Note: Descriptions are shown in the official language in which they were submitted.
CA 02351059 2001-05-09
WO 00/34295 PCT/US99/Z7777
ACYLATED BENZYLMALTOSIDES AS INHIBITORS OF SMOOTH
MUSCLE CELL PROLIFERATION
10
BACKGROUND OF THE INVENTION
This invention relates to the use of substituted acylated benzylmaltosides as
smooth muscle cell proliferation inhibitors and as therapeutic compositions
for
treating diseases and conditions which are characterized by excessive smooth
muscle
proliferation such as restenosis.
All forms of vascular reconstruction such as angioplasty and vein bypass
procedures effect a response to injury that ultimately leads to smooth muscle
cell
(SMC) proliferation and subsequently, deposition of profuse amounts of
extracellular matrix (Clowes, A. W.; Reidy, M. A. J. Vasc. Surg 1991, 13,
885).
These events are also central processes in the pathogenesis of atherosclerosis
(Raines
E.W.; Ross R. Br. Heart J. 1993, 69 (Supplement), S. 30) as well as transplant
arteriosclerosis (Isik, F.F.; McDonald, T. O.; Ferguson, M.; Yamanaka, E.;
Gordon
Am. J. Pathol. 1992, 141, 1139). In the case of restenosis following
angioplasty,
clinically relevant solutions for controlling SMC proliferation through
pharmacological intervention have remained elusive to date (Herrman, J. P. R.;
Hermans, W.R.M.; Vos, J.; Serruys P. W. Drugs 1993, 4, 18 and 249). Any
successful approach to selective SMC proliferation inhibition must not
interfere with
endothelial cell repair or the normal proliferation and function of other
cells
(Weissberg, P.L.; Grainger, D.J.; Shanahan C.M.; Metcalfe, J.C. Cardiovascular
Res. 1993, 27, 1191 ).
The glycosaminoglycans heparin and heparan sulfate are endogenous
inhibitors of SMC proliferation, yet are able to promote endothelial cell
growth
(Castellot, J.J. Jr.; Wright, T. C.; Karnovsky, M.J. Seminars in Thrombosis
and
Hemostasis 1987, 13, 489). However, the full clinical benefits of heparin,
heparin
fragments, chemically modified heparin, low molecular weight heparins, and
other
heparin mimicking anionic polysaccharides may be compromised due to other
pharmacological liabilities (excessive bleeding arising from anticoagulation
effects, in
CA 02351059 2001-05-09
WO 00/34295 PCTNS99/27777
-2-
particular) coupled with heterogeneity of the various preparations (Borman, S.
Chemical and Engineering News, 1993, June 28, 27).
WO 96/14325 discloses acylated benzylglycosides as smooth muscle cell
proliferation inhibitors. The compounds of the present invention differ in
that the
substituents on the carbohydrate backbone are different.
Zehavi, U.; Herchman, M. in Carbohyd Res. 19$6, 151, 371, disclosed 4-
carboxy-2-nitrobenzyl 4-O-a-D-glucopyranosyl-~3-D-glucopyranoside which is
attached to a polymer for study as an acceptor in the glycogen synthase
reaction. The
compounds of the present invention differ in that the substituents on the
benzyl
groups are different and the use (smooth muscle antiproliferation) is
different.
Patent numbers US 5,498,775, W096/14324, and US 5,464,827 describe
polyanionic benzylglycosides or cyclodextrins as smooth muscle cell
proliferation
inhibitors for treating diseases and conditions which are characterized by
excessive
smooth muscle proliferation. ~i-cyclodextrin tetradecasulfate has been
described as a
smooth muscle cell proliferation inhibitor and as an effective inhibitor of
restenosis
(Reilly, C.F.; Fujita, T.; McFall, R. C.; Stabilito, I. L; Wai-se E.; Johnson,
R. G.
Drug Development Research 1993, 29, 137). US 5019562 discloses anionic
derivatives of cyclodextrins for treating pathological conditions associated
with
undesirable cell or tissue growth. WO 93/09790 discloses antiproliferative
polyanionic derivatives of cyclodextrins bearing at least 2 anionic residues
per
carbohydrate residues. Meinetsberger (EP 312087 A2 and EP 312086 A2) describes
the antithrombotic and anticoagulant properties of sulfated bis-aldonic acid
amides.
US 4431637 discloses polysulfated phenolic glycosides as modulators of the
complement system. The compounds of the present invention differ from all of
the
prior art in that the compounds (a) are benzylmaltosides which bear no
structural
resemblance to heparin, sulfated cyclodextrins, or to sulfated lactobionic
acid dimers,
(b) contain no more than two contiguous sugar residues (disaccharide), (c) are
of a
defined structure, (d) and are not sulfated.
CA 02351059 2001-05-09
WO 00/34295 PCTNS99/27777
-3-
DESCRIPTION OF THE INVENTION
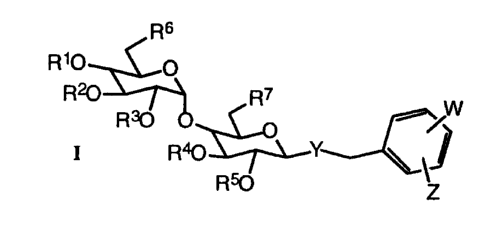
This invention provides benzylmaltosides of formula I
,~s
R
wherein
Rl, R2, R3, R4, and RS are each, independently, hydrogen, acyl of 2-7 carbon
atoms,
haloacyl of 2-7 carbon atoms, nitroacyl of 2-7 carbon atoms, cyanoacyl of 2-
7 carbon atoms, trifluoromethylacyl of 3-$ carbon atoms, or benzoyl in which
the phenyl moiety is substituted with Rg;
R6 and R~ are each, independently, -OH, -OR9, O-tent-butyldimethylsilyl, O-
trialkylsilyl of 1-6 carbon atoms per alkyl moiety, O-triphenylsilyl,
~o » g°2 Rio
'~ -~-° ~ ~/1 - _° ~'
-R~2 ~ -R~1
i'~ n ~ ~ ,
__
/ ~ .~'~' .
or I ,
R14
Rg, Rl~, R11, and RI2 are each, independently, hydrogen, -CN, -N02, halogen,
CF3,
alkyl of 1-6 carbon atoms, acetyl, benzoyl, or alkoxy of 1-6 carbon atoms;
CA 02351059 2001-05-09
WO 00/34295 PCTNS99/27777
-4-
R9 is acyl of 2-7 carbon atoms, haloacyl of 2-7 carbon atoms, nitroacyl of 2-7
carbon
atoms, cyanoacyl of 2-7 carbon atoms, trifluoromethylacyl of 3-8 carbon
atoms, or benzoyl in which the phenyl moiety is substituted with Rg;
Y is O, S, NH, NMe, or CH2;
W is halogen, -CN, CFg, alkyl of 1-6 carbon atoms, haloalkyl of 1-6 carbon
atoms,
nitroalkyl of 1-6 carbon atoms, cyanoalkyl of 1-6 carbon atoms, alkoxyalkyl
of 2-12 carbon atoms, alkoxy of 1-6 carbon atoms, or phenyl mono-, di-, or
tri-substituted with Rg;
Z is -N02, -NH2, -NHRt3, or -NHCO-Het;
R13 is acyl of 2-7 carbon atoms, haloacyl of 2-7 carbon atoms, nitroacyl of 2-
7
carbon atoms, cyanoacyl of 2-7 carbon atoms, trifluoromethylacyl of 3-8
carbon atoms, benzoyl in which the phenyl moiety is substituted with Rg, or
R13 is an a-amino acid in which the a carboxyl group forms an amide with the
nitrogen of Z, wherein if said amino acid is glutamic acid or aspartic acid,
the
non-a carboxylic acid is an alkyl ester in which the alkyl moiety contains
from 1-6 carbon atoms;
Het is pyridyl substituted with Rg, thienyl substituted with Rg, furyl
substituted with
Rg, oxazolyl substituted with Rg, pyrazinyl substituted with Rg, pyrimidinyl
substituted with Rg, or thiazolyl substituted with Rg;
R14 is Rg, -NH2, -C02H, or -NH-acyl of 2-7 carbon atoms;
n = 0-3;
with the proviso that when Z is -NHR13 and Y is O, at least one of R1, R2, R3,
R4,
and RS is hydrogen, or at least one of R6 and R~ is OH, or a pharmaceutically
acceptable salt thereof.
Alkyl includes both straight chain as well as branched moieties. Halogen
means bromine, chlorine, fluorine, and iodine. When R13 is an a-amino acid,
the
carboxyl moiety exists as an amide with the amide nitrogen being bonded to the
phenyl ring of the compound of formula I. The following exemplifies the
resulting
structure when Rt3 is alanine:
CA 02351059 2001-05-09
WO 00/34295 PCT/US99/27777
-5-
y ~W ~TH2
NH ~
'1( 'CH3
~~O
When the amino acid contains a second carboxyl moiety, the moiety is an
alkyl ester of the free acid. The following example shows aspartic acid methyl
ester.
_~ Y .a ~DTH2
OCH3
O
Preferred amino acids include alanine, arginine, aspartic acid, cystine,
glutamic acid, glycine, histidine, isoleucine, leucine, lysine, methionine,
phenylalanine, proline, serine, threonine, tryptophan, tyrosine, and valine.
The
amino acids defined by R13 include both the D and L amino acids.
Pharmaceutically acceptable salts can be formed from organic and inorganic
acids, for example, acetic, propionic, lactic, citric, tartaric, succinic,
fumaric, malefic,
malonic, mandelic, malic, phthalic, hydrochloric, hydrobromic, phosphoric,
nitric,
sulfuric, methanesulfonic, napthalenesulfonic, benzenesulfonic,
toluenesulfonic,
camphorsulfonic, and similarly known acceptable acids. Salts may also be
formed
from organic and inorganic bases, preferably alkali metal salts, for example,
sodium,
lithium, or potassium. Acid addition salts can be prepared when Y contains a
nitrogen or the compound of formula I contains a basic nitrogen, and base
addition
salts can typically be prepared when the compound of formula I contains a
hydroxyl
group.
The compounds of this invention may contain an asymmetric carbon atom or
sulfoxide moiety and some of the compounds of this invention may contain one
or
more asymmetric centers and may thus give rise to optical isomers and
diastereomers.
While shown without respect to stereochemistry in Formula I, the present
invention
includes such optical isomers and diastereomers; as well as the racemic and
resolved,
enantiomerically pure R and S stereoisomers; as well as other mixtures of the
R and S
stereoisomers and pharmaceutically acceptable salts thereof.
CA 02351059 2001-05-09
WO 00/34295 PCTNS99/27777
-6-
Preferred compounds of this invention are benzylmaltosides of formula I
R6
R
wherein
R1, R2, R3, R4, and RS are each, independently, hydrogen or acyl of 2-7
carbon;
R6 and R~ are each, independently, -OH, -OR9, O-ten-butyldimethylsilyl,
R1~ R11 ~S02 Rio
/~ R12 ~ O I ~~ R11
n '
~ N ,~ N-''~
_~_O I. ~ ~N
or ~~ ,
R14
Rg, Ri~, R11, and R12 are each, independently, hydrogen, -CN, -N02, halogen,
CF3,
alkyl of 1-6 carbon atoms, acetyl, benzoyl, or alkoxy of 1-6 carbon atoms;
R9 is acyl of 2-7 carbon atoms, or benzoyl in which the phenyl moiety is
substituted
with Rg;
YisOorS;
W is halogen, or alkyl of 1-6 carbon atoms, haloalkyl of 1-6 carbon atoms,
nitroalkyl
of 1-6 carbon atoms, cyanoalkyl of 1-6 carbon atoms, alkoxyalkyl of 2-12
carbon atoms, alkoxy of 1-6 carbon atoms, or phenyl mono-, di-, or tri-
substituted with Rg;
Z is -N02, -NH2, -NHR13, or -NHCO-Het;
R13 is acyl of 2-7 carbon atoms, or benzoyl in which the phenyl moiety is
substituted
with Rg, or
r'_v Z
CA 02351059 2001-05-09
WO 00/34295 PCT/US99/27777
R13 is an a-amino acid in which the a carboxyl group forms an amide with the
nitrogen of Z, wherein if said amino acid is glutamic acid or aspartic acid,
the
non-a carboxylic acid is an alkyl ester in which the alkyl moiety contains
from 1-6 carbon atoms;
Het is pyridyl substituted with Rg, thienyl substituted with Rg, furyl
substituted with
Rg, oxazolyl substituted with Rg, pyrazinyl substituted with Rg, pyrimidinyl
substituted with Rg, or thiazolyl substituted with Rg;
R14 is Rg, -NH2, -C02H, or -NH-acyl of 2-7 carbon atoms;
n = 0-3;
with the proviso that when Z is -NHR13 and Y is O, at least one of Ri, R2, R3,
R4,
and RS is hydrogen, or at least one of R6 and R~ is OH, or a pharmaceutically
acceptable salt thereof.
