Note: Descriptions are shown in the official language in which they were submitted.
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
INHIBITORS OF THE LECTIN COMPLEMENT PATHWAY (LCP) AND THEIR USE
Field of the Invention
The present invention relates to methods and products for regulating lectin
complement pathway (LCP) associated complement activation. In particular, the
invention
relates to methods for inhibiting LCP associated complement activation by
contacting a
mammalian cell having a mannose binding lectin (MBL) ligand (alternatively
referred to
to herein as an "MBL receptor") with an antagonist of MBL. The invention also
relates to novel
complement inhibitors which are MBL receptor antagonists, such as plant
lectins and
functional equivalents thereof.
Background Of The Invention
The immune system functions to defend the body against pathogenic bacteria,
viruses
and parasites. Immunity against foreign pathogens usually involves the
complement system.
The complement system is a cascade of 18 sequentially activated serum proteins
which
function to recruit and activate other cells of the _ immune system, effect
cytolysis of target
cells and induce opsonization of foreign pathogens. Complement can be
activated by the
2o presence of either antibody/antigen complexes, as in the classical
complement pathway, or
microbial surfaces, as in the alternative complement pathway. Complement
activation can
also occur via the lectin complement pathway (LCP). Lectins are carbohydrate-
binding
proteins that recognize oligosaccharide structures present on cell surfaces,
the extracellular
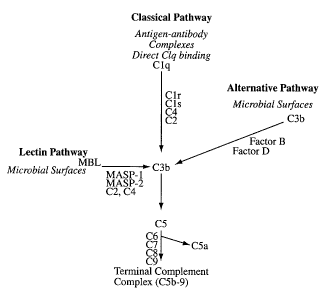
matrix, and secreted glycoproteins. As shown in Figure l, these distinct
activation pathways
ultimately converge at the common enzymatic step of serum protein C3 cleavage
to C3b and
C3a. This in turn initiates the terminal steps of complement function
including the cleavage
of CS to CSb and CSa and subsequent deposition of C5b-C9 onto the target cell
membrane.
The LCP is an antibody-independent cascade that is initiated by binding of
mannan- (or
mannose) binding lectin (MBL) to cell surface carbohydrates on bacteria,
yeasts, parasitic
protozoa, and viruses (Turner MW, "Mannose-binding lectin: The pluripotent
molecule of the
innate immune system", Immunol. Today, 1996;17:532-540). MBL (-~-600 kDa) is a
member
of the collectin protein family and is structurally related to the classical
complement C 1
subcomponent, C 1 q. Associated with MBL are two serine proteases, Mannose
binding lectin
1
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
associated serine protease, MASP-1 and MASP-2, which show striking homology to
the two
C 1 q-associated serine proteases of the classical complement pathway, C 1 r
and C 1 s (Thiel S,
et al., "A second serine protease associated with mannan-binding lectin that
activates
complement", Nature 1997;386:506-510). The selectivity of MBL sugar binding
is: N-
acetyl-D-glucosamine (GIcNAc) > mannose > N-acetylmannosamine and fucose >
maltose >
glucose » galactose and N-acetylgalactosamine (Thiel S, et al., "A second
serine protease
associated with mannan-binding lectin that activates complement", Nature
1997;386:506-
510; Turner MW, "Mannose-binding lectin: The pluripotent molecule of the
innate immune
system", Immunol. Today, 1996;17:532-540). Binding of the MBL/MASP complex to
cell
surface carbohydrates activates the LCP, which in turn activates the classical
complement
pathway independently of C 1 q, C 1 r, C 1 s or antibodies (Fig. 1 ). Most if
not all the
carbohydrate moieties to which MBL binds are not normally expressed by
unperturbed
human tissue.
t 5 Summary Of The Invention
The present invention relates to methods and products for regulating lectin
complement pathway (LCP) associated complement activation. Prior to the
instant invention,
it was known that LCP associated complement activation was a mechanism used by
the body
to recognize and destroy an invading microorganism. LCP activation normally
occurs
2o through the binding of mannan-binding lectin (MBL) and its two associated
serine proteases,
MASP-1 and MASP-2, to carbohydrates on the surface of microorganisms. Once MBL
and
MASP-1 and MASP-2 are localized to the surface of the microorganism,
complement begins
to assemble, ultimately killing the microorganism. These prior art teachings
demonstrate that
MBL is an important cellular component in the process of the eradication of
infectious
25 microorganisms. In fact, MBL deficiencies can result in medical disorders.
A disease known
as MBL deficiency, in which children that are deficient in MBL, renders the
children prone to
the development of infectious diseases.
Recent findings from our laboratory have demonstrated that the mechanism of
complement activation following oxidative stress of human endothelial cells is
initiated by
30 mannose-binding lectin (MBL) deposition to an endothelial cell ligand.
Thus, specific
inhibition of MBL or its counter-ligand on endothelial cells appeared to us to
be an attractive
therapeutic strategy for treating conditions characterized by activation of
the MBL
2
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
complement activation pathway. In view of our findings, we have investigated
the molecular
mechanism of complement activation during oxidative stress of endothelial
cells using novel
therapeutics directed against the endothelial MBL ligand. Thus, the
experiments described
herein were undertaken with the general aim of characterizing the molecular
mechanisms
governing complement activation following oxidative stress of endothelial
cells and to
develop novel, small molecular weight complement inhibitors directed towards
inhibition of
MBL deposition on endothelial cells. To this end, we have identified several
legume-derived
lectins [e.g., Ulex europaeus (UEA)-II and Laburnum alpinum (LAA)-I] that bind
to the
MBL ligand and inhibit complement activation.
to Additionally, we have designed several peptide-based MBL receptor
antagonists that
display molecular mimicry to N-acetylglucosamine (i.e., a specific inhibitor
of MBL), and/or
have similar functions. Some of these peptides were designed by using the
known amino
acid sequence of cytokeratin (K) 14 and 17, which belong to the larger
cytokeratin family
members of which we have identified as likely MBL ligands on stressed
endothelial cells.
Although not wishing to be bound to any particular theory or mechanism, we
believe that
Cytokeratin (K) expression is responsible for complement activation following
oxidative
stress of human endothelial cells. Accordingly, we believe that inhibition of
K with naturally
occurring legume lectins, functional equivalents of these lectins, or other
specific anti-
cytokeratin inhibitors, e.g. antibodies, antibody fragments, binding peptides,
is useful for
2o inhibiting MBL deposition and the resulting complement activation following
oxidative stress
in vitro and in vivo.
Our understanding of the role of cytoskeletal filaments in cellular function
has
significantly advanced in recent years. In addition to providing structural
support, it is now
clear that intermediate filaments play a key role in a variety of cellular
functions, including
cell-cell and cell-extracellular matrix interactions, cell motility, receptor-
ligand interactions,
and receptor internalization (Pavalko and Otey, Proc. Soc. Exxp. Biol. Med,
205, 282-293,
1994 and Fuches and Cleveland, Cell Motif Cytoskelaton, 17, 291-300, 1990).
Although
various intermediate filaments exists in human endothelial cells, their non-
structural roles
have not been fully elucidated. Intermediate filaments were previously
reported to activate
3o the "classical" complement pathway in an antibody independent fashion
(Linder et al.,
Nature, 278, 176-177, 1979; Linder et al., Clin. Imm. and Immunopath., 40, 265-
275, 1986).
Recently, the intermediate filament cytokeratin 1 (CK1) was cloned from a
human
3
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
endothelial cell library and identified as a kininogen binding protein (Joseph
et al., Clin.
Immunol. 92, 246-255, 1999; Joseph et al., Immunopharmacology, 43, 203-210,
1999;
Shariat-Madar et al., J. Biol. Chem. 274, 7137-7145, 1999; Hasan et al., Proc.
Natl. Acad. Sci
USA 95:3615-3620, 1998), suggesting that endothelial cytokeratins may function
as
extracellular binding proteins.
Thus, in one aspect, the present invention is based upon the surprising
discovery that
certain plant lectins (e.g., UEA-II, LAA-1, Cytisus Sessilifolius anti-H(O)
Lectin 1 (CSA-1),
and functional equivalents thereof) have a similar binding profile as MBL with
respect to
recognizing specific carbohydrates or peptides on the surface of mammalian
endothelial cells
and competitively inhibit MBL deposition and subsequent complement activation
of
HUVECs following oxidative stress and thus are MBL receptor antagonists.
Surprisingly, it
has also been discovered that certain plant lectins function as receptor
antagonists of MBL,
thereby inhibiting MBL deposition on the surface of mammalian cells and
inhibiting the
development of diseased or damaged tissue. In another aspect, the invention is
based on the
discovery that antibodies, antibody fragments and other keratin binding
molecules are also
MBL receptor antagonists.
In another aspect, the invention is a method for inhibiting LCP-associated
complement activation. The method includes the step of contacting a mammalian
cell having
a surface exposed MBL ligand with an effective amount of an MBL receptor
antagonist to
2o inhibit cellular MBL deposition and LCP-associated complement activation.
In one
illustrative embodiment, the method is an in vitro screening assay.
In yet another aspect, the invention is a method for inhibiting a cellular
injury
mediated by LCP-associated complement activation. The method includes the step
of
administering to a subject in need thereof an effective amount of an MBL
receptor antagonist
to inhibit LCP-associated complement activation.
In one embodiment of the methods of the invention, the MBL receptor antagonist
is
an isolated molecule that selectively binds to a mannose binding lectin (MBL)
ligand
(alternatively referred to herein as an "MBL receptor") on a mammalian cell
and inhibits
MBL binding thereto. In an illustrative embodiment, the isolated MBL receptor
antagonist is
3o a binding molecule such as a peptide mimetic of the carbohydrate
recognition domain (CRD)
of the plant lectins disclosed herein or their functional variants. In another
embodiment, the
MBL receptor antagonist is a keratin binding molecule.
4
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
It is believed that the cellular injury mediated by LCP-associated complement
activation contributes to the development of injured tissue associated with a
variety of
disorders. In one embodiment, the cellular injury is associated with
atherosclerosis. In
another embodiment, the cellular injury is associated with arthritis,
myocardial infarction,
ischemia and reperfusion, transplantation, CPB, stroke, ARDS, SLE, Lupus, or
dialysis.
The MBL receptor antagonist may be administered to the subject by any route
known
in the art. When the cellular injury is associated with the pulmonary system,
the MBL
receptor antagonist may be administered to the subject by an aerosol route of
delivery. When
the cellular injury is due to ischemia or reperfusion, the MBL receptor
antagonist may be
to locally administered to the heart or arteries that have been subjected to
ischemia or
reperfusion conditions.
According to another aspect of the invention, an MBL receptor antagonist is
provided.
The MBL receptor antagonist is an isolated molecule that selectively binds to
a human MBL
receptor and inhibits LCP-associated complement activation. Although not
wishing to be
bound to a particular theory or mechanism, it is believed that the MBL
receptor antagonists of
the invention competitively inhibit MBL binding to its receptor (also referred
to herein as its
ligand), thereby. inhibiting LCP-associated complement activation.
According to still another aspect of the invention, a method for optimizing a
selected
MBL receptor antagonist for inhibiting LCP-associated complement activation is
provided.
The method involves identifying molecular mimics of the naturally-occurring
peptides and
functional equivalents disclosed herein which bind to a human MBL receptor and
inhibit
LCP-associated complement activation. Such molecular mimics can be identified,
for
example, by generating a library of closely related compounds and screening
the library for
compounds which possess the functional characteristics of the MBL receptor
antagonists
disclosed herein. (See, Gold, L. et al. (1995) Ahn. Rev. Biochem. 64:763-797
which describes
the selection of compounds from a combinatorial library using SELEX
technology).
According to yet another aspect, the invention is a composition of an MBL
receptor
antagonist, wherein the MBL receptor antagonist is an isolated binding
molecule that
selectively binds to a human MBL receptor and that inhibits LCP-associated
complement
3o activation. In an illustrative embodiment the composition is a
pharmaceutical composition
including an effective amount of the isolated MBL receptor antagonist for
treating an MBL
5
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
mediated disorder and a pharmaceutically acceptable carrier. In certain
embodiments, the
compositions also include one or more drugs for the treatment of an MBL
mediated disorder.
A method for screening a subject for susceptibility to treatment with MBL
receptor
antagonist is provided in another aspect of the invention. The method includes
the steps of
contacting a mammalian cell from a subject with a labeled isolated MBL
receptor antagonist,
and detecting the presence of an MBL on the surface of the mammalian cell,
wherein the
presence of the MBL indicates that the cell is susceptible to LCP-associated
complement
activation and that the subject is susceptible to treatment with an MBL
receptor antagonist.
In one embodiment, the mammalian cell is an endothelial cell; in yet other
embodiments, the
1 o mammalian cell is an epithelial cell.
Each of the limitations of the invention can encompass various embodiments of
the
invention. It is, therefore, anticipated that each of the limitations of the
invention involving
any one element or combinations of elements can be included in each aspect of
the invention.
Brief Description Of The Drawings
This application may include drawings which illustrate various aspects of the
invention; however, the drawings are not required for enablement of the
claimed invention.
Figure 1 is a schematic depicting the antigen/antibody-dependent classical
complement pathway and the antibody-independent alternative and lectin
complement
2o pathways. All three pathways merge at C3 and lead to the formation of the
terminal
complement complex (CSb-9).
Figure 2 is lectin complement pathway activation on keratin-coated plates.
3F8,
GIcNAc, or GLUPEP, but not 1 C 10 (non-functional anti-MBL mAb) inhibited C3
deposition.
P<0.05 compared to A. N=3.
Figure 3 is an MBL deposition to BSA-coupled GLUPEP. MBL deposit was
increased on GLUPEP coupled to BSA (Vehicle) compared to BSA only. GIcNAc (100
mmol/L), 3F8 (1 mg/ml) or GLUPEP (50 ug/ml) attenuated/inhibited MBL
deposition to
GLUPEP coupled to BSA. Data are the means of 4 wells from one experiment.
Figure 4 is LAA-I inhibiting C3 deposition on HUVECs following oxidative
stress.
3o Similar to observations made with UEA-II, this legume lectin decreased C3
deposition in a
dose-dependent manner. N=3.
Figure 5 is a graph depicting UEA-II inhibition of C3 deposition on keratin.
6
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
Figure 6 is graph depicting GLUPEP inhibition of MBL binding to GIcNAc-BSA.
Detailed Description Of The Invention
The invention relates to methods and products for regulating and manipulating
lectin
complement pathway (LCP)-associated complement activation. As discussed above,
the
invention is based on the finding that LCP-associated complement activation
plays a role in
complement induced cellular injury of mammalian cells and that MBL receptor
antagonists
inhibit LCP-associated complement activation. It was discovered according to
an aspect of
the invention that MBL interacts with carbohydrates or peptides on the surface
of mammalian
~ o cells in vitro and in vivo and that certain plant lectins and functional
equivalents thereof
(disclosed herein) function as MBL receptor antagonists. It was also
discovered that keratin
binding molecules function as MBL receptor antagonists. Accordingly, the MBL
receptor
antagonists of the invention inhibit the accumulation of surface associated
MBL which leads
to the accumulation of complement on the surface of the cell, thereby
inhibiting cell injury or
~ 5 death. According to the prior art, LCP-associated complement activation
was predominantly
associated with infectious microorganisms, suggesting that MBL deposition
should be
promoted in order to enhance the killing of infectious microorganisms. It was
discovered,
according to the invention, that in mammals it is preferable to block MBL
cellular association
and that such blockage could be achieved using the MBL receptor antagonists
disclosed
2o herein. The LCP is not necessary for eradication of infectious
microorganisms in adult
mammals, and it is believed that it contributes to cellular injury associated
with several types
of disorders, such as atherosclerosis, arthritis, myocardial infarction,
ischemia and
reperfusion injury, transplantation, CPB, stroke, ARDS, SLE, Lupus, and
dialysis.
In one aspect, the invention is a method for inhibiting LCP-associated
complement
25 activation. The method includes the steps of contacting a mammalian cell of
a subject having
surface exposed MBL ligand (alternatively referred to herein as an "MBL
receptor") with an
effective amount of an MBL receptor antagonist to inhibit LCP-associated
complement
activation.
A "subject" as used herein includes humans, non-human primates, dogs, cats,
horses,
3o sheep, goats, cows, rabbits, pigs and rodents.
A "human MBL ligand" or "human MBL receptor" as used herein is a receptor
expressed on a mammalian cell which when contacted with an MBL activates LCP-
7
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
associated complement activation. An MBL ligand is any cell surface molecule
capable of
interacting with MBL. One example of an MBL ligand is a cytokeratin.
Cytokeratins, such
as cytokeratin 1 (CK1), are intermediate filaments found on the cell surface.
The methods of the invention are useful for inhibiting LCP-associated
complement
activation on the surface of a mammalian cell having surface exposed MBL
ligand
(carbohydrate or peptide groups) recognized by MBL. The mammalian cell may be
any cell
in which the cell surface carbohydrates or peptides interact with MBL. In
certain illustrative
embodiments, the mammalian cell is an endothelial cell or an epithelial cell
having a surface
exposed MBL ligand. For instance, vascular endothelial cells have been shown
in subjects
1 o that have sustained ischemic/reperfusion injury to express an MBL ligand.
Mammalian cells
having MBL ligands can easily be identified. For instance, an MBL binding
assay (e.g., such
as those described below) can be used to identify MBL ligands.
The method for inhibiting LCP-associated complement activation may be used for
a
variety of in vitro and in vivo purposes. The method may be used, for
instance, as an in vitro
~ s screening assay. The in vitro screening assay may be used to identify
compounds which
function as an MBL receptor antagonist, such as the assay described above, to
identify
mammalian cells having surface exposed MBL ligands, or to detect
susceptibility of a subject
to treatment with an MBL receptor antagonist. In order to screen a subject for
susceptibility
to treatment with an MBL receptor antagonist, a cell is isolated from the
subject and the
2o presence of MBL or the ability of MBL to bind to the surface is detected.
If MBL is present
on the surface of a cell or is able to bind to the surface of a cell, then the
cell is susceptible to
LCP-associated complement activation. If this is the case, then the subject is
susceptible to
treatment with an MBL receptor antagonist.
The methods of the invention are also useful in vivo when it is desirable to
inhibit
2s MBL deposition on a mammalian cell surface. For instance, the methods of
the invention are
useful for treating an MBL mediated disorder. The MBL receptor antagonists can
be used
alone as a primary therapy or in combination with other therapeutics as an
adjuvant therapy
to enhance the therapeutic benefits of other medical treatments.
The mammalian cell is contacted with an MBL receptor antagonist. The step of
30 "contacting" as used herein refers to the addition of the MBL receptor
antagonist to a
medium containing a mammalian cell. The medium may be an in vitro tissue
culture or a
biological specimen, an ex vivo sample, or in vivo. The step of contacting
refers to the
8
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
addition of the MBL receptor antagonist in such a manner that it will prevent
LCP-associated
complement activation associated with the mammalian cell.
An "MBL mediated disorder" as used herein is a disorder which involves
cellular
injury caused by LCP-associated complement activation. MBL disorders include,
for
instance, atherosclerosis, arthritis, myocardial infarction, ischemia and
reperfusion injury,
transplantation, CPB, stroke, ARDS, (Systemic lupus erythematosus) SLE, Lupus,
and
dialysis. Each of these disorders is well-known in the art and is described,
for instance, in
Harrison's Principles of Internal Medicine (McGraw Hill, Inc., New York),
which is
incorporated by reference.
1 o Atherosclerosis and myocardial infarction can lead to ischemia-reperfusion
(I/R)
injury. One of the underlying mechanisms for I/R-induced injury is the hypoxic
and
reoxygenated environments created in affected tissues. Fluctuations in oxygen
content as
observed in these instances can create oxygen free radicals which have been
reported to,
among other things, modulate endothelial cell surface profile.
Thus, in one aspect, the invention involves a method for treating or
preventing
myocardial infarction in a subject. "Myocardial infarction" is a focus of
necrosis resulting
from inadequate perfusion of the cardiac tissue. Myocardial infarction
generally occurs with
the abrupt decrease in coronary blood flow that follows a thrombotic occlusion
of a coronary
artery previously narrowed by atherosclerosis. Generally, infarction occurs
when an
2o atherosclerotic plaque fissures, ruptures, or ulcerates, and a mural
thrombus forms leading to
coronary artery occlusion.
A number of laboratory tests, well known in the art for diagnosis of
myocardial
infarction, are described, for example, in Harrison's: Principles of Internal
Medicine
(McGraw Hill, Inc., New York). Generally, the tests may be divided into four
main
categories: (1) nonspecific indexes of tissue necrosis and inflammation, (2)
electrocardiograms, (3) serum enzyme changes (e.g., creatine phosphokinase
levels), and (4)
cardiac imaging. A person of ordinary skill in the art could easily apply any
of the foregoing
tests to determine when a subject is at risk, is suffering, or has suffered, a
myocardial
infarction. A positively identified subject would thus benefit from a method
of treatment of
3o the invention.
According to the invention, the method involves administering to a subject
having or
at risk of having a myocardial infarction an MBL receptor antagonist in an
amount effective
9
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
to inhibit cardiac tissue necrosis in the subject. It is believed that
immediate administration
of an MBL receptor antagonist would greatly benefit the subject by inhibiting
complement
activation and associated tissue damage prior to, at the same time as, or
following the infarct.
In one embodiment, when MBL receptor antagonists are used in the treatment of
diseases (e.g., myocardial infarction), a growth factor may be co-
administered. In some
embodiments, Insulin-like Growth Factor-1 (IGF-1) is the growth factor of
choice.
The co-administered growth factor or other medicament for the treatment of the
MBL
mediated disorder can act cooperatively, additively or synergistically with
the MBL receptor
antagonist of the invention to inhibit complement activation.
to Optionally, in other embodiments of the invention for treating myocardial
infarction,
an isolated MBL receptor antagonist of the invention is administered to a
subject in need of
such treatment in combination with a method for treating an arteriosclerotic
condition. An
arteriosclerotic condition, as used herein, is a term of art that refers to
classical
atherosclerosis, accelerated atherosclerosis, atherosclerotic lesions and
other physiological
conditions characterized by undesirable vascular smooth muscle cell
proliferation. See, e.g.,
Harrisons, Principles of Internal Medicine (McGraw Hill, Inc., New York) for a
more
detailed description of these conditions. The method for treating an
arteriosclerotic condition
may be a surgical method, an agent for treating restenosis, a method involving
a drug therapy
(e.g., gene therapy) or a combination of the foregoing.
