Note: Descriptions are shown in the official language in which they were submitted.
CA 02367040 2001-09-14
WO 00/56336 PCT/EP00/01940
1
PHARMACEUTICAL COMPOSITIONS COMPRISING A PYR1MIDINE DERIVATIVE AND
CYCLODEXTRIN
The present invention relates to pharmaceutical compositions comprising a
1,2,4-triazolo[1,5-a]pyrimidine derivative and a cyclodextrin, and to their
use in the
treatment and/or prophylaxis of migraine, seizures, neurological disorders
such as
epilepsy and/or conditions in which there is neurological damage such as
stroke,
brain trauma, head injuries and haemorrhage.
Cerebral ischaemia is a major cause of morbidity and mortality in the
Western world, but drug therapy for this condition remains inadequate and
prevention is still the most effective treatment (De Deyn, P.' P., De Reuck,
J.,
Deberdt, W., Vlietinck, R., Orgogozo, J. M. (1997) Treatment of acute ischemic
stroke with piracetam. Stroke, 28, 2347-2352.). Drugs which provide
neuroprotection
in stroke victims suffering progressing, rather than threatened ischaemic
damage
would have profound implications for the treatment of this disease.
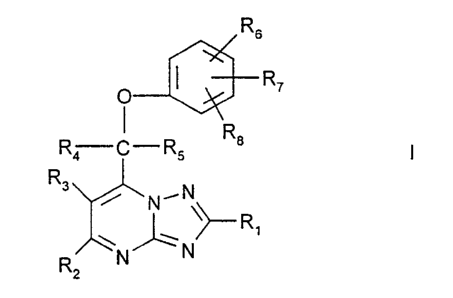
Compounds of formula A
Rs
R~
Rs
R4 C-RS A
R3
N~N
~~-- R
~N
R N
2
in which:R~ represents H or one of the following groups (optionally
substituted with
one or more of halo, cyano, hydroxy or amino): C»alkyl, C»alkoxy or
C~_salkanoyl;
R2 and R3 independently represent H or one of the following groups (optionally
substituted with one or more of halo, cyano, hydroxy or amino): C~~alkyl,
C~_salkoxy,
C»alkanoyl, C~_salkylthio, C~_salkylsulphinyl, or C~~alkylsulphonyl ; R4 and
R5
independently represent H, C~_salkyl or R4 and R5 combined together with the
carbon
atom to which they are attached represent C3~cycloalkylidene (each alkyl or
cycloalkylidene being optionally substituted with one or more of halo, cyano,
hydroxy,
CA 02367040 2001-09-14
WO 00/56336 PCT/EP00/01940
2
amino or C~_6alkyl); and R6, R~ and R8 independently represent H, halo,
hydroxy,
mercapto, nitro, cyano or one of the following groups (optionally substituted
with one
or more of halo, cyano, hydroxy or amino; and any nitrogen atom being
optionally
substituted with one or more C» alkyl): C~_6alkyl, C»alkanoyl, C~_salkoxy,
C2_salkoxycarbonyl, carboxy, C~_fialkanoyioxy, C~_salkylthio,
C~_salkylsulphinyl,
C~_salkylsulphonyl, C~_salkyl-sulphonylamino, sulphamoyl, carbamoyl, C2 salkyl-
carbamoyl or C~_salkanoylamino; processes for their preparation, and their use
in the
treatment and/or prophylaxis of seizures, neurological disorders such as
epilepsy
and/or conditions in which there is neurological damage such as stroke, brain
trauma, head injuries and haemorrhage are described in W095/10521 (Knoll AG).
A
process for preparing these compounds is disclosed in W098/07724 (Knoll AG).
However, the compounds of formula A suffer from the disadvantages that
they. are insoluble in water and have a short plasma half-life in humans after
solid
oral dosing. These findings would suggest that these compounds would be of
limited
use in the treatment of stroke. Surprisingly the present invention solves
these
problems and provides a potential method for the effective treatment of
stroke.
The present invention provides a pharmaceutical composition comprising
a) a compound of formula I
Rs
R~
R
R4 C-Rs s I
R3
N~N
~~--- R
~N
R N
z
including pharmaceutically acceptable salts, solvates, racemates, enantiomers,
diastereoisomers and mixtures thereof in which:
R~ represents H or one of the following groups (optionally substituted with
one or
more of halo, cyano, hydroxy or amino): C~_salkyl, C~~alkoxy or C~_salkanoyl;
CA 02367040 2001-09-14
WO 00/56336 PCT/EP00/01940
3
R2 and R3 independently represent H or one of the following groups (optionally
substituted with one or more of halo, cyano, hydroxy or amino): C~_salkyl,
C~_salkoxy,
C~_salkanoyl, C~_salkylthio, C~_6alkylsulphinyl, C~_salkylsulphonyl or
hydroxy;
R4 and R5 independently represent H, C~_salkyl or R4 and R5 combined together
with
the carbon atom to which they are attached represent C3_6cycloalkylidene (each
alkyl
or cycloalkylidene being optionally substituted with one or more of halo,
cyano,
hydroxy, amino or C~_salkyl); and
R6, R7 and R$ independently represent H, halo, hydroxy, mercapto, nitro, cyano
or
one of the following groups (optionally substituted with one or more of halo,
cyano,
hydroxy or amino; and any nitrogen atom being optionally substituted with one
or
more C1_6 alkyl): C~_salkyl, C~_salkanoyl, C~_fialkoxy, CZ_salkoxycarbonyl,
carboxy,
C~_salkanoyloxy, C~_fialkylthio, C~_salkylsulphinyl, C~_salkylsulphonyl,
C~_salkyl-
sulphonylamino, sulphamoyl, carbamoyl, C2_6alkylcarbamoyl or
C~_salkanoylamino;
and
b) a cyclodextrin.
