Note: Descriptions are shown in the official language in which they were submitted.
CA 02356345 2001-06-26
WO 00/44711 PCT/US00/01865
-1-
ACETYLENIC (3-SULFONAMIDO AND PHOSPHINIC ACID AMIDE
HYDROXAMIC ACID TACE INHIBITORS
FIELD OF INVENTION
S
This invention relates to acetylenic aryl sulfonamide and phosphinic acid
amide hydroxamic acids which act as inhibitors of TNF-a converting enzyme
(TACE). The compounds of the present invention are useful in disease
conditions
mediated by TNF-a, such as rheumatoid arthritis, osteoarthritis, sepsis, AIDS,
ulcerative colitis, multiple sclerosis, Crohn's disease and degenerative
cartilage loss.
BACKGROUND OF THE INVENTION
TNF-a converting enzyme (TACE) catalyzes the formation of TNF-a from
1 S membrane bound TNF-a precursor protein. TNF-a is a pro-inflammatory
cytokine
that is believed to have a role in rheumatoid arthritis [Shire, M. G.; Muller,
G. W.
Exp. Opin. Ther. Patents 1998, 8(S), 531; Grossman, J. M.; Brahn, E. J.
Women's
Health 1997, 6(6), 627; Isomaki, P.; Punnonen, J. Ann. Med 1997, 29, 499;
Camussi,
G.; Lupia, E. Drugs, 1998, 55(S), 613.] septic shock [Mathison, et. al. J.
Clin. Invest.
1988, 81, 1925; Miethke, et. al. J. Exp. Med. 1992, 175, 91.], graft rejection
[Piguet,
P. F.; Grau, G. E.; et. al. J. Exp. Med. 1987, 166, 1280.], cachexia [Beutler,
B.;
Cerami, A. Ann. Rev. Biochem. 1988, 57, 505.], anorexia, inflammation
[Ksontini,
R,; MacKay, S. L. D.; Moldawer, L. L. Arch. Surg. 1998, 133, 558.], congestive
heart
failure [Packer, M. Circulation, 1995, 92(6), 1379; Ferrari, R.; Bachetti, T.;
et. al.
Circulation, 1995, 92(6), 1479.], post-ischaemic reperfusion injury,
inflammatory
disease of the central nervous system, inflammatory bowel disease, insulin
resistance
[Hotamisligil, G. S.; Shargill, N. S.; Spiegelman, B. M.; et. al. Science,
1993, 259,
87.] and HIV infection [Peterson, P. K.; Gekker, G.; et. al. J. Clin. Invest.
1992, 89,
574; Pallares-Trujillo, J.; Lopez-Soriano, F. J. Argiles, J. M. Med Res.
Reviews,
1995, 15(6), 533.]], in addition to its well-documented antitumor properties
[Old, L.
Science, 1985, 230, 630.]. For example, research with anti-TNF-a antibodies
and
transgenic animals has demonstrated that blocking the formation of TNF-a
inhibits
the progression of arthritis [Rankin, E.C.; Choy, E.H.; Kassimos, D.;
Kingsley, G.H.;
Sopwith, A.M.; Isenberg, D.A.; Panayi, G.S. Br. J. Rheumatol. 1995, 34, 334;
Pharmaprojects, 1996, Therapeutic Updates 17 (Oct.), au197-M2Z.]. This
CA 02356345 2001-06-26
WO 00/44711 PCT/US00/01865
-2-
observation has recently been extended to humans as well as described in "TNF-
a in
Human Diseases", Current Pharmaceutical Design, 1996, 2, 662.
It is expected that small molecule inhibitors of TACE would have the
potential for treating a variety of disease states. Although a variety of TACE
inhibitors are known, many of these molecules are peptidic and peptide-like
which
suffer from bioavailability and pharmacokinetic problems. In addition, many of
these
molecules are non-selective, being potent inhibitors of matrix
metalloproteinases and,
in particular, MMP-1. Inhibition of MMP-1 (collagenase 1) has been postulated
to
cause joint pain in clinical trials of MMP inhibitors [Scrip, 1998, 2349, 20].
Long
acting, selective, orally bioavailable non-peptide inhibitors of TACE would
thus be
highly desirable for the treatment of the disease states discussed above.
Examples of sulfonamide hydroxamic acid MMP/TACE inhibitors in which a
2 carbon chain separates the hydroxamic acid and the sulfonamide nitrogen, as
shown
below, are disclosed in WIPO international publications W09816503, W09816506,
W09816514 and W09816520 and U. S. patent 5,776,961.
R
H~ ~ SOzAr
U. S. patents 5,455,258, 5,506,242, 5,552,419, 5,770,624, 5,804,593 and
5,817,822 as well as European patent application EP606,046A1 and WIPO
international publications W09600214 and W09722587 disclose non-peptide
inhibitors of matrix metalloproteinases and/or TACE of which the aryl
sulfonamide
hydroxamic acid shown below, in which 1 carbon separates the hydroxamic acid
and
the sulfonamide nitrogen, is representative. Additional publications
disclosing
sulfonamide based MMP inhibitors which are variants of the sulfonamide-
hydroxamate shown below, or the analogous sulfonamide-carboxylates, are
European
patent applications EP-757037-A1 and EP-757984-A1 and WIPO international
publications W09535275, W09535276, W09627583, W09719068, W09727174,
W09745402, W09807697, and W09831664, W09833768, W09839313,
W09839329, W09842659 and W09843963. The discovery of this type of MMP
inhibitor is further detailed by MacPherson, et. al. in J. Med. Chem., (1997),
40, 2525
and Tamura, et. al. in J. Med. Chem. (1998), 41, 640.
CA 02356345 2001-06-26
WO 00/44711 PCT/US00/O1$65
-3-
O ~ ~ 4
HO~
"2
Ri R2
Publications disclosing (3-sulfonamide-hydroxamate inhibitors of MMPs
and/or TACE in which the carbon alpha to the hydroxamic acid has been joined
in a
S ring to the sulfonamide nitrogen, as shown below, include U. S. patent
5,753,653,
WIPO international publications W09633172, W09720824, W09827069,
W09808815, W09808822, W09808823, W09808825, W09834918, W09808827,
Levin, et. al. Bioorg. & Med Chem. Letters 1998, 8, 2657 and Pikul, et. al. J.
Med.
Chem. 1998, 41, 3568.
/Ar
O2$
HO, IN
H
The patent applications DE19,542,189-A1, W09718194, and EP803505
disclose additional examples of cylic sulfonamides as MMP and/or TACE
inhibitors.
In this case the sulfonamide-containing ring is fused to a aromatic or
heteroaromatic
1 S ring.
~Ar
R
Analogous to the sulfonamides are the phosphinic acid amide hydroxamic
acid MMP/TACE inhibitors, exemplified by the structure below, which have been
disclosed in WIPO international publication W09808853.
3
HO~
R5
Ri R2 R4
CA 02356345 2001-06-26
WO 00/44711 PCT/US00/01865
-4-
Sulfonamide MMP/TACE inhibitors in which a thiol is the zinc chelating
group, as shown below, have been disclosed in WIPO international application
9803166.
R3
HS
~~/ Ra
Ri 1~ RS ~
It is an object of this invention to provide aryl sulfonamide and phosphinic
acid amide hydroxamic acid MMP/TACE inhibitors in which the Y (the sulfonyl or
phosphinyl aryl) is para-substituted with a substituted butynyl moiety or a
propargylic
ether, amine or sulfide. These compounds provide enhanced levels of inhibition
of the
activity of TACE in vitro and in a cellular assay and/or selectivty over MMP-
1. These
compounds may therefore be used in the treatment of diseases mediated by TNF.
SUMMARY OF THE INVENTION
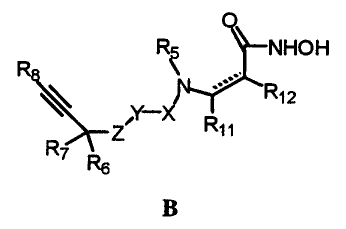
The TACE and MMP inhibiting ortho-sulfonamido hydroxamic acids of the
present invention are represented by the formula:
Rs NHOH
R8 \
' Rt2
R~ ~
R Z
Rs
B
where the C(=O)NHOH moiety and the -NRS moiety are bonded to adjacent
carbons;
wherein
X is SOZ or -P(O)R,o;
Y is 5-10 membered heteroaryl ring having from 1-3 heteroatoms selected
from N, NRg, S and O, phenyl or naphthyl; with the proviso that X and Z may
not be bonded to adjacent atoms of Y;
Z is O, NH, CHZ or S;
CA 02356345 2001-06-26
WO 00/44711 PCT/US00/01865
-5-
RS is hydrogen or alkyl of 1-6 carbon atoms;
R6 and R, are each, independently, hydrogen or methyl;
Re is hydrogen, alkyl of 1-6 carbon atoms, alkenyl of 2-6 carbon atoms,
alkynyl of 2-6 carbon atoms, cycloalkyl of 3-6 carbon atoms, a 5-7
membered heteroaryl having 1-3 heteroatoms selected from N, NRq, S
and O, a 5-7 membered heterocycloalkyl having 1 or 2 heteroatoms
selected from N, NR9, S and O, or phenyl;
Rgis hydrogen, alkyl of I-6 carbon atoms, cycloalkyl of 3-6 carbon atoms, or
phenyl;
R,ois alkyl of 1-6 carbon atoms, cycloalkyl of 3-6 carbon atoms, phenyl, or 5-
7 membered heteroaryl, having 1-3 heteroatoms selected from N, NRg,
S and O;
R" and R,Z are, independently, hydrogen, alkyl of 1-6 carbon atoms,
cycloalkyl of 3-6 carbon atoms, a 5-7 membered heteroaryl having I-3
heteroatoms
selected from N, NR9, S and O, a S-7 membered heterocycloalkyl having 1 or 2
heteroatoms selected from N, NRg, S and O, or phenyl, and the optional double
bond
represented by the dotted line is present; or
R" and R,2, together with the carbons to which they are attached, form a 5-10
membered saturated or unsaturated mono or bicyclic alkyl ring optionally fused
to
one of a 5 to 7 membered saturated or unsaturated cycloalkyl ring, a 5-7
membered
heteroaryl having 1-3 heteroatoms selected from N, NRg, S and O, a 5-7
membered
heterocycloalkyl having 1 or 2 heteroatoms selected from N, NRg, S and O,
phenyl or
napthyl rings; or
R,~ and R,2, together with the carbons to which they are attached form a 5-10
membered saturated or unsaturated mono- or bicyclic heterocycloalkyl having 1-
2
heteroatoms selected from N, NR9, S and O, optionally fused to one of a 5-7
membered mono or bi-cyclic heteroaryI having 1-3 heteroatoms selected from N,
NR9, S and O, a 5-7 membered saturated or unsaturated cycloalkyl ring or a
phenyl or
napthyl ring;
the dotted line represents an optional double bond;
and n = 0-2; or a pharmaceutically acceptable salt thereof.
CA 02356345 2001-06-26
WO 00/44711 PCT/US00/01865
-6-
Preferred compounds of this invention include compounds of structure B
wherein X is SO2.
