Note: Descriptions are shown in the official language in which they were submitted.
CA 02387660 2002-04-15
WO 01/32602 PCT/US00/30074
DIARYL-ENYNES
The present invention relates to a class of diaryl-enynes, to pharmaceutical
compositions
containing them and to methods of treating neurological and neuropsychiatric
disorders
using such compounds.
Background of the Invention
Synaptic transmission is a complex form of intercellular communication that
involves a
considerable array of specialized structures in both the pre- and post-
synaptic terminal
and surrounding glial cells (Kanner and Schuldiner, CRC Critical Reviews in
Biochemistry, 22, 1987:1032). Transporters sequester neurotransmitter from the
synapse, thereby regulating the concentration of neurotransmitter in the
synapse, as well
as its duration therein, which together influence the magnitude of synaptic
transmission.
Further, by preventing the spread of transmitter to neighbouring synapses,
transporters
maintain the fidelity of synaptic transmission. Lastly, by sequestering
released
transmitter into the presynaptic terminal, transporters allow for transmitter
reutilization.
Neurotransmitter transport is dependent upon extracellular sodium and the
voltage
difference across the membrane; under conditions of intense neuronal firing,
as, for
example, during a seizure, transporters can function in reverse, releasing
neurotransmitter in a calcium-independent non-exocytotic manner (Attwell et
al.,
Neuron, 11, 1993:401-407). Pharmacologic modulation of neurotransmitter
transporters
thus provides a means for modifying synaptic activity, which provides useful
therapy for
the treatment of neurological and psychiatric disturbances.
The amino acid glycine is a major neurotransmitter in the mammalian central
nervous
system, functioning at both inhibitory and excitatory synapses. By nervous
system, both
the central and peripheral portions of the nervous system are intended. These
distinct
functions of glycine are mediated by two different types of receptor, each of
which is
associated with a different class of glycine transporter. The inhibitory
actions of glycine
are mediated by glycine receptors that are sensitive to the convulsant
alkaloid strychnine,
and are thus referred to as ''strychnine-sensitive". Such receptors contain an
intrinsic
CA 02387660 2002-04-15
WO 01/32602 - 2 - PCT/US00/30074
chloride channel that is opened upon binding of glycine to the receptor; by
increasing
chloride conductance, the threshold for firing of an action potential is
increased.
Strychnine-sensitive glycine receptors are found predominantly in the spinal
cord and
brainstem, and pharmacological agents that enhance the activation of such
receptors will
thus increase inhibitory neurotransmission in these regions.
Glycine also functions in excitatory transmission by modulating the actions of
glutamate,
the major excitatory neurotransmitter in the central nervous system (Johnson
and Ascher,
Nature, 325, 1987:529-531; Fletcher et al., Glycine Transmission, Otterson and
Storm-
Mathisen, eds., 1990:193-219). Specifically, glycine is an obligatory co-
agonist at the
class of glutamate receptor termed N-methyl-D-aspartate (NMDA) receptor.
Activation
of NMDA receptors increases sodium and calcium conductance, which depolarizes
the
neuron, thereby increasing the likelihood that it will fire an action
potential.
NMDA receptors in the hippocampal region of the brain play an important role
in a
model of synaptic plasticity known as long-term potentiation (LTP), which is
integral in
certain types of learning and memory (Hebb, D.O (1949) The Organization of
Behavior;
Wiley, NY; Bliss and Collingridge (1993) Nature 361: 31-39; Morris et al.
(1986)
Nature 319: 774-776). Enhanced expression of selected NMDA receptor sub-units
in
transgenic mice results in increased NMDA-receptor-mediated currents, enhanced
LTP,
and better performance in some tests of learning and memory (Tang et al. (
1999) Nature
401: 63).
Conversely, decreased expression of selected NMDA receptor sub-units in
transgenic
mice produces behaviors similar to pharmacologically-induced animal models of
schizophrenia, including increased locomotion, increased stereotypy, and
deficits in
social/sexual interactions (Mohn et al. (1999) Cell 98:427-436). These
aberrant
behaviors can be ameliorated using the antipsychotics haloperidol and
clozapine.
NMDA receptors are widely distributed throughout the brain, with a
particularly high
density in the cerebral cortex and hippocampal formation.
CA 02387660 2002-04-15
WO 01/32602 - 3 - PCT/US00/30074
Molecular cloning has revealed the existence in mammalian brains two classes
of glycine
transporters, termed GIyT-1 and GIyT-2. GIyT-1 is found throughout the brain
and
spinal cord, and it has been suggested that its distribution corresponds to
that of
glutamatergic pathways and NMDA receptors (Smith, et al., Neuron, 8, 1992:927-
935).
Molecular cloning has further revealed the existence of three variants of GIyT-
1, termed
GIyT-1 a, GIyT-1 b and GIyT-1 c. Two of these variants ( 1 a and 1 b) are
found in rodents,
each of which displays a unique distribution in the brain and peripheral
tissues
(Borowsky et al., Neuron, 10, 1993:851-863; Adams et al., J. Neuroscience, 15,
1995:2524-2532). The third variant, lc, has only been detected in human
tissues (Kim,
et al., Molecular Pharmacology, 45, 1994:608-617). These variants arise by
differential
splicing and exon usage, and differ in their N-terminal regions. GIyT-2, in
contrast, is
found predominantly in the brain stem and spinal cord, and its distribution
corresponds
closely to that of strychnine-sensitive glycine receptors (Liu et al., J.
Biological
Chemistry, 268, 1993:22802-22808; Jursky and Nelson, J. Neurochemistry, 64,
1995:1026-1033). Another distinguishing feature of glycine transport mediated
by
GIyT-2 is that it is not inhibited by sarcosine as is the case for glycine
transport mediated
by GIyT-1. These data are consistent with the view that, by regulating the
synaptic
levels of glycine, GIyT-1 and GIyT-2 selectively influence the activity of
NMDA
receptors and strychnine-sensitive glycine receptors, respectively.
Compounds which inhibit or activate glycine transporters would thus be
expected to alter
receptor function and, thus, provide therapeutic benefits in a variety of
disease states.
For example, compounds which inhibit GIyT-1 mediated glycine transport will
increase
glycine concentrations at NMDA receptors, which receptors are located in the
forebrain,
among other locations. This concentration increase elevates the activity of
NMDA
receptors, thereby alleviating schizophrenia and enhancing cognitive function.
