Note: Descriptions are shown in the official language in which they were submitted.
CA 02350069 2001-05-09
WO 00/33161 PCT/US99128582
METHODS TO REDUCE VARIANCE IN TREATMENT STUDIES
USING GENOTYPING
s CROSS-REFERENCES TO RELATED APPLICATIONS
This application claims the benefit of U.S. Provisional Application No.
60/110,668, filed December 2, 1998, which is incorporated by reference in its
entirety for
all purposes.
1o FIELD OF INVENTION
The present invention resides in the fields of medicine, genetics and
statistics.
BACKGROUND OF THE INVENTION
~ s The conduct and design of studies for investigating treatment efficacy
such
as clinical trials aims to eliminate the bias that can arise from "random"
biological influence
be they genetic or environmental, as well as bias introduced by the
investigator wittingly or
otherwise. One approach for reducing bias is to randomize individuals to
either treatment or
control groups with the view that if the individuals in the two groups are
unrelated
2o genetically and live independent of one another, then both genetic and
environmental
influences on the trial will be balanced in the two arms of the study. An
immediate
consequence of randomization in this way, however, is that the variance of the
biological
condition measured is greater than if each case is matched for genetic and
known
environmental influences.
2s One method for determining genetic variability is by assessment of
polymorphic profile. Polymorphisms refer to the coexistence of multiple forms
of a
sequence in a population. Several different types of polymorphisms have been
reported. A
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
restriction fragment length polymorphism (RFLP), for example, means a
variation in DNA
sequence that alters the length of a restriction fragment (see, e.g., Botstein
et al., Am. J.
Hum. Genet. 32:314-331 (1980)). Short tandem repeats (STRs), as the name
implies, are
short tandem repeats that consist of tandem di-, tri- and tetra-nucleotide
repeat motifs. Such
polymorphisms are also sometimes referred to as variable number tandem repeat
(VNTR)
polymorphisms (see, e.g., U.S. Patent No. 5,075,217; Armour et al., FEBS Lett.
307:113-
115 (1992); and Hom et al., WO 91/14003).
By far the most common form of polymorphisms are those involving single
nucleotide variations between individuals of the same species; such
polymorphisms are
to called single nucleotide polymoiphisms, or simply SNPs. Some SNPs that
occur in protein
coding regions give rise to the expression of variant or defective proteins,
and thus are
potentially the cause of a genetic disease. Even SNPs that occur in non-coding
regions can
nonetheless result in defective protein expression (e.g., by Causing defective
splicing).
Other SNPs have no phenotypic effects.
SUMMARY OF THE INVENTION
Certain methods of the invention are designed to provide an assessment of
the efficacy of a treatment procedure. In general, such methods involve
selecting treated
and control subpopulations from treated and control populations of subjects,
wherein the
2o treated population has been treated with a treatment procedure and the
control population
has been treated with a control procedure. The subjects in both the treated
and control
populations have been characterized for polymorphic profile and are selected
because
they have similar polymorphic profiles. A determination is then made whether
there is a
statistically significant difference in a test parameter between the treated
and control
subpopulations. In general, such a statistically significant difference
indicates that there is
a correlation between the type of treatment and one or more polymorphic forms
within the
polymorphic profile for which the treated and control subpopulations were
selected.
In some instances, especially when a significant difference is not found,
the selecting and determining steps are repeated one or more times. In such
additional
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
cycles, the polymorphic profile for which the treated and control groups are
selected
differs from the polymorphic profile selected for in previous cycles. The
polymorphic
profile for which the subpopulations are selected can vary in terms of numbers
of
polymorphic forms within the profile and the extent of similarity in profiles
between
treated and control groups. For example, the polymorphic profile can include a
single
polymorphic form, but more typically includes a plurality of polymorphic forms
(e.g., 10
or up to 100 polymorphic forms or more). In most instances, the polymorphic
profiles of
the subpopulations have at least 10%, SO%, 75% or up to 100% identity.
Certain methods of the invention are directed towards methods for
to performing clinical trials. Some of these methods initially involve
treating a treated
population and a control population of patients having the same disease with a
drug and a
control procedure (e.g., treating with a placebo or with a different amount of
the drug or
according to a different treatment schedule), respectively. A subpopulation of
patients is
then selected from each of the treated and control populations for similarity
in a
15 polymorphic profile. A determination is then made whether treatment With
the drug
correlates with status of the disease in the subpopulations to assess the
efficacy of the
drug in treating the disease. With these methods too, a carrelation indicates
that at least
one or more polymorphic forms within the polymorphic profiles correlates with
treatment
efficacy.
2o Some methods of the invention are computerized methods. For example,
certain methods of the invention include providing a database capable of
storing: (1)
designations for each member of a treated population treated according to a
treatment
procedure and designations for each member of a control population treated
according to
a control procedure, (2) designations for a polymorphic profile for each
member of the
25 treated and control populations, and (3) designations for a test parameter
for each
member of the treated and control populations. Using the database,
subpopulations from
each of the treated and control populations are selected for similarity in
polymorphic
profile. A determination is then made to ascertain whether there is a
statistically
significant difference in the test parameter between the subpopulations. The
output from
3o the determining step is then displayed on an output device (e.g., a monitor
or video
CA 02350069 2001-05-09
WO 00/33161 PC'T/US99/28582
display).
In another aspect, the invention provides various computer systems and
programs. For instance, certain computer products for assessing a treatment
procedure
are provided. Some systems include program products that generally include
code for
providing or receiving data, wherein the data includes: ( 1) designations for
each member
of a treated population treated according to a treatment procedure and for
each member of
a control population treated according to a control procedure, (2)
designations for a
polymorphic profile for each member of the treated and control populations,
and (3)
designations for a test parameter for each member of the treated and control
populations.
to The program also includes code for selecting a subpopulation from each of
the treatment
and control populations that have a similar polymorphic profile, code for
determining
whether there is a statistically significant difference in the test parameter
between the
subpopulations and code for displaying an output that indicates whether a
statistically
significant difference was found between the subpopulations. The code is
typically stored
15 on a computer readable storage medium.
The invention further provides a computerized system for assessing
treatment procedures. Some systems generally include a memory, a system bus
and a
processor. The processor is operatively disposed to provide or receive data,
wherein the
data includes: (1) designations for each member of a treated population having
been
2o treated according to a treatment procedure and for each member of a control
population
treated according to a control procedure, (2) designations for a polymorphic
profile for
each member of the treated and control populations, and (3) designations for a
test
parameter for each member of the treated and control populations. The
processor is
further disposed to select a subpopulation from each of the treatment and
control
2s populations that have a similar polymoiphic profile and determine whether
there is a
statistically significant difference in the test parameter between the
subpopulations. The
microprocessor is also capable of displaying an output indicating whether a
statistically
significant difference was found between the subpopulations.
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
BRIEF DESCRIPTION OF THE DRAWINGS
FIGS. 1 and 2 depict computer systems for implementing the methods of
the invention.
FIG. 3 is a flow chart for a method of assessing a treatment procedure
according to the present invention.
DETAILED DESCRIPTION
I. Definitions
A "treatment procedure" refers to methods or processes that are performed
on a member of a treated or treatment population. In general the treatment
procedure is a
process performed on a subject to affect some biological condition,
susceptibility, or
resistance of the subject. Examples of treatment procedures include, but are
not limited
to, treatment with pharmaceutical compounds or other biologics (including, for
example,
recombinantly produced proteins), surgical procedures and various behavioral
therapies
(e.g., prescribed diet and/or exercise regimes). Treatment procedures can be
prophylactic
or therapeutic. For example, a treatment procedure can include treating
members of a
treatment population with a vaccine.
In like manner, a "control procedure" refers to methods that are performed
on a member of a control population. The control procedure can differ from the
treatment
procedure in quantitative or qualitative aspects. For example, the members of
a treatment
population can receive a pharmaceutical composition, whereas the control
population
receives a placebo (i.e., no pharmaceutical composition). In other instances,
the control
procedure involves administration of a drug at different concentrations than
the treatment
procedure or can involve a different schedule for administering the
pharmaceutical
composition relative to the treatment procedure.
A "clinical study" is an inquiry into the cause and sometimes treatment of
a particular phenotype that is represented by at least one random variable.
This phenotype
may often be a disease state or a measure of disease severity. A clinical
study can take
5
CA 02350069 2001-05-09
WO 00/33161 PCTNS99/28582
the form of a case-control study (for a discrete random variable, the groups
being affected
and unaffected individuals) or a single population study where the cause of
the degree or
severity of the phenotype is being investigated (for example, a quantitative
study can
examine blood pressure, blood glucose, etc.).
A "treatment study" is an inquiry into the effect or influence a particular
treatment procedure has on a biological condition, biological susceptibility
or biological
resistance of a subject. The study can be quite structured, formal and
extensive in scope,
or can be relatively unstructured and of limited scope. For example, a
treatment study can
be a formal clinical trial or study performed on a relatively large group of
subjects
tp wherein the study is performed according to set guidelines (e.g.,
governmental
regulations). However, the treatment study can also be a preclinical study, a
field trial of
a plant population or even an informal study by a scientist, veterinarian or a
physician of
the effects of a treatment on relatively few subjects. In a treatment study,
the subjects are
divided into several (though often just two) groups. These may represent
different doses
is ranges or simply the treated and the untreated subjects. In the study, the
random variable
is measured after treatment. It may also be measured before treatment if it is
a change in
the variable over time that is being investigated (e.g., bone mineral density
or blood
pressure). It is preferable that subjects are not undergoing any other
treatments for their
pathological condition. However, if such a constraint is unreasonable, the
study should be
20 designed so that subjects in both treated and untreated groups are
undergoing the same
alternative treatment. The subjects of the treatment study can be conducted
with any type
of organism, including, for example, animals (including humans), plants,
bacteria and
mruses.
A "biological condition" refers to the condition, susceptibility or resistance
25 of the organism upon which the study seeks to determine whether the
treatment procedure
has an effect. Typically, the biological condition is a physical or
physiological condition
of the organism. For example, in some instances the biological condition is a
pathological condition (i.e., a physiological state that normally does not
exist, such as a
disease for example). Pathological conditions typically studied with the
methods of the
3o invention are those with a minimal environmental variance (e.g., high
cholesterol levels in
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
serum), although this is not required. Examples of pathological conditions
include AIDS,
arteriosclerosis, cancer, and diabetes, elevated blood pressure, elevated
serum cholesterol
level or psychosis. A biological condition can be the biological
susceptibility or
resistance of a subject. For example, the treatment study can involve an
analysis of the
effect of certain treatments on the susceptibility of a plant to an herbicide
or susceptibility
of a plant to frost damage. Alternatively, the study can be directed towards
an organism
defense response (i.e., resistance) to some type of insult, for example.
A "random variable," either discrete or continuous, can be any biological,
physiological or biochemical endpoint measured or observed, particularly in
the setting of
1 o a clinical study. This includes measured and observed effects of
treatments, the changes in
those observations and measurements over the course of time (the so -called
natural
history), or any other intervention that may alter traits, signs or symptoms.
Examples of
random variables include pathological conditions susceptible to treatment
with, e.g.,
pharmaceutical compounds; biologics, including recombinantly produced
proteins;
15 surgical techniques; restrictive diets; and behavioral therapy. For
example, serum
concentrations of cholesterol, height, body mass, are all continuous random
variables.
This notion can extend to discrete variables such as the presence or absence
of a physical
trait or symptom which include, for example, nevi on the skin, cysts in the
liver, particular
antibodies in the serum, the degree of swollenness of the joints, the number
of affected
2o joints, or the number of hallucinations in a psychotic episode.
A "test parameter" is the characteristic that is measured or observed to
determine the effect or efficacy (or lack thereof) of the treatment procedure
being
evaluated and is utilized to determine whether there is a statistically
significant difference
in the treatment and control protocols. The test parameter can be a random
variable.
25 Typically, the test parameter is expressed in quantitative terms, although
in some
instances the test parameter can be evaluated in qualitative terms. The nature
of the test
parameter varies according to the biological condition being studied. If the
biological
condition is a disease, the test parameter provides a measure for the status
of the disease.
For example, if the biological condition is AIDS, the test parameter can be
the
3o concentration of HIV in the blood of a subject. If the biological condition
is
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
arteriosclerosis, the test parameter can be serum cholesterol concentration.
