Note: Descriptions are shown in the official language in which they were submitted.
CA 02383184 2002-03-13
WO 01/19849 PCT/SE00/01747
A PROCESS FOR THE PREPARATION OF H-TYR-D-ALA-PHE(F)-PHE-NHZ
FIELD OF THE INVENTION
The present invention is directed to a new process for the preparation of a
tetrapeptide,
more specifically the tetrapeptide H-Tyr-D-Ala-Phe(pF)-Phe- NH2, or a
pharmaceutically
acceptable salt thereof. In further aspects, the present invention also
relates to new
intermediates used in the process.
BACKGROUND AND PRIOR ART
io
WO 97/07129 discloses a process for producing inter alia the peptide H-Tyr-D-
Ala-
Phe(pF)-Phe-NH~. The peptide H-Tyr-D-Ala-Phe(pF)-Phe-NHS is also disclosed in
WO
97/07130. Said peptide exhibits peripheral analgesic activity and selectivity
for the ~.-
subtype of opioid receptors, and is particularly suitable in pain therapy.
Furthermore, it is
~s prepared using solid phase synthesis according to procedures well
established in the art.
The drawback with solid phase synthesis is that it is difficult to use in
large-scale
production, in addition to being expensive.
W099/47548 discloses a process for the preparation of the tetrapeptide H-Tyr-D-
Ala-
zo Phe(pF)-Phe- NHS using stepwise synthesis.
The process of the present invention provides the tetrapeptide H-Tyr-D-Ala-
Phe(pF)-Phe-
NH2 in a simpler manufacturing process with, e.g. easier purification of the
final product.
zs Thus, the object of the present invention is to provide a novel process
suitable for use in
large-scale synthesis. A further object of the present invention is to provide
a process
containing as few reaction steps as possible.
CA 02383184 2002-03-13
WO 01/19849 PCT/SE00/01747
OUTLINE OF THE INVENTION
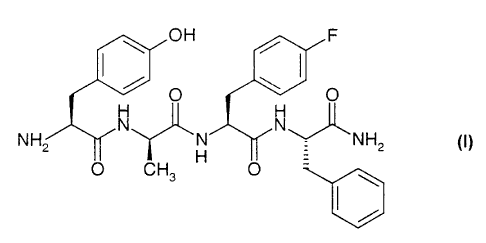
The present invention provides a new process for large-scale preparation of
the peptide H-
Tyr-D-Ala-Phe(pF)-Phe-NH~, which is the peptide of formula (I)
\ OH / F
/ \
O v O
2 ~"~ H 2
NH N N N~NH
O CH3 O \
/
or a pharmaceutically acceptable salt thereof.
The process according to the present invention for preparing the compound of
formula (I)
is a fragment synthesis (2+2). In a fragment synthesis a plurality of
intermediate
~o compounds, are prepared in parallel and then coupled together to give the
key
intermediates) or the final compound. This strategy should be compared to a
traditional
stepwise synthesis wherein a number of synthetic steps are performed
sequential. The
different approaches in a stepwise vs. fragment synthesis are schematically
shown in
Figure 1 below.
CA 02383184 2002-03-13
WO 01/19849 PCT/SE00/01747
3
.C__ _ D E _ F _. ..G . ~ product
Stepwise synthesis
A --~ 8 ~ C
G ~ product
D ~ E ~ F
Fragment synthesis
Figure 1. Stepwise vs. fragment synthesis
The process of the present invention can be described as comprising the steps
shown in
s Figure 2 below;
step 1 step z
+ ~ 4 ---~ 5
'3
step 3 step 4
----~ 1
6 step 1' step 2'
+--~ 8~ 9
7
Figure 2. Fragment synthesis according to the present invention
io St_ ep 1
A coupling step wherein an activated tyrosine derivative (II),
CA 02383184 2002-03-13
WO 01/19849 PCT/SE00/01747
4
ORZ
(II)
A_N R
H O
wherein
A is an amino protecting group, and
s R is an activating agent residue group, and
R2 is H or a benzyl-like group;
previously prepared by a pre-activation step or generated in situ, is reacted
with the amino
group of D-alanine, wherein the carboxyl group is protected as an ester, i. e.
