Note: Descriptions are shown in the official language in which they were submitted.
CA 02348540 2001-04-27
WO 00/25822 PCT/US98/23014
-1-
TRANSDERMAL DELIVERY OF MEDICATIONS USING
AACCOMBINATION OFCPENETRATION ENHANCERS
BACKGROUND OF THE INVENTION
Field of the Invention:
The invention herein relates to the transdermal delivery of medications to
a patient. More particularly it relates to compositions which allow medication
molecules to be solubilized and delivered transdermally and to methods for
formation of such compositions and for their therapeutic use.
(For convenience herein the terms "drug" and "medication" may be used
interchangeably. We wish to emphasize, however, that this invention is
applicable to the delivery of any type of compound or molecular species which
is intended to be administered to a patient transdermally for a therapeutic or
physiological purpose. Whether the material happens to meet a particular
specific definition of a "drug" or "medication" or other applicable term is
not
critical for the purposes of this invention, and the invention should not be
limited
by the particular term applied to the material being administered.)
Description of the Prior Art:
In the past the delivery of medications transdermally to a patient has been
limited to administration by transcutaneous injection or by transdermal
migration
from a patch placed on the outer surface of the patient's skin. The
deficiencies
of administration by injection are obvious. With only a few exceptions
injections
must be administered by trained and qualified medical personnel. The injection
itself causes a break in the skin which can lead to infection, despite
precautions;
an injection needle may itself be contaminated causing infection to the
patient;
and, course, it is a simple fact that injections are uncomfortable to almost
all
patients. Further, an injection is normally not "location specific." Rather
the
injection is made at a location on the body remote from the affected area, and
the injected medication must be transported through the body to that location.
CA 02348540 2001-04-27
WO 00/25822 PCT/US98/23014
-2-
This results in losses in transport, so that to administer an effective amount
of
medication to the affected area, and excess of medication must be injected.
In view of these deficiencies of injection administration, significant effort
has been spent in the last few years in seeking alternative methods of
transdermal administration of medications. It has been necessary to meet two
requirements. First, the method must provide for extended containment of the
drug and any carrier while in place on the patient's skin (in effect analogous
to
containment of the medication and carrier in the reservoir vial of the
injection
syringe), in a form that does not lend itself either to contamination of the
medication and carrier or to loss of the medication and carrier. Second, the
systems employed must provide for a regulated and predictable rate of transfer
of the medication (with or without the carrier) from the containment device
into
and through at least some layers of skin to where the medication will be
dispersed throughout the affected area of the body.
The only workable prior art embodiment of such a device has been what
is commonly known as a "patch." A patch is generally a flat hollow device with
a permeable membrane on one side and also some form of adhesive to maintain
the patch in place on the patient's skin, with the membrane in contact with
the
skin so that the medication can permeate out of the patch reservoir and into
and
through the skin. The outer side the patch is formed of an impermeable layer
of
material, and the membrane side and the outer side are joined around the
perimeter of the patch, forming a reservoir for the medication and carrier
between the two layers.
Numerous kinds of medications have been administered through the use
of a patch, notably scopolamine for preventing motion sickness, nicotine
derivatives intended to discourage an addicted smoker from continuing the
smoking habit and estrogen hormones.
Patches have their own set of disadvantages. A principal disadvantage
is that, not withstanding the presence of a penetration enhancer, the delivery
of
CA 02348540 2001-04-27
WO 00/25822 PCT/US98/23014
-3-
the medication is necessarily limited by the rate of passage of the medication
through the patch membrane to the skin. Since the medication is not in contact
with the skin while it is enclosed in the patch, whatever length of time is
required
for the medication to permeate through the skin itself to become effective is
necessarily lengthened by the time needed for the medication first to exit
from
the patch through the membrane. In many cases the membrane permeation rate
is the significant rate limiting step of speed of effectiveness of a
particular
medication, and can render patch administration essentially ineffective
because
the medication cannot reach the patient's system rapidly enough to be
efficacious. In addition, the adhesive which is intended to secure the patch
to
the patient's skin can fail, so that the patch disengages from the skin before
completion of the transfer of the medication, resulting in loss of that
quantity of
medication which remains within the patch's reservoir.
Various methods have been used to increase skin permeation of
medications, including penetration enhancers, pro drugs, superfluous vehicles,
iontophoresis, phonophoresis and thermophoresis. For the purposes of this
invention, only the penetration enhancers are relevant. Ideal enhancers have
no irritancy and toxicity to the skin, and the whole body, together with
having
high enhancing effects. Enhancers themselves should be phisiochemically
stable and not have pharmacologic effects, and preferably should not have
smell, color, or taste. A typical example of an enhancer is disclosed in U.S.
Patent No. 4,783,450 (to Fawzi et al.) in which lecithin is used for
penetration
enhancement.
The stratum corneum provides the principal barrier to the percutaneous
penetration of topically applied substances. It is the most superficial
cutaneous
layer and is a horny. layer that consists of flat, scalelike "squames" made up
of
the fibrous protein keratin. The squames are continually being replaced from
below by epidermal cells that die in the process of manufacturing keratin. It
is
unlikely that the emulsified fat on the skin surface greatly affects
permeability.
