Note: Descriptions are shown in the official language in which they were submitted.
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
1
GONADOTROPIN-RELEASING HORMONE RECEPTOR ANTAGONISTS
AND METHODS RELATING THERETO
TECHNICAL FIELD
This invention relates generally to gonadotropin-releasing hormone
(GnRH) receptor antagonists, and to methods of treating disorders by
administration of
such antagonists to a warm-blooded animal in need thereof.
BACKGROUND OF THE INVENTION
Gonadotropin-releasing hormone (GnRH), also known as luteinizing
hormone-releasing hormone (LHRH), is a decapeptide (pGlu-His-Trp-Ser-Tyr-Gly
Leu-Arg-Pro-Gly-NHZ) that plays an important role in human reproduction. GnRH
is
released from the hypothalamus and acts on the pituitary gland to stimulate
the
biosynthesis and release of luteinizing hormone (LH) and follicle-stimulating
hormone
(FSH). LH released from the pituitary gland is responsible for the regulation
of
gonadal steroid production in both males and females, while FSH regulates
spermatogenesis in males and follicular development in females.
Due to its biological importance, synthetic antagonists and agonists to
GnRH have been the focus of considerable attention, particularly in the
context of
prostate cancer, breast cancer, endometriosis, uterine leiomyoma, and
precocious
puberty. For example, peptidic GnRH agonists, such as leuprorelin (pGlu-His-
Trp-Ser-
Tyr-n-Leu-Leu-Arg-Pro-NHEt), have been used to treat such conditions. Such
agonists
appear to function by binding to the GnRH receptor in the pituitary
gonadotropins,
thereby inducing the synthesis and release of gonadotropins. Chronic
administration of
GnRH agonists depletes gonadotropins and subsequently down-regulates the
receptor,
resulting in suppression of steroidal hormones after some period of time
(e.g., on the
order of 2-3 weeks following initiation of chronic administration).
In contrast, GnRH antagonists are believed to suppress gonadotropins
from the onset, and thus have received the most attention over the past two
decades. To
date, some of the primary obstacles to the clinical use of such antagonists
have been
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
2
their relatively low bioavailability and adverse side effects caused by
histamine release.
However, several peptidic antagonists with low histamine release properties
have been
reported, although they still must be delivered via sustained delivery routes
(such as
subcutaneous injection or intranasal spray) due to limited bioavailability.
In view of the limitations associated with peptidic GnRH antagonists, a
number of nonpeptidic compounds have been proposed. For example, Cho et al.
(J.
Med. Chem. 41:4190-4195, 1998) discloses thieno[2,3-b]pyridin-4-ones for use
as
GnRH receptor antagonists; U.S. Patent Nos. 5,780,437 and 5,849,764 teach
substituted
indoles as GnRH receptor antagonists (as do published PCTs WO 97/21704,
98/55479,
98/55470, 98/55116, 98/55119, 97/21707, 97/21703 and 97/21435); published PCT
WO 96/38438 discloses tricyclic diazepines as GnRH receptor antagonists;
published
PCTs W097/14682, 97/14697 and 99/09033 disclose quinoline and thienopyridine
derivatives as GnRH antagonists; published PCTs WO 97/44037, 97/44041,
97/44321
and 97/44339 teach substituted quinolin-2-ones as GnRH receptor antagonists;
and
published PCT WO 99/33831 discloses certain phenyl-substituted fused nitrogen-
containing bicyclic compounds as GnRH receptor antagonists.
While significant strides have been made in this field, there remains a
need in the art for effective small molecule GnRH receptor antagonists. There
is also a
need for pharmaceutical compositions containing such GnRH receptor
antagonists, as
well as methods relating to the use thereof to treat, for example, sex-hormone
related
conditions. The present invention fulfills these needs, and provides other
related
advantages.
SUMMARY OF THE INVENTION
In brief, this invention is generally directed to gonadotropin-releasing
hormone (GnRH) receptor antagonists, as well as to methods for their
preparation and
use, and to pharmaceutical compositions containing the same. More
specifically, the
GnRH receptor antagonists of this invention are compounds having the following
general structure (I):
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
3
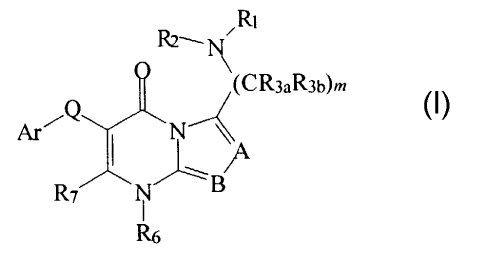
R~
(CR3aR36)m
Ar~Q N
R7 N B
R6
(I)
including stereoisomers, prodrugs and pharmaceutically acceptable salts
thereof,
wherein Ar, A, B, Q, R,, Rz, R3a, R3b, Rb, R~ and m are as defined below.
The GnRH receptor antagonists of this invention have utility over a wide
range of therapeutic applications, and may be used to treat a variety of sex-
hormone
related conditions in both men and women, as well as a mammal in general (also
referred to herein as a "subject"). For example, such conditions include
endometriosis,
uterine fibroids, polycystic ovarian disease, hirsutism, precocious puberty,
gonadal
steroid-dependent neoplasia such as cancers of the prostate, breast and ovary,
gonadotrophe pituitary adenomas, sleep apnea, irritable bowel syndrome,
premenstrual
syndrome, benign prostatic hypertrophy, contraception and infertility (e.g.,
assisted
reproductive therapy such as in vitro fertilization). The compounds of this
invention
are also useful as an adjunct to treatment of growth hormone deficiency and
short
stature, and for the treatment of systemic lupus erythematosis. The compounds
are also
useful in combination with androgens, estrogens, progesterones, and
antiestrogens and
antiprogestogens for the treatment of endometriosis, fibroids, and in
contraception, as
well as in combination with an angiotensin-converting enzyme inhibitor, an
angiotensin
II-receptor antagonist, or a renin inhibitor for the treatment of uterine
fibroids. In
addition, the compounds may be used in combination with bisphosphonates and
other
agents for the treatment and/or prevention of disturbances of calcium,
phosphate and
bone metabolism, and in combination with estrogens, progesterones and/or
androgens
for the prevention or treatment of bone loss or hypogonadal symptoms such as
hot
flashes during therapy with a GnRH antagonist.
The methods of this invention include administering an effective amount
of a GnRH receptor antagonist, preferably in the form of a pharmaceutical
composition,
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
4
to a mammal in need thereof. Thus, in still a further embodiment,
pharmaceutical
compositions are disclosed containing one or more GnRH receptor antagonists of
this
invention in combination with a pharmaceutically acceptable carrier and/or
diluent.
These and other aspects of the invention will be apparent upon reference
to the following detailed description. To this end, various references are set
forth
herein which describe in more detail certain background information,
procedures,
compounds and/or compositions, and are each hereby incorporated by reference
in their
entirety.
DETAILED DESCRIPTION OF THE INVENTION
As mentioned above, the present invention is directed generally to
compounds useful as gonadotropin-releasing hormone (GnRH) receptor
antagonists.
The compounds of this invention have the following structure (I):
~Ri
Rz
CR3aR3b)m
R6
(I)
including stereoisomers, prodrugs and pharmaceutically acceptable salts
thereof,
wherein:
A is independently selected from N or CR4;
B is independently selected from N or CRS;
Q is a direct bond or -(CR$aR$b)r Z-(CRloaRiob)s ;
m, r and s are the same or different and selected from an integer from 0
to 6;
Z is a direct bond or -O-, -S-, -NR9-, -SO-, -S02-, -OS02-, -SO20-,
-SO2NR9-, -NR9SOZ-, -CO-, -COO-, -OCO-, -CONR9-, -NR9CO-, -NR9CONR9a,
-OCONR9- Or -NR9COO-;
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
R~ is hydrogen, alkyl, substituted alkyl, aryl, substituted aryl, arylalkyl,
substituted arylalkyl, heteroaryl, substituted heteroaryl, heteroarylalkyl,
substituted
heteroarylalkyl, -C(R,a)(=NR~b), or -C(NRIaRa)(=NR,b);
RZ is hydrogen, alkyl or substituted alkyl;
5 or Rl and RZ taken together with the nitrogen atom to which they are
attached form a heterocycle ring or a substituted heterocycle ring;
R3a and R3b are independently selected from hydrogen, alkyl, substituted
alkyl, aryl, substituted aryl, arylalkyl, heterocycle, heterocyclealkyl,
hydroxy, alkoxy,
alkylthio, alkylamino, CONR~4R~5, or -COORIa;
or R3a and R36 taken together with the carbon atom to which they are
attached form a 3-6 membered homocyclic ring, substituted homocyclic ring,
heterocyclic ring or substituted heterocyclic ring;
or R3a and R36 taken together form =NR3~;
R4 is hydrogen, halogen, cyano, nitro, alkyl, substituted alkyl, arylalkyl,
substituted arylalkyl, heterocyclealkyl, substituted heterocyclealkyl, -COR>
>, -COORI,,
-CONR12R13, -ORIn -OCORIU -OSOZRIa -SRS,, -SOZRIU -NRI2R13~ -NR11CORI2,
-NR11CONR12R~3, -NR11SOZRlZ or -NR11S02NR,zRl3; or R4 and Rl, together with
the
atoms to which they are attached, form a 5-7 member heterocyclic ring or
substituted
heterocyclic ring;
or R4 and R3a, together with the atoms to which they are attached, form a
5-7 membered homocyclic ring, substituted homocyclic ring, heterocyclic ring
or
substituted heterocyclic ring;
RS is hydrogen, halogen, lower alkyl, arylalkyl, alkoxy, alkylthio,
alkylamino, cyano or nitro;
Rb is hydrogen, alkyl, substituted alkyl, aryl, substituted aryl, arylalkyl,
substituted arylalkyl, heteroaryl, substituted heteroaryl, heteroarylalkyl or
substituted
heteroarylalkyl;
R~ is hydrogen, halogen, cyano, alkyl, substituted alkyl, alkoxy,
alkylthio, alkylsulfonyl or alkylamino;
Ar is aryl, heteroaryl, substituted aryl or substituted heteroaryl; and
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
6
Rta, Rtb~ Rtc, R3c~ R8a, Rsb, R9, R9a~ RtOa, Rtob~ Rtt, Rt2, Rt3, Rt4 arid Rt5
are the same or different and at each occurrence independently hydrogen, acyl,
alkyl,
substituted alkyl, aryl, substituted aryl, arylalkyl, substituted arylalkyl,
heterocycle,
substituted heterocycle, heterocyclealkyl or substituted heterocyclealkyl;
or Rta and Rtb, R8a and Rgb, Rtoa and Rtob, or Rt2 and Rt3 taken together
with the atom or atoms to which they are attached form a homocyclic ring,
substituted
homocyclic ring, heterocyclic ring or substituted heterocyclic ring.
As used herein, the above terms have the following meaning:
"Alkyl" means a straight chain or branched, noncyclic or cyclic,
unsaturated or saturated aliphatic hydrocarbon containing from 1 to 10 carbon
atoms,
while the term "lower alkyl" has the same meaning as alkyl but contains from 1
to 6
carbon atoms. Representative saturated straight chain alkyls include methyl,
ethyl, n
propyl, n-butyl, n-pentyl, n-hexyl, and the like; while saturated branched
alkyls include
isopropyl, sec-butyl, isobutyl, tent-butyl, isopentyl, and the like.
Representative
saturated cyclic alkyls include cyclopropyl, cyclobutyl, cyclopentyl,
cyclohexyl, and
the like; while unsaturated cyclic alkyls include cyclopentenyl and
cyclohexenyl, and
the like. Cyclic alkyls are also referred to herein as "homocyclic rings."
Unsaturated
alkyls contain at least one double or triple bond between adjacent carbon
atoms
(referred to as an "alkenyl" or "alkynyl", respectively). Representative
straight chain
and branched alkenyls include ethylenyl, propylenyl, 1-butenyl, 2-butenyl,
isobutylenyl, 1-pentenyl, 2-pentenyl, 3-methyl-1-butenyl, 2-methyl-2-butenyl,
2,3-
dimethyl-2-butenyl, and the like; while representative straight chain and
branched
alkynyls include acetylenyl, propynyl, 1-butynyl, 2-butynyl, 1-pentynyl, 2-
pentynyl, 3-
methyl-1 butynyl, and the like.
"Aryl" means an aromatic carbocyclic moiety such as phenyl or
naphthyl.
"Arylalkyl" means an alkyl having at least one alkyl hydrogen atoms
replaced with an aryl moiety, such as benzyl, -(CH2)zphenyl, -(CH2)3phenyl,
-CH(phenyl)2, and the like.