Surgical methods for treating an arteriosclerotic condition include procedures
such as
bypass surgery, atherectomy, laser procedures, ultrasonic procedures, and
balloon
angioplasty. In a preferred embodiment of the invention, the isolated MBL
receptor
antagonist is administered to a subject in combination with a balloon
angioplasty procedure.
A balloon angioplasty procedure involves inserting a catheter having a
deflated balloon into
an artery. The deflated balloon is positioned in proximity to the
atherosclerotic plaque and is
inflated such that the plaque is compressed against the vascular wall. As a
result, the balloon
surface is in contact layer of vascular endothelial cells on the surface of
the vessel. The
isolated MBL receptor antagonist may be attached to the balloon angioplasty
catheter in a
manner which permits release of the isolated MBL receptor antagonist at the
site of the
3o atherosclerotic plaque. The isolated MBL receptor antagonist may be
attached to the balloon
angioplasty catheter in accordance with standard procedures known in the art.
For example,
the isolated MBL receptor antagonist may be stored in a compartment of the
balloon
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
angioplasty catheter until the balloon is inflated, at which point it is
released into the local
environment. Alteratively, the isolated MBL receptor antagonist may be
impregnated on the
balloon surface, such that it contacts the cells of the arterial wall as the
balloon is inflated.
The MBL receptor antagonist also may be delivered in a perforated balloon
catheter such as
those disclosed in Flugelman, et al., Circulation, v. 85, p. 1110-1117 (1992).
See, also, e.g.,
published PCT Patent Application WO 95/23161, for an exemplary procedure for
attaching a
therapeutic protein to a balloon angioplasty catheter. This procedure can be
modified using
no more that routine experimentation to attach a therapeutic nucleic acid or
polypeptide to the
balloon angioplasty catheter.
Additionally, the MBL receptor antagonist may be co-administered with a
medicament for the treatment of the MBL mediated disease, e.g., anti-
atherosclerotic agent
for treating or preventing clinically significant atherosclerosis. The term
"co-administered,"
means administered substantially simultaneously with another agent. By
substantially
simultaneously, it is meant that an MBL receptor antagonist of the invention
is administered
to the subject close enough in time with the administration of the other agent
(e.g., an anti-
atherosclerotic agent, growth factor, etc.).
Preferred anti-atherosclerotic agents used in combination with the MBL
receptor
antagonist of the invention, include but are not limited to, the following
drugs: HMG-CoA
reductase inhibitors, diuretics, antiadrenergic agents, vasodilators, calcium
channel
antagonists, angiotensin-converting enzyme (ACE) inhibitors, angiotensin II
antagonists, and
clot dissolvers.
"HMG-CoA reductase (3-hydroxy-3-methylglutaryl-coenzyme A)" is the microsomal
enzyme that catalyzes the rate limiting reaction in cholesterol biosynthesis
(HMG-CoA6Mevalonate). An "HMG-CoA reductase inhibitor" inhibits HMG-CoA
2s reductase, and therefore inhibits the synthesis of cholesterol. There is a
large number of
compounds described in the art that have been obtained naturally or
synthetically, which have
been seen to inhibit HMG-CoA reductase, and which form the category of agents
useful for
practicing the present invention. Traditionally these agents have been used to
treat
individuals with hypercholesterolemia. Examples include some which are
commercially
3o available, such as simvastatin (U.S. Patent No. 4, 444,784), lovastatin
(U.S. Patent No.
4,231,938), pravastatin sodium (U.S. Patent No. 4,346,227), fluvastatin (U.S.
Patent No.
4,739,073), atorvastatin (U.S. Patent No. 5,273,995), cerivastatin, and
numerous others
11
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
described in U.S. Patent No. 5,622,985, U.S. Patent No. 5,135,935, U.S. Patent
No.
5,356,896, U.S. Patent No. 4,920,109, U.S. Patent No. 5,286,895, U.S. Patent
No. 5,262,435,
U.S. Patent No. 5,260,332, U.S. Patent No. 5,317,031, U.S. Patent No.
5,283,256, U.S. Patent
No. 5,256,689, U.S. Patent No. 5,182,298, U.S. Patent No. 5,369,125, U.S.
Patent No.
5,302,604, U.S. Patent No. 5,166,171, U.S. Patent No. 5,202,327, U.S. Patent
No. 5,276,021,
U.S. Patent No. 5,196,440, U.S. Patent No. 5,091,386, U.S. Patent No.
5,091,378, U.S. Patent
No. 4,904,646, U.S. Patent No. 5,385,932, U.S. Patent No. 5,250,435, U.S.
Patent No.
5,132,312, U.S. Patent No. 5,130,306, U.S. Patent No. 5,116,870, U.S. Patent
No. 5,112,857,
U.S. Patent No. 5,102,911, U.S. Patent No. 5,098,931, U.S. Patent No.
5,081,136, U.S. Patent
~o No. 5,025,000, U.S. Patent No. 5,021,453, U.S. Patent No. 5,017,716, U.S.
Patent No.
5,001,144, U.S. Patent No. 5,001,128, U.S. Patent No. 4,997,837, U.S. Patent
No. 4,996,234,
U.S. Patent No. 4,994,494, U.S. Patent No. 4,992,429, U.S. Patent No.
4,970,231, U.S. Patent
No. 4,968,693, U.S. Patent No. 4,963,538, U.S. Patent No. 4,957,940, U.S.
Patent No.
4,950,675, U.S. Patent No. 4,946,864, U.S. Patent No. 4,946,860, U.S. Patent
No. 4,940,800,
U.S. Patent No. 4,940,727, U.S. Patent No. 4,939,143, U.S. Patent No.
4,929,620, U.S. Patent
No. 4,923,861, U.S. Patent No. 4,906,657, U.S. Patent No. 4,906,624 and U.S.
Patent No.
4,897,402, the disclosures of which patents are incorporated herein by
reference.
Diuretics include thiazides, e.g., hydrochlorothiazide; loop acting diuretics,
e.g.,
furosemide; potassium-sparing, e.g., spironolactone, triamterene, and
amiloride.
2o Antiadrenergic agents include clonidine; guanabenz; guanfacine; methyldopa;
trimethapajn; Rauwolfia alkaloids, e.g., reserpine; guanethidine; guanadrel;
phentolamine;
phenoxybenzamine; prazosin; terazosin; propranolol; metoprolol; nadolol;
atenolol; timolol;
timdolol; acebutolol; and labetalol.
Vasodilators include hydralazine; minoxidil; diazoxide; and nitroprusside.
Calcium channel antagonists include nisadipine; diltiazen; and verapamil.
Angiotensin II antagonists are compounds which interfere with the activity of
angiotensin II by binding to angiotensin II receptors and interfering with its
activity.
Angiotensin II antagonists are well known and include peptide compounds and
non-peptide
compounds. Most angiotensin II antagonists are slightly modified congeners in
which
3o agonist activity is attenuated by replacement of phenylalanine in position
8 with some other
amino acid; stability can be enhanced by other replacements that slow
degeneration in vivo.
Examples of angiotensin II antagonists include: peptidic compounds (e.g.,
saralasin,
12
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
[(San~)(Vals)(Ala$)] angiotensin -(1-8) octapeptide and related analogs); N-
substituted
imidazole-2-one (US Patent Number 5,087,634); imidazole acetate derivatives
including 2-N-
butyl-4-chloro-1-(2-chlorobenzile) imidazole-5-acetic acid (see Long et al.,
J. Pharmacol.
Exp. Ther. 247(1), 1-7 (1988)); 4, 5, 6, 7-tetrahydro-1H-imidazo [4, 5-c]
pyridine-6-
carboxylic acid and analog derivatives (US Patent Number 4,816,463); N2-
tetrazole beta-
glucuronide analogs (US Patent Number 5,085,992); substituted pyrroles,
pyrazoles, and
tryazoles (US Patent Number 5,081,127); phenol and heterocyclic derivatives
such as 1, 3-
imidazoles (US Patent Number 5,073,566); imidazo-fused 7-member ring
heterocycles (US
Patent Number 5,064,825); peptides (e.g., US Patent Number 4,772,684);
antibodies to
1o angiotensin II (e.g., US Patent Number 4,302,386); and aralkyl imidazole
compounds such as
biphenyl-methyl substituted imidazoles (e.g., EP Number 253,310, January 20,
1988);
ES8891 (N-morpholinoacetyl-(-1-naphthyl)-L-alanyl-(4, thiazolyl)-L-alanyl (35,
45)-4-
amino-3-hydroxy-5-cyclo-hexapentanoyl-N-hexylamide, Sankyo Company, Ltd.,
Tokyo,
Japan); SKF108566 (E-alpha-2-[2-butyl-1-(carboxy phenyl) methyl] 1H-imidazole-
5-
yl[methylane]-2-thiophenepropanoic acid, Smith Kline Beecham Pharmaceuticals,
PA);
Losartan (DUP753/MK954, DuPont Merck Pharmaceutical Company); Remikirin (R042-
5892, F. Hoffman LaRoche AG); AZ agonists (Marion Merrill Dow) and certain non-
peptide
heterocycles (G.D.Searle and Company).
ACE, is an enzyme which catalyzes the conversion of angiotensin I to
angiotensin II.
2o ACE inhibitors include amino acids and derivatives thereof, peptides,
including di- and tri-
peptides and antibodies to ACE which intervene in the renin-angiotensin system
by inhibiting
the activity of ACE, thereby reducing or eliminating the formation of pressor
substance
angiotensin II. ACE inhibitors have been used medically to treat hypertension,
congestive
heart failure, myocardial infarction and renal disease. Classes of compounds
known to be
useful as ACE inhibitors include acylmercapto and mercaptoalkanoyl prolines
such as
captopril (U.S. Patent Number 4,105,776) and zofenopril (U.S. Patent Number
4,316,906),
carboxyalkyl dipeptides such as enalapril (U.S. Patent Number 4,374,829),
lisinopril (U.S.
Patent Number 4,374,829), quinapril (U.S. Patent Number 4,344,949), ramipril
(U.S. Patent
Number 4,587,258), and perindopril (U.S. Patent Number 4,508,729),
carboxyalkyl dipeptide
3o mimics such as cilazapril (U.S. Patent Number 4,512,924) and benazapril
(U.S. Patent
Number 4,410,520), phosphinylalkanoyl prolines such as fosinopril (U.S. Patent
Number
4,337,201) and trandolopril.
13
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
Renin inhibitors are compounds which interfere with the activity of renin.
Renin
inhibitors include amino acids and derivatives thereof, peptides and
derivatives thereof, and
antibodies to renin. Examples of renin inhibitors that are the subject of
United States patents
are as follows: urea derivatives of peptides (U.5. Patent Number 5,116,835);
amino acids
connected by nonpeptide bonds (U.5. Patent Number 5,114,937); di- and tri-
peptide
derivatives (U.5. Patent Number 5,106,835); amino acids and derivatives
thereof (U.5. Patent
Numbers 5,104,869 and 5,095,119); diol sulfonamides and sulfinyls (U.5. Patent
Number
5,098,924); modified peptides (U.S. Patent Number 5,095,006); peptidyl beta-
aminoacyl
aminodiol carbamates (U.S. Patent Number 5,089,471); pyrolimidazolones (U.5.
Patent
1o Number 5,075,451); fluorine and chlorine statine or statone containing
peptides (U.5. Patent
Number 5,066,643); peptidyl amino diols (U.S. Patent Numbers 5,063,208 and
4,845,079);
N-morpholino derivatives (U.S. Patent Number 5,055,466); pepstatin derivatives
(U.5. Patent
Number 4,980,283); N-heterocyclic alcohols (U.5. Patent Number 4,885,292);
monoclonal
antibodies to renin (U.S. Patent Number 4,780,401); and a variety of other
peptides and
analogs thereof (U.5. Patent Numbers 5,071,837, 5,064,965, 5,063,207,
5,036,054,
5,036,053, 5,034,512, and 4,894,437).
The invention also is useful for treating cellular injury arising from
ischemia/reperfusion, e.g., associated with atherosclerosis and/or cardio-
vascular remodeling.
Injury to the vascular system can lead to a number of undesirable health
conditions,
2o including, for example, forms of atherosclerosis and arteriosclerosis that
are associated with
unwanted vascular smooth muscle cell proliferation. A common injury to the
vascular
system occurs as a side effect of a medical procedure for treating ischemic
heart disease.
Ischemia refers to a lack of oxygen due to inadequate perfusion of blood.
Ischemic
heart disease is characterized by a disturbance in cardiac function due to an
inadequate supply
0
z5 of oxygen to the heart. The most common form of this disease involves a
reduction in the
lumen of coronary arteries, which limits coronary blood-flow. Under these
conditions the
carbohydrate or peptide residues of the cell surface become exposed, the cells
present a
microbial carbohydrate (e.g., foam cell chlamydia), or an MBL ligand is
synthesized,
allowing MBL to associate with the cell surface and initiate the LCP
associated complement
30 activation.
When ischemic heart disease becomes very serious, then management must be
invasive. Until recently, ischemic heart disease was treated by coronary-
artery, bypass
14
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
surgery. Less invasive procedures, however, now have been developed. These
procedures
involve the use of catheters introduced into the narrowed region of the blood
vessel ("the
stenosis") for mechanically disrupting, laser ablating or dilating the
stenosis.
An "ischemic disease or condition" as used herein refers to a condition
characterized
by local inflammation resulting from an interruption in the blood supply to a
tissue due to a
blockage or hemorrhage of the blood vessel responsible for supplying blood to
the tissue such
as is seen for myocardial or cerebral infarction. A cerebral ischemic attack
or cerebral
ischemia is a form of ischemic condition in which the blood supply to the
brain is blocked.
This interruption in the blood supply to the brain may result from a variety
of causes,
1o including an intrinsic blockage or occlusion of the blood vessel itself, a
remotely originated
source of occlusion, decreased perfusion pressure or increased blood viscosity
resulting in
inadequate cerebral blood flow, or a ruptured blood vessel in the subarachnoid
space or
intracerebral tissue.
The methods of the invention are useful also for treating cerebral ischemia.
Cerebral
ischemia may result in either transient or permanent deficits and the
seriousness of the
neurological damage in a patient who has experienced cerebral ischemia depends
on the
intensity and duration of the ischemic event. A transient ischemic attack is
one in which the
blood flow to the brain is interrupted only briefly and causes temporary
neurological deficits,
which often are clear in less than 24 hours. Symptoms of TIA include numbness
or weakness
of face or limbs, loss of the ability to speak clearly andlor to understand
the speech of others,
a loss of vision or dimness of vision, and a feeling of dizziness. Permanent
cerebral ischemic
attacks, also called stroke, are caused by a longer interruption in blood flow
to the brain
resulting from either a thromboembolism or hemorrhage. A stroke causes a loss
of neurons
typically resulting in a neurologic deficit that may improve but that does not
entirely resolve.
Thromboembolic stroke is due to the occlusion of an extracranial or
intracranial blood vessel
by a thrombus or embolus. Because it is often difficult to discern whether a
stroke is caused
by a thrombosis or an embolism, the term "thromboembolism" is used to cover
strokes
caused by either of these mechanisms. The term thromboembolism will be used
throughout
this patent application to describe thrombotic and embolic strokes.
Hemorrhagic stroke is
3o caused by the rupture of a blood vessel in a subarachnoid space or
intracerebral tissue.
The methods of the invention in some embodiments are directed to the treatment
of
acute thromboembolic stroke. An acute stroke is a medical syndrome involving
neurological
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
injury resulting from an ischemic event, which is an interruption in the blood
supply to the
brain. Acute stroke may be thromboembolic or hemorrhagic.
An effective amount of an MBL receptor antagonist alone or in combination with
another therapeutic for the treatment of stroke is that amount sufficient to
reduce in vivo brain
injury resulting from the stroke. A reduction of brain injury is any
prevention of injury to the
brain which otherwise would have occurred in a subject experiencing a
thromboembolic
stroke absent the treatment of the invention. Several physiological parameters
may be used
to assess reduction of brain injury, including smaller infarct size, improved
regional cerebral
blood flow, and decreased intracranial pressure, for example, as compared to
pretreatment
to patient parameters, untreated stroke patients or stroke patients treated
with thrombolytic
agents alone.
The pharmaceutical preparation of the MBL receptor antagonist also may be used
alone or in combination with a therapeutic agent for treating an ischemic
disease or condition.
Therapeutics for treating ischemic diseases or conditions are described in
medical textbooks
~ 5 such as Harrisons, Principles of Internal Medicine (McGraw Hill, Inc., New
York ). The
particular therapeutic used depends on the nature of the disease or condition.
Examples of
therapeutics useful in the treatment of ischemic diseases or conditions
include anticoagulation
agents, antiplatelet agents, and thrombolytic agents.
Anticoagulation agents prevent the coagulation of blood components and thus
prevent
20, clot formation. Anticoagulants include, but are not limited to, heparin,
warfarin, coumadin,
dicumarol, phenprocoumon, acenocoumarol, ethyl biscoumacetate, and indandione
derivatives.
Antiplatelet agents inhibit platelet aggregation and are often used to prevent
thromboembolic stroke in patients who have experienced a transient ischemic
attack or
25 stroke. Antiplatelet agents include, but are not limited to, aspirin,
thienopyridine derivatives
such as ticlopodine and clopidogrel, dipyridamole and sulfinpyrazone, as well
as RGD
mimetics and also antithrombin agents such as, but not limited to, hirudin.
Thrombolytic agents lyse clots which cause the thromboembolic stroke.
Thrombolytic agents have been used in the treatment of acute venous
thromboembolism and
3o pulmonary emboli and are well known in the art (e.g. see Hennekens et al,
JAm Coll Cardiol;
v. 25 (7 supp), p. 18S-22S (1995); Holmes, et al, JAm Coll Cardiol; v.25 (7
supply, p. 10S-
16
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
17S(1995)). Thrombolytic agents include, but are not limited to, plasminogen,
az-
antiplasmin, streptokinase, antistreplase, tissue plasminogen activator (tPA),
and urokinase.
In a preferred embodiment of the invention tPA is the thrombolytic agent. The
mature tPA polypeptide has 527 amino acids, at least 17 (Asn) of which have
been shown to
be linked with carbohydrate structures. Spellman et al., have identified
several of these
carbohydrates, including a high-mannose structure on amino acid 117, and di-
tri-and tetra-
antennary N-acetyllactosamine-type structures on amino acids 184 and 448 ~J.
Biol. Chem.
264(24) 14100-14111 (1989)].
"tPA" as used herein includes native tPA and recombinant tPA, as well as
modified
1o forms of tPA that retain the enzymatic or fibrinolytic activities of native
tPA. The enzymatic
activity of tPA can be measured by assessing the ability of the molecule to
convert
plasminogen to plasmin. The fibrinolytic activity of tPA may be determined by
any in vitro
clot lysis activity known in the art, such as the purified clot lysis assay
described by Carlson,
et. al., Anal. Biochem. 168, 428-435 (1988) and its modified form described by
Bennett, W.
F. Et al., 1991, Supra, the entire contents of which are hereby incorporated
by reference.
Recombinant tPA has been described extensively in the prior art. Several forms
of
recombinant tPA are commercially available such as ACTIVASE ~.
Modified forms of tPA ("modified tPA") have been characterized and are known
to
those skilled in the art. Modified tPA includes, but is not limited to,
variants having deleted
or substituted amino acids or domains, variants conjugated to other molecules,
and variants
having modified glycosylation. Several preferred modified tPAs have been
described in PCT
Publication No. W093/24635; EP 352,119; EP382174; and Suzuki et al., J.
Cardiovasc.
Pharmacal. 22, 834-840 (1993). Each of these references is hereby incorporated
by
reference.
Briefly, PCT Publication No. W093/24635 discloses tPA variants having an extra
glycosylation site at any of the amino acid positions 103-105 and the native
glycosylation site
removed at position 117 of the native human tPA. The amino acid number refers
to the
amino acid in that position of the mature, wild-type tPA polypeptide as
disclosed in US Pat.
No. 4,766,075. These variants have extended circulatory half lives and exhibit
substantially
3o the same or improved fibrin binding affinity and fibrinolytic potency as
compared to wild-
type human tPA. The disclosed variants may also include at least one amino
acid substituted
in the 296-299 position with alanine and/or a substitution of the amino acids
at positions 274-
17
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
277 of wild type tPA (phenylalanine, arginine, isoleucine, lysine) with
leucine, histidine,
serine, and threonine, respectively. One particularly effective type of
variant disclosed in the
reference is a triple mutant variant of wild type tPA. The first mutation in a
triple mutant is
the addition of one glycosylation site at least one of the amino acid
positions 103-105 by e.g.,
substituting the native amino acid sequence 103 with an asparagine as part of
an Asn-X-Ser
or Asn-X-Thr tripeptidyl sequence, wherein X is any amino acid except proline.
The second
mutation involves the removal of a glycosylation site at native amino acid
site 117 (Asn) and
replacing it with another amino acid, preferably glutamine. The third mutation
is the
replacement of native amino acids 296-302 with other amino acids. The most
effective of the
1o triple mutant variants is the specific molecule, T103N, N117Q, KHRR (296-
299) AAAA tPA
(TNK tPA).
EP 352,119 discloses Vampire Bat tPA's (Bat-Pa (H), (I), and (L)). Vampire bat-
Pa's
are variants of native tPA having a variety of sequence modifications.
Although the Bat-Pa
variants are structurally distinct from tPA because they lack the Kringle 2
domain and
plasmin-sensitive processing site, these variants are functionally similar to
native tPA. They
are however, more potent than native tPA.
Suzuki et al., J. Cardiovasc. Pharmacal. 22, 834-840 (1993) disclose tPA
variants in
which a cysteine at position 84 of the growth factor domain of native tPA is
replaced by
serine (C84S tPA). Although this variant retains the functional activity of
native tPA, it has
been shown to have a longer in vivo half life than native tPA.
The MBL receptor antagonists of the invention are directed to a specific
epitope (e.g.,
present on endothelial cells) that mediates complement activation.
