Note: Descriptions are shown in the official language in which they were submitted.
CA 02372586 2001-11-O1
WO 00/66163 PCT/US00/11642
1
CONTRACEPTIVE COMPOSITIONS CONTAINING 2,1-BENZISOTHIAZOLINE 2,2-DIOXIDES AND
PROGESTATIONALS
Background of the Invention
Intracellular receptors (IR) form a class of struo~_11~' rP~ated gene
regulators
known as "ligand dependent transcription factors" (R. M. Evans, Science 240,
889,
1988). The steroid receptor family is a subset of the IR family, including
progesterone
receptor (PR), estrogen receptor (ER), androgen receptor (AR), glucocorticoid
receptor (GR), and mineralocorticoid receptor (MR).
The natural hormone, or ligand, for the PR is the steroid progesterone, but
synthetic compounds, such as medroxyprogesterone acetate or levonorgestrel,
have
been made which also serve as ligands. Once a ligand is present in the fluid
surrounding a cell, it passes through the membrane via passive diffusion, and
binds to
the IR to create a receptor/ligand complex. This complex binds to specific
gene
promoters present in the cell's DNA. Once bound to the DNA the complex
modulates
the production of mRNA and protein encoded by that gene.
A compound that binds to an IR and mimics the action of the natural hormone
is termed an agonist, whilst a compound that inhibits the effect of the
hormone is an
antagonist.
PR agonists (natural and synthetic) are known to play an important role in the
health of women. PR agonists are used in birth control formulations, typically
in the
presence of an ER agonist. ER agonists are used to treat the symptoms of
menopause,
but have been associated with a proliferative effect on the uterus that can
lead to an
increased risk of uterine cancers. Co-administration of a PR agonist reduces
or ablates
that risk.
PR antagonists may also be used in contraception. In this context they may be
administered alone (Ulmann et al, Ann. N. Y. Acad. Sci. 261, 248, 1995), in
combination with a PR agonist (Kekkonen et al, Fertility and Sterility 60,
610, 1993)
or in combination with a partial ER antagonist such as tamoxifen (WO 96/19997
Al
July 4, 1996).
CA 02372586 2001-11-O1
WO 00/66163 PCT/US00/11642
2
PR antagonists may also be useful for the treatment of hormone dependent
breast cancers (Horwitz et al, Horm. Cancer, 283, pub: Birkhaeuser, Boston,
Mass.,
ed. Vedeckis) as well as uterine and ovarian cancers. PR antagonists may also
be
useful for the treatment of non-malignant chronic conditions such as fibroids
(Murphy
et al, J. Clin. Endo. Metab. 76, 513, 1993) and endometriosis (Kettel et al,
Fertility
and Sterility 56, 402, 1991).
PR antagonists may also be useful in hormone replacement therapy for post
menopausal patients in combination with a partial ER antagonist such as
tamoxifen
(US 5719136).
PR antagonists, such as mifepristone and onapristone, have been shown to be
effective in a model of hormone dependent prostate cancer, which may indicate
their
utility in the treatment of this condition in men (Michna et al, Ann. N. Y.
Acad. Sci.
761, 224, 1995).
Jones et al (US 5,688,810) is the PR antagonist dihydroquinoline A.
N
\\
Ma
S
~Me
H' \M a
A
Jones et al described the enol ether B (US 5,693,646) as a PR ligand.
F
Me
B
CA 02372586 2001-11-O1
WO 00/66163 PCT/US00/11642
3
Jones et al described compound C (US 5,696,127) as a PR ligand.
Me CI
C Me
F
Me
H Me
C
Zhi et al described lactones D, E and F as PR antagonists (J. Med. Chem. 41,
291, 1998).
0
~ O O Me I ~ O Me I ~ Me
~ i ~ ~
Me ~ ~ Me ~ ~ Me
Me H Me O O H Me
D E F
Zhi et al described the ether G as a PR antagonist (J Med. Chem. 41, 291,
1998).
i
~O Me
i
Me
Me
G
Combs et al disclosed the amide H as a ligand for the PR (J. Med. Chem. 38,
4880, 1995).
CA 02372586 2001-11-O1
WO 00/66163 PCT/US00/11642
4
/ F
I
~N.N ~ gr
/ O
H
Penman et al described the vitamin D analog I as a PR ligand (Tetrahedron.
Lett. 35, 2295, 1994).
O
cH2
H O''
I
Hamann et al described the PR antagonist J (Ann. N.Y Acad. Sci. 761, 383,
1995).
OMe
Br ~HZC ~ Me
/
OAc Me Me
J
Chen et al described the PR antagonist K (Chen et al, POI-37, 16'h Int. Cong.
Het. Chem., Montana, 1997).
CA 02372586 2001-11-O1
WO 00/66163 PCT/US00/11642
CI
\ CI
,." N
H
a
N
H
K
Kurihari et al described the PR ligand L (J. Antibiotics 50, 360, 1997).
OAc
O
O \ \ OH
5
L
There are several examples of 2,1-benzisothiazoline 2,2-dioxides ('sultams')
in the chemical and patent literature which contain no reference to
progesterone
activity, and do not carry the correct substitution pattern for PR modulator
activity.
Chiarino et al described the preparation of the parent 2,1-benzisothiazoline
2,2-dioxide, i.e., M (and derivatives, e.g., N), that was used in the present
invention
(J. Heterocycl. Chem. 23(6), 1645-9, 1986).
SO ~N
N 2 ~ 502
H ~ N
H
M N
Skorcz et al described a series of 5-(2-morpholinyl)-2,1-benzisothiazolines,
e.g., O, which are useful as central nervous depressants (U.5. 3,635,964).
CA 02372586 2001-11-O1
WO 00/66163 PCT/US00/11642
6
CN
O
~~~S O2
O
Kamireddy et al disclosed a series of cyclic sulfonamides, e.g., P and Q,
useful for
controlling undesired vegetation (WO 95/33746).