More preferred compounds of this invention include compounds of structure
B wherein X is SOZ and Y is a phenyl ring substituted at the 1- and 4-
positions by X
and Z, respectively.
More preferred compounds of this invention include compounds of swcture
B wherein X is SOZ,Y is a phenyl ring substituted at the 1- and 4-positions by
X and
Z, respectively, and Z is oxygen.
More preferred compounds of this invention include compounds of structure
B wherein X is S02,Y is a phenyl ring substituted at the 1- and 4-positions by
X and
Z, respectively, Z is oxygen and R6 and R, are hydrogen.
More preferred compounds of this invention include compounds of structure
B wherein X is SOZ,Y is a phenyl ring substituted at the 1- and 4-positions by
X and
Z, respectively, Z is oxygen, R6 andR, are hydrogen and R$ is -CHzOH or
methyl.
Still more preferred compounds of the present invention are (1R,2R)-2-[{[4-
(2-Butynyloxy)phenyl]sulfonyl } (methyl)amino]-N-
hydroxycyclohexanecarboxamide;
(1R, 2R)-2-({[4-(2-Butynyloxy)phenyl]sulfonyl}amino)-N-hydroxycyclo-
hexanecarboxamide;
3-({ [4-(2-Butynyloxy)phenyl]sulfonyl }amino)-N-hydroxypropanamide;
3-( { [4-(2-Butynyloxy)phenyl]sulfonyl } (methyl) amino)-N-hydroxypropan-
amide;
(1R, 2S)-2-[{[4-(2-Butynyloxy)phenyl]sulfonyl}amino)-N-hydroxycyclo-
pentanecarboxamide;
( 1R, 2S)-2-[ { [4-(2-Butynyloxy)phenyi]sulfonyl } (methyl) amino] N-hydroxy-
cyclopentanecarboxamide;
(Cis)-2-[ { [4-(2-butynyloxy)phenyl] sulfonyl } amino)-N-hydroxycyclohexane-
carboxamide;
(Cis)-2-[{ [4-(2-butynyloxy)phenyl]sulfonyl } (methyl) amino]-N-hydroxy-
cyclohexanecarboxamide;
CA 02356345 2001-06-26
WO 00/44711 PCT/US00/01865
_'7_
(1R, 2R, 3S, 4R)-(Cis)-3-({[4-(2-butynyloxy)phenyl]sulfonyl}amino)-N-
hydroxybicyclo [2.2.1) heptane-2-carboxamide; and
(1R, 2R, 3S, 4R)-(Cis)-3-({[4-(2-butynyloxy)phenyl]sulfonyl}~ (methyl)
amino)-N-hydroxybicyclo [2.2.1 ] heptane-2-carboxamide.
Heteroaryl, as used throughout, is a S-10 membered mono- or bicyclic ring
having
from 1-3 heteroatoms selected from N, NRg, S and O. Heteroaryl is preferably
K K K K
I I /
~~N ~ N ,
, N R Ni
s R
s
~ QK~ ~~~ (,~
wherein K is NRq, O or S and Rq is hydrogen, alkyl of 1-6 carbon atoms,
cycloalkyl of 3-6 carbon atoms, or phenyl. Preferred heteroaryl rings
include pyrrole, furan, thiophene, pyridine, pyrimidine, pyridazine,
pyrazine, triazole, pyrazole, imidazole, isothiazole, thiazole, isoxazole,
oxazole, indole, isoindole, benzofuran, benzothiophene, quinoline,
isoquinoline, quinoxaline, quinazoline, benzotriazole, indazole,
benzimidazole, benzothiazole, benzisoxazole, and benzoxazole.
Heteroaryl groups of the present invention may optionally be mono- or di-
substituted.
Heterocycloalkyl as used herein refers to a 5 to 10 membered saturated
or unsaturated mono or bi-cyclic ring having 1 or 2 heteroatoms selected from
CA 02356345 2001-06-26
WO 00/44'711 PCT/US00/01865
-g_
N, NR4, S or O. Heterocycloalkyl rings of the present invention are preferably
selected from
Ra ~ M
n - ~N~ ~'~ .
M ~ M M
~NRa ~ ~ ' N N
Ra
wherein M is NR4, O or S and RQ is hydrogen, alkyl of 1-6 carbon
atoms, cycloalkyl of 3-6 carbon atoms, phenyl, naphthyl, heteroaryl, -
S(O)oR2, -COORZ, -CONRZR3, -SOZNR2R3 or -CORZ. Preferred
heterocycloalkyl rings include piperidine, piperazine, morpholine,
tetrahydropyran, tetrahydrofuran or pyrrolidine. Heterocycloalkyl
groups of the present invention may optionally be mono- or di-
substituted.
Aryl, as used herein refers to phenyl or naphthyl which may, optionally be
mono-, di- or tri-substituted.
Alkyl, alkenyl, alkynyl, and perfluoroalkyl include both straight chain as
well
as branched moieties. Alkyl, alkenyl, alkynyl, and cycloalkyl groups may be
unsubstituted (carbons bonded to hydrogen, or other carbons in the chain or
ring) or
may be mono- or poly-substituted. Cycloalkyl groups may be mono or bicyclic.
Examples of monocyclic cycloalkyl groups include cyclopentyl and cyclohexyl.
Examples of bicyclic cycloalkyl groups include bicycloheptane and adamantyl.
Halogen means bromine, chlorine, fluorine, and iodine.
CA 02356345 2001-06-26
WO 00/44711 PCT/US00/01865
-9-
Suitable substituents of aryl, heteroaryl, alkyl, alkenyl, alkynyl, and
cycloalkyl include, but are not limited to halogen, alkyl of 1-6 carbon
atoms, alkenyl of 2-6 carbon atoms, alkynyl of 2-6 carbon atoms,
cyclocalkyl of 3-6 carbon atoms, -ORZ, -CN, -COR2, perfluoroalkyl of
1-4 carbon atoms, -O-pertluoroalkyl of 1-4 carbon atoms, -CONRZR3,
-S(O)nR2 -OPO(ORZ)OR3, -PO(ORz)R3, -OC(O)NRZR,, -C(O)NRZOR3,
-cooR2, -so,H, -NRZR,, -Nycl~)Zl2NRz, -NRZcoR,, -NRZcooR3,
-SOZNRZR3, -NO2, -N(RZ)SOZR3, -NRZCOIVR2R3,
-~C(=~3)~~~-~C(=~)N(SOZ)~~,
NRZC(=NR3)N(C=O)RZR3, -S02NHCOR4, -CONHS02R,, -tetrazol-S-
yl, -SOZNHCN, -S02NHCONRZR3, phenyl, naphthyl, heteroaryl or
heterocycloalkyl;
wherein -NRZR3 may form a pyrrolidine, piperidine, morpholine,
thiomorpholine, oxazolidine, thiazolidine, pyrazolidine, piperazine, or
azetidine ring;
RZ and R3 are each, independently, hydrogen, alkyl of 1-6 carbon atoms,
cycloalkyl of 3-6 carbon atoms, phenyl, naphthyl, heteroaryl or
heterocycloalkyl;
R, is hydrogen, alkyl of 1-6 carbon atoms, cycloalkyl of 3-6 carbon atoms,
phenyl, naphthyl, heteroaryl, -S(O)nR2, -COOR2, -CONR2R3, -SOZNRZR3 or -COR2 ;
and n is 0-2.
Suitable substituents of heterocycloalkyl groups of the present invention
include, but are not limited to alkyl of 1-6 carbon atoms, cycloalkyl of 3-6
carbon
atoms, phenyl, naphthyl, heteroaryl and heterocycloalkyl.
When a moiety contains more than one substituent with the same designation
each of those substituents may be the same or different.
Pharmaceutically acceptable salts can be formed from organic and inorganic
acids, for example, acetic, propionic, lactic, citric, tartaric, succinic,
fumaric, malefic,
CA 02356345 2001-06-26
WO 00/44711 PCT/US00/01865
-10-
malonic, mandelic, malic, phthalic, hydrochloric, hydrobromic, phosphoric,
nitric,
sulfuric, methanesulfonic, naphthalenesulfonic, benzenesulfonic,
toluenesulfonic,
camphorsulfonic, and similarly known acceptable acids when a compound of this
invention contains a basic moiety. Salts may also be formed from organic and
S inorganic bases, preferably alkali metal salts, for example, sodium,
lithium, or
potassium, when a compound of this invention contains an acidic moiety.
The compounds of this invention may contain an asymmetric carbon atom and
some of the compounds of this invention may contain one or more asymmetric
centers and may thus give rise to optical isomers and diastereomers. While
shown
without respect to stereochemistry, the present invention includes such
optical
isomers and diastereomers; as well as the racemic and resolved,
enantiomerically pure
R and S stereoisomers; as well as other mixtures of the R and S stereoisomers
and
pharmaceutically acceptable salts thereof. It is recognized that one optical
isomer,
including diastereomer and enantiomer, or stereoisomer may have favorable
properties over the other. Thus when disclosing and claiming the invention,
when one
racemic mixture is disclosed, it is clearly contemplated that both optical
isomers,
including diastereomers and enantiomers, or stereoisomers substantially free
of the
other are disclosed and claimed as well.
The compounds of this invention are shown to inhibit the enzymes MMP-1,
MMP-9, MMP-13 and TNF-a converting enzyme (TALE) and are therefore useful in
the treatment of arthritis, tumor metastasis, tissue ulceration, abnormal
wound
healing, periodontal disease, graft rejection, insulin resistance, bone
disease and HIV
infection. In particular, the compounds of the invention provide enhanced
levels of
inhibition of the activity of TACE in vitro and in cellular assay and/or
enhanced
selectivity over MMP-1 and are thus particularly useful in the treatment of
diseases
mediated by TNF.
Accordingly this invention provides a process for preparing compounds of
formula I ,
as defined above, which comprises one of the following:
a) reacting a compound of formula V:
CA 02356345 2001-06-26
WO 00/44711 PCT/US00/01865
-11-
R8 RS O
Y-X' N Q
Z
R~ R6 R~~ R~2
(V)
wherein RS R6, R,, R8, and R" R,2, X, Y, Z and the dotted line are defined
above, and
Q is OH or a reactive derivative thereof, with hydroxylamine to give a
corresponding
compound of formula B;
b) deprotecting a compound of formula VI:
R8
R5 O
\' ,Y x'N NHOR3o
Z
R~ R6 R~ ~ R~2
(VI)
wherein RS R6, R,, Re, R" R,Z, X, Y, Z and the dotted line are defined above,
and R3o
is a suitable protecting group such as t-butyl, benzyl, and trialkylsilyl, to
give a
corresponding compound of formula B
c) resolving a mixture (e.g. racemate) of optically active isomers of a
compound
of formula B to isolate one enantiomer or diastereomer substantially free of
the other
enantiomer or diastereomers;
d) acidifying a basic compound of formula B with a pharmaceutically acceptable
acid to give a pharmaceutically acceptable salt.
With regards to process a) the reaction can be carried out by processes known
in the
art e.g. by reaction of the acid chloride reactive derivative with
hydroxylamine.