Accordingly, the
development of MBL receptor antagonists which inhibit MBL binding to this
complement
activating epitope on cells offers several advantages over inhibiting a
specific complement
component. First, selective inhibition of an endothelium epitope requires
small amounts of
MBL receptor antagonist to be given versus inhibiting a specific circulating
complement
component (e.g., most complement components circulate at concentrations in the
range of 50
pg/ml of plasma and higher). Second, specific inhibition of
ischemia/reperfusion induced
complement activation at the epitope allows complement to be activated at
distal sites for
other host immune responses. Third, inhibition of a specific complement
component may
compromise the protective aspects of complement activation in other situations
(e.g.,
opsonization). Thus, in view of the above-noted factors, selective. inhibition
of complement
18
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
at the site of complement activation is believed to be advantageous. See,
Mulligan MS,
Warner RL, Rittershaus CW, Thomas LJ, Ryan US, Foreman KE, Crouch LD, Till GO,
Ward
PA: Endothelial targeting and enhanced anti-inflammatory effects of complement
inhibitors
possessing sialyl Lewis moieties. J. Immunol. 1999;162:4952-4959, which
reports that
coupling of sCRl with sialyl Lewis x provides site-directed inhibition of
complement
activation and tissue protection following ischemia/reperfusion injury by
directing sCRI to
selectin ligands upregulated during ischemia/reperfusion injury.
The MBL receptor antagonists of the invention may be administered alone or in
combination with other therapeutic treatments. For instance, the MBL receptor
antagonist
1o may be delivered with a medicament for the treatment of an MBL-mediated
disorder.
The MBL receptor may be administered alone or may be delivered in a mixture
with
other medicaments, such as those disclosed herein and others known in the art.
In some
embodiments, a common administration vehicle (e.g., pill, tablet, implant,
injectable solution,
etc.) would contain both the MBL receptor antagonist useful in this invention
and the
therapeutic. Thus, the present invention also provides pharmaceutical
compositions, for
medical use, which comprise the MBL receptor antagonist of the invention
together with one
or more pharmaceutically acceptable carriers thereof and optionally other
therapeutic
ingredients.
An "MBL receptor antagonist" as used herein is a compound that prevents LCP-
2o associated complement activation by inhibiting MBL binding to an MBL ligand
(alternatively
referred to herein as an "MBL receptor") expressed on the surface of a
mammalian cell. The
ability of an MBL receptor antagonist to block MBL deposition can be detected
using routine
in vitro binding assays, such as those described in the Examples as well as
the following
assay.
MBL deposition can be measured by ELISA on normoxic HUVECs and HUVECs
subjected to 24 hr of hypoxia followed by 3 hr of reoxygenation in the
presence of 30%
human serum (HS) or 30% HS treated with 3, 30, or 300 mmol/L of N-acetyl-D-
glucosamine
(GIcNAc) or with the putative MBL receptor antagonist to inhibit competitively
MBL
deposition.
3o C3 and MBL specific cell surface ELISAs can be performed using peroxidase-
conjugated polyclonal goat anti-human C3 antibody (Cappel, West Chester, PA)
and
monoclonal anti-human MBL antibody (Biodesign, Kennebunk, ME, clone #131-1),
19
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
respectively. HUVECs are grown to confluence on 0.1% gelatinized 96-well
plastic plates
(Corning Costar, Cambridge, MA). The plates are then subjected to 0 (normoxia)
or 24 hr of
hypoxia. Hypoxic stress is maintained using a humidified sealed chamber (Coy
Laboratory
Products, Inc., Grass Lake, MI) at 37 °C gassed with 1% Oz, 5% COZ,
balance NZ (Collard
CD, et al., "Reoxygenation of hypoxic human umbilical vein endothelial cells
activates the
classical complement pathway", Circulation, 1997;96:326-333). Following the
specified
period of normoxia or hypoxia, the cell media are aspirated and 100 ~.1 of one
the following is
added to each well: 1) 30% HS, 2) Hank's balanced salt solution, 3) 30% HS +
3, 30, or 300
mmol/L GIcNAc, 4) 30% HS + 3, 30, or 300 mmol/L D-mannose, 5) 30% HS + 3, 30,
300
1o mmol/L L-mannose, 6) 30% MBL-depleted HS 7) 30% MBL-depleted HS + 0.6 ~g/ml
MBL
or 8) 30% HS + 3, 30, or 300 mmol/L putative MBL binding peptide.
Additionally, 100
g.1 of purified MBL (3-300 ng/ml) is added to select wells to form a standard
curve for
quantitative analysis of MBL deposition. The cells are then reoxygenated for 3
hr at 37°C in
95% air and 5% COZ. The cells are washed and then fixed with 1%
paraformaldehyde
~ 5 (Sigma Chemical Co., St. Louis, MO) for 30 min. The cells are then washed
and incubated at
4 °C for 1.5 hr with 50 p.1 of peroxidase-conjugated polyclonal goat
anti-human C3 antibody
(1:1000 dilution) or monoclonal anti-human MBL antibody (1:1000 dilution). The
MBL
ELISA plates are then washed and incubated for 1 hr at 4 °C with 50 ~,l
of peroxidase-
conjugated polyclonal goat anti-mouse IgG secondary antibody (1:1000
dilution). After
2o washing the cells, the plates are developed with 50 p,1 of ABTS (2,2'-azino-
bis(3-
ethylbenzthiazoline-6-sulfonic acid)), and read (Molecular Devices, Sunnyvale,
CA) at 405
nm. Background controls for the C3 ELISA consist of cells to which only the
anti-human C3
antibody is added (i.e., no HS) or cells incubated with 30% heat-inactivated
HS. Background
controls for the MBL ELISA consist of cells to which only secondary antibody
and an
25 isotype control monoclonal antibody to porcine CSa are added. Background
optical density is
subtracted from all groups. All ELISA experiments are performed 3 times using
6 wells per
experimental group. Endothelial C3 and MBL deposition on normoxic vs. hypoxic
HUVECs
is analyzed by two-way analysis of variance (ANOVA).
The MBL receptor antagonist prevents LCP-associated complement activation.
30 Whether a particular compound can inhibit LCP-associated complement
activation can also
be assessed using routine in vitro screening assays. For instance, a
complement hemolytic
assay (CHSO) can be performed on MBL-depleted HS in order to demonstrate that
MBL
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
depletion does not inhibit classical complement activation. The assay may be
performed,
however, using MBL containing HS and adding an MBL receptor antagonist and/or
a control
peptide to demonstrate specificity of the MBL inhibition.
In one illustrative embodiment, the MBL receptor antagonist is an isolated MBL
binding peptide. An "isolated MBL receptor antagonist" as used herein is a
peptide which
binds to an MBL ligand ("MBL receptor") and inhibits LCP associated complement
activation. It is believed that the MBL receptor antagonists inhibit LCP
associated
complement activation by binding to the MBL ligand (receptor) and inhibiting
MBL
association with surface exposed MBL ligands.
to The preferred compositions of the invention include an MBL receptor
antagonist
which is an isolated binding molecule that selectively binds to a human MBL
receptor and
that inhibits LCP-associated complement activation. The MBL receptor
antagonists of the
invention can be identified using routine assays, such as the binding and LCP
complement
activation assays described above and elsewhere throughout this patent
application.
The molecules of the invention are isolated molecules, e.g. isolated peptides.
As used
herein, with respect to molecules, the term "isolated molecules" means that
the peptides are
substantially pure and are essentially free of other substances with which
they may be found
in nature or in vivo systems to an extent practical and appropriate for their
intended use. In
particular, the molecules are sufficiently pure and are sufficiently free from
other biological
2o constituents of their hosts cells so as to be useful in, for example,
producing pharmaceutical
preparations or sequencing. Because an isolated molecule of the invention may
be admixed
with a pharmaceutically acceptable carrier in a pharmaceutical preparation,
the molecule may
comprise only a small percentage by weight of the preparation. The molecule is
nonetheless
substantially pure in that it has been substantially separated from the
substances with which it
2s may be associated in living systems. The term isolated refers to molecules
which are either
naturally occurring or synthetic. Thus, in some embodiments the isolated
molecules are
derived from natural sources.
In other embodiments the isolated molecules are synthetic MBL receptor
antagonists
may easily be synthesized or produced by recombinant means by those of skill
in the art.
3o Methods for preparing or identifying molecules which bind to a particular
target are well
known in the art. Molecular imprinting, for instance, may be used for the de
novo
construction of macromolecular structures such as peptides which bind to a
particular
21
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
molecule. See for example Kenneth J. Shea, Molecular Imprinting of Synthetic
Network
Polymers: The De Novo synthesis of Macromolecular Binding and Catalytic Sites,
TRIP
Vol. 2, No. 5, May 1994; Klaus Mosbach, Molecular Imprinting, Trends in
Biochem. Sci.,
19(9) January 1994; and Wulff, G., in Polymeric Reagents and Catalysts (Ford,
W. T., Ed.)
ACS Symposium Series No. 308, pp 186-230, American Chemical Society (1986).
One
method for preparing mimics of MBL receptor antagonists involves the steps of:
(i) polymerization of functional monomers around a known MBL receptor
antagonist that
exhibits a desired activity; (ii) removal of the template molecule; and then
(iii) polymerization of a second class of monomers in the void left by the
template, to provide
a new molecule which exhibits one or more desired properties which are similar
to that of the
template. In addition to preparing peptides in this manner other MBL receptor
antagonists
such as polysaccharides, nucleosides, drugs, nucleoproteins, lipoproteins,
carbohydrates,
glycoproteins, steroids, lipids, and other biologically active materials can
also be prepared.
This method is useful for designing a wide variety of biological mimics that
are more stable
t 5 than their natural counterparts, because they are typically prepared by
the free radical
polymerization of functional monomers, resulting in a compound with a
nonbiodegradable
backbone. Other methods for designing such molecules include for example drug
design
based on structure activity relationships which require the synthesis and
evaluation of a
number of compounds and molecular modeling.
2o Molecules which bind to the MBL receptor may also be identified by
conventional
screening methods such as phage display procedures (e.g., methods described in
Hart, et al.,
J. Biol. Chem. 269:12468 (1994)). Hart et al. report a filamentous phage
display library for
identifying novel peptide ligands for mammalian cell receptors. In general,
phage display
libraries using, e.g., M13 or fd phage, are prepared using conventional
procedures such as
25 those described in the foregoing reference. The libraries display inserts
containing from 4 to
80 amino acid residues. The inserts optionally represent a completely
degenerate or a biased
array of peptides. Receptor antagonists that bind selectively to MBL receptor
are obtained by
selecting those phage which express on their surface a peptide that binds to
the MBL
receptor. These phage then are subjected to several cycles of reselection to
identify the
3o peptide ligand-expressing phage that have the most useful binding
characteristics. Typically,
phage that exhibit the best binding characteristics (e.g., highest affinity)
are further
characterized by nucleic acid analysis to identify the particular amino acid
sequences of the
22
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
peptides expressed on the phage surface and the optimum length of the
expressed peptide to
achieve optimum binding to the MBL receptor. Alternatively, such peptide
ligands can be
selected from combinatorial libraries of peptides containing one or more amino
acids. Such
libraries can further be synthesized which contain non-peptide synthetic
moieties which are
less subject to enzymatic degradation compared to their naturally-occurring
counterparts.
Thus, according to another aspect of the invention, a method for optimizing a
selected
MBL receptor antagonist for its ability to bind to a MBL receptor and/or
inhibit LCP-
associated complement activation is provided. "Optimizing" as used herein
refers to an
iterative process of introducing changes to an existing system or compound and
evaluating
the functional significance of each change, followed by selecting the
resulting system or
compound associated with a functional outcome that is most improved; these
steps are
repeated until a desired endpoint is achieved or it appears further changes
will not improve
the functional outcome. The same objective can be achieved in a parallel
manner by
generating a library of closely related compounds and screening the library
for the compound
or compounds possessing the most favorable embodiment of the characteristic
being
optimized. In this particular instance, optimizing a selected MBL receptor
antagonist for
MBL receptor binding activity involves testing a panel of structurally related
MBL receptor
antagonists for their ability to bind to MBL receptor. The screening method
involves
contacting at least one candidate optimized MBL receptor antagonist selected
from a group
of candidate optimized MBL receptor antagonists with an MBL receptor under
conditions
which, in the absence of a competitor, permit a reference MBL receptor
antagonist to bind or
remain bound to the MBL receptor. The candidate optimized MBL receptor
antagonist is
contacted with the MBL receptor before, after, or simultaneously with contact
between the
labeled reference MBL receptor antagonist and the MBL receptor. The residual
binding of
the labeled reference MBL receptor antagonist to the MBL receptor is then
detected.
Detection of a decrease in binding of the reference MBL receptor antagonist
indicates that the
candidate optimized MBL receptor antagonist interferes with the binding of the
reference
MBL receptor antagonist to the MBL receptor. Candidate optimized MBL receptor
antagonist can be generated as members of a combinatorial library of
compounds, for
3o example using SELEX technology. Gold L et al. (1995) Annu Rev Biochem
64:763:797.
This assay can involve the separation of both unbound unlabeled candidate
optimized
MBL receptor antagonists and unbound labeled reference MBL receptor
antagonists from the
23
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
sample. The separation step can be accomplished in any way known in the art,
in a manner
similar to the separation method described above. Likewise, the detection of
the remaining
bound labeled reference MBL receptor antagonist can be accomplished in any way
known in
the art, in a manner similar to the detection method described above.
The screening assay can also be performed as a competition between labeled
candidate optimized MBL receptor antagonists and unlabeled reference MBL
receptor
antagonists for the MBL receptor. In this format, binding of the labeled
optimized MBL
receptor antagonist to the MBL receptor is then detected. Detection of bound
optimized
MBL receptor antagonist indicates that the candidate optimized MBL receptor
antagonist
to interferes with the binding of the reference MBL receptor antagonist to the
MBL receptor.
The screening assay can also be performed by contacting labeled MBL receptor
to
immobilized MBL receptor antagonist. In this format a panel of candidate
optimized MBL
receptor antagonists can be presented in an array fashion on a silicon chip or
in a plastic
multiwell microtiter or microarray plate. Alternatively, each candidate
optimized MBL
receptor antagonist can be separately coupled to a bead, a resin, a
nitrocellulose filter, a slide,
or a biomolecular interaction analysis (BIA) chip. After contacting the MBL
receptor with
the immobilized candidate MBL receptor antagonists and, if indicated, washing
away
unbound MBL receptor, detection of complexes formed between the immobilized
MBL
receptor antagonist and the MBL receptor provides the basis for selecting
particular MBL
receptor antagonists as optimized. Exemplary molecular mimics of MBL which
bind to an
MBL receptor and/or inhibit LCP associated complement activation are provided
in Example
7 (See, e.g., Table 1).
The above-described and other methods disclosed herein also can be used to
identify
molecular mimics of GIcNAc which bind to MBL. For example, the peptide
referred to
herein as "GLUPEP," potently inhibits iC3b deposition on HUVECS following
oxidative
stress in a dose-related manner. Accordingly, the screening and optimization
methods
disclosed herein can be used to identify molecular mimics of GLUPEP which bind
to MBL
and/or inhibit LCP associated complement activation. Exemplary molecular
mimics of
GIcNAc are provided in Example 1 (see, e.g., Table 2).
3o To determine whether a putative MBL receptor antagonist binds to an MBL
receptor,
any known binding assay may be employed. For example, the antagonist may be
immobilized on a surface and then contacted with a labeled MBL receptor. The
amount of
24
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
MBL receptor which interacts with the antagonist or the amount which does not
bind to the
antagonist may then be quantitated to determine whether the antagonist binds
to the MBL
receptor.
Screening of molecules of the invention, also can be carried out utilizing a
competition assay. If the molecule being tested competes with MBL for binding
to an MBL
receptor, as shown by a decrease in binding of the MBL to the receptor, then
it is likely that
the molecule and the MBL bind to the same, or a closely related, epitope on
the MBL
receptor. Using routine procedures known to those of ordinary skill in the
art, one can
determine whether a molecule which binds to MBL receptor is useful according
to the
to invention by determining whether the molecule is one which blocks MBL from
binding to an
MBL receptor. Such assays are described above and in the Examples section.
Other assays
will be apparent to those of skill in the art, having read the present
specification, which are
useful for determining whether a molecule which binds to MBL receptor also
inhibits LCP
associated complement activation.
Activation assays also can be used to assess the relative inhibitory
concentrations of a
molecule in an activation assay and to identify those molecules which inhibit
activation by at
least, e.g., 75%.
Other assays will be apparent to those of skill in the art, having read the
present
specification, which are useful for determining whether an MBL receptor
antagonist which
binds to an MBL receptor also inhibits MBL activation.
The procedures for the identification of a human MBL receptor expressed on
endothelial cells are described in the Examples. The same strategy can be used
to identify
MBL receptors expressed in human and other cell types using no more than
routine
experimentation.
As discussed above the MBL receptor antagonists of the present invention
encompass
in some embodiments MBL receptor binding which include an MBL receptor binding
region
which specifically binds to a human MBL ligand and inhibits LCP associated
complement
activation, e.g., by preventing MBL from interacting with MBL ligands.
In certain embodiments, the MBL receptor antagonists are peptides which are
derived
3o from plant lectins. The MBL receptor antagonist is a legume derived lectin
or a functional
equivalent thereof that binds to the MBL ligand and that inhibits complement
activation. A
legume derived lectin is an isolated peptide (naturally occurring or
synthetic) derived from a
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
legume. The legume derived lectin in some embodiments is Ulex europaeus (UEA)-
II,
Laburnum alpinum (LAA)-I, or Cytisus Sessilifolius anti-H(O) Lectin 1 (CSA-1).
A
functional equivalent of a legume derived lectin is a molecule, peptide or non-
peptide that has
an equivalent function to the legume derived lectins, such as a molecule
having conservative
substitutions.
As used herein, "conservative substitution" refers to an amino acid
substitution which
does not alter the relative charge or size characteristics of the peptide in
which the amino acid
substitution is made. Conservative substitutions of amino acids include
substitutions made
amongst amino acids with the following groups: (1) M,I,L,V; (2) F,Y,W; (3)
K,R,H; (4) A,G;
to (5) S,T; (6) Q,N; and, (7) E,D. Such substitutions can be made by a variety
of methods
known to one of ordinary skill in the art to known MBL receptor antagonists of
the invention
to define novel peptide antagonists of the invention. For example, amino-acid
substitutions
may be made by PCR-directed mutation, site-directed mutagenesis according to
the method
of Kunkel (Kunkel, Proc. Nat. Acad. Sci. U.S.A. 82: 488-492, 1985), or by
chemical synthesis
t 5 of a gene encoding the CDR3 region. These and other methods are known to
those of
ordinary skill in the art and may be found in references which compile such
methods, e.g.
Sambrook. et al., Molecular Cloning: A Laboratory Manual, 2nd edition, Cold
Spring Harbor
Laboratory Press, 1989. The activity of functionally equivalent variants of
the MBL receptor
antagonists of the invention can be tested by the binding and activity assays
discussed above.
2o As used herein, a "vector" may be any of a number of nucleic acids into
which a
desired sequence may be inserted by restriction and ligation for transport
between different
genetic environments or for expression in a host cell. Vectors are typically
composed of
DNA although RNA vectors are also available. Vectors include, but are not
limited to,
plasmids and phagemids. A cloning vector is one which is able to replicate in
a host cell, and
25 which is further characterized by one or more endonuclease restriction
sites at which the
vector may be cut in a determinable fashion and into which a desired DNA
sequence may be
ligated such that the new recombinant vector retains its ability to replicate
in the host cell. In
the case of plasmids, replication of the desired sequence may occur many times
as the
plasmid increases in copy number within the host bacterium or just a single
time per host
3o before the host reproduces by mitosis. In the case of phage, replication
may occur actively
during a lytic phase or passively during a lysogenic phase. An expression
vector is one into
which a desired DNA sequence may be inserted by restriction and ligation such
that it is
26
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
operably joined to regulatory sequences and may be expressed as an RNA
transcript. Vectors
may further contain one or more marker sequences suitable for use in the
identification of
cells which have or have not been transformed or transfected with the vector.
Markers
include, for example, genes encoding proteins which increase or decrease
either resistance or
sensitivity to antibiotics or other compounds, genes which encode enzymes
whose activities
are detectable by standard assays known in the art (e.g., 13-galactosidase or
alkaline
phosphatase), and genes which visibly affect the phenotype of transformed or
transfected
cells, hosts, colonies or plaques. Preferred vectors are those capable of
autonomous
replication and expression of the structural gene products present in the DNA
segments to
which they are operably joined.
The expression vectors of the present invention include regulatory sequences
operably
joined to a nucleotide sequence encoding one of the peptides of the invention.
As used
herein, the term "regulatory sequences" means nucleotide sequences which are
necessary for
or conducive to the transcription of a nucleotide sequence which encodes a
desired peptide
and/or which are necessary for or conducive to the translation of the
resulting transcript into
the desired peptide. Regulatory sequences include, but are not limited to, 5'
sequences such
as operators, promoters and ribosome binding sequences, and 3' sequences such
as
polyadenylation signals. The vectors of the invention may optionally include
5' leader or
signal sequences, 5' or 3' sequences encoding fusion products to aid in
protein purification,
2o and various markers which aid in the identification or selection of
transformants. The choice
and design of an appropriate vector is within the ability and discretion of
one of ordinary skill
in the art. The subsequent purification of the peptides may be accomplished by
any of a
variety of standard means known in the art.
A preferred vector for screening peptides, but not necessarily preferred for
the mass
production of the peptides of the invention, is a recombinant DNA molecule
containing a
nucleotide sequence that codes for and is capable of expressing a fusion
peptide containing,
in the direction of amino- to carboxy-terminus, ( 1 ) a prokaryotic secretion
signal domain, (2)
a peptide of the invention, and, optionally, (3) a fusion protein domain. The
vector includes
DNA regulatory sequences for expressing the fusion peptide, preferably
prokaryotic
regulatory sequences. Such vectors can be constructed by those with skill in
the art and have
been described by Smith et al. (Science 228:1315-1317, 1985), Clackson et al.
