Note: Descriptions are shown in the official language in which they were submitted.
CA 02387646 2002-04-15
WO 01/29225 PCT/US00/08477
A GENERAL METHOD FOR OPTIMIZING THE EXPRESSION
OF HETEROLOGOUS PROTEINS
Technical Field
This invention is related to methods and compositions for obtaining stable
expression of
a protein of interest, to obtain mutant proteins having enhanced stability as
compared to wild
type proteins, and to stabilize unstable proteins associated with disease by
expressing the protein
of interest as a chimeric protein composed of a selection marker and the
protein of interest under
selective conditions. The invention is examplified by preparation of highly
fluorescent GFP
mutants by expressing the GFP as a C-terminal fusion with chloramphenicol
acetyltransferase.
INTRODUCTION
~5 Background
Natural proteins have three fundamental properties in vivo which can be
exploited to
obtain stable expression of heterologous proteins in the absence of any
distinguishing
phenotype of the protein itself.
20 1. Natural proteins have unique minimum energy conformations.
In its broadest sense, a fold is simply a minimum energy conformation or
ground state.
The vast majority of sequences in protein sequence space do not have unique
folds, but rather
have multiple, inter-convertible minimum energy conformations (Li et al.,
Science ( 1996)
273:666-669; Godzik, TIBTECH ( 1997) 15:147-1 S 1; Sauer, Folding and Design (
1996)
?5 1:827-R30; Govindarajan and Goldstein, Proc. Natl. Acad. Sci USA (1996)
93:3341-3345).
However, protein function is so exquisitely dependent on specific tertiary
structure that it is
generally not possible for a protein to be functional in more than one fold.
For this reason
evolution has selected sequences which fold cooperatively in the intracellular
milieu into
unique minimum energy conformations with high energy barriers between them and
all
to kinetically accessible alternatives (Govindarajan and Goldstein, Proc.
Natl. Acad. Sci USA
(1996) 93:3341-3345). A general feature of these folds is a compact
hydrophobic core. If,
under adverse conditions of temperature, pH, ionic strength, etc., such as
might occur during
CA 02387646 2002-04-15
WO 01/29225 PCT/L1S00/08477
2
physical or metabolic stress, the hydrophobic cores are disrupted or fail to
form properly or in
a timely fashion inside the cell, the exposed hydrophobic surfaces have a
strong tendency to
initiate intermolecular aggregation, which is generally toxic to cells. In
fact, the aggregation of
misfolded proteins has become increasingly recognized as a common etiologic
component of
disease (Wetzel, Cell (1996) 86:699-702). For this and other regulatory
reasons cells have
evolved highly efficient proteolytic mechanisms to detect exposed hydrophobic
structure and
prevent misfolded proteins from accumulating (Weissman et al., Science (1995)
268:523-524;
Coux et al. , Ann. Rev. Biochem. ( 1996) 65: 801; Tamura et al. , Science (
1996) 274:1385-
1389; Lowe et al., Science (1996) 268:533-539).
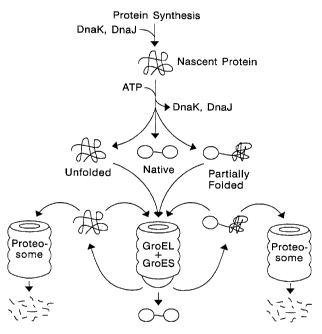
~ o Nascent proteins partition among three fates in vivo: ( 1 ) folding into
the soluble,
functional, native ground state, (2) amorphous aggregation into insoluble
inclusions, (3)
proteolysis. Fate ( 1 ) is generally proportional to the folding rate. The
faster a protein folds
the more of it wilt reach the native ground state before succumbing to fates
(2) and (3), which
are essentially irreversible. With respect to stability, the most important
effect of folding is to
t 5 sequester hydrophobic surface, since exposed hydrophobic surface is the
driving force for both
aggregation and proteolysis. From these properties of natural proteins in
vivo, it may be
inferred that most natural proteins can only be stable in vivo in their native
conformations in
which they are also likely to be functional. Thus, most mutations which
stabilize a natural
protein in a heterologous host, will generally favor the native conformation,
and will restore at
20 least some measure of native functionality.
2. A mufti-domain protein is only as stable in vivo as its least stable
domain.
Inside the cell, nascent proteins first encounter the hsp40 and hsp70 classes
of
chaperone proteins and their associates which bind to any exposed hydrophobic
sequence to
25 protect the nascent protein in its unfolded state (Hartl, Nature ( 1996)
381:571-580). The new
protein is then released from the chaperones by a cooperative, energy-
dependant mechanism.
Many proteins then fold with two-state kinetics, collapsing rapidly into their
native folds
without discernable intermediates. The remainder, however, may accumulate as
'molten
globule' intermediates while searching for conformation space for their native
folds. Since in
this state they are vulnerable to aggregation or proteolysis, the hsp60
chaperonin system has
evolved to provide a protective environment for slower folders (Hartl, 1996).
Misfolded
CA 02387646 2002-04-15
WO 01/29225 PCT/US00/08477
3
proteins are drawn into the multimeric Hsp60 cylindrical complexes
(illustrated in Figure 1),
where they are bound to the inner surface in a fully extended state (Zahn et
al. , Nature ( 1994)
368:261-265; Buckle et al., Proc. Natl. Acad. Sci. USA (1997) 94:3571-3575).
Each protein
then undergoes cooperative, energy-dependant release into the cavity of the
complex where it
can attempt to fold without risk of aggregation or proteolysis. Thus, the
folding machinery
acts not by catalyzing or accelerating folding, but by protecting nascent
proteins from
alternative fates.
Any protein, new or old, which undergoes sufficient transient thermal or
chemical
denaturation to expose hydrophobic surface may be bound and unfolded by the
chaperonin
complex. Each protein may then undergo multiple rounds of binding, unfolding,
release, and
refolding until its native fold is achieved. However, after each round in
which a protein still
fails to achieve its native fold, it may either be rebound by the folding
complex, or it may be
bound by the protein turnover machinery, which also recognizes exposed
hydrophobic
surfaces. These alternative fates for nascent proteins are illustrated in
Figure 1. Thus, the
~s longer it takes a protein to fold, the more vulnerable it is to proteolysis
or aggregation.
Proteins which incur significant delays in folding in heterologous hosts may
fail to accumulate
for this reason.
There are many apparent parallels between mechanisms of protein folding and
turnover
in cells (Weissman et al., Science (1995) 268:523-524). In both cases exposed
hydrophobic
2o surfaces are recognized and bound cooperatively, leading to unfolding of
the entire protein
within multi-subunit complexes having similar cylindrical architectures
(Weissman et al.,
Science ( 1995) 268:523-524; Zahn et al. , Nature ( 1994) 368:261-265; Buckle
et al. , Proc.
Natl. Acad. Sci. USA ( 1997) 94:3571-3575). However, whereas in the folding
process the
bound protein is released to fold again, in the proteosome, the bound protein
is proteolyzed
25 (see Fig. 1). Thus, the ability of proteins to accumulate in cells depends
on both the rate of
folding and the stability of the final fold, though recent work has suggested
that these two may
in fact be related (Scalley and Baker, Proc. Natl. Acad. Sci. USA (1997)
94:10636-40). From
the foregoing it may be surmised that if any single domain in a mufti-domain
protein is
unstable, the entire protein may be subject to proteosomal proteolysis. This
is logical given
3o that any surviving stable fragments from proteolysis of natural proteins
would likely be either
useless or deleterious, especially if they retained unregulated activity.
Indeed, stable
CA 02387646 2002-04-15
WO 01/29225 PCT/US00/08477
4
fragments of natural proteins are rarely detected in cells unless they have
functional
significance. From this it follows that the strength of a selectable cellular
phenotype should be
proportional to the "foldability" of any domain to which the phenotype-
conferring domain is
fused, and that this proportionality could have many useful applications in
protein engineering.
3. Mutations which stabilize proteins in vivo promote the native fold and
function.
As discussed above, the vast majority of sequences in protein sequence space
are not
"foldable", i.e., they do not specify unique minimum energy conformations. The
tiny fraction
that do specify unique stable folds have been selected by evolution to serve
as scaffolds for
protein function. Interestingly, computational experiments have suggested that
the most stable
folds are also the most "designable" (Li et al., Science ( 1996) 273:666-669).
That is, they
may be specified by the largest number of different sequences. This makes them
evolutionarily stable as well as thermodynamically stable. Among natural
proteins a number
of "superfolds" have been observed, such as the immunoglobulin fold, a - 12
kDa sandwich
~ 5 of two 3-S-strand (3-sheets, which has been adapted repeatedly to many
different functions,
specified by many different sequences (Padlan, Molecular Immunology ( 1994)
31:169-217).