Specifically preferred compounds of this invention are:
4-Chloro-3-nitro-benzyl- p-D-maltoside heptaacetate or a pharmaceutically
acceptable
salt thereof;
N-{ 5-[(Hepta-O-acetyl-(3-D-maltosyloxy)-methyl]-2-chloro-phenyl }-L-
aspartamide-y-
tert- butyl ester or a pharmaceutically acceptable salt thereof;
N { 2-Chloro-5-[(2,2',3,3',4',6,6')-hepta-O-acetyl-p-D-maltosyl-oxymethyl]-
phenyl }-
(9H-fluoren-9-ylmethoxycarbonyl)-L-alaninamide or a pharmaceutically
acceptable
salt thereof;
4-Benzoyl-N { 2-chloro-5-[(2,2',3,3',4',6,6'-hepta-O-acetyl-p-D-maltosyl)-oxy-
methyl]- phenyl }-benzamide or a pharmaceutically acceptable salt thereof;
(4-Chloro-3-nitro-benzyl) -hepta-O-acetyl-1-thio-p-D-maltoside or a
pharmaceutically
acceptable salt thereof;
CA 02351059 2001-05-09
WO 00/34295 PCT/US99/27777
_g_
(3-Amino-4-chloro-benzyl) hepta-O-acetyl-1-thio-p-D-maltoside or a
pharmaceutically acceptable salt thereof;
N {2-chloro-5-[ hepta-O-acetyl-p-D-maltosyl-1-thio)-methyl]-phenyl}-acetamide
or a
pharmaceutically acceptable salt thereof;
5-[(Hepta-O-acetyl-p-D-maltosyl)-oxy-methyl]-2-cyano-1-nitrobenzene or a
pharmaceutically acceptable salt thereof.
N [2-Chloro-5-(p-D-maltosyl-oxymethyl)-phenyl]-acetamide or a pharmaceutically
acceptable salt thereof;
N { 5-[6,6'-Di-O-(tert-butyl-dimethyl-silyl)-p-D-maltosyloxy-methyl]-2-methyl-
phenyl}- acetamide or a pharmaceutically acceptable salt thereof;
N-{ 2-Chloro-5-[6,6'-di-O-(tert-butyl-dimethyl-silyl)-p-D-maltosyloxy-methyl]-
phenyl}- acetamide or a pharmaceutically acceptable salt thereof;
N-{2-Chloro-5-[([6,6'-di-O-benzoyl-p-D-maltosyl]oxy)methyl)phenyl}-acetamide
or
a pharmaceutically acceptable salt thereof;
N-{ 2-Chloro-5-[([6,6'-di-O-benzoyl-2,2',3,3',4'-penta-acetyl-p-D-
maltosyl)oxy)-
methyl)phenyl }-acetamide or a pharmaceutically acceptable salt thereof;
(4-Chloro-3-nitrophenyl)methyl-4-O-[6-O-(3-pyridinylcarbonyl)-a-D-gluco-
pyranosyl]-p-D- glucopyranoside-6-(3-pyridinecarboxylate) or a
pharmaceutically
acceptable salt thereof;
(4-Chloro-3-nitrophenyl)methyl-4-D-[6-O-(3-pyridinylcarbonyl)-«-D-gluco-
pyranosyl]-p-D- glucopyranoside or a pharmaceutically acceptable salt thereof;
CA 02351059 2001-05-09
WO 00/34295 PCT1US99/27777
-9-
N [2-Chloro-5-[[(4-O-«-D-glucopyranosyl-p-D-glucopyranosyl)oxy]methyl] phenyl]-
3- pyridinecarboxamide or a pharmaceutically acceptable salt thereof;
Benzoic acid 6-{4-chloro-3-[(pyridine-3-carbonyl)-amino]-benzyloxy}-4,5-
dihydoxy-3- (3,4,5-trihydroxy-6-hydroxymethyl-tetrahydro-pyran-2-yloxy)-
tetrahydro-pyran-2- ylmethyl ester or a pharmaceutically acceptable salt
thereof;
(4-Chloro-3-nitro-benzyl)-I-deoxy-I-thio-p-D-maltoside or a pharmaceutically
acceptable salt thereof;
N {2-chloro-5-[p-D-maltosyl-I-thio)-methyl]-phenyl}-acetamide or a
pharmaceutically acceptable salt thereof;
5-{[6,6'-Bis-O-(4-toluenesulfonyl)- p-maltosyl]-oxy-methyl}-2-methyl-1-
nitrobenzene or a pharmaceutically acceptable salt thereof;
5-{[2,2',3,3',4'-Penta-O-acetyl-6,6'-bis-O-(4-toluenesulfonyl)- p-maltosyl]-
oxy-
methyl}- 2-methyl-1-nitrobenzene or a pharmaceutically acceptable salt
thereof;
5-{[6,6'-Dideoxy-6,6'-bis(4-nitro-imidazol-1-yl)-p-maltosyl]-oxy-methyl}-2-
methyl-1- nitrobenzene or a pharmaceutically acceptable salt thereof; and
5-{[2,2',3,3',4'-Penta-O-acetyl-6,6'-dideoxy-6,6'-bis(4-vitro-imidazol-I-yl)-
[i-
maltosyl]- oxy-methyl}-2-methyl-I-nitrobenzene or a pharmaceutically
acceptable
salt thereof.
The compounds of this invention were be prepared according to the following
schemes from commercially available starting materials or starting materials
which
can be prepared using literature procedures. These schemes show the
preparation of
representative compounds of this invention.
Acetobromomaltose 1 is coupled with a benzyl alcohol 2 in the presence of a
catalyst such as a mercuric bromide, mercuric cyanide, silver triflate, or
silver
CA 02351059 2001-05-09
WO 00/34295 PCT/US99/27777
- 10-
perchlorate in an aprotic solvent such as acetonitrile, dichloromethane,
ether, toluene
or nitromethane at temperatures ranging from -40 °C to reflux to yield
glycoside 3
(Scheme 1). This glycosidation can also be accomplished using Schmidt's
trichloroacetimidate coupling with zinc bromide in a solvent such as
dichloromethane. Reduction of the nitro group of 3 can be accomplished with a
reducing agent such as stannous chloride in a polar aprotic solvent such as
ethyl
acetate at ambient temperature to reflux to afford an anilino compound 4.
Coupling of
4 with an acid chloride can be completed in the presence of an amine base such
as
triethylamine or diisopropylethylamine or using a stronger base such as sodium
hydride (for sterically hindered systems) in an aprotic solvent such as
dichloromethane or tetrahydrofuran at temperatures ranging from 0 °C to
ambient
temperature to yield the target compound 5. The peracetylated compound 5 can
be
converted to the heptahydroxy compound 6 with catalytic sodium methoxide in
methanol or aqueous sodium hydroxide in methanol at temperatures ranging from
ambient temperature to reflux.
As illustrated in Scheme 2, the C-6 and C-6' positions can be selectively
protected as a silyl ether (7) using t-butyldimethylchlorosilane, a tertiary
base such as
triethylamine, and a catalytic amount of 4-dimethylaminopyridine. In addition,
the h-
and 6'-position primary alcohols can be selectively acylated (Scheme 3) using
an
appropriate acid chloride in a 1:1 mixture of tetrahydrofuran and the hindered
base
2,4,6-collidine at -40 °C initially to ambient temperature overnight.
The remaining
five secondary alcohols of the disaccharide can then be protected with acetic
anhydride and triethylamine in a solvent such as dichloromethane to afford the
peracetylated compound 8.
In Scheme 4 the two primary alcohol positions (C-6 and C-6') are first
converted to tosylates using tosyl chloride and pyridine in a solvent such as
dichloromethane; the resulting intermediate is then peracetylated as mentioned
above
to generate compound 9. Through displacement of the tosylates of 9,
heterocyclic
ring systems can be incorporated at the 6 and 6' positions. Finally, the five
secondary
acetates are removed with catalytic sodium methoxide in methanol or aqueous
sodium
hydroxide in methanol at temperatures ranging from ambient temperature to
reflux to
afford compound 10.
CA 02351059 2001-05-09
WO 00/34295 PC'1'/US99/27777
-11-
Scheme 1
Ac
Ac0 V
Ac0 Ac
A c0 O ,+ HY
Ac Ac0 2 N02
A ci
A
1
reduction
OAc
Ac0 O
Ac0 Ac0 O c j
Ac0 Y
A c0 NHZ
acylation
~OAc
A c0 0
A c0 A c0 O c
Ac0 Y
5 Ac0
Z
hydrolysis
OH
HO O
HO HO O %
HO Y
HO Z
Br
glycosidation
CA 02351059 2001-05-09
WO 00/34295 PCT/US99/27777
- 12-
If Rz = OH or OTBDMS:
Scheme 2
OH
HO O
HO HO H W
HO Y
Ho 1/
Z
silylation
O'B D MS
HO O
HO HO OTBDMS W
HO Y
HO
Z
If RZ = ester linkage:
CA 02351059 2001-05-09
WO 00/34295 PCTNS99/27777
- 13-
Scheme 3
H
HO
HO HO O H
HO Y
6 HO Z
1) G6,G6'-diacytation
2) peracetylation
R2
RIO
R10 R2
RIO O
RIO ~ Y
R O
Z
If R~ = tosylate or imidazole:
CA 02351059 2001-05-09
WO 00/34295 PCT/US99/27777
- 14-
Scheme 4
H
HO "'~~
HO HO O H
HO Y
HO
Z
1 ) tosylation
2) peracetylation
1
CH3
I
O~~ CH3
R R O O~
O
RIO Y
R10
Z
1 ) displacement
2) hydrolysis
R2
1
R10
Rt0 Rt0 O R2
R10 Y
RIO Z
5
The compounds of this invention are useful as andproliferative agents. The
following procedures show the evaluation of representative compounds of this
invention in standard pharmacological test procedure which measured ability of
the
10 evaluated compound to inhibit smooth muscle cell proliferation
CA 02351059 2001-05-09
WO 00/34295 PCTNS99/27777
-15-
Effects of Compounds on Cell Proliferation Usin 3g H Thymidine Incorporation
Human and porcine smooth muscle cells were tested in early passage (generally
passage 3 - 7) at sub-confluent conditions. Cultures were grown in 16 mm (24
well)
multi-well culture dishes in medium 199 supplemented with 10% fetal bovine
serum
and 2% antibiotic / antimycotic. At sub-confluence, the cells were placed in a
defined serum free medium (AIM-V; Gibco) for 24 - 48 h prior to initiating the
experimental protocol.
Although compounds were found to be more effective with longer pre-
incubations,
in general, the procedures were initiated with the addition of compound,'H
thymidine
and serum / growth factor to serum deprived synchronized cells and results are
reported accordingly.
Compounds were added to each well at 50 fold dilution (20 pL / well) and the
plates
were incubated for 24 - 36 h at 37 °C in 5% COZ. Compounds were
initially
dissolved in 50% ethanol and serially diluted into media. Compounds were
routinely
evaluated at concentrations from 1 to 100 pM. As a control, grade II porcine
intestinal mucosal heparin (sodium salt) was routinely evaluated in all cell
preparations at concentrations from 0.1 to 100 pg/mL.
At the completion of the test procedure, plates were placed on ice, washed
three
times with ice cold phosphate buffered saline (PBS) and incubated in ice cold
10%
trichloroacetic acid (TCA) got 30 min to remove acid soluble proteins.
Solution was
transferred to scintillation vials containing 0.4 N HCl (S00 uL/ vial to
neutralize
NaOH) and each well was rinsed two times with water (S00 pL,) for a total
volume of
2 mL / vial.
Data was obtained, in triplicate, for both control and experimental samples.