C F3
N/
O
N O O N ~O
F / I F
N SOZ ~ ~ N SOZ
H H
CI CI
P Q
Description of the Invention
This invention provides combination therapies and dosing regimens utilizing
antiprogestational agents in combination with one or more progestational
agents. This
invention further provides methods of treatment and dosing regimens further
utilizing
in combination with these antiprogestins and progestins, an estrogen, such as
ethinyl
estradiol.
These regimens and combinations may be administered to a mammal to
induce contraception or for the treatment and/or prevention of secondary
amenorrhea,
dysfunctional bleeding, uterine leiomyomata, endometriosis; polycystic ovary
syndrome, carcinomas and adenocarcinomas of the endometrium, ovary, breast,
colon, prostate. Additional uses of the invention include stimulation of food
intake.
The uses herein for the treatment and/or prevention of the conditions or
diseases
CA 02372586 2001-11-O1
WO 00/66163 PCT/US00/11642
7
described above includes the continuous administration or periodic
discontinuation of
administration of the invention to allow for minimization of effect dose or
minimization of side effects or cyclic menstrual bleeding.
The use of this invention for contraception includes administration,
preferably
orally, to a female of child bearing age an antiprogestin in combination with
an
estrogen or progestin or both. These administration regimens are preferably
carried
out over 28 consecutive days, with a terminal portion of the cycle containing
administration of no progestins, estrogens or anti-progestins.
The progestins of these combinations may be administered alone or in
combination with an estrogen for the first 14 to 24 days of the cycle, the
progestins
being administered at a dosage range equal in progestational activity to about
35 pg to
about 150 pg levonorgestrel per day, preferably equal in activity to from
about 35 pg
to about 100 ~g levonorgestrel per day. An antiprogestin may then be
administered
alone or in combination with an estrogen for a period of 1 to 11 days to begin
on any
cycle day between day 14 and 24. The anti-progestin in these combinations may
be
administered at a dose of from about 2pg to about 50 pg per day and the
estrogen may
be administered at a dose of from about 10 ~g to about 35 pg per day. In an
oral
administration, a package or kit containing 28 tablets will include a placebo
tablet on
those days when the antiprogestin or progestin or estrogen is not
administered.
In a preferred embodiment of this invention, the progestins of this invention
may be administered alone or in combination with estrogen for the initial 18
to 21
days of a 28-day cycle, followed by administration of an antiprogestin, alone
or in
combination with an estrogen, for from 1 to 7 days.
The estrogen to be used in the combinations and formulations ofthis invention
is preferably ethinyl estradiol.
Progestational agents useful with this invention include, but are not limited
to,
levonorgestrel, norgestrel, desogestrel, 3-ketodesogestrel, norethindrone,
gestodene,
norethindrone acetate, norgestimate, osaterone, cyproterone acetate,
trimegestone,
dienogest, drospirenone, nomegestrol, or (17-deacetyl)norgestimate. Among the
CA 02372586 2001-11-O1
WO 00/66163 PCT/US00/11642
8
preferred progestins for use in the combinations of this invention are
levonorgestrel,
gestodene and trimegestone.
Examples of orally administered regimens of this invention over a 28 day
cycle include administration of a progestational agent solely for the first 21
days at a
daily dose equal in progestational activity to from about 35 to about 100 pg
of
levonorgestrel. An antiprogestin compound of this invention may then be
administered at a daily dose of from about 2 to 50 mg from day 22 to day 24,
followed by no administration or administration of a placebo for days 25 to
28. It is
most preferred that the daily dosages of each relevant active ingredient be
incorporated into a combined, single daily dosage unit, totaling 28 daily
units per 28-
day cycle.
In another regimen, a progestational agent may be coadministered for the first
21 days at a daily dose equal in progestational activity to from about 35 to
about 150
~g levonorgestrel, preferably equal in activity to from about 35 to about 100
pg
levonorgestrel, with an estrogen, such as ethinyl estradiol, at a daily dose
range of
from about 10 to about 35 pg. This may be followed as described above with an
antiprogestin administered at a daily dose of from about 2 to 50 mg from day
22 to
day 24, followed by no administration or administration of a placebo for days
25 to
28.
Still another regimen within the scope of this invention will include
coadministration from days 1 to 21 of a progestational agent, the
progestational agent,
preferably levonorgestrel, being administered at a daily dose equal in
progestational
activity to from about 35 to about 100 ~g levonorgestrel, and an estrogen,
such as
ethinyl estradiol, at a daily dose range of from about 10 to about 35 pg. This
will be
followed on days 22 to 24 by coadministration of an antiprogestin (2 to 50
mg/day)
and an estrogen, such as ethinyl estradiol, at a daily dose of from about 10
to about 35
fig. From day 25 to day 28, this regimen may be followed by no administration
or
administration of a placebo.
This invention also kits or packages of pharmaceutical formulations designed
for use in the regimens described herein. These kits are preferably designed
for daily
CA 02372586 2001-11-O1
WO 00/66163 PCT/US00/11642
9
oral administration over a 28-day cycle, preferably for one oral
administration per
day, and organized so as to indicate a single oral formulation or combination
of oral
formulations to be taken on each day of the 28-day cycle. Preferably each kit
will
include oral tablets to be taken on each the days specified, preferably one
oral tablet
will contain each of the combined daily dosages indicated.
According to the regimens described above, one 28-day kit may comprise:
a) an initial phase of from 14 to 21 daily dosage units of a
progestational agent equal in progestational activity to about 35 to about 150
ug
levonorgestrel, preferably equal in progestational activity to about 35 to
about 100 pg
levonorgestrel;
b) a second phase of from 1 to 11 daily dosage units of an antiprogestin
compound of this invention, each daily dosage unit containing an antiprogestin
compound at a daily dosage of from about 2 to 50 mg; and
c) optionally, a third phase of an orally and pharmaceutically
acceptable placebo for the remaining days of the cycle in which no
antiprogestin,
progestin or estrogen is administered.