CA 02348540 2001-04-27
WO 00/25822 PCT/US98/23014
-4-
However, vehicles can control, to a great extent, the rate of penetration of
drugs
that are applied to the skin. The intercellular lipids may be important for
the
permeability barrier in skin.
It is known that some combinations of enhancers and vehicles act
synergistically, such as the combination of ethanol as a vehicle for the
enhancer
laurocapram. However, many combinations are not synergistic; for instance, n-
decylmethylsulfoxide lowers the zeta potential of the skin, and thus
enhancement due to conduction flow (iontophoresis) is minimized. in the past,
synergism of combinations could not be predicted.
Further, one must differentiate between penetration enhancer which act
to improve the ability of the medication to pass through a patch membrane to
reach the skin, and those which act to enhance the separation of the
medication
from its carrier matrix or to enhance the diffusion of the medication into and
through the skin.
However, notwithstanding the various deficiencies mentioned,
administration by injection or by patch remain only by viable transdermal
administration techniques known to the prior art.
SUMMARY OF THE INVENTION
We have now developed a system that provides for a convenient,
efficacious and simple system for transdermal administration of medications in
which the medication is present in a composition for direct application to the
skin,
commonly in the form of a cream or similar material. The transdermal
administration of the drug is therefore not hindered by having to penetrate a
patch membrane, since the cream and its medication content are directly in
contact with the skin and the medication needs only to separate from the cream
in order to be available for transdermal migration. In addition, since the
composition is in the form of a cream or other viscous moldable and spreadable
material, the drug may be effectively administered by application of the cream
CA 02348540 2007-08-17
73529-294
-5-
to many bodily areas where a patch either will not fit or cannot be shaped to
conform to the skin contours.
(As with the use of the terms "medication" and "drug," our invention is not
to be limited by the term used to describe the physical properties of the
composition herein. We will for convenience use the term "cream," but other
terms such as "gel," "lotion," "paste" and the like also could be applicable.
As
will be seen from the description below, the physical nature of the
composition
containing the medication and to be applied to the patient's skin will be
defined
by functional parameters, rather than being limited by an arbitrary
descriptive
term.)
A key element in the success of the present invention is our discovery that
the use of at least two separate penetration enhancers of defined function
results
in a synergism which provides rapid but controllable separation of the
medication
from the cream and its penetration into and within or through the skin. At
least
one of the penetration enhancers acts to facilitate the separation of drug
from the
carrier within the cream and at least a second penetration enhancer alters the
structure of the outer layers of skin, particularly the stratum corneum, such
that
migration of the drug through the stratum corneum is enhanced and expedited.
The medication is thus taken up-by the patient's system and is efficacious
much
more rapidly than would be the case for administration of the medication by
means of the prior art patch system. Further, although permeation of the skin
does not provide for as rapid administration by the medication as would result
from direct injection, the use of the present invention avoids the problems
associated with injection administration.
CA 02348540 2010-04-23
53869-1
6 -
Therefore, in one principal embodiment, the
invention provides a composition for diffusional transdermal
delivery of medication to a patient, wherein said
composition comprises: a. a medication capable of being
administered transdermally, wherein said medication is
selected from non-steroidal antiinflammatory agents; b. a
solvent for said medication; c. a first penetration enhancer
which improves diffusion of said medication into and within
said patient's skin; and d. a second penetration enhancer
which improves diffusion of said medication out of said
composition for transdermal migration; said composition (A)
having a viscosity in a range such that it may be applied
topically and conform to and adhere to said patient's skin
for a period of time sufficient for a significant portion of
said medication to be delivered transdermally to said
patient, and (B) being the product of a process comprising
the steps of: (i) providing a medication/organogel mixture
by mixing (a) a substantially uniform organogel that
comprises said first penetration enhancer with (b) a
composition comprised of said medication solubilised in said
solvent, wherein said organogel is a fatty acid phospholipid
emulsifying agent and a fatty acid or ester thereof; (ii)
providing a composition comprised of said second penetration
enhancer and a carrier therefor; and (iii) mixing said
medication/organogel mixture with the composition from step
(ii), producing said composition for diffusional transdermal
delivery.
In another principal embodiment, the invention
provides a method for the preparation of a therapeutic
composition to be transdermally administered, wherein said
method comprises the following steps: a. solubilizing a
medication capable of being administered transdermally,
wherein said medication is selected from non-steroidal
CA 02348540 2010-04-23
53869-1
- 6a -
antiinflammatory agents; b. forming an organogel comprising
a first penetration enhancer which comprises a fatty acid
phospholipid emulsifying agent and a fatty acid or ester
thereof and which improves diffusion of said medication into
and within said patient's skin, and a solvent for said
solubilized medication; c. forming a polymeric component
comprising a second penetration enhancer which improves
diffusion of said medication out of said composition for
transdermal migration; and d. blending said solubilized
medication, organogel and polymeric component to form said
composition having a viscosity in a range such that when
applied topically, it will conform to and adhere to said
patient's skin for a period of time sufficient for a
significant portion of said medication to be delivered
transdermally to said patient.
