Note: Descriptions are shown in the official language in which they were submitted.
CA 02359967 2008-11-14
- 1 -
METHOD FOR PREPARING AN AZA-MACROLIDE WITH 4"(R) NH2.
The subject-matter of the present invention is
a process of use in converting the 40(S)-OH functional
group of the cladinose unit of an azamacrolide to
41 (R) -NH2.
The present invention relates more particularly
to the field of macrolide antibiotics of erythromycin
type and more particularly their azamacrolide
derivatives which form the subject-matter of Patent
EP 508,699 and which correspond to the following
general formula:
NMe2
QH3
= HO,..,.
R. 2'
CH sa CH3 / 0
HO',.f ,a0 H3
HO,.... OH
H3
0 O CC
CH:'
Me
CH3 CH3 4.. `''CH3
0 0
NH2
CH3
in which R is a hydrogen atom or a C1-C10 alkyl, C2-Cl0
alkenyl or C6-C12 arylsulphonyl group, which are, if
appropriate, substituted.
These compounds are obtained from erythromycin
and their synthesis involves two major stages:
- the creation of the Ba-azalide rnacrocycle
starting from the (Z) oxime, which is subjected to a
=stereospecific Beckmann rearrangement, and
- the modification of the cladinose group at
the 4" position, which consists of the conversion of
the 4" (S) -OH to 4" (R) -NH21 that is to say with inversion
of configuration, which can be illustrated as follows:
CA 02359967 2008-11-14
2 -
RO õOMe RO OMe
=,,,
O } 0
OH "NH2
In fact, the route currently used to provide
for this conversion of the 4" (S) -OH to 4" (R) -NH2 is not
completely suitable for production on an industrial
scale.
It involves,. successively, an oxidation of the
hydroxyl functional group at the 4" position to a
ketone functional group and.then the conversion of this
ketone to an oxime, which, by reduction, results in an
approximately 1 to 1 mixture of the expected amino
derivative and of its 4" epimer.
This synthetic route consequently has the major
disadvantage of requiring the formation of sp` C-4"
intermediates and thus of losing the stereochemical
information initially present at the spa C-4" of the
cladinose unit. This result is all the more of a
nuisance since the isomers, acquired on conclusion of
this synthetic route, are obtained with a low yield of
about 20% and are in addition difficult to separate.
Thus, for a crude reaction yield of about 20%, only
approximately 7% of the amino derivative with inversion
of configuration is obtained.
The object of the present invention is
specifically to provide a new access route to these
derivatives, aminated at the 4" position, which
advantageously , makes it possible to retain a
significant stereoselectivity and provides =a
satisfactory yield.
More specifically, a first subject-matter of
the present invention is a process for the preparation
of a compound of general formula I
CA 02359967 2010-02-24
30754-40
3
H3C"N..CH3
CH3 HO,,,M
Z
CH3 ea CH3
HO'""' ,.,110 CH3
HO"'-- OH
CH"'tV O O CH3 0
~CH3 t~)
""CH3
H3 CH30 4"
O
NA2
in which: CH3
- R is a hydrogen atom or a C1-Cl0 alkyl, C2-Clo
alkenyl or C6-C12 arylsulphonyl group, which are, if
appropriate, substituted, and
A, which are identical or different, are
= a hydrogen atom',
= a nitrogen atom, if appropriate substituted,
20' = -a -. C1-C4 alkyl group, which is . optionally
substituted by one or more aryl groups, which are, if
appropriate, substituted,
= an R2CO or R2SO2 group, with R2 being a
hydrogen atom, a Cl-C8 alkyl group or an . aryl group,
which are, if appropriate, substituted,
V means that the C in the 4" position has
undergone an inversion of configuration with respect to
the formula II,
CA 02359967 2010-02-24
30754-40
- 3a -
H3 CIII N"ICH3
CH3
P1O
z
R\ Ns, CH3
CH3 HO,,,= O O CH3
HOB,,,, OH
CH3 (II)
CH3 O O~ CH3
3
CH~ CH3 4 ""'CH
O
OH
CH3
CA 02359967 2008-11-14
4 -
with: - -
- R as defined in general formula I and
- P1 being a protective group for the hydroxyl
functional group at the 2' position,
characterized in that it comprises at least the stages
consisting in:
activating the hydroxyl functional group at the
4" position in the compound of general formula II, in
order to obtain a compound of general formula III
H3C,,.CH3
QH3 PlO'"
y
R,. 2'
CH3 8a CH3
O CH3
Ha..,. OH
CH3 C CH3 O ,CH3
CH3 CH ~~~'CH
3 e
O
OR1
CH3
in which:
R and P1 are as defined in general formulae I
and II and
OR1 is a leaving group,
bringing the said compound of general formula
III thus obtained into contact with a nitrogenous
nucleophilic derivative under conditions which are
sufficient to allow the stereoselective displacement of
the hydroxyl functional group activated by the said
nitrogenous nucleophile, and
- deprotecting the hydroxyl functional group at
the 2' position,
in order to result in the expected compound of general
formula I.