"Heteroaryl" means an aromatic heterocycle ring of 5- to 10 members
and having at least one heteroatom selected from nitrogen, oxygen and sulfur,
and
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
7
containing at least 1 carbon atom, including both mono- and bicyclic ring
systems.
Representative heteroaryls are furyl, benzofuranyl, thiophenyl,
benzothiophenyl,
pyrrolyl, indolyl, isoindolyl, azaindolyl, pyridyl, quinolinyl, isoquinolinyl,
oxazolyl,
isooxazolyl, benzoxazolyl, pyrazolyl, imidazolyl, benzimidazolyl, thiazolyl,
benzothiazolyl, isothiazolyl, pyridazinyl, pyrimidinyl, pyrazinyl, triazinyl,
cinnolinyl,
phthalazinyl, and quinazolinyl.
"Heteroarylalkyl" means an alkyl having at least one alkyl hydrogen
atom replaced with a heteroaryl moiety, such as -CH2pyridinyl, -
CHZpyrimidinyl, and
the like.
"Heterocycle" (also referred to herein as a "heterocycle ring") means a
5- to 7-membered monocyclic, or 7- to 10-membered bicyclic, heterocyclic ring
which
is either saturated, unsaturated, or aromatic, and which contains from 1 to 4
heteroatoms independently selected from nitrogen, oxygen and sulfur, and
wherein the
nitrogen and sulfur heteroatoms may be optionally oxidized, and the nitrogen
heteroatom may be optionally quaternized, including bicyclic rings in which
any of the
above heterocycles are fused to a benzene ring. The heterocycle may be
attached via
any heteroatom or carbon atom. Heterocycles include heteroaryls as defined
above.
Thus, in addition to the heteroaryls listed above, heterocycles also include
morpholinyl,
pyrrolidinonyl, pyrrolidinyl, piperidinyl, hydantoinyl, valerolactamyl,
oxiranyl,
oxetanyl, tetrahydrofuranyl, tetrahydropyranyl, tetrahydropyridinyl,
tetrahydroprimidinyl, tetrahydrothiophenyl, tetrahydrothiopyranyl,
tetrahydropyrimidinyl, tetrahydrothiophenyl, tetrahydrothiopyranyl, and the
like.
"Heterocyclealkyl" means an alkyl having at least one alkyl hydrogen
atom replaced with a heterocycle, such as -CHZmorpholinyl, and the like.
"Homocycle" means a saturated or unsaturated (but not aromatic)
carbocyclic ring containing from 5-7 carbon atoms, such as cyclopentane,
cyclohexane,
cycloheptane, cyclohexene, and the like.
The term "substituted" as used herein means any of the above groups
(i.e., alkyl, aryl, arylalkyl, heteroaryl, heteroarylalkyl, homocycle,
heterocycle and
heterocyclealkyl) wherein at least one hydrogen atom is replaced with a
substituent. In
the case of a keto substituent ("-C(=O)-") two hydrogen atoms are replaced.
When
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
8
substituted, "substituents" within the context of this invention include
halogen,
hydroxy, cyano, nitro, amino, alkylamino, dialkylamino, alkyl, alkoxy,
alkylthio,
haloalkyl, aryl, substituted aryl, arylalkyl, substituted arylalkyl,
heteroaryl, substituted
heteroaryl, heteroarylalkyl, substituted heteroarylalkyl, heterocycle,
substituted
heterocycle, heterocyclealkyl, substituted heterocyclealkyl, -NRaRb, -
NRaC(=O)Rb, -
NRaC(-O)NRaNRb , -NRaC(-O)ORb -NRaSO2Rb, -C(=O)~ -C(-O)ORa~ -
C(-O)NRaRb, -OC(=O)NRaRb, -SH, -SORa, -S(=O)2Ra, -OS(=O)2Ra, -S(=O)20Ra,
wherein Raand Rb are the same or different and independently hydrogen, alkyl,
substituted alkyl, aryl, substituted aryl, arylalkyl, substituted arylalkyl,
heteroaryl,
substituted heteroaryl, heteroarylalkyl, substituted heteroarylalkyl,
heterocycle,
substituted heterocycle, heterocyclealkyl or substituted heterocyclealkyl.
"Halogen" means fluoro, chloro, bromo and iodo.
"Haloalkyl" means an alkyl having at least one hydrogen atom replaced
with halogen, such as trifluoromethyl and the like.
"Alkoxy" means an alkyl moiety attached through an oxygen bridge
(i. e., -O-alkyl) such as methoxy, ethoxy, and the like.
"Alkylthio" means an alkyl moiety attached through a sulfur bridge (i.e.,
-S-alkyl) such as methylthio, ethylthio, and the like.
"Alkylsulfonyl" means an alkyl moiety attached through a sulfonyl
bridge (i.e., -S02-alkyl) such as methylsulfonyl, ethylsulfonyl, and the like.
"Alkylamino" and "dialkylamino" mean one or two alkyl moiety
attached through a nitrogen bridge (i. e., -N-alkyl) such as methylamino,
ethylamino,
dimethylamino, diethylamino, and the like.
In one embodiment of this invention, Q is a direct bond and
representative GnRH receptor antagonists of this invention include compounds
having
the following structure (II):
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
9
~Rl
Rz~~
~CR3aR3b)m
Rb
(II)
In a further embodiment of structure (II), Ar is phenyl, substituted
phenyl, heteroaryl or substituted heteroaryl.
In another embodiment, Q is -(CRBaRgb)r Z-(CRIOaRIOb)S , r and s are
both zero, and representative GnRH receptor antagonists of this invention
include
compounds having the following structure (III):
~Rl
R2
CR3aR3b)m
~,/
R
(III)
In another embodiment of structures (II), A is CR4 and B is N, as
represented by the following structures (IV):
R1
,(CR3aR3b)m
2 It4
I
R7 N N
(IV)
Similarly, in another embodiment of structures (II), A is CR4 and B is
CRS, as represented by the following structure (V):
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
R1
' i
(CR3aR3b)m
Ar 6 ~ N
z R4
Q ~3
R7 N
Rs
(V)
In still further embodiments of structure (II), A is N and B is Rs or both
A and B are N, as represented by the following structures (VI) and (VII),
respectively:
Rz~~ Rz
:R3aR3b)m ~(CR3aR3b)m
s 4 N
z~j
6 ~~ 1/
R7 N N
~s
5
(VI) (VII)
In yet further embodiments of structure (I) of this invention, Q is a direct
bond, Rb is benzyl (substituted or unsubstituted), A is CR4 and B is N or CRs,
as
represented by the following structures (VIII) and (IX), respectively (wherein
X
10 represents one or more optional substituents as defined above):
N RI ~'~ Rl
.3aR3b)m .3aR3b)m
X X
(VIII) (IX)
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
11
In a more specific embodiment of structures (VIII) and (IX), m is 1 and
both R3a and R3b are hydrogen, as represented by the following structures (X)
and (XI):
R~
N
~' R4 R4
~N
R7 N
X
(X) (XI)
In still more specific embodiments, R~ in structures (X) and (XI) is
methyl, as represented by the following structures (XII) and (XIII),
respectively:
~Rl ,R1
Ar
CH3 ~N N
X~ ~~ X
(XII) (XIII)
One class of representative compounds having structures (XII) or (XIII)
include those compounds wherein Ar is aryl, substituted aryl, heteroaryl or
substituted
heteroaryl, such as substituted or unsubstituted phenyl, pyridyl, pyrimidinyl,
and the
like.
The compounds of the present invention may be prepared by known
organic synthesis techniques, including the methods described in more detail
in the
Examples. However in general, the compounds of structure (I) above may be made
by
the following Reaction Schemes. Specifically, compounds wherein A is CR4 and B
is N
may be made by Reaction Scheme A, compounds wherein A is CR4 and B is CRS may
be made by Reaction Scheme B, compounds wherein A is N and B is CRS may be
made
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
12
by Reaction Scheme C, and compounds wherein A is N and B is N may be made by
Reaction Scheme D. All substituents in the following Reaction Schemes are as
defined
above unless indicated otherwise. To this end, R3 and Rg in the following
Reaction
Schemes are the same or different and independently hydrogen, a substituent as
defined
above, or the moiety -Q-Ar.
Reaction Scheme A
O OH R4 O O
R Br + N ~ R$ NaH/DMF ~ Rg
a~ ~ ~ ~ R3 N
R3 HZN N R~
(i) (iia) HZN N R~
R OH R3 O
3
Rs TBAF/R6X N R8
R4~~ ~ Ra~~ I
N %~ N R~ N N R~
(iiia) (va)
Rs R3 ~C(=O)CRBCOOEt
NH R6NHz ~ NH
R4~~ ~ Ra
N S(O)nMe N NH
i
(iva) (iv')
Cyclization of a-bromocarbonyl or a,-bromocarboxylate (i) with 2-
aminopyrimid-4-one (iia) in the presence of a base such as sodium hydride,
tetrabutylammonium fluoride, potassium carbonate in an inert solvent such as
DME,
dimethylformamide, ethanol at a temperature of 25-100°C for a period of
12-24 hours
gives the imidazolo[1,2-a]pyrimidone (iiia). Compound (iiia) can be modified
by
alkylation with an alkyl halide in the presence of a base such as TBAF, sodium
hydride
or silver oxide in an inert solvent such as DME, THF or DMF at a temperature
of 0-
100°C for a period of 1-24 hours to give the 7-alkylated imidazolo[1,2-
a]pyrimid-4-one
(va).
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
13
Alternatively, the imidazolopyrimidone (va) can be synthesized from
imidazole (iva). Reaction of compound (iva) with a primary amine with or
without a
solvent such as DMF, DMSO, toluene or glycol at a temperature of 25-
180°C for a
period of 2-24 hours gives the 2-aminoimidazole (iv'), which reacts with a
acetoacetate
derivative in a solvent such as dioxane, ethanol or DMF at a temperature of 25-
120°C
for a period of 2-24 hours gives the desired compound (va).
Reaction Scheme B
O OH R3 O
R Br + N . ~ R8 NaH/DMF R4 ~ N Ra
/ O
R3 ~ N R~ ~ N R~
R
( ) Rs (iib) s (iiib)
OH R3 O
R3 R
8
NaOEt ~ R / N ~ Rg TBAF/R6X R4 / N
4 ~
N R~ N R~
Rs Rs R6
(ivb) (vb)
Reaction of a-bromocarbonyl (i) with 4-hydroxypyridine (iib) in the
presence of a base such as sodium hydride, TBAF, or potassium carbonate in an
inert
solvent such as DMF, THF or ethanol at a temperature of 25-100°C for a
period of 1-24
hours gives the N-alkylated pyrimidone (iiib). Compound (iiib) can be cyclized
in the
1 S presence of a base such as sodium ethoxide, sodium methoxide or potassium
t-butoxide
in an inert solvent such as ethanol, THF or DMF at a temperature of 25-
100°C for a
period of 0.5 to 16 hours gives the pyrrolo[1,2-a]pyrimidone (ivb). Alkylation
of
compound (ivb) can be accomplished with an alkyl halide in the presence of a
base
such as sodium hydride, TBAF or potassium carbonate at a temperature of 25-
100°C for
a period of 1-24 hours give the alkylated compound (vb).
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
14
Reaction Scheme C
R3 R3 R3
HZ/Pd-C ~ NH EtOCR~=CRACOOEt ~ N~ NH R~
N ~ COOEt
N~ NOZ N~ NHZ H
Rs Rs Rs Rs
(ic) (iic) (iiic)
OH R3 O
R3 R
s
Ph20/heat _ N~ N ~ R8 TBAF/R6X N~ N
i
N R~ ~ N R~
Rs Rs
(ivc) (vc)
Reduction of the 4-nitroimidazole (ic) with hydrogenation catalyzed by a
catalyst such as palladium on carbon, under hydrogen atmosphere, in a solvent
such as
methanol, acetic acid or ethyl acetate at room temperature for a period of 1-
16 hours
gives the 4-aminoimidazole (iic). Reaction of compound (iic) with beta-
ethoxyacrylate
in a solvent such as benzene or methanol with or without a catalyst ' such as
toluenesulfonic acid at a temperature of 25-100°C for a period of 1-16
hours gives the
enamine (iiic). When the compound (iiic) is heated at a temperature of 200-
280°C in a
solvent such as phenyl ether for a period of 0.25 to 2 hours the cyclized
product (ivc) is
obtained. Compound (ivc) can be modified by alkylation in the presence of a
base such
as sodium hydride, TBAF or potassium carbonate in an inert solvent such as
THF,
DME or DMF at a temperature of 25-100°C for a period of 1-16 hours
to give
compound (vc).