Control
( 100%) data was obtained from maximally stimulated cells, as the result of
growth
factor or serum stimulation. Experimental data was obtained from cells
maximally
stimulated with growth factor or serum and treated with compound. Data are
expressed as an ICsp or percent inhibition in Table I below.
CA 02351059 2001-05-09
WO 00/34295 PCT/US99/27777
- 16-
Table 1.
Porcine Smooth Muscle
Com ound of Exam Cell
le _A__nti roliferation IC50
~ ~
1 0.850 M
2 4.61 M
3 1.71 M
4 0.164 M
1.14 M
6 0.667 M
7 7.97
8 2.05 M
9 0% @ 500 M
6.10 M
11 3.90 M
12 0.390-2.20
13 0.360 M
14 6.56 M
1 S 73.2
16 18%a @ 50 M
17 0.245
18 25% @ 50
19 28% @ 50 M
0.740
21 67.4 M
22 23% @ 100 M
23 4.70
The compounds of this invention are useful in treating or inhibiting diseases
5 which are characterized by excessive smooth muscle cell proliferation
(smooth
muscle cell hyperproliferation). The compounds are particularly useful in
treating
hyperproliferative vascular diseases which are characterized by smooth muscle
cell
hyperproliferation, such as restenosis, which most frequently arises from
vascular
reconstructive surgery and transplantation, for example, balloon angioplasty,
vascular
10 graft surgery, coronary artery bypass surgery, and heart transplantation.
Other
disease states in which there is unwanted "cellular" vascular proliferation
include
hypertension, asthma, and congestive heart failure. The compounds of this
invention
are also useful as inhibitors of angiogenesis. Angiogenesis
(neovascularization), the
process by which new capillaries are formed, is of principal importance for a
number
CA 02351059 2001-05-09
WO 00/34295 PCTNS99/27777
-17-
of pathological events including chronic inflammation and malignant processes.
The
compounds of this invention are therefore useful as antineoplastic agents.
The compounds of this invention can be formulated neat or with a
pharmaceutical carrier for administration, the proportion of which is
determined by
the solubility and chemical nature of the compound, chosen route of
administration
and standard pharmacological practice. The pharmaceutical carrier may be solid
or
liquid.
A solid carrier can include one or more substances which may also act as
flavoring agents, lubricants, solubilizers, suspending agents, fillers,
glidants,
compression aids, binders or tablet-disintegrating agents; it can also be an
encapsulating material. In powders, the carrier is a finely divided solid
which is in
admixture with the finely divided active ingredient. In tablets, the active
ingredient is
mixed with a carrier having the necessary compression properties in suitable
proportions and compacted in the shape and size desired. The powders and
tablets
preferably contain up to 99% of the active ingredient. Suitable solid carriers
include,
for example, calcium phosphate, magnesium stearate, talc, sugars, lactose,
dextrin,
starch, gelatin, cellulose, methyl cellulose, sodium carboxymethyl cellulose,
polyvinylpyrrolidine, low melting waxes and ion exchange resins.
Liquid carriers are used in preparing solutions, suspensions, emulsions,
syrups, elixirs and pressurized compositions. The active ingredient can be
dissolved
or suspended in a pharmaceutically acceptable liquid carrier such as water, an
organic ,
solvent, a mixture of both or pharmaceutically acceptable oils or fats. The
liquid
carrier can contain other suitable pharmaceutical additives such as
solubilizers,
emulsifiers, buffers, preservatives, sweeteners, flavoring agents, suspending
agents,
thickening agents, colors, viscosity regulators, stabilizers or osmo-
regulators.
Suitable examples of liquid carriers for oral and parenteral administration
include
water (partially containing additives as above, e.g. cellulose derivatives,
preferably
sodium carboxymethyl cellulose solution), alcohols (including monohydric
alcohols
and polyhydric alcohols, e.g. glycols) and their derivatives, lethicins, and
oils (e.g.
fractionated coconut oil and arachis oil). For parenteral administration, the
Garner
can also be an oily ester such as ethyl oleate and isopropyl myristate.
Sterile liquid
carriers are useful in sterile liquid form compositions for parenteral
administration.
CA 02351059 2001-05-09
WO 00/34295 PCT/US99/27777
-18-
The liquid Garner for pressurized compositions can be halogenated hydrocarbon
or
other pharmaceutically acceptable propellant.
Liquid pharmaceutical compositions which are sterile solutions or suspensions
can be utilized by, for example, intramuscular, intraperitoneal or
subcutaneous
injection. Sterile solutions can also be administered intravenously. The
compounds
of this invention can also be administered orally either in liquid or solid
composition
form.
l0
The compounds of this invention may be administered rectally or vaginally in
the form of a conventional suppository. For administration by intranasal or
intrabronchial inhalation or insufflation, the compounds of this invention may
be
formulated into an aqueous or partially aqueous solution, which can then be
utilized
in the form of an aerosol. The compounds of this invention may also be
administered
transdermally through the use of a transdermal patch containing the active
compound
and a carrier that is inert to the active compound, is non toxic to the skin,
and allows
delivery of the agent for systemic absorption into the blood stream via the
skin. The
Garner may take any number of forms such as creams and ointments, pastes,
gels, and
occlusive devices. The creams and ointments may be viscous liquid or semisolid
emulsions of either the oil-in-water or water-in-oil type. Pastes comprised of
absorptive powders dispersed in petroleum or hydrophilic petroleum containing
the
active ingredient may also be suitable. A variety of occlusive devices may be
used to
release the active ingredient into the blood stream such as a semipermeable
membrane covering a reservoir containing the active ingredient with or without
a
carrier, or a matrix containing the active ingredient. Other occlusive devices
are
known in the literature.
The dosage requirements vary with the particular compositions employed, the
route of administration, the severity of the symptoms presented and the
particular
subject being treated. Based on the results obtained in the standard
pharmacological
test procedures, projected daily dosages of active compound would be 0.1 to 10
mglkg administered parenterally (intravenous preferred), with projected daily
oral
dosage being approximately ten-fold higher. Anticipated intravenous
administration
would last for approximately 5-30 days following acute vascular injury (i.e.,
balloon
angioplasty or transplantation) and for a longer duration for the treatment of
chronic
disorders. Treatment will generally be initiated with small dosages less than
the
optimum dose of the compound. Thereafter the dosage is increased until the
optimum effect under the circumstances is reached; precise dosages for oral,
parenteral, nasal, or intrabronchial administration will be determined by the
CA 02351059 2001-05-09
WO 00/34295 PCT/US99/27777
- 19-
administering physician based on experience with the individual subject
treated.
Preferably, the pharmaceutical composition is in unit dosage form, e.g. as
tablets or
capsules. In such form, the composition is sub-divided in unit dose containing
appropriate quantities of the active ingredient; the unit dosage forms can be
packaged
compositions, for example, packaged powders, vials, ampoules, pre filled
syringes or
sachets containing liquids. The unit dosage form can be, for example, a
capsule or
tablet itself, or it can be the appropriate number of any such compositions in
package
form.
The following provides the preparation of representative compounds of this
invention.
Example 1
4-Chloro-3-nitro-benzyl-~i-D-maltoside hentaacetate
To a stirred solution of 4-chloro-3-nitrobenzyl alcohol (6.70 g, 35.7 mmol)
and HgBrz ( 14.2 g, 39.3 mmol) in freshly distilled CH3CN (239 mL) was added
in
one portion Hg(CN)Z (9.02 g, 35.7 mmol). After 0.5 h, acetobromomaltose (25.0
g,
35.7 mmol) was added, and the mixture stirred for 18 h at rt. The reaction was
then
quenched with a mixture of HZO:brine (1:1, 100 mL) and extracted with 10%
CH2C12:EtOAc. The combined organic extracts were dried (MgSO,) and
concentrated.
Purification by flash chromatography (10:90 to 80:20 EtOAc:petroleum ether
gradient) gave 51.9 g (90%) of the title compound as a glassy oil which was
recrystallized from EtzO:petroleum ether to afford a glassy white solid, mp
107-111
°C; 'H NMR (CDC13) s 2.00 (s, 3H), 2.02 (s, 3H), 2.03, (s, 3H), 2.04
(s, 6H), 2.11 (s,
3H), 2.15 (s, 3H), 3.70 (ddd, J = 2.9, 4.2, 9.7 Hz, 1H), 3.94-3.98 (m, 1H),
4.01-4.07
(m, 2H), 4.20-4.28 (m, 2H), 4.54 (dd, J = 2.9, 12.3 Hz, 1H), 4.63-4.68 (m,
2H), 4.84-
4.94 (m, 3H), 5.06 {t, J = 10.1 Hz, 1H), 5.26 (t, J = 9.2 Hz, 1H), 5.36 (dd, J
= 9.7,
10.3 Hz, 1 H), 5.42 {d, J = 4.2 Hz, 1 H), 7.43 (dd, J = 2.2, 8.3 Hz, 1 H),
7.53 (d, J = 8.3
Hz, 1H), 7.83 (d, J = 2.0 Hz, 1H); IR {KBr) 3450, 2950, 1755, 1550, 1375, 1230
and
1050 cm~'; mass spectrum [(+) ESI], m/z 823/825 (M + NH,'), 828/830 (M + Na)';
Anal. Calcd. for C33H4aC1NOZO: C, 49.17; H, 5.00; N, 1.74, Found: C, 49.16; H,
4.88; N, 1.71.
CA 02351059 2001-05-09
WO 00/34295 PCTNS99/27777
-20-
Example 2
N-15-f(Hepta-O-acetyl-D-D-maltosvlox )-~vll-2-chloro-phenvll-L-aspartamide-v-
tert-but,1
step 1
S 2-Chioro-5-(hepta-O-acetyl-~-D-maltosyl-oxymethyl)-phenylamine
A solution containing 4-chloro-3-nitro-benzyl-p-n-maltoside heptaacetate
(Example 1, 19.3 g, 23.9 mmol) and tin (II) chloride dehydrate (37.7 g, 167
mmol) in
EtOAc (479 mL) was refluxed for 2 h. The reaction was cooled to rt, carefully
quenched with sat. aq. NaHCO3 (until basic), diluted with EtOAc (250 mL),
stirred
for 0.5 h and filtered. The biphasic filtrate was separated and the aqueous
phase
extracted with EtOAc. The combined organic extracts were dried (NazS04) and
concentrated. Purification by flash chromatography (0 to 12% acetone/CHC13
gradient) gave 17.8 g (96%) 2-Chloro-S-(hepta-O-acetyl-p-D-maltosyl-oxymethyl)-
phenylamine as a glassy solid, mp 78-79 °C; 'H NMR (CDC13) s 2.00 (s,
9H), 2.026
(s, 3H), 2.032 (s, 3H), 2.11 (s, 3H), 2.16 (s 3H), 3.00-5.00 (bs, 2H), 3.64-
3.68 (m,
1H), 3.97 (ddd, J = 2.4, 4.2, 10.1 Hz, 1H), 4.02-4.07 (m, 2H), 4.24 (dd, J =
2.2, 3.7,
1 H), 4.27 (dd, J = 2.6, 4.0 Hz, 1 H), 4.50-4.57 (m, 3H), 4.74 (d, J = 12.1
Hz, 1 H),
4.83-4.90 (m, 2H), 5.05 (t, J = 10.1 Hz, 1H), 5.22 (t, J = 9.2 Hz, 1H), 5.35
(dd, J =
9.7, 10.5 Hz, 1 H), 5.42 (d, J = 4.0 Hz, 1 H), 6.62 (dd, J = 2.0, 8.1 Hz, 1
H), 6.76 (d, J
= 2.0 Hz, 1H), 7.21 (d, J = 8.1, 1H); IR (KBr) 3450, 3350, 2950, 1755, 1650,
1425,
1375, 1230 and 1050 cm~'; mass spectrum [(+) ESI], m/z 776/778 (M + H)',
798/800
(M + Na)'; Anal. Calcd. for C33H42C1NO'e: C, 51.07; H, 5.45; N, 1.80, Found:
C,
50.94; H, 5.52; N, 1.60.
step 2
N {5-[(Hepta-O-acetyl-p-D-maltosyloxy)-methyl]-2-chloro-phenyl}-(9H-fluoren-
9-ylmethyoxycarbonylamino)-L-aspartamide-4-tert-butyl ester
To a stirred solution of N (9H-fluoren-9-ylmethyoxycarbonylamino)-L
aspartic acid-4-tert-butyl ester (0.117 g, 0.284 mmol) and DMF (cat. amt.) in
CHZCl2
(3 mL) at rt was added oxalyl chloride (24.8 ~L, 0.284 mmol) dropwise. After 5
min.
at this temperature, it was heated to 40 °C for an additional 10 min.
