Note: Descriptions are shown in the official language in which they were submitted.
CA 02348893 2001-05-O1
WO 00!32601 PCT/US99/27316
PROCESS FOR PREPARING CROSS-BR)DGED
TETRAAZA MACROCYCLES
FIELD OF THE INVENTION
The present invention relates to an improved process for preparing cross-
bridged tetraaza
macrocycles, said macrocycles suitable as ligands for use in preparing
transition metal
complexes. The present invention provides a process which is well suited for
use in industrial
and other commercial preparations of the herein described crossed-bridged
macrocycles.
BACKGROUND OF THE INVENTION
Tetraaza macrocyclics, for example, cyclam, have been prepared in numerous
ways,
however, there is a paucity of information relating to the preparation of
cross-bridged tetraaza
macrocyclics inter alia bis N-substituted tetraaza macrocyclics inter alia
5,12 dialkyl 1,5,8,12-
tetraaza-bicyclo[6.6.2]hexadecanes which have recently found wide
applicability as ligands
especially in the area of transition metal catalysts inter alia bleach
catalysts.
WO 98/39335 A1 "Improved Methods of Making Cross-Bridged Macropoly-cycles"
discloses a rational procedure for preparing cross bridged macropolycyclic
ligands which is
amenable to high yields necessary for industrial scale-up. However, the
reductive ring cleavage
step which results in bicyclo bridged-ring formation utilizes a borohydride
reducing agent. This
type of reducing agent can place constraints on the formulator. For example,
the need to break up
the amine/borohydride complex during work-up and the proper recovery and
disposal of boron
waste products adds cost to the process. Also, if excess borohydride is
needed, this requires
neutralization which involves the use of acid and the evolution of large
quantities of hydrogen
gas.
Therefore, a need exists for a highly quantitative, preferably catalytic,
process for
preparing cross-bridged macropolycyclic ligands which is adaptable to either
continuous flow
processes or batch preparations.
SUMMARY OF THE INVENTION
The present invention meets the aforementioned needs in that it has been
surprisingly
discovered that select bis N-substituted tetraaza macrocyclics can be
converted to cross-bridged
macropolycyclic ligands via catalytic hydrogenation.
CA 02348893 2001-05-O1
WO 00/32601 PCT/US99/27316
A first aspect of the present invention relates to a process for preparing a
tetraaza
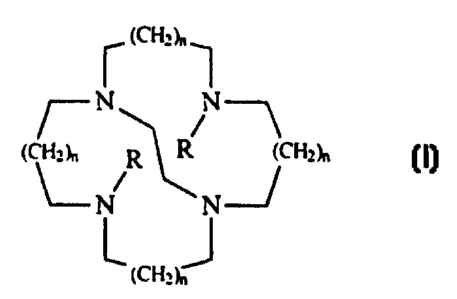
macrocyclic ligand having the formula:
(CHz~,~
~-N
(CH~~, R R {CHz~,
~N N
~ (~Hz~J
wherein each R is independently C,-C8 linear or branched alkyl, -(CH,)rCO~M,
and mixtures
thereof, preferably both of the R units are not methyl; M is hydrogen or a
salt forming cation; x is
from 1 to 6; each index n is independently from 0 to 3; said process
comprising the steps of:
a) hydrogenating a tetraaza macrocyclic ligand precursor having the formula:
(CHz~,
R
~N H N
(CHz~, (CHz~,
\ / 2X-
~N H N
R J
/ ~ (CHz~,~
wherein X is an anion which provides charge neutrality, with from about 1 ppm
of a
transition metal hydrogenation catalyst at a pH of at least $ to form a
tetraaza
macrocyclic ligand; and
b) optionally isolating said ligand.
These and other objects, features, and advantages will become apparent to
those of
ordinary skill in the art from a reading of the following detailed description
and the appended
claims. All percentages, ratios and proportions herein are by weight, unless
otherwise specified.