It will be understood that any group mentioned herein which contains a chain
of three or more carbon atoms signifies a group in which the chain may be
straight or
branched. For example, an alkyl group may comprise propyl which includes n-
propyl
and isopropyl and butyl which includes n-butyl, sec-butyl, isobutyl and tert-
butyl. The
total number of carbon atoms is specified herein for certain substituents, for
example
C,_s signifies an alkyl group having from 1 to 6 carbon atoms. The term '
halo' as
used herein signifies fluoro, chloro, bromo and iodo. The term 'optionally
substituted'
as used herein, unless immediately followed by a list of substituent groups,
means
optionally substituted with one or more group or groups selected from halo,
cyano,
hydroxy and amino. When the phenyl ring substituents R6, R~ and Re are other
than
H, the substituent may replace any H attached to a carbon atom in the ring and
may
be located at any such position of the ring, ie up to three of positions 2, 3,
4 andlor 5.
Suitably the composition is a mixture of the compound of formula I and a
cyclodextrin. Preferably the composition comprises a complex of the compound
of
CA 02367040 2001-09-14
WO 00/56336 PCT/EP00/01940
4
formula I and the cyclodextrin. Suitably the ratio of the compound of formula
I to the
cyclodextrin in the complex is in the range from 0.1-1 to 1-0.1.
Suitably the complex may be prepared by forming a mixture of the compound
of formula I or a pharmaceutically acceptable salt thereof and the
cyclodextrin and
water at a temperature in the range of 10-100°C to form a suspension or
a solution
and to then remove the water by a technique such as spray granulation, spray
drying, drum drying or freeze drying. Spray drying is preferred as this may
produce
a free-flowing granular solid. Fluid bed spray drying is particularly
preferred.
A mixture of the compound of formula I with a cyclodextrin or the complex of
the compound of formula I with a cyclodextrin may be formulated into any of
the well-
known solid pharmaceutical formulations, for example tablets, capsules,
granules,
powders, etc. The compositions may be formulated with an effervescent couple,
a
buffer system, sweetners, flavourings, colours, etc. and diluents are carriers
such as
water soluble saccharides, such as dextrose, lactose or sucrose.
In therapeutic use, the compositions of the present invention may be
formulated into pharmaceutical compositions for oral, rectal or parenteral
administration. Thus the therapeutic compositions of the present invention may
take
the form of any of the known pharmaceutical compositions for such methods of
administration. The compositions may be formulated in a manner known to those
skilled in the art, to give a controlled release, for example rapid release or
sustained
release, of the active compound. Pharmaceutically acceptable carriers suitable
for
use in such compositions are well known in the art of pharmacy. The
compositions
may contain from about 0.1 % to about 99% by weight of active compound and are
generally prepared in unit dosage form. Preferably the unit dosage of active
ingredient is from about 1 mg to about 1000 mg, more preferably from about 1
mg to
about 500 mg. The excipients used in the preparation of these compositions are
the
excipients known in the pharmacist's art.
Preferably the compositions of the invention are administered orally in the
known pharmaceutical forms for such administration. Dosage forms suitable for
oral
administration may comprise tablets, pills, capsules, caplets,
multiparticulates
CA 02367040 2001-09-14
WO 00/56336 PCT/EP00/01940
including: granules, beads, pellets and micro-encapsulated particles; powders,
elixirs, syrups, suspensions and solutions.
