Note: Descriptions are shown in the official language in which they were submitted.
CA 02364253 2001-08-29
WO 00/53173 PCT/iIS00/02626
TITLE OF THE INVENTION
DIHYDROXY OPEN-ACID AND SALTS OF HMG-CO-A REDUCTASE
INHIBITORS
FIELD OF THE INVENTION
The instant invention relates to the use of dihydroxy open acid statins
and salts and esters thereof, which are inhibitors of 3-hydroxy-3-
methylglutaryl
coenzyme A (HMG-CoA) reductase, in such a way so as to minimize their in vivo
lactonization, and to a particular crystalline hydrated form of the calcium
salt of
dihydroxy open acid simvastatin referred to herein as compound I.
BACKGROUND OF THE INVENTION
It has been clear for several decades that elevated blood cholesterol is a
major risk factor for coronary heart disease (CHD), and many studies have
shown that
the risk of CHD events can be reduced by lipid-lowering therapy. Prior to
1987, the
lipid-lowering armamentarium was limited essentially to a low saturated fat
and
cholesterol diet, the bile acid sequestrants (cholestyramine and colestipol),
nicotinic
acid (niacin), the fibrates and probucol. Unfortunately, all of these
treatments have
limited efficacy or tolerability, or both. With the introduction of lovastatin
(MEVACOR~; see US Patent No. 4,231,938), the first inhibitor of HMG-CoA
reductase to become available for prescription in 1987, for the first time
physicians
were able to obtain comparatively large reductions in plasma cholesterol with
very
few adverse effects.
In addition to the natural fermentation products, mevastatin and
lovastatin, there are now a variety of semi-synthetic and totally synthetic
HMG-CoA
reductase inhibitors, including simvastatin (ZOCOR~; see US Patent No.
4,444,784),
pravastatin sodium salt (PRAVACHOL~; see US Patent No. 4,346,227), fluvastatin
sodium salt (LESCOL~; see US Patent No. 5,354,772), atorvastatin calcium salt
(LIPITOR~; see US Patent No. 5,273,995) and cerivastatin sodium salt (also
known
as rivastatin; see US Patent No. 5,177,080). The structural formulas of these
and
additional HMG-CoA reductase inhibitors, are described at page 87 of M.
Yalpani,
"Cholesterol Lowering Drugs", Chemistry & Industry, pp. 85-89 (5 February
1996).
The HMG-CoA reductase inhibitors described above belong to a structural class
of
compounds which contain a moiety which can exist as either a 3-hydroxy lactone
ring
or as the corresponding ring opened dihydroxy open-acid, and are often
referred to as
-1-
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
"statins." An illustration of the lactone portion of a statin and its
corresponding open-
acid form is shown below.
HO p H
'COON
O OH
Lactone Dihydroxy Open-Acid
Salts of the dihydroxy open-acid can be prepared, and in fact, as noted above,
several
of the marketed statins are administered as the dihydroxy open acid salt
forms.
Lovastatin and simvastatin are marketed worldwide in their lactonized form.
Lovastatin is shown as structural formula II, and simvastatin is shown as
structural
formula III, below.
HO O HO O
O ~O O O
O H ~'~' ~ H
II III
The lactonized forms of the statins are not active inhibitors of HMG-
CoA reductase, but the dihydroxy open acid forms are. It is known that
condensation
of the dihydroxy open acid form of statins to the corresponding lactonized
form
occurs under acidic conditions, that is at about pH 4 or under. Therefore, due
to the
low gastric pH of the stomach, a statin conventionally administered by oral
dosing in
its lactone form will remain largely in its lactone form in the stomach. The
vast
majority of the drug will still be in the lactone form at the time of
absorption from the
intestine following oral dosing with the lactose. After absorption, the
lactose enters
the liver and it is in the hepatocytes that the lactose can be metabolized to
the active
-2-
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
open acid form, a reaction catalyzed by two hepatic esterases or "lactonases,"
one
which is in the cytosolic and the other in the microsomal fraction. Once in
the blood
there is an additional plasma esterase which can also hydrolyze the lactone to
the open
acid. There may be some minimal chemical, i.e., non-enzymatic, hydrolysis that
occurs in blood or in the liver; however, at the pH in blood and liver, there
should not
be any lactonization, i.e., conversion of open acid back to the lactone.
A statin conventionally administered by oral dosing in its dihydroxy
open acid form or a pharmaceutically acceptable salt or ester thereof will
tend to
convert to its lactone form in the acidic environment of the stomach, so that
a mixture
of the open ring and the corresponding closed ring forms will co-exist there.
For
example, see M.J. Kaufman, International Journal of Pharmaceutics, 1990,
66(Dec 1 ),
p. 97-106, which provides hydrolysis data that are used to simulate the extent
of drug
degradation that occurs in acidic gastric fluids following oral administration
of several
structurally related hypocholesterolemic agents, including simvastatin and
lovastatin,
and also see A.S. Kearney, et al., Pharmaceutical Research, 1993, 10(10), p.
1461-
1465, which describes the interconversion kinetics and equilibrium of CI-981
(atorvastatin in its free acid form). Therefore, even after conventional oral
dosing
with a dihydroxy open acid statin or a salt or ester thereof, a mixture of the
open acid
and the corresponding lactone form of the drug could exist by the time of
absorption
from the intestine.
The preparation of the naturally occurring compound lovastatin and the
semi-synthetic analog simvastatin leads to a mixture of the lactone and the
open-ring
dihydroxy acid forms. Several procedures have been published describing ways
to
make simvastatin from lovastatin, and most proceed through a lactone ring
opening
step at some point in the process and sometimes formation of a salt at the
resulting
carboxy acid, and end with a ring-closing step in order to make the final
simvastatin
product. For example, U.S. Patent No. 4,820,850 describes a process for making
simvastatin which involves opening the lactone ring of lovastatin and forming
an
alkyl-amide at the resulting carboxy acid, followed by protection of the two
hydroxy
groups and methylation of the 8' acyl sidechain. After the methylation step,
the
hydroxy protecting groups are removed, the amide is hydrolyzed to the free
acid and
an ammonium salt of the free acid is formed, followed by a step to re-
lactonize the
ring. In U.S. Patent No. 4,444,784, the 8'-acyl sidechain of lovastatin is
removed and
the lactone ring opened in the first step, followed by re-lactonization of the
ring and
protection of its hydroxy group. Next, the 8' position is acylated to
introduce the
-3-
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
simvastatin sidechain and a deprotection step is performed to obtain the
simvastatin
final product. In another process disclosed in U.S. Patent No. 4,582,915, the
potassium salt of the ring opened form of lovastatin is methylated at the 8'
acyl
sidechain, the free acid is then re-generated, and the dihydroxy open acid
moiety is re-
lactonized.
Since becoming available, millions of doses of simvastatin have been
administered and these drugs have developed an excellent safety record.
However, as
noted in the Physician's Desk Reference (PDR), in rare instances myopathy has
been
associated with the use of all statins, including simvastatin. The mechanism
for
statin-related myopathy is currently poorly understood. It is also known that
many
drugs, including certain statins such as simvastatin, are metabolised in the
liver and
intestine by the cytochrome P450 3A4 (CYP 3A4) enzyme system. As also noted in
the PDR, there are adverse drug interaction concerns if a potent inhibitor of
CYP3A4,
such as itraconazole, and a CYP3A4-metabolized statin are used together, and
some
cases of myopathy were found to have occurred in patients taking such a drug
combination. Simvastatin has been administered to over 20 million patients
worldwide in the past 11 years and has been demonstrated to be remarkably
safe.
However, the very low risk of myopathy is substantially increased when
simvastatin is
given together with potent inhibitors of CYP3A4While the overall safety record
for
simvastatin is exceptional, it would be desirable to further optimize its
safety profile
by reducing the potential for drug interactions with inhibitors of CYP3A4. It
would
also be desirable to further reduce the already low rate of occurrence of
myopathy
associated with the use of all statins. Statins are among the most widely used
drugs in
the world, and therefor the benefit of any further optimization of their
safety profile
would be significant.
SUMMARY OF THE INVENTION
One object of this invention is to minimize or eliminate the in vivo
lactonization of a dihydroxy open-acid statin. For oral administration, the
dihydroxy
open-acid statin or a pharmaceutically acceptable salt or ester thereof is to
be
administered so as to minimize formation of lactonized statin and thereby
minimize
the amount of lactonized statin that is absorbed from the intestine while
maximizing
the amount of dihydroxy open-acid statin that is absorbed from the intestine.
Accordingly, this invention involves a method of inhibiting HMG-CoA
reductase with an effective inhibitory amount of an orally dosed statin
comprising
-4-
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
delivering at least 90% of the dosed statin in its dihydroXy open acid form to
the
intestinal mucosa of a patient in need of such treatment.
The instant invention further provides a method for inhibiting HMG-
CoA reductase, as well as for treating and/or reducing the risk for diseases
and
conditions affected by inhibition of HMG-CoA reductase, comprising orally
administering a therapeutically effective amount of a compound selected from a
dihydroxy open acid statin and a pharmaceutically acceptable salt or ester
thereof in a
delayed-release pharmaceutical dosage form to a patient in need of such
treatment
wherein substantial release of the compound from the dosage form is delayed
until
after passage of the dosage form through the stomach.
One embodiment of the first object is to provide the above-described
method wherein the delayed-release pharmaceutical dosage form is a gel
extrusion
module (GEM) drug delivery device.
Another embodiment of the first object is to provide the above-
described method wherein the delayed-release pharmaceutical dosage form is an
enterically coated pharmaceutical dosage form. An additional aspect of this
embodiment is to provide the above-described method wherein the delayed-
release
pharmaceutical dosage form is selected from an enterically coated rapid-
release
pharmaceutical dosage form and an enterically coated time controlled-release
pharmaceutical dosage form. Yet another aspect of this embodiment is to
provide the
above-described method wherein the delayed-release pharmaceutical dosage form
is
an enterically coated gel extrusion module (GEM) drug delivery device.
The compound used for the above-described obj ect and embodiments
may particularly be dihydroxy open acid simvastatin and the pharmaceutically
acceptable salts thereof, and mare particularly a pharmaceutically acceptable
salt
thereof.
A second object of the instant invention is to provide novel HMG-CoA
reductase inhibitors which are crystalline hydrated forms of the calcium salt
of
dihydroxy open acid simvastatin, and particularly the compound referred to
herein as
compound I.
Additional objects are to provide the use of the crystalline hydrated
forms of the calcium salt of dihydroxy open acid simvastatin, particularly
compound
I, for inhibiting HMG-CoA reductase, as well as for treating and/or reducing
the risk
for diseases and conditions affected by inhibition of HMG-CoA reductase, and
also to
provide pharmaceutical formulations, including conventional rapid-release,
delayed-
-5-
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
release and time controlled-release formulations, including the GEM drug
delivery
device and enterically coated dosage forms, that can be used with the
compounds. A
further object is to provide a process for making compound I. Additional
objects will
be evident from the following detailed description.