In yet another principal embodiment, the invention
is of a method for the transdermal administration of a
medication which comprises solubilizing a medication capable
of being administered transdermally; forming an organogel
comprising a first penetration enhancer which improves
diffusion of the medication into and within the patient's
skin, and a carrier for the solubilized medication; forming
a polymeric component comprising a second penetration
enhancer which improves diffusion of the medication out of
the composition for transdermal migration; blending the
solubilized medication, organogel and polymeric component to
form the composition having a viscosity in a range such that
it may be applied topically and conform to and adhere to the
patient's skin for a period of time sufficient for a
significant portion of the medication to be delivered
transdermally to the patient; and applying the composition
to the skin
CA 02348540 2001-04-27
WO 00/25822 PCT/US98/23014
-7-
of a patient for the period of time and allowing the medication to diffuse out
of the
composition and through the skin, such that the medication is taken up by the
body of the patient and acts therapeutically on the patient.
In preferred embodiments the first penetration enhancer is a lecithin
organogel formed with isopropyl palmitate or isopropyl myristate, and the
second
penetration enhancer is a polyoxymer, preferably a polyoxyalkylene derivative
of propylene glycol. A wide variety of medications can be delivered by this
invention. Further, while the invention herein is described in terms of the
minimum number of synergistically acting penetration enhancers (i.e., two), it
will
be understood that additional penetration enhancers can also be present. Thus
there may be more than one enhancer which operates with a specific
mechanism, or there may be additional enhancers which provide yet other
modes of operation, or both.
The methods and compositions described herein provide a unique and
highly effective technique for administering medication directly to an
affected
area of the body with the minimum amount of medication and with the avoidance
of unwanted side effects. Unlike administration by injection or orally, the
transdermal administration herein is site specific; the cream is applied to
the skin
directly at the affected area of the body. There are therefore no losses of
medication during transport from a remote application site. Similarly, the
long
delays in having an effective quantity of the medication reach the affected
area
of the body, which are inherent in injection and oral administration, are
entirely
eliminated in the present invention.
The present method also avoids unwanted side effects. For instance, in
oral administration of a medication, the medication itself can adversely
affect the
gastrointestinal tract as it is swallowed and dissolved for assimilation into
the
circulatory system. Those skilled in the art are well familiar with the common
caution required for many oral medications that they must be administered only
in conjunction with a meal, or, conversely, that they cannot be administered
in
CA 02348540 2001-04-27
WO 00/25822 PCT/US98/23014
-8-
the presence of specific types of food products, such as dairy products. These
cautions are necessary since the orally administered medication's efficacy
will
be adversely affected by certain foods, or the person's gastrointestinal tract
will
be irritated by the medication if the latter is not diluted by the presence of
food
in the gastrointestinal tract. Such considerations are, of course, entirely
absent
in the present invention, where the same medications can be easily and
conveniently administered transdermally without incurring such side effects.
Further, the transdermal administration avoids the "first pass effect,"
which often results when a medication is administered orally and thus has to
pass through various organs, including the liver, before reaching the affected
area of the body. These organs can absorb or chemically alter significant
quantities of the passing medication, thus requiring that large excess
quantities
of the medication by administered initially to insure that an effective
quantity of
the medication will ultimately reach the affected area of the body. Since in
this
method the medication commonly passes through the skin directly to the
affected site, there is no problem of loss in intermediate organs, and
therefore
excessive quantities of medication do not need to be delivered to counter such
losses. (As an example, ketoprofen is commonly administered orally in
quantities of about 50-75 mg per dose for the desired efficacy. In the present
invention, however, an equally effective dose of ketoprofen can be delivered
by
topical transdermal administration of only 3 mg.)
Finally, since the present invention is site specific, the depth of delivery
of the medication can be readily controlled, as contrasted to injection
delivery.
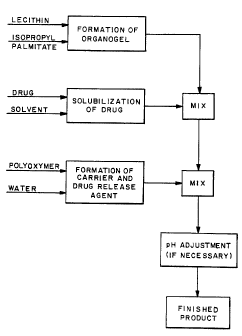
BRIEF SUMMARY OF THE DRAWING
The single Figure of the drawing is a flow chart illustrating schematically
formulation of a preferred embodiment of a composition of this invention.
CA 02348540 2001-04-27
WO 00/25822 PCT/US98/23014
-9-
DETAILED DESCRIPTION AND PREFERRED EMBODIMENTS
The unique compositions of the present invention require a specific
sequence of steps in their formation if a therapeutically effective and
pharmaceutically compatible composition is to be obtained. This is best
understood by reference to the Figure of the drawing.
The basic composition of this invention is a mixture of an organogel, a
solubilized medication or drug and a.carrier combined with a drug release
agent.
Penetration enhancement is provided by the organogel and by the release
agent.