Solid oral dosage forms, for example tablets, may be prepared by mixing the
5 pharmaceutical composition of the present invention with one or more of the
following
ingredients or mixtures thereof:
inert diluents, for example calcium carbonate, calcium sulphate, compressible
sugar,
confectioner's sugar, dextrates, dextrin, dextrose, dibasic calcium phosphate
dihydrate, glyceryl palmitostearate, hydrogenated vegetable oil, kaolin,
lactose,
magnesium carbonate, magnesium oxide, maltodextrin, mannitol, microcrystalline
cellulose, polymethacrylates, potassium chloride, powdered cellulose,
pregelatinized
starch, sodium chloride, sorbitol, starch, sucrose, sugar spheres, talc and
tribasic
calcium phosphate;
disintegrating agents, for example alginic acid, carboxymethylcellulose
calcium,
carboxymethylcellulose sodium, colloidal silicon dioxide, croscarmellose
sodium,
crospovidone, guar gum, magnesium aluminium silicate, methylcellulose,
microcrystalline cellulose, polacrilin potassium, powdered cellulose,
pregelatinized
starch, sodium alginate, sodium starch glycolate, starch including maize
starch and
agar;
lubricating agents, for example calcium stearate, glyceryl monostearate,
glyceryl
palmitostearate, hydrogenated castor oil, hydrogenated vegetable oil, light
mineral
oil, magnesium stearate, mineral oil, polyethylene glycol, sodium benzoate,
sodium
lauryl sulphate, sodium stearyl fumarate, stearic acid, talc and zinc
stearate;
binders, for example acacia, alginic acid, carbomer, carboxymethylcellulose
sodium,
dextrin, ethylcellulose, gelatin, guar gum, hydrogenated vegetable oil,
hydroxyethyl
cellulose, hydroxypropyl cellulose, hydroxypropyl methylcellulose, liquid
glucose,
magnesium aluminium silicate, maltodextrin, methylcellulose,
polymethacrylates,
povidone, pregelatinized starch, sodium alginate, starch including maize
starch, zein,
sugars (such as sucrose, molasses and lactose), and natural and synthetic gums
(such as extract of Irish moss, polyethylene glycol, waxes, microcrystalline
cellulose
and polyvinylpyrrolidone);
colouring agents, for example conventional pharmaceutically acceptable dyes;
sweetening and flavouring agents;
preservatives;
CA 02367040 2001-09-14
WO 00/56336 PCT/EP00/01940
6
one or more pharmaceutically acceptable couple or couples (such as those
comprising an acid and a carbonate or bicarbonate salt), which effervesces to
aid
dissolution when the solid dosage form is added to water; and
other optional ingredients known in the art to permit production of oral
dosage forms
by known methods such as tabletting.
Solid oral dosage forms may be formulated in a manner known to those
skilled in the art so as to give a sustained release of the compounds of the
present
invention. Film coated, solid oral dosage forms comprising compositions of the
invention may be advantageous, depending on the nature of the active compound.
Various materials, for example shellac and/or sugar, may be present as
coatings, or
to otherwise modify the physical form of the oral dosage form. For example
tablets
or pills may, if desired, be provided with enteric coatings by known methods,
for
example by the use of cellulose acetate phthalate and/or hydroxy propyl
methylcellulose phthalate.
Capsules or caplets (for example hard or soft gelatin capsules) comprising
the composition of the present invention (with or without added excipients
such as a
fatty oil), may be prepared by conventional means and, if desired, provided
with
enteric coatings in a known manner. The contents of the capsule or caplet may
be
formulated using known methods to give sustained release of the active
compound.
A particularly preferred oral liquid dosage form is a pharmaceutical
formulation which is an aqueous solution comprising a therapeutically
effective
amount of a compound of formula I including pharmaceutically acceptable salts,
solvates, racemates, enantiomers, diastereoisomers and mixtures thereof as
defined
above and an amount of cyclodextrin which is sufficient to cause the
dissolution of
the compound of formula I. Preferably the cyclodextrin is a (3-cyclodextrin.
Liquid oral
dosage forms may also comprise one or more sweetening agent, flavouring agent,
preservatives and mixtures thereof.
The compositions of the present invention may be formulated into granules or
powders with or without additional excipients. The granules or powders may be
ingested directly by the patient or they may be added to a suitable liquid
carrier (for
CA 02367040 2001-09-14
WO 00/56336 PCT/EP00/01940
7
example water) before ingestion. The granules or powders may contain
disintegrants (for example a pharmaceutically acceptable effervescent couple
formed
from an acid and a carbonate or bicarbonate salt) to facilitate dispersion in
the liquid
medium.
Preferably each of the above oral dosage forms may contain from about 1 mg
to about 500 mg (for example 10 mg, 50 mg, 100 mg, 200 mg, 400 mg or 500 mg)
of
the active compound.
Compositions of the invention may be administered rectally in the known
pharmaceutical forms for such administration, for example, suppositories with
hard
fat, semi-synthetic glyceride, cocoa butter or polyethylene glycol bases.
Compositions of the invention may also be administered parenterally, for
example by intravenous injection, in the known pharmaceutical forms for such
administration, for example sterile solutions in a suitable solvent.
The compositions of the invention may also be administered by continuous
infusion either from an external source (for example by intravenous infusion)
or from
a source of the compound placed within the body. Internal sources include
implanted
reservoirs containing the compound to be infused which is continuously
released (for
example by osmosis) or implants. The amount of active compound present in an
internal source should be such that a therapeutically effective amount of the
compound is delivered over a long period of time.
A particularly preferred embodiment of the invention for parenteral
administration, especially intravenous injection, is a pharmaceutical
formulation
which is an aqueous solution comprising a therapeutically effective amount of
a
compound of formula I including pharmaceutically acceptable salts, solvates,
racemates, enantiomers, diastereoisomers and mixtures thereof as defined above
and an amount of cyclodextrin which is sufficient to cause the dissolution of
the
compound of formula I.
CA 02367040 2001-09-14
WO 00/56336 PCT/EP00/01940
8
In some compositions it may be beneficial to use the active compound in the
form of particles of very small size, for example as obtained by fluid energy
milling.
In the above compositions the active compound may, if desired, be
associated with other compatible pharmacologically active ingredients.
The compositions of the present invention are indicated for use as
neuroprotective agents to protect against conditions such as stroke, and for
the
treatment of seizures and neurological disorders such as epilepsy. The
therapeutic
activity of the compositions has been demonstrated by means of various in vivo
pharmacological tests in standard laboratory animals.