(Nature
352:624-628, 1991); Kang et al. (in "Methods: A Companion to Methods in
Enzymology:
27
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
Vol. 2", R.A. Lerner and D.R. Burton, ed. Academic Press, NY, pp 111-
118,1991); Barbas et
al. (Proc. Natl. Acad. Sci. (USA) 88:7978-7982, 1991), Roberts et al. (Proc.
Natl. Acad. Sci.
(USA) 89:2429-2433, 1992)
A fusion peptide may be useful for purification of the peptides of the
invention. The
fusion domain may, for example, include a poly-His tail which allows for
purification on Ni+
columns or the maltose binding protein of the commercially available vector
pMAL (New
England BioLabs, Beverly, MA). A currently preferred, but by no means
necessary, fusion
domain is a filamentous phage membrane anchor. This domain is particularly
useful for
screening phage display libraries of monoclonal antibodies but may be of less
utility for the
to mass production of antibodies. The filamentous phage membrane anchor is
preferably a
domain of the cpIII or cpVIII coat protein capable of associating with the
matrix of a
filamentous phage particle, thereby incorporating the fusion peptide onto the
phage surface,
to enable solid phase binding to specific antigens or epitopes and thereby
allow enrichment
and selection of the binding peptides or fragments encoded by the phagemid
vector.
The secretion signal is a leader peptide domain of a protein that targets the
protein
membrane of the host cell, such as the periplasmic membrane of gram negative
bacteria. A
preferred secretion signal for E. coli is a pelB secretion signal. The
predicted amino acid
residue sequences of the secretion signal domain from two pelB gene producing
variants
from Erwinia carotova are described in Lei, et al. (Nature 381:543-546, 1988).
The leader
2o sequence of the pelB protein has previously been used as a secretion signal
for fusion
proteins (Better, et al., Sciehce 240:1041-1043, 1988; Sastry, et al., Proc.
Natl. Acad. Sci
(USA) 86:5728-5732, 1989; and Mullinax, et al., Proc. Natl. Acad. Sci. (USA)
87:8095-8099,
1990). Amino acid residue sequences for other secretion signal peptide domains
from E. coli
useful in this invention can be found in Oliver, In Neidhard, F.C. (ed.),
Escherichia coli and
Salmonella Typhimurium, American Society for Microbiology, Washington, D.C.,
1:56-69
(1987).
To achieve high levels of gene expression in E. coli, it is necessary to use
not only
strong promoters to generate large quantities of mRNA, but also ribosome
binding sites to
ensure that the mRNA is efficiently translated. In E. coli, the ribosome
binding site includes
3o an initiation codon (AUG) and a sequence 3-9 nucleotides long located 3-11
nucleotides
upstream from the initiation codon (Shine, et al., Nature 254:34, 1975). The
sequence,
AGGAGGU, which is called the Shine-Dalgarno (SD) sequence, is complementary to
the 3'
2s
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
end of E. coli 16S rRNA. Binding of the ribosome to mRNA and the sequence at
the 3' end
of the mRNA can be affected by several factors:
(i) The degree of complementarity between the SD sequence and 3' end of the
16S rRNA.
(ii) The spacing and possibly the DNA sequence lying between the SD sequence
and the AUG (Roberts, et al., Proc. Natl. Acad. Sci. (USA) 76:760.,1979a:
Roberts, et al., Proc. Natl. Acad. Sci. (USA) 76:5596, 1979b; Guarente, et
al.,
Science 209:1428, 1980; and Guarente, et al., Cell 20:543, 1980).
Optimization is achieved by measuring the level of expression of genes in
plasmids in which this spacing is systematically altered. Comparison of
different mRNAs shows that there are statistically preferred sequences from
positions -20 to +13 (where the A of the AUG is position 0) (Gold, et al.,
Annu. Rev. Microbiol. 35:365, 1981). Leader sequences have been shown to
influence translation dramatically (Roberts, et al., 1979a, b supra).
(iii) The nucleotide sequence following the AUG, which affects ribosome
binding
(Taniguchi, et al., J. Mol. Biol_, 118:533, 1978).
The 3' regulatory sequences define at least one termination (stop) codon in
frame with and
operably joined to the heterologous fusion peptide.
In preferred embodiments with a prokaryotic expression host, the vector
utilized
2o includes a prokaryotic origin of replication or replicon, i.e., a DNA
sequence having the
ability to direct autonomous replication and maintenance of the recombinant
DNA molecule
extra-chromosomally in a prokaryotic host cell, such as a bacterial host cell,
transformed
therewith. Such origins of replication are well known in the art. Preferred
origins of
replication are those that are efficient in the host organism. A preferred
host cell is E. coli.
For use of a vector in E coli, a preferred origin of replication is ColEl
found in pBR322 and
a variety of other common plasmids. Also preferred is the plSA origin of
replication found
on pACYC and its derivatives. The ColEl and plSA replicons have been
extensively utilized
in molecular biology, are available on a variety of plasmids and are described
by Sambrook.
et al., Molecular Cloning: A Laboratory Manual, 2nd edition, Cold Spring
Harbor Laboratory
3o Press, 1989).
In addition, those embodiments that include a prokaryotic replicon preferably
also
include a gene whose expression confers a selective advantage, such as drug
resistance, to a
29
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
bacterial host transformed therewith. Typical bacterial drug resistance genes
are those that
confer resistance to ampicillin, tetracycline, neomycin/kanamycin or
chloramphenicol.
Vectors typically also contain convenient restriction sites for insertion of
translatable DNA
sequences. Exemplary vectors are the plasmids pUC 18 and pUC 19 and derived
vectors such
as pcDNAII available from Invitrogen, (San Diego, CA).
The receptor antagonist peptides of the present invention may also, of course,
be
produced by eukaryotic cells such as CHO cells, human hybridomas, immortalized
B-
lymphoblastoid cells, and the like. In this case, a vector is constructed in
which eukaryotic
regulatory sequences are operably joined to the nucleotide sequences encoding
the peptide.
l0 The design and selection of an appropriate eukaryotic vector is within the
ability and
discretion of one of ordinary skill in the art. The subsequent purification of
the peptides may
be accomplished by any of a variety of standard means known in the art.
In another embodiment, the present invention provides host cells, both
prokaryotic
and eukaryotic, transformed or transfected with, and therefore including, the
vectors of the
present invention.
As used herein, a coding sequence and regulatory sequences are said to be
"operably
joined" when they are covalently linked in such a way as to place the
expression or
transcription of the coding sequence under the influence or control of the
regulatory
sequences. If it is desired that the coding sequences be translated into a
functional peptide,
two DNA sequences are said to be operably joined if induction of a promoter in
the 5'
regulatory sequences results in the transcription of the coding sequence and
if the nature of
the linkage between the two DNA sequences does not (1) result in the
introduction of a
frame-shift mutation, (2) interfere with the ability of the promoter region to
direct the
transcription of the coding sequences, or (3) interfere with the ability of
the corresponding
RNA transcript to be translated into a protein. Thus, a promoter region would
be operably
joined to a coding sequence if the promoter region were capable of effecting
transcription of
that DNA sequence such that the resulting transcript might be translated into
the desired
peptide.
The precise nature of the regulatory sequences needed for gene expression may
vary
3o between species or cell types, but shall in general include, as necessary,
5' non-transcribing
and 5' non-translating sequences involved with initiation of transcription and
translation
respectively, such as a TATA box, capping sequence, CART sequence, and the
like.
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
Especially, such 5' non-transcribing regulatory sequences will include a
promoter region
which includes a promoter sequence for transcriptional control of the operably
joined gene.
Regulatory sequences may also include enhancer sequences or upstream activator
sequences,
as desired.
In other embodiments the MBL receptor antagonist is a keratin binding
molecule. . A
keratin binding molecules may be isolated from natural sources or synthesized
or produced
by recombinant means. Methods for preparing or identifying molecules which
bind to a
particular target are well-known in the art and are described above. Binding
peptides, such as
antibodies, may easily be prepared by generating antibodies to keratin (or
obtained from
t o commercial sources) or by screening libraries to identify peptides or
other compounds which
bind to the keratin.
Many keratin antibodies are commercially available. These include but are not
limited to those antibodies commercially available from Research Diagnostics,
Inc., e.g.,
RDI-CYTOKIabr (Cytokeratin 1 rabbit polyclonal); RDI- (mouse cytokeratins
rabbit
polyclonals); RDI-CBL222 (Cytokeratin 1-3 AE8 + mIgGl); RDI-PR061808
(Cytokeratin
1,10/11 K8.60 + mIgGl); RDI-PR065177 (Cytokeratin 2E Ks 2.398.3.1 mIgGl); RDI-
PR065191 (Cytokeratin 2E Ks2.342.7.1 + mIgGl); RDI-CBL218 (Cytokeratin 3
(bovine &
rabbit) AES mIgGl); RDI-PR010525 (Cytokeratin 4 (most mammals) 6B10 mIgGl);
RDI-
CBL234 (Cytokeratin 4,5,6,8,10,13,18 C11 mIgG); RDI-CBL232 (Cytokeratin 5,8
C50 +
mIgGl); RDI-PR010521 (Cytokeratin 5+8 RCK102 + mIgGl); RDI-PR061531
(Cytokeratin 5+8 RCK12+ Biotin conj); RDI-PR061431 (Cytokeratin 5+8 RCK102+
FITC
conj); RDI-PR061031 (Cytokeratin 5+8(pan epithelial) C22 mIgGl); RDI-PR065190
(Cytokeratin 6 Ks6.Kal2+ mIgGl); RDI-CBL194 (Cytokeratin 7 LPSK mIgG2b); RDI-
CBL 184 (Cytokeratin 7 Lds68 IgM); RDI-PR061025 (Cytokeratin 7 (bovine, sheep,
pig)
KS7.18+ mIgGl); RDI-PR010522 (Cytokeratin 7 (hamster; mouse) RCK105 mIgGl);
RDI-
CBL195 (Cytokeratin 8 LP3K mIgGl); RDI-CBL195FT (Cytokeratin 8 LP3K mIgGl
FITC);
RDI-PR061038 (Cytokeratin 8 (mouse, rat, pig, hamster) Ks8.7+* mIgGl); RDI-
PR065130
(Cytokeratin 8(phosphorylated) Ks 8-17.2 mIgGl); RDI-CBL170 (Cytokeratin 8,18
5D3 +
mIgG2a); RDI-PR010526 (Cytokeratin 8 (most mammals) M20 mIgGl); RDI-PR0651104
(Cytokeratin 9 KS9.7 & KS 9.216 mIgGl/mIgG3); RDI-CBL196 (Cytokeratin 10 LH2
mIgG); RDI-PRO10501 (Cytokeratin 10 (rat, mouse, bovine, rabbit) RKSE60
mIgGl); RDI-
PR011414 (Cytokeratin 10 DE-K10 + mIgGl); RDI-CBL217 (Cytokeratin 10,11,1 & 2
AE2
31
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
+ mIgG 1 ); RDI-PR061007 (Cytokeratin 13 (bovine & rat) Ks 13.1 + mIgG 1 );
RDI-
PRO 10523 (Cytokeratin 13 (human, mouse, rabbit) 1 C7 mIgG2a); RDI-PRO 10524
(Cytokeratin 13 (human, mouse, rabbit) 2D7 mIgG2b); RDI-CBL 197 (Cytokeratin
14 LL002
+ mIgG3); RDI-CBL197FT (Cytokeratin 14 LL002 + mIgG3 FITC); RDI-PR010003
(Cytokeratin 14 RCK 107 mIgGl); RDI-PR061036 (Cytokeratin 17 (rat 46kDa polyp)
KS
17.E3+ mIgG2b); RDI-CBL236 (Cytokeratin 18 C04 + mIgGl); RDI-PR061028
(Cytokeratin 18 (mouse, rat, pig, dog, sheep, hamster, bovine, trout) Ks 18.04
+ mIgG 1 );
ARDI-PR011416 (Cytokeratin 18 RCK 106 mIgGl); RDI-PRO10500 (Cytokeratin 18 RGE
53 mIgGl); RDI-CBL185 (Cytokeratin 18 DC10 + mIgGl); RDI-CBL178 (Cytokeratin
19
to Ks19.1 + mIgG2a); RDI-CBL198 (Cytokeratin 19 BA17 + mIgGl); RDI-PR061029
(Cytokeratin 19 (rat, fish, bovine) KS 19.2 mIgG2b); RDI-CBL199 (Cytokeratin
19 LAS86
mIgM); RDI-CBL247 (Cytokeratin 19 A53-B/A2.26 mIgG2a); RDI-CBL208 (Cytokeratin
20
Ks20.8 + mIgGl); RDI-PR061054 (Cytokeratin 20 (rat, pig) mouse IT-KS20.10 +
mIgGl);
RDI-PR061033 (Cytokeratin 20 Ks20.5 mIgG2a); RDI-CBL215 (Cytokeratin Type I
(monkey, rabbit, mouse, rat, bovine, chicken, monkey) AE1 + mIgGl); RDI-CBL216
(Cytokeratin Type II monkey, rabbit, mouse, rat, bovine, chicken, monkey) AE3
+ mIgGl);
RDI-PR061031 (Cytokeratin (Pan epithelial) C22 mIgGl); RDI-PR061006
(Cytokeratin
TYPE II Ks pan 1-8 mIgG2a); RDI-PR061056 (Cytokeratin TYPE II " " Biotin
labeled);
RDI-PR061406 (Cytokeratin Type II " " FITC conjugated); and RDI-PR061835
(Cytokeratin Type I & II AE1/AE3 mIgGl). These antibodies also include anti-
pan-
cytokeratin (human, bovine rat and mouse, catalog number 250400) from
CALBIOCHEM,
and product numbers C7034 (anti-cytokeratin 8.12); C6909 (anti-cytokeratin
8.13); C7284
(anti-cytokeratin 8.60); C7785 (anti-cytokeratin CKS); C8791 (anti-cytokeratin
peptide 14);
and C1399 (anti-cytokeratin peptide 18) from Sigma-Aldrich.
Thus, as described above, a template, such as a keratin binding antibody can
be used
to identify keratin binding molecules. It is now routine to produce large
numbers of
molecules having inhibitory functions based on one or a few peptide sequences
or sequence
motifs. (See, e.g., Bromme, et al., Biochem. J. 315:85-89 (1996); Palmer, et
al., J. Med.
Chem. 38:3193-3196 (1995)). For example, if keratin is known to interact with
protein X
3o i.e. MBL) at position Y, an inhibitor of keratin-MBL interactions may be
chosen or designed
as a polypeptide or modified polypeptide having the same sequence as protein
X, or structural
similarity to the sequence of protein X, in the region adjacent to position Y.
In fact, the
32
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
region adjacent to the cleavage site Y spanning residues removed by 10
residues or, more
preferably 5 residues, N-terminal and C-terminal of position Y, may be defined
as a
"preferred protein X site" for the choice or design of keratin-MBL interaction
inhibitors.
Thus, a plurality of these compounds chosen or designed to span the preferred
protein X
s binding site around position Y, may be produced, tested for inhibitory
activity, and
sequentially modified to optimize or alter activity, stability, and/or
specificity.
The method is useful for designing a wide variety of biological mimics that
are more
stable than the natural counterpart, because they are typically prepared by
the free radical
polymerization of functional monomers, resulting in a compound with a non-
biodegradable
1o backbone. Thus, the created molecules would have the same binding
properties as the keratin
antibody but be more stable in vivo, thus preventing keratin from interacting
with components
normally available in its native environment. Other methods for designing such
molecules
include, for example, drug design based on structure activity relationships
which require the
synthesis and evaluation of a number of compounds and molecular modeling.
I5 Binding molecules may also be identified by conventional screening methods,
such as
those described above. Additionally, keratin binding molecules can be
identified from
combinatorial libraries. Many types of combinatorial libraries have been
described. For
instance, U.S. Patent Nos. 5,712,171 (which describes methods for constructing
arrays of
synthetic molecular constructs by forming a plurality of molecular constructs
having the
2o scaffold backbone of the chemical molecule and modifying at least one
location on the
molecule in a logically-ordered array); 5, 962, 412 (which describes methods
for making
polymers having specific physiochemical properties); and 5, 962, 736 (which
describes
specific arrayed compounds).
By using the known keratin monoclonal antibodies, it is also possible to
produce anti-
25 idiotypic antibodies which can be used to screen other antibodies to
identify whether the
antibody has the same binding specificity as the known monoclonal antibody.
Such anti-
idiotypic antibodies can be produced using well-known hybridoma techniques
(Kohler and
Milstein, Nature, 256:495, 1975). An anti-idiotypic antibody is an antibody
which
recognizes unique determinants present on the known monoclonal antibodies.
These
3o determinants are located in the hypervariable region of the antibody. It is
this region which
binds to a given epitope and, thus, is responsible for the specificity of the
antibody. An anti-
idiotypic antibody can be prepared by immunizing an animal with the known
monoclonal
33
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
antibodies. The immunized animal will recognize and respond to the idiotypic
determinants
of the immunizing known monoclonal antibodies and produce an antibody to these
idiotypic
determinants. By using the anti-idiotypic antibodies of the immunized animal,
which are
specific for the known monoclonal antibodies, it is possible to identify other
clones with the
same idiotype as the known monoclonal antibody used for immunization.
Idiotypic identity
between monoclonal antibodies of two cell lines demonstrates that the two
monoclonal
antibodies are the same with respect to their recognition of the same epitopic
determinant.
Thus, by using anti-idiotypic antibodies, it is possible to identify other
hybridomas expressing
monoclonal antibodies having the same epitopic specificity.
l0 It is also possible to use the anti-idiotype technology to produce
monoclonal
antibodies which mimic an epitope. For example, an anti-idiotypic monoclonal
antibody
made to a first monoclonal antibody will have a binding domain in the
hypervariable region
which is the image of the epitope bound by the first monoclonal antibody.
In one embodiment the binding molecules useful according to the invention are
antibodies or functionally active antibody fragments. Antibodies are well
known to those of
ordinary skill in the science of immunology. Many of the binding molecules
described herein
are available from commercial sources as intact functional antibodies, as
described above.
As used herein, the term "antibody" means not only intact antibody molecules
but also
fragments of antibody molecules retaining specific binding ability. Such
fragments are also
well known in the art and are regularly employed both in vitro and in vivo. In
particular, as
used herein, the term "antibody" means not only intact immunoglobulin
molecules but also
the well-known active fragments F(ab')2, and Fab. F(ab')2, and Fab fragments
which lack the
Fc fragment of intact antibody, clear more rapidly from the circulation, and
may have less
non-specific tissue binding of an intact antibody (Wahl et al., J. Nucl. Med.
24:316-325
(1983)).
As is well-known in the art, the complementarity determining regions (CDRs) of
an
antibody are the portions of the antibody which are largely responsible for
antibody
specificity. The CDR's directly interact with the epitope of the antigen (see,
in general,
Clark, 1986; Roitt, 1991 ). In both the heavy chain and the light chain
variable regions of IgG
immunoglobulins, there are four framework regions (FR1 through FR4) separated
respectively by three complementarity determining regions (CDR1 through CDR3).
The
framework regions (FRs) maintain the tertiary structure of the paratope, which
is the portion
34
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
of the antibody which is involved in the interaction with the antigen. The
CDRs, and in
particular the CDR3 regions, and more particularly the heavy chain CDR3
contribute to
antibody specificity. Because these CDR regions and in particular the CDR3
region confer
antigen specificity on the antibody these regions may be incorporated into
other antibodies or
peptides to confer the identical specificity onto that antibody or molecule.
According to one embodiment, the molecule is an intact soluble monoclonal
antibody
in an isolated form or in a pharmaceutical preparation. An intact soluble
monoclonal
antibody, as is well known in the art, is an assembly of polypeptide chains
linked by disulfide
bridges. Two principle polypeptide chains, referred to as the light chain and
heavy chain,
make up all major structural classes (isotypes) of antibody. Both heavy chains
and light
chains are further divided into subregions referred to as variable regions and
constant regions.
As used herein the term "monoclonal antibody" refers to a homogenous
population of
immunoglobulins which specifically bind to an epitope (i.e. antigenic
determinant) , e.g., of
keratin.
The molecule useful according to the methods of the present invention may be
an
intact humanized a monoclonal antibody. A "humanized monoclonal antibody" as
used
herein is a human monoclonal antibody or functionally active fragment thereof
having human
constant regions and a binding CDR3 region from a mammal of a species other
than a human.
Humanized monoclonal antibodies may be made by any method known in the art.
Humanized monoclonal antibodies, for example, may be constructed by replacing
the non-
CDR regions of a non-human mammalian antibody with similar regions of human
antibodies
while retaining the epitopic specificity of the original antibody. For
example, non-human
CDRs and optionally some of the framework regions may be covalently joined to
human FR
and/or Fc/pFc' regions to produce a functional antibody. There are entities in
the United
States which will synthesize humanized antibodies from specific murine
antibody regions
commercially, such as Protein Design Labs (Mountain View California). For
instance, a
humanized form of the anti-keratin antibody used in the attached Examples
could be easily
prepared and used according to the methods of the invention.
European Patent Application 0239400, the entire contents of which is hereby
3o incorporated by reference, provides an exemplary teaching of the production
and use of
humanized monoclonal antibodies in which at least the CDR portion of a murine
(or other
non-human mammal) antibody is included in the humanized antibody. Briefly, the
following
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
methods are useful for constructing a humanized CDR monoclonal antibody
including at least
a portion of a mouse CDR. A first replicable expression vector including a
suitable promoter
operably linked to a DNA sequence encoding at least a variable domain of an Ig
heavy or
light chain and the variable domain comprising framework regions from a human
antibody
and a CDR region of a murine antibody is prepared. Optionally a second
replicable
expression vector is prepared which includes a suitable promoter operably
linked to a DNA
sequence encoding at least the variable domain of a complementary human Ig
light or heavy
chain respectively. A cell line is then transformed with the vectors.
Preferably the cell line is
an immortalized mammalian cell line of lymphoid origin, such as a myeloma,
hybridoma,
trioma, or quadroma cell line, or is a normal lymphoid cell which has been
immortalized by
transformation with a virus. The transformed cell line is then cultured under
conditions
known to those of skill in the art to produce the humanized antibody.
As set forth in European Patent Application 0239400 several techniques are
well
known in the art for creating the particular antibody domains to be inserted
into the replicable
vector. (Preferred vectors and recombinant techniques are discussed in greater
detail below.)