A preferred embodiment of this kit may comprise:
a) an initial phase of 21 daily dosage units of a progestational agent
equal in progestational activity to about 35 to about 150 pg levonorgestrel,
preferably
equal in progestational activity to about 35 to about 100 pg levonorgestrel;
b) a second phase of 3 daily dosage units for days 22 to 24 of an
antiprogestin compound of this invention, each daily dosage unit containing an
antiprogestin compound at a daily dosage of from about 2 to SO mg; and
c) optionally, a third phase of 4 daily units of an orally and
pharmaceutically acceptable placebo for each of days 25 to 28.
Another 28-day cycle packaging regimen or kit of this invention comprises:
a) a first phase of from 18 to 21 daily dosage units of a progestational
agent equal in progestational activity to about 35 to about 150 pg
levonorgestrel,
preferably equal in activity to from about 35 to about 100 pg levonorgestrel,
and, as
CA 02372586 2001-11-O1
WO 00/66163 PCTNS00/11642
an estrogen, ethinyl estradiol at a daily dose range of from about 10 to about
35 pg;
and
b) a second phase of from 1 to 7 daily dosage units of an antiprogestin
of this invention at a daily dose of from about 2 to SO mg; and
5 c) optionally, an orally and pharmaceutically acceptable placebo for
each of the remaining 0 to 9 days in the 28-day cycle in which no
progestational
agent, estrogen or antiprogestin is administered.
A preferred embodiment of the kit described above may comprise:
a) a first phase of 21 daily dosage units of a progestational agent equal
10 in progestational activity to about 35 to about 150 pg levonorgestrel,
preferably equal
in activity to from about 35 to about 100 pg levonorgestrel, and, as an
estrogen,
ethinyl estradiol at a daily dose range of from about 10 to about 35 pg; and
b) a second phase of 3 daily dosage units for days 22 to 24 of an
antiprogestin administered at a daily dose of from about 2 to 50 mg; and
c) optionally, a third phase of 4 daily dose units of an orally and
pharmaceutically acceptable placebo for each of days 25 to 28.
A further 28-day packaged regimen or kit of this invention comprises:
a) a first phase of from 18 to 21 daily dosage units, each containing a
progestational agent of this invention at a daily dose equal in progestational
activity to
about 35 to about 150 pg levonorgestrel, preferably equal in activity to from
about 35
to about 100 pg levonorgestrel, and ethinyl estradiol at a daily dose range of
from
about 10 to about 35 pg;
b) a second phase of from 1 to 7 daily dose units, each daily dose unit
containing an antiprogestin of this invention at a concentration of from 2 to
50 mg;
and ethinyl estradiol at a concentration of from about 10 to about 35 pg; and
c) optionally, an orally and pharmaceutically acceptable placebo for
each of the remaining 0 to 9 days in the 28-day cycle in which no
progestational
agent, estrogen or antiprogestin is administered.
A preferred embodiment of the package or kit just described comprises:
CA 02372586 2001-11-O1
WO 00/66163 PCTNS00/11642
11
a) a first phase of 21 daily dosage units, each containing a
progestational agent of this invention at a daily dose equal in progestational
activity to
about 35 to about 150 pg levonorgestrel, preferably from about 35 to about 100
pg
levonorgestrel, and ethinyl estradiol at a daily dose range of from about 10
to about 35
fig;
b) a second phase of 3 daily dose units for days 22 to 24, each dose
unit containing an antiprogestin of this invention at a concentration of from
2 to 50
mg; and ethinyl estradiol at a concentration of from about 10 to about 35 fig;
and
c) optionally, a third phase of 4 daily units of an orally and
pharmaceutically acceptable placebo for each of days 25 to 28.
In each of the regimens and kits just described, it is preferred that the
daily
dosage of each pharmaceutically active component of the regimen remain fixed
in
each particular phase in which it is administered. It is also understood that
the daily
dose units described are to be administered in the order described, with the
first phase
followed in order by the second and third phases. To help facilitate
compliance with
each regimen, it is also preferred that the kits contain the placebo described
for the
final days of the cycle. It is further preferred that each package or kit
comprise a
pharmaceutically acceptable package having indicators for each day of the 28-
day
cycle, such as a labeled blister package or dial dispenser packages known in
the art.
In this disclosure, the terms anti-progestational agents, anti-progestins and
progesterone receptor antagonists are understood to be synonymous. Similarly,
progestins, progestational agents and progesterone receptor agonists are
understood to
refer to compounds of the same activity.
These dosage regimens may be adjusted to provide the optimal therapeutic
response. For example, several divided doses of each component may be
administered daily or the dose may be proportionally increased or reduced as
indicated by the exigencies of the therapeutic situation. In the descriptions
herein,
reference to a daily dosage unit may also include divided units that are
administered
over the course of each day of the cycle contemplated.