Accordingly, a further aspect of the present invention provides a method of
treating seizures and/or neurological disorders such as epilepsy and/or a
method of
neuroprotection to protect against conditions such as stroke, in animals
including
human beings, which comprises the administration to a patient in need thereof
a
therapeutically effective amount of a pharmaceutical composition containing a
therapeutically effective amount of the active compound of a compound of
formula I
and a cyclodextrin. Thus the compositions of the present invention are useful
for the
inhibition of seizures and/or neurological disorders such as epilepsy and/or
as
neuroprotective agents to protect against conditions such as stroke, in
animals
including human beings. The compositions of the present invention are also
useful in
the treatment and prevention of migraine.
Whilst the precise amount of the active compound administered in the
treatments outlined above will depend on a number of factors, for example the
severity of the condition, the age and past medical history of the patient,
and always
lies within the sound discretion of the administering pharmacist, physician or
veterinary a suitable daily dose of the active compound for administration to
human
beings, is generally from about 1 mg to about 5000 mg, more usually from about
5 mg to about 1000 mg, given in a single dose or in divided doses at one or
more
times during the day. Oral administration or intravenous infusion is
preferred.
CA 02367040 2001-09-14
WO 00/56336 PCT/EP00/01940
9
The compositions of the present invention may be used in adjunctive therapy
with one or more other compound or compounds having activity in the treatment
of
seizures and/or neurological disorders such as epilepsy and/or as
neuroprotective
agents to protect against conditions such as stroke, and in the treatment and
prevention of migraine in animals including human beings. It will be
appreciated that
the term therapy as used herein includes prophylactic use of the active
compound
and pharmaceutical ~ composition or compositions comprising a therapeutically
effective amount of the active compound, for example to prevent the onset of
neurological disorders such as epileptic seizures, or as neuroprotective
agents to
protect against conditions such as stroke, in animals including human beings.
The
active compound and/or a pharmaceutical composition or compositions comprising
a
therapeutically effective amount of the active compound may be used to provide
a
local and/or systemic therapeutic effect.
A still further aspect of the present invention provides use of the
compositions
of the present invention in the preparation of a medicament for the treatment
of
seizures and/or neurological disorders such as epilepsy and/or for
neuroprotection to
protect against conditions such as stroke and/or the treatment and prevention
of
stroke, in animals including human beings.
Specific compounds of formula I are:-
7-[1-(4-fluorophenoxy)ethyl]-1,2,4-triazolo(1,5-a]pyrimidine;
7-[1-(4-chlorophenoxy)ethyl]-1,2,4-triazolo(1,5-a]pyrimidine;
7-[1-(4-bromophenoxy)ethyl]-1,2,4-triazolo(1,5-a]pyrimidine;
7-[1-(4-cyanophenoxy)ethyl]-1,2,4-triazolo[1, 5-a]pyrimidine;
7-[1-(4-trifluoromethylphenoxy)ethyl]-1,2,4-triazolo[1,5-a]pyrimidine;
7-[1-(4-methoxyphenoxy)ethyl]-1,2,4-triazolo(1, 5-a]pyrimidine;
7-[1-(4-trifluoromethoxyphenoxy)ethyl]-1,2,4-triazolo[1,5-a]pyrimidine;
7-[1-(4-acetylphenoxy)ethyl]-1,2,4-triazolo(1, 5-a]pyrimidine;
7-{1-[4-(methylthio)phenoxy]ethyl}-1,2,4-triazolo[1,5-a]pyrimidine;
CA 02367040 2001-09-14
WO 00/56336 PCT/EP00/01940
7-[1-(4-methylsulphinylphenoxy)ethyl]-1,2,4-triazolo[1,5-a]pyrimidine;
7-[1-(4-methylsulphonylphenoxy)ethyl]-1,2,4-triazolo[1,5-a]pyrimidine;
5 7-{1-[4-(ethylthio)phenoxy]ethyl}-1,2,4-triazolo[1,5-a]pyrimidine;
7-[1-(3-chlorophenoxy)ethyl]-1,2,4-triazolo[1, 5-a]pyrimidine;
7-(1-(2,4-difluorophenoxy)ethyl]-1,2,4-triazolo[1,5-a]pyrimidine;
7-[1-(2,4-dichlorophenoxy)ethyl]-1,2,4-triazolo[1; 5-a]pyrimidine;
7-[1-(3,4-dichlorophenoxy)ethyl]-1,2,4-triazolo[1,5-a]pyrimidine;
7-[1-(2-chloro-4-fluorophenoxy)ethyl]-1,2,4-triazolo[1,5-a]pyrimidine;
7-[1-(4-chlorophenoxy)ethyl]-2-methyl-1,2,4-triazolo[1,5-a]pyrimidine;
7-(4-chlorophenoxymethyl)-1,2,4-triazolo[1,5-a]pyrimidine;
7-(1-(4-chlorophenoxy)-1-methylethyl]-1,2,4-triazolo[1,5-a]pyrimidine;
7-[1-(4-chlorophenoxy)propyl]-1,2,4-triazolo[1,5-a]pyrimidine; and
7-[1-(4-chlorophenoxy)ethyl]-1,2,4-triazolo[1,5-a]pyrimidin-5-of
their stereoisomers; and pharmaceutically acceptable salts thereof.