BRIEF DESCRIPTION OF THE DRAWINGS
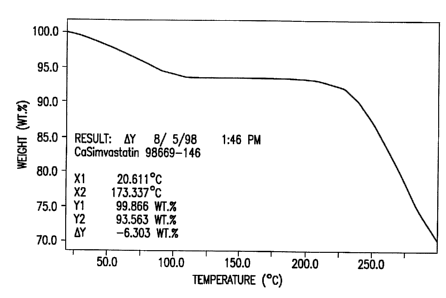
Figure 1 is a thermogravimetry (TG) weight loss curve for compound I
obtained under a nitrogen flow at a heating rate of 10°C/minute.
Figure 2 is a differential scanning calorimetry (DSC) curve for
compound I obtained under a nitrogen flow bubbled through 16.0°C water
at a heating
rate of 10°C/minute in an open cup.
Figure 3 is an x-ray powder diffraction (XRPD) pattern for compound
I. The XRPD pattern was obtained using CuKa, radiation. The ordinate or Y-axis
is
x-ray intensity (cpm) and the abscissa or X-axis is the angle two-theta (28)
in degrees.
Figure 4 is a solid-state 13C nuclear magnetic resonance spectrum for
Compound I.
DETAILED DESCRIPTION OF THE INVENTION
Applicants have now discovered that dihydroxy open acid statins may
be less reliant on CYP3A4 metabolism than their closed ring lactonized
counterparts.
The instant invention involves methods and pharmaceutical compositions for
orally
administering open-ring dihydroxy open acid statins and salts and esters
thereof,
which are HMG-CoA reductase inhibitors, in such a way so as to minimize
conversion to their lactonized counterparts. This allows for delivery of a
dihydroxy
open acid statin without its lactone counterpart directly to the absorptive
mucosa of
the small intestine thus allowing for absorption of the open acid statin into
the portal
circulation, penetration by active open acid statin into hepatocytes to
achieve
enhanced efficacy, and systemic exposure consisting of open acid moieties.
More
particularly, delayed-release of an orally administered dihydroxy open acid
statin or a
pharmaceutically acceptable salt or ester thereof, for example dihydroxy open
acid
simvastatin or a salt thereof, until after passage through the stomach reduces
the
amount of lactone formed and absorbed in the body. Maintaining the statin in
its open
acid form in the body thereby reduces the potential for drug interactions
between
statins whose metabolism is CYP3A4-mediated and other active agents which
inhibit
the CYP3A4 enzymatic pathway, and also can provide enhanced efficacy.
Moreover,
-6-
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
maintaining the statin in its open acid form in the body may have additional
clinical
benefits for all statins, even for those statins that are not significantly
metabolized by
the CYP3A4 enzymatic pathway.
In addition, a novel crystalline hydrated form of the calcium salt of
dihydroxy open acid simvastatin has now been discovered to be a
pharmaceutically
suitable salt form having desirable physical properties for formulation into
an anti-
hypercholesterolemic drug composition.
The term "statin(s)" as used herein is intended to be defined as
inhibitors of HMG-CoA reductase which belong to a structural class of
compounds
that contain a moiety which can exist as either a 3-hydroxy lactone ring or as
the
corresponding ring opened dihydroxy open acid, wherein the lactone portion of
the
statin and its corresponding dihydroxy open-acid form is shown below.
HO O H
'COOH
O OH
Lactone Dihydroxy Open-Acid
All hydrates, solvates and polymorphic crystalline forms of HMG-CoA reductase
inhibitors having the above-described lactone/dihydroxy open acid moiety are
included within the scope of the term "statin(s)". Pharmaceutically acceptable
salts
and esters of the dihydroxy open-acid statins are included within the scope of
the term
"statin(s)".
Statins inhibit HMG-CoA reductase in their dihydroxy open acid form.
Compounds which have inhibitory activity for HMG-CoA reductase can be readily
identified by using assays well-known in the art. For example, see the assays
described or cited in U.S. Patent 4,231,938 at col. 6, and WO 84/02131 at pp.
30-33.
The term "dihydroxy open acid statin(s)" is intended to be defined as
statins which contain the dihydroxy open acid moiety, including
pharmaceutically
acceptable salts and esters thereof. The phrases "dihydroxy open acid
statin(s)" and
"dihydroxy open acid statin(s) and the pharmaceutically acceptable salts and
esters
thereof' are used interchangeably herein and are both intended to encompass
the open
acid and salt and ester forms of the open acid of the statin, unless otherwise
indicated.
7_
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
All hydrates, solvates and polymorphic crystalline forms are encompassed
within the
the scope of the term "dihydroxy open acid statin(s)."
In the broadest embodiment, any dihydroxy open acid statin or a
pharmaceutically acceptable salt or ester thereof may be used with the present
S invention. Examples of dihydroxy open acid statins that may be used with the
present
invention include but are not limited to the dihydroxy open acid forms and
pharmaceutically acceptable salts and esters thereof o~ lovastatin (see US
Patent No.
4,342,767); simvastatin (see US Patent No. 4,444,784), pravastatin,
particularly the
sodium salt thereof; fluvastatin particularly the sodium salt thereof;
atorvastatin,
particularly the calcium salt thereof; cerivastatin, particularly the sodium
salt thereof,
nisvastatin also referred to as NK-104 (see PCT international publication
number WO
97/23200) and ZD-4522 (see US Patent No. 5,260,440, and Drugs of the Future,
1999, 24(5), pp. 511-513).
In a narrower embodiment, any dihydroxy open acid statin or a
pharmaceutically acceptable salt or ester thereof may be used with the present
invention, such as those listed above, provided the statin is not pravastatin
or
fluvastatin. In a class of this embodiment, the open acid statin includes
dihydroxy
open acid lovastatin, simvastatin, atorvastatin, cerivastatin, nisvastatin and
pharmaceutically acceptable salts thereof. Pharmaceutically acceptable salts
of
dihydroxy open acid simvastatin, particularly the ammonium and calcium salt
forms
thereof, are preferred for use in the methods and compositions of this
invention. More
particularly, the calcium salt of dihydroxy open acid simvastatin includes the
crystalline hydrated forms of the calcium salt of dihydroxy open acid
simvastatin, and
more particularly the hydrated calcium salt of dihydroxy open acid simvastatin
referred to herein as compound I.
Herein, the term "pharmaceutically acceptable salts" shall mean non-
toxic salts of the compounds employed in this invention which are generally
prepared
by reacting the free acid with a suitable organic or inorganic base,
particularly those
formed from cations such as sodium, potassium, aluminum, calcium, lithium,
magnesium, zinc and tetramethylammonium, as well as those salts formed from
amines such as ammonia, ethylenediamine, N-methylglucamine, lysine, arginine,
ornithine, choline, N,N'-dibenzylethylenediamine, chloroprocaine,
diethanolamine,
procaine, N-benzylphenethylamine, 1-p-chlorobenzyl-2-pyrrolidine-1'-yl-
methylbenzimidazole, diethylamine, piperazine, morpholine, 2,4,4-trimethyl-2-
pentamine and tris(hydroxymethyl)aminomethane. Pharmaceutically acceptable
_g-
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
esters at the carboxylic acid group can be made by treating a dihydroxy open
acid
statin with an alcohol. Examples of pharmaceutically acceptable esters of
dihydroxy
open acid statins include, but are not limited to, -C 1 _q. alkyl and - C 1
_q. alkyl
substituted with phenyl-, dimethylamino-, and acetylamino. "C1-q. alkyl"
herein
includes straight or branched aliphatic chains containing from 1 to 4 carbon
atoms, for
example methyl, ethyl, n-propyl, n-butyl, iso-propyl, sec-butyl and tent-
butyl.
The instant invention involves methods and pharmaceutical
compositions for orally administering open-ring dihydroxy open acid statins
and salts
and esters thereof, which are HMG-CoA reductase inhibitors, in such a way so
as to
minimize conversion to their lactonized counterparts. This allows for delivery
of a
dihydroxy open acid statin without its lactone counterpart directly to the
absorptive
mucosa of the small intestine thus allowing for absorption of the open acid
statin into
the portal circulation, penetration by active open acid statin into
hepatocytes to
achieve enhanced efficacy, and systemic exposure consisting of open acid
moieties.
More particularly, delayed-release of an orally administered dihydroxy open
acid
statin or a pharmaceutically acceptable salt or ester thereof, for example
dihydroxy
open acid simvastatin or a salt thereof, until after passage through the
stomach
reduces the amount of lactone formed and absorbed in the body. Maintaining the
statin in its open acid form in the body thereby reduces the potential for
drug
interactions between statins whose metabolism is CYP3A4-mediated and other
active
agents which inhibit the CYP3A4 enzymatic pathway, and also can provide
enhanced
efficacy. Administering a statin in its open acid form in such a way so as to
minimize
conversion to its lactonized counterpart, for example by using an oral delayed
release
dosage form, should reduce the potential for drug interaction compared to the
conventional administration of an open acid statin or its lactonized
counterpart, for
example by using an oral rapid release dosage form.
An object of this invention is to provide methods for reducing the
amount of lactonized statin formed and absorbed in the body after oral
administration
of a dihydroxy open acid statin in order to achieve enhanced clinical
benefits. A way
to achieve this is to administer the dihydroxy open acid statin in a delayed-
release
pharmaceutical dosage form. A delayed-release pharmaceutical dosage form as
defined herein is an orally administerable pharmaceutical dosage form or
device that
does not release a substantial amount of the active compound, i.e., the
dihydroxy
open-acid statin, until after the dosage form has passed through the stomach.
Therefore, substantial release of the active compound would initially occur
after entry
-9-
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
into the duodenum. By "substantial release," it is intended that 90% or more
by
weight of the active compound in the delayed-release dosage form is released
after
entry into the duodenum, and that 10% or less by weight of the active compound
in
the delayed-release dosage form is released in the stomach, i.e., the
geometric mean
ratio of the plasma AUC (area under the curve) of active vs. total HMG-CoA
reductase inhibitory activity will be greater than or equal to 90%.
Particularly, the
amount of active compound released in the stomach before entry into the
duodenum is
5% or less by weight, i.e., the geometric mean ratio of the plasma AUC of
active vs.
total HMG-CoA reductase inhibitory activity will be greater than or equal to
95%, and
more particularly the amount of active compound released in the stomach before
entry
into the duodenum is 1% or less by weight, i.e., the geometric mean ratio of
the
plasma AUC of active vs. total HMG-CoA reductase inhibitory activity will be
greater
than or equal to 99%.
It is to be understood that metabolism of the dihydroxy open acid
statins will occur, primarily in the liver, after orally dosing in a delayed-
release dosage
form. However, since lactonization of the dihydroxy open acid statin would
have
been substantially avoided by use of a delayed release dosage form, the active
and
inactive metabolites that are formed will also be in the dihydroxy open acid
form. In
essence, if the dihydroxy open acid statin is administered in a delayed
release dosage
form, the internal exposure to lactonized parent compound and also to
lactonized
active and inactive metabolites will be minimized.