a compound of
the formula D-Ala-Rl (III), wherein Rl is the ester group, e.g. OMe, in the
presence of a
io solvent, providing a protected dipeptide derivative (IV)
OR2
w/
O
A-N N R' (I~
H
O
CH3
wherein
~s A is an amino protecting group,
Rl is an ester residue group, and
R2 is H or a benzyl-like group;
Step 2 A deprotection step wherein a protected dipeptide derivative (IV)
prepared in the
~o previous step, is deprotected by treatment with aqueous base or acid to
give the dipeptide
CA 02383184 2002-03-13
WO 01/19849 PCT/SE00/01747
derivative (V),
~ORZ
O
A-N N OH
H
O
CH3
5
wherein
A is an amino protecting group, and
R2 is H or a benzyl-like group.
io However, if the activated tyrosine derivative is an activated ester or
urethane protected N-
carboxyanhydride (UNCA) of the structure (II')
OR2
O
/ N Q (II')
A
O
wherein
is A is an amino protecting group, and
R2 is H or a benzyl-like group
CA 02383184 2002-03-13
WO 01/19849 PCT/SE00/01747
6
then the carboxyl group of D-alanine needs no protection, i.e. can be a
compound of the
formula D-Ala-OH, and the coupling reaction thereby provides a protected
dipeptide
derivative V, which can be used directly in the step 3 without any further
deprotection.
s The preparation and use of UNCA-derivatives is discussed by Fehrentz et al.
( 1990. "The
use of N-urethane protected N-carboxyanhydrides (UNCAs) in amino acid and
peptide
synthesis." J. Pept. Sci., 1 (2), 124-131; and by Fuller et al, ( 1996).
"Urethane-protected a-
aminoacid N-carboxanhydrides and peptide synthesis." Biopolymers (Peptide
Science),
40(2), 183-205 which are incorporated herein by reference.
~o
Alternatively, the activated tyrosine derivative (II) can be reacted with the
amino group of
non-protected D-Ala, i.e. H-D-Ala-OH, providing dipeptide derivative (V)
directly.
St. ep 1'
i; A coupling step wherein an activated p-fluorophenylalanine derivative (VI),
F
(VI)
A-N
H O
wherein
~o A is an amino protecting group, and
R is an activating agent residue group;
previously prepared by a pre-activation step or generated in situ, is reacted
with the amino
group of phenylalanine, wherein the carboxyl group is protected as an ester or
amide, i. e. a
compound of the formula Phe-R1 (VII), wherein R1 is -NHS or an ester residue
group, e.g.