Alternatively, compounds that interact directly with the glycine receptor
component of
the NMDA receptor can have the same or similar effects as increasing or
decreasing the
availability of extracellular glycine caused by inhibiting or enhancing GIyT-1
activity,
respectively. See, for example, Pitkanen et al., Eur. J. Pharmacol., 253, 125-
129 (1994);
Thiels et al., Neuroscience, 46, 501-509 (1992); and Kretschmer and Schmidt,
J.
Neurosei., 16, 1561-1569 (1996).
CA 02387660 2002-04-15
WO 01/32602 - 4 - PCT/US00/30074
The present invention provides compounds that affect glycine transport. The
invention
also provides compositions useful to treat medical conditions for which a
glycine
transport modulator, and particularly glycine uptake inhibitors, are
indicated.
Summary of the Invention
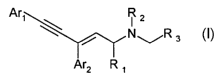
According to one aspect of the invention, there are provided compounds of
Formula I:
Are R 2
\ I
\ / N ~/R s
Ar2 R ~
Formula I
wherein:
Are and Ar2 are independently selected aryl groups, optionally substituted
with up to five
substituents independently selected from the group consisting of alkyl,
alkoxy,
cycloalkyl, cycloalkyloxy, heterocycloalkyl, heterocycloalkyloxy, alkanoyl,
thioalkyl,
aralkyl, aralkyloxy, aryloxyalkyl, aryloxyalkoxy, cycloalkyl-substituted
alkyl,
cycloalkyloxy-substituted alkyl, cycloalkyl-substituted alkoxy, cycloalkyloxy-
substituted
alkoxy, heterocycloalkyl-substituted alkyl, heterocycloalkyloxy-substituted
alkyl
heterocycloalkyl-substituted alkoxy, heterocycloalkyloxy-substituted alkoxy,
thioaryl,
aralkylthio, thioaryl-alky, aralkylthioalkyl, halo, NO2, CF3, CN, OH,
alkylenedioxy,
SOZNRR', NRR', COZR (where R and R' are independently selected from the group
consisting of H and alkyl) and a second aryl group, which may be substituted
as above ;
R, is selected from the group consisting of H and alkyl ;
RZ is selected from the group consisting of H, alkyl and benzyl ;
R3 is selected from the group consisting of COZR, CONRR', CONH(OH), COSR,
SOZNRR', PO(OR)(OR') and tetrazolyl, wherein R and R' are independently
selected
from the group consisting of H and alkyl;
CA 02387660 2002-04-15
WO 01/32602 - 5 - PCT/US00/30074
and a salt, solvate or hydrate thereof.
It has been found that compounds of Formula I inhibit glycine transport (or
reuptake) via
the GIyT-1 transporter, or are precursors (for example, pro-drugs) of such
compounds
and, thus, are useful in the treatment of schizophrenia, as well as other CNS-
related
disorders such as cognitive dysfunction, dementia (including that related to
Alzheimer's
disease), attention deficit disorder and depression.
According to another aspect of the invention, there is provided a
pharmaceutical
composition comprising a compound of Formula I in an amount effective to
inhibit
glycine transport, and a pharmaceutically acceptable carrier.
In another aspect of the invention there are provided compositions containing
the present
compounds in amounts for pharmaceutical use to treat medical conditions for
which a
glycine transport inhibitor is indicated. Preferred are those compositions
containing
compounds useful in the treatment of medical conditions for which GIyT-1-
mediated
inhibition of glycine transport is needed, such as the treatment of
schizophrenia or
cognitive dysfunction.
Definitions
The term aryl as used herein means a monocyclic aromatic group such as phenyl,
pyridyl, furyl, thienyl, and the like, or a benzo-fused aromatic group such as
naphthyl,
indanyl, quinolinyl, fluorenyl and the like.
The term alkyl as used herein means straight- and branched-chain alkyl
radicals
containing from one to six carbon atoms and includes methyl, ethyl and the
like.
The term cycloalkyl as used herein means a carbocyclic ring containing from
three to
eight carbon atoms and includes cyclopropyl, cyclohexyl and the like.
Similarly, the
term "cycloalkyloxy" refers to such a carbocycle that is coupled through an
oxygen to
another group, and includes cyclohexyloxy and the like.
CA 02387660 2002-04-15
WO 01/32602 - 6 - PCT/US00/30074
The term heterocycloalkyl as used herein means a three- to eight-membered ring
containing up to two heteroatoms selected from the group consisting of N, S
and O, and
includes piperidinyl, piperazinyl, thiopyranyl and the like. Such rings
coupled to another
group through an oxygen, such as piperidinyloxy and the like, are referred to
as
heterocycloalkyloxy groups.
The terms aralkyl, aryloxyalkyl, aralkyloxy and aryloxyalkoxy as used herein
refer to an
alkyl or alkoxy radical substituted with an aryl or aryloxy group and includes
benzyl,
phenethyl, benzyloxy, 2-phenoxyethyl and the like. Similarly, the terms
cycloalkyl-
substituted alkyl, cycloalkyl-substituted alkoxy, heterocycloalkyl-substituted
alkyl and
heterocycloalkyl-substituted alkoxy mean groups such as 2-cyclohexyl-ethyl and
the
like. Further, substituents in which an alkyl or alkoxy group is substituted
by another
group through a bridging oxygen, are groups referred to herein as
cycloalkyloxy-
substituted alkyl, cycloalkyloxy-substituted alkoxy, heterocycloalkyloxy-
substituted
alkyl and heterocycloalkyloxy-substituted alkoxy.
The terms alkylene (e.g., -CH2-CH2-), alkenylene (e.g., -CH=CH-) and
alkynylene (e.g.,
-CH---CH-) as used herein means straight- and branched-chain bivalent radicals
containing from one to six carbon atoms, such as methylene, ethylene,
vinylene,
propenylene and ethynylene.
The terms alkylene (e.g., -CH2-CH2-), alkenylene (e.g., -CH=CH-) and
alkynylene (e.g.,
-CH---CH-) as used herein means straight- and branched-chain bivalent radicals
containing from one to six carbon atoms, such as methylene, ethylene,
vinylene,
propenylene and ethynylene.
The term alkoxy as used herein means straight- and branched-chain alkoxy
radicals
containing from one to six carbon atoms and includes methoxy, ethoxy and the
like.
The term thioalkyl as used herein means straight- and branched-chain alkyl
radicals
containing from one to six carbon atoms and includes thiomethyl (CH3-S-),
thiopropyl
and the like.