The term "variance" refers to variation, scatter, spread or dispersion about
the arithmetic mean. Schematically, the variance is the mean value of the
squared
deviations (Armitage, P., STATISTICAL METHODS IN MEDICAL RESEARCH, Blackwell
Scientific, Oxford, United Kingdom (1971)). A large variance indicates large
deviations
from the arithmetic mean. For example, if cholesterol level is the test
parameter being
measured, a mean cholesterol level is determined. The variance represents the
average
squared deviation of all cholesterol levels relative to the mean. Other
statistical measures
of spread or dispersion about a mean can also be used. Typically, the
distribution of the
Io test parameter takes the shape of a bell-shaped or a normal (Gaussian)
curve. Pictorially,
the invention decreases the variance and thus narrows the bell-shape of the
normal curve
or, described mathematically, the distribution becomes leptokurtic.
Typically, the variance is due to dissimilar effects on the subjects that
influence the biological condition being analyzed by statistical methods,
e.g., genetic,
15 environmental and measurement variables. For example, in most treatment
studies,
because the same methods are used to measure the biological condition among
the entire
population being studied, the variance is due to genetic differences between
individual
subjects and the environment in which the subjects live. Examples of
environmental
influences include diet, sleep patterns, geographical location and culture.
2o A "polymorphism" refers to the occurrence of two or more genetically
determined alternative sequences or alleles in a population generally said to
be occurring
at a frequency of greater than 0.1 %. A polymorphic marker or site is the
locus at which
genetic divergence occurs. Preferred markers have at least two alleles, each
occurnng at
frequency of greater than 1 % in a selected population. A polymorphic locus
can be as
2s small as one base pair. Such a locus is referred to as a single nucleotide
polyrnorphism or
simply SNP.
Polymorphic markers include restriction fragment length polymorphisms,
variable number of tandem repeats (VNTR's), hypervariable regions,
minisatellites,
dinucieotide repeats, trinucleotide repeats, tetranucleotide repeats, simple
sequence
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
repeats, and insertion elements such as Alu. The first identified allelic form
is arbitrarily
designated as the reference form and other allelic forms are designated as
alternative or
variant alleles. The allelic form occurring most frequently in a selected
population is
sometimes referred to as the wildtype form or allele and the other forms
referred to as
mutant forms or alleles. Diploid organisms can be homozygous or heterozygous
for
allelic forms. A diallelic polymorphism has two forms. A triallelic
polymorphism has
three forms.
A "single nucleotide polymorphism" occurs at a polymorphic site that is
occupied by a single nucleotide, which is the site of variation between
allelic sequences.
to The site is usually preceded by and followed by highly conserved sequences
of the allele
(e.g., sequences that vary in less than 1/100 or 1/1000 members of the
populations).
A single nucleotide polymorphism (SNP) usually arises due to substitution
of one nucleotide for another at the polymorphic site. A transition is the
replacement of
one purine by another purine or one pyrimidine by another pyrimidine. A
transversion is
is the replacement of a purine by a pyrimidine or vice versa. Single
nucleotide
polymorphisms can also arise from a deletion of a nucleotide or an insertion
of a
nucleotide relative to a reference allele.
A "polymorphic profile" refers to one or more polymorphic forms for
which a subject is characterized. A polymorphic form is characterized by
identifying
2o which nucleotides) is (are) present at a polymorphic site in a nucleic acid
sample
acquired from a subject. The profile includes at least one polymorphic form
and
preferably includes a plurality of polymorphic forms, such as at least 5, 10,
20, 30, 40, S0,
60, 70, 80, 90 or 100 polymorphic forms or more. Polymorphic profiles are
similar when
the polymorphic profiles being compared share at least one polymorphic form at
least one
2s polymorphic site. Typically, similar polymorphic profiles share identity of
polymorphic
forms in at least 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90% or 100% in at
least
10, 20, 30, 40, 50, 60, 70, 100, or 500 polymorphic sites. Polymorphic forms
are identical
if the nucleotides) at a particular polymorphic site are the same. Thus, two
polymorphic
profiles each including 10 polymorphic forms are SO% identical if five of the
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
polymorphic forms in the two profiles are identical. If the organism is
diploid, then the
polymorphic forms at each polymorphic site are considered to be identical in
two
individuals if both individuals have the same two alleles at the polymorphic
site. For
example, an individual having alleles al and a2 at polymorphic site A is
considered to
have the same profile as an individual having alleles al and a2 but not to an
individual
i
having alleles al and al, or a2 and a2, or al and a3 and so forth.
The term "linkage" describes the tendency of genes, alleles, loci or genetic
markers to be inherited together as a result of their location on the same
chromosome, and
can be measured by percent recombination between the two genes, alleles, loci
or genetic
ro markers.
"Linkage disequilibrium" or "allelic association" means the preferential
association of a particular allele or genetic marker with a specific allele,
or genetic marker
at a nearby chromosomal location more frequently than expected by chance (see,
far
example, Weir, B., Genetic Data Analysis, Sinauer Associate Inc., 1996). For
example, if
15 locus X has alleles a and b, which occur equally frequently, and linked
locus Y has alleles
c and d, which occur equally frequently, one would expect the combination ac
to occur
with a frequency of 0.25. If ac occurs more frequently, then alleles a and c
are in linkage
disequilibrium. Linkage disequilibrium may result from natural selection of
certain
combination of alleles or because an allele has been introduced into a
population too
2o recently to have reached equilibrium with linked alleles.
A marker in linkage disequilibrium can be particularly useful in detecting
susceptibility to disease (or other phenotype) notwithstanding that the marker
does not
cause the disease. For example, a marker (X) that is not itself a causative
element of a
disease, but which is in linkage disequilibrium with a gene (including
regulatory
25 sequences) (Y) that is a causative element of a phenotype, can be detected
to indicate
susceptibility to the disease in circumstances in which the gene Y may not
have been
identified or may not be readily detectable.
to
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
"Haplotype" refers to a collection of polymorphic markers either in close
physical proximity on a single chromosome, or unlinked physically but
associated
together, which confers a biologic property or association with a phenotype.
A "nucleic acid" is a deoxyribonucleotide or ribonucleotide polymer in
either single- or double-stranded form, including known analogs of natural
nucleotides
unless otherwise indicated.
The term "genotype" as used herein broadly refers to the genetic
composition of an organism, including, for example, whether a diploid organism
is
heterozygous or homozygous for one or more alleles of interest.
to The term "primer" refers to a single-stranded oIigonucleotide capable of
acting as a point of initiation of template-directed DNA synthesis under
appropriate
conditions (i.e., in the presence of four different nucleoside triphosphates
and an agent for
polymerization, such as, DNA or RNA polymerase or reverse transcriptase) in an
appropriate buffer and at a suitable temperature. The appropriate length of a
primer
1s depends on the intended use of the primer but typically ranges from 15 to
30 nucleotides,
although shorter or longer primers can also be used. Short primer molecules
generally
require cooler temperatures to form sufficiently stable hybrid complexes with
the
template. A primer need not reflect the exact sequence of the template but
must be
sufficiently complementary to hybridize with a template. The term "primer
site" refers to
2o the area of the target DNA to which a primer hybridizes. The term "primer
pair" means a
set of primers including a 5' upstream primer that hybridizes with the 5' end
of the DNA
sequence to be amplified and a 3', downstream primer that hybridizes with the
complement of the 3' end of the sequence to be amplified.
The term "nucleic acid probe" refers to a nucleic acid molecule that binds
25 to a specific sequence or sub-sequence of another nucleic acid molecule. A
probe is
preferably a nucleic acid molecule that binds through complementary base
pairing to the
full sequence or to a sub-sequence of a target nucleic acid. Probes can bind
target
sequences lacking complete complementarity with the probe sequence depending
upon
the stringency of the hybridization conditions. The probes are typically
directly labeled as
11
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
with isotopes, chromophores, lumiphores, chromogens, or indirectly labeled
such as with
biotin to which a streptavidin complex can later bind. By assaying for the
presence or
absence of the probe, the presence or absence of the select sequence or sub-
sequence can
be detected.
A "label" is a composition detectable by spectroscopic, photochemical,
biochemical, immunochemical, or chemical means. For example, useful labels
include
'zP, fluorescent dyes, electron-dense reagents, enzymes (e.g., as commonly
used in an
ELISA), biotin, dioxigenin, or haptens and proteins for which antisera or
monoclonal
antibodies are available (e.g., by incorporating a radio-label into the
peptide, and used to
t0 detect antibodies specif cally reactive with the peptide). A label often
generates a
measurable signal, such as radioactivity, fluorescent light or enzyme
activity, which can
be used to quantitate the amount of bound label.
A "labeled nucleic acid probe" is a nucleic acid probe that is hound, either
covalently, through a linker, or through ionic, van der Waals or hydrogen
bonds to a label
15 such that the presence of the probe can be detected by detecting the
presence of the label
bound to the probe.
The phrase "selectively hybridizes to" refers to the binding, duplexing, or
hybridizing of a molecule only to a particular nucleotide sequence under
stringent
hybridization conditions when that sequence is present in a complex mixture
(e.g., total
2o cellular) DNA or RNA. The phrase "stringent hybridization conditions"
refers to
conditions under which a probe hybridizes to its target subsequence, but to no
other
sequences. Stringent conditions are sequence-dependent and are different in
different
circumstances. Longer sequences hybridize specifically at higher temperatures.
An
extensive guide to the hybridization of nucleic acids is found in Tijssen,
Techniques in
2s Biochemistry and~Molecular Biology--Hybridization with Nucleic Probes,
"Overview of
principles of hybridization and the strategy of nucleic acid assays" ( 1993).
Generally,
stringent conditions are selected to be about 5-10 °C lower than the
thermal melting point
(Tm) for the specific sequence at a defined ionic strength pH. The Tm is the
temperature
(under defined ionic strength, pH, and nucleic concentration) at which 50% of
the probes
12
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
complementary to the target hybridize to the target sequence at equilibrium
(as the target
sequences are present in excess, at Tm, 50% of the probes are occupied at
equilibrium).
Stringent conditions are those in which the salt concentration is less than
about 1.0
sodium ion, typically about 0.01 to 1.0 M sodium ion concentration (or other
salts) at pH
7.0 to 8.3 and the temperature is at least about 30 °C for short probes
(e.g., 10 to 50
nucleotides) and at least about 60 °C for long probes (e.g., greater
than 50 nucleotides).
Stringent conditions can also be achieved with the addition of destabilizing
agents as
formamide. If degenerate hybridization is desired, less than stringent
conditions are
necessary. For example, if single nucleotide mismatching is preferred,
hybridization
1o conditions will be relaxed with lower temperatures and higher salt content.
The term "statistical correlation" refers to a statistical association between
two variables or parameters as measured by any statistical test including, for
example,
chi-squared analysis, ANOVA or multivariate analysis. The correlation between
a
polymorphic form of DNA and a random variable or test parameter is considered
statistically significant if the probability of the result happening by chance
(the P-value) is
less than some predetermined level (e.g., 0.05). The tenor "statistically
significant
difference" refers to a statistical confidence level, P, that is < 0.05,
preferably < 0.01, and
most preferably < 0.001.
A "drug" or "pharmaceutical agent" means any substance used in the
2o prevention, diagnosis, alleviation, treatment or cure of a disease. The
terms include a
vaccine, for example.
The term "subject" or "individual" typically refers to humans, but also to
mammals and other animals, multicellular organisms such as plants, and single-
celled
organisms or viruses. "Tissue" means any sample taken from any subject,
preferably a
human. Tissues include blood, saliva, urine, biopsy samples, skin or buccal
scrapings,
and hair.
The term "patient" refers to both human and veterinary subjects.
13
CA 02350069 2001-05-09
WO 00/33161 PCTNS99/28582
II. General
The present invention provides methods, computer programs and
computerized systems useful for designing treatment studies and for evaluating
the
efficacy of various types of treatment procedures (e.g., clinical trials) as a
function of the
s genotype of a subject. The methods of the invention are designed to control
for underlying
genetic factors that may influence the response to a treatment. The present
invention is
based, in part, on the insight that controlling, either directly or
indirectly, genetic factors that
influence a patient's response to treatment can greatly increase the power of
the clinical
trial. Some methods are designed to reduce the genetic diversity of the
patient population so
to as to increase the probability of individuals sharing the same alleles at
genes involved in
response to the treatment. In cases where polymorphisms (usually in genes) are
known to
be associated with or cause differences in response to the treatment, these
polymorphisms
can be used directly in the design of the clinical trial.