An increasing number of other studies have shown that extensive sequence
alterations may be
made in the cores of model proteins without substantially altering the native
fold or function
(Axe et al. , Proc. Natl. Acad. Sci. USA ( 1996) 93:5590-5594; Sauer, Folding
and Design
20 ( 1996) 1: R27-R30).
Many of the known protein folds have been observed in both prokaryotic and
eukaryotic proteins (Netzer and Hartl, Nature (1997) 388:343-349). This is
consistent with the
fact that the intracellular milieus of prokaryotic and eukaryotic cells are
quite similar with
respect to bulk properties such as pH, ionic strength, protein concentration,
etc. In spite of
25 this, many natural proteins are unstable in heterologous hosts in that they
either fail to
accumulate to detectable levels, or when over-expressed they accumulate only
as insoluble
aggregates. Protein synthesis is ten times faster in prokaryotes than in
eukaryotes ( 15 sec vs 2-
3 min for a 40 kDa protein; Netzer and Hartl, Nature (1997) 388:343-349). This
parallels cell
division rates and allows each domain of a mufti-domain protein to fold as
it's made in
3o eukaryotes, free from interference by simultaneously folding downstream
domains. This
adaptation accommodates the rise of mufti-domain proteins in eukaryotes, which
facilitate
CA 02387646 2002-04-15
WO 01/29225 PCT/US00/08477
compartmentalization of complex metabolic networks. In prokaryotes, protein
synthesis is so
fast that multiple domains are synthesized before they have time to fold, and
may therefore
interfere with the folding of each other. For this reason, prokaryotes have
fewer multi-domain
proteins, and many examples exist of prokaryotic single-domain proteins the
eukaryotic
homologs of which are linked into continuous polypeptides. Thus, inter-domain
interference
during folding may be a major factor contributing to the high failure rate for
heterologous
expression of eukaryotic multi-domain proteins in prokaryotes.
If most natural proteins have highly "designable" folds, then for each such
protein
there should be many sequences capable of specifying efficient, stable folding
of the functional
i o protein in heterologous environments. Natural proteins cannot be expected
to fold any more
efficiently than necessary in their natural milieus. In general, eukaryotic
proteins may fold
more slowly than prokaryotic proteins because the risk of aggregation is much
greater in the
prokaryotic cytoplasm. Because of the ten-fold higher prokaryotic protein
synthesis rate, local
concentrations of nascent proteins are much higher and nascent proteins have
little chance to
~ 5 fold while still tethered to the ribosome. Folding is essentially a uni-
molecular reaction and
therefore independent of concentration, whereas, aggregation is effectively bi-
molecular, and
is therefore strongly favored by high concentrations of the protein in
question. Since the
insoluble fraction does not turn over, it grows monotonically, providing an
ever increasing
substrate for aggregation. As a result, the aggregation rate may rise
exponentially, rapidly
2o reaching a point where little nascent protein escapes. Thus, aggregation is
a threshold-like
phenomenon which
is exquisitely sensitive to small changes in parameters such as the folding
rate and synthesis
rate, which affect the initial aggregation kinetics.
The sampling of conformation space to find associations which nucleate
cooperative
25 assembly of the final fold is generally the rate-limiting step in folding,
and the rate of this
process is sharply limited by the energies of off pathway interactions.
Mutations which cause
even modest destabilization of off pathway interactions may accelerate folding
sufficiently to
increase soluble protein by several orders of magnitude. Thus, single
mutations have been
observed to accelerate folding up to 1000-fold, with double and triple mutants
having even
30 larger effects (Jackson, Folding and Design (1998) 3:881-R91). For unstable
heterologous
proteins with selectable phenotypes, it should be possible to access such
mutations by
CA 02387646 2002-04-15
WO 01/29225 PCT/US00/08477
6
combinatorial mutagenesis, selecting for restoration of the phenotype. For
proteins which do
not have selectable phenotypes, there is currently no reliable method to
select for mutations
which accelerate folding in a heterologous host. However, our demonstration of
a tight
correlation between the folding rate of proteins of interest and the strength
of an artificially-
linked phenotype, now makes it possible to identify such mutations in proteins
of interest
which lack selectable phenotypes. Often, over-expression of a misfolding
protein as a fusion
with an otherwise stable protein will improve the soluble yield of the
misfolder. However,
this works best when the stable partner is N-terminal, and has a chance to
fold before the
misfolder can aggregate or turn over. When the stable partner is C-terminal,
as in our system,
i o aggregation or proteolysis of the misfolder can commence before the stable
partner can fold.
In bacteria, the N-terminal stable partner may sterically hinder aggregation
of the misfolding
partner, and the local concentration of nascent misfolder is reduced by a
factor approximately
equal to 1 minus its proportion of the total molecular weight. Nevertheless,
the strength of the
selectable phenotype is still proportional to the solubility of the fusion
protein, which in turn is
~5 limited by the aggregation rate of the slowest folding component.
We have shown that after random mutagenesis of a protein of interest using a
low
mutational operator, such as error-prone PCR (Cadwell and Joyce, in PCR Primer
A
Laboratory Manual, Dieffenbach and Dveksler (eds.) (1995) Cold Spring Harbor
Press, Cold
Spring Harbor, NY, pp. 583-590), DNA shuffling (Crameri et al., Nature
Biotechnology
20 ( 1996) 14:315-9), random-priming recombination (RPR), (Shao et al. ,
Nucleic Acids Research
(1998) 26:681-3), or the staggered extension process (StEP), (Zhao et al.,
Nature
Biotechnology ( 1998) 16:258-261 ) faster-folding variants can be selected by
screening for
higher levels of the selectable phenotype. Furthermore, as variants are
selected which fold
faster than the marker, the marker folding rate becomes limiting because even
stable proteins
25 will aggregate to some extent when over-expressed. However, as explained
above, the marker
is generally less prone to aggregation as the fusion than alone, so the
maximum phenotype
level produced by fusion of the marker with fast-folding variants may be even
higher than that
produced by the marker alone. Since the solubility of the fusion protein may
become limited
by the folding rate of the marker domain, the solubility of the optimized
protein of interest
3o may be even higher when expressed alone, without the marker fusion domain.
Also, we have
found that substantial improvements in expression can be achieved with single
mutations, even
CA 02387646 2002-04-15
WO 01/29225 PCT/US00/08477
for proteins which already express well. We attribute this to the fact that
rate-limiting
intermediates may be readily destabilized by single mutations. This means that
in mutagenic
libraries with mutation frequencies on the order of one per molecule, the
frequency of faster
folders may be greater than ~ one-tenth of the inverse of the chain length, or
-- one in 2500
for a 250-residue protein. Thus, large libraries are not needed to find high-
expressing variants
of poor expressors. The fact that folding can be optimized with few mutations
also minimizes
the likelihood of introducing immunogenic epitopes into therapeutic proteins.
Optimization of bacterial expression of pharmaceuticallindustrial proteins.
i o The industrial and pharmaceutical utility of many proteins is limited by
prohibitive
production costs, due to the difficulty of producing stable, functional
protein in quantity. Most
such proteins are not abundant either in their native sources, or in
heterologous hosts.
Combinatorial optimization of the expression of most such proteins has
previously been limited
to those which confer selectable phenotypes on the production host. However,
with the subject
~ 5 invention it is now possible to optimize the expression of any protein in
any heterologous host
by mutagenizing the protein and expressing the mutant library as a C-terminal
fusion with a
selectable marker. Mutations which accelerate folding are selected on the
basis of the strength
of the marker phenotype. Selected clones should be enriched for native
activity such that only
a modest number will have to be screened to recover the desired activity. The
benefits of
optimized expression are manifold. Not only are yields increased and
production and
purification costs lowered, but higher levels of purity are often possible
when the desired
product is a higher proportion of the starting material.
Since proteins have evolved to fold only as efficiently as necessary in their
native
environments, it is reasonable to expect that most proteins could be mutated
to fold more
efficiently. The absolute limits of folding efficiency in vivo are not known,
but with the subject
invention, it may be possible to test those limits. First, selectable markers
can be optimized by
mutagenesis and selection for maximum strength of phenotype. If such folding-
optimized
markers have any remaining tendency to aggregate when over-expressed, it will
be even
3o further reduced when they are expressed as fusions to mutagenized proteins
of interest. Thus,
folding-optimized markers should place no limit on the optimization of
proteins of interest.
CA 02387646 2002-04-15
WO 01/29225 PCT/US00/08477
8
This could allow valuable proteins to be produced in higher yields with higher
activities and
purity than previously possible. Also, fast-folding variants are likely to be
more
thermodynamically stable as well, since recent experimental (Plaxco et al., J.
Mol. Biol.
(1997) 270:763-70; Mines et al., Chem. Biol. (1996) 3:491-7) and theoretical
(Gutin et al.,
s Proc. Natl. Acad. Sci. USA ( 1995) 92:1282-6; Wolynes et al. , Chem. Biol. (
1996) 3:425-32)
work suggests that folding rates are closely correlated with stability of the
native state. This is
not unexpected if mutations which accelerate folding by destabilizing off
pathway
intermediates also stabilize the native conformation by reducing the ensemble
of kinetically-
accessible alternatives. Thus, it will be important to compare the thermal
stabilities of fast-
io folding variants and their wild-type precursors.
There are two types of stability in proteins. The first relates to tolerance
of extreme
conditions, and the second relates to half life under favorable conditions.
They are not
necessarily mutually inclusive. The reason for this is that activity is often
lost reversibly
before it is lost irreversibly, but the reverse is not possible. In fact, if
loss of activity under
~5 extreme conditions were entirely reversible, it would have little to do
with the half-life of the
protein, which is primarily a function of the rate of irreversible
aggregation. Each trait is
potentially valuable for industrial proteins. Proteins which work better under
extreme
conditions, and/or last longer will fetch a premium on the market, in addition
to savings
realized from reduced production costs, and possible premiums for higher
purity. Folding
20 optimization selects primarily for reduced tendency to aggregate. Since
aggregation is the
principal route of irreversible inactivation, this should prolong the half
life. So long as this is
accomplished by destabilizing aggregation-prone intermediates without undue
effect on the
enthalpy or entropy of the ground state, reversible stability should not be
adversely affected.
25 Efficient searching of protein libraries in vivo will require pre-selection
for stability.
Current efforts to accelerate the discovery and validation of new therapeutic
and
diagnostic targets and reagents to meet growing health care and pharmaceutical
industry
demands depend heavily on continuing development of new and improved
recombinant DNA-
based protein engineering methods. For example, to realize the potential of
genomics for the
3o identification of new therapeutic targets, high-throughput methods for the
functional analysis
of expressed sequences of interest will be required. One important approach to
rapid
CA 02387646 2002-04-15
WO 01/29225 PCT/US00/08477
9
functional analysis will be to use protein-protein interaction traps to
identify networks of
interactions within and between the proteomes of human cells, tissues, and
pathogenic
organisms, using cDNA expression libraries. However, current methods for the
construction
of cDNA libraries for fusion expression, as required for interaction trapping
are so inefficient
that recovery of only the most abundant and robust ligands can be expected.
The vast majority
of cDNA sequences in such libraries are not stably expressed, either because
the reading frame
is incorrect, or because the encoded fragment is not in register with a
foldable domain.
Current recombinant DNA methods allow the construction of cDNA libraries in
bacteria containing up to 10y independent clones, or > 10~ times the average
number of
~ o expressed genes in mammalian tissues. If the frequencies of the rarest
genes are assumed to
be in the 10'6 range, and on average only one in a hundred clones of a gene
make a stable
interaction-competent product, there would still be ten such clones for each
rare expressed
sequence in a library of 109. Thus, the initial size of the library is not
theoretically limiting.
What limits recovery from these libraries with respect to the expression host
is the
~5 transformation efficiency, and with respect to the screen recovery is
limited by throughput and
signal-to-noise ratio. In yeast, libraries are limited by transformation
efficiency to -- 106-10'
clones. In mammalian cells the limit is --- 105-106. Thus, comprehensive
searching of such
libraries in eukaryotic hosts will require not only enrichment for stable
expressors, but also
normalization to bring the frequencies of rare expressors within range of the
library size
20 limits.
Throughput is limited by the ability to detect positive clones in the presence
of an
excess of negative clones. Clone-by-clone screens have the lowest throughput,
color screens
have intermediate throughputs, and viability screens generally have the
highest throughputs.
However, even when neither transformation nor screening is limiting, as with
biopanning or
25 viability selection in a bacterial expression host, recovery is still
limited by the "needle-in-a-
haystack" problem, whereby the discriminating power of the screen, or "signal-
to-noise" ratio
determines the minimum product of frequency and affinity which can be
selectively enriched
above background. Thus, if on average, only one randomly-primed cDNA fragment
in a
hundred expresses a stable, functional domain in the heterologous host (a
conservative
o estimate), then the frequencies of all such clones could rise by up to two
orders of magnitude
if a way could be found to eliminate the non-expressors from the libraries
before screening.
CA 02387646 2002-04-15
WO 01/29225 PCT/L1S00/08477
Such an enrichment could make the critical difference for recovery of many
important
mteractors.
Relevent Literature
5 Albano et al. , Biotechnol Prog ( 1998) 14:351-4 describes the use of the
correlation
between the fluorescent intensity of the reporter GFP and the functional
activity of a protein to
which it is fused. Similarly, Waldo et al., Nature Biotechnology, (199) 17:691-
695, describes
the use of GFP to predict solubility of a protein of interest by expressing it
as a chimeric with
the protein of interest. See also PCT/US98/25862.
~ltMMARY
Methods are provided for obtaining host cells expressing a mutant of a desired
protein
optimized for expression in the host cells, for obtaining a protein with
enhanced stability as
~5 compared to a wild type of the desired protein, and for identifying
peptides that can
stabilize an unstable protein, in each case by expressing the protein linked
to a selector
protein that confers a selectable phenotype on the host cell. In the method
for identifying
stabilizing peptides, the unstable protein is coexpressed with members of a
random peptide
library. To obtain optimized expression of a protein in a host cell, the
method includes the
2o steps of preparing a library of mutagenized coding sequences for the
protein of interest,
purifying the members of the library of mutagenized coding sequences, ligating
each
member of the library into an expression cassette in frame with the coding
sequence for a
selector protein, transforming a multiplicity of host cells with the
expression cassettes,
growing the resulting transformed host cells under conditions for which the
selector protein
25 confers ability for the transformed cells to grow to produce the mutant
proteins joined to
the selector protein, identifying cells that express mutant proteins at a
selective pressure
higher than that of cells expressing an unmutagenized protein. Proteins with
enhanced
stability can be obtained by cleavage from the selector protein, or expressing
the mutant
protein as a free protein in the host cells for which it is optimized. The
invention finds use
3o for example in optimizing mammalian peptides for improved expression in
prokaryotic
CA 02387646 2002-04-15
WO 01/29225 PCT/US00/08477
11
cells and for identifying peptides that can be used for treating diseases that
are
characterized by production of an unstable variant of a wild type protein.
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1. An illustration of the alternative fates for nascent proteins when
expressed in
cells at normal levels. When overexpressed, aggregation is an additional fate
(not shown).
DnaK and DnaJ are bacterial Hsp70 and Hsp40 proteins, respectively. GroEL is
the bacterial
Hsp60 complex, and GroES is the companion HsplO complex. For 2-domain proteins
like
i o GFP-CAT, we hypothesize that misfolding of a single domain leads to
turnover of the entire
protein.
Figure 2. Expression construct for GFP-CAT fusions. T7prom, phage T7 promoter;
(GaS)s, flexible spacer between the GFP and CAT domains; Hiss, hexa-histidine
tail for
affinity purification; T7t, phage T7 transcription terminator; ori, origin of
replication; bla,
~5 ampicillin resistance. Arrow denotes start of translation.
Figure 3. Chloramphenicol resistance of E. coli NovaBlue DE3 cells expressing
CAT,
wtGFP-CAT, and GFPuv-CAT. Cells expressing each construct were plated at 1000
cells per
plate onto solid LB medium containing 0.02 mM IPTG and icreasing
concentrations of
chloramphenicol. After overnight growth at 37° C colonies per plate
were scored and plotted
2o against cam concentration.
Figure 4. Correlation of chloramphenicol resistance with fluorescence
intensity in cells
expressing mutagenized GFP as the GFP-CAT fusion. Mutant library transformants
were
seeded at 1000 per plate on increasing concentrations of cam. The percentage
of colonies
fluorescing brighter than wtGFP-CAT was determined visually and plotted
against cam
25 concentration.
Figure S. Fluorescence emission spectra for cells expressing four GFP-CAT
constructs. GFP-CAT expression was induced during log phase growth in
suspension, after
which the cells were washed and adjusted to a density of O.D.bcx> = 0.1.
Emission spectra
were then taken at an excitation wavelength of 390 nm.
3o Figure 6. Selection of protein-stabilizing peptides from random peptide
libraries (RPL)
using the Fold Selector system. Figure A. Genes for unstable extra-cellular
proteins of choice
CA 02387646 2002-04-15
WO 01/29225 PCT/US00/08477
12
(p.o.c.), such as amyloid (3 protein (A(3), fused to the N-terminus of (3-
lactamase via a flexible
linker, (G~S)3, may be transcribed from the trp-lac fusion promoter (trc prom)
in a plSA
replicon (p 15A ori) with kanamycin resistance (kan) for plasmid retention. N-
terminal signal
peptides (SP) on this and the RPL fusion protein allow export of the gene
products to the E.
coli periplasm. The RPL genes, encoding random peptides fused to the N-
terminus of
thioredoxin via a GaS linker, may be transcribed from the lac promoter in a
pUC phagemid
with chloramphenicol resistance (cat) for piasmid retention. The phagemid
origin of
replication (fl ori) allows the RPL construct to be packaged in phage and
quantitatively
introduced into cells expressing the unstable protein by infection at high
multiplicity (m.o.i.).
io Peptides are selected by their ability to stabilize the p.o.c. and thereby
confer growth on non-
permissive antibiotic concentrations. Figure B. Expression of unstable intra-
cellular proteins
is similar except that chloramphenicol acetyl transferase (CAT) is used as the
C-terminal
fusion to allow selection of stabilizing peptides for chloramphenicol
resistance. Also, SPs are
eliminated to allow retention of expressed proteins in the cytoplasm, and
ampicillin resistance
~ 5 (amp) is used for p 15A plasmid retention.
BRIEF DESCRIPTION OF THE SPECIFIC EMBODIMENTS
Methods for obtaining a protein of interest that is optimized for expression
in a host
2o cell, particularly a prokaryotic cell to which the protein of interest is
heterologous are
provided. To obtain a protein that is optimized for expression in a particular
host cell, such as
E. coli, members of a library of mutagenized coding sequences for the protein
of interest
joined to the coding sequence for a selector protein are transformed into the
host cells which
are then grown under conditions for which the selector protein confers ability
for the
25 transformed cells to grow. Generally the coding sequence for the chimeric
protein contains a
coding sequence for a linker peptide between the mutangenized library member
and the coding
sequence for the selector protein; prefereably the linker is a flexible
linker. The selector
protein can be any protein that provides for a selectable phenotype, such as
antibiotic
resistance. The protein of interest need not have a selectable phenotype.
Cells that express
3o the mutant proteins joined to the selector protein at a selective pressure
that is nonpermissive
for host cells expressing an unmutagenized protein are those that contain a
protein optimized
for expression in the host cell. The mutant proteins can be further screened
to identify those
CA 02387646 2002-04-15
WO 01/29225 PCT/US00/08477
13
that have retained one or more wild type function, and also can be screened to
identify those
that have one or more altered characteristic, such as increased solubility,
increased half life
and decreased temperature sensitivity.
Also provided are methods for screening for peptides, particularly peptides of
from
about 3 to 20, generally of about 12 or less amino acids, that can stabilize
unstable proteins
such as those associated with particular disease states. A chimeric protein
that includes the
defective protein and a selector protein is coexpressed with a member of a
tethered random
peptide library in a host cell grown under selective conditions. The members
of the library are
each a linear chain of about 3 to 20 amino acids, preferably a linear chain of
12 or less amino
acids, fused via a tlexible linker to a stable carrier. Growth of cells under
selective conditions
is indicative of cells that contain a peptide which stabilizes the defective
protein, which can
then be identified and screened to assess whether it can also stabilize a free
defective protein.
Description of the Invention
t 5 What is demonstrated is the feasibility of using a surrogate marker domain
is
demonstrated in a two-domain fusion with a protein of interest in E. coli to
select mutagenic
variants of the protein of interest with improved expression as a function of
accelerated folding
kinetics. We accomplished this goal by demonstrating a high degree of
correlation between
the functional stabilities of a selectable phenotype, chloramphenicol
resistance, and a test
o protein, GFP, in E. coli. Chloramphenicol resistance is conferred by
chloramphenicol acetyl
transferase (CAT), which has been stably and functionally expressed as both N-
and C-
terminal fusions with many heterologous proteins (e.g., Dekeyzer et al.,
Protein Engineering
(1994) 7:125-130; Zelazny and Bibi, Biochemistry (1996) 35:10872-10878).
Functional GFP
and folding variants thereof (Stemmer, Proc. Natl. Acad. Sci. USA (1994b)
91:10747-10751)
25 emit a bright green fluorescence in blue or uv light. The fluorescent
chromophore of GFP is
formed autocatalytically in the folded protein by cyclization of the peptide
backbone of Ser65,
Tyr66, and G1y67 (Cubitt et al., Trends inBiochem. Sci. (1995) 20:448-455).
GFP has also
been stably and functionally expressed as both N- and C-terminal fusions with
many
heterologous proteins (Cubitt et al., Trends inBiochem. Sci. (1995) 20:448-
455).
3o Previously, we had attempted to isolate spectral variants of GFP from
mutagenic
libraries for use in a fluorescence energy transfer-based protein-protein
interaction trap
CA 02387646 2002-04-15
WO 01/29225 PCT/US00/08477
14
(Delagrave et al., BiolTechnology (1995) 13:151-154; Mitra et al., Gene (1996)
173:13-17).
In the course of that work, many non-fluorescent GFP variants were observed
during mutant
library screening. Western blot analyses revealed that for most of these
variants, soluble,
recombinant GFP protein failed to accumulate in cells under conditions in
which soluble wild
s type GFP was readily detectable (Mitra and Balint, unpublished). However, in
most cases
full-length GFP protein could be recovered from the insoluble fraction of both
mutant and wt
GFP-expressing cells in comparable amounts. These observations suggested that
fluorescence-
null GFP variants arise from destabilizing structural or folding mutations
more frequently than
from active site mutations, i.e., mutations which inhibit chromophore
formation but not
folding. This is consistent with the fact that in most proteins, active sites
are smaller
mutational targets than structure-determining sites, and this can be estimated
for GFP from the
known x-ray crystal structures (Ormo et al. , Science ( 1996) 273:1392-1395;
Yang et al. ,
Nature Biotechnology (1996) 14:1246-1251). The availability of x-ray
structures and known
~ 5 folding mutations in a protein which is otherwise stably and functionally
expressed in E. coli,
make GFP an useful tool for testing the concept that the correlation of the
stability of one
domain with the activity of another domain in a multi-domain protein can be
used to isolate
stable variants of proteins which do not have selectable phenotypes. While the
invention is
exemplified with GFP as the protein to be stabilized, and neomycin as the
selectable marker,
2o any protein which is desired to stabilize can be substituted for GFP and
any selectable marker
can be substituted for neomycin. Likewise, while the invention has been
exemplified in E.
coli, any other host cell of interest either prokaryotic or eukaryotic, can be
substituted for E.
coli.
25 The following examples are offered by way of illustration of the present
invention, not
limitation.
CA 02387646 2002-04-15
WO 01/29225 PCT/US00/08477
EXAMPLES
Example 1
5 Correlation of functional GFP with chloramphenicol resistance for wild-type
GFP
and a fast-folding variant expressed as C-terminal fusions with CAT in E.
coli.
The coding sequences for wild-type (wt) GFP and a highly-expressing variant of
GFP
(GFPuv), (Crameri et al., Nature Biotechnology (1996) 14:315-9) were inserted
into the
pET23a vector (Novagen, Inc.) between Nhel and BamHI (see Figure 2). pET23a is
an
i o ampicillin-resistant pBR322 derivative in which transcription of inserted
coding sequences is
controlled by the bacteriophage T7 promoter and transcription terminator
(Moffatt and Studier,
J. Mol. Biol. (1986) 189:113-130). Expression is restricted to hosts, such as
NovaBlue (DE3)
(Novagen, Inc.), which have been transformed to express the T7 RNA polymerase.
GFP
fluorescence can be readily observed in colonies of these cells harboring the
pET-GFP
~5 construct by illuminating with long-wave uv light. The spectrum, quantum
yield, and
extinction coefficient of GFPuv do not differ appreciably from wtGFP,
consistent with a
difference of only three of 238 amino acids (Crameri et al. , Nature
Biotechnology ( 1996)
14:315-9). However, when expressed from identical constructs in E. coli cells
GFPuv
produces 30-45 times more steady state fluorescence than does wtGFP. Since the
specific
2o fluorescence of both proteins is comparable, it may be concluded that the
higher fluorescence
intensity of cells expressing uvGFP is due to a comparable increment in the
steady state
amount of soluble, functional uvGFP protein. This is supported by data
indicating that the
total amount of GFP protein in the cells is comparable for both, but that
proportionally more
GFPuv is present in the soluble pool. Thus, the mutations in GFPuv appear to
have increased
its steady-state activity in E. coli by specifically reducing its tendency to
aggregate,
presumably as a result of an increased folding rate.
To use a surrogate marker to select for more stable variants of a protein of
interest, the
selectable marker coding sequence must be inserted downstream from that of the
protein of
interest to insure that selection is not favored by premature termination of
the protein of
3o interest. Thus, the CAT coding sequence was inserted into the XhoI site of
pET23a in the
same reading frame as the upstream GFPs. Between the two a 15-residue
flexible, hydrophilic
linker, (GIyaSer)3, was encoded with convenient restriction sites for facile
replacement of both
CA 02387646 2002-04-15
WO 01/29225 PCT/US00/08477
16
GFP and CAT sequences. The CAT sequence terminates in a Hisb tail for facile
purification.
This construct, pET23a-GFP-CAT is shown in Figure 2.
Table I and Figure 3 show the results of comparisons of the chloramphenicol
resistance
and fluorescence characteristics of wtGFP and GFPuv expressed alone and as C-
terminal
s fusions with CAT from pET23a in E. coli strain BL21(DE3). When expressed
alone, GFPuv
produces -- 30 times more steady state fluoresence than wtGFP, as determined
by fluorometry
of suspensions of equal numbers of cells from overnight growth on solid
medium. Maximum
transcription normally requires induction of T7 polymerase expression with
IPTG, but a low
level of transcription occurs even in the absence of IPTG. Interestingly,
induction of wtGFP
i o by IPTG produces no detectable increment in fluorescence over the
uninduced level, though
the level of wtGFP protein is considerably higher, as determined by gel
electrophoresis. This
suggests that at the uninduced expression level, aggregation is minimal,
probably due to sub-
threshold concentrations of nascent GFP. This is supported by the fact that
under uninduced
conditions, GFPuv fluorescence is comparable to that produced by wtGFP, which
would be
~ 5 expected if wtGFP fluorescence is not limited by aggregation. At the
induced expression
level, however, substantial insoluble material forms in the wtGFP-expressing
cells,
presumably due to much higher nascent wtGFP concentrations. Under the same
conditions
GFPuv produces --- 30-fold higher fluorescence intensity. From this we
conclude that GFPuv
is much less prone to aggregation at similar expression levels, i.e., nascent
protein
2o concentrations, presumably due to a higher folding rate.
30
CA 02387646 2002-04-15
WO 01/29225 PCT/US00/08477
17
Table I. Comparison of Relative Steady-State Fluorescence Intensity and
Chloramphenicol Resistance (Cam') for CAT Fusions of wtGFP and GFPuv in E.
coli.
Expression Product IPTG Cam' w Fluorescence
n'
0 NA 1 x
wtGFP
0.02 mM NA lx
0 NA 1 x
GFPuv (3 mutations)
0.02 mM NA 30x
0 34 p,g/ml NA
CAT
0.02 mM 306 p.g/ml NA
0 34 u,g/ml lx
wtGFP-CAT
0.02 mM 238 p.g/ml 2x
0 34 pg/ml lx
GFPuv-CAT
I 0.02 mM 340 Etg/ml 4x
s a. Chloramphenicol resistance (CamR) was determined as the highest
concentration
in solid LB medium on which at least 50% of cells plated formed visible
colonies after overnight growth.
b. Fluorescence was determined by fluorometry at ,excite = 395 nm and ,emit =
508 nm of cell suspensions of 0.1 OD600 after overnight growth on solid LB
medium.
Interestingly, when induced, cells expressing CAT alone were resistant to less
chloramphenicol than cells expressing GFPuv-CAT. This suggests that GFPuv,
derived from
~s a jellyfish protein adapted to life at -- 13°C, is less prone to
aggregation when over-expressed
CA 02387646 2002-04-15
WO 01/29225 PCT/US00/08477
18
in bacteria at 37°C than is CAT, a native bacterial protein. Consistent
with this, cells
expressing induced GFPuv-CAT are much less fluorescent than cells expressing
GFPuv alone,
suggesting that the stability of GFPuv-CAT is limited by the stability of CAT.
The
improvement in CAT expression in GFPuv-CAT is probably due to its reduced
concentration
in nascent protein (CAT is less than half the size of GFPuv-CAT and their
synthesis rates
should be similar), and/or GFPuv may sterically interfere with CAT
aggregation. On the other
hand, wtGFP appears predictably to be less stable than CAT, since induced
wtGFP-CAT
confers less chloramphenicol resistance than CAT alone, but more fluorescence
than wtGFP
alone. In general, SDS PAGE analyses of soluble and total extracts were
consistent with the
n> fluoresence and chloramphenicol resistance phenotypes, i.e., higher soluble
protein levels
correlated with higher tluorescence and higher cam resistance, though total
protein remained
more or less constant.
~5
Example 2
Isolation of super-stable variants of GFP from mutagenic library expressed as
fusion with CAT and selected for increased chloramphenicol resistance.
The GFP coding sequence may be subjected to random mutagenesis by any of
several
methods, including error-prone PCR (Cadwell and Joyce, in PCR Primer A
Laboratory
2o Manual, Dieffenbach and Dveksler (eds.) (1995) Cold Spring Harbor Press,
Cold Spring
Harbor, NY, pp. 583-590), DNA shuffling (1994a,b), random-priming
recombination (RPR),
(Shao et al., Nucleic Acids Research (1998) 26:681-3), or the staggered
extension process
(StEP), (Zhao et al., Nature Biotechnology (1998) 16:258-261). The wtGFP
coding sequence
may be amplified from pET-wtGFP using primers containing the NdeI site at the
translation
start, and at the unique EcoRI site just beyond the GFP C-terminus in pET-GFP-
CAT. The
error-prone amplification reaction is carried out in the presence of excess
Mg++ and excess
deoxynucleoside triphosphates to encourage mis-incorporation. An excess of
primers may also
be used, since the final mutation frequency is proportional to the number of
cycles, so long as
3o the primers are not exhausted. Under standard error-prone conditions
(Cadwell and Joyce, in
PCR Primer A Laborator Manual, Dieffenbach and Dveksler (eds.) (1995) Cold
Spring
Harbor Press, Cold Spring Harbor, NY, pp. 583-590), a mutation frequency of -
0.7% is
produced in the final product after 25-30 cycles. Since 75 % of coding
mutations produce
CA 02387646 2002-04-15
WO 01/29225 PCT/US00108477
19
coding changes, we would expect - 3-4 amino acid substitutions per GFP clone
from this
protocol. The GFP coding sequence in plasmid pGFP (Clontech Laboratories,
Inc.) was used
as template for error-prone amplification using the end-specific primers
AGCAGTCGCTTCACGTTCGCTCGC and GCATTCATCAGGCGGGCAAGAATG. The
template GFP sequence was that reported by Chalfie et al. ( 1994) with the
following changes:
the starting triplet MGK has been replaced by MASK derived from the pET23a
multiple
cloning site, and the final SG has been replaced by NS. The same changes were
also
introduced into the GFPuv sequence (Clontech Laboratories, Inc.).
Additionally, a Q80R
mutation derived from a PCR error has been retained. The error-prone PCR
product was gel-
o purified, and ligated back into the GFP-CAT fusion expression construct
shown in Figure 2.
The ligation product was then introduced into cells of E. coli strain NovaBlue
(DE3) by high-
voltage electroporation. Transformants were then plated onto solid Luria-
Bertani medium
containing increasing amounts of chloramphenicol, ranging from 34 pg/ml to 544
ltg/ml, and
incubated at 37°C overnight.
~ 5 An initial assessment of the correlation of cam resistance with
fluorescence intensity
was made by a visual estimation of the percentage of colonies which fluoresced
more intensely
than wtGFP-CAT as a function of cam concentration. The results are illustrated
in Figure 4.
As the concentration of cam increased, the frequency of brighter colonies also
increased. At
cam concentrations above 270 ~tg/ml, the probability of visually identifying a
brighter colony
2o rose above 10 % . When these brighter clones were restreaked, they
invariably remained
brighter than wtGFP-CAT-expressing cells. Interestingly, when colonies
resistant to high cam
were replated onto 34 pg/ml cam, the percentage of brighter colonies rose to --
- 30 % . Thus,
many clones expressing brighter GFP variants were masked by the high
concentrations of cam
used for selection which probably inhibited protein synthesis somewhat.
?5 From the first round of mutagenesis colonies were recovered from cam
concentrations
of up to 408 pg/ml, exceeding the cam resistance of the GFPuv-CAT construct.
For the
purpose of phenotypic screening, ten clones were picked from plates containing
between 306
and 408 ~tg/ml cam. These clones were selected solely for their ability to
grow on cam
concentrations which were non-permissive for the parental wtGFP-CAT construct.
In ambient
3o room light GFP fluorescence was not visible. For a second round of
mutagenesis forty clones
from the first round were pooled in ambient light from cam concentrations
which were non-
CA 02387646 2002-04-15
WO 01/29225 PCT/US00/08477
permissive for wtGFP-CAT. Plasmid DNA was purified from the pooled clones and
used as
template for mutagenesis by the staggered extension process (StEP) for in
vitro recombination
of mutations selected from the first round (Zhao et al., Nature Biotechnology
(1998) 16:258-
261). This method employs a template switch recombination mechanism, in which
a short
5 extension time is used to allow only partial replication of the sequence
during each cycle.
Thus, each full-length copy is generated over several cycles with the template
being switched
between each cycle. The second round product was ligated back into the vector
as before and
plated onto the same range of cam concentrations as used for the first round.
Colonies were
observed on cam concentrations of up to 510 pg/ml. Again, for phenotypic
screening, 10
i o clones were picked in ambient room light from plates containing between
306 and 510 ltg/ml
cam.
The 20 total clones selected from cam plates in rounds one and two were grown
in
suspension in 5 ml of LB containing either 100 pglml ampicillin or 34 p.g/ml
chloramphenicol.
At an O.D~oo of 0.4, protein expression was induced with 0.4 mM IPTG. After 16
hours of
~ 5 overnight growth, cells were washed and resuspended in phosphate buffered
saline (PBS) at
OD~o of 0.1, and the whole cell fluorescence was measured by excitation at 395
nm and
emission at 508 nm. Fluorescence emission spectra were then determined for the
brightest
mutants from each round, designated GFPR1-CAT and GFPR2-CAT respectively. Each
had
an emission maximum at 510 nm when excited at 390 nm. The emission spectra of
these
2o clones are compared to those of wtGFP-CAT and GFPuv-CAT in Figure 5. To
assess the
effect of the CAT gene on GFP expression, stop codons were inserted at the C-
termini of the
coding sequences of GFPwt, GFPuv, GFPRI, and GFPR2 in the GFP-CAT fusion
constructs
to allow expression of the free GFPs. The relative fluorescence intensities
for GFP expression
with and without CAT are summarized in Table II.
~o
CA 02387646 2002-04-15
WO 01/29225 PCT/US00/08477
21
Table II. Relative Fluorescence Intensities of GFP Constructs
CONSTRUCT Relative Emission
Intensity at 510 nm
I GFPwt-CAT 1
s ~ GFPuv-CAT 4
I GFPR1-CAT 14
~ GFPR2-CAT 56
I GFPwt 1
o ~ GFPuv
~ GFPR1
I GFPR2 40
is
The brightest mutant from round one, GFPR1-CAT, was 14 times brighter than
wtGFP-CAT and 3.5 times brighter than GFPuv-CAT. GFPR1-CAT was also resistant
to at
least 408 ~g/ml cam, whereas GFPuv-CAT could resist only 340 pg/ml. The
brightest mutant
from round two, GFPR2-CAT, was 56 times brighter than GFPwt and 14 times
brighter than
2o GFPuv-CAT, and could grow in 510 pg/ml cam. Interestingly, the dramatic
increases in
expression seen with GFPR1-CAT and with GFPR2-CAT over GFPuv-CAT essentially
vanished when CAT was removed. All three expressed at comparable levels, 30-40
times that
of wtGFP. Whereas GFPuv expression was inhibited by fusion to CAT by 7-8-fold,
expression of GFPR1 was inhibited only -~-~ 3-fold, and expression of GFPR2
was actually
25 enhanced slightly by fusion to CAT. Thus, not only were mutations selected
which enhanced
expression of the free GFP protein, but mutations were also selected which
specifically
CA 02387646 2002-04-15
WO 01/29225 PCT/L1S00/08477
22
enhanced expression of GFP as fusions with other proteins. The reduced
expression of
GFPuv-CAT relative to free GFPuv is probably not due to dominant weaker
expression of
CAT because CAT does not have the same effect on GFPR2. Rather the reduced
expression
of GFPuv-CAT is probably due to mutual steric interference with the folding of
the two
proteins, and the same is probably true to a lesser extent for GFPR1-CAT. SDS-
PAGE
confirmed that the GFPR1-CAT, GFPR2-CAT, GFPwt-CAT, and GFPuv-CAT all
comprised
over 50% of the total cell protein. The increase in brightness for the GFPR1-
CAT and
GFPR2-CAT mutants over that of wtGFP-CAT and GFPuv-CAT is reflected by the
difference
in the amount of protein in the soluble fraction. For example, about 25 % of
GFPR2-CAT
protein is soluble, whereas only about 1-2% of wtGFP-CAT protein was soluble.
DNA sequences were determined for the entire open reading frames of GFPR1 and
GFPR2, and compared to those of wtGFP and GFPuv (see Table III). In addition,
the
reported mutations in GFPuv were confirmed. Surprisingly, one mutation was
shared by all
three improved GFPs, V 164A. Even more surprisingly, this was the only
mutation present in
~5 GFPR1. Since GFPR1 expresses as well or better than GFPuv as the free
protein, this
suggests the other mutations in GFPuv are not necessary. GFPuv had originally
been
"evolved" by repeated rounds of recombinatorial mutagenesis by DNA shuffling
and
phenotypic selection, followed by back-crossing to eliminate deleterious
mutations (Crameri et
al. , Nnture Biotechnology ( 1996) 14:315-9). However, it appears that only
one of the three
2o remaining mutations is actually required for the complete phenotype. Thus,
not only was
recombination unnecessary, but the required mutation could have been recovered
easily from a
few thousand clones of a standard Taq polymerase amplification of the wtGFP
coding
sequence. Under standard conditions - 25 cycles of PCR with a non-proofreading
polymerase
such as Taq produces one mutation per - 700 bp, which is roughly the size of
the GFP coding
25 sequence. Since only a single nucleotide change with a frequency of 1/3 is
required for the
V 164A mutation, the expected frequency would be one in only - 2100 clones.
~o
CA 02387646 2002-04-15
WO 01/29225 PCT/US00/08477
23
Table III. Sequence Comparison of Three High-Expressing GFP Variants and wtGFP
Amino Acid Residue GFPwt GFPuv GFPR1 GFPR2
100 TTT (F) TCT (S) TTT (F) 'I'I'I' (F)
l05 AAC (N) AAC (N) AAC (N) AGC (S)
154 ATG (M) ACG (T) ATG (M) ATG (M)
GTT (V) GCT (A) GCT (A) GCT (A)
Since the V 164A mutation could account for all of the increase in free GFP
expression
for all three improved GFPs, we wished to see if any other independently
adaptive mutations
could be recovered. Before embarking on the arduous task of sequencing a large
number of
additional clones, however, we first examined the eighteen other independent
clones selected
i o from rounds one and two, which grew on non-permissive cam, for the
presence of the V 164A
mutation. This mutation was present in all eighteen clones. Thus, V 164A
appeared to be the
only single-hit mutation capable of destabilizing the aggregation-prone
intermediate in GFP
folding. Indeed, such a mutation would be expected to reduce the
hydrophobicity at that
position, and it is hydrophobicity which would be expected to drive
aggregation. Any other
i 5 independently adaptive mutation of comparable frequency should have
appeared at least once.
Of course, it is possible, even likely, that combinations of two or more
mutations could have
had a comparable effect, but their frequency would have been too low to be
readily selected
from our library.
2o Neither of the other two mutations present in GFPuv appeared in any of the
twenty
selected clones, consistent with their apparent dispensibility for increased
cam resistance.
More revealing, however, is the fact that GFPRI showed 3.5-fold higher
expression as the
CA 02387646 2002-04-15
WO 01/29225 PCT/US00/08477
24
CAT fusion than GFPuv-CAT. This suggests that at least one of the two other
mutations in
GFPuv is responsible for mutual interference with CAT folding. The fact that
GFPR1 itself is
still somewhat inhibited as the CAT fusion relative to its expression as the
free protein,
suggests that the wtGFP sequence is still somewhat inhibited by the folding or
presence of
CAT. The folding of GFPR2, however, was not inhibited at all by the presence
of CAT.
GFPR2 contains only one mutation in addition to V 164A, namely N LOSS. Thus,
this mutation
is apparently responsible for the complete elimination of folding interference
between GFP and
CAT. It is not likely that the combination of mutations in GFPR2 arose by
recombination
because V 164A is apparently indispensable. Rather, the combination probably
arose by simple
i o addition of the N lOSS mutation to V 164A. We have confirmed that the N l
OSS mutation by
itself is not sufficient to confer a selectable increment in cam resistance on
GFP-CAT.
The ability of the surrogate marker fold selection system to select mutations
which
specifically enhance fusion protein expression in addition to those that
enhance independent
folding is an added benefit for proteins like GFP, which have important
applications as fusion
~ 5 proteins. One question is whether fold selection in the context of fusion
proteins could, on
occasion, select only mutations which accelerated folding only in the context
of the fusion, and
did not accelerate folding of the free protein. Such mutations would be highly
unlikely
because in addition to protecting the protein of interest from interference by
the fusion partner,
such mutations would also have to in effect convert the fusion partner into a
chaperone, for
2o which there is no precedent. Mutations which specifically improve fusion
expression, like
GFP-NlOSS, can only be selected in proteins which already fold independently,
like GFPR1.
In preliminary tests with other fusion partners, GFPR2 has continued to fold
independently of
the fusion partner, neither inhibiting nor being inhibited by it. For example,
as a C-terminal
fusion with neomycin phosphotransferase (GFPR2-NPT) both fluorescence and
kanamycin
25 resistance were at least as high as those of the free GFPR2 and the free
NPT, respectively,
whereas both functions were inhibited in the GFPuv-NPT fusion. Since GFPuv was
reported
to express at 30-fold higher levels than wtGFP in mammalian cells, it may
exhibit the same
sensitivity to fusion expression in these cells as it does in bacteria. GFPR2
is the subject of US
Patent Application 60/160,461.
~o
CA 02387646 2002-04-15
WO 01/29225 PCTlLTS00/08477
Example 3
Unstable proteins can be stabilized by peptides selected from random peptide
libraries.
5 Many diseases are caused by unstable proteins, which fail to accumulate in
biologically
active form in cells or tissues due to one or more mutations which cause a
delay in folding or
which destabilize the active conformation such that the protein is prone to
insoluble
aggregation and/or proteolysis. There are two main types of unstable
proteinopathies: those
which cause disease by forming toxic insoluble aggregates, and those which
cause disease by
o loss of function. The former are represented by amyloidogenic polypeptides
such as the
amyloid (3 protein (A~3), which forms insoluble amyloid fibrils in the brain
(Li et al., J.
Leukocyte Biol. ( 1999) 66:567-74; Cappai and White, Int. J. Biochem. Cell
Biol. ( 1999)
31:885-9). Amyloid deposits can induce chronic inflammation and tissue damage,
which are
major etiologic components of Alzheimer's disease. There is currently much
interest in the
~5 development of chemo-therapeutic strategies to counter the progress of
amyloidogenesis and
resultant tissue degeneration in Alzheimer's and other amyloidogenic
proteinopathies. Drugs
which could interfere directly with the aggregation of the A(3 protein would
be highly
desirable. However, no reliable method currently exists for screening chemical
libraries for
such activities.
2o We have demonstrated that some unstable proteins can be stabilized by
interaction with
small peptides obtained from a random sequence library using the "Fold
Selector" technology
described above. It is expected that such peptides may be used
therapeutically, or may be used
as leads for the development of therapeutic agents, which can be used to
inhibit, or perhaps
even reverse the formation of amyloid or other types of toxic insoluble
protein deposits. It is
25 further expected that similar peptides could be selected for their ability
to stabilize intra-
cellular proteins responsible for loss-of function proteinopathies. Most
mutations which lead to
loss of physiological function do not disable the active site of a protein per
se, but rather they
destabilize the active conformation, or they interfere with the folding
pathway so that the
protein gets trapped in meta-stable intermediates which are prone to
aggregation or
3o proteolysis. The reason for this is that the target size for structural
mutations in a natural (i.e.,
highly evolved) protein is typically much greater than the active site(s).
Thus a high
proportion of inborn errors of metabolism and other genetic disorders are
caused by proteins
which do not remain properly folded and/or do not fold properly to begin with.
Peptides which
CA 02387646 2002-04-15
WO 01/29225 PCT/LTS00/08477
26
stabilize such proteins in vitro could be used to develop cell-penetrating
peptido-mimetics
which in turn could be used to restore the missing functions by stabilizing
the proteins in vivo.
Because amyloidogenic proteins such as the A~3 peptide do not produce
screenable or
selectable phenotypes, there is no conventional method to select for
stabilization of these
proteins. However, as we have demonstrated, when unstable proteins are
expressed as C-
terminal fusions to a protein with a selectable phenotype, the selectable
phenotype is
destabilized and can be used to select for stabilization of the amyloidogenic
protein. Extra-
cellular amyloidogenic proteins may be expressed in the E. coli periplasm as C-
terminal
fusions to TEM-1 (3-lactamase with an intervening flexible linker such as
(GlyaSer)s. TEM-1
o (3-lactamase is an E. coli plasmid-born enzyme which confers resistance to
the penicillin class
of (3-lactam antibiotics (Genbank Accession no. J01749; Sutcliffe, Proc. Natl.
Acad. Sci. USA
(1978) 75:3737-3741). The coding sequence for the 42-amino acid A(3 peptide
(Glenner and
Wong, Biochem. Biophys. Res. Commun. ( 1984) 3:885-90; Kang et al. , Nature (
1987)
325:733-736) was sub-cloned for expression in the E. coli periplasm as a
fusion to the N-
~ 5 terminus of (3-lactamase as illustrated in Figure 6. When expressed in E.
coli strain DHSa,
the tendency of A(3 to form insoluble aggregates reduced (3-lactamase activity
such that the
cells would not grow on ampicillin concentrations above 200 ~tg/ml, whereas,
when A(3 was
removed or replaced with a stable domain of comparable size (c-fos), the host
cells grew with
quantitative efficiency (i.e., >0.5 colonies per cell) on ampicillin
concentrations up to 800
2o pg/ml. Polyacrylamide gel electrophoresis (PAGE) confirmed that
proportionally more (3-
lactamase partitioned into the insoluble fraction as the A~3 fusion than as
either the free enzyme
or as the c-fos fusion. Thus, under these expression conditions ampicillin
resistance could be
used to select for stabilization of the human A(3 protein.
A random peptide-encoding library (RPL) was constructed using synthetic
25 oligonucleotides to encode a chain of 12 randomly selected amino acids at
the N-terminus of
E. coli thioredoxin (trxA; Genbank accession no. M54881) with an intervening
flexible linker
(GlyaSer). The expression constructs for this library and A(3-(3-lactamase are
illustrated in
Figure 6. The expression cassette for the RPL-trx fusion library was assembled
in a pUC-
based phagemid (Sambrook et al., in Molecular Cloning A Laboratory Manual, 2"d
ed., (1989)
3o Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 4.17-4.19)
to allow
rescue as phage, which could then be used to transfect the construct
quantitatively into cells
CA 02387646 2002-04-15
WO 01/29225 PCT/US00/08477
27
harboring the A(3-(3-lactamase expression construct. At least 10g clones of
the RPL were
rescued as filamentous bacteriophage by infection with helper phage M13K07
(Sambrook et
al. , in Molecular Cloning A Laboratory Manual, 2"~ ed. , ( 1989) Cold Spring
Harbor
Laboratory Press, Cold Spring Harbor, NY, pp. 4.19-4.50). At least 10y DHSa
cells bearing
the A(3-(3-lactamase construct were infected with a 100-fold excess of RPL
phage to insure
quantitative infection. At least 10~ independent transfectants were then
plated onto solid
medium containing 400, 600, and 800 ug/ml ampicillin. 110 colonies were
recovered after
overnight growth on 400 ltg/ml, 19 were recovered from 600 ~tg/ml, and 4 were
recovered
from 800 pg/ml. For negative controls, ten clones were selected at random from
the
i o unselected RPL/A(3-(3-lactamase co-transformants, and 10~ cells of each
were plated onto 400,
600, and 800 pg/ml ampicillin. After overnight growth no colonies appeared for
any clone on
any ampicillin concentration. By contrast most of the selected clones replated
onto 400 pg/ml
ampicillin, 13 replated onto 600 ~tg/ml, and 2 replated onto 800 pg/ml. Thus,
12-mer
peptides were selected which substantially reduced the tendency of A(3 to form
insoluble
i 5 aggregates, as judged by the increased ~3-lactamase activity. These
peptides can be produced
synthetically and modified by known methods to increase their stability in
vivo. It is expected
that this can be accomplished for at least some of the selected peptides
without sacrificing their
ability to stabilize A(3 against amyloid formation both in vitro and in vivo.
Interestingly, when a similar 12-mer RPL was constrained between the disulfide-
2o forming cysteines in the active site of thioredoxin, and co-expressed with
A~3-(3-lactamase
fusion, fewer clones were recovered at 400 pg/ml ampicillin, and none were
recovered at
higher concentrations. It is instructive to consider why such a library might
be so much
poorer a source of stabilizing peptides than the tethered N-terminal library
we used. The
thioredoxin active site-constrained RPL and others like it have been widely
used to obtain
25 artificial ligands for antibodies, receptors and other proteins of interest
(Colas et al., Nature
(1996) 380:548-550). The peptides in this RPL are constrained into a closed
loop on the
surface of the protein by the disulfide, and are therefore expected to be
somewhat more rigid
than the N-terminal peptides in our library. Rigidity is important for high-
affinity protein-
protein interactions because it minimizes the entropy cost of binding.
However, flexibility
o may be more important for protein stabilization. Conventional protein-
protein interactions are
surface interactions, whereas stabilizing peptides may need to interact with
residues which
CA 02387646 2002-04-15
WO 01/29225 PCT/US00/08477
28
become iternalized in the active conformation. Also, the diversity of
constructive interactions
which can occur between unstable proteins and flexible peptides should be much
greater than
interactions with the surfaces of rigid proteins. Thus, some of the
stabilizing peptides may
extend into the interior of the stabilized A~i protein, and/or may interact
with non-contiguous
regions of the protein. It is even possible that in cases where instability is
due to folding
intermediates, and not to a loss of stability of the active conformation, that
stabilizing peptides
may, in effect, catalyze the folding reaction without remaining structural
components of the
folded protein.
We also expect that at least some of the A(3-stabilizing peptides will not
require all 12
amino acids for activity, and may be equally active as smaller peptides. We
have stabilized
other proteins with smaller peptides. For example, we have identified several
linear tri-
peptides, which when tethered to a carrier protein can stabilize an unstable
fragment of (3-
lactamase. The peptide-stabilized ~3-lactamase fragment can then complement a
second
fragment to form active [3-lactamase. We believe that the methods described
herein can be
~5 broadly used to isolate peptides which can stabilize desired proteins,
particularly those which
do not produce screenable or selectable phenotypes. Such methods are not
currently available,
and since there are few if any reports in the literature of protein-
stabilizing peptides, it is
generally not appreciated that unstable proteins can be stabilized at will
with appropriate
peptides, and possibly small molecules derived therefrom.
2o The foregoing results suggest a general procedure for selecting protein-
stabilizing
peptides from unconstrained terminal RPLs, which begins with expressing the
unstable protein
of choice as a C-terminal fusion with a flexible hydrophilic linker and a
selectable marker in
the appropriate compartment of E. coli, as illustrated in Figure 6. If the
protein of choice is a
secreted protein, as in the case of the A(3 peptide, ~i-lactamase may be used
as the C-terminal
25 fusion partner to allow periplasmic selection for (3-lactam antibiotic
resistance. A signal
peptide must be encoded at the N-terminus for export of the fusion protein to
the bacterial
periplasm. It is preferable to use the p 15A replicon with chloramphenicol
resistance, so that
universal RPLs can be constructed in the pUC phagemid with kanamycin
resistance. If the
protein of choice is cytoplasmic, as in the case of GFP described above, CAT
(Genbank
3o accession no. X06403; Rose, Nucleic Acids Research (1988) 16:355) may be
used as the
fusion partner to allow selection for chloramphenicol resistance (Dekeyzer et
al., Protein
CA 02387646 2002-04-15
WO 01/29225 PCT/US00/08477
29
Engineering (1994) 7:125-130; Zelazny and Bibi, Biochemistry (1996) 35:10872-
10878). In
this case it is preferable to use the plSA replicon with ampicillin
resistance, so that the
universal RPLs in the pUC phagemid with kanamycin resistance can be used.
The first requirement which must be met is that the unstable protein must
cause a
substantial quantitative reduction in the selectable phenotype. This must be
quantified and the
minimum stringency must be established for quantitative selection, as was done
for the use of
ampicillin resistance to select for stabilization of the A~i protein. One or
more universal RPLs
may then be quantitatively introduced into cells expressing the fusion of the
desired protein
with the selector, and the transfectants are then plated onto the minimum
concentration of
i o antibiotic which is quantitatively non-permissive for growth of the fusion
protein. The number
of independent transformants plated should be equivalent to or greater than
the size of the
RPL, and the minimum non-permissive concentration of antibiotic should allow
no colonies to
grow from the same number of cells expressing the fusion protein alone. The
RPL used was a
12-mer on the N-terminus of thioredoxin, but the RPL may vary in length from 3
to 20 or
~5 more residues on either end of the carrier. However, the proportion of
unstable peptides in
the RPL rapidly increases when the length exceeds -12 amino acids. The carrier
may be any
stable protein which tolerates terminal fusions well. Selected peptides may be
verified by co-
expression with the free protein of choice. A substantial increase in the
proportion of the
protein which partitions into the soluble fraction should be observed in the
presence of the
2o selected peptide only and not in the presence of a non-selected peptide.
Example 4
Unstable proteins can be stabilized by tri-peptides selected from random
peptide
25 libraries
The most common cause of instability in proteins is the tendency of folding
intermediates, which may be accessed either on the folding pathway or by
partial unfolding of
the folded protein, to form inter-molecular associations between structural
elements which
normally associate intra-molecularly in the native conformation. Such inter-
molecular
3o associations may initiate polymerization reactions which lead to the
formation of insoluble
aggregates such as amyloid fibrils (Dobson, 1999, Trends Biochem. Sci. 24, 329-
32). We
wished to test the hypothesis that small peptides could be selected from
random sequence
libraries for their ability to protect unstable proteins from aggregation. To
accomplish this we
CA 02387646 2002-04-15
WO 01/29225 PCT/US00/08477
utilized a fragment complementation system, which we had developed for the
enzyme (3-
lactamase. E. coli TEM-I (3-lactamase (Sutcliffe, 1978, Proc Nntl Acad Sc. USA
75, 3737-41)
may be separated into two fragments at E 197-L 198 which can complement to
form active
enzyme with the aid of interacting domains such as hetero-dimerizing helixes
which are fused to
5 the break-point termini of the fragments (Balint and Her, US Patent
Application 60/124,339).
The activity of the (3-lactamase fragment complementation system is limited,
however,
by the stability of the N-terminal fragment, denoted a197. When a197 and the
stable C-terminal
fragment, cn198 were co-expressed in the E. coli periplasm as fusions to the
hetero-dimerizing
helixes of the c-fos and c-jun subunits of the transcription factor AP-1
(Karin et al.. 1997, Curr
Opin Cell Biol 9. ?40-6), only enough ~3-lactamase activity was produced to
confer a plating
efficiency of -1% on 50 ~g/ml ampicillin. However, when the fragment fusions
were co-
expressed with a library of random tri-peptides at the N-terminus of a carrier
protein, E. coli
thioredoxin (trrA; Genbank accession no. M54881) with an intervening Gly4Ser
linker, four tri-
peptides were independently selected which specifically increased ~3-lactamase
activity to confer
I5 100% plating efficiency on the host cells. These tri-peptides all turned
out to have the same
sequence, Gly-Arg-Glu (GRE). The GRE tripeptide conferred no resistance to
ampicillin in the
absence of the interacting helixes, thus it does not stabilize the re-folded
fragment complex, but
rather it must stabilize the a197 fragment since activity is limited by the
amount of soluble
a197. Since the GRE tri-peptide had the same stabilizing effect on x197
fragment when a
different carrier was used, its activity must be context independent. Thus, an
18 kDa enzyme
fragment could be stabilized at least 100-fold by a tri-peptide selected from
a random sequence
library.
Interestingly, though the GRE tri-peptide could inhibit aggregation of a197,
it apparently
did not interfere with re-folding of the fragment complex. Since aggregate
formation proceeds
~g exponentially, it is exquisitely sensitive to small shifts in the inter-
molecular association rate
constants (Dobson, 1999). Thus, even weak binding of an excess of the tri-
peptide to the
interacting surfaces could effectively defeat inter-molecular aggregation. On
the other hand,
cooperative folding of the fragment complex should readily displace the weakly
bound tri-
peptides because the effective intra-molecular concentrations of interacting
structural elements
relative to one another would be much higher than the tri-peptide
concentration. In this way the
general ability of small peptides to stabilize large proteins without
interfering with protein
CA 02387646 2002-04-15
WO 01/29225 PCT/US00/08477
31
folding may be understood. We believe this phenomenon is not widely
appreciated, and in fact
this may be the first demonstration that a functional protein could be
deliberately stabilized by
something as small as a tri-peptide.
All publications and patent applications mentioned in this specification are
indicative of
the level of skill of those skilled in the art to which this invention
pertains. All publications and
patent applications are herein incorporated by reference to the same extent as
if each individual
publication or patent application was specifically and individually indicated
to be incorporate by
reference.
The invention now having been fully described, it will be apparent to one of
ordinary
m skill in the art that many changes and modifications can be made thereto
without departing from
the spirit or scope of the appended claims.
~5
?o
30