Specific examples of the stereoisomers of compounds of formula I are:-
(+)-7-[1-(4-fluorophenoxy)ethyl]-1,2,4-triazolo[1,5-a]pyrimidine;
(-)-7-[1-(4-fluorophenoxy)ethyl]-1, 2,4-triazolo(1, 5-a]pyrimidine
(+)-7-[1-(4-chlorophenoxy)ethyl]-1,2,4-triazolo[1,5-a]pyrimidine;
(-)-7-[1-(4-chlorophenoxy)ethyl]-1,2,4-triazolo[1,5-a]pyrimidine;
(+)-7-[1-(4-chlorophenoxy)ethyl]-1,2,4-triazolo[1,5-a]pyrimidin-5-ol; and
(-)-7-[ 1-(4-chlorophenoxy)ethyl]-1,2,4-triazolo[1,5-a]pyrimidin-5-ol;
and pharmaceutically acceptable salts thereof.
Preferred compound of formula I are 7-[1-(4-chlorophenoxy)ethyl]-1,2,4-
triazolo[1,5-a]pyrimidine and 7-[1-(4-chlorophenoxy)ethyl]-1,2,4-triazolo(1,5-
a]-
pyrimidin-5-of including the racemates, enantiomers and mixtures thereof, and
CA 02367040 2001-09-14
WO 00/56336 PCT/EP00/01940
11
pharmaceutically acceptable salts thereof. More preferred compounds of formula
I
are (R)-7-[1-(4-chlorophenoxy)ethyl]-1,2,4-triazolo[1,5-a]pyrimidine and (S)-7-
[1-(4-
chlorophenoxy)ethyl]-1,2,4-triazolo[1,5-a]pyrimidine and pharmaceutically
acceptable
salts thereof. A most preferred compound of formula I is (R)-7-[1-(4-chloro-
phenoxy)ethyl]-1,2,4-triazolo[1,5-a]pyrimidine and pharmaceutically acceptable
salts
thereof. The free base of (R)-7-[1-(4-chlorophenoxy)ethyl]-1,2,4-triazolo[1,5-
a]pyrimidine has a negative optical rotation.
The compounds of formula I may be prepared as described in W095/10521
(Knoll AG) and W098/07724 (Knoll AG).
Suitably the cyclodextrin is selected from an a-cyclodextrin, (3-cyclodextrin
or
a y-cyclodextrin. Suitable (3-cyclodextrins include (3-cyclodextrin, methyl-R-
cyclodextrin, 2-hydroxyethyl-~i-cyclodextrin, 2-hydroxypropyl-[3-cyclodextrin,
3-hydroxypropyl-(3-cyclodextrin, 2,3-dihydroxypropyl-(3-cyclodextrin,
trimethyl-(3-
cyclodextrin, 6-O-a-D-glucosyl-[3-cyclodextrin, 6-O-a-D-maltosyl-[i-
cyclodextrin,
dimaltosyl-~-cyclodextrin, diglucosyl-[3-cyclodextrin, maltotriosyl-~i-
cyclodextrin,
carboxymethyl-(3-cyclodextrin, carboxyethyl-(3-cyclodextrin, heptakis-(2,6-di-
O-
methyl)- (3-cyclodextrin, heptakis-(2,3,6-tri-O-methyl)- (3-cyclodextrin, (3-
cyclodextrin
sulphobutyl ether (described in US 5,376,645 and US 5,134,127). Suitable y-
cyclodextrins include 2-hydroxypropyl-y-cyclodextrin. Preferably the
cyclodextrin is a
(3-cyclodextrin. More preferably the cyclodextrin is methyl-(3-cyclodextrin or
2-hydroxypropyl-(3-cyclodextrin. Most preferably the cyclodextrin is 2-
hydroxypropyl-
(3-cyclodextrin.
A preferred embodiment of the present invention is a pharmaceutical
composition in the form of an aqueous solution comprising a) a therapeutically
effective amount of a compound of formula I as defined above and b) a
(3-cyclodextrin.
Surprisingly a composition comprising a compound of formula I, and in
particular (R)-7-[1-(4-chlorophenoxy)ethyl]-1,2,4-triazolo[1,5-a]pyrimidine,
and 2-
hydroxypropyl-(3-cyclodextrin is particularly advantageous as a result of
increased
solubility and reduced adverse toxicity compared to compositions containing
other
CA 02367040 2001-09-14
WO 00/56336 PCT/EP00/01940
12
cyclodextrins. Such compositions provide aqueous solutions which may be
administered by intravenous injection or by continuous infusion to patients
suffering
from stroke. Such compositions may provide a major advance in the treatment of
stroke where rapid treatment is essential. These compositions may be
considered to
be a particular selection from the possible combinations of the compounds of
formula
I with cyclodextrins with unexpected advantageous properties not shared by the
other possible combinations.
A more preferred embodiment of the present invention is a pharmaceutical
composition in the form of an aqueous solution comprising a) a therapeutically
effective amount of a compound of formula I as defined above and b) 2-
hydroxypropyl-[i-cyclodextrin.