CA 02387660 2002-04-15
WO 01/32602 - ~ - PCT/US00/30074
The term thioaryl refers to an aryl group that is bridged to another group
through a
sulfur. Similarly, a thioarylalkyl group is a thioaryl group bridged to
another group
through an alkylene group. Also, an aralkythio group is an aralkyl group, such
as benzyl,
which is bridged to another group through a sulfur atom. Further, an
arylalkylthioalkyl
group is an arylalkyl group that is bridged to another group through a
thioalkyl group.
The term alkanoyl as used herein means straight- and branched-chain radicals
containing
from one to six carbon atoms and includes acetyl, propionyl and the like.
The term halo as used herein means halogen and includes fluoro, chloro, bromo
and the
like. The term haloalkyl refers to an alkyl group substituted by one or more
independently selected halo atoms, such as -CF3. Similarly, the term
haloalkoxy refers
to an alkoxy group substituted by one or more independently selected halo
atoms, such
as -OCF3.
The term alkylenedioxy refers to a group of the formula -O-(CH2)n-O-, in which
the
terminal oxygen typically are fused to atoms on an aryl group to form a
bicyclic ring
system, and includes methylenedioxy, ethylenedioxy and the like.
The term hetero atom as used herein means atoms other carbon and includes N, S
and O.
Detailed Description and Preferred Embodiments
The geometry about the double bond of the compounds of Formula I is as drawn.
That
is, group Ar2 and the carbon atom to which group RI is attached are cis to
each other.
Compounds of Formula I include those in which Are and Ar2 are, independently,
optionally-substituted aryl groups.
Substitution sites on rings Are and Ar2 will be limited in practice to the
carbon atoms on
the ring not bound to the core of the molecule. For example, a benzene ring
can be
substituted with up to 5 substituents; pyridine and pyran can accommodate up
to 4
CA 02387660 2002-04-15
WO 01/32602 - g - PCT/US00/30074
substituents pyrole furan and thiophene can accommodate up to 3 substituents;
imidazole
2 substituents and triazole can accommodate only one substituent.
In embodiments of the invention Arl is an optionally monocyclic aromatic group
such as
benzene, pyridine, pyran, thiophene, furan, pyrole, imidazole and triazole.
Arl suitably
accomodates 1, 2 or 3 substituents on the aromatic ring and these can be
chosen from
such groups as alkyl, alkoxy, cycloalkyl, cycloalkyloxy, heterocycloalkyl,
heterocycloalkyloxy, alkanoyl, thioalkyl, aralkyl, aralkyloxy, aryloxyalkyl,
aryloxyalkoxy, cycloalkyl-substituted alkyl, cycloalkyloxy-substituted alkyl,
cycloalkyl-
substituted alkoxy, cycloalkyloxy-substituted alkoxy, heterocycloalkyl-
substituted alkyl,
heterocycloalkyloxy-substituted alkyl heterocycloalkyl-substituted alkoxy,
heterocycloalkyloxy-substituted alkoxy, thioaryl, aralkylthio, thioaryl-alky,
halo, NO2,
CF3, CN, OH, methylenedioxy, ethylenedioxy, S02NRR', NRR', C02R (where R and
R'
are independently selected from the group consisting of H and alkyl) or an
aryl group
optionally substituted as stated above.
In suitable embodiments of the invention, Are is selected from benzene,
pyridine, pyran,
thiophene, furan and pyrole, optionally substituted with 1, 2 or 3
substituents selected
from halo, N02, CF3, CN, OH, alkyl, alkoxy, aryl, aralkyl, and R"(X)n. where n
is 0 or
1; X is CH2 or a heteroatom; and R" is H, alkyl or aryl substituted optionally
with up to
three substituents selected from alkyl, halo, N02, CF3, CN, OH, S02NRR', NRR',
and
C02R (where R and R' are independently selected from the group consisting of H
and
alkyl).
In particular embodiments, Ar1 is phenyl optionally substituted with 1, 2 or 3
substituents
selected from halo, N02, CF3, CN, OH, and R"(X)n. where n is 0 or 1; X is CH2
O, S, or
NR; and R" is H, alkyl or aryl substituted optionally with up to three
substituents
selected independently from alkyl, halo, N02, CF3, CN, OH, SOZNRR', NRR', COZR
(where R and R' are independently selected from the group consisting of H and
alkyl).
In more particular embodiments, Are is phenyl optionally substituted with 1 or
2
substituents selected from alkyl, thioalkyl, alkoxy, halo, haloalkyl,
haloalkoxy,
CA 02387660 2002-04-15
WO 01/32602 - 9 - PCT/US00/30074
substituted or unsubstituted aryl, substituted or unsubstituted aryloxy, and
substituted or
unsubstituted aralkyl.
In specific embodiments, Are is mono-substituted phenyl where the substituent
is
located at the 4 position and is selected from methyl, ethyl, n-propyl, i-
propyl, n-butyl, 3-
furyl, and 3-thienyl.
In other embodiments, Are is an optionally substituted benzofused aromatic
group such
as naphthalene, quinoline, indole, anthracene, fluorenyl, alkylenedioxyphenyl
and the
like, where the substituents can be selected from halo, N02, CF3, CN, OH,
alkyl,
alkoxy, aryl, aralkyl, and R"(X)". where n is 0 or 1; X is CHZ or a
heteroatom; and R" is
H, alkyl or aryl substituted optionally with up to three substituents selected
from alkyl,
halo, N02, CF3, CN, OH, S02NRR', NRR', C02R (where R and R' are independently
selected from the group consisting of H and alkyl).
In particular embodiments, Arl can be naphthyl, quinolinyl, indanyl, or
alkylenedioxyphenyl, optionally substituted with 1 or 2 substituents selected
from alkyl,
alkoxy, thioalkyl and aryl.
In a specific embodiment, Are is selected from unsubstituted naphthalene and
methylenedioxyphenyl.
In other embodiments of the invention, Ar2 is an optionally substituted aryl,
where aryl,
is a monocyclic aromatic group such as benzene, pyridine, pyran, furan,
thiophene,
pyrrolidine and the like, or is a benzofused aromatic ring system such as
naphthalene,
quinoline, indole, anthracene, fluorenyl, alkylenedioxyphenyl and the like.