~s OMe, in the presence of a solvent, providing a protected dipeptide
derivative (VIII)
CA 02383184 2002-03-13
WO 01/19849 PCT/SE00/01747
7
F
1
A~ ~R
N ~N
H
(VIII)
wherein
A is an amino protecting Group, and
Rl is -NH2 or an ester residue group;
Step 2' A deprotection step wherein a protected dipeptide derivative (VIII)
prepared in the
previous step, is deprotected by either catalytic hydrogenation, base or acid
treatment,
io depending on the amino protecting group used, to give the dipeptide
derivative (IX),
F
O
~R1
H2N II H
O (IX)
i s wherein
Rl is -NHS or an ester residue group;
CA 02383184 2002-03-13
WO 01/19849 PCT/SE00/01747
8
St_ ep 3
A coupling step wherein an activated dipeptide derivative (V'),
OR2
I/
O
A-N N R (V')
H
O
CH3
s
wherein
A is an amino protecting group,
R is an activating agent residue group, and
R2 is H or a benzyl-like group
io previously prepared by a pre-activation step or generated in situ from
compound (V), is
reacted with the amino group of compound (IX) in the presence of a solvent,
providing a
protected tetrapeptide derivative (X)
ORz ~ F
~J
O O
A-N ~ H N
H O O R (X)
CH3
is wherein
A is an amino protecting group,
Rl is -NHS or an ester residue group, and
R2 is H or a benzyl-like group
CA 02383184 2002-03-13
WO 01/19849 PCT/SE00/01747
9
An additional transformation step is performed if the protected tetrapeptide
derivative (X),
prepared in the previous step, is an ester. Thus the ester compound (X)
wherein R ~ is an
ester residue group, e.g. OMe, is reacted with an amine in an organic alcohol,
preferably
ammonia in methanol, providing the protected dipeptide derivative (X'),
s
~ORz ~F
/~
O O
A_~ N H N~ 1
O O R (X')
CH3
/
wherein
A is an amino protecting group,
Rl is -NHS, and
io R2 is H or a benzyl-like group
Optionally, the additional step described above may be prepared on the
protected dipeptide
derivative (VIII), if Rl is an ester, whereby the ester compound (VIII) is
reacted with an
amine in an organic alcohol, preferably ammonia in methanol, providing the
protected
is dipeptide derivative (VIII'),
F
1
A\N R
H N
H
O (VIII')
CA 02383184 2002-03-13
WO 01/19849 PCT/SE00/01747
wherein
A is an amino protecting group, and
Rl is -NHS.
5
Stet A deprotection step wherein the protected tetrapeptide derivative (X) is
deprotected
either by catalytic hydrogenation, or treatment with acid or base, depending
on the amino
protecting group used, providing the final tetrapeptide (I), which optionally
may be
converted to a pharmaceutically acceptable salt thereof.
~o
A person skilled in the art will appreciate suitable amino, carboxyl and
tyrosine side-chain
protecting groups, which may be used in the present invention. The Na-amino
protecting
group may be selected from any protecting group suitable in peptide synthesis,
such as tert-
butoxycarbonyl (Boc), 9-fluorenylmetoxycarbonyl (Fmoc) or benzyloxycarbonyl,
often
~s abbreviated Z-, just to mention three possible amino protecting groups.
However,
benzyloxycarbonyl is particularly preferred to be used in the present
invention since it is
easily removed by catalytic hydrogenation, and contrary to the protecting
group Boc, it
does not require neutralization of the liberated amine. C 1-C6 alkyl esters
and alkylaryl
ester, such as benzyl, are preferred carboxyl protecting groups. Methyl esters
are
zo particularly preferred carboxyl protecting groups. Benzyl-like protecting
groups are
suitable tyrosine side-chain protecting groups to be used in the present
invention.
Preferably no tyrosine side-chain protecting group is used. Reference is made
to J.
Meienhofer in The Peptides, yol.l, Eds: E. Gross & J. Meienhofer, Academic
Press. Inc.
London 1979, pp. 26=l-309; The peptides, Vol. 1-9, E. Gross & J. Meienhofer.
Eds.,
as Academic Press Inc., London, 1979-1987: Houben-Weyl, Methoden der
organischen
Chemie, E. Muller, ed , Iiol. 1 ~. Part I-II> Thieme, Stuttgart 197=1; and M.
Bodans~kv,
Principles ofpeptide Synthesis. Springer Verlag, Berlin 198-1.
The pre-activation step preceding steps 1, 1' and 3, or the in situ generation
of the activated
3o amino acid derivatives (II), (VI) and (V'), is achieved by reacting an
amino acid, wherein
CA 02383184 2002-03-13
WO 01/19849 PCT/SE00/01747
the amino function has been protected by a suitable protecting group, such as
tert-
butoxycarbonyl (Boc), 9-fluorenylmetoxycarbonyl (Fmoc) or benzyloxycarbonyl
(Z),
which are either commercially available or available by techniques known in
the art, with
an activating agent in the presence of a suitable amine and an organic
solvent, to give the
activated amino acid derivative. A schematic representation of a pre-
activation step is
shown below;
F
F activating agent
O
_ ~R
O A ~ O
aminoacid derivative activated aminoacid derivative
wherein
io A is an amino protecting group, and
R is an activating agent residue group;
For the coupling step, in Steps l, 1'and 3 described above, a variety of
powerful solvents
may be used, as long as the amino component is essentially soluble and
available for
i s immediate reaction with the activated peptide derivative. Examples of
suitable solvents for
the coupling step are acetone, acetonitrile, DMF, N-methyl pyrrolidone (NMP),
EtOAc,
and mixtures thereof.