This completed
the preparation of the acid chloride starting material. At this point, to a
second stirred
CA 02351059 2001-05-09
WO 00/34295 PCT/US99/27777
-21-
solution of NaH (0.0103 g, 0.258 mmol) and CHZCl2 (4 mL) at rt was added 2-
chloro-
5-(hepta-O-acetyl-p-D-maltosyl-oxymethyl)-phenylamine (0.200 mg, 0.258 mmol).
After 10 min., the acid chloride solution was added to this solution dropwise.
The
reaction was stirred at rt for 1 h and then diluted with EtOAc ( 100 mL). This
layer
was washed with 1 N HCl ( 10 mL), sat. NaHC03 ( 10 mL), and brine ( 10 mL) and
then dried (MgSO,). After concentration, the oilly residue was purified by
flash
chromatography (10:90 to 70:30 EtOAc:petroleum ether gradient) to afford the
product (0.157 g, 52%) as a white foam, mp 103-105 °C; 'H NMR (CDCl3) s
1.46 (s,
9H), 1.99 (s, 3H), 2.00 (s, 3H), 2.01 (s, 3H), 2.02 (s, 3H), 2.03 (s, 3H),
2.10 {s, 3H),
2.16 (s, 3H), 2.70 (dd, J = 5.9, 17.4 Hz, 1H), 2.96-3.06 (m, 1H), 3.67 (ddd, J
= 2.6,
4.2, 9.7 Hz, 1H), 3.96 (ddd, J = 2.4, 3.7, 10.3 Hz, 1H), 4.00-4.06 (m, 2H),
4.22-4.28
(m, 3H), 4.42-4.48 (m, 1H), 4.48-4.56 (m, 2H), 4.58 (dd, J = 2.2, 10.1 Hz,
2H), 4.68-
4.76 (m, 1H), 4.81-4.91 (m, 3H), 5.05 (t, J = 10.1 Hz, 1H), 5.22 (t, J = 9.2
Hz, 1H),
5.35 (dd, J = 9.7, 10.5 Hz, 1 H), 5.41 (d, J = 4.0 Hz, 1 H), 6.07-6.15 (m, 1
H), 7.00 (dd,
J = 2.0, 8.1 Hz, 1H), 7.28-7.36 (m, 3H), 7.40 (t, J = 7.2 Hz, 2H), 7.57-7.62
(m, 2H),
7.77 (d, J = 7.5 Hz, 2H), 8.31 (d, J = 1.8 Hz, 1H), 8.86 (s, 1H); IR (KBr)
3380, 2960,
1755, 1600, 1540, 1440, 1420, 1375, 1230, I 160, and 1050 cm'; mass spectrum
[(+)
FAB], m/z I 169 (M + H)', 1191 {M + Na)+; Anal. Calcd. for C~H65C1N~O23 ~ 2.0
H20:
C, 55.79; H, 5.77; N, 2.32, Found: C, 55.89; H, 5.45; N, 2.25.
step 3
N-{5-[(Hepta-O-acetyl-p-n-maltosyloxy)-methyl]-2-chloro-phenyl}-L-
aspartamide-~-tert-butyl ester
To a stirred solution of 20% piperidine (2.00 mL, 20.2 mmol) in DMF (10
mL) at rt was added N {S-[(hepta-O-acetyl-p-D-maltosyloxy)-methyl]-2-chloro-
phenyl}-(9H-fluoren-9-ylmethyoxycarbonylamino)-L-aspartamide-4-tert-butyl
ester
(0.300 g, 0.256 mmol). After 1 h at this temperature, the solution was
concentrated
on the high vacuum. At this point, the residue was diluted with cold Hz0 (20
mL) and
then extracted with EtzO (50 mL). This layer was dried (NazSO,) and after
concentration, the resulting oil was purified by flash chromatography (20:80
to 90:10
EtOAc:petroleum ether gradient) to afford the product (0.186 g, 77%) as a
white
solid, mp 85-87 °C; 'H NMR (CDCl3) s 1.45 (s, 9H), 1.89 (s, 2H), 1.99
(s, 6H), 2.01
CA 02351059 2001-05-09
WO 00/34295 PCT/US99/27777
-22-
(s, 3H), 2.02 (s, 3H), 2.03 (s, 3H), 2.10 (s, 3H), 2.16 (s, 3H), 2.68 (dd, J =
8.1, 16.7
Hz, 1 H), 2.91 (dd, J = 3.7, 16.7 Hz, 1 H), 3.64-3.69 (m, 1 H), 3.80 (dd, J =
3.7, 8.3 Hz,
1H), 3.93-3.98 (m, 1H), 3.99-4.05 (m, 2H), 4.21-4.27 (m, 2H), 4.50 (dd, J =
2.6, 12.1
Hz, 1H), 4.56 (d, J = 3.7 Hz, 1H), 4.59 (d, J = 8.1 Hz, 1H), 4.81-4.91 (m,
3H), 5.05
(t, J = 10.1 Hz, 1 H), 5.22 (t, J = 9.4 Hz, 1 H), 5.34 (dd, J = 9.4, 10.3 Hz,
1 H), 5.40 (d,
J = 4.0 Hz, 1 H), 6.98 (dd, J = 2.0, 8.1 Hz, 1 H), 7.34 (d, J = 8.1 Hz, 1 H),
8.42 (d, J =
2.0 Hz, 1H), 10.28 (s, 1H); IR (KBr) 3380, 2960, 1755, 1600, 1540, 1440, 1420,
1375, 1235, 1140, and 1040 cm~'; mass spectrum [(+) FAB], m/z 947/949 (M +
H)',
969/971 (M + Na)'; Anal. Calcd. for C4'H55C1NZOz': C, 51.98; H, 5.85; N, 2.96,
Found: C, 51:62; H, 5.89; N, 2.95.
Example 3
N (2-Chloro-5-f12.2'.3.3'.4'.6.6')-hepta-O-acetyl-a-D-maltosvl-oxvmethvll-
phenvll-
l9H-fluoren-9-ylmethoxycarbonyl)-L-alaninamide
The title compound was prepared as a white foam (2.50 g, 36%) from 2-
chloro-5-(hepta-O-acetyl-p-D-maltosyl-oxymethyl)-phenylamine using N (9H
fluoren-9-ylmethyoxycarbonylamino)-L-alanine and a procedure similar to step 2
of
Example 2, mp >96 °C (decomp.); 'H NMR (DMSO-d6) s 1.33 (dd, J = 7.2
Hz, 3H),
1.918 (s, 3H), 1.919 (s, 3H), 1.94 (s, 3H), 1.966 (s, 3H), 1.97 (s, 3H), 2.01
(s, 3H),
2.07 (s, 3H), 3.91-4.02 (m, 4H), 4.12-4.24 (m, 3H), 4.24-4.34 (m, 3H), 4.34-
4.40 (m,
1 H), 4.53 (d, J = 12.7 Hz, 1 H), 4.68-4.75 (m, 2H), 4.84 (d, J = 4.0 Hz, 1
H), 4.86 (d, J
= 2.6 Hz, 1H), 4.97 (t, J = 9.7 Hz, 1H), 5.21 (t, J = 9.7 Hz, 1H), 5.27 (d, J
= 3.7 Hz,
1 H), 5.27-5.32 (m, 1 H), 7.08 (dd, J = 1.8, 8.1 Hz, 1 H), 7.32 (t, J = 7.2
Hz, 2H), 7.40
(t, J = 7.5 Hz, 2H), 7.47 (d, J = 8.1 Hz, 1 H), 7.69-7.78 (m, 4H), 7.88 (d, J
= 7.5 Hz,
2H), 9.42 (s, 1H); IR (KBr) 3360, 3010, 2950, 1755, 1590, 1535, 1440, 1420,
1370,
1230, 1050, and 755 cm '; mass spectrum [(+) ESI], m/z 1069.2 (M + H)',
1086.2/1088.2 (M + NH,)'; Anal. Calcd. for CS,H"C1N20z' ~ 3.5 H20: C, 54.09;
H,
5.70; N, 2.47, Found: C, 53.67; H, 5.11; N, 2.34.
CA 02351059 2001-05-09
WO 00/34295 PCTNS99/27777
-23-
Example 4
4-Benzoyl-N 12-chloro-5-f(2.2',3.3',4',6,6'-hepta-O-acetyl-a-~-maltosvl)-ox~
methyll-phenyl l-benzamide
The title compound was prepared as a white foam (0.240 g, 94%) from 2-
chloro-5-(hepta-O-acetyl-p-D-maltosyl-oxymethyl)-phenylamine using p-
benzoylbenzoic acid and a procedure similar to step 2 of Example 2, mp >84
°C
(decomp.); 'H NMR (DMSO-db) s 1.93 (s, 3H), 1.94 (s, 6H), 1.97 (s, 6H), 2.01
(s,
3H), 2.08 (s, 3H), 3.93-4.03 (m, 4H), 4.15 (dd, J = 4.6, 12.3 Hz, 1H), 4.21
(dd, J =
4.6, 12.1 Hz, 1 H), 4.39 (dd, J = 2.2, 11.9 Hz, 1 H), 4.70 (ABq, J = 12.7 Hz,
es = 0.14,
2H), 4.74 (dd, J = 8.1, 9.7 Hz, IH), 4.86 (dd, J = 4.0, 10.5 Hz, 1H), 4.90 (d,
J = 8.1
Hz, 1 H), 4.98 (t, J = 9.7 Hz, 1 H), 5.21 (dd, J = 9.7, 10.5 Hz, 1 H), 5.28
(d, J = 4.0 Hz,
I H), 5.31 (dd, J = 8.6, 9.4 Hz, 1 H), 7.22 {dd, J = 2.0, 8.3 Hz, 1 H), 7.52
(d, J = 2.0
Hz, 1H), 7.55-7.62 (m, 3H), 7.69-7.74 (m, 1H), 7.76-7.80 (m, 2H), 7.85-7.88
(m,
2H), 8.11-8.14 (m, 2H), 10.30 (s, 1 H); IR (KBr) 3400, 3010, 2950, 1755, 1675,
1650,
1590, 1530, 1440, 1420, 1370, 1230, 1130, and 1040 cm'; mass spectrum [(+)
FAB],
m/z 984/986 (M + H)', 1006/1008 (M + Na);; Anal. Calcd. for C"HSOCINOZO: C,
57.35; H, 5.12; N, 1.42, Found: C, 57. I 1; H, 5.03; N, 1.32.
Example 5
(4-Chloro-3-nitro-benzyl)-hepta-O-acetyl-1-thio-t~-D-maltoside
To a stirred solution of hepta-O-acetyl-I-thio-p-maltose (2.0 g, 3.065 mmol)
[P. L. burette; T. Y. Shen. Carb. Res. 1978, 67, 484-490.] in acetone (20 ml)
were
added 4-chloro-3-nitrobenzyl bromide (0.844 mg, 3.37 mmol) and a solution of
potassium carbonate (0.423 mg, 3.065 mmol) in water (10 ml). The mixture was
boiled under reflux for 30 min, cooled and concentrated. The residue was
extracted
with dichloromethane, and the combined extracts were washed with water, and
brine,
dried (MgSO,) and concentrated. Purification by flash chromatography (40%-60%
EtOAc/petroleum ether gradient) afforded 1.588 g (63%) of the title compound
as a
white solid, mp 73-75 °C; 'H NMR (CDC13) s 1.99 (s, 3H), 2.00 {s, 3H),
2.02 (s, 3H),
2.03 (s, 6H), 2.11 (s, 3H), 2.15 (s, 3H), 3.61-3.64 (m, 1H), 3.80 (d, J = 13.6
Hz, 1H),
3.94 - 4.00 (m, 3H), 4.08 (dd, J = 12.3, 2.4 Hz, 1 H), 4.18-4.27 (m, 2H), 4.36
(d, J =
9.9 Hz, 1H), 4.50 (dd, J = 12.1, 2.6 Hz, 1H), 4.85 (dd, J = 10.5, 4.0 Hz, 1H),
4.90
CA 02351059 2001-05-09
WO 00/34295 PCTNS99/27777
-24-
(apparent t, J = 9.9 Hz, 1H), 5.05 (apparent t, J = 9.9 Hz, 1H), 5.23
(apparent t, J =
9.2 Hz, 1 H), 5.34 (apparent t, J = 9.7 Hz, 1 H), 5.40 (d, J = 4.0 Hz, 1 H),
7.47 (dd, J =
8.4, 2.0 Hz, 1H), 7.51 (d, J = 8.4 Hz, IH), 7.87 (d, J = 2.0, Hz, 1H); IR
(KBr) 3500,
2950, 1750, 1250 and 1050 cm-'; mass spectrum [(+) FAB], m/z 822 (M + H)', 844
(M + Na)'; Anal. Calcd. for C33H.oC1NO,9S: C, 48.21; H, 4.90; N, 1.70, Found:
C,
47.75; H, 4.86; N, 1.65.
