Note: Descriptions are shown in the official language in which they were submitted.
CA 02361561 2001-07-31
WO 00/45800 I PCT/EP00/00938
IMMtJNOSUPPRESSIVE EFFECTS OF PTERIDINE DERIVATIVES
The invention relates to a pharmaceutical
composition for the treatment of autoimmuno disorders
and/or the treatment or prevention of transplant-
rejections comprising pteridine derivatives.
The invention further relates to combined
pharmaceutical preparations comprising one or more
pteridine derivates and one or more known
immunosuppressant, and to a group of novel pteridine
derivates as such.
Further the invention is also related to a
method for the treatment of autoimmuno disorders and/or
of transplant-rejections.
Several pteridine derivates are known in nature
and used in the preparation of medicines, for example as
described in EP-A-108 890. Other medical uses of
derivatives of pteridine are described in WO 95-31987 as
NO-synthase inhibitors, for example for the treatment of
diseases caused by a high nitrogen monoxide level.
Further, WO-95-32203 describes also the use of
tetrahydropteridine derivatives as NO-synthase
inhibitors.
Both above-mentioned WO publications disclose
also the use of these specific pteridine derivatives in
the treatment of pathologically low blood pressure, in
particular septic shock and combined with cytokines in
tumor therapy and in transplant-rejection diseases.
Although some of these pteridine derivatives
are claimed as potentially active for the treatment of
transplant-rejection diseases, direct evidence for their
effectiveness is lacking. Thus there still is a need for
specific and highly active immunosuppressive compounds,
in particular immunosupressive compounds active in the
cosignal pathway.
A first object of the invention is to provide a
pharmaceutical composition having high immunosuppressive
activity. Another object of the invention is to provide a
CONFIRMATION COPY
CA 02361561 2001-07-31
WO 00/45800 PCT/EP00/00938
2
combined immunosuppressive preparation which causes a
superadditive effect, comprising a pteridine derivative
of the invention and other known immunosuppressants.
Another further object of the invention is to
provide immunosuppressive compounds, which are active in
a minor dose, in order to decrease the considerable
treatment costs.
Known immunosuppressive compounds are for
example cyclosporine A, subsituted xanthines, tacrolimus
(FK 506), rapamycine (RPM), leflunomide, mofetil,
adrenocortical steroids, cytotoxic drugs and antibody
preparations.
The immunosuppressive effect of cyclosporine A
(CyA) is already known since 1972. However, due to its
nephrotoxicity and several other side effects CyA has not
been able to establish itself as the optimal and final
drug of choice.
Methylxanthines, for example pentoxifylline
(PTX), are known having immunosuppressive effects in
vitro.
Recently (Lin Y. et al, Transplantation 63
(1997) it has been found that the co-medication of an
immunosuppressive compound such as cyclosporine A (CyA)
or FK506 or RPM (rapamycine) with a methylxanthine
derivative, in particular A802715 (7-propyl-1(5-hydroxy-
5-methylhexyl)-3-methylxanthine) leads to a superadditive
increase in the immunosuppressive action.
Likewise, other substituted, in particular
substituted 8-phenylxanthines have been found to possess
immunosuppressive effects in vitro (application EP
98.201323.7).
The present invention relates in particular to
the application of a group pteridine derivatives and
their pharmaceutical salts, possessing unexpectedly
desirable pharmaceutical properties, i.c. are highly
active immunosuppressive agents.
The invention demonstrates the
CA 02361561 2001-07-31
WO 00/45800 PCT/EP00/00938
3
immunosuppressive effects of pharmaceutical composition
for the treatment of autoimmuno disorders and/or for the
treatment or prevention of transplant-rejections
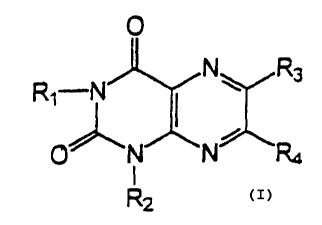
comprising a pteridine derivative of general fornula:
0
~N~ Rs
Ri-N II
0"N~N
to
R2
wherein:
R1 and R~ are ir_dependently hydrogen; aliphatic
saturated or unsaturated; straight or branched carbon
chain with 1 to 7 carbon atoms; substituted or
unsubstituted aryl or alk_,rlaryl substituents, whereby the
carbon atoms may be oxidized represented by alcohol or
carbonyl function or carboxylic acids and their esters;
R3 and R; are ir_deper_dently hydrogen, hydroxyl ,
halogen,alkyl, haloalkyl, alkoxy, wherein alkyl and the
alkyl group may be branched or straight and contains one
or four carbon atoms, fornyl and derivatives such as
hydroxylamino conjugates and acetals, cyano, carboxylic
acids and carboxyl acid derivatives such as esters and
amides, sulfhydryl, amino, al.'~ylamino, cycloalkylamino,
alkenylamino, alkynylamino, benzylamino,
hydroxylalkylamino, morfolinoalkylamino, fenylhydrazino,
morfoline, piperidine, mercaptobenzyl, mercaptoalkyl,
cysteinyl ester, styryl, substituted or unsubstituted
aromatic ring; aromatic or heterocyclic substituent
substituted with an aliphatic spacer between the
pteridine ring and the aromatic substituent of 2 to 4
carbon atoms, whereby said spacer may contain an alcohol
function, carbonyl function, halogen, ether, and may be
saturated or unsaturated; branched or straight, saturated
or unsaturated aliphatic chain of 1 to 7 carbon atoms
CA 02361561 2001-07-31
WO 00/45800 PCT/EP00/00938
4
which may contain one or more functions chosen from the
group comprising carbonyl, alcohol, ether, carboxyester,
nitro, thioalkyl, halogen or a pharmaceutically
acceptable salt thereof; and
X and Y are independently oxygen or sulfur
or a pharmaceutical salt thereof, and a
pharmaceutically acceptable carrier.
Preferred pteridine derivatives comprising
compositions are given in claims 2-9. Particularly
preferred are the compositions according to claim 10.
The invention further relates to a combined
preparation having synergetic effects containing 1)
cyclosporine A, substituted xanthines, tacrolimus
(FK506), Rapamycin (RPM), Leflunomide, Mofetil,
adrenocortical steriods, cytotoxic drugs and antibody
compositions and 2) at least one pteridine derivative of
formula (I) defined above, and optionally a
pharmaceutical excipient, for simultaneous, separate or
sequential use in (auto)immune disorders and/or in the
treatment of transplant-rejections.