Removal of protecting groups, as illustrated by process b) can be carried out
by
processes known in the art to provide the hydroxamic acid.
With regard to process c) standard separation techniques may be used to
isolate
particular enantiomeric or diastereomeric forms. For example a racemic mixture
may
CA 02356345 2001-06-26
WO 00/44711 PCT/US00/01865
-12-
be converted to a mixture of optically active diastereoisomers by reaction
with a
single enantiomer of a 'resolving agent' (for example by diastereomeric salt
formation or formation of a covalent bond). The resulting mixture of optically
active
diastereoisomers may be separated by standard techniques (e.g. crystallisation
or
S chromatography) and individual optically active diastereoisomers then
treated to
remove the 'resolving agent' thereby releasing the single enantiomer of the
compound of the invention. Chiral chromatography (using a chiral support,
eluent or
ion pairing agent) may also be used to separate enantiomeric mixtures
directly.
The compounds of formula B may be isolated in the form of a salt of a
pharmaceutically acceptable acid e.g. an organic or inorganic acid by
treatment with
an acid such as described above.
The invention is further directed to a process for making compounds of
structure B involving one or more reactions as follows:
1 ) alkylating a compound of formula I, or a salt or solvate thereof,
into a compound of formula II
Rs
R
I~
Rs II
2) reacting a compound of formula II above, or a salt or solvate thereof, with
a
chlorinating agent such as thionyl chloride, chlorosulfonic acid, oxalyl
chloride,
phosphorus pentachloride, or other halogenating agents such as fluorosulfonic
acid or
thionyl bromide to a compound of formula III:
CA 02356345 2001-06-26
WO 00/44711 PCT/US00/01865
-13-
Rs
R
I
R8 III
wherein J is fluorine, bromine, chlorine.
The resultant sulfonyl chloride, fluoride or bromide, may be further converted
into triazolide, imidazolide or benzothiazolide derivatives, where J is 1,2,4-
triazolyl,
imidazol-yl or benzotriazolyl, by reacting the compound with 1,2,4-triazole,
imidazole or benzotriazole, respectively. R6, R, and R$ are as defined above.
The invention is still further directed to a process for making compounds of
structure B involving one or more reactions as follows:
1) alkylating phenol, or a salt or solvate thereof, into a compound of formula
IV:
R6~Rs
IV
2) reacting a compound of formula IV above, or a salt or solvate thereof with
chlorosulfonic acid to prepare a compound of formula II above.
Particularly preferred intermediates are compounds of formulae II and III,
with the proviso that R6 is not hydrogen.
The invention compounds are prepared using conventional techniques known
to those skilled in the art of organic synthesis. The starting materials used
in
preparing the compounds of the invention are known, made by known methods or
are
commercially available.
Those skilled in the art will recognize that certain reactions are best
carried
out when other potentially reactive functionality on the molecule is masked or
CA 02356345 2001-06-26
WO 00/44711 PCT/US00/01865
-14-
protected, thus avoiding undesirable side reactions and/or increasing the
yield of the
reaction. To this end, those skilled in the art may use protecting groups.
Examples of
these protecting group moieties may be found in T. W. Greene, P. G. M. Wuts
"Protective Groups in Organic S nthesi ", 2"° Edition, 1991, Wiley &
Sons, New
York. Reactive side chain functionalities on amino acid starting materials are
preferably protected. The need and choice of protecting groups for a
particular
reaction is known to those skilled in the art and depends on the nature of the
functional group to be protected (hydroxy, amino, carboxy, etc.), the
structure and
stability of the molecule of which the substituent is part and the reaction
conditions.
When preparing or elaborating compounds of the invention containing aryl,
heteroaryl or heterocyclic rings, those skilled in the art recognize that
substituents on
that ring may be prepared before, after or concomitant with construction of
the ring.
For clarity, substituents on such rings have been omitted from the schemes
herein
below.
Those skilled in the art will recognize that the nature and order of the
synthetic steps presented may be varied for the purpose of optimizing the
formation
of the compounds of the invention.
The hydroxamic acid compounds of the invention, 1, are prepared according
to Scheme 1 by converting a carboxylic acid, 2 wherein A = R" and R,z, into
the
corresponding acid chloride or anhydride, or by reacting it with a suitable
peptide
coupling reagent, followed by reaction with hydroxylamine to give 1, or with a
protected hydroxylamine derivative to give 3. Compounds 3, wherein R3o is a t-
butyl,
benzyl, trialkylsilyl or other suitable masking group may then be deprotected
by
known methods to provide the hydroxamic acid 1.
CA 02356345 2001-06-26
WO 00/44711 PCT/US00/01865
-15-
Scheme 1:
y
R~ ~ R~
H - "' I I 30
R OH ~ I
A Rs A Ra
3
2
I~' R~
HOH I
A Ra
1
Carboxylic acids 2 may be prepared as shown in Scheme 2. Amino acid
derivative 4, in which R,o is hydrogen or a suitable carboxylic acid
protecting group,
may be sulfonylated or phosphorylated by reacting with compounds 5, in which J
is a
suitable leaving group including, but not limited to chlorine. The N-H
compound 6
may then be alkylated with R3J and a base such as potassium carbonate or
sodium
hydride in a polar aprotic solvent such as acetone, N,N-dimethylformamide
(DMF),
or tetrahydrofuran (THF) to provide sulfonamide 7. Compound 7 is also
available
through direct reaction of 5 with an N-substituted amino acid derivative, 8.
Conversion of 7 into the carboxylic acid is performed by acid, base
hydrolysis, or
other method consistent with the choice of protecting group R4o and the
presence of a
carbon-carbon triple bond.
CA 02356345 2001-06-26
WO 00/44711 PCTNS00/01865
-16-
Scheme 2:
s
H R~
I H2 5 Ra
RaoO >
A A Rs
6
4
RSJ
r
I H 5 Or
R4o0 > Rao
p Ra
T
1
2
Methods of preparation of sulfonylating agents 5 are shown in Scheme 3.
Thus, sulfonic acid salts 9, where ZRsa is a hydroxy, thiol or substituted
amino moiety
may be alkylated with acetylenes 10, where J is a suitable leaving group such
as
halogen mesylate, tosylate, or triflate to give 11. Acetylenes ZO are
commercially
available or known compounds, or they may be synthesized by known methods by
those skilled in the art. The sulfonic acid salts 11 may be converted into the
corresponding sulfonyl chloride or other sulfonylating agent 5 by known
methods,
such as reaction with oxalyl chloride or other reagent compatible with
substituents R6,
R, and R8 and the acetylene. Alternatively, the disulfide 12 may be converted
into di-
acetylene 13 by reaction with compounds 10, followed by reduction of the
disulfide
bond to provide the analogous thiols which may be converted into 5 by known
methods. Alkylation of the phenol, thiophenol, aniline or protected aniline 14
with 10
to give 15, followed by reaction with chlorosulfonic acid provide sulfonic
acids 16
which are readily converted into 5 with oxalyl chloride or similar reagents.
CA 02356345 2001-06-26
WO 00/44711 PCT/US00/01865
-17-
Thiophenols 17 are also precursors to 5 via protection of the thiol,
alkylation of ZH,
where Z is O, N or S, and deprotection of the sulfur followed by oxidation to
the
sulfonic acid 16.
Scheme 3:
Rs Rs
RB~R~ R
S43Na ~ ~ ( S43Na
g 10
s 11
R5~-~-S'S-~-.ZR50
12
r
R ~_S~ R s 5
8 13 R$
R~
SH gR~ SRsp
~S03H
-~ r -/'
s
H ZH ~~ 16
R~
17 18 ciso,H
19
R$
~ + 10 ----~ ~ I R~
~ZRso
14 15
The phosphorus containing analogs of 8 may be prepared using similar
methodology, as shown in Scheme 4.
CA 02356345 2001-06-26
WO 00/44711 PCT/US00/01865
-18-
Scheme 4:
R Rs i I > R Rs Z
~I~OII 4
Rio
Rs
i
IR5 ~Z R~
8 ~
RaoO ~R~o IRa
A
7
R5 ZR$0
RSOZ RSOZ
> RIO ~R~o
R A 'O
~o
The acetylenic side chain may also be appended after sulfonylation or
phosphorylation of the amino acid derivative, as shown in Scheme 5. Thus, the
amino
acid derivatives 4 and 8 can be sulfonylated or phosphorylated with compounds
20,
where ZRso is hydroxy or protected hydroxy, thiol or amine, and, if necessary,
alkylated with R,J as in Scheme 2, to give 21. Removal of the Rso masking
group to
give 22 and subsequent alkylation of the resulting phenol, thiol or amine with
10
provides 7. in the case where ZRso is equal to OH, no deprotection step is
required to
give 22.
CA 02356345 2001-06-26
WO 00/44711 PCT/US00/01865
-19-
Scheme 5:
) ZR5° ~ Zoo
J~ '~w
20 ~ '~X~'~ i 20
4 g
2) RsJ A
21
Deprotect
ZH
,~ t
T s ~noo ,. _
A
22
The propargylic amine analogs of 7 can be synthesized as shown in Scheme 6
5 starting from the amino acid derivatives 4 and/or 8. Sulfonylation or
phosphoryladon
with para-nitro aryl compound 23, for example 4-nitrobenzenesulfonyl chloride,
followed by alkylation with RSJ (for 4) using a base such as potassium
carbonate or
sodium hydride in DMF provides 24. Reduction of the nitro moiety with hydrogen
and palladium on carbon, tin chloride or other known method to give aniline 25
and
10 subsequent alkyladon with 10 then provides 7. Aniline 25 may be derivatized
with a
suitable nitrogen protecting group, such as t-butoxycarbonyl, to give 26 prior
to
alkylation with 10 subsequent deprotection after the alkylation step.
CA 02356345 2001-06-26
WO 00/44711 PCT/US00/01865
-20-
Scheme 6:
.,. N02 R5 ~ N02
1) ~ O
JX ~ ~ N,X ~ 23
4 R ~0 8
2) Rel A
24
1) Reduce
0 ~5 i NHR~ 0 ~s ~ NHZ
N,X ~ ~ N,x
R,~O R,~p
A A
26 2S
~) 10
7
2) Deprotect
Acetylenic derivatives 7 are also accessible via the fluoro compounds 27,
5 readily prepared from the amino acid derivatives 4 and/or 8 by reaction with
fluoraryl
26, as shown in Scheme 7. Displacement of the fluorine of 27 in the presence
of a
base such as sodium hydride with a masked hydroxy, thiol, or amino group
(HZR,o,
where R,o is a suitable protecting group) in a polar aprotic solvent such as
DMF,
followed by deprotection gives 28, which can then be alkylated with 10 to
provide 7.
10 Conversion of 27 to 28, where Z is sulfur, might also be accomplished with
NazS,
KZS, NaSH or KS(C=S)OEt. The fluorine of 27 can also be displaced in a polar
aprotic solvent with the propargylic derivative 29, where Z is O, S or NH, in
the
presence of a base such as sodium hydride, to give 7 directly.