Example 6
(3-Amino-4-chloro-benzvl) hepta-O-acetyl-1-thio-!~-D-maltoside
The title compound was prepared as a white solid from (4-chloro-3-nitro-
benzyl)-hepta-O-acetyl-1-thio-p-D-maltoside using a procedure similar to step
1 of
Example 2, mp 78 °C; 'H NMR (CDCl3) s 1.99 (s, 9H), 2.02 (s, 3H), 2.03
(s, 3H),
2.11 (s, 3H), 2.18 (s, 3H), 3.57-3.60 (m, 1H), 3.73 (ABq, J = 13.0 Hz, ns =
0.16, 2H),
3.95-4.08 (m, 3H), 4.17 (bs, 2H), 4.23 (d, J = 4.2, Hz, 2H), 4.27 (dd, J =
7.7, 4.4 Hz,
1H), 4.31 (d, J = 4.4, Hz, 1H), 4.34 (d, J = 10.1, Hz, 1H), ), 4.44 (dd, J =
12.1, 3.3
Hz, 1H), ), 4.84 (dd, J = 10.5, 4.0 Hz, 1H), 4.88 (apparent t, J = 9.9 Hz,
1H), 5.03
(apparent t, J = 9.9 Hz, 1H), 5.35 (apparent t, J = 9.0 Hz, 1H), 5.35
(apparent t, J =
9.4 Hz, 1H), 5.40 (d, J = 4.0, Hz, 2H), 6.60 (dd, J = 8.1, 2.0 Hz, 1H), 6.73
(d, J = 2.0
Hz, 1H), 7.18 (d, J = 8.13 Hz, 1H); IR (KBr) 3500, 2950, 1750, 1245 and 1050
cm';
mass spectrum [{-) FAB], m/z 790 (M - H)'; Anal. Calcd. for C33H,~C1NO": C,
50.03;
H, 5.34; N, 1.77, Found: C, 49.55; H, 5.21; N, 1.71.
Example 7
N 12-Chloro-5-f henta-D-acetyl-a-D-maltos 1-~0~ methyl-phenyl l-acetamide
The title compound was prepared as a white solid from (3-amino-4-chloro-
benzyl) hepta-O-acetyl-1-thio-p-D-maltoside using a procedure similar to step
1 of
Example 9, mp 80-81 °C; 'H NMR (CDC13) s 1.99 (s, 9H), 2.03 (s, 6H),
2.11 (s, 3H),
2.16 (s, 3H), 2.25 (s, 3H), 3.61-3.65 (m, 1H), 3.82 {ABq, J = 13.2 Hz, os =
0.14, 2H),
3.94-4.11 (m, 3H), 4.34 (d J = 10.1 Hz, 1 H), 4.53 (dd, J = 12.3, 2.6 Hz, 1
H), 4.83-
4.91 (m, 2H), 5.05 (apparent t, J = 9.7 Hz, 1H), 5.20 (apparent t, J = 9.0 Hz,
1H),
5.34 (apparent t, J = 10.3 Hz, 1H), 5.39 (d, J = 4.2 Hz, 1H), 7.00 (dd, J =
8.1, 2.2 Hz,
1H), 7.33 (d, J = 8.4 Hz, 1H), 7.62 (s, IH), 8.31 (s, 1H); IR (KBr) 3400,
2955, 1750,
CA 02351059 2001-05-09
WO 00/34295 PCT/US99/27777
-25-
1245 and 1050 cm''; mass spectrum [(+) FAB], m/z 834 (M + H)+, 856 (M + Na)';
Anal. Calcd. for C35H"CiN0,8 S: C, 50.39; H, 5.32; N, 1.68, Found: C, 49.99;
H,
5.07; N, 1.59 .
Example 8
5-fyHepta-O-acetyl-a-D-maltosyl)-oxy-meth 1~-~ano-1-nitrobenzene
step 1
a-Bromo-2-vitro p-tolunitrile
A stirred mixture containing 4-methyl-2-nitrobenzonitrile (2.04 g, 12.6
mmol), N-bromosuccinimide (2.24 g, 12.6 mmol) and azobisisobutyronitrile
(0.103
g, 0.630 mmol) in CCl4 (50 mL) was irradiated with a 300 watt flood light for
2 h.
The reaction was diluted with CHZC12 (50 ml), filtered and concentrated.
Purification
by flash chromatography (35 and 40% ether/pet. ether gradient) gave 1.44 g
(47%) of
the title compound as a yellow oil. 'H NMR (DMSO-db) s 4.90 (s, 2H), 8.05 (dd,
J =
8.0, 1.5 Hz, 1H), 8.18 (d, J = 8.0, 1H), 8.52 (s, 1H).
step 2
a-Hydroxy-2-vitro p-tolunitrile
A stirred solution containing a-bromo-2-vitro p-tolunitrile ( 1.228 g, 5.095
mmol) and sodium formate (0.8664 g, 12.74 mmol) in ethanol:water (4:1, 25 mL)
was refluxed for 2 h. The reaction was cooled to room temperature, diluted
with 20%
CHZCIz/EtOAc, washed with H20 (3x), dried (MgSO,) and concentrated.
Purification
by flash chromatography (1,2 and 3% MeOH/CHCl3 gradient) gave 0.695 g (77%) of
the title compound as a white solid. 'H NMR (DMSO-db) s 4.71 (d, 2H), 5.75 (t,
1H),
7.89 (dd, J = 7.9 Hz, 1H), 8.14 (d, J = 7.9 Hz, 1H), 8.32 (s, 1H).
step 3
5-[(Hepta-O-acetyl-p-D-maltosyl)-oxy-methyl)-2-cyano-1-nitrobenzene
At ambient temperature, to a stirred solution of acetobromomaltose (2.39 g,
3.41 mmol), a-hydroxy-2-vitro p-tolunitrile (0.789 g, 4.43 mmol) and HgBr2
(1.60 g,
4.43 mmol) in freshly distilled CH3CN (34 mL) was added in one portion Hg(CN)Z
( 1.12 g, 4.43 g, mmol}. After 16 h, brine (50 mL) was added and the mixture
was
CA 02351059 2001-05-09
WO 00/34295 PCT/US99/27777
-26-
extracted with 10% CHZClz/EtOAc. The combined organic extracts were washed
with brine (3x), dried (MgS04) and concentrated. Purification by flash
chromatography (1,2 and 3% MeOH/CHC13 gradient) gave 1.941 g (71%) of the
title
compound as a foam. An analytical sample was obtained by recrystallization
from
EtOAc/Hexane followed by recrystallization from EtOH to give a white solid, mp
155-157 °C; 'H NMR (DMSO-d6) s 1.93 (s, 3H), 1.94 (s, 3H), 1.97 (s,
3H), 1.99 (s,
3H), 2.01 (s, 3H), 2.06 (s, 3H), 3.93-4.01 (m, 4H), 4.36 (d, J = 11.0 Hz, 1H),
4.77
(dd, J = 9.6, 8.0 Hz, 1 H), 4.83-4.88 (m, 2H), 4.93-5.00 (m, 3H), 5.21 (dd, J
= 10.3,
9.7 Hz, 1 H), 5.27 (d, J = 3.7 Hz, 1 H), 5.30-5.34 (m, I H), 7.84 (dd, J =
7.9, 1.5 Hz,
1H), 8.18 (d, J = 7.9 Hz, 1H), 8.27 (s, 1H); IR (KBr) 3450, 2950, 2225, 1750,
1225
and 1050 cm-'; mass spectrum [(+) FAB], m/z 797 (M + H)'; Anal. Calcd. for
C34H40N2020' C~ 51.26; H, 5.06; N, 3.52, Found: C, 51.06; H, 5.02; N, 3.31.
Example 9
N-f2-Chloro-5 ~3-D-maltosyl-oxvmethyl)-phenvll-acetamide
step 1
N-[2-Chloro-5-(hepta-O-acetyl-~-n-maltosyl-oxymethyl)-phenylJ-
To a stirred solution of 2-chloro-5-(hepta-O-acetyl-a-D-maltosyl-oxymethyl)-
phenylamine (20.6 g, 26.5 mmol) and triethylamine (8.13 mL, 58.3 mmol) in THF
(265 mL) at 0 °C was added dropwise acetyl chloride (2.26 mL, 31.8
mmol). After
0.5 h at this temperature, it was warmed to rt and stirred an additional 6 h.
At this
point, the reaction was concentrated and taken up in EtOAc (700 mL). This
organic
solution was washed with 1 N HCl (70 mL), sat. aq. NaHC03 (70 mL), and brine
(70
mL) and then dried (MgSO,). After concentration, the residue was purified by
flash
chromatography (20:80 to 100:0 EtOAc:petroleum ether gradient) to afford the
product (16.2 g, 75%) as a glassy solid, mp 84-86 °C; 'H NMR (CDCI,) s
2.00 (s,
6H), 2.020 (s, 3H), 2.027 (s, 3H), 2.03 (s, 3H), 2.11 (s, 3H), 2.16 {s, 3H),
2.24 (s,
3H), 3.66-3.69 (m, 1H), 3.94-3.98 (m, 1H), 4.00-4.06 (m, 2H), 4.22-4.28 (m,
2H),
4.50-4.61 (m, 3H), 4.80-4.91 (m, 3H), 5.05 (t, J = 10.1 Hz, 1 H), 5.22 (t, J =
9.2 Hz,
1H), 5.35 (dd, J = 9.4, 10.5 Hz, 1H), 5.41 (d, J = 4.0 Hz, 1H), 6.99 (dd, 3 =
2.0, 8.1
Hz, 1H), 7.34 (d, J = 8.1 Hz, 1H), 7.62 (s, 1H), 8.32 (s, 1H); IR (KBr) 3400,
2950,
1750, 1690, 1600, 1540, 1425, 1375, 1230 and 1050 cm-'; mass spectrum ((+)
ESI],
CA 02351059 2001-05-09
WO 00/34295 PCT/US99/27777
-27-
m/z 818/820 (M + H)', 840 (M + Na)+; Anal. Calcd. for C35H,~C1N0,9: C, 51.38;
H,
5.42; N, 1.71, Found: C, 51.03; H, 5.36; N, 1.59.
step 2
N-[2-Chloro-5-(p-D-maltosyl-oxymethyl)-phenyl]-acetamide
A solution containing N [2-chloro-5-(hepta-O-acetyl-p-D-maltosyl-
oxymethyl)-phenyl]-acetamide (0.945 g, 1.12 mmol) and 25 weight % NaOMe in
MeOH (19.2 ~L, 0.336 mmol) in MeOH (27.6 ml) was refluxed for 2.5 h. The
reaction was cooled to room temperature and concentrated, and the resulting
residue
was triturated with EtzO to afford the product (0.583 g, 99%) as a foam; 'H
NMR
(DMSO-d6) s 2.07 (s, 3H), 3.03-3.16 (m 2H), 3.19-3.49 (m, 7H), 3.55-3.62 (m,
2H),
3.67-3.73 (m, 1H), 4.28 (d, J = 7.7 Hz, 1H), 4.33-5.76 (bs, 7H), 4.67 (ABq, J
= 12.5
Hz, es = 0.22, 2H), 5.01 (d, J = 3.7 Hz, 1H), 7.21 (dd, J = 1.8, 8.1 Hz, 1H),
7.44 (d, J
= 8.1 Hz, 1H), 7.64 (d, J = 1.5 Hz, 1H), 9.33-9.69 (bs, 1H); IR (ICBr) 3400,
2900,
1680, 1600, 1540, 1430, 1375, 1310, 1150 and 1035 cm-'; mass spectrum [(+)
ESI],
m/z 524/526 (M + H)', 546 (M + Na)'; Anal. Calcd. for CZ,H3oC1N0,2 ~ 1.0 MeOH:
C, 47.53; H, 6.16; N, 2.52, Found: C, 47.94; H, 6.34; N, 2.42.