One example of a suitable delayed-release dosage form is a pH-
dependent enterically coated dosage form. The enteric coating will not
dissolve in the
acidic gastric environment, but will dissolve in the higher pH environment of
the
duodenum. An enterically coated dosage form will therefore not permit release
of any
significant amount of the active compound from the dosage fornl in the
stomach, but
once the enteric coating dissolves in the duodenum, the active compound will
be
released. Suitable compositions for enteric coatings that can be used with the
present
invention are known to those of ordinary skill in the pharmaceutical arts; for
example,
see L. Lachman, The Theory and Practice of Industrial Pharmacy, 3rd Ed., H.
Liebermann and J. Kanig contributors (Lea & Febiger, 1986). An example of a
suitable enteric coating includes but is not limited to SURETERIC WHITE sold
by
Colorcon which is composed of polyvinyl acetate phthalate, titanium dioxide,
talc,
colloidal silicon dioxide, triethyl acetate, polyethylene glycol, sodium
bicarbonate,
purified stearic acid, and sodium alginate. Many other suitable enteric
coating
-10-
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
materials are commercially available and are known in the art. Additional
coatings
employed in the preparation of the dosage form, such as those used to provide
an
elegant, aesthetically pleasing final product or for other purposes, may be
applied
before or after, or before and after, application of the enteric coating.
Suitable enterically coated pharmaceutical dosage forms for use with
this invention include enterically coated conventional rapid-release (also
referred to as
immediate-release) pharmaceutical dosage forms wherein the drug is relatively
rapidly released once the enteric coating is breached, and enterically coated
time-
controlled release dosage forms such as but not limited to an enterically
coated GEM
delivery device, described below. Time controlled-release dosage forms are
also well
known in the pharmaceutical art, and are designed so as to slowly release the
active
compound in the body over a period of time, for example over a period of from
about
6 to 24 hours. Use of an enteric coated time controlled-release dosage form is
preferred with more potent dosage amounts of a dihydroxy open acid statin so
as to
lower the systemic exposure to the active statin. Whether the dosage form is
an
enterically coated rapid-release or time-controlled release dosage form, the
enteric
coating will prevent release of any substantial amount of the active compound
from
the dosage form in the stomach.
Enterically coated pharmaceutical dosage forms also include but are
not limited to those wherein the dosage form or unit is comprised of the
dihydroxy
open acid statin in a tablet, capsule or the like that is surrounded by an
enteric
overcoating, and those wherein the dosage form or unit is a tablet, capsule or
the like
comprised of enterically coated granules of the dihydroxy open acid statin.
Where the
dosage form is surrounded by an enteric overcoat, the enteric coating may be
the
outer-most external coating on the dosage form, or there may be one or more
additional finish coatings applied over the enteric coat. In a more limited
embodiment, when the delayed-release dosage unit contains enterically coated
granules of the drug, the drug is selected from the dihydroxy open-acid form
of
lovastatin and simvastatin and the pharmaceutically acceptable salt and ester
forms
thereof, and is more preferably a salt of dihydroxy open acid simvastatin, and
most
preferably the calcium or ammonium salt thereof. In an alternative embodiment,
any
dihydroxy open acid statin or pharmaceutically acceptable salt or ester
thereof may be
used with the present invention, such as those described herein, provided that
the
statin is not dosed in a single pharmaceutical dosage form or unit comprised
of enteric
-11-
CA 02364253 2001-08-29
WO 00/53173 PCT/~JS00/02626
coated granules of the statin and enteric coated or non-enteric coated
granules of
aspirin.
An example of a delayed-release dosage form that also functions as a
time controlled-release dosage form is described in U.S Patent No. 5,366,738,
herein
incorporated by reference in its entirety. The controlled-release drug
delivery device
described in U.S Patent No. 5,366,738 is known as a gel extrusion module (GEM)
delivery device. The GEM device is a dmg delivery device for the controlled in
situ
production and release of a dispersion containing a beneficial agent such as a
pharmaceutical drug comprising:
(A) a compressed core prepared from an admixture comprising:
(i) a therapeutically effective amount of the beneficial agent; and
(ii) a polymer which upon hydration forms gelatinous microscopic
particles; and
(B) a water insoluble, water impermeable polymeric coating comprising a
polymer and a plasticizes, which surrounds and adheres to the core, the
coating having
a plurality of formed apertures exposing between about 1 and about 75% of the
core
surface;
and wherein the release rate of the beneficial agent from the device is a
function of the
number and size of the apertures.
In the GEM device, the polymer inside the compressed core is
preferably selected from sodium polyacrylate, carboxypolymethylenes and the
pharmaceutically acceptable salts thereof such as a sodium salt, wherein the
carboxypolymethylenes are prepared from acrylic acid crosslinked with
allylethers of
sucrose or pentaerythritol, and more preferably it is selected from
carboxypolymethylenes prepared from acrylic acid crosslinked with allylethers
of
sucrose or pentaerythritol, and the pharmaceutically acceptable salts thereof.
Most
preferably, CARBOPOL~ 974P and pharmaceutically acceptable salts thereof,
particularly the sodium salt, is used as the polymer inside the compressed
core. In
addition, the compressed core may also contain one or more polymer hydration
modulating agents, anti-oxidants, lubricants, fillers and excipients. An
optional
subcoating may be applied to the compressed core prior to application of the
water
insoluble coating as an aid in the manufacturing process. The subcoating may
be
comprised of, for example, hydroxypropyl cellulose and
hydroxypropylmethylcellulose. Additional coatings may be applied for aesthetic
or
functional purposes.
-12-
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
The water insoluble, water impermeable polymeric coating is
preferably comprised of ( 1 ) a polymer selected from polyvinyl chloride,
cellulose
acetate, cellulose acetate butyrate, ethylcellulose and combinations of these
polymers;
and (2) a plasticizer selected from diethylphthalate, dibutylsebacate and
triethylcitrate.
More preferably, the polymeric coating is comprised of cellulose acetate
butyrate and
triethyl citrate. The GEM device does not function as an osmotic drug delivery
device,
hence the release function of the device depends on passage of fluids from the
external environment of the body to the internal environment of the compressed
core
through the formed apertures. It is intended that the terms "water insoluble,
water
impermeable" used to describe the polymeric coating define a coating which is
essentially water insoluble and water impermeable, meaning that the polymeric
coating allows minimal to no passage of water through the coating from the
external
environment of the body to the internal environment of the compressed core,
except
for the fluid passage that occurs through the drilled apertures, during the
period of
time the drug is being released from the GEM device in the body. Any minimal
amount of water that does pass through the water insoluble, water impermeable
polymeric coating is insubstantial and does not significantly contribute to
the function
of the GEM device, i.e. the release rate of the drug through the apertures.
Rather the
release rate of the drug from the GEM device is primarily a function of the
number
and size of the apertures on the device.
For an elegant, aesthetically pleasing final product, an nutter finish coat
may finally be applied to the GEM delivery device containing colorants, waxes,
and
the like. The GEM device can also be enterically coated, either before or
after the
application of additional finish coatings. Even without enteric coating,
extrusion of
the polymer which carries the drug out from inside the compressed core of the
GEM
device does not occur to a substantial extent in the acidic pH of the stomach,
therefore
substantial release of the drug should not occur in the stomach. Further
details and
examples of the GEM delivery device are described in US Patent No. 5,366,738.
The
compound employed with the GEM device may particularly be a pharmaceutically
acceptable salt of dihydroxy open acid simvastatin, and more particularly the
ammonium salt of dihydroxy open acid simvastatin.
The term "patient" includes mammals, especially humans, who use the
instant active agents for the prevention or treatment of a medical condition.
Administering of the drug to the patient includes both self administration and
administration to the patient by another person. The patient may be in need of
-13-
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
treatment for an existing disease or medical condition, or may desire
prophylactic
treatment to prevent or reduce the risk for diseases and medical conditions
affected by
inhibition of HMG-CoA reductase.
The term "therapeutically effective amount" is intended to mean that
S amount of a drug or pharmaceutical agent that will elicit the biological or
medical
response of a tissue, a system, animal or human that is being sought by a
researcher,
veterinarian, medical doctor or other clinician. The term "prophylactically
effective
amount" is intended to mean that amount of a pharmaceutical drug that will
prevent or
reduce the risk of occurrence of the biological or medical event that is
sought to be
prevented in a tissue, a system, animal or human by a researcher,
veterinarian, medical
doctor or other clinician. Particularly, the dosage a patient receives can be
selected so
as to achieve the amount of LDL (low density lipoprotein) cholesterol lowering
desired; the dosage a patient receives may also be titrated over time in order
to reach a
target LDL level. The dosage regimen utilizing a dihydroxy open acid statin is
selected in accordance with a variety of factors including type, species, age,
weight,
sex and medical condition of the patient; the severity of the condition to be
treated;
the potency of the compound chosen to be administered; the route of
administration;
and the renal and hepatic function of the patient. A consideration of these
factors is
well within the purview of the ordinarily skilled clinician for the purpose of
determining the therapeutically effective or prophylactically effective dosage
amount
needed to prevent, counter, or arrest the progress of the condition.
The novel compounds of this invention are the crystalline hydrated
forms of the calcium salt of dihydroxy open acid simvastatin. A particular
crystalline
hydrated calcium salt of dihydroxy open acid simvastatin is the one having an
x-ray
powder diffraction (XRPD) pattern obtained using CuKa radiation characterized
by
refelections at d-spacings of 30.7, 24.6, 15.9, 11.2, 8.58, 7.31, 6.74, 6.06,
5.35, 5.09,
4.93, 4.60, 3.93, 3.84, 3.67, 3.51 and 3.28. For convenience, the crystalline
hydrated form of the calcium salt of dihydroxy open acid simvastatin having
the
above-defined XRPD pattern will also be referred to herein as compound I.
Compound I can be represented two-dimensionally as a hydrated form of the
following structural formula Ia:
-14-
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
HO _ Ca++
COO
O OH
,,; .
O H
/ /
2
Ia
In addition to the XRPD pattern described above, compound I of the
instant invention is further characterized by the thermogravimetry (TG) curve
shown
S in Figure I. The TG curve for compound I was obtained under a nitrogen flow
at a
heating rate of 10°C/minute and is characterized by a 6.3% weight loss
from ambient
room temperature to a stable weight loss plateau at about 175°C.
Additional weight
losses due to the onset of decomposition of the compound are observed above
about
190°C.
Compound I is still further characterized by the differential scanning
calorimetry (DSC) curve shown in Figure 2. The DSC curve for compound I was
obtained under a nitrogen flow bubbled through 16.0°C water at a
heating rate of
10°C/minute in an open cup, and is characterized by three lower
temperature
endotherms with peak temperatures of 52, 77 and 100°C and associated
heats of 48,
90 and 21 J/g, respectively, and two higher temperature endotherms due to
decomposition with peak temperatures of 222 and 241 °C and associated
heats of 163
and 92 J/g.
Compound I of the instant invention is still further characterized by the
'H nuclear magnetic resonance (NMR) spectral data,'3C NMR and mass spectral
(MS) data as given in Example 1 herein.