In the exemplary process as illustrated in the Figure, an organogel is
formed, in this example from lecithin and isopropyl palmitate. These two
materials are thoroughly blended and mixed until a substantially uniform gel
structure forms. The organogel, which is the base for the cream composition,
should be formed at the time that the composition is to be formulated. The
drug
or medication is solubilized with a solvent, such as water, alcohol or other
appropriate solvent, again by mixing in a known manner. When it is desired to
start formation of the actual composition, the solubilized drug is mixed
thoroughly
into the organogel matrix, again by conventional mixing techniques. The
technique used will of course be such that the organogel's structure is not
broken down. Finally, a carrier, such as water or alcohol, and a drug release
agent, such as a polyoxymer, are blended. The carrier/release agent mixture
can be made up in large lots and stored under refrigerator until needed, at
which
time an appropriate quantity can be taken for and the remainder retained in
refrigerated storage. The carrier/release agent mixture is then mixed with the
drug/organogel mixture to produce the final "cream" composition. Details will
be
provided below.
Considering first the organogel, the blend of the two components will be
in the range of from about 25% to 75% of the lecithin component, the remainder
being the fatty acid ester component. (Unless stated otherwise, all
percentages,
CA 02348540 2001-04-27
WO 00/25822 PCT/US98/23014
-10-
parts and concentrations are by weight.) The "lecithin component" may be
lecithin, any comparable fatty acid phospholipid emulsifying agent, such as
fatty
acids and their esters, cholesterol, tri-glycerides, gelatin, acacia, soybean
oil,
rapeseed oil, cottonseed oil, waxes or egg yolk, or any other material which
acts
in the same manner as lecithin.
The other component is an organic solvent/emollient, particularly including
fatty acid esters, of which the esters of the saturated alkyl acids are
preferred.
The preferred solvent/emollient in the present invention is isopropyl
palmitate or
isopropyl myristate. However, there are numerous compounds available which
exist in liquid form at ambient temperatures and will function in a manner
equivalent to the fatty acid esters. These are all quite well known and
described.
They include, but are not limited to, the following:
Ethanol Laurocapram (azone)
Propylene glycol (1; 1 -dodecylazacycloheptan-2-
Water one)
Sodium oleate Acetonitrile
Leucinic acid 1-decanol
Oleic acid 2-pyrrolidone
Capric acid N-methylpyrrolidone
Sodium caprate N-ethyl-l-pyrrolidone
Lauric acid 1-methyl-2-pyrrolidone
Sodium laurate 1 -lauryl-2-pyrrolidone
Neodecanoic acid Sucrose monooleate
Dodecylamine Dimethylsulfoxide
Cetyl lactate Decylmethylsulfoxide
Myristyl lactate Acetone
Lauryl lactate Polyethylene glycol (100-400mw)
Methyl laurate Dimethylacetamide
Phenyl ethanol Dimethylformamide
Hexamthhylene lauramide Dimethylisosorbide
Urea and derivatives Sodium bicarbonate
Dodecyl n,n-dimethylamino acetate Various C7 to C16 alkanes
Hydroxyethyl lactamide Mentane
Phyophatidylcholine Menthone
Sefsol-318 (a medium chain glyceride) Menthol
Isopropyl myristate Terpinene
Isopropyl palmitate D-terpinene
Surfactants (including): Dipentene
polyoxyethylene (10) lauryl ether N-nonalol
diethyleneglycol lauryl ether Limonene
Ethoxy diglycol
CA 02348540 2001-04-27
WO 00/25822 PCT/US98/23014
-11-
This combination of the phospholipid emulsifying agent and the fatty acid
or fatty acid ester or equivalent thereof forms an organogel. In the example
referred to in the Figure, the organogel will be a lecithin organogel, which
is both
isotropic and thermally reversible. At temperatures greater than about 40 C
the
organogel will become a liquid and its viscosity will be greatly reduced.
Water
can be also be added to control the viscosity of the organogel. The organogel
serves as one of the penetration enhancers in the cream, and acts on the
stratum corneum of the skin to promote interaction between the phospholipids
of the cream and the phospholipids of the skin. This causes a disruption in
the
normal regular arrangement of layers in lipids in the stratum corneum so that
openings are created which then allow the drug to pass more easily through the
skin. The organogel will be compatible with a wide variety of lipophilic,
hydrophilic and amphoteric drugs and medications.
Using the above-described lecithin organogel and its components as an
example, the properties needed for inclusion of a components in this invention
will be evident. The various compounds, polymers, etc. comprising the
organogel, the solubilized drug and the carrier/polyoxymer components must all
be compatible with each other, so that chemical reactions do not occur which
would adversely affect the efficacy or safety of the cream composition; they
must
be mutually soluble so that they can be mixed and blended to a uniform
consistency; they must be such that the resulting cream composition has a
viscosity under ambient conditions which is low enough to allow it to be
applied
easily and smoothly to the skin, but not so low that the cream acts as at
least in
part like a liquid and cannot be retained on the skin where it is applied;
they must
not be toxic, irritating or otherwise harmful to the patient; they must be
sufficiently stable that the overall composition will have a reasonable shelf
life
and service life; and, as a practical matter, they must be available at
reasonable
cost. Thus, it will generally be found that the characteristics of a drug or
medication which make it difficult to administer transdermally through the
present
CA 02348540 2001-04-27
WO 00/25822 PCT/US98/23014
-12-
system include its having low stability, particularly at ambient temperatures;
not
being soluble in the composition; having high molecular weight resulting in
difficulty penetrating the stratum corneum, even with the enhanced openings;
and/or causing an adverse reaction with the one or more skin layers.