All temperatures are in degrees Celsius (o C) unless otherwise specified. All
documents cited are
in relevant pan, incorporated herein by reference.
DETAILED DESCRIPTION OF THE INVENTION
The present invention relates to the catalytic hydrogenation of tetraaza
macrocycles
having the formula:
CA 02348893 2001-05-O1
WO 00/32601 PCT/US99/27316
~(cH,~,~ /R
NI H IN
(CH~~, (CH=~, _
~ / 2X
'-N H N
R
/ ~(CH.~,J
wherein the covalent bond between the N-substituted quaternary ring nitrogen
and the bridging
carbon is broken and a cross-bridged macrocyclic ligand having the formula:
(CH:~,
~--N i -~
(cH~~, R R (CH:~,
~N/ N
~ (~H~~J
is the resulting product.
In the above formula each R is independently C,-Ca linear or branched alkyl, -
(CH,)rCO~M, and mixtures thereof, preferably both of the R units are not
methyl; more
preferably one R unit is methyl and the other R unit is selected from the
group consisting of ethyl,
propyl, butyl, pentyl, hexyl, and mixtures thereof of the methyl/alkyl R unit
mixtures preferably
one R is methyl and the other R is ethyl or propyl. A most preferred ligand
consists of a unit
wherein one R unit is methyl and the other R unit is ethyl. A preferred ligand
is a macrocyclic
ring wherein each R unit is ethyl. M is hydrogen or a salt forming cation, non-
limiting examples
of which are sodium, potassium, calcium, ammonium. When R is -(CH~)xCO~M the
index x has
the value from 1 to about 6, preferably x is 1. The index n defines the size
of the macrocyclic
ring. The index n has the value of from 0 to about 3. A preferred macrocyclic
ring has one
opposite set of n indices equal to 1 and the other set of n indices equal to 0
as in the general
formula:
CA 02348893 2001-05-O1
WO 00/32601 PCT/US99/27316
4
N N
R R
N N
wherein R is the same as defined herein above.
Preferred ligands according to the present invention comprise R unit pairs
which are
selected from the group consisting of methyl and ethyl, diethyl, methyl and
propyl, ethyl and
propyl, methyl and butyl. ethyl and butyl, and mixtures thereof.
The starting materials for the process of the present invention are tetraaza
macrocyclic
ligand precursors, or alternatively, bis-quaternary cis tetracycles, having
the formula:
(CH:~,
R
~N H N
~+
(CFiz~, (CHz~, 2 X -
'--.- N H N
R
~ (~H~~J
wherein R and n are the same as defined herein above. X is an anion which
serves to provide
electronic neutrality to the bis-quaternary cis tetracycle. Those of ordinary
skill in the art
recognize that the term "electronic neutrality" refers to "a sufficient amount
of an anionic species
which satisfies the molecular charge balance requirements" and that a mixture
of mono-, di-, tri-.
etc. electronic species may be use herein. X preferably has unit negative
charge, for example,
1 S halogen, tosylate, methylsulfate, ethylsulfate. However, X may have more
than one negative
charge, for example, sulfate, in which case the formulator requires only half
the amount
necessary when using a unit negative-charged anion. Preferred X is chloride,
bromide, iodide,
sulfate, ethyl sulfate, methyl sulfate, tosylate, mesylate, triflate, and
mixtures thereof.
STEP (a) Hydrogenation - Reductive Cleavage
Step (a) comprises the reductive cleavage via catalytic hydrogenation as
outlined in the
following scheme:
CA 02348893 2001-05-O1
WO 00/32601 PCT/US99/27316
(CH=~, ~ ~ (CH=~,
R
~N H N
/~ N N
(CH=~, fCH=~, H~ (C~ R R ~ H:~
, / 2X- /
~N H N~ catalyst ~N/ y
R
(CH~~,~
(CH=~,~
wherein the cis-tetracycle bis quaternary salt is converted to the cross-
bridged tetraaza
macrocycle.