Either l, 2,
or 3 substituents may be present, and these may be independently selected from
halo,
haloalkyl, alkyl, haloalkoxy, and alkoxy.
In a particular embodiment, A is a monocyclic aromatic ring bearing up to
three
substituents selected independently from halo, haloalkyl, alkyl, haloalkoxy,
and alkoxy.
In more particular embodiments, A is selected from mono or di-substituted
phenyl,
where the substituents are selected from halo, haloalkyl, alkyl, haloalkoxy,
and alkoxy.
CA 02387660 2002-04-15
WO 01/32602 - 1 ~ - PCT/US00/30074
In specific embodiments, Ar2 is a phenyl group that is either unsubstituted or
has one
substituent selected from halo and alkoxy.
In more specific embodiments, Ar2 is selected from unsubstituted or mono
substituted
phenyl, where the substituent is selected from chloro and flouro.
In other embodiments of the invention, R3 is selected from the group
consisting of -
C02R, -CONRR', -CONH(OH), -COSR, -S02NRR', -PO(OR)(OR') and tetrazolyl,
wherein R and R' are independently selected from the group consisting of H and
alkyl.
In particular embodiments, R3 is COOR. In preferred embodiments of the
invention, R3
is COON.
The compounds of Formula I include those in which R1 is selected from the
group
consisting of H and alkyl. Preferably, R, is H.
The compounds of Formula I include those in which R~ is selected from the
group
consisting of H, alkyl and benzyl. Suitably, RZ alkyl; more preferably, R2 is
methyl.
In preferred embodiments, compounds of Formula I are those in which R~ is H,
R2 is
methyl, R3 is COOH. In this context, Are and Ar2 are desirably substituted or
unsubstituted phenyl. Preferably, Are is either phenyl or 4-(substituted)-
phenyl. When
substituted, Arl is desirably a 4-(alkyl)-phenyl group, particularly where the
alkyl group
is a straight-chain alkyl group, including 4-isopropyl-phenyl, 4-ethyl-phenyl,
and 4-n-
propyl-phenyl. Either in combination therewith or independently thereof, Ar2
is
preferably is chloro or fluoro substituted phenyl.
In another preferred embodiment, R~ is H, RZ is methyl, R3 is COOH, Ar2 is
unsubstituted phenyl and Are is 4-alkyl substituted phenyl where alkyl is C»
straight
chain.
CA 02387660 2002-04-15
WO 01/32602 - 11 - PCT/US00/30074
In another preferred embodiment R~ is H, R2 is methyl, R3 is COOH, Ar2 is 2-
chloro-
phenyl and Arl is 4-alkyl phenyl where the alkyl substituent is selected from
ethyl and
propyl.
In another preferred embodiment of the invention R~ is H, R2 is methyl, R3 is
COOH, Arl
is naphthyl, especially 2-naphthyl, and Ar2 is phenyl.
In yet another preferred embodiment of the invention Rl is H, R2 is methyl, R3
is COON,
Are is 3,4-methylenedioxyphenyl and Ar2 is 3-fluoro-phenyl.
In still another preferred embodiment of the invention R~ is H, R2 is methyl,
R3 is
COOH, Ar2 is phenyl and Are is an optionally substituted aryl substituted
phenyl.
In a more preferred embodiment of the invention R~ is H, RZ is methyl, R3 is
COOH, Ar2
is phenyl and Ar, is phenyl substituted by a 5-membered heteroaryl that is
optionally
substituted.
In a most preferred embodiment of the invention R1 is H, R2 is methyl, R3 is
COOH, Ar2
is phenyl and Ar, is 4-(3-furyl)phenyl.
Specific compounds of Formula I include:
N-(5-(4-Fluorophenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine
N-(5-(2-Fluorophenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine
N-(5-(2,4-Difluorophenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine
N-(5-(3-Nitrophenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine
N-(5-(4-Nitrophenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine
N-(3-Phenyl-5-(2-thiomethylphenyl)-2-penten-4-yn-1-yl)-sarcosine
N-(5-(4-Chlorophenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine
N-(5-(4-Isopropylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine,
N-(5-(3,5-Bis(trifluoromethyl)phenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine
N-(3,5-biphenyl-2-penten-4-yn-1-yl)-sarcosine
N-(5-(4-diphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine
N-(5-(4-trifluoromethylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine
CA 02387660 2002-04-15
WO 01/32602 - 12 - PCT/US00/30074
N-(S-(4-benzylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine
N-(5-(4-ethylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine
N-(5-(4-°propylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine
N-(5-(4-°butylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine
N-(5-(4-°pentylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine
N-(5-(4-phenoxyphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine
N-(5-( 1-naphthyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine
N-(5-(4-methyphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine
N-(5-(3-isopropylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine
N-(5-(2-naphthyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine
N-(5-(3,4-dimethylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine
N-(5-(2-isopropylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine
N-(5-(3,4-methylenedioxyphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine
N-(5-(4-pyrrolylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine
N-(5-(4-trifluoromethoxyphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine
N-(5-(3,4-dimethoxyphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine
N-(3-Phenyl-5-(4-thiomethylphenyl)-2-penten-4-yn-1-yl)-sarcosine
N-(5-(4-Methylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine
N-(3-Phenyl-5-(3-thiophene)-2-penten-4-yn-1-yl)-sarcosine
N-(3-Phenyl-5-(4-tbutylphenyl)-2-penten-4-yn-1-yl)-sarcosine
N-(5-(4-(3-furyl)-phenyl)-3-phenyl-2-penten-4yn-1-yl)-sarcosine
N-(5-(4-(3-thiophene)-phenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine
N-(5-(4-Isopropylphenyl)-3-(4-(trifluoromethyl)phenyl)-2-penten-4-yn-1-yl)-
sarcosine
N-(5-(4-Isopropylphenyl)-3-(4-fluorophenyl)-2-penten-4-yn-1-yl)-sarcosine
N-(5-(4-Isopropylphenyl)-3-(2-fluorophenyl)-2-penten-4-yn-1-yl)-sarcosine
N-(5-(4-t-Butylphenyl)-3-(2-fluorophenyl)-2-penten-4-yn-1-yl)-sarcosine
N-(5-(4-Isopropylphenyl)-3-(4-chlorophenyl)-2-penten-4-yn-1-yl)-sarcosine
N-(5-(4-t-Butylphenyl)-3-(4-chlorophenyl)-2-penten-4-yn-1-yl)-sarcosine
N-(5-(4-Isopropylphenyl)-3-(2-chlorophenyl)-2-penten-4-yn-1-yl)-sarcosine
N-(5-(4-t-Butylphenyl)-3-(2-chlorophenyl)-2-penten-4-yn-1-yl)-sarcosine
N-(5-(4-Isopropylphenyl)-3-(3-fluorophenyl)-2-penten-4-yn-1-yl)-sarcosine
N-(5-(4-Isopropylphenyl)-3-(3-thienyl)-2-penten-4-yn-1-yl)-sarcosine
N-(5-(4-Isopropylphenyl)-3-(4-methoxyphenyl)-2-penten-4-yn-1-yl)-sarcosine
CA 02387660 2002-04-15
WO 01/32602 - 13 - PCT/US00/30074
N-(5-(3,4-Methylenedioxyphenyl)-3-(3-fluorophenyl)-2-penten-4-yn-1-yl)-
sarcosine
N-(5-(4-Ethylphenyl)-3-(2-chlorophenyl)-2-penten-4-yn-1-yl)-sarcosine
N-(5-(4-Propylphenyl)-3-(2-chlorophenyl)-2-penten-4-yn-1-yl)-sarcosine
Compounds of Formula I can be considered to be amino acids or derivatives
thereof.