The claimed process thus has the significant
advantage of not requiring the formation of the sp2 C-4"
intermediate necessarily generated in the prior
CA 02359967 2008-11-14
-
synthetic route discussed above. It involves only an
inversion of configuration at the 4" position and this
inversion is obtained efficiently by displacement by a
nitrogenous nucleophile of the activated alcohol
5 functional group present at this 4" position.
Consequently, the claimed process proves to be
particularly advantageous for preparing with a very
satisfactory yield, a 4"(R)-NA2 derivative of general
formula 1'
C_ H
3 FL,
N 8a CH3 CH3
CH3 O'n~'
H
H01"s- OH
CHi O O CH3 0*eCH3 (p)
CH3 "" CH
4 3
C
0
NA2
CH3
with A and R as defined above
from a 4" (S) -OH azamacrolide derivative of general
formula II'
H3C` N ,CH3
CH3
P104".
FL, 2'
00- CH3 N88 CH3 0
HO''1~~ ,....0 CH3
OHO,t'.H
CH0 O CC
H3 OCH3 (II')
CH3 CH3 ' 6161CH3
0 4I
O OH
CH3
with R and P. as defined above.
As regards the leaving group represented by OR1
in general formula III, it is preferably selected from
CA 02359967 2008-11-14
6
C1-C10 alkyl sulphonates, C5-C6 aryl or heteroaryl
sulphonates or C. to C26 alkylaryl sulphonates, which
are substituted, if appropriate, by one or more halogen
atoms, preferably fluorine, and/or a nitro, cyano or
trifluoromethyl group.
The leaving group represented by OR, in general
formula III is preferably a group selected from
mesylate, triflate and tosylate and is more preferably
a triflate group.
Use may in particular be made according to the
invention, as nitrogenous nucleophilic compound, of
compounds of the following types: ammonia, amines which
may or may not be substituted by deprotectable groups,
such as a benfyl group or one of its derivatives,
amides, imides, sulphonamides, sulphonimides,
hydrazines or azides.
According to a preferred alternative form of
the claimed process, it is more preferably an organic
.20 organosoluble azide which can be generated in situ.
The leaving groups deriving from the activation
of the hydroxyl functional group at the 4" position in
the general formula II by a compound of formula IVA or
IVB
BSO2X or (BS02) 20
IVA IVB
with:
- X being a halogen atom or a nitrogenous
heterocycle, preferably an imidazole ring,
- and
- B being a C1-C20 alkyl, C5-C6 aryl or
heteroaryl or C6-C26 alkylaryl group, which
are or are not substituted by one or more
halogen atoms, preferably fluorine, and/or a
nitro, cyano or trifluoromethyl group,
are very particularly suitable for the invention.
The compound of general formula III thus
obtained is preferably brought into contact with an
organosoluble azide in order to result, by
CA 02359967 2008-11-14
- 7 -
stereoselective nucleophilic displacement, in a
compound of general formula V - -
H3C\ CH3
N
Q H3
R 21
CH 8a CH3 lot$ 3 O CH3
HO,..,. OH
CH3=' 0 CH3 0`1 CH3 N)
CH3 CH30 4n
O
N3
CH3
in which R and Fl are as defined in general formula I
and V means that the C in the 411 position has undergone
an inversion of configuration with respect to the
formula II,
The C-4" carbon of the compound II preferably
has a 'S configuration and the C-4" carbon of the
compound V a R configuration.
According to this alternative form of the
claimed process, a reduction of the said compound of
formula V can additionally be carried out, prior or
otherwise to the deprotection of the hydroxyl
functional group at the 2' position, so as to obtain a
compound of general formula I in which A is a hydrogen
atom. This reduction of the azide functional group can
be carried out by any conventional method, such as
those described by E.F.V. Scriven et al., Chem. Rev.