CA 02372586 2001-11-O1
WO 00/66163 PCT/US00/11642
12
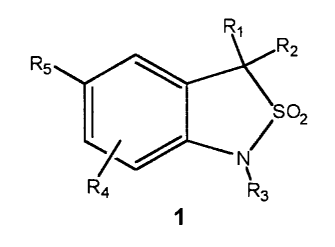
Antiprogestin compounds which may be used in the kits, methods and
regimens herein are those of the Formula 1:
1
wherein
Rl and RZ are each, independently, hydrogen, alky, substituted alkyl, hydroxy,
alkoxy,
substituted alkoxy, aryl, substituted aryl, heteroaryl, substituted heteroary,
arylalkyl, heteroarylalkyl, and alkynyl; or
Rl and R2 are taken together form a ring and together contain -CHZ(CHZ)"CHZ- ,
-CHzCHzCMezCH2CH2-, -O(CHZ)pCH2-, O(CHZ)q0-, -CHZCHzOCH2CH2-,
-CHZCH2NR7CHZCH2-; or
RI and R2 are a double bond, said double bond having two methyl groups bonded
to
the terminal end, having a cycloalkyl group bonded to the terminal end, having
an oxygen bonded to the terminal end, or having a cyc:cJe~her bonded to the
terminal end;
R~ is hydrogen or alkyl of 1-6 carbon atoms;
n = 1-5;
p = 1-4;
q = 1-4;
R3 is hydrogen, hydroxyl, NH2, alkyl, substituted alkyl, alkenyl, alkynyl,
substituted
or, COR'';
RA is hydrogen, alkyl, substituted alkyl, alkoxy, substituted alkoxy,
aminoalkyl, or
substituted aminoalkyl;
R4 is hydrogen, halogen, -CN, -NHZ, alkyl, substituted alkyl, alkoxy, alkoxy,
aminoalkyl, or substituted aminoalkyl;
CA 02372586 2001-11-O1
WO 00/66163 PCT/US00/11642
13
RS is a trisubstituted phenyl ring having the structure,
Y~~/ Z
I ,
X
X is halogen, OH, -CN, alkyl, substituted alkyl, alkoxy, substituted alkoxy,
thioalkyl, substituted thioalkyl, S(O)alkyl, S(O)zalkyl, aminoalkyl,
substituted aminoalkyl, -NOz, perfluoroalkyl, 5 or 6 membered
heterocyclic ring containing 1 to 3 heteroatoms, thioalkoxy, -CORD,
-OCORB, or -NR~CORB;
RB is hydrogen, alkyl, substituted alkyl, aryl, substituted aryl, alkoxy,
substituted alkoxy, aminoalkyl, or substituted aminoalkyl;
R~ is hydrogen, alkyl, or substituted alkyl;
Y and Z are each, independently, hydrogen, halogen, -CN, -NOz, alkoxy,
alkyl, or thioalkyl; or
RS is a five or six membered heteroaryl ring containing l, 2, or 3 heteroatoms
selected
from the group consisting of O, S, SO, SOz and NR6 with said ring carbons
being optionally substituted with one or two substituents independently
selected from the group consisting of hydrogen, halogen, CN, NOz alkyl,
alkoxy, aminoalkyl, CORD, and NRECORD;
RD is hydrogen, alkyl, substituted alkyl, aryl, substituted aryl, alkoxy,
substituted alkoxy, aminoalkyl, or substituted aminoalkyl;
RE is hydrogen, alkyl, or substituted alkyl;
R6 is hydrogen, alkyl, alkoxycarbonyl, or is absent when the nitrogen of NR6
is bonded to a ring double bond;
or pharmaceutically acceptable salt thereof.
Preferred antiprogestin compounds for use with the methods and regimens this
invention are those having the structure:
CA 02372586 2001-11-O1
WO 00/66163 PCT/US00/11642
14
R5
~4 Rg
1
wherein
Rl and Rz are taken together form a ring and together contain -CHZ(CHZ)"CHz- ;
n = 2-3;
R3 is hydrogen;
R4 is hydrogen;
RS is a trisubstituted phenyl ring having the structure,
Y~'~~ Z
I ;
X
X is halogen, OH, -CN, alkyl, alkoxy, thioalkyl, substituted thioalkyl,
S(O)alkyl, S(O)zalkyl, aminoalkyl, substituted aminoalkyl, -NOZ,
perfluoroalkyl, 5 or 6 membered heterocyclic ring containing 1 to 3
heteroatoms, or thioalkoxy;
Y and Z are each, independently, hydrogen, halogen, -CN, -NOZ, alkoxy,
alkyl, or thioalkyl; or
RS is a five or six membered heteroaryl ring containing 1, 2, or 3 heteroatoms
selected
from the group consisting of O, S, and NR6 with said ring carbons being
optionally substituted with one or two substituents independently selected
from the group consisting of hydrogen, halogen, CN, NOz , alkyl, or alkoxy;
R6 is hydrogen, alkyl, alkoxycarbonyl, or is absent when the nitrogen of NR6
is bonded to a ring double bond;
or pharmaceutically acceptable salt thereof.
CA 02372586 2001-11-O1
WO 00/66163 PCT/US00/11642
More preferred compounds of this invention are those having the structure
Rs
1
5 wherein
Ri and RZ are taken together form a ring and together contain -CHZ(CHZ)nCHz- ;
n = 2-3;
R3 is hydrogen;
R4 is hydrogen;
10 RS is a disubstituted phenyl ring having the structure,
X
Y
X is halogen, -CN, or -NOz ;
15 Y is hydrogen, halogen, -CN, -NO2, alkoxy, alkyl, or thioalkyl; or
RS is a five or six membered heteroaryl ring containing a heteroatom selected
from the
group consisting of O, S, and NR6 with said ring carbons being optionally
substituted with one or two substituents independently selected from the group
consisting of hydrogen, halogen, CN, or NOz ;
R6 is hydrogen, or is absent when the nitrogen of NR6 is bonded to a ring
double bond;
or pharmaceutically acceptable salt thereof.
CA 02372586 2001-11-O1
WO 00/66163 PCT/US00/11642
16
The antiprogestin compounds of this invention may contain an asymmetric
carbon atom and some of the compounds of this invention may contain one or
more
asymmetric centers and may thus give rise to optical isomers and
diastereoisomers.
While shown without respect to stereochemistry in Formula 1, the present
invention
includes such optical isomers and diastereoisomers; as well as the racemic and
resolved, enantiomerically pure R and S stereoisomers; as well as other
mixtures of
the R and S stereoisomers and pharmaceutically acceptable salts thereof.
The term "alkyl" is used herein to refer to both straight- and branched-chain
saturated aliphatic hydrocarbon groups having 1 to 6 carbon atoms; "alkenyl"
includes both straight- and branched-chain alkyl group of 2 to 6 carbon atoms
containing at least one carbon-carbon double bond; "alkynyl" group includes
both
straight- and branched-chain alkyl group of 2 to 6 carbon atoms with at least
one
carbon-carbon triple bond.
The terms "substituted alkyl", "substituted alkenyl", and "substituted
alkynyl"
refer to alkyl, alkenyl, and alkynyl as containing one or more substituents
from the
group including halogen, CN, OH, NO2, amino, aryl, heterocyclic, substituted
aryl,
substituted heterocyclic, alkoxy, aryloxy, substituted alkyloxy,
alkylcarbonyl,
alkylcarboxy, alkylamino, arylthio. These substituents may be attached to any
carbon
of alkyl, alkenyl, or alkynyl group provided that the attachment constitutes a
stable
chemical moiety.