Example 10
N -( 5-f6,6'-Di-O-(tert-butyl-dimethYl-sil,yl)-a-D-maltosvloxy-meth l~methyl=
phenyl ~-acetamide
To a stirred solution of N [5-(p-n-maltosyl-oxymethyl)-2-methyl-phenyl]-
acetamide {prepared from 4-methyl-3-nitrobenzyl alcohol and acetobromomaltose
using procedures similar to Example 1, Example 2-step 1, and Example 9)(1.5 g,
2.98 mmol) in CHZC12:DMF (1:1, 12 mL) at rt was added DMAP (0.109 g, 0.892
mmol) followed by triethylamine ( 1.66 mL, 11.9 mmol), and then finally
TBDMSCI
( 1.35 g, 8.96 mmol). After 18, the solvent was removed on high vac and the
residue
diluted with EtOAc (200 mL). This organic layer was washed with 1 N HCl (20
mL),
sat. aq. NaHC03 (20 mL), and brine (20 mL) and then dried (MgSO,). After
concentration, the residue was purified by flash chromatography {0:100 to
25:75
MeOH:CHC13 gradient) to afford the product (16.2 g, 75%) as a white foam, mp
111-
114 °C; 'H NMR (CDC13) s 0.067 (s, 6H), 0.080 (s, 3H), 0.084 (s, 3H),
0.89 (s, 9H),
CA 02351059 2001-05-09
WO 00/34295 PCTNS99/27777
-28-
0.90 (s, 9H), 2.07 (s, 3H), 2.16 (s, 3H), 3.25-3.28 (m, 1H), 3.34-3.38 (m,
1H), 3.45-
3.52 (m, 3H), 3.65-3.78 (m, 4H), 3.84-3.94 (m, 4H), 4.28 (d, J = 7.7 Hz, 1H),
4.64
(ABq, J = 12.1 Hz, os = 0.27, 2H), 4.65-4.69 (bs, 1H), 4.74-4.78 (m, 1H), 4.97
(d, J
= 3.3 Hz, 1H), 5.25-5.30 (m, 1H), 5.63-5.68 (bs, 1H), 6.99 (d, J = 7.9 Hz,
1H), 7.07
(d, J = 7.7 Hz, 1H), 7.54 (s, 1H), 7.59-7.63 (bs, 1H); IR (KBr) 3400, 2930,
2870,
1670, 1600, 1550, 1450, 1375, 1255, 1125, 1050, 840, and 790 cm-'; mass
spectrum
[(+) FAB], m/z 754 (M + Na)'; Anal. Calcd. for C~,H6~NO,ZSi2 ~ 0.5 H20: C,
55.11;
H, 8.43; N, 1.89, Found: C, 54.91; H, 8.36; N, 1.85.
Example 11
N -(2-Chloro-5-f6.6'-di-O-(tert.-butyl-dimeth~il-silyl)-Q-D-maltosyl_oxy-
methyll-
phenyl )-acetamide
The title compound was prepared as a white foam (0.845 g, 59%) from N [2-
chloro-5-(p-D-maltosyl-oxymethyl)-phenyl]-acetamide using a procedure similar
to
Example 10, mp 93-98 °C; 'H NMR (CDCl3) s 0.07 (s, 12H), 0.88 (s, 9H),
0.89 (s,
9H), 2.18 (s, 3H), 3.25-3.31 (m, IH), 3.41-3.46 (m, 1H), 3.49-3.58 (m, 3H),
3.69-
3.79 (m, 4H), 3.85 (d, J = 4.0 Hz, 1H), 3.87-3.92 (m, 2H), 3.92 (d, J = 2.6
Hz, 1H),
4.32 (d, J = 7.7 Hz, 1H), 4.53-4.58 (m, 1H), 4.68 (ABq, J = 12.5 Hz, os =
0.25, 2H),
4.71-4.75 (m, 1 H), 5.00 (d, J = 3.5 Hz, 1 H), 5.25 (dd, J = 2.0, 6.4 Hz, 1
H), 5.69 (s,
1H), 6.98-7.04 (m, 1H), 7.25-?.29 (m, 1H), 7.66 (s, 1H), 8.25 (s, 1H); IR
(KBr)
3400, 2930, 2880, 1675, 1600, 1550, 1460, 1420, 1365, 1250, 1050, 850, and 800
cm
'; mass spectrum [(+) FAB], m/z 774/776 {M + Na)i; Anal. Calcd. for
C33HS8C1NO,~Si2 ~ 0.5 H20: C, 52.05; H, 7.81; N, 1.84, Found: C, 52.16; H,
7.82; N,
1.80.
Example 12
N-12-Chloro-5-f(16.6'-di-O-benzoyl-p-D-maltosyllox )v meth,~~ll,phenyl)-
acetamide
To a stirred solution of N [2-chloro-5-(p-D-maltosyl-oxymethyl)-phenyl]-
acetamide (1.00 g, 1.91 mmol) in THF (20.0 mL) at -40 °C was added
collidine (20.0
mL, 151 mmol) followed by dropwise addition of benzoyl chloride (0.532 mL,
4.58
mmol). After 2 h at this temperature, it was warmed to rt and stirred an
additional 18
h. At this point, the solvent was distilled off using the high vac, and the
residue was
CA 02351059 2001-05-09
WO 00/34295 PCT/US99/27777
-29-
purified by flash chromatography (2% to 20% MeOH:CHCl3 gradient) to afford the
product (0.500 g, 36%) as a glassy white solid, mp 99-100 °C; 'H NMR
(CDC13) s
2.12 (s, 3H), 3.41-3.51 (m, 3H), 3.59-3.68 (m , 2H), 3.77 (t, J = 9.2 Hz, 1H),
3.85 (t,
J = 9.2 Hz, 1H), 4.06-4.12 (m, 1H), 4.30-4.34 (m, 2H), 4.40- 5.35 (bs, 3H),
4.51 (dd,
J = 5.3, 12.1 Hz, 1 H), 4.57 (ABq, J = 12.5 Hz, es = 0.22, 2H), 4.59 (d, J =
10.5 Hz,
1H), 4.80 (d, J = I1.0 Hz, 1H), 5.07 (d, J = 3.1 Hz, 1H), 5.45-5.66 (bs, 1H),
5.75-
5.95 (bs, 1H), 6.86 (dd, J = 1.3, 8.3 Hz, 1H), 7.12 (d, J = 8.3 Hz, 1H), 7.23
(t, J =7.7
Hz, 2H), 7.33-7.52 (m, 4H), 7.70 (s, 1H), 7.91-7.96 (m, 4H), 8.14 (s, IH); IR
(KBr)
3400, 2900, 1720, 1620, 1600, 1550, 1450, 1425, 1375, 1320, 1225, 1100, and
1060
cm-'; mass spectrum [(+) FAB], m/z 732 (M + H);, 754 (M + Na)'; Anal. Calcd.
for
C35H38C1NO" ~ 0.5 H20: C, 56.72; H, 5.30; N, 1.89, Found: C, 56.62; H, 5.04;
N,
1.90.
Example 13
N 12-Chloro-5-f1C6.6'-di-O-benzovl-2.2'.3.3'.4'-nenta-acetyl-a-D-
malto~lloxvlmethvllphenvl )-acetamide
To a stirred solution of N ( 5-[([6,6'-di-O-benzoyl-p-n-maltosyl]oxy)methyl)-
2-chlorophenyl }-acetamide (0.122 g, 0.167 mmol) and triethylamine (0.256 mL,
1.84
mmol) in CHZCIZ (5.0 mL) at rt was added dropwise acetic anhydride (().0865
mL,
0.916 mmol) followed by a catalytic amount of DMAP (0.0102 g, 0.0835 mmol).
After 18 h, the mixture was diluted with EtOAc (50 mL). This layer was washed
with
1 N HCl (5 mL), sat. aq. NaHC03 (5 mL), and brine (5 mL) and then dried
(MgS04).
After concentration, the residue was purified by flash chromatography (2% to
20%
acetone:CHCl3 gradient) to afford the product { 1.68 g, 94%) as a fine white
powder
(0.120 g, 76%), mp 97-100 °C; 'H NMR (CDCI3) s 1.96 (s, 3H), 1.99 (s,
3H), 2.01 (s,
3H), 2.03 (s, 3H), 2.04 (s, 3H), 2.22 (s, 3H), 3.80-3.84 (m, 1H), 4.06-4.10
(m, 1H),
4.18-4.22 (m, 3H), 4.53 (dd, J = 4.0, 12.3 Hz, 1H), 4.58 (d, J = 9.7 Hz, 1H),
4.61 (d,
J = 5.1 Hz, 1H), 4.76-4.79 (m, 1H), 4.79-4.82 (m, 1H), 4.86 (dd, J = 4.0, 10.5
Hz,
1 H), 4.92 (dd, J = 7.7, 9.2 Hz, 1 H), 5.15 (t, J = 9.9 Hz, 1 H), 5.27 (t, J =
9.0 Hz, 1 H),
5.41 (dd, J = 9.7, 10.3 Hz, 1H), 5.47 (d, J = 4.2 Hz, 1H), 6.95 (dd, J = 1.8,
8.1 Hz,
1H), 7.29 (d, J = 8.3 Hz, 1H), 7.41-7.50 (m, 4H), 7.52-7.62 (m, 3H), 7.98-8.01
(m,
2H), 8.07-8.09 (m, 2H), 8.30 (s, 1H); IR (KBr) 3400, 2950, 1760, 1725, 1620,
1600,
CA 02351059 2001-05-09
WO 00/34295 PCT/US99/27777
-30-
1550, 1450, 1425, 1375, 1275, 1245, 1120, 1050, and 710 cm'; mass spectrum
[{+)
FAB], m/z 942 (M + H)', 964 (M + Na)'; Anal. Calcd. for C45H48C1N0,9 ~ 2.0
H20:
C, 55.25; H, 5.36; N, 1.43, Found: C, 55.18; H, 4.87; N, 1.36.
Example 14
L4-Chloro-3-nitrophenyl)methyl-4-O-f6-O-(3-p rv idinvlcarbonvl)-a-D-
glucopyranos ly 1-a-~- lg~ucopvranoside-6-f3-pvridinecarboxylate)
To a stirred solution of N-[2-chloro-5-(~-D-maltosyl-oxymethyl)-phenyl]-
acetamide (0.500 g, 0.977 mmol) in THF (9.8 mL) at -40 °C was added
coIlidine (9.8
mL, 74.3 mmol) dropwise followed by nicotinoyl chloride hydrochloride (0.417
g,
2.34 mmol). After 2 h at this temperature, it was warmed to rt and stirred an
additional 42 h. At this point, the reaction was concentrated on high vacuum
and then
diluted with EtOAc (250 mL). The solid was filtered off and washed with
additional
EtOAc (50 mL). The filtrate was concentrated, and the resulting oilly residue
purified
by flash chromatography (40:2:1 to 10:2:1 EtOAc:EtOH:HzO gradient) to afford
the
product (0.159 g, 23%) as a white solid, mp 145-147 °C;'H NMR (DMSO-d6)
s 3.15-
3.25 (m, 2H), 3.32-3.35 (m, 1H), 3.43 (dd, J = 5.3, 9.2 Hz, 1H), 3.45-3.52 (m,
1H),
3.54 (t, J = 8.8 Hz, 1H), 3.70-3.76 (m, 1H), 3.91 (ddd, J = 1.3, 5.5, 9.7 Hz,
1H), 4.29-
4.38 (m, 2H), 4.41 (d, J = 7.7 Hz, 1H), 4.53-4.58 (m, 1H), 4.67-4.72 (m, 1H),
4.72
(ABq, J = 13.6 Hz, es = 0.07, 2H), 5.11 (d, J = 4.8 Hz, 2H), 5.35 (d, J = 5.7
Hz, 1H),
5.42 (d, J = 5.1 Hz, 1H), 5.70 (d, J = 2.6 Hz, 1H), 5.74 (d, J = 5.9 Hz, 1H),
7.42 (ddd,
J = 0.6, 4.8, 7.9 Hz, 1H), 7.50 (ddd, J = 0.7, 4.8, 7.9 Hz, 1H), 7.62 (dd, J =
1.8, 8.3
Hz, 1H), 7.67 (d, J = 8.3 Hz, 1H), 8.01 (d, J = 1.8 Hz, 1H), 8.16 (tt, J =
2.0, 8.6 Hz,
2H), 8.66 ( dd, J = 1.8, 4.8 Hz, 1H), 8.77 (dd, J = 1.8, 4.8 Hz, 1H), 8.97
(dt, J = 1.3,
9.2 Hz, 2H); IR (KBr) 3480, 3390, 3110, 2900, 1725, 1590, 1550, 1475, 1420,
1390,
1360, 1340, 1285, 1175, 1140, 1090, 1050, and 1015 cm-'; mass spectrum [(+)
FAB],
m/z 722/724 (M + H)', 744/746 (M + Na)'; Anal. Calcd. for C3,H3~C1N3O,5 ~ 1.5
H20:
C, 49.71; H, 4.71; N, 5.61, Found: C, 49.68; H, 4.53; N, 5.59.
