USA 89:392-396; and ibid., 1992, Nucleic
Acids Res. 20:1691-1696).
As used herein, the term "gene" is well known in the art and
relates to a nucleic acid sequence defining a single protein or polypeptide. A
"structural gene" defines a DNA sequence which is transcribed into RNA and
translated into a protein having a specific amino acid sequence thereby giving
rise to a specific polypeptide or protein. It will be readily recognized by
the
person of ordinary skill, that the nucleic acid sequence of the present
invention
can be incorporated into anyone of numerous established kit formats which are
well known in the art.
The term "vector" is commonly known in the art and defines
a plasmid DNA, phage DNA, viral DNA and the like, which can serve as a DNA
vehicle into which DNA of the present invention can be cloned. Numerous types
of vectors exist and are well known in the art.
The term "allele" defines an alternative form of a gene which
occupies a given locus on a chromosome.
As commonly known, a "mutation" is a detectable change in
25 the genetic material which can be transmitted to a daughter cell. As well
known,
a mutation can be, for example, a detectable change in one or more
deoxyribonucleotide. For example, nucleotides can be added, deleted,
substituted for, inverted, or transposed to a new position. Spontaneous
mutations and experimentally induced mutations exist. The result of a
mutations
CA 02346146 2001-04-04
WO 00/20630 PCT/CA99/00933
20
of nucleic acid molecule is a mutant nucleic acid molecule. A mutant
polypeptide
can be encoded from this mutant nucleic acid molecule.
As used herein, the term "purified" refers to a molecule
having been separated from a cellular component. Thus, for example, a
"purified
5 protein" has been purified to a level not found in nature. A "substantially
pure"
molecule is a molecule that is lacking in most other cellular components.
The present invention also relates to a kit comprising the
oligonucleotide primers of the present invention. For example, a
compartmentalized kit in accordance with the present invention includes any
kit
10 in which reagents are contained in separate containers. Such containers
include
small glass containers, plastic containers or strips of plastic or paper. Such
containers allow the efficient transfer of reagents from one compartment to
another compartment such that the samples and reagents are not
cross-contaminated and the agents or solutions of each container can be added
15 in a quantitative fashion from one compartment to another. Such containers
will
include a container which will accept the test sample (DNA, RNA or cells), a
container which contains the primers used in the assay, containers which
contain enzymes, containers which contain wash reagents, and containers
which contain the reagents used to detect or isolate the extension products.
20 Of course, cDNA cloning kits could be adapted by inserting
thereinto the primers of the present invention.
BRIEF DESCRIPTION OF THE DRAWINGS
Having thus generally described the invention, reference will
25 now be made to the accompanying drawings, showing by way of illustration a
preferred embodiment thereof, and in which:
Figure 1 (PRIOR ART) shows an example of the steps
involved in generating cDNA libraries from mRNA. Although a number of
strategies can be used for cDNA library generation, of which only one is
shown,
30 all libraries require as a first step, a primer from which the reverse
transcriptase
CA 02346146 2001-04-04
WO 00/20630 PCT/CA99/00933
21
(RT) can prime. In the case of full-length cDNA libraries, an oligo d(T)
primer is
used because it anneals to the 3' poly (A) tail of the eukaryotic mRNAs. In
the
case of prokaryotic, some viral, or other eukaryotic mRNAs which lack a poly
(A) tail, a homopolymeric stretch of nucleoside 5'-monophosphates can be
5 added to the 3' end of the mRNA. For example, poly (A) polymerase can be
used to add a poly (A) tail to mRNAs which lack one. An oligonucleotide which
contains complementary nucleotides (e.g. oligo d(T)) is then annealed to the
mRNA and serves as primer for the RT.