Compound I is also characterized by the solid-state 13C nuclear
magnetic resonance spectrum shown in Figure 4, which was completed using a
Bruker
DSX 400WB NMR system operating at 100.6 MHz for 13C and 400.1 MHz for 1H
using a Bruker MAS 400WB BL7 double-resonance probe with a spinning module
-15-
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
housing a 7 mm zirconia rotor with Kel-f end caps. The solid-state 13C nuclear
magnetic resonance (NMR) spectrum was acquired using cross polarization (CP),
magic-angle spinning (MAS), and high-power (~59 kHz) decoupling with variable-
amplitude cross-polarization and total sideband suppression. Proton and carbon
90°
pulse widths were 4.25 psec with a contact time of 2.0 msec. The sample was
spun at
7.0 kHz and a total of 1024 scans were collected for the spectrum with a
recycle delay
of 3.0 seconds. A line broadening of 10 Hz was applied to the spectrum before
FT
was performed. Chemical shifts are reported on the TMS scale using the
carbonyl
carbon of glycine ( 176.03 ppm) as a secondary reference.
In the methods of treatment and prophylaxis described herein, as well
as the pharmaceutical compositions and medicaments, a dihydroxy open acid
statin or
a pharmaceutically acceptable salt or ester thereof is employed. Preferably
the
compound employed is a pharmaceutically acceptable salt of a dihydroxy open
acid
statin, more preferably it is a pharmaceutically acceptable salt of dihydroxy
open acid
simvastatin such as an ammonium salt or calcium salt, and particularly a
crystalline
hydrated form of the calcium salt of dihydroxy open acid simvastatin such as
compound I. All hydrates, solvates and polymorphic crystalline forms of the
above-
described compounds and their use are encompassed within scope of the instant
invention.
The instant invention provides methods for inhibiting HMG-CoA
reductase, and for treating lipid disorders including hypercholesterolemia,
hypertriglyceridemia and combined hyperlipidemia, comprising administering a
therapeutically effective amount of a dihydroxy open acid statin to a person
in need of
such treatment. Further provided are methods for preventing or reducing the
risk of
developing atherosclerosis, as well as for halting or slowing the progression
of
atherosclerotic disease once it has become clinically evident, comprising the
administration of a prophylactically or therapeutically effective amount, as
appropriate, of a dihydroxy open acid statin to a mammal who is at risk of
developing
atherosclerosis or who already has atherosclerotic disease.
Atherosclerosis encompasses vascular diseases and conditions that are
recognized and understood by physicians practicing in the relevant fields of
medicine.
Atherosclerotic cardiovascular disease including restenosis following
revascularization procedures, coronary heart disease (also known as coronary
artery
disease or ischemic heart disease), cerebrovascular disease including multi-
infarct
-16-
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
dementia, and peripheral vessel disease including erectiIe dysfunction are all
clinical
manifestations of atherosclerosis and are therefore encompassed by the terms
"atherosclerosis" and '.'atherosclerotic disease."
A dihydroxy open acid statin may be administered to prevent or reduce
the risk of occurrence, or recurrence where the potential exists, of a
coronary heart
disease event, a cerebrovascular event, and/or intermittent claudication.
Coronary
heart disease events are intended to include CHD death, myocardial infarction
(i.e., a
heart attack), and coronary revascularization procedures. Cerebrovascular
events are
intended to include ischemic or hemorrhagic stroke (also known as
cerebrovascular
accidents) and transient ischemic attacks. Intermittent claudication is a
clinical
manifestation of peripheral vessel disease. The term "atherosclerotic disease
event" as
used herein is intended to encompass coronary heart disease events,
cerebrovascular
events, and intermittent claudication. It is intended that persons who have
previously
experienced one or more non-fatal atherosclerotic disease events are those for
whom
the potential for recurrence of such an event exists.
Accordingly, the instant invention also provides a method for
preventing or reducing the risk of a first or subsequent occurrence of an
atherosclerotic disease event comprising the administration of a
prophylactically
effective amount of a dihydroxy open acid statin to a patient at risk for such
an event.
The patient may or may not have atherosclerotic disease at the time of
administration,
or may be at risk for developing it.
The instant invention also provides a method for preventing and/or
treating inflammatory diseases or disorders alone or in conjunction with the
treatment
of conditions described above, comprising the administration of a dihydroxy
open-
acid statin to a patient in need of such treatment. This includes, for
example, the
treatment of inflammatory conditions susceptible to treatment with a non-
steroidal
anti-inflammatory agent, arthritis including rheumatoid arthritis, and
degenerative
joint diseases (osteoarthritis), dementia, Alzheimer's disease, multiple
sclerosis,
inflammatory bowel disease, asthma, psoriasis, systemic lupus erythematosis,
vasculitis, gout, adrenoleukodystrophy, diabetic retinopathy, nephropathy and
diabetes
mellitus type II.
Persons to be treated with the instant therapy include those at risk of
developing atherosclerotic disease and of having an atherosclerotic disease
event.
Standard atherosclerotic disease risk factors are known to the average
physician
practicing in the relevant fields of medicine. Such known risk factors include
but are
-17-
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
not limited to hypertension, smoking, diabetes, low levels of high density
lipoprotein
(HDL) cholesterol, and a family history of atherosclerotic cardiovascular
disease.
Published guidelines for determining those who are at risk of developing
atherosclerotic disease can be found in: National Cholesterol Education
Program,
Second report of the Expert Panel on Detection, Evaluation, and Treatment of
High
Blood Cholesterol in Adults (Adult Treatment Panel II), National Institute of
Health,
National Heart Lung and Blood Institute, NIH Publication No. 93-3095,
September
1993; abbreviated version: Expert Panel on Detection, Evaluation, and
Treatment of
High Blood Cholesterol in Adults, Summary of the second report of the national
cholesterol education program (NCEP) Expert Panel on Detection, Evaluation,
and
Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel II),
JAMA,
1993, 269, pp. 3015-23. People who are identified as having one or more of the
above-noted risk factors are intended to be included in the group of people
considered
at risk for developing atherosclerotic disease. People identified as having
one or more
of the above-noted risk factors, as well as people who already have
atherosclerosis,
are intended to be included within the group of people considered to be at
risk for
having an atherosclerotic disease event.
The oral dosage amount of the dihydroxy open acid statin, particularly
a salt of a dihydroxy open acid statin such as simvastatin, and more
particularly the
ammonium salt or a crystalline form of the calcium salt of dihydroxy open acid
simvastatin such as compound I, is from about 1 to 200 mg/day, and more
preferably
from about 5 to 160 mg/day. However, dosage amounts will vary depending on
factors as noted above, including the potency of the particular compound.
Although
the active drug of the present invention may be administered in divided doses,
for
example from one to four times daily, a single daily dose of the active drug
is
preferred. As examples, the daily dosage amount may be selected from, but not
limited to, 5 mg, 10 mg, 15 mg, 20 mg, 25 mg, 40 mg, 50 mg, 75 mg, 80 mg, 100
mg,
150 mg and 160 mg.
The active drug employed in the instant therapy can be administered in
such oral forms as tablets, capsules, pills, powders, granules, elixirs,
tinctures,
suspensions, syrups, and emulsions. Oral formulations are preferred.
For the crystalline hydrated forms of the calcium salt of dihydroxy
open acid simvastatin, for example compound I, administration of the active
drug can
be via any pharmaceutically acceptable route and in any pharmaceutically
acceptable
dosage form. This includes the use of oral conventional rapid-release, time
-18-
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
controlled-release and delayed-release (such as described above)
pharmaceutical
dosage forms. An oral delayed-release dosage formulation of the instant drug
is
preferred, and particularly an enteric overcoat surrounding a rapid-release
dosage unit,
or the GEM controlled-release drug delivery device with an enteric overcoat
surrounding the dosage unit, and most particularly an enteric overcoat
surrounding a
rapid-release dosage unit. Additional suitable pharmaceutical compositions for
use
with the present invention are known to those of ordinary skill in the
pharmaceutical
arts; for example, see Remington's Pharmaceutical Sciences, Mack Publishing
Co.,
Easton, PA.
In the methods of the present invention, the active drug is typically
administered in admixture with suitable pharmaceutical diluents, excipients or
carriers
(collectively referred to herein as "carrier" materials) suitably selected
with respect to
the intended form of administration, that is, oral tablets, capsules, elixirs,
syrups and
the like, and consistent with conventional pharmaceutical practices.
For instance, for oral administration in the form of a tablet or capsule,
the active drug component can be combined with a non-toxic, pharmaceutically
acceptable, inert carrier such as lactose, starch, sucrose, glucose, modified
sugars,
modified starches, methyl cellulose and its derivatives, dicalcium phosphate,
calcium
sulfate, mannitol, sorbitol and other reducing and non-reducing sugars,
magnesium
stearate, steric acid, sodium stearyl fumarate, glyceryl behenate, calcium
stearate and
the like. For oral administration in liquid form, the drug components can be
combined with non-toxic, pharmaceutically acceptable inert carrier such as
ethanol,
glycerol, water and the like. Moreover, when desired or necessary, suitable
binders,
lubricants, disintegrating agents and coloring and flavoring agents can also
be
incorporated into the mixture: Stabilizing agents such as antioxidants, for
example
butylated hydroxyanisole (BHA), 2,6-di-tert-butyl-4-methylphenol (BHT), propyl
gallate, sodium ascorbate, citric acid, calcium metabisulphite, hydroquinone,
and 7-
hydroxycoumarin, particularly BHA, propyl gallate and combinations thereof,
can
also be added to stabilize the dosage forms; the use of at least one
stabilizing agent is
preferred with the instant composition. Preferably an antioxidant is employed
with
dihydroxy open acid simvastatin or a salt thereof, and particularly compound
I. Other
suitable components include gelatin, sweeteners, natural and synthetic gums
such as
acacia, tragacanth or alginates, carboxymethylcellulose, polyethylene glycol,
waxes
and the like.
-19-
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
The active drug can also be administered in the form of liposome
delivery systems, such as small unilamellar vesicles, large unilamellar
vesicles and
multilamellar vesicles. Liposomes can be formed from a variety of
phospholipids,
such as cholesterol, stearylamine or phosphatidylcholines.
Active drug may also be delivered by the use of monoclonal antibodies
as individual carriers to which the compound molecules are coupled. Active
drug
may also be coupled with soluble polymers as targetable drug carriers. Such
polymers
can include polyvinyl-pyrrolidone, pyran copolymer, polyhydroxy-propyl-
methacrylamide-phenol, polyhydroxy-ethyl-aspartamide-phenol, or
polyethyleneoxide-polylysine substituted with palmitoyl residues. Furthermore,
active drug may be coupled to a class of biodegradable polymers useful in
achieving
controlled release of a drug, for example, polylactic acid, polyglycolic acid,
copolymers of polylactic and polyglycolic acid, polyepsilon caprolactone,
polyhydroxy butyric acid, polyorthoesters, polyacetals, polydihydropyrans,
polycyanoacrylates and cross linked or amphipathic block copolymers of
hydrogels.
The instant invention also encompasses a process for preparing a
pharmaceutical composition comprising combining a dihydroxy open acid statin
with
a pharmaceutically acceptable Garner. Also encompassed is the pharmaceutical
composition which is made by combining a dihydroxy open acid statin with a
pharmaceutically acceptable Garner.