The term "aryl" is used herein to refer to an aromatic system of 6 to 14
carbon
atoms, which may be a single ring or multiple aromatic rings fused or linked
together
as such that at least one part of the fused or linked rings forms the
conjugated
aromatic system. Preferred aryl groups include phenyl, naphthyl, biphenyl,
anthryl,
tetrohydronaphthyl, phenanthryl groups.
The term "substituted aryl" refers to aryl substituted by one or more
substituents from the group including halogen, CN, OH, N02, amino, alkyl,
cycloalkyl, alkenyl, alkynyl, alkoxy, aryloxy, substituted alkyloxy,
alkylcarbonyl,
alkylcarboxy, alkylamino, or arylthio.
CA 02372586 2001-11-O1
WO 00/66163 PCT/US00/11642
17
The term "heterocyclic" is used herein to describe a stable 4 to 14 membered
monocyclic or multicyclic heterocyclic ring which is saturated, partially
unsaturated,
or unsaturated, and which consists of carbon atoms and from one to four
heteroatoms
selected from the group including N, O, and S atoms. The N and S atoms may be
oxidized, as an N-oxide, sulfoxide, or sulfone. The heterocyclic ring also
includes
any multicyclic ring in which any of above defined heterocyclic rings is fused
to an
aryl ring. The heterocyclic ring may be attached at any heteroatom or carbon
atom
provided the resultant structure is chemically stable. Such heterocyclic
groups
include, for example, tetrahydrofuran, piperidinyl, piperazinyl, 2-
oxopiperidinyl,
azepinyl, pyrrolidinyl, imidazolyl, pyridyl, pyrazinyl, pyrimidinyl,
pyridazinyl,
oxazolyl, isoxazolyl, morpholinyl, indolyl, quinolinyl, thienyl, furyl,
benzofuranyl,
benzothienyl, thiamorpholinyl, thiamorpholinyl sulfoxide, and isoquinolinyl.
The term "substituted heterocyclic" is used herein to describe a heterocyclic
having one or more substituents selected from the group which includes
halogen, CN,
OH, NOZ, amino, alkyl, substituted alkyl, cycloalkyl, alkenyl, substituted
alkenyl,
alkynyl, alkoxy, aryloxy, substituted alkyloxy, alkylcarbonyl, alkylcarboxy,
alkylamino, or arylthio. The term "thioalkyl" is used herein to refer to the
SR group,
where R is alkyl or substituted alkyl. The term "alkoxy" is used herein to
refer to the
OR group, where R is alkyl or substituted alkyl. The term "aryloxy" is used
herein to
refer to the OR group, where R is aryl or substituted aryl. The term
"alkylcarbonyl" is
used herein to refer to the RCO group, where R is alkyl or substituted alkyl.
The term
"alkylcarboxy" is used herein to refer to the COOR group, where R is alkyl or
substituted alkyl. This term is also referred to as alkoxycarbonyl. The term
"aminoalkyl" refers to both secondary and tertiary amines wherein the alkyl or
substituted alkyl groups may be either same or different and the point of
attachment is
on the nitrogen atom. The term "halogen"is defined as Cl, Br, F, and I.
Pharmaceutically acceptable salts can be formed of these antiprogestin
compounds from organic and inorganic acids, for example, acetic, propionic,
lactic,
citric, tartaric, succinic, fumaric, malefic, malonic, mandelic, malic,
phthalic,
hydrochloric, hydrobromic, phosphoric, nitric; sulfuric, methanesulfonic,
CA 02372586 2001-11-O1
WO 00/66163 PCT/US00/11642
18
napthalenesulfonic, benzenesulfonic, toluenesulfonic, camphorsulfonic, and
similarly
known acceptable acids. Salts may also be formed from inorganic bases,
preferably
alkali metal salts, for example, sodium, lithium, or potassium, and organic
bases,
such as ammonium, mono-, di-, and trimethylammonium, mono-, di- and
triethylammonium, mono-, di- and tripropylammonium (iso and normal), ethyl-
dimethylammonium, benzyldimethylammonium, cyclohexylammonium, benzyl-
ammonium, dibenzylammonium, piperidinium, morpholinium, pyrrolidinium,
piperazinium, 1-methylpiperidinium, 4-ethylmorpholinium, 1-
isopropylpyrrolidinium,
1,4-dimethylpiperazinium, 1-n-butyl piperidinium, 2-methylpiperidinium, 1-
ethyl-2-
methylpiperidinium, mono-, di- and triethanolammonium, ethyl
diethanolammonium,
n-butylmonoethanolammonium, tris(hydroxymethyl)methylammonium, phenylmono-
ethanolammonium, and the like.
The antiprogestin compounds of this invention were be prepared according to
the following schemes from commercially available starting materials or
starting
materials which can be prepared using literature procedures. These schemes
show the
preparation of representative antiprogestin compounds of this invention.
CA 02372586 2001-11-O1
WO 00/66163 PCT/US00/11642
19
Scheme 1
Soa2
OCIz
/ \ \
> ~ -> ~ %s~ ->
CI ~ H
4 5 6
= i
Rz Rz
\ B \
O2 > ~ ~z > / Oz
1
PG pG
9
Ri R~
\ Rz Rz
~Oz ~ SOz
PG H
11
According to Scheme 1, commercially available sulfonyl chloride 4 is
converted via the sulfonamide 5 to the 2,1-benzisothiazoline 2,2-dioxide 6 as
5 described in the literature (Chiarino et al, J Heterocycl. Chem. 23(6), 1645-
9, 1986).
The nitrogen atom of sultam 6 is then protected by a suitable protecting
group, e.g.,
trimethyl silyl ethyl.