For example, the DNA sequence encoding the domain may be prepared by
oligonucleotide
synthesis. Alternatively a synthetic gene lacking the CDR regions in which
four framework
regions are fused together with suitable restriction sites at the junctions,
such that double
stranded synthetic or restricted subcloned CDR cassettes with sticky ends
could be ligated at
2o the junctions of the framework regions. Another method involves the
preparation of the
DNA sequence encoding the variable CDR containing domain by oligonucleotide
site-
directed mutagenesis. Each of these methods is well known in the art.
Therefore, those
skilled in the art may construct humanized antibodies containing a murine CDR
region
without destroying the specificity of the antibody for its epitope.
Human monoclonal antibodies may be made by any of the methods known in the
art,
such as those disclosed in US Patent No. 5,567,610, issued to Borrebaeck et
al., US Patent
No. 565,354, issued to Ostberg, US Patent No. 5,571,893, issued to Baker et
al, Kozber, J.
Immunol. 133: 3001 (1984), Brodeur, et al., Monoclonal Antibody Production
Techniques
and Applications, p. 51-63 (Marcel Dekker, Inc, new York, 1987), and Boerner
et al., J.
3o Immunol., 147: 86-95 (1991). In addition to the conventional methods for
preparing human
monoclonal antibodies, such antibodies may also be prepared by immunizing
transgenic
animals that are capable of producing human antibodies (e.g., Jakobovits et
al., PNAS USA,
36
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
90: 2551 (1993), Jakobovits et al., Nature, 362: 255-258 (1993), Bruggermann
et al., Year in
Immuno., 7:33 (1993) and US Patent No. 5,569,825 issued to Lonberg).
The binding peptides may also be functionally active antibody fragments.
Significantly, as is well-known in the art, only a small portion of an
antibody molecule, the
paratope, is involved in the binding of the antibody to its epitope (see, in
general, Clark, W.R.
(1986) The Experimental Foundations of Modern Immunology Wiley & Sons, Inc.,
New
York; Roitt, I. (1991) Essential Immunology, 7th Ed., Blackwell Scientific
Publications,
Oxford). The pFc' and Fc regions of the antibody, for example, are effectors
of the
complement cascade but are not involved in antigen binding. An antibody from
which the
pFc' region has been enzymatically cleaved, or which has been produced without
the pFc'
region, designated an F(ab')2 fragment, retains both of the antigen binding
sites of an intact
antibody. An isolated F(ab')2 fragment is referred to as a bivalent monoclonal
fragment
because of its two antigen binding sites. Similarly, an antibody from which
the Fc region has
been enzymatically cleaved, or which has been produced without the Fc region,
designated an
Fab fragment, retains one of the antigen binding sites of an intact antibody
molecule.
Proceeding further, Fab fragments consist of a covalently bound antibody light
chain and a
portion of the antibody heavy chain denoted Fd (heavy chain variable region).
The Fd
fragments are the major determinant of antibody specificity (a single Fd
fragment may be
associated with up to ten different light chains without altering antibody
specificity) and Fd
fragments retain epitope-binding ability in isolation.
The terms Fab, Fc, pFc', F(ab')Z and Fv are used consistently with their
standard
immunological meanings [Klein, Immunology (John Wiley, New York, NY, 1982);
Clark,
W.R. (1986) The Experimental Foundations of Modern Immunology (Whey & Sons,
Inc.,
New York); Roitt, I. (1991) Essential Immunology, 7th Ed., (Blackwell
Scientific
Publications, Oxford)].
According to the methods of the invention, the compositions may be
administered in a
pharmaceutically acceptable composition. In general, pharmaceutically-
acceptable carriers
for peptides and structurally-related small molecules are well-known to those
of ordinary skill
in the art. As used herein, a pharmaceutically-acceptable carrier means a non-
toxic material
that does not interfere with the effectiveness of the biological activity of
the active
ingredients, i.e., the ability of the MBL receptor antagonist to inhibit LCP
associated
complement activation. Pharmaceutically acceptable carriers include diluents,
fillers, salts,
37
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
buffers, stabilizers, solubilizers and other materials which are well-known in
the art.
Exemplary pharmaceutically acceptable carriers for peptides in particular are
described in
U.S. Patent No. 5,211,657. The receptor antagonist peptides of the invention
may be
formulated into preparations in solid, semi-solid, liquid or gaseous forms
such as tablets,
capsules, powders, granules, ointments, solutions, depositories, inhalants
(e.g., aerosols) and
injections, and usual ways for oral, parenteral or surgical administration.
The invention also
embraces locally administering the compositions of the invention, including as
implants.
According to the methods of the invention the compositions can be administered
by
injection by gradual infusion over time or by any other medically acceptable
mode. The
administration may, for example, be intravenous, intraperitoneal,
intramuscular, intracavity,
subcutaneous or transdermal. Preparations for parenteral administration
includes sterile
aqueous or nonaqueous solutions, suspensions and emulsions. Examples of
nonaqueous
solvents are propylene glycol, polyethylene glycol, vegetable oil such as
olive oil, an
injectable organic esters such as ethyloliate. Aqueous carriers include water,
alcoholic/aqueous solutions, emulsions or suspensions, including saline and
buffered media.
Parenteral vehicles include sodium chloride solution, Ringer's dextrose,
dextrose and sodium
chloride, lactated Ringer's or fixed oils. Intravenous vehicles include fluid
and nutrient
replenishers, electrolyte replenishers, (such as those based on Ringer's
dextrose), and the like.
Preservatives and other additives may also be present such as, for example,
antimicrobials,
2o antioxidants, chelating agents, and inert gases and the like. Those of
skill in the art can
readily determine the various parameters for preparing these alternative
pharmaceutical
compositions without resort to undue experimentation. When the compositions of
the
invention are administered for the treatment of pulmonary disorders the
compositions may be
delivered for example by aerosol.
The compositions of the invention are administered in therapeutically
effective
amounts. As used herein, an "effective amount" of the inhibitor of the
invention is a dosage
which is sufficient to inhibit the increase in, maintain or even reduce the
amount of
undesirable LCP associated complement activation. The effective amount is
sufficient to
produce the desired effect of inhibiting associated cellular injury until the
symptoms
3o associated with the MBL mediated disorder are ameliorated or decreased.
Preferably an
effective amount of the peptide is an effective amount for preventing cellular
injury.
Generally, a therapeutically effective amount may vary with the subject's age,
condition, and
38
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
sex, as well as the extent of the disease in the subject and can be determined
by one of skill in
the art. The dosage may be adjusted by the individual physician or
veterinarian in the event
of any complication. A therapeutically effective amount typically will vary
from about 0.01
mg/kg to about 500 mg/kg, were typically from about 0.1 mg/kg to about 200
mg/kg, and
often from about 0.2 mg/kg to about 20 mg/kg, in one or more dose
administrations daily, for
one or several days (depending of course of the mode of administration and the
factors
discussed above). A preferred concentration of the MBL receptor antagonist is
a
concentration which is equimolar to the concentration of MBL in the plasma of
a subject.
The normal plasma concentration of MBL can be assessed clinically. A normal
range of
to MBL is 1-2mg/L MBL/plasma.
One of skill in the art can determine what an effective amount of an MBL
receptor
antagonist is by screening the ability of the antagonist to inhibit the LCP
associated
complement activation in an in vitro assay. The activity of the antagonist can
be defined in
terms of the ability of the antagonist to inhibit LCP associated complement
activation. An
exemplary assay for measuring the ability of a putative MBL receptor
antagonist of the
invention to inhibit LCP associated complement activation is provided in the
Examples and
has been discussed above. The exemplary assay is predictive of the ability of
an MBL
receptor antagonist to inhibit LCP associated complement activation in vivo
and, hence, can
be used to select such antagonists for therapeutic applications.
2o The MBL receptor antagonists may be administered in a physiologically
acceptable
carrier. The term "physiologically-acceptable" refers to a non-toxic material
that is
compatible with the biological systems of a tissue or organism. The
physiologically
acceptable carrier must be sterile for in vivo administration. The
characteristics of the carrier
will depend on the route of administration. The characteristics of the carrier
will depend on
the route of administration.
The invention further provides detectably labeled, immobilized and toxin
conjugated
forms of the peptides of the invention, as well as fragments and functional
equivalents
thereof. The MBL receptor antagonists of the invention may be labeled using
radiolabels,
fluorescent labels, enzyme labels, free radical labels, avidin-biotin labels,
or bacteriophage
labels, using techniques known to the art (Chard, Laboratory Techniques in
Biology, "An
Introduction to Radioimmunoassay and Related Techniques," North Holland
Publishing
Company (1978).
39
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
Typical fluorescent labels include fluorescein isothiocyanate, rhodamine,
phycoerythrin, phycocyanin, allophycocyanin, and fluorescamine.
Typical chemiluminescent compounds include luminol, isoluminol, aromatic
acridinium esters, imidazoles, and the oxalate esters.
Typical bioluminescent compounds include luciferin, and luciferase. Typical
enzymes include alkaline phosphatase, 13-galactosidase, glucose-6-phosphate
dehydrogenase,
maleate dehydrogenase, glucose oxidase, and peroxidase.
The invention also includes methods for screening a subject for susceptibility
to
treatment with an MBL receptor antagonist. In one aspect, the method is
accomplished by
1o isolating a mammalian cell from a subject and detecting the presence of an
MBL or an MBL
ligand on a surface of the mammalian cell. The presence of the MBL indicates
that the cell is
susceptible to LCP-associated complement activiation, and that the subject is
susceptible to
treatment with an MBL receptor antagonist. The mammalian cell may be isolated
by any
method known in the art, for instance by a biopsy. Another method for
accomplishing the
screening assay involves the steps of contacting a mammalian cell from the
subject with a
labeled isolated MBL receptor antagonist and detecting the presence of an MBL
receptor
antagonist the surface of the mammalian cell. This assay may be performed in
vitro, ex vivo,
or in vivo. Many labels which can be used to observe the MBL receptor
antagonist
interacting with the mammalian cell are known in the art under each of these
conditions. For
2o instance, radioactive compounds can be used in vitro, and other
biocompatible labels can be
used ex vivo or in vivo. Once the subjects are identified who are susceptible
to treatment with
an MBL receptor antagonist, the subjects can then be treated according to the
methods of the
invention.
The following examples are provided to illustrate specific instances of the
practice of
the present invention and are not to be construed as limiting the present
invention to these
examples. As will be apparent to one of ordinary skill in the art, the present
invention will
find application in a variety of compositions and methods. The following
abbreviations have
been used throughout the Examples: MBL=mannose binding lectin; K=cytokeratin;
CRD=carbohydrate recognition domain; MASP=MBL associated serine protease;
PMN=polymorphonuclear leukocytes; mAb=monoclonal antibody; HUVECs=human
umbilical vein endothelial cells; GIcNAc=N-acetylglucosamine; ROS=reactive
oxygen
species; HS=human serum/sera; I/R=ischemia/reperfusion;
H/R=hypoxia/reoxygenation;
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
NFKB=nuclear factor K B; AA=amino acid; UEA= Ulex europaeus; LAA= Laburnum
alpinum; DEF=desferrioxamine; DMTU=dimethylthiourea.
Examples
Introduction to the Examples:
The experiments described herein investigate the molecular mechanisms of
complement activation during or following ischemia/reperfusion and are useful
for
developing novel therapeutics to inhibit complement activation during
reperfusion. More
particularly, the experiments disclosed herein 1) evaluate novel legume-based
complement
l0 inhibitors directed against an endothelial MBL ligand; 2) describe
structure/activity
relationships of novel functionally inhibitory peptides that block MBL binding
to endothelial
cells following oxidative stress; 3) characterize the regulation of
endothelial cell K expression
during oxidative stress; and 4) characterize the action and/or the mechanism
of UEA-II
induced tissue protection following ischemia/reperfusion injury in vivo.
Example 1. The Role of MBL in activating complement on hvnoxic/reoxv~enated
endothelial cells.
Previous published findings from our lab demonstrate that reoxygenation/
reperfusion
of hypoxic/ischemic endothelial cells generates intracellular reactive oxygen
species which
activate NF-KB, leading to transcription and translation of a novel ligand for
MBL (Collard
CD, Agah A, Stahl GL: Complement activation following reoxygenation of hypoxic
human
endothelial cells: role of intracellular reactive oxygen species, NF- kappaB
and new protein
synthesis. Immunopharmacology 1998;39:39-50). The intracellular response to
hypoxia/
reoxygenation leads to upregulation of a ligand(s) for MBL, complement
activation and
iC3b/CSb-9 deposition on the extracellular membrane. We have developed a panel
of mAbs
that bind to the CRD of MBL and thus functionally inhibit MBL binding to the
endothelial
MBL ligand. (USSN 60/112,390, entitled "Anti-Mannose (Mannan) Binding Lectin
Therapy
for Ischemia/Reperfusion Injury.") A significant increase in C3 and MBL
deposition on
endothelial cells was observed following oxidative stress (in the presence of
30% HS)
3o compared to normoxic cells. Treatment of the human sera with a non-
functionally inhibitory
mAb (clone 1C10; 50 ~.g/ml) to MBL did not attenuate MBL or C3 deposition. In
contrast,
clone 3F8 (5 pg/ml) significantly attenuated MBL and C3 deposition. Co-
localization of
41
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
MBL and C3 was also demonstrated in this study. These data coupled with other
data
demonstrating that depletion of MBL from sera, treatment with a specific MBL
inhibitory
sugar (i.e., N-acetylglucosamine) and use of purified complement components
conclusively
demonstrated that MBL is responsible for complement activation on endothelial
cells
following oxidative stress.
Example 2. Characterization of the MBL ligand present on endothelial cells
following
oxidative stress.
Recognition and characterization of a MBL ligand and generation of
functionally
inhibitory molecules to the ligand represent a novel therapeutic approach for
complement
inhibition. The principal binding ligands of MBL are oligosaccharide
oligomers, particularly
oligomers of GIcNAc and mannose. We believe that the novel epitope for MBL
binding on
hypoxic/reoxygenation HUVECs is a glycoprotein (Collard CD, Agah A, Stahl GL:
Complement activation following reoxygenation of hypoxic human endothelial
cells: role of
intracellular reactive oxygen species, NF- kappaB and new protein synthesis.
Immunopha~macology 1998;39:39-50). Since MBL purification is tedious and the
yield from
1 L of human plasma varies from 100-200 pg, immunoprecipitation of the ligand
with
purified MBL would be very costly and time consuming. We therefore developed
another
strategy. There are many legume lectins that display "MBL-like" binding
characteristics
(e.g., calcium-dependent and inhibited by specific sugars). Thus, we screened
the binding
characteristics of several known legume lectins that are competitively
inhibited by GIcNAc or
complex oligomers of GIcNAc and display calcium-dependent binding. After
screening
eleven different lectins, the lectin Ulex europaeus II(UEA-II) showed an
increase in
deposition on hypoxic (1% O2; 24 hrs)/ reoxygenated (3 hrs, room air, in
buffer) HUVECs
compared to normoxic cells. Deposition of UEA-II to hypoxic/reoxygenated cells
was
similar to that of MBL as it: 1) was inhibited by DMTU treatment; 2) was
inhibited by DEF,
but not iron-loaded DEF, 3) displayed calcium-dependent binding, and 4) was
inhibited by
cycloheximide treatment (n=3 for each group; data not shown). Further, when
UEA-II was
bound to hypoxic HUVECs, MBL competitively inhibited UEA-II binding in a dose-
dependent manner. Similarly, MBL binding to H/R HUVECs was competitively
inhibited by
UEA-II in a dose-dependent manner. Thus, it appeared that UEA-II and MBL bound
to the
same ligand.
42
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
UEA-II (CY Laboratories) was coupled to Sepharose. Cell membranes were
prepared
by nitrogen cavitation from 1500 cm2 of either normoxic or
hypoxic/reoxygenated HUVECs.
The membranes were solubilized and pre-cleared with Sepharose. UEA-II coupled
Sepharose was used for immunoprecipitation. The resulting immunoprecipitate
was resolved
under reducing conditions by 9% SDS-PAGE. A single Coomassie stainable diffuse
band
(MW ~49-54 kDa) was cut from the gel and sent to our Core Facility (Bill Lane;
Harvard
MicroChemistry) for tryptic digestion and MALDI-TOF MS analysis. A peptide
(QIEGLKEELAYLR/K, SEQ ID NO. 1) was sequenced that displayed a high degree of
homology to human K14, K15, K16, K17 and K19. In order to demonstrate that the
peptide
1o was present on the cell surface, we biotinylated the cell surface proteins,
immunoprecipitated
with UEA-II and the immunoprecipitate was resolved under reducing conditions
by 9% SDS-
PAGE. The gel was electroblotted onto nitrocellulose, the membrane blocked and
resolved
with HRP-streptavidin. HRP-streptavidin was visualized with the ECL system
(Amersham).
A single band (~50 kDa) from H/R cells was observed that was not present on
normoxic
cells. The approximate MW of 50 kDa is consistent with the known MW of K14 and
K17,
but not K15, K16 or K19. Searching the literature we found that anti-
endothelial antibodies
from patients with accelerated transplant coronary artery disease recognize a
50 kDa keratin-
like protein on human endothelial cells (Ationu A: Identification of
endothelial antigens
relevant to transplant coronary artery disease from a human endothelial cell
cDNA expression
library. Int.J.Mol.Med. 1998;1:1007-1010). Further, a previous publication
demonstrated
that a peptide AA sequence within K14 (SFGSGFGGGY, SEQ ID NO. 2) mimics N-
acetylglucosamine (GIcNAc) in reactions with anti-N-acetylglucosamine
antibodies and some
plant lectins(Shikhman AR, Greenspan NS, Cunningham MW: Cytokeratin peptide
SFGSGFGGGY, SEQ ID NO. 2 mimics N-acetyl-beta-D- glucosamine in reaction with
antibodies and lectins, and induces in vivo anti-carbohydrate antibody
response. J.Immunol.
1994;153:5593-5606). This was an important finding, as GIcNAc is a potent
inhibitor of
MBL when it is present in the fluid phase. However, a BLAST search
demonstrated that this
AA sequence (i.e., SFGSGFGGGY, SEQ ID NO. 2) is also present in K17, CK1 and
similar
sequences are present in many cytokeratins. If this AA sequence does indeed
mimic N-
3o acetylglucosamine (i.e., a specific ligand of MBL), then MBL may be binding
to this specific
AA sequence of K14 or K17. We had the SFGSGFGGGY, SEQ ID NO. 2 peptide
synthesized (referred to herein as "GLUPEP"). GLUPEP potently inhibits iC3b
deposition
43
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
on HUVECs following oxidative stress in a dose-related manner. These data are
consistent
with the hypothesis that cytokeratins are responsible for MBL deposition and
complement
activation following oxidative stress. It is well recognized in the literature
that MBL binds to
carbohydrate groups, however this is the first demonstration of a peptide
inhibiting the MBL
pathway.
To verify that keratin will activate complement via the MBL pathway, we
developed
two solid phase ELISAs for the functional characterization of MBL and its
associated serine-
proteases (i.e., MASP1 and MASP2) based on a published manuscript(Super M,
Levinsky
RJ, Turner MW: The level of mannan-binding protein regulates the binding of
complement-
to derived opsonins to mannan and zymosan at low serum concentrations.
Clin.Exp.lmmunol.
1990;79:144-150). In these assays, keratin or GLUPEP coupled to BSA was coated
onto 96
well microtiter RIA/EIA plates. Human serum (2%) was then added to the wells,
incubated
for 30 min, the plates are washed and HRP-conjugated anti-C3 polyclonal
antibodies was
used to assess C3 deposition. Positive and negative controls consist of wells
receiving
vehicle and GIcNAc (100 mmol/L) or our functionally inhibitory mAb to hMBL
(clone 3F8,
deposited with the ATCC on December 15, 1998 under Accession number HB-
126621),
respectively. As shown in figure 2, keratin coated plates activated complement
and deposited
C3 that was significantly inhibited by either GIcNAc or 3F8 (i.e., MBL pathway
activation).
Further, GLUPEP or UEA-II inhibited keratin-induced complement activation in a
dose-
dependent manner. When GLUPEP was coupled to BSA, we demonstrated that MBL
bound
to GLUPEP-BSA, but not to BSA and that GIcNAc, 3F8, or GLUPEP inhibited MBL
binding
(Figure 3). These data demonstrate that keratin activates the MBL pathway and
that MBL
binds to GLUPEP. Based upon these results, we believe that purified K
activates the MBL
pathway. In view of this discovery, we believe that novel peptide inhibitors
of the MBL
pathway can be designed since previous results have reported that
structure/activity
relationships of GLUPEP, as specific substitutions of the AA sequence have
been increased
affinity of anti-GIcNAc antibodies to GLUPEP by as much as 350% (Shikhman AR,
Greenspan NS, Cunningham MW: Cytokeratin peptide SFGSGFGGGY, SEQ ID NO. 2
mimics N-acetyl-beta-D- glucosamine in reaction with antibodies and lectins,
and induces in
3o vivo anti-carbohydrate antibody response. J.Immunol. 1994;153:5593-5606).
44
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
Example 3 Oxidative Stress Increases Human Endothelial Cell Surface CK1
protein
Expression
A significant increase in endothelial cell surface CK1 expression following
oxidative
stress was observed. The data are consistent with the hypothesis that MBL may
bind to
many different keratins since the amino and hydroxy terminals of cytokeratins
are highly
homologous. Interestingly, exons 1 and 9 of CK1 contain sequences highly
homologous to
the amino acid sequence SFGSGFGGGY, SEQ ID NO. 2 which is present in K17.
Human endothelial cell CK1 protein expression following oxidative stress was
determined by both ELISA and confocal microscopy. ELISA measurements indicate
a
1o significant increase in HUVEC CK1 expression following oxidative stress was
observed as
compared to the normoxic HUVEC (OD4os = 0.43 ~ 0.05 vs 0.14 ~ 0.01,
respectively;
p<0.05). Immunofluorescent confocal microscopy studies confirmed that
oxidative stress
caused a significant increase in extracellular HUVEC CK1 expression. The
immunofluorescent experiments were performed three times (n=3).