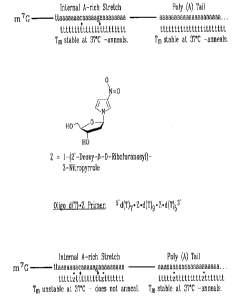
Figure 2A shows a hybridization of oligo d(T},5 primer to the
bona fide poly (A) tail of an mRNA (right) or to an internal A-rich stretch
(left)
within the mRNA by conventional oligo d(T) primer used in current cDNA library
construction. Although the length of the primer used can differ, and the two
3'
most nucleotides are sometimes (A,C,G,T) and (A,G,C) to "lock" the
oligonucleotide in place at the junction of the body of the mRNA and the poly
(A)
15 tail, neither of these modifications prevent the misannealing of the oligo
d(T)
primer to internal A-rich stretches. The asterisks denotes mispairing
resulting in
destabilization of the duplex. Figure 2B shows the chemical structure of 3-
nitropyrrole. Figure 2C shows the structure of oligo d(T)~Z primer. Figure 2D
shows the expected discrimination between the poly (A) tail (right) and
internal
20 A-rich stretches (left) when hybridizing to oligo d(T)~Z. The asterisks
denote
mispairing resulting in destabilization of the duplex and circles represent 3-
nitropyrrole artificial mismatches.
Figure 3A shows the structure ofthe eIF-4611 cDNA construct
used to analyze mispriming at the 3' end. The location of four internal A-rich
25 sequences are shown - all of which generated 3' truncated clones when eI F-
4611
was isolated from a cDNA library. The plasmid was linearized with Asp 718 and
T7 RNA polymerase used to generate a 2400 nt 3H-test transcript. Figure 3B
shows the integrity of the in vitro generated transcript following
fractionation on
a formaldehyde 1.2% agarose gel, treatment with EN3HANCE, and
30 autoradiography of the dried gel. Figure 3C shows the alkaline agarose
analysis
CA 02346146 2001-04-04
WO 00/20630 PCT/CA99100933
22
of RT products generated by priming synthesis with oligo d(T) (lane 1 ) or
oligo
d(T)~Z (lane 2) using MMLV RT. Complementary DNA was labeled with a 3zP-
dCTP. The position of migration of truncated products are indicated by a
filled
circle and full length product by an arrow. These results directly demonstrate
5 correction of 3' mispriming by utilizing oligo d(T)~Z as primer during first
strand
synthesis.
Figure 4A shows the structure of eIF-4611 construct used to
demonstrate mispriming at the 3' end. The location of five oligonucleotides
(a,
b, c, d, e) used in the hybridization assay to map the sites of 3' mispriming
by
10 oligo d(T) are shown. The nucleotide targets of the oligonucleotides on eIF-
4611
are:
Oligo a, 5~'GAAATTGACTCAGTACTATT55g';
Oligo b, s4,sGAAGGAAATGCTGTGGACCss3s.
Oligo c, 5'~'TGTATAATAGAAAAGCAGAG5z,3;
15 Oligo d, lose-~TTTAAACAAGGACTCATACS°$'; and
Oligo e, 4'8'AAGAGGAGTCTGAGGATAAC°8°o
Figure 4B shows the Southern blot of the alkaline agarose gel
of RT products generated by priming synthesis with either oiigo d(T) or oligo
d(T)~Z. Marker lane refers to the 1 kb size ladder from GIBCO and sizes (in
bp)
20 are indicated to the left of the diagram. eIF-4611 DNA refers to a DNA
fragment
of eIF-4611 used as a positive control for DNA hybridizations.
Oligonucleotides
used as probes on each blot are indicated below each panel. The asterisks on
the left denotes the cDNA product obtained by priming at the correct poly (A)
site. The filled circle denotes the cDNA product obtained by priming from the
A-
25 rich stretch between nucleotides 5550-5575, whereas the arrow denoted the
cDNA obtained by priming from nucleotides 5085-5120.
Figure 5 shows an example illustrating mispriming events at
the 5' end of cDNAs during cDNA library construction of 5' RACE analysis to
extend the sequence of known genes.