A most preferred embodiment of the present invention is a pharmaceutical
composition in the form of an aqueous solution comprising a) a therapeutically
effective amount of a compound of formula I as defined above at a
concentration in
the range of 0.1-30% w/v and b) 2-hydroxypropyl-[3-cyclodextrin at a
concentration in
range of 5-75% w/v. It will be understood that the remainder of the
composition is
normally water to 100% but that additional pharmaceutically acceptable
additives, for
example preservatives, anti-oxidants, anti-microbials, flavourings etc. may
also be
present at levels known to those skilled in the art. Preferably the compound
of
formula I is present at a concentration in the range of 1-30% w/v, for example
10-20% w/v and 12-18% w/v, more preferably at a concentration in the range of
0.5-5% w/v and most preferably at a concentration in the range of 1-3% w/v.
Preferably the concentration of 2-hydroxypropyl-(3-cyclodextrin is in the
range of
20-50% w/v and more preferably is in the range of 30-40% w/v.
An especially preferred embodiment of the present invention is a
pharmaceutical composition in the form of an aqueous solution comprising:
a) (R)-7-[1-(4-chlorophenoxy)ethyl]-1,2,4-triazolo[1,5-a]pyrimidine at a
concen-
tration in the range of 0.1-30% w/v and
b) 2-hydroxypropyl-(3-cyclodextrin at a concentration in range of 5-75% w/v.
CA 02367040 2001-09-14
WO 00/56336 PCT/EP00/01940
13
Preferably the concentration of (R)-7-[1-(4-chlorophenoxy)ethyl]-1,2,4-
triazolo(1,5-
a]pyrimidine is in the range of 1-30% w/v, for example 10-20% w/v and most
preferably is in the range of 12-18% w/v, more preferably at a concentration
in the
range of 0.5-5% w/v and most preferably at a concentration in the range of 1-
3% w/v.
Preferably the concentration of 2-hydroxypropyl-[i-cyclodextrin is in the
range
20-50% w/v and more preferably is in the range 30-40% w/v.
1~0 The efficacy of the compositions of the present invention was demonstrated
by the following study.
METHODS
i) Middle Cerebral Artery (MCA) occlusion
Male Sprague-Dawley rats (300 - 350g) were anaesthetised with halothane in
oxygen (1000 ml/min). Anaesthesia was induced with 5% (v/v) halothane and
maintained using a face mask at a concentration of 2.25 - 2.5%. Body
temperature
of the animals was maintained at 37 t 0.5°C throughout the procedure
using a
thermostatically-controlled heated operating blanket. A scalp incision was
made at
the mid-point between the right eye and right ear. The temporalis muscle was
separated in the plane of its fibre bundles and retracted to expose the zygoma
and
squamosal bones. Muscle and connective tissue was scraped from the bone to
locate the drill point. Using microsurgical techniques, a craniotomy was made
using
a No. 5 round-head dental burr, rostral to the fusion point of the two cranial
bones.
The skull was cooled by regular swabbing with sterile saline to avoid causing
thermal
injury to the cerebral cortex. The dura mater and pia-arachnoid membranes were
incised with a 25 g hypodermic needle bent at the tip to form a hook and were
gently
pulled from the surface of the cortex with fine forceps. The exposed middle
cerebral
artery was electrocauterised using bipolar diathermy (GU Manufacturing
Company,
London) below the rhinal fissure and at a second point below the bifurcation
of the
artery into parietal and frontal cortical branches. Animals which haemorrhaged
on
occlusion of the MCA were discarded from the study. The MCA was severed at
both
CA 02367040 2001-09-14
WO 00/56336 PCT/EP00/01940
14
points with a pair of micro-scissors to ensure separation of the vessel. Care
was
taken to avoid causing non-specific damage to the surface of the cerebral
cortex.
Finally, the temporalis muscle and overlying skin were placed back into
position and
sutured. Following operation, animals were placed in cages on absorbent paper
and
allowed to regain consciousness under gentle warming from a red, 60 W light
bulb.
After recovery, animals were grouped together (n = 4 - 6) in cages. The
duration of
the operation was 15 - 20 min and animals regained consciousness within
another
- 15 min after arterial occlusion.
10 ii) Drug preparation
Two different cyclodextrin solutions (Research Biochemicals Inc.) were
initially used for solubilisation of (-)-7-[1-(4-chlorophenoxy)ethyl]-1,2,4-
triazolo[1,5-
a]pyrimidine (hereinafter referred to as the test compound); 2-hydroxypropyl-
[3-
cyclodextrin and methyl-(3-cyclodextrin. The test compound was added to each
cyclodextrin solution (35% w/v in water) and sonicated for 5 - 10 minutes in a
sonicating water bath, avoiding warming of the solution. The compound was
soluble
to approximate concentrations of 15 mg/ml in the 2-hydroxypropyl-[3-
cyclodextrin
solution (that is from 0.1 to 15 mg/ml) and 40 mg/ml in methyl-[3-
cyclodextrin. 2-
Hydroxypropyl-[3-cyclodextrin was selected for the study as animals were
unable to
tolerate intraperitoneal injection of methyl-[i-cyclodextrin.
iii) Treatment of animals
Two groups of animals were used; MCA occlusion plus test compound (n=11)
and MCA occlusion plus vehicle (n=12). Animals were weighed immediately before
MCA occlusion and after 6 days of recovery. The test compound was dissolved to
a
concentration of 15 mg/ml in 35% w/v 2-hydroxypropyl-(3-cyclodextrin in
sterile saline
by placing in a sonicating water bath. The pH of the vehicle and drug
solutions was
adjusted to 7.4 and they were stored at 4°C for the duration of the
experiment. Sixty
minutes after coagulation and severance of the MCA, the animals were injected
intraperitoneally with 35 mg/kg of test compound or equivalent volume of
vehicle.