CA 02356345 2001-06-26
WO 00/44711 PCT/US00/01865
-21-
Sch a me 7:
F
Rs F
1) JX ~ ( 26 0 ~ ~ 26
I N.X
4 R~0
2) ~ A
27
Re
HZ R~
29
1) HZR~
R~ Base
2) Deprotect
0 i 5 ~, ZH
7 1~ I N,X ". I
R ~0
A
2$
Compound 7, wherein Z is a methylene group, is available via 30, as shown in
Scheme 8. Benzylic bromination of 30 with N-bromosuccinimide in a chlorinated
hydrocarbon solvent provides bromide 31. This is followed by displacement of
the
bromide with the appropriate propynyl cuprate to provide sulfonamide 8.
CA 02356345 2001-06-26
WO 00/44711 PCT/US00/01865
-22-
Scheme 8:
1) H3C ' 0 ~~ ~ CH3
I X~ i N,
4 RnoO X 8
2) R5J A
0 R3 ~ CHZBr
___ 7 ~ N,X ''
'-' R ~0
A
31
Compounds of the invention can also be prepared by modifying substituents
on the acetylenic side chain at any stage after sulfonylation or
phosphorylation of the
5 starting amino acid derivatives 4 or 8. Functional groups such as halogen,
hydroxy,
amino, aldehyde, ester, ketone, etc. may be manipulated by standard methods to
form
the moieties defined by R,-R8 of compounds 1. It is recognized by those
skilled in the
art of organic synthesis that the successful use of these methods is dependent
upon the
compatibility of substituents on other parts of the molecule. Protecting
groups and/or
10 changes in the order of steps described herein may be required.
Some of the methods available for the derivadzation of compounds of
structure 32 (equivalent to compound 7 wherein R,2 is hydrogen) are shown in
Scheme 9. Metallation of the terminal acetylene 32 followed by addition of an
aldehyde or alkyl halide, sulfonate or triflate provides derivatives 33 and
34. Reaction
15 of 32 with formaldehyde and an amine provides the Mannich addition product
35.
Cyanogen bromide addition to 35 gives the propargylic bromide 36 which may be
displaced with a variety of nucleophiles to give, for example, ethers,
thioethers and
amines 37. Palladium catalyzed coupling reactions of 32 provide the aryl or
heteroaryl acetylenes 38. It is recognized by those skilled in the art of
organic
CA 02356345 2001-06-26
WO 00/44711 PCT/US00/O1$65
-23-
synthesis that the successful use of these methods is dependent upon the
compatibility
of substituents on other parts of the molecule. Protecting groups and/or
changes in the
order of steps described herein may be required, and R35, R45, RSS, R65 and
R,5 are
alkyl, e.g. methyl;
Scheme 9:
Rs
0 Rs Z R7 R~ ~ Z Re
N ~ ~ I 0 N . ~ ~ R~
R Ldp I ~ X
R3s R ao0
HO Raa A
34
33
R~R~CHOI
R~CH2J
R Re
Re ~ ~~Z R
p Ng ~ I Z R~ c~Hz~, R 0~ ~ X I
_ -'~ ~o
R~0 x ~ ~ ~uc~sR~s
A H aooH A 35 NR~m
32
BrCN
Arx
Pd( Ij
C a I R8
TEA ~ R
0 ~5 ~Z R~
R ~0
R
R$ ~ Z R7 A Br
36
R CIO
A Ar
38
Rs
0 R~ .'~ I Z R~
R~0
A ZR
37
CA 02356345 2001-06-26
WO 00/44711 PCTNS00/01865
-24-
The following specific examples illustrate the preparation of representative
compounds of this invention. The starting materials, intermediates, and
reagents are
either commercially available or can be readily prepared following standard
literature
procedures by one skilled in the art of organic synthesis.
Example 1
(trans)-2-(4-Methoxybenzenesulfonyl)aminocyclohexanecarboxylic acid
To a room temperature solution of lg (6.8 mmol) of trans-2-amino-1
cyclohexylcarboxylic acid in 50 ml of dioxane:H20 ( 1:1 ) containing 1.7 ml (
12.2
mmol) of triethylamine was added 1.54g ( 7.46 mmol) of 4-
methoxybenzenesulfonyl
chloride. The mixture was stirred at 25 °C for 18 h. The resulting
mixture was diluted
with pentane to afford 1.119 g (51 %) of the desired sulfonamide product as a
white
solid. 1H NMR(DMSO-d(): 7.7 ppm (dd, 2H, Ar), 7.4 ppm (d, 1H, NH), 7.0 ppm
(dd, 2H, Ar), 3.8 ppm (s, 3H, OMe), 3.5 ppm (m, 1H, N-CH), 1.0-1.7 ppm (m, 9H,
hydrocarbon).
Example 2
(cis)-2-(4-Methoxybenzenesulfonyl)aminocyclohexanecarboxylic a~d
In the same manner as described in Example 1, 2.5 g (17 mmol) of cis-2
amino-1-cyclohexylcarboxylic acid provided 3.283 g (60%) of the desired
carboxylic
acid. Electrospray Mass Spec 314.1 (M+H)~.
Example 3
(traps)-2-(4-Methoxybenzenesulfonyl)aminocyclohexane
carboxylic acid t-butyl ester
To a solution of 0.313g (1 mmol) of the product from Example 1 in 5.0 mL of
toluene was added 1 mL (4 mmol) of N,N-dimethylformamide di-tert-butyl acetal.
The resulting mixture was heated at 110 ~C for 4h under nitrogen and then
allowed to
cool to room temperature. The solution was then poured on top of a silica gel
column. Chromatography on silica gel eluting with 10-20% ethyl acetate/hexane
gave 353 mg (96%) of the desired ester as a white solid. 1H NMR(CDC13): 7.8
ppm
(dd, 2H, Ar), 7.0 ppm (dd, 2H, Ar), 5.7 ppm (d, 1H, NH), 3.9 ppm (s, 3H, OMe),
3.4
ppm (m, 1 H, N-CH), 2.5 ppm (m, 1 H, CH-C02-), 1.0-2.0 ppm (m, 17H,
hydrocarbon).
CA 02356345 2001-06-26
WO 00/44711 PCTNS00/01865
-25-
Example 4
(cis)-2-(4-Methoxy-benzenesulfonylamino)-cyclohexanecarboxylic acid
tert-butyl ester
In the same manner as described in Example 3, 1.438 g (4.59 mmol) of the
product from Example 2 provided 0.739 g (44%) of the desired tert-butyl ester
as a
colorless oil. Electrospray Mass Spec 370.1 (M+H)'.
Example 5
(traps)-2-[Benzyl-(4-methoxybenzenesulfonyl )amino]-cyclohexanecarboxylic
acid t-butyl ester
To a solution of 1.146 g (3.1 mmol) of the product from Example 3 in 31 mL
of DMF was added 0.137g (3.42 mmol) of 60% sodium hydride. The resulting
mixture was stirred for 30 min at 25~C and then 0.42 mL (3.50 mmol) of benzyl
bromide was added all at once. This reaction mixture was stirred for 10 hr at
55 ~C
and then poured into water and extracted with ether. The combined organics
were
washed with water and brine, dried over MgS04, filtered and concentrated in
vacuo
to provide a white solid which was recrystallized from ethyl acetate/Hexanes
to
provide 1.364 g (95%) of the desired product. 1H NMR(CDC13): 7.7 ppm (dd, 2H,
Ar), 7.1-7.4 (m, SH, Ar), 6.9 ppm (dd, 2H, Ar), 4.5-4.7 ppm (AB quartet, 2H,
CH2-
Ar), 3.9 ppm (s, 3H, OMe), 4.0 ppm (m, 1H, N-CH), 2.9 ppm (m, 1H, CH-C02-),
1.0-2.3 ppm (m, 17H, hydrocarbon protons).
Example 6
(cis)-2-[Benzyl-(4-methoxy-benzenesulfonyl)-amino]-cyclohexanecarboxylic acid
tert-butyl ester
In the same manner as described in Example S, 0.600 g ( 1.62 mmol) of the
product from Example 4 provided 0.310 g (42%) of the desired benzylated ester
as a
colorless oil. Electrospray Mass Spec 460.1 (M+H)'.
Example 7
(traps)-2-[Benzyl-(4-methoxy-benzenesulfonyl)-amino]- cyclohexanecarboxylic
acid
To a solution of 1.364 g (2.97 mmol) of the product from Example 5 in lOmL
of dichloromethane was added IOmL of trifluoroacetic acid and the mixture was
stirred for 4h at room temperature. The solvent was then concentrated in vacuo
and
the residue was chromatographed on silica gel eluting with 10-100% ethyl
CA 02356345 2001-06-26
WO 00/44711 PCT/US00/O1$65
-26-
acetate/hexane to provide 1.092 g (73%) of the desired product as a white
solid.
Electrospray Mass Spec 404.2 (M+H)'
Example 8
(cis)-2-[Benzyl-(4-methoxy-benzenesulfonyl)-amino]-cyclohexanecarboxylic acid
In the same manner as described in Example 7, 0.240 g (0.522 mmol) of the
product from Example 6 provided 0.207 g (98%) of the desired carboxylic acid
as a
white solid. Electrospray Mass Spec 404.0 (M-H)'.
Example 9
4-But-2-ynyloxy-benzenesulfonic acid sodium salt
To a solution of 52.35g (U.225 mol) of 4-hydroxybenzenesulfonate sodium
salt in l L of isopropanol and 225 mL of a 1.ON solution of sodium hydroxide
was
added 59.96g (0.45 mol) of 1-bromo-2-butyne. The resulting mixture was heated
to
i 5 70° for 15h and then the isopropanol was removed by evaporation in
vacuo. The
resulting white precipitate was collected by filtration, washed with
isopropanol and
ether and dried in vacuo to give 56.Og (100%) of the butynyl ether as a white
solid.
Example 10
4-But-2-ynyloxy-benzenesulfonyl chloride
To a 0° solution of 43.8 mL (0.087 mol) of 2M oxalyl
chloride/dichloro-
methane solution in 29 mL of dichloromethane was dropwise added 6.77 mL (0.087
mol) of DMF followed by 7.248 (0.029 mol) of the product of Example 9. The
reaction mixture was stirred for 10 minutes at 0° then let warm to room
temperature
and stirred for 2 days. The reaction was then poured into ice and extracted
with 150
mL of hexanes. The organics were washed with water and brine, dried over
Na2S04,
filtered and concentrated in vacuo to provide 6.23g (88%) of the sulfonyl
chloride as
a yellow solid; m.p. 63-65°C. EI Mass Spec: 243.9 (M').
CA 02356345 2001-06-26
WO 00/44711 PCT/US00/01865
-27-
Example 11
But-2-ynyloxy-benzene
To a solution of 6.14g (0.023 mol) of triphenylphosphine dissolved in 100 mL
of benzene and 40 mL of THF was added 1.75 mL (0.023 mol) of 2-butyn-1-ol.
After
five minutes 2.00 (0.023 mol) phenol, dissolved in 10 mL of THF, was added to
the
reaction followed by 3.69 mL (0.023 mol) of diethyl azodicarboxylate. The
resulting
reaction mixture was stirred for 18h at room temperature and then concentrated
in
vacuo. The residue was chromatographed on silica gel eluting with ethyl
acetate/hexanes (1:10) to provide 2.18g (70%) of the butynyl ether as a clear
liquid.