3 Step (a) is conducted in the presence of a catalyst, preferably a supported
catalyst. Non-
limiting examples of catalysts include platinum on carbon, palladium on carbon
(Pd/C),
palladium hydroxide on carbon (Pd(OH),iC), rhodium on carbon (Rh/C), Raney
nickel, and
mixtures thereof. A preferred catalyst is Pd(OH)Z/C. The supported catalysts
may comprise from
about 1% to about 50% by weight of transition metal, however, the pure metal,
i.e. palladium, can
be used without the need for a "support", i.e., carbon. A "catalytic amount"
of catalyst is
sufficient to provide the reduction of step (a). For the purposes of the
present invention the term
catalytic amount is defined as "from about 1 ppm of a S% by weight transition
metal catalyst".
However, the formulator, due to poisoning of the catalyst surface by reaction
products may use
more than a catalytic amount of a catalyst. The amount of catalyst used in
step (a) of the present
invention is preferably from about 10 ppm of a catalyst which contains from
about 5% to 50% by
weight, of a transition metal, more preferably from about 100 ppm, yet more
preferably from
about 0.1% by weight, of a transition metal supported catalyst. A reaction
solution which
comprises 0.1% by weight, of a transition metal supported catalyst, said
catalyst comprising, for
example, 10% by weight.of palladium on carbon, has 0.01 % transition metal or
100 ppm
transition metal present.
The amount of hydrogen gas present in step (a) of the present invention need
only be
enough to sufficiently saturate the catalyst surface, preferably the hydrogen
pressure is from 200
psi, more preferably from 400 psi, most preferably from 800 psi to about 2000
psi, more
preferably to 1000 psi.
Step (a) of the present process can be conducted at a temperature of from
20° C,
preferably from about 40° C, more preferably from about 60° C to
about 100° C, preferably to
about 90° C, more preferably to about 80° C, most preferably to
about 65° C.
CA 02348893 2001-05-O1
WO 00/32601 PCT/US99/27316
6
The pH under which Step (a) must be conducted is at least 8, preferably at
least 10, more
preferably at least about 11. Preferably the base which is used to adjust the
pH is in the form of
an aqueous solution. Preferred bases are selected from the group consisting of
potassium
carbonate, sodium carbonate, sodium hydroxide, potassium hydroxide, and
mixtures thereof. As
a non-limiting example, it is satisfactory to use a sufficient amount of 1 M
(molar) aqueous base
to adjust the pH to the required level. A convenient and preferred base is
potassium carbonate.
As a suitable alternative, Step (a) can be conducted in the presence of a
solvent other
than water, or in the absence of water. A mixture of a suitable solvent and
water is a suitable
means for conducting the hydrogenation process of the present invention. In
the case where
water is absent, sufficient base must be present to stabilize the transition
state of the reactants and
products during hydrogenation. Non-limiting examples of solvents include
methanol, ethanol, n-
propanol, isopropanol, N,N-dimethyl formamide, n-butanol, iso-butanol, tert-
butanol, and
mixtures thereof; preferred solvents are selected from the group consisting of
ethanol, n-
propanol, N,N-dimethyl formamide, and mixtures thereof. VVhen a solvent is
present and the
base is in the form of an aqueous solution, the ratio of said volume of an
aqueous base, to said
solvent is from about 1:10 to about 1:1, preferably the ratio of the volume of
aqueous base to
solvent is 1:4. It is desirable, but not a requirement, that the aqueous base
and solvent form a two
phase system.
The process of the present invention comprises an optional, although
preferred, Step (b)
which is an isolation step. Typically this step involves filtration of the
reaction solution to
remove the catalyst. In addition, this step may comprise a neutralization
step, however, the
product can be isolated or removed from the reaction matrix in any manner
which the formulator
desires.
When filtration of the catalyst is a desired step, the reaction solution
containing the cross-
bridged ligand is filtered to remove the catalyst to form a crude filtrate.
This crude filtrate can be
neutralized or the tetraaza macrocyclic ligand can be isolated by extraction
or crystallization
directly from the preferably aqueous solution.
Steps {a) and (b) and any optional extensions thereto, for example, a
crystallization step,
a solvent drying step, a purging step, may be suitably adapted for either
batch processes or
continuous processes, for example, continuous flow processes. As mentioned
herein, the process
of the present invention may comprise other optional steps as deemed necessary
and/or desirable
by the formulator. These optional steps may include but are not limited to,
pre-saturation of the
catalyst with hydrogen, drawing a vacuum on the system, and recovery of the
catalyst and
solvents.
CA 02348893 2001-05-O1
WO 00/32601 PCT/US99/27316
7
Preferably the ligands formed by the process of the present invention are
converted into
manganese containing transition metal catalysts in a subsequent, however
optional, process step.
The bleach catalysts comprise a central manganese atom and a cross-branched
ligand formed by
the process of the present invention. The final bleach catalyst may comprise
one or more other
compatible ligands inter alia chlorine atom. The preferred catalysts are
suitable as bleaching
agents.
The following is a non-limiting example of the process of the present
invention.
Preparation of 5,12-diethyl-1,5,8,12-tetraaza-bicvclo(6.6.21hexadecane
To a thick walled, glass autoclave sleeve is added the bis-quaternary cis
tetracycle having
the formula:
~ HzCH3
N H N
2 Br -
N H N
CH3CH2
(3.0 g, 6.8 mmol) and a 1M aqueous solution of KZC03 (30 mL). The solution is
agitated to
dissolve the substrate then 20°.o Pd(OH)~/carbon (0.7 g, 1.0 mmol) is
added. The glass sleeve is
placed into a rocking autoclave and hydrogenated at 65° C with 1900
psig hydrogen for 4 hours.
The reaction is cooled and the solution filtered through glass fiber filter
paper to remove the
catalyst and the filtrate is reduced to a white solid under vacuum. The white
solid is suspended in
refluxing ethanol and the un-dissolved inorganic salts are collected by
filtration. After
concentrating the filtrate under vacuum, the resulting oily residue is
dissolved in aqueous 4M
KOH (4 mL) and extracted three times with 25 mL portions of toluene. The
toluene extracts are
combined and concentrated in vacuo to afford 5,12-diethyl-1,5,8,12-tetraaza-
bicyclo[6.6.2Jhexadecane (1.56 g) in 81% yield as a clear oil.
The following is an example of an optional, but preferred step of the process
of the
present invention which is conversion to a manganese transition metal bleach
catalyst.
Preparation of dichloro 5,12-diethyl-1,5,8,12-tetraaza-
bicvclof6.6.Zlhexadecane maneanese
To a 100 mL reaction flask is charged anhydrous acetonitrile (50 mL) and 5,12-
diethyl-
1,5,8,12-tetraaza-bicyclo[6.6.2]hexadecane (1.4 g, 5 mmol). The resulting
suspension is
degassed under vacuum with subsequent re-filling with argon. This process is
repeated six times.
Manganese(II) chloride (0.590 g, 4.7 mmol) is added and the reaction is
refluxed for 3 hours.
CA 02348893 2001-05-O1
WO 00/32601 PCTNS99/27316
The resulting solution is filtered through glass-fiber filter paper. The
resulting filtrate is
concentrated under reduced pressure at 45° C to afford a solid. The
solid is suspended in toluene
(~0 mL) and the resulting dark supernatant is discarded. Treatment with
toluene is repeated five
times. The resulting solid is dried under vacuum to yield dichloro 5,12-
diethyl-1,5,8,12-tetraaza-
bicyclo[6.6.2]hexadecane manganese (1.48 g, 73% yield).