As used herein, the term " benzyl-like group " denotes any substituted or un-
substituted
zo benzyl group that is hydrogenolyzed under similar reaction conditions as
the
benzyloxycarbonyl group.
The term "pF" denotes apara-fluoro substituent.
CA 02383184 2002-03-13
WO 01/19849 PCT/SE00/01747
12
The term "C~-C6 alkyl" denotes a cyclic or linear, straight or branched,
substituted or
unsubstituted alkyl group having from 1 to 6 carbon atoms. Examples of said
alkyl include,
but are not limited to methyl, ethyl, n-propyl, iso-propyl, n-butyl, iso-
butyl, sec-butyl, tert.-
butyl, cyclohexyl, and cyclopentyl.
The term "substituted" denotes a Group that is substituted by one or more C,-
C6 alkyl, C,-
C6 alkoxy, halogen, amino, thiol, nitro, hydroxy, C,-C6 acyl or cyano groups.
io Possible as well as preferred reagents and reaction conditions in each step
are the
following.
The pre-activation or in situ Generation step
Suitable activating agents may be selected from those that generates any of
the commonly
~s used activated amino acid derivatives including, but not limited to,
carbodiimides,
activated esters, azide, or anhydrides. Isobutylchloroformiate (iBuOCOCI) and
2-(1H-
Benzotriazole-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate (TBTU) are
the preferred
activating agents together with UNCA-derivatives. The amount of activating
agent is
between 0.9-1.2 molar equivalents, preferably 0.95-1.05 equivalents. From a
practical
~o point of view the amount of activating agent shall be as close to 1.0 as
possible. When
isobutylchloroformiate (iBuOCOCI) is the activating agent, the activated
peptide derivative
will have the following structure, exemplified on D-alanine,
H3C -iBu
O
A'N \\ O
H O
The suitable amine may be selected from any tertiary amine. However, NMM
(N-methylmorpholine), di-isopropylethylamine and triethylamine are preferred.
The
amount of amine is between 0.9-2.0 molar equivalents, compared to the acid,
and
CA 02383184 2002-03-13
WO 01/19849 PCT/SE00/01747
13
preferable between 0.95 to 1.5 molar equivalents. From a practical point of
view the
amount of suitable amine shall be at least equal to the molar amount of
activating agent
used.
The organic solvent may be any organic solvent known to a person skilled in
the art to be
suitable in peptide chemistry. However, ethyl acetate, acetonitrile, acetone,
tetrahydrofurane, DMF as well as mixtures thereof are preferred solvents in
the pre-
activation step.
io The coupling step; Steps 1, 1' and 3
The solvent used for the coupling step may be selected from a variety of
solvents, as long
as the amino component is essentially soluble and available for immediate
reaction with
the activated amino acid residue. Examples of suitable solvents for the
coupling steps are
is acetone, acetonitrile, DMF, N-methyl pyrrolidone (NMP), EtOAc, and mixtures
thereof, of
which acetone, EtOAc, NMP and DMF are preferred.
Any temperature where the activated amino acid derivative is not degraded or
the reaction
rate is too slow may be used. The preferred range when isobutyl chloroformate
is used as
zo the activating agent is from 0°C to -20°C, and particularly
preferred is from -5°C to -15°C.