Compounds which contain, instead of a carboxylate group, a "carboxylate
equivalent"
group, such as hydroxamic acids, phosphonic acids, phosphinic acids, sulfonic
acids,
sulfinic acids, amides or tetrazoles, are also considered embodiments of the
present
invention.
In another embodiment of the invention, the compound of Formula I is provided
in
labeled form, such as radiolabeled form (e.g. labeled by incorporation within
its structure
3H or'4C or by conjugation to ~25I). In a preferred aspect of the invention,
such
compounds, which bind preferentially to GIyT-1, can be used to identify GIyT-1
receptor
ligands by techniques common in the art. This can be achieved by incubating
the
receptor or tissue in the presence of a ligand candidate and then incubating
the resulting
preparation with an equimolar amount of radiolabeled compound of the
invention.
GIyT-1 receptor ligands are thus revealed as those that significantly occupy
the GIyT-1
site and prevent binding of the radiolabeled compound of the present
invention.
Alternatively, GIyT-1 receptor ligand candidates may be identified by first
incubating a
radiolabeled form of a compound of the invention then incubating the resulting
preparation in the presence of the candidate ligand. A more potent GIyT-1
receptor
ligand will, at equimolar concentration, displace the radiolabeled compound of
the
invention.
Acid addition salts of the compounds of Formula I are most suitably formed
from
pharmaceutically acceptable acids, and include for example those formed with
inorganic
acids e.g. hydrochloric, sulphuric or phosphoric acids and organic acids e.g.
succinic,
malefic, acetic or fumaric acid. Other non-pharmaceutically acceptable salts
e.g. oxalates
may be used for example in the isolation of compounds of Formula I for
laboratory use,
or for subsequent conversion to a pharmaceutically acceptable acid addition
salt. Also
included within the scope of the invention are base addition salts (such as
sodium,
CA 02387660 2002-04-15
WO 01/32602 - 14 - PCT/US00/30074
potassium and ammonium salts), solvates and hydrates of compounds of the
invention.
Base salts are preferred and sodium and potassium salts are especially
preferred.
The conversion of a given compound salt to a desired compound salt is achieved
by
applying standard techniques, well known to one skilled in the art.
The compounds of the present invention can be prepared by processes analogous
to those
established in the art. For example, compounds of Formula I are readily
prepared by the
method shown in Scheme l, below. Intermediate C was prepared according to the
method of Trost (Trost, B. M.; Sorum, M. T.; Chan, C.; Harms, A. E.; Ruther,
G. J. Am.
Chem. Soc. 1997,119, 698-708 ; Trost, B. M.; Hachiya, L; McIntosh, M. C.
Tetrahedron
Lett. 1998, 39, 6445-6448) by coupling an arylpropiolic ester such as A with
trimethylsilylacetylene B in the presence of palladium acetate and tris(2,6-
dimethoxyphenyl)phosphine. Reduction of the ester to the alcohol, and
treatment with
N-Bromosuccinimide gave bromide D. Treatment of D with a sarcosine ester (such
as
tbutyl sarcosine) in the presence of base gave the intermediate sarcosine
derivative E.
Removal of the trimethylsilyl group (for example, by treatment with potassium
carbonate
in methanol), followed by introduction of the second aryl group by a
Sonogashira
coupling (Sonogashira, K.; Yohda, Y. and Hagihara, N.; Tetrahedron Lett.,
1975, 4467),
gave the diaryl species G which, upon deprotection with, for example, formic
acid, gave
the final product H.
This route is an attractive one for the parallel synthesis of a series of
related compounds
in which group Ar2 is constant, but group Ar, represents a number of different
aryl
groups. Common intermediate F can be prepared in bulk, and simply treated with
the
appropriate aryliodide under Sonogashira conditions to yield the desired
products.
CA 02387660 2002-04-15
WO 01/32602 _ 15 ~ PCT/US00/30074
OZMe H OsMe Br
I '~ I
Ar2 ~ Ar2
Ar2 SiMe3 Me3Si Me3Si
C D
p B
iii
I i ~cozt-B~ " N~co2t-Bu N~co t-Bu
Ar ~ I I iv I ' I z
Ar ~ 2 / Ar2 ~ ~ Ar2
G H
p Me3Si
vii
i ~COzH
'Arz
Are
Reagents : (i) Pd(OAc)2 , phosphine ligand ; (ii) (a) DIBAL-H , (b) NBS , PPh3
; (iii) t-Bu sarcosine , KZC03 , KI ; (iv) KZC03, MeOH ;
(v) Ar,-I , Cul , Pd(PPh3), , Et3N ; (vi) formic acid , 50°C
Scheme 1
Alternatively, such compounds may also be prepared according to the route
shown in
Scheme 2, below. This route complements that shown above, in that it allows
the
parallel synthesis of a series of related compounds in which group Arl is
constant, but
group Ar2 represents a number of different aryl groups. In this case, common
intermediate L can be prepared in bulk, and simply treated with the
appropriate
arylpropiolic ester O (readily accessible from aryliodide M by treatment with
propiolic
ester N in the presence of Cul and Pd(PPh3)4), under the conditions outlined
above, to
yield, after deprotection, products H.