The protected sultam 7 next is treated with a strong organo-metallic base
(e.g.,
butyl lithium, lithium diisopropylamide, potassium hexamethyldisilylazide) in
an inert
10 solvent (e.g., THF, diethyl ether) under nitrogen at reduced temperature
(ca. -20°C)
(Kende et al, Synth. Commun. 12, 1, 1982). The resulting di-anion then is
treated with
CA 02372586 2001-11-O1
WO 00/66163 PCT/US00/11642
excess electrophile such as an alkyl halide, preferably the iodide. If R1 and
RZ are to
be joined such as the product contains a spirocycle at position 3, then the
electrophile
should be bifunctional, i.e., a diiodide. Subsequent bromination of the sultam
8
proceeds regioselectively at room temperature with bromine in acetic acid (an
organic
5 co-solvent such as dichloromethane may be added as required) in the presence
of
sodium acetate, to give the aryl bromide 9. Judicious choice of reaction
conditions
may facilitate simultaneous removal of the protecting group at this step.
The bromide 9 then is reacted with a palladium salt (e.g.,
tetrakis(triphenylphoshine)palladium(0)), in a suitable solvent (e.g., THF,
10 dimethoxyethane, ethanol, toluene) under an inert atmosphere (argon,
nitrogen). The
mixture then is treated with an arylboronic acid or arylboronic acid ester and
a base
(sodium carbonate, triethylamine, potassium phosphate) in water or fluoride
source
(cesium fluoride) under anhydrous conditions at elevated temperature to give
the
biphenyl sultam 10. Finally, the protecting group is removed under appropriate
15 conditions and the final product 11 is isolated and purified by standard
means.
If Rl and Rz are different then the intermediate is prepared by reacting the
dianion of 7 with one equivalent of the electrophile Rl-X (X = leaving group,
e.g.,
iodide). The resultant mono-alkylated compound may be then isolated and re-
subjected to the reaction conditions using RZ-X, or alternativel~,~ used in
situ for the
20 second alkylation with RZ-X. Alternatively, if the desired product is to
contain RZ = H,
then the isolated mono-alkylated intermediate is taken though the subsequent
steps.
Br \ R1,.'R2 Ar ~ R~~-_R2
N SOz ~ I / N SOz
PG PG
9 10
Scheme 2
Other methodologies also are available for coupling the aryl group, Ar, to the
sultam platform: for example, reaction of the bromide 9 with an aryl stannane,
aryl
zinc, or aryl magnesium halide in the presence of a palladium or nickel
catalyst
CA 02372586 2001-11-O1
WO 00/66163 PCT/US00/11642
21
(Scheme 2). The required aryl-metallic species are formed via standard
techniques.
Furthermore, the bromide 9 may be converted to an aryl boronic acid via
standard
procedures (treatment with n-butyllithium followed by addition of trimethyl
borate
and subsequent boronic ester hydrolysis) that will then undergo the range of
previously described coupling procedures with a suitable aryl bromide.
The antiprogestational activity of the compounds of this invention was
demonstrated in an in vitro standard pharmacological test procedure which
evaluated
the antiprogestational potency of a representative compound of this invention
by
measuring its effect on PRE-luciferase reporter activity in CV-1 cells co-
transfected
with human PR and PRE-luciferase plasmids. When evaluated in the above-
described
test procedure, the compound of Example 1 had an IC50 of 900 nM. The IC50 is
the
concentration of test compound that gives half maximal decrease in 3 nM
progesterone induced PRE-luciferase activity.
The results obtained in this standard pharmacological test procedure
demonstrate that the compounds of this invention are progestational
antagonists, and
are therefore useful as oral contraceptives (male and female), in hormone
replacement
therapy (particularly when combined with an estrogen), in the treatment of
endometriosis, luteal phase defects, benign breast and prostatic diseases and
prostatic,
breast, ovarian, uterine and endometrial cancers.
The antiprogestin compounds of this invention can be used alone as a sole
therapeutic agent or can be used in combination with other agents, such as
other
estrogens, progestins, or androgens.
The antiprogestational compounds of this invention can be formulated neat or
with a pharmaceutical carrier for administration, the proportion of which is
determined by the solubility and chemical nature of the compound, chosen route
of
administration and standard pharmacological practice. The pharmaceutical
carrier
may be solid or liquid.
A solid carrier can include one or more substances which may also act as
flavoring agents, lubricants, solubilizers, suspending agents, fillers,
glidants,
compression aids, binders or tablet-disintegrating agents; it can also be an
encapsulating material. In powders, the carrier is a finely divided solid that
is in
admixture with the finely divided active ingredient. In tablets, the active
ingredient is
mixed with a carrier having the necessary compression properties in suitable
proportions and compacted in the shape and size desired. The powders and
tablets
CA 02372586 2001-11-O1
WO 00/66163 PCT/US00/11642
22
preferably contain up to 99% of the active ingredient. Suitable solid carriers
include,
for example, calcium phosphate, magnesium stearate, talc, sugars, lactose,
dextrin,
starch, gelatin, cellulose, methyl cellulose, sodium carboxymethyl cellulose,
polyvinylpyrrolidine, low melting waxes and ion exchange resins.
Liquid carriers are used in preparing solutions, suspensions, emulsions,
syrups, elixirs and pressurized compositions. The active ingredient can be
dissolved
or suspended in a pharmaceutically acceptable liquid carrier such as water, an
organic
solvent, a mixture of both or pharmaceutically acceptable oils or fats. The
liquid
carrier can contain other suitable pharmaceutical additives such as
solubilizers,
emulsifiers, buffers, preservatives, sweeteners, flavoring agents, suspending
agents,
thickening agents, colors, viscosity regulators, stabilizers or osmo-
regulators.
Suitable examples of liquid carriers for oral and parenteral administration
include
water (partially containing additives as above, e.g. cellulose derivatives,
preferably
sodium carboxymethyl cellulose solution), alcohols (including monohydric
alcohols
and polyhydric alcohols, e.g. glycols) and their derivatives, lethicins, and
oils (e.g.
fractionated coconut oil and arachis oil). For parenteral administration, the
carrier can
also be an oily ester such as ethyl oleate and isopropyl myristate. Sterile
liquid
carriers are useful in sterile liquid form compositions for parenteral
administration.