The preferred range when TBTU is used as the activating agent is around room
temperature. The rate of addition is in both cases adjusted so that the
preferred temperature
is maintained in the reaction mixture.
zs The deprotection step; Step 2' and 4
The catalyst used for hydrogenation may be selected from a great variety of
catalysts as
will be appreciated by a person skilled in the art. However 5% Pd on carbon is
preferred.
Any solvent that can dissolve at least some of the peptide is possible to use
except ketones,
such as acetone, or those solvents that poison the catalyst or react with the
components of
CA 02383184 2002-03-13
WO 01/19849 PCT/SE00/01747
14
the reaction. A person skilled in the art will appreciate the choice of
solvent. DMF and
NMP are the preferred solvents.
The deprotection step; Step 2
Hydrolysis of the ester residue group in compound (IV) can be achieved by any
method
known to the skilled person, e.g. aqueous acid, base treatment or
hydrogenolyzis,
depending on the carboxyl protecting group used. C1-C6 alkyl esters are
preferred esters
and treatment with aqueous base under standard conditions is the preferred
method for
io ester hydrolysis.
In a preferred embodiment of the present invention the protected amino acid,
preferably
using benzyloxycarbonyl- as Na-amino protecting group, is activated as a mixed
anhydride
with isobutyloxycarbonylchloride, or a similar type of chloroformate. The
method
is employed is based on the general method reviewed by J. Meienhofer in The
Peptides,
Vol.l, Eds: E. Gross & J. Meienhofer, Academic Press, Inc, London 1979, pp.
264-309.
We have surprisingly found that the activation time can be extended to at
least 30 min at a
temperature about 0 - -15°C, contrary to the recommended 1-2 min at -
15°C. We also
zo found that strictly anhydrous conditions are not necessary as otherwise is
recommended.
This allows the present method to be used for large-scale production where the
longer
reaction times allow a safe and reproducible process to be carried out. The
stereochemical
integrity has been completely maintained and the chemical purity as well as
yields have
been typically over 90%. The generated mixed anhydride is coupled with the
slow addition
zs of the amino component (amino acid/ peptide amide or ester) at about 0 - -
IS°C and the
reaction mixture is then allowed to reach 20-30°C in about 30-60 min.
or longer before
crystallization of the product is initiated directly from the reaction
mixture.
We have also surprisingly found that when using the present method, if
appropriately
3o selected solvent combinations are used, there is no need for a separate
washing step prior
CA 02383184 2002-03-13
WO 01/19849 PCT/SE00/01747
to crystallization. DMF, acetonitrile, EtOAc and water are preferably used as
solvents. A
controlled crystallization achieves an excellent purification, shortens the
filtering or
centrifugation time during work up, as well as shortens the drying time, if
dry
intermediates are required. One important factor is to generate sufficiently
large crystals
s with a relatively narrow size distribution not to block the filter medium or
centrifugation
cloth. It is very common for peptides in particular to generate gels or
amorphous crystals
that are almost impossible to filter.
We have further surprisingly found that isobutylchloroformiate can be used in
the key step
io in the fragment synthesis of the present invention, i.e. coupling step 3,
without any
substantial racemization of the D-alanine aminoacid fragment.
INTERMEDIATES
is Another object of the present invention is to provide new intermediates
that can be used in
the preparation of compound of formula I.
Accordingly, a further aspect of the present invention is a compound of the
general
formula X'
OR2 ~ F
J
0 0
A_ N ~ N
O R
CH3
zo (X' )
wherein
CA 02383184 2002-03-13
WO 01/19849 16 PCT/SE00/01747
A is tert-butoxycarbonyl (Boc), 9-fluorenylmethoxycarbonyl (Fmoc),
benzyloxycarbonyl
(Z) or a substituted benzyloxycarbonyl derivative;
R' is -NHZ,C,-C6 alkyl ester, benzyl ester, OH, 9-fluorenylmetyl ester or a
substituted
benzyl ester derivative:
RZ is H, a benzyl-like group, tert. butyl group, or 9-flourenylmethyl group,
as intermediates for use according to the present invention.