The drug or medication which is to be administered usually must be
solubilized in a solvent to enable it be blended properly with the organogel
and
the carrier/release agent. Typical solvents for such use include water, the
low
molecular weight alcohols and other low molecular weight organic solvents.
Solvents such as water, methanol, ethanol and the like are preferred. The
purpose of solubilizing is to enable the medication to become properly
dispersed
in the final cream. It is possible that a few drugs or medications might
themselves be sufficiently soluble in the cream that a solvent, and therefore
a
separate solubilizing step, would not be needed. For the purpose of this
description, therefore, the term "solubilized" drug or medication shall be
considered to include those drugs or medications which can be dispersed or
dissolved into the cream with or without the presence of a separate solvent.
Usually the amount each of medication and solvent which will be present, based
on the entire composition, will be in the range of up to <1%-20%, with the
preferred concentration of each being about 10%. The concentrations of both
need not be identical.
A wide variety of drugs may be transported by this method and through
this type of composition. Typical of the various drugs which can be
successfully
incorporated into the present composition and transdermally transported
include
the following classes of substances:
CA 02348540 2001-04-27
WO 00/25822 PCT/US98/23014
-13-
Antidiabetic Agents Multivitamin Preparations
Sulfonylureas Vitamin Combinations
Acetohexamide
Chlorpropamide Antihyperlipidemic Agents
Tolazamide Fluvastatin
Tolbutamide Lovastatin
Glipizide Pravastatin
Glyburide Simvastatin
Glimepiride Probucol
Metformin Niacin
Acarbose Dexothyroxine
Insulin Clofibrate
Gemfibrozil
Glucose Elevating Agents
Diazoxide Cardiac Drugs
Glucose Cardiac Glycosides
Digitoxin
Thyroid Hormones Digoxin
Levothyroxine Antianginal Agents
Liothyronine Nitroglycerin
Thyroid USP Isosorbide Dinitrate
Thyroglobulin Isosorbide Mononitrate
Liotrix Antiarrhythmic Agents
Moricizine
Thyroid Drugs Quinidine
Iodine Procainamide
Propylthiouracil Disopyramide
Methimazole Lidocaine
Tocainide
Parathyroid Drugs Mexiletine
Calcitonin Flecanide
Etidronate Encainide
Pamidronate Amiodarone
Alendronate
Gallium Nitrate Respiratory Drugs
Bronchodilators
Vitamins Albuterol
Vitamin A Metaproterenol
Vitamin D Terbutaline
Vitamin E isoproterenol
Vitamin B1 Ephedrine
Vitamin B2 Theophylline
Vitamin B3 Dyphylline
Vitamin B6
Vitamin B12
Vitamin C (con't. next column)
CA 02348540 2001-04-27
WO 00/25822 PCT/US98/23014
-14-
Nasal Decongestants Antirheumatic Agents
Phenylpropanolamine Gold Compounds
Pseudoephedrine Penicillamine
Phenylephrine Azathioprine
Ephedrine Methotrexate
Naphazoline
Oxymetazoline Agents for Gout
Tetrahydrozoline Probenecid
Xylometazoline Sulfinpyrazone
Propylhexedrine Allopurinol
Colchicine
Gastrointestinals
Sucralafate Agents for Migraine
Metoclopramide Sumatriptan
Cisapride Methysergide
Laxatives Ergotamine Derivatives
Mesalamine
Olsalazine Sedatives and Hypnotics
Antidiarrheals Zolpidem
Famotidine Paraldehyde
Nizatidine Chloral Hydrate
Cimetadine Acetylcarbromal
Rantadine Glutethimide
Omeprazol Ethchlorvynol
Cifapride Ethimate
Temazepam
Miscellaneous Estazolam
Finasteride Flurazepam
Lamsoprazole Quazepam
Papaverine Triazolam
Prostaglandins Phenobarbital
Mephobarbital
Amphetamines Amobarbital
Dextroamphetamine Butabarbital
Secobarbital
Anorexiants Pentobarbital
Phentermine
Benzphetamine Antianxiety Agents
Phendimetrazine Meprobamate
Diethylpropion Alprazolam
Mazindol Chlordiazepoxide
Fenfluramine Clonazepam
Dexfenfluramine Clorazepate
Diazepam
Halazepam
Lorazepam
Oxazepam (con't. next page)
CA 02348540 2001-04-27
WO 00/25822 PCT/US98/23014
-15-
Prazepam Antipsychotic Agents
Buspirone Chlorpromazine
Hydroxyzine Promazine
Doxepin Triflupromazine
Chlormezanone Thioridazine
Mesoridazine
Anticonvulsants Acetophenazine
Phenytoin Perphenazine
Mephenytoin Fluphenazine
Ethotoin Trifluoperazine
Ethosuximide Chlorprothixene
Methsuximide Thiothixene
Phensuximide Haloperidol
Paramethadione Molindone
Trimethadione Loxapine