(1988), 88, 297-368. A catalytic reduction with
hydrogen or hydrazine in the presence of palladium-on-
charcoal, for example, or of Raney nickel can in
particular be carried out.
On conclusion of this reduction, the expected
4,"(R)-NH2 amino derivative, that is to say with
inversion of configuration, is thus recovered with a
satisfactory yield.
CA 02359967 2010-02-24
30754-40
- 8 -
Consequently,- this alternative form of the
claimed process is very particularly of use in the
preparation of the compounds of general formula I"
H3C~N. CH3
CH3
- R - 2'
CH3 N 8a CH3 O
HO ''1'* ,....0 CH3
H01111. OH
CH3 0 O CH3 O~CH3 (~")
CH3 4õ CH3
0
'-NH2
CH3
in which:
R is a hydrogen atom or a C1-C10 alkyl, C2-C10
alkenyl or C6--C12 arylsulphonyl group, which are, if
appropriate, substituted, .
from a compound of general formula II' as defined above.
Mention may very particularly be made,. as
illustration of the azides which are suitable for the
present invention, of tetra (C1 to C20 alkyl) ammonium or
-phosphonium azide, substituted or unsubstituted
triarylsulphoniums and hexa(C1 to C20 alkyl)guani-
diniums.
According to a preferred alternative form of
the invention, it is a tetraaltylammonium azide and
more particularly tetrabutyl- or - tetraoctylammonium
azide.
In a specific embodiment of the invention, the
azide derivative is formed in a two-phase medium and
more specifically in solid/liquid phase transfer. In
this case, the organosoluble azide is generated in situ
from an inorganic azide, such as sodium azide, and from
a phase transfer agent in the presence of the compound
of general formula III in an organic solvent. The phase
CA 02359967 2008-11-14
9 -
transfer agent is - preferably a tetra (Cl to C20
alkyl)ammonium or -phosphonium methanesulphonate
As regards the compound of general formula II,
it is generally obtained beforehand by protection of
the hydroxyl functional group at the 2' position in the
corresponding derivative. Of course, this protection is
carried out conventionally using a conventional
protective group for the hydroxyl functional group;
such as those which appear in "Prot=ective Groups in
Organic Synthesis", Second Edition, Theodora W. Greene
and P. G. Wuts, Wiley Intersciences, p. 10-142. The
procedures for carrying out the protecting and
deprotecting operations are also described in the work
referred to above.
Following this protection of the. hydroxyl.
functional group at the 2' position, the hydroxyl
functional group at the 4" position is activated. This
activation of the compound of general formula II is
also carried out under conventional operating
.20 conditions, such as those described in "Protective
Groups in Organic Synthesis", Second Edition, Theodora
W. Greene and P. G. M. Wuts, Wiley Intersciences, p.
117-118. The examples submitted below describe a
detailed procedure for the activation of the 4"
hydroxyl functional group with triflic anhydride.
As regards the nucleophilic substitution
.reaction, it is carried out in an organic solvent,
preferably an anhydrous organic solvent. In the
preferred alternative form of the invention employing
an organosoluble azide, aromatic solvents, such as
benzene and toluene, or ethers, such as THE or methyl
tert-butyl ether, are suitable in particular as
solvents.
The nitrogenous nucleophilic compound,
preferably the azide, is used in a proportion of
approximately 1 to 30 equivalents with respect to the
compound of formula III and preferably in a proportion
of approximately 1 to 5 equivalents.
CA 02359967 2008-11-14
-
The temperature is conventionally between -20
and 180 C. As a general rule, it is adjusted so as to
favour the kinetics of the reaction without harming the
stability of the compounds.
5 According to a preferred alternative form of
the invention, in the first stage, the hydroxyl
functional group at the 4" position is activated by a
trifluoromethanesulphonate group and the nucleophilic
substitution is carried out with inversion of
10 configuration with tetrabutyl- or tetraoctylammonium
azide in toluene at room temperature.
According to a preferred alternative form of
the invention, R is a methyl group in the general
formulae I, I', II', II", III and V and A a hydrogen
atom in the general formula I and If.
Another subject-matter of the present invention
is the compounds of general formula VI
H3C\ CH3
N
'20 CH3 P2011,., ~N 8a 2,
CH3 0
CH3 HO''~' .,u0 CH3
H0,119. OH
~.~
CH3 0 0 CH3 0,CH3 (VI)
H3 ~~ "11CH3
C H30 4
O OR1
CH3
in which
- P2 is a hydrogen atom or a protective group,
- R is a hydrogen atom or a Cl-C0 o alkyl, C2-C10
alkenyl or C6-C12 arylsulphonyl group, which are, if
appropriate, substituted, and
- OR1 is a leaving group,
as intermediates in the preparation of a compound of
general formula I.