EI Mass Spec: 146.0 MH+
Example 12
4-But-2-ynyloxy-benzenesulfonyl chloride
To a solution of 0.146g (1.0 mmol) of the product of Example 11 in 0.3 mL
of dichloromethane in an acetone/ice bath under NZ was dropwise added a
solution of
0.073 mL (1.1 mmol) of chlorosulfonic acid in 0.3 mL of dichloromethane. After
the
addition was complete, the ice bath was removed and the reaction was stirred
at room
temperature for 2h. To the reaction was then dropwise added 0.113 mL (1.3
mmol)
of oxalyl chloride, followed by 0.015 mL DMF. The reaction was heated to
reflux for
2h and then diluted with hexane and poured into ice water. The organic layer
was
washed with brine, dried over sodium sulfate, and concentrated in vacuo to
provide
0.130mg (53%) of the desired product as a light brown solid.
Example 13
(1R,2R)-2-({[4-(2- Butynyloxy)phenyl]sulfonyl}amino)-
cyclohexanecarboxylic acid
To a room temperature solution of l.Sg (10.2 mmol) of traps-2-amino-1-
cyclohexylcarboxylic acid in 75 ml of dioxane:H20 ( l : l ) containing 2.55 ml
( 18.3
mmol) of triethylamine was added 3.0g (11.2 mmol) of 4-
butynyloxybenzenesulfonyl
chloride. The mixture was stirred at 25°C for 18 h. The resulting
mixture was diluted
with ethyl acetate and washed with 1N aqueous hydrochloric acid (3X). The
organic
phase was dried over anhydrous magnesium sulfate and concentrated in vacuo to
CA 02356345 2001-06-26
WO 00/44711 PCT/US00/01865
-28-
afford (1R,2R)-2-({[4-(2-
butynyloxy)phenyl]sulfonyl}amino)cyclohexanecarboxylic
acid as a white solid. Electrospray Mass Spec 352.2 (M+H)'
Example 14
tent-Butyl (1R,2R)-2-({[4-(2- butynyloxy)phenyl]sulfonyl}amino)-
cyclohexanecarboxylate
To a solution of 2.1 g (6 mmol) of (1R,2R)-2-({[4-(2-butynyloxy)phenyl]-
sulfonyl}amino) cyclohexanecarboxylic acid in 30 mL of toluene was added 6 mL
(24 mmol) of N,N-dimethylformamide di-tert-butyl acetal. The resulting mixture
was
heated at 110°C for 4h under nitrogen and then allowed to cool to room
temperature.
The solution was then poured on top of a silica gel column. Chromatography on
silica gel eluting with 10-20% ethyl acetate/hexane gave 1.7g of tert-
butyl(1R,2R)-2-
( { [4-(2-butynyloxy)phenyl]sulfonyl } amino)cyclohexane-carboxylate as a
white solid.
Electrospray Mass Spec 408.3 (M+H)'
Example 15
tent-Butyl (1R,2R)-2-[{[4-(2- butynyloxy)phenyl]sulfonyl}(methyl)amino]
cyclohexanecarboxylate
To a solution of 1.38 g (3.4 mmol) of tert-butyl (IR,2R)-2-({[4-(2- butynyl-
oxy)phenyl]sulfonyl} amino) cyclohexanecarboxylate in 20 mL of DMF was added
0.164g (4.1 mmol) of 60% sodium hydride. The resulting mixture was stirred for
30
min at 25~C and then 0.26 mL (4.1 mmol) of iodomethane was added all at once.
This reaction mixture was stirred for 0.5 hr at 25°C and then water and
ethyl acetate
were added. The organics were washed with water, dried over anhydrous
potassium
carbonate and concentrated in vacuo to provide tert-butyl ( 1 R,2R)-2-[ { [4-
(2- butynyl-
oxy)phenyl]sulfonyl } (methyl)amino] cyclohexanecarboxylate as a white solid.
Electrospray Mass Spec 422.2 (M+H)
Example 16
(1R,2R)-2-[{[4-(2-Butynyloxy)phenyl]sulfonyl}(methyl)amino]-
cyclohexanecarboxylic acid
To a solution of tert-butyl ( 1 R,2R)-2-[ { [4-(2-butynyloxy)phenyl]sulfonyl }-
(methyl)amino]cyclohexanecarboxylate in 20 mL of dichloromethane was added 5
mL of trifluoroacetic acid and the mixture was stirred for 3h at room
temperature.
The solvent was then removed in vacuo and the residue was chromatographed on
silica gel eluting with methanoUdichloromethane. Trituration with ethyl
acetate/-
CA 02356345 2001-06-26
WO 00/44711 PCT/US00/01865
-29-
hexane provided 1.04 g of ( 1 R,2R)-2-[ { (4-(2-butynyloxy)phenyl]sulfonyl }
(methyl)-
amino]cyclohexanecarboxylic acid as a white solid. Electrospray Mass Spec
364.3
(M-H)
Example 17
(1R,2R)-2-[{[4-(2-Butynyloxy)phenyl]sulfonyl}(methyl)amino]-N
hydroxycyclohexanecarboxamide
To oxalyl chloride (1.42 mL of a 2 M solution in dichloromethane) in
dichloromethane at 0°C was added dimethylformamide (0.22 mL). After 1 S
min a
solution of ( 1 R,2R)-2-[ { [4-(2-butynyloxy)phenyl]sulfonyl }
(methyl)amino]cyclo
hexanecarboxylic acid in dimethylformamide was added and the resulting
reaction
mixture was stirred at room temperature for lh.
In a separate flask, 3 mL of triethylamine was added to a 0°C
mixture of
0.987 g of hydroxylamine hydrochloride in 7.6 mL of THF and 3.2 mL of water.
After this mixture had been stirred for lSmin at 0°C, the acid chloride
solution was
added to it in one portion and the resulting solution was allowed to warm to
room
temperature and stirred for another 18h. Ethyl acetate and aqueous sodium
bicarbonate were then added to the reaction flask. The organic phase was
washed
with aqueous sodium bicarbonate and dried over anhydrous potassium carbonate.
Concentration in vacuo and trituration with diethyl ether gave (1R, 2R)-2-[{[4-
(2-
butynyloxy)phenyl]sulfonyl ) (methyl)amino]-N- hydroxycyclohexanecarboxamide
as
a white powder (485 mg). Electrospray Mass Spec 381.2 (M+H) '
Example 18
(1R, 2R)-2-({[4-(2-Butynyloxy)phenyl]sulfonyl}amino)-N-
hydroxycyclohexanecarboxamide
In the same manner as described in Example 17, 0.50 g (1.42 mmol) of
(1R,2R)-2-({[4-(2- butynyloxy)phenyl)sulfonyl} amino) cyclohexanecarboxylic
acid
provided 0.32 g of (1R, 2R)-2-({[4-(2-butynyloxy)phenyl]sulfonyl}amino)-N-
hydroxycyclohexanecarboxamide as a white solid. Electrospray Mass Spec 367.2
(M+H)'.
Example 19
tent-Butyl 3-({[4-(2-butynyloxy)phenyl]sulfonyl}amino)propanoate
To tert-butyl-2-aminopropanoate (2.Og, 11.0 mmol) in 20 mL of
dichloromethane at 0°C was added triethylamine (6.75 mL, 48.4 mmol)
followed by
CA 02356345 2001-06-26
WO 00/44711 PCT/US00/01865
-30-
4-(2-butynyloxy)phenylsulfonyl chloride (2.94 g, 12.1 mmol). An additional 10
mL
of dichloromethane was added to the thick slurry. The mixture was stirred
overnight
then diluted with dichloromethane and washed sequentially with water, 2N
aqueous
citric acid and brine, then dried over anhydrous sodium sulfate. Filtration
and
concentration in vacuo gave a solid that was triturated with hexane/ethyl
acetate to
give tert-butyl 3-({[4-(2-butynyloxy)phenyl]sulfonyl}amino)propanoate (3.88 g)
as
an off-white solid, mp 63 - 65°C. Analysis for C"H23NOSS: Calc'd: C,
57.77; H,
6.56; N, 3.96. Found: C, 57.68; H, 6.42; N, 3.90. Electrospray Mass Spec 354.2
(M+H) '.
Example 20
N-{[4-(2-Butynyloxy)phenyl]sulfonyl }-beta-alanine
In the same manner as described in Example 16 tent-butyl 3-({[4-(2-
butynyloxy) phenylJsulfonyl}amino) propanoate (1.0 g, 2.83 mmol) gave N-{[4-(2-
butynyloxy)phenyl]sulfonyl}-beta-alanine (1.12 g) as a white solid.
Electrospray
Mass Spec 296.2 (M-H) -.
Example 21
3-({[4-(2-Butynyloxy)phenyl]sulfonyl}amino)-N-hydroxypropanamide
To N-{[4-(2-butynyloxy)phenyl]sulfonyl}-beta-alanine (0.80 g, 2.69 mmol)
in dimethylformamide (5 mL) was added 1-hydroxybenzotriazole (0.436 g, 3.23
mmol), and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide (0.67 g, 3.5 mmol).
After one hour 50% hydroxylamine in water ( 1.3 mL) was added. The reaction
mixture was stirred overnight and concentrated in vacuo. Ethyl acetate was
added
and the organic phase washed with water (2X) and brine then dried over
anhydrous
sodium sulfate. Filtration and concentration in vacuo gave a white solid that
was
triturated with ethyl acetate to give 3-({[4-(2-
butynyloxy)phenyl]sulfonyl}amino)-N-
hydroxypropanamide (0.30 g) as a white solid,. mp 118 - 128°C.
Electrospray Mass
Spec 313.3 (M+H) '.
CA 02356345 2001-06-26
WO 00/44711 PCT/US00/01865
-31-
Example 22
tent-Butyl-3-[{[4-(2-butynyloxy)phenyl]sulfonyl}(methyl)amino]propanoate
To a solution of tert-butyl 3-( { [4-(2-butynyloxy)phenyl]-sulfonyl } amino)-
propanoate (1.0 g, 2.83 mmol) in dimethylformamide (5 mL) at 0°C was
added
sodium hydride (3.39 mmol) followed by iodomethane {211 uL, 3.39 mmol). After
72 hours the reaction mixture was diluted with ethyl acetate and washed with
water
and brine and dried over anhydrous sodium sulfate. Filtration and
concentration in
vacuo gave tert-butyl-3-[{[4-(2-butynyloxy)phenyl]-sulfonyl}(methyl)amino]-
propanoate (1.0 g) as a colorless oil. Electrospray Mass Spec 368.2 (M+H)'.
Example 23
N-{[4-(2-Butynyloxy)phenyl]sulfonyl}-N-methyl-beta-alanine
In the same manner as described in Example 16 tert-butyl-3-[ { [4-(2-
butynyloxy)phenyl]sulfonyl } (methyl)amino]propanoate (0.863 g, 2.34 mmol)
gave
1 S N-{ [4-(2-butynyloxy)phenyl]sulfonyl }-N-methyl-beta-alanine (0.760 g) as
a white
solid, mp 90-110°C. Electrospray Mass Spec 312.1 (M+H)+.