For example, the invention provides methods for reducing the variance in
15 the biological condition or phenotype of interest by controlling for
genetic factors
influencing that phenotype. In the context of a clinical trial, the phenotype
of interest is
the response to a treatment. Genetic factors can be controlled in a number of
different
ways but the principle underlying the methods of the invention can be
illustrated by an
example. If the test parameter is measured in two groups, the first (which is
of size n) is
2o treated and the second (of size m) is untreated, the mean and variance of
these samples
can be calculated in the standard way (see Armitage & Berry, Statistical
Methods in
Medical Research, Blackwell Science, 1995.) Thus, for instance, in an example
the mean
and variance of the treated group are ,u, and s; respectively, and the mean
and variance
of the untreated group are fcz and s2 , respectively. Then an approximate
confidence
25 interval at a% (where, for example a = 0.95 ) for the difference in
response between the
two groups is given as,
z z
S1 S2
fy - fez ~ Zarz -n + m
14
CA 02350069 2001-05-09
WO 00/33161 PCTNS99/28582
where Za,2 is the value of the standard normal distribution that is exceeded
by chance in
a / 2% of cases.
Hence, any method that decreases the variance in either sample (i.e., which
decreases s; or s2 ) necessarily decreases the size of the confidence
interval.
Alternatively, when the variance of one of both of the samples is decreased,
the size of the
confidence interval can be held constant with fewer patients enrolled in the
trial (i.e., n
and/or m can be reduced). Thus, reducing the variance in response can lead
either to
greater certainty of a difference (here encapsulated by a smaller confidence
interval) or in
1'o a reduced sample size for the same statistical power. The variance can be
reduced in a
number of different ways as described in the following sections.
A. Selective enrollment of patients One approach is to control for potentially
confounding factors by increasing the homogeneity of the population. In the
context of
genetics, a set of polymorphic markers can be examined in a large group of
subjects and
15 those with similar polymorphic profiles enrolled in the treatment study.
Incorporating
genetic factors (represented by the polymorphic profile) into the
inclusion/exclusion criteria
of a treatment study allows an experimenter to reduce the variance in response
due to
underlying genetic factors.
B. Division of patient population into ~enetically homo;~enous subsets A
2o second approach is to categorize individuals into subsets depending on how
similar the
polymorphic profiles are to one another. Within each subset, subjects are
randomly
allocated into treatment or control subpopulations, as they are in a standard
clinical trial for
example. This method of dividing the subjects creates subsets that are
genetically more
homogenous than a random sample of the same size. This design is equivalent to
2s conducting several, small, independent treatment studies each of which
contains patients that
have more similar polymorphic profiles than expected by chance. Many
environmental
variables can be manifestations of underlying genetic factors. By examining
genetic
polymorphisms directly, it is possible not only to reduce variance due to
genetic factors that
are not directly observable, but also to improve the stratification based on
environmental
3o factors that are acting as surrogates for the underlying genetics factors
that control them. As
is
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
used herein, stratification refers to the division of the sample into subsets
that are more
similar than expected~by chance for a given factor.
C. Matching of patients for their polymorphic profiles. An alternative
approach
is to match the subjects in the treatment and control groups. That is, pairs
of individuals
with similar polymorphic profiles are sought and one is allocated to the
treatment group
while the other is placed in the control group. In this way, the difference in
response of each
pair can be examined where the pairs have been matched for their underlying
genetics. In
the same manner as described in section B above, matching on the basis of
genetic factors
can control new, previously unknown causes of variance due to genetic factors
and also
provide greater discriminatory power when matching by environmental factors
that have an
underlying genetic cause.
D. Using Qenes known to influence response in the design of clinical trials.
When one or more known polymorphisms is known to be associated with the
response to
treatment, these may be used directly to allocate patients into treatment and
control groups.
In the simplest case where a subject's polymorphic profile indicates whether
or not they will
respond to the treatment, this information can be used as an
exclusion/inclusion criterion at
the time of enrollment, thus reducing the sample size needed to observe a
given level of
response. Alternatively, all subjects can be enrolled in the treatment study
with the
treatment non-randomly assigned. For example, those known to be non-responders
by their
2o polymorphic profile can be treated according to a control procedure (e.g.,
administered a
placebo), while those who deemed responders from their polymorphic profile can
be given
the treatment procedure (e.g., administered a drug). This maximizes the
difference in
response between treatment and control groups. Conversely, non-responders can
be given
the treatment and responders the treatment. In this scenario, the minimum
difference
between treated and untreated subjects can be evaluated.
E. Use of genes known to influence response to determine dosine. When one
or more known polymorphisms is known to be associated with response to
treatment, this
information may be used to allocate the most appropriate dose to subjects
enrolled in a
treatment study such as a clinical trial. The polymorphic profiles of patients
can determine
3o the degree of response of individuals to the treatment. In this way, it may
be possible to
16
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
allocate different doses to different patient depending on their polymorphic
prof les. For
example, if a treatment potentially has side effects, it will be desirable to
administer the
minimum efficacious dose. This can vary for subjects with different
polymorphic profiles.
F. Post-trial analysis and sub-division The present invention can also be
used after the completion of a treatment study such as a clinical trial. Data
obtained from
such a treatment study are re-analyzed on subsets of the treated and control
populations
selected for similarity of a polymorphic profile to each other. The reanalysis
of data is
carried out on subsets of individuals sharing a similar polymorphic profile
and indicates
whether the treatment reaches statistical significance on individuals having
that profile. If
1.o the profile contains one or more polymorphic forms associated in some way
with the
biological condition of interest (e.g., disease), the treatment may reach
statistical
significance on the subpopulations when it does not on the initial treatment
populations.
If the profile does not contain such polymorphic DNA forms, then the re-
analysis of data
also shows a lack of statistical significance.
At this point, a further re-analysis is performed in which further
subpopulations of individuals from treated and control populations are
selected for
similarity to a second polymorphic profile. Because the individuals have
already been
characterized for polymorphic profile, the second re-analysis can be performed
without
further experimental work in a highly automated and iterative fashion. Again,
the second
2o analysis indicates whether the treatment reaches statistical significance
on the individuals
having similarity to the polymorphic profile by which subpopulations are
selected in the
second analysis.
Subsequent rounds of analysis can be performed according to the same
principles without further experimental work. A suitably programmed computer
can
perform thousand, millions or billions of cycles of analysis in which
different
subpopulations of individuals are selected based on similarity to different
polymorphic
profiles. Performing multiple tests typically requires a re-evaluation of the
p-value at
which a result is declared to be statistically significant to control the rate
of false positive
results. If after exhaustive analysis, statistical significance is not reached
for any
3o polymorphic profile, one can conclude with increased confidence that the
treatment
CA 02350069 2001-05-09
WO 00/33161 PCTNS99/28582
procedure (e.g., administration of a drug) being tested is unlikely to be
effective in any
significant portion of the population, and that further research is not
justified. If,
however, statistical significance is reached for a particular polymorphic DNA
profile, at
least two conclusions follow. First, in the case of a clinical trial on a drug
that the drug is
effective in at least a portion of the population, and further development of
the drug may
well be justified. Second, one knows the portion of the general population in
which the
drug is effective, this portion being defined by a polymorphic profile. This
profile can be
used as a diagnostic to identify patients appropriate for treatment when the
decision to
treat or a choice of treatments is made.
As an example of a method of the invention, a clinical trial can be carned
out as follows:
1. Identification and choice of polvmorphisms.
A set of polymorphisms is identified that allow the division of the patient
cohort into sub-groups. These polymorphisms may be known to be involved in the
test
parameter (e.g., the phenotype or endpoint) that is to be measured or can be
chosen at
random. (In the latter case, the genetic sub-groups may show identical results
with
respect to the phenotype of interest. This implies the method of grouping does
not
2o decrease the variance in the endpoint and the population can be re-analyzed
as a whole.
Thus, stratification by using genetic data does not have a deleterious effect
on the
experiment or trial, even in cases where it does not influence the outcome).
2. Genotyping of the cohort.
Some or all of the markers are genotyped in the entire cohort of patients
enrolled in the clinical trial. These data are then used either as
inclusion/exclusion criteria
(see 3a below) or to divide the cohort into subgroups (see 3b below).
3a. Inclusion/exclusion of patients using_~enetic information.
If some or all of the polymorphisms are known to influence the test
18
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
parameter that is to be measured, it may be appropriate to exclude individuals
when it is
known, a priori, they will present a particular phenotype or endpoint. In the
context of a
clinical trial, this can represent excluding those individuals who, by
information gained
from the set of polymorphisms examined, will not respond to the therapy.
3b. Division of the clinical trial into sub rgroups.
A metric is used to determine the genetic similarity of patients in the
cohort. This information is used to divide the population into subgroups that
have greater
genetic similarity than might be expected by chance. That is, the subgroups
are
t0 genetically more homogenous than a random subset of the same size.
The precise method of measuring similarity will depend on the number and
type of markers used. In the simplest case, the number of markers at which two
individuals have the same alleles can be used to determine similarity. Many
other more
complex metrics can be employed that, for example, giving extra weight to
markers
15 known to be particularly informative or that influence the test parameter
of interest.
By altering the method of determining genetic similarity, an experimenter
can control the number of subgroups that need to be formed. For N individuals,
this can
range from 1 (the entire population) to N (each individual is in a separate
subgroup).
Practical as well as scientific reasons are considered in determining how many
subgroups
20 are optimal for a given experiment or trial. With the methods of the
invention, groups can
be merged at a later time.
4. Allocation of treatment within the Genetic subgroups.
When the patients have been grouped into genetic subgroups based on
25 information from the set of polymorphism described in 1, several strategies
are available
for conducting a treatment study such as a clinical trial.
One method is to randomize the treatment and placebo within each
subgroup. This is similar to treating each subgroup as a separate experiment
or clinical
trial. Results of each subgroup may be analyzed separately or may be pooled
and then
3o analyzed.
19
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
Alternatively, treatment can be non-randomly allocated within the
subgroups. This may be appropriate, for example, when the polymorphisms are
known to
be associated with the outcome or endpoint of interest. For example, in the
context of a
clinical trial, if there are only two subgroups and one of the subgroups is
known to
contain high responders and the other low responders to a treatment,
allocating the
treatment to the first group and the placebo to the second group maximizes the
difference
between response for treated and untreated individuals. Conversely, allocating
the
placebo to the first group and the treatment to the second group shows the
minimum
difference between treated and untreated individuals. Which of these
approaches is most
1o appropriate depends on the exact objective of the experiment or clinical
trial.
S. Use of information from one experiment in the desi n of
subsequent studies.
The utility of stratifying by using a set of genetic polymorphisms can be
15 re-assessed through successive experiments of clinical trials.
Uninformative
polymorphisms can be dropped and new polymorphisms added to increase the
usefulness
of the set as a whole. Use of these polymorphisms in subsequent treatment
studies or
clinical trials leads to greater reproducibility of results and the need for
enrolling fewer
subjects in replication studies.
2o By identifying and correlating polymorphisms to a particular effect of a
drug, and thus reducing the variance due to genetic factors, a clinician can
devise clinical
trials that involve fewer subjects, decrease the confidence intervals, or
increase the
precision or discriminatory power of a given trial. The clinician can decide
which of
these three aspects of trial design or analysis to change while keeping the
other two
25 constant.
In addition to altering the statistic of variance which in turn can affect
subject number, precision or power of a study, using analysis of polymorphic
markers in a
clinical trial population in a manner as disclosed herein permits, upon
analysis, the
identification of subsets of polymorphic markers that may correlate with
either a
3o salubrious response, unresponsiveness or excessive response to a treatment,
an unwanted
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
or toxic response to a treatment, and may identify by virtue of
unresponsiveness, a clinical
subset of patients that define a "different" disease. In short, a post facto
genetic analysis
correlated with a specific clinical phenotype such as drug responsiveness or
unresponsiveness can reveal different etiologic mechanisms for the disease
being treated.