CA 02346146 2001-04-04
WO 00/20630 PCT/CA99/00933
23
Figure 6 shows an autoradiograph following oligo d(T) primed
first strand synthesis on eIF-4611 mRNA. Lane 2 oligo d(T) control; lane 3
oligo
d(T)~Z; and lane 4 oligo d(T)~I. A molecular mass standard ladder is shown in
lane 1.
Other objects, advantages and features of the present
invention will become more apparent upon reading of the following
non-restrictive description of preferred embodiments with reference to the
accompanying drawing which is exemplary and should not be interpreted as
limiting the scope of the present invention.
DESCRIPTION OF THE PREFERRED EMBODIMENT
The demonstration of the destabilization effect of non-specific
or artifactual duplex formation and of its concurrent effect on mismatch
and/or
mispriming events was carried out with an oligo d(T) primer, modified with one
15 or two universal analogues. Whether such an introduction could result in
increased discrimination between the perfectly matched target of that primer
(i.e.
the 3' poly (A) tail of the mRNA) and an imperfect matched sequenced (internal
A-rich stretches) was analyzed.
More specifically, an ofigo d(T) primer, called oligo d(T)~Z
20 was generated, in which two of the thymine bases were substituted by 3
nitropyrrole (Fig. 2C). General teachings on 3-nitropyrrole, the synthesis
thereof
and the like can be found for example in U.S. 5,438,131.
To test whether this primer can reduce mispriming from
internal A-rich sequences (Fig. 2D), a cDNA clone from eIF-4611, a eukaryotic
25 translation factor, was obtained. When cDNA clones to this gene were
initially
isolated, only one of 5 clones had the correct 3' end. Sequence
characterization
of these clones demonstrated that all the truncated clones were the result of
internal priming by oligo d(T) at four different sites (denoted as leftward
arrows
in Fig. 3A). In vitro transcribed RNA generated from this clone thus serves as
an
30 excellent test reagent to determine the ability of the 3-nitropyrrofe
substituted
CA 02346146 2001-04-04
WO 00/20630 PCT/CA99/00933
24
oligo d(T) to decrease the number of mispriming events. The quality of the in
vitro transcribed RNA is shown in Fig. 3B and demonstrates that the test
template is intact. This RNA was then annealed to oligo d(T) or oligo d(T)~Z,
and reverse transcription performed with MMLV RT. As shown in Fig. 3C, use
5 of oligo d(T) on this template resulted in shorter than full-length products
(>95%}
generated as a result of internal priming (Fig. 3C, lane 1 ). However, use of
oligo
d(T)~Z as primer on the same template resulted in the majority (>95%) of
products being full-length (Fig. 3C, lane 2).
These results demonstrate that use of oligo d(T}~Z in reverse
10 transcription reactions significantly improves the specificity for the 3'
poly (A) tail
and demonstrates the usefulness of this procedure in destabilizing non-
specific
duplex formation and more particularly for generating full length cDNAs.
The sites of mispriming with oligo d(T) on the control eIF-4611
template were identified (Fig. 4). This was done by fractionating the products
of
15 RT reactions performed with either oligo d(T) or oligo d(T}~Z on an
alkaline
agarose gel followed by transfer to a nylon membrane. This membrane was then
probed, by hybridization, with oligonucleotides designed to target various
regions of the 3' untranslated region of eIF-4611 (oligonucleotides are
labelled
a, b, c, d and a in Fig. 4A). As shown in Fig. 4B, hybridization with
20 oligonucleotide "a" detected correctly primed cDNA when both oligo d{T) and
oligo d(T}~Z were used as primer. Hybridization with oligonucleotides b and c,
detected a novel truncated product when the RT reaction was primed with oligo
d(T), indicating mispriming from an internal A-rich stretch with this primer
(Fig.