Example 24
3-({[4-(2-Butynyloxy)phenyl]sulfonyl} (methyl) amino)-N-hydroxypropanamide
N-{ [4-(2-Butynyloxy)phenyl]sulfonyl }-N-methyl-beta-alanine (0.7 g, 2.25
mmol) was convened to 3-({ [4-(2-butynyloxy)phenyl]sulfonyl } (methyl) amino)-
N-
hydroxypropanamide (0.525 g white solid) as described in Example 21. Analysis
for
C"H,$NZOSS: Calc'd: C, 51.52; H, 5.56; N, 8.58. Found: C, 51.38; H, 5.16; N,
8.28.
Electrospray Mass Spec 327.2 (M+H)'.
Example 25
(1R, 2S)-2-[{[4-(2-Butynyloxy)phenyl]sulfonyl}amino cyclopentanecarboxylic
acid
To a solution of cis-2-amino-1-cyclopentane (1.0 g, 7.74 mmol) in 1:1
water:dimethylformamide (10 mL) at 0°C was added sodium carbonate (2.7
g, 25.5
mmol) followed by 4-(2-butynyloxy)phenyl sulfonyl chloride (2.08 g, 8.5 mmol).
The reaction mixture was allowed to warm to room temperature. After stirring
overnight water and ethyl acetate were added and the mixture acidified to pH =
1
with 6N aqueous hydrochloric acid. The organic phase was washed with water and
CA 02356345 2001-06-26
WO 00/44711 PCTNS00/01865
-32-
brine and dried over anhydrous sodium sulfate. Filtration and concentration in
vacuo
gave (1R, 2S)-2-[{[4-(2-butynyloxy)phenyl]sulfonyl}amino
cyclopentanecarboxylic
acid ( 1.58 g) as a white solid, mp 105-135°C. Electrospray Mass Spec
336.4
(M+H)'.
Example 26
(1R, 2S)-2-({[4-(2-Butynyloxy)phenyl]sulfonyl)amino)-N
hydroxycyclopentanecarboxamide
( 1 R,2S)-2-[ { [4-(2-Butynyloxy)phenyl] sulfonyl } aminocyclopentane-
carboxylic acid (0.506g, 1.5 mmol) was converted to (1R,2S)-2-[{[4-(2-
butynyloxy)-
phenyl]sulfonyl}amino)-N-hydroxycyclopentanecarboxamide (0.28 g) as described
in
Example 21 to give an off-white solid (0.185 g), mp 140-145°C.
Electrospray Mass
Spec 353.4 (M+H)'.
I S Example 27
ten-Butyl (1R, 2S)-2-[{[4-(2-butynyloxy)phenyl]sulfonyl}amino
cyclopentanecarboxylate
(1R, 2S)-2-[{[4-(2-Butynyloxy)phenyl]sulfonyl}amino cyclopentane
carboxylic acid (0.80 g) was converted to tert-butyl (IR, 2S)-2-[{[4-(2-
butynyloxy)
phenyl]sulfonyl}amino cyclopentanecarboxylate as described in Example 14 to
give
white crystals (0.60g), mp 97-100°C. Analysis for C2oHZ,N05: Calc'd: C,
61.05; H,
6.92; N, 3.56. Found: C, 61.04; H, 6.79; N, 3.72. Electrospray Mass Spec 394.2
(M+H)'.
Example 28
tent-Butyl (1R, 2S)-2-({[4-(2-butynyloxy)phenyl]sulfonyl} (methyl) amino
cyclopentanecarboxylate
tert-Butyl (1R, 2S)-2-({[4-(2-butynyloxy)phenyl]sulfonyl}aminocyclo-
pentanecarboxylate (0.50 g, 1.27 mmol) in dimethylformamide (4 mL) was treated
with potassium carbonate (0.527g, 3.81 mmol) and iodomethane (95 uL, 1.53
mmol).
After 18 hours the reaction mixture was concentrated in vacuo, diluted with
ethyl
acetate and washed with water and brine then dried over anhydrous sodium
sulfate.
Filtration and concentration in vacuo gave tert-butyl (1R, 2S)-2-[{[4-(2-
butynyl-
oxy)phenyl]sulfonyl} (methyl) amino cyclopentanecarboxylate as a white solid
(0.495 g), mp 131-133°C. Electrospray Mass Spec 408.2 (M+H) +.
CA 02356345 2001-06-26
WO 00/44711 . PCT/US00/01865
-33-
Example 29
(1R, 2S)-2-[{[4-(2-Butynyloxy)phenyl]sulfonyl} (methyl) amino]
cyclopentanecarboxylic acid
tent-Butyl (1R, 2S)-2-[{(4-(2-butynyloxy)phenyl]sulfonyl}amino cyclo-
pentanecarboxylate (0.44 g) was converted to (1R, 2S)-2-[{[4-(2-butynyloxy)-
phenyl]sulfonyl}(methyl)amino]cyclopentanecarboxylic acid as described in
Example I6 to give a white solid (0.375 g). Electrospray Mass Spec 352.2
(M+H)'.
Example 30
(1R, 2S)-2-j{(4-(2-Butynyloxy)phenyl]sulfonyl} (methyl) amino] N-
hydroxycyclopentanecarboxamide
(1R,2S)-2-[{[4-(2-Butynyloxy)phenyl]sulfonyl}(methyl)amino] cyclopentane-
carboxylic acid (0.320 g) was converted to (1R,2S)-2-[{[4-(2-
butynyloxy)phenyl]-
sulfonyl}(methyl)amino]N-hydroxycyclopentanecarboxamide as described in
Example 21 to give white crystals (0.105 g), mp 160-164°C.
Analysis for
C"HZZNZOSS: Calc'd: C, 55.72; H, 6.05; N, 7.64. Found: C, 55.40; H, 6.15; N,
7.50.
Electrospray Mass Spec 367.25 (M+H)'.
Example 31
(Cis)-2-[{[4-(2-butynyloxy)phenyl]sulfonyl}amino cyclohexanecarboxylic acid
Cis-2-amino-1-cyclohexane (1.2 g, 8.38 mmol) was converted to (cis)-2-[{[4-
(2-butynyloxy)phenyl]sulfonyl}amino cyclohexanecarboxylic acid as described in
Example 25 to give a white solid (0.825 g), mp 172-175°C. Analysis
for
C"HZ,NOSS: Calc'd: C, 58.10; H, 6.02; N, 3.99. Found: C, 58.32; H, 5.92; N,
3.87.
Electrospray Mass Spec 350.1 (M-H) ~.
Example 32
(Cis)-2-[{[4-(2-butynytoxy)phenyl]sulfonyl}amino)-N-
hydroxycyclohexanecarboxamide
(Cis)-2-[{[4-(2-butynyloxy)phenyl]sulfonyl}aminocyclohexanecarboxylic
acid (0.3008, 0.854 mmol) was converted to (cis)-2-[{ [4-(2-butynyloxy)phenyl]-
sulfonyl } amino)-N-hydroxycyclohexanecarboxamide as described in Example 21
to
give an off-white foam (0.210 g). Electrospray Mass Spec 367.2 (M+H)'.
CA 02356345 2001-06-26
WO 00/44711 PCT/US00/01865
-34-
Example 33
(Cis)-tent-butyl-2-[{[4-(2-butynyloxy)phenyl]sulfonyl}amino)
cyclohexanecarboxylate
(Cis)-2-[{[4-(2-butynyloxy)phenyl]sulfonyl}aminocyclohexanecarboxylic
acid (0.38 g, 1.08 mmol) was converted to (cis)-tert-butyl-2-[{[4-(2-
butynyloxy)-
phenyl]sulfonyl}amino)cyclohexanecarboxylate as described in Example 14 to
give a
white solid (0.450 g). Electrospray Mass Spec 408.2 (M+H)'.
Example 34
(Cis)-tent-butyl-2-[{[4-(2-butynyloxy)phenyl]sulfonyl} (methyl) amino)
cyclohexanecarboxylate
(Cis)-tert-butyl-2-[{ [4-(2-butynyloxy)phenyl]sulfonyl }amino)cyclohexane-
carboxylate (0.390 g, 0.956 mmol) was converted to (cis)-tert-butyl-2-[ { [4-
(2-
butynyloxy)phenyl]sulfonyl } (methyl) amino) cyclohexanecarboxylate as
described
in Example 28 to give a colorless oil (0.260 g). Electrospray Mass Spec 422.2
(M+H)'.
Example 35
(Cis)-2-[{[4-(2-butynyloxy)phenyl]sulfonyl} (methyl) amino)
cyclohexanecarboxylic acid
(Cis)-tert-butyl-2-[{ [4-(2-butynyloxy)phenyl]sulfonyl } (methyl)amino)cyclo-
hexanecarboxylate (0.220 g, 0.522 mmol) was converted to (cis)-2-[ { [4-(2-
butynyl-
oxy)phenyl]sulfonyl } (methyl)amino)cyclohexanecarboxylic acid as described in
Example 16 to give a white solid (0.190 g). Electrospray Mass Spec 366.2
(M+H)'.
Example 36
(Cis)-2-[{[4-(2-butynyloxy)phenyl]sulfonyl} (methyl) amino]-N-
hydroxycyclohexanecarboxamide
(Cis)-2-[ { [4-(2-butynyloxy)phenyl]sulfonyl } {methyl) amino)cyclohexane-
carboxylic acid (0.165 g, 0.45 mmol) was converted to (cis)-2-[ { [4-(2-
butynyloxy)-
phenyl]sulfonyl} (methyl) amino]-N-hydroxycyclohexanecarboxarnide as described
in Example 21 to give an off white solid (0.50 g). Electrospray Mass Spec
381.2
(M+H)'.
CA 02356345 2001-06-26
WO 00/44711 PCTNS00/01865
-35-
Example 37
(1R, 2R, 3S, 4R)-(Cis)-3-({[4-(2-butynyloxy)phenyl]sulfonyl}amino)bicyclo
[2.2.1] heptane-2-carboxylic acid
S 3-Exo-aminobicyclo [2.2.1 ] heptane-2-exo carboxylic acid ( 1.0 g, 6.44
mmol)
was converted to (1R, 2R, 3S, 4R)-(cis)-3-({[4-(2-butynyloxy)phenyl]-sulfonyl}-
amino)bicyclo[2.2.1)heptane-2-carboxylic acid as described in Example 25 to
give a
white solid (1.32 g), mp 195-215°C. Electrospray Mass Spec 364.3
(M+H)'.
Example 38
(iR, 2R, 3S, 4R)-(Cis)-3-({[4-(2-butynyloxy)phenyl]sulfonyl}amino)-N-
hydroxybicyclo [2.2.1] heptane-2-carboxamide
(1R, 2R, 3S, 4R)-(Cis)-3-({[4-(2-butynyloxy)phenyl]sulfonyl}amino)bicyclo-
[2.2.1]heptane-2-carboxylic acid (0.363 g, 1 mmol) was converted to
(1R,2R,3S,4R)-
(cis)-3-({[4-(2-butynyloxy)phenyl)sulfonyl}amino)-N-
hydroxybicyclo[2.2.1]heptane-
2-carboxamide as described in Example 21 to give a white solid (0.30 g).
