Note: Descriptions are shown in the official language in which they were submitted.
CA 02364730 2001-12-06
1
COMPOUND
The present invent'ron relates to a compound.
One aspect of gene therapy involves the introduct'ron of foreign nucleic acid
(such as DNA)
into cells, so that its expressed protein may carry out a desired therapeutic
function.
Examples of this type of therapy include the insertion of TK, TSG or ILG genes
to treat
cancer; the insertion of the CFTR gene to treat cystic fibrosis; the insertion
of NGF, TH or
LDL genes to treat neurodegenerative and cardiovascular disorders; the
insertion of the IL-
1 antagonist gene to treat rheumatoid arthritis; the insertion of HIV antigens
and the TK
gene to treat AIDS and CMV infections; the insertion of antigens and cytokines
to act as
vaccines; and the insertion of p-globin to treat haemoglobinopathic
conditions, such as
thalassaemias.
Many current gene therapy studio utilise,adenoviral gene vectors - such as Ad3
or Ad5 - or
other gene vectors. However, serious problems have been associated with their
use. This
has prompted the development of less hazardous, n on-viral approaches to gene
transfer.
A non-viral transfer system of great potential involves the use of cationic
liposomes. In this
regard, cationic liposomes - which usually consist of a neutral phospholipid
and a cationic
lipid - have been used to transfer DNA, mRNA, antisense oligonucleotides,
proteins, and
drugs into cells. A number of ~nic liposomes are commercially available and
many n~v
cationic lipids have recently been synthesised. The efficacy of these
liposomes has been
illustrated by both in vitro and in vivo.
A cytofectin useful in the preparation of a cationic liposome is 111 [1-(2,3-
dioleoytoxy)propyl]-
N,N,N trimethyl ammonium chloride, otherwise known as "DOTMA".
One of the most commonly used cationic liposome systems consists of a mixture
of a
neutral phospholipid dioleoylphosphatidylethanolamine (commonly known as
"DOPE") and
a cationic lipid, 3[i-[(N,N-dimethylaminoethane)carbamoyl]cholesterol
(commonly known as
"DC-Chol").
CA 02364730 2001-12-06
2
Despite the efficacy of the known cationic liposomes there is still a need to
optimise the
gene transfer efficiency of cationic liposomes in human gene therapy. With the
near
completion of the human genome project, the use of genes for therapeutic
purposes,
described as gene therapy is increasingly expected to revolutionise medicine.
In this
context, even though still less effective than viral technology, non-viral
delivery is
increasingly recognised by the scientific community as the safest option for
human
applications.
This field has evolved considerably in the last decade with the apparition of
complex
macromolecular constructs including many elements of different existing
technologies
(viral proteins or peptides, liposomes, polymers, targeting strategies and
stealth
properties).
Our copending application PCT/GB00/04767 teaches a system based on a triplex
composed of a viral core peptide Mu, plasmid DNA and cationic Liposome (LMD).
This
platform technology gave us good success in vitro and promising results in
vivo. But as
for all existing non-viral technology more development is needed to achieve a
therapeutic
level in vivo.
To this end, formulation must achieve stability of the particle in biological
fluids (serum,
lung mucus) and still maintain efficient transfection abilities.
This n3quirement is one of the main hurdles of all existing technology.
Current stable
formulations achieve little transfection and most present efficient
transfecting agents are
drastically limited in the scope of their application due to this instability.
After administration (in blood for systemic application or in mucus for lung
topical
administration), the charged complexes are exposed to salt and biological
macromolecules leading to strong colloidal aggregation and adsorption of
biological
active elements (opsoni ns) at their surface. The gene vehicles undergo
drastic changes
that could include precipitation, binding of proteins leading to particle
elimination by
macrophages and surface perturbation resulting in its destruction.
With the aim of generating drug and gene delivery systems for cell specific
targeting in
vitro and in vivo, protocols are required for the production of biological
fluid- stable
CA 02364730 2001-12-06
3
delivery systems with sufficient activity to exhibit therapeutic benefits.
Therefore, a
balance between stability and activity must be found for an efficient
drug/gene delivery
vehicle.
Our copending applications PCTIGB00/04767 teaches a system based on modified
lipid
wherein the lipid carries a carbohydrate moiety. These modified lipids have
been found
to stable and have low toxicity. Such systems require the linking an
additional moiety to
the lipid to assist in the provision of a modified lipid which is stable and
has low toxicity.
There is a desire in the art to provide lipids comprising groups to which
additional
moieties may be readily linked.
The present invention alleviates the problems of the prior art.
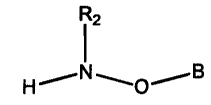
According to one aspect of the present invention there is provided a compound
of the
formula
Rz
~ i'B
H O
wherein B is a lipid; and wherein RZ is H or a hydrocarbyl group.
According to one aspect of the present invention there is provided a process
for preparing
a modified lipid of the formula
Rz
A/.X~.C/N..~O..~-.B
R~
comprising reacting (i) a compound of the formula; and
O
A'
X R~
(ii) a compound of the formula
Rz
w
H O
CA 02364730 2001-12-06
4
wherein B is a lipid and A is a moiety of interest (M01); wherein X is an
optional linker
group; wherein R ~ is H or a hydrocarbyl group; and wherein R 2 is a lone pair
or R4,
wherein R4 is a suitable substituent.
According to one aspect of the present invention there is provided a
composition
comprising (i) a compound of the formula
O
A~
X R~
(ii) a compound of the formula
R2
H O
wherein B is a lipid and A is a moiety of interest (M01); wherein X is an
optional linker
group; wherein R, is H or a hydrocarbyl group; and wherein R2 is a lone pair
or a suitable
substituent.
According to another aspect of the present invention there is provided a
compound, a
composition or a compound when prepared by the process of the present
invention for use
in therapy.
According to another aspect of the present invention there is provided the use
of a
compound, a composition or a compound when prepared by the process of the
present
invention in the manufacture of a medicament for the treatment of a genetic
disorder or a
condition or a disease.
According to another aspect of the present invention there is provided a Ii
posome formed
from a compound, a composition or a compound when prepared by the process of
the
present invention.
According to another aspect of the present invention there is provided a
method of
preparing a liposome comprising forming the liposome from a compound, a
compos~ion or
a compound when prepared by the process of the present invention.
CA 02364730 2001-12-06
According to another aspect of the present invention there is provided a
liposome according
to the present invention or a liposome as prepared by the method of the
present invention
for use in therapy.
5 According to another aspect of the present invention there is provided the
use of a liposome
according to the present invention or a liposome as prepared by the method of
the present
invention in the manufacture of a medicament for the treatment of genetic
disorder or
condition or disease.
According to another aspect of the present invention there is provided a
combination of a
nucleotide sequence or a pharmaceutically active agent and any one or more of:
a
compound, a composition, a compound when prepared by the process of the
present
invention, a liposome of the present invention, or a liposome as prepared by
the method of
the present invention.
According to another aspect of the present invention there is provided a
combination
according to the present invention for use in therapy.
According to another aspect of the present invention there is provided the use
of a
combination according to the present invention in the manufacture of a
medicament for the
treatment of genetic disorder or condition or disease.
According to another aspect of the present invention there is provided a
pharmaceutical
composition comprising a compound, a composition or a compound when prepan3d
by the
process of the present invention ad mixed with a pharmaceutical and,
optionally, admixed
with a pharmaceutically acceptable diluent, carrier or excipient.
According to another aspect of the present invention there is provided a
pharmaceutical
composition comprising a liposome according to t he present invention or a
liposome as
prepared by the method of the present invention admixed with a pharmaceutical
and,
optionally, admixed with a pharmaceutically acceptable diluent, carrier or
excipient.
Some further aspects of the invention are define d in the appended claims.
We have found the provision of a lipid comprising an aminoxy group allows for
simple
CA 02364730 2001-12-06
6
linking of further moieties to the lipid via the aminoxy group. When reacted
with a moiety
(M01) comprising an aldehyde or ketone group, a compound is provided in which
the
MOI and lipid are linked via an amide group. Such a linkage may be simple
prepared in a
"one-pot" reaction. This methodology avoids extensive purification procedures
by simple
dialysis or excess, non-reacted reagents.
15
The post-coating one-pot methodology of the present process is based on
selective and
high reactivity of the aminoxy-linker to react with aldehydes and ketones to
form -C=N-
(Schiff-base like) covalent linkages. Importantly, the reaction can be carried
out in
aqueous environment at basic or acidic pH. Furthermore, there is no partial
breakdown
of the reactive group when exposed to aqueous conditions as it is the case for
NHS -
activated carboxyls and other esters. Therefore, the stability of the reactive
species, e.g.
the aldehyde/ketone and the aminoxy allows total control of the surface
reaction without
loss of reactive species due to hydrolysis/degradation.
PREFERRED ASPECTS
The compound of the present invention is of the formula
Rz
./
H O
wherein B is a lipid; and wherein R2 is H or a hydrocarbyl group.