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
Reaction Scheme D
O O
NHZ R~C(=O)CHRRCOOEt~ N Rs NI-t2NHz N R8
S ~ N _ R6 I ~ HzN
H HS N R~ ~ H N R~
(id) R6 R6
(iiid)
(iid)
R O
3
R3C(OEt)~ N~ N Rg
R3C0
,N~N R~
(vd)
O R3 O
s
R~ N Rg TBAF/R6X N~ N I R
I N
NN~N I R7 ~N R
H R6
(ivd) (vd)
5 Cyclization of thiourea (1d) with an acetoacetate derivative in the
presence of a base such as sodium methoxide or potassium t-butoxide in an
inert
solvent such as methanol, ethanol at a temperature of 25-120°C for a
period of 2-24
hours gives the pyrimidone (iid) as one of the two isomers. Reaction of
compound (iid)
with hydrazine in an appropriate solvent such as ethanol or water at a
temperature of
10 25-100°C for a period of 1-24 hours give the hydazinopyrimidine
(iiid), which is
cyclized upon treatment of triethoxyalkane or a carboxylic acid derivative at
a
temperature of 25-120°C for a period of 2-24 hours to give the 1,2,4-
triazolo[1,2-
a]pyrimidone (vd).
Alternatively, compound (vd) can be obtained by alkylation of
15 compound (ivd) with an alkyl halide in the presence of a base such as
sodium hydride,
TBAF, potassium carbonate at a temperature of 25-100°C for a period of
1-24 hours.
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
16
Reaction Scheme E
H O N R3a O
A~N I Rg R~RZNH Ri R
R CHO/AcOH ~ N~
'B~N~R7 3a A'B
N R~
(via) R6
(vii)
R3b R2 R3b
R3a O N R3a O
N R$ i) NBS Rl N Rs
I
A,B~ I ii) RIRzNH _ A
N R~ 'B N R~
R6 R6
(vib) (viii)
The amino compound (vii) can be prepared by treatment of the starting
material (via) with a secondary amine and an aldehyde such as formaldehyde or
acetaldehyde in an appropriate solvent such as ethanol, dioxane or acetic acid
at a
temperature of 25-100°C for a period of 1-24 hours.
The pyrimidone (vib) can be modified by treatment with a halogenating
reagent such as N-bromosuccinamide in an inert solvent such as
tetrachloromethane or
DMF at a temperature of 50-100°C for a period of 2-16 hours to give a
halide
compound, which reacts with a primary or secondary amine with or without an
amine
base such as triethylamine, diisopropylethylamine or pyridine in an inert
solvent such
as chloroform, tetrachloromethane or tetrahydrofuran at a temperature of 25-
80°C for a
period of 1-16 hours to give the amino analog (viii).
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
17
Reaction Scheme F
R O R3 O
3 Ar
N Br ~'B(OH)z ~ ~ N
A I ~ Pd(0) A ~ I
'B~N R~ 'B N R~
R6 R6
(ix)
(x)
R3 O R3 O
~N I COOEt NaOH ~ COOH B~yCOX2 _
A ' I or C1CHX~COX2
N R~ ,B N R~
R6
(xi) (xii)
Xz
R3 O O X1 O N
/ _N O X2 NHaOAc R3 ~ ~Xt
A/ I - ~N O
O HOAc A I
N R~ 'B~ N R~
R6
R6
(xiii) (xiv)
The bromopyrimidone (ix) can be converted to the corresponding aryl
(Ar = phenyl or substituted phenyl) or heteroaryl (heterocyclic aromatic ring
with or
without substituents) compound (x) by a palladium-catalyzed cross coupling
reaction of
compound (ix) with an organoboronic acid in the presence of a base such as
sodium
carbonate, cesium fluoride, or sodium acetate in an inert solvent such as
benzene,
ethanol, water or mixture thereof at a temperature of 25-120°C for a
period of 1-72
hours. Alternatively, heteroaryl compound such as oxazole (xiv) can be
prepared by a
cyclization of an ester (xiii) with an ammonium moiety such as ammonium
acetate in
an appropriate solvent such as acetic acid at a temperature of 25-120°C
for 1-24 hours.
The ester (xiii) can be synthesized from alkylation of an acid (xii) with a
alpha-
bromoketone in the presence of a base such as potassium carbonate or
triethylamine in
an inert solvent such as DMF at a temperature of 25-100°C for a period
of 1-24 hours
(wherein X' and XZ are substituents as defined herein).
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
18
Reaction Scheme G
X'
R O R O NH
O N'C
A~N CN NHZOH~ A~N I H.OH X~C(pEt)3_ R3 NO
,B~ ANN I
N R~ N R~
N R~
(xv) 6 R6 (xvi) RG
(xvii)
NHZNHXZ
X1
R3 O NH H R3 O NH R3 O
H' N'XZ and/or A// ' N I X NHZ X ~ C~ A~ N I N Xz
.B Z2
N R~ B~N R~ 'B~N R
~S ~S i
R6
(xviii) (xix) (xx)
Cyano compound (xv) can be converted to the corresponding heteroaryl
analogs such as oxodiazole or triazole. Reaction of compound (xv) with
hydroxylamine in a solvent such as methanol, ethanol, water, dioxane or the
mixture
thereof at a temperature of 25-120°C for a period of 1-24 hours gives
the N-
hydroxylamidine (xvi), which is treated with triethoxyalkane with or without a
solvent
such as ethanol, dioxane or an appropriate carboxylic acid at a temperature of
25-120°C
for a period of 1-24 hours to give the 1,2,3-oxodiazole (xvii) (wherein X1 is
a
substituent as defined herein). Reaction of (xv) with hydrazine, including
alkylhydrazine and arylhydrazine in an appropriate solvent such as ethanol,
dioxane,
water or a mixture thereof for a period of 1-24 hours gives N-aminoamidine
(xviii) or
(xix) (wherein X' is a substituent as defined herein), which is converted to
the
corresponding 1,2,3-triazole (xx) by reaction with triethoxyalkane (wherein X2
is a
substituent as defined herein) with or without a solvent such as dioxane,
ethano,
toluene, or an appropriate carboxylic acid at a temperature of 25-120°C
for a period of
1-24 hours.
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
19
Reaction Scheme H
O OMe R O O Xs
A~N Br X1 / SnBu3 A//'N I X' R3 O ~ X2
'B~ N I R Pd(0) ~B~ N R~ A ~ ~ Xt
i ~ then H+ ~ B N R~
R6 R6 RS
(xxi) (xxii)
(xxiv)
X2C(OMe)2NMe2 ~ ~ X3NHNH
2 X3
R3 O O~ X2 R3 O O NMe2 X3 R3 O N~N
p// _ NI I \X i N~ A//' N I ~ X2 H2N' '-NH A// ' N I ~ X2
'B~N R ' ~ X~ ' ~ X1
i ~ B N R~ B N R7
(xxv) (xxiii) (xxvi)
The bromo pyrimidone (xxi) can be modified by a palladium catalyzed
cross coupling with a vinyltin in a solvent such as THF, toluene or DMF at a
temperature of 25-120°C for a period of 1-24 hours to give the ketone
(xxii) (wherein
X' is a substituent). Condensation of the ketone (xxii) with triethoxyalkane
or
dimethoxydimethylaminoalkane with or without a solvent such as toluene,
dioxane or
DMF at a temperature of 25-120°C for a period of 1-24 hours gives the
a,[3-unsaturated
ketone (xxiii) (wherein X2 is a substituent). Cyclization of compound (xxiii)
with
hydrazine including alkylhydrazine and arylhydrazine in a solvent such as
ethanol,
dioxane, water or a mixture thereof at a temperature of 25-120°C for a
period of 1-24
hours gives pyrazole (xxiv) (wherein X3 is a substituent). Cyclization of
compound
(xxiii) with hydroxylamine in a solvent such as ethanol, dioxane, water or a
mixture
thereof at a temperature of 25-120°C for a period of 1-24 hours gives
isoxazole (xxv).
Cyclization of compound (xxiii) with amidine, urea, thiourea or guanidine in a
solvent
such as ethanol, dioxane, water or a mixture thereof at a temperature of 25-
120°C for a
period of 1-24 hours gives pyrimidine (xxvi) (wherein X3 is a substituent).
Reaction Scheme I
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
OH
R O O NMe2 CN
R / N ~ X2 NCCH2CONH2 R3 O \ I 2
a~~ ~ X ~ / N X
N N R~ Ra~~ ~ Xt
R6 N N R~
xxvii
xxviii
'-'' Nu
R O N, CN O N ~ CN
R3
POC13 _ N ~ Xz Nu- ~ 2
i
Ra~~ I X t Ra~ N ~ I X
N N R2 N~N R7X
R
6 R
XX1X
XXX
Cyclization of compound (xxvii) (wherein X1 and X2 are substituents)
with cyanoacetamide in the presence of a base such as potassium carbonate,
potassium
5 tert-butoxide, sodium methoxide in an appropriate solvent such as dioxane,
ethanol,
DMF at a temperature of SO-150°C for a period of 1-24 hours gives the
pyridone
(xxviii). Compound (xxviii) can be converted to the corresponding chloride
(xxix) by
reaction with POC13 with or without a solvent such as acetonitrile or toluene
at a
temperature of 25-120°C for a period of 1-24 hours. Compound (xxix) can
be modified
10 by reaction with a variety of nucleophiles ("Nu ") such as mono or
dialkylamine,
alkoxide, thiol or carbon nucleophile to give compound (xxx).
Reaction Scheme J
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
21
R3 O R3 O
~N COO(CR~oaR~Ob)sAr Ar(CR10aR10b)sR9NH ~N CONR9(CR10aR10b)sAr
A/,/B~ I EtgAl A, ~ I
N R~ B N R~
R6 R6
(xxxii) (xxxiii)
Pd(0)/CO Pd(0)/CO
Ar(CR~paRlOb)sOH ~(RIOaRIOb)sR9NH
Et3N/DMF Et3N/DMF
R3 O R3 O / Ar R3 O
A~ N I Br ~ A ~ I HZ/pd/BaS04_ A ~ I \ Ar
/'/B~N R~ Pd(0)/CuI B N R~ B N R
(xxxi)~ (xxxiv~ (xxxv)~
ArB(OH)2 HgS04 ~ HZ/Pd-C
CO/Pd(o) HZO
R3 O O R3 O O R3 O R3 O
// _N I Ar A~N I Ar ~N Ar A~N
A ~ ' ~ and/or A' ~ I O
'B N R~ B N R~ B N R~ B N R~
i i i i
R6 R6 R6 R5
(xxxvi) (xxxvii) (xxxviii) (xxxix)
The bromo compound (xxxi) (see Reaction Scheme H) can be converted
to the corresponding carbon analogs by various palladium catalyzed cross
coupling
reactions. Palladium catalyzed reaction of compound (xxxi) with carbon
monoxide in
the presence of an alcohol in a solvent such as alcohol or DMF at a
temperature of 25-
60°C for a period of 1-24 hours gives the ester (xxxii). Reaction of
the ester (xxxii)
with a primary or secondary amine and triethylaluminum in a solvent such as
dichloromethane or toluene at a temperature of 0-100°C for a period of
1-16 hours gives
the amide (xxxiii).
Palladium catalyzed coupling of compound (xxxi) with arylacetylene in
the presence of CuI and a base such as triethylamine in a solvent such as
triethylamine,
dioxane or DMF at a temperature of 25-120°C for a period of 1-16 hours
gives the
alkyne (xxxiv). Selective hydrogenation of the alkyne (xxxiv) catalyzed by
palladiumBaS04 under hydrogen atmosphere in a solvent such as methanol, ethyl
acetate or DMF at room temperature for a period of 1-24 hours gives the alkene
(xxxv).
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
22
Further reduction with a catalyst such as palladium on carbon under hydrogen
atmosphere in a solvent such as methanol, ethanol or ethyl acetate at room
temperature
for a period of 1-24 hours gives the corresponding alkane (xxxix).
Palladium catalyzed cross coupling of compound (xxxi) with an
arylboronic acid in the presence of a base such as sodium carbonate, cesium
carbonate
or cesium fluoride in the carbon monoxide atmosphere in a solvent such as
benzene,
ethanol, water, DME or a mixture thereof at a temperature of 25-100°C
for a period of
1-24 hours gives the ketone (xxxvi).
Reaction of the alkyne xxxiv with HgS04 in water at a temperature of
25-120°C for a period of 1-24 hours gives the corresponding ketone
(xxxvii) and/or
(xxxviii).