Analysis
for C,BHZZNZOSS: Calc'd: C, 57.13; H, 5.86; N, 7.4. Found: C, 57.76; H, 6.12;
N, 7.6.
Electrospray Mass Spec 379.3 (M+H)+.
Example 39
tert-Butyl (1R, 2R, 3S, 4R)-(cis)-3-({[4-(2-
butynyloxy)phenyl]sulfonyi}amino)bicyclo [2.2.1] heptane-2-carboxylate
(1R, 2R, 3S, 4R)-(Cis)-3-({[4-(2-butynyloxy)phenyl]sulfonyl}amino)bicyclo-
[2.2.1) heptane-2-carboxylic acid (0.80 g, 2.2 mmol) was converted to tert-
butyl
(1R,2R,3S,4R)-(cis)-3-({[4-(2-butynyloxy)phenyl)sulfonyl}amino)bicyclo[2.2.1]-
heptane-2-carboxylate as described in Example 14 to give a white solid (0.69
g),
mp 94 - 99°C. Analysis for CnHz9NOSS: Calc'd: C, 62.98; H, 6.97; N,
3.34. Found:
C, 62.65; H, 6.95; N, 3.7. Electrospray Mass Spec 420.3 (M+H) ~.
Example 40
tent-Butyl (1R, 2R, 3S, 4R)-(cis)-3-({[4-(2-butynyloxy)phenyl]sulfonyl}
(methyl)
amino)bicyclo [2.2.1] heptane-2-carboxylate
tert-Butyl (1R, 2R, 3S, 4R)-(cis)-3-({[4-(2-butynyloxy)phenyl]-sulfonyl}-
amino)bicyclo[2.2.1]heptane-2-carboxylate (O.SSg, 1.31 mmol) was converted to
tert-
CA 02356345 2001-06-26
WO 00/44711 PCT/US00/01865
-36-
butyl (1R, 2R, 3S, 4R)-(cis)-3-({[4-(2-
butynyloxy)phenyl]sulfonyl}(methyl)amino)-
bicyclo[2.2.1]heptane-2-carboxylate as described in Example 28 to give a white
solid
(0.54 g), mp 120-125°C. Analysis for C~H3,NOSS: Calc'd: C, 63.72; H,
7.21; N,
3.23. Found: C, 63.34; H, 7.11; N, 3.55. Electrospray Mass Spec 434.2 (M+H)'.
Example 41
(1R, 2R, 3S, 4R)-(Cis)-3-({[4-(2-butynyloxy)phenyl]sulfonyl} (methyl)
amino)bicyclo (2.2.1] heptane-2-carboxylic acid
tert-Butyl( 1 R,2R,3S,4R)-(cis)-3-({ [4-(2-butynyloxy)phenyl]sulfonyl }-
(methyl)amino)bicyclo[2.2.1]heptane-2-carboxylate (0.45 g, 1.04 mmol) was
converted to (1R,2R,3S,4R)-(cis)-3-({[4-(2-butynyloxy)phenyl]sulfonyl}(methyl)
amino)bicyclo [2.2.1 ) heptane-2-carboxylic acid as described in Example 16 to
give
a white solid (0.37 g), mp 153-158°C. Analysis for C,9H~NOSS: Calc'd:
C, 60.46; H,
6.14; N, 3.71. Found: C, 60.71; H, 5.94; N, 3.97. Electrospray Mass Spec 378.1
(M+H)f.
Example 42
(1R, 2R, 3S, 4R)-(Cis)-3-({[4-(2-butynyloxy)phenyl]sulfonyl} (methyl) amino)-N
hydroxybicyclo [2.2.1] heptane-2-carboxamide
(1R,2R,3S,4R)-(Cis)-3-({[4-(2-butynyloxy)phenyl]sulfonyl}(methyl)
amino)bicyclo-
[2.2.1]heptane-2-carboxylic acid (0.30 g, 0.79 mmol) was converted to (1R, 2R,
3S,
4R)-(cis)-3-({ [4-(2-butynyloxy)phenyl]sulfonyl } (methyl) amino)-N-
hydroxybicyclo-
[2.2.1]heptane-2-carboxamide (0.137 g) as described in Example 21.
Electrospray
Mass Spec 393.2 (M+H) '.
PharmacoloQv
The ability of the compounds of the invention, or their pharmaceutically
acceptable salts, to inhibit matrix metalloproteinases or TALE and,
consequently,
demonstrate their effectiveness for treating diseases modulated by matrix
metalloproteinases or TACE is shown by the following in vitro assays.
Test Procedures for Measuring MMP-1 MMP-9 and MMP-13 Inhibition
These standard pharmacological test procedures are based on the cleavage of a
thiopeptide substrates such as Ac-Pro-Leu-Gly(2-mercapto-4-methyl-pentanoyl)-
Leu-
Gly-OEt by the matrix metalloproteinases MMP-1, MMP-13 (collagenases) or MMP-
CA 02356345 2001-06-26
WO 00/44711 PCTNS00/01865
-37-
9 (gelatinase), which results in the release of a substrate product that
reacts
colorimetrically with DTNB (5,5'-dithiobis(2-nitro-benzoic acid)). The enzyme
activity is measured by the rate of the color increase. The thiopeptide
substrate is
made up fresh as a 20 mM stock in 100% DMSO and the DTNB is dissolved in 100%
S DMSO as a 100 mM stock and stored in the dark at room temperature. Both the
substrate and DTNB are diluted together to 1 mM with substrate buffer (50 mM
HEPES pH 7.5, 5 mM CaCl2) before use. The stock of enzyme is diluted with
buffer (50 mM HEPES, pH 7.5, 5 mM CaCl2, 0.02%o Brij) to the desired final
concentration. The buffer, enzyme, vehicle or inhibitor, and DTNB/substrate
are
added in this order to a 96 well plate (total reaction volume of 200 pl) and
the
increase in color is monitored spectxophotometrically for S minutes at 405 nm
on a
plate reader and the increase in color over time is plotted as a linear line.
Alternatively, a fluorescent peptide substrate is used. In this test
procedure,
the peptide substrate contains a fluorescent group and a quenching group. Upon
cleavage of the substrate by an MMP, the fluorescence that is generated is
quantitated
on the fluorescence plate reader. The assay is run in HCBC assay buffer (SOmM
HEPES, pH 7.0, 5 mM Ca+2, 0.02% Brij, 0.5% Cysteine), with human recombinant
MMP-1, MMP-9; or MMP-13. The substrate is dissolved in methanol and stored
frozen in 1 mM aliquots. For the assay, substrate and enzymes are diluted in
HCBC
buffer to the desired concentrations. Compounds are added to the 96 well plate
containing enzyme and the reaction is started by the addition of substrate.
The
reaction is read (excitation 340 nm, emission 444 nm) for 10 min. and the
increase in
fluorescence over time is plotted as a linear line.
For either the thiopeptide or fluorescent peptide test procedures, the slope
of
the line is calculated and represents the reaction rate. The linearity of the
reaction
rate is confirmed (r2 >0.85). The mean (x~sem) of the control rate is
calculated and
compared for statistical significance (p<0.05) with drug-treated rates using
Dunnett's
multiple comparison test. Dose-response relationships can be generated using
multiple doses of drug and ICSp values with 95% CI are estimated using linear
regression.
CA 02356345 2001-06-26
WO 00/44711 PCT/US00/01865
-38-
Test Procedure for Measuring TACE Inhibition
Using 96-well black microtiter plates, each well receives a solution composed
of 10 pL, TACE (final concentration lpg/mL), 70~I. Tris buffer, pH 7.4
containing
10% glycerol (final concentration 10 mM), and 10 pL, of test compound solution
in
DMSO (final concentration 1~M, DMSO concentration <1%) and incubated for 10
minutes at room temperature. The reaction is initiated by addition of a
fluorescent
peptidyl substrate (final concentration 100 pM) to each well and then shaking
on a
shaker for 5 sec.
The reaction is read (excitation 340 nm, emission 420 nm) for 10 min. and the
increase in fluorescence over time is plotted as a linear line. The slope of
the line is
calculated and represents the reaction rate.
The linearity of the reaction rate is confirmed (r2 >0.85). The mean (x~sem)
of the control rate is calculated and compared for statistical significance
(p<0.05)
with drug-treated rates using Dunnett's multiple comparison test. Dose-
response
relationships can be generate using multiple doses of drug and ICSp values
with 95%
CI are estimated using linear regression.
Human Monocvtic THP-1 Cell Differentiation As~y For Soluble Proteins
ITHP-1 Soluble Protein Assav)
Mitogenic stimulation of THP-1 cells cause differentiation into macrophage
like cells with concomitant secretion of tumor necrosis factor (TNF-a and TNF
receptor {TNF-R p75/80 and TNF-R p55/60) and Interleukin-8 (IL-8), among other
proteins. In addition, non-stimulated THP-1 cells shed both the p75/80 and the
p55/60 receptors over time. The release of membrane bound TNF-a and possibly
TNF-R p75/80 and TNF-R p55/60, but not IL-8, is mediated by an enzyme called
TNF-a converting enzyme or TACE. This assay can be used to demonstrate either
an
inhibitory or a stimulatory compound effect on this TALE enzyme and any
cytotoxic
consequence of such a compound.
THP-1 cells (from ATCC) are a human monocytic cell Iine which were
obtained from the peripheral blood of a one year old male with acute monocytic
leukemia. They can be grown in culture and differentiated into macrophage like
cells
by stimulation with mitogens.
For the assay, THP-1 cells are seeded from an ATCC stock which was
previously grown and frozen back at 5 x 106/ml/vial. One vial is seeded into a
T25
flask with 16 mls of RPMI-1640 with glutamax (Gibco) media containing 10 %
fetal
bovine serum, 100 units/ml penicillin, 100 pg/ml streptomycin, and 5 x 105 M 2-
CA 02356345 2001-06-26
WO 00/44711 PCT/US00/01865
-39-
mercapto-ethanol (THP-1 media). Each vial of cells are cultured for about two
weeks
prior to being used for an assay and then are used for only 4 to 6 weeks to
screen
compounds. Cells are subcultured on Mondays and Thursdays to a concentration
of
1 x 105/ml.
To perform an assay, the THP-1 cells are co-incubated in a 24 well plate with
50 ml/well of a 24 mg/mI stock of Lipopolysacharide (LPS) (Calbiochem Lot#
B 13189) at 37; C in 5% COZ at a concentration of 1.091 x 106 cells/ml ( 1. I
ml/well)
for a total of 24 hours. At the same time, 50 mUwell of drug, vehicle or THP-1
media is plated in appropriate wells to give a final volume of 1.2 ml/well.