The term "hydrocarbyl group" as used herein means a group comprising at least
C and H
and may optionally comprise one or more other suitable substituents. Examples
of such
substituents may include halo, alkoxy, vitro, an alkyl group, a cyclic group
etc. In
addition to the possibility of the substituents being a cyclic group, a
combination of
substituents may form a cyclic group. If the hydrocarbyl group comprises more
than one
C then those carbons need not necessarily be linked to each other. For
example, at
least two of the carbons may be linked via a suitable element or group. Thus,
the
hydrocarbyl group may contain hetero atoms. Suitable hetero atoms will be
apparent to
those skilled in the art and include, for instance, sulphur, nitrogen and
oxygen. A non-
limiting example of a hydrocarbyl group is an acyl group.
A typical hydrocarbyl group is a hydrocarbon group. Here the term
"hydrocarbon" means
CA 02364730 2001-12-06
7
any one of an alkyl group, an alkenyl group, an alkynyl group, which groups
may be
linear, branched or cyclic, or an aryl group. The term hydrocarbon also
includes those
groups but wherein they have been optionally substituted. If the hydrocarbon
is a
branched structure having substituent(s) thereon, then the substitution may be
on either
the hydrocarbon backbone or on the branch; alternatively the substitutions may
be on the
hydrocarbon backbone and on the branch.
Preferably the reaction of the present invention is performed in an aqueous
medium.
OPTIONAL LINKER X
In a preferred aspect optional linker X is present.
In a preferred aspect X is a hydrocarbyl group.
In a preferred aspect the linker X comprises or is linked to the lipid via a
polyamine group.
It is believed that the polyamine group is advantageous because it incr~ses
the DNA
binding abilit)r and efficiency of gene transfer of the resultant liposome.
In one embodiment, preferably the polyamine group is a unnaturally occurring
polyamine. It
is believed that the polyamine head -group is advantageous because the
increased amino
functionality increases the overall positive charge of the liposome. In
addifron, polyamines
are known to both strongly bind and stabilise DNA. In addition, polyamines
occur naturally
in cells and so it is believed that toxicological problems are minimised.
In another embodiment, preferably two or more of the amine groups of the
polyamine group
of the present invention are separated by one or more groin which are not
found in nature
that separate amine groups of naturally occurring polyamine compounds (i.e.
preferably the
polyamine group of the present invention has un -natural spacing).
Preferably the polyamine group contains at least iwo amines of the polyamine
group that
are separated (spaced from each other) from each other by an ethylene ( -
CH2CH~-) group.
CA 02364730 2001-12-06
8
Preferably each of the amines of the polyamine group are separated (s~ced from
each
other) by an ethylene (-CH2CHr) group.
Typical examples of suitable polyamines include spermidine, spermine,
caldopentamine,
norspermidine and norspermine. Preferably the polyamine is spermidine or
spermine as
these polyamines are known to interact with single or double stranded DNA. An
alternative
preferred polyamine is caldopentamine.
R~
In a preferred aspect R~ is H
C=N
The C=N bond may be acid labile or acid resistant.
In one aspect the C=N bond is acid labile.
In one aspect the C=N bond is acid resistant.
MOI
The moiety of interest (M01) may be any moiety which one wishes to link to a
lipid.
The MOI may be a carbohydrate moiety.
In a preferted aspect the carbohydrate moiety is a mono -saccharide.
In a preferred aspect the carbohydrate moiety is a sugar moiety.
Preferably the carbohydrate moiety is selected from mannose, glucose (D-
glucose),
galactose, glucuronic acid, lactose, maltose, maltotriose, maltotetraose,
maltoheptaose
and mixtures thereof. More preferably the carbohydrate moiety is D-glucose.
CA 02364730 2001-12-06
9
In one aspect the compound of the present invention comprises from 1 to 7
carbohydrate
moieties. Preferably the compound comprises one carbohydrate moiety.
LIPID
In a preferred aspect the lipid is or comprises a cholesterol group or a
glycerol/ceramide
backbone. Any lipid-like structure or polyamine is suitable.
Preferably the cholesterol group is cholesterol.
Preferably the cholesterol group is linked to X via a carbamoyl linkage.
The cholesterol group can be cholesterol or a derivative thereof. Examples of
cholesterol
derivative include substituted derivatives wherein one or more of the cydic
CHz or CH
groups and/or one or more of the straight- chain CH2 or CH groups islare
appropriately
substituted. Alternatively, or in addition, one or more of the cyclic groups
and/or one or
more of the straight-chain groups may be unsaturated.
In a preferred embodiment the cholesterol group is cholesterol. It is believed
that
cholesterol is advantageous as it stabilises the resultant liposomal bilayer.
Preferably the cholesterol group is linked to the optional linker group via a
carbamoyl
linkage. It is believed that this linkage is advantageous as the resultant
liposome has a low
or minimal cytotoxicity.
Further Aspects
Preferably R2 is H or a hydrocarbyl group.
In a preferred aspect the R2 hydrocarbyl group contains optional heteroatoms
selected
from O, N and halogens.
In a preferred aspect R2 is H.
CA 02364730 2001-12-06
Preferably the process of the present invention is an aqueous medium or in a
wholly
aqueous medium.
The present invention further provide a compound prepared by a process of the
pn3sent
5 invention defined herein, a compound obtained by a process of the present
invention
defined herein, and/or a compound obtainable by a process of the present
invention defined
herein.
Preferably the compound is in admixture with or associated with a nucleo tide
sequence.
The nucleotide sequence may be part or all of an expression system that may be
useful in
therapy, such as gene therapy.
In a preferred aspect the compound of the present invention is in admixture
with a
condensed polypeptide/ nucleic acid complex to provide a non-viral nucleic
acid delivery
vector. The condensed polypeptide/ nucleic acid complex preferably include
those
disclosed in our copending application PCT/GBOOI04767. Preferably the
polypeptides or
derivatives thereof are capable of binding to the nucleic acid complex.
Preferably the
polypeptides or derivatives thereof are capable of condensing the nucleic acid
complex.
Preferably the nucleic acid complex is heterologous to the polypeptides or
derivatives
thereof.
Preferably the process comprises the use of a molecular sieve.
Preferably, the cationic liposome is formed from the compound of the present
invention and
a neutral phospholipid - such as DOTMA or DOPE. Preferably, the neutral
phospholipid is
DOPE.
The present invention will now be described in further detail by way of
example only with
reference to the accompanying frgures in which:-
Figure 1 - Scheme 1 Synthesis of Hydroxylamine lipid 11. Reagents: (a) CHZCI2,
Et3N,
BoczO, rt, 5h, 98%; (b) EtOAc, N-hydroxysuccinimide (1 eq.), DCC (1 eq.), 10
h., rt; (c)
(8), EtOACITHF [95/5], Et3N (pH = 8), 2 h., r.t, 90%; (d) CHZ CI2, TFA (15
eq), 0°C, Na, 5
h, 86%.
CA 02364730 2001-12-06
11
Figure 2 - Principle of chemioselective giycosylation of O -substituted
hydroxylamine with
D-Glucose (Although the [i-anomer is shown, mutarotation does occur and a-
anomer is
produced as well).
Figure 3 - Possible structures of neoglycolipid obtained from mannose.
Figure 4 - Result of analysis of differents lipoplexes size by photon
correlation
spectroscopy (PCS). The size was measured after 30 min for lipoplexes at
[DNA]=1
p,g/ml in Optimem +/- 10~o FCS, 37°C. The comparison of standard LMD
formulation
(LMD) and LMD modified by addition of 7.5 molar °~ of product 12h and
12i was made in
Optimem (white) and 10% Serum ( black) and expressed in percent of size
increase
over the original measured size of 180 nm .
Figure 5 - A comparison between the transfection efficiencies of basic LMD and
LMD
glycomodified with 7.5 molar % of product 12h and 12i onto Hela Cells in
0%(white),
50%(black and white) and 100 % Serum ( black) conditions. The results are
expressed
as relative light units per milligram of protein (n = 4).
Figure 6 - A structure of an aminoxy lipid
The present invention will now be described in further detail in the f
ollowing examples.
EXAMPLES
Experimental Section
Synthesis of neoglycolipids
General:' H NMR spectra were recorded at ambient temperature on either Brucker
DRX400, DRX300 or Jeol GX-270Q spectrometers, with residual nonisotopicaly
labeled
solvent (e.g. CHCI3, S,,=7.26) as an internal reference. '3C-NMR spectra were
recorded
on the same range of spectrometers at 100, 75 and 68.5 MHz respectively, also
with
residual nonisotopicaly labelled solvent (e.g. CHCI3, ~=77.2) as an internal
reference.
Infrared Spectra were recorded on Jasco FT/IR 620 using NaCI plates and Mass
spectra
CA 02364730 2001-12-06
12
(Positive ions electrospray) were recorded using VG -70708 or JEOL SX-102
instruments. Chromatography refers to flash column chromatography, which was
performed throughout on Mer ck-Kieselgel 60 (230-400 mesh) with convenient
solvent.
Thin layer chromatography (Tlc) was performed on pre -coated Merck-Kieselgel
60 F254
aluminium backed plated and revealed with ultraviolet light, iodine, acidic
ammonium
molybdate(IV), acidic ethanolic vanilin, or other agents as appropriate.