CA 02387660 2002-04-15
WO 01/32602 - I 6 - PCT/US00/30074
i TMs ii
Ark I + = TMS ---~
Are Are
I J K L
i iii
Ar2 I + - COZEt ~ Arz - COzEt
M N O
Br OH COzEt
v ~ iv
Ar2 ~ / Ar2 / Arz
Are R Are Q Are
vi
N ~ CO2tBu N ~ CO2H
I Ar2 I vu ...~ / I Arz
Are Are
G H
Reagents : (i) Cul, Pd(PPh3), , Et3N ; (ii) KzC03 , MeOH ; (iii) Pd(OAc)2 ,
phosphine ligand, PhMe ;
(iv) DIBAL-H , PhMe, -78~C ; (v) NBS , PPh3; (vi) t-Bu sarcosine , KZC03 , KI
, MeCN : (vii) formic acid , SO~C
Scheme 2
To prepare compounds in which Arl is Aryl-substituted phenyl (Ar3-phenyl), the
following synthesis (Scheme 3) is useful. Intermediate F can be prepared
according to
Scheme 1, then coupled to bromoiodobenzene via Sonogashira coupling to yield
species
S. The arylbromide of species S can then be reacted with a boronic acid (Ar3-
boronic
acid) under Suzuki coupling conditions to give intermediate G'. (G' is
equivalent to G,
Scheme 1, Where Artis Ar3-phenyl). G' can then be deprotected as in Scheme 1
to give
H' .
CA 02387660 2002-04-15
WO 01/32602 - 1 ~ - PCT/LTS00/30074
~N~COztBu CO tBu
z
ii
Arz -.
H
F
a
~COzli
rz
H'
(i) 4-bromoiodobenune, Pd(PPh3),, CuI, Et3N, r.t, overnight; (ii) Ar3-boronic
acid, Pd(PPh3),, 2M
NazCO~, DME, 110 °C, 1 hour, (iii) formic acid, 40 °C,
overnight.
Scheme 3
Compounds which inhibit GIyT-1 mediated glycine transport will increase
glycine
S concentrations at NMDA receptors, which receptors are located in the
forebrain, among
other locations. This concentration increase elevates the activity of NMDA
receptors,
thereby alleviating schizophrenia and enhancing cognitive function.
Alternatively,
compounds that interact directly with the glycine receptor component of the
NMDA
receptor can have the same or similar effects as increasing or decreasing the
availability
of extracellular glycine caused by inhibiting or enhancing GIyT-1 activity,
respectively.
See, for example, Pitkanen et al., Eur. J. Pharmacol" 253, 125-129 (1994);
Thiels et al.,
Neuroscience, 46, 501-509 (1992); and Kretschmer and Schmidt, J. Neurosci.,
16, 1561-
1569 (1996).
The compounds of the invention are, for instance, administered orally,
sublingually,
rectally, nasally, vaginally, topically (including the use of a patch or other
transdermal
CA 02387660 2002-04-15
WO 01/32602 - 18 - PCT/US00/30074
delivery device), by pulmonary route by use of an aerosol, or parenterally,
including, for
example, intramuscularly, subcutaneously, intraperitoneally, intraarterially,
intravenously or intrathecally. Administration can be by means of a pump for
periodic or
continuous delivery. The compounds of the invention are administered alone, or
are
combined with a pharmaceutically-acceptable carrier or excipient according to
standard
pharmaceutical practice. For the oral mode of administration, the compounds of
the
invention are used in the form of tablets, capsules, lozenges, chewing gum,
troches,
powders, syrups, elixirs, aqueous solutions and suspensions, and the like. In
the case of
tablets, carriers that are used include lactose, sodium citrate and salts of
phosphoric acid.
Various disintegrants such as starch, and lubricating agents such as magnesium
stearate
and talc, are commonly used in tablets. For oral administration in capsule
form, useful
diluents are lactose and high molecular weight polyethylene glycols. If
desired, certain
sweetening and/or flavoring agents are added. For parenteral administration,
sterile
solutions of the compounds of the invention are usually prepared, and the pHs
of the
solutions are suitably adjusted and buffered. For intravenous use, the total
concentration
of solutes should be controlled to render the preparation isotonic. For ocular
administration, ointments or droppable liquids may be delivered by ocular
delivery
systems known to the art such as applicators or eye droppers. Such
compositions can
include mucomimetics such as hyaluronic acid, chondroitin sulfate,
hydroxypropyl
methylcellulose or polyvinyl alcohol, preservatives such as sorbic acid, EDTA
or
benzylchromium chloride, and the usual quantities of diluents and/or carriers.
For
pulmonary administration, diluents and/or carriers will be selected to be
appropriate to
allow the formation of an aerosol.
Suppository forms of the compounds of the invention are useful for vaginal,
urethral and
rectal administrations. Such suppositories will generally be constructed of a
mixture of
substances that is solid at room temperature but melts at body temperature.
The
substances commonly used to create such vehicles include theobroma oil,
glycerinated
gelatin, hydrogenated vegetable oils, mixtures of polyethylene glycols of
various
molecular weight and fatty acid esters of polyethylene glycol. See,
Remington's
Pharmaceutical Sciences, 16th Ed., Mack Publishing, Easton, PA, 1980, pp. 1530-
1533
for further discussion of suppository dosage forms. Analogous gels or creams
can be
used for vaginal, urethral and rectal administrations.
CA 02387660 2002-04-15
WO 01/32602 - 19 - PCT/US00/30074
Numerous administration vehicles will be apparent to those of ordinary skill
in the art,
including without limitation slow release formulations, liposomal formulations
and
polymeric matrices.
Examples of pharmaceutically acceptable acid addition salts for use in the
present
invention include those derived from mineral acids, such as hydrochloric,
hydrobromic,
phosphoric, metaphosphoric, nitric and sulfuric acids, and organic acids, such
as tartaric,
acetic, citric, malic, lactic, fumaric, benzoic, glycolic, gluconic, succinic,
p-
toluenesulphonic and arylsulphonic acids, for example. Examples of
pharmaceutically
acceptable base addition salts for use in the present invention include those
derived from
non-toxic metals such as sodium or potassium, ammonium salts and organoamino
salts
such as triethylamine salts. Numerous appropriate such salts will be known to
those of
ordinary skill.
The physician or other health care professional can select the appropriate
dose and
treatment regimen based on the subject's weight, age, and physical condition.
Dosages
will generally be selected to maintain a serum level of compounds of the
invention
between about 0.01 ~g/cc and about 1000 ~g/cc, preferably between about 0.1
~g/cc and
about 100 ~g/cc. For parenteral administration, an alternative measure of
preferred
amount is from about 0.001 mg/kg to about 10 mg/kg (alternatively, from about
0.01
mg/kg to about 10 mg/kg), more preferably from about 0.01 mg/kg to about 1
mg/kg
(from about 0.1 mg/kg to about 1 mg/kg), will be administered. For oral
administrations,
an alternative measure of preferred administration amount is from about 0.001
mg/kg to
about 10 mg/kg (from about 0.1 mg/kg to about 10 mg/kg), more preferably from
about
0.01 mg/kg to about 1 mg/kg (from about 0.1 mg/kg to about 1 mg/kg). For
administrations in suppository form, an alternative measure of preferred
administration
amount is from about 0.1 mg/kg to about 10 mg/kg, more preferably from about
0.1
mg/kg to about 1 mg/kg.
For use in assaying for activity in inhibiting glycine transport, eukaryokic
cells,
preferably QT-6 cells derived from quail fibroblasts, have been transfected to
express
CA 02387660 2002-04-15
WO 01/32602 - 20 - PCT/US00/30074
one of the three known variants of human GIyT-1, namely GIyT-la, GIyT-lb or
GIyT-
1 c, or human GIyT-2. The sequences of these GIyT-1 transporters are described
in Kim
et al., ll~lolec. Pharm. 45: 608-617, 1994, excepting that the sequence
encoding the
extreme N-terminal of GIyT-1 a was merely inferred from the corresponding rat-
derived
sequence. This N-terminal protein-encoding sequence has now been confirmed to
correspond to that inferred by Kim et al. The sequence of the human GIyT-2 is
described
by Albert et al., U.S. Application No. 08/700,013, filed August 20, 1996,
which is
incorporated herein by reference in its entirety. Suitable expression vectors
include
pRc/CMV (Invitrogen), Zap Express Vector (Stratagene Cloning Systems, LaJolla,
CA;
hereinafter "Stratagene"), pBk/CMV or pBk-RSV vectors (Stratagene), Bluescript
II SK
+/- Phagemid Vectors (Stratagene), LacSwitch (Stratagene), pMAM and pMAM neo
(Clontech), among others. A suitable expression vector is capable of fostering
expression of the included GIyT DNA in a suitable host cell, preferably a non-
mammalian host cell, which can be eukaryotic, fungal, or prokaryotic. Such
preferred
host cells include amphibian, avian, fungal, insect, and reptilian cells.
Examples
Example 1: 1-Methoxycarbonyl-2-phenyl-4-trimethylsilyl-1-buten-4-yne (C).
To a solution of palladium acetate (28 mg, 0.125 mmol) in anhydrous toluene (5
mL)
was added tris(2,6-dimethoxyphenyl)phosphine (55 mg, 0.125 mmol). After 15
minutes
a solution of methyl phenylpropiolate (1.00 g, 6.24 mmol) in anhydrous toluene
(5 mL)
was added. After an additional 5 minutes trimethylsilylacetylene (0.88 mL,
0.61 g, 6.24
mmol) was added. After 16 hours the reaction mixture was concentrated. Column
chromatography (10% ethyl acetate/hexanes) provided enyne C (1.39 g, 86%) as a
yellow oil. C: 1H NMR (CDC13, 300 MHz) 0.21 (s, 9H), 3.62 (s, 3H), 6.34 (s,
1H),
7.33-7.44 (m, SH).
Example 2: 1-Hydroxy-3-phenyl-5-trimethylsilyl-2-penten-4-yne.
A solution of the ester C (1.30 g, 5.03 mmol) in anhydrous toluene (20 mL) was
chilled
in a dry ice/acetone bath. A 1.0 M solution of diisobutylaluminum hydride in
toluene
( 12.6 mL, 12.6 mmol) was added. After 5 minutes the chilling bath was
removed. After
a further 15 minutes the reaction mixture was re-chilled in an ice bath. The
reaction was
quenched with the addition of celite and sodium sulphate decahydrate. The
slurry was
CA 02387660 2002-04-15
WO 01/32602 - 21 - PCT/US00/30074
diluted with ethyl acetate and filtered through celite. The filter cake was
washed 3 times
with ethyl acetate. The filtrate was washed with water and brine, dried
(sodium
sulphate), filtered, and concentrated to provide the intermediate alcohol
(0.821 g, 71 %)
as a yellow oil. : 'H NMR (CDC13, 300 MHz) 0.20 (s, 9H), 1.40 (t, 1 H), 4.31
(dd, 2H),
6.37 (t, 1H), 7.33-7.37 (m, SH).
Example 3: 1-Bromo-3-phenyl-5-trimethylsilyl-2-penten-4-yne (D).
A solution of the above alcohol (0.82 g, 3.56 mmol) in anhydrous
dichloromethane (20
mL) was chilled in a dry ice /acetonitrile bath. Triphenylphosphine (1.40 g,
5.34 mmol)
and N-bromosuccinimide (0.95 g, 5.34 mmol) were added. After 30 minutes the
reaction
was quenched with saturated sodium bicarbonate. The reaction mixture was
partitioned
between saturated sodium bicarbonate and dichloromethane. The organic phase
was
washed with brine, dried (sodium sulphate), filtered, and concentrated to
provide crude
allylic bromide D, used directly in the next step.
Example 4: N-(3-Phenyl-5-(trimethylsilyl)-2-penten-4-yn-1-yl)-sarcosine,
tbutyl
ester (E).
To a solution of the above bromide in anhydrous acetonitrile ( 15 mL) was
added t-butyl
sarcosine hydrochloride (0.71 g, 3.90 mmol), potassium carbonate (4.91 g, 35.5
mmol),
and potassium iodide (2.95 g, 17.8 mmol). After 16 hours the reaction mixture
was
filtered through celite. The filter cake was washed with ethyl acetate. The
filtrate was
poured into water and extracted with ethyl acetate. The organic phase was
washed with
water and brine, dried (sodium sulphate), filtered, and concentrated. Column
chromatography (25% ethyl acetate/hexanes) provided product E (0.74 g, 58%
over 2
steps) as a pale yellow oil. E: 'H NMR (CDC13, 300 MHz) 0.19 (s, 9H), 1.41 (s,
9H),
2.32 (s, 3H), 3.10 (s, 2H), 3.31 (d, 2H), 6.33 (t, 1H), 7.26-7.38 (m, SH).