CA 02351059 2001-05-09
WO 00/34295 PCT/US99/27777
-3I-
Example 15
(4-Chloro-3-nitrophenvl)methyl-4-O-f 6-O-l3-pyridinvlcarbonyl)-«-D-
lg ucopyranosvll-a-D- lg ucopyranoside
The title compound was prepared as a white glass (0.070 g, 10°!0)
from N [2-
chloro-5-(p-D-maltosyl-oxymethyl)-phenyl]-acetamide using a procedure similar
to
Example 14, mp >85 °C (decomp.); 'H NMR (DMSO-d6) s 3.07-3.14 (m,
1H), 3.14-
3.23 (m, 1H), 3.25-3.36 (m, 2H), 3.36-3.47 (m, 3H), 3.51-3.58 (m, 1H), 3.67-
3.73
(m, 1 H), 3.83-3.89 (m, 1 H), 4.29-4.34 (m, 2H), 4.54-4.62 (m, 2H), 4.79 (ABq,
J =
13.4 Hz, es = 0.17, 2H), 5.05-5.08 (m, 2H), 5.31 (d, J = 5.7 Hz, 1H), 5.34 (d,
J = 5.1
Hz, 1 H), 5.56 (d, J = 6.4 Hz, 1 H), 5.59 (d, J = 3.1 Hz, 1 H), 7.56 (ddd, J =
0.9, 4. 8,
8.1 Hz, IH), 7.71 (dd, J = 2.0, 8.3 Hz, 1H), 7.75 (d, J = 8.3 Hz, 1H), 8.09
(d, J = I.5
Hz, 1 H), 8.33 (dt, J = 1.8, 7.9 Hz, 1 H), 8.81 (dd, J = 1.8, 4.8 Hz, 1 H),
9.10 (dd, J =
0.7, 2.0 Hz, IH); IR (KBr) 3390, 2910, 1730, 1625, 1600, 1540, 1475, 1410,
1360,
1290, 1140, 1120, and 1040 cm~'; mass spectrum [(+) FAB], mlz 617/619 (M +
H)';
Anal. Calcd. for CZSH29C1NZO~4 ~ 1.5 HZO: C, 46.63; H, 5.01; N, 4.35, Found:
C,
46.58; H, 4.88; N, 4.26.
Example 16
N f2-Chloro-5-ff(4-O-«-D- Ig ucopyranos 1-~ t3-D- lucop r~yl)oxylmethYll
phenvll-
3-pyridinecarboxamide
step 1
N-[2-Chloro-5-[[[2,3,6-tri-O-acetyl-4-O-(2,3,4,6-tetra-O-acetyl-«-D-
glucopyranoysl)-p-D-glucopyranosyl]oxy]methyl]phenyl]-3-pyridinecarboxamide
To a stirred solution of 2-chloro-5-(hepta-O-acetyl-p-D-maltosyl-oxymethyl)-
phenylamine (0.200 g, 0.258 mmol) and triethylamine (0.119 mL, 0.851 mmol) in
THF (3 mL} at 0 °C was added nicotinoyl chloride hydrochloride (0.0551
mg, 0.310
mmol). After 0.5 h at this temperature, it was warmed to rt and stirred an
additional
18 h. At this point, the solid was filtered off and washed with additional THF
( 10
mL). The filtrate was then concentrated and taken up in EtOAc (100 mL). This
organic solution was washed with H20 (10 mL) and brine (10 mL) and then dried
(Na~SO,). After concentration, the residue was purified by preparatory plate
chromatography (10:90 MeOH:CHCl3) to afford the product (0.183 g, 80%) as a
CA 02351059 2001-05-09
WO 00/34295 PCT/US99/27777
-32-
white foam, mp 83-86 °C; 'H NMR (CDC13) s 1.99 (s, 3H), 2.~ (s, 3H),
2.02 (s, 3H),
2.03 (s, 3H), 2.04 {s, 3H), 2.10 (s, 3H), 2.16 (s, 3H), 3.67-3.72 (m, 1H),
3.93-3.98
(m, 1H), 4.04 {dd, J = 2.2, I 1.9 Hz, 2H), 4.25 (dt, J = 3.7, 12.5 Hz, 2H),
4.53 (dd, J =
2.9, 12.3 Hz, 1 H), 4.60 (d, J = 7.7 Hz, 1 H), 4.64 (d, J = 12.5 Hz, 1 H),
4.83-4.93 (m,
3H), 5.05 (t, J = 10.1 Hz, 1 H), 5.23 (t, J = 9.4 Hz, 1 H), 5.34 (dd, J = 9.7,
10.5 Hz,
1 H), 5.41 (d, J = 4.2 Hz, 1 H), 7.07 (dd, J = 2.0 Hz, 1 H), 7.41 (d, J = 8.1
Hz, 1 H),
7.48 (ddd, J = 0.9, 4.8, 7.9 Hz, 1 H), 8.23 (ddd, J = 1.5, 2.2, 7.9 Hz, 1 H),
8.43 {s, 1 H),
8.48 (d, J = 2.0 Hz, 1 H), 8.82 (dd, J = 1.5, 4.8 Hz, 1 H), 9.15 (dd, J = 0.7,
2.2 Hz,
1H); IR (KBr) 3400, 2950, 1755, 1675, 1600, 1550, 1420, 1375, 1235, and 1050
cm-';
mass spectrum [{+) FAB], m/z 881 (M + H)', 903 (M + Na)'; Anal. Calcd. for
C39H45C1NZO,9 ~ 2.0 H20: C, 51.07; H, 5.38; N, 3.05, Found: C, 50.80; H, 4.83;
N,
2.89.
step 2
N [2-Chloro-5-[[(4-O-a-D-glucopyranosyl-p-v-glucopyranosyl)oxy]methyl]
phenyl]-3-pyridinecarboxamide
The title compound was prepared as a white foam (1.97 g, 57%) from N [2-
chloro-5-[[[2,3,6-tri-O-acetyl-4-O-(2,3,4,6-tetra-O-acetyl-«-D-glucopyranoysl)-
p-D-
glucopyranosyl]oxy]methyl]phenyl]-3-pyridinecarboxamide using a procedure
similar to step 2 of Example 9, mp >106 °C {decomp.); 'H NMR (DMSO-db)
s 3.02-
3.13 (m, 2H), 3.19-3.29 (m, 2H), 3.31-3.39 (m, 1H), 3.39-3.50 (m, 3H), 3.55-
3.63
(m, 2H), 3.70-3.76 (m, 1 H), 4.09 (q, J = 5.3 Hz, 1 H), 4.31 (d, J = 7.9 Hz, 1
H), 4.49-
4.55 (m, 2H), 4.60 (d, J = 12.5 Hz, 1H), 4.84-4.91 (m, 3H), 5.01 (d, J = 3.7
Hz, 1H),
5.26 (d, J = 5.1 Hz, 1H), 5.43 (d, J = 6.4 Hz, iH), 5.52 (d, J = 3.1 Hz, 1H),
7.35 (dd,
J = 2.0, 8.3 Hz, 1H), 7.54 (d, J = 8.1 Hz, 1H), 7.56-7.60 (m, 2H), 8.31 (dt, J
= 2.0,
7.9 Hz, 1 H), 8.77 (dd, J = 1.5, 4.8 Hz, 1 H), 9.12-9.14 (m, 1 H), 10.34 (s, 1
H); IR
(KBr) 3390, 2910, 2320, 1660, 1590, 1525, 1475, 1450, 1420, 1360, 1310, 1190,
1140, 1080, and 1030 cm-'; mass spectrum [(+) FAB], mlz 587 (M + H)', 609 (M +
Na)'; Anal. Calcd. for CZSH3,CINZO,2 ~ 1.5 HZO: C, 48.90; H, 5.58; N, 4.56,
Found:
C, 49.18; H, 5.52; N, 4.32.
CA 02351059 2001-05-09
WO 00/34295 PCT/US99/27777
-33-
Example 17
Benzoic acid 6-(4-chloro-3-f(pyridine-3-carbonyl)-aminol-benz~y)-4,5-
dihydoxx-3-(3.4,5-trihydroxv-6-hvdroxymethvl-tetrah~pvran-2-vloxv)-
rPtrahvdro ~vran-2-vlmeth l~h~drochloride
S To a stirred solution of (R)-N (5-[[(6-O-benzoyl-4-O-(4,6-O-
(phenylmethylene)-o~.-D-glucopyranosyl]-p-D-glucopyranosyl]oxy]methyl]-2-
chlorophenyl]-3-pyridinecarboxamide (check other invention record for
preparation)
(0.275 g, 0.353 mmol) in MeOH (10 mL) at 0 °C was added 1.0 M HCl in
EtzO
(0.388 mL, 0.388 mmol). After 10 min. at this temperature, it was warmed to rt
and
stirred an additional 1 S min. The mixture was concentrated to a thin oil and
then
triturated with EtzO (5 mL). At this point, the solid which formed was
filtered off and
washed with additional Et20 (4x, 2 mL). The solid was then dried on the high
vacuum to afford the product (0.200 g, 70%) as an off white solid (86% pure,
contaminated with 14% HCl salt of SM), mp >157 °C (decomp.); 'H NMR
(DMSO-
db) s 3.09-3.19 (m, 2H), 3.24 (dd, J = 3.7, 9.4 Hz, 1H), 3.34-3.41 (m, 1H),
3.41-3.65
(m, SH), 3.71 (dd, J = 6.8, 8.1 Hz, 1H), 4.20-5.04 (m, 12H), 5.05 (d, J = 3.7
Hz, 1H},
7.28 (dd, J = 2.0, 8.3 Hz, I H), 7.46-7.55 (m, 4H), 7.60-7.66 (m, 1 H), 7.82
(dd, J =
5.1, 7.9 Hz, 1H}, 7.94-7.99 (m, 2H), 8.58 (d, J = 7.7 Hz, 1H), 8.90 (dd, J =
1.5, 5.3
Hz, 1H), 9.23 (d, J = 1.5 Hz, 1H), 10.54 (s, 1H); IR (KBr) 3400, 2910, 2850,
2110,
1720, 1690, 1630, 1590, 1530, 1440, 1420, 1320, 1275, 1120, 1070, 1050, 1025,
and
715 crn-'; mass spectrum [(+) ESI], m/z 691.2 (M - HCl + H)'; Anal. Calcd. for
C32H35C1Nz0" ~ HCl ~ 0.5 HZO: C, 52.89; H, 5.06; N, 3.74, Found: C, 52.99; H,
5.27; N, 3.46.