This is especially likely in the case of ethnic differences among patients
where each
ethnic group has a distinctive response to a treatment. Finally, analysis of
phenotypic
markers can provide insight into genetic diversity of the subjects being
treated allowing
the clinician to alter enrollment in a drug trial to accommodate more or less
genetic
diversity as is scientifically prudent.
to
III. Methods
A. General
In the methods of the invention, members of a treated and control
(untreated) population having a biological condition of interest (e.g., a
disease) are
characterized for polymorphic profile and a test parameter that is a measure
of the
biological condition, assuming the members have not already been so
characterized. The
members in the treated population have been (or are) treated according to a
treatment
procedure, whereas the members of the control population have been (or are)
treated
according to a control procedure.
2o To reduce total variance in the treatment assessment or study,
subpopulations fram the treated and control populations are selected for
similar genetic
composition such that the members in the two populations have similar or
identical
polyrnorphic profiles. The polymorphic profile of the subpopulations includes
one or
more polymorphic forms. Typically, the polymorphic profile includes a
plurality of
polymorphic forms, generally at least 5, in other instances at least 10, and
in still other
instances at least 100, or any number there between.
To minimize genetic variance between the treated and control
subpopulations, the polymorphic profiles for the two groups are selected to be
similar.
21
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
This means that there is at least one common polymorphic form between the two
subpopulations, although typically there are more. The polymorphic profiles
for the two
subpopulations are typically at least 10% identical, in some instances at
least SO%
identical, in still other instances greater than 75% identical, in yet other
instances 90%
identical or more, and in still other instances 100% identical. The analysis
of phenotypic
markers can provide additional insight into the genetic diversity of the
subjects being
treated and allows the researcher to alter the members in a study to
accommodate more or
less genetic diversity.
Polymorphisms can be selected in three distinct ways. First, they can be
to chosen at random. Second, only those polymoiphisms known or suspected to be
involved
in the phenotype of interest (response to treatment in the case of a clinical
trial) can be
selected. Third, DNA polymorphism selection can be driven by identifying
polymorphisms that reside in regions of the genome that have previously been
shown to
harbor a genetic linkage with the observable traits) under study. In the first
case, random
15 polymorphisms are unlikely to be directly involved in response. However,
they can be
used to determine the genetic similarity of patients and hence can be used to
form
subgroups that are more genetically homogenous than expected by chance. This
strategy
is particularly effective when a large number of (usually unknown) genes are
involved in
determining an individual's response to treatment. If there are polymorphisms
known to
2o be involved in response or to be associated with (possibly unknown) genes
involved in
response, then these can preferentially be used to determine subgroups of
patients.
Not all polymorphisms will be equally informative or useful in
determining subgroups. Factors such as the allele frequencies, whether the
polymorphism
is protein coding or non-coding, whether the polymorphism is in linkage
disequilibrium
25 with another polymorphism already in the polymorphic profile being used,
whether the
polymorphism is in linkage disequilibrium with a gene or genes known to be
involved in
response to treatment can all influence the utility of a given polymorphism.
Note that in
some cases, it may be desirable to give more weight to some polymorphisms than
others
in the formation of subgroups. That is, polymorphisms known to be associated
with
22
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
responsiveness may be more important (and hence given more weight) than random
polymorphisms.
The polymorphisms can be in genomic DNA, RNA or cDNA. While any
polymorphisms can be used, those of particular import are polymorphisms in
genes that
encode proteins that directly or indirectly influence a biochemical pathway
that is
correlated with the biological condition being measured or observed. Thus, for
example,
if a study involves assessing the efficacy of methods for treating patients
having elevated
blood cholesterol levels, the polymorphic profile can be tailored to include
polymorphisms located in genes known to be involved in cholesterol synthesis
and
to metabolism.
Once appropriate subpopulations have been selected such that the
subpopulations have the desired level of similarity in polymorphic profile, a
determination is made whether there is a correlation between the polymorphic
profile and
the efficacy (or lack thereof) of the treatment method by ascertaining whether
there is a
15 statistically significant difference in a test parameter between the
treated and control
subpopulations, where the test parameter is a measure or is representative of
the efficacy
of the treatment for the biological condition shared by members of the
subpopulation. A
finding of a statistically significant difference, indicates that the
polymorphic forms in the
polymorphic profile of the treated subpopulation correlate with the biological
condition
20 (e.g., the polymorphic profile is correlated with a particular disease) and
that the treatment
method under study is useful (or not beneficial) for treating subjects with
the biological
condition.
As noted above, such correlations are particularly important, for example,
in clinical trials on a drug. In some instances, the correlation identifies a
set of genetic
25 markers associated with the disease and thus has diagnostic value. In other
instances, the
correlation identifies markers that are associated with a positive treatment
result and thus
are important from a therapeutic standpoint.
A statistically significant difference in a test parameter between the
treatment and control subpopulations can be determined using standard methods
of
23
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
statistical analysis. Methods include, for example, the analysis of variance,
logistic
regression, cluster analysis, non-parametric statistics, contingency table
test and other
standard statistical tests.
B. Repetition of Method
The polymorphic profile of the subpopulation initially selected, often do
not correlate with a statistically significant difference in the test
parameter that is used to
measure the efficacy of treatment. In such instances, the method can be
repeated with
different subpopulations created by using an alternative definition or measure
of genetic
to similarity, or by dividing the population into greater or fewer sub-
populations. This
reflects the fact that there will rarely be a single unique way to group
patients. Indeed, for
a study with N individuals, it will often be possible to form any number of
sub-
populations from 1 (the entire population) to N (each individual in its own
sub-
population). Repeating the process is often an effective way of detecting
which
15 polymorphisms within the polymorphic profile are particularly informative
with respect
to the test parameter of interest. Once a correlation is identified,
additional cycles can be
repeated using, for example, a subset of the polymotphic forms utilized in an
earlier cycle
to determine whether the subset might show an even greater correspondence with
the test
parameter and thus treatment efficacy.
2o Typically the polymorphic forms within a polymorphic profile evolve over
time to account for a greater proportion of the genetic component of the
variance.
However, these polymorphic forms generally do not contribute equally. Some
account
for more variance than others; markers that do not correlate with differences
in the
treatment and control procedures are discarded from the analysis. The set of
markers as a
2s collection have value distinct from the individual markers. This collection
has enduring
value for understanding the genetic contribution to a distinct biological
condition of
interest. Individual markers can have diagnostic utility, as can the
collection.
24
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
The analysis of treatment or trial data involving groups and subgroups is
amenable to both parametric and non-parametric (distribution-free methods)
statistical
methods.
C. Treatment and Control Groups
The members of the treatment and control groups all share some biological
condition upon which the study is designed to determine whether the treatment
procedure
has a statistically significant different effect relative to the control
procedure. The
members of the treatment and control groups can be essentialiy any type of
organism
including, for example, humans, non-human animals, plants, bacteria and
viruses. In
1o some instances, the members are mammals (e.g., humans, primates) that are
part of a
clinical trial involving the testing of a pharmaceutical agent or behavioral
therapy for
example. The number of members in the subpopulations selected from the
treatment and
control group is at least one hut generally is more than one, typically at
least 5, in other
instances at least 10, and in still other instances at least 100 or more,
although any number
of members between these numbers can also be selected.
In some instances, the members of the subpopulation are selected not only
to have similar polymorphic profiles, but also to have other common features.
Selecting
on the basis of other commonalties can further reduce total variance beyond
that achieved
by reducing the variance attributable to genetic factors. Hence, for example,
members of
2o the treatment and control subpopulations can also be selected to have been
similarly
exposed to an environmental factor. Examples of such environmental factors
include, but
are not limited to, exposure to various agents such as radiation, chemicals,
and second
hand smoke; geographical location; and life style characteristics such as
sleep patterns,
diet, and amount of exercise. In some instances, it is useful to conduct
studies using
subpopulations that have not been similarly exposed to an environmental
factors; such
studies can be serve as a counterpoint to studies wherein the subpopulations
have been
selected for similar exposure to certain environmental factors. Besides
environmental
factors, members can also be selected to be from the same ethnic group or to
share a
common phenotypic trait (e.g., visual acuity, height, weight, physical
abnormality).
CA 02350069 2001-05-09
WO 00/33161 PCTNS99/28582
When the methods of the invention are utilized in clinical trials, typically
subjects in the two groups are not undergoing any other treatments for their
pathological
condition. In other instances, the study is designed so that subjects in both
treated and
untreated groups are undergoing the same alternative treatment.
D. Treatment and Control Procedures
The types of treatment and control procedures vary according to the
biological condition to which the treatment is directed. As noted above, the
biological
. conditions can be any of a number of conditions, such as a pathological
condition or
simply a biological susceptibility, for example. A variety of different
procedures can be
performed when the biological condition is a pathological condition. In many
instances,
the procedures involve administering a pharmaceutical agent, including, for
example:
1) administering a pharmaceutical agent to members of the treated population
and giving
members in the control population a placebo or nothing at all, 2) giving
members of the
treated population one pharmaceutical agent (or combination of pharmaceutical
agents)
and a different pharmaceutical agent (or combination of pharmaceutical agents)
to the
control members; 3) providing one quantity of a pharmaceutical agent to the
treated
population and a different amount to the control population, or 4)
administering a
pharmaceutical agent to the treatment and control populations according to
different
schedules.
Instead of administering a pharmaceutical agent, the treatment procedure
can include some type of behavioral therapy. Examples of such therapy include,
but are
not limited to, a particular diet regime (e.g., low fat, low sodium, high
protein, or a
restricted calorie diet), a prescribed exercise regime (e.g., exercising for a
certain time
2s period a certain number of times a week, performing low-impact exercises,
exercising to
reach a target heart rate, therapies that work certain muscle groups),
meditation, yoga, and
stress reduction techniques. Of course, the treatment procedure can include
combinations
of the foregoing procedures as well. Members in control groups may not undergo
therapy
at all or may be treated in opposing fashion (or may already be engaged in
contrary
26
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
behaviors). For example, if the treatment group is placed on a low caloric
diet, members
in the control group can be placed on a high caloric diet or can simply be
selected for
those whose normal diet already is a high caloric diet and thus is not
altered.
The treatment procedure can also be directed towards a biological
s susceptibility or resistance rather than a pathological condition. Thus, for
example, in the
case of plants, plants can be treated with various agricultural agents used to
affect plant
growth or health (e.g., fertilizer or other growth stimulants, herbicides,
insecticides, and
pH altering agents) to assess the effect of such agents on various
susceptibilities or
resistances of plants (e.g., susceptibility to frost or freeze damage and
resistance to
to herbicides). In like manner, humans or other organisms can also be treated
with various
agents, for example vaccines, to determine the effect of the agents on various
susceptibilities or resistances.
E. Utility
15 The reduction in variance achieved by the methods of the invention
enables researchers to selectively optimize treatment studies. For example, as
the genetic
variance decreases, the confidence level of the statistical analysis
increases. Thus, with
the methods of the invention, researchers can more confidently attribute
differences in
effects as seen between the treated subjects and the control subjects to the
treatment
2o administered, rather than being consequences of genetic differences between
patients.
Furthermore, differences between control and test groups can be appreciated
sooner. This
allows smaller, less costly studies to be performed that have the same
statistical power as
much larger studies that do not match for the underlying genetics.
Alternatively, a study
in which patients are matched for genetic factors will be able to detect much
smaller
25 difference in response between treated and untreated individuals than a
study of the same
size that ignores genetics factors. This allows for less costly studies, more
rapid
assessment regarding the feasibility and desirability of additional treatment
studies, and
ultimately, in the case of clinical trials on pharmaceutical compounds for
example, allows
for more rapid marketing of the pharmaceuticals.
27
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
The methods also enable more efficient treatment studies to be designed.
For instance, once polymorphisms that correlate with pathological conditions
have been
identified, subjects that have the polymorphisms as well as the biological
condition can be
identified and enrolled in additional studies to analyze the effect that other
treatments
have on the biological condition of interest. Because subjects that will not
respond to the
treatment are not enrolled, fewer subjects need to be enrolled. Alternatively,
if a set of
polymorphisms emerges that, when matched between patients in a control and
test arms
of a trial, is highly correlative with the biological condition being studied,
subsequent
trials of the efficacy of a treatment can be tested with fewer patients
regardless of
1o response rate if the biological condition being measured has a genetic
component. In
addition, when polymorphisms associated with differential response are
identified, it may
be possible to tailor the dose a specific patient receives to be optimal given
their
polymorphic profile. This will be particularly important when there are
unwanted side
effects of the treatment and it is desirable to give the minimum efficacious
dose.
Furthermore, as noted above, the treatment methods described herein
permit the identif cation of subsets of polymorphic forms that correlate with
either a
favorable response or unresponsiveness to treatment, or an unwanted or toxic
response to
a treatment. Clinical trials on the efficacy of certain phanmaceutical
treatments can
identify individuals that are unresponsive to treatment and, in so doing, can
in some
2o instances result in the identif cation of a clinical subset of patients
that define a "different"
disease. Such correlations can also be used as a prognostic and/or diagnostic
toot to
identify subjects having or likely to acquire a disease or to select
appropriate treatment
procedures for a subject based upon the particular genetic composition of the
subject.
Information gained from clinical trials in which patients are genotyped for
a set of polymorphic genetics markers can also be used in other stages of drug
discovery
and development. For example, genes shown to be associated with response via
the
polymorphic profile of the patients may be amenable to intervention and hence
represent
potential drug targets. Furthermore, identification of treatments that show
low efficiency
(i.e., many non-responders) or that have high rates of adverse events can be
identified by
3o examining the polymorphism profile of patients in early phase trials. This
information
28
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
can then be used in the decision whether to take a treatment forward into
large and more
costly trials. For example, if non-response is associated with a polymorphism
profile that
is common in the general population, it may be inappropriate to use the
treatment in a
larger trial.
IV. Computer Systerns and Pro rg ams
FIG. 1 depicts a representative computer system 10 suitable for
implementing certain methods of the present invention. As shown in FIG. 1,
computer
system 10 typically includes a bus 12 that interconnects major subsystems such
as a
1o central processor 14, a system memory 16, an input/output controller 18, an
external
device such as a printer 23 via a parallel port 22, a display screen 24 via a
display adapter
26, a serial port 28, a keyboard 30, a fixed disk drive 32 via storage
interface 34, and a
floppy disk drive 33 operative to receive a floppy disk 33A. Many other
devices can be
connected such as a scanner 60 via I/O controller 18, a mouse 36 connected to
serial port
28, a CD ROM player 40 operative to receive a CD ROM 42, or a network
interface 44.
Source code to implement the present invention can be operably disposed in
system
memory 16 or stored on storage media such as a fixed disk 32 or a floppy disk
33A.
Other devices or subsystems can be connected in a similar manner. All of the
devices
shown in FIG. 1 are not required to practice the present invention. The
devices and
2o subsystems can also be interconnected in different ways from that shown in
FIG. 1. The
operation of a computer system 10 such as that shown in FIG. 1 is readily
known in the
art; hence, operations of the system are not described in detail herein.
FIG. 2 is an illustration of a representative computer system 10 of FIG. 1
suitable for performing the methods of the present invention; however, FIG. 2
depicts but
one example of many possible computer types or configurations capable of being
used
with the present invention. As depicted in FIG. 2, computer system 10 can
include
display screen 24, cabinet 20, keyboard 30, a scanner 60, and mouse 36. Mouse
36 and
keyboard 30 are examples of "user input devices." Other examples of user input
devices
include, but are not limited to, a touch screen, light pen, track ball and
data glove.
29
CA 02350069 2001-05-09
WO 00/33161 PCTNS99/28582
Mouse 36 can have one or more buttons such as buttons 37. Cabinet 20
houses familiar computer components such as floppy disk drive 33, a processor
14 and a
storage means (see FIG. 1 ). As used in this specification "storage means"
includes any
storage device capable of storing data that can be used in connection with a
computer
system. Examples of such devices include, but are not limited to, disk drives,
magnetic
tape, solid-state memory and bubble memory. Cabinet 20 can include additional
hardware such as input/output (I/O) interface for connecting computer system
10 to
external devices such as a scanner 60, external storage, other computers or
additional
peripheral devices.
1o In some instances, system 10 includes a computer having a Pentium~
microprocessor 14 that runs 1~VINDOWS~ Version 3.1, WINDOWS95~ or
WINDOWS98~ operating system by Microsoft Corporation. However, the methods of
the invention can easily be adapted to other operating systems (e.g., UNIX)
without
departing from the scope of the present invention.
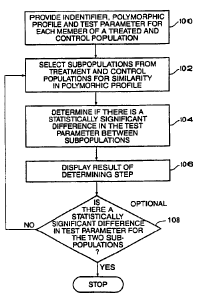
FIG. 3 is a flowchart of simplified steps in one computerized method of
the invention for assessing a treatment procedure. In step 100 a database is
provided that
contains a plurality of designations for each member of a population that has
either been
treated according to a treatment procedure or a control procedure and that
shares a
common biological condition (the database is described further below). Hence,
the
2o population includes both treatment and control populations. One group of
designations is
to identify each member of the two populations. There are also typically
separate
designations for a polymorphic profile and a test parameter for each member of
the
population. Subpopulations from the treated and control populations are
selected in a
selection step 102 for similarity in polymorphic profile. In determining step
104, a
determination is made whether there is a statistically significant difference
in the test
parameter between the subpopulations. A statistically significant difference
indicates that
the polymorphic profile of the subpopulations correlates with the biological
condition and
effect of treatment. Finally, in a displaying step 106, an output of the
result of the
determining step is displayed on an output device to facilitate the analysis.
In an optional
3o decisional step 108, if there is not a statistically significant difference
in the test parameter
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
for the two subpopulations, the selecting step 102, the determining step 104,
and the
displaying step 106 are repeated using subpopulations that have a polymorphic
profile
that is different from that in earlier cycles.
Hence, the microprocessor in the computer system of the present invention
is operatively disposed relative to the system memory, the system bus and the
input/output so as to perform the foregoing functions. For example, the
processor
provides or receives data that comprises designations for each member of the
treated and
control populations, as well as designations for a polymorphic profile and a
test parameter
for each member of the two populations. The microprocessor is also operatively
disposed
to to select a subpopulation from each of the treatment and control
populations for similarity
in polymorphic profile, determine whether there is a statistically significant
difference in
the test parameter between the subpopulations and display an output of the
result
obtained.
The computer program of the invention includes code for providing or
receiving data comprising the various designations for the identity of the
members of the
test and control populations, their polymorphic profiles and test parameter
results. The
program also includes code necessary to perform the selecting, determining and
displaying steps set forth above.
2o V. Methods for Determining Pol ry norphic Profiles
A. Preparation of Samples
Polymorphisms are detected in a target nucleic acid from an individual
being analyzed. For assay of genomic DNA, virtually any biological sample
(other than
pure red blood cells) is suitable. For example, convenient tissue samples
include whole
blood, semen, saliva, tears, urine, fecal material, sweat, buccal, skin and
hair. For assay
of cDNA or mRNA, the tissue sample must be obtained from an organ in which the
target
nucleic acid is expressed. For example, if the target nucleic acid is a cDNA
encoding
cytochrome P450, the liver is a suitable source.
31
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
Many of the methods described below require amplification of DNA from
target samples. This can be accomplished by e.g., PCR. See generally, PCR
TECHNOLOGY: PRINCIPLES AND APPLICATIONS FOR DNA AMPLIFICATION (ed. H.A.
Erlich,
Freeman Press, NY, NY, 1992); PCR PROTOCOLS: A GUIDE TO METHODS AND
APPLICAT10NS (eds. Innis, et al., Academic Press, San Diego, CA, 1990);
Mattila, et al.,
Niccleic Acids Res. 19:4967 (1991); Eckert, et al., PCR Methods and
Applications 1:17
(1991); PCR (eds. McPherson, et al., IRL Press, Oxford); and U.S. Patent
4,683,202
(each of which is incorporated by reference for all purposes).
Other suitable amplification methods include the ligase chain reaction
to (LCR) (see Wu & Wallace, Genomics 4:560 (1989), Landegren, et al., Science
241:1077
(1988), transcription amplification (Kwoh, et al., Proc. Nat'I Acad. Sci. USA
86:1173
(1989)), and self sustained sequence replication (Guatelli, et al., Proc.
Nat'I Acad. Sci.
USA 87:1874 (1990)) and nucleic acid based sequence amplification (NASBA). The
latter two amplification methods involve isothermal reactions based on
isothermal
15 transcription, which produce both single stranded RNA (ssRNA) and double
stranded
DNA (dsDNA) as the amplification products in a ratio of about 30 or 100 to 1,
respectively.
B. Detection of Pol ryorphisms in Target DNA
2o There are two distinct types of analysis depending whether a
polymorphism in question has already been characterized. The first type of
analysis is
sometimes referred to as de novo characterization. This analysis compares
target
sequences in different individuals to identify points of variation, i.e.,
polymorphic sites.
The second type of analysis involves determining which fonm(s) of a
characterized
25 polymorphism are present in individuals under test. There are a variety of
suitable
procedures for determining polymorphic forms and thus polymorphic profiles,
including,
for example, the methods that follow.
32
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
Allele-Specific Hybridization (ASH)
ASH technology is based on the stable annealing of a short, single-
stranded, oligonucleotide probe to a completely complementary single-strand
target
nucleic acid. Hybridization is detected from a radioactive or non-radioactive
label on the
probe. For each polymorphism, two or more different probes are designed to
have
identical DNA sequences, except at the polymorphic nucleotides. Each probe has
exact
homology with one allele sequence so that the complement of probes can
distinguish all
the alternative allele sequences. With appropriate probe design and stringency
conditions,
a single-base mismatch between the probe and target DNA prevents
hybridization. In this
to manner, only one of the alternative probes hybridizes to a target sample
that is
homozygous for an allele (an allele is defined by the DNA homology between the
probe
and target). Samples containing DNA that is heterozygous for two alleles
hybridize to
both of two alternative probes. Details regarding ASH are described, for
example, by
Saiki, et al., Nature 324:163-166 (1986); Dattagupta, EP 235,726; Saiki, WO
89/11548;
15 and in U.S. Patent 5,468,613.
2. Restriction Fragment Len tg-h Pol~rphisms (RFLP)
The phrase "restriction fragment length polymorphism" or "RFLP" refers
to inherited differences in restriction enzyme sites (for example, caused by
base changes
in the target site), or additions or deletions in the region flanked by the
restriction enzyme
20 site that result in differences in the lengths of the fragments produced by
cleavage with a
relevant restriction enzyme. A point mutation leads to either longer fragments
if the
mutation is within the restriction site or shorter fragments if the mutation
creates a
restriction site. Additions and transposable elements lead to longer fragments
and
deletions lead to shorter fragments.
25 An RFLP can be used as a genetic marker in the determination of
segregation of alleles with quantitative phenotypes. In one embodiment of the
invention,
the restriction fragments are linked to specific phenotypic traits. More
specifically, the
presence of a particular restriction fragment can be used to predict the
prevalence of a
specific phenotypic trait.
33
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
3. Direct-Sequencing
Polymorphisms can be analyzed directly using the traditional dideoxy-
chain termination method or the Maxam -Gilbert method (see Sambrook, et al.,
MOLECULAR CLONING, A LABORATORY MANUAL (2nd Ed., CSHP, New York 1989); and
Zyskind et al., RECOMBINANT DNA LABORATORY MANUAL, (Acad. Press, 1988)). Other
nucleic acid sequencing methods, including, but not limited to, fluorescence-
based
techniques (U.S. Patent No. 5,171,534), mass spectroscopy (U. S. Patent No.
S,I74,962)
and capillary electrophoresis (U.S. Patent No. 5,728,282) can also be used.
4. Drag-TaQ~in~ Oli~onucleotides for Electrophoresis
IO Oligonucleotides having additional chemical moieties that cause
differential mobilities in an electrophoretic separation system can be used in
analyzing
polymorphisms. The addition of a molecular species increases the apparent
molecular
weight of a piece of amplified DNA, even DNA having only a single nucleotide
polymorphism present. The added species, or drag tags, can be attached
covalently or
15 non-covalently to the nucleic acid, either before or after labeling (for
visualization of the
electrophoretic band). Any charge associated with the added species is blocked
or
neutralized so that nucleic acid mobility remains dependent on size and not
charge.
Any of a number of different moieties can be attached to a nucleic acid to
form a plurality of different sized amplification products. Examples of such
moieties
2o include, but are not limited to, phosphate monomers, acrylamide and
polypeptides. Thus,
for instance, prior to amplification of a polymorphic stretch of DNA,
phosphate monomer
units can be attached to each nucleotide monomer that is to be used in the PCR
reaction.
For example, assume one phosphate monomer is added to dATP; two phosphate
monomers
are added to dCTP;. three phosphate monomers are added to dGTP; and four
phosphate
25 monomers are added to dTTP. The resulting amplified polymorphic nucleic
acids contain
different amounts of phosphate monomers depending on the nucleotide content.
Hence,
while the amplified products have the same numbers of base pairs, the
different
polymorphic forms nonetheless may be size separated on an electrophoretic gel
due to
differences in phosphate monomer content. In a variation of this general
approach, the
34
CA 02350069 2001-05-09
WO 00/33161 PCTNS99/28582
monomer units are added after amplification to specific nucleotides or to non-
amplified
nucleic acids prior to separation on the basis of size (e.g., by capillary
electrophoresis).
5. Isozyme Markers
Other embodiments include identification of isozyme markers and allele-
specific hybridization. Isozymes are a group of enzymes that catalyze the same
reaction
but vary in physical properties resulting from differences in amino acid
sequence (and
hence nucleic acid sequence). Some isozymes are multimeric enzymes containing
slightly different subunits. Other isozymes are either multimeric or monomeric
but have
been cleaved from the proenzyme at different sites in the amino acid sequence.
Nucleic
1o acid variation of isozymes can be determined by hybridizing primers that
flank a variable
portion of an isozyme nucleic acid sequence to target nucleic acids contained
in a sample
obtained from an organism. The variable region is amplified and sequenced.
From the
sequence, the different isozymes are determined and linked to phenotypic
characteristics.
6. Amplified Variable Se,~uences
15 Amplified variable sequences of the genome and complementary nucleic
acid probes also can be used as polyrnorphic markers. The phrase "amplified
variable
sequences" refers to amplified sequences of the genome that exhibit high
nucleic acid
residue variability between members of the same species. All organisms have
variable
genomic sequences and each organism (with the exception of a clone) has a
different set
20 of variable sequences. The presence of a specific variable sequence can be
used to predict
phenotypic traits. A variable sequence of DNA can be amplified (e.g.,
utilizing the
amplification techniques listed above) by template-dependent extension of
primers that
hybridize to flanking regions of the DNA obtained from a subject. The
amplified
products can then be sequenced.
25 7. Allele-Specific Primers and Hybridization
An allele-specific primer hybridizes to a site on target DNA overlapping a
polymorphism and only primes amplification of an allelic form to which the
primer
exhibits perfect complementarity. This primer is used in conjunction with a
second
CA 02350069 2001-05-09
WO 00/33161 PCTNS99/28582
primer that hybridizes at a distal site. Amplification proceeds from the two
primers and
produces a detectable amplified product that can be characterized for the
particular allelic
form present in a nucleic acid sample. See, e.g., Gibbs, Nucleic Acid Res.
17:2427-2448
(1989) and WO 93/22456.
8. Single-Strand Conformation Polymorphism Analysis
Alleles of target sequences can be differentiated using single-strand
conformation polymorphism analysis, which identifies base differences by
alteration in
electrophoretic migration of single-stranded PCR products, (see, e.g., Orita,
et al., Proc.
Nat'1 Acad. Sci. USA 86:2766-2770 (1989). Typically, amplified PCR products
are
io denatured (e.g., according to known chemical or thermal methods) to form
single-
stranded amplification products that can refold or form secondary structures,
depending in
part upon the base sequence of the product. The different electrophoretic
mobilities of
single-stranded amplification products can be related to base-sequence
difference between
alleles of target sequences.
I s 9. Self sustained Sequence Replication
Polymorphisms can also be identified by self sustained sequence
replication. In this approach, target nucleic acid sequences are amplified
(replicated)
exponentially in vitro under isothermal conditions using three enzymatic
activities
involved in retroviral replication: (1) reverse transcriptase, (2) RNase H,
and (3) a DNA-
2o dependent RNA polymerise (Guatelli, et al., Proc. Natl. Acid. Sci. USA
87:1874 (1990)).
By mimicking the retroviral strategy of RNA replication by means of cDNA
intermediates, cDNA and RNA copies of the original target are accumulated.
10. Arbitrary Fragment Length Polymorphisms ~AFLP)
Arbitrary fragment length polymorphisms (AFLP) can also be used as
2s polymorphisms (Vos, et al., Nucl. Acids Res. 23:4407 (1995)). The phrase
"arbitrary
fragment length polymorphism" refers to selected restriction fragments that
are amplified
before or after cleavage by a restriction endonuclease. The amplification step
permits
36
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
easier detection of specific restriction fragments as compared to determining
the size of
all restriction fragments and comparing the sizes to a known control.
AFLP allows the detection of a large number of polymorphic markers (see,
szrpra) and has been used for genetic mapping of plants (Becker, et al., Mol.
Gen. Genet.
s 249:65 (1995); and Meksem, et al., Mol. Gen. Genet. 249:74 (1995)) and to
distinguish
among closely related bacterial species (Huys, et al., Int'1 J. Systematic
Bacteriol. 46:572
( 1996)).
11. Simple Sequence Repeats (SSR,
SSR methods are based upon high levels of di-, tri- or tetra-nucleotide
Io tandem repeats within a genome. Dinucleotide repeats have been reported to
occur in the
human genome as many as 50,000 times with n varying from 10 to 60 (Jacob, et
al., Cell
67:213 (1991)). The dinucleotide repeats have also been found in higher plants
(Condit &
Hubbell, Genome 34:66 (1991)).
SSR data is generated by hybridizing primers to conserved regions of the
15 genome that flank the SSR region. The dinucleotide repeats between the
primers are
amplified by PCR. The resulting amplified sequences are then electrophoresed
to
determine the size, and therefore the number, of di-, tri- and tetra-
nucleotide repeats.
12. Denaturine Gradient Gel Electrophoresis
Amplification products generated using the polymerise chain reaction can
2o be analyzed through the use of denaturing gradient gel electrophoresis.
Different alleles
are identified based on the different sequence-dependent melting properties
and
electrophoretic migration of DNA in solution. Erlich, ed., PCR TECHNOLOGY,
PRINCIPLES AND APPLICATIONS FOR DNA AMPLIFICATION, (W.H. Freeman and Co, New
York, 1992), Chapter 7.
25 13. Single base extension methods
Polymorphisms can also be detected by single base extension. A primer is
designed to hybridize to a target sequence so that the 3' end of the primer
immediately
abuts but does not overlap a polymorphic site. The target sequence is then
contacted with
37
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
primer and at least one nucleotide (typically labelled), that is complementary
to the base
occupying the polymorphic site in one allelic form. If that allelic form is
present, then the
primer is extended and becomes labelled. In some methods, biallelic
polymorphic sites
are analyzed by including two differentially labelled dideoxynucleotides
respectively
complementary to bases occupying the polymorphic site in first and second
allelic forms
of the target. Analysis of label present in the extended primer indicates
whether one or
both of the allelic forms are present in a target sample.
C. High Throughput Screening
In some instances, identification of polymorphisms is done by high
throughput screening. In one embodiment, high throughput screening involves
providing
a library of polymorphic forms of DNA including RFLPs, AFLPs, isozymes,
specific
alleles and variable sequences, including SSR. Such "libraries" are then
screened against
genoinic DNA from the subjects in the treatment study. Once the polymorphic
alleles of
15 a subject have been identified, a link between the polymorphic DNA and the
treatment
effect can be determined through statistical associations.
Such high throughput screening can be performed in many different
formats. For example, for those methods involving hybridization reactions,
hybridization
can be performed in a 96-, 324-, or a 1024-well format or in a matrix on a
silicon chip. In
2o a well-based format, a dot blot apparatus is used to deposit samples of
fragmented and
denatured genomic DNA on a nylon or nitrocellulose membrane. After cross-
linking the
nucleic acid to the membrane, either through exposure to ultra-violet light if
nylon
membranes are used or by heat if nitrocellulose is used, the membrane is
incubated with a
labeled hybridization probe. The membranes are washed extensively to remove
non-
25 hybridized probes and the presence of the label on the probe is determined.
The labels are incorporated into the nucleic acid probes by any of a number
of methods well known to those of skill in the art. In some instances, a label
is
simultaneously incorporated during the amplification procedure in the
preparation of the
nucleic acid probes. Thus, for example, polymerase chain reaction (PCR) with
labeled
38
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
primers or labeled nucleotides provide labeled amplification product. In
another
embodiment, transcription amplification using a labeled nucleotide (e.g.,
fluorescein-
labeled UTP and/or CTP) incorporates a label into the transcribed nucleic acid
probes.
Detectable labels suitable for use in the present invention include any
composition detectable by spectroscopic, radioisotopic, photochemical,
biochemical,
immunochemical, electrical, optical or chemical means. Useful labels in the
present
invention include biotin for staining with labeled streptavidin conjugate,
magnetic beads,
fluorescent dyes (e.g., fluorescein, Texas red, rhodamine, green fluorescent
protein, and
the like), radiolabels (e.g., 'H, 'ZSI,'sS, ~"C, or'ZP), enzymes (e.g., horse
radish peroxidase,
to alkaline phosphatase and others commonly used in an ELISA), and
colorimetric labels
such as colloidal gold or colored glass or plastic (e.g., polystyrene,
polypropylene, latex,
etc.) beads. Patents teaching the use of such labels include U.S. Patent Nos.
3,817,837;
3,850,752; 3,939,350; 3,996,345; 4,277,437; 4,275,149; and 4,366,241.
Methods for detecting such labels are also well known to those of skill in
the art. Thus, for example, radiolabels are detected using photographic film
or
scintillation counters and fluorescent markers are detected using a
photodetector to detect
emitted light. Enzymatic labels are typically detected by providing the enzyme
with a
substrate and detecting the reaction product produced by the action of the
enzyme on the
substrate, and colorimetric labels are detected by simply visualizing the
colored label.
2o A number of well-known robotic systems have been developed for high
throughput screening, particularly in a 96 well format. These systems include
automated
workstations like the automated synthesis apparatus developed by Takeda
Chemical
Industries, LTD. (Osaka, Japan) and many robotic systems utilizing robotic
arms (Zymate
II, Zymark Corporation, Hopkinton, Mass.; Orca, Hewlett-Packard, Palo Alto,
Calif.) that
mimic the manual 'synthetic operations performed by a chemist. Any of the
above devices
are suitable for use with the present invention. The nature and implementation
of
modifications to these devices (if any) so that they can operate as discussed
herein will be
apparent to persons skilled in the relevant art.
39
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
In addition, high throughput screening systems themselves are
commercially available (see, e.g., Zymark Corp., Hopkinton, MA; Air Technical
Industries, Mentor, OH; Beckman Instruments, Inc. Fullerton, CA; Precision
Systems,
Inc., Natick, MA, etc.). These systems typically automate entire procedures
including all
sample and reagent pipetting, liquid dispensing, timed incubations, and final
readings of
the microplate or membrane in detectors) appropriate for the assay. These
configurable
systems provide high throughput and rapid start up as well as a high degree of
flexibility
and customization. The manufacturers of such systems provide detailed
protocols the
various high throughput.
to
D. Solid-Phase Arrays
Polymorphic forms of DNA can also be identified by hybridization to
nucleic acid arrays, some examples of which are described by WO 95/11995
(incorporated by reference in its entirety for all purposes). In one variation
of the
1s invention, solid phase arrays are adapted for the rapid and specific
detection of multiple
polymorphic nucleic acids. Typically, a nucleic acid probe is linked to a
solid support
and a target nucleic acid is hybridized to the probe. Either the probe, or the
target, or
both, can be labeled, typically with a fluorophore. If the target is labeled,
hybridization is
detected by detecting bound fluorescence. If the probe is labeled,
hybridization is
2o typically detected by quenching of the label by the bound nucleic acid. If
both the probe
and the target are labeled, detection of hybridization is typically performed
by monitoring
a color shift resulting from proximity of the two bound labels.
The construction and use of solid phase nucleic acid arrays to detect target
nucleic acids has been described extensively in the literature. See, Fodor, et
al., Science
25 251:767 (1991); Sheldon, et al., Clin. Chem. 39(4):718 (1993); Kozal, et
al., Nature
Medicine 2(7):753 (1996) and Hubbell, U.S. Pat. No. 5,571,639. See also,
Pinkel, et al.,
PCT/L1S95/16155 (WO 96/17958). In addition to being able to design, build and
use
probe arrays using available techniques, one of skill is also able to order
custom-made
CA 02350069 2001-05-09
WO 00/33161 PCTNS99/28582
arrays and array-reading devices from manufacturers specializing in array
manufacture.
For example, Affymetrix in Santa Clara CA manufactures DNA VLSIPTM arrays.
It will be appreciated that probe design is influenced by the intended
application. For example, where several probe-target interactions are to be
detected in a
single assay, e.g.; on a single DNA chip, it is desirable to have similar
melting
temperatures for all of the probes. Accordingly, the lengths of the probes are
adjusted so
that the melting temperatures for all of the probes on the array are closely
similar
(different lengths for different probes may be needed to achieve a particular
Tm where
different probes have different GC contents). Although melting temperature is
a primary
to consideration in probe design, other factors are optionally used to further
adjust probe
construction.
E. Capillary and Microchannel Plate Electrophoresis
As described above, certain methods for identifying polymorphisms
15 involve size based separations (e.g., RFLP and SSR). In such cases,
capillary
electrophoresis can be used to analyze polymorphism. Such techniques are
described in
detail in U.S. Patent Nos. 5,534,123 and 5,728,282, which are incorporated
herein by
reference. Briefly, capillary electrophoresis tubes are filled with the
separation matrix.
The separation matrix contains hydroxyethyl cellulose, urea and optionally
fonmamide.
2o The RFLP or SSR samples are loaded onto the capillary tube and
electrophoresed.
Because of the small amount of sample and separation matrix required by
capillary
electrophoresis, the run times are very short. The molecular sizes and
therefore the
number of nucleotides present in the nucleic acid sample are determined by
techniques
described herein.
25 Electrophoresis can also be performed in microchannel plates. These
plates have channels less than 100 p in diameter etched on a solid substrate.
By virtue of
their smaller dimensions, they allow for even faster separations of nucleic
acids in a
separation matrix. Using these etched plates, samples can be evaluated with a
high
throughput format.
41
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
In another high throughput format, multiple capillary tubes are placed in a
capillary electrophoresis apparatus. Samples are loaded onto the tubes and
electrophoresis of the samples is run simultaneously. See, for example,
Mathies &
Huang, Nature 359:167 (1992). Because the separation matrix is of low
viscosity, after
each run, the capillary tubes can be emptied and reused.
The following examples are offered to further illustrate specific aspects of
the present invention and are not to be interpreted so as to limit the scope
of the present
invention.
EXAMPLE 1
to Effect of Genetic Matching on Sample Size and Confidence
The invention can be illustrated by the example of studying serum
cholesterol and the effects drugs may have on this biological condition. It
has been
established that up to 80% of the variance in serum cholesterol can be
attributed to
genetics (see, for example, R. A. King, J. h Rotter & A. (i. Motulsky, THE
GENETICS
BASIS OF COMMON DISEASE, Oxford University Press, 1992).
The utility of matching patients at genetic loci is that the confidence,
sample
size or discriminating power of a given study can be favorably affected by
genetic matching.
For example, if a study is required to have 80% power to detect a difference
of 20mg/dl at
the 5% level and za =1.645 (representing the value from the standard normal
distribution
2o which is exceeded in S% of cases) and zb = 0.842 (the value of the standard
normal
distribution that is exceeded in 80% of cases) the minimum sample size may be
calculated
as follows:
za <- qnorm (al) [.95 @ 1.645]
zb <- qnorm (be) [.8 @.842]
If the genetic contribution to the variance of a variable x is 80% and the
variance is 1600
(the standard deviation squared for cholesterol) then the sample size is:
2 x (za + zb)Z x variance
(difference)2
42
CA 02350069 2001-05-09
WO 00/33161 PCTNS99/28582
or
or
2 x (2.49)z x 1600 = [ 1600 ]
(20)2 [400 ]
SO
Thus 50 patients per arm of the study are needed to see a difference of
cholesterol of 20mg/dl. If the patients are genetically matched (i.e., the 80%
contribution to
1 o variance is eliminated), the number of patients for each arm is reduced to
10.
[1600 x .80% = 1280, 1600-1280 = 320; 320 400 = .8]
The power of genetic matching can be realized in two other ways. For
15 example, using the same number of patients (50) in each arm, with genetic
matching the
difference that can be resolved with the same power drops from 20 to 8.8.
Likewise, the
power of the study increases from .8 to greater than 0.99 assuming genetic
matching and a
difference of 20. If genetic matching cannot be fully achieved, some degree of
matching
can still have a favorable impact on such studies. This is illustrated in
Table 1 below.
2o TABLE 1
Matching % Variance ReductionNew Variance n=Patients
128 1472 45
256 1344 41
384 1216 37
512 1088 33
640 960 2g
43
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/285$2
EXAMPLE 2
Use of genetic analysis to reduce the sample size
necessary in clinical trials
The following example is illustrative of the method of identifying
underlying genetic factors that influence the response to treatment and the
use of this
information in the design of clinical trials.
1o A. Genetic Sub-Populations
In the instance of two distinct genetic sub-populations A and B, associated
with a low response and high response to treatment, respectively, the response
of treated
individuals from the first sub-population, A, has mean /CA and variance, QA .
In the
second sub-population, the mean and variance of response are given by ~cB and
QB ,
respectively. No assumption is made about the shape of either distribution
(i.e. they do
not have to be normal). In control individuals who do not receive the
treatment (instead
receiving a placebo), the mean and variance of response is given by (~,t,Q,q )
and
(,uB,og) for populations A and B, respectively. When a sample is taken from
one of
these populations, the distribution of the sample mean is normally
distributed. For
2o example, if a sample of size N of treated individuals is drawn from sub-
population A, the
sample mean has distribution Norm[~A, azq l N] .
If the genetic background of the sub-populations is ignored, both the
treated (case) and untreated (control) populations include a mixture of
individuals
sampled from the two distributions. When the probability of an individual
being chosen
from genetic sub-population A is p and the probability of an individual being
selected
from genetic sub-population B is q =1- p , the distribution of the mean
response of a
sample selected from the two populations can be described.
For example, when N is the total population size and {x1, x2 ,..., x; ,...xN )
44
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/Z8582
is a set of random variables, each describing an individual in the sample, the
expectation
of the mean response in the sample is given by,
N (1)
E[x] = N ~ E[xi ]
~=1
where E[xt ] _ ~,q with probability p and E[x~ ] =,uB with probability q =1- p
. So
the mean of the distribution of the sample mean is,
E[x] = Pf~A 't' 9f~B ~ (2)
The variance of the mean response in such a case depends on both the
variances of each of the two distributions (A and B) and on the difference
between the
means of these distributions. This variance can be expressed in terms of the
sum of the
variances of the individuals,
N
V [x] = 12 ~ V [xl ] = V N ]
N i=t
When a random variable Y is defined such that when Y = 0 , V[x~ ~ Y = 0] = aA
and
when Y =1, V[x; ~ Y =1] = aB , the expectation of this variance is then,
E[V [xi ( Y]] = PEA + q~B
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
Further, E[xl ~ Y = 0] _ ~~ and E[xl ~ Y =1] _ ~B , hence,
V [E[xi ~ Y]] = Pf~A + 9f~B - (Pf~A + ~'f~B ) 2 = Pq(f~A - f~B ) 2
By standard theory, V[x~ ] = E[V[x~ ~ Y]] + V[E[x; ~ Y]] so,
V[x]= PEA +9~B +Pq(f~A -f~B)2 ,
N
Importantly, V[x] > Min[6A l N,6g l N] . That is, the variance when the sub-
populations
io are ignored is always larger than the variance of one of the sub-
populations. This can
intuitively be seen from the shape of equation 6. The variance is the weighted
sum of the
two population specific variances (~A, aB ) plus a term representing the
difference
between the two population means (PA -,uB ) 2 . Thus, the variance consists of
both the
within population variance and the between population variance.
The distribution of the sample's mean response (when the ratio of
individuals from sub-populations A and B is p : q ) is normal (by the Central
Limit
Theorem) and has mean and variance (puA + qpg, p6A + q~B + pq(,uA - ~B )2 ) .
N
B. Marker Informativeness
2o The determination of which of the two genetic sub-populations (A and B)
an individual belongs to can be made by examining genetic markers. Generally,
the more
markers that are genotyped, the greater the probability of assigning an
individual to the
correct group. In the absence of genetic information, a sample represents a
random mix
of the two populations with ratio p : q of individuals from sub-populations A
and B. If
46
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
the two genetic backgrounds have the same frequency, then p = q = 0.5 and the
distribution of the sample mean is characterized as,
(,uA +,uB 6~ '+' 6B + 0.5(,uA -,uB )2 ) .
2 ' 2N
s
In some instances, all individuals from the sub-population are genotyped for k
equally
informative markers. This is sometimes the case when markers are chosen at
random (i.e.
. if nothing is known about genes involved in responsiveness). Additional
markers will
usually provide decreasing information (i.e. though the k + 1 th marker
increases the
1o probability of correctly assigning an individual to a sub-population, it
provides less
information than the k th marker); this does not necessarily have to be the
case but often
is the case. For example, if there is a priori knowledge of the genes involved
in response,
these are typically examined first. Alternatively, if there is no information
about the
underlying genetics of response, then the genetic matching is based on
relatedness (i.e.,
15 the overall degree of genetic similarity in the genome) and hence the first
few markers
will be highly informative with diminishing information from each additional
markers.
Consider a simple model where the probability of assigning an individual
to the correct genetic sub-population when k markers have been genotyped is
given by,
20 P[correct ~ k] = 1 (1 + k ) . (8)
2 k+1
As k tends to infinity, the probability of correct assignment asymptotes to 1.
This
probability can be used in the equations above to determine the mixture of the
sampled
population. For k -~ o~, p -~ 1 in equations 2 and 6, so the mean and variance
of the
25 sample mean is given by (~A, a-A l N) for population A and (~tB, ag l N)
for population
B (using the information to set p = 0 and so select non-responders) as
expected.
47
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
C. Power of the clinical trial with ~~enetic matching
In a clinical trial that consists of a cohort of patients, half of whom are
given the treatment and half who are given a placebo, wherein the two genetic
sub-
s populations are equi-frequent, then the response in the treated sample is
normally
distributed with mean and variance given by,
(Pf~A + 9f~B ~ p~A + q~B + P9(f~A - f~B ) 2 ) .
N
to In the control (placebo) sample, the distribution of the mean is again
normal with mean
and variance,
(PEA + qf~B ~ p6A + q6B + P9(~A - f~B )2 ) . (10)
N
15 For the sake of simplicity, the two samples are selected to be of equal
size, but this does
not have to be so. When N is reasonably large (>30), standard theory of normal
distributions can be used to show that the necessary sample size to detect a
difference in
response between the treated and placebo groups at the a% level with power ~3
is,
(Za~P6A '~9~B ~' P~1(~A -f~B)2 +Zt_Q pa',Zg +qcrB + pq(f;A -f~B)2 )2
20 (11)
(P(f~A - f~A ) + 9(f~B - f~B )) 2
where N is the number in each arm of the trial, giving 2N as the total number
of
individuals. From this equation it can be seen that the sample size increases
as the
difference between the means of the cases and controls decreases (i.e., when
the means
48
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
are identical faA = ,u,g and ~B = ~B , the sample size is infinity). The
sample size also
increases as the variances of the sub-populations increase.
D. Example of the sample size for matched and unmatched populations
In one instance, the two genetic sub-populations have the same response
characteristics when no treatment is administered ( ~c,q = ~B = 0 , aA = o~g =
8 ) and the
mean response to treatment of individuals from group A is described by ,u,q =
S, Q,t = 8 .
Response to treatment for individuals from group B is the same as'for the
placebo (i.e.,
they are non-responders) with ~.tB = O,QB = 8. Further, in this example, a S%
to significance level (a = 0.05 ) is used and the sample size represents the
minimum number
of individuals needed for 80% power (~3 = 0.8) . Table 2 below gives the
number of
markers (k ), the probability of the selected individual coming from group A
(i.e., being
correctly identified as a responder) ( p ), the variance of the sample mean
for the treated
population ( V[x] ) and the sample size required in each arm of the trial ( N
).
TABLE 2
k p V[ ] N
0 0.50 70 g3
1 0.75 69 36
5 0.92 66 24
10 0.95 65 22
00 1.00 64 20
2o In this example, the within population variance is fixed at 64 for both
genetic subpopulations and in both treated and untreated samples. Table 2
shows that,
when no markers are genotyped, the variance is 70. This increase in the
variance is
entirely due to the difference in the response due to the underlying genotype.
When this
49
CA 02350069 2001-05-09
WO 00/33161 PCTNS99/28582
is accounted for (when p -~ 1 ), the variance returns to the expected value of
64. This
inflation in variance has a marked effect on the necessary sample size (note
in equation
11, the sample size does not increase linearly increasing variance, but rather
with the
square of the variance). Where 83 individuals are needed in each arm of the
trial if no
genetic information is available, only 20 individuals are needed if
individuals can be
correctly assigned as responders using their polymorphic profile.
EXAMPLE 3
Effects of linkage disequilibrium and marker allele frequencies
to on the power of a clinical trial
In this example, there are two genetic sub-populations A and B, and a
single bi-allelic SNP (single nucleotide polymorphism) that is present in both
sub-
populations. One of the alleles, labeled g , has frequency p[g ~ A] = pA in
sub-
population A and p[g ~ B] = pB in sub-population B. In this particular
example, the two
sub-populations have equal frequency (i.e. p(A) = p(B) = 0.5 ). In this
situation, the
population-wide frequency of the rare allele (i.e., the allele of the pair
that has the lower
frequency in the general population) is then, p(g) _ ( pA + pB ) l 2 . In this
example, it has
been shown that the allele g is associated with increased response to the
treatment. The
2o increased response can be due to the function of the allele itself, but
more usually arises
due to the allele being in linkage disequilibrium with some (unknown) genetic
factor or
mutation. The magnitude of linkage disequilibrium, d , is defined as
d A = p(A & g) - p(A) p(g) for sub-population A and d~ = p(B & g) - p(B) p{g)
for
sub-population B. If the factor causing increased response is more common in
sub-
population A and the SNP marker is in close proximity, it is expected that d,~
> dB . In
this sense, the sub-populations can be regarded as carrying the high-response
(A) and
low-response (B) alleles of the unknown gene that influences an individual's
response to
treatment.
In some instances, an SNP lies in a region that is known to harbor a gene
so
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
involved in response to treatment and sub-population A consists of high
responders
whereas sub-population B consists of low responders. In instances such as
this, the SNP
can then be used to determine which population an individual comes from and
hence
whether or not they are enrolled in a clinical trial. For example, when p,~ _
.6 and
pB = .2 , this implies that individuals carrying the g allele are three times
as likely to
come from sub-population A and from sub-population B. As such, this marker is
informative about whether an individual will respond well to the treatment
(i.e., whether
they come from sub-population A). When the two sub-population are equi-
frequent, then
p(A & g) = 0.3 and p(B & g) = 0.1, hence
dA =p(A&g)-p(A)p(g)=0.3-0.5x0.4=0.1, (1)
and
dB = p(B & g) - p(B) p(g) = 0.1- 0.5 x 0.4 = -0.1. (2)
That is, there is a positive association between the g allele and an
individual belonging to
sub-population A. Conversely, there is negative association between g and sub-
population B. With these measures of linkage disequilibrium, it is possible to
calculate
the conditional probability of an individual being a high responder (coming
from sub-
population A) given the individual carries the g allele. This is given as,
PEA ~ g~ = P g) + P(A) - 0.75 , (3)
and similarly,
51
CA 02350069 2001-05-09
WO 00/33161 PCTNS99/28582
PIB I g~ - P g) + P(B) = 0.25 .
Note that by substituting for d,~ and dB , these conditional probabilities can
also be
expressed (using Bayes' theorem) as,
PEA I g~ = PEA & g~ __ P,, (S)
Pig) P~ + Pe
and
PCB I g~ = PCB & g~ __ Pa
PLg~ P~ + Pe
to That is, the strength of the association is encapsulated in the difference
between the allele
frequencies in the sub-populations.
Using information on this SNP increase, the probability of correctly
choosing high responders from the null of 50% (given the two sub-populations
are
equally frequent) to 75%. This probability is a function of the frequency of
the associated
marker allele, the frequency of the allele influencing response and the
strength of linkage
disequilibrium between the two alleles. The necessary sample size can then be
calculated
as described in the previous section with p = 0.5 representing random sampling
of
individuals from the two populations and p = 0.75 representing sampling using
the
information from the genetic marker.
2o Note that in the example above, it is assumed that the allele frequencies
in
the two populations are known. This will often be the case if the polymorphism
is already
known to be involved in response but may not generally be known. However, it
may often
be the case that previous trials can be used to estimate allele frequencies in
responders and
non-responders.
This method extends naturally to multiple markers. In this case,
52
CA 02350069 2001-05-09
WO 00/33161 PCTNS99/28582
G E {g, ,..., gk } represents the genotype of an individual at a set of bi-
allelic markers with
frequencies {p,,..., pk } in sub-population A and {q"..., qk } in sub-
population B. Then, by
Bayes' Theorem,
k
p[A I G) = P[G I A)P[A) _ ~ P~ 7
r=i
P[G I A)P[A) + P[G I B)P[B) ~k k C )
~P. +~9a
r=~ r=i
where sub-populations A and B are equally frequent. Similarly, p[B ( G) can be
expressed as,
k
l0 p[B I G) - PEG I B)P[B) 1 1. qa
P[G I A)P[A) + P[G I B)P[B) ~k k (8)
~P~ +~qr
r=~
These values can then be used as in the single locus case to determine the
probability of
allocating an individual to the correct sub-population and hence to calculate
the sample
size.
For example, suppose five markers are genotyped and it is known that for
all of them the rare allele has frequency 0.4 in sub-population A and 0.5 in
sub-population
B. The study is conducted with patients from sub-populations A and individuals
from
sub-population B, the latter individuals being known not to respond to
treatment. Patients
are enrolled dependent on the outcome of the 5 markers. Using equations 7 and
8, the
2o probability of an individual belonging to sub-population A when the
individual has the
rare allele at n of the S markers is as shown in Table 3.
53
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
TABLE 3
Number of markers at which the Probability of belonging to
rare allele sub-population
is seen. A
0 0.713
1 0.620
2 0.525
3 0.424
4 0.330
0.247
Table 3 shows that the probability of correctly assigning an individual to
the correct sub-population increases rapidly with the number of markers
genotyped. In
5 this example, the markers are randomly chosen (not known to be associated
with a
specific gene) and the difference in the frequency of the rare allele between
the two sub-
populations is chosen to be 0.1.
Information of the type given in Table 3 can be used directly to categorize
individuals into sub-populations or as a criterion for enrollment into a
trial. For example,
individuals having the rare allele at 1 or 0 of the 5 polymorphisms can be
included in a
clinical trial. This increases the probability of the trial containing
individuals that will
respond to the treatment (where it is assumed that'sub-population A responds
to treatment
and sub-population B does not). In the case of individuals with no rare
alleles, the
probability of belonging to sub-population A is 0.713 and for individuals with
1 rare
allele, the probability of belonging to sub-population A is 0.620. As such,
this selection
criterion greatly increases the proportion of individuals from sub-population
A enrolled in
the clinical trial. This simple method of selection is only illustrative and
many other more
complex procedures (for example, cluster analysis) can be employed depending
on the
number of polymorphisms and their respective allele frequencies.
54
CA 02350069 2001-05-09
WO 00/33161 PCTNS99I28582
EXAMPLE 4
Effects of the number of markers and their allele frequencies on the power of
the
polymorphic profile to discriminate between distinct groups of patients.
In this example, there are two types of markers, those with common alleles
(i.e., the two alleles are at similar frequency) and those with a rare allele.
For the first
marker-type, one allele has frequency p, = 0.5 in the sub-population A
(responders) and
q, = 0.4 in the sub-population B (non-responders). For the second type of
markers, one
allele has frequency pz = 0.1 in sub-population A and q2 = 0.08 in sub-
population B. In
1 o both cases, the rare allele has a 20% lower frequency in sub-population B
compared to its
frequency in sub-population A. A set of markers, K in number, are genotyped in
a sample
of 2000 patients who are known to belong either to sub-population A or sub-
population B
(from a previous clinical trial). For each of these individuals there are k
observations
x,, x2, x3,..., xk which take the value 0 if the rare allele is present and 1
otherwise. These
individuals can be classified into sub-populations with y = 0 if they come
from sub-
population A and y =1 if they belong to sub-population B. Using such training
data, 2000
individuals were generated and assigned to one sub-population or the other
using a linear
logistic model (Christensen, Log Linear Models and Logistic Regression,
Springer Verlag,
New York, 1997) of the form,
log[ P(y =1) ~ _ ~o + ~~X'i + IQZxz + ... + ~3k xk
P(y = 0)
Other statistical methods (such as described in section those in Example 3)
can also be used.
This linear logistic model was chosen to illustrate another method of
classification.
Table 4 gives the probability of assigning an individual to the correct sub-
population for 2, 5, 10, 20 and 50 markers. Values are given for both types of
markers and
for a mixture of the two. In this example, all markers are assumed to be
independent of one
another. If this were not the case, other, more powerful, statistical methods
can be applied
CA 02350069 2001-05-09
WO 00/33161 PCT/US99/28582
(for example, methods of classification trees (Breiman et al., Classification
and Regression
Trees, CRC Press, 1984).
TABLE 4
Number
of
markers
(k)
0 2 5 10 20 50 100
Common allele 0.50 0.58 0.58 0.62 0.66 0.75 0.84
p, =O.S,q, =0.4
Rare allele 0.50 0.50 0.53 0.55 0.56 0.57 0.62
pz = 0.1, qz
= 0.08
Equal mix of 0.50 0.56 0.58 0.57 0.62 0.70 0.77
p, =O.S,q, =0.4
and
Pz = 0.1, qz
= 0.08
In these simulations, the markers for which the two alleles have similar
frequencies are more effective in determining which group and individual
belongs to, than
are markers with very different allele frequencies. An equal mixture of
markers from these
two classes provides, as expected, intermediate results.
In many cases, data from previous clinical trials is available, but there is
no
information about which of the two sub-populations the responders and non-
responders are
to drawn from. Such a scenario is more amenable to analysis by clustering
methods. Data for
2000 individuals was simulated using the same allele frequencies as described
above. These
data were analyzed using K-means clustering (with K=2) to investigate how well
the sub-
populations can be defined by the markers alone. Because less information is
available,
there is little power in this method when very few markers (k < 10) are used.
Results are
shown in table 5 for 10 or more markers.
56
CA 02350069 2001-05-09
WO 00/33161 PCTNS99/28582
TABLE 5
Number
of
markers
(k)
10 20 50 100
Common allele 0.58 0.52 0.69 0.77
p, =O.S,q, =0.4
Rare allele 0.53 0.50 0.57 0.53
Pz = 0.1, qz =
0.08
Equal mix of 0.52 0.52 0.68 0.75
p, =O.S,q, =0.4
and
pz = 0.1, qz =
0.08
In this example, markers for which both alleles are common perform better
than those for which one allele is very rare. These results using cluster
analysis correctly
assign individuals with a lower probability than in Table 4. This is to be
expected since less
information was available a priori for these simulations.
As illustrated in part by the foregoing examples and the above description,
the present invention has a number of uses with regard to treatment studies
generally, and
l0 clinical trials in particular. For example, the invention includes the use
of a polymorphic
profile to conduct a clinical trial on a population of patients having the
same disease,
wherein the polymorphic profile includes at least one polymorphic site not
known to be
associated with the disease. The invention also includes the use of a
polymorphic profile
to conduct a reanalysis of data from a clinical trial in which statistical
significance is
is determined on subpopulations of original treated and control groups
selected for
similarity of polymorphic profile. The invention also includes the use of a
polymorphic
profile to divide a population of individuals subject to a clinical trial into
a plurality of
subsets, the members of a subset showing greater similarity of polymorphic
profile to
each other than members in different subsets.
57
CA 02350069 2001-05-09
WO 00/33161 PCTNS99/28582
It is understood that the examples and embodiments described herein are for
illustrative purposes only and that various modifications or changes in light
thereof will be
suggested to persons skilled in the art and are to be included within the
spirit and purview of
this application and scope of the appended claims. All publications, patents,
and patent
applications cited herein are hereby expressly incorporated by reference in
their entirety for
all purposes to the same extent as if each individual publication, patent or
patent application
were specifically and individually indicated to be so incorporated by
reference.
58