In order to demonstrate the specificity of the anti-human CK1 pAb used in
these
experiments, HUVEC CK1 was immunoprecipitated (67 kDa band, reduced 9% linear
SDS-
PAGE gel) and confirmed by protein sequencing (human keratin, Type II
cytoskeletal 1).
Example 4 Purified Human CK1 Activates the LCP
2o Endothelial cytokeratins induce complement activation following oxidative
stress.
Human MBL and C3 deposition on purified human dermal CK1 was determined by
ELISA.
Treatment of 2% HS with GIcNAc (100 mmol/L Sigma) or the functionally
inhibitory anti-
human MBL mAB, 3F8 (10 ~g/ml) significantly inhibited MBL deposition on
purified CK1-
coated plates by 78 ~ 4% and 64 ~ 6%, respectively compared to untreated HS
(vehicle).
Further, treatment with GIcNAc (100 mmol/L) or 3F8 (10 ~.g/ml) significantly
inhibited C3
deposition on CK1-coated plates by 70 ~ 1% and 69 ~ 1%, respectively compared
to
untreated HS. (n=3; data normalized to vehicle).
These data suggest that MBL binds human CK1 and activates the complement
pathway. Furthermore, these data demonstrate that MBL inhibition attenuates
CK1-induced
3o complement activation.
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
Example 5 Human MBL Recognizes Endothelial CK1 Following Oxidative Stress.
In order to determine whether human MBL binds endothelial CK1, purified human
MBL was used to immunoprecipitate HUVEC CK1. Western blot of the
immunoprecipitates
using a monospecific anti-human CK1 antibody revealed a 67-kDa band consistent
with
human CK1. Interestingly, the 67-kDa band was observed following endothelial
oxidative
stress, but not in normoxic HUVEC or in the control lanes. These data strongly
suggest that
human MBL recognizes and binds to endothelial CK1 following oxidative stress.
To further confirm that human MBL binds endothelial CK1 following oxidative
stress, MBL and CK1 were co-immunoprecipitated from hypoxic HUVEC reoxygenated
in
HS. Western blot of HUVEC lysates immunoprecipitated with a monospecific anti-
human
CK1 pAb Convance/BAbCO (Richmond, CA) revealed a 32-kDa band consistent with
reduced purified human MBL. Further, the 32-kDa band was observed following
endothelial
oxidative stress, but not in normoxic HUVEC or in the control lanes.
The data that MBL binds endothelial CK1 are consistent with the earlier
studies
indicating that MBL may bind other cytokeratins. As already noted earlier,
exons 1 and 9 of
CK1 contain sequences highly homologous to the amino acid sequence SFGSGFGGGY,
SEQ ID NO. 2 which is present in K17 and other cytokeratins.
Example 6 Anti human Keratin Treatment Attenuates MBL and C3 Deposition
Following
Endothelial Oxidative Stress.
Oxidative stress increases MBL deposition on HUVEC and activates the lectin
complement pathway. In the present set of experiments it is demonstrated that
anti-human
keratin antibodies attenuate endothelial MBL and C3 deposition.
HUVEC MBL and C3 deposition following oxidative stress were measured by
ELISA. Consistent with the previous findings, a significant increase in MBL
(OD4o5 = 0.05 ~
0.01) and C3 (OD4o5 = 0.21 t 0.02) deposition was observed following oxidative
stress
compared to normaxic HUVEC (OD4os = 0.01 ~ 0.01 and 0.07 ~ 0.01, respectively;
p<0.05).
Treatment of 30% HS with GIcNAc (100 mmol/L) or anti-human keratin pAb (50
pg/ml)
significantly inhibited MBL deposition by 66 ~ 11% and 53 ~ 9%, respectively,
compared to
3o untreated HS (vehicle). Further, treatment with GIcNAc (100 mmol/L) or anti-
human keratin
Fab fragments (20 p,g/ml synthesized in Stahl laboratory) significantly
inhibited C3
deposition by 46 ~ 7% and 48 ~ 6%, respectively, compared to untreated HS.
These data
46
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
demonstrate that anti-MBL or keratin treatment significantly attenuates
endothelial MBL and
C3 deposition following oxidative stress.
To further confirm these findings, HUVEC MBL and C3 deposition following
oxidative stress was determined by immunofluorescent confocal microscopy.
Normoxic and
hypoxic HUVEC were reoxygenated in 30% HS treated with and without anti-human
keratin
Fab fragments (20 ~g/ml). Small amounts of MBL and C3 staining were observed
under
normoxic conditions, confirming our previous finding of low level C3
deposition under
normoxic conditions. MBL and C3 staining following HUVEC oxidative stress was
increased compared to normoxic cells. Incubation of human sera with anti-human
keratin Fab
fragments attenuated MBL and C3 staining. These data further demonstrate that
anti-keratin
treatment inhibits MBL deposition and complement activation following
endothelial
oxidative stress.
Example 7 UEA-II Inhibits C3 Deposition on Keratin.
~ 5 As shown above, UEA-II recognizes a MBL ligand on human endothelial cells
following oxidative stress. We hypothesized that UEA-II may also inhibit
complement
activation by acting as a "receptor/ligand antagonist". Confocal micrographs
of HUVECs
following oxidative stress (in the presence of 30% HS) demonstrated co-
localization of MBL
and C3. We observed complete inhibition of C3 and MBL deposition at a UEA-II
2o concentration of 100 pmol/L (10 ng/ml). Significant reductions in C3 and
MBL deposition
were also observed at 100 fmol/L (10 pg/ml) and 0.10 fmol/L (10 fg/ml). These
data
demonstrate that UEA-II is a potent complement inhibitor on endothelial cells
following
oxidative stress.
An experiment was performed in which keratin (2 pg/ml) or BSA (2 pg/ml) in
sodium
25 carbonate buffer was plated to 96-well plastic plates. The plates were then
blocked with
BSA. Human sera (30%) was applied to the wells in the presence of GVB
(vehicle), GIcNAc
(100 mmol/L) or UEA-II (E-Y laboratories, Manson, CA) (10 ng/ml). The plates
were
incubated at 37C for 1 hour, washed and incubated with a HRP-anti-human C3 Pab
(YCN,
Aurora, Ohio) for detection of C3 deposition to the plastic. Both GIcNAc or
UEA-II
3o inhibited the activation of the LCP and the resultant C3 deposition to the
keratin coated
plastic. These data further demonstrate that keratin does indeed activate the
lectin
complement pathway. The results are shown in Figure 5.
47
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
It was possible that UEA-II inhibited complement activation by activating
complement and thus produced complement-depleted sera. In order to rule this
out, UEA-II
at 100 ~g/ml (i.e., ~1 ~.mol/L) was incubated with human sera and hemolytic
assays using
sensitized chicken RBCs were performed. UEA-II did not activate complement or
attenuate
classical pathway activation. Thus, the mechanism of UEA-II mediated
complement
inhibition on HUVECs following oxidative stress is not a result of complement
depletion, but
a result of specific inhibition of MBL binding to its ligand. Further, UEA-II
deposition on
H/R HUVECs is inhibited by GLUPEP. These data demonstrate that GLUPEP inhibits
UEA-
II deposition on HUVECs following oxidative stress or MBL-dependent complement
activation on keratin (Figure 2). Since GLUPEP inhibits MBL and UEA-II
deposition, these
data suggest that GLUPEP mimics GIcNAc.
The lectins UEA-II, Cytisus sessilifolius (CSA-I) and Laburnum alpinum (LAA-I)
share a similar CRD peptide sequence (Yamamoto K, Konami Y, Osawa T, Irimura
T:
Carbohydrate-binding peptides from several anti-H(O) lectins. J.Biochem.
(Tokyo.)
is 1992;111:436-439) (see Table 1). Like UEA-II, LAA-I inhibited MBL mediated
C3
deposition on HUVECs following oxidative stress in a dose-dependent manner
(Figure 4).
Other lectins (e.g., CSA-I and LAA-I, and functional equivalents thereof such
as fragments
and/or peptides containing conservative amino acid substitutions) can be
evaluated for
inhibiting MBL binding to keratin and H/R HUVECs using the methods disclosed
herein.
Table 1. Amino acid sequences for the CRD of legume lectins.
I: DTYFGKTYNPW, SEQ ID NO. 3
LAA-I: DTYFGKAYNPW, SEQ ID NO. 4
UEA-II: DSYFGKTYNPW, SEQ ID NO. 5
Derived from a published manuscript (Yamamoto K, Konami Y, Osawa T, Irimura T:
Carbohydrate-binding peptides from several anti-H(O) lectins. J.Biochem.
(Tokyo.)
1992;111:436-439). Highlighted amino acids
represent divergent amino acids.
We have demonstrated that activation of the MBL pathway mediates the
complement
activation and deposition on human endothelial cells following oxidative
stress and have
developed antibody technology to inhibit MBL directly (USSN 60/112,390,
entitled "Anti-
48
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
Mannose (Mannan) Binding Lectin Therapy for Ischemia/Reperfusion Injury). We
disclose
herein the discoveries of an MBL ligand present on human endothelial cells and
that MBL,
which is known to bind carbohydrate groups, binds to specific peptides in the
absence of
carbohydrate groups. Thus, the further experiments presented below describe
the further
investigation of the role of cytokeratin in activating the MBL pathway. These
experiments
also are directed to developing and conducting studies relating to
structure/activity
relationships of novel potent peptide based MBL inhibitors based on the MBL
recognition of
cytokeratin and the development of specific functionally inhibitory mAbs/Fabs
to cytokeratin
in order to inhibit MBL deposition and lectin pathway activation.
to
Example 8 Characterization of K expression following oxidative stress.
The above-described data strongly supports the upregulation of keratins and
particularly CK1 as a novel MBL ligand on hypoxic/reoxygenated HUVECs. The
experiments disclosed herein evaluate the expression of other keratins
following oxidative
15 stress to assess their role in the physiological results of oxidative
stress (similar ro the studies
performed on CK1). A recent report demonstrated that anti-endothelial
antibodies from
patients with accelerated transplant coronary artery disease recognize a 50
kDa keratin-like
protein on human endothelial cells (Ationu A: Identification of endothelial
antigens relevant
to transplant coronary artery disease from a human endothelial cell cDNA
expression library.
2o Int.J.Mol.Med. 1998;1:1007-1010). Data from our lab has demonstrated that
intracellular
ROS, new protein synthesis and NFKB translocation is necessary for the MBL
ligand to be
upregulated. The experiments presented herein establish that K is a ligand for
MBL on
HUVECs following oxidative stress.
Several strategies are used to identify the specific keratin present on
HUVECs. First,
25 we have designed specific primers to K17 and K14 based on their cDNA and
conserved
regions using commercially available software (Sequencer). RT-PCR, northern
and RNAse
protection assays are performed to establish an increase in mRNA levels in
HUVECs
following oxidative stress using K specific probes. These data establish that
mRNA is present
and upregulated by oxidative stress. Oxidative stress is induced by H/R and
also by H202, as
3o we have described above. A time course for K expression is done. Full
length cDNAs or a
cell line (i.e., T84 or HeLa cells) is used as a positive control for all
experiments. The RT-
49
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
PCR products for K14 in HUVECs are cloned/sequenced and all positive RT-PCR
products
are sequenced to demonstrate specificity.
Rabbits and mice are immunized for Pab and mAb production against GLUPEP.
Antibodies to keratins are made and purified. Cell surface proteins are
immunoprecipitated
from HUVECs following normoxic or oxidative stress conditions using our
monospecific Pab
to GLUPEP. This immunoprecipitate recovers proteins containing this AA
sequence. A
western blot is performed using the commercially available monospecific Pab to
K14
(BAbCo) or a mAb to K17 (clone E3; Dako) to determine which K is present.
These
experiments establish which K is present on the HUVECs. Additionally, cell
surface proteins
t o are biotinylated on HUVECs and immunoprecipitate with commercially
available mAbs and
detected which K is present on the cell surface with streptavidin-HRP.
In situ hybridization and confocal microscopy is performed to demonstrate
localization of K to endothelial cells as we have described previously
(Collard CD,
Bukusoglu C, Agah A, Colgan SP, Reenstra WR, Morgan BP, Stahl GL: Hypoxia-
induced
expression of complement receptor type 1 (0R1, CD35) in human vascular
endothelial cells.
Am.J.Physiol. 1999;276:0450-0458). Localization of the K protein to the cell
surface also is
confirmed by the use of antibodies and confocal microscopy.
We have identified 3 potential cAMP response elements binding (CREB) sites in
the
5' promoter region of K17, based on a BLAST search of the published promoter
region of
K17. Further, a previous study has demonstrated that K17 expression can be
enhanced by y-
interferon under the direct activation of the transcription factor STAT 1
(Komine M,
Freedberg IM, Blumenberg M: Regulation of epidermal expression of keratin K17
in
inflammatory skin diseases. J.Invest.Dermatol. 1996;107:569-575). However,
preliminary
data demonstrate that y-interferon does not induce K17 in HUVECs by RT-PCR.
Since
hypoxia is known to induce CREB (Taylor CT, Fueki N, Agah A, Hershberg RM,
Colgan SP:
Critical Role of cAMP Response Element Binding Protein Expression in Hypoxia-
elicited
Induction of Epithelial Tumor Necrosis Factor-alpha. J.Biol.Chem.
1999;274:19447-19454),
the following experiments are performed to investigate K17 expression. We have
shown that
complement activation is decreased in the presence of intracellular ROS
production and
3o NFxB translocation. As STAT1 activation is associated with ROS and
increased K
expression (Komine M, Freedberg IM, Blumenberg M: Regulation of epidermal
expression
of keratin K17 in inflammatory skin diseases. J. Invest.Dermatol. 1996;107:569-
575), K
so
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
protein expression is evaluated under oxidative stress conditions known to
increase
complement activation. HUVECs undergo H/R and/or H202 (1-1000 ~.mol/L) are
given to
HUVECs as previously described (Collard CD, Agah A, Stahl GL: Complement
activation
following reoxygenation of hypoxic human endothelial cells: role of
intracellular reactive
oxygen species, NF- kappaB and new protein synthesis. Immunopharmacology
1998;39:39-
50). NFKB translocation is inhibited by administration of SN50 (SM50 as a
control), using
NFoB decoys (Kupatt C, Habazettl H, Goedecke A, Wolf DA, Zahler S, Boekstegers
P, Kelly
RA, Becker BF: Tumor necrosis factor-alpha contributes to ischemia- and
reperfusion-
induced endothelial activation in isolated hearts. Circ.Res. 1999;84:392-400),
or MG132
(proteasome inhibitor). A cell permeable cAMP stable analogue (i.e., dinitro-
cAMP) or
forskolin is used to pretreat endothelial cells prior to hypoxia to prevent
CRE
phosphorylation and formation of the CREB/CPB complex and resulting
transcription.
Protein phosphorylation events are known to regulate K17 expression in
keratinocytes
(Komine M, Freedberg IM, Blumenberg M: Regulation of epidermal expression of
keratin
t5 K17 in inflammatory skin diseases. J.Invest.Dermatol. 1996;107:569-575).
Thus,
staurosporine and genistein should inhibit, whereas okadaic acid should
augment, K
expression.
The above-described experiments are used to identify K as the MBL ligand, and
that
K expression is enhanced by hypoxia/reoxygenation. We expect to observe an
increase in K
2o mRNA and protein expression following oxidative stress. These experiments
lay the
groundwork for understanding K regulation in HUVECs. The actions of pro-
inflammatory
cytokines and K regulation also are investigated. It is possible that the K
present on
HUVECs is a K-like, 50 kDa protein as previously described (Ationu A:
Identification of
endothelial antigens relevant to transplant coronary artery disease from a
human endothelial
25 cell cDNA expression library. Int.J.Mol.Med. 1998;1:1007-1010). With the
tools we have
developed (antibodies), immunoprecipitation is performed using the anti-GLUPEP
antibodies
and the protein is sequenced (e.g., the Harvard MicroChemistry Core facility
for micro-
sequencing) to confirm its identity.
51
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
Example 9 Development and characterization of small molecular weight
inhibitors of
MBL based on its interaction site with cytokeratin (Structure/activity
relationship analysis)
Our preliminary data support an important role for K in mediating complement
activation and MBL binding to HUVECs following oxidative stress. UEA-II and
GLUPEP
inhibit MBL deposition and the resulting complement activation in several
assays, as shown
above. A previous study has shown that specific AA substitutions within GLUPEP
increase
and decrease the affinity of anti-GIcNAc antibodies and lectins to the new
peptides
(Shikhman AR, Greenspan NS, Cunningham MW: Cytokeratin peptide SFGSGFGGGY,
SEQ ID NO. 2 mimics N-acetyl-beta-D- glucosamine in reaction with antibodies
and lectins,
and induces in vivo anti-carbohydrate antibody response. J.Immunol.
1994;153:5593-5606).
Our data are the first to demonstrate that MBL interacts with peptides or
proteins in the
absence of carbohydrate groups. The following prophetic experiments describe
the
evaluation and characterization of the actions of these peptides on MBL
binding, MBL
inhibition and complement activation. The data collected and analyzed in this
section are
useful for establishing the rank order potency of each peptide. These data are
then used to
evaluate the energy minimization of these conformations. These data are then
used to aid in
the development of small molecular weight inhibitors based on total organic
synthesis.
A determination of the structure/activity relationship for GLUPEP to interact
and
inhibit MBL is performed as follows. A previous study has shown that specific
AA
2o substitutions within GLUPEP increase and decrease the affinity of anti-
GIcNAc antibodies
and lectins (Shikhman AR, Greenspan NS, Cunningham MW: Cytokeratin peptide
SFGSGFGGGY, SEQ ID NO. 2 mimics N-acetyl-beta-D- glucosamine in reaction with
antibodies and lectins, and induces in vivo anti-carbohydrate antibody
response. J.Immunol.
1994;153:5593-5606). Table 2 summarizes the data obtained in that study. This
experiment
is performed to repeat these studies and to determine the functional activity
of these peptides
in relationship to MBL activity, binding and complement activation. As seen in
Table 2,
specific AA substitutions within the GLUPEP backbone either increases,
decreases or does
not alter the binding of a monoclonal antibody against GIcNAc. Specifically,
introduction of
lysine (K) at position 1, 2, 6 or 10 significantly increases antibody binding.
Similarly, WGA,
a lectin with specificity similar to GIcNAc, binding was increased or
decreased with the same
amino acid substitutions as outlined in Table 2. Aromatic substitution in
position 2 with
hydrophobic nonaromatic alanine significantly decreased binding. These
peptides are
52
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
synthesized (>95% pure) and tested in the functional assays disclosed herein.
First, the
peptides are screened for their ability to inhibit MBL-dependent complement
activation in the
keratin solid phase ELISA. IC50's are obtained for each peptide. Second, the
peptides are
coupled to BSA and their ability to activate complement via the lectin pathway
using purified
complement components (e.g., MBL, C2, C3 and C4) and/or HS is evaluated.
Preliminary
data using GLUPEP coupled to BSA demonstrated specific MBL binding that was
inhibited
by 3F8. Specific interactions of selected peptides with MBL are observed by
surface
plasmon resonance (Biacore). Selected peptides also are evaluated for their
ability to inhibit
complement activation following oxidative stress of HUVECs. The peptide
interaction with
to UEA-II also is evaluated.
Table 2. Reaction of an anti-GIcNAc mAb (clone CKB-1) to
GLUPEP and its substituted variants (adapted from4g). Similar
binding observations were observed with the legume lectin WGA
(i.e., a lectin inhibited by GIcNAc).
Peptide Amino Acid Sequence %Binding
1................10
GLUPEP SFGSGFGGGY, SEQ ID No. 2 100
1 TFGSGFGGGY, SEQ ID No. 6 81
2 AFGSGFGGGY, SEQ ID No. 7 77
3 DFGSGFGGGY, SEQ ID No. 8 28
4 KFGSGFGGGY, SEQ ID No. 9 222
SYGSGFGGGY, SEQ ID No. 10 100
6 SAGSGFGGGY, SEQ ID No. 11 25
7 SDGSGFGGGY, SEQ ID No. 12 7
g SKGSGFGGGY, SEQ ID No. 13 213
9 SFGSGKGGGY, SEQ ID No. 14 175
10 SFGSGFGGGF, SEQ ID No. 15 105
11 SFGSGFGGGA, SEQ ID No. 16 58
12 SFGSGFGGGD, SEQ ID No. 17 16
14 SFGSGFGGGK, SEQ ID No. 18 347
Specific utions are in BOLD.
substit
~e~e experiments tides
establish and
the structure/activity
relationship
of these
pep
MBL inhibition. The assays described herein are used to determine which of
these peptides
2o display "GIcNAc mimicry" and functionally inhibit MBL. These data then are
used to
evaluate the energy minimization of these AA conformations. These data are
then used to
develop small molecular weight inhibitors based on total organic synthesis.
IC50 values are
obtained for each peptide in each assay.
53
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
Example 10 Determination of other peptides that bind to the CRD region of MBL
Historically, MBL is known to only bind to carbohydrate groups. We have made
the
novel observation that MBL binds to a peptide (GLUPEP). Binding of MBL to
GLUPEP in a
solid phase ELISA was inhibited by excess GLUPEP, 3F8 or GIcNAc in the fluid
phase
(Figure 3). These data demonstrate that peptides bind to the CRD of MBL. Thus,
it is
possible that other families of peptides may bind to the CRD region of MBL and
inhibit its
function/binding. Identification of these peptide families will aid in the
development of small
molecular weight inhibitors derived by total organic synthesis.
1 o Other peptides binding to the CRD of MBL are mapped by using commercially
available phage display peptide libraries (New England Biolabs). Three
separate phage
display peptide libraries (New England Biolabs) displaying linear 7-mer (2x109
independent
clones), 7-mer disulfide constrained (3.7x109 independent clones) and linear
12-mer (1.9x109
independent clones) are used according to the manufacturers instructions.
These two 7-mer
libraries are sufficiently complex to contain most if not all of the 20'
possible 7-mer
sequences. The phage are plated and two plaque lifts are performed. The
membranes are
blocked and purified, functionally, active human MBL are incubated with the
membranes.
The membranes are washed in calcium and magnesium sufficient buffer and then
incubated
with our non-functional anti-human MBL mAb, 1 C 10, conjugated with HRP to
identify
MBL-positive plaques (ECL system). The second plaque lift is screened with
human MBL
that is functionally blocked with our other functionally inhibitory mAb to
human MBL (clone
3F8). Alternatively, GLUPEP is used to block the MBL CRD. Comparison of the
first
screen with the second screen eliminates those phage that bind to regions of
MBL other than
to the CRD. The positive colonies are picked, amplified and sequenced. The
corresponding
peptides encoded by these phage are synthesized and tested in our screening.
assays for
functional activity against MBL and binding to MBL. Binding of these peptides
with MBL is
confirmed with surface plasmon resonance (Biacore).
Alternatively, another approach is used if MBL binds to the bacteria used in
this
system. 1C10 is covalently coupled to protein A Sepharose with a commercially
available kit
(Pierce). This approach allows specific orientation of the mAb to allow for
efficient coupling
of functionally active MBL to 1 C 10. Coupling of MBL in this fashion allows
MBL's CRD
to be available for direct binding of the phage display peptide libraries.
Previous work by
54
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
Drs. Stahl and Klickstein have demonstrated by Biacore that the Kd of 1 C 10
is very low.
Saturating concentrations of 3F8 are then added to block the MBL's CRD and the
"column"
washed. The phage are biopanned across the Sepharose-1C10-MBL-3F8 "column".
Again,
GLUPEP can be used to block the CRD instead of 3F8. This may be a reasonable
approach,
as GLUPEP inhibits MBL (above-described results). Those phage that don't bind
to the
"column" are passed over another Sepharose-1C10-MBL "column" (i.e., no 3F8).
The phage
that bind to the MBL CRD (i.e., absence of 3F8 ~ functional CRD on MBL) are
eluted with
100 mmol/L GIcNAc, amplified and biopanned 3-4 more rounds to select those
peptides with
increased specificity of binding. Colonies then are picked and sequenced.
Example 11 Development of functionally inhibitory Fabs.
We have strong data demonstrating that MBL binds to a specific AA sequence
conserved in K. Development of functionally inhibitory Fabs (or recombinant
scFV) is
another therapeutic strategy for the functional inhibition of MBL binding to
this peptide
sequence. Based on the above-described experiments, additional Fabs to these
peptide
sequences, relevant to known inflammatory proteins, also are made.
We have coupled GLUPEP to BSA and KLH. Mice are immunized with purified K
and/or KLH-GLUPEP and hybridomas are made as we have described (Tofukuji M,
Stahl
GL, Agah A, Metais C, Simons M, Sellke FW: Anti-CSa monoclonal antibody
reduces
cardioplegia-induced coronary endothelia dysfunction. J. Thorac. Cardiovasc.
Surg.
1998;116:1060-1068). The supernatants of GLUPEP hybridomas are screened
against BSA-
GLUPEP to isolate only those clones that recognize GLUPEP. Monoclonal
antibodies from
IgG containing parent lines are obtained by limiting dilution. Fabs are made
for GLUPEP
mAbs. Fabs are used in complement inhibition assays, as classical complement
pathway
activation would be expected with the use of whole antibodies. Fabs and/or
whole antibodies
are assayed for their ability to 1) bind to keratin (i.e., K14/17), 2)
purification of K14/17 by
affinity chromatography from gamma-interferon stimulate human HeLa cells
(ATCC) or T84
cells (Dr. Colgan), 3) immunoprecipitation of K from HUVECs following
oxidative stress, 4)
inhibition (Fabs only) of MBL deposition to keratin and/or HUVECs following
oxidative
3o stress, and 5) confocal microscopy of keratin expression on HUVECs.
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
Kinetics, specificity and affinity of the Fabs are obtained against GLUPEP, K.
The
ability of the anti-K mAbs to block MBL binding to K (Biacore and solid-phase
ELISA) is
used as a screening tool to identify functionally inhibitory antibodies.
Once functionally inhibitory antibodies to GLUPEP (e.g., Fabs) are prepared
and
characterized, humanized scFv's are prepared for potential clinical use. (See,
e.g., USSN
60/112,390, entitled "Anti-Mannose (Mannan) Binding Lectin Therapy for
Ischemia/Reperfusion Injury" for a description of the methods useful for such
purposes.).
Since K17 is known to play an important role in psoriasis, allergic reactions
and other
inflammatory conditions that also involve complement activation, development
of specific
l0 inhibitors to cytokeratin should have broad utilization in many human
diseases.
Example 12 Complement activation during gastrointestinal ischemia and
renerfusion is
responsible for upre~ulation of the neutr~hil adherence molecule ICAM-1.
Adherence of
PMN within the gastrointestinal vasculature is dependent on MBL deposition and
C5
cleavage but not C3 degradation products.
We have previously demonstrated in vivo that inhibition of complement at the
level of
CS (inhibition of CSa and CSb-9 formation) decreases infarct size, apoptosis
and PMN
accumulation (Vakeva A, Agah A, Rollins SA, Matis LA, Li L, Stahl GL:
Myocardial
infarction and apoptosis after myocardial ischemia and reperfusion. Role of
the terminal
complement components and inhibition by anti-C5 therapy. Circulation
1998;97:2259-
2267). A recent study demonstrated that the C1 esterase inhibitor prevented
the upregulation
of P-selectin and ICAM-1 in the ischemic/reperfused rat heart in vivo (Buerke
M, Prufer D,
Dahm M, Oelert H, Meyer J, Darius H: Blocking of classical complement pathway
inhibits
endothelial adhesion molecule expression and preserves ischemic myocardium
from
reperfusion injury. Journal of Pharmacology and Experimental Therapeutics
1998;286:429-
438). However, these authors recognized that the C1 esterase inhibitor also
inhibits the MBL
pathway and conclusive data on the role of MBL in their study could not be
made (Buerke M,
Prufer D, Dahm M, Oelert H, Meyer J, Darius H: Blocking of classical
complement pathway
inhibits endothelial adhesion molecule expression and preserves ischemic
myocardium from
reperfusion injury. Journal of Pharmacology and Experimental Therapeutics
1998;286:429-
438). Others have reported that CSb-9 directly induces ICAM-1, MCP-1, ELAM and
IL-8 in
HUVECs (Kilgore KS, Flory CM, Miller BF, Evans VM, Warren JS: The membrane
attack
56
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
complex of complement induces interleukin-8 and monocyte chemoattractant
protein-1
secretion from human umbilical vein endothelial cells. Am.J.Pathol.
1996;149:953-961;
Kilgore KS, Schmid E, Shanley TP, Flory CM, Maheswari V, Tramontini NL, Cohen
H,
Ward PA, Friedl HP, Warren JS: Sublytic concentrations of the membrane attack
complex of
complement induce endothelial interleukin-8 and monocyte chemoattractant
protein-1
through nuclear factor-kappaB activation. Am.J.Pathol. 1997;150:2019-2031;
Kilgore KS,
Shen JP, Miller BF, Ward PA, Warren JS: Enhancement by the complement membrane
attack
complex of tumor necrosis factor-a-induced endothelial cell expression of E-
selectin and
ICAM-1. J.Immunol. 1995;155:1434-1441). CSb-9 and CSa also directly upregulate
CD62P
l0 on vascular endothelial cells (Mulligan MS, Schmid E, Till GO, Hugli TE,
Friedl HP, Roth
RA, Ward PA: CSa-dependent up-regulation in vivo of lung vascular P-selectin.
J.Immunol.
1997;158:1857-1861; Hattori R, Hamilton KK, McEver RP, Sims PJ: Complement
proteins
CSb-9 induce secretion of high molecular weight multimers of endothelial von
Willebrand
factor and translocation of granule membrane protein GMP-140 to the cell
surface.
J.Biol.Chem. 1989;264:9053-9060). Complement activation by immune complexes
induces
two rat chemokines, macrophage inflammatory protein-2 (MIP-2) and cytokine-
induced
neutrophil chemoattractant (CINC) (Shanley TP, Schmal H, Warner RL, Schmid E,
Friedl
HP, Ward PA: Requirement for C-X-C chemokines (Macrophage inflammatory protein-
2 and
cytokine-induced neutrophil chemoattractant) in IgG immune complex-induced
lung injury.
2o J.Immunol. 1997;158:3439-3448). PMN sequestration in the isolated
ischemic/reperfused rat
heart is dependent on complement and is inhibited by anti-CD18 or sCRI (Lefer
DJ,
Shandelya SML, Serrano CV, Jr., Becker LC, Kuppusamy P, Zweier JL:
Cardioprotective
actions of a monoclonal antibody against CD-18 in myocardial ischemia-
reperfusion injury.
Circulation 1993;88:1779-1787). We have demonstrated recently that CSb-9
induces
VCAM-1 protein expression by decreasing intracellular cGMP and inducing
nuclear
translocation of NFxB. The experiments presented below are intended to
investigate the
biochemical and molecular aspects of UEA-II treatment and CS inhibition on PMN
adherence
and the induction of ICAM-1 in the ischemic/reperfused rat GI tract.
The SMA of rats are occluded for 1 hr and reperfused (2-6 hrs). The rats are
3o separated into the following groups: 1) sham; 2) vehicle control; 3) anti-
CS mAb (18A; 20
mg/kg); 4) UEA-II (0.01 -1 mg/kg); and 5) GLUPEP mimetic (dose and structure
to be
determined by the in vitro findings outlined above). (16C is used as the
isotype and
57
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
nonfunctional control for 18A as we have described (Vakeva A, Agah A, Rollins
SA, Matis
LA, Li L, Stahl GL: Myocardial infarction and apoptosis after myocardial
ischemia and
reperfusion. Role of the terminal complement components and inhibition by anti-
CS therapy.
Circulation 1998;97:2259-2267). Scrambled peptides are used as controls for
the GLUPEP
mimetic.)
At the end of the reperfusion period, the area at risk (blue dye negative) is
removed
and assessed for ICAM-1 mRNA expression by semi-quantitative RT-PCR. GAPDH or
(3-
actin is used as a housekeeping gene and control for mRNA loading. These
results are
confirmed by northern analysis and in situ hybridization coupled to confocal
microscopy as
1o we described (Collard CD, Bukusoglu C, Agah A, Colgan SP, Reenstra WR,
Morgan BP,
Stahl GL: Hypoxia-induced expression of complement receptor type 1 (0R1, CD35)
in
human vascular endothelial cells. Am.JPhysiol. 1999;276:0450-0458).
Commercially
available primer sets are used or designed from published sequences.
Immunohistochemical
staining for CD62P and ICAM-1 will be done as previously described (Buerke M,
Prufer D,
t 5 Dahm M, Oelert H, Meyer J, Darius H: Blocking of classical complement
pathway inhibits
endothelial adhesion molecule expression and preserves ischemic myocardium
from
reperfusion injury. Journal of Pharmacology and Experimental Therapeutics
1998;286:429-
438) . MPO activity is assessed as an index of PMN infiltration as described
(Vakeva A,
Agah A, Rollins SA, Matis LA, Li L, Stahl GL: Myocardial infarction and
apoptosis after
20 myocardial ischemia and reperfusion. Role of the terminal complement
components and
inhibition by anti-CS therapy. Circulation 1998;97:2259-2267).
We expect to observe complement activation (05b-9 deposition) and PMN
adherence
within the gastrointestinal tract in vehicle treated animals, but not in UEA-
II or the anti-CS
antibody (i.e., 18A) treated animals. We also expect to observe significant
increases in
25 ICAM-1 mRNA expression in the splanchnic region of vehicle treated rats and
decreased
expression within UEA-II or anti-CS treated animals. These data support our
previous
findings for a role of CS cleavage products in the upregulation of PMN
adherence molecules
and extend these findings to show the effectiveness of UEA-II in vivo (Vakeva
A, Agah A,
Rollins SA, Matis LA, Li L, Stahl GL: Myocardial infarction and apoptosis
after myocardial
30 ischemia and reperfusion. Role of the terminal complement components and
inhibition by
anti-CS therapy. Circulation 1998;97:2259-2267). We expect to observe
decreased C3
deposition in UEA-II treated animals, but do not expect iC3b to play a major
role as a PMN
ss
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
ligand as we demonstrated previously in the myocardium (Vakeva A, Agah A,
Rollins SA,
Matis LA, Li L, Stahl GL: Myocardial infarction and apoptosis after myocardial
ischemia and
reperfusion. Role of the terminal complement components and inhibition by anti-
CS therapy.
Circulation 1998;97:2259-2267). Experiments using the anti-CS antibody and UEA-
II
demonstrate that MBL activates complement and the terminal CS molecule is
necessary for
adherence molecule expression in the ischemic/reperfused gut. It is possible
that the
production of reactive oxygen species in these experiments increases adherence
molecule and
cytokine expression (Gasic AC, McGuire G, Krater S, Farhood AI, Goldstein MA,
Smith
CW, Entman ML, Taylor AA: Hydrogen peroxide pretreatment of perfused canine
vessels
to induces ICAM-1 and CD18-dependent neutrophil adherence. Circulation
1991;84:2154-
2166). Indeed, we have demonstrated that hypoxia enhances cytokine mediated
ICAM
expression in human endothelial cells (Zund G, Uezono S, Stahl GL, Dzus AL,
McGowan
FX, Hickey PR, Colgan SP: Hypoxia enhances induction of endothelial ICAM-1:
role for
metabolic acidosis and proteasome activation. Am.J.Physiol. 1997;273:01571-
01580). We
believe that the complement system however, will amplify this response and
increase the
overall inflammatory condition. This finding is consistent with our previous
in vitro findings
(Zund G, Uezono S, Stahl GL, Dzus AL, McGowan FX, Hickey PR, Colgan SP:
Hypoxia
enhances induction of endothelial ICAM-1: role for metabolic acidosis and
proteasome
activation. Am.J.Physiol. 1997;273:01571-01580). Thus, inhibition of
complement at CS
or UEA-II treatment attenuates complement-mediated increases in the
inflammatory process.
0D47 is involved in PMN trafficking in the gastrointestinal system (Parkos CA,
Colgan SP,
Liang TW, Nusrat A, Bacarra AE, Carnes DK, Madara JL: CD47 mediates post-
adhesive
events required for neutrophil migration across polarized intestinal
epithelia. J. Cell Biol.
1996;132:437-450). We have also shown that CSb-9 induces CD47 expression in
epithelial
cells. We also investigate the effect of anti-CS and UEA-II treatment on this
important PMN
adherence molecule. At least 6-10 rats are used in each group to establish a
mean and SEM.
Appropriate statistical analysis is done.
Through different experimental techniques, we are able to establish the
affinities of
GLUPEP (this is possible for other conjugates and/or amino acid sequences
related to
3o GLUPEP too). Further, with this new experimental application (i.e., GIcNAc-
BSA and
BIAcore) we are able to ligand fish either chemical, phage display and other
libraries for
those molecules that either bind to GIcNAc-BSA and inhibit MBL binding or bind
to MBL
59
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
and inhibit MBL binding to the GIcNAc-BSA chip. This new technique will allow
us to
rapidly screen hybridomas that we make to keratin or other MBL ligands to
identify those
antibodies that specifically bind to the ligand and inhibit MBL binding. As
shown in Figure
6, GLUPEP inhibited MBL binding to GIcNAc-BSA in a concentration related
manner.
Specific affinities of these novel inhibitors will be calculated using the
BIAcore 3000.
(GLUPEP inhibited native MBL (1 Op.g/ml) binding in a concentration dependent
manner,
The beginning and ending of the MBL injection on the chip is denoted in the
figure with
arrows. Time is in seconds.)
1o Example 13 UEA-II Si~nificantly Decreases Oxidative Stress Induced
Neutronhil
Chemotaxis in Endothelial Cells.
As previously demonstrated, UEA-II recognizes a MBL ligand on human
endothelial
cells following oxidative stress. By selectively blocking MBL binding to its
ligand, UEA-II
inhibits complement activation in HUVEC cells. The activation of the
complement pathway
on endothelial cells leads to the generation of potent chemoattractants
including the
anaphylatoxin CSa, IL-8, and monocyte chemoattractant protein (Kilgore, K.S.,
et al., Am. J.
Pathol. 149, 953-961 (1996), Saadi, S., Circulation 101, 1867-1873)).
Neutrophil chemotaxis
following oxidative stress was therefore utilized as an assay of UEA-II
activity in HUVEC
cells. We hypothesized that UEA-11 would decrease complement-mediated
neutrophil
chemotaxis following endothelial oxidative stress.
Reoxygenation of hypoxic HUVEC in HS significantly (p < 0.05) increased
neutrophil chemotaxis compared to normoxic cells bathed in HS. As predicted,
treatment
with UEA-II (100 nmol/L). significantly attenuated neutrophil chemotaxis
following
endothelial oxidative stress compared to vehicle-treated cells. Neutrophil
chemotaxis to
HUVEC following oxidative stress was measured by analysis of myeloperoxidase
levels and
transformed to a neutrophil count with a standard curve using known numbers of
neutrophils.
This experiment was performed 3 times, with 3 wells per experimental group
(n=3).
Treatment of normoxic cells with UEA-II had no effect on fMLP driven
chemotaxis
compared to untreated normoxic HUVEC (data not shown), which demonstrates that
the
3o effect of UEA-II on chemotaxis is not a non-specific effect on PMN
function. These data
demonstrate that in endothelial cells UEA-II attenuates complement-mediated
neutrophil
chemotaxis induced by oxidative stress,
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
Methods
HUVEC Studies
HUVEC isolation. HUVECs will be isolated, cultured and purity established as
we
previously demonstrated from human umbilical veins (Collard CD, Vakeva A,
Bukusoglu C,
Zund G, Sperati CJ, Colgan SP, Stahl GL: Reoxygenation of hypoxic human
umbilical vein
endothelial cells (HUVECs) activates the classic complement pathway.
Circulation
1997;96:326-333; Collard CD, Agah A, Stahl GL: Complement activation following
reoxygenation of hypoxic human endothelial cells: role of intracellular
reactive oxygen
1o species, NF- kappaB and new protein synthesis. Immunopharmacology
1998;39:39-50; Zund
G, Uezono S, Stahl GL, Dzus AL, McGowan FX, Hickey PR, Colgan SP: Hypoxia
enhances
induction of endothelial ICAM-1: role for metabolic acidosis and proteasome
activation.
Am.J.Physiol. 1997;273:01571-01580) . Cells will be used during passage 1-3.
Cells will be
grown in flasks, petri dishes and 96-well plates as required for the
experiments outlined in
15 this proposal.
Hypoxia induction. Hypoxia will be induced as we have previously demonstrated
(Collard CD, Vakeva A, Bukusoglu C, Zund G, Sperati CJ, Colgan SP, Stahl GL:
Reoxygenation of hypoxic human umbilical vein endothelial cells (HUVECs)
activates the
classic complement pathway. Circulation 1997;96:326-333; Collard CD, Agah A,
Stahl GL:
2o Complement activation following reoxygenation of hypoxic human endothelial
cells: role of
intracellular reactive oxygen species, NF- kappaB and new protein synthesis.
Immunopharmacology 1998;39:39-50; Zund G, Uezono S, Stahl GL, Dzus AL, McGowan
FX, Hickey PR, Colgan SP: Hypoxia enhances induction of endothelial ICAM-1:
role for
metabolic acidosis and proteasome activation. Am.J.Physiol. 1997;273:01571-
01580) .
2s Briefly, the cells are placed in a commercially available, microprocessor
controlled, sealed,
humidified, temperature controlled and gloved chamber (Coy). Oxygen
concentration is
regulated to 1 % 02 and maintained for the experimental period. Cells are
placed in the
chamber for the specified period of time, removed and assayed as described in
the Research
Plan outlined above. This amount of oxygen decreases media P02 to 14-16 mm Hg
within 8
3o hr (unpublished observations). Our previous findings demonstrate that
complement
activation during hypoxia is augmented by reoxygenation in a time-dependent
manner
(Collard CD, Vakeva A, Bukusoglu C, Zund G, Sperati CJ, Colgan SP, Stahl GL:
61
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
Reoxygenation of hypoxic human umbilical vein endothelial cells (HUVECs)
activates the
classic complement pathway. Circulation 1997;96:326-333; Collard CD, Agah A,
Stahl GL:
Complement activation following reoxygenation of hypoxic human endothelial
cells: role of
intracellular reactive oxygen species, NF- kappaB and new protein synthesis.
Immunopharmacology 1998;39:39-50) .
Cell surface biotinylation. Cell surface biotinylation of proteins will be
done as we
previously described (Collard CD, Vakeva A, Bukusoglu C, Zund G, Sperati CJ,
Colgan SP,
Stahl GL: Reoxygenation of hypoxic human umbilical vein endothelial cells
(HUVECs)
activates the classic complement pathway. Circulation 1997;96:326-333).
to MBL ELISA. Turner and colleagues have previously described the MBL ELISA
(Super M, Levinsky RJ, Turner MYV.~ The level of mannan-binding protein
regulates the
binding of complement- derived opsonins to mannan and zymosan at low serum
concentrations. Clin.Exp.Immunol. 1990; 79:144-150). Briefly, 96-well
microtiter plates are
coated with mannan (500 p.g/ml; 50 p1 each well). Human sera (1-4%) with or
without
GIcNAc (30 mmol/L) is added to the wells and incubated for 30 min at
37°C. The plates are
then washed and developed with a polyclonal antibody to human C3 (Cappel) as
previously
described (Collard CD, hakeva A, Bukusoglu C, Zund G, Sperati CJ, Colgan SP,
Stahl GL:
Reoxygenation of hypoxic human umbilical vein endothelial cells (HUVECs)
activates the
classic complement pathway. Circulation 1997; 96: 326-333). MBL-dependent C3
deposition
2o to plastic is inhibited by GIcNAc or our functionally inhibitory mAbs
against MBL in this
assay. We will use this assay to screen functionally inhibitory peptides, to
evaluate
pharmacokinetic and pharmacodynamic properties of the peptides, and to assess
therapeutic
doses of MBL inhibitors ex vivo.
Keratin ELISA. We have developed this assay along the same lines as the MBL
ELISA. Briefly, 96-well microtiter plates are coated with keratin (2 ~.g/ml;
50 ~,1 each well).