Clonazepam Clozapine
Clorazepate Riperidone
Valproic Acid Pimozide
Lamotrigine Prochlorperazine
Primidone
Gabapentin Other Psychotherapeutic Agents
Phenacemide Lithium
Carbamazepine Methylphenidate
Phenobarbitol Tacrine
Pemoline
Antidepressants
Amitryptyline Antimicrobials
Clornipramine Antibacterials
Doxepin Penicillins
Imipramine Cephalosporins
Trimipramine Carbapenems
Amoxapine Monobactams
Desipramine Chloramphenicoi
Nortriptyline Fluoroquinolones
Protriptyline Tetracyclines
Venlafaxine Macrolides
Maprotiline Spectinomycin
Trazodone Vancomycin
Bupropion Lincosamides
Fluoxetine Aminoglycosides
Paroxetine Colistin
Sertraline Polymixin B
Fluvoxamine Bacitracin
Tranylcypromine Novobiocin
Pheneizine Metronidazoie
Nefazodone
CA 02348540 2001-04-27
WO 00/25822 PCT/US98/23014
-16-
Antifungals Amantadine
Flucytosine Foscarnet
Nystatin Didanosine
Miconazole Acyclovir
Ketoconazole Ganciclovir
Amphotericin B Zalcitabine
Griseofulvin Rimantadine
Fluconazole Miscellaneous Anti-infectives
Itraconazole Trimethoprim
Sulfonamides Trimethoprim-
Sulfadiazine Sulfamethoxazole
Sulfacytine Erythromycin-
Sulfamethoxazole Sulfisoxazole
Suflamethiazole Furazolidone
Antimalarials Pentamidine
Quinine Sulfate Eflornithine
Mefloquine Atovaquone
Quinacrine Trimetrexate Glucuronate
Doxycycline Leprostatics
4-Aminoquinolone Dapsone
Compounds Clofazime
8-Aminoquinolone Antihelmintics
Compounds Mebendazole
Folic Acid Antagonists Diethylcarbamazine
Antituberculous Drugs Citrate
Isoniazid Pyrantel
Rifampin Thiabendazole
Rifabutin Piperazine
Ethambutol HCI Quinacrine
Pyrazinamide Niclosamide
Aminosalicylate Sodium Oxamniquine
Ethionamide Praziquantel
Cycloserine
Streptomycin Sulfate Antihistamines
Capreomycin Diphenhydramine
Amebicides Chlorpheniramine
Paromomycin Pyrilamine
lodoquinol Doxepin
Metronidazole Carbinoxamine
Emetine Clemastine
Chloroquine Tripelennamine
Antivirals Brompheniramine
Famciclovir Dexchlorpheniranune
Stavudine Triprolidine
Zidovudine Methdilazine
Ribavarin (con't. next Promethazine
column) Trimeprazine (con't. next page)
CA 02348540 2001-04-27
WO 00/25822 PCT/US98/23014
-17-
Hydroxyzine HCl Desoximetasone
Azatadine Fluocinolone
Cyproheptadine Halcinonide
Phenindamine Clocortolone
Astemizole Flurandrenolide
Loratadine Fluticasone
Terfenadine Mometasone
Cetirizine Aclometasone
Desonide
Antimetabolites Fludrocortisone
5-Fluorouracil
6-Mercaptopurine Local Anesthetics
Mycophenolic Acid Dibucaine
Methotrexate Lidocaine
Cytarabine Benzocaine
Floxuridine Butamben Picrate
Thioguanine Tetracaine
Dyclonine
Anticholinergics Pramoxine
Atropine Prilocaine
Scopolamine
Homatropine Antiplatelet Drugs
Tropicamide Dipyridamole
Pirenzepine Ticlopidine
Isopropamide Warfarin
Propantheline Coumarin
Methscopolamine
Methantheline Non-steroidal Antiinflammatory Agents
Trihexyphenidyl Fenoprofen
Benztropine Ibuprofen
Biperiden Flurbiprofen
Ketoprofen
Steroidal Antiinflammatory Agents Naproxen
Cortisone Oxaprozin
Hydrocortisone Diclofenac
Hydrocortisone Acetate Etodalac
Prednisone Indomethacin
Prednisolone Ketorolac
Triamcinolone Nabumetone
Methylprednisolone Sulindac
Dexamethasone Tolmentin
Betamethasone Meclofenamate
Clobetasol Flufenamic Acid
Diflorasone Mefenamic Acid
Halobetasol Meclofenamic Acid
Amicinonide (con't. next column) Piroxicam
Salicylates (con't. next page)
CA 02348540 2001-04-27
WO 00/25822 PCT/US98/23014
-18-
Diflunisal Carteolol
Indomethacin Nadolol
Phenylbutazone Penbutolol
Oxyphenbutazone Pindolol
Sulfinpyrazone Sotalol
Allopurinol Timolol
Penicillamine Labetalol
Colchicine Ace Inhibitors
Probenicid Benazepril
Captopril
Sunscreen Agents Enalapril
Oxybenzone Fosinopril
Dioxybenzone Lisinopril
p-Aminobenzoic Acid Moexipril
Ethyl Dihydroxy Propyl PABA Quinapril
Padimate 0 Ramipril
Glyceryl PABA Calcium Channel Blockers
Cinoxate Diltiazem
Ethylhexyl p-methoxycinnamate Verapamil
Octocrylene Nifedipine
Octyl Methoxycinnamate Felodipine
Ethylhexyl salicylate Nicardipine
Homosalate Nimodipine
Octyl Salicylate Nisoldipine
Menthyl Anthranilate Isradipine
Digalloyl Trioleate Bepridii
Avobenzone Amlodipine
Nisoldipine
Muscle Relaxants Alpha Blockers
Carisoprodol Methyldopa
Chiorphenesin Clonidine
Chlorzoxazone Phentolamine