Reaction Scheme K
O O
R3 O O R3 NaOMe R3
A~ N I mCPBA A~ N I OAc ~.Cl A~ N I O~Ar
~B~N R7' ,B~N R~ ,B~N R~
R6
(x~ (xli) NaOMe (xlii)
NaOEt I
Ar(CR~ par. Ob)sBr~ Ar(CR~ paRl ob)ss02C1
R3 O R3 O
~N O(CRloaRlob)s~' N OSOz(CRIOaR~ob)s~
A ~ I
B N R~ 'B N R~
R6 R6
(xliii) (xliv)
Oxidation of the ketone (x1) with m-chloroperoxybenzoic acid in a
solvent such as dichloromethane or chloroform at a temperature of 0-
60°C for a period
of 1-72 hours gives the ester (xli). Reaction of compound (xli) with a
reactive aryl
halide, such as substituted or unsubstituted fluoronitrobenzene, 2- or 4-
chloropyridine
or 2- or 4-chloropyrimidine, in the presence of a base such as sodium
methoxide or
potassium t-butoxide in an inert solvent such as THF, DME or DMF at a
temperature of
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
23
25-120°C for a period of 1-24 hours gives the aryloxy compound (xlii).
Alkylation of
compound (xli) with an aryl alkyl halide in the presence of a base such as
sodium
ethoxide or potassium hydroxide in as solvent such as THF, DMF or DMSO at a
temperature for a period of 1-24 hours gives the corresponding alkoxy compound
(xliii). Treatment of the ester (xli) with a base such as sodium methoxide in
a solvent
such as THF or DMF at room temperature for a period of 0.5 to 2 hours,
followed by a
sulfonyl chloride at a temperature of 0-60°C for a period of 1-16 hours
gives the
sulfonate (xliv).
Reaction Scheme L
O R3 O
z
N NOz Raney Ni/Hz A~~~NH
A,
B N R~ B N R~
R6
(xlvi)
(xlv)
~Alkylation
R O Ry R3 O R O R9
~ N~ AtCI ~N~NHRg 3 N (CRloaRlOb)s~
~N Ar ~ A I Acylation ~N
A' ~ I ,B~N R A'
B N R~ R ~ B N R~
6
R~ (xlmi) isoc anate R6 (1i)
(xlviii) Alkyaltion sulfonyl chloride y
R3 O R3 O R3 O R9
A ~~~9(CRIOaRIOb)sAr A~N ~9SOz(CR~oaRtob)sAr A~N I N~NR98 (CR~oaR~ob)SAr
I ~ ~~ O
B '
N R~ B N R~ B N R~
i i
R6 (xlix) R6 (1) R6 (lii)
The nitro compound (xlv) can be reduced to the corresponding amine
(xlvi) by 1) hydrogenation in the presence of a catalyst such as Raney nickel
in a
solvent such as methanol, ethanol or ethyl acetate at room temperature for a
period of
1-24 hours; 2) chemical reduction such as SnClz in a solvent such as water at
a
temperature of 25-100°C for a period of 1-24 hours. The amine (xlvi)
can be alkylated
with 1) an alkyl halide in the presence of a base such as potassium carbonate,
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
24
triethylamine or sodium methoxide in an appropriate solvent such as
acetonitrile,
ethanol or chloroform at a temperature of 25-100°C for a period of 1-24
hours; 2) an
aldehyde in the presence of a reducing agent such as sodium cyanoborohydride
in a
solvent such as methanol, dichloromethane or a mixture thereof at a
temperature of 25-
80°C for a period of 1-72 hours to give compound (xlvii). Reaction of
the primary (R9 =
H) or secondary amine (xlvii) with a reactive aryl chloride such as
chloronitrobenzene,
chloropyridine or chloropyrimidine with or without a base such as potassium
carbonate,
triethylamine or sodium hydride in an appropriate solvent such as THF, DMF or
dioxane at a temperature of 25-120°C for a period of 1-24 hours give
the arylamino
compound (xlviii). Further alkylation of (xlvii) with an arylalkyl halide with
or without
a base such as potassium carbonate, triethylamine or sodium hydride in an
appropriate
solvent such as acetonitrile, THF or DMF at a temperature of 25-100°C
for a period of
1-24 hours give the amino compound (xlix). Reaction of amino compound (xlvii)
with
sulfonyl chloride in the presence of a base such as triethylamine, pyridine or
potassium
carbonate in a solvent such as dichloromethane, chloroform or benzene at a
temperature
of 25-100°C for a period of 1-24 hours gives the sulfonamide (1).
Acylation of the
amine (xlvii) with 1 ) an acid chloride in the presence of a base such as
triethylamine,
pyridine or potassium carbonate in an appropriate solvent such as
dichloromethane,
THF or DMF at a temperature of 25-100°Cfor a period of 1-16 hours; 2)
an carboxylic
acid with a coupling reagent such as DCC or EDC in an appropriate solvent such
as
chloroform or DMF for a period of 1-16 hours gives the corresponding amide
(1i).
Reaction of the amine (xlvii) with an isocyanate in a solvent such as benzene,
chloroform or dioxane at a temperature of 25-120°C for a period of 1-24
hours gives the
urea compound (lii).
Reaction Scheme M
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
O R O R O
3
~N Br2/NH4SCN ~N SCN Ar(CRtpaRtob)sOH ~N S(CRtoaRt06)sAr
A I A,~ I A,
. ~ B n-Bu3P
B N R~ N R~ B N R~
R6 R6 R6
(liii) (liv) (lv)
R3 O R3 O
mCPBA ~N SO(CRtpaRtob)sAr ~N SOz(CRto~Rtob)s~
A ~ I and/or A,
N R~ B N R~
R6 R6
(lvi) (lvii)
Treatment of compound (liii) with bromine in the presence of
ammonium thioisocyanate or potassium isothiocyanate in a solvent such as
acetic acid,
5 chloroform or ethanol at a temperature of 0-80°C for a period of 1-24
hours gives the
thioisocyanate (liv). Alkylation of compound (liv) with an alcohol in the
presence of
tributylphosphine in a solvent such as benzene, chloroform or dioxane at a
temperature
of 25-120°C for a period of 1-24 hours gives the sulfide (1v).
Oxidation of the sulfide
(1v) with an oxidation reagent such as mCPBA or hydrogen peroxide in an
appropriate
10 solvent such as dichloromethane, ethanol, water or a mixture thereof at a
temperature of
0-60°C for a period of 1-48 hours gives the corresponding sulfoxide
(lvi) and sulfone
(lvii).
Reaction Scheme N
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
26
Rt
O R OHC O N'RO
s
A~ N I DMF/POC13 _ / N Rs reductive ~ Rs
/ 'N
'B~N R~ ,B~N I R7 ammation
R6 i B N R~
(Iviii) R3bCH2N02 (lix) R6 (lxv) R6
NH40Ac/Ac0
~2
R3b R3b R t- N
NOZ ~ R3b
O R Al/N~CIz O R reductive O R
A/ N s A/ N s amination A/ N I s
,B~N R7 ,B~N R'7 ,B~N R7
(lx) ~
(Ixi) R6 (lxii)
LiAlH4 or Alkylation~
hydrogenation
R3b R3b ~ 1
NHZ
O O
/ N Rs Alkylation ~. / N Rs
A ~ I or Reductive
'B N R~ amination 'B N R~
(lxiv)
(lxiii)~
Compound (hiii) can be converted to the corresponding aldehyde (lix)
by reaction with POC13 and DMF at a temperature of 0-120°C for a period
of 1-24
hours. Reductive amination of the aldehyde (lix) with a primary or secondary
amine in
the presence of a reducing reagent such as sodium cyanoborohydride in an
appropriate
solvent such as methanol, dichloromethane, THF or a mixture thereof at a
temperature
of 0-80°C for a period of 1-24 hours gives the amine (lxv).
Condensation of the
aldehyde (lix) with nitroalkane in the presence of a base such as ammonium
acetate in a
solvent such as acetic acid at a temperature of 40-100°C for a period
of 1-24 hours
gives the nitroolefin (lx), which can be reduced to the corresponding carbonyl
compound (lxi) with a reducing agent such as aluminum in the presence of a
catalyst
such as nickel chloride in a solvent such as DMF, THF or ethanol at a
temperature of
25-100°C for a period of 1-24 hours. Reductive amination of carbonyl
(lxi) with an
1 S amine and a reducing agent such as sodium cyanoborohydride in a solvent
such as
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
27
methanol, dichloromethane or mixture thereof at a temperature of 0-80°C
for a period
of 1-16 hours gives the amino compound (lxii). Alternatively, the nitroolefin
(lx) can
be reduced to the corresponding amine (lxiii) by using a reducing agent such
as lithium
aluminum hydride in an appropriate solvent such as ether or THF at a
temperature of 0-
80°C for a period of 1-16 hours, or by hydrogenation with a catalyst
such as palladium
on carbon under hydrogen atmosphere in a solvent such as methanol or ethyl
acetate at
room temperature for a period of 1-16 hours. The amine (lxiii) can be
converted to the
secondary amine (lxiv) or tertiary amine (lxii) by 1) alkylation with an alkyl
halide in a
solvent such as dichloromethane or ethyl acetate at a temperature of 0-
80°C for a period
of 1-16 hours; or 2) reductive amination with an aldehyde and a reducing agent
such as
sodium cyanoborohydride in a solvent such as methanol, dichloromethane or
mixture
thereof at a temperature of 0-80°C for a period of 1-16 hours.
Reaction Scheme O
Rlc
~1b
H O R3b R3a O ~N'~ R3b
Rla N O
N R8 R3aR3bCH0 H ~ N R8 R R3a
A~ ~ I AcOH A~ ~ I 2 ~ N R8
B~N~R~ B~N~R~ A'B
N R~
(lxv) (lxvi)
i) MsCI/Py (lxix)
ii) RZNHz RlaRIcNC(-~1b)SMe
~(/ X16
R3b HN R3b O Rla~ R3b O
R3a O
~ R R3a R R3a
8
//'N i)NBS N R8 RIaC(=NRIb)OEt Z N R8
A~B~ I ii) RZNHz A~ ~ ~ A,
N R~ B N R~ B N R~
R6 R6
1 S (lxvii)
(lxviii) (lxx)
Compound (lxv) can be converted to the corresponding alcohol (lxvi) by
reaction with an aldehyde with or without an acid catalyst such as
hydrochloric acid,
toluenesulfonic acid in an inert solvent such as ethanol, dioxane or acetic
acid at a
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
28
temperature of 25-120°C for a period of 1-24 hours. Alternatively, the
alcohol (lxvi)
may be formed first by condensation of an appropriate acid or acid chloride
with
starting (lxv) to give a ketone followed by reaction with a Grignard or
organolithium
reagent. The alcohol (lxvi) can be further modified to the corresponding amine
(lxviii)
S by reaction first with methanesulfonyl chloride in the presence of a base
such as
pyridine, triethylamine in an inert solvent such as dichloromethane,
chloroform or
pyridine at a temperature of 25-60°C for a period of 1-24 hours,
followed by reaction
with ammonium or a primary amine. Alternatively, bromination of compound
(lxvii)
with a brominating reagent such as N-succinamide in an inert solvent such as
carbon
tetrachloride or DMF at a temperature of 25-100°C for a period of 2-16
hours gives the
corresponding bromide (Ixviii) which reacts with an amine to give the amino
compound
(lxviii). The amino compound (lxviii) can be converted to guanidine derivative
(lxix)
by reaction with a S-methylthiourea in an appropriate solvent such as DMF,
THF,
ethanol or acetonitrile at a temperature of 25-120°C for a period of 1-
24 hours. The
amino compound (lxviii) can also be converted to the corresponding amidine
derivative
(lxx) by reaction with an imidate in an appropriate solvent such as ethanol,
acetonitrile
or DMF for a period of 2-24 hours.
Reaction Scheme P
o _ ~ sa
R OHC O O~C O
A~ N I 8 DMF/POC13 _ ~ N R8 1. RgaM ~ N R8
,B~N R A, ~ I 2.I0~ A,
i ~ B N R~ B N R~
R6
(hiii) (fix) R6 (lxxi)R6
RgbCHZN02
amination a 1 NH40Ac/AcOH
R3a N RO
R3b
NOZ
N R8 R / O
A~B~N I R~ A/ N Rs
R6 'B~N I R~
(lxxii)
(lxxiii) R6
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
29
Compound (hiii) can be converted to the corresponding aldehyde (lix)
by reaction with POC13 and DMF at a temperature of 0-120°C for a period
of 1-24
hours. Aldehyde (lix) may then form ketone (lxxi) first through reaction with
an
appropriate Grignard or organolithium reagent in a solvent such as THF or
ethyl ether
at a temperature of -78-60°C followed by an oxidation using Swern
conditions or MnOz
or PCC in a solvent such as methylene chloride at a temperature from 0-
75°C..
Reductive amination of the ketone (lxxi) with a primary or secondary amine in
the
presence of a reducing reagent such as sodium cyanoborohydride in an
appropriate
solvent such as methanol, dichloromethane, THF or a mixture thereof at a
temperature
of 0-80°C for a period of 1-24 hours gives the amine (lxxii).