Standard
and test compounds are dissolved in DMSO at a concentration of 36 mM and
diluted
from here to the appropriate concentrations in THP-1 media and added to the
wells at
the beginning of the incubation period to give final concentrations of 100 mM,
30
mM, 10 mM, 3 mM, .1 mM, 300 nM, and 100 nM. Cell exposure to DMSO was
limited to 0.1 % final concentration. Positive control wells were included in
the
experiment which had mitogen added but no drug. Vehicle control wells were
included as well, which were identical to the positive control wells, except
that
DMSO was added to give a final concentration of 0.083%. Negative control wells
were included in the experiment which had vehicle but no mitogen or drug added
to
the cells. Compounds can be evaluated for their effect on basal (non-
stimulated)
shedding of the receptors by replacing the LPS with 50 ml/well of THP-1 media.
Plates are placed into an incubator set at 5% C02 and at 37° C. After 4
hours of
incubation, 300 ml/well of tissue culture supernatant (TCS) is removed for use
in an
TNF-oc ELISA. Following 24 hours of incubation, 700 ml/well of TCS is removed
and used for analysis in TNF-R p75/80, TNF-R p55/60 and IL-8 ELISAs.
In addition, at the 24 hours timepoint, and the cells for each treatment group
are collected by resuspension in 500 pl/well of THP-1 media and transferred
into a
FACS tube. Two ml/tube of a 0.5 mg/rnl stock of propidium iodide (PI)
(Boerhinger
Mannheim cat. # 1348639) is added. The samples are run on a Becton Dickinson
FaxCaliber FLOW cytometry machine and the amount of dye taken up by each cell
is
measured in the high red wavelength (FL3). Only cells with compromised
membranes (dead or dying) can take up PI. The percent of live cells is
calculated by
the number of cells not stained with PI, divided by the total number of cells
in the
sample. The viability values calculated for the drug treated groups were
compared to
the viability value calculated for the vehicle treated mitogen stimulated
group
("vehicle positive control") to determine the "percent change from control".
This
"percent change from control" value is an indicator of drug toxicity.
CA 02356345 2001-06-26
WO 00/44711 PCT/US00/01865
-40-
The quantity of soluble TNF-a, TNF-R p75/80 and TNF-R p55/60 and IL-8
in the TCS of the THP-1 cell cultures are obtained with commercially available
ELISAs from R&D Systems, by extrapolation from a standard curve generated with
kit standards. The number of cells that either take up or exclude PI are
measured by
the FLOW cytometry machine and visualized by histograms using commercially
available Cytologic software for each treatment group including all controls.
Biological variability in the magnitude of the response of THP-1 cell cultures
requires that experiments be compared on the basis of percent change from
"vehicle
positive control" for each drug concentration. Percent change in each soluble
protein
evaluated from the "vehicle positive control" was calculated for each compound
concentration with the following formula:
% Change _ Dg/ml (compound) - pg ml (veh pos control) x 100 _
pg/ml (veh pos control) - pg/ml (veh peg control)
For the soluble protein (TNF-a, p75/80, p55/60, IL-8) studies under
stimulated conditions, the mean pg/ml of duplicate wells were determined and
the
results expressed as percent change from "vehicle positive control". For the
soluble
protein (p75/80 and p55/60 receptors) studies under non-stimulated conditions,
the
mean pg/ml of duplicate wells were determined and the results expressed as
percent
change from "vehicle positive control" utilizing the following formula:
% Change = nQ/ml (compound ne;~ control) -~g/ml (veh peg control x 100
pg/ml (veh peg control)
ICso values for each compound are calculated by non-linear regression analysis
using customized software utilizing the JUMP statistical package.
For the cell viability studies, the viabilities (PI exclusion) of pooled
duplicate
wells were determined and the results expressed as % change from "vehicle
positive
control". The viability values calculated for the compound treated groups were
compared to the viability value calculated for the "vehicle positive control"
to
determine "percent change from control" as below. This value "percent change
from
control" is an indicator of drug toxicity.
% Change = % live cells (compound) -1 X I00
% live cells (veh pos control)
CA 02356345 2001-06-26
WO 00/44711 PCT/US00/01865
-41-
References:
Bjornberg, F., Lantz, M., Olsson, L, and Gullberg, U. Mechanisms involved in
the
processing of the p55 and the p75 tumor necrosis factor (TNF) receptors to
soluble
receptor forms. Lymphokine Cytokine Res. 13:203-211, 1994.
Gatanaga, T., Hwang, C., Gatanaga, M., Cappuccini, F., Yamamoto, R., and
Granger,
G. The regulation of TNF mRNA synthesis, membrane expression, and release by
PMA- and LPS-stimulated human monocytic THP-I cells in vitro. Cellular Immun.
138:1-10, 1991.
Tsuchiya, S., Yamabe, M., Yamagughi, Y., Kobayashi, Y., Konno, T., and Tada,
K.
Establishment and characterization of a human acute monocytic leukemia cell
line
(THP-1). Int. J. Cancer. 26:1711-176, 1980.
Results of the above in vitro matrix metalloproteinase inhibition, TACE
inhibition, and THP standard pharmacological test procedures are given in
Table 1.
Table 1:
Exampe # MMP-1 MMP-9a MMP-13a TACE THPb
17 >10 uM >10 uM >10 uM 28 14
18 >10 uM >10 uM >10 uM 61 1
21 >10 uM >10 uM >10 uM 145 0
24 >10 uM -10 uM -10 uM 73 9
26 >1 uM 5056 672 15 23
383 308 14 65
32 >10 uM ~10 uM 1738 36 2g
36 4229 1319 1820 53 23
38 >10 uM >10 uM -10 uM 30 9
42 -10 uM - 1142 62 77
a) IC,~ (nM)
b) % Inhibition @ 3rtM
Based on the standard pharmacological test procedures described above, the
compounds of this invention are useful in the treatment of disorders such as
arthritis,
tumor metastasis, tissue ulceration, abnormal wound healing, periodontal
disease,
graft rejection, insulin resistance, bone disease and HIV infection.
The compounds of this invention are also useful in treating or inhibiting
pathological changes mediated by matrix metalloproteinases such as
atherosclerosis,
atherosclerotic plaque formation, reduction of coronary thrombosis from
atherosclerotic plaque rupture, restenosis, MMP-mediated osteopenias,
inflammatory
diseases of the central nervous system, skin aging, angiogenesis, tumor
metastasis,
CA 02356345 2001-06-26
WO 00/44711 PCTNS00/01865
-42-
tumor growth, osteoarthritis, rheumatoid arthritis, septic arthritis, corneal
ulceration,
proteinuria, aneurysmal aortic disease, degenerative cartilage loss following
traumatic
joint injury, demyelinating diseases of the nervous system, cirrhosis of the
liver,
glomerular disease of the kidney, premature rupture of fetal membranes,
infammatory
bowel disease, age related macular degeneration, diabetic retinopathy,
proliferative
vitreoretinopathy, retinopathy of prematurity, ocular inflammation,
keratoconus,
Sjogren's syndrome, myopia, ocular tumors, ocular
angiogenesis/neovascularizadon
and corneal graft rejection.
Compounds of this invention may be administered neat or with a
pharmaceutical carrier to a patient in need thereof. The pharmaceutical
carrier may
be solid or liquid.
Applicable solid Garners can include one or more substances which may also
act as flavoring agents, lubricants, solubilizers, suspending agents, fillers,
glidants,
compression aids, binders or tablet-disintegrating agents or an encapsulating
material.
1 S In powders, the Garner is a finely divided solid which is in admixture
with the finely
divided active ingredient. In tablets, the active ingredient is mixed with a
carrier
having the necessary compression properties in suitable proportions and
compacted in
the shape and size desired. The powders and tablets preferably contain up to
99% of
the active ingredient. Suitable solid carriers include, for example, calcium
phosphate,
magnesium stearate, talc, sugars, lactose, dextrin, starch, gelatin,
cellulose, methyl
cellulose, sodium carboxymethyl cellulose, polyvinylpyrrolidine, low melting
waxes
and ion exchange resins.
Liquid carriers may be used in preparing solutions, suspensions, emulsions,
syrups and elixirs. The active ingredient of this invention can be dissolved
or
suspended in a pharmaceutically acceptable liquid carrier such as water, an
organic
solvent, a mixture of both or pharmaceutically acceptable oils or fat. The
liquid
carrier can contain other suitable pharmaceutical additives such a
solubilizers,
emulsifiers, buffers, preservatives, sweeteners, flavoring agents, suspending
agents,
thickening agents, colors, viscosity regulators, stabilizers or osmo-
regulators.
Suitable examples of liquid carriers for oral and parenteral administration
include
water (particularly containing additives as above, e.g., cellulose
derivatives,
preferable sodium carboxymethyl cellulose solution), alcohols (including
monohydric
alcohols and polyhydric alcohols, e.g., glycols) and their derivatives, and
oils (e.g.,
fractionated coconut oil and arachis oil). For parenteral administration the
carrier can
also be an oily ester such as ethyl oleate and isopropyl myristate. Sterile
liquid
carriers are used in sterile liquid form compositions for parenteral
administration.
CA 02356345 2001-06-26
WO 00/44711 PC'TNS00/01865
-43-
Liquid pharmaceutical compositions which are sterile solutions or suspensions
can be utilized by, for example, intramuscular, intraperitoneal or
subcutaneous
injection. Sterile solutions can also be administered intravenously. Oral
administration may be either liquid or solid composition form.
The compounds of this invention may be administered rectally in the form of
a conventional suppository. For administration by intranasal or intrabronchial
inhalation or insufflation, the compounds of this invention may be formulated
into an
aqueous or partially aqueous solution, which can then be utilized in the form
of an
aerosol. The compounds of this invention may also be administered
transdermally
through the use of a transdermal patch containing the active compound and a
carrier
that is inert to the active compound, is non-toxic to the skin, and allows
delivery of
the agent for systemic absorption into the blood stream via the skin. The
earner may
take any number of forms such as creams and ointments, pastes, gels, and .
occlusive -
devices. The creams and ointments may be viscous liquid or semi-solid
emulsions of
either the oil in water or water in oil type. Pastes comprised of absorptive
powders
dispersed in petroleum or hydrophilic petroleum containing the active
ingredient may
also be suitable. A variety of occlusive devices may be used to release the
active
ingredient into the blood stream such as a semipermeable membrane covering a
reservoir containing the active ingredient with or without a carrier, or a
matrix
containing the active ingredient. Other occlusive devices are known in the
literature.
The dosage to be used in the treatment of a specific patient suffering a MMP
or TACE dependent condition must be subjectively determined by the attending
physician. The variables involved include the severity of the dysfunction, and
the
size, age, and response pattern of the patient. Treatment will generally be
initiated
with small dosages less than the optimum dose of the compound. Thereafter the
dosage is increased until the optimum effect under the circumstances is
reached.
Precise dosages for oral, parenteral, nasal, or intrabronchial administration
will be
determined by the administering physician based on experience with the
individual
subject treated and standard medical principles.
Preferably the pharmaceutical composition is in unit dosage form, e.g., as
tablets or capsules. In such form, the composition is sub-divided in unit dose
containing appropriate quantities of the active ingredient; the unit dosage
form can be
packaged compositions, for example packed powders, vials, ampoules, prefilled
syringes or sachets containing liquids. The unit dosage form can be, for
example, a
capsule or tablet itself, or it can be the appropriate number of any such
compositions
in package form.