Neoglycolipids
purity was assessed using analytical high- pressure liquid chromatography
(HPLC) on a
Hitachi system using a Purospher~ RP -18 endcapped column (5 pm). Elution was
pertormed at an isocratic flow rate of 1 mUmin with CH3 CN/HZO (60:40) and
fraction
were detected at 205 nm wavelength before collection and Mass Analysis. Dried
CH2Cl2 was distilled with phosphorous pentoxide before use. All other dry
solvents and
chemicals were purchased from Sigma-Ald rich Company LTD (Poole, Dorset, UK).
Abbreviations: Boc: tort-butoxycarbonyl ; br: broad ; Chol: cholesteryl ; DMF:
N, N-
dimethyl formamide ; DMSO: dimethyl sulfoxide ; TFA: trifluoroacetic acid ;
THF:
tetrahydrofuran.
2-(Cholesteryloxycarbonyl)aminoetha nol (2): A solution of cholesteryl
chloroformate
(99.898, 0.218 mol) in CHZ CI2 (600 mL) was added to a stirred solution of 2-
aminoethanol (29.5 mL, 0.489 mol, 2.2 equiv) in CH2CI 2 (450 mL) at 0°C
over a period of
2 hours. The reaction was allowed to warm to room temperature and stirring
continued
for a further 14h. The reaction mixture was washed with saturated NaHC03
(2*200mL),
water (2*200mL), dried (MgS04) and the solvents removed under reduced. The
solid
obtained was recrystallised (CH2 CI2/MeOH) to give 2 as a white solid. Yield:
99.678
(97%) ; m.p. : 180°C; R,= 0.26 (acetonelether 1:9); IR (CHZ CI2): vm,~=
3353, 2942, 2870,
1693, 1674, 1562, 1467, 1382, 1264 crri ';'H NMR (270 MHz, CDCI3): X5.35 (d,
J=6.5
Hz, 1 H, H6'), 5.25-5.29 (m, 1 H, NH), 4.42-4.57 (1 H, m, H3'), 3.70-3.62 (m,
2H, H1 ), 3.25-
3.35 (m, 2H, H2), 3.12 (s, 1H, OH), 2.28-2.38 (m, 2H, H4'), 1.77-2.03 (m, 5H,
H2', HT,
H8'), 1.59-0.96 (m, 21H, H1', H9', H11', H12', H14'-H1T, H22'-H25'), 1 (3H, s,
H-19'),
0.9(d, J = 6.5 Hz, 3H, H21'), .87 (d, J = 8.5 Hz, 6H, H26'&H2T) and .67 (s,
3H, H18'); MS
(FAB'"): m/z = 496 [M+Na]+, 474 [M+H] ', 369[Chol]'",
255,175,145,105,95,81,43.
2-((Cholesterylouycarbonyl)amino~sthyl methanesulfonate (3): To a solution of
2
(258, 52.3 mmol) and triethylamine (22 mL, 0.16 mol, 3 equiv) in CH ZCIZ (500
mL) at 0°C,
was added dropwise a solution of methanesulfonyl chloride (10.5 mL, 0.13 mol,
2.5
CA 02364730 2001-12-06
13
equiv). The reaction mixture was allowed to warm at room temperature and
stirred for
1 h30. After Tlc analysis has indicated that the reaction had gone to
completion, ice was
added to quench the reaction. The reaction mixture was added to saturated
aqueous
NH4CI (600 mL), and extracted with ether (3*300 mL). The combined organic
layers
were washed with water (2*300mL), brine (250 mL) and dried (Na2S0 4). The
solvent
was remove under reduced pressure to give a white solid, which on purification
by
chromatography (ether) gave 3. Yield: 28.3g (98 %); IR (CHZ CI2): vmeX= 3453,
3342,
1716, 1531, 1377, 1137 8 798 cm''; 'H NMR (270 MHz, CDCI3): ~ 5.34 (d, J=6.5
Hz,
1H, H6'), 5-5.1 (m, 1H, NH), 4.41-4.53 (1H, m, H3'), 4.29-4.25 (t, J=5 Hz, 2H,
H1), 3.47-
3.52 (m, 2H, H2), 3.01 (s, 3H, H3), 2.24-2.36 (m, 2H, H4'), 1.74-2 (m, 5H,
H2', HT, H8'),
0.9-1.6 (m, 21H, H1', H9', H11', H12', H14'-H1T, H22'-H25'), 0.98 (3H, s, H-
19'), 0.84(d,
J = 6.5 Hz, 3H, H21'), .83 (d, J = 6.5 Hz, 6H, H26'&H2T) and .65 (s, 3H,
H18'); MS
(FAB'~: m/z = 1104[2M+H]+, 574 [M+Na]+, 552 [M+H]'", 369[Chol]+,
255,175,145,95,81.
4-aza-N6(cholesteryloxyca~bonylamino) hexsnol (4): To a stirred solution of 3
(28,3 g,
51 mmol) dissolved in a minimum amount of THF, was added amino -propanol (160
mL,
2 mol, 39 equiv). Once Tlc indicated reaction completion (12h), CHCI3 (350 mL)
and
K2C03 (20 g) were added and the solution was vigorously stirred for 30 min.
The
suspension was then filtered through a short pad of Celite~ , washing
thoroughly with
CHCI3. This was washed with a saturated solution of Sodium Hydrogenocarbonate
and
dried (NaZC03). The solvent was removed to give 4 as a white solid. Yield:
26.1 g (96
%); IR (CH2CI2): v",~=3350-3210, 2937, 2850, 1531, 1460, 1380, 1220, 1120,
1040 crri';
'H NMR (270 MHz, CDCI3): ~ 5.33-5.35 (m, 1H, H6'), 4.92-4.96 (m, 1H, NH), 4.42-
4.51
(1H, m, H3'), 3.7-3.83. (m, 2H, H5), 3.23-3.29 (m, 2H, H1), 2.73-2.57 (m, 6H,
H2, H3,
H4), 2.2-2.36 (m, 2H, H4'), 1.7-2 (m, 5H, H2', HT, H8'), 0.85-1.58 (m, 21H,
H1', H9',
H11', H12', H14'-H1T, H22'-H25'), 0.98 (3H, s, H-19'), 0.84 (d, J = 6.5 Hz,
3H, H21'), .8
(d, J = 6.5 Hz, 6H, H26'&H2T) and 0.61 (s, 3H, H18'); MS (FAB~): rn/z = 543
[M+Na]+,
530 [M+H]+, 485 [M-C02] +, 369[Chol]'', 144 [M-Ochol]+,69,55.
4-aza-(Boc)-N°(cholesteryloxycarbonyl amino) hexs~nol (5): To a
solution of 4 (28.1 g,
49 mmol), was added Et3N (8.3 mL, 1.1 equiv) and Bocz O (10.7g, 1 equiv) in
CH2CIZ
(200 mL) and the resulting solution followed by tlc. On completion, the
reaction mixture
was poured into NH4CI (100 mL), and was washed with water and dried (Na2S04 ).
The
solvent was removed in vacuo to give the white solid 5. The solvent was remove
under
reduced pressure to give a white solid, which on purification by
chromatography
CA 02364730 2001-12-06
14
(CHzCI2/MeOH/NH3 92:7:1) gave 3. Yield (27.9 g, 90%); IR (CHZCI 2): vm~= 3352,
3054,
2937, 1675, 1530, 1455, 1380, 1220, 1120; 'H NMR (270 MHz, CDCI3): ~ 5.33-5.35
(m,
1 H, H6'), 4.86 (m, 1 H, NH), 4.42-4.5 (1 H, m, H3'), 3.62-3.7 (m, 2H, H5),
3.27- 3.38 (m,
6H, H1, H2, H3), 2.18-2.33 (m, 2H, H4'), 1.73-2 (m, 5H, H2', HT, H8'), 1.45
(s, 9H, Boc),
1-1.65 (m, 23H, H4, H1', H9', H11', H12', H14' -H1T, H22'-H25'), 0.97 (3H, s,
H-19'), 0.93
(d, J = 6.5 Hz, 3H, H21'), 0.8 (d, J = 6.5 Hz, 6H, H26'&H2T) and 0.65 (s, 3H,
H18'); MS
(FAB+): m/z = 654 [M+Na]+, 543 [M-Boc]', 369[Chol]+, 145, 121, 95, 89,57.
4-aza-(Boc)-Ne(cholesteryloxycarbonylamino) hexyl methane-sulfonate (6): This
experiment was carried out in a similar way as the preparation of 2-
[(Cholesteryloxycarbonyl)amino]ethyl methanesulfonate 3 on 44 mmol scale
giving 6.
Yield (28g, 90%); IR (CHZCIZ): v""x= 3305, 2980, 2900, 2865, 1675, 1530, 1455,
1350,
1150;'H NMR (270 MHz, CDCI9): ~ 5.33-5.35 (m, 1H, H6'), 4.86 (m, 1H, NH), 4.35-
4.55
(m, 1 H, H3'), 4.22 (t, 2H, J = 6.5 Hz, H5), 3.2-3.4 (m, 8H, H 1, H2, H3),
3.01 (s, 3H, H6),
2.15-2.33 (m, 2H, H4'), 1.73-2 (m, 5H, H2', HT, H8'), 1.44 (s, 9H, Boc), 1-
1.67 (m, 23H,
H4, H1', H9', H11', H12', H14'-H1T, H22'-H25'), 0.97 (3H, s, H-19'), 0.94 (d,
J = 6.5 Hz,
3H, H21'), 0.8 (d, J = 6.5 Hz, 6H, H26'&H2T) and 0.65 (s, 3H, H18'); MS
(FAB+): m/z =
722 [M+Na]', 609 [M-Boc]', 369[Chol]+, 145, 121, 95, 69,55.