The animals were dosed thereafter at 20 mg/kg at 12 hourly intervals for 36
hours.
CA 02367040 2001-09-14
WO 00/56336 PCT/EP00/01940
iv) Histopathological assessment of ischaemic damage
After 6 days of recovery, animals were anaesthetised with Sagatal (85 mg/kg
5 intraperitoneally; Rhone Merieux) and perfused traps-cardially with cold
(4°C)
phosphate-buffered saline (Oxoid) for 3 min, followed by cold 4% formaldehyde
in
phosphate-buffered saline (pH 7.4) for 15 min. The brains were removed and
immersion-fixed for at least 48 hours at 4°C .
10 The brains were sectioned in the coronal plane at 250 Nm intervals and
30 pm thickness using a vibratome (General Scientific, Series 1000) and were
stained with 0.2% toluidine blue. The extent of ischaemic damage was assessed
in
13 planes at 500Nm intervals from +2.7 mm to -3.3 mm relative to bregma.
Slides
were placed onto a slide illuminator (Carl Zeiss; 17.5x magnification) and
areas of
15 damage were delineated onto stereotaxic brain maps. Lesion areas were
measured
using a SeeScan image analyser and lesion volumes calculated using
computational
analysis (GraphPad Prism). All sections were recoded and lesion sizes were
measured by an operator unaware of treatment procedures.
v) Statistical analysis of data
Histological damage assessment and changes in rat weights were analysed
using two-way ANOVA. All data are quoted as mean t standard error of the mean.
RESULTS
Animals did not show gross neurological deficits, such as seizures or
hemiparesis due to MCA occlusion and those treated with the test compound
showed only mild sedation, particularly in the first 5 - 6 hours after the
initial dosing.
Two rats out of a total of 25 were killed shortly after their recovery from
the
anaesthetic because of haemorrhaging due to incomplete sealing of the MCA by
the
electrocoagulator. None of the animals treated with either vehicle or the test
compound died.
CA 02367040 2001-09-14
WO 00/56336 PCT/EP00/01940
16
i) Histopathological damage after MCA occlusion
Ischaemic injury in toluidine blue-stained brain sections was manifest as
areas of pallor with sharply demarcated margins between healthy and damaged
tissue. Damage from MCA occlusion was present mainly in the parietal, insular,
frontal and forelimb cortical regions. Damage was also found in the outer
region of
the caudate-putamen. This was caused by secondary damage produced by
compression, due to the presence of oedema in the cortex and corpus callosum.
This may be concluded as there is no arterial supply to the caudate-putamen
distal to
the occlusion site.
When assessed 6 days after the ischaemic insult, treatment of rats with test
compound (60 min post-MCAO; 35 mg/kg ip + 20 mglkg x 3) resulted in a 40%
reduction of lesion volume when compared with animals given vehicle alone. The
lesion volumes were 72.0 t 7.9 mm3 for vehicle treated rats and 43.6 ~ 4.9 mm3
for
test compound-treated animals.
In a comparative study rats were treated with a suspension of the test
compound (15 min post-MCAO; 50 mg/kg x 3) by oral gavage. The test compound
provided a 31 % reduction of lesion volume (56.9 ~ 6.1 mm3) when compared with
vehicle-treated controls (82.6 ~ 6.47 mm3).
These results demonstrate that the compositions of the present invention are
advantageous over previously known compositions. In particular, solutions of
the
compounds of formula I are advantageous as they are more efficacious than
suspensions, i.e. greater reduction in lesion volume, with lower doses and
administered at a later time-point after MCA occlusion. Solution formulations
according to the present invention are also likely to overcome the short half-
life of the
compounds of formula I in humans. Additionally such solution formulations can
be
administered by continuous intravenous infusion which is particularly
beneficial in the
treatment of stroke.
The invention is illustrated by the following Example which is given by way of
example only. The final product of each of this Examples was characterised by
the
CA 02367040 2001-09-14
WO 00/56336 PCT/EP00/01940
17
following procedures: gas-liquid chromatography; high performance liquid
chromatography; elemental analysis; nuclear magnetic resonance spectroscopy
and
infrared spectroscopy.
Other compounds of the present invention are prepared as described in
W095/10521 (Knoll AG) and W098/07724 (Knoll AG) which are incorporated herein
by reference. Cyclodextrins are commercially available or may be prepared by
methods disclosed in Chemical Reviews (1998) 98,1474-2076 and references cited
therein.