Condensation of the
aldehyde (lxxi) with nitroalkane in the presence of a base such as ammonium
acetate in
a solvent such as acetic acid at a temperature of 40-100°C for a period
of 1-24 hours
gives the nitroolefin (lxxiii).
The compounds of the present invention may generally be utilized as the
free base. Alternatively, the compounds of this invention may be used in the
form of
acid addition salts. Acid addition salts of the free amino compounds of the
present
invention may be prepared by methods well known in the art, and may be formed
from
organic and inorganic acids. Suitable organic acids include malefic, fumaric,
benzoic,
ascorbic, succinic, methanesulfonic, acetic, trifluoroacetic, oxalic,
propionic, tartaric,
salicylic, citric, gluconic, lactic, mandelic, cinnamic, aspartic, stearic,
palmitic,
glycolic, glutamic, and benzenesulfonic acids. Suitable inorganic acids
include
hydrochloric, hydrobromic, sulfuric, phosphoric, and nitric acids. Thus, the
term
"pharmaceutically acceptable salt" of structure (I) is intended to encompass
any and all
acceptable salt forms.
In addition, prodrugs are also included within the context of this
invention. Prodrugs are any covalently bonded carriers that release a compound
of
structure (I) in vivo when such prodrug is administered to a patient. Prodrugs
are
generally prepared by modifying functional groups in a way such that the
modification
is cleaved, either by routine manipulation or in vivo, yielding the parent
compound.
Prodrugs include, for example, compounds of this invention wherein hydroxy,
amine or
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
sulfhydryl groups are bonded to any group that, when administered to a
patient, cleaves
to form the hydroxy, amine or sulfllydryl groups. Thus, representative
examples of
prodrugs include (but are not limited to) acetate, formate and benzoate
derivatives of
alcohol and amine functional groups of the compounds of structure (I).
Further, in the
5 case of an carboxylic acid (-COOH), esters may be employed, such as methyl
esters,
ethyl esters, and the like.
With regard to stereoisomers, the compounds of structure (I) may have
chiral centers and may occur as racemates, racemic mixtures and as individual
enantiomers or diastereomers. All such isomeric forms are included within the
present
10 invention, including mixtures thereof. Furthermore, some of the crystalline
forms of
the compounds of structure (I) may exist as polymorphs, which are included in
the
present invention. In addition, some of the compounds of structure (I) may
also form
solvates with water or other organic solvents. Such solvates are similarly
included
within the scope of this invention.
15 The effectiveness of a compound as a GnRH receptor antagonist may be
determined by various assay methods. Suitable GnRH antagonists of this
invention are
capable of inhibiting the specific binding of GnRH to its receptor and
antagonizing
activities associated with GnRH. For example, inhibition of GnRH stimulated LH
release in immature rats may be measured according to the method of Vilchez-
Martinez
20 (Endocrinology 96:1130-1134, 1975). Briefly, twenty-five day old male
Spraque-
Dawley rats are administered an GnRH antagonist in saline or other suitable
formulation by oral gavage, subcutaneous injection, or intravenous injection.
This is
followed by subcutaneous injection of 200 ng GnRH in 0.2 ml saline. Thirty
minutes
after the last injection, the animals are decapitated and trunk blood
collected. After
25 centrifugation, the separated plasma is stored at -200°C until
determination of the LH
and FSH by radioimmunoassay. Other techniques for determining the activity of
GnRH receptor antagonists are well known in the field, such as the use of
cultured
pituitary cells for measuring GnRH activity (Vale et al., Endocrinology 91:562-
572,
1972), and a technique for measuring radioligand binding to rat pituitary
membranes
30 (Perrin et al., Mol. Pharmacol. 23:44-51, 1983).
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
31
Activity of GnRH receptor antagonists are typically calculated from the
ICSO as the concentration of a compound necessary to displace 50% of the
radiolabeled
ligand from the GnRH receptor, and is reported as a "K;" value calculated by
the
following equation:
_ IC50
K' 1+L/KD
where L = radioligand and KD = affinity of radioligand for receptor (Cheng and
Prusoff,
Biochem. Pharmacol. 22:3099, 1973). GnRH receptor antagonists of this
invention
have a K; of 100 pM or less. In a preferred embodiment of this invention, the
GnRH
receptor antagonists have a K; of less than 10 pM, and more preferably less
than 1 pM.
More preferred compounds include: 1-1, 1-2-5, 12, 13-15, 19, 21, 27, 31, 32,
34, 37,
S5, 58, 64, 73, 2B, 2D, 3A, 3B, 3D, 4A, 4B, 4C, 4D, 4G, 4H, SA and SB (see
Examples
below).
Representative GnRH receptor antagonists of this invention include the
following compounds:
(a) 3-(N-Benzyl-N-methyl)aminomethyl-2-(tert-butyl)-6-methyl-7-
(2-fluorobenzyl)-5-(3-methoxyphenyl)imidazolo[ 1,2-a]pyrimid-4-one
(b) 3-(N-(2-Pyridylmethyl))aminomethyl-2-(tert-butyl)-6-methyl-7-
(2-fluorobenzyl)-5-(3-methoxyphenyl)imidazolo[ 1,2-a]pyrimid-4-one
(c) 3-(N-(2-Pyridylmethyl)-N-methyl)aminomethyl-2-(tert-butyl)-6-
methyl-7-(2-fluorobenzyl)-5-(3-methoxyphenyl)imidazolo[ 1,2-a]pyrimid-4-one
(d) 3-[N-Methyl-N-(2-pyridylethyl)]aminomethyl-2-(tert-butyl)-6-
methyl-7-(2-fluorobenzyl)-5-(3-methoxyphenyl)imidazolo[ 1,2-a]pyrimid-4-one
(e) 3-[N-(2-Furanmethyl)-N-methyl]aminomethyl-2-(tert-butyl)-6-
methyl-7-(2-fluorobenzyl)-5-(3-methoxyphenyl)imidazolo[1,2-a]pyrimid-4-one
(f) 3-[N-Methyl-N-(2-pyridylethyl)]aminomethyl-2-
(ethoxycarbonylmethyl)-6-methyl-7-(2-fluorobenzyl)-5-(3-
methoxyphenyl)imidazolo[ 1,2-a]pyrimid-4-one
(g) 1-(N-Benzyl-N-methyl)aminomethyl-2-(tert-butyl)-4-(2-
fluorobenzyl)-6-(3-phenylpropylaminocarbonyl)pyrrolo[1,2-a]pyrimid-7-one
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
32
(h) 1-[(N-Methyl-N-(2-pyridylethyl)]aminomethyl-2-(tert-butyl)-4-
(2-fluorobenzyl)-6-(3-phenylpropylaminocarbonyl)pyrrolo[ 1,2-a]pyrimid-7-one
(i) 1-[(N-Methyl-N-(2-pyridylethyl)]aminomethyl-2-(tert-butyl)-4-
(2-fluorobenzyl)-5-methyl-6-(3-methoxyphenyl)pyrrolo[ 1,2-a]pyrimid-7-one
(j) 1-(N-Benzyl-N-methyl)aminomethyl-2-(t-butyl)-4-(2-
fluorobenzyl)-S-methyl-6-(3-methoxyphenyl)imidazolo[3,4-a]pyrimid-7-one
(k) 1-[N-Methyl-N-(2-pyridylethyl)]aminomethyl-4-(2-
fluorobenzyl)-5-methyl-6-(3-methoxyphenyl)imidazolo[3,4-a]pyrimid-7-one
(1) 1-[N-(2-Furanmethyl)-N-methyl]aminomethyl-4-(2-
fluorobenzyl)-5-methyl-6-(3-methoxyphenyl)imidazolo[3,4-a]pyrimid-7-one
(m) 3-(N-Benzyl-N-methyl)aminomethyl-6-methyl-7-(2-
fluorobenzyl)-5-(3-methoxyphenyl)-1,2,4-triazolo[4,3-a]pyrimid-4-one
(n) 3-[N-Methyl-N-(2-pyridylethyl)]aminomethyl-6-methyl-7-(2-
fluorobenzyl)-5-(3-methoxyphenyl)-1,2,4-triazolo[4,3-a]pyrimid-4-one
(o) 3-[N-(2-Furanmethyl)-N-methyl]aminomethyl-6-methyl-7-(2-
fluorobenzyl)-5-(3-methoxyphenyl)-1,2,4-triazolo[4,3-a]pyrimid-4-one
(p) 1-(N-Benzyl-N-methyl)aminomethyl-2-(1-methoxycarbonyl-1-
methylethyl)-7-(2-fluorobenzyl)-6-methyl-5-(3-methoxyphenyl)imidazolo[ 1,2-
a]pyrimid-4-one
(~ 1-[N-(2-Pyridylethyl)-N-methyl]aminomethyl-2-(1-
methoxycarbonyl-1-methylethyl)-7-(2-fluorobenzyl)-6-methyl-5-(3-
methoxyphenyl)imidazolo[ 1,2-a]pyrimid-4-one
As mentioned above, the GnRH receptor antagonists of this invention
have utility over a wide range of therapeutic applications, and may be used to
treat a
variety of sex-hormone related conditions in both men and women, as well as
mammals
in general. For example, such conditions include endometriosis, uterine
fibroids,
polycystic ovarian disease, hirsutism, precocious puberty, gonadal steroid-
dependent
neoplasia such as cancers of the prostate, breast and ovary, gonadotrophe
pituitary
adenomas, sleep apnea, irritable bowel syndrome, premenstrual syndrome, benign
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
33
prostatic hypertrophy, contraception and infertility (e.g., assisted
reproductive therapy
such as in vitro fertilization).
The compounds of this invention are also useful as an adjunct to
treatment of growth hormone deficiency and short stature, and for the
treatment of
systemic lupus erythematosis.
In addition, the compounds are useful in combination with androgens,
estrogens, progesterones, and antiestrogens and antiprogestogens for the
treatment of
endometriosis, fibroids, and in contraception, as well as in combination with
an
angiotensin-converting enzyme inhibitor, an angiotensin II-receptor
antagonist, or a
renin inhibitor for the treatment of uterine fibroids. The compounds may also
be used
in combination with bisphosphonates and other agents for the treatment and/or
prevention of disturbances of calcium, phosphate and bone metabolism, and in
combination with estrogens, progesterones and/or androgens for the prevention
or
treatment of bone loss or hypogonadal symptoms such as hot flashes during
therapy
with a GnRH antagonist.
In another embodiment of the invention, pharmaceutical compositions
containing one or more GnRH receptor antagonists are disclosed. For the
purposes of
administration, the compounds of the present invention may be formulated as
pharmaceutical compositions. Pharmaceutical compositions of the present
invention
comprise a GnRH receptor antagonist of the present invention and a
pharmaceutically
acceptable carrier and/or diluent. The GnRH receptor antagonist is present in
the
composition in an amount which is effective to treat a particular disorder--
that is, in an
amount sufficient to achieve GnRH receptor antagonist activity, and preferably
with
acceptable toxicity to the patient. Typically, the pharmaceutical compositions
of the
present invention may include a GnRH receptor antagonist in an amount from 0.1
mg to
250 mg per dosage depending upon the route of administration, and more
typically
from 1 mg to 60 mg. Appropriate concentrations and dosages can be readily
determined by one skilled in the art.
Pharmaceutically acceptable carrier and/or diluents are familiar to those
skilled in the art. For compositions formulated as liquid solutions,
acceptable carriers
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
34
and/or diluents include saline and sterile water, and may optionally include
antioxidants, buffers, bacteriostats and other common additives. The
compositions can
also be formulated as pills, capsules, granules, or tablets which contain, in
addition to a
GnRH receptor antagonist, diluents, dispersing and surface active agents,
binders, and
lubricants. One skilled in this art may further formulate the GnRH receptor
antagonist
in an appropriate manner, and in accordance with accepted practices, such as
those
disclosed in Remington's Pharmaceutical Sciences, Gennaro, Ed., Mack
Publishing Co.,
Easton, PA 1990.
In another embodiment, the present invention provides a method for
treating sex-hormone related conditions as discussed above. Such methods
include
administering of a compound of the present invention to a warm-blooded animal
in an
amount sufficient to treat the condition. In this context, "treat" includes
prophylactic
administration. Such methods include systemic administration of a GnRH
receptor
antagonist of this invention, preferably in the form of a pharmaceutical
composition as
discussed above. As used herein, systemic administration includes oral and
parenteral
methods of administration. For oral administration, suitable pharmaceutical
compositions of GnRH receptor antagonists include powders, granules, pills,
tablets,
and capsules as well as liquids, syrups, suspensions, and emulsions. These
compositions may also include flavorants, preservatives, suspending,
thickening and
emulsifying agents, and other pharmaceutically acceptable additives. For
parental
administration, the compounds of the present invention can be prepared in
aqueous
injection solutions which may contain, in addition to the GnRH receptor
antagonist,
buffers, antioxidants, bacteriostats, and other additives commonly employed in
such
solutions.
Rat Anterior Pituitary Cell Culture Assay of GnRH Antagonists
Anterior pituitary glands are collected from 7-week-old female Sprague-
Dawley rats and the harvested glands digested with collagenase in a dispersion
flask for
1.5 hr at 37°C. After collagenase digestion, the glands are further
digested with
neuraminidase for 9 min at 37°C. The digested tissue is then washed
with 0.1%
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
BSA/McCoy's SA medium, and the washed cells suspended in 3% FBS/0.1
BSA/McCoy's SA medium and plated into 96-well tissue culture plates at a cell
density
of 40,000 cells per well in 200 ~1 medium. The cells are then incubated at
37°C for 3
days. One pituitary gland normally yields one 96-well plate of cells, which
can be used
5 for assaying three compounds. For assay of an GnRH antagonist, the incubated
cells
are first washed with 0.1% BSA/McCoy's SA medium once, followed by addition of
the
test sample plus 1nM GnRH in 200 ~l 0.1% BSA/McCoy's SA medium in triplicate
wells. Each sample is assayed at 5-dose levels to generate a dose-response
curve for
determination of its potency on the inhibition of GnRH stimulated LH and/or
FSH
10 release. After 4-hr incubation at 37°C, the medium is harvested and
the level of LH
and/or FSH secreted into the medium determined by RIA.
RIA of LH and FSH
For determination of the LH levels, each sample medium is assayed in
duplicates and all dilutions are done with RIA buffer (0.01M sodium phosphate
15 buffer/O.15M NaCI/1% BSA/0.01% NaN3, pH 7.5) and the assay kit is obtained
from
the Nation Hormone and Pituitary Program supported by NIDDK. To a 12x75 mm
polyethylene test tube is added 100 ~1 of sample medium diluted 1:5 or rLH
standard in
RIA buffer and 100 ~1 of [ 125I]-labeled rLH (30,000 cpm) plus 100 ~1 of
rabbit anti-
rLH antibody diluted 1:187,500 and 100 ~1 RIA buffer. The mixture is incubated
at
20 room temperature over-night. In the next day, 100 ~1 of goat anti-rabbit
IgG diluted
1:20 and 100 ~1 of normal rabbit serum diluted 1:1000 are added and the
mixture
incubated for another 3 hr at room temperature. The incubated tubes are then
centrifuged at 3,000 rpm for 30 min and the supernatant removed by suction.
The
remaining pellet in the tubes is counted in a gamma-counter. RIA of FSH is
done in a
25 similar fashion as the assay for LH with substitution of the LH antibody by
the FSH
antibody diluted 1:30,000 and the labeled rLH by the labeled rFSH.
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
36
Radio-iodination of GnRH peptide
The GnRH analog is labeled by the chloramine-T method. To 10 ~g of
peptide in 20 ~1 of O.SM sodium phosphate buffer, pH 7.6, is added 1 mCi of
Na125I,
followed by 22.5 wg chloramine-T and the mixture vortexed for 20 sec. The
reaction is
stopped by the addition of 60 ~g sodium metabisulfite and the free iodine is
removed by
passing the iodinated mixture through a C-8 Sep-Pak cartridge (Millipore
Corp.,
Milford, MA). The peptide is eluted with a small volume of 80%
acetonitrile/water.
The recovered labeled peptide is further purified by reverse phase HPLC on a
Vydac C-
18 analytical column (The Separations Group, Hesperia, CA) on a Beckman 334
gradient HPLC system using a gradient of acetonitrile in 0.1 % TFA. The
purified
radioactive peptide is stored in 0.1 % BSA/20% acetonitrile/0.1 % TFA at -800C
and can
be used for up to 4 weeks.
GnRH receptor membrane binding assay
Cells stably, or transiently, transfected with GnRH receptor expression
vectors are harvested, resuspended in 5% sucrose and homogenized using a
polytron
homogenizer (2x15 sec). Nuclei are removed by centrifugation (3000 x g for 5
min.),
and the supernatant centrifuged (20,000 x g for 30 min, 4° C) to
collect the membrane
fraction. The final membrane preparation is resuspended in binding buffer
(IOmM
Hepes (pH 7.5), 150 mM NaCI, and 0.1% BSA) and stored at -70°C. Binding
reactions
are performed in a Millipore MultiScreen 96-well filtration plate assembly
with
polyethylenimine coated GF/C membranes. The reaction is initiated by adding
membranes (40 ug protein in 130 u1 binding buffer) to SOuI of ~l2sl]_labeled
GnRH
peptide (100,000 cpm), and 20u1 of competitor at varying concentrations. The
reaction is terminated after 90 minutes by application of vacuum and washing
(2X)
with phosphate buffered saline. Bound radioactivity is measured using 96-well
scintillation counting (Packard Topcount) or by removing the filters from the
plate and
direct gamma counting. K; values are calculated from competition binding data
using
non-linear least squares regression using the Prism software package (GraphPad
Software).
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
37
The following examples are provided for purposes of illustration, not
limitation. In summary, the GnRH receptor antagonists of this invention may be
assayed by the methods disclosed above, while Examples 1-4 disclose the
synthesis of
representative compounds of this invention.
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
38
EXAMPLE 1
SYNTHESIS OF 2-(TERT-BUTYL)-3-[ N-METHYL-N-(2-PYRIDYLETHYL)AMINOMETHYL~-S-
(3-METHOXYPHENYL)-6-METHYL-7-(2-FLUOROBENZYL)IMMIDAZOLO[ 1,2-A~PYRIMID-4-
ONE
' I ~N
~O
N, O i
N N w
Step 1A S-Bromo-6-meth ~~1-2-(tert-butyl)-7H imidazolo[1,2-alpyrimid-4-one
Under nitrogen atmosphere, to a solution of 2-amino-S-bromo-4-
hydroxy-6-methylpyrimidine (4.08 g, 20 mmol.) in dry DMF (80 ml), sodium
hydride
(800 mg, 20 mmol., 60% in mineral oil) was carefully added. The mixture was
stirred
at room temperature for O.S hour, followed by addition of bromomethyl tert-
butyl
ketone (20 mmol.). The mixture was then stirred at room temperature overnight.
The
resultant mixture was concentrated in vacuo, and the residue was dissolved in
acetone
(SO ml) and diluted with water (100 ml). Slowly concentration under reduced
pressure
resulted in a precipitation. The solid was collected by filtration, washing
with water,
1 S ether, and dried to yield the desired product as a white powder (4.16 g,
80% yield); MS
:284/286 (M+H)+.
Step 1 B S ~3-methoxyphenXl)-6-methyl-2-(tert-butyl)-7H imidazolo [ 1,2-
a]pyrimid-4-one
To a pressure vessel , potassium carbonate (1.38 g, 10 mmol.) , 3-
methoxyphenylboronic acid (1.06 g, 7.0 mmol.), S-bromo-6-methyl-2-(tert-butyl)-
7H
imidazolo[1,2-a]pyrimid-4-one (1.42 g, S mmol) , toluene (20 ml), water (S ml)
were
added. The mixture was bubbled by nitrogen gas for 10 minutes to remove the
air.
Then Pd(PPh3)4 (S00 mg) was added and the vessel was sealed immediately and
heated
at 110°C for 6 hours. After cooled to room temperature, the mixture was
extracted
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
39
with ethyl acetate (100 ml). The ethyl acetate layer was washed with water,
dried and
concentrated to give a yellow solid. It was stirred with a mixture of ether
/ethyl acetate
(SOmI/Sml) and solid was collected by filtration to give the crude product
(0.74 g); MS
312 (M+H)+.
Step 1 C 5-(3-methoxyphenyl)-6-methyl-2-(tert-butyl)-7-(2-fluorophenylmethyl)-
imidazolo[ 1,2-a]pyrimid-4-one
To a solution of 5-(3-methoxyphenyl)-6-methyl-2-(tert-butyl)-7H
imidazolo[1,2-a]pyrimid-4-one (0.74 g, 2.4 mmol.) in DME (5 ml),
tetrabutylammonium fluoride (4 ml, 1.0 M in THF) was added and followed by
addition of 2-fluorobenzyl bromide (0.45 ml, 1.5 eq.). The mixture was stirred
at room
temperature overnight and then concentrated and purified by silica gel
chromatography
(hexane/ethyl acetate) to give the desired product (0.55 g) as a white powder;
MS:
420(M+H)+; NMR (CDC13, 8): 7.38-6.77 (9H, 4m), 5.69 (2H, s ), 3.80(3H, s),
2.20 (3H,
s), 1.32 (9H, s).
Step 1D 3- f N-methyl-N-[2-(2-pyridyl)ethyl~aminomethyl}-5-(3-
methoxyphenyl)-6-methyl-2-(tent-butyl)-7-(2-fluorophenylmethyl)-
imidazolo[ 1,2-a~pyrimid-4-one
To a solution of N-[2-(2-pyridylethyl)]-N-methylamine (136 mg, 1.0
mmol.) and aqueous formaldehyde (0.1 ml) in acetic acid (2 ml), 5-(3-
methoxyphenyl)
6-methyl-2-(tert-butyl)-7-(2-fluorophenylmethyl)-imidazolo[1,2-a]pyrimid-4-one
(210mg, O.Smmol) was added. The solution was stirred at room temperature for 1
hours and directly purified by prep HPLC to give the pure product (240 mg) as
a
trifluoroacetic acid salt; MS :568(M+H)+, 432. NMR (DMSO-d6, 8): 9.16 (1H,
brs),
8.21 (1H, d), 7.80 (1H, t), 7.39-6.86 (10H, m), 5.65 (2H, s), 4.85 (2H, s),
3.78 (3H, s),
3.66 (2H, brs), 3.28 (2H, t), 2.97 (3H, s), 2.27 (3H, s), 1.35 (9H; s).
Following the procedure similar to that described above, the following
compounds were prepared:
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
-Ar
R
Example R1NR2 R4 -Q-Ar MS(M+H)+
1-1 2-PyCH2CH2NMe t-Bu 3-OMe- 568
Ph
1-2 BnNMe t-Bu 3-OMe- 553
Ph .
1-3 FuryICHZNme t-Bu 3-OMe- 543
Ph
1-4 MeZNCH2CH2NMe t-Bu 3-OMe- 534
Ph
1-S MeOCHZCH2NMe t-Bu 3-OMe- 521
Ph
1-6 - N t-Bu 3-OMe- 532
Ph
1-7 2-PyCHzCH2NMe i-Bu 3-OMe- 568
Ph
1-8 BnNMe i-Bu 3-OMe- 553
Ph
1-9 2-PyCHZCH2NMe Me 3-OMe- 526
Ph
1-10 BnNMe Me 3-OMe- 511
Ph
1-11 ~~ ~ ~ t-Bu 3-OMe- 593
~ Ph
\ H ~
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
41
1-12 2-PyCHZNme t-Bu 3-OMe- 554
Ph
1-13 2-PyCHZCH2NMe 2-OH-t-Bu 3-OMe-
Ph
1-14 PhCHZNme t-Bu Ph 523
1-15 2-PyCHZCH2NMe t-Bu 2-Py 538
1-16 PhCH2Nme t-Bu Ph 524
1-17 BuNMe t-Bu Ph
1-18 ~ ~ t-Bu Ph
N
y
1-19 2-FuryICHZNMe t-Bu Ph 513
1-20 "~ t-Bu Ph 502
N
1-21 PhCH2Nme ~ 3-OMe- 595
Ph
1-22 PhCHZNCH2CH2CN ~ 3-OMe- 634
Ph
1-23 ~o i ~ ~1 ~ 3-OMe- 694
~
Ph
1-24 ~ ~ H ~ 3-OMe- 685
'. N' ,
Ph
1-25 F'' ~ ~ N ~ 3-OMe- 719
N
7 Ph
1-26 F'' ~ ~ 3-OMe- 749
i
. O
N
"
~"
Ph
0 0
-
~
~
1-27 2-FuryICHZNMe ~ 3-OMe- 585
Ph
1-28 Me2NCHZCH2NMe ~ 3-OMe- 576
Ph
1-29 2-PyCH2NH ~ 3-OMe- 582
Ph
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
42
1-30 PhCHZNme t-Bu 3-Py 524
1-31 PhCHzNme t-Bu 3-AcPh 565
1-32 2-PyCH2CHzNMe t-Bu 3-AcPh 580
1-33 2-FurylCH2NMe t-Bu 3-AcPh 555
1-34 2-PyCH2NH t-Bu 3-OMe- 552
Ph
1-35 Me2NCHZCH2NMe t-Bu 3-OMe- 546
Ph
1-36 ;~ t-Bu 3-OMe- 567
.~"~ Ph
1-37 "'_~ t-Bu 3-OMe- 567
.~"~ Ph
1-38 ' ~ t-Bu 3-OMe- 617
.~"~ Ph
1-39 \ ~ t-Bu 3-OMe- 553
.~" Ph
1-40 ~ i t-Bu 3-OMe- 567
,~" Ph
1-41 ' ~ t-Bu 3-OMe- 432, 603
.~"~ Ph
1-42 ' ~ t-Bu ' v 456, 577
\
N~
1-43 \ ~ t-Bu 2-F-Bn 434, 555
N
1-44 \ i t-Bu 2-F-Ph 541
N
1-45 ~ ~ t-Bu 2-F-Ph 5 83
~Nw
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
43
1-46 ~~ t-Bu 2-F-Ph 523
N
1-47 (~ t-Bu 2-F-Ph 533
N
1-48 N'~ t-Bu 2-F-Ph 564
~
,~
~
1-49 ~ i t-Bu 2-F-Ph 531
N
1-50 ~ t-Bu 2-F-Ph 564
~
N
1-51 ~ t-Bu 2-F-Ph 537
N~
1-52 ~'1 t-Bu 2-F-Ph 509
N
1-53 ~N~ t-Bu 2-F-Ph 536
N~
1-54 2-PyCH2CH2NMe t-Bu ' v 582
OH
1-55 2-PyCH2CHZNMe t-Bu 2-F-Ph 556
1-56 ' i t-Bu 2-F-Ph 420
Ho N,
1-57 2-PyCH2CH2NMe t-Bu r ~ 7 446
1-58 2-PyCH2Nme t-Bu r ~ 7 446
1-59 ~ ~N I ~ t-Bu r ~ 7 446
1-60 (~ t-Bu r ~ 7 559
O
~N
1-61 \ ~ t-Bu r ~ 7 446
~Nw
1-62 ~~ t-Bu r ~ 7 446
O
N
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
44
1-63 N t-Bu r ~ 7 562
~
~
~N~
1-64 ~ t-Bu r ~ 7 446
~ _
1-65 ~ t-Bu r ~ 7 446
0
N
1-66 ~ t-Bu r ~ 7 590
~
~Nv
1-67 ~ t-Bu r ~ 7 446
~
1-68 "1 t-Bu r ~ 7 446
~Nv O
1-69 ~ ~ ~ t-Bu r ~ 7 446
Ny
1-70 ~ ~ t-Bu r ~ 7 446
~ j
y
1-71 \ ~ t-Bu r ~ 7 446
~
y
1-72 N N -t-Bu r ~ 7 446
~I
\N
~
1-73 EtzNCH2CHZNMe t-Bu r ~ 7 576
EXAMPLE 2.1
SYNTHESIS OF 1-~N-BENZYL-N-METHYLAMINOMETHYL~-2-~TERT-BUTYL-4-~2-
FLUOROBENZYL~-6-ETHOXYCARBONYLPYRROL0~1,2-A~PYRIMID-7-ONE ~INTERMEDIATE~
N~ O O
N~O~
JN
F
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
Step 2A S-Ethoxycarbonyl-2-methyl-3-(2-oxo-3,3-dimethyl-butyl)pyrimid-4-one
To a suspension of 5-ethoxycarbonyl-2-methylpyrimid-4-one (2.7g,
14.75 mmol.) in DME (20 ml), tetrabutylammonium fluoride (22 ml, 22.0 mmol.)
was added . The solution was stirred at room temperature until solids
dissolved , then 1-
S bromopinacolone (2.2 ml, 1.1 eq., 16.22 mmol) was added. The solution was
stirred
overnight and concentrated to a brown oil. The crude mixture was purified by
silica gel
column chromatography (hexane /ethyl acetate, 100/0 to 0/100). A less polar O-
alkylated by-product was eluted first (1.8 g) and then the desired N-alkylated
product (1.1 g); MS (281, M+H)+. NMR (CDC13 ,8): 8.57 (1H, s) , 5.06( 1H, s),
10 4.35(2H, q), 2.42 (3H, s), 1.34 (3H, t), 1.30 (9H,s ).
Step 2B 6-Ethoxycarbon~tert-butyl)-4H pyrrolo[1,2-alpyrimid-7-one
5-Ethoxycarbonyl-2-methyl-3-(2-oxo-3,3-dimethyl-butyl)pyrimid-4-one
(1.1 g, 3.9 mmol.) was added into sodium ethoxide solution, made in situ from
sodium
(200 mg) and dry ethanol (50 ml). After stirred for 2 hours, the mixture was
acidified
15 slowly with 6N HCl resulting a precipitation. The precipitates were
collected by
filtration and washed with water (20 ml x2), ether (20 ml x3), dried to give
the desired
product (0.8 g); MS (263, M+H)+.
Step 2C 6-Ethoxycarbonyl-2-(tent-butyl)-4-(2-fluorobenzyl)pyrrolo[1,2-
a]pyrimid-7-one
20 To 6-ethoxycarbonyl-2-(tert-butyl)-4H pyrrolo[1,2-a]pyrimid-7-one (0.5
g, 1.9 mmol.) in DME (5 ml), tetrabutylammonium fluoride (4 ml, 4.0M in THF)
was
added, and a white foamy material formed. 2-Fluorobenzyl bromide (0.38 ml, 3
mmol.)
was added and the mixture was stirred at room temperature for 2 days.
Concentration of
the reaction mixture in vacuo produced an oil, which was dissolved in acetone
(20 ml)
25 and diluted with water until the solution turned a slight cloudy. Partially
concentration
to remove acetone by nitrogen flow resulting precipitation. The precipitates
were
collected by filtration and washed with water (20 ml x2), ether (20 ml x3) and
dried.
The desired product (0.57g) was obtained with excellent purity; MS: 371
(M+H)+, 325;
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
46
NMR (CDC13, 8): 8.29 (1H, s), 7.43-7.14(SH, 2m), 5.99 (1H, s) 5.18 (2H,s ),
4.35 (2H,
q), 1.37(3H, s), 1.26 (9H, s).
Step 2D 6-Ethoxycarbonyl-3-(N-benzyl-N-methylaminomethyl)-2-(tert-butyl)-7-
(2-fluorobenzyl)pyrrolo[ 1,2-a]pyrimid-7-one
Formaldehyde in water ( 1 drop) and N-benzyl-N-methylamine (2
drops) were added to acetic acid ( 1 ml) and stirred for 5 minutes. The 6-
ethoxycarbonyl-2-(tert-butyl)-7-(2-fluorobenzyl)pyrrolo[1,2-a]pyrimid-7-one
(20 mg,
0.05 mmol) was added. The solution was stirred at room temperature for 1 hour
and
concentrated to an oil. It was then neutralized by potassium carbonated
(saturated) and
the crude product was purified by prep TLC plate using CHC13/MEOH/NH40H
(400/50/2) to give the desired compound (11.1 mg); MS: 504(M+H)+, 383. NMR
(CDCl3, 8): 8.18 (1H, s), 7.41-7.13 (9H, m), 5.93 (1H, s), 5.10(2H, s), 4.47
(2H, s), 4.36
( 2H, q), 3.62 (2H, s), 2.08 (3H, s), 1.34 (9H, s). 1.34 (3H, t).
Following a procedure similar to that described above, the following
intermediates were prepared:
R2~.R1
O O
/ N ~ ~OEt
N
F
Example RINRz MS (M+H~
2A BnNMe 504
2B 2-PyCH2CHZNMe S 19
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
47
EXAMPLE 2.2
SYNTHESIS OF 1-(N-BENZYL-N-METHYLAMINOMETHYL)-2-(TERT-BUTYL)-4-(2
FLUOROBENZYL)-6-[(3-PHENYLPROPYLAMINO)CARBONYL]PYRROLO[ 1,2-A]PYRIMID-7
ONE
N O O
N~H I ,
N
I~
F
Step 2E 6-(3-Phenylpropylaminocarbonyl)-2-(tert-butyl)-7-(2-
fluorobenzyl)pyrrolo[ 1,2-a]pyrimid-7-one
To a solution of 3-phenyl-1-propylamine (0.27 g, 2.0 mmol.) in DME (3
ml) under nitrogen atmosphere, triethylaluminum (0.5 ml, 1.9 M in toluene) was
added. The solution was stirred at room temperature for 0.5 hour, followed by
addition
of 6-ethoxycarbonyl-2-(tent-butyl)-7-(2-fluorobenzyl)pyrrolo[1,2-a]pyrimid-7-
one
(135mg, 0.5 mmole) . The solution was then heated at 50°C overnight and
poured into a
6N HCl solution (5 ml). The crude product was extracted out by ethyl acetate
(50 ml).
The organic layer was filtered through a silica pad (2 g) and concentrated to
give the
desired product (100 mg) which was pure based on TLC (hexane/ethyl
acetate=1/1)
and used for the next step; MS: 460 (M+H)+.
Step 2F 6-(3-Phenylpropylaminocarbonyl)-2-(tent-butyl)-3-(N-benzyl-N-
methylaminomethyl)-7-(2-fluorobenzyl)pyrrolo[ 1,2-a]pyrimid-7-one
To a solution of formaldehyde (37% aqueous, 1 drop) and N-benzyl-N-
methylamine ( 1 drop) in acetic acid ( 1 ml) was added 6-(3-
phenylpropylaminocarbonyl)-2-(tert-butyl)-4-(2-fluorobenzyl)pyrrolo[ 1,2-
a]pyrimid-7-
one (l4mg, 0.03 mmol). The mixture was stirred at room temperature for 1 hour
and
concentrated in vacuo. The crude product was purified on prep-TLC plates using
CHC13/MEOH/NH40H (400/50/2) to give the desired product (13 mg); MS: 593
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
48
(M+H)+, 472; NMR (CDC13, 8) 9.29 (1H, t), 8.43 (1H, s), 7.34-7.11 (14H, m),
S.9S
(1H, s), 5.13 (2H, s), 4.42 (2H, s), 3.60 (2H, s), 3.46 (2H, m), 2.73 (2H, t),
2.23 (3H, s),
2.01 (2H, m), 1.38 (9H, s).
Following a procedure similar to that described above, the following
S compounds were prepared:
R~.RI
O O
/ N ~ H ~ i
N
F
Example R1NR2 MS (M+I~+
2C BnNMe S93
2D 2-PyCH2CHZNMe 608
EXAMPLE 3
1 O SYNTHESIS OF 2-(TERT-BUTYL)-3-[N-(2-FLUOROPHENYL)ETHYL]AMINOMETHYL-S-(3-
METHOXYPHENYL)-6-METHYL-7-(2-FLUOROBENZYL)IMMIDAZOLO[ 1,2-A]PYRIMID-4-ONE
/ F
~O
NH ~
/ N
N~N
F
Step 3A. 2-(tert-Butyl)-3-formyl-S-(3-methoxyphenyl)-6-methyl-7-(2-
fluorobenzyl)imidazolo[ 1,2-a]pyrimid-4-one
1 S To a solution of 2-(tert-butyl)-S-(3-methoxyphenyl)-6-methyl-7-(2-
fluorobenzyl)imidazolo[1,2-a]pyrimid-4-one (8S0 mg, 2.02 mmol.) in dry DMF (2
ml),
POC13(lml) was added . The mixture was heated at SO °C for 10 minutes
and ethyl
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
49
acetate (200 ml ) was added, followed by addition of saturated sodium
bicarbonate
slowly until it is neutral. The organic layer was dried and concentrated to
give the
crude product (910 mg) as a yellow solid; MS: 448 (M+H)+.
Step 3B 2-(tert-Butyl)-3-[N-(2-fluorophenyl)ethyl]aminomethyl-5-(3-
methoxyphenyl)-6-methyl-7-(2-fluorobenzyl)imidazolo[ 1,2-a]pyrimid-
4-one
To a solution of 2-(tert-butyl)-3-formyl-5-(3-methoxyphenyl)-6-methyl-
7-(2-fluorobenzyl)imidazolo[1,2-a]pyrimid-4-one (20 mg, 0.045 mmol.) in 1.2-
dichloroethane ( 1 ml), 2-(2-fluorophenyl)-ethylamine ( 12.5 mg, 2.0 eq.) was
added,
followed by addition of sodium triacetoxyborohydride (48 mg, 5 eq.). The
mixture was
stirred at room temperature overnight. The product (12.9 mg) was isolated by a
prep-
TLC plate ( 0.5 mm thickness, 20x20 cm size) using CHCl3/MeOH/NH40H (400/50/2)
as elution solvents; MS: 571(M+H)+, 432. NMR (CDC13, 8) : 7.36-6.72 (12H, m),
5.63(2H,s), 4.28(2H,s), 3.81(3H,s), 3.05-2.87 (4H,m), 2.16(3H,s), 1.40(9H,s).
Following a procedure similar to that described above, the following
compounds were prepared:
R
O
/ ~ ~ w O~
N
i
F
Example R1 MS (M+H)+
3A (2-FPh)CHZCHZ 571
3B (2-Py)CH2CH2 554
3C PhCHzCH2CHzCHz 581
3D 2-PyCH2 540
3E EtOCH2CHZCH2 535
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
SO
EXAMPLE 4.1
SYNTHESIS OF 2-(1-METHOXYCARBONYL-1-METHYLETHYL)-3-{N-[(2
PYRIDYL)ETHYL]AMINOMETHYL}-S-BROMO-6-METHYL-7-(2
FLUOROBENZYL)IMMIDAZOLO[1,2-A]PYRIMID-4-ONE (INTERMEDIATE)
1
I ~N
NH O
p~~ N Br
1. ~\ %~ ~
-O N
F
S
Step 4.1A S-Bromo-6-methyl-2-(1-methoxycarbonyl-1-methylethyl)-7H
imidazolo[ 1,2-a]pyrimid-4-one
Under nitrogen atmosphere, to a solution of 2-amino-S-bromo-4-
hydroxy-6-methylpyrimidine (4.08 g, 20 mmol.) in dry DMF (80 ml), sodium
hydride
(800 mg, 20 mmol., 60% in mineral oil) was carefully added. The mixture was
stirred
at room temperature for O.S hour, followed by addition of methyl 4-bromo-2,2-
dimethyl
acetoacetate (20 mmol.). The mixture was then stirred at room temperature
overnight.
The resultant mixture was concentrated in vacuo, and the residue was dissolved
in
acetone (SO ml) and diluted with water (100 ml). Slowly concentration under
reduced
1 S pressure resulted a precipitation. The solid was collected by filtration,
washing with
water, ether, and dried to yield the desired product; MS: 328 (M+H); proton
NMR
(CDC13): 1.64 (s, 6H), 2.SS (s, 3H), 3.77 (s, 3H), 7.40 (s, 1H).
Step 4.2B S-Bromo-6-methyl-2-(1-methoxycarbonyl-1-methylethyl)-7-(2-
fluorophenylmethyl)-imidazolo[ 1,2-a]pyrimid-4-one
To a solution of S-bromo-6-methyl-2-(1-methoxycarbonyl-1-
methylethyl)-7H imidazolo[1,2-a]pyrimid-4-one (2.4 mmol.) in DME (S ml),
tetrabutylammonium fluoride (4 ml, 1.0 M in THF) was added and followed by
addition of 2-fluorobenzyl bromide (0.45 ml, 1.S eq.). The mixture was stirred
at room
temperature overnight and then concentrated and purified by silica gel
chromatography
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
S1
(hexane/ethyl acetate) to give the desired product as a white powder; MS: 436
(M+H);
NMR (CDC13, 8): 1.59 (s, 6H), 2.60 (s, 3H), 3.64 (s, 3H), 5.68 (s, 2H), 7.00-
7.38 (m,
4H), 7. S 1 (s, 1 H).
Step 4.1C 3-{N-[2-(2-Pyridyl)ethyl]aminomethyl}-S-bromo-6-methyl-2-(1-
S methoxycarbonyl-1-methylethyl)-7-(2-fluorophenylmethyl)-
imidazolo[ 1,2-a]pyrimid-4-one
To a solution of 2-(2-pyridyl)ethylamine (12 mg, 0.1 mmol.) and
aqueous formaldehyde (0.01 ml) in acetic acid (1 ml), S-bromo-6-methyl-2-(1-
methoxycarbonyl-1-methylethyl)-7-(2-fluorophenylmethyl)-imidazolo[ 1,2-
a]pyrimid-4
one (22 mg, O.OS mmol.) was added. The solution was stirred at room
temperature for 1
hours and directly purified by prep HPLC to give the product; MS :570 (M+H).
EXAMPLE 4.2
SYNTHESIS OF 2-(1-METHOXYCARBONYL-1-METHYLETHYL)-3-(N-METHYL-N-
BENZYLAMINOMETHYL)-S-(3-METHOXYPHENYL)-6-METHYL-7-(2-
1 S FLUOROBENZYL)IMIDAZOLO[ 1,2-A]PYRIMID-4-ONE
Step 4.2A 2-(1-Methoxycarbonyl-1-methylethyl)-S-(3-methoxyphenyl)-6-methyl-
7-(2-fluorobenzyl)imidazolo[ 1,2-a]pyrimid-4-one
Into a mixture of 2-(1-methoxycarbonyl-1-methylethyl)-S-(bromo)-6-
methyl-7-(2-fluorobenzyl)imidazolo[1,2-a]pyrimid-4-one (200 mg, 4S8 pmol), 3-
methoxyphenylboronic acid (139 mg, 916 pmol), potassium carbonate (190 mg, 1.4
mmol) in toluene (4 ml) and HZO (2 ml) were added under NZ
tetrakis(triphenylphosphine)palladium(0) (26 mg, 23 ~mol). The resulting
solution was
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
52
stirred and refluxed in a ChemGlass pressure tube under NZ for 2.5 hours. The
solution
was extracted with EtOAc and purified using Flash silica chromatography
(hexane to
hexane/EtOAc, 7/3) to give the desired product as an ecru solid in 83% yield;
MS: 464
(M+H).
Step 4.2B 2-(1-Methoxycarbonyl-1-methylethyl)-3-(N-methyl-N-
benzylaminomethyl)-5-(3-methoxyphenyl)-6-methyl-7-(2-
fluorobenzyl)imidazolo[ 1,2-a]pyrimid-4-one
2-( 1-Methoxycarbonyl-1-methylethyl)-5-(3-methoxyphenyl)-6-methyl-
7-(2-fluorobenzyl)imidazolo[1,2-a]pyrimid-4-one (17.7 mg, 38 pmol) was added
to a
stirring solution of aqueous formaldehyde (one drop) and N-methylbenzylamine
(O.lml,
775 ~mol) in acetic acid ( 1 ml), and then stirred at room temperature
overnight. The
solution was dried under N2, made basic with NaHC03, extracted with
dichloromethane
and purified using prep silica TLC (hexane/EtOAc, 6/4) to give the product as
an oil in
94% yield. H'-NMR (CDC13): 1.69(s,6H), 2.07(s,3H), 2.16(s,3H), 3,58(s, 2H),
3.61 (s,3H), 3.81 (s,3H), 4.26(s,2H), 5.61 (s,2H), 6.75(m,13H).
Following a procedure similar to that described above, the following
compounds were prepared:
~-Ar
Example R~NRZ Q-Ar MS (M+I~+
4A BnNMe 3-OMe-Ph 597
4B 2-PyCHZCHZNMe 3-OMe-Ph 611
4C PhCH2CHZNMe 3-OMe-Ph 612
4D 2-FuranCH2NMe 3-OMe-Ph 587
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
53
4E / \ 3-OMe-Ph 651
N-
4F F 3-OMe-Ph 763
/ \
U
/ \
F
4G 2-PyCHzNH 3-OMe-Ph 584
4H 2-PyCHZCH2NMe 2-F-3- 630
OMePh
EXAMPLE 5
SYNTHESIS OF 2-ETHOXYCARBONYLMETHYL-3-{N-METHYL-N-[2-(2-
PYRIDYL)ETHYL] AMINOMETHYL } -S-(3-METHOXYPHENYL)-6-METHYL-7-(2-
FLUOROBENZYL)IMIDAZOLO[ 1,2-A]PYRIMID-4-ONE
Step SA 2-Ethoxycarbonylmethyl-5-bromo-6-methyl-7H-imidazolo[1,2-
a]pyrimid-4-one.
A solution of 2-amino-5-bromo-6-methylpyrimid-4-one (8.16 g, 40
mmol.) in DMF (30 ml) was cooled down with ice-water bath. NaH (60% in oil,
1.76 g,
44 mmol.) was added in portions and the reaction mixture stirred at 0
°C for 30 min and
then warmed to ambient temperature for 1h. Ethyl 4-chloroacetoacetate (6.91 g,
42
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
54
mmol) in 50 ml DMF was added dropwise in 3 h and the reaction mixture was
stirred at
ambient temperature overnight. Reaction mixture was quenched with saturated .
NH4C1/HZO, and precipitates were filtered. The filtrate was concentrated in
vacuo and
the residue was partitioned between water and dichloromethane. The aqueous
phase
was extracted with dichloromethane (3 x 100 ml). The organic phases were
combined,
dried over sodium sulfate and concentrated to give the tilted compound (3.05g,
24%);
MS 314 (M+H)+.
Step SB 2-Ethoxycarbonylmethyl-5-bromo-6-methyl-7-(2-fluorobenzyl)-
imidazolo[ 1,2-a]pyrimid-4-one.
A solution of 2-ethoxycarbonylmethyl-5-bromo-6-methyl-7H-
imidazolo[1,2-a]pyrimid-4-one. (3.00 g, 9.55 mmol.) in DME (35 ml) was treated
with
1M TBAF/THF (14.3 mL, 14.3 mmol.) and stirred at ambient temperature for 30
min.
2-Fluorobenzyl bromide (2.71 g, 14.3 mmol.) was introduced. The reaction
mixture
was stirred at ambient temperature overnight, concentrated in vacuo, and the
residue
was purified by flash chromatography (silica, 40% EtOAc/hexanes) to give the
designed compound (356 mg, 9%). MS 424 (M+H)+.
Step SC 2-Ethoxycarbonylmethyl-5-(3-methoxyphenyl)-6-methyl-7-(2-
fluorobenzyl)imidazolo[ 1,2-a]pyrimid-4-one.
2-Ethoxycarbonylmethyl-5-bromo-6-methyl-7-(2-
fluorobenzyl)imidazolo[1,2-a]pyrimid-4-one (190 mg, 0.45 mmol.) in
benzene/water (8
m1/5 ml) was added KZC03 (155 mg, 1.12 mmol.), 3-methoxyphenylboronic acid (86
mg, 0.56 mmol.), and tetrakis(triphenylphosphine)palladium(0) (52 mg, 0.045
mmol.).
The reaction mixture was deoxygenated with NZ and heated at 90 °C for
16 h. The
reaction mixture was partitioned between brine and EtOAc. The organic layer
was dried
(sodium sulfate), evaporated, purified by flash chromatography (silica, 45%
EtOAc/hexane) to give the title compound. (93 mg, 46%); MS 450 (M+H)+.
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
Step SD 2-Ethoxycarbonylmethyl-3-{N-methyl-N-[2-(2-
pyridyl)ethyl]aminomethyl}-5-(3-methoxyphenyl)-6-methyl-7-(2-
fluorobenzyl)imidazolo[ 1,2-a]pyrimid-4-one.
A mixture of aqueous formaldehyde (37%, 1 drop) and amine (1 drop) in
5 acetic acid ( 1.5 ml) was added 2-ethoxycarbonylmethyl-5-(3-methoxyphenyl)-6
methyl-7-(2-fluorobenzyl)imidazolo[1,2-a]pyrimid-4-one (20 mg, 0.045 mmol.)
and the
reaction mixture was stirred at ambient temperature for 12 h. The solvent was
evaporated and the residue was partitioned between dichloromethane and
saturated
NaHC03/water, the organic layer was dried (sodium sulfate), evaporated and
purified
10 by prep. TLC (silica, 5% MeOH/dichloromethane) to give the titled compound;
MS 598
(M+H)+.
Replacing 2-(2-pyridyl)ethyl at the R~ position with benzyl, gave 2-
ethoxycarbonylmethyl-3- {N-methyl-N-benzyl)-S-(3-methoxyphenyl)-6-methyl-7-(2-
fluorobenzyl)imidazolo[1,2-a]pyrimid-4-one. MS 583 (M+H)+; H'-NMR (CDC13):
15 1.23 (t,3H), 2.15 (s,3H), 2,47 (s,3H), 3.79 (s,2H), 3.83 (2,2H), 3.87
(s,3H), 4.14 (q, 2H),
4.30 (s,2H), 5.62 (s,2H), 6.78-7.65 (m,13H).
Following a procedure similar to that described above, the following
compounds were prepared:
Example R1NR2 MS (M+H)
SA 2-PyCH2CH2NMe 598
SB BnNMe 583
SC BuNMe 549
SD ~N Bn 637
SUBSTITUTE SHEET (RULE 26)
CA 02386942 2002-04-09
WO 01/29044 PCT/US00/28677
56
SE ~ n - 562
a
It will be appreciated that, although specific embodiments of the
invention have been described herein for purposes of illustration, various
modifications
may be made without departing from the spirit and scope of the invention.
Accordingly, the invention is not limited except as by the appended claims.
SUBSTITUTE SHEET (RULE 26)