The invention further relates to a method for
treating auto-immuno disorders or transplant-rejections
in a subject by administering an effective amount of a
pharmaceutical composition of claims 1-11, to the
compounds as such as defined above, to the use of these
compounds for the treatment of autoimmuno disorders
and/or the treatment and/or prevention of transplant
rejections, and to a method for selecting potent
immunosuppressive agents based on the determination of
the three parameters MLC, ACD3 and ACDZe.
Hereunder the effects of the pteridine
derivatives on the lymphocyte activation are elucidated
and are compared with standard reference compounds (see
table I, compound 4, 6, 7, 11, 13, 19, 20, 21, 22, 25,
26, 28, 30, 34, 35).
Table I summarizes the tested compounds. These
pteridine derivatives were obtained as follows:
CA 02361561 2001-07-31
WO 00/45800 PCT/EP00/00938
S
6-Bromomethyl-1,3-dimethyllumazine:
To a solution of 1,3,6-trimethyllumazine [1] (2.06 g,
0.01 moles) in glacial AcOH (60 ml) was added dropwise
bromine (3.2 g, 0.02 moles) in AcOH (10 ml) and then
heated under reflux for 1 hour. After cooling was
evaporated, the residue dissolved in CHC13 (100 ml),
washed with Hz0 (3 x 70 ml), the organic layer dried over
Na2S04 and again evaporated. The residue was purified by
silica gel column chromatography starting with
toluene/EtOAc 9/1 to elute first 6-dibromomethyl-1,3-
dimethyllumazine (1.49 g, 4l0) and followed by
toluene/EtOAc 4/1 to get 6-bromomethyl-1.3-
dimethyllumazine. Yield: 1.2 g (42%). M.p. 228°C
(decomp. ) . W (MeOH) : 244 (4.16) ; [264 (400) ] ; 337
(3.86) .
7-Bromomethyl-1,3-dimethyllumazine:
Analogous to the preceding procedure with 1,3,7-
trimethyllumazine [1] (2.06 g, 0.01 moles) and bromine
(3.2 g, 0.02 moles) by heating for 2 hours. Isolation by
silica gel column chromatography with toluene/EtOAc 9/1
to elute first 7-dibromomethyl-1,3-dimethyliumazine (2.07
g, 57%) and second 7-bromomethyl-1,3-dimethyllumazine.
Yield: 0.97 g (340). M.p.165-166°C. W (MeOH): 241
(4.23); 338 (4.02).
1,3-Dimethyllumazin-6-ylmethyl-triphenylphosphonium
bromide:
To a suspension of 6-bromomethyl-1,3-dimethyllumazine
(1.0 g, 3.5 mmoles) in toluene (20 ml) triphenylphosphane
(1.1 g, 4.2 mmoles) was added and then heated at 80°C in
an oilbath with stirring for 8 hours. After cooling the
precipitate was collected, washed with EtOAc and dried at
100°C to give 1.8 g (940) of a colorless powder of m.p.
289°C. UV (MeOH) : 204 (4.74) ; 227 (4.52) ; [243 (4.42) ] ;
[262 (4.21) ] ; 338 (3 .88) .
CA 02361561 2001-07-31
WO 00/45800 PCT/EP00/00938
6
1,3-Dimethyllumazin-7-ylmethyl-triphenylphosphonium
bromide:
Analogous to the preceding procedure from 7-bromomethyl-
1,3-dimethyllumazine and triphenylphosphane in toluene by
heating under reflux for 1 day. Yield: 1.86 g (970). M.p.
261°C. UV (MeOH): 204 (4.76); [221 (4.54)]; 342 (4.09);
414 (4.38) .
General synthesis of 1,3-dimethyl-6-(E)-styryllumazines
1, 2, 3, 5:
To a solution of 1,3-dimethyllumazin-6-ylmethyl-
triphenylphosphonium bromide and 1,3-dimethyllumazin-7-
ylmethyl-triphenylphosphonium bromide (0.547 g, 1 mmole),
respectively, in MeOH (5 ml) was added sodium methoxide
(0.108 g, 2 mmoles) and stirred at room temperature for
30 min. Then 1.5 mmoles of the aromatic or heteroaromatic
aldehyde were added and stirring continued for 5 hours.
The resulting precipitate was filtered off, washed with
MeOH and purified by recrystallization from DMF/H20 to
give a yellowish powder.
1,3-Dimethyl-6-(E)-styryllumazine (1):
According to the general procedure with benzaldehyde
(0.16 g). Yield: 0.124 g (420). M.p. 238°C. W (MeOH):
[220 (4.17) ] ; 308 (4.42) ; 372 (4.03) .
1,3-Dimethyl-6-[(E)-2-(pyrid-3-yl)vinyl]lumazine (2):
According to the general procedure with pyridine-3-
carboxaldehyde (0.162 g). Yield: 0.195 g (660). M.p.
210°C. UV (MeOH): [236 (3.92)]; 308 (4.29); 370 (3.97).
1, 3-Dimethyl-6- [ (E) -2- (pyrid-4-yl)vinyl] lumazine (3)
According to the general procedure with pyridine-4-
carboxaldehyde (0.162 g). Yield: 0.156 g (530). M.p.
262°C. W (MeOH) : 202 (4.20) ; [238 (3.92) ] ; 307 (4.51) ;
370 (4.20) .
6-(1,2-Dibromo-2-phenylethyl)-1,3-dimethyllumazine (4):
CA 02361561 2001-07-31
WO 00/45800 PCT/EP00/00938
7
To a solution of 1,3-dimethyl-6-(E)-styryllumazine (1)
(0.735 g, 2.5 mmoles) in CHC13 (20 ml) was added bromine
(0.8 g, 5 mmoles) dissolved in CHC13 (5 ml) and then the
mixture stirred at room temperature for 4 hours. It was
evaporated to dryness and the residue treated with MeOH
to give a colorless precipitate. The solid was collected,
washed with MeOH and dried in a vacuum desiccator. Yield:
1.067 g (94a). M.p. 176°C. UV (MeOH): 245 (4.18); [260
(4.10) ] ; 341 (3 .83) .