4-aza-(Boc)-Ns(cholesteryioxycarbonylamino) hexanamine (7): To 6 (25g, 35
mmol),
sodium azide (11.49, 175.7 mmol, 5 equiv), and sodium iodine (5g, 35 mmol, 1
equiv)
under nitrogen was added anhydrous DMF (200 mL), with stirring. Equipped with
a
reflux condenser, heating at 80°C for 2h resulted in completion of
reaction. The reaction
mixture was allowed to cool to room temperature, the DMF removed under reduced
pressure and the residue dissolved in EtOAc. This was washed with water (2*100
mL),
brine (100 mL) and dried (NaZS04) to give after purification by chromatography
(hexane/ether 1:1) 7 as a white solid. Yield (22g, 95 %); 'H NMR (270 MHz,
CDCI3): c~
5.34-5.36 (m, 1 H, H6'), 4.35-4.55 (m, 1 H, H3'), 4.25 (t, 2H, J = 6.5 Hz,
H5), 3.2-3.5 (m,
6H, H 1, H2, H3), 2.25-2.33 (m, 2H, H4'), 1.7- 2.05 (m, 5H, H2', HT, H8'),
1.45 (s, 9H,
Boc), 1-1.72 (m, 23H, H4, H1', H9', H11', H12', H14'-H1T, H22' -H25'), 0.98
(3H, s, H-
19'), 0.94 (d, J = 6.5 Hz, 3H, H21'), 0.83 (d, J = 6.5 Hz, 6H, H26'8~H2T) and
0.64 (s, 3H,
H18'); MS (FAB~): m/z = 568 [M+Na-Boc]'", 556 [M-Boc]+, 369[Chol]'", 145, 121,
95,
69,57.
CA 02364730 2001-12-06
4-aza-(Boc)-N°(cholesteryloxycarbonylamino) hexylamins (8): To a round
bottomed
flask charged with 7 (22.75 g, 34,6 mmol) in THF (230 mL) was added
trimethylphosphine in THF (1 M, 40 mL, 1.15 equiv), and the reaction was
monitored by
tlc. On the completion the reaction was stirred with water (3 mL) and aqueous
ammonia
5 (3mL) for 1 h and the solvent was remove under reduce pressure. After
chromatography
(CHZCIZ/MeOH/NH3 92:7:1 to 75:22:3) 8 was obtained as a white crystal. Yield
(19.1 g,
88 %); IR (CHxCIz): vm~= 3689, 3456, 3155, 2948, 2907, 2869, 2253, 1793 ,
1709, 1512,
1468, 1381, 1168; 'H NMR (270 MHz, CDCI3): ~ 5.32-5.35 (m, 1H, H6'), 4.35-4.51
(m,
1H, H3'), 3.45-3.05 (m, 8H, H1, H2, H3, H5), 2.18-2.4 (m, 2H, H4'), 1.8-2.1
(m, 5H, H2',
10 HT, H8'), 9.46 (s, 9H, Boc), 1.01-1.72 (m, 23H, H4, H1', H9', H11', H12',
H14'-H1T, H22'-
H25'), 0.97 (3H, s, H-19'), 0.85 (d, J = 6.5 Hz, 3H, H21'), 0.82 (d, J = 6.5
Hz, 6H,
H26'&H2T) and 0.64 (s, 3H, H18'); MS (FAB'): m/z = 630 [M+H]+, 530 [M-Boc]'',
369[Chol]+, 145, 121, 95, 69,57.
15 (Boc)aminooxyacetic acid (9): O-(Carboxymethyl)hydroxylamine
hemihydrochloride
(1.16 g, 5.3 mmol) was dissolved in CHzCl2 (40 mL) and the pH was adjusted to
9 by
addition of triethylamine (3 mL). Then di -tert-butyl Bicarbonate (2.36 g,
10.6 mmol, 2.0
equiv) was added and the mixture was stirred at room temperature until tlc
indicated
completion of reaction. The pH was lowered to 3 by addition of diluted HCI.
The
reaction mixture was partitioned between saturated aqueous NH4CI (20 mL) and
CH2CIz
(30 mL). The aqueous phase was extracted with CHZ CI2 (3x100 mL). The combined
organic extracts were washed with H20 (2x100 mL) and dried (Na2 S04). The
solvent
was removed in vacuo to afford 9 as a white solid. Yield (1.86 g, 97%); IR
(CHZCIZ ):
v",ax= 3373, 2983, 2574, 2461, 1724, 1413, 1369, 1235; 'H NMR (270 MHz,
CDC13): ~
4.48 (s, 2H, CH2), 1.48 (s, 9H, Boc); MS (FAB'"): m/z =214 [M+Na]+, 192
[M+H]+, 135,
123, 109, 69.
(Boc)aminooxy compound (10): N-hydroxysuccinimide (0.36 g, 3.13 mmoi, 1
equiv), 9
(0.6 g, 3.13 mmol, 1 equiv), and N,N'-dicyclohexylcarbodiimide (0.68 g, 3.13
mmol, 1
equiv) were dissolved in EtOAc (90 mL), and the heterogeneous mixture was
allowed to
stir at room temperature overnight. The mixture was then filtered through a
pad of
Celite~ to remove the dicyclohexylurea, which was formed as a white
precipitate (rinsed
with 60 mL of EtOAc), and added to a solution of 8 (1.97 g, 3.13 mmol, 1
equiv) in THF
(10 mL). A pH of 8 was maintained far this heterogeneous reaction by addition
of
triethylamine (6 mL). The resulting mixture was allowed to stir at room
temperature
CA 02364730 2001-12-06
16
overnight. On completion the mixture was filtered and the solvent was removed
under
reduced pressure to give after purification by flash-chromatography (CH
ZCI2/MeOH/NH3
92:7:1 ) 10 as a white solid. Yield (2.3 g, 90 %); ' H NMR (270 MHz, CDCI3 ):
~ 5.33-5.35
(m, 1 H, H6'), 4.4-4.52 (m, 1 H, H3'), 4.3 (s, 2H, H90, 3.2-3.42 (m, 8H, H 1,
H2, H4, H6),
2.23-2.35 (m, 2H, H4'), 1.7-2.1 (m, 7H, H2', HT, H8', H5), 1.44-1.46 (m, 18H,
2 Boc), 1-
1.73 (m, 21H, H1', H9', H11', H12', H14'-H1T, H22'- H25'), 0.98 (3H, s, H-
19'), 0.85 (d, J
= 6.5 Hz, 3H, H21'), 0.83 (d, J = 6.5 Hz, 6H, H26'8H2T) and 0.65 (s, 3H,
H18'); MS
(FAB'): m/z =803 [M+H]+, 703 (M-Boc]+, 647, 603 [M-2Boc]', 369, 279, 255, 235,
204,
145, 95, 69.
Hydroxylamine (11 ): To a solution 10 (1.1 g, 1.36 mmol, 1 equiv) in CH2C1 Z
(10 mL) was
added TFA (2 mL, 20.4 mmol, 15 equiv) at 0°C. The solution was allowed
to stir at room
temperature for 5 hours. On completion toluene was added to azeotrope TFA from
the
reaction mixture. The solvents were removed in vacuo to afford after
purification by
chromatography (CHZCI2IMeOH/NH3 92:7:1 to 75:22:3) 11 as a white solid (709
mg,
Yield: 86 %); IR (CHCI3): vm~= 3306, 2948, 2850, 2246, 1698, 1647, 1541, 1467,
1253,
1133;'H NMR (270 MHz, CDCI3): X5.26-5.4 (m, 1H, H6'), 4.4-4.52 (m, 1H, H3'),
4.12 (s,
2H, H9), 3.34-3.41 (m, 2H, H2), 3.15-3.3 (m, 2H, H4), 2.6-2.74 (m, 4H, H1 &
H6), 2.14-
2.39 (m, 2H, H4'), 1.62-2.1 (m, 7H, H2', HT, H8', H5), 1.02-1.6 (m, 21H, H1',
H9', H11',
H 12', H 14'-H 17', H22' -H25'), 0.96 (3H, s, H-19'), 0.86 (d, J = 6.5 Hz, 3H,
H21'), 0.83 (d, J
= 6.5 Hz, 6H, H26'&H2T) and 0.66 (s, 3H, H18'); MS (FAB+): m/z = 603 [M+H]+,
369[Chol]+, 160, 137, 109, 95, 81, 69, 55.
Mannosyl compound (12a): A solution of D-mannose (266 mg, 4.8 mmol) in Acetic
aqueous Buffer (sodium acetatelacetic acid 0.1 M, pH 4, 7mL) and a solution of
11 (290
mg, 0.48 mmol, 10 equiv) in DMF (7 mL) was mixed and stirred for 3 days at
room
temperature. The solvent was removed in vacuo by freeze drying and
chromatography
(CH2CI21MeOH/NH3 75:22:3) afforded the product 21 a white solid (233 mg, Yield
: 65
%). The purity was further confirmed by HPLC. The final pnxluct contained of
the ,~
pyranose (82 %) form and cr-pyranose (18 9~6) form that were not isolated but
characterized in the mixture. MS (FAB+): m/z = 765 [M+H]+, 787 [M+Na]+, 397,
369[Chol]+, 322, 240, 121, 109, 95, 81, 69,57. ~pyranose form.'H NMR (400 MHz,
CD30D/CDC13 [75/25]): ~ 7.64-7.62 (d, 3J,~.2, = 7 Hz, 1H, H1a), 5.35-5.36 (m,
1H, H6'),
4.45-4.5 (s, 2H, H9), 4.35-4.5 (m, 1 H, H3'), 4.19-4.24 (dd, 1 H, H2,, 3J~~z,
= 7.4 Hz, 3J28.a.
= 7.7 Hz), 3.81-3.9 (m, 1 H, H3a), 3.73- 3.8 (m, 2H, H4a, H68,~a), 3.63-3.71
(m, 2H, H5a,
CA 02364730 2001-12-06
17
H~6a), 3.34-3.42 (m, 2H, H2), 3.27-3.30 (m, 2H, H4), 3-3.08 (m, 2H, H1), 2.9-
2.98 (m,
2H, H6), 2.25-2.35 (m, 2H, H4'), 1.78-2.07 (m, 7H, H2', HT, H8', H5), 1.03-
1.65 (m, 21 H,
H1', H9', H11', H12', H14'-H1T, H22'-H25'), 1.01 (3H, s, H-19'), 0.91 (d, J =
6.5 Hz, 3H,
H21'), 0.85 (d, J = 6.5 Hz, 6H, H26'8~H2T) and 0.69 (s, 3H, H18'); '3C NMR
(400MHz,
CDCh/ CD30D [25/75]): 12.33 (C18'), 19.20 (C21'), 19.74 (C19'), 21.91 (C11'),
22.91
(C2T), 23.17 (C26'), 24.67 (C23'), 25.07 (C15'), 27.37 (C5), 28.85 (C25'),
28.96 (C2'),
29.07 (C12'), 32.76 (CT), 32.87 (C8'), 36,38 (C2), 36.78 (C20'), 37.09 (C1)
37.76
(C22'),37.95 (C1'), 38.4 (C4), 39.36 (C4'), 40.41 (C24'), 40.76 (C16'), 46.16
(C6), 51.19
(C9'), 57.19 (C1T), 57.75 (C14'), 64.62 (C6a), 70.19 (C2a), 70.58 (C4a), 72.12
(C3a),
72.37 (C5a), 73.11 (C9), 75.91 (C3'), 123.39 (C6'), 140.72 (C5'), 155.02
(C1a), 158.69
(NHCOOChoI), 173.1 (C8); a-pyranose form : identical data except, 'H NMR (400
MHz,
CD30D/CDCI3 [75/25]): ~ 6.90-6.88 (d, 3J,~.2, = 7 Hz, 1 H, H1 a), 5-5.05 (dd,
1 H, H2a,
3J~8-ze = 7.3 Hz, 3Jza.ae -- 7.6 Hz); '~C NMR (400 MHz, CDCh/ CD30D [25/75]):
65.33
(C2a), 155.79 (C1a).'H NMR (400 , CD30D/CDCI3 [75!25]): (m, 1H, H3') missing,
underneath solvent peak; confirmed by'H NMR (300 MHz, DMSO): s= 4.67-x.82 (m,
1 H,
H3'). '3C NMR (400 MHz, CDCI~I CD30D [25/75]): C1 missing, underneath MeOH
peak
confirmed by'H/'3C correlation at 400 MHz, around 49. Proton resonance
assignments
were confirmed using 'H gradient type DQF-COSY and TOCSY; 'HI'3C correlation
and
DEPT 135 were used to assign unambiguously the carbon resonances. a pyrannose
form gave ' J'3C 1 a-H 1 a = 177 Hz and ~ pyrannose form gave ' J' 3C 1 a-H 1
a = 167 Hz. ' H
phase-sensitive NOESY confirmed conformation.
Glucoayl compound (12b): This was prepared with a solution of D-glucose (150
mg,
0.82 mmol) and 11 (100 mg, 0.16 mmol) in a similar way to the preparation of
12a,
stirred for 1 day and purified by chromatography (CHz CI2IMeOH/NH3 75:22:3) to
afford
the product 12b as a white solid (103 mg, Yield: 82 %). The purity was further
confirmed
by HPLC. The final product contained of the a-pyranose (11 %) anomer and ~-
pyranose
(89 %) anomer that were not isolated but characterized in the mixture. (FAB+):
m/z = 765
[M+H]+, 787 [M+Na]'', 391, 369 [Chol]+, 309, 290, 171, 152, 135, 123, 109, 95,
81, 69 ;
pyranose form. (300 MHz, CDCI~ICD30D [90/10]): ~ 7.53-7.56 (d, J = 5.6 Hz, 1H,
H1a),
5.26-5.36 (m, 1 H, H6'), 4.2-4.45 (m, 3H, H9, H3'), 4.05-4.15 (m, 1 H, H2a),
3.45-3.85 (m,
5H, H6a, H3a, HSa, H4a), 2.9-3.4 (m, H2, H4, MeOH), 2.9- 3.15 (m, 4H, H1, H6),
2.15-
2.3 (m, 2H, H4'), 1.65-2 (m, 5H, H2', HT, H8'), 0.95-1.55 (m, 23H, H5, H1',
H9', H11',
H12', H14'-H1T, H22'-H25'), 0.93 (3H, s, H-19'), 0.84 (d, J = 6.5 Hz, 3H,
H21'), 0.78 (d, J
= 6.5 Hz, 6H, H26'&H2T) and 0.62 (s, 3H, H18'); a-pyranose form : identical
data except,
CA 02364730 2001-12-06
18
'H NMR (300 MHz, CDCI~/CD30D [90/10]): ~ 7.22-7.24 (d, J = 6,61 Hz, 1H, H1a),
4.95-
5.07 (m, 1 H, H2a); ' H NMR (300 MHz, CDR OD): (m, 1 H, H3') missing,
presumably
underneath solvent peak; confirmed by 'H NMR (300 MHz, DMSO): ~ 4.7-4.86 (m,
1H,
H3')
Galactosyl compound (12c): This was prepared with a solution of D -galactose
(50 mg,
0.27 mmol) and 11 (40 mg, 0.066 mmol in a similar way to the preparat ion of
12a, stirred
for 1 day and purified by chromatography (CH 2Ch/MeOH/NH3 75:22:3) to afford
the
product 12c as a white solid (35 mg, Yield: 70 %). The purity was further
confirmed by
HPLC. The final product contained of the c~pyranose (15 %) form and
,l~pyranose (85
%) form that were not isolated but characterized in the mixture. MS (FAB'~:
m/z = 765
[M+H]*, 588, 391, 369 [Chol]*, 322, 290, 165, 152, 135, 121, 109, 95, 81, 69 ;
/fpyranose
form.'H NMR (270 MHz, DMSO): ~ 7.78-7.82 (m, 1H, NHCO of C8), 7.55-7.58 (d, J
=
7.2 Hz, 1 H, H1 a), 6.95-7.1 (m, 1 H, NHCOOChoI), 5.25-5.37 (m, 1 H, H6'), 4.2-
4.43 (m,
3H, H9, H3'), 3.2-3.9 (m, H2a, H6a, H3a, H5a, H4a, OH), 2.9-3.18 (m, 4H, H2,
H4), 2.4-
2.65 (m, 4H, H1, H6), 2.15-2.3 (m, 2H, H4'), 1.67-2 (m, 5H, H2', HT, H8'),
0.92-1.6 (m,
23H, H5, H1', H9', H11', H12', H14'-H1T, H22'-H25'), 0.96 (3H, s, H-19'), 0.89
(d, J = 6.5
Hz, 3H, H21'), 0.84 (d, J = 6.5 Hz, 6H, H26'&H2T) and 0.65 (s, 3H, H18'). a-
pyranose
form : identical data except,'H NMR (270 MHz, DMSO): 6.86-6.88 (d, J = 6 Hz,
1H, H1a)
Glucuronic compound (12d): This was prepared with a solution of D- glucuronic
acid,
sodium salt monohydrate (30 mg, 0.128 mmol, 1.5 equiv) and 11 (50 mg, 0.08
mmol) in
a similar way to the preparation of 12a, stirred for 1 day, purified by
chromatography
(CH2Ch/MeOH/NH3 75:22:3) to afford the sodium salt of 12d as a white solid (41
mg,
Yield: 60 %). The purity was further confirmed by HPLC. The final product
contained of
the c~pyranose (85 %) form and ~3-pyranose (15 %) form that were not isolated
but
characterized in the mixture. MS (FAB *): m/z = 779 [M+H]*, 733, 588, 411,
369[Chol]*,
336, 290, 240, 214, 159, 145, 135, 121, 109, 95, 81, 69,55. ~3-pyranose form.
'H NMR
(300 MHz, CDCI~/CD30D [75/25]) : ~ 7.51-7.53 (d, J = 5.9 Hz, 1H, H1a), 5.25-
5.33 (m,
1 H, H6'), 4.2-4.45 (m, 3H, H9, H3'), 3.8-4.1 (m, 3H, H2a, H3a, H4a), 3.6-3.75
(m, 1 H,
H5a), 3.2-3.55 (m, H2, H4, MeOH), 2.7- 3.15 (m, 4H, H1, H6), 2.18-2.32 (m, 2H,
H4'),
1.62-2 (m, 5H, H2', HT, H8'), 0.9-1.6 (m, 23H, H5, H1', H9', H11', H12', H14'-
H1T, H22'-
H25'), 0.93 (3H, s, H-19'), 0.83 (d, J = 6.5 Hz, 3H, H21'), 0.77 (d, J = 6.5
Hz, 6H,
H26'&H2T) and 0.6 (s, 3H, H18') ); cr-pyranose form: identical data except, 'H
NMR (300
MHz, CD30D): ~ 7.22-7.24 (d, J = 6.3 Hz, 1 H, H1 a), 5-5.1 (m, 1 H, H2a).
CA 02364730 2001-12-06
19
,~D-lactosyl compound (12e): A solution of /3~D-Lactose, ~ntaining 25-30 % of
a (1.13
g, 3.3 mmol) and 11 (200 mg, 0.33 mmol) in 14 mL of DMF/Acetic aqueous Buffer
was
stirred for 4 days at room temperature. The solvent was removed in vacuo by
freeze-
drying and chromatography (CHZCh/MeOH/NH 3 75:22:3) afforded the product 12e
as a
white solid (145 mg, Yield: 47 ~). The purify was further confirmed by HPLC.
The final
product contained of the a-pyranose (15 %) form and /fpyranose (85 %) form
(containing itself around 25 % of a lactose) that were not isolated but
characterized in
the mixture. MS (FAB+): m/z = 927 [M+H]+, 588, 482, 369[Chol]', 290, 243, 216,
178,
152, 135, 121, 109, 95, 81, 69,55 ; ,B-pyranose fiorm. 'H NMR (400 MHz, CDChI
CD~OD
[20180]): ~,= 7.69-7.71 (d,'J~~.2a = 5.8 Hz, 1H, H1a of,Blactose), 7.66-7.68
(d, 3J~~2,= 6.2
Hz, 1 H, H1 a of a lactose), 5.35-5.37 (m, 1 H, H6'), 4.37-4.6 (m, 4H, H9,
H3', H2a), 4.2-
4.37 (m, 1 H, H1 b), 3.65-4.05 (m, 7 H, H3a, H4a, H5a, H4b, HSb, H6b), 3.25-
3.6 (m, SH,
H2, H4, H6a, H2b, H3b,MeOH), 3-3.2 (m, 4H, H1, H6), 2.25-2,42 (m, 2H, H4'),
1.8-2.15
(m, 5H, H2', HT, H8'), 1-1.65 (m, 23H, H5, H1', H9', H11', H12', H14'-H1T,
H22'-H25'),
1.01 (3H, s, H-19'), 0.91 (d, J = 6.5 Hz, 3H, H21'), 0.85 (d, J = 6.5 Hz, 6H,
H26'&H2T)
and 0.69 (s, 3H, H18'); '3C NMR (400 MHz, CDCI~/ CD30D [20180]): '3C NMR (400
MHz,
CDCI~/ CD~OD [20/80]): 12.32 (C18'),19.2 (C21'), 19.76 (C19'), 21.94 (C11'),
22.91
(C2T), 23.17 (C26'), 24.7 (C23'), 25.1 (C15'), 27.22 (C5), 28.89 (C25'), 29
(C2'), 29.1
(C12'), 32.8 (CT), 32.92 (C8'), 36.29 (C22'),36.81 (C10'), 37.12 (C1'), 37.99
(C6), 38.11
(C1), 39.48 (C2), 40.45 (C24'), 40.80 (C16'), 46.13 (C4'), 51.23 (C9'), 57.22
(C1T),
57.80 (C14'), 62.41 (C6a), 63.4 (C6a), 70.02 (C 5b), 70.63 (C2a), 72.8 (C3a),
73 (C3'),
73.18 (C9), 74,75 (C2b), 76.8 (C3a), 81 (C4b), 92.39 (C1b), 105.2 (C3'),
123.42 (C6'),
140.72 (C5'), 154.8 (C1a), 156.2 (NHCOOChoI), 173.17 (C8). c~pyranose form :
identical
data except, 'H NMR (400 MHz, CD30D/CDCI3 [80/20]): Ei.,= 7.04-7.05 (d, 3J~~2o
= 5.6
Hz, 1 H, H1 a), 5.05-5.07 (m, 1 H, H2a), 4.09-4.11 (m, 1 H, H3a); 'H NMR (270
MHz,
CD30D): (m, 1H, H3') missing, presumably underneath solvent peak; confirmed by
'H
NMR (300 MHz, DMSO): ~ 4.7-4.85 (m, 1 H, H3'). Proton resonance assignments
were
confirmed using 'H gradient type DQF-COSY and TOCSY; 'H/'3C correlation and
DEPT
135 were used to assign unambiguously the carbon resonances. 'H phase-
sensitive
NOESY confirmed conformation.
Maltosyl compound (12f): This was prepared with a solution of D Maltose
monohydrate
(30 mg, 1.8 mmol, 5 equiv) and 11 (100 mg, 0.16 mmol) ) in a similar way to
the
preparation of 12e, stirred for 1 day and purified by chromatography (CHz
CIZ/MeOH/NH3
75:22:3) to afford 12f as a white solid (100 mg, Yield : 65 %). The purity was
further
CA 02364730 2001-12-06
zo
confirmed by HPLC. The final product contained of the a pyranose (87 %) form
and ~3-
pyranose (13 °~) form that were not isolated but characterized in the
mixture. MS
(FAB+): m/z = 927 [M+H]'', 765, 588, 559, 484, 369[Chol]'', 322, 290, 213,
167, 161, 143,
135, 121, 109, 95, 81, 69,55. ~pyranose form. 'H NMR (300 MHz, CDCI~/CD30D
[80/20]): ~ 7.55-7.57 (d, 3J,a2, = 5.3 Hz, 1 H, H 1 a), 5.3 (s, 1 H, H6'),
4.85-5.02 (m, 1 H,
H3'), 4.094.22 (m, 1 H, H1 b), 3.57-4 (m, 7 H, H3a, H4a, HSa, H4b, HSb, H6b),
3.2-3.6
(m, 8H, H2, H4, H6a, H2b, H3b,MeOH), 2.8-3.1 (m, 4H, H1, H6), 2.1-2.36 (m, 2H,
H4'),
1.6-2.05 (m, 5H, H2', HT, H8'), 1-1.6 (m, 23H, H5, H1', H9', Hi1', H12', H14'-
H1T, H22'-
H25'), 0.93 (3H, s, H-19'), 0.83 (d, J = 6.5 Hz, 3H, H21'), 0.78 (d, J = 6.5
Hz, 6H,
H26'&H2T) and 0.6 (s, 3H, H18'); a-pyranose form : identical data except, 'H
NMR (300
MHz, CD30D/CDCI3 [80/20]): ~ 6.92-6.94 (d, J = 4.62 Hz, 1 H, H1 a), 5.02-5.15
(m, 1 H,
H2a), 4.04.08 (m, 1 H, H3a)
Maltotriosyl compound (12g): This was prepared with a solution of maltotriose
(246.4
mg, 0.46 mmol, 7 equiv) and 11 (40 mg, 0.066 mmol) in a similar way to the
preparation
of 12e, stirred for 5 days and purified by chromatography (CH2 Ch/MeOH/NH3
75:22:3) to
afford 12f as a white solid (61 mg, Yield: 85 %). The purity was further
confirmed by
HPLC. The final product contained of the a-pyranose (15 %) form and ~t-
pyranose (85
%) form that were not isolated but characterized in the mixture. MS (FAB +):
m/z = 1111
[M+Na]+, 1089 [M+H]+, 588, 423, 391, 369 [Chol]'', 240, 171, 159, 145, 121,
105, 95, 81,
69; ,l3-pyranose form.: 'H NMR (300 MHz, CDCI~/MeOH[20/80]): ~ 7.56-7.58 (d, J
= 6
Hz, 1 H, H 1 a), 5.2-5.27 (m, 1 H, H8'), 4.9-4.95 (m, 1 H, H3'), 4.2-4.45 (m,
4H, H9, H3',
H2a), 4.05-4.2 (m, 2H, H1 b, H1 c), 2.95-4 (m, 21 H, H2, H4, H6a, H3a, HSa,
H4a, H2b- 6b,
H2c-6c, MeOH), 2.85-2.95 (m, 4H, H1, H6), 2.2-2.3 (m, 2H, H4'), 1.8-2.1 (m,
5H, H2',
HT, H8'), 0.98-1.6 (m, 23H, H5, H1', H9', H11', H12', H14'-H1T, H22'-H25'),
0.94 (3H, s,
H-19'), 0.84 (d, J = 6.5 Hz, 3H, H21'), 0.78 (d, J = 6.5 Hz, 6H, H26':%H2T)
and 0.61 (s,
3H, H18'); cr-pyranose form: identical data except, 'H NMR (300 MHz,
CDChIMeOH[20/80]): 8=6.85 (d, J = 5.6 Hz, 1 H, H 1 a).