CA 02359967 2008-11-14
- 11 -
More preferably, R is a methyl group and OR1 is
a triflate group and more preferably the C-4" carbon
has a S configuration.
The present invention also relates to the
compounds of general formula VII
H3C,~ N ,CH3
CH3
R, P201144. 2'
N Sa CH3
olle,
CH3 O CH3
Hoge, Opto H01114- OH
CH~'~=, 0 0 CH3 CH3 N+1)
H3 CH3 4õ "CH3
O 0
NA2
CH3
in which
- P2 is a hydrogen atom or a protective group,
- R is a hydrogen atom or a C1-C10 alkyl, C2-C10
alkenyl or CG-C12 arylsulphonyl group, which are, if
appropriate, substituted, and
A, which are identical or different, are
= a nitrogen atom, if appropriate substituted,
3.0 = a C1-C4 alkyl group, which. is optionally
substituted by one or more aryl groups, which are, if
appropriate, substituted,
as intermediates in the preparation of a
compound of general formula I.
More preferably, R is a methyl group and NA2 an
N3 group and more preferably, the C-4" carbon has a R
configuration.
CA 02359967 2008-11-14
30754-40
- lla -
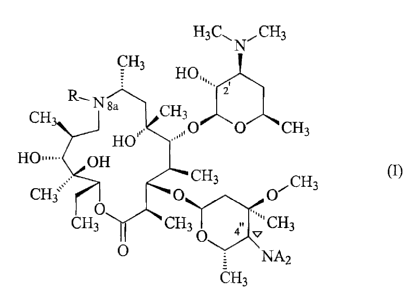
According to one aspect of the present invention,
there is provided a process for the stereoselective
preparation of a compound of general formula I
H3C,, N"CH3
CH3
HO,,,
". CH R, Nsa CH3
"" O O CH3
HO""
H& OH
(I)
CH. CH3
0.11 CH3
3 CH3 CH3 O a'v CH3
NA2
0
CH3
in which:
- R is a hydrogen atom or a C1-C10 alkyl, C2-C10
alkenyl or C6-C12 arylsulphonyl group, which are, if
appropriate, substituted;
A, which are identical or different, are
= a hydrogen atom,
= a nitrogen atom, if appropriate substituted,
= a C1-C4 alkyl group, which is optionally
substituted by one or more C6-C12 aryl groups, which are, if
appropriate, substituted,
= an R2CO or R2S02 group, with R2 being a hydrogen
atom, a C1-C8 alkyl group or a C6-C12 aryl group, which are,
if appropriate, substituted; and
CA 02359967 2008-11-14
30754-40
- lib -
- V means that the C in the 4" position has
undergone an inversion of configuration with respect to the
formula II,
from a compound of general formula II
H3C,, N~CH3
CH3 7
1
'' z
R, N 8a CH3
HO 1.
CH3
"'* .1O O CH3
HO...... OH (II)
CH3 CH3
CH3 O O
CH3 CH3 O a CH3
O OII
CH3
with:
- R as defined in general formula I; and
- P1 being a protective group for the hydroxyl
functional group at the 2' position,
wherein said process comprises at least the stages
consisting in:
(a) - activating the hydroxyl functional group at
the 4" position in the compound of general formula II, in
order to obtain a compound of general formula III
CA 02359967 2008-11-14
30754-40
- llc -
H3C,, N~CH3
CH3
P 10,,
''
N )8a
CH O O CH3
HO...... O(III)
CH3 CH3
CH3 O
CH3 3 a'~ CH3
O O ORi
CH3
in which:
- R and P1 arc as defined in general formulae I and
II; and
- OR1 is a leaving group,
(b) - bringing said compound of general formula
III thus obtained into contact with a nitrogenous
nucleophilic derivative under conditions which are
sufficient to allow the stereoselective displacement of the
hydroxyl functional group activated by said nitrogenous
nucleophile, and
(c) - deprotecting the hydroxyl functional group
at the 2' position,
in order to result in the expected compound of general
formula I.
According to another aspect of the present
invention, there is provided a compound of general formula
VI
CA 02359967 2008-11-14
30754-40
- lid -
H3C,, N"CH3
CH3
R P20'''' 2
CH )8a N O O CH3
HO...... OH (VI)
CH3
CH3 U01 3
CH3 3 0 a'- CH3
O OR1
CH3
in which
- P2 is a hydrogen atom or an acetyl group;
- R is a hydrogen atom or a C1-C10 alkyl, C2-C10
alkenyl or C6-C12 arylsulphonyl group, which is, if
appropriate, substituted; and
- OR1 is a Cl-C20 alkyl sulfonate, a C5-C6 aryl or
heteroaryl sulfonate, or C6-C26 alkylaryl sulfonate, which is
substituted, if appropriate, by one or more halogen atoms
and/or a nitro, cyano, or trifluoromethyl group,
as intermediate in the preparation of a compound
of general formula I as defined herein.
CA 02359967 2008-11-14
12 -
The examples which appear below are presented
by way of illustration and without implied limitation
of the present invention.
EXAMPLE 1
Preparation of the compound 4"-dehvdroxy-4" (R)-amino-
2'-acetoxy-9-deoxo-8a-aza-8a-methyl-8a-homoerythromycin
A:
The synthetic scheme used is as follows:
CA 02359967 2008-11-14
13 -
2
O C
z
p ...nu
0
O
2 N
O ~ O Q z lunn
z= _- O N W
= O 0
p 0
Ib=+ \ j
PIE
4 Q
z p Q 1111111
O p ..,.II
n ~ Z
O n
-ii
_O D O
p
Inure \
O
Ilan.
z
z
(D
CA 02359967 2008-11-14
14 -
All the tests are carried out under an inert
atmosphere.
1) Formation of 4"(S)-trifluoromethylsulnhonyl-2'-
acetoxy-9-deoxo-Ba-aza-8a-methyl-8a-homoerythromvcin A:
Pyridine (39.5 mg, 0.51 mmol, 5 equiv.) is
added to a solution of alcohol 2'-acetoxy-9-deoxo-Ba-
aza-8a-methyl-8a-homoerythromycin A (0.1 g,' 0.12 mmol,
1 equiv.) in anhydrous dichloromethane (0.4 ml). The
solution is cooled to 0 C and then a solution of
triflic anhydride (42.3 mg, 0.15 mmol, 1.2 equiv.) is
added dropwise. The solution is stirred for 1 h at 0 C
and then 30 min at room temperature. After diluting the
reaction mixture with anhydrous dichloromethane
(10 ml), the reaction mixture is cooled to 0 C and then
hydrolysed by addition of a saturated. aqueous sodium
bicarbonate solution (10 ml). The organic phase is
separated and then washed with distilled water (10 ml),
dried over magnesium sulphate and evaporated. The crude
product is taken up in heptane (10 ml) in order to
remove any trace of residual pyridine by azeotropic
distillation. 110.4 mg of 4"(S)-trifluoromethyl-
sulphonyl-2'-acetoxy-9-deoxo-8a-aza-8a-methyl-8a-homo-
erythromycin A are obtained with a purity greater of
than or equal to 90%. The structure is confirmed by NMR
and MS analysis.
2) Formation of 4"-dehydroxy-4"(R)-azido-2'-acetoxy-9-
deoxo-8a-aza-8a--methyl-8a-hornoerythromycin A:
A 0.58M solution of tetrabutylammonium azide in
toluene (4.5 ml; app. 1.3 equiv.) is added to
unpurified 4"(S)-trifluoromethylsulphonyl-2'-acetoxy-9-
deoxo-8a-aza-8a-methyl-8a-homoerythromycin A from. the
preceding stage (1.84 g, 2.0 mmol, 1 equiv.) at room
temperature. The reaction mixture is stirred for 3 days
at room temperature and then diluted with toluene
(25 ml). This solution is washed three times with
distilled water (3 x 10 ml), then dried over magnesium
sulphate and evaporated. 1.63 g of 4"-dehydroxy-4"(R)-
azido-2'-acetoxy-9-deoxo-8a-aza-8a-methyl-8a-homo-
CA 02359967 2008-11-14
- 15 -
erythromycin A are obtained with a purity of 70%. The
structure is confirmed by NMR and MS analysis.
3) Formation of the compound 4"-dehydroxy-4" (R)-amino-
2'-acetoxy-9-deoxo-8a-aza-8a-methyl-8a-homoerythromycin
A:
Raney nickel (200 mg) is added to a solution in
isopropanol (5 ml) of unpurified 4"-dehydroxy-4"(R)-
azido-2'-acetoxy-9-deoxo-8a-aza-8a-methyl-8a-homo-
erythromycin A from the preceding stage (250.0 mg,
0.30 mmol, 1 equiv.). Hydrazine monohydrate
(30 microlitres, 0.6 mmol, 2 equiv.) is added every 30
minutes. The reaction time is 2 h. The reaction mixture
is diluted with ethyl acetate (10 ml) and filtered. The
filtrate is washed with a saturated aqueous sodium
bicarbonate solution (10 ml) and then with water
(10 ml). After drying over magnesium sulphate, the
filtrate is evaporated. 230 mg of 4"-dehydroxy-4"(R)-
amino-2'-acetoxy-9-deoxo-8a-aza-8a-methyl-8a-
homoerythromycin A are obtained with a purity of 60%.
The structure is confirmed by NMR and MS analysis.
EXAMPLE 2
Tetraoctylammonium azide (190.3 ml, 0.5 mmol,
5 equiv.) is added at room temperature to a solution of
4"(S)-trifluoromethylsulphonyl-2'-acetoxy-9-deoxo-8a-
aza-8a-methyl-8a-homoerythromycin A (92.3 mg, 0.1 mmol,
1 equiv.) in toluene (0.2 ml) . After stirring for two
days at room temperature, tetraoctylammonium azide
(58 mg, 0.15 mmol, 1.5 equiv.) is again added. After
stirring for an additional two days at room
temperature, the reaction mixture is diluted with
toluene (10 ml) and washed with water (10 ml). The
organic phase is separated and dried over sodium
sulphate. After evaporating the solvents, 1H NMR
analysis shows the predominant presence of the compound
4"-dehydroxy-4"(R)-azido-2'-acetoxy-9-deoxo-8a-aza-8a-
methyl-8a-homoerythromycin A.
EXAMPLE 3
Tetrabutylphosphonium methanesulphonate
(355 ma, 1 mmol, 5 eciuiv.) and then sodium azide
CA 02359967 2008-11-14
- 16 -
(325 mg, 5 mmol, 25 equiv.) are successively added to a
solution of 4"(S)-trifluoromethylsulphonyl-2'-acetoxy-
9-deoxo-8a-aza-8a-methyl-8a-homoerythromycin A (185 mg,
0.2 mmol, 1 equiv.) in toluene (0.4 ml) at room
temperature. After stirring for three days at room
temperature, the reaction mixture is diluted with
toluene (10 ml) and washed with water (10 ml). The
organic phase is. separated and dried over sodium
sulphate. After evaporating the solvents, 1H NMR
analysis shows the predominant presence of the compound
4"-dehydroxy-4"(R)-azido-2'-acetoxy-9-deoxo-8a-aza-8a-
methyl-8a-homoerythromycin A.
EXAMPLE 4
Tetraoctylammonium methanesulphonate (217 mg,
0.38 mmol, 3.8 equiv.) and then LeLrabutylammonium
azide (158 mg, 2.5 mmol, 25 equiv.) are successively
added to a solution of 4"(S)-trifluoromethylsulphonyl-
2'-acetoxy-9-deoxo-8a-aza-8a-methyl-8a-homoerythromycin
A (92 mg, 0..1 mmol, 1 equiv.) in toluene (0.25 ml) at
room temperature. After-reacting for 4 days at room.
temperature, the reaction mixture is diluted with
toluene (10 ml) and washed with water (10 ml). The
organic phase is separated and dried over sodium
sulphate. After evaporating the solvents, 1H NMR
analysis shows the predominant presence of the compound
4"-dehydroxy-4"(R)-azido-2'-acetoxy-9-deoxo-8a-aza-8a-
methyl-8a-homoerythromycin A.
EXAMPLE 5
A solution of 4"(S)-trifluoromethylsulphonyl-
2'-acetoxy-9-deoxo-8a-aza-8a-methyl-8a-homoerythromycin
A (21.4 mg, 0.023 mmol) in N-methylpyrrolidinone is
saturated with gaseous ammonia. This solution is
stirred for 48 h at room temperature. The reaction
mixture is subsequently diluted with ethyl acetate
(10 ml) and washed with water (15 ml) The organic
phase is separated, dried over sodium sulphate and
evaporated. LC/MS analysis shows the formation of 220,
by internal standardization, of 4"-dehydroxy-4"(R)-
CA 02359967 2008-11-14
17 -
amino-2'-acetoxy-9-deoxo-8a-aza-8a-methyl-8a-homo-
erythromycin A.