Example 5: N-(3-Phenyl-2-penten-4-yn-1-yl)-sarcosine, tbutyl ester (F).
To a solution of the above compound (0.74 g, 2.06 mmol) in methanol ( 10 mL)
was
added potassium carbonate ( 1.42 g, 10.3 mmol). After 20 minutes the reaction
mixture
was poured into water and extracted 2 times with ethyl acetate. The combined
organic
extracts were washed with brine, dried (sodium sulphate), filtered, and
concentrated to
provide terminal acetylene F (0.58 g, 99%) as an off white solid. F: 'H NMR
(CDC13,
CA 02387660 2002-04-15
WO 01/32602 - 22 - PCT/US00/30074
300 MHz) 1.41 (s, 9H), 2.33 (s, 3H), 2.96 (s, 1H), 3.10 (s, 2H), 3.33 (d, 2H),
6.37 (t, 1H),
7.26-7.39 (m, SH).
Example 6-l: N-(5-(4-Fluorophenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine,
tbutyl
ester, (G).
To a solution of the terminal acetylene F (50 mg, 0.175 mmol) in triethylamine
(2 mL)
was added 4-fluoroiodobenzene (26 ~.L, 51 mg, 0.228 mmol),
tetrakis(triphenylphosphine)palladium(0) (20 mg, 0.0175 mmol), and copper(I)
iodide
(10 mg, 0.0525 mmol). After 16 hours the reaction mixture was diluted with
dichloromethane and filtered. The filtrate was concentrated. Column
chromatography
(25% ethyl acetate/hexanes) provided acetylene G (51 mg, 77%) as a yellow oil.
G: 'H
NMR (CDCl3, 300 MHz) 1.42 (s, 9H), 2.35 (s, 3H), 3.13 (s, 2H), 3.36 (d, 2H),
6.37 (t,
1 H), 7.00 (dd, 2H), 7.26-7.44 (m, 7H).
In a similar fashion the following compounds were prepared from intermediate F
and 1.3
equivalents of the corresponding aryliodide treated under the conditions
described
above.:
6-2: N-(5-(2-Fluorophenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, tbutyl
ester, (G).
Prepared in a similar fashion from 2-fluoroiodobenzene to provide 45 mg (68%)
of a
yellow oil. 'H NMR (CDCl3, 300 MHz) 1.42 (s, 9H), 2.36 (s, 3H), 3.14 (s, 2H),
3.39 (d,
2H), 6.43 (t, 1H), 7.06 (dd, 2H), 7.24-7.44 (m, 7H).
6-3: N-(5-(2,4-Difluorophenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, butyl
ester,
(G).
Prepared in a similar fashion from 2,4-difluoroiodobenzene to provide 49 mg
(70%) of a
yellow oil. 1H NMR (CDCI;, 300 MHz) 1.42 (s, 9H), 2.36 (s, 3H), 3.13 (s, 2H),
3.38 (d,
2H), 6.42 (t, 1 H), 6.83 (dd, 2H), 7.26-7.44 (m, 6H).
6-4: N-(5-(3-Nitrophenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, 'butyl
ester, (G).
Prepared in a similar fashion from 3-nitroiodobenzene to provide 73 mg (102%)
of a
yellow oil. 1H NMR (CDCI;, 300 MHz) 1.42 (s, 9H), 2.36 (s, 3H), 3.13 (s, 2H),
3.38 (d,
CA 02387660 2002-04-15
WO 01/32602 - 23 - PCT/US00/30074
2H), 6.45 (t, 1 H), 7.26-7.40 (m, SH), 7.48 (dd, 1 H), 7.72 (d, 1 H), 8.13 (d,
1 H), 8.27 (s,
1 H).
6-5: N-(5-(4-Nitrophenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, butyl ester,
(G).Prepared in a similar fashion from 4-nitroiodobenzene to provide 31 mg
(44%) of a
yellow oil. 1H NMR (CDCl3, 300 MHz) 1.42 (s, 9H), 2.36 (s, 3H), 3.14 (s, 2H),
3.38 (d,
2H), 6.47 (t, 1H), 7.34-7.43 (m, SH), 7.57 (d, 2H), 8.17 (d, 2H).
6-6: N-(3-Phenyl-5-(2-thiomethylphenyl)-2-penten-4-yn-1-yl)-sarcosine, tbutyl
ester,
(G).
Prepared in a similar fashion from 2-thiomethyliodobenzene to provide 19 mg
(26%) of a
yellow oil. 'H NMR (CDCl3) 1.42 (s, 9H), 2.36 (s, 3H), 2.46 (s, 3H), 3.14 (s,
2H), 3.39
(d, 2H), 6.45 (t, 1 H), 7.06 (dd, 1 H), 7.14 (d, 1 H), 7.24-7.40 (m, 6H), 7.46
(d, 1 H).
6-7: N-(5-(4-Chlorophenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, tbutyl
ester,
(G).
Prepared in a similar fashion from 4-chloroiodobenzene to provide 52 mg (75%)
of a
yellow oil. 'H NMR (CDC13, 300 MHz) 1.42 (s, 9H), 2.35 (s, 3H), 3.13 (s, 2H),
3.36 (d,
2H), 6.38 (t, 1H), 7.26-7.39 (m, 9H).
6-8: N-(5-(4-Isopropylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, butyl
ester,
(G).
Prepared in a similar fashion from 4-isopropyliodobenzene to provide 38 mg
(53%) of a
yellow oil. 'H NMR (CDC13, 300 MHz) 1.23 (d, 6H), 1.42 (s, 9H), 2.36 (s, 3H),
2.89
(hept, 1H), 3.13 (s, 2H), 3.36 (d, 2H), 6.36 (t, 1H), 7.16 (d, 2H), 7.26-7.42
(m, 7H).
6-9: N-(5-(3,5-Bis(trifluoromethyl)phenyl)-3-phenyl-2-penten-4-yn-1-yl)-
sarcosine,
tbutyl ester, (G).
Prepared in a similar fashion from 3,5-Bis(trifluoromethyl)iodobenzene to
provide 40 mg
(46%) of a yellow oil. 'H NMR (CDