Note: Descriptions are shown in the official language in which they were submitted.
CA 02387529 2002-03-20
- 1 -
Biphenyl derivatives as NHE-3 inhibitors
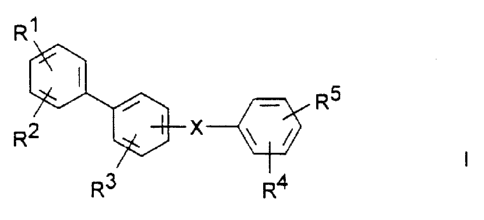
The invention relates to compounds of the formula I
R1
RS
R2 ~~ X \ /
I
R3 R4
in which
R1, Rq in each case independently of one another are
-C(=NH)-NHz, which can also be monosubsti-
tuted by -COA, -CO- [C (R6) z] n-Ar, -COOA, -OH or
by a conventional amino protective group,
NH-C (=NH) -NHz, -CO-N=C (NHz) z,
~~N.O ~~N,O
HN~ or N=
0 CHI
R2, R3, RS in each case independently of one another are
H, A, OR6, N (R6) z, NOz, CN, Hal, NHCOA,
NHCOAr, NHSOzA, NHSOzAr, COOR6, CON (R6) 2.
CONHAr, CORE, COAr, S (0) nA, ~ S (0) nAr,
2 0 -0- [ C ( R6 ) 2 ] m-COOR6, - [ C ( R6 ) z ] p-COOR°,
-~- [ C ( R6 ) 2 ] m-CON ( R6 ) 2. - [ C ( R6 ) 2 ] p-CON ( R6 ) z .
-0- [ C ( R6 ) z ] m-CONHAr or - [ C ( R6 ) z ] p-CONHAr,
X is - [C (R6) 2] n-i -CR°=CR6-, - [C (R6) 2] WOW
2 5 -0- [ C ( R6 ) 2 ] n-, -C00-, -OOC-, -CONR6- or
-NR6C0-,
R6 is H, A or benzyl,
30 A is alkyl having 1-20 C atoms, in which one or
two CHI groups can be replaced by 0 or S
CA 02387529 2002-03-20
- 2 -
atoms or by -CR6=CR°- groups and/or 1-7 H
atoms can be replaced by F,
Ar is phenyl or naphthyl, which is unsubstituted
or mono-, di- or trisubstituted by A, Ar',
OR6, OAr' , N (R6) z, NOz, CN, Hal, NHCOA,
NHCOAr' , NHSOzA, NHSOzAr' , COOR6, CON (R6) z,
CONHAr' , CORE, COAr' , S ( 0 ) nA or S ( 0 ) nAr' ,
Ar' is phenyl or naphthyl, which is unsubstituted
or mono-, di- or trisubstituted by A, OR6,
N (R6) z, NOz, CN, Hal, NHCOA, COOR6, CON (R6) z.
CORE Or S (0) nA,
Hal is F, Cl, Br or I,
n is 0, 1 or 2,
m is 1 or 2,
p is 1 or 2,
and their salts and solvates as NHE-3 inhibitors.
Other inhibitors of the sodium/proton exchanger
subtype 3 are described, for example, in EP 0 825 178.
The invention was based on the object of finding novel
compounds having valuable properties, in particular
those which can be used for the production of
medicaments.
DE 19819548 describes that the compounds of the
formula I and their salts have factor Xa-inhibiting
properties and can therefore be employed for the
control and prevention of thromboembolic disorders such
as thrombosis, myocardial infarct, arteriosclerosis,
inflammation, apoplexy, angina pectoris, restenosis
after angioplasty and intermittent claudication.
*.
1
CA 02387529 2002-03-20
- 3 -
Surprisingly, it has been found that the compounds of
the formula I and their salts inhibit the sodium/proton
exchanger subtype 3 and have good tolerability.
The compounds of the formula I can be employed as
pharmaceutical active compounds in human and veterinary
medicine.
It is known that the Na+/H+ exchanger is a family having
at least 6 different isoforms (NHE-1 to NHE-6), which
have now all been cloned. While the subtype NHE-1 is
distributed ubiquitously in all tissues in the whole
body, the other NHE subtypes are expressed selectively
in specific organs such as in the kidney or in the
lumen wall and contraluminal wall of the small
intestine. This distribution reflects the specific
functions served by the various isoforms, namely on the
one hand the regulation of the intracellular pH and of
the cell volume by the subtype NHE-1 and on the other
hand the Na+ absorption and reabsorption in the
intestine and kidney by the isoforms NHE-2 and NHE-3.
The isoform NHE-4 was mainly found in the stomach. The
expression of NHE-5 is restricted to the brain and
neuronal tissue. NHE-6 is the isoform which forms the
sodium proton exchanger in the mitochrondria.
The isoform NHE-3 is expressed in particular in the
apical membrane of the proximal renal tubuli; an NHE-3
inhibitor therefore exerts, inter alia, a renal protec
tive action.
The therapeutic use of a selective inhibitor for NHE-3
isoforms is varied. NHE-3. inhibitors inhibit or reduce
tissue damage and cell necroses after pathophysio-
logical hypoxic and ischaemic events which lead to an
activation of the NHE activity, such as is the case
during renal ischaemia or during the removal, transport
and reperfusion of a kidney in kidney transplantation.
A
CA 02387529 2002-03-20
- 4 -
The compounds of the formula I have a hypotensive
action and are suitable as pharmaceutical active
compounds for the treatment of hypertension. They are
furthermore suitable as diuretics.
The compounds of the formula I have an anti-ischaemic
action on their own or in combination with NHE inhibi-
tors of different subtype specificity and can be used
in thromboses, atherosclerosis, vasospasms, for the
protection of organs, e.g. the kidney and liver, before
and during operations, as well as chronic or acute
kidney failure.
They can furthermore be used for the treatment of
stroke, of cerebral oedema, ischaemia of the nervous
system, various forms of shock as well as for improving
the respiratory drive in, for example, the following
conditions: central sleep apnoea, sudden infant death,
post-operative hypoxia and other respiratory disorders.
The respiratory activity can be further improved by
combination with a carboanhydrase inhibitor.
The compounds of the formula I have an inhibitory
action on the proliferation of cells, for example
fibroblast cell proliferation and the proliferation of
smooth vascular muscle cells and can therefore be used
for the treatment of diseases in which cell prolifera
tion is a primary or secondary cause.
The compounds of the formula I can be used against
diabetic late complications, carcinomatous disorders,
fibrotic disorders, endothelial dysfunction, organ
hypertrophy and hyperplasia, in particular in prostate
hyperplasia or prostate hypertrophy.
They are further suitable as diagnostics for the
determination and differentiation of certain forms of
hypertension, atherosclerosis, diabetes and prolifera-
tive disorders.
Since the compounds of the formula I also advanta-
geously influence the level of the serum lipoproteins,
they can be employed on their own or in combination
with other medicaments for the treatment of an
increased blood fat level.
CA 02387529 2002-03-20
- 5 -
The invention relates to the use of compounds of the
formula I according to Claim 1 and their physio-
logically acceptable salts and/or solvates for the
production of a medicament for the treatment of
thromboses, ischaemic conditions of the heart, of the
peripheral and central nervous system and of stroke,
ischaemic conditions of peripheral organs and limbs and
for the treatment of states of shock.
The invention further relates to the use of compounds
of the formula I according to Claim 1 and their physio-
logically acceptable salts and/or solvates for the pro-
duction of a medicament for use in surgical operations
and organ transplantation and for the preservation and
storage of transplants for surgical measures.
The invention also relates to the use of compounds of
the formula I according to Claim 1 and their physio-
logically acceptable salts and/or solvates for the pro-
duction of a medicament for the treatment of diseases
in which cell proliferation is a primary or secondary
cause, for the treatment or prophylaxis of disorders of
the lipid metabolism or disturbed respiratory drive.
The invention further relates to the use of compounds
of the formula I according to Claim 1 and their physio-
logically acceptable salts and/or solvates for the pro-
duction of a medicament for the treatment of ischaemic
kidney, ischaemic intestinal disorders or for the
prophylaxis of acute or chronic kidney disorders.
Methods for the identification of substances which
inhibit the sodium/proton exchanger subtype 3 are des
cribed, for example, in US 5,871,919.
For all radicals in the compounds of the formula I
which occur a number of times, such as, for example,
R6, it is a condition that their meanings are indepen-
CA 02387529 2002-03-20
- 6 -
dent of one another.
Hydrates and solvates are understood as meaning, for
example, the hemi-, mono- or dehydrates, solvates are
understood as meaning, for example, alcohol addition
compounds such as, for example, with methanol or
ethanol.
In the above formulae, A is alkyl, is linear or
branched, and has 1 to 20, preferably 1, 2, 3, 4, 5, 6,
7, 8, 9, 10, 11 or 12 C atoms. A is preferably methyl,
furthermore ethyl, propyl, isopropyl, butyl, isobutyl,
sec-butyl or tert-butyl, furthermore also pentyl, 1-,
2- or 3-methylbutyl, 1,1-, 1,2- or 2,2-dimethylpropyl,
1-ethylpropyl, hexyl, 1-, 2-, 3- or 4-methylpentyl,
1,1-, 1,2-, 1,3-, 2,2-, 2,3- or 3,3-dimethylbutyl, f-
or 2-ethylbutyl, 1-ethyl-1-methylpropyl, 1-ethyl-
2-methylpropyl, 1,1,2- or 1,2,2-trimethylpropyl,
heptyl, octyl, nonyl or decyl.
A is furthermore, for example, trifluoromethyl, penta-
fluoroethyl, allyl or crotyl.
CORE is acyl and is preferably formyl, acetyl,
propionyl, furthermore also butyryl, pentanoyl or
hexanoyl.
COOR6 is preferably methoxycarbonyl, ethoxycarbonyl,
propoxycarbonyl or butoxycarbonyl.
Hal is preferably F, C1 or Br, but also I.
R2, R3 and RS are, in each case independently of one
another, preferably H, fluorine, chlorine, bromine,
iodine, hydroxyl, methoxy, ethoxy, propoxy, nitro,
amino, methylamino, dimethylamino, ethylamino, diethyl-
amino, acetamido, sulfonamido, methylsulfonamido,
phenylsulfonamido, methylthio, ethylthio, methyl-
sulfinyl, ethylsulfinyl, methylsulfonyl, ethylsulfonyl,
phenylsulfinyl, phenylsulfonyl, cyano, carboxyl,
methoxycarbonyl, ethoxycarbonyl, carboxymethoxy,
CA 02387529 2002-03-20
_ 7 _
methoxycarbonylmethoxy, carboxymethyl, methoxycarbonyl-
methyl, aminocarbonylmethoxy, aminocarbonylmethyl,
N-phenylaminocarbonylmethoxy, N-phenylaminocarbonyl-
methyl, furthermore also acyl or benzoyl.
In particular, R', R5 are H.
R3 is, in particular, for example, H, CODA or
-OCHZCOOR6, where R6 is H or alkyl having 1-4 C atoms.
R6 is H, A or benzyl, but in particular H or alkyl
having 1-4 C atoms.
X is preferably, for example, -CHZ-, -CH=CH-, -CH20-,
-0-CHZ-, -C00-, -OOC-, -CONH- or -NHCO-; -CH20-, -0-CHZ-
or -CHz-CHz- is very particularly preferred.
Ar is preferably unsubstituted phenyl or naphthyl, fur-
thermore preferably, for example, phenyl or naphthyl,
which is mono-, di- or trisubstituted by A, fluorine,
chlorine, bromine, iodine, hydroxyl, methoxy, ethoxy,
propoxy, butoxy, pentyloxy, hexyloxy, benzyloxy,
phenethyloxy, methylthio, ethylthio, methylsulfinyl,
ethylsulfinyl, methylsulfonyl, ethylsulfonyl, phenyl-
sulfinyl, phenylsulfonyl, vitro, amino, methylamino,
ethylamino, dimethylamino, diethylamino, formamido,
acetamido, propionylamino, butyrylamino, methylsulfon-
amido, ethylsulfonamido, propylsulfonamido, butyl-
sulfonamido, phenylsulfonamido, (4-methylphenyl)sulfon-
amido, carboxymethoxy, carboxyethoxy, methoxycarbonyl-
methoxy, methoxycarbonylethoxy, hydroxymethoxy,
hydroxyethoxy, methoxyethoxy, carboxyl, methoxy-
carbonyl, ethoxycarbonyl, cyano, phenylaminocarbonyl,
acyl or benzoyl, furthermore also biphenyl.
Ar is therefore preferably, for example, o-, m- or
p-tolyl, o-, m- or p-ethylphenyl, o-, m- or p-propyl-
phenyl, o-, m- or p-isopropylphenyl, o-, m- or p-tert-
butylphenyl, o-, m- or p-hydroxyphenyl, o-, m- or
p-nitrophenyl, o-, m- or p-aminophenyl, o-, m- or
p-(N-methylamino)phenyl, o-, m- or p-acetamidophenyl,
o-, m- or p-methoxyphenyl, o-, m- or p-ethoxyphenyl,
CA 02387529 2002-03-20
-
o-, m- or p-carboxyphenyl, o-, m- or p-methoxy-
carbonylphenyl, o-, m- or p-(N,N-dimethylamino)phenyl,
o-, m- or p-(N-ethylamino)phenyl, o-, m- or p-(N,N-di-
ethylamino)phenyl, o-, m- or p-acetylphenyl, o-, m- or
p-formylphenyl, o-, m- or p-fluorophenyl, o-, m- or
p-bromophenyl, o-, m- or p-chlorophenyl, o-, m- or
p-methylsulfonylphenyl, o-, m- or p-(phenylsulfon-
amido)phenyl, o-, m- or p-(methylsulfonamido)phenyl,
o-, m- or p-methylthiophenyl, furthermore preferably
2,3-, 2,4-, 2,5-, 2,6-, 3,4- or 3,5-difluorophenyl,
2,3-, 2,4-, 2,5-, 2,6-, 3,4- or 3,5-dichlorophenyl,
2, 3-, 2, 4-, 2, 5-, 2, 6-, 3, 4- or 3, 5-dibromophenyl, 2, 4-
or 2,5-dinitrophenyl, 2,5- or 3,4-dimethoxyphenyl,
3-nitro-4-chlorophenyl, 3-amino-4-chloro-, 2-amino-
3-chloro-, 2-amino-4-chloro-, 2-amino-5-chloro- or
2-amino-6-chlorophenyl, 2-nitro-4-N,N-dimethylamino- or
3-nitro-4-N,N-dimethylaminophenyl, 2,3-diaminophenyl,
2,3,4-, 2,3,5-, 2,3,6-, 2,4,6- or 3,4,5-trichloro-
phenyl, 2,4,6-trimethoxyphenyl, 2-hydroxy-3,5-dichloro-
phenyl, p-iodophenyl, 3,6-dichloro-4-aminophenyl,
4-fluoro-3-chlorophenyl, 2-fluoro-4-bromophenyl,
2,5-difluoro-4-bromophenyl, 3-bromo-6-methoxyphenyl,
3-chloro-6-methoxyphenyl, 3-chloro-4-acetamidophenyl,
3-fluoro-4-methoxyphenyl, 3-amino-6-methylphenyl,
3-chloro-4-acetamidophenyl or 2,5-dimethyl-4-chloro-
phenyl.
Ar' is in particular, for example, phenyl or naphthyl,
furthermore preferably, for example, o-, m- or p-tolyl,
0-, m- or p-ethylphenyl, o-, m- or p-propylphenyl, o-,
m- or p-isopropylphenyl, o-, m- or p-tert-butylphenyl,
o-, m- or p-hydroxyphenyl, o-, m- or p-nitrophenyl, o-,
m- or p-aminophenyl, o-, m- or p-(N-methylamino)phenyl,
o-, m- or p-acetamidophenyl, o-, m- or p-methoxyphenyl,
0-, m- or p-ethoxyphenyl, o-, m- or p-carboxyphenyl,
o-, m- or p-methoxycarbonylphenyl, o-, m- or p-(N,N-di-
methylamino)phenyl, o-, m- or p-(N-ethylamino)phenyl,
o-, m- or p-(N,N-diethylamino)phenyl, o-, m- or
p-acetylphenyl, o-, m- or p-formylphenyl, o-, m- or
CA 02387529 2002-03-20
- 9 -
p-fluorophenyl, o-, m- or p-bromophenyl, o-, m- or
p-chlorophenyl or o-, m- or p-methylsulfonylphenyl.
Accordingly, the invention relates in particular to the
use of those compounds of the formula I in which at
least one of the radicals mentioned has one of the pre-
ferred meanings indicated above. Some preferred groups
of compounds can be expressed by the following sub-
formulae Ia to Ii, which correspond to the formula I
and in which the radicals not designated in greater
detail have the meaning indicated in the formula I, but
in which
in Ia R1, R4 in each case independently of one
another are -C(=NH)-NHz, which can also
be monosubstituted by OH or
-CO-N=C ( NHz ) 2 ;
in Ib R', RS are H;
in Ic R1, R4 in each case independently of one
another are -C(=NH)-NH2, which can also
be monosubstituted by OH or
-CO-N=C (NHZ) 2,
R2, RS are H and
R3 is H or COOR6;
in Id R1, Rq in each case independently of one
another are -C(=NH)-NHZ, which can also
be monosubstituted by OH or
-CO-N=C ( NHZ ) a ,
RZ, RS are H and
R3 is H, COOR6 or -0- (CHZ) COOR6;
in Ie X is -CHz-0- or -0-CHz-;
in If R1, Rq in each case independently of one
another are -C(=NH)-NH2, which can also
be monosubstituted by OH or
3 5 -CO-N=C ( NHZ ) 2 ,
R2 R5 are H
, ,
R3 is H or COOR6 and
X is -CHZ-0- or -0-CHz-;
in Ig R1, R4 in each case independently of one
CA 02387529 2002-03-20
- 10 -
another are -C(=NH)-NHz, which can also
be monosubstituted by OH or
-CO-N=C ( NHz ) ~,
R2, R5 are H,
R3 is H, COOR6 or -0- (CH2) COOR6, and
X is -CHZ-0-, -0-CHZ- or -CHZ-CHZ-;
in Ih R1, Rq in each case independently of one
another are -C(=NH)-NH2, which can also
be monosubstituted by OH or
-CO-N=C ( NHz ) ~ ,
R', RS are H,
R3 is H, COOR6, -0-CHZ-COOR6, CHZ-COOR6,
-0-CH2-CON ( R6 ) 2 , CHZ-CON ( R6
) 2.
-0-CH2-CONHAr or CHz-CONHAr,
X is -CHz-0-, -0-CH2- or -CHZ-CHZ-,
R6 is H or A,
A is alkyl having 1-4 C atoms;
in Ii R1, R4 in each case independently of one
another are -C(=NH)-NH2, which can also
be monosubstituted by OH or
-CO-N=C ( NH2 ) ~,
R2, R5 are H,
RJ is H, COOR6, -0-CHZ-COOR6, CHZ-COOR6,
-0-CHZ-CON (R6) Z or CHz-CON (R6)
2,
X is -CHZ-0-, -0-CHz- or -CHZ-CHZ-,
Rb is H or A,
A is alkyl having 1-4 C atoms.
The compounds of the formula I and also the starting
substances for their preparation are otherwise prepared
by methods known per se, such as are described in the
literature (e.g. in the standard works such as
Houben-Weyl, Methoden der organischen Chemie [Methods
of Organic Chemistry]., Georg-Thieme-Verlag, Stuttgart),
namely under reaction conditions which are known and
suitable for the reactions mentioned. Use can also be
made in this case of variants which are known per se,
but not mentioned here in greater detail.
CA 02387529 2002-03-20
- 11 -
If desired, the starting substances can also be formed
in situ such that they are not isolated from the
reaction mixture, but are immediately reacted further
to give the compounds of the formula I.
S
Compounds of the formula I can preferably be obtained
by liberating compounds of the formula I from one of
their functional derivatives by treating them with a
solvolysing or hydrogenolysing agent.
Preferred starting substances for the solvolysis or
hydrogenolysis are those which otherwise correspond to
the formula I, but instead of one or more free amino
and/or hydroxyl groups contain corresponding protected
amino and/or hydroxyl groups, preferably those which,
instead of an H atom which is bonded to an N atom,
carry an amino protective group, in particular those
which, instead of an HN group, carry an R'-N- group, in
which R' is an amino protective group, and/or those
which, instead of the H atom of a hydroxyl group, carry
a hydroxyl protective group, e.g. those which
correspond to the formula I, but instead of a group
-COOH carry a group -COOK" in which R" is a hydroxyl
protective group.
Preferred starting substances are also the oxadiazole
derivatives which can be converted into the correspond-
ing amidino compounds.
The introduction of the oxadiazole group is carried
out, for example, by reaction of the cyano compounds
with hydroxylamine and reaction with phosgene, dialkyl
carbonate, chloroformic acid ester, N,N'-carbonyl-
diimidazole or acetic anhydride.
It is also possible for a number of - identical or dif-
ferent - protected amino and/or hydroxyl groups to be
present in the molecule of the starting substance. If
the protective groups present are different from one
another, in many cases they can be removed selectively.
CA 02387529 2002-03-20
- 12 -
The expression "amino protective group" is generally
known and relates to groups which are suitable for
protecting (or blocking) an amino group from chemical
reactions, but which are easily removable after the
desired chemical reaction has been carried out at other
positions in the molecule. Typical groups of this type
are in particular unsubstituted or substituted acyl,
aryl, aralkoxymethyl or aralkyl groups. Since the amino
protective groups are removed after the desired reac-
tion (or reaction sequence), their size and nature are
otherwise not critical; however, those having 1-20, in
particular 1-8, C atoms are preferred. The expression
"acyl group" is to be interpreted in the widest sense
in connection with the present process. It includes
acyl groups derived from aliphatic, araliphatic,
aromatic or heterocyclic carboxylic acids or sulfonic
acids, and, in particular, alkoxycarbonyl, aryloxy-
carbonyl and especially aralkoxycarbonyl groups.
Examples of acyl groups of this type are alkanoyl such
as acetyl, propionyl, butyryl; aralkanoyl such as
phenylacetyl; aroyl such as benzoyl or toluyl; aryloxy-
alkanoyl such as POA; alkoxycarbonyl such as methoxy-
carbonyl, ethoxycarbonyl, 2,2,2-trichloroethoxy-
carbonyl, BOC (tert-butyloxycarbonyl), 2-iodoethoxy-
carbonyl; aralkyloxycarbonyl such as CBZ ("carbo-
benzoxy"), 4-methoxybenzyloxycarbonyl, FMOC; aryl-
sulfonyl such as Mtr. Preferred amino protective groups
are BOC and Mtr, furthermore CBZ, Fmoc, benzyl and
acetyl.
The expression "hydroxyl protective group" is likewise
generally known and relates to groups which are suit-
able for protecting a hydroxyl group from chemical
reactions, but which are easily removable after the
desired chemical reaction has been carried out at other
positions in the molecule. Typical groups of this type
are the abovementioned unsubstituted or substituted
aryl, aralkyl or acyl groups, furthermore also alkyl
' CA 02387529 2002-03-20
- 13 -
groups. The nature and size of the hydroxyl protective
groups are not critical, as they are removed again
after the desired chemical reaction or reaction
sequence; groups having 1-20, in particular 1-10, C
atoms are preferred. Examples of hydroxyl protective
groups are, inter alia, benzyl, p-nitrobenzoyl,
p-toluenesulfonyl, tert-butyl and acetyl, benzyl and
tert-butyl being particularly preferred.
The liberation of the compounds of the formula I from
their functional derivatives is carried out - depending
on the protective group used - for example using strong
acids, expediently using TFA or perchloric acid, but
also using other strong inorganic acids such as hydro-
chloric acid or sulfuric acid, strong organic
carboxylic acids such as trichloroacetic acid or
sulfonic acids such as benzene- or p-toluenesulfonic
acid. The presence of an additional inert solvent is
possible, but not always necessary. Suitable inert
solvents are preferably organic solvents, for example
carboxylic acids such as acetic acid, ethers such as
tetrahydrofuran or dioxane, amides such as DMF,
halogenated hydrocarbons such as dichloromethane,
furthermore also alcohols such as methanol, ethanol or
isopropanol, and also water. In addition, mixtures of
the abovementioned solvents are possible. TFA is
preferably used in an excess without addition of a
further solvent, perchloric acid in the form of a
mixture of acetic acid and 70°s perchloric acid in the
ratio 9:1. The ru ction temperatures for the cleavage
are expediently between approximately 0 and
approximately 50°; the reaction is preferably carried
out between 15 and 30° (room temperature).
The groups BOC, OBut and Mtr can preferably be removed,
for example, using TFA in dichloromethane or using
approximately 3 to 5 N HC1 in dioxane at 15-30°; the
Fmoc group using an approximately 5 to 50% solution of
dimethylamine, diethylamine or piperidine in DMF at
CA 02387529 2002-03-20
- 14 -
15-30°.
Hydrogenolytically removable protective groups (e. g.
CBZ, benzyl or the liberation of the amidino group from
its oxadiazole derivative) can be removed, for example,
by treating with hydrogen in the presence of a catalyst
(e. g. of a noble metal catalyst such as palladium,
expediently on a support such as carbon or such as
moist Raney nickel with addition of, for example,
acetic acid). Suitable solvents here are those
indicated above, in particular, for example, alcohols
such as methanol or ethanol or amides such as DMF. As a
rule, the hydrogenolysis is carried out at temperatures
between approximately 0 and 100° and pressures between
approximately 1 and 200 bar, preferably at 20-30° and
1-10 bar. Hydrogenolysis of the CBZ group is readily
carried out, for example, on 5 to 10% Pd/C in methanol
or on Pd/C in methanol/DMF at 20-30° using ammonium
formate (instead of hydrogen).
Compounds of the formula I in which R1 and R9 are
-C(=NH)-NH2 can preferably be obtained from the corres-
ponding cyano compound.
A cyano group is converted into an amidino group by
reaction with, for example, hydroxylamine and subse
quent reduction of the N-hydroxyamidine with hydrogen
in the presence of a catalyst such as, for example,
Pd/C or Raney nickel.
For the preparation of an amidine of the formula I
(R1 - -C (=NH) -NHZ) , it is also possible to add ammonia
to a nitrite of the formula I (R1 - CN). The addition
is preferably carried out in a number of stages by, in
a manner known per se, a) converting the nitrite using
HZS into a thioamide which is converted using an
alkylating agent, e.g. CH3I, into the corresponding
S-alkyl imidothioester, which for its part reacts with
NH3 to give the amidine, b) converting the nitrite
using an alcohol, e.g. ethanol in the presence of HCl,
into the corresponding imidoester and treating this
CA 02387529 2002-03-20
- 15 -
with ammonia, or c) reacting the nitrile with lithium
bis(trimethylsilyl)amide and then hydrolysing the
product.
Preparation of the cyano compounds is carried out by
methods known per se.
Compounds of the formula I in which R1 and Rq are
-CON(=NH)-NHZ can preferably be obtained from the
corresponding alkoxycarbonyl compounds by reacting with
guanidine.
It is furthermore possible to convert a compound of the
formula I into another compound of the formula I by
converting one or more radicals R1, Rz, R3, R4 and/or R5
into one or more radicals R1, R2, R3, Rq and/or R5, a . g.
by acylating an amino group or reducing nitro groups to
amino groups (for example by hydrogenation on Raney
nickel or Pd/carbon in an inert solvent such as
methanol or ethanol).
Esters can be hydrolysed, for example, with acetic acid
or with NaOH or KOH in water, water/THF or water/
dioxane at temperatures between 0 and 100°.
It is further possible to acylate free amino groups in
a customary manner using an acid chloride or anhydride
or to alkylate them using an unsubstituted or substi-
tuted alkyl halide, expediently in an inert solvent
such as dichloromethane or THF and/or in the presence
of a base such as triethylamine or pyridine at tempera-
tures between -60 and +30°.
As a rule, the reaction is carried out in an inert
solvent, in the presence of an acid-binding agent,
preferably of an alkali metal or alkaline earth metal
hydroxide, carbonate or bicarbonate or of another salt
of a weak acid of the alkali or alkaline earth metals,
preferably of potassium, sodium, calcium or caesium.
CA 02387529 2002-03-20
- 16 -
The addition of an organic base such as triethylamine,
dimethylaniline, pyridine or quinoline or an excess of
the amine component of the formula II or of the
alkylating derivative of the formula III can also be
favourable. Depending on the conditions used, the reac-
tion time is between a few minutes and 14 days, the
reaction temperature between approximately 0° and 150°,
normally between 20° and 130°.
Suitable inert solvents are, for example, hydrocarbons
such as hexane, petroleum ether, benzene, toluene or
xylene; chlorinated hydrocarbons such as trichloro-
ethylene, 1,2-dichloroethane, carbon tetrachloride,
chloroform or dichloromethane; alcohols such as
methanol, ethanol, isopropanol, n-propanol, n-butanol
or tert-butanol; ethers such as diethyl ether, diiso-
propyl ether, tetrahydrofuran (THF) or dioxane; glycol
ethers such as ethylene glycol monomethyl or monoethyl
ether (methyl glycol or ethyl glycol), ethylene glycol
dimethyl ether (diglyme); ketones such as acetone or
butanone; amides such as acetamide, dimethylacetamide,
N-methylpyrrolidone (NMP) or dimethylformamide (DMF);
nitrites such as acetonitrile; sulfoxides such as
dimethyl sulfoxide (DMSO); carbon disulfide; carboxylic
acids such as formic acid or acetic acid; nitro
compounds such as nitromethane or nitrobenzene; esters
such as ethyl acetate or mixtures of the solvents
mentioned.
A base of the formula I can be converted with an acid
into the associated acid addition salt, for example by
reaction of equivalent amounts of the base and of the
acid in an inert solvent such as ethanol and subsequent
evaporation. Suitable acids for this reaction are in
particular those which yield physiologically acceptable
salts. Thus inorganic acids can be used, e.g. sulfuric
acid, nitric acid, halohydric acids such as hydro-
chloric acid or hydrobromic acid, phosphoric acids such
as orthophosphoric acid, sulfamic acid, furthermore
CA 02387529 2002-03-20
17 -
organic acids, in particular aliphatic, alicyclic,
araliphatic, aromatic or heterocyclic mono- or poly-
basic carboxylic, sulfonic or sulfuric acids, e.g.
formic acid, acetic acid, propionic acid, pivalic acid,
diethylacetic acid, malonic acid, succinic acid,
pimelic acid, fumaric acid, malefic acid, lactic acid,
tartaric acid, malic acid, citric acid, gluconic acid,
ascorbic acid, nicotinic acid, isonicotinic acid,
methane- or ethanesulfonic acid, ethanedisulfonic acid,
2-hydroxyethanesulfonic acid, benzenesulfonic acid,
p-toluenesulfonic acid, naphthalenemono- and
-disulfonic acids and laurylsulfuric acid. Salts with
physiologically unacceptable acids, e.g. picrates ,
can
be used for the isolation and/or purification o f
the
compounds of the formula I.
On the other hand, compounds of the formula I can be
converted into the corresponding metal salts, in
particular alkali metal salts or alkaline earth metal
salts, or into the corresponding ammonium salts using
bases (e.g. sodium or potassium hydroxide or
carbonate).
Physiologically acceptable organic bases, such as, for
example, ethanolamine, can also be used.
The invention furthermore relates to the use of the
compounds of the formula I as NHE-3 inhibitors and/or
their physiologically acceptable salts for the produc-
tion of pharmaceutical preparations, in particular by a
non-chemical route. In this context, they can be
brought into a suitable dose form together with at
least one solid, liquid and/or semi-liquid vehicle or
excipient and if appropriate in combination with one or
more further active compounds.
The invention furthermore relates to pharmaceutical
preparations comprising at least one NHE-3 inhibitor of
the formula I and/or one of its physiologically
acceptable salts and solvates.
CA 02387529 2002-03-20
- 18 -
These preparations can be used as medicaments in human
or veterinary medicine. Possible vehicles are organic
or inorganic substances which are suitable for enteral
(e. g. oral) or parenteral administration or topical
application and do not react with the novel compounds,
for example water, vegetable oils, benzyl alcohols,
alkylene glycols, polyethylene glycols, glycerol
triacetate, gelatin, carbohydrates such as lactose or
starch, magnesium stearate, talc and petroleum jelly.
In particular, tablets, pills, coated tablets, cap-
sules, powders, granules, syrups, juices or drops are
used for oral administration, suppositories are used
for rectal administration, solutions, preferably oily
or aqueous solutions, furthermore suspensions, emul-
sions or implants, are used for parenteral administra-
tion and ointments, creams or powders are used for
topical application, or transdermally in patches.
The novel compounds can also be lyophilized and the
lyophilizates obtained can be used, for example, for
the production of injection preparations. The
preparations indicated can be sterilized and/or can
contain excipients such as lubricants, preservatives,
stabilizers and/or wetting agents, emulsifiers, salts
for influencing the osmotic pressure, buffer
substances, colourants, flavourings and/or one or more
further active compounds, e.g. one or more vitamins.
Pharmaceutical preparations which are suitable for
administration in the form of aerosols or sprays are,
for example, solutions, suspensions or emulsions of the
active compound of the formula I in a pharmaceutically
acceptable solvent.
The compounds of the formula I and their physio-
logically acceptable salts and solvates can be used for
the treatment and/or prophylaxis of the diseases or
disease conditions indicated above.
In this context, the substances according to the
CA 02387529 2002-03-20
- 19 -
invention are as a rule preferably administered in
doses between approximately 0.1 and 100 mg, in particu-
lar between 1 and 10 mg, per dose unit. The daily dose
is preferably between approximately 0.001 and 10 mg/kg
of body weight. The specific dose for each patient,
however, depends on all sorts of factors, for example
on the efficacy of the specific compound employed, on
the age, body weight, general state of health, sex, on
the diet, on the time and route of administration, on
the excretion rate, pharmaceutical combination and
severity of the particular disorder to which the
therapy applies. Oral administration is preferred.
Above and below, all temperatures are indicated in °C.
In the following examples, "customary working up"
means: if appropriate, water is added, the mixture is
adjusted, if necessary, depending on the constitution
of the final product, to a pH of between 2 and 10 and
extracted with ethyl acetate or dichloromethane, the
organic phase is separated off, dried over sodium
sulfate and evaporated, and the residue is purified by
chromatography on silica gel and/or by crystallization.
Mass spectrometry (MS): EI (electron impact ionization) M+
FAB (fast atom bombardment) (M+H)'
Example 1
A solution of 2.06 g of 3-bromobenzonitrile and 1.50 g
of 3-tolylboronic acid in 50 ml of dimethoxyethane is
treated with 247 mg of palladium(II) acetate, 335 mg of
tri-o-tolylphosphine, 20 ml of water and 954 mg of
anhydrous sodium carbonate and heated at 100°C for
18 hours with stirring. The mixture is worked up in the
customary manner, the residue is chromatographed on a
silica gel column using petroleum ether/ethyl acetate
9:1 and 3'-methylbiphenyl-3-carbonitrile is obtained as
a colourless solid ("A"), EI 193.
A solution of 1.17 g of "A" in 10 ml of carbon tetra-
chloride is treated with 1.09 g of N-bromosuccinimide
CA 02387529 2002-03-20
- 20 -
(NBS) and 60 mg of azobisisobutyronitrile and heated at
70°C for 18 hours. It is worked up in the customary
manner, the residue is chromatographed on a silica gel
column using petroleum ether/ethyl acetate 9:1 and
3'-bromomethylbiphenyl-3-carbonitrile is obtained as a
colourless solid ("B" ) .
A solution of 500 mg of "B" and 238 mg of 3-hydroxy-
benzonitrile in 10 ml of acetonitrile is treated with
652 mg of caesium carbonate and stirred at room tem-
i0 perature for 40 hours. After customary working up, the
residue is chromatographed on a reversed-phase column
using acetonitrile/water 65:35. 3'-(3-Cyanophenoxy-
methyl)biphenyl-3-carbonitrile ("C") is obtained as a
colourless solid, FAB 311.
A solution of 90 mg of "C" and 125 mg of hydroxyl-
ammonium chloride in 10 ml of ethanol is treated with
1.2 g of polymer-bound dimethylaminopyridine (DMAP) and
stirred at room temperature for 18 hours. The polymer
is filtered off, the solvent is removed and N-hydroxy-
3'-[3-(N-hydroxycarbamimidoyl)phenoxymethyl]biphenyl-
3-carboxamidine ("D") is obtained as a colourless
solid, FAB 377.
A solution of 76 mg of "D" in 10 ml of methanol is
treated with 100 mg of water-moist Raney nickel and
30 mg of acetic acid and hydrogenated at room tempera
ture and normal pressure for 18 hours. The catalyst is
filtered off, the solvent is removed and
3'-(3-carbamimidoylphenoxymethyl)biphenyl-3-carbox-
amidine, acetate, is obtained, EI 327 (M+ - NH3), 310
(M+ - 2 NH3)
HN w ~ / O I / NHZ
I
NH2 ~ NH
The following compounds are obtained analogously
3'-(3-carbamimidoylphenoxymethyl)biphenyl-4-car-
CA 02387529 2002-03-20
- 21 -
boxamidine, diacetate, FAB 345;
3'-(4-carbamimidoylphenoxymethyl)biphenyl-4-car-
boxamidine, diacetate, FAB 345;
3'-(4-carbamimidoylphenoxymethyl)biphenyl-3-car-
boxamidine, diacetate, FAB 345;
4'-(4-carbamimidoylphenoxymethyl)biphenyl-4-car-
boxamidine,
4'-(4-carbamimidoylphenoxymethyl)biphenyl-3-car-
boxamidine,
4'-(3-carbamimidoylphenoxymethyl)biphenyl-3-car-
boxamidine and
4'-(3-carbamimidoylphenoxymethyl)biphenyl-4-car-
boxamidine.
Example 2
Analogously to Example 1, the compound 3'-methoxy-
biphenyl-3-carbonitrile is obtained by reaction of
3-bromobenzonitrile with 3-methoxyphenylboronic acid.
By subsequent ether cleavage using aluminium triiodide
in acetonitrile and reaction with 3-bromomethylbenzo-
nitrile, 3'-(3-cyanobenzyloxy)biphenyl-3-carbonitrile
is obtained.
By reaction with hydroxylamine and reduction with
hydrogen under Ra Ni catalysis, 3'-(3-carbamimidoyl
benzoyloxy)biphenyl-3-carboxamidine is obtained
w
HN w ~ ~ O I i NHZ
I
NH2 ~ NH
The following are obtained analogously
4'-(4-carbamimidoylbenzyloxy)biphenyl-4-carboxamidine,
diacetate, FAB 345;
4'-(3-carbamimidoylbenzyloxy)biphenyl-4-carboxamidine,
diacetate, FAB 345.
CA 02387529 2002-03-20
- 22 -
Example 3
Analogously to Example 1, the compound methyl 3'-cyano-
5-methylbiphenyl-3-carboxylate is obtained by reaction
of 3-cyanophenylboronic acid with methyl 3-bromo-
5-methylbenzoate. By bromination with NBS and reaction
with 3-hydroxybenzonitrile, methyl 3'-cyano-5-(3-cyano-
phenoxymethyl)biphenyl-3-carboxylate is obtained.
Reaction with hydroxylamine and reduction with Hz/Ra Ni
affords the compound methyl 3'-carbamimidoyl-
5-(3-carbamimidoylphenoxymethyl)biphenyl-3-carboxylate.
By hydrolysis of the ester using aqueous NaOH,
3'-carbamimidoyl-5-(3-carbamimidoylphenoxymethyl)bi-
phenyl-3-carboxylic acid is obtained therefrom.
H ~ ~ i NH2
NH
By chromatography on a reversed-phase column using an
acetonitrile/water/TFA mixture, 3'-carbamimidoyl-
5-(3-carbamimidoylphenoxymethyl)biphenyl-3-carboxylic
acid, bistrifluoroacetate, is obtained.
The following compounds are obtained analogously
methyl 4'-carbamimidoyl-4-(4-carbamimidoylphenoxy-
methyl)biphenyl-3-carboxylate, FAB 403;
methyl 4'-carbamimidoyl-4-(3-carbamimidoylphenoxy-
methyl)biphenyl-2-carboxylate, FAB 403;
methyl 3'-carbamimidoyl-4-(4-carbamimidoylphenoxy-
methyl)biphenyl-2-carboxylate, FAB 403;
hi U U
CA 02387529 2002-03-20
- 23 -
methyl 3'-carbamimidoyl-4-(3-carbamimidoylphenoxy-
methyl)biphenyl-2-carboxylate, FAB 403;
methyl 4'-carbamimidoyl-5-(3-carbamimidoylphenoxy-
methyl)biphenyl-4-carboxylate, FAB 403;
methyl 3'-carbamimidoyl-5-(3-carbamimidoylphenoxy-
methyl)biphenyl-4-carboxylate, FAB 403.
Example 4
Analogously to Example 1, methyl 3'-methylbiphenyl-
3-carboxylate is obtained by reaction of methyl
3-bromobenzoate with 3-tolylboronic acid. By bromina-
tion with NBS and reaction with methyl 3-hydroxy-
benzoate, methyl 3'-(3-methoxycarbonylphenoxymethyl)-
biphenyl-3-carboxylate is obtained therefrom. By
reaction with guanidine hydrochloride in methanolic
sodium methoxide solution, N-[3'-(3-guanidinocarbonyl-
phenoxymethyl)biphenyl-3-carbonyl]guanidine is obtained
therefrom
O w ~ ~ I .~ N~ NHZ
H2N ~ N w I O NHZ
NH2
The compound N-[4'-(4-guanidinocarbonylphenoxymethyl)-
biphenyl-4-carbonyl]guanidine is obtained analogously.
Example 5
A solution of 7.0 g of 3-bromo-5-methylphenol and
5.97 g of methyl bromoacetate and also 13 g of caesium
carbonate in 100 ml of acetonitrile is stirred
overnight at room temperature. After customary working
up, 9.70 g of methyl (3-bromo-5-methylphenoxy)acetate
CA 02387529 2002-03-20
- 24 -
"AB" ) are obtained.
A suspension of 2.0 g of "AB", 100 mg of tetrakis-
(triphenylphosphine)palladium and 0.85 g of sodium
carbonate in 50 ml of toluene is heated to boiling. A
solution of 2.94 g of 3-cyanophenylboronic acid in
30 ml of methanol is then added dropwise and the
mixture is heated under reflux for 14 hours. It is
worked up in the customary manner and 2.17 g of methyl
(3'-cyano-5-methylbiphenyl-3-yloxy)acetate ("AC") are
obtained.
A solution of 1.2 g of "AC" and 0.765 g of NBS in 50 ml
of carbon tetrachloride is irradiated at room
temperature using UV light. After customary working up,
1.54 g of methyl (3'-cyano-5-bromomethylbiphenyl
3-yloxy)acetate ("AD") are obtained.
A solution of 185 mg of "AD", 63.1 mg of 4-hydroxy-
benzonitrile and 172.7 mg of caesium carbonate in 10 ml
of acetonitrile is stirred at room temperature for
4 days. After customary working up, methyl [3'-cyano-
5-(4-cyanophenoxymethyl)biphenyl-3-yloxy]acetate ("AE")
is obtained.
A solution of 60 mg of "AE", 69.5 mg of hydroxyl-
ammonium chloride and 101 mg of triethylamine in 10 ml
of ethanol is heated under reflux for 14 hours. After
removal of the solvent, the residue is taken up in
water. The solid is separated off and 70 mg of methyl
[3'-N-hydroxyamidino-5-(4-N-hydroxyamidinophenoxy-
methyl)biphenyl-3-yloxy]acetate ("AF") are obtained. By
reduction using H2/Raney nickel, methyl [3'-amidino-
5-(4-amidinophenoxymethyl)biphenyl-3-yloxy]acetate,
FAB 433, is obtained.
CA 02387529 2002-03-20
- 25 -
NH
"2N ~
NHz
The following compounds are obtained analogously
methyl [4'-amidino-5-(4-amidinophenoxymethyl)-
biphenyl-3-yloxy]acetate, FAB 433
methyl [3'-amidino-5-(3-amidinophenoxymethyl)-
biphenyl-3-yloxy]acetate, FAB 433
methyl [4'-amidino-5-(3-amidinophenoxymethyl)-
biphenyl-3-yloxy]acetate, FAB 433.
If methyl bromoacetate is replaced in the first stage
by tert-butyl bromoacetate, the tert-butyl esters
obtained in the last stage can be cleaved using
trifluoroacetic acid and the corresponding carboxylic
acids
[3'-amidino-5-(4-amidinophenoxymethyl)biphenyl-
3-yloxy]acetic acid, bistrifluoroacetate, FAB 419;
[4'-amidino-5-(4-amidinophenoxymethyl)biphenyl-
3-yloxy]acetic acid;
[3'-amidino-5-(3-amidinophenoxymethyl)biphenyl-
3-yloxy]acetic acid;
[4'-amidino-5-(3-amidinophenoxymethyl)biphenyl-
O O
CA 02387529 2002-03-20
- 26 -
3-yloxy]acetic acid
are obtained.
Example 6
A solution of 5.0 g of 3'-bromomethylbiphenyl-
3-carbonitrile and 5 ml of triethyl phosphite are com-
bined and slowly heated to 150°. The mixture is stirred
at 150° for 6 h and 6.05 g of diethyl (3'-cyano-
biphenyl-3-ylmethyl)phosphonate ("BA") are obtained
after customary working up.
150 mg of sodium hydride are added with ice-cooling and
under nitrogen to a solution of 1.0 g of "BA" and
3-cyanobenzaldehyde in 20 ml of ethylene glycol
dimethyl ether. The mixture is subsequently stirred for
4 hours, worked up in the customary manner and 0.93 g
of 3'-[2-(3-cyanophenyl)vinyl]biphenyl-3-carbonitrile
(" BB" ) is obtained.
After hydrogenation of 360 mg of "BB" using Pd-C 5% in
methanol, 360 mg of 3'-[2-(3-cyanophenyl)ethyl]-
biphenyl-3-carbonitrile ("BC") are obtained. After
reaction with hydroxylammonium chloride and hydrogena-
tion using Raney nickel, the compound 3'-[2-(3-amidino-
phenyl)ethyl]biphenyl-3-carboxamidine, FAB 343, is
obtained analogously to Example 1
H2r
H
The compound 3'-[2-(4-amidinophenyl)ethyl]biphenyl
3-carboxamidine, FAB 343, is obtained analogously.
Example 7
CA 02387529 2002-03-20
- 27 -
The compound N-[3'-(4-guanidinocarbonylbenzyloxy)-
biphenyl-3-carbonyl]guanidine, dihydrochloride, FAB 431
O NH2
N NH2
O \ / O
H2N ~ N
NHZ
is obtained e.g. according the following reaction
scheme:
CA 02387529 2002-03-20
- 28 -
\ Br / Pd(PPh3)3lNaZC03 N \ /
/ ~ \ I / w \ \ O/
(HO)ZB O
AII~JAcetonitril /
N ~ 1. MeOHIHCI O /
\ OH ---~. ~O \ \ ~ OH
/ 2. HZO
Br
O~
O /
O
~0 \ \ O \
Cs2CO~lAcetonitril ~ / ~ / O~
O
NaOHIMeOH O
Ho \ \ I o \
off
O
O NHZ
~O~N~NHZ
1-Chlor-1-methylpyridiniumiodide
N-Methylpyrrolidone
N-Ethyldiisopropylamine
(Mukaiyama)
O NH O
~O~N~N \ \ O \
H H I ~ N N O
U /
O NH O
25% HCI NHz O /
HZN~N ~ \ \ p ~ \
N"NHZ
O ~~N H2
Dihydrochloride
CA 02387529 2002-03-20
- 29 -
The compounds
N-[3'-(3-guanidinocarbonylbenzyloxy)biphenyl-
3-carbonyl]guanidine, FAB 431;
N-[3'-(4-guanidinocarbonylbenzyloxy)biphenyl-
4-carbonyl]guanidine, FAB 431;
N-[3'-(3-guanidinocarbonylbenzyloxy)biphenyl-
4-carbonyl]guanidine, FAB 431;
are obtained analogously.
CA 02387529 2002-03-20
- 30 -
Pharmacological tests
The methodology which was used for the characterization
of the compounds of the formula I as NHE-3 inhibitors
is presented below.
The compounds of the formula I were characterized with
respect to their selectivity to the isoforms NHE-1 to
NHE-3. The three isoforms were stably expressed in
mouse fibroblast cell lines. The inhibitory action of
the compounds was assessed by determination of the
EIPA-sensitive z'Na+ absorption into the cells after
intracellular acidosis.
For the characterization of the Na+/H+ exchange inhibi
torn with respect to their isoform selectivity, we
investigated the compounds for their inhibition of the
NHE isoforms NHE-l, -2 and -3, which were stably
expressed in a mouse fibroblast cell line (see
procedure section) by determining the EIPA-sensitive
zZNa+ absorption into the cells after intracellular
acidosis.
Material and methods
LAP1 cell lines which express the different NHE
isoforms
The LAP1 cell lines which express the isoforms NHE-1,
-2 and -3 (a mouse fibroblast cell line), were obtained
from Prof. J. Pouyssegur (Nice, France). The transfec-
tions were carried out by the procedure of Franchi et
al. (1986). The cells were cultured in Dulbecco's
modified Eagle medium (DMEM) with 10o inactivated
foetal calf serum (FCS). For the selection of the
NHE-expressing cells, the so-called placid destruction
procedure" of Sardet et al. (1989) was used. The cells
were first removed to an NHqCl-containing bicarbonate-
and sodium-free buffer by washing with a bicarbonate-,
NH4C1- and sodium-free buffer and incubation was
CA 02387529 2002-03-20
- 31 -
carried out with a bicarbonate-free NaCl-containing
buffer. Only those cells which functionally express NHE
were able to survive in the intracellular acidification
to which they were exposed.
Characterization of NHE inhibitors with respect to
their isoform selectivity
Using the abovementioned mouse fibroblast cell lines
which express the isoforms NHE-1, NHE-2 and NHE-3,
compounds were tested for selectivity to the isoforms
according to the procedure described by Counillon et
al. (1993) and Scholz et al. (1995). The cells were
acidified intracellularly by the NHqCl prepulse
procedure and then by incubation in a bicarbonate-free
z2Na+-containing buffer. On account of the intracellular
acidification, NHE was activated and sodium was
absorbed into the cells. The effect of the test
compound was expressed as the inhibition of the EIPA
(ethylisopropylamiloride)-sensitive z2Na+ absorption.
The cells which expressed NHE-l, NHE-2 and NHE-3 were
transferred to microtitre plates having 24 wells by
inoculation at a density of 5-7.5 x 104 cells/well and
cultured to confluence for 24 to 48 hours. The medium
was aspirated and the cells were incubated at 37°C for
60 minutes in the NH9C1 buffer (50 mM NH4C1, 70 mM
choline chloride, 15 mM MOPS, pH 7.0). The buffer was
then removed and the cells were rapidly covered twice
with a layer of the choline chloride wash buffer
(120 mM choline chloride, 15 mM PIPES/tris, 0.1 mM
ouabain, 1 mM MgCl2, 2 mM CaCl2, pH 7.4); the cells were
incubated in this buffer for 6 minutes. After the
incubation time was over, the incubation buffer was
aspirated. For the purpose of removal of extracellular
radioactivity, the cells were washed rapidly four times
with ice-cold phosphate-buffered saline solution (PBS).
The cells were then solubilized by addition of 0.3 ml
of 0.1 N NaOH per well. The cell-fragment-containing
solutions were transferred to scintillation tubes. Each
CA 02387529 2002-03-20
- 32 -
well was additionally washed twice with 0.3 ml of 0.1 N
NaOH and the wash solutions were likewise added to the
corresponding scintillation tubes. The tubes containing
the cell lysate were mixed with scintillation cocktail
and the radioactivity absorbed into the cells was
determined by determination of the (3 radiation.
Inhibition of the ZzNa+ absorption in rabbit
erythrocytes
The Na+/H+ exchange activity was also determined by
observation of the absorption of ZzNa+ ions in acidified
rabbit erythrocytes. Rabbit erythrocytes have found
wide application in investigations on Na+/H+ exchange
activity (Escobales & Fugueroa, 1991: Morgan & Canessa,
1990). The EIPA-sensitive portion of the 22Na+
absorption in acidified erythrocytes was regarded as
Na+/H+-dependent 'zNa+ absorption.
Cell preparation
The preparation of the red blood corpuscles and the
internal acidification of the red blood corpuscles were
carried out closely following the procedures of Morgan
and Canessa (1990).
The blood was obtained from rabbits (e. g. New Zealand
White). It was collected in 50 ml Falcon centrifuge
tubes which contained 5 ml of sodium heparin solution
(250 U/ml). The blood and the heparin solution were
well mixed. The red blood corpuscles were recovered by
centrifugation at 2000 x g at 4°C; the plasma and huffy
coat were removed. The remaining solution was filtered
through 200 ~m gauze. The filtrate was again suspended
in the original volume with wash buffer (140 mM KCl,
0.15 mM MgClz, 10 mM TRIS/MOPS, pH 7.4). The red blood
corpuscles were again recovered by centrifugation
(2000 x g, 4°C). The washing process was repeated
twice.
CA 02387529 2002-03-20
- 33 -
Intracellular acidification
For intracellular acidification, 5 ml of the deposited
collected red blood corpuscles were again suspended
using 45 ml of acidification buffer (170 mM KC1,
0.15 mM MgCl2, 0.1 mM ouabain, 10 mM glucose, 10 mM
sucrose, 20 mM tris/Mes, pH 6.2). The suspension of the
red blood corpuscles was incubated at 37°C for
minutes (with occasional mixing). In order to fix
10 the internal pH, treatment is carried out with up to
200 ~M and 1 mM DIDS or DIAMOX (acetazolamide). The
mixture was additionally incubated at 37°C for
30 minutes.
The red blood corpuscles were then recovered by
centrifugation (4 minutes at 2000 x g, 4°C); they were
again suspended using ice-cold unbuffered wash solution
(170 mM KCl, 40 mM sucrose, 0.15 mM MgClz) and washed
four times therewith.
Incubation and measurement of the ZZNa+ absorption
The incubation was carried out in macrowell tube strips
in a format of 8 x 12. The incubation was begun by
mixing 200 ~l of incubation buffer (160 mM KCl, Z2NaCl
(37 MBq/well), 10 mM NaCl, 0.15 mM MgCl2, 0.1 mM
ouabain, 10 mM glucose, 40 mM sucrose, 10 mM tris/MOPS,
pH 8.0, 0.5 mM Diamox, 1o DMSO) with 20 ~,l of the
(prewarmed) acidified solution of the red blood
corpuscles. The test substances were first dissolved
using 1000 DMSO and the solution was then diluted to
the corresponding concentrations using incubation
buffer. Incubation was carried out at 37°C for
5 minutes. (In preliminary experiments it was shown
that under these incubation conditions the ZZNa absorp-
tion rate was linear during the 5-minute incubation
period). The incubation was stopped by addition of
800 ~l of ice-cold stop solution (112 mM MgClZ, 0.1 mM
ouabain). The tubes were briefly stored on ice. The
tubes were then covered with Parafilm and the red blood
CA 02387529 2002-03-20
- 34 -
corpuscles were recovered by centrifugation at 2000 x g
and 4°C for 7 minutes. The supernatant was aspirated
with a home-made aspiration apparatus, with which it
was possible to aspirate 4 tubes next to one another
simultaneously; spacer rings on the further ends of the
tips prevented the tips dipping too deeply into the
tubes and the pellet of the red blood corpuscles being
aspirated. All 'ZNa-containing supernatants and wash
solutions were stored and disposed of as radioactive
waste.
The red blood corpuscles were washed three times with
900 ~1 of ice-cold stop solution, namely by repeating
the suspension/centrifugation step described above.
After the last wash, the pellet of the red blood
corpuscles was mixed with 200 ~1 of water. The tubes
were then treated with ultrasound for 2 x 30 minutes.
The macrowell tube strips were then taken apart and
each tube was put headfirst into an individual
scintillation tube; the haemolysed solution of the red
blood corpuscles emptied into the scintillation vial as
a result of gentle shaking. Each vial was treated with
3 ml of the scintillation fluid Aquasafe 300 PS, and
the vials were provided with closures and well mixed.
The radioactivity absorbed into the red blood
corpuscles was determined in a scintillation counter by
monitoring the (3 decomposition.
Per substance concentration, the determination was
carried out in triplicate. The mean of the count
determination in the presence of 10 ~tM EIPA was
subtracted from each value in order to include the non-
Na+/H+-dependent Z2Na+ absorption into the erythrocytes.
The mean of the remaining counts in the absence of a
substance was used as a 1000 control; the mean values
in the presence of the test compounds were expressed as
a percentage of this control value. The percentage
absorption data were plotted semilogarithmically; IC50
values were obtained by fitting the values to a non-
linear curve using the equation f(x) -
100/ (1 + (IC50/x) "n) .
CA 02387529 2002-03-20
- 35 -
References:
Counillon et al. (1993) Mol. Pharmacol. 44: 1041-1045
Escobales and Figueroa (1991) J. Membrane Biol. 120,
41-49
Franchi et al. (1986) Proc. Natl. Acad. Sci. USA 83:
9388-9392
Morgan and Canessa (1990) J. Membrane Biol. 118,
193-214
Sardet et al. (1989) Cell 56: 271-280
Scholz et al. (1995) Cardiovasc. Res. 29: 260-268
CA 02387529 2002-03-20
- 36 -
Test results
1.
HN \ I O ~ / NH
~/ / 2
NH2 \ I NH
Code EMD 221960
iC50 (NHE-3) = 1-2 ~M
2.
N H.,
H
O / NH2
NH
Diacetate;
Code EMD 221963
IC50 (NHE-3) - 1 ~tM
3.
NH
I ~ ''. ,NHz
HZN ~. ~ O
/O
Diacetate;
NH O
Code EMD 246326
IC50 (NHE-3) - 3 ~M
' CA 02387529 2002-03-20
- 37 -
4.
H2N ~ / ~ / NH
NH O '~. I NHZ
~O
Diacetate;
Code EMD 246327
IC50 (NHE-3) - 3-4 ~M.
CA 02387529 2002-03-20
- 38 -
The following examples relate to pharmaceutical
preparations:
Example A: Injection vials
A solution of 100 g of an NHE-3 inhibitor of the
formula 1 and 5 g of disodium hydrogenphosphate is
adjusted to pH 6.5 using 2N hydrochloric acid in 3 1 of
double-distilled water, sterile filtered, dispensed
into injection vials, lyophilized under sterile
conditions and aseptically sealed. Each injection vial
contains 5 mg of active compound.
Example B: Suppositories
A mixture of 20 g of an NHE-3 inhibitor of the
formula I is fused with 100 g of soya lecithin and
1400 g of cocoa butter, poured into moulds and allowed
to cool. Each suppository contains 20 mg of active
compound.
Example C: Solution
A solution is prepared from 1 g of an NHE-3 inhibitor
of the formula I, 9.38 g of NaHZPOq ~ 2 HzO, 28.48 g of
NazHPOq ~ 12 H20 and 0.1 g of benzalkonium chloride in
940 ml of doubled-distilled water. It is adjusted to
pH 6.8, made up to 1 1 and sterilized by irradiation.
This solution can be used in the form of eye drops.
Example D: Ointment
500 mg of an NHE-3 inhibitor of the formula I are mixed
with 99.5 g of petroleum jelly under aseptic
conditions.
Example E: Tablets
A mixture of 1 kg of an NHE-3 inhibitor of the
CA 02387529 2002-03-20
- 39 -
formula I, 4 kg of lactose, 1.2 kg of potato starch,
0.2 kg of talc and 0.1 kg of magnesium stearate is
compressed in a customary manner to give tablets such
that each tablet contains 10 mg of active compound.
Example F: Coated tablets
Tablets are pressed analogously to Example E and then
coated in a customary manner with a coating of sucrose,
potato starch, talc, tragacanth and colourant.
Example G: Capsules
2 kg of an NHE-3 inhibitor of the formula I are filled
into hard gelatin capsules in a customary manner such
that each capsule contains 20 mg of the active
compound.
Example H: Ampoules
A solution of 1 kg of NHE-3 inhibitor of the formula I
in 60 1 of double-distilled water is sterile filtered,
dispensed into ampoules, lyophilized under sterile
conditions and aseptically sealed. Each ampoule
contains 10 mg of active compound.