Example 18
f4-Chloro-3-nitro-benzy_1 -l-deoxy-1-thio-a-D-maltoside
The title compound was prepared as a white solid from (4-chloro-3-nitro-
benzyl)-hepta-O-acetyl-1-thio-p-D-maltoside using a procedure similar to step
2 of
Example 9, mp 90-93 °C; 'H NMR (DMSO-db) s 3.03-3.74 (m, , 11H), 3.80
(d, J =
6.2 Hz, 1H), 3.86 (d, J = 13.4 Hz, 1H), 4.01-4.08 (m, 2H), 4.58 (bd, 2H), 4.98
(bd,
3H), 5.20-5.67 (bs, 3 H), 7.65-7.72 (m, 2H), 8.03 (d, J = 1.76 Hz, 1H).IR
(KBr)
3400, 2930, 1550, 1300 and1075 cm~'; mass spectrum [(-)FAB], mlz 526 (M - H)~;
CA 02351059 2001-05-09
WO 00/34295 PCT/US99/27777
-34-
Anal. Calcd. for C,9Hz6C1NO,2S ~ HZO: C, 41.80; H, 5.13; N, 2.56, Found: C,
41.35;
H, 4.89; N, 2.40.
Example 19
N-{2-Chloro-5-fti-D-maltosyl-1-thin)-methvll-nhen_yl~E-acetamide
The title compound was prepared as a white solid from N {2-chloro-5-[ hepta-
O-acetyl-p-D-maltosyl-1-thio)-methyl]-phenyl}-acetamide using a procedure
similar
to step 2 of Example 9, mp 120-125 °C; 'H NMR (CD30D-d4) s 2.17 (s,
3H), 3.23-
3.33 (m, 3H), 3.41 (dd, J = 9.9, 3.7, Hz, 1H), 3.52-3.83 (m, 8H), 3.89 (dd, J
= 12.3,
2.0, Hz, 1 H), 4.00 (d, J = 13.2 Hz, 1 H), 4.20 (d, J = 9.9 Hz, 1 H), 5.15 (d,
J = 4.0 Hz,
1 H), 7.20 (dd, J = 8.4, 1. 8, Hz, 1 H), 7.37 (d, J = 8.4 Hz, 1 H), 7.74 (s, 1
H); IR (ICBr)
3400, 2900, 1600, 1550 and 1050 cm~'; mass spectrum [(-)FAB)], m/z 538 (M - H)-
;
Anal. Calcd. for CZ,H3oC1N0"S ~ 1.0 H20: C, 45.20; H, 5.78; N, 2.51, Found: C,
45.43; H, 5.62; N, 2.43.
Example 20
5- ( f 6.6'-Bis-O-(4-toluenesulfonvl )-a-maltos,~ll-oxv-methvl 1-2-methyl- I -
nitrobenzene
step 1
5-[(p-Maltosyl)-oxy-methyl]-2-methyl-1-nitrobenzene
A stirred solution containing 5-[(hepta-O-acetyl-p-maltosyl)-oxy-methyl]-2-
methyl-1-nitrobenzene (prepared from 4-methyl-3-nitrobenzyI alcohol and
acetobromomaltose using procedures similar to Example 1 )(0.835 g, 1.06 mmol)
and 25 weight% NaOMe/MeOH (0.115 g, 0.531 mmol) in MeOH (25 mL) was
refluxed for 4h. The reaction was cooled to ambient temperature and
concentrated in
vacuo. Purification by reverse phase HPLC (C18, 25% CH3CN:HZO) gave 0.380 g
(73%) of the title compound as a white foam; 'H NMR (DMSO-d6) s 2.50 (s, 3H),
3.60-3.21 (m, 2H), 3.22-3.57 (m, 7H), 3.59-3.65 (m, 2H), 3.71-3.73 (m, 2H),
4.31 (d,
1H), 4.51-4.54 (m, 2H), 4.67 (d, 1H), 4.87-4.91 (m, 3H), 5.02 (d, 1H), 5.29
(d, 1H),
5.44 (d, 1H), 5.53 (d, 1H), 7.48 (d, 1H), 7.64 (dd, 1H), 8.01 (s, 1H).
CA 02351059 2001-05-09
WO 00/34295 PCTNS99/27777
-35-
step 2
5-{[6,6'-Bis-O-(4-toluenesulfonyl)-p-maltosyl]-oxy-methyl}-2-methyl-1-
nitrobenzene
At 0 °C, to a stirred solution of 5-[(p-maltosyl)-oxy-methyl]-2-
methyl-1-
nitrobenzene (0.380 g, 0.774 mmol) in pyridine (4.5 mL) was added portionwise
a
solution of p-toluenesulfonyl chloride (0.812 g, 4.25 mmol) in CHZC12 (4.5
mL), over
a period of 5 h. The reaction was quenched with Hz0 (30 mL) and extracted with
EtOAc. The combined organic extracts were washed successively with sat. aq.
NaHC03 (3x), with aq. CuSO, (3x), brine(3x), dried (NazSO,)
sat. with and
concentrated.Purificationby preparative (C18,65% CH3CN:H20)
HPLC gave
0.373 g (59%) of the compound as a solid,mp 85-92 C; 'H
title white NMR
(DMSO-d6) 8 2.34 (s, 3H), 2.40 (s, 3H), 2.48 (s, 3H), 3.01-3.10 (m, 2H), 3.12-
3.17
(m, 1H), 3.23-3.40 (m, 3H), 3.51-3.55 (m, 2H), 4.02-4.14 (m, 3H), 4.23-4.29
(m,
2H), 4.63 (ABq, J = 13.1 Hz, ns = 0.05, 2H), 4.92 (d, 1 H), 5.01 (d, 1 H),
5.22 (d,
1 H), 5.36 (d, 1 H), 5.48 (d, 1 H), 5.54 (d, 1 H), 7.39 (d, 2H), 7.45-7.48 (m,
3H), 7.60
(dd, 1H), 7.71-7.77 (m, 4H), 7.96 (s, 1H); IR (KBr) 3380, 2920, 1600, 1530,
1360
and 1175 cm''; mass spectrum [(+) FAB], m/z 822 (M + Na)'; Anal. Calcd. for
C3,H"NO"SZ ~ H20: C, 49.93; H, 5.17; N, 1.75, Found: C, 49.77; H, 4.94; N,
1.70.
Example 21
5-( f 2.2' ,3.3' .4'-Penta-O-acetyl-6.6'-bis-O-(4-toluenesulfonyl)-D-maltosvll-
oxv-
methyl 1-2-methyl-1-nitrobenzene
At 0 °C, to a stirred solution containing 5-( [6,6'-bis-O-(4-
toluenesulfonyl)-p-
maltosyl]-oxy-methyl}-2-methyl-1-nitrobenzene (1.79 g, 2.24 mmol), pyridine
(5.43
mL, 67.1 mmol) and 4-dimethylaminopyridine (1.25 g, 11.2 mmai) was added
dropwise acetic anhydride (2.09 mL, 22.4 mmol). After 3 h, the reaction
eventually
warmed to room temperature. The solution was diluted with diethyl ether ( 100
mL),
washed successively with HZO (2x), sat. aq. NaHC03 (2x), sat. aq. CuSO, (2x),
brine
(2x), dried (NazS04) and concentrated. Purification by flash chromatography
(3, 4 and
5% MeOH:CHC13 gradient) gave 1.785 g, (?9%), of the title compound as a white
solid after crystallization from EtOAc:hexane, mp 83 °C; 'H NMR (DMSO-
d6)
s 1.910 (s, 3H), 1.918 (s, 3H), 1.920 (s, 3H), 1.924 (s, 3H), 1.932 (s, 3H),
2.36 (s,
CA 02351059 2001-05-09
WO 00/34295 PCT/US99/27777
-36-
3H), 2.42 (s, 3H), 2.51 (s, 3H), 3.66 (t, 1H), 3.86 (dd, 1H), 3.92-3.96 (m,
1H), 4.06
(dd, 1H), 4.16-4.29 (m, 3H), 4.51-4.60 (m, 2H), 4.64-4.72 (m, 2H), 4.79 (d,
1H),
4.89 (t, 1H), 5.06-5.13 (m, 2H), 5.24 (t, 1H), 7.43 (d, 2H), 7.47-7.50 (m,
4H), 7.74-
7.77 (m, 4H), 7.84 (s, 1H); mass spectrum [(+) FAB], m/z 1010 (M + H)'; Anal.
Calcd. for C44HS,NO~Sz: C, 52.32; H, 5.09; N, 1.39, Found: C, 52.46; H, 5.15;
N,
1.41.
Example 22
5-(fb 6'-Dideoxv-6.6'-bis(4-nitro-imidazol-1-vl)- D-maltosyll-oxv-methvli-2-
methyl-1-nitrobenzene
At ambient temperature, to a stirred solution of 4-nitroimidazole (0.478 g,
4.23 mmol) in DMF (8 mL) was added KZC03 (0.278 g, 2.01 mmol). After 0.5h, to
the reaction was added a solution of 5-{ [6,6'-bis-O-(4-toluenesulfonyl)-ø-
maltosyl]-
oxy-methyl}-2-methyl-1-nitrobenzene (1.61 g, 0.201 mmol) in DMF (20 mL) and
the
reaction was heated at 100 °C for 24h. The reaction was concentrated in
vacuo.
Purification by reverse phase HPLC (C18, 30% CH,CN:H20) gave 0.40 g (29%) of
the title compound as a white solid, mp 146 °C; 'H NMR (DMSO-d6) s 2.48
(s, 3H),
2.89 (t, 1H), 3.08 (t, 1H), 3.20-3.29 (m, 2H), 3.40-3.47 (m, 2H), 3.64 (dt,
1H), 3.88
(dd, 1H), 3.97-4.03 (m, 2H), 4.21-4.27 (m, 2H), 4.43-4.47 (m, 2H), 4.59 (d,
1H),
5.11 (d, 1H), 5.17 (d, 1H), 5.40 (br. s, 1H), 5.57 (br. s, 1H), 5.68 (br. s,
1H), 5.84 (br.
s, 1 H), 7.41-7.46 (m, 2H), 7.65 (d, 1 H), 7.74 (d, 1 H), 7. 85 (s, 1 H), 8.22
(d, 1 H), 8.25
(d, 1H); mass spectrum [(+) FAB], m/z 682 (M + H);; Anal. Calcd. for
CZ6H3,N,O,s ~ 2
HZO: C, 43.52; H, 4.92; N, 13.66, Found: C, 43.90; H, 4.72; N, 13.31.
Example 23
5-f f2 2' 3 3' 4'-Penta-O-acetyl-6.6'-dideoxv-6.6'-bis(4-nitro-imidazol-1-vl)-
a-
maltosyll-oxv-methyl 1-2-methyl-1-nitrobenzene
At ambient temperature, to a stirred solution of 4-nitroimidazole (0.177 g,
1.57 mmol) in DMF (3 mL) was added KZCO, (0.103 g, 0.747 mmol). After O.Sh, to
the reaction was added a solution of 5-{ [2,2',3,3',4'-penta-O-acetyl-6,6'-bis-
O-(4-
toluenesulfonyl)-~-maltosyl]-oxy-methyl}-2-methyl-1-nitrobenzene (0.754 g,
0.747
mmol) in DMF (7 mL) and the reaction was heated at 100 °C for 4h. At
ambient
CA 02351059 2001-05-09
WO 00/34295 PCT/US99/27777
-37-
temperature, the reaction was quenched with ice cold HZO (50 mL) and extracted
with
EtOAc. The organic extracts were dried (NaZSO,) and concentrated. Purification
by
flash chromatography (5, 6 and 7% MeOH:CHC13 gradient) gave 0.315 g,
(47°l0), of
the title compound as a white solid after crystallization from EtOAc:hexane,
mp 140
°C; 'H NMR (DMSO-d6) s 1.92 (s, 3H), 1.93 (s, 3H), 1.94 (s, 6H), 2.08
(s, 3H),
2.49 (s, 3H), 3.64 (dd, 1 H), 3.91 (t, 1 H), 4.00-4.06 (m, 1 H), 4.27-4.40 (m,
SH), 4.54
(d, 1H), 4.74-4.79 (m, 3H), 4.96 (dd, 1H), 5.22-5.30 (m, 3H), 7.33 (dd, 1H),
7.43 (d,
1 H), 7.69 (d, 1 H), 7.72 (d, 1 H), 7.77 (d, 1 H), 8.26 (d, 1 H), 8.34 (d, 1
H); mass
spectrum [(+) FAB], m/z 892 (M + H)'; Anal. Calcd. for C36H4~N,OZO: C, 48.49;
H,
4.63; N, 11.00, Found: C, 48.32; H, 4.52; N, 10.90.