Human sera (4-30%) with or without GIcNAc (30 mmol/L) or 3F8 ( 2 ~g/ml) is
added to the
wells and incubated for 30 min at 37°C. The plates are then washed and
developed with a
polyclonal antibody to human C3 (Cappel) as previously described (Collard CD,
l~akeva A,
Bukusoglu C, Zund G, Sperati CJ, Colgan SP, Stahl GL: Reoxygenation of hypoxic
human
umbilical vein endothelial cells (HUVECs) activates the classic complement
pathway.
Circulation 1997; 96: 326-333). MBL-dependent C3 deposition to plastic is
inhibited by
GIcNAc or 3F8 (our functionally inhibitory mAb against MBL) in this assay. We
will use
62
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
this assay to screen functionally inhibitory peptides and Fabs, to evaluate
pharmacokinetic
and pharmacodynamic properties of the peptides, and to assess therapeutic
doses of MBL
inhibitors ex vivo.
Antibody production. Antigenic material is purified and emulsified with Titer
Max.
(NOTE: We have unpublished observations that the immune response to Titer Max
is greater
than Freund's adjuvant, giving higher antibody titers in a shorter time span
and a
predominance of IgG subclasses and virtually no IgM.) The mice or rabbits are
immunized
first with the Titer Max suspension and boosted weekly for 3-4 weeks to
produce the immune
response. Rabbits are then bled and the polyclonal antibodies purified by
protein G
1 o purification. Mice spleens are removed and fused to produce hybridomas as
previously
described (Tofukuji M, Stahl GL, Agah A, Metais C, Simons M, Sellke FW: Anti-
CSa
monoclonal antibody reduces cardioplegia-induced coronary endothelia
dysfunction.
J. Thorac. Cardiovasc. Surg. 1998;116:1060-1068). Hybridomas are then screened
for those
producing only IgG isotypes and recognizing the antigen. The secondary screen
is a
functional screen and those clones of interest are limited diluted to produce
a monoclonal cell
line. Antibodies are isotyped (Gibco) and purified from tissue culture
supernatant by Protein
A/G affinity chromatography.
Neutrophil Chemotaxis Assay. HUVEC were grown to confluence on 24-well plates
and then subjected to 0 or 24 hour of hypoxia. Following the specified period
of normoxia or
2o hypoxia, the media was aspirated and the cells reoxygenated (3 hours) in
the presence of 30%
HS or 30% HS treated with UEA-II (100 nmol/L). During the reoxygenation
period, human
neutrophils were harvested and isolated as previously described (Henson, P.M.
et al., J. Clin.
Invest. 56, 1053-1061 (1975)). Five-micron transwell inserts (Corning Costar,
Cambridge,
MA) were then placed in each well of the reoxygenated HUVEC. Human neutrophils
(2 x
106 cells/well) were added to each transwell and incubated for 90 min at
37° C. The
supernatant covering the HUVEC was removed and centrifuged at 150 x g for 10
minutes.
The resulting pellet was resuspended in 1 ml of HBSS, solubilized with 50 ~1
of 10% Triton x
-100 and acidified with 100 ~1 of citrate buffer (1 mol/L pH 6.5). The
myeloperoxidase
(MPO) content of the wells was then assayed as previously described (Parkos,
C.A., et al., .l.
3o Cell Biol. 117, 757-764 (1992)).
63
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
Molecular Biology
RT-PCR. Rat tissue will be removed and frozen in liquid nitrogen. Total RNA
from
homogenized rat tissue or HUVECs is extracted using a commercially available
product
containing guanidinium isothiocyanate/chloroform (TRIzoI, Life Technologies).
The poly A
mRNA is purified by oligo(dT) cellulose (Promega). First strand cDNA synthesis
is
constructed using reverse transcription, 1 ~g poly A mRNA and a commercially
available kit
(Promega). cDNA amplification of rat ICAM-1 will be done using primers sets as
described
(Beck-Schimmer B, Schimmer RC, Schmal H, Flory CM, Friedl HP, Pasch T, Ward
PA:
Characterization of rat lung ICAM-1. Inflamm.Res. 1998;47:308-315; Lee SK,
Park JY,
1o Chung SJ, Yang WS, Kim SB, Park SK, Park JS: Chemokines, osteopontin, ICAM-
1 gene
expression in cultured rat mesangial cells. J.Korean.Med.Sci. 1998;13:165-170)
. Rat (3-
actin or GAPDH (housekeeping genes) will be used to control for RNA loading
conditions
and run in the gels with the other PCR products. Northern analysis and in situ
hybridization
for will be used to validate the PCR findings as we have described (Collard
CD, Bukusoglu
C, Agah A, Colgan SP, Reenstra WR, Morgan BP, Stahl GL: Hypoxia-induced
expression of
complement receptor type 1 (0R1, CD35) in human vascular endothelial cells.
Am.J.Physiol.
1999;276:0450-0458). Controls for RT-PCR will include PCR in the absence of
RT. Primer
sets for human keratin are listed below.
Human keratin 14: Forward: TTCTGAACGAGATGCGTGAC, SEQ ID NO. 19
(product size: 448 bp)
Reverse: AGAACTGGGAGGAGGAGAG, SEQ ID NO. 20
Human keratin 17: Forward: ATTGGCAGCGTGGGAGGA, SEQ ID NO. 21
(product size: 332 BP)
Reverse: AGACTGTGGGGCAGATGG, SEQ ID NO. 22
Peptide phage display library. To map potential binding sites of peptides to
MBL,
we will use phage display peptide libraries from New England Biolabs (NEB)
according to
their instructions. NEB and others have used these libraries to identify
consensus peptide
binding sequences against streptavidin, monoclonal and polyclonal antibodies,
RNase A,
3o PAP kinase and cell-surface receptors (communication from NEB). Specific
methods are
given by the manufacture and will be modified as we have described in the
Research Design
64
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
Section 3b. It takes a minimum of 4-6 weeks for a single round of biopanning
and
sequencing for each screening.
MBL purification. We have successfully purified human, rat and porcine MBL
using
a modification of published procedures (Tan SM, Chung MCM, Kon OL, Thiel S,
Lee SH,
Lu J: Improvements on the purification of mannan-binding lectin and
demonstration of its
CaZ+-independent association with a Cls- like serine protease. Biochem.J.
1996;319:329-
332). This method isolates MBL associated with MASP1 and MASP2. Briefly,
plasma is
precipitated with PEG3500 (10%; w:v). The pellet is then dissolved in a
calcium buffer and
applied to a mannan affinity column (Sigma). Bound material is eluted from the
mannan
to column with EDTA containing buffer. Protein positive tubes are collected
and re-calcified
and applied to a small maltose column. MBL is eluted from the maltose column
with
GIcNAc (100 mmol/L). The MBL is dialyzed against PBS containing 0.5 mmol/L
NaCI,
sterile filtered and stored at 4°C. Western analysis of this material
demonstrates MBL and
the lack of contaminating IgG or IgM.
Isolation and purification of human CKl. Human CK1 was purified from human
dermal keratin (Sigma, St. Louis, MO) using a monospecific rabbit anti-human
CK1
polyclonal antibody (pAb) (Convance/BAbCO, Richmond, CA) conjugated to protein
G
Sepharose (ImmunoPure Protein G IgG Plus orientation Kit; Pierce, Rockford
IL). After
equilibrating the protein G column with binding buffer (10 mmol/L TRIS, pH
7.5), human
2o keratin was loaded onto the column. The column was then washed extensively
with binding
buffer and eluted with 0.1 mol/L glycine-HCl buffer, pH 2.8, with the eluent
being collected
in 0.5 ml fractions containing 1 mol/L TRIS-HCl buffer (1:10; v:v), pH 9.5.
The protein-
containing fractions were pooled, dialyzed overnight in 10 mmol/L TRIS buffer,
pH 7.5, and
the protein concentration determined.
Generation of anti-human keratin antibodies and Fab fragments. Male NZW
rabbits (Harlan, Indianapolis, IN) were immunized initially with human keratin
(100 ~.g, s.c.)
in TiterMax (Sigma, St. Louis, MO) and then with human keratin (50 pg, s.c.)
in PBS on a
biweekly basis for 6 weeks. Two weeks after the last immunization, the animals
were bled
and the resultant pAb purified by protein G affinity chromatography. All pAb
were dialyzed
3o against PBS, concentrated and sterile filtered.
Polyclonal anti-human keratin Fab fragments were generated by digesting anti-
human
keratin pAb with papain (Sigma, St. Louis, MO) for 16 hr at 37 °C. The
reaction was
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
terminated with iodoacetamide (Sigma, St. Louis, MO). The resulting mixture
was then
dialyzed in PBS, pH 8.0 overnight at 4 °C. Any remaining whole IgG and
the Fc portion of
the anti-human keratin pAb were removed from the mixture by protein A affinity
chromatography. Fab fragment generation was confirmed by SDS-PAGE.
HUVEC CKI ELISA. Confluent HUVEC were subjected to 0 or 24 hr of hypoxia
(1% OZ). The cell media were aspirated and 100 ~,1 of gelatin-veronal buffer
(GVB)
containing Caz+/Mg2+ added to each well. The cells were then reoxygenated for
3 hr at 37 °C,
washed and fixed with 1% paraformaldehyde (Sigma, St. Louis, MO) for 30 min.
After
washing, the cells were incubated with 50 ~1 of rabbit anti-human CK1 pAb
(1:500 dilution;
1 o Convance/BAbCO, Richmond, CA) or anti-porcine C7 pAb (20 ~,g/ml; isotype
control) for 1
hr at 4 °C. After washing, 50 ~1 of peroxidase-conjugated goat anti-
rabbit pAb was added to
each well and incubated for 1 hr at 4 °C. The plates were washed,
developed with 50 p1 of
ABTS [2,2'-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid] and read
(Molecular Devices,
Sunnyvale CA) at 405 nm. Background optical density was determined from cells
to which
15 only the secondary antibody was added, and was subtracted from all groups.
This experiment
was performed 3 times using 3 wells per experimental group (n=3).
Immunoprecipitation and Sequencing of HUVEC CKI. To confirm the specificity
of the anti-human CK1 pAb used in these experiments, HUVEC CK1 was
immunoprecipitated and sequenced. Confluent HUVEC cultures grown in 100 mm
Petri
2o dishes were subjected to 24 hr of hypoxia followed by 3 hr of reoxygenation
in the presence
of GVB. The cells were then washed with ice cold GVB and incubated with lysing
buffer
(150 mmol/L NaCI, 25 mmol/L Tris, 1 mmol/L MgCL2, 1% Triton X-100, 1% Nonidet
P-40,
mmol/L EDTA, 5 ~g/ml chymostatin, 2 pg/ml aprotinin, and 1.25 mmol/L PMSF, pH
7.4,
all from Sigma Chemicals). Cell debris was removed by centrifugation (10,000 x
g; 5 min).
25 Cell lysates were pre-cleared with 50 ~1 of pre-equilibrated protein-G
Sepharose (Pharmacia,
Uppsala, Sweden) overnight at 4 °C. CK1 immunoprecipitation was
performed by addition
of rabbit anti-human CKl pAb (4 ~.g/ml; Convance/BAbCO, Richmond, CA).
Following
centrifugation (10,000 x g; 5 min) and washing, the immunoprecipitates were
boiled in
reducing sample buffer and separated by SDS-PAGE. After staining with
Coomassie blue,
3o the resultant protein band was cut from the gel and sent to the Harvard
University Core
Microchemistry Facility for microsequencing.
66
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
C3 and MBL Deposition (ELISA) on Purified CKl. Purified human CK1 (50 ~,1; 2
~.g/ml in 15 mmol/L sodium carbonate, pH 9.6) was added to 96-well microtiter
plates for 12-
16 hr at 4 °C. After washing, the plates were blocked for 2 hr at room
temperature with 3%
bovine serum albumin (BSA) and washed again. HS (2% final concentration) was
incubated
with a) 100 mmol/L GIcNAc; b) 20 ~g/ml anti-human MBL mAb, 3F8 (Collard et
al., 2000);
or c) vehicle [veronal buffered saline (VBS) containing Ca2+/Mg2+] for 30 min
at room
temperature. The plates were then inoculated with 100 ~l of treated or
untreated HS, and
incubated for 30 min at 37 °C. The plates were washed and 50 ~1 of HRP-
conjugated goat
anti-human C3 pAb (1:2000 dilution; ICN, Aurora, OH) or rabbit anti-human MBL
pAb
~0 (R2.2; 1:500 dilution) added for 1 hr at room temperature. The plates were
then washed and
developed as described above. Background optical density was determined from
wells coated
with BSA only and was subtracted from all groups. This experiment was
performed 3-4
times using 3 wells per experimental group (n =3-4).
Immunoprecipitation and Western Blot of human CKl and MBL. Confluent
~5 HUVEC cultures grown on 100 mm Petri dishes were subjected to 0 or 24 hr of
hypoxia
followed by 3 hr of reoxygenation in the presence of GVB (for CK1 analysis) or
30% HS (for
MBL analysis). The cells were then washed with ice cold GVB and incubated with
lysing
buffer. Cell debris was removed by centrifugation (10,000 x g; 5 min). Cell
lysates were
pre-cleared with 50 ~.l of pre-equilibrated protein-G Sepharose. The lysates
were then
2o immunoprecipitated by addition of human MBL (90 fig) and 50 ~1 of anti-
human MBL mAb
( 1 C 10) (Collard et al., 2000) or anti-human CK 1 pAb (Convance/BAbCO,
Richmond; CA)
conjugated to protein-G Sepharose (ImmunoPure Protein G IgG Plus orientation
Kit; Pierce,
Rockford IL). Following centrifugation (10,000 x g; 5 min) and washing, the
immunoprecipitates were boiled in reducing sample buffer and separated by SDS-
PAGE.
25 The gel was electroblotted to nitrocellulose and blocked with 10% non-fat
dry milk (NFDM)
overnight at 4°C.
For CK1 analysis, anti-human CK1 pAb (1:500 dilution) was incubated with the
nitrocellulose in 3% NFDM for 1 hr at 4°C. The nitrocellulose was then
washed and
incubated with HRP-conjugated goat anti-rabbit pAb (1:1000 dilution; ICN,
Aurora, OH) for
3o 1 hr at 4°C. For MBL analysis, HRP-conjugated anti-human MBL mAb
(2A9; 1:2000
67
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
dilution) was incubated with the nitrocellulose in 3% NFDM for 1 hr at
4°C. The membranes
were then washed and developed with the ECL system (Amersham) and x-ray films
(Kodak).
HUVEC C3 and MBL ELISA. HUVEC C3 and MBL deposition following oxidative
stress was measured by ELISA as previously described (Collard et al., 2000).
HUVEC were
grown to confluence and then subjected to 0 (normoxia) or 24 hr of hypoxia (1
% 02). The
cell media were aspirated and 100 ~,l of one of the following was added to
each well: 1) 30%
HS, 2) GVB, 3) 30% HS + 100 mmol/L GIcNAc, 4) 30% HS + 50 pg/ml anti-human
keratin
pAb or 5) 30% HS + 20 p.g/ml anti-human keratin Fab fragments. The cells were
then
reoxygenated for 3 hr at 37 °C, washed and fixed with 1%
paraformaldehyde for 30 min.
to After washing, the cells were incubated with HRP-conjugated goat anti-human
C3 pAb
( 1:1000 dilution; Cappel, West Chester, PA) or anti-human MBL mAb, 1 C 10 (
1:1000
dilution) for 1 hr at 4 °C. The plates were then washed and developed
as described above.
Background optical density was determined from cells to which only the anti-
human C3 or
MBL antibody was added, and was subtracted from all groups. These experiments
were
t 5 performed 3 times using 4-6 wells per experimental group (n=3).
Immunofluorescent confocal microscopy. HUVEC were grown on LabTech tissue
culture microscope slides (NUNC) were subjected to 0 or 24 hr of hypoxia and
then
reoxygenated for 3 hr in 30% HS treated with PBS (vehicle), anti-human keratin
Fab
fragments (20 pg/ml) or GIcNAc (100 mmol/L). The slides were then washed in
PBS
2o containing calcium and magnesium and fixed in 4% paraformaldehyde for 15
min, washed
again and blocked with 10% goat serum. Human MBL deposition (green) was
identified
using biotinylated 1 C 10 and streptavidin-conjugated FITC (Jackson
Immunoresearch, West
Grove, PA). Human C3 deposition (green) was evaluated with a FITC-conjugated
goat anti-
human C3 F(ab')2 antibody (ICN, Aurora, OH). Following incubation with the
appropriate
25 antibodies, the slides were washed (x3; 10 min each) and incubated with
propidium iodide
(10 ~g/ml; Sigma). The slides were , then coated with anti-fade mounting media
(Molecular
Probes, Eugene, OR), covered and analyzed with a Zeiss confocal microscope as
previously
described (Collard et al., 1999). Controls with streptavidin-conjugated FITC
only were
processed as above, omitting the primary antibody to determine nonspecific
binding. All
3o analyses were conducted at the same pinhole, voltage and laser settings.
This experiment was
performed three times (n=3).
68
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
Statistical Analysis. All data presented represent the mean and SEM for n
determinations. Data analyses were performed using Sigma Stat (Jandel
Scientific, San
Rafael, CA). A p value of <0.05 was considered significant. Endothelial CK1
expression
and MBL / C3 deposition on purified CK1-coated plates were analyzed by one-way
analysis
of variance (ANOVA). Endothelial C3 and MBL deposition on normoxic vs. hypoxic
HUVEC were analyzed by two-way ANOVA. All pairwise multiple comparisons were
made
using the Student-Newman-Keuls test. MBL and C3 deposition on purified CK1-
coated
plates (ELISA; Fig. 3) were normalized to untreated 2% HS. Means ~ SEM of the
raw data
used for normalization are presented in the results and/or figure legends.
to
69
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
-1-
SEQUENCE LISTING
<110> The Brigham and Women's Hospital, Inc.
Stahl, Gregory
Lekowski, Robert
<120> COMPLEMENT INHIBITORS
<130> A0752/7003W0/HCL
<150> US 60/148,815
<151> 1999-08-13
<160> 22
<170> FastSEQ for Windows Version 3.0
<210> 1
<211> 13
<212> PRT
<213> Artificial Sequence
<220>
<223> Cytisus sessilifolius
<221> VARIANT
<222> (13)...(13)
<223> Arg OR Lys
<400> 1
Gln Ile Glu Gly Leu Lys Glu Glu Leu Ala Tyr Leu Xaa
1 5 10
<210> 2
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Cytisus sessilifolius
<400> 2
Ser Phe Gly Ser Gly Phe Gly Gly Gly Tyr
1 5 10
<210> 3
<211> 11
<212> PRT
<213> Artificial Sequence
<220>
<223> Cytisus sessilifolius
<400> 3
Asp Thr Tyr Phe Gly Lys Thr Tyr Asn Pro Trp
1 5 10
<210> 4
<211> 11
<212> PRT
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
-2-
<213> Artificial Sequence
<220>
<223> Cytisus sessilifolius
<400> 4
Asp Thr Tyr Phe Gly Lys Ala Tyr Asn Pro Trp
1 5 10
<210> 5
<211> 11
<212> PRT
<213> Artificial Sequence
<220>
<223> Cytisus sessilifolius
<400> 5
Asp Ser Tyr Phe Gly Lys Thr Tyr Asn Pro Trp
1 5 10
<210> 6
<211> 10
<212> PRT
<213> Cytisus sessilifoliusArtificial Sequence
<220>
<223> Cytisus sessilifolius
<400> 6
Thr Phe Gly Ser Gly Phe Gly Gly Gly Tyr
1 5 10
<210> 7
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Cytisus sessilifolius
<400> 7
Ala Phe Gly Ser Gly Phe Gly Gly Gly Tyr
1 5 10
<210> 8
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Cytisus sessilifolius
<400> 8
Asp Phe Gly Ser Gly Phe Gly Gly Gly Tyr
1 5 10
<210> 9
<211> 10
<212> PRT
<213> Artificial Sequence
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
-3-
<220>
<223> Cytisus sessilifolius
<400> 9
Lys Phe Gly Ser Gly Phe Gly Gly Gly Tyr
1 5 10
<210> 10
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Cytisus sessilifolius
<400> 10
Ser Tyr Gly Ser Gly Phe Gly Gly Gly Tyr
1 5 10
<210> 11
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Cytisus sessilifolius
<400> 11
Ser Ala Gly Ser Gly Phe Gly Gly Gly Tyr
1 5 10
<210> 12
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Cytisus sessilifolius
<400> 12
Ser Asp Gly Ser Gly Phe Gly Gly Gly Tyr
1 5 10
<210> 13
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Cytisus sessilifolius
<400> 13
Ser Lys Gly Ser Gly Phe Gly Gly Gly Tyr
1 5 10
<210> 14
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Cytisus sessilifolius
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
-4-
<400> 14
Ser Phe Gly Ser Gly Lys Gly Gly Gly Tyr
1 5 10
<210> 15
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Cytisus sessilifolius
<400> 15
Ser Phe Gly Ser Gly Phe Gly Gly Gly Phe
1 5 10
<210> 16
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Cytisus sessilifolius
<400> 16
Ser Phe Gly Ser Gly Phe Gly Gly Gly Ala
1 5 10
<210> 17
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Cytisus sessilifolius
<400> 17
Ser Phe Gly Ser Gly Phe Gly Gly Gly Asp
1 5 10
<210> 18
<211> 10
<212> PRT
<213> Artificial Sequence
<220>
<223> Cytisus sessilifolius
<400> 18
Ser Phe Gly Ser Gly Phe Gly Gly Gly Lys
1 5 10
<210> 19
<211> 20
<212> DNA
<213> Artificial Sequence
<220>
<223> Homo Sapiens
<900> 19
CA 02380979 2002-02-07
WO 01/12212 PCT/US00/22123
-5-
ttctgaacga gatgcgtgac 20
<210> 20
<211> 19
<212> DNA
<213> Artificial Sequence
<220>
<223> Homo Sapiens
<400> 20
agaactggga ggaggagag 19
<210> 21
<211> 18
<212> DNA
<213> Artificial Sequence
<220>
<223> Homo Sapiens
<400> 21
attggcagcg tgggagga 18
<210> 22
<211> 18
<212> DNA
<213> Artificial Sequence
<220>
<223> Homo sapiens
<400> 22
agactgtggg gcagatgg 18