Cyclobenzaprine Guanabenz
Metaxalone Phenoxybenzamine
Methocarbamol Guanfacine
Orphenadrine Yohimbine
Diazepam Reserpine
Baclofen Guanethidine
Guandrel
Antihypertensives Doxazosin
Beta-Blockers Prazosin
Propranolol Terazosin
Acebutolol Vasodilators
Betaxolol Hydralazine
Bisoprolol Minoxidil
Esmolol (con't. next Nitroglycerin (con't. next
column) page)
CA 02348540 2001-04-27
WO 00/25822 PCT/US98/23014
-19-
Isosorbide Dinitrate Oxymorphone
Isosorbide Mononitrate Oxycodone
Papaverine Meperidine
Methadone
Diuretics Propoxyphene
Thiazides Tramadol
Loop Diuretics Acetaminophen
Spironolactone Pentazocine
Triamterene Fentanyl
Acetazolamide Salicylates
Methazolamide
Dichlorphenamide Sex Hormones
Estogens
Antiemetics Estriol
Chlorpromazine Estradiol
Triflupromazine Estrone
Perphenazine Testosterone
Prochlorperazine Methyltestosterone
Promethazine Progesterone
Thiethylperazine Medroxyprogesterone
Metoclopramide Hydroxyprogesterone
Cyclizine Norethindrone
Meclizine Megesterol
Buclizine
Dimenhydrinate Pituitary Hormones
Trimethobenzamide DDAVP
Scopolamine Methylergonovine
Diphenidol
Benzquinamide Uterine Hormones
Hydroxyzine Carboprost
Dinoprostone
Analgesics
Codeine Adrenal Steroid Inhibitors
Hydrocodone Aminoglutethimide
Hydromorphone
Morphine (can't. next column)
In one preferred embodiment, the drug is ketoprofen.
Finally, the carrier and drug release agent form a polymeric composition
which provides the separate penetration enhancement of facilitating the rapid
release of the medication from the cream upon topical application to the
patient.
The purpose of this combination of materials is to provide for penetration
enhancement of a different type than that of the organogel, i.e., by effecting
CA 02348540 2001-04-27
WO 00/25822 PCT/US98/23014
-20-
rapid release of the drug from the cream and transport by the carrier out of
the
cream and into the skin through the enhanced openings in the stratum corneum.
The drug release agent may be any of a variety of polyoxymers, i.e.,
polyoxyalkylene derivatives of propylene glycol. Preferred are those which
contain mixtures of polyoxyethylene and polyoxypropylene polymeric derivatives
of propylene glycol or methyl oxirane polymers. By acting essentially as an
emulsifier, stabilizer and dispersing agent, the polyoxymer facilitates the
separation of the drug or medication from the other components of the cream
and transfers it to the carrier, which will normally be water or a low
molecular
weight alcohol or organic solvent. Useful polyoxymers are available under the
trademark "Pluronic" from Wyandotte Chemical Company.
The concentration of the carrier provided with the drug release agent as
a mixture in the cream will determine the particular diffusion coefficient of
the
drug. With higher concentrations of the carrier, the diffusion coefficient
will be
lower and the drug will be absorbed more slowly and produce more local
effects.
Conversely, lowering the concentration of the carrier will speed the
absorption
of the drug and enhance the ability of the drug to be absorbed systemically.
The
normal concentration of the drug release agent in the mixture with the cream
will
be approximately 20% to 30%, with the balance being the carrier, during the
formation of the carrier/drug release agent mixture.
The overall concentrations of the various components in the composition
will generally be in the ranges of:
Medication <1 %-20%
Solvent for medication <1 %-20%
Organogel 20%-40%
Carrier/release agent 40%-70%
CA 02348540 2001-04-27
WO 00/25822 PCTNS98/23014
-21-
It will of course be understood that these ranges represent the typical
ranges for the specific example upon which the Figure is based, i.e., an
example
with a lecithin organogel, ketoprofen as the drug, and a "Pluronic NF-127"
polyoxymer as the drug release agent. In general the ranges for other
compositions of this invention in which other suitable organogels, drugs,
carriers
and release agents are used will be similar, and those skilled in the art will
have
no difficulty formulating suitable compositions from the description herein.
Other factors will need to be considered in preparing specific formulations.