15
1,3-Dimethyl-6-[(E)-4-(phenyl)butadienyl]lumazine (5):
According to the general procedure with cinnamaldehyde
(0.2 g) . Yield: 0.138 g (43%) . M.p. 252°C (decomp:) . UV
(MeOH) : 228 (4.02) ; [244 (3.97) ] ; 330 (4.66) ; 389 (4.23) .
6-[(E)-2-methoxycarbonylethenyl]lumazine:
To a suspension of methoxycarbonylmethyl-
triphenylphosphonium bromide (0.415 g, 1 mmole) in
dioxane (3 ml) was added DBU (0.23 g, 1.5 mmoles) and
stirred at room temperature for 30 min. Then 1,3-
dimethyllumazine-6-carboxaldehyde [1] (0.2 g, 0.91
mmoles) was added and stirring continued for 5 hours. The
precipitate was collected, washed with MeOH and dried to
give a colorless crystal powder. Yield: 0.158 g (630).
M.p. 211-213°C (decomp. ) . UV (MeOH) : 202 (4.46) ; [256
(4.14) ] ; 286 (4.21) ; 348 (4.08) .
6-(1,2-Dibromo-2-(methoxycarbonyl)ethyl)-1,3-
dimethyllumaz ine ( 6 )
To a solution of 6-[(E)-2-methoxycarbonylethenyl]lumazine
(0.7 g, 2.53 mmoles) in CHC13 (20 ml) was added bromine
(0.64 g, 4 mmoles) dissolved in CHC13 (5 ml) and then the
mixture stirred at room temperature for 6 hours. It was
evaporated to dryness and the residue treated with MeOH
to give a colorless precipitate. The solid was collected,
washed with MeOH and dried in a vacuum desiccator. Yield:
0.97 g (88%) . M.p. 163°C. W (MeOH) : 247 (4.16) ; [260
(4.08) ] ; 339 (3 .88) .
CA 02361561 2001-07-31
WO 00/45800 PCT/EP00/00938
6-(2-Bromo-2-methoxycarbonyl-ethenyl)-1,3-
dimethyllumazine (7):
To a solution of 6 (0.1 g, 0.23 mmoles) in dioxane (20
ml) was added DBU (70 MG, 0.43 g, 0.43 mmoles) and then
stirred at room temperatur for 2 hours. It was diluted
with ethyl acetate (100 ml), washed with HZO (3 x 50 ml),
the organic layer separated, dried over Na2S04 and then
evaporated. The residue was treated with MeOH, the solid
collected and purified by recrystallization from DMF to
give a yellowish powder. Yield: 0.055 g (68%). M.p.
204°C. W (MeOH) : [254 (4.08) ) ] ; 285 (4.25) ; 360 (4.01) .
6-Chlorocarbonyl-1,3-dimethyllumazine:
A suspension of 1,3-dimethyllumazine-6-carboxylic acid
[2] (3.0 g, 12.7 mmoles) in dry toluene (80 ml) was
treated with freshly destilled thionyl chloride (50 ml)
under reflux for 3 hours. It was evaporated to dryness,
the residue treated with dry ether, the solid collected,
washed with ether and dried in a vaccuum desiccator.
Yield: 3.13 g (93%). M.p. 262-264°C. UV (dioxane): 256
(4.08) ; [280 (4.00) ] ; 333 (4.03) .
6-[(2-Acetyl-2-ethoxycarbonyl)acetyl]-1,3-
dimethyllumazine ( 8 )
A solution of ethyl acetoacetate ethoxy-magnesium salt
[3] (0.8 g, 4 mmoles) in THF (8 ml) was added dropwise to
a suspension of 6-chlorocarbcnyl-1,3-dimethyllumazine
(0.51 g, 2 mmoles) in THF (10 ml) and then the mixture
stirred at room temperature for 3 days. It was evaporated
and the residue treated with cold 1 N HC1 (20 ml, 0-5°C).
The precipitate was collected, washed with H20 and dried
in a vacuum desiccator. Purification was achieved by
column chromatography (silica gel 3.5 x 12 cm) with
CHC13/MeOH 95/5 and the first main fraction collected.
After evaporation was recrystallized from toluene (12 ml)
to give colorless crystals. Yield: 0.247 g (36%). M.p.
153-156°C. W (pH 2.0) : 251 (4.09) ; 293 (4.10) ; 330
(4.11) .
CA 02361561 2001-07-31
WO 00/45800 PCT/EP00/00938
9
6-[2,2-(Diethoxycarbonyl)acetyl]-1,3-dimethyllumazine
(9)
To a solution of ethylmalonate ethoxy-magnesium salt [4]
(0.685 g, 3 mmoles) in THF (12 ml) was added 6-
chlorocarbonyl-1,3-dimethyllumazine (0.51 g, 2 mmoles)
and then the mixture stirred at room temperature for 20
hours. It was evaporated, the residue treated with 1 N
HC1 (20 ml) and the resulting solid collected.
Recyrstallization from EtOH (40 ml) gave yellowish
crystals. Yield: 0.585 g (78%). M.p. 124-126°C. UV (pH
2.0): 253 (4.05); 291 (4.08); 332 (4.04).
6-(1-Methoxy-2-methoxycarbonyl)ethenyl)-1,3-
dimethyllumazine (10):
A suspension of 6 (0.2 g, 0.46 mmoles) in dry MeOH (8 ml)
was treated with a solution of sodium (0.046 g, 2 mmoles)
in MeOH (2 ml) at room temperature with stirring for 15
min. Then NH4C1 (0.1 g) and H20 (10 ml) were added and the
mixture extracted with CHC13 (2 x 50 ml). The organic
layer was dried over Na2S04, evaporated and the residue
crystallized from CHC13/n-hexane. Yield: 0.085 g (60%).
M.p. 160°C. W (MeOH): 204 (4.20); 245 (4.15); 288
(4 . 23 ) ; 350 (3 . 99 ) .
1, 3-Dimethyl-6- [ (2-nitro) ethenyl] lumazine (11)
A solution of 12 (0.562 g, 2 mmoles) in pyridine (10 ml)
was cooled to 0°C and then acetic anhydride (4- ml)
dropwise added. Cooling was removed and the mixture
stirred at room temperature for 3 hours. The resulting
precipitate was collected, washed with Hz0 and dried in a
vacuum desiccator to give a chromatographically pure
product. Yield: 0.515 g (98a). Crystallization from CHC13.
M.p. 232-234°C. W (MeOH): [239 (3.63)]; 309 (3.87); 365
(3.95) .
6-[(1-Hydroxy-2-vitro)ethyl]-1,3-dimethyllumazine (12):
To a solution of nitromethane (0.61 g, 10 mmoles) and
triethylamine (1.44 g, 10 mmoles) in MeOH (20 ml) was
CA 02361561 2001-07-31
WO 00/45800 PCT/EP00/00938
added 6-formyl-1,3-dimethyllumazine [1] (2.0 g, 9 mmoles)
and then the mixture stirred at room temperature for 5
hours. The precipitate was collected, washed with MeOH
and ether and dried. Yield: 2.22 g (78%).
5 Recrystallization from CHC13. M.p. 166-167°C. UV (MeOH):
240 (4.38) ; 336 (3.98) ; [347 (3.89) ] .
6-[(1-Ethylthio-2-vitro)ethyl]-1,3-dimethyllumazine (13):
To a suspension of compound 11 (0.263 g, 1 mmole) in MeOH
10 (5 ml) and H20 (5 ml) was,added ethylmercaptane (0.093 g,
1.5 mmoles) and DBU (0.2 g) and then the mixture stirred
at room temperature for 1 hour. The precipitate was
collected, washed and dried. Yield: 0.25 g (770). M.p.
88°C. Uu (MeOH): 203 (4.28); 240 (4.26); [262 (4.05)];
341 (3.89) .
6-Hydroxymethyl-1,3-dimethyllumazine (14) [1]:
1,3-Dimethyl-7-[(E)-2-(pyrid-2-yl)vinyl]lumazine (15):
According to the general procedure with pyridine-2-
carboxaldehyde (0.162 g). Yield: 0.233 g (79%). M.p. 282-
283°C. UV (MeOH): 203 (4.14); 238 (4.23); 312 (3.95); 375
(4.36) .
1, 3-Dimethyl-7- [ (E) -2- (pyrid-3-yl)vinyl] lumazine (16)
According to the general procedure with pyridine-3-
carboxaldehyde (0.162 g). Yield: 0.195 g (660). M.p. 264-
265°C. UV (MeOH) : 208 (4.45) ; 234 (4.43) ; [274 (4.09) ] ;
307 (4.08) ; 375 (4.48) .
1, 3-Dimethyl-7- [ (E) -2- (pyrid-4-yl)vinyl] lumazine (17)
According to the general procedure with pyridine-4-
carboxaldehyde (0.162 g). Yield: 0.215 g (73%). M.p. 307-
310°C. UV (MeOH): 207 (4.12); 229 (4.01); 282 (3.79);
(296 (3 .76) ] ; 372 (4.00) .
1,3-Dimethyl-7-[(E)-4-(phenyl)butadienyl]lumazine (18):
CA 02361561 2001-07-31
WO 00/45800 PCT/EP00/00938
According to the general procedure with cinnamaldehyde
(0.2 g). Yield: 0.195 g (610). M.p. 277-287°C (decomp.).
UV (MeOH): 239 (3.79); 299 (3.66); 402 (4.15).
7-[1,2-Dibromo-2-(methoxycarbonyl)ethyl)-1,3-
dimethyllumazine (19):
To a suspension of 7-[(E)-2-methoxycarbonylethenyl]luma-
zine (1.79 g, 6.5 mmoles) in CHC13 (70 ml) was added
bromine (0.7 g, 14 mmoles) dissolved in CHC13 (10 ml) and
then the mixture stirred at room temperature for 3 hours.
It was evaporated to dryness and the residue treated with
MeOH to give a colorless precipitate. The solid was
collected, washed with MeOH and dried in a vacuum
desiccator. Yield: 2.34 g (770). Crystallization from
EtOAc/n-hexane. M.p. 144-145°C. UV (MeOH): 240 (4.14);
343 (3.90) .
7-[(E)-2-methoxycarbonylethenyl]lumazine (20):
To a suspension of methoxycarbonylmethyl-
triphenylphosphonium bromide (0.415 g, 1 mmole) in
dioxane (5 ml) was added DBU (0.23 g, 1.5 mmoles) and
stirred at room temperature for 30 min. Then 1,3-
dimethyllumazine-7-carboxaldehyde [1] (0.2 g, 0.91
mmoles) was added and stirring continued for 20 hours.
The precipitate was collected, washed with MeOH and dried
to give a colorless crystal powder. Recrystallization
from DMF. Yield: 0.15 g (60%)~. M.p. 242-245°C (decomp.).
UV (MeOH): 201 (4.21); 225 (4.29); 252 (4.20); 364
(4.11) .
7-(1,2-Dibromo-2-phenylethyl)-1,3-dimethyllumazine (21):
To a solution of 1,3-dimethyl-7-(E)-styryllumazine (23)
(0.735 g, 2.5 mmoles) in CHC13 (20 ml) was added bromine
(0.48 g, 3 mmoles) dissolved in CHC13 (5 ml) and then the
mixture stirred at room temperature for 3 hours. It was
evaporated to dryness and the residue treated with MeOH
to give a colorless precipitate. The solid was collected,
washed with MeOH and dried in a vacuum desiccator. Yield:
CA 02361561 2001-07-31
WO 00/45800 PCT/EP00/00938
12
1.08 g (95%). M.p. 187-188°C. UV (MeOH): 241 (4.25); 341
(4.06) .
7-(1-Bromo-2-phenyl)ethenyl-1,3-dimethyllumazine (22):
To a suspension of 21 (0.2 g, 0.44 mmoles) in dry MeOH (4
ml) was added a solution of sodium (0.05 g, 2.2 mmoles)
in MeOH (1 ml) and then the mixture stirred at room
temperature for 3 hours. The precipitate was collected,
washed with MeOH and dried in vacuum. Yield: 0.117 g
(710). Yellowish powder from DMF. M.p. 245-246°C. UV
(MeOH) : 243 (4.15) ; 372 (4.15) .
7-Benzyl-1,3-dimethyllumazine:
A solution of 5,6-diamino-1,3-dimethyluracil
monohydrochloride [5] (2.06 g, 0.01 mole) in H20 (50 ml)
was treated with benzylglyoxal [6] (2.22 g, 0.015 moles)
in EtOH (20 ml) and heated under reflux for 1 hour. It
was diluted with Hz0 (50 ml) and then extracted with CHC13
(5 x 100 ml). The organic layer was dried over NaZS04,
evaporated and the residue purified by silica gel column
chromatography with toluene/EtOAc 10/1. The main fraction
was collected, evaporated and crystallized from EtOH.
Yield: 1.7 g (61%). M.p. 147-148°C. W (MeOH): 238
(4.22); 332 (4.07).
30
1,3-Dimethyl-6-(E)-styryllumazine (23):
According to the general procedure with benzaldehyde
(0.16 g). Yield: 0.223 g (76%). M.p. 259-260°C. UV
(MeOH): 203 (4.17); 237 (4.11); 379 (4.29).
7-Benzoyl-1,3-dimethyllumazine (24):
A suspension of 7-benzyl-1,3-dimethyllumazine (0.56 g, 2
mmoled) in HZO (30 ml) was treated with KMn04 (0.6 g) and
heated under reflux for 30 min. After cooling was
extracted with CHC13 (3 x 100 ml), the organic layer dried
over Na2S04, filtered and evaporated to dryness.
Crystallization from dioxane/HzO. Yield: 0.5 g (84%). M.p.
190-191°C. W (MeOH) : 233 (4.23) ; [255 (4.10) ] ; 347 (3.97) .
CA 02361561 2001-07-31
WO 00/45800 PCT/EP00/00938
~3
7-Chloro-1,3-dimethyllumazine (25) [7].
1,3-Dimethyl-7-mercaptolumazine (26) [8].
1,3-Dimethyl-6,7-diphenyllumazine (27) [9].
1,3-Dimethyl-6-phenyl-7-mercaptolumazine (28):
A mixture of 7-hydroxy-1,3-dimethyl-6-phenyllumazine [5J
(2.84 g, 0.01 mole) and P4Slo (3.3 g) was heated in
pyridine (75 ml) under reflux for 1 hour. After cooling
was diluted with H20 (50 ml) and after standing for
several hours the yellow precipiptate (28-pyridinium
salt, 3.3 g, 87%). The salt was dissolved in hot H20 (100
ml) and acidified by HCl to pH 0. The resulting yellow
crystals were collected, washed and dried in the oven.
Yield: 2.22 g (740). M.p. 145°C (decomp.). W (MeOH): 203
(4.37) ; 227 (4.36) ; [283 (3 .86) ] ; 370 (4.05) .
7-Methoxy-1,3-dimethyl-6-phenyllumazine (29):
A solution 7-hydroxy-1,3-dimethyl-6-phenyllumazine [5]
(1.42 g, 0.005 moles) in 0.5 N NaOH (20 ml) and MeOH (10
ml) was treated with dimethyl sulfate (1 ml) and stirred
for 1 hour at room temperature. The resulting precipitate
was collected, washed and dried in the oven. Yield: 1.26
g (81%). M.p. 194°C. W (MeOH): 205 (4.53); [240 (4.08)];
281 (4.22) ; 343 (4.23) .
7-Chloro-1,3-dimethyl-6-phenyllumazine (30):
A mixture of 7-hydroxy-1,3-dimethyl-6-phenyllumazine [5]
(2.84 g, 0.01 mole) and NH4C1 (1 g) was heated in POC13
under reflux for 36 hours. It was evaporated to a syrup,
ice was added and stirred with a glasrod till a
precipitate was formed. The solid was collected, washed
with H20, dried and then recrystallized from MeOH. Yield:
2.36 g (78%). M.p. 180°C. W (MeOH): 204 (4.47); 249
(4.23) ; 273 (4.24) ; 350 (4.05) .
CA 02361561 2001-07-31
WO 00/45800 PCT/EP00/00938
14
6-Benzoyl-7,8-dihydro-1,3-dimethyl-7-(4-methoxyphenyl)lu-
mazine (31a)
A solution of 6-benzoyl-1,3-dimethyllumazine (0.2 g, 0.68
mmoles) in dry 1,2-dichloroethane (20 ml) was treated
with AlCl3 (0.4 g, 3 mmoles) and freshly distilled anisol
(10 ml, 92 mmoles) at room temperature and stirred for 24
hours. Then ice (50 g) was added, the aquous phase
extracted with CHC13 (3 x 50 ml), the organic phase washed
with 2°s-NaHC03 solution (50 ml) and Hz0 (50 ml) , dried
over Na2S04 and evaporated in high vacuum to remove excess
of anisol. The residue was treated with toluene (50 ml)
to obtain a yellow precipitate. Recrystallization from
EtOH/H20 1/1 gave yellow crystals. Yield: 0.176 g (65%).
M.p. 240-244°C (decomp.). UV (MeOH): 254 (4.25); [270
(4.21) ] ; 406 (4.08) .
6-Benzoyl-1,3-dimethyl-7-(4-methoxyphenyl)lumazine (31):
A suspension of 6-benzoyl-7,8-dihydro-1,3-dimethyl-7-(4-
methoxyphenyl)lumazine (31a) (0.3 g, 0.74 mmoles) in
dioxane (40 ml) was treated at room temperature with 1%-
KMn04 solution (10 ml) by dropwise addition with stirring.
After 30 min the excess of KMn04 was reduced by NaHS03,
the Mn02 filtered off, washed with warm EtOH (3 x 20 ml)
and then the united organic phases evaporated to dryness.
The residue was purified by silica gel chromatography
with CHC13/MeOH (25/1). The main fraction was collected,
evaporated and the solid rec~ystallized from EtOAc with
charcoal. Yield: 0.175 g (59%). M.p. 255-257°C. W
(MeOH) : 253 (4.24) ; 367 (4.10) .
6-Benzoyl-7,8-dihydro-1,3-dimethyl-7-phenyllumazine
(32a)
Analogous to procedure 31a from 6-benzoyl-1,3-
dimethyllumazine (0.2 g, 0.68 mmoles) and benzene (15
ml). Yield: 0.21 g (83%). W (MeOH): 254 (4.26); 407
(4.12) .
6-Benzoyl-1,3-dimethyl-7-phenyllumazine (32):
CA 02361561 2001-07-31
WO 00/45800 PCT/EP00/00938
Analogous to procedure 31 from 6-benzoyl-7,8-dihydro-1,3-
dimethyl-7-phenyllumazine (32a)(0.3 g, 0.78 mmoles).
Yield: 0.18 g (620). M.p. 185-187°C. UV (MeOH): 252
(4.39) ; [290 (4.08) ] ; 349 (4.16) .
5
7-Methoxy-1,3-dimethyl-6-styryllumazine (33):
To a suspension of compound 4 (0.2 g, 0.44 mmoles) in dry
MeOH (6 ml) was added DBU (0.2 ml, 1.34 mmoles) and then
stirred at room temperature for 2 hours. The precipitate
10 was filtered off, washed with MeOH and dried in a vacuum
desiccator. Yield: 0.134 (940). Crystallization from DMF.
M.p. 271-272°C. UV (MeOH): [232 (4.11)]; 306 (4.36); 375
(4.38) .
15 1-Methyl-6,7-diphenyllumazine (34) [10].
7-Hydroxy-3-methyl-6-phenyllumazine (35) [5].
7-Hydroxy-1,6-diphenyllumazine (36).
To a suspension of 6-diamino-5-nitroso-1-phenyluracil
[11] (2.32 g, 0.01 moles) in H20 (50 ml) and EtOH (20 ml)
was reduced catalytically with PtOz/HZ in a shaking
apparatus till about 450 ml of hydrogen was consumed. The
mixture was heated, the catalyst filtered off and the
filtrate treated with ethyl phenylglyoxylate (2.5 g,
0.014 mmoles) by heating under reflex for 30 min. The
warm solution was acidified by HC1 to pH 0 and the
resulting precipitate collected after cooling.
Recrystallization from DMF. Yield: 2.59 (78%). M.p.
330°C. UV (MeOH) : 204 (4.54) ; [222 (4.37) ] ; 284 (4.17) ;
346 (4.25) .
7-Hydroxy-3,6-dimethyl-1-phenyllumazine (37) [12]:
7-Hydroxy-6-phenyl-1,3-di-n-propyllumazine (38):
A suspension of 5,6-diamino-1,3-di-n-propyluracil (1.13
g, 0.005 moles) in H20 (30 ml), EtOH (5 ml) and AcOH (2
ml) was treated with ethyl phenylglyoxylate (1.25 g,
CA 02361561 2001-07-31
WO 00/45800 PCT/EP00/00938
16
0.007 mmoles) and heated under reflux for 30 min forming
a brownish oil. After cooling was acidified by HC1 to pH
0 whereby the oil solidified. Filtration and
recrystallization from EtOH/H20 gave yellowish needles.
Yield: 1.28 g (750) . M.p. 245°C. W (MeOH) : 212 (4.30) ;
[243 (4.01)]; 284 (4.02); 349 (4.19).
Ref erences
[1] Y. Kang, R. Soyka, W. Pfleiderer, J Heterocvcl.
Chem. 1987, 24, 597.
[2] R. Eisele, K. Aritomo, W. Pfleiderer, Pteridines
1993, 4, 178.
[3] M. Viscontini, K. Adank, Helv. Chim. Acta 1952, 35,
1342 .
[4] R.E. Bowman, J. Chem. Soc. 1950, 324.
[5] W. Pfleiderer, W. Hutzenlaub, Chem. Ber. 1973, 106,
3149.
[6] -H.D. Dakin, H.W. Dudley, J. Biol. Chem. 1914, 18,
42.
[7] H. Steppan, J. Hammer, R. Baur, R. Gottlieb, W.
Pfleiderer, Liebias Ann. Chem. 1982, 2135.
[8] Z. Kazimierczuk, W. Pfleiferer, Chem. Ber. 1979,
112, 1499.
[9J -F.F. Blicke, H.C. Godt, J. Am. Chem. Soc. 1954, 76,
2798.
[10] H. Fink, W. Pfleiderer,,Chem. Ber. 1963 , 96, 2950.
[11) -J. Litschitz, Ber. Deist. Chem. Ges. 1922, 55, 1619.
[12] W. Hutzenlaub, H. Yamamoto, G.B. Barlin, W.
Pfleiderer, Chem. Ber. 1973, 106, 3203.
CA 02361561 2001-07-31
WO 00/45800 PCT/EP00/00938
17
Materials and methods
Various models may be used for testing an
immunosuppressive effect. In vivo, for example, different
transplantation models are available. They are strongly
influenced by different immunogenicities, depending on
the donor and recipient species used and depending on the
nature of the transplanted organ. The survival time of
transplanted organs can thus be used to measure the
suppression of the immune response. In vitro, there exist
also various models. The most used are lymphocyte
activation tests. Usually activation is measured via
lymphocyte proliferation. Inhibition of proliferation
thus always means immunosuppression under the
experimental conditions applied. There exist different
stimuli for lymphocyte activation:
- coculture of lymphocytes of different species (MLR
- mixed lymphocyte reaction): lymphocytes expressing
different minor and major antigens of the HLA-DR type (_
alloantigens) activate each other non-specifically.
- CD3 assay: here there is an activation of the T-
lymphocytes via an exogenously added antibody (OKT3).
This antibody reacts against the CD3 molecule located on
the lymphocyte membrane. This molecule has a
costimulatory function. The interaction anti-CD3 (_
OKT3)-CD3 results in T-cell activation which proceeds via
the Ca2+/calmodulin/calcineurin system and can be
inhibited by CyA.
- CD28 assay: here specific activation of the T
lymphocyte goes also via an exogenously added antibody
against the CD28 molecule. This molecule is also located
on the lymphocyte membrane, and delivers strong
costimulatory signals. This activation is Ca2+-independent
and thus cannot be inhibited by CyA.
Reagents
All derivatives were dissolved in 0.5 ml DMSO and
further diluted in culture medium before use in in vitro
CA 02361561 2001-07-31
WO 00/45800 PCT/EP00/00938
1~
experiments. The culture medium consisted of RPMI-1640 +
loo FCS.
Mixed Lvmphocvte -Reaction
Peripheral blood mononuclear cells (PBMC) were
isolated from heparinized peripheral blood by density
gradient centrifugation over Lymphoprep (Nycomed,
Maorstua, Norway). Allogeneic PBMC or EBV-transformed
human B cells [RPMI1788 (ATCC name CCL156)] which
strongly express B7-1 and B7-2 were used as stimulator
cells after irradiation with 30 Gy. MLR was performed in
triplicate wells. After 5 days incubation at 37°C, 1 ~Ci
['H]-thymidine was added to each cup. After a further 16
hours incubation, cells were harvested and counted in a
i~-counter.
The percent suppression of proliferation by drugs
was counted using the formula:
Percent inhibition = (cpm+drugs) - cpm Cult. Med
___________________________ x 100
(cpm-drugs) - cpm Cult. Med
_T cell purification
T cells were purified by removing non-T cells.
Briefly, monocytes were removed by cold agglutination.
The resulting lymphoid cells were further purified by a
cell enrichment immunocolumn [Celled Human T (Biotex,
Edmonton, Alberta, Canada)] by a process of negative
selection. More than 95% of the B cells were removed with
this procedure. After depletion, the resulting T cell
preparation was highly purified explaining these cells
could not be activated by PHA or rIL-2 alone at
concentrations capable of stimulating RBMC prior to
deletion.
Measurements of T cell ,p~roliferations induced by anti-CD3
mAb + PMA or anti-CD28 mPb + PMA
CA 02361561 2001-07-31
WO 00/45800 PCT/EP00/00938
19
Highly purified T cells (106/ml) were stimulated
by immobilized anti-CD3 or anti-CD28 mAb in the presence
of PMA. Anti-CD3 mAb (CLB-CD3; CLB, Amsterdam, The
Netherlands) were fixed on the 96-microwell plates by
incubating the wells with 50 ~.1 of mAb solution (1/800
dilution in culture medium). Anti-CD28 mAb (CLB-CD28;
CLB, Amsterdam, The Netherlands) 50 ~.1 (1/650 dilution in
culture medium) was added directly to the wells. Further,
20 ~.1 PMA (Sigma, St. Louis, M0, USA) solution (final
concentration: 0.5 ng/ml) was added. Subsequently, 20 ~.1
of immunosuppressants were added by serial dilution in
triplicate wells. Finally 100 ~1 of the T cell suspension
(106/ml) was added. After 48-hour incubation at 37°C in 5%
C02 20 ~.l BrdU (100 ~.M solution) (Cell Proliferation
Elisa, Boehringer-Mannheim Belgium) was added to each
well. After a further overnight incubation the T cell
proliferation was measured using a colorimetric
immunoassay for qualification of cell proliferation based
on measurements of the incorporation of BrdU during DNA
synthesis. The optical density (OD) was measured by a
Behring EL311 plate reader at 450 nm (reference
wavelength: 690 nm). The percent suppression of
proliferation by drugs was counted using the formula:
Percent inhibition = (OD+drugs) - (OD Cult. Med.)
___________________________- x 100
(OD-drugs) - (OD Cult. Med. )
In vitro immunosu~~ressive effect of Pteridine Derivati-
ves as measured with the MLR and with tests involvincr
golyclonal T cell proliferation induced by anti-CD3 mAb +
PMA or anti-CD28 mAb + PMA (table II)
- In the table II column II shows the IC50 values of
the various substances in the MLR. The IC50 value
represents the lowest concentration of the substances
that resulted in a 50% suppression of the MLR.
- Column III shows the IC50 value of the various
substances for the anti-CD3 mAb + PMA pathway and row IV
the IC50 values of the various substances for the
CA 02361561 2001-07-31
WO 00/45800 PCT/EP00/00938
anti-CD28 mAb + PMA pathway.
- As a comparison the values of other
immunosuppressants: CsA, FK506, Rapamycin, Leflunomide
and Mycophenolic acid methatroxate (MTX) and 5-
5 Fluoro-uracil (5-FU) in table III are given as well.
Whole Blood Assay fWBA)
WBA is a lymphoproliferation assay performed in vitro but
using lymphocytes present in whole blood, taken from
10 animals that were previously given test substances in
vivo. Hence it reflects the in vivo effect of substances
as assessed with an in vitro read-out assay.
Rats: inbred, male 6- to 8-weeks old R/A rats
weighing ~ 200 g were used as recipients.
15 Drug administration: Pteridine derivatives were
dissolved in DMSO and further diluted with PBS. Products
were given orally in different concentrations 2 times a
day for 2 days. To perform the experiments, 6-8 hours
after the last administration 1 ml of blood is taken by
20 heart puncture after ether anesthesia and anticoagulated
with 100 U/ml of preservative free heparine.
Whole Blood Assay: This assay was performed as we
described previously [Use of the Methylxanthine
Derivatives A802715 in Transplantation Immunology. II In
vitro Experiments. (Yuan Lin, et al., Transplantation
1997, 63, No. 12, 1734-1738)].
Heparinized whole blood was diluted (1:25) with
complete RPMI medium and stimulated with 15 ~,g/ml of
concanavalin A (Con A) in triplicate wells in 96-well
microtiter plates at 37 °C and 5% C02. After 96-h culture,
proliferation was determined by measuring the
incorporation (cpm) of [3H]-thymidine.
The Con A induced proliferation of lymphocytes
taken from rats receiving the test substances (exp) was
compared with that from rats receiving only the solvent
(con). The percent suppression was calculated as
follows:
CA 02361561 2001-07-31
WO 00/45800 PCT/EP00/00938
21
Results
No. %suppress AdministrationBlood taken
ion of drugs after:
28 57% 80 n'b/kg/d 8 h
34 63% 2x/d 2d 8 h
40 mg/kg/d
2x/d 2d
First, most of the pteridine classes (I) according to the
invention contain compounds with a clear suppressive
effect in the MLR (mixed lymphocyte reaction). The MLR is
considered as an in vitro analogue of the transplant
rejection as it is based on the recognition of allogeneic
MHC (major histocompatibility antigens) on the stimulator
leucotyes, by responding lymphocytes. Various established
immunosuppressive drugs are known to suppress the MLR,
and were also shown in this description.
From these data it can be deduced that the
pteridine derivatives are effective in clinical
situations where other immunosuppressants are active as
well.
These include the prevention and/or treatment of
organ transplant rejection, the prevention and/or
treatment of both rejection and the occurrence of graft-
versus-host-disease after BM transplantation; the
prevention and/or treatment of autoimmune diseases
including diabetes mellitus, multiple sclerosis,
glomerulonephritis, rheumatoid arthritis, psoriasis
systemic diseases such as vasculitis; scleroderma,
polymyositis, autoimmune endocrine disorders
(thyroiditis), ocular diseases (uveitis), inflammatory
bowel diseases (Crohn~s disease, colitis ulcerosa),
autoimmune liver diseases (autoimmune hepatitis, primary
CA 02361561 2001-07-31
WO 00/45800 PCT/EP00/00938
22
biliary cirrhosis) autoimmune pneumonitis and auto-immune
carditis.
Whereas cyclosporine A and FK506 are only active
in the anti-CD3 + PMA test, the pteridine derivatives
according to the invention were active, not only in the
anti-CD3 + PMA but also in the anti-CD28 + PMA test. It
has been shown that the latter is Ca-calmodulin
resistant, and resistant to CsA and FK506. The anti-CD28
+ PMA pathway has also been called the cosignal pathway
and is important to induce energy and even tolerance in T
cells. Moreover, representative compounds have been found
to be active in an whole blood assay.
Under the term "organ" in the description is
understood all organs or parts of organs (even several)
in mammals, in particular humans, for example kidney,
heart, skin, liver, muscle, cornea, bone, bone marrow,
lung, pancreas, intestine or stomach.
After organ transplantation, rejection of the
transplanted organ by the recipient occurs (host-
versus-graft reaction). After bone marrow
transplantation, also rejection of the host by the
grafted cell may occur (graft-versus-host reaction).
Rejection reactions mean all reactions of the recipient
body or of the transplanted organ which in the end lead
to cell or tissue death in the transplanted organ or
adversely affect the functional ability and viability of
the transplanted organ or adversely affect the functional
ability and viability of the transplanted organ or the
recipient. In particular, this means acute and chronic
rejection reactions.
Auto-immune disorders include, inter alia,
systemic lupus erythematosus, rheumatoid arthritis,
psoriasis, pemphigus, atopic dermatitis, myositis,
multiple sclerosis, nephrotic syndrome (in particular
glomerulonephritis), ulcerative colitis or juvenile
diabetes.
An additive or synergetic effect of pteridine
derivatives and other immunosuppressants may be
CA 02361561 2001-07-31
WO 00/45800 PCT/EP00/00938
23
anticipated. This may be especially, although not
exclusively the case for combinations with CyA or FK 506
as the latter are not suppressive in the aCD28 pathway of
T cell activation (table III) whereas most pteridine
derivatives are.
The invention further relates to the use of
cyclosporin A or FK506 or Rapamycine and at least one
pteridine derivative according to the invention for the
production of a pharmaceutical composition for inhibiting
the replication of viruses such as picorna-, toga-,
bunya-, orthomyxo-, paramyxo-, rhabdo-, retro-, arena-,
hepatitis B-, hepatitis C-, hepatitis D-, adeno-,
vaccinia-, papilloma-, herpes-, varicella-zoster-virus or
human immunodeficiency virus (HIV); or for treating of
cancer such as lung cancers, leukaemia, ovarian cancers,
sarcoma, Kaposi's sarcoma, meningioma, colon cancers,
lymp node tumors, glioblastoma multiforme, prostate
cancers or skin carcinoses.
The invention further relates to the use of
cyclosporin A or FK506 or rapamycin and at least one
pteridine derivative of the general formula (I) for the
production of a pharmaceutical composition for the
treatment of human after organ transplantation or of
(auto)immune disorders.
Hence, the advantage to associate pteridine with
other immunosuppressants may be that, first, the
therapeutic spectrum of action of the individual
components is quantitatively and qualitatively broadened.
Secondly that it allows, by means of a dose reduction
without reduced efficacy but with increased safety, that
the treatment of immune disorders which were hitherto no
indication for immunosuppressive therapy as a result of
side effects may be considered. At the same time, the
therapy costs can be decreased to an appreciable extent.
As a comparison, known pteridine derivatives are
submitted to the same test conditions as the pteridine
derivatives of the invention. These compounds and the
results thereof are given in table IV and show no
CA 02361561 2001-07-31
WO 00/45800 PCT/EP00/00938
24
particular ittununosuppressive activity.
In all tables
0: concentration > 151 ~.M
+: concentration range 16-150 ~.M
++: concentration range 1-15 ~.M
+++: concentration range < 1 ~.M
The skilled person will appreciate that the
preparation according to the invention may contain the
pteridine compounds over a broad content range depending
on the contemplated use of the preparation. Generally,
the content of the preparation is within the range of
0.01-50 wt.o, preferably within the range of 0.01-10
wt.%, more preferably within the range of 0.1-10 wt. o,
and most preferably within the range of 0.1-5 wt. o.
Accordingly, the preparation may be used in a dosing
regime which is suitable for most contemplated
pharmaceutical utilities.
The preparation according to the invention may be
used as such or in combination with any acceptable
carrier material, excipient or diluent.
The preparation according to the invention may be
administared orally or in any other suitable fashion.
Oral administration is preferred and the preparation may
have the form of a tablet, aqueous dispersion,
dispersable powder or granule, emulsion, hard or soft
capsule, syrup-, elixir or gel. The dosing fortes may be
prepared using any method known in the art for
manufacturing these pharmaceutical compositions and may
comprise as additives sweeteners, flavoring agents,
coloring agents, preservatives and the like. Carrier
materials and excipients may include calcium carbonate,
sodium carbonate, lactose, calcium phosphate or sodium
phosphate; granulating and disintegrating agents, binding
agents and the like. The preparation may be included in a
gelatin capsule mixed with any inert solid diluent or
carrier material, or has the form of a soft gelatin
capsule, in which the ingredient is mixed with a water or
CA 02361561 2001-07-31
WO 00/45800 PCT/EP00/00938
oil medium. Aqueous dispersions may comprise the
preparation in combination with a suspending agent,
dispersing agent or wetting agent. Oil dispersions may
comprise suspending agents such as a vegetable oil.