Maltot~ett'aosyl compound (12h): This was prepared with a solution of D
Maltotetraose
(200 mg, 0.3030 mmol) and 11 (80 mg, 0.133 mmol, stirred for 5 days and
purified by
chromatography (CHZCI2IMeOHINH3 75:22:3) to afford 12h as a white solid (67.5
mg,
Yield : 41 %). The purity was further confirmed by HPLC. The final product
contained of
the a pyranose (15 %) form and /.~pyranose (85 %) form that were not isolated
but
characterized in the mixture. MS (FAB+); m/z = 1273 [M+Na]+, 1251 [M+H]+, 588,
369
CA 02364730 2001-12-06
21
[Chol]+, 159, 145, 121, 109, 95, 81, 69; HRMS (FAB +) CS9H,aZN4O24Na: [M+Na]+
calcd
1273.6782, found 1273.6821. /fpyranose form.: 'H NMR (300 MHz,
CDChIMeOH[20/80]): S= 7.56-7.58 (d, 1 H, H1 a), 5.15-5.25 (m, 1 H, H6'), 4.95-
5.1 (m, 1 H,
H3'), 4.38-4.5 (m, 4H, H9, H3', H2a), 4.04-4.22 (m, 3H, H1b, H1c, H1d), 3.1-
3.95 (m,
27H, H2, H4, H6a, H3a, HSa, H4a, H2b-6b, H2c -6c, H2d-6d, MeOH), 2.85-3.1 (m,
4H,
H1, H6), 2.2-2.33 (m, 2H, H4'), 1.75-2.1 (m, 5H, H2', HT, H8'), 1-1.6 (m, 23H,
H5, H1',
H9', H11', H12', H14'-H1T, H22'-H25'), 0.92 (3H, s, H-19'), 0.82 (d, J=6.5 Hz,
3H, H21'),
0.78 (d, J = 6.5 Hz, 6H, H26'8~H27') and 0.68 (s, 3H, H18'); c~pyranose form:
identical
data except,'H NMR (300 MHz, CDCI~IMeOH[20/80]): ~ 7 (d, 1H, H1a).
Maltoheptaosyl compound (12i): This was prepared with a solution of D
Maltoheptaose
(100 mg, 0.08673 mmol) and 11 (30 mg, 0.0497 mmol) stirred for 7 days and
purified by
chromatography (CH2CI2/MeOH/NH3 75:22:3) to afford 12i as a white solid (46mg,
Yield
53 %). The purity was further confirmed by HPLC. The final product contain ed
of the c~-
pyranose (15 %) form and ~-pyranose (85 %) form that were not isolated but
characterized in the mixture. MS (FAB+): m/z = 1759 [M+Na]+, 1737 [M+H]', 369
[Chol]+,
145, 121, 109, 95, 81. /3-pyranose form.: 'H NMR (300 MHz, CDCI~IMeOH[20/80]):
c~
7.53-7.58 (d, 1 H, H 1 a), 5.35-5.37 (m, 1 H, H6'), 4.97-5.12 (m, 1 H, H3'),
4.45-4.6 (m, 4H,
H9, H3', H2a), 4~.5 (m, 6H, H1b, H1o-g), 3.1 -3.9 (m, 45H, H2, H4, H6a, H3a,
HSa, H4a,
H2b-6b, H2c-6c, H2d-6d, H2e-6e , H2f-6f , H2g-6g, MeOH), 2.7-3 (m, 4H, H1,
H6), 2.15-
2.35 (m, 2H, H4'), 1.7-2.1 (m, 5H, H2', HT, H8'), 1-1.6 (m, 23H, H5, H1', H9',
H11', H12',
H14'-H1T, H22'-H25'), 0.94 (3H, s, H-19') 0.84 (d, J = 6.5 Hz, 3H, H21'), 0.77
(d, J = 6.5
Hz, 6H, H26'&H2T) and 0.63 (s, 3H, H18'); a-pyranose fom~: identical data
except, 'H
NMR (300 MHz, CDCh/MeOH[20/80]): 8=6.9 (d, 1H, H1a).
Biological and biophysical evaluation:
General: Dioleoylphosphatidyl-ethanolamine (DOPE) was purchased from Avanti
Lipid
(Alabaster, AL, USA). Plasmid pCMV[3 was produced by Bayou Biolabs (Harahan,
LA,
USA). DC-Chol was synthesised in our Laboratory ~2'~. Mu-peptide was
synthesised by
M. Keller by standard Fmoc based Memifield solid phase peptide chemistry on
Wang
resine ~~~. All other chemicals were reagent grade.
Preparation of Liposomes: DC-Chol (7.5 mg, 15 pmol) and DOPE (7.5 mg, 10
p,mol)
were combined in dichloromethane. The solution was transferred to a round-
bottomed
CA 02364730 2001-12-06
22
flask (typically 50 ml) and organic solvent removed under reduced pressure
(rotary
evaporator) giving a thin-lipid film that was dried for 2-3 h in vacuo.
Following this, 4 mM
HEPES buffer, pH 7.2 (3 ml) was added to the round-bottomed flask so as to
hydrate the
thin-lipid film. After brief sonication (2-3 min.) under argon, the resulting
cationic
liposome suspension (lipid concentration of 5 mg/ml) was extruded by means of
an
extruder device (Northern lipid). Initially, three times through two stacked
polycarbonate
filters (0.2 Vim) and then ten times through two stacked polycarbonate filters
(0.1 p,m) to
form small unilamellar cationic liposomes (average diameter 105 nm according
to PCS
analysis). Lipid concentrations (approx. 4-4.8 mg/ml) were determined by
Stewart assay
~~'~.
Preparation of Liposome:Mu:DNA (LMD) and Liposome:DNA (LD) complexes:
Initially, mu:DNA (MD) particles were prepared by mixing as follows. Plasmid
DNA stock
solutions (typically 1.2 mg/ml) were added to a vortex -mixed, dilute solution
of mu
peptide (1 mg/ml) in 4mM HEPES buffer, pH 7.2. The final mu:DNA ratio was
0.6:1 wlw,
unless otherwise stated, and final plasmid DNA concentration was 0.27 mglml.
MD
containing solutions were then added slowly under vortex conditions to
suspensions of
extruded cationic liposomes (typically app rox. 4.5mg/ml), prepared as
described above,
resulting in the formation of small LMD particles with narrow size
distribution (120 t 30
nm) as measured by PCS. Final Iipid:mu:DNA ratio 12:0.6:1 wlw/w. A solution of
sucrose (100 %, w/v) in 4mM HEPES buffer, pH 7.2, was then added to obtain LMD
particle suspensions in 4mM HEPES buffer, pH 7.2 containing 10% wlv sucrose at
the
desired DNA concentration (final DNA concentration typically 0.14 mg/ml) and
the whole
stored at -80°C. Liposome:DNA (LD) complexes (lipoplexes) were prepared
for
experiments with a Iipid:DNA ratio of 12:1 (wlw) following the same protocol
without the
addition of Mu peptide.
Particle size measurements: The sizes of the lipoplexes were evaluated after
30 min
exposure at 37° C to biological media by Photon Correlation
Spectroscopy (N4 plus,
Coulter). The chosen DNA particular concentration was compatible with in vitro
condition
(1 pg/ml of DNA). The parameters used were: 20° C, 0.089 cP, reflexive
index of 1.33,
angle of 90° C, 632.8 nm. Unimodal analysis was used to evaluate the
mean particle
size in Optimem. Size distribution program using the CONTIN algorithm was
utilised to
separate the sub-population of small serum particle of less than 50nm and to
extracted
the calculated size of lipoplexes in Optimem + 10% FCS.
CA 02364730 2001-12-06
23
Transfection of HeLa cells: Cells were seeded in a 24 wells culture plate in
DMEM
supplemented w~h 10% FCS and grown to approximately 70% confluence for 24h at
37°C in the presence of 5% C02. The cells were washed in PBS before the
transfection
media was administered to each well (0.5 ml of solution of 0, 50 or 100 %
Foetal Calf
Serum in Dube~o OptiMem). 5 p,1 at 100 pg/ml DNA (nls (igal) of LMD were
transfected
onto Hella Cells for 30 min. Cells were then rinsed 3 tim es with PBS and
incubated for a
further 48h in DMEM supplemented with 10% FCS prior to processing for ~3-Gal
expression by using standard chemiluminescent reporter gene assay kit (Roche
Diagnostics, GmbH Cat No. 1 758 241).
Results and Discussion:
Synthesis of Neoglycotipids: Each member of the targeted family of
neoglycolipids
consisted of a cholesterol bearing lipid and an oligosaccharide molecule bound
together
via a linker. The whole synthetic approach was divided in two parts; firstly
the synthesis
of a lipid containing the linker and secondly the chemioselective coupling of
this lipid with
chosen sugar molecules. The key to this strategy is the formation of a
hydroxylamine
(Figure 1 ).