In a broad embodiment, any suitable additional active agent or agents
may be used in combination with the dihydroxy open acid statin in a single
dosage
formulation, or may be administered to the patient in a separate dosage
formulation,
which allows for concurrent or sequential administration of the active agents.
One or
more additional active agents may be administered with a dihydroxy open acid
statin.
The additional active agent or agents can be lipid lowering compounds or
agents
having other pharmaceutical activities, or agents that have both lipid-
lowering effects
and other pharmaceutical activities. Examples of additional active agents
which may
be employed include but are not limited to HMG-CoA synthase inhibitors;
squalene
epoxidase inhibitors; squalene synthetase inhibitors (also known as squalene
synthase
inhibitors), acyl-coenzyme A: cholesterol acyltransferase (ACAT) inhibitors
including
selective inhibitors of ACAT-1 or ACAT-2 as well as dual inhibitors of ACAT-1
and
-2; microsomal triglyceride transfer protein (MTP) inhibitors; probucol;
niacin;
cholesterol absorption inhibitors such as SCH-58235, which is described in
U.S.
Patent No.'s 5,767,115 and 5,846,966; bile acid sequestrants; LDL (low density
-20-
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
lipoprotein) receptor inducers; platelet aggregation inhibitors, for example
glycoprotein IIb/IIIa fibrinogen receptor antagonists and aspirin; human
peroxisome
proliferator activated receptor gamma (PPARy) agonists including the compounds
commonly referred to as glitazones for example troglitazone, pioglitazone and
rosiglitazone and, including those compounds included within the structural
class
known as thiazolidinediones as well as those PPARy agonists outside the
thiazolidinedione structural class; PPARa agonists such as clofibrate,
fenofibrate
including micronized fenofibrate, and gemfibrozil; PPAR dual a/y agonists;
vitamin
B6 (also known as pyridoxine) and the pharmaceutically acceptable salts
thereof such
as the HCl salt; vitamin B I 2 (also known as cyanocobalamin); folic acid or a
pharmaceutically acceptable salt or ester thereof such as the sodium salt and
the
methylglucamine salt; anti-oxidant vitamins such as vitamin C and E and beta
carotene; beta-blockers; angiotensin II antagonists such as losartan;
angiotensin
converting enzyme inhibitors such as enalapril and captopril; calcium channel
Mockers such as nifedipine and diltiazam; endothelian antagonists; agents that
enhance ABCI gene expression; FXR and LXR ligands including both inhibitors
and
agonists; bisphosphonate compounds such as alendronate sodium; and
cyclooxygenase-2 inhibitors such as rofecoxib and celecoxib. Additionally, the
dihydroxy open acid statins of this invention, for example compound I, may be
used
in combination with anti-retroviral therapy in AIDS infected patients to treat
lipid
abnormalities associated with such treatment, for example but not limited to
their use
in combination with HN protease inhibitors such as indinavir, nelfinavir,
ritonavir
and saquinavir.
A therapeutically or prophylactically effective amount, as appropriate,
of a crystalline hydrated form of the calcium salt of dihydroxy open acid
simvastatin,
for example compound I, can be used for the preparation of a medicament useful
for
inhibiting HMG-CoA reductase, as well as for treating and/or reducing the risk
for
diseases and conditions affected by inhibition of HMG-CoA reductase, such as
treating lipid disorders, preventing or reducing the risk of developing
atherosclerotic
disease, halting or slowing the progression of atherosclerotic disease once it
has
become clinically manifest, and preventing or reducing the risk of a first or
subsequent occurrence of an atherosclerotic disease event. For example, the
medicament may be comprised of about 1 mg to 200 mg of a crystalline hydrated
form of the calcium salt of dihydroxy open acid simvastatin, or more
particularly
about 5 mg to 160 mg.
-21 -
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
A therapeutically or prophylactically effective amount, as appropriate,
of a dihydroxy open acid statin can be used for the preparation of an oral
medicament
adapted for delayed-release, wherein substantial release of the statin after
oral
administration does not occur until after passage of the medicament through
the
stomach, or alternately wherein at least 90% of the statin is delivered in its
dihydroxy
open acid form to the intestinal mucosa of a patient after oral
administration. Said
oral medicaments are also useful for inhibiting HMG-CoA reductase, as well as
for
treating and/or reducing the risk for diseases and conditions affected by
inhibition of
HMG-CoA reductase, as described above.
The medicament comprised of a dihydroxy open acid statin, for
example compound I, may also be prepared with one or more additional active
agents,
such as those described supra.
Simvastatin is a semi-synthetic product which can be made from the
natural product lovastatin. Processes for preparing lovastatin and simvastatin
are well
documented in the published literature. For example, U.S. Patent No.
4,231,938,
herein incorporated by reference, describes a fermentation and isolation
process for
obtaining lovastatin using the microorganism Aspergillus terreus. U.S. Patent
No.'s
4,444,784, 4,820,850, 4,916,239 and 4,582,915, herein all incorporated by
reference,
describe methods for making dihydroxy open-acid and lactonized forms of
simvastatin.
Compound I of the instant invention can generally be prepared as
follows. Simvastatin and its dihydroxy open acid counterpart, including
compound I,
tend to form oxidative by-products; therefore, to minimize the formation of
such by-
products, it is preferred that the procedures used to make compound I are
performed
under an inert atmosphere such as nitrogen. Although compound I can be
obtained
without using an inert atmosphere, the purity of the desired product will not
be
optimized.
Hydrolysis of the lactone ring of simvastatin can be accomplished by
treating simvastatin with at least one equivalent, and preferably a slight
excess of one
equivalent, of an aqueous base. If more than a slight excess of base is used,
the
excess base is preferably neutralized before proceeding to the salt formation
step in
order to prevent formation of insoluble calcium hydroxide or calcium carbonate
by-
product. The base employed for the hydrolysis can be an aqueous solution of a
metal
hydroxide or metal carbonate, for example but not limited to sodium hydroxide,
potassium hydroxide, lithium hydroxide, sodium carbonate and potassium
carbonate.
-22-
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
Sodium hydroxide is preferred. The hydrolysis can be performed in water, an
aqueous-protic organic solvent mixture, or an aqueous-aprotic organic solvent
mixture. Suitable protic organic solvents include but are not limited to
methanol
(MeOH), ethanol (EtOH), isopropyl alcohol, n-propyl alcohol (propanol).
Examples
of suitable aprotic organic solvents include but are not limited to
acetonitrile,
dimethyl sulfoxide (DMF), N,N-dimethylformamide (DMSO), tetrahydrofuran (THF),
tent-butyl methyl ether (MTBE) and toluene. Particularly, an aqueous ethanol
or n-
propyl alcohol solvent mixture may be used, and more particularly an aqueous n-
propyl alcohol solvent mixture.
After the hydrolysis reaction is complete, the pH of the reaction
mixture is adjusted to about 6 to 11, particularly 6 to 9, and more
particularly 7 to 8.5,
by addition of an acid. In this pH range, the dihydroxy open acid simvastatin
will
exist as a metal salt, for example as the sodium salt if the base used in the
hydrolysis
step is sodium hydroxide or sodium carbonate. Any acid that is capable of
forming a
soluble calcium salt such as calcium chloride or calcium citrate, is suitable.
A soluble
calcium salt is intended to be a salt that is soluble in the solvent sytem
employed in
the instant process. Preferably an acid such as acetic acid (HOAc) or a
mineral acid
is employed, particularly HCI.
The resulting pH-adjusted reaction mixture containing the metal salt of
dihydroxy open acid simvastatin is next combined with a solution of about 0.50
to
0.55 equivalents of calcium acetate hydrate [Ca(OAc),~xHzO] in water or an
aqueous-
organic solvent mixture, such as aqueous EtOH, MeOH, i-PrOH, n-PrOH,
acetonitrile, DMF, DMSO, THF, and particularly aqueous EtOH or aqueous n-
propyl
alcohol. The pH-adjusted reaction mixture can be added to the calcium acetate
hydrate solution, or the calcium acetate hydrate solution can be added to the
pH-
adjusted reaction mixture. The addition can occur all at once, or optionally
it can be
performed in portions over time with periods of aging. For example, a small
portion,
e.g., about one-quarter, of the calcium acetate hydrate solution can be added
to the
pH-adjusted reaction mixture over a short period of time, for example over
about 30
minutes, and then the resulting mixture can be allowed to age for an
additional short
period of time at room temperature, optionally followed by a further period of
aging at
a temperature up to about 50°C, for example from about 10°C up
to about 50°C,
particularly from room temperature up to about 50°C, more particularly
from about 30
to 40°C, and most particularly from about 30 to 35°C, after
which the remaining
calcium acetate hydrate solution can be added in portions over several hours
at a
- 23 -
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
temperature up to about 50°C, for example from about 10°C up to
about 50°C,
particularly from room temperature up to about 50°C, and more
particularly from
about 30 to 40°C, and. most particularly from about 30 to 35°C.
Optionally, the
reaction mixture can be seeded with crystalline Compound I.
Whether the pH-adjusted reaction mixture and the calcium acetate
hydrate solution are combined at once or in portions, the resulting slurry
must be aged
until turnover of the resulting amorphous calcium salt of dihydroxy open acid
simvastatin to the crystalline product is complete, usually for at least
several hours.
Complete turnover to the crystalline product can be assessed by standard
techniques in
the art, for example, by examining a sample of the product under a microscope.
This
aging step can be performed at a temperature up to about 50°C, for
example from
about 10°C up to about 50°C, particularly from room temperature
up to about 50°C,
and more particularly from about 30 to 40°C, and most particularly from
about 30 to
35°C. During the aging period or periods, the use of lower temperatures
will lead to
crystallized product; however, it has been found that as the temperature
drops, the rate
of crystal turnover becomes slower, making the procedure less time-efficient.
If necessary, the slurry is then allowed to cool to room temperature and
is collected by suction filtration. The recovered solid is suction dried under
a moist
atmosphere (about 30 to 70% relative humidity, particularly 40 to 70%),
preferably a
moist inert atmosphere such as nitrogen, and particularly at a temperature
from about
10 to 40°C, and more particularly 25 to 35°C. The final step of
suction filtration in
the recovery of compound I should be done under a moist atmosphere, and
preferably
a moist inert atmosphere in order to minimize oxidative by-products. If an
additional
step of adding an antioxidant to compound I is performed, as described below,
then
the final suction filtration is the one performed after combining the
antioxidant with
compound I.
As noted above, compound I has a tendency to oxidize upon contact
with air, and one way to minimize oxidation is to perform the reaction
sequence under
an inert atmosphere. Additionally, one or more anti-oxidants such as BHA, BHT,
propyl gallate, ascorbic acid, calcium metabisulphite, hydroquinone,
nordihydroguaiaracetic acid (NDGA) or 7-hydroxycoumarin can be combined with
compound I. This is done by agitating a slurry of compound I with one or more
of the
antioxidants and recovering the resulting solid by suction filtration.
Alternatively, the ammonium salt of dihydroxy open acid simvastatin
can be used as the starting material to be combined with the calcium acetate
hydrate,
-24-
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
thus avoiding the hydrolysis and pH adjustment steps needed when starting with
lactonized simvastatin. The other reaction conditions described above, such as
solvents, temperatures, etc., can otherwise be employed.
Abbreviations which may appear herein are as follows: MeOH is
methanol; EtOH is ethanol; PrOH is propanol; HOAc is acetic acid; MeCN is
acetonitrile; DMF is dimethyl sulfoxide; DMSO is N,N-dimethylformamide;
Ca(OAc)2 is calcium acetate; HPLC is high performance liquid chromatography;
min.
is minutes; h is hour(s); D.I. is de-ionized; NMR is nuclear magnetic
resonance; EI
MS is electron impact mass spectrum; HR-EI MS is high resolution electron
impact
mass spectrum; RH is relative humidity. The "seed" used in the examples is
Compound I.
EXAMPLE 1
Preparation of crystalline hydrated calcium salt of dihydroxy open acid
simvastatin
(Compound I)
A 22 L four-necked round bottom flask was equipped with a
temperature probe, a N2 inlet, an addition funnel, and an overhead stirrer.
8.0 L of
15% EtOH-H20 was added and the solution was purged with N2 for 10 min.
Simvastatin (396 g, 0.946 mol) was added, and the slurry was purged with N2
for 5
min. Then SN NaOH ( 198 mL) was added at room temperature. After about 1 hour,
the hydrolysis reaction was complete as analyzed by HPLC (> 99.9% conversion).
The pH of the reaction solution was adjusted to 7 to 8.5 by addition of 1 N
HCI
(approx. 65 mL). A solution of Ca(OAc)2~H20(116.6 g, 0.662 mol) in 4.0 L of
60%
EtOH-H20 was purged with nitrogen for 5 min. A 1.0 L portion of this solution
was
added to solution of the sodium salt over 30 min. The resulting slurry was
aged at
room temperature for 30 min, and then at 30 to 35° C for 1-2 h.
The rest of Ca(OAc)2 in EtOH-water was added over approx. 30 min
at 30 to 35°C. The slurry was aged at 30 to 35 °C for 5 hours
under atmosphere of N2.
The slurry was cooled to room temperature and was collected by suction-
filtration.
The wet cake was washed with 4 L 30% EtOH-H20, 4 L 20% EtOH-H20, followed
by 6 L x 3 of D.I. water. The solid was suction dried under an atmosphere of
moist
N2 (40 to 70% relative humidity) at room temperature for 4 days. Crystalline
- 25 -
CA 02364253 2001-08-29
WO 00/53173 PCT/~JS00/02626
hydrated calcium salt of dihydroxy open acid simvastatin was obtained as a
white
powder.
The calcium salt was delumped with a single pass through a cleaned
QUADROTM COMIL~ (Model 1975).
HPLC CONDITIONS
Column: YMC Basic 4.6 mm x 25 cm
Detector: ABS 757 lAU/volt output
Sample solvent: EtOH/CH3CN/H20 (1:1:1)
Column temp: 25 °C (Anal. Dept. runs samples at 5 °C to
prevent formation of
simvastatin on column).
Flow rate: 1 .5 mL / min.
Wavelength: 238 and 210 nm
Gradient: %H20 (10 mM 2HP04-
Time min. %CH3CN
KH2P04LpH
=
6.5)
0.00 30 70
20.00 45 55
34.00 70 30
39.00 70 30
39.50 30 70
43.00 30 70
Retention time of simvastatin 17.07 min.
open acid:
Retention time of simvastatin: 32.90 min.
SPECTRAL DATA
1H NMR (400 MHz, CD30D), ~ 5.97 (d, J = 9.6 Hz, 1 H), 5.77 (dd, J = 9.6, 5.2
Hz, 1
H), 5.49 (m, 1 H), 5.33 (m, 1 H), 4.17 (m, 1 H), 3.70 (m, 1 H), 2.44 - 2.35
(m, 2 H),
2.42 (dd, J = 15.7, 3.6 Hz, 1 H), 2.31 - 2.27 (m, 1 H), 2.29 (dd, J = 15.7,
8.4 Hz, 1 H),
2.00 (ddd, J = 15.7, 7.6, 2.4 Hz, 1 H), 1.93 (m, 1 H), 1.68 (m, 1 H), 1.61 -
1.55 (m, 2
-26-
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
H), 1.55 (m, 2 H), 1.42 (m, 1 H), 1.32 (m, 1 H), 1.19(m, 1 H), 1.12 (s, 6 H),
1.08 (d, J
= 7.2 Hz, 3 H), 0.89 (d, J = 7.2 Hz, 3 H), 0.84 (t, J = 7.6 hz, 3 H) ppm.
13C ~R (100.55 MHz, CD30D), 8 182.3, 179.3, 134.1, 133.2, 130.3, 129.6, 72.5,
69.8, 45.1, 44.4, 44.1, 38.8, 38.3, 36.4, 34.3, 33.9, 32.0, 28.6, 25.9, 25.37,
25.36, 23.7,
14.3, 9.9 ppm
EI MS: m/e: 437 (M+H), 419(M+H-H20), 303.
HR-EI MS: Calcd. for C25H3805 418.2719; Found: 418.2712.
EXAMPLE 2
Preparation of crystalline hydrated calcium salt of dihydroxy open acid
simvastatin
(Compound I) with BHA
A 22 L three-necked round bottom flask was equipped with a
temperature probe, a NZ inlet, an additional funnel, and an overhead stirrer.
8.0 L of
15% EtOH-HBO was added and purged with N, for 10 min. Simvastatin (396 g,
0.946
mol) was added, and the slurry purged with N, for 5 min. 198 mL of SN NaOH
(0.993 mol, 1.05 equiv.) was then added at room temperature. The hydrolysis
reaction
is usually complete in 1 h. as analyzed by HPLC (> 99.9% conversion). The pH
of the
reaction solution was adjusted to 7 to 8.5 by addition of 1 N HCl (about 65
mL).
A solution of Ca(OAc)2~H20(91.7 g, 0.520 mol, 0.55 equiv.) in 4.0 L
of 60% EtOH- HBO was purged with nitrogen for 5 min. 1.0 L of this solution
was
added to reaction solution over 30 min. The slurry was aged at room
temperature for
30 min, and then at 30 to 35 °C for 1-2 h. The rest of the Ca(OAc), in
EtOH-water
was added in portions over 3h hours at 30-35°C. The slurry was allowed
to age at 30
to 35 °C for 5 h under an atmosphere of Nz. The slurry was allowed to
cool to room
temperature and was collected by suction-filtration. The wet cake was washed
with 4
L 30% EtOH-H20, 4 L 20% EtOH-HzO, followed by 6 L x 3 of D.I. water. The solid
was suction dried under atmosphere of moist NZ (40 to 70% RH) at room
temperature
to give 1.7 Kg of wet cake.
The above wet cake was placed in a clean 20 L three necked flask
under atmosphere of nitrogen. A solution of BHA (2.603 g, 0.2 wt % equiv) in
degassed 15% EtOH HZO (8.5 L) was added, followed by addition of degassed
water
-27-
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
(2.55 L), and the slurry was agitated at room temperature for 1 to 2 h. The
solid was
collected by suction filtration under atmosphere of moist NZ with no washing
to give
1.49 Kg wet cake. The solid was suction dried under atmosphere of moist NZ (40
to
70% RH) at room temperature for 4 days. The calcium salt title product was
obtained
as a white powder (94% yield. 99.4%A at 238 nm, 0.2wt% BHA, KF = 7.3 %wt).
EXAMPLE 3
Preparation of crystalline hydrated calcium salt of dihydroxy open acid
simvastatin
(Compound I) in agueous nPrOH: Conventional addition mode
Step l: Hydrolysis
A 72 L three-necked round bottom flask was equipped with a
temperature probe, a NZ inlet, an additional funnel, and an overhead stirrer.
28.5 L of
D.I. H20 was added and purged with NZ for 10 min. 1.5 Kg simvastatin was
added,
followed by 788mL of SN NaOH in one portion at room temperature. The
hydrolysis
reaction is usually complete in 2 h. as analyzed by HPLC (> 99.9% conversion).
1.SL
of nPrOH was added and the pH of the reaction solution was adjusted to 9.5 to
I1.0
by adding 2 N HOAc (about 170 mL).
St-e~2: Salt Formation
150 g of seed (Compound I) was added to the above solution and the
resulting slurry was allowed to warm up to 35 to 40 °C. A solution of
Ca(OAc)z~H~O
(347 g) in 15 L of 20% nPrOH was purged with nitrogen for Smin. and added to
slurry
over 3 h. The resulting slurry was aged at 35 to 40 °C for 5 h. under
an atmosphere of
NZ and then cooled to room temperature. The solid was collected by filtration
and was
washed with 10% nPrOH-H20 (15 L x 3).
Step 3: BHA loading
The above wet cake (9.1 kg) was transferred into a clean 72 L three
necked flask under an atmosphere of nitrogen. A solution of BHA (7.6 g) in
degassed
10% nPrOH (45 L) was added and the slurry was agitated at room temperature for
1 h.
and filtered under atmosphere of NZ , and then suction dried under atmosphere
of
moist NZ (30 to 70% RH) at room temperature for 7 days. 1.78 Kg of Ca salt
title
-28-
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
product was obtained as a white powder (94% yield. 99.4%A at 238 nm, 0.2wt%
BHA, KF = 6.6 %wt).
EXAMPLE 4
Preparation of hydrated crystalline calcium salt of dihydroxy open acid
simvastatin in
aqueous PrOH: Simultaneous addition mode
The process described in this example allows for keeping half of the
batch in the vessel at all times as seed in a semi-continuous process.
100 g of simvastatin was hydrolyzed in 1.9 L water as described in
Example 3. Then, 100 ml nPrOH was added and the solution pH was adjusted to 9
to
11 with 1 N HOAc. The resulting solution and a solution of Ca(OAc)Z~H,O (23.2
g)
in 1.0 L of 20% nPrOH were added separately but simultaneously to a suspension
of
10-50 wt% Ca salt in 10% nPrOH (30 volume 10% PrOH relative to the amount of
the seed) at 30 to 40 °C over 3 h. After 5 h age at 30 to 40 °C,
the slurry was cooled
to room temperature, filtered, and loaded with anti-oxidant and dried as
described in
conventional addition mode process. 95% yield.
EXAMPLE 5
Preparation of crystalline hydrated calcium salt of dihydroxy open acid
simvastatin
(Compound I) in aqueous PrOH: Loading BHA throug-h co-crystallization
Method A: 100 g of simvastatin was hydrolyzed in 1.9 L water as
described in Example 3. Then, 100 ml nPrOH was added and the solution pH was
adjusted to 9 to 11 with 1 N HOAc. l Owt% seed was added and the slurry was
allowed to warm up to 35 to 40 °C. A solution of Ca(OAc)Z~HZO (23.2 g)
and BHA
(540 mg) in 1.0 L of 20% nPrOH were added to the slurry at 35 to 40 °C
over 3 h.
After 5 h age at 30 to 40 °C, the slurry was cooled to room
temperature, filtered and
washed with a solution ofBHA (O.lg/L) in 10% nPrOH (1L x 3). The wet cake was
dried under moist NZ as described in conventional addition mode process. The
final
dried Ca salt title product contained 0.2wt% BHA. 95% yield.
Method B: The procedure of Method A was employed with the
following change. Instead of adding BHA to a Ca(OAc)z solution, the same
amount of
BHA was added into the pH adjusted solution of hydrolyzed simvastatin at room
temperature. The solution was warmed to 35 to 40 °C to dissolve BHA.
Then, l Owt%
-29-
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
seed was introduced. The rest of the steps were as described in Method A. The
final
dried Ca salt title product contained 0.2wt% BHA. 95% yield.
EXAMPLE 6
Preparation of crystalline hydrated calcium salt of dihydroxy open acid
simvastatin
(Compound I) in aqueous PrOH: Loading BHA/propyl ~allate
Starting with 2.0 Kg of simvastatin, the calcium salt of dihydroxy open
acid simvastatin was crystallized, isolated, and washed as described in
Example 3.
The first wet cake was transferred to a clean 100L vessel under atmosphere of
Nz. A
solution of BHA (9.2g) and propyl gallate (11.2 g) in 50 L 10% nPrOH was added
to
above vessel. The slurry was aged at room temperature for 1 h. The slurry was
filtered
with no wash. The wet cake was dried under moist N~. 95% yield. The dried salt
was
loaded with 0.07wt% propyl gallate and 0.2 wt% BHA.
EXAMPLE 7
Preparation of crystalline hydrated calcium salt of dihydroxy open acid
simvastatin
(Compound I) in aqueous PrOH: Loading_prop~gallate
Starting with 2.0 Kg of simvastatin, the calcium salt of dihydroxy open
acid simvastatin was crystallized, isolated, and washed as described in
Example 3.
Then, the wet cake was washed with l OL of a solution of propyl gallate in 10%
nPrOH (propyl gallate concentration = 0.224 g/L). Then, 20 L of propyl gallate
solution in 10% nPrOH (propyl gallate concentration = 0.224 g/L) was added and
the
wet cake was mixed in the filtration pot before filtration. The wet cake was
dried
under moist N2. 95% yield. The dried salt was loaded with 0.07wt% propyl
gallate.
EXAMPLE 8
Preparation of crystalline hydrated calcium salt of dihydroxy open acid
simvastatin
(Compound I) Loading BHA, BHA/Vitamin E, and Vitamin E in heptane
100 g of simvastatin was hydrolyzed in 1.9 L water as described in
Example 3. Then, 100 ml nPrOH was added and the solution pH was adjusted to 9
to
11 with 1 N HOAc. l Owt% seed was added and the slurry was allowed to warm up
to
-30-
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
35 to 40 °C. A solution of Ca(OAc)2~H20 (23.2 g) in 1.0 L of 20% nPrOH
was added
to a slurry at 35 to 40 °C over 3 h. After 5 h age at 30 to 40
°C, the slurry was cooled
to room temperature. The calcium salt slurry was filtered and washed with 10%
nPrOH (500 mL x 1 ), followed by water ( 1 L x 3). The wet cake (KF = 75 to 80
wt%
water) was then washed with 1 L of heptane, to displace most of the water.
This wet
cake was washed with a solution of BHA or Vitamin E or BHA/Vitamin E (conc. _
1.38 g/ L, 800 mL) and dried under moist Nz.
EXAMPLE 9
Preparation of crystalline hydrated calcium salt of dihydroxy open acid
simvastatin
(Compound I) in agueous MeCN
A 7.2 L three-necked round bottom flask was equipped with a
temperature probe, a Nz inlet, an additional funnel, and an overhead stirrer.
2.1 L of
D.I. HBO was added and purged with NZ for 10 min. 150 g simvastatin was added,
followed by 78.8mL of SN NaOH in one portion at room temperature. The
hydrolysis
reaction is usually complete in 2 h. as analyzed by HPLC (> 99.9% conversion).
900
mL of MeCN was added and the pH of the reaction solution was adjusted to 9.5
to
11.0 by adding 2 N HOAc (about 17 mL).
30.0 g crystalline seed was added to above solution and the resulting
slurry was allowed to warm up to 30 to 35 °C. A solution of
Ca(OAc)2~H~O (34.7 g)
in 1.5 L of 30% MeCN was purged with nitrogen for 5 min. and added to reaction
slurry over 3 h. The slurry was at 3~ to 40 "C for 5 h. under atmosphere of
N~. The
slurry was allowed to cool to room temperature and the solid was collected by
filtration. The wet cake was washed with 30% MeCN ( 1.5 L) and 10% MeCN ( 1.0
L),
and rinsed/washed with a solution of BHA (0.9 g/L) in degassed 10% MeCN (1.0 L
x
2). The solid was suction dried under atmosphere of moist NZ (30 to 70% RH) at
room
temperature for S days. 1.67 Kg of the title compound was obtained as a white
powder (88% yield. 99.4%A at 238 nm, 0.2wt% BHA, KF = 6.6 %wt).
EXAMPLE 10
Preparation of crystalline hydrated calcium salt of dihydroxy open acid
simvastatin
(Compound I) in agueous MeOH
-31 -
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
50 g simvastatin was hydrolyzed in 850 mL water as described in
Example 3. Then, 150 ml MeOH was added and the solution pH was adjusted to 7
to
11 with 1N HOAc. l Owt% seed was added and the slurry was allowed to warm up
to
30-35 °C. A solution of Ca(OAc)z~Hz0 (11.6 g) in 500 mL of 30% MeOH was
added
to the slurry at 30-35 °C over 3 h. After 5 h age at 30-35 °C,
the slurry was cooled to
room temperature. The dihydroxy open acid simvastatin calcium salt slurry was
filtered and washed with 20% MeOH (200 ml) and water (500 ml x 3). The wet
cake
was dried under moist N2. The final dried Ca salt title product was isolated
in 96%
yield.
EXAMPLE 11
Preparation of crystalline hydrated calcium salt of dihydroxy open acid
simvastatin
(Compound I) in agueous i-PrOH DMF DMSO
50 g simvastatin was hydrolyzed in 850 mL water as described in
Example 3. Then, 150 ml i-PrOH was added and the solution pH was adjusted to 7
to
11 with 1 N HOAc. l Owt% seed was added and the slurry was allowed to warm up
to
30-35 °C. A solution of Ca(OAc)Z~HZO (11.6 g) in 500 mL of 30% i-PrOH
was added
to the slurry at 30-35 °C over 3 h. After 5 h age at 30-35 °C,
the slurry was cooled to
room temperature. Ca salt slurry was filtered and washed with 20% ml i-PrOH
(200
ml) and with water (500 ml x 3). The wet cake was dried under moist N2. The
final
dried Ca salt title product was isolated in 96% yield.
The same procedure could be applied to prepare Compound I in DMF,
DMSO, and similar solvents.
EXAMPLE 12
Preparation of crystalline hydrated calcium salt of dihydroxy open acid
simvastatin
(Compound I) in water
50 g of simvastatin was hydrolyzed in 1.0 L water as described in
Example 3. Then, the solution pH was adjusted to 11 with 1 N HOAc. lOwt% seed
was added and the slurry was allowed to warm up to 35-40 °C. A solution
of
Ca(OAc)Z~H20 (11.6 g) in 500 mL of water was added to the slurry at 35-40
°C over 5
h. After 10 h age at 35-40 °C, the slurry was cooled to room
temperature. Ca salt
-32-
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
slurry was filtered and washed with water (500 ml x 3). The wet cake was dried
under
moist N2. The final dried Ca salt title product was isolated in 96% yield.
EXAMPLE 13
Preparation of crystalline hydrated calcium salt of dihydroxy open acid
simvastatin
(Compound I) from dihydroxy open acid simvastatin ammonium salt
Method A: 50 g of dihydroxy open acid simvastatin ammonium salt
was dissolved into 800 ml of 25% nPrOH which was then added dropwise to a
solution of Ca(OAc)~~H20 (10.7 g) in 75 ml of water at room temperature over
2h.
The resulting slurry was aged at 30 to 35 °C 5 h. After cooling to room
temperature,
the slurry was isolated by filtration. The wet cake was washed with 10% nPrOH
(500
ml x 3). The wet cake was loaded with antioxidants and dried under moist NZ as
described above to give the title product.
Method B: 50 g of dihydroxy open acid simvastatin ammonium salt
was added into a solution of Ca(OAc)Z~HZO ( 10.7 g) in 1.5 L of 10% nPrOH in
one
portion at room temperature. The resulting slurry was aged at 30 to 35
°C 5 h. After
cooling to room temperature, the slurry was isolated by filtration. The wet
cake was
washed with 10% nPrOH (500 ml x 3). The wet cake was loaded with antioxidants
and dried under moist Nz as described above to give the title product.
By using both Methods A and B described as above, the title product
could also prepared from ammonium salt in the following aqueous solvents:
acetone,
MeOH, EtOH, iPrOH, , MeCN, neat water, DMF, DMSO, and similar solvents.
EXAMPLE 14
Recrystallization Procedure Using nPrOH-HZO
Dried Compound I (21 g) was dissolved in 150m1 of 40% nPrOH at
35°C and line filtered. This solution was added dropwise to a slurry of
10 wt% seed in
480 ml of 4% PrOH at 35 to 40°C over 3 to 5 h. After aging overnight at
35 to 40°C,
the slurry was allowed to cool to room temperature. The solid was filtered and
washed
with 10% nPrOH (200m1 x 2). The wet cake was dried under moist N2. 95% yield.
EXAMPLE 15
-33-
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
Recrystallization Process Using EtOH-HZ_O
Method A: 25 g of Compound I was dissolved into 425m1 of 95%
EtOH at 40 °C and line filtered. The filtered solution was added
dropwise to 825m1 of
water in the presence of 10% wt seed at 30 to 35 °C over 3 to 5 h. The
slurry was aged
overnight and cooled to 0 to 5 °C before filtration. The wet cake was
washed with 250
ml of 30% EtOH and dried under moist NZ at room temperature. 92% yield.
Method B: 25 g of Compound I was dissolved into 625m1 of 95%
EtOH at 30 to 40 °C and line filtered. 525 ml of water was added at 30
to 40 °C. After
adding l Owt% seed, 825 ml of water was added dropwise at 30 to 40 °C
over 3 h. The
slurry was aged overnight and cooled to 0 to 5 "C before filtration. The wet
cake was
washed with 250 ml of 30% EtOH and dried under moist NZ at room temperature.
92% yield.
EXAMPLE 1 G
An open, randomized, four-period, crossover study to compare the effect of
itraconazole on the single-dose pharmacokinetics of intraduodenally
administered
dihydroxy open acid simvastatin versus orally administered simvastatin in
healthy
male subiects
Objectives: (1) To determine the effect of itraconazole, a potent CYP3A
inhibitor,
on the plasma AUC of active and of total HMG-CoA reductase inhibitory activity
following a single intraduodenal dose of a solution containing 5 mg dihydroxy
open
acid simvastatin; (2) to determine and compare the dose-adjusted plasma AUC of
active HMG-CoA reductase inhibitory activity following a single intraduodenal
dose
of a solution containing 5 mg dihydroxy open acid simvastatin versus a single
oral
administration of simvastatin 20-mg film coated tablet (FCT); (3) to determine
the
effect of itraconazole on the plasma AUC's of dihydroxy open acid simvastatin
and of
simvastatin lactone concentration following a single intraduodenal dose of a
solution
containing 5 mg dihydroxy open acid simvastatin.
-34-
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
Study Design: This study was designed in an open, four-period crossover
randomized
fashion. Twelve healthy male subjects received four treatments (A, B, C and
D). In
Treatment A, subjects received itraconazole 200 mg (2 x 100-mg capsule) for 4
days
followed by a single dose of 5-mg dihydroxy open acid simvastatin solution
administered intraduodenally on day 4, 1 hour after the fourth daily dose of
itraconazole. In Treatment B, subjects were given a single dose of 5-mg
dihydroxy
open acid simvastatin solution administered intraduodenally on day 1.
Intraduodenal
administration was accomplished via a nasoduodenal tube placed under
fluoroscopic
guidance by an experienced gastroenterologist just prior to dosing and removed
following the 1-hr postdose measurements. Treatments C and D were similar to
those
of Treatments A and B, except that orally dosed simvastatin 20-mg conventional
film
coated tablet was used. The wash out between treatment periods was at least 7-
days
following a treatment containing itraconazole or at least 3 days following a
treatment
without itraconazole. Plasma samples were collected at appropriate time
intervals for
up to 24 hours following simvastatin or dihydroxy open acid simvastatin
administration, for analysis of total and active HMG-CoA reductase inhibitory
activities as well as for simvastatin and dihydroxy open acid simvastatin
concentrations.
Analytical Methodology: Plasma concentrations of simvastatin and dihydroxy
open acid simvastatin acid were determined simultaneously by an improved
liquid
chromatography/tandem mass spectrometry (LC/MS/MS) method using lovastatin and
dihydroxy open acid lovastatin acid as internal standards. An enzymatic assay
method
was used to determine plasma concentrations of active and total (active plus
potentially active) HMG-CoA reductase inhibitory activity.
Pharmacokinetics: The area under the plasma concentration-time profile from
time
zero to the last sampling time (AUCO-last) was calculated using linear
trapezoidal
rule. The apparent elimination rate constant (k) of simvastatin and dihydroxy
open
acid simvastatin was estimated by least-squares regression analysis of the log-
linear
portion of the simvastatin and dihydroxy open acid simvastatin concentration-
time
data, and the apparent elimination half life (t~i2) was calculated as tli2 =
0.693/k. All
calculations were based on designated sampling times or actual sampling times
when
they differed from the designated times by more than 10 minutes.
-35-
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
Discussion of Results: This was an open, randomized, four-period crossover
study in twelve healthy male subj ects. The results showed that intraduodenal
administration of dihydroxy open acid simvastatin 5-mg solution yielded higher
(~4-
fold) dose-adjusted plasma AUC of the active HMG-CoA reductase inhibitory
activity
than oral administration of simvastatin 20-mg tablet (Table 1). Following
dihydroxy
open acid simvastatin administration, the unchanged dihydroxy open acid
simvastatin
was the major component (~60%), while simvastatin was a minor component (<10%)
contributing to plasma HMG-CoA reductase inhibitory activity. As evident by
comparable AUC values for both the total and active inhibitors (see Table 1)
as well
as low plasma levels of simvastatin in plasma (AUC <10% of dihydroxy open acid
simvastatin AUC) (see Table 3), lactonization of either dihydroxy open acid
simvastatin or its active metabolites occurred minimally following
intraduodenal
administration of dihydroxy open acid simvastatin. Pretreatment with
itraconazole
caused minimal changes (1.3-1.5-fold) in the systemic exposure as measured by
AUC
and Cmax of HMG-CoA reductase inhibitory activity (total or active) following
administration of dihydroxy open acid simvastatin 5-mg intraduodenally, as
compared
to that observed following oral administration of simvastatin 20-mg tablet
(1.3-3.8-
fold) (see Table 2) When measured as the unchanged drug, the effect of
itraconazole
observed following dihydroxy open acid simvastatin administration was also
minimal
(1.5-fold) and was much less than the corresponding measure obtained following
simvastatin administration (19-fold) (see Table 3). A moderate effect (3-4-
fold
increase) was noted for the AUC and Cmax of simvastatin following treatment
with
itraconazole prior to dihydroxy open acid simvastatin administration (see
Table 3).
However, apparent tl/2 values for dihydroxy open acid simvastatin or
simvastatin
were essentially unchanged by itraconazole(see Table 3). Overall, these
results
indicate that the pharmacokinetics of dihydroxy open acid simvastatin is less
prone to
alteration by itraconazole, a potent CYP3A inhibitor, than that of simvastatin
in
humans. From these results it appears that dihydroxy open acid simvastatin,
although
a substrate for CYP3A, is metabolized with a much lower intrinsic clearance
than that
of simvastatin in human liver microsomes.
-36-
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
TABLE 1
Pharmacokinetic parameters for total and active HMG-CoA reductase inhibitors
following administration of S-mg dihydroxy open acid simvastatin (SVA)
intraduodenally or 20-mg simvastatin (SV) tablet orally to 12 healthy male
volunteers.
Results are means from 12 subjects. Values in parentheses are SD.
Drug Administered5 mg SVA (ID) 20 mg SV (FCT)
12 12
Total 56.0 (28.6) 180.7 (57.2)
AUC Active 54.1 (28.5) 59.7 (18.1)
(ng eq. hr/ml) SV 1.02 (0.5) 16.9 (10.5)
SVA 33.9 (16.7) 7.7 (4.9)
Total 6.2 (3.6) 67.1 (27.2)
Cmax Active 5.7 (3.0) 16.1 (4.4)
(ng eq./ml) SV
0.13 (0.05) 6.84 (4.6)
SVA 3.78 (1.93) 0.92 (0.58)
Total 3.3 (2.3) 1.2 (0.5)
Active 3.6 (2.5) 1.4 (0.5)
Tmax (h
r)
SV 4.9 (2.0) 1.1 (0.5)
SVA 4.1 (2.1) 3.7 (2.1)
i
SV 6.7 (1.7) ~ 4.2 (1.9)
t 1 /2 (hr) n=8
SVA 2.3 (0.5) 3.5 (1.0)
-37-
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
TABLE 2
Pharmacokinetic parameters for total HMG-CoA reductase inhibitor following
administration of 5-mg dihydroxy open acid simvastatin (SVA) intraduodenally
or 20-
mg simvastatin (SV) tablet orally with or without pretreatment with
itraconazole
(2x 100-mg capsule, qd) for 4 days to 12 healthy male volunteers. Results are
means
from 12 subjects. Values in parentheses are SD.
Treatment AtJC Cmax Tmax
n a .hr/mLn a /mL hr
Total Inhibitors
Single 5-mg SVA (ID) + 4 Days of 86.1 8.2 ~ 4.3 t
Daily 200-mg Itraconazole f 47 4.0 2.2
Single 5-mg SVA (ID) 56.0 6.2 t 3.3 f
t 28.6 3.6 2.3
Geometric Mean Ratio 1.5 1.3
Single 20-mg SV (CT) + 4 Days of 683 f 126 f 1.9 ~
Daily 200-mg ltraconazole 232 54.8 0.9
Single 20-mg SV (CT) 180.7 67.1 t 1.2 t
t 57.2 27.2 0.5
a me ric Mean Ratio ,g 1,
Active Inhibitors
Single 5-mg SVA (ID) + 4 Days of 75.0 7.4 t 5.5 +
Daily 200-mg Itraconazole t 39.6 3.9 1.7
Single 5-mg SVA (ID) 54. I 5.7 ~ 3.6 ~
f 28.5 3.0 2.5
Geometric Mean Ratio 1.3 1.3
Single 20-mg SV (CT) + 4 Days of 195 t 23.0 ~ 3.1 f
Daily 200-mg Itraconazole 104 13.6 0.9
Single 20-mg SV (CT) 59.7 16.1 f 1.4 f
t 18.1 4.4 0.5
Geometric Mean Ratio 3.1 1.3
-38-
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
TABLE 3
Pharmacokinetic parameters for simvastatin or dihydroxy open acid simvastatin
following administration of 5-mg dihydroxy open acid simvastatin (SVA)
intraduodenally or 20-mg simvastatin (SV) tablet orally with or without
pretreatment
with itraconazole (2x100-mg capsule, qd) for 4 days to 12 healthy male
volunteers.
Results are means from 12 subjects. Values in parentheses are SD.
Treatment AUC Cmax tl/2 Tmax
no a n a hr hr
.hr/mL /mL
Simvastatin
Single 5-mg SVA (ID) + 4 Days 5.18 0.43 6.7 t 2.5 5.3
of Daily 200-mg Itraconazole t 3.15 f 0.2 (n=11 t
) 1.9
Single 5-mg SVA (ID) 1.02 0.13 6.7 t 1.7 4.9
t 0.5 t 0.05 (n=8) t
2.0
Geometric Mean Ratio 4.4 3.2 1.0
Single 20-mg SV (CT) + 4 Days 316 t 77.2 5.2 t 1.5 1.8
of Daily 200-mg Itraconazole 142 t 50.7 t
1.1
Single 20-mg SV (CT) 16.9 6.84 4.2 t 1.9 1.1
t 10.5 t 4.6 t
0.5
Simvastatin Acid
Single 5-mg SVA (ID) + 4 Days 56.8 5.78 2.8 t 1.1 4.7
of Daily 200-mg Itraconazole t 33.5 t 3.27 t
2.4
Single 5-mg SVA (ID) 33.9 3.78 2.3 t 0.5 4.1
t 16.7 t 1.93 t
2.1
Geometric Mean Ratio 1.5 L5 1.1
Single 20-mg SV (CT) + 4 Days 86.2 9.76 4.5 t I 4.0
of Daily 200-mg Itraconazole t 65.6 f 7.38 .3 t
1.4
Single 20-mg SV (CT) 7.70 0.92 3.5 t I.0 3.7
t 4.9 t 0.58 t
2.1
Geometric Mean Ratio 1 I.0 10.7 1.3
-39-
CA 02364253 2001-08-29
WO 00/53173 PCT/US00/02626
While the invention has been described and illustrated with reference
to certain particular embodiments thereof, those skilled in the art will
appreciate that
various changes, modifications and substitutions can be made therein without
departing from the spirit and scope of the invention. For example, effective
dosages
other than the particular dosages as set forth herein above may be applicable
as a
consequence of variations in the responsiveness of the mammal being treated
for any
of the indications for the active agents used in the instant invention as
indicated
above. Likewise, the specific pharmacological responses observed may vary
according to and depending upon the particular active compound selected or
whether
there are present pharmaceutical carriers, as well as the type of formulation
employed,
and such expected variations or differences in the results are contemplated in
accordance with the objects and practices of the present invention. It is
intended,
therefore, that the invention be defined by the scope of the claims which
follow and
that such claims be interpreted as broadly as is reasonable.
-40-