4B). Hybridization with oligonucleotides d and e, detected an additional
novel,
25 more abundant truncated product (denoted by arrowheads in Fig. 4B) when the
RT reaction was primed with oligo d(T), indicating mispriming from a second
internal A-rich stretch with this primer but not with oligo d(T)~Z (Fig. 4B).
Mispriming event are common in Rapid Amplification of cDNA
ends (RACE). An example of mispriming at the 5' end of cDNAs during 5' RACE
30 analysis is shown in Figure 5. Such mispriming events could be resolved by
CA 02346146 2001-04-04
WO 00/20630 PCT/CA99/00933
25
incorporating a universal nucleoside into the oligo d(C) primer to increase
the
discrimination between the homologous target (e.g. - the 5' end G tail) and an
internal G-rich sequence. It is expected that incorporation of at least one
universal base (e.g. 3-nitropyrrole) in the homopolymeric oligo d(C) primer
5 should significantly reduce such mispriming.
The present invention is illustrated in further detail by the
following non-limiting example.
EXAMPLE 1
10 Destabilization of mispriming and reduction
of mispriming using an oligo d(T)~I primer
To demonstrate that other "modifications" of nucleotides that
destabilize hydrogen bonding between mismatched sequences could be used
in accordance with the present invention, the oligo d(T) primer was modified
by
15 inserting thereinto deoxynucleotide deoxyinosine (I).
An oligonucleotide (called oligo d(T)~I] having the sequence
5'TTTTTTTI*TTTTTTTTTI*TTTTT3' was thus synthesized (McGill University
Sheldon Biotechnology Center), where I* represents the position where the
2'deoxyinosine was incorporated into the oligonucleotide. Reverse
transcription
20 reactions were performed on in vitro generated eIF-4G mRNA templates (1 Ng)
with Superscript IIT"" (LifeTechnologies) under conditions recommended by
LifeTechnologies. Oligonucleotide primers that were utilized to prime the
first
strand synthesis were 0.1 Ng of either Oligo d(T),5, oligo d(T)~Z, or oligo
d(T)~I.
The radioisotope a 32P-dCTP (New England Nuclear) was used as a tracer to
25 monitor the quality of the cDNA product. Following the generation of cDNA
products at 42°C for 1 hr, the mixture was extracted with
phenol/chloroform,
back extracted with an equal volume of water, passed through a G50T"" spun
column, and precipitated with 2M ammonium acetate and 2.5 volumes of
ethanol. The precipitate was washed with 70% ethanol, dried and resuspended
30 in 20 Nl of water. An aliquot (5 ul) was loaded onto a 1.2% alkaline
agarose gel
CA 02346146 2001-04-04
WO 00/20630 PCT/CA99/00933
26
and electrophoresis performed at 78 volts for 6.5 hours. The gel was
neutralized
in 7% trichloroacetic acid for 30 minutes, dried, and exposed to X-GMAT X-ray
film (Kodak) at -70°C for 10 hrs with an intensifying screen.
An photograph of the autoradiograph is presented in Figure
5 6. A molecular mass standard ladder is shown in lane 1 (purchased from
LifeTechnologies). The cDNA product obtained by priming with oligo d(T),5 is
shown in lane 2. Clearly, the major cDNA product is shorter than full-length
and
arises due to internal mispriming at an internal A-rich site. As shown
previously,
priming with oligo d(T)~Z is able to correct the mispriming phenomenon, and in
10 this particular experiment over 50% of the cDNA is correctly primed from
the
poly (A) tail of the mRNA (lane 3, full-length product indicated with an
arrow).
Priming the cDNA reaction with oligo d(T)~I also efficiently corrected the
mispriming reaction observed with oligo d(T),5 primer and resulted in a
significant proportion of cDNAs being full-length (lane 4, full length product
15 indicated with an arrow).
Although the present invention has been described
hereinabove by way of preferred embodiments thereof, it can be modified,
without departing from the spirit and nature of the subject invention as
defined
in the appended claims.