If the carrier concentration in the cream lies above the useful range, it
becomes
relatively stiff and difficult to apply, or, conversely, if the concentration
falls below
the suitable level, the cream will have a tendency to separate. Further, the
pH
of the cream must be adjusted to match the pH of the solubilized medication
component to maximize the amount of non-ionized drug present in the cream.
All suitable medications have acid/base characteristics that can be altered by
adjustment of the pH of the composition. The greater proportion of non-ionized
drug present, the greater the drug's solubility and the greater the ability
for larger
quantities of the drug to be transported transdermally. The control of the pH
can
also be used to determine whether the drug is likely to become absorbed
systemically or to be absorbed locally, since the speed of transdermal
transport
will be dependent on the pH.
The physical properties of the cream will also be important. As noted the
viscosity must be such that it can be applied topically and conform to and
adhere
to the patient's skin for a period of time sufficient for a significant
portion of the
medication to be delivered transdermally to the patient. It must also be
capable
of being removed from the patient's skin with ordinary physiologically
acceptable
cleansers or solvents, so that the cream may be removed if medically
necessary,
CA 02348540 2001-04-27
WO 00/25822 PCT/US98/23014
-22-
or the residue may be removed once the treatment time period for each
administration has been completed. The components must be capable of being
blended into a smooth, homogenous mixture with a cream- or lotion-like
consistency and appearance, which either has a natural light colored
appearance or can be lightly tinted if flesh-compatible tones are desired. The
cream must also be capable of being covered with a light gauze or other type
of
dressing if desired, particularly where the cream would otherwise be in
contact
with the patient's clothing.
Adjustment of pH, effects of concentration and achievement of suitable
physical properties in compositions containing polyoxymers have been studied
and reported by Chi et al., J. Pharm. Sci., 80 (3): 280-283 (1991). Reference
is
made to that article, and the prior references reported therein, for guidance
in
determining practical limits of pH, concentration, viscosity and the like when
varying the specific materials herein. The techniques and methods reported
there are quite suitable for use in the present invention.
Examples of the formation of different components are given below:
EXAMPLE 1
Formation of a Lecithin Organogel
A number of different lecithin organogels were formed by mixing different
quantities of granular lecithin soya with isopropyl palmitate and a solvent .
In
three different typical compositions the respective amounts of lecithin soya
and
isopropyl palmitate were 25%/75%, 50%/50%, and 75%/25%. The first
composition can be characterized as a thin oil, the second as a medium oil and
the third as a heavy oil. In all cases the lecithin granules and isopropyl
palmitate
were allowed to sit for several hours, commonly overnight, by the end of which
a liquid of oil or syrup consistency had formed. Alternatively one can mix the
CA 02348540 2001-04-27
WO 00/25822 PCT/US98/23014
-23-
lecithin soya and the isopropyl palmitate at 500 to 60 C until the dissolution
is
complete.
At any point during formation of the mixture one can also add the drug or
medication. If the latter is soluble in alcohol it may be previously dissolved
in the
alcohol and the alcohol/drug mixture incorporated into the lecithin soya and
isopropyl palmitate mixture.
EXAMPLE 2
Formation of a Carrier/Drug Release Agent Component
A polymeric gel for use as a carrier was formed by mixing 20 grams of a
commercial polyoxymer designated as "Pluronic NF-127" with 0.2 g of pure
potassium sorbate and adding sufficient refrigerated purified water to bring
to
volume of 100 ml. Other similar compositions were formed with 30 g and 40 g
of the "Pluronic NF-127" respectively. A typical commercial mixer was used to
mix the material. Once all of the granules of the polymeric material had been
wetted the gel was refrigerated so that dissolution took place upon cooling in
the
refrigerator. The compositions must be maintained under refrigeration because
at ambient conditions they will solidify, since (as opposed to water)
polyoxymer
mixtures as prepared herein solidify when heated and liquefy when cooled.
Stock solutions of these materials may be made and kept in refrigerated
storage
for repeated use in the formulation of the compositions of the present
invention.
EXAMPLE 3
Mixture of a Cream Containing Medication,
Lecithin Organogel and Carrier/Drug Release Agent
In a typical procedure equivalent weights of the lecithin soya and the
isopropyl palmitate are combined and a small quantity of sorbic acid is
incorporated to control pH. The mixture is stirred until a syrup or oil
consistency
is obtained. Large quantities may be prepared and kept as a stock solution.
The
CA 02348540 2001-04-27
WO 00/25822 PCT/US98/23014
-24-
drug or medication, e.g., ketoprofen, is dissolved in water, alcohol or an
equivalent solvent by using a the minimal amount of solvent necessary to
obtain
complete solubilizing. The dissolved drug is added to a small portion of the
lecithin organogel and stirred to disperse the drug in the gel. The mixture of
the
carrier and the polyoxymer is then added to bring the entire formulation to
the
desired volume, and, if necessary, the pH of the cream is adjusted.
It will be evident that there are numerous embodiments of this invention
which, while not expressly described above, are clearly within the scope and
spirit of the invention. The above description is therefore intended to be
exemplary only, and the scope of the invention is to be limited solely by the
appended claims.
WE CLAIM: