Note: Descriptions are shown in the official language in which they were submitted.
CA 02362380 2001-08-07
WO 00/56706 PCT/US00/07262
1
TITLE
AMINO-THIO-ACRYLONITRILES AS MEK INHIBITORS
FIELD OF THE INVENTION
This invention relates generally to amino-thio-
acrylonitriles as MEK inhibitors, pharmaceutical
compositions containing the same, and methods of using the
same as for treatment and prevention of inflammatory
disorders, cancer or other proliferative diseases or as a
radiosensitizing agents against cancer or other
proliferative disorders.
BACKGROUND OF THE INVENTION
The mitogen activated protein kinase (MAPK) signaling
pathways are involved in cellular events such as growth,
differentiation and stress responses (J. Biol. Chem. (1993)
268, 14553-14556). Four parallel pathways have been
identified to date ERK1/ERK2, JNK, p38 and ERK5. These
pathways are linear kinase cascades in that MAPKKK
phosphorylates and activates MAPKK that phosphorylates and
activates MAPK. To date, there are 7 MAPKK homologs (MEK1,
MEK2, MKK3, MKK4/SEK, MEK5, MKK6, and MKK7) and 4 MAPK
families (ERK1/2, JNK, p38, and ERK5). The MAPKK family
members are unique in that they are dual-specific kinases,
phosphorylating MAPKs on threonine and tyrosine. Activation
of these pathways regulates the activity of a number of
substrates through phosphorylation. These substrates
include transcription factors such as TCF, c-myc, ATF2 and
the AP-1 components, fos and Jun; the cell surface
components EGF-R; cytosolic components including PHAS-I,
p90rsk, cpLA2 and c-Raf-1; and the cytoskeleton components
such as tau and MAP2.
The prototypical mitogen activated protein kinase
cascade is reflected by the ERK pathway (Biochem J. (1995)
309, 361-375). The ERK pathway is activated primarily in
response to ligation of receptor tyrosine kinases (RTKs)
(FEBS Lett. (1993) 334, 189-192). Signal propagation from
CA 02362380 2001-08-07
WO 00/56706 PCT/US00/07262
2
the RTKs occurs down the Ras pathway through sequential
phosphorylation of Raf, MEK and ERK. This pathway has not
been typically viewed of as an important contributor to the
inflammatory response, but rather involved in growth and
differentiation processes. This view stems from the profile
of typical activators of this pathway, which include growth
factors (PDGF, NGF, EGF), mitogens (phorbol esters), and
polypeptide hormones (insulin, IGF-1). Evidence for ERK
pathway involvement in inflammatory and immune responses
has, however, gained some support in recent years (Proc.
Natl. Acad. Sci. USA. (1995) 92, 1614-1618; J. Immunol.
(1995) 155, 1525-1533; and J. Biol. Chem. (1995) 270, 27391-
27394). Cytokines such as TNFa and IL-1b, the bacterial
cell wall mitogen, LPS, and chemotactic factors such as
fMLP, CSa, and IL-8 all activate the ERK pathway. In
addition, the ERK pathway is activated as a result of T cell
receptor ligation with antigen or agents such as
PMA/ionomycin or anti-CD3 antibody, which mimic TCR ligation
in T cells (Pros. Natl. Acad. Sci. USA (1995) 92, 7686-
7689). These findings indicate that inhibitors of the ERK
pathway should function as anti-inflammatory and immune
suppressive agents.
Small molecule inhibitors of the Raf/MEK/ERK pathway
have been identified. A series of benzoquinones has been
disclosed by Parke-Davis, which is exemplified by PD 098059
that inhibits MEK activity (J. Biol. Chem. (1995) 46, 27498-
27494). Recently, we identified a MEK inhibitor, U0126 (J.
Biol. Chem. (1998) 29, 18623-18632). Comparative kinetic
analysis showed that U0126 and PD 098059 were non-
competitive inhibitors of activated MEK (J. Biol. Chem.
(1998) 29, 18623-18632). These MEK inhibitors have been
used to investigate the role of the ERK activation cascade
in a wide variety of systems including inflammation, immune
suppression and cancer. For example, PD 098059 blocks
thymidine incorporation into DNA in PDGF-stimulated Swiss
3T3 cells (J. Biol. Chem. (1995) 46, 27498-27494). PD
098059 also prevents PDGF-BB-dependent SMC (Smooth Muscle
Cell) chemotaxis at concentrations which inhibit ERK
CA 02362380 2001-08-07
WO 00/56706 PCT/US00/07262
3
activation (Hypertension (1997) 29, 334-339). Similarly,
U0126 prevents PDGF-dependent growth of serum starved SMC.
We have also shown that U0126 blocks keratinocyte
proliferation in response to a pituitary growth factor
extract, which consists primarily of FGF. These data
coupled with those obtained with PD 098059 above indicate
that MEK activity is essential for growth factor-stimulated
proliferation.
The role of the MEK/ERK pathway in inflammation and
immune suppression has been examined in a number of systems,
including models of T cell activation. The T cell antigen
receptor (TCR) is a non-RTK receptor whose intracellular
signaling pathways have been elucidated (Proc. Natl. Acad.
Sci. USA (1995) 92, 7686-7689). DeSilva et al. have
generated a great deal of information with U0126 in T cell
systems (J. Immunol. (1998) 160, 4175-4181). Their data
showed that U0126 prevents ERK activation in T cells in
response to PMA/ionomycin, Con A stimulation, and antigen in
the presence of costimulation. In addition, T cell
activation and proliferation in response TCR engagement is
blocked by U0126 as is IL-2 synthesis. These results
indicate that MEK inhibition does not result in a general
antiproliferative effect in this IL-2-driven system, but
selectively blocks components of the signaling cascades
initiated by T cell receptor engagement.
PD 098059 has also been shown to inhibit T cell
proliferation in response to anti-CD3 antibody, which is
reversed by IL-2 (J. Immunol. (1998) 160, 2579-2589.). PD
098059 also blocked IL-2 production by T cells stimulated
with anti-CD3 antibody in combination with either anti-CD28
or PMA. In addition, the MEK inhibitor blocked TNFa, IL-3
GM-CSF, IFN-g, IL-6 and IL-10 production. In contrast, PD
098059 enhanced production of IL-4, IL-5 and IL-13 in
similarly stimulated T cell cultures. These differential T
cells effects with MEK inhibition suggest that therapeutic
manipulations may be possible.
Neutrophils show ERK activation in response to the
agonists N-formyl peptide (fMLP), IL-8, C5a and LTB9, which
CA 02362380 2001-08-07
WO 00/56706 PCT/US00/07262
4
is blocked by PD 098059 (Biochem. Biophy. Res. Commun.
(1997) 232, 474-477). Additionally, PD 098059 blocks
neutrophil chemotaxis in response to all agents, but does
not alter superoxide anion production. However, fMLP-
stimulated superoxide generation was inhibited by PD098059
in HL-60 cells (J. Immunol. (1997) 159, 5070-5078),
suggesting that this effect may be cell-type specific.
U0126 blocks ERK activation in fMLP- and LTB9-stimulated
neutrophils, but does not impair NADPH-oxidase activity or
bacterial cell killing. U0126 at 10 mM blunts up regulation
of b2 integrin on the cell surface by 50% and blocks
chemotaxis through a fibrin gel >80% in response to IL-8 and
LTB4. Thus, neutrophil mobility is affected by MEK
inhibition although the acute functional responses of the
cell remain intact.
Eicosanoids are key mediators of the inflammatory
response. The proximal event leading to prostaglandin and
leukotriene biosynthesis is arachidonic acid release from
membrane stores, which is mediated largely through the
action of cytosolic phospholipase A2 (cPLA2). Activation of
cPLA2 requires Ca2' along with phosphorylation on a consensus
MAP kinase site, Ser505, which increases catalytic
efficiency of the enzyme (J. Biol. Chem. (1997) 272, 16709-
16712). In neutrophils, mast cells, or endothelial cells,
PD 098059 blocks arachidonic acid release in response to
opsonized zymosan, aggregation of the high affinity IgG
receptor, or thrombin, respectively. Such data support a
role for ERK as the mediator of cPLA2 activation through
phosphorylation (FEBS Lett. (1996) 388, 180-184. Biochem J.
(1997) 326, 867-876 and J. Biol. Chem. (1997) 272, 13397-
13402). Similarly, U0126 is able to block arachidonic acid
release along with prostaglandin and leukotriene synthesis
in keratinocytes stimulated with a variety of agents. Thus,
the effector target, cPLA2, is sensitive to MEK inhibition
in a variety of cell types.
MEK inhibitors also seem to affect eicosanoid
production through means other than inhibition of
CA 02362380 2001-08-07
WO 00/56706 PCT/US00/07262
arachidonic acid release. PD 098059 partially blocked LPS-
induced Cox-2 expression in RAW 264.7 cells, indicating ERK
activation alone may not be sufficient to induce expression
of this key enzyme mediating inflammatory prostanoid
5 production (Biochem J. (1998) 330, 1107-1114). Similarly,
U0126 inhibits Cox-2 induction in TPA-stimulated
fibroblasts, although it does not impede serum induction of
the Cox-2 transcript. PD 098059 also inhibits Cox-2
induction in lysophosphatidic acid (LPA)-stimulated rat
mesangial cells, which further supports a role for ERK
activation in production of prostaglandins (Biochem J.
(1998) 330, 1107-1114). Finally, 5-lipoxygenase
translocation from the cytosol to the nuclear membrane along
with its activation as measured by 5-HETE production can be
inhibited by PD 098059 in HL-60 cells (Arch. Biochem.
Biophys. (1996) 331, 141-144).
Inflammatory cytokines such as TNFa and IL-1b are
critical components of the inflammatory response. Cytokine
production in response to cell activation by various stimuli
as well as their activation of downstream signaling cascades
represent novel targets for therapeutics. Although the
primary effect of IL-1b and TNF-a is to up regulate the
stress pathways (Nature (1994) 372, 729-746), published
reports (Proc. Natl. Acad. Sci. USA (1995) 92, 1614-1618.
J. Immunol. (1995) 155, 1525-1533. J. Biol. Chem. (1995)
270, 27391-27394. Eur. J.). Cytokines such as TNFa and IL-
1b, the bacterial cell wall mitogen, LPS, and chemotactic
factors such as fMLP, CSa, and IL-8 all activate the ERK
pathway. In addition, the ERK pathway is activated as a
result of T cell receptor ligation with antigen or agents
such as PMA/ionomycin or anti-CD3 antibody, which mimic TCR
ligation in T cells (Proc. Natl. Acad. Sci. USA (1995) 92,
7686-7689) and clearly show that the ERK pathway is also
affected. U0126 can block MMP induction by IL-1b and TNF-a
in fibroblasts (J. Biol. Chem. (1998) 29, 18623-18632),
demonstrating that ERK activation is necessary for this
proinflammatory function. Similarly, lipopolysaccharide
(LPS) treatment of monocytes results in cytokine production
CA 02362380 2001-08-07
WO 00/56706 6 PCT/US00/07262
that has been shown to be MAP kinase-dependent being blocked
by PD 098059 (J. Immunnol. (1998) 160, 920-928). Indeed, we
have observed similar results in freshly isolated human
monocytes and THP-1 cells where LPS-induced cytokine
production is inhabitable by U0126 (J. Immunol. (1998)
161:5681-5686).
The proximal involvement of RAS in the activation of
the ERK pathway suggests that MEK inhibition might show
efficacy in models where oncogenic RAS is a determinant in
the cancer phenotype. Indeed, PD 098059 (J. Biol. Chem.
(1995) 46, 27498-27494) as well as U0126 are able to impede
the growth of RAS-transformed cells in soft agar even though
these compounds show minimal effects on cell growth under
normal culture conditions. We have further examined the
effects of U0126 on the growth of human tumor cell lines in
soft agar. We have shown that U0126 can prevent cell growth
in some cells, but not all, suggesting that a MEK inhibitor
may be effective in only certain kinds of cancer. In
addition, PD 098059 has been shown to reduce urokinase
secretion controlled by growth factors such as EGF, TGFa and
FGF in an autocrine fashion in the squamous cell carcinoma
cell lines UM-SCC-1 and MDA-TV-138 (Cancer Res. (1996) 56,
5369-5374). In vitro invasiveness of UM-SCC-1 cells through
an extracellular matrix-coated porous filter was blocked by
PD 098059 although cellular proliferation rate was not
affected. These results indicate that control of the tumor
invasive phenotype by MEK inhibition may also be a
possibility. The observed effects with PD 098059 and U0126
suggest that MEK inhibition may have potential for efficacy
in a number of disease states. Our own data argue strongly
for the use of MEK inhibitors in T-cell mediated diseases
where immune suppression would be of value. Prevention of
organ transplant rejection, graft versus host disease, lupus
erythematosus, multiple sclerosis, and rheumatoid arthritis
are potential disease targets. Effects in acute and chronic
inflammatory conditions are supported by the results in
neutrophils and macrophage systems where MEK inhibition
blocks cell migration and liberation of proinflammatory
CA 02362380 2001-08-07
WO 00/56706 PCT/US00/07262
7
cytokines. A use in conditions where neutrophil influx
drives tissue destruction such as reperfusion injury in
myocardial infarction and stroke as well as inflammatory
arthritis may be warranted. Blunting of SMC migration and
inhibition of DNA replication would suggest atherosclerosis
along with restenosis following angioplasty as disease
indications for MEK inhibitors. Skin disease such as
psoriasis provides another potential area where MEK
inhibitors may prove useful since MEK inhibition prevents
skin edema in mice in response to TPA. MEK inhibition also
blocks keratinocyte responses to growth factor cocktails,
which are known mediators in the psoriatic process.
Finally, the use of a MEK inhibitor in cancer can not
be overlooked. Ionizing radiation initiates a process of
apoptosis or cell death that is useful in the treatment
solid tumors. This process involves a balance between pro-
apoptotic and anti-apoptotic signal (Science 239, 645-647),
which include activation of MAP kinase cascades. Activation
of the SAPK pathway delivers a pro-apoptotic signal
(Radiotherapy and Oncology (1998) 47, 225-232.), whereas
activation of the MAPK pathway is anti-apoptotic (Nature
(1996) 328, 813-816.). Interference with the anti-apoptotic
MAPK pathway by dominant negative MEK2 or through direct
inhibition of MEK with synthetic inhibitors sensitizes cells
to radiation-induced cell death (J. Biol. Chem. (1999) 274,
2732-2742; and Oncogene (1998) 16, 2787-2796).
W098/37881 describe MEK inhibitors useful for treating
or preventing septic shock. The inhibitors include 2-(2-
amino-3-methoxyphenyl)-4-oxo-4H-[1]benzopyran and a compound
of the formula:
Z
R5
R1~_ ~~~\R
R3
Br or I
The above diphenyl amines are not considered to be part of
the presently claimed invention.
CA 02362380 2001-08-07
WO 00/56706 ~ PCT/US00/07262
Therefore, efficacious and specific MEK inhibitors are
needed as potentially valuable therapeutic agents for the
treatment of inflammatory disorders, cancer or other
proliferative diseases or as a radiosensitizing agents
against cancer or other proliferative disorders. It is thus
desirable to discover new MEK inhibitors.
SUMMARY OF THE INVENTION
Accordingly, one object of the present invention is to
provide novel amino-thio-acrylonitriles which are useful as
MEK inhibitors or pharmaceutically acceptable salts or
prodrugs thereof.
It is another object of the present invention to
provide pharmaceutical compositions comprising a
pharmaceutically acceptable carrier and a therapeutically
effective amount of at least one of the compounds of the
present invention or a pharmaceutically acceptable salt or
prodrug form thereof.
It is another object of the present invention to
provide a method for treating a disorder involving MEK,
comprising: administering to a host in need of such
treatment a therapeutically effective amount of at least one
of the compounds of the present invention or a
pharmaceutically acceptable salt or prodrug form thereof.
It is another object of the present invention to
provide a novel method of using the compounds of the present
invention as a radiosensitizing agent for the treatment of
cancers or proliferative diseases, comprising:
administering to a host in need of such treatment a
therapeutically effective amount of a compound of the
present invention, or a pharmaceutically acceptable prodrug
or salt form thereof.
It is another object of the present invention to
provide a novel method of treating a condition or disease
wherein the disease or condition is referred to as
rheumatoid arthritis, osteoarthritis, periodontitis,
gingivitis, corneal ulceration, solid tumor growth and tumor
invasion by secondary metastases, neovascular glaucoma,
CA 02362380 2001-08-07
WO 00/56706 PCT/US00/07262
9
multiple sclerosis, or psoriasis in a mammal, comprising:
administering to the mammal in need of such treatment a
therapeutically effective amount of a compound of formula
(I) or a pharmaceutically acceptable salt form thereof.
It is another object of the present invention to
provide a novel method of treating a condition or disease
wherein the disease or condition is referred to as fever,
cardiovascular effects, hemorrhage, coagulation, cachexia,
anorexia, alcoholism, acute phase response, acute infection,
shock, graft versus host reaction, autoimmune disease or HIV
infection in a mammal comprising administering to the mammal
in need of such treatment a therapeutically effective amount
of a compound of formula (I) or a pharmaceutically
acceptable salt form thereof.
It is another object of the present invention to
provide novel amino-thio-acrylonitriles or salts or prodrugs
thereof for use in therapy.
It is another object of the present invention to
provide the use of novel amino-thio-acrylonitriles or salts
or prodrugs thereof for the manufacture of a medicament for
the treatment of an inflammatory disease.
It is another object of the present invention to
provide the use of novel amino-thio-acrylonitriles or salts
or prodrugs thereof for the manufacture of a medicament for
the treatment of cancer.
These and other objects, which will become apparent
during the following detailed description, have been
achieved by the inventors' discovery that compounds of
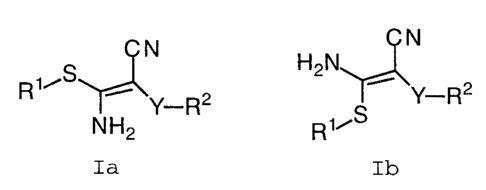
formula Ia or Ib:
N N
S H2N
3 0 NH2 R1~S
Ia Ib
or pharmaceutically acceptable salt or prodrug forms
thereof, wherein R1 and RZ are defined below, are effective
MEK inhibitors.
CA 02362380 2001-08-07
WO 00/56706 PCT/US00/07262
DETAILED DESCRIPTION OF PREFERRED EMBODIMENTS
Thus, in a first embodiment, the present invention
provides a novel compound of formula Ia or Ib:
N N
S H2N
R1~ ~ Y-R2 Y-R2
5 NH2 R1~S
Ia Ib
or stereoisomer or pharmaceutically acceptable salt form
thereof, wherein;
10 R1 is phenyl, naphthyl, 2,3-dihydroindol-5-yl or a 5-6
membered heteroaryl ring with 1-4 heteroatoms selected
from N, NH, O, and S, and R1 is substituted with 0-2
Ra;
Ra is selected from H, C1, F, Br, I, C1-g alkyl, C1-4
alkoxy, OH, CH20H, NH2, (C1-3 alkyl)NH, (C1-3 alkyl)2N,
(H2NCH2C(O))NH, (H2NCH(CH3)C(O))NH, (CH3NHCH2C(O))NH,
((CH3)2NCH2C(0))NH, CF3, OCF3, -CN, N02, C(O)NH2, and
CH3C(O)NH;
Y is selected from phenyl substituted with 0-5 Rb, naphthyl
substituted with 0-5 Rb, and CHR3;
Rb is selected from H, Cl, F, Br, I, C1-4 alkyl, OH, C1-4
alkoxy, CH20H, CH(OH)CH3, CF3, OCF3, -CN, N02, NH2,
(C1-3 alkyl)NH, (C1-3 alkyl)2N, and C(O)O-C1-4 alkoxy;
R2 is selected from H, R2a, C(O)R2a, CH(OH)R2a, CH2R2a,
OR2a, SR2a, and NHR2a;
CA 02362380 2001-08-07
WO 00/56706 11 PCT/US00/07262
R2a is selected from phenyl, naphthyl, and a 5-6 membered
heteroaryl ring with 1-4 heteroatoms selected from N,
NH, O, and S, and R2a is substituted with 0-5 Rb;
R3 is phenyl substituted with 0-2 RC or naphthyl substituted
with 0-2 Rc; and,
Rc is selected from H, C1, F, Br, I, C1-4 alkyl, OH, C1-4
alkoxy, CH20H, CH(OH)CH3, CF3, OCF3, -CN, N02, NH2,
(C1-3 alkyl)NH, (C1-3 alkyl)2N, and C(O)O-C1-4 alkoxy.
In a preferred embodiment, the present invention
provides a novel compound, wherein:
R1 is phenyl or a 5-6 membered heteroaryl ring with 1-2
heteroatoms selected from N, NH, O, and S, and R1 is
substituted with 0-2 Ra;
Ra is selected from H, C1, F, C1-g alkyl, C1-g alkoxy, OH,
CH20H, NH2, (C1-3 alkyl)NH, (C1-3 alkyl)2N,
(H2NCH2C(O))NH, (H2NCH(CH3)C(O))NH, (CH3NHCH2C(O))NH,
((CH3)2NCH2C(O))NH, and CH3C(O)NH;
Y is selected from phenyl substituted with 0-5 Rb, naphthyl
substituted with 0-5 Rb, and CHR3;
Rb is selected from H, Cl, F, Br, C1-4 alkyl, OH, C1-4
alkoxy, CH20H, CH(OH)CH3, CF3, -CN, N02, NH2, and (C1-3
alkyl)NH, (C1-3 alkyl)2N;
R2 is selected from H, R2a, C(O)R2a, CH(OH)R2a, CH2R2a, and
OR2a;
CA 02362380 2001-08-07
WO 00/56706 PCT/US00/07262
12
R2a is selected from phenyl, naphthyl, and a 5-6 membered
heteroaryl ring with 1-4 heteroatoms selected from N,
NH, O, and S, and R2a is substituted with 0-5 Rb;
R3 is phenyl substituted with 0-2 RC or naphthyl substituted
with 0-2 Rc; and,
Rc is selected from H, Cl, F, Br, I, C1_4 alkyl, OH, C1-4
alkoxy, CH20H, CH(OH)CH3, CF3, -CN, N02, NH2, (C1-3
alkyl)NH, and (C1-3 alkyl)2N.
In a more preferred embodiment, the present invention
provides a novel compound, wherein:
R1 is phenyl or a 5-6 membered heteroaryl ring with 1-2
heteroatoms selected from N, NH, 0, and S, and R1 is
substituted with 0-2 Ra;
Ra is selected from H, OH, and NH2;
Y is selected from phenyl substituted with 0-2 Rb, naphthyl
substituted with 0-2 Rb, and CHR3;
Rb is selected from H, Cl, F, Br, C1-g alkyl, OH, C1-4
alkoxy, CH20H, CH(OH)CH3, CF3, -CN, N02, NH2, and (C1-3
alkyl)NH, (C1-3 alkyl)2N;
R2 is selected from H, R2a, C(O)R2a, CH(OH)R2a, CH2R2a, and
OR2a;
R2a is selected from phenyl, naphthyl, and a 5-6 membered
heteroaryl ring with 1-4 heteroatoms selected from N,
NH, O, and S, and R2a is substituted with 0-5 Rb;
CA 02362380 2001-08-07
WO 00/56706 PCT/US00/07262
13
R3 is phenyl substituted with 0-2 Rc or naphthyl substituted
with 0-2 Rc; and,
RC is selected from H, Cl, F, Br, I, C1-4 alkyl, OH, C1-4
alkoxy, CH20H, CH(OH)CH3, CF3, -CN, N02, NH2, (C1-3
alkyl)NH, and (C1-3 alkyl)2N.
In an even more preferred embodiment, the present
invention provides a novel compound selected from:
E- and Z-oc-[amino[(2-aminophenyl)thio]methylene]-4-chloro-2-
methyl-(3-phenylbenzenepropanenitrile;
E- and Z-oc-[amino[(2-aminophenyl)thio]methylene]-3-[(2,4
dinitrophenyl)hydroxymethyl]benzeneacetonitrile;
E- and Z-oc-[amino[(2-aminophenyl)thio]methylene]-3-[(4-
carbomethoxyphenyl)hydroxymethyl]benzeneacetonitrile;
E- and Z-oc-[amino[(2-aminophenyl)thio]methylene]-3-[(4
nitrophenyl)hydroxymethyl]benzeneacetonitrile;
E- and Z-oc-[amino[(2-aminophenyl)thio]methylene]-3-
[(pentafluorophenyl)hydroxymethyl]benzeneacetonitrile;
E- and Z-oc-[amino[(2-aminophenyl)thio]methylene]-2-methyl-3-
[(4-pyridyl)hydroxymethyl]benzeneacetonitrile;
E- and Z-oc-[amino[(2-aminophenyl)thio]methylene]-2-methyl-3-
[(phenyl)hydroxymethyl]benzeneacetonitrile;
E- and Z-oc-[amino[(2-aminophenyl)thio]methylene]-2-methyl-3-
[(4-pyridyl)hydroxymethyl]benzeneacetonitrile;
E- and Z-oc-[amino[(2-hydroxyphenyl)thio]methylene]-3-[(4
cyanophenyl)hydroxymethyl]benzeneacetonitrile;
E- and Z-oc-[amino[(2-aminophenyl)thio]methylene]-3-[(3-
nitrophenyl)hydroxymethyl]benzeneacetonitrile;
E- and Z-oc-[amino[(2-aminophenyl)thio]methylene]-2-methyl-3-
[(pentafluorophenyl)hydroxymethyl]benzeneacetonitrile;
CA 02362380 2001-08-07
WO 00/56706 PCT/US00/07262
14
E- and Z-oc-[amino[(2-hydroxyphenyl)thio]methylene]-3-[(4-
pyridyl)hydroxymethyl]benzeneacetonitrile;
E- and Z-oc-[amino[(2-aminophenyl)thio]methylene]-3-[(2-
trifluoromethylphenyl)hydroxymethyl]benzeneacetonitrile
E- and Z-oc-[amino[(4-aminophenyl)thio]methylene]-3-[(4-
pyridyl)hydroxymethyl]benzeneacetonitrile;
E- and Z-a,-[amino[(4-hydroxyphenyl)thio]methylene]-3-[(4-
pyridyl)hydroxymethyl]benzeneacetonitrile;
E- and Z-oc-[amino[(2-aminophenyl)thio]methylene]-3-[(3-
cyanophenyl)hydroxymethyl]benzeneacetonitrile;
E- and Z-a-[amino[(4-aminophenyl)thio]methylene]-3-[(4
cyanophenyl)hydroxymethyl]benzeneacetonitrile;
E- and Z-oc-[amino(phenylthio)methylene]-3-[(4-
cyanophenyl)hydroxymethyl]benzeneacetonitrile;
E- and Z-oc-[amino(phenylthio)methylene]-3-[(4-
pyridyl)hydroxymethyl]benzeneacetonitrile;
E- and Z-oc-[amino[(4-aminophenyl)thio]methylene]-2-
bromobenzeneacetonitrile;
E- and Z-oc-[amino[(2-aminophenyl)thio]methylene]-3-[(2,4-
dimethylphenyl)hydroxymethyl]benzeneacetonitrile;
E- and Z-a-[amino[(2-aminophenyl)thio]methylene]-3-
[(phenyl)hydroxymethyl]benzeneacetonitrile;
E- and Z-oc-[amino[(2-aminophenyl)thio]methylene]-3-[(2
thienyl)hydroxymethyl]benzeneacetonitrile;
E- and Z-oc-[amino[(2-aminophenyl)thio]methylene]-4-chloro-(3-
phenylbenzenepropanenitrile;
E- and Z-oc-[amino[(2-thienyl)thio]methylene]-3-
[(phenyl)hydroxymethyl]benzeneacetonitrile;
E- and Z-oc-[amino[(2,4-diaminophenyl)thio]methylene]-1-
naphthyleneacetonitrile;
E- and Z-oc-[amino[(2-aminophenyl)thio]methylene]-2-methyl-(3-
(4-pyridyl)benzenepropanenitrile;
E- and Z-a,-[amino[(4-aminophenyl)thio]methylene]-3-
(benzyl)benzeneacetonitrile;
CA 02362380 2001-08-07
WO 00/56706 PCT/US00/07262
E- and Z-a-[amino[(2-naphthyl)thio]methylene]-1-
naphthyleneacetonitrile;
E- and Z-oc-[amino[(2-aminophenyl)thio]methylene]-3-
5 (benzoyl)benzeneacetonitrile;
E- and Z-oc-[amino[(2-aminophenyl)thio]methylene]-~3-(1-
methyl-2-pyrrolyl)benzenepropanenitrile;
10 E- and Z-oc-[amino[(2-aminophenyl)thio]methylene]-3-
phenoxybenzeneacetonitrile;
E- and Z-oc-[amino[(2-aminophenyl)thio]methylene]-2-
bromobenzeneacetonitrile;
E- and Z-oc-[amino[(2-aminophenyl)thio]methylene]-3-[(2-
furanyl)hydroxymethyl]benzeneacetonitrile;
E- and Z-a-[amino[(2-thienyl)thio]methylene]-3-[(2,3,4,5,6-
pentafluorophenyl)hydroxymethyl]benzeneacetonitrile;
E- and Z-oc-[amino[(2-aminophenyl)thio]methylene]-3-[(3-
methyl-2-pyridyl)hydroxymethyl]benzeneacetonitrile;
E- and Z-oc-[amino[(4-aminophenyl)thio]methylene]-2-
methylbenzeneacetonitrile;
E- and Z-oc-[amino[(4-aminophenyl)thio]methylene]-4-(1,1-
dimethylethyl)benzeneacetonitrile;
E- and Z-oc-[amino[(4-aminophenyl)thio]methylene]-1-
naphthyleneacetonitrile;
E- and Z-oc-[amino[(2-aminophenyl)thio]methylene]-3-
(trifluoromethyl)benzeneacetonitrile;
E- and Z-oc-[amino[(2-aminophenyl)thio]methylene]-1-
naphthyleneacetonitrile;
E- and Z-oc-[amino[(2-aminophenyl)thio]methylene]-2
(trifluoromethyl)benzeneacetonitrile;
E- and Z-oc-[amino[(4-aminophenyl)thio]methylene]-4-
methylbenzeneacetonitrile;
E- and Z-oc-[amino[(2-aminophenyl)thio]methylene]-2-
methylbenzeneacetonitrile;
E- and Z-oc-[amino[(2-fluorophenyl)thio]methylene]-1-
naphthyleneacetonitrile; and,
CA 02362380 2001-08-07
WO 00/56706 PCT/US00/07262
16
E- and Z-oc-[amino[(2-aminophenyl)thio]methylene]-3-phenyl
benzeneacetonitrile;
or a pharmaceutically acceptable salt form thereof.
In a further preferred embodiment, the present
invention provides a novel compound selected from:
E-oc-[amino[(2-aminophenyl)thio]methylene]-4-chloro-2-methyl-
(3-phenylbenzenepropanenitrile;
E-oc-[amino[(2-aminophenyl)thio]methylene]-3-[(2,4-
dinitrophenyl)hydroxymethyl]benzeneacetonitrile;
E-oc-[amino[(2-aminophenyl)thio]methylene]-3-[(4-
carbomethoxyphenyl)hydroxymethyl]benzeneacetonitrile;
E-oc-[amino[(2-aminophenyl)thio]methylene]-3-[(4-
nitrophenyl)hydroxymethyl]benzeneacetonitrile;
E-oc-[amino[(2-aminophenyl)thio]methylene]-3-
[(pentafluorophenyl)hydroxymethyl]benzeneacetonitrile;
E-oc-[amino[(2-aminophenyl)thio]methylene]-2-methyl-3-[(4
pyridyl)hydroxymethyl]benzeneacetonitrile;
E-oc-[amino[(2-aminophenyl)thio]methylene]-2-methyl-3
[(phenyl)hydroxymethyl]benzeneacetonitrile;
E-oc-[amino[(2-aminophenyl)thio]methylene]-2-methyl-3-[(4-
pyridyl)hydroxymethyl]benzeneacetonitrile;
E-oc-[amino[(2-hydroxyphenyl)thio]methylene]-3-[(4-
cyanophenyl)hydroxymethyl]benzeneacetonitrile;
E-oc-[amino[(2-aminophenyl)thio]methylene]-3-[(3-
nitrophenyl)hydroxymethyl]benzeneacetonitrile;
E-oc-[amino[(2-aminophenyl)thio]methylene]-2-methyl-3-
[(pentafluorophenyl)hydroxymethyl]benzeneacetonitrile;
E-oC-[amino[(2-hydroxyphenyl)thio]methylene]-3-[(4-
pyridyl)hydroxymethyl]benzeneacetonitrile;
E-oc-[amino[(2-aminophenyl)thio]methylene]-3-[(2-
trifluoromethylphenyl)hydroxymethyl]benzeneacetonitrile
CA 02362380 2001-08-07
WO 00/56706 17 PCT/US00/07262
E-Ol,-[amino[(4-aminophenyl)thio]methylene]-3-[(4-
pyridyl)hydroxymethyl]benzeneacetonitrile;
E-oc-[amino[(4-hydroxyphenyl)thio]methylene]-3-[(4-
pyridyl)hydroxymethyl]benzeneacetonitrile;
E-oc-[amino[(2-aminophenyl)thio]methylene]-3-[(3-
cyanophenyl)hydroxymethyl]benzeneacetonitrile;
E-oc,-[amino[(4-aminophenyl)thio]methylene]-3-[(4-
cyanophenyl)hydroxymethyl]benzeneacetonitrile;
E-oc-[amino(phenylthio)methylene]-3-[(4-
cyanophenyl)hydroxymethyl]benzeneacetonitrile;
E-oc-[amino(phenylthio)methylene]-3-[(4-
pyridyl)hydroxymethyl]benzeneacetonitrile;
E-oc-[amino[(4-aminophenyl)thio]methylene]-2-
bromobenzeneacetonitrile;
E-oc-[amino[(2-aminophenyl)thio]methylene]-3-[(2,4-
dimethylphenyl)hydroxymethyl]benzeneacetonitrile;
E-oc-[amino[(2-aminophenyl)thio]methylene]-3-
[(phenyl)hydroxymethyl]benzeneacetonitrile;
E-oc-[amino[(2-aminophenyl)thio]methylene]-3-[(2-
thienyl)hydroxymethyl]benzeneacetonitrile;
E-oc-[amino[(2-aminophenyl)thio]methylene]-4-chloro-~3-
phenylbenzenepropanenitrile;
E-oc-[amino[(2-thienyl)thio]methylene]-3-
[(phenyl)hydroxymethyl]benzeneacetonitrile;
E-a-[amino[(2,4-diaminophenyl)thio]methylene]-1-
naphthyleneacetonitrile;
E-oc-[amino[(2-aminophenyl)thio]methylene]-2-methyl-(3-(4-
pyridyl)benzenepropanenitrile;
E-oc-[amino[(4-aminophenyl)thio]methylene]-3-
(benzyl)benzeneacetonitrile;
E-oc-[amino[(2-naphthyl)thio]methylene]-1-
naphthyleneacetonitrile;
E-oc-[amino[(2-aminophenyl)thio]methylene]-3-
(benzoyl)benzeneacetonitrile;
CA 02362380 2001-08-07
WO 00/56706 PCT/US00/07262
18
E-oc-[amino[(2-aminophenyl)thio]methylene]-(3-(1-methyl-2-
pyrrolyl)benzenepropanenitrile;
E-oc-[amino[(2-aminophenyl)thio]methylene]-3-
phenoxybenzeneacetonitrile;
E-a-[amino[(2-aminophenyl)thio]methylene]-2-
bromobenzeneacetonitrile;
E-oc-[amino[(2-aminophenyl)thio]methylene]-3-[(2-
furanyl)hydroxymethyl]benzeneacetonitrile;
E-oc-[amino[(2-thienyl)thio]methylene]-3-[(2,3,4,5,6-
pentafluorophenyl)hydroxymethyl]benzeneacetonitrile;
E-oc-[amino[(2-aminophenyl)thio]methylene]-3-[(3-methyl-2-
pyridyl)hydroxymethyl]benzeneacetonitrile;
E-oc-[amino[(4-aminophenyl)thio]methylene]-2-
methylbenzeneacetonitrile;
E-a-[amino[(4-aminophenyl)thio]methylene]-4-(1,1-
dimethylethyl)benzeneacetonitrile;
E-oc-[amino[(4-aminophenyl)thio]methylene]-1-
naphthyleneacetonitrile;
E-a-[amino[(2-aminophenyl)thio]methylene]-3-
(trifluoromethyl)benzeneacetonitrile;
E-oc-[amino[(2-aminophenyl)thio]methylene]-1-
naphthyleneacetonitrile;
E-a-[amino[(2-aminophenyl)thio]methylene]-2-
(trifluoromethyl)benzeneacetonitrile;
E-oc-[amino[(4-aminophenyl)thio]methylene]-4-
methylbenzeneacetonitrile;
E-oc-[amino[(2-aminophenyl)thio]methylene]-2-
methylbenzeneacetonitrile;
E-oc-[amino[(2-fluorophenyl)thio]methylene]-1-
naphthyleneacetonitrile; and,
E-oc-[amino[(2-aminophenyl)thio]methylene]-3-phenyl
benzeneacetonitrile;
or a pharmaceutically acceptable salt form thereof.
CA 02362380 2001-08-07
WO 00/56706 19 PCT/US00/07262
In a further preferred embodiment, the present
invention provides a novel compound selected from:
Z-oc-[amino[(2-aminophenyl)thio]methylene]-4-chloro-2-methyl-
~3-phenylbenzenepropanenitrile;
Z-a-[amino[(2-aminophenyl)thio]methylene]-3-[(2,4-
dinitrophenyl)hydroxymethyl]benzeneacetonitrile;
Z-a-[amino[(2-aminophenyl)thio]methylene]-3-[(4-
carbomethoxyphenyl)hydroxymethyl]benzeneacetonitrile;
Z-oc-[amino[(2-aminophenyl)thio]methylene]-3-[(4-
nitrophenyl)hydroxymethyl]benzeneacetonitrile;
Z-oc-[amino[(2-aminophenyl)thio]methylene]-3-
[(pentafluorophenyl)hydroxymethyl]benzeneacetonitrile;
Z-a-[amino[(2-aminophenyl)thio]methylene]-2-methyl-3-[(4-
pyridyl)hydroxymethyl]benzeneacetonitrile;
Z-oc-[amino[(2-aminophenyl)thio]methylene]-2-methyl-3
[(phenyl)hydroxymethyl]benzeneacetonitrile;
Z-a-[amino[(2-aminophenyl)thio]methylene]-2-methyl-3-[(4
pyridyl)hydroxymethyl]benzeneacetonitrile;
Z-a-[amino[(2-hydroxyphenyl)thio]methylene]-3-[(4-
cyanophenyl)hydroxymethyl]benzeneacetonitrile;
Z-oc-[amino[(2-aminophenyl)thio]methylene]-3-[(3-
nitrophenyl)hydroxymethyl]benzeneacetonitrile;
Z-oc-[amino[(2-aminophenyl)thio]methylene]-2-methyl-3
[(pentafluorophenyl)hydroxymethyl]benzeneacetonitrile;
Z-a-[amino[(2-hydroxyphenyl)thio]methylene]-3-[(4-
pyridyl)hydroxymethyl]benzeneacetonitrile;
Z-oc-[amino[(2-aminophenyl)thio]methylene]-3-[(2-
trifluoromethylphenyl)hydroxymethyl]benzeneacetonitrile
Z-oc-[amino[(4-aminophenyl)thio]methylene]-3-[(4-
pyridyl)hydroxymethyl]benzeneacetonitrile;
Z-a-[amino[(4-hydroxyphenyl)thio]methylene]-3-[(4-
pyridyl)hydroxymethyl]benzeneacetonitrile;
Z-oc-[amino[(2-aminophenyl)thio]methylene]-3-[(3-
cyanophenyl)hydroxymethyl]benzeneacetonitrile;
CA 02362380 2001-08-07
WO 00/56706 2~ PCT/US00/07262
Z-oc-[amino[(4-aminophenyl)thio]methylene]-3-[(4-
cyanophenyl)hydroxymethyl]benzeneacetonitrile;
Z-oc-[amino(phenylthio)methylene]-3-[(4-
cyanophenyl)hydroxymethyl]benzeneacetonitrile;
Z-oc-[amino(phenylthio)methylene]-3-[(4-
pyridyl)hydroxymethyl]benzeneacetonitrile;
Z-oc-[amino[(4-aminophenyl)thio]methylene]-2-
bromobenzeneacetonitrile;
Z-oc-[amino[(2-aminophenyl)thio]methylene]-3-[(2,4
dimethylphenyl)hydroxymethyl]benzeneacetonitrile;
Z-oc-[amino[(2-aminophenyl)thio]methylene]-3-
[(phenyl)hydroxymethyl]benzeneacetonitrile;
Z-oc-[amino[(2-aminophenyl)thio]methylene]-3-[(2-
thienyl)hydroxymethyl]benzeneacetonitrile;
Z-oc-[amino[(2-aminophenyl)thio]methylene]-4-chloro-~3-
phenylbenzenepropanenitrile;
Z-a-[amino[(2-thienyl)thio]methylene]-3-
[(phenyl)hydroxymethyl]benzeneacetonitrile;
Z-a-[amino[(2,4-diaminophenyl)thio]methylene]-1-
naphthyleneacetonitrile;
Z-oc-[amino[(2-aminophenyl)thio]methylene]-2-methyl-(3-(4-
pyridyl)benzenepropanenitrile;
Z-oc-[amino[(4-aminophenyl)thio]methylene]-3-
(benzyl)benzeneacetonitrile;
Z-oc-[amino[(2-naphthyl)thio]methylene]-1-
naphthyleneacetonitrile;
Z-oc-[amino[(2-aminophenyl)thio]methylene]-3-
(benzoyl)benzeneacetonitrile;
Z-oc-[amino[(2-aminophenyl)thio]methylene]-(3-(1-methyl-2-
pyrrolyl)benzenepropanenitrile;
Z-oc,-[amino[(2-aminophenyl)thio]methylene]-3-
phenoxybenzeneacetonitrile;
Z-oc-[amino[(2-aminophenyl)thio]methylene]-2-
bromobenzeneacetonitrile;
CA 02362380 2001-08-07
WO 00/56706 21 PCT/US00/07262
z-oc-[amino[(2-aminophenyl)thio]methylene]-3-[(2-
furanyl)hydroxymethyl]benzeneacetonitrile;
Z-oc,-[amino[(2-thienyl)thio]methylene]-3-[(2,3,4,5,6
pentafluorophenyl)hydroxymethyl]benzeneacetonitrile;
Z-oc-[amino[(2-aminophenyl)thio]methylene]-3-[(3-methyl-2-
pyridyl)hydroxymethyl]benzeneacetonitrile;
Z-oc-[amino[(4-aminophenyl)thio]methylene]-2-
methylbenzeneacetonitrile;
Z-oc-[amino[(4-aminophenyl)thio]methylene]-4-(1,1-
dimethylethyl)benzeneacetonitrile;
Z-a-[amino[(4-aminophenyl)thio]methylene]-1-
naphthyleneacetonitrile;
Z-oc-[amino[(2-aminophenyl)thio]methylene]-3-
(trifluoromethyl)benzeneacetonitrile;
Z-oc-[amino[(2-aminophenyl)thio]methylene]-1-
naphthyleneacetonitrile;
Z-a-[amino[(2-aminophenyl)thio]methylene]-2-
(trifluoromethyl)benzeneacetonitrile;
Z-oc-[amino[(4-aminophenyl)thio]methylene]-4-
methylbenzeneacetonitrile;
Z-oc-[amino[(2-aminophenyl)thio]methylene]-2-
methylbenzeneacetonitrile;
z-a,-[amino[(2-fluorophenyl)thio]methylene]-1-
naphthyleneacetonitrile; and,
Z-oC-[amino[(2-aminophenyl)thio]methylene]-3-phenyl
benzeneacetonitrile;
or a pharmaceutically acceptable salt form thereof.
In another embodiment, the present invention provides
novel pharmaceutical compositions, comprising: a
pharmaceutically acceptable carrier and a therapeutically
effective amount of a compound of formula Ia or Ib or a
pharmaceutically acceptable salt form thereof.
In another embodiment, the present invention provides a
novel method for treating or preventing a disorder related
CA 02362380 2001-08-07
WO 00/56706 PCT/US00/07262
22
to MEK, comprising: administering to a patient in need
thereof a therapeutically effective amount of a compound of
formula Ia or Ib or a pharmaceutically acceptable salt form
thereof.
In another embodiment, the present invention provides
novel compounds of formula Ia or Ib or a pharmaceutically
acceptable salt form thereof for use in therapy.
In another embodiment, the present invention provides
novel compounds of formula Ia or Ib or a pharmaceutically
acceptable salt form thereof for the manufacture of a
medicament for the treatment of an inflammatory disease.
In another embodiment, the present invention provides
novel compounds of formula Ia or Ib or a pharmaceutically
acceptable salt form thereof for the manufacture of a
medicament for the treatment of cancer.
In another embodiment, the present invention provides a
novel method of treating a condition or disease wherein the
disease or condition is referred to as rheumatoid arthritis,
osteoarthritis, periodontitis, gingivitis, corneal
ulceration, solid tumor growth and tumor invasion by
secondary metastases neovascular glaucoma, multiple
sclerosis, or psoriasis in a mammal, comprising:
administering to the mammal in need of such treatment a
therapeutically effective amount of a compound of formula
(I) or a pharmaceutically acceptable salt form thereof.
In another embodiment, the present invention provides a
novel method of treating a condition or disease wherein the
disease or condition is referred to as fever, cardiovascular
effects, hemorrhage, coagulation, cachexia, anorexia,
alcoholism, acute phase response, acute infection, shock,
graft versus host reaction, autoimmune disease or HIV
infection in a mammal comprising administering to the mammal
in need of such treatment a therapeutically effective amount
CA 02362380 2001-08-07
WO 00/56706 PCT/US00/07262
23
of a compound of formula (I) or a pharmaceutically
acceptable salt form thereof.
DEFINITIONS
The compounds herein described may have asymmetric
centers. Compounds of the present invention containing an
asymmetrically substituted atom may be isolated in optically
active or racemic forms. It is well known in the art how to
prepare optically active forms, such as by resolution of
racemic forms or by synthesis from optically active starting
materials. Many geometric isomers of olefins, C=N double
bonds, and the like can also be present in the compounds
described herein, and all such stable isomers are
contemplated in the present invention. Cis and trans
geometric isomers of the compounds of the present invention
are described and may be isolated as a mixture of isomers or
as separated isomeric forms. All chiral, diastereomeric,
racemic forms and all geometric isomeric forms of a
structure are intended, unless the specific stereochemistry
or isomeric form is specifically indicated. All processes
used to prepare compounds of the present invention and
intermediates made therein are considered to be part of the
present invention.
"Substituted" is intended to indicate that one or more
hydrogens on the atom indicated in the expression using
"substituted" is replaced with a selection from the
indicated group(s), provided that the indicated atom's
normal valency is not exceeded, and that the substitution
results in a stable compound. When a substituent is keto
(i.e., =0) group, then 2 hydrogens on the atom are replaced.
The present invention is intended to include all
isotopes of atoms occurring in the present compounds.
Isotopes include those atoms having the same atomic number
but different mass numbers. By way of general example and
without limitation, isotopes of hydrogen include tritium and
deuterium. Isotopes of carbon include C-13 and C-14.
When any variable (e. g., R6) occurs more than one time
in any constituent or formula for a compound, its definition
CA 02362380 2001-08-07
WO 00/56706 PCT/US00/07262
24
at each occurrence is independent of its definition at every
other occurrence. Thus, for example, if a group is shown to
be substituted with 0-2 R6, then said group may optionally
be substituted with up to two R6 groups and R6 at each
occurrence is selected independently from the definition of
R6. Also, combinations of substituents and/or variables are
permissible only if such combinations result in stable
compounds.
When a bond to a substituent is shown to cross a bond
connecting two atoms in a ring, then such substituent may be
bonded to any atom on the ring. When a substituent is
listed without indicating the atom via which such
substituent is bonded to the rest of the compound of a given
formula, then such substituent may be bonded via any atom in
such substituent. Combinations of substituents and/or
variables are permissible only if such combinations result
in stable compounds.
As used herein, "alkyl" is intended to include both
branched and straight-chain saturated aliphatic hydrocarbon
groups having the specified number of carbon atoms. C1_,
alkyl is intended to include C1, C~, C;;, and C= alkyl.
Examples of alkyl include, but are not limited to, methyl,
ethyl, n-propyl, i-propyl, n-butyl, s-butyl, t-butyl,
n-pentyl, and s-pentyl. "Alkoxy" represents an alkyl group
as defined above with the indicated number of carbon atoms
attached through an oxygen bridge. C,_; alkoxy is intended to
include C.,, C~, C; , and C; alkoxy. Examples of alkoxy
include, but are not limited to, methoxy, ethoxy, n-propoxy,
i-propoxy, n-butoxy, s-butoxy, t-butoxy, n-pentoxy, and
s-pentoxy.
As used herein, the term "aromatic heterocyclic system"
or "heteroaryl" is intended to mean a stable 5 or 6 membered
monocyclic aromatic ring which consists of carbon atoms and
1, 2, 3, or 4 heterotams independently selected from the
group consisting of N, NH, 0 and S. It is to be noted that
that the total number of S and O atoms in an aromatic
heterocycle is not more than 1.
CA 02362380 2001-08-07
WO 00/56706 25 PCT/US00/07262
Examples of heterocycles include, but are not limited
to, 2H,6H-1,5,2-dithiazinyl, furanyl, imidazolyl,
isothiazolyl, isoxazolyl, oxadiazolyl, 1,2,3-oxadiazolyl,
1,2,4-oxadiazolyl, 1,2,5-oxadiazolyl, 1,3,4-oxadiazolyl,
oxazolidinyl, oxazolyl, pyrimidinyl, pyranyl, pyrazinyl,
pyrazolidinyl, pyrazolinyl, pyrazolyl, pyridazinyl, pyridyl,
2H-pyrrolyl, pyrrolyl, tetrazolyl, 6H-1,2,5-thiadiazinyl,
1,2,3-thiadiazolyl, 1,2,4-thiadiazolyl, 1,2,5-thiadiazolyl,
1,3,4-thiadiazolyl, thiazolyl, thienyl, triazinyl, 1,2,3-
triazolyl, 1,2,4-triazolyl, 1,2,5-triazolyl, and 1,3,4-
triazolyl. Preferred heterocycles include, but are not
limited to, pyridinyl, furanyl, thienyl, pyrrolyl,
pyrazolyl, and imidazolyl.
The phrase "pharmaceutically acceptable" is employed
herein to refer to those compounds, materials, compositions,
and/or dosage forms which are, within the scope of sound
medical judgment, suitable for use in contact with the
tissues of human beings and animals without excessive
toxicity, irritation, allergic response, or other problem or
complication, commensurate with a reasonable benefit/risk
ratio.
As used herein, "pharmaceutically acceptable salts"
refer to derivatives of the disclosed compounds wherein the
parent compound is modified by making acid or base salts
thereof. Examples of pharmaceutically acceptable salts
include, but are not limited to, mineral or organic acid
salts of basic residues such as amines; alkali or organic
salts of acidic residues such as carboxylic acids. The
pharmaceutically acceptable salts include the conventional
non-toxic salts or the quaternary ammonium salts of the
parent compound formed, for example, from non-toxic
inorganic or organic acids. For example, such conventional
non-toxic salts include those derived from inorganic acids
such as hydrochloric, hydrobromic, sulfuric, sulfamic,
phosphoric, and nitric; and the salts prepared from organic
acids such as acetic, propionic, succinic, glycolic,
stearic, lactic, malic, tartaric, citric, ascorbic, pamoic,
malefic, hydroxymaleic, phenylacetic, glutamic, benzoic,
CA 02362380 2001-08-07
WO 00/56706 26 PCT/US00/07262
salicylic, sulfanilic, 2-acetoxybenzoic, fumaric,
toluenesulfonic, methanesulfonic, ethane disulfonic, oxalic,
and isethionic.
The pharmaceutically acceptable salts of the present
invention can be synthesized from the parent compound which
contains a basic or acidic moiety by conventional chemical
methods. Generally, such salts can be prepared by reacting
the free acid or base forms of these compounds with a
stoichiometric amount of the appropriate base or acid in
water or in an organic solvent, or in a mixture of the two;
generally, nonaqueous media like ether, ethyl acetate,
ethanol, isopropanol, or acetonitrile are preferred. Lists
of suitable salts are found in Remington's Pharmaceutical
Sciences, 17th ed., Mack Publishing Company, Easton, PA,
1985, p. 1418, the disclosure of which is hereby
incorporated by reference.
"Prodrugs" are intended to include any covalently
bonded carriers which release the active parent drug
according to formula Ia or Ib in vivo when such prodrug is
administered to a mammalian subject. Prodrugs of a compound
of formula Ia or Ib are prepared by modifying functional
groups present in the compound in such a way that the
modifications are cleaved, either in routine manipulation or
in vivo, to the parent compound. Prodrugs include compounds
of formula Ia or Ib wherein a hydroxy, amino, or sulfhydryl
group is bonded to any group that, when the prodrug or
compound of formula Ia or Ib is administered to a mammalian
subject, cleaves to form a free hydroxyl, free amino, or
free sulfhydryl group, respectively. Examples of prodrugs
include, but are not limited to, acetate, formate and
benzoate derivatives of alcohol and amine functional groups
in the compounds of formula Ia or Ib.
"Stable compound" and "stable structure" are meant to
indicate a compound that is sufficiently robust to survive
isolation to a useful degree of purity from a reaction
mixture, and formulation into an efficacious therapeutic
agent.
CA 02362380 2001-08-07
WO 00/56706 2~ PCT/US00/07262
"Therapeutically effective amount" is intended to
include an amount of a compound of the present invention or
an amount of the combination of compounds claimed effective
to inhibit MEK or treat the symptoms of MEK over production
in a host. The combination of compounds is preferably a
synergistic combination. Synergy, as described for example
by Chou and Talalay, Adv. Enzyme Regul. 22:27-55 (1984),
occurs when the effect (in this case, MEK inhibition) of the
compounds when administered in combination is greater than
the additive effect of the compounds when administered alone
as a single agent., In general, a synergistic effect is most
clearly demonstrated at suboptimal concentrations of the
compounds. Synergy can be in terms of lower cytotoxicity,
increased antiviral effect, or some other beneficial effect
of the combination compared with the individual components.
The term "radiosensitize", as used herein refers to a
process whereby cells are made susceptible to radiation-
induced cell death, or the cells that result from the
process.
SYNTHESIS
The compounds of the present invention can be prepared
in a number of ways known to one skilled in the art of
organic synthesis. The compounds of the present invention
can be synthesized using the methods described below,
together with synthetic methods known in the art of
synthetic organic chemistry, or by variations thereon as
appreciated by those skilled in the art. Preferred methods
include, but are not limited to, those described below. The
reactions are performed in a solvent appropriate to the
reagents and materials employed and suitable for the
transformations being effected. It will be understood by
those skilled in the art of organic synthesis that the
functionality present on the molecule should be consistent
with the transformations proposed. This will sometimes
require a judgment to modify the order of the synthetic
steps or to select one particular process scheme over
another in order to obtain a desired compound of the
CA 02362380 2001-08-07
WO 00/56706 2g PCT/US00/07262
invention. It will also be recognized that another major
consideration in the planning of any synthetic route in this
field is the judicious choice of the protecting group used
for protection of the reactive functional groups present in
the compounds described in this invention. An authoritative
account describing the many alternatives to the trained
practitioner is Greene and Wuts (Protective Groups In
Organic Synthesis, Wiley and Sons, 1991). All references
cited herein are hereby incorporated in their entirety
herein by reference.
Compounds of the present invention (3) may be
synthesized by the route described in Scheme 1. A thiol 1,
such as a thiophenol, may be treated with a malononitrile
such as malononitrile 2 in the presence of a base catalyst
such as triethylamine, DBU, Hunig's base, or aqueous base
(for example, 10% NaOH), etc., in a nonreactive solvent such
as THF, acetone, etc., to yield the vinylogous cyanamide 3.
The reaction medium can be degassed to eliminate the
presence of oxygen which can facilitate disulfide formation
via the dimerization of thiol 1. The vinylogous cyanamide
is frequently isolated as a mixture of z- and E-isomers and
the melting point varies significantly with isomer
composition. A crystalline single isomer or material
enriched in one isomer may sometimes be obtained by
spontaneous crystallization of one isomer,
recrystallization, or stirring solid in a solvent which
dissolves only part'of the material. Alternatively, isomers
may sometimes be separated by chromatography. However, the
double bond in 3 isomerizes very easily. NMR spectroscopy
of a single isomer in DMSO-d6 shows that an equilibrium
mixture of Z- and E-isomers is generated faster than the
spectrum could be obtained (about 5 minutes). Isomerization
also takes place in other solvents such as water, acetone,
methanol, and chloroform, but more slowly than in DMSO.
Rapid NMR in one of these solvents may be used to establish
isomeric composition. For in vitro assays, the compounds
may be dissolved in DMSO to ensure that an equilibrium
mixture of isomers is tested.
CA 02362380 2001-08-07
WO 00/56706 PCT/US00/07262
29
Scheme 1: Preparation of vinylogous cyanamides
N base R~S~ N
R1SH + N ~C
Y-R2 H2N Y-R2
1 2 or 2a
Many thiols (1) are commercially available.
Alternatively, there are many methods for their synthesis
familiar to one skilled in the art. For example, aryl or
heterocyclic anions may be quenched with sulfur to yield
thiols CChem. Pharm. Bull. 1989, 37 (1), 36). Displacement
of aryldiazonium salts with EtOCS2K leads to aryl thiols
(Collect. Czech. Chem. Commun. 1990, 55, 1266). The Newman
rearrangement of phenols via their dimethylthiocarbamates
leads to thiophenols (Organic Syntheses VI, (1988) 824).
When the Y group in Scheme 1 is substituted phenyl or
naphthyl, the malononitrile precursors (2) to the compounds
of this invention may be prepared by one of the three routes
shown in Scheme 2. In the first route, aryl iodides 4 may
be treated with malononitrile in the presence of a copper
catalyst to yield arylmalononitriles 2 (J. Org. Chem. 1993
(58) 7606-7). Malononitrile can also be coupled to aryl
halides 4 (X=halide) using (Ph3P)2PdC12 or Pd(Ph3P)4 in THF
(J. Chem. Soc. Chem. Comm. 1984, 932-3). The aryl iodides
needed for these methods are commercially available or
prepared by methods familiar to one skilled in the art. In
particular, aryl iodides may be prepared by iodination with
a source of electrophilic iodine, such as iodine
monochloride, or by diazotization of anilines.
Scheme 2: Preparation of malononitriles 2 when Y is a
substituted phenyl or naphthyl
CA 02362380 2001-08-07
WO 00/56706 3~ PCT/US00/07262
X Cul, K2C03 / DMSO CN
Y-R2 NC--C
or Pd(II) or Pd(0) / THF Y-R2
4 2
1 ) LDA CN
NCB 2) 2-CI-C6H4-CH2-SCN NC-
Y-R2 Y-R2
CsHs
2
CN n-BuLi Li CN CN
NC-~ NC--~ ~ NC--~
Y-X Y-Li Y-R2
6 7 2
Arylmalononitriles may also be prepared from aryl
acetonitriles as shown the second route in Scheme 2. Aryl
5 acetonitriles 5 may be deprotonated with a base, such as
LDA, and quenched with a electrophilic source of cyanide,
such as cyanogen chloride (J. Org. Chem. 1966, 21, 919) or
2-chlorobenzylthiocyanate (J. Org. Chem. 1983, 48, 2774-5)
to yield malononitrile 2. Along the same lines,
acetonitrile 5 can also be acylated in the presence of NaOMe
with dimethyl carbonate to form the methyl cyanoacetate (not
shown in Scheme 2). Conversion of the methyl ester to a
nitrile group via procedures familiar to one skilled in the
art leads to malononitrile 2 (J. Am. Chem. Soc. 1904, 32,
119). The aryl acetonitriles needed for these methods are
commercially available or prepared by methods familiar to
one skilled in the art, for example, from aryl acetamides or
from toluenes. When R2 is an optionally substituted phenoxy
group, the initial step in the preparation of the compounds
of this invention may be an Ullmann condensation between an
aryl halide and a phenol. (For useful protocols, see: U.S.
Patent No. 4,288,386; and Tetrahedron (1961), 15, 144-153.)
A methyl substituent on either of these substrates may be
subsequently converted to a -CH2CN group by free radical
halogenation, with a reagent such as N-bromosuccinimide,
followed by displacement with cyanide.
CA 02362380 2001-08-07
WO 00/56706 PCT/US00/07262
31
As shown in the third route shown in Scheme 2,
arylmalononitriles 2 may also be synthesized from simpler
bromo- or iodoarylmalononitriles. These bromo- or iodo-
substituted arylmalononitriles may be prepared by either of
the first two routes indicated in Scheme 2 for the
preparation of malononitriles. Bromo- or iodo-substituted
arylmalononitriles undergo halogen-metal exchange in the
presence of two or more equivalents of an alkyllithium
reagent, such as n-butyllithium, to form dianion
intermediate 7. This process may be carried out in an
ethereal solvent such as THF at a temperature of -78 to 0
oC. The dianion may be quenched in situ with one equivalent
of an electrophile, such as an aldehyde, alkyl halide,
disulfide, ester, or ketone, to yield a substituted
malononitrile 2 with a new R2 group attached to the former
site of the bromine or iodine atom. This is process is
illustrated in more detail in Scheme 3 for the case where Y
is a 1,3-disubstituted phenyl group.
3-Bromophenylmalononitrile (6) may be converted to dianion
7a
Scheme 3: An illustration of the use of arylmalononitrile
dianions
CA 02362380 2001-08-07
WO 00/56706 PCT/US00/07262
32
CN 2 eq n-BuLi, -78 ~C CN
NC \ Br THF
NC ~ ~ \ ~ 2 Li+
6
_ 7
dianion intermediate
RzCHO
CN O CN OH
[O]
NC ~ ~Rz NC \ ~Rz
/
9 8
[H~ [Hl
CN
NC ~~ ~ Rz
by deprotonation and halogen-metal exchange with 2
equivalents of n-butyllithium in THF at -78 °C. The dianion
5 may be treated in situ with an aldehyde to produce
hydroxy-phenylmalononitriles 8.
Hydroxy-phenylmalononitriles 8 may be oxidized to the
corresponding keto-phenylmalononitrile 9 using Mn02 or a
variety of other oxidizing agents familiar to one skilled in
10 the art. Compounds 8 and 9 may be reduced to the
corresponding CH2R2-substituted phenylmalononitriles 10
using hydrogen and a noble metal catalyst, NaBHg and TFA
(Synthesis 1978, 763-5), or other procedures familiar to one
skilled in the art. Malononitriles 8, 9, and 10 may be
treated with thiols 1 to yield the compounds of this
invention. It must be noted that although only the meta-
bromo isomer of 6 is pictured in Scheme 3, one trained in
the art may apply this methodology using other aryl halides
CA 02362380 2001-08-07
WO 00/56706 33 PCT/US00/07262
and electrophiles to prepare isomers and compounds with
different Y groups.
Scheme 4: Preparation of Malononitriles when Y is CHR3
O\/H CH2(CN)2 CN R2Mggr CN
'R ~ NCH NC~R2
3
R3 R3
11 12 2a
O~R2 CH2(CN)2 C\ R reduction CN R
'( NC~ 2 NC~ 2
R3
R3 R3
13 14 2a
X R CH2(CN)2, base CN
2 NC~R2
3 R
3
15 2a
When Y is CHR3, malononitrile precursors useful for
preparation of the compounds of this invention have
structure 2a and may be prepared as shown in Scheme 4.
Knoevenagel condensation (Organic Reactions 15, 204-509
(1967)) between an aldehyde 11 or a ketone 13 may be used to
produce alkylidene malononitriles 12 or 14. Conjugate
addition of a Grignard or organolithium reagent to 12
affords the malononitrile prescursors 2 used in Scheme 1.
Alternatively, alkylidene malononitriles 14 may be reduced
to malononitriles 2a with sodium borohydride, catalytic
hydrogenation or other reducing agents familiar to one
skilled in the art. A third alternative is to alkylate
malononitrile with an alkyl halide 15 (X=halide).
Other features of the invention will become apparent in
the course of the following descriptions of exemplary
CA 02362380 2001-08-07
WO 00/56706 34 PCT/US00/07262
embodiments which are given for illustration of the
invention and are not intended to be limiting thereof.
EXAMPLES
Abbreviations used in the Examples are defined as
follows: "1 x" for once, "2 x" for twice, "3 x" for thrice,
"°C" for degrees Celsius, "eq" for equivalent or
equivalents, "g" for gram or grams, "mg" for milligram or
milligrams, "mL" for milliliter or milliliters, "1H" for
proton, "h" for hour or hours, "M" for molar, "min" for
minute or minutes, "MHz" for megahertz, "MS" for mass
spectroscopy, "NMR" for nuclear magnetic resonance
spectroscopy, "rt" for room temperature, "tlc" for thin
layer chromatography; "v/v" for volume to volume ratio.
"a,", "(3", "R" and "S" are stereochemical designations
familiar to those skilled in the art.
Example 1
Z- and E-oc-faminof(4-aminophenyl)thiolmethylenel-2-
(trifluoromethyl)benzeneacetonitrile
CN CF3
NH2 /
Hz N
Part A. Preparation of 2-[(2-
trifluoromethyl)phenyl]malononitrile.
CN CF3
NC
A mixture of 2-trifluoromethyl-1-iodobenzene (21.76 g,
0.08 mol, 1 eq), malononitrile (10.56 g, 0.16 mol, 2 eq),
copper(I) iodide (1.52 g, 0.008 mol, 0.1 eq), potassium
carbonate (11.04 g, 0.32 mol, 4 eq), and 200 mL DMSO was
stirred and heated at 120 oC for 21 h. The reaction mixture
was cooled and poured into 1.2 L of 0.5 M HCl. The mixture
was filtered and extracted with ethyl acetate. The organic
CA 02362380 2001-08-07
WO 00/56706 35 PCT/US00/07262
layer was dried (MgS04) and the solvent removed in vacuo to
yield an oil. This oil was purified by flash chromatography
on silica gel with 3:1 hexane/ethyl acetate to yield 4.46 g
(27%) of 2-[(2-trifluoromethyl)phenyl]malononitrile as a
yellow oil. 1H-NMR (CDC13) 8: 8.05-7.10 (m, 4H); 5.30 (s,
1H ) .
Part B. Preparation of oc-[amino[(4-
aminophenyl)thio]methylene]-2-
(trifluoromethyl)benzeneacetonitrile.
2-[(2-Trifluoromethyl)phenyl]malononitrile (the product
from Part A) (3.07 g, 14.6 mmol, 1.1 eq), freshly distilled
4-aminothiophenol (1.66 g, 13.3 mmol, 1 eq), and THF (25 mL)
were mixed. The reaction flask was then degassed by placing
under vacuum followed by flushing with N2 several times to
prevent disulfide formation. After cooling to -78 °C,
triethylamine (1.85 mL, 13.3 mmol, 1 eq) was added via
syringe and the flask degassed once more. The contents were
allowed to warm to room temperature and the mixture was
stirred overnight. TLC the following morning showed no
malononitrile present, only thiol. Therefore, another 0.2
equivalents of malononitrile were added followed by
degassing, followed by 0.5 equivalents of triethylamine,
followed by degassing. TLC after a few hours no starting
material was present. The reaction was worked up after
stirring over the weekend at room temperature. The solvent
was removed in vacuo and the residue was purified by flash
chromatography on silica gel with 25-1000 ethyl acetate in
hexane. Two fractions were isolated. The faster eluting
fraction yielded 1.63 g of a tan oily solid. The slower
eluting fraction yielded 2.61 g of a tan oily solid. Both
compounds were recrystallized from n-butylchloride. The
faster eluting compound yielded 274 mg of a white solid
(m.p. 147.0-148.0 °C). This compound proved to be the E
isomer of the titled compound through NMR NOE experiments.
The slower eluting compound yielded 1.85 g of a white solid
CA 02362380 2001-08-07
WO 00/56706 36 PCT/US00/07262
(m.p. 130.0-130.5 oC). This compound proved to be the Z
isomer of the titled compound through NMR NOE experiments.
Anal. calcd. for C16H12F3N3S (faster eluting isomer): C,
57.31; H, 3.62; F, 17.00; N, 12.53; S, 9.56. Found: C,
57.19; H, 3.75; F, 16.83; N, 12.24; S, 9.50. Anal. calcd.
for C16H12F3N3S (slower eluting isomer): C, 57.31; H, 3.62;
F, 17.00; N, 12.53; S; 9.56. Found: C, 57.28; H, 3.80; F,
16.96; N, 12.37; S, 9.22. 1H-NMR (faster eluting isomer)
(CDC13) 8 7.75 (d, 1H, J= 7 Hz) ; 7.57 (t, 1 H, J = 7 Hz) ;
7.49 (t, 1H, J = 7 Hz); 7.47 (d, 1H, J = 7 Hz); 7.24 (d, 2H,
J= 7 Hz); 6.66 (d, 2H, J = 7 Hz). 1H-NMR (slower eluting
isomer) (CDC13) 8 7.75 (d, 1H, J= 7 Hz); 7.58 (t, 1 H, J = 7
Hz) ; 7.48 (t, 1H, J = 7 Hz) ; 7.43 (d, 1H, J = 7 Hz) ; 7.40
(d, 2H, J= 7 Hz); 6.68 (d, 2H, J = 7 Hz).
Example 2
Z- and E-oc-(aminof(2-aminophenyl)thiolmethylenel-3-((4
cyanophenLrl)hydroxymethyllbenzeneacetonitrile
CN OH
\ s / \ \
NH2 ~ / ~ /
2 O HZN v ~CN
Part A. Preparation of 2-(3-bromophenyl)malononitrile.
CN
NC ~ \ Br
To a flame dried 5L 3-neck flask equipped with a
mechamical overhead stirrer under nitrogen was added
diisopropylamine (78.60 mL, 0.56 mol, 2.2 eq) and 2 L of
benzene. After cooling to 0-5 °C, 1.6 M n-BuLi (351.0 mL,
0.56 mol, 2.2 eq) was added dropwise via addition funnel
while keeping the temperature at 0-5 oC. The LDA was
stirred for 45 min. at 0-5 oC. 3-Bromophenylacetonitrile
(50.0 g, 0.26 mol, 1.0 eq) dissolved in 200 mL of benzene
CA 02362380 2001-08-07
WO 00/56706 PCT/US00/07262
37
was added dropwise via addition funnel keeping the
temperature at 0-5 °C. The mixture was stirred an
additional 15 min at this temperature. 2-
Chlorobenzylthiocyanate (J. Am. Chem. Soc., 1954, 76, 585)
(103.0 g, 0.56 mol, 2.2 eq) dissolved in 200 mL benzene was
added dropwise via addition funnel keeping the temperature
at 0-5 °C. During the addition, a precipitate formed. The
reaction was allowed to warm to room temperature and the
mixture stirred overnight. The reaction was quenched by
adding water and 200 mL 10% NaOH. The layers were
separated, and the benzene layer extracted with 10o NaOH (3
X 1L). The basic layers were collected and acidified with
conc. HCl to pH 1-2. A precipitate formed. Methylene
chloride was added to dissolve the precipitate. The layers
were separated and the aqueous layer reextracted with
methylene chloride (2X). The methylene chloride layers were
collected, dried (MgS04) and the solvent removed in vacuo to
yield 65.32 g of 2-(3-bromophenyl)malononitrile as a yellow
solid. Recrystallization from methylcyclohexane yielded two
crops: crop 1, 42.86 g of orange crystals, m.p. 99.5-101.5
°C; crop 2, 2.18 g of orange crystals, m.p. 97.0-99.0 oC.
Combined yield 79.9 %. 1H-NMR (CDC13) 8: 7.67 (s, 1H); 7.63
(d, 1H, J=7 Hz); 7.47 (d, 1H, J=7 Hz); 7.39 (t, 1H, J=7 Hz);
5.08 (s, 1H).
Part B. Preparation of 2-[3-[(4-
cyanophenyl)hydroxymethyl]phenyl]malononitrile
CN OH
NC \ \
CN
2-(3-Bromophenyl)malononitrile (the product from part
A) (1.00 g, 4.52 mmol, 1 eq) was dissolved in dry THF (50
mL) under N2 and cooled to -70 oC. 1.6 M n-BuLi (5.94 mL,
9.50 mmol, 2.1 eq) was then added dropwise via syringe
maintaining the temperature at -65 to -70 oC. An orange
CA 02362380 2001-08-07
WO 00/56706 PCT/US00/07262
38
slurry formed. The temperature was maintained for 20 min
after which 4-cyanobenzaldehyde (0.59 g, 4.52 mmol, 1 eq)
was added via syringe. After two hours, the reaction was
complete. The reaction was added to water and the pH was
adjusted to 3 with 1 N HCl. The mixture was extracted with
ethyl acetate (3X), the organic layers combined, dried
(MgS04) and the solvent removed in vacuo to yield 1.84 g an
an amber oil. Flash chromatography on silica gel with 7:3
to 1:1 hexane/ethyl acetate yielded 2-(3-
bromophenyl)malononitrile (0.88 g) as an amber oil. 1H-NMR
(CDC13) ~: 7.66 (d, 2H, J=7 Hz); 7.60-7.15 (m, 6H); 5.95 (s,
1H); 5.07 (s, 1H); 2.63 (br s, 1H). NH4-CI MS: 291
(M+NH4)+.
Part C. Preparation of Z- and E-oc-[amino[(2-
aminophenyl)thio]methylene]-3-[(4-
cyanophenyl)hydroxymethyl]benzene-acetonitrile
2-[3-[(4-Cyanophenyl)hydroxymethyl]phenyl]malononitrile
(the product from part B) (250 mg, 0.915 mmol, 1 eq), 2-
aminothiophenol (0.10 mL, 0.915 mmol, 1 eq), triethylamine
(0.13 mL, 0.915 mmol, 1 eq), and THF were reacted by the
procedure described in Example 1, part B. After 4 hours,
the solvent was then removed in vacuo and the residue
purified by flash chromatography on silica gel with 1:1
hexane/ethyl acetate to yield the title compound (200 mg) as
a mixture of isomers. HRMS calcd. for C23H18N40S: 399.1264;
Found: 399.1280. 1H-NMR (CDC13) 8: (major isomer) 7.61 (d,
2H, J= 7 Hz); 7.60-7.10 (m, 8H); 6.90-6.70 (m, 2H); 5.86 (br
s, 1H); 4.71 (br s, 2H); 4.44 (br s, 2H); 2.51 (br s, 1H).
Example 3
Z- and E-oC-~amino~(2-aminophenyl)thiolmethylenel-3-f(4
pyridyl)hydroxymethyllbenzeneacetonitrile
CA 02362380 2001-08-07
WO 00/56706 39 PCT/US00/07262
CN OH
S / \ \
NHz ~ / ~ / N
H2 N v
Part A. Preparation of 2-[3-[(4-
pyridyl)hydroxymethyl]phenyl]malononitrile
CN OH
NC ~ \
/ ~N
2-(3-Bromophenyl)malononitrile (the product from
Example 2, part A) (2.00 g, 9.05 mmol, 1 eq) was dissolved
in dry THF (100 mL) under N2 and cooled to -70 oC. 1.6 M n-
BuLi (11.87 mL, 19.0 mmol, 2.1 eq) was then added dropwise
via syringe maintaining the temperature at -65 to -70 oC.
An orange slurry formed. The temperature was maintained for
min after which 4-pyridinecarboxaldehyde (0.86 mL, 9.05
mmol, 1 eq) was added via syringe. After one hour, the
reaction was essentially complete. It was worked up by
15 adding water and adjusting the pH to 3 with 1 N HCl. The
mixture was extracted with ethyl acetate (3X), the organic
layers combined, dried (MgS04) and the solvent removed in
vacuo to yield an an amber oil. Flash chromatography with
1:1 hexane/ethyl acetate to 100% ethyl acetate yielded 0.95
20 g of an orange glass as product. 1H-NMR (DMSO-d6)8: 8.41 (d,
2H, J = 7 Hz); 7.26 (br s, 1H); 7.35-7.20 (m, 6H); 6.03 (br
s, 1H); 5.65 (s, 1H).
Part B. Preparation of a-[amino[(2-
aminophenyl)thio]methylene]-3-[(4-pyridyl)hydroxymethyl]benz
eneacetonitrile.
2-[3-[(4-Pyridyl)hydroxymethyl]phenyl]malononitrile
(the product from Part A) (850 mg, 3.41 mmol, 1 eq), 2-
aminothiophenol (0.36 mL, 3.41 mmol, 1 eq), triethylamine
(0.48 mL, 3.41 mmol, 1 eq), and THF (20 mL) were reacted by
the procedure described in Example 1, part B. As soon as
CA 02362380 2001-08-07
WO 00/56706 PCT/US00/07262
the triethylamine was added, a precipitate began to form.
More THF was added (50 mL) but the precipitate did not
dissolve. The mixture was stirred overnight and the
precipitate dissolved. TLC showed the reaction to be
5 complete. The solvent was then removed in sracuo and the
residue was purified by flash chromatography on silica gel
with 1:1 hexane/ethyl acetate to 100% ethyl acetate to yield
950 mg of a white solid. The solid was stirred in THF and
filtered to yield 462 mg of a white solid (mp 97.5-101.0
10 °C). NMR shows a mixture of isomers. An analytical sample
was prepared by recrystallization (50 mg) from ethyl
acetate. The recrystallized solids were filtered, rinsed
with ether, and dried under high vacuum to yield 23 mg of a
white solid (mp 150.0-151.0 °C). NMR showed the presence of
15 mainly one isomer. Anal calcd.for C21H18N40S~0.4 H20: C,
66.09; H, 4.96; N, 14.68; S, 8.40. Found: C, 66.16; H,
5.03; N, 14.46; S, 8.35. 1H-NMR (major isomer) (acetone-d6)
8 8.49 (d, 2H, J= 7 Hz); 7.52 (s, 1H); 7.50-7.20 (m, 7H);
7.00-6.80 (m, 1H); 6.70 (t, 1H, J = 7 Hz); 5.84 (d, 1H, J= 6
20 Hz); 5.80-5.50 (m, 2H); 5.45-5.30 (m, 2H); 5.20 (d, 1H, J =
6 Hz). The above procedure was repeated several times on
larger scale to yield 112.12 g of the title compound. This
material was stirred overnight at room temperature in 900 mL
of ethyl acetate. The solids were filtered, rinsed with
25 ether (1 L), and dried under high vacuum to yield 86.11 g of
a white solid (mp 146.5-147.5 oC). Anal calcd.for
C21H18N40S: C, 67.36; H, 4.86; N, 14.96; S, 8.56. Found: C,
67.39; H, 4.94; N, 14.76; S, 8.84.
30 Example 4
Z- and E-oc-faminof(2-aminophenyl)thiolmethylenel-2-methyl-3
phenoxybenzeneacetonitrile.
NH2 CN CH3
S , ~ O
~ I NH2
CA 02362380 2001-08-07
WO 00/56706 PCT/US00/07262
41
Part A. Preparation of 2,3-dimethyldiphenylether.
CH3
H3C ~ O
~ i
2,3-dimethylphenol (10 g, 82 mmol), sodium hydroxide
(3.28 g, 82 mmol), water (1.8 mL) and chlorobenzene (70 mL)
were refluxed for 3 h under nitrogen in a flask equipped
with a Dean-Stark trap. Water and chlorobenzene removed
from the trap several times (100 mL total) while adding an
equal volume of chlorobenzene to the flask. The resulting
suspension was dried further by refluxing through a soxhlet
extractor filled with 3A molecular sieves for 30 min.
Cuprous iodide (0.81 g, 0.082 mmol) and tris[2-(2-
methoxyethoxy)ethyl]amine (1.5 mL, 4.1 mmol) were added and
the reaction was refluxed overnight. The solution was
decanted from the solid. Additional cuprous iodide (0.81 g,
0.082 mmol) and tris[2-(2-methoxyethoxy)ethyl]amine (1.5 mL,
4.1 mmol) were added to the solution and the reaction was
refluxed overnight with mechanical stirring. The reaction
mixture was absorbed onto silica gel and eluted with hexane
to afford the title compound (1.4 g). GC-MS: Calcd, 199;
Found, 199. 1H-NMR (CDC13) S: 7.28 (t, 2H); 7.04 (m, 3H);
6.88 (d, 2H); 6.78 (d, 1H); 2.32 (s, 3H); 2.25 (s, 3H).
Part B. Preparation of 2-(3-phenoxy-2-
methylphenyl)malononitrile.
CN CH3
NC
A solution of 2,3-dimethyldiphenylether (2.2 g, 11
mol), N-bromosuccinimide (1.76 g, 11 mmol) and benzoyl
peroxide (0.27 g, 1 mmol) in carbon tetrachloride (60 mL)
CA 02362380 2001-08-07
WO 00/56706 42 PCT/US00/07262
was refluxed for 2.5 h. The reaction mixture was added to
methylene chloride and extracted with saturated aqueous
sodium bisulfite, water (twice), and brine. The organic
layer was dried over sodium sulfate and the residue was
purified by chromatography on silica gel with hexane to
afford a mixture 3-bromomethyl-2-methyldiphenylether and 2-
bromomethyl-3-methyldiphenylether (2.0 g).
A solution of the above bromination products (2.0 g,
7.2 mmol) and tetraethylammonium cyanide (1.2 g , 7.7 mmol)
in dichloromethane (60 mL) was refluxed for 1 h. The
reaction was added to dichloromethane and extracted with 10%
aqueous sodium hydroxide (three times) and brine (twice).
After concentrating the organic layer, the residue was
purified by chromatography on silica gel with toluene and 5%
ethyl acetate in toluene to afford a 1:3 mixture of 2-(3-
phenoxy-2-methylphenyl)acetonitrile and 2-(2-phenoxy-6-
methylphenyl)acetonitrile (1.08 g). 1H-NMR (CDC13) 8: 6.7-
7.2 (m, 8H); 3.78 (s, 1.5H); 3.74(s, 0.5H); 2.48 (s, 2.25H);
2.28(s, 0.75H).
Methyllithium (4.3 mL of a 2.5 M solution, 10.7 mmol)
was added to a solution of diisopropylamine (1.5 mL, 10.7
mmole) in dry benzene (70 mL) cooled in an ice-water bath.
After stirring for 1 h, a solution of the above mixture of
phenylacetonitriles in dry benzene (30 mL) was added
dropwise. After stirring for 1 h at 0 °C, a solution of
2-chlorobenzylthiocyanate in dry benzene was added. After
stirring for 1 h while the reaction mixture warmed to room
temperature, the reaction mixture was added to benzene and
extracted with 10o aqueous sodium hydroxide (4X). The
combined aqueous layers were acidified to pH 1 with
concentrated hydrochloric acid and extracted with ethyl
acetate. The organic layer was dried over sodium sulfate,
concentrated, and purified twice by silica gel
chromatography with 0-10% ethyl acetate in toluene and then
10% ether in hexane, removing the high Rf major isomer and
affording isomerically pure title compound (61 mg) as a pale
yellow solid. 1NMR(CDC13) b: 7.2-7.4 (m, 4H); 7.13 (t, 1H);
CA 02362380 2001-08-07
WO 00/56706 43 PCT/US00/07262
6.9-7.0 (m, 3H); 5.10 (s, 1H); 2.40 (s, 3H). HRMS:
Calculated for C16H12N2~ (M): 248.0959; Found: 248.0950.
Part C. Preparation of Z- and E-OC-[amino[(2-
aminophenyl)thio]methylene]-2-methyl-3-
phenoxybenzeneacetonitrile.
A solution of 2-(3-phenoxy-2-methylphenyl)malononitrile
(50 mg, 0.20 mmol), 2-aminothiophenol (21 uL, 0.20 mmol) and
triethylamine (28 uL) in tetrahydrofuran was stirred under
nitrogen overnight. The reaction was added to water and
extracted with ethyl acetate. The combined organic layers
were washed with brine, dried over sodium sulfate, and
concentrated. The residue was purified by chromatography on
silica gel with 20-50% ether in hexanes to afford the title
compound as a colorless oil (30 mg). 1H-NMR (CDC13) was
consistent with the presence of a 1:1 mixture of isomers. 8:
7.49 (d, 0.5H); 6.7-7.4 (m, 11.5H); 4.83 (br s, 1H); 4.51
(br s, 1H); 4.41 (br s, 1H); 4.33 (br s, 1H); 2.31 (s,
1.5H); 2.25 (s, 1.5H). HRMS: Calculated for C22H20N3~S
(M+H): 374.1327; Found: 374.1307.
Example 5
Z- and E-oc-~aminof(2-amino~henyl)thiolmethylenel-4-chloro-(3-
(2-methylphenyl)benzenepro~anenitrile
NH2 CN / CI
H2N ~CH3
Part A. Preparation of 2-(2-
methylbenzylidene)malononitrile.
CA 02362380 2001-08-07
WO 00/56706 PCT/US00/07262
44
CN
NC \
CH3
A solution of 2-methylbenzaldehyde (9.6 mL, 83 mmol),
malononitrile (5.5 g, 83 mmol) and 3.5 M ammonium acetate in
acetic acid (2.4 mL, 8.4 mmol) in isopropanol (83 mL) was
stirred overnight at room temperature. A precipitate
formed. Water (100 mL) was added and the precipitate was
collected. The precipitate was washed with water (50 mL)
and vacuum dried to afford of 2-(2-
methylbenzylidene)malononitrile (12.7 g) as a white solid
(mp 105-106°). 1H-NMR (CDC13) 8: 8.10 (s 1H); 8.08 (d, 1H);
7.50 (dd, 1H); 7.30-7.40 (m, 2H); 2,45 (s, 3H).
Part B. Preparation of 2-[oc-(2-methylphenyl)-4-
chlorobenzyl]malononitrile.
CI
N
4-Chlorophenylmagnesium bromide (2.1 mL of a 1.0 M
solution in ether, 2.1 mmol) was added dropwise to a
solution of 2-(2-methylbenzylidene)malononitrile (0.32 g,
1.9 mmol) in dry tetrahydrofuran (7.5 mL) at 0 °C and
stirred for 1 h. Saturated aqueous ammonium chloride was
added and the layers were separated. The aqueous layer was
extracted twice with dichloromethane and the combined
organics layers were dried over magnesium sulfate. After
concentrating, the residue was purified by flash
chromatography on silica gel with 12.5% ethyl acetate in
hexanes to afford the title compound (0.25 g) as a white
solid (mp 132-140°). 1H-NMR (CDC13):8: 7.20-7.39 (m 8H);
4.78 (d, 1H); 2.25 (s, 3H). GC-MS: m/e = 281/283 (M+H).
CA 02362380 2001-08-07
WO 00/56706 PCT/US00/07262
Part C. Preparation of Z- and E-OC-[amino[(2-
aminophenyl)thio]methylene]-4-chloro-(3-(2-
methylphenyl)benzenepropanenitrile.
A solution of 2-[a,-(2-methylphenyl)-4-
chlorobenzyl]malononitrile (0.20 g, 0.71 mmol), 2-
aminothiophenol (0.11 mL, 1.00 mmol) and triethylamine
(0.14 mL, 1.00 mmol) in tetrahydrofuran (1.4 mL) was
stirred under nitrogen for 78 h. The reaction mixture
was absorbed onto silica gel and eluted with 20-30% ethyl
acetate in hexanes to afford the title compound (253 mg)
as a white foam (mp 63.5-72°). 1H-NMR (CDC13) was
consistent with the presence of a 4:6 mixture of isomers:
b 7.44 (d, 0.4H); 7.10-7.36 (m, 9.6H); 6.71-6.79 (m, 2H);
5.34 (s, 0.6H); 4.97 (s, 0.4H); 4.65 (br s, 1.2H); 4.44
(br s, 0.8H); 4.12 (br s, 2H); 2.33 (s, 1.8H); 2.22 (s,
1.2H). HRMS: Calcd for C23H21N3SC1 (M+H), 406.1145;
Found, 406.1130. Elem. Anal. Calcd for C23H20N3SC1: C,
68.05, H, 4.98; N, 10.35; S, 7.91. Found: C, 68.03, H,
5.09; N, 10.20; S, 7.97.
Examples 6-51.
The following compounds were prepared by procedures
similar to those described above.
Ex Nee
.
6 Z- and E-a-[amino[(2-aminophenyl)thio]
methylene]-4-chloro-2-methyl-~3-phenylbenzene-
propanenitrile
7 Z- and E-oC-[amino[(2-aminophenyl)thio]
methylene]-3-[(2,4-dinitrophenyl)hydroxy-
methyl]benzeneacetonitrile
8 Z- and E-oc-[amino[(2-aminophenyl)thio]
methylene]-3-[(4-carbo-methoxyphenyl)hydroxy-
methyl]benzeneaceto-nitrile
SUBSTITUTE SHEET (RULE 26)
CA 02362380 2001-08-07
WO 00/56706 PCT/US00/07262
46
~Z- and E-a-[am-ino[(2-aminophenyl)thio]
thylene]-3-[(4-nitrophenyl)hydroxymethyl]
benzeneacetonitrile
SUBSTITUTE SHEET (RULE 26)
CA 02362380 2001-08-07
WO 00/56706 4~ PCT/US00/07262
Z- and E-a,-[amino[(2-aminophenyl)thio]
methylene]-3-[(pentafluorophenyl)hydroxy-
methyl]benzeneacetonitrile
11 Z- and E-oc-[amino[(2-aminophenyl)thio]
methylene]-2-methyl-3-[(4-pyridyl)hydroxy-
methyl]benzeneacetonitrile
12 Z- and E-oc-[amino[(2-aminophenyl)thio]
methylene]-2-methyl-3-[(phenyl)hydroxymethyl]
benzeneacetonitrile
13 Z- and E-oc-[amino[(2-aminophenyl)thio]
methylene]-2-methyl-3-[(4-pyridyl)hydroxy-
methyl]benzeneacetonitrile
14 Z- and E-oc-[amino[(2-hydroxyphenyl)thio]
methylene]-3-[(4-cyanophenyl)hydroxy-
methyl]benzeneacetonitrile
Z- and E-a,-[amino[(2-aminophenyl)thio]
methylene]-3-[(3-nitrophenyl)hydroxy-
methyl]benzeneacetonitrile
16 Z- and E-oc-[amino[(2-aminophenyl)thio]
methylene]-2-methyl-3-[(pentafluorophenyl)
hydroxymethyl]benzeneacetonitrile
1~ Z- and E-oc-[amino[(2-hydroxyphenyl)thio]
methylene]-3-[(4-pyridyl)hydroxymethyl]
benzeneacetonitrile
18 Z- and E-oc-[amino[(2-aminophenyl)thio]
methylene]-3-[(2-trifluoromethylphenyl)
hydroxymethyl]benzeneacetonitrile
19 Z- and E-oc-[amino[(4-aminophenyl)thio]
methylene]-3-[(4-pyridyl)hydroxymethyl]
benzeneacetonitrile
Z- and E-oc-[amino[(4-hydroxyphenyl)thio]
methylene]-3-[(4-pyridyl)hydroxymethyl]
benzeneacetonitrile
SUBSTITUTE SHEET (RULE 26)
CA 02362380 2001-08-07
WO 00/56706 PCT/US00/07262
48
21 z- and E-oc-[amino[(2-aminophenyl)thio]
methylene]-3-[(3-cyanophenyl)hydroxymethyl]
benzeneacetonitrile
22 z- and E-oc-[amino[(4-aminophenyl)thio]
methylene]-3-[(4-cyanophenyl)hydroxymethyl]
benzeneacetonitrile
23 z- and E-oc-[amino(phenylthio)methylene]-3-
[(4-cyanophenyl)hydroxymethyl]benzene-
acetonitrile
24 z- and E-oc-[amino(phenylthio)methylene]-3-
[(4-pyridyl)hydroxymethyl]benzeneacetonitrile
25 z- and E-oc-[amino[(4-aminophenyl)thio]
methylene]-2-bromobenzeneacetonitrile
26 z- and E-oc-[amino[(2-aminophenyl)thio]
methylene]-3-[(2,4-dimethylphenyl)hydroxy-
methyl]benzeneacetonitrile
SUBSTITUTE SHEET (RULE 26)
CA 02362380 2001-08-07
WO 00/56706 PCT/US00/07262
49
2~ Z- and E-a-[amino[(2-aminophenyl)thio]
methylene]-3-[(phenyl)hydroxymethyl]benzene-
acetonitrile
28 Z- and E-oc-[amino[(2-aminophenyl)thio]
methylene]-3-[(2-thienyl)hydroxymethyl]
benzeneacetonitrile
29 Z- and E-a-[amino[(2-aminophenyl)thio]
methylene]-4-chloro-~3-phenylbenzenepropane-
nitrile
30 Z- and E-oc-[amino[(2-thienyl)thio]methylene]-
3-[(phenyl)hydroxymethyl]benzeneacetonitrile
31 Z- and E-a,-[amino[(2,4-diaminophenyl)thio]
methylene]-1-naphthyleneacetonitrile
32 Z- and E-a,-[amino[(2-aminophenyl)thio]
methylene]-2-methyl-(3-(4-pyridyl)benzenepro-
panenitrile
33 Z- and E-oc-[amino[(4-aminophenyl)thio]
methylene]-3-(benzyl)benzeneacetonitrile
34 Z- and E-a,-[amino[(2-naphthyl)thio]
methylene]-1-naphthyleneacetonitrile
35 Z- and E-a,-(amino[(2-aminophenyl)thio]
methylene]-3-(benzoyl)benzeneacetonitrile
36 Z- and E-a,-[amino[(2-aminophenyl)thio]
methylene]-(3-(1-methyl-2-pyrrolyl)benzene-
propanenitrile
3~ Z- and E-oc-[amino[(2-aminophenyl)thio]
methylene]-3-phenoxybenzeneacetonitrile
38 Z- and E-oc-[amino[(2-aminophenyl)thio]
methylene]-2-bromobenzeneacetonitrile
39 Z- and E-a-[amino[(2-aminophenyl)thio]
methylene]-3-[(2-furanyl)hydroxymethyl]
benzeneacetonitrile
SUBSTITUTE SHEET (RULE 26)
CA 02362380 2001-08-07
WO 00/56706 5~ PCT/US00/07262
40 z- and E-ot,-[amino[(2-thienyl)thio]methylene]-
3-[(2,3,4,5,6-pentafluorophenyl)hydroxy-
methyl]benzeneacetonitrile
41 Z- and E-oc-[amino[(2-aminophenyl)thio]
methylene]-3-[(3-methyl-2-pyridyl)hydroxy-
methyl]benzeneacetonitrile
42 z- and E-oc-[amino[(4-aminophenyl)thio]
methylene]-2-methylbenzeneacetonitrile
SUBSTfTUTE SHEET (RULE 26)
CA 02362380 2001-08-07
WO 00/56706 51 PCT/US00/07262
43 Z- and E-oc-[amino[(4-aminophenyl)thio]
methylene]-4-(1,1-dimethylethyl)benzeneaceto-
nitrile
44 Z- and E-a-[amino[(4-aminophenyl)thio]
methylene]-1-naphthyleneacetonitrile
45 Z- and E-a-[amino[(2-aminophenyl)thio]
methylene]-3-(trifluoromethyl)benzeneaceto-
nitrile
46 Z- and E-oc-[amino[(2-aminophenyl)thio]
methylene]-1-naphthyleneacetonitrile
47 Z- and E-oc-[amino[(2-aminophenyl)thio]
methylene]-2-(trifluoromethyl)benzeneaceto-
nitrile
48 Z- and E-a-[amino[(4-aminophenyl)thio]
methylene]-4-methylbenzeneacetonitrile
49 Z- and E-a-[amino[(2-aminophenyl)thio]
methylene]-2-methylbenzeneacetonitrile
50 Z- and E-a-[amino[(2-fluorophenyl)thio]
methylene]-1-naphthyleneacetonitrile
51 Z- and E-oc-[amino[(2-aminophenyl)thio]
methylene]-3-phenyl benzeneacetonitrile
Example FORMULA HRMS HRMS
Calculated* Found*
6 C23H20C1N3S 406.1145 406.1148
7 C22H17N5O5S 464.1029 464.1024
8 C24H21N3O3S 432.1382 432.1364
9 C22H18N4O3S 419.1178 419.1168
C22H14F5N30S 464.0856 464.0827
11 C22H20N40S 389.1436 389.1428
12 C23H21N30S 388.1484 388.1471
13 C22H20N40S 388.1436 388.1436
14 C23H17N3O2S 400.1120 400.1105
C22H18N4O3S 419.1179 419.1156
16 C23H16F5N30S 478.1013 478.1002
SUBSTITUTE SHEET (RULE 263
CA 02362380 2001-08-07
WO 00/56706 52 PCT/US00/07262
17 C21H18N40S 376.1120 376.1103
18 C23H18F3N30S 442.1201 442.1200
SUBSTITUTE SHEET (RULE 26~
CA 02362380 2001-08-07
WO 00/56706 53 PCT/US00/07262
19 C21H18N40S ~ 375.1280 375.1268
20 C21H17N30S 376.1120 376.1104
21 C23H18N40S 399.1280 399.1273
22 C23H18N40S 399.1280 399.1265
24 C21H17N30S 360.1171 360.1172
25 C15H12BrN3S 360.1171 360.1172
26 C24H23N30S 402.1640 402.1648
27 C22H19N30S 374.1327 374.1327
28 C20H17N30S2 379.0813 (M+) 379.0799 (M+)
29 C22H18C1N3S 392.0988 392.0982
30 C20H16N20S2 365.0782 365.0763
31 C19H16N4S 333.1174 333.1166
32 C22H20N4S 373.1496 373.1487
33 C22H19N3S 358.1378 358.1393
34 C23H17N3S 368.1221 368.1227
35 C22H17N30S 372.1171 372.1160
36 C21H20N4S 361.1487 361.1479
37 C21H17N30S 360.1171 360.1144
39 C20H17N302S 364.1120 364.1097
40 C20H11F5N20S2 455.0311 455.0305
41 C22H20N40S 389.1436 389.1431
43 C19H21N3S 324.1534 324.1524
44 C19H15N3S 318.1065 318.1076
45 C16H12F3N3S 336.0782 336.0782
46 C19H15N3S 318.7065 318.1048
47 C16H12F3N3S 336.0782 336.0776
51 C21H17N3S 344.1221 334.1208
_..,- _, r
w.ui~.u.i.cmcu 1Vt 1~1'rn 11i11C5S IlOLeQ.
Example Solvent Chemical Shift
23 CDC13 7.15-7.65 (m, 13H); 5.85&2.88 (2 d, 1H);
4.61&4.88 (2 br s, 2H); 2.50&2.59 (2 d,
2H) .
25 CDC13 6.64-7.68 (m, 8H); 3.95-4.81 (3 br s,
4H) .
SUBSTITUTE SHEET (RULE 26)
CA 02362380 2001-08-07
WO 00/56706 PCT/US00/07262
54
38 CDC13 6.54-7.71 (m, 8H); 4.37-4.90 (4 br s,
4H) .
42 CDC13 6.56-7.43 (m, 8H); 3.75-4.70 (4 br s,
4H); 2.37&2.40 (2 s, 3H).
SUBSTfiTUTE SHEET (RULE 2G~
CA 02362380 2001-08-07
WO 00/56706 PCT/US00/07262
48 CDC13 6.57-7.41 (m, 8H); 3.75-4.74 (4 br s, 4H);
2.33&2.34 (2 s, 3H).
49 CDC13 6.72-7.54 (m, 8H); 4.26-4.80 (3 br s, 4H);
2.36&2.43 (2 s, 3H).
SUBSTITUTE SHEET (RULE 26)
CA 02362380 2001-08-07
WO 00/56706 56 PCT/US00/07262
UTILITY
Compounds of the present invention are inhibitors of
the dual-specificity kinase MEK1/MEK2 (Mapk or Erk kinase,
where Mapk=mitogen-activated protein kinase) and are
expected to be useful for treating proliferative diseases,
e.g. cancer, psoriasis, restenosis or atherosclerosis, and
also autoimmune diseases. The presently claimed MEK
inhibitors are also expected to have utility as
radiosensitizers for the treatment of solid tumors. In
addition, the presently claimed compounds are expected to
have utility for the treatment of chronic pain or for
inhibiting memory acquisition. Assays for chronic pain are
found Science and Medicine (1996), Nov./Dec., 22-31. Assays
for mammalian associative learning are found in Nature
Neuroscience (1998) 1 (1) 602-609.
The ERK signal transduction pathway includes two very
similar forms of MEK, MEK1 and MEK2, and two similar forms
of ERK, ERK1 and ERK2. To block signal transduction via the
ERK pathway, a MEK inhibitor must prevent ERK1 and/or ERK2
from being phosphorylated (and thereby activated) by the
kinases MEK1 or MEK2. The different roles played by MEK1
and MEK2 and by ERK1 and ERK2 are not currently understood
well understood and they may be redundant under some or all
circumstances. MEK1 and MEK2 are phosphorylated (and
thereby activated) by an upstream kinase, RAF. Since MEK1
or MEK2 have little ability to phosphorylate ERK1 and ERK2
until they have been phosphorylated and since they are
usually isolated in their unphosphorylated state, it is
difficult to obtain adequate quantities of phosphorylated
MEK1 or MEK2 suitable for assaying many compounds. To make
assays more practical, a constitutively active mutant of
MEK1 (e. g., 2X-MEK1)(see J. Biol. Chem. (1998) 29, 18623-
18632) was initially used to characterize the MEK inhibitors
of this invention. This mutant enzyme has negatively-
charged residues at the residues which are normally
phosphorylated by RAF. Selected inhibitors of 2X-MEK1
disclosed herein have been shown to be inhibitors of
phosphorylated (i.e., active) wild-type MEK1 and MEK2.
CA 02362380 2001-08-07
WO 00/56706 5~ PCT/US00/07262
Furthermore, many of the MEK inhibitors of this invention
have been shown to be capable of blocking phosphorylation of
ERK induced by treatment of Jurkat cells with TPA (see J.
Biol. Chem. (1998) 29, 18623-18632).
Selected MEK inhibitors from this invention have also
been shown to block the upregulation of AP-1 expression in
Cos-7 cells induced by stimulation with TPA (see J. Biol.
Chem. (1998) 29, 18623-18632). AP-1 in turn regulates the
expression of a number of pro-inflammatory and growth-
stimulating genes including. These experiments prove that
inhibitors of 2X-MEK1 function as inhibitors of MEK and ERK
signal transduction in cell culture.
DOSAGE AND FORMULATION
The compounds of this invention can be administered in
such oral dosage forms as tablets, capsules (each of which
includes sustained release or timed release formulations),
pills, powders, granules, elixirs, tinctures, suspensions,
syrups, and emulsions. They may also be administered in
intravenous (bolus or infusion), intraperitoneal,
subcutaneous, or intramuscular form, all using dosage forms
well known to those of ordinary skill in the pharmaceutical
arts. They can be administered alone, but generally will be
administered with a pharmaceutical carrier selected on the
basis of the chosen route of administration and standard
pharmaceutical practice.
The dosage regimen for the compounds of the present
invention will, of course, vary depending upon known
factors, such as the pharmacodynamic characteristics of the
particular agent and its mode and route of administration;
the species, age, sex, health, medical condition, and weight
of the recipient; the nature and extent of the symptoms; the
kind of concurrent treatment; the frequency of treatment;
the route of administration, the renal and hepatic function
of the patient, and the effect desired. A physician or
veterinarian can determine and prescribe the effective
amount of the drug required to prevent, counter, or arrest
the progress of the thromboembolic disorder.
CA 02362380 2001-08-07
WO 00/56706 5g PCT/US00/07262
By way of general guidance, the daily oral dosage of
each active ingredient, when used for the indicated effects,
will range between about 0.001 to 1000 mg/kg of body weight,
preferably between about 0.01 to 100 mg/kg of body weight
per day, and most preferably between about 1.0 to 20
mg/kg/day. Intravenously, the most preferred doses will
range from about 1 to about 10 mg/kg/minute during a
constant rate infusion. Compounds of this invention may be
administered in a single daily dose, or the total daily
dosage may be administered in divided doses of two, three,
or four times daily.
Compounds of this invention can be administered in
intranasal form via topical use of suitable intranasal
vehicles, or via transdermal routes, using transdermal skin
patches. When administered in the form of a transdermal
delivery system, the dosage administration will, of course,
be continuous rather than intermittent throughout the dosage
regimen.
The compounds are typically administered in admixture
with suitable pharmaceutical diluents, excipients, or
carriers (collectively referred to herein as pharmaceutical
carriers) suitably selected with respect to the intended
form of administration, that is, oral tablets, capsules,
elixirs, and syrups, and consistent with conventional
pharmaceutical practices.
For instance, for oral administration in the form of a
tablet or capsule, the active drug component can be combined
with an oral, non-toxic, pharmaceutically acceptable, inert
carrier such as lactose, starch, sucrose, glucose, methyl
callulose, magnesium stearate, dicalcium phosphate, calcium
sulfate, mannitol, and sorbitol; for oral administration in
liquid form, the oral drug components can be combined with
any oral, non-toxic, pharmaceutically acceptable inert
carrier such as ethanol, glycerol, and water. Moreover,
when desired or necessary, suitable binders, lubricants,
disintegrating agents, and coloring agents can also be
incorporated into the mixture. Suitable binders include
starch, gelatin, natural sugars such as glucose or beta-
CA 02362380 2001-08-07
WO 00/56706 59 PCT/US00/07262
lactose, corn sweeteners, natural and synthetic gums such as
acacia, tragacanth, or sodium alginate,
carboxymethylcellulose, polyethylene glycol, and waxes.
Lubricants used in these dosage forms include sodium oleate,
sodium stearate, magnesium stearate, sodium benzoate, sodium
acetate, and sodium chloride. Disintegrators include,
without limitation, starch, methyl cellulose, agar,
bentonite, and xanthan gum.
The compounds of the present invention can also be
administered in the form of liposome delivery systems, such
as small unilamellar vesicles, large unilamellar vesicles,
and multilamellar vesicles. Liposomes can be formed from a
variety of phospholipids, such as cholesterol, stearylamine,
or phosphatidylcholines.
Compounds of the present invention may also be coupled
with soluble polymers as targetable drug carriers. Such
polymers can include polyvinylpyrrolidone, pyran copolymer,
polyhydroxypropylmethacrylamide-phenol,
polyhydroxyethylaspartamidephenol, or polyethyleneoxide-
polylysine substituted with palmitoyl residues.
Furthermore, the compounds of the present invention may be
coupled to a class of biodegradable polymers useful in
achieving controlled release of a drug, for example,
polylactic acid, polyglycolic acid, copolymers of polylactic
and polyglycolic acid, polyepsilon caprolactone, polyhydroxy
butyric acid, polyorthoesters, polyacetals,
polydihydropyrans, polycyanoacylates, and crosslinked or
amphipathic block copolymers of hydrogels.
Dosage forms (pharmaceutical compositions) suitable for
administration may contain from about 1 milligram to about
100 milligrams of active ingredient per dosage unit. In
these pharmaceutical compositions the active ingredient will
ordinarily be present in an amount of about 0.5-95% by
weight based on the total weight of the composition.
Gelatin capsules may contain the active ingredient and
powdered carriers, such as lactose, starch, cellulose
derivatives, magnesium stearate, and stearic acid. Similar
diluents can be used to make compressed tablets. Both
CA 02362380 2001-08-07
WO 00/56706 6~ PCT/US00/07262
tablets and capsules can be manufactured as sustained
release products to provide for continuous release of
medication over a period of hours. Compressed tablets can
be sugar coated or film coated to mask any unpleasant taste
and protect the tablet from the atmosphere, or enteric
coated for selective disintegration in the gastrointestinal
tract.
Liquid dosage forms for oral administration can contain
coloring and flavoring to increase patient acceptance.
In general, water, a suitable oil, saline, aqueous
dextrose (glucose), and related sugar solutions and glycols
such as propylene glycol or polyethylene glycols are
suitable carriers for parenteral solutions. Solutions for
parenteral administration preferably contain a water soluble
salt of the active ingredient, suitable stabilizing agents,
and if necessary, buffer substances. Antioxidizing agents
such as sodium bisulfate, sodium sulfite, or ascorbic acid,
either alone or combined, are suitable stabilizing agents.
Also used are citric acid and its salts and sodium EDTA. In
addition, parenteral solutions can contain preservatives,
such as benzalkonium chloride, methyl- or propyl-paraben,
and chlorobutanol.
Suitable pharmaceutical carriers are described in
Remington's Pharmaceutical Sciences, Mack Publishing
Company, a standard reference text in this field.
Representative useful pharmaceutical dosage-forms for
administration of the compounds of this invention can be
illustrated as follows:
Capsules
A large number of unit capsules can be prepared by
filling standard two-piece hard gelatin capsules each with
100 milligrams of powdered active ingredient, 150 milligrams
of lactose, 50 milligrams of cellulose, and 6 milligrams
magnesium stearate.
Soft Gelatin Capsules
A mixture of active ingredient in a digestable oil such
as soybean oil, cottonseed oil or olive oil may be prepared
and injected by means of a positive displacement pump into
CA 02362380 2001-08-07
WO 00/56706 61 PCT/US00/07262
gelatin to form soft gelatin capsules containing 100
milligrams of the active ingredient. The capsules should be
washed and dried.
Tablets
Tablets may be prepared by conventional procedures so
that the dosage unit is 100 milligrams of active ingredient,
0.2 milligrams of colloidal silicon dioxide, 5 milligrams of
magnesium stearate, 275 milligrams of microcrystalline
cellulose, 11 milligrams of starch and 98.8 milligrams of
lactose. Appropriate coatings may be applied to increase
palatability or delay absorption.
In~ectable
A parenteral composition suitable for administration by
injection may be prepared by stirring 1.5o by weight of
active ingredient in 10% by volume propylene glycol and
water. The solution should be made isotonic with sodium
chloride and sterilized.
Suspension
An aqueous suspension can be prepared for oral
administration so that each 5 mL contain 100 mg of finely
divided active ingredient, 200 mg of sodium carboxymethyl
cellulose, 5 mg of sodium benzoate, 1.0 g of sorbitol
solution, U.S.P., and 0.025 mL of vanillin.
Where the compounds of this invention are combined with
other anticoagulant agents, for example, a daily dosage may
be about 0.1 to 100 milligrams of the compound of Formula I
and about 1 to 7.5 milligrams of the second anticoagulant,
per kilogram of patient body weight. For a tablet dosage
form, the compounds of this invention generally may be
present in an amount of about 5 to 10 milligrams per dosage
unit, and the second anti-coagulant in an amount of about 1
to 5 milligrams per dosage unit.
Where the compounds of Formula I are administered in
combination with an anti-platelet agent, by way of general
guidance, typically a daily dosage may be about 0.01 to 25
milligrams of the compound of Formula I and about 50 to 150
milligrams of the anti-platelet agent, preferably about 0.1
to 1 milligrams of the compound of Formula I and about 1 to
CA 02362380 2001-08-07
WO 00/56706 62 PCT/US00/07262
3 milligrams of antiplatelet agents, per kilogram of patient
body weight.
Where the compounds of Formula I are adminstered in
combination with thrombolytic agent, typically a daily
dosage may be about 0.1 to 1 milligrams of the compound of
Formula I, per kilogram of patient body weight and, in the
case of the thrombolytic agents, the usual dosage of the
thrombolyic agent when administered alone may be reduced by
about 70-80o when administered with a compound of Formula I.
Where two or more of the foregoing second therapeutic
agents are administered with the compound of Formula I,
generally the amount of each component in a typical daily
dosage and typical dosage form may be reduced relative to
the usual dosage of the agent when administered alone, in
view of the additive or synergistic effect of the
therapeutic agents when administered in combination.
Particularly when provided as a single dosage unit, the
potential exists for a chemical interaction between the
combined active ingredients. For this reason, when the
compound of Formula I and a second therapeutic agent are
combined in a single dosage unit they are formulated such
that although the active ingredients are combined in a
single dosage unit, the physical contact between the active
ingredients is minimized (that is, reduced). For example,
one active ingredient may be enteric coated. By enteric
coating one of the active ingredients, it is possible not
only to minimize the contact between the combined active
ingredients, but also, it is possible to control the release
of one of these components in the gastrointestinal tract
such that one of these components is not released in the
stomach but rather is released in the intestines. One of
the active ingredients may also be coated with a material
which effects a sustained-release throughout the
gastrointestinal tract and also serves to minimize physical
contact between the combined active ingredients.
Furthermore, the sustained-released component can be
additionally enteric coated such that the release of this
component occurs only in the intestine. Still another
CA 02362380 2001-08-07
WO 00/56706 63 PCT/IJS00/07262
approach would involve the formulation of a combination
product in which the one component is coated with a
sustained and/or enteric release polymer, and the other
component is also coated with a polymer such as a low-
s viscosity grade of hydroxypropyl methylcellulose (HPMC) or
other appropriate materials as known in the art, in order to
further separate the active components. The polymer
coating serves to form an additional barrier to interaction
with the other component.
These as well as other ways of minimizing contact
between the components of combination products of the
present invention, whether administered in a single dosage
form or administered in separate forms but at the same time
by the same manner, will be readily apparent to those
skilled in the art, once armed with the present disclosure.
Obviously, numerous modifications and variations of the
present invention are possible in light of the above
teachings. It is therefore to be understood that within the
scope of the appended claims, the invention may be practiced
otherwise that as specifically described herein.