The liquid Garner for pressurized compositions can be halogenated hydrocarbon
or
other pharmaceutically acceptable propellant.
Liquid pharmaceutical compositions that are sterile solutions or suspensions
can be utilized by, for example, intramuscular, intraperitoneal or
subcutaneous
injection. Sterile solutions can also be administered intravenously. The
compounds
of this invention can also be administered orally either in liquid or solid
composition
form.
The compounds of this invention may be administered rectally or vaginally in
the form of a conventional suppository. For administration by intranasal or
intrabronchial inhalation or insufflation, the compounds of this invention may
be
formulated into an aqueous or partially aqueous solution, which can then be
utilized in
the form of an aerosol. The compounds of this invention may also be
administered
transdermally through the use of a transdermal patch containing the active
compound
and a carrier that is inert to the active compound, is non toxic to the skin,
and allows
delivery of the agent for systemic absorption into the blood stream via the
skin. The
carrier may take any number of forms such as creams and ointments, pastes,
gels, and
CA 02372586 2001-11-O1
WO 00/66163 PCT/US00/11642
23
occlusive devices. The creams and ointments may be viscous liquid or semisolid
emulsions of either the oil-in-water or water-in-oil type. Pastes comprised of
absorptive powders dispersed in petroleum or hydrophilic petroleum containing
the
active ingredient may also be suitable. A variety of occlusive devices may be
used to
release the active ingredient into the blood stream such as a semipermeable
membrane
covering a reservoir containing the active ingredient with or without a
carrier, or a
matrix containing the active ingredient. Other occlusive devices are known in
the
literature.
The dosage requirements vary with the particular compositions employed, the
route of administration, the severity of the symptoms presented and the
particular
subject being treated. Based on the results obtained in the standard
pharmacological
test procedures, projected daily dosages of active compound would be 0.02
p,g/kg -
750 ~g/kg. Treatment will generally be initiated with small dosages less than
the
optimum dose of the compound. Thereafter the dosage is increased until the
optimum
effect under the circumstances is reached; precise dosages for oral,
parenteral, nasal,
or intrabronchial administration will be determined by the administering
physician
based on experience with the individual subject treated. Preferably, the
pharmaceutical composition is in unit dosage form, e.g. as tablets or
capsules. In such
form, the composition is sub-divided in unit dose containing appropriate
quantities of
the active ingredient; the unit dosage forms can be packaged compositions, for
example, packaged powders, vials, ampoules, pre filled syringes or sachets
containing
liquids. The unit dosage form can be, for example, a capsule or tablet itself,
or it can
be the appropriate number of any such compositions in package form.
The following provides the preparation of a representative compound of this
invention.
Example 1
5-(3-chlorophenyl)s~irof2.l-benzisothiazole-3(1H).1'-cyclohexane] 22-dioxide
To 1,3-dihydro-2,1-benzisothiazoline 2,2-dioxide (Chiarino et al, J
Heterocycl. Chem. 23(6), 1645-9, 1986) (0.74 g, 4.4 mmol) in anhydrous
dichloromethane (minimum amount) at room temperature was added sequentially
N,N-diisopropylethylamine (0.76 mL, 4.4 mmol) and 2-
(trimethylsilyl)ethoxymethyl
chloride (0.77 mL, 4.4 mmol). After 30 min, the reaction was poured into water
(50
CA 02372586 2001-11-O1
WO 00/66163 PCTNS00/11642
24
mL), the layers were separated, and the aqueous phase was extracted with ethyl
acetate (2 x 50 mL). The organic layers were combined, washed with brine (30
mL),
dried over magnesium sulfate, filtered and concentrated in vacuo to give 1, 3-
dihydro-
1-(2'-trimethylsilylethyl)-2,1-benzisothiazoline 2,2-dioxide (1.3 g, 99%) as
an off
white solid. 1H NMR (CDC13, 300MHz) 8 0.02 (s, 9 H), 0.97 (dd, 2 H, J = 8.3,
8.2
Hz), 3.73 (dd, 2 H, J= 8.2, 8.3 Hz), 4.40 (s, 2 H), 5.08 (s, 2 H), 7.05 (d, 1
H, J= 7.4
Hz), 7.07 (dd, 1 H), 7.26 (d, 1 H, J= 7.4 Hz), 7.35 ('t', 1 H, J= 7.6, 7.6
Hz). MS ((+)
APCI m/z 317 [M+NH4]+.
To 1,3-dihydro-1-(2'-trimethylsilylethyl)-2,1-benzisothiazoline 2,2-dioxide
(1.3 g, 4.3 mmol) in anhydrous tetrahydrofuran (13 mL) at room temperature was
added 1,5-diiodopentane (1.29 mL, 8.6 mmol). The mixture was cooled to -78
°C and
lithium bis(trimethylsilyl)amide (1.0 M solution in tetrahydrofuran, 17.3 mL,
17
mmol) was added. After 15 min, the reaction mixture was poured into water (50
mL),
the layers were separated, and the aqueous phase was extracted with ethyl
acetate (3 x
50 mL). The organic layers were combined, washed with brine (30 mL), dried
over
magnesium sulfate, filtered and concentrated in vacuo. Purification by flash
column
chromatography (5% ethyl acetate/hexane) on silica gel gave 1,3-dihydro-3-
spirocyclohexyl-1-(2'-trimethylsilylethyl)-2,1-benzisothiazoline 2,2-dioxide
(0.8 g,
51 %) as an off white solid. 'H NMR (CDC13, 300 MHz) 8 0.00 (~:, 9 H), 0.95
(dd, 2
H, J= 8.3, 8.2 Hz), 1.18-2.36 (m, 10 H), 3.72 (dd, 2 H, J= 8.2, 8.3 Hz), 5.06
(s, 2 H),
7.03 ('t', 1 H, J = 7.8 Hz), 7.06 (dd, 1 H, J = 1, 7.6 Hz), 7.18 (dd, 1 H, J =
1.1, 7.6
Hz), 7.28 (dt, 1 H, J= 1.3, 7.7 Hz). MS (EI) mlz 367 [M]+.