Example 1
1 a) A mixture of 2-(4-chlorophenoxy)propionic acid (30.0 g) in toluene (300
ml)
was added to a solution of thionyl chloride (22.0 ml) and dimethylformamide
(2 ml) in toluene (150 ml) at 60-70°C with stirring. The mixture was
stirred for
18 hours at 70-80°C and then evaporated under reduced pressure to give
the
acid chloride. A solution of Meldrum's acid (23.5 g) in dichloromethane
(90 ml) was cooled to 0-5°C under nitrogen and pyridine (33 ml) was
added at
0-5°C. To this mixture was added a solution of the acid chloride
prepared
above in dichloromethane (90 ml) dropwise keeping the temperature below
5°C. The mixture was stirred for 1 hour at 0-5°C and then at
ambient
temperature for 18 hours. The mixture was diluted with dichloromethane
(150 ml) and washed with 2M hydrochloric acid and then with saturated
sodium bicarbonate solution and then water. The bicarbonate washes were
acidified with 5M hydrochloric acid and extracted into dichloromethane.
These dichloromethane extracts were combined with the original
dichloromethane solution and dried and evaporated to give the crude
intermediate Meldrum's acid derivative. This derivative was boiled under
reflux with methanol (400 ml) for 6 hours and then left to stand at ambient
temperature for 66 hours with methanolic hydrogen chloride solution (10 ml).
The mixture was evaporated under reduced pressure and the residue was
partitioned between ethyl acetate and water. The ethyl acetate layer was
separated off, washed with saturated sodium bicarbonate solution, brine and
then dried and evaporated under reduced pressure to give an oil which was
CA 02367040 2001-09-14
WO 00/56336 PCT/EP00/01940
18
distilled under high vacuum. The distillate was purified by flash column
chromatography on silica using petroleum ether, b.p. 60-80°C I ethyl
acetate
20:1 as the mobile phase to give methyl 4-(4-chlorophenoxy)-3-
oxopentanoate as an oil.
b) Guanidine hydrochloride (1.87 g) was added with stirring to a solution of
sodium (0.41 g) in ethanol (15 ml). The mixture was stirred for 15 minutes
and then to this mixture was added a solution of methyl 4-(4-
chlorophenoxy)-3-oxopentanoate (5.0 g) in ethanol (15 ml). The mixture
was stirred and boiled under reflux for 16 hours. The mixture was cooled
and then evaporated to dryness under reduced pressure to give a solid.
The solid was triturated with water (10 ml) containing glacial acetic acid
(2 ml) and dichloromethane (20 ml) for 1 hour and then filtered. The
residue obtained was washed with water (20 ml) and then with
dichloromethane to give 2-amino-4-[1-(4-chlorophenoxy)ethyl-6-
hydroxypyrimidine, m.p. 125°C.
c) A mixture of 2-amino-4-[1-(4-chlorophenoxy)ethyl-6-hydroxypyrimidine
(0.5 g), dimethylformamide dimethyl acetal (0.5 ml) and toluene (5 ml) was
stirred and boiled under reflux for 8 hours. The mixture was evaporated for
8 hours. The mixture was allowed to stand at ambient temperature for 24
hours and then evaporated to dryness under reduced pressure to give a
solid which was triturated with ether and filtered to give the amidine as a
solid.
d) Sodium hydride (62 mg of a 60% dispersion in mineral oil from which the
mineral oil had been removed by washing with petrol) was dissolved in
methanol (10 ml) and hydroxylamine hydrochloride (0.12 g) was added.
The mixture was stirred for 5 minutes and then added to the
amidinopyrimidine (0.5 g) obtained in c). The mixture was stirred at ambient
temperature for 72 hours and then evaporated to dryness under reduced
pressure to give a residue which was washed with water and dried to give
the formamidoxine.
CA 02367040 2001-09-14
WO 00/56336 PCT/EP00/01940
19
e) The product from d) (5.0 g) and polyphosphoric acid (100 g) were stirred
and
heated on a steam bath for 4 hours. The mixture was allowed to cool to
ambient temperature and then ice (100 g) and ethyl acetate (100 ml) were
added. A solution of potassium carbonate (110 g) in water (total volume
100 ml) was added dropwise over 15 minutes to neutralise the mixture. The
mixture was separated and the aqueous layer was extracted with ethyl
acetate. The combined ethyl acetate layers were dried and evaporated to
give a solid which was purified by flash column chromatography on silica
using dichloromethane / methanol (75:3) as the mobile phase to give 7-[1-(4-
chlorophenoxy)ethyl]-1,2,4-triazolo[1,5-a]pyrimidin-5-ol, m.p. 190°C.
Example 2
(-)-7-[1-(4-Chlorophenoxy)ethyl]-1,2,4-triazolo[1,5-a]pyrimidine was prepared
as described in W095/10521.
Solubility Determinations
Example 1 Example 2
Water < 33 p.g/ml 125-170 p.g/ml
35% w/v methyl-(3-cyclodextrin2 mg/ml 40 mg/ml
in water
35% w/v 2-hydroxypropyl-(3-cyclodextrin1 mg/ml 15 mglml
in water
35% w/v 2-hydroxypropyl-y-cyclodextrin< 62 p.g/ml 6 mg/ml
in water