This synthesis of the Boc-protected lipid (8) was originally designed based on
a
convergent methodology using readily available aminoalcohols as starting
materials with
a complementary blocking group strategy for the amine group. This previously
published
methodology allowed the preparation of this polyamide- based lipid for gene
transfer with
little modification ~2'~.
As mentioned, the glycosylation of hydroxylamino derivatives offers an elegant
solution
to our synthetic requirements. The commercially available O-
(Carboxymethyl)hydroxyl-
amine hydrochloride was first Boc-protected and then reacted with N-
hydroxysuccinimide
and N,N'-dicyclohexylcarbodiimide (DCC) resulting in the corresponding
activated ester.
This compound was treated immediately in situ with lipid (8) in THF at pH 8,
affording a
protected hydroxylamine. After a very straightforward deprotection with
aqueous
trifluoroacetic acid, the synthesis of the hydroxylamino lipid (11) was
completed.
CA 02364730 2001-12-06
24
At this stage, we investigated the potential of our chemoselective coupling by
reacting
the lipid (11) with a number of commercially available non- protected
oligosaccharides.
This reaction was conducted under mild conditions using a solvent system of
DMF and
aqueous acetic acid pH 4 Buffer (1:1) which facilitates solubility of both
sugar and lipi d.
As shown in Figure 2 the reactants are in dynamic equilibrium with the open
chair
protonated intermediate. In order to force the equilibrium to product
formation, an
excess of sugar was added. Due to the amphiphilic nature of the neoglycolipid
product,
isolation during workup was found to be difficult as a result of micelle and
foam
formation. Solubility problems also hampered the isolation, purification and
analytical
process. Reaction times and yields varied depending on the carbohydrate used
(Tab !e
1).
Table 1 Yields, reaction times and diastereoselectivity of glycosylation of
product 11 .
Product Sugar Times (days)Yield (%) li/a
12a Mannose 3 65 82J18
12b Glucose 1 80 89/11
12c Galactose 1 70 85/15
12d Glucuronic 1 60 85115
acid
12e Lactose 4 50 85/15
12f Maltose 1 65 87!13
12g Maltotriose 5 85 85/15
12h Maltotetraose5 40 85/15
12i Maltoheptaose7 55 85115
Neoglycoiid Conformation: Carbohydrate conformations can be ascertained by NMR
in
solution 1281. The most useful data for conformation at the anomeric centre
(C1a) is
probably'J'3C1a-H1a ~~'~ ~l. The absolute value of this coupling constant
depends upon
the orientation of the carbon-hydrogen bond relative to the lone pairs of the
ring oxygen,
the electronegativity of the substituent at C1 and the nature of
electronegative
substituents attached to the rest of the molecule. The difference of 'J'3C1-H1
between a
and ~ anomer of pyranoses can be used to determine the anomeric configuration.
It is
firmly established that 'J(C1-H1eq) > 'J(C1-H1ax) with an approximate
difference of 10
Hz. 'J(C1-H1eq) is usually around 170 Hz and 'J(C1-H1ax) approximately 160 Hz.
Higher values are observed when O -1 is exchanged with more electronegative
element
as chlorine or fluorine but a 10 Hz difference is usually observed t3gl.
Carbon-hydrogen
CA 02364730 2001-12-06
coupling constants of furanosides have been investigated and 'J(C1-H1eq) >
'J(C1-
H1 ax) but the difference is much smaller (1-3 Hz).
The characterization will be discussed based on the mannose example but the
same
5 analysis procedure was used for the other saccharides when NMR analysis
conditions
were favourable. Four distinct ring structures can be envisaged (Figure 3).
The
pyranose forms can be reasonably expected to be favoured over the furanose
rings for
steric reasons. So out of the two observed compounds in NMR, the main one is
probably a pyranose. The secondary observed compound could not be attributed
to
10 mutarotation equilibrium because phase sensitive NOESY did not show a cross
peak
between the two C1a signals (proving it is a distinctive molecule). Therefore,
this
compound was not attributed to a furanose form because no shift of '3C5a was
observed
and '3C1a was not deshiekfed as has been demonstrated for related substituted
furanose equivalents ~~~.
We measured 'J'3C1a-H1a = 167 Hz for the main compound and 'J'3Cla-H1a = 177
Hz
for the secondary one. The absolute value of those 'J'gC1-H1 is 10 Hz higher
than
expected for classical 4C~ conformation but this is explained by the extreme
electronegativity of the O-substituted hydroxylamine group that could slightly
deform the
chair structure. For pyranose rings it has been established that ['J(C1-H1eq) -
'J(C1-
H1ax)] ~ 10 Hz, therefore it can be easily concluded that the main compound is
the (i
anomer (H1ax) and the secondary compound is the a anomer (H1eq).
'H phase sensitive NOESY confirmed this conclusion. Nuclear Overhauser effect
was
observed between H1a and H2a & H3a for t he main compound. Considering the
above
detailed structure, this compound could not be the a pyranosyl anomer because
the
equatorial H1 cannot interact in space with H3, whereas the ~3 anomer is
perfectly able to
generate such interactions. No nuclear overhauser effect was observed for H1 a
of the
secondary compound but this could be due to a lack of sensitivity. Hence, in
accordance
with data from 'J'3C1-H1 and NOESY analyses, we concluded that two mannose
pyranose a1(3 forms (20/80) were produced.
The very similar anomeric ((3/a) isomers ratio obtained for the neoglycolipids
is not
surprising (Table 1 ), all the sugars having an equatorial hydroxyl in C2 but
mannose.
The ratio obtained for this last compound is surprising because the ~i anomer
is usually
CA 02364730 2001-12-06
26
reported as sterically less favourable than the a one. A possible explanation
is that this
reaction could be driven by some secondary interactions (Hydrogen bonding)
between
the sugar and the hydroxylamine linker, stabilizing the ~3 anomer (this is
consistent with
the observation that the NMR signal of the ~i anomer is always much more
deshielded
than the a one). This anomeric mixture of synthesized gtycolipids are not
expected to
affect greatly the researched biological properties of the liposomal
constructs, therefore
we did not attempt the tedious separation of those diasteroisomers by
preparative high
pressure liquid chromatography.
Biologicai application:The glyco-mod~cation of LMD was based on the natural
ability
of misceilar suspension to incorporate into lipid membranes ~"~3e~. Firstly
LMD were
formulated following standard protocol and secondly a suspension of
synthesized
neoglycolipids miscelles in Hepes Buffer 4mM pH 7 was added to the LMD and
incubated for 30min at room temperature before usual -80°C storage.
Different percents
of all the neoglycolipids produced were tested for stabilization effect but
only the longer
chain (maltotetraose 12h and maltoheptaose 12i) exhibited significant
properties at less
than 10 % (data not shown).
The stabilisation effect of neoglycolipid modified LMD was demonstrated by
incorporation
of 7.5 molar % of compound 12h or 12i. Lipid layers of liposomes based
formulation are
known to aggregate after salt or serum exposure ~".3s,4o~. This phenomenon can
be
followed by measuring the average particle size increase after a fixed time;
any
stabilization of the LMD particle should be reflected in a reduction of this
parameter. It
was chosen to measure the size of the lipoplexes by Photon Correlation
Spectroscopy
(PCS) after 30 min exposure at 37°C to OptiMem or OptiMem + 10% FCS to
mimic
standard in vitro conditions. It was not possible to analyse the effect with
PCS at higher
serum percentages, the conditions being too extreme to allow for the taking of
meaningful measurements. Figure 4 describes the percentage of size increase of
those
lipoplexes.
The results indicate a clear stabilisation of the particle between LMD and
standard
liposome formulation. Neoglycolipids introduction at 7.5% proved significantly
beneficial
in OptiMem and 10% serum. 12i incorporation proved to be the most efficient.
This
result indicates the need of long carbohydrate chains to create efficient
molecular
brushes on top of those cat'ronic lipid layers ~°'~
s''°"'°, i°°, *"s~.
CA 02364730 2001-12-06
z~
Even if some degree of stabilization is demonstrated, usually it results in a
reduction of
the affinity of the positively charged LMD for the negatively charged cell
membrane,
inducing a drop in the transfection a bility of the construct. However in this
case, the in-
s vitro transfection results indicated an enhancement of the transfection
efficiency due to
neoglycolipid modification in both 0°r6 and 50% Serum condition (Figure
5). This result
was attributed to a short range protective effect due to these neoglycolipids
hindering
short range van der waals based interactions between lipid bilayers of similar
polarities
but not affecting the longer range charge interactions between oppositely
charged
membranes. The aggregation induced by serum being based primarily on
interaction of
LMD with negatively charged proteins ~'2~, the beneficial effect of our
neoglycolipids was
also lowered with an increasing percentage of serum (no significant benefit in
100%
serum).
All publications mentioned in the above specification are herein incorporated
by
reference. Various modifications and variations of the described methods and
system of
the invention will be apparent to those skilled in the art without departing
from the scope
and spirit of the invention. Akhough the invention has been described in
connection with
specific preferred embodiments, it should be understood that the invention as
claimed
should not be unduly limited to such specific embodiments. Indeed, various
modifications of the described modes for carrying out the invention which are
obvious to
those skilled in biology, chemistry or related fields are intended to be
within the scope of
the following claims