To a stirred solution of 1,3-dihydro-3-spirocyclohexyl-I-(2'-
trimethylsilylethyl)-2,1-benzisothiazoline 2,2-dioxide (0.8 g, 2.2 mmol) in
glacial
acetic acid (5 mL) at room temperature was added dropwise a solution of
bromine
(0.11 mL, 2.2 mmol) in glacial acetic acid (2.2 mL) After stirring for 10 min,
anhydrous sodium acetate (0.18 g, 2.2 mmol) was added and the solution was
concentrated in vacuo. The residue was dissolved in ethyl ether (50 mL) and
washed
sequentially with water (50 mL), aqueous saturated sodium bicarbonate solution
(50
mL), water (50 mL) and brine (30 mL). The organic layer was dried over
magnesium
CA 02372586 2001-11-O1
WO 00/66163 PCTNS00/11642
sulfate, filtered and concentrated in vacuo. Purification by flash column
chromatography (20% ethyl acetate/hexane) on silica gel gave a complex mixture
of
products (0.56 g) with identical TLC characteristics as a white foam. The
mixture
was used without further purification.
5 A solution of the mixture containing 5-bromo-1,3-dihydro-3-spirocyclohexyl-
1-(2'-trimethylsilylethyl)-2,1-benzisothiazoline 2,2-dioxide (0.56 g, 1.25
mmol) and
tetrakis(triphenylphosphine)palladium(0) (100 mg) in toluene (25 mL) was
stirred
under a flow of nitrogen for 25 min. To the solution was added sequentially
solutions
of 3-chlorophenylboronic acid (0.4 g, 2.5 mmol) in absolute ethanol (5 mL) and
10 potassium carbonate (0.35 g, 2.5 mmol) in water (5 mL). The mixture was
heated to
80 °C for 16 h and allowed to cool. The reaction mixture was poured
into aqueous
saturated sodium bicarbonate solution (50 mL) and the layers were separated.
The
aqueous phase was extracted with ethyl acetate (3 x 50 mL). The organic layers
were
combined, washed with water (50 mL) and brine (30 mL) and dried over magnesium
15 sulfate. The solution was filtered, concentrated in vacuo, and the residue
was
purified by flash column chromatography on silica gel (2% ethyl
acetate/toluene) and
then by HPLC to give the title compound (65 mg) as a low melting yellow foam.
HPLC conditions: Zorbax PRO, C18, 10u, 15A, 50 x 250 mm; mobile phase
composition and gradient program, 70% water/ 30% AcCN; flow rate, 100 mL/min;
20 injection volume, 120 mg/3 mL MeOH; detection wavelength, 280 nm, 500 PSI;
temperature, amb. 1H NMR (DMSO-d6, 300 MHz), 8 1.47-2.19 (m, 10 H), 6.87 (d, 1
H , J= 8.2 Hz), 7.38 ('d', 1 H, J= 8.1 Hz), 7.46 ('t', 1 H, J= 7.9, 7.7 Hz),
7.56 (dd, 1
H, J = 1.7, 8.2 Hz), 7.62 ('d', 1 H, J = 7.7 Hz), 7.71, ('d', 1 H, J = 1.7
Hz), 7.75 (bs,
1H), 10.55 (bs, 1 H). MS (EI) m/z 347 [M]+. Anal. Calcd for ClgHIgCINOzS: C,
25 62.15; H, 5.22; N, 4.03. Found: C, 59.84; H, 5.30; N, 3.57.
Example 2 - Biological Activity
The antiprogestational activity of the compound of Example 1 was
demonstrated in a conventional pharmacological test.
CA 02372586 2001-11-O1
WO 00/66163 PCT/US00/11642
26
Reagents
Growth medium: DMEM (BioWhittaker) containing 10% (v/v) fetal
bovine serum (heat inactivated), 0.1 mM MEM non-essential amino acids,
100U/ml penicillin, 100mg/ml streptomycin, and 2 mM GIutaMax (GIBCO,
BRL).
Experimental medium: DMEM (BioWhittaker), phenol red-free,
containing 10% (v/v) charcoal-stripped fetal bovine serum (heat-inactivated),
0.1 mM
MEM non-essential amino acids, 100U/ml penicillin, 100mg/ml streptomycin, and
2
mM GlutaMax (GIBCO, BRL).
Test Procedure
Stock CV-1 cells were maintained in growth medium. Co-transfection
was done using 1.2x10' cells, 5 mg pLEM plasmid with hPR-B inserted at Sphl
and
BamHl sites, 10 mg pGL3 plasmid with two PREs upstream of the luciferase
sequence, and 50 mg sonicated calf thymus DNA as carrier DNA in 250 ml.
Electroporation was carried out at 260 V and 1,000 mF in a Biorad Gene Pulser
II.
After electroporation, cells were resuspended in growth medium and plated in
96-well
plate at 40,000 cells/well in 200 ml. Following overnight incubation, the
medium was
changed to experimental medium. Cells were then treated with reference or test
compounds in experimental medium. Compounds were tested for antiprogestational
activity in the presence of 3 nM progesterone. Twenty-four hours after
treatment, the
medium were discarded, cells were washed three times with D-PBS (GIBCO, BRL).
Fifty ml of cell lysis buffer (Promega, Madison, WI) was added to each well
and the
plates were shaken for 15 min in a Titer Plate Shaker (Lab Line Instrument,
Inc.).
Luciferase activity was measured using luciferase reagents from Promega.
When evaluated in the above-described test procedure, the compound of
Example 1 had an ICsp of 900 nM. The ICSO is the concentration of test
compound
that gives half maximal decrease in 3 nM progesterone induced PRE-luciferase
activity.
CA 02372586 2001-11-O1
WO 00/66163 PCT/US00/11642
27
All publications cited in this specification are incorporated herein by
reference herein. While the invention has been described with reference to a
particularly preferred embodiment, it will be appreciated that modifications
can be
made without departing from the spirit of the invention. Such modifications
are
intended to fall within the scope of the appended claims.