DETAILED DESCRIPTION OF THE INVENTION
The preparation of the peptide H-Tyr-D-Ala-Phe(pF)-Phe-NH, or a
pharmaceutically
acceptable salt thereof, will now be described in more detail by the following
Examples,
which should not be construed as limiting the invention. Furthermore, Scheme 1
below
provides a detailed overview of the synthetic route followed for the
preparation of the
peptide of the formula (I) according to the present invention using an alanine
derivative,
wherein the carboxyl group is protected as an amid. The compound numbers
referred to
in the Examples below corresponds to the compound numbering in Scheme 1.
/ \
F CHs ~ / ~ F +H2N
HsC \ / , O
O CN~ NHZ
/ \ O~N OH +H3 O~C~ O O O~O~H3 H_Phe-NH
H O EtOAc p H O O CH3
Z-Phe(pF)-OH iBuOCOCI -10°C (7)
(s) 20 min. Z-Phe(pF)-OCOOiBu
-10°C - +25°C
EtOAc + DMF
i ~ F F
O /
H2N N~NH2 H2 ~ / ~ O NHZ
O H Pd/C O N N
H O H
/ \
DMF / \
H-Phe(pF}-Phe-NHZ 40°C
(9) Z-Phe(pFrPhe-NH2
Scheme 1 ($)
SLJ~~T6 T U i ~ SFE~ET (l~~ILE 2~)
CA 02383184 2002-03-13
WO 01/19849 PCT/SE00/01747
17
OH ~~
\ / \ o
O O TBTU
OH \ DIEA i
\ O C -~ HzN -'~ / \ O C~ CHs
-~I \O CHs EtOAc ~ ~H O
O O (4)
(3)
(2)
/ NaOH
tan/H20
TBTU
DIEA
DMF
H ~ F
\/
O
~~----NHZ
CHs \
(10)
/
1. HZ Pd/C
2. HCI
\ F
/
CI O
HsN H ~~-NHz
O
(1)
Scheme 1, cont'd
CA 02383184 2002-03-13
WO 01/19849 PCT/SE00/01747
18
EXAMPLES
Step 1
s Preparation of Z-Tvr-D-Ala-OMe (Compound 4 in Scheme 1)
Example 1
2,85g (22mmo1) DIEA was added dropwise to an ice-cool solution of Z-Tyr-OH
(2), H-D-
Ala-OMe (3) and TBTU in lOml of EtOAc (lOmmol each). The reaction mixture was
left
io to assume room temperature after 25min at 4°C. The solvents were
then evaporated and the
residue partitioned between EtOAc and water. The organic phase was washed with
3 x 1M
Na~C03, 3x 1M KHSOa and brine before drying over Na~SO:~. The product was
crystallized by the addition of light petroleum ether to yield 3.2g (80%) and
99% pure.
~s Step 2
Preparation of Z-Tvr-D-Ala-OH (Compound ~ in Scheme 1)
Example 2
Z-Tyr-D-Ala-OMe ( 1.0g, 2.46mmol, of purity 95%) was dissolved in dioxane
(8mL). 1M
~o NaOH~aq~ (~.2mLl was then charged and the reaction left over night. The
solvents were
removed by vacuum distillation. The residue was dissolved in EtOAc (250mL) and
extracted, first with brine (4x75mL) followed by 1M KHS04 (3x75mL). The
organic layer
was then dried over MgS04 ~a"nya> for several hours before filtration. The
filtrate was
evaporated to dryness by vacuum distillation. The residual oil was dissolved
in
zs Acetone/EtOAc (2:1, SmL) and then isopropyl ether (30mL) was charged slowly
in order
to precipitate the product. The solid was filtered off and dried under reduced
pressure at
30°C, yielding 0.8g (86%) of the product with a purity of 99% (HPLC).
Example 3
CA 02383184 2002-03-13
WO 01/19849 PCT/SE00/01747
19
Z-Tyr-OH (63,1 a, 200 mmol) and H-(D)-Ala-OMe x HC1 (31,0 g, 222 mmol) were
charged to a one liter reactor under nitrogen. Acetone (400 ml) was added and
the slurry
cooled to -20°C. Isobutylchloroformiat (30,1 g, 220 mmol) was then
added, quickly
followed by N-Methylmorpholine (47,9 g, 472 mmol) while maintaining the temp
at about
s -10°C. Upon completion of the NMM-charge the temp was allowed to
reach 20°C, the
precipitate filtered off and washed with Acetone (100m1). The product was
slowly charged
to NaOH (20 g, 500 mmol) in water (600 ml) at RT. HCI, 32% (60 ml) was added
followed
by evaporation of half the reaction volume at 70°C. Upon cooling and
seeding the product
crystallised. The product was isolated by filtration and dried in vacuum to
yield 19.3 g Z-
io Tyr-(D)-Ala-OH (25%).
Example 4
A 250 mL flask was charged with H-D-Alanine-OH (4,52 g; 50,2 mmol; 2 eq),
potassium
carbonate anhydrate (7,05 g; 50,2 mmol; 2 eq) and polyethylene glycol 200 (50
mL; 11,1
is mL/g Alanine), which was stirred at ambient temperature. Another 250 mL
flask was
charged with Z-Tyrosine (8,00 g; 25,1 mmol) and EtOAc ( 100 mL; 12,6 mL/g Z-
Tyrosine). After cooling (jacket temperature -17°C) to -9°C
Isobutylchloroformate (3,55
mL; 30,1 mmol; 1,2 eq) was charged. N-Methylmorpholine (3,35 mL; 30,1 mmol;
1,2 eq)
was charged keeping the temperature below -7°C. The cool mixture was
charged to the
~o first 250 mL flask containing the alanine via a peristaltic pump. The
conversion was 90%
by HPLC. The product was not isolated, but ready to use in the next step.
Step 1'
Preparation of Z-Phe(pF)-Phe-NHS (Compound 8 in Scheme 1)
Example 5
Z-Phe(pF)-OH ( 1 eq.) is first dissolved in acetonitrile (MeCN)( 1.7L/mole)
and cooled
before addition of i-Butylchloroformiate ( 1.05 eq). The reaction is then
controlled by the
rate of addition, (about 20 minutes) 15 min actual, of N-Methylmorpholine (
l.4eq). A
~o reaction temperature between 0 and -15°C is recommended where the
reaction occurs
CA 02383184 2002-03-13
WO 01/19849 PCT/SE00/01747
immediately upon addition of l~'-Methylmorpholine, yet prevents the mixed
anhydride
from decomposing to rapidly.
H-Phe-NHS x HCl ( 1.04 eq) is meanwhile dissolved in DMF (4.OL/mole),
neutralized with
s N-Methylmorpholine (1.04eq) and cooled to about -10°C. This slurry is
upon completion
of the activation added at a rate that maintains the temperature around -
10°C for about 1~
minutes.
After completion of the coupling the product was crystallized from the
reaction mixture by
slow addition of 50% Ethanol/water (3.6L/mole). After 30 min a total of
2.85L/mole water
io in three portions were charged with about 25 min wait between each addition
and at
temperature of about 20°C. The crystals can after about 17 hours be
filtered or centrifuged
and washed with 50% Ethanol/water followed by several portions of acetonitrile
before
drying under vacuum at 20-60°C. Yield 90% and 99.9% purity.
is Step 2'
Preparation of H-Phe(pF)-Phe-NH~ (compound 9 in Scheme 1)
Example 6
Z-Phe(pF)-Phe-NHS prepared in the previous step is mixed with DMF (3.SL/mole)
and a
zo Pd/C catalyst (5% Pd) is added 5%, by weight and the resulting mixture
hydrogenated for
more than 0.5 hours at 25-30°C and about 3bar H~. The reaction mixture
is then filtered
and cooled to about -15°C before the next step. 99.6% purity in
solution and >99%
conversion of starting material.
zs Step 3
Preparation of Z-Tyr-D-Ala- Phe(pF)-Phe-NHZ (compound 10 in Scheme 1)
Example 7
The two building blocks Z-Tyr-D-Ala-OH ( 1.29mmo1, 1.00 eq) and H-Phe(pF)-Phe-
NH~x
~o HCl ( 1.29mmo1, l .00eq) were added to a round bottom flask containing
solvent (DMF,
CA 02383184 2002-03-13
WO 01/19849 PCT/SE00/01747
21
SmL) and the coupling reagent TBTU ( 1.28mmo1, 0.99eq). The base (DIEA,
2.58mmol,
2.OOeq) was charged to this slurry. The reaction was monitored by HPLC and was
terminated after 4h. The solvent was then evaporated. 22mL of a solvent
mixture
(EtOAc/Acetone/MeOH 5:10:1 ) was charged to 2.4g of the remaining oil to yield
the
s product as a solid, which was filtered and washed with acetone (3x~mL). The
moist
crystals were dried under reduced pressure at 30°C over night. The
overall yield was 0.74g
(80%) corrected for the purity 97% (HPLC).
Example 8
~o Z-Tyr-(D)-Ala-OH ( 15.0 g, 39 mmol) and H-Phe(pF)-Phe-NH2xHCl ( 12.8 g, 35
mmol)
were mixed with aceton (450 ml) in a one liter reactor. The slurry temperature
was reduced
to -10°C prior to addition of isobutylchloroformate (4.5~ ml, 3~ mmol).
NMM (8.45 ml,
77 mmol) was slowly charged to maintain the temp at about -10°C. Upon
completed
addition the temp was increased to room temperature and 2M HCl (40 ml, 80
mmol) added
is followed by water (365 ml). The mixture was heated to reflux, ca
70°C, and acetone
removed. The precipitate was filtered off and washed with acetonitril/water
3:2 (250 ml x
2) and dried to yield 18.9 g (77 %) of the title compound.
Step 4
zo Preparation of H-Tyr-D-Ala-Phe(pF)-Phe-NH, (compound I in Scheme 1)
Example 9
Compound 4 is mixed with DMF (2-2.6L/mole) and a 5 % PdIC catalyst is added (6-
7%,
by weight) and the resulting mixture hydrogenated for more than 0.5 hours at
20-50°C and
~s 3bar H~. The reaction mixture is then filtered to remove the Pd/C before
crystallizing the
product by addition of EtOAc until all substance has crystallized (typically
lOL/mole). The
solid is separated by filtration or centrifugation and washed with EtOAc prior
to drying
under vacuum at 20-60°C.
CA 02383184 2002-03-13
WO 01/19849 PCT/SE00/01747
22
Preparation of H-Tyr-D-Ala-Phe(pF)-Phe-NH, hydrochloride
Example 10
The free base compound I is dissolved in a mixture of water and acetone with
one
s equivalent HCl added and clear filtered (146g/mole 25% HCl/H~O, 2L
Acetone/mole in
actual run). The salt has a limited solubility in acetone and therefore the
filter is washed
once with an additional amount of the acetone/water (95:5) mixture
(O.SL/mole). The
crystallization is initiated by a slow addition of acetone (3.4L/mole) at high
agitation rate
and then 1 % w/w of seeding crystals is optionally added. After 30 minutes the
first amount
~o of MIBK (3L/mole) is slowly charged and left with slow stirring until the
batch clearly
thickens. MIBK (3L/mole) is charged three additional times separated by 30-60
minutes
while maintaining the reactor inner temperature at about 20°C. The
solid is then separated
by centrifugation or filtration and washed with MIBK before drying under
vacuum at 20-
50°C for more than 16 hours or until the solvent levels are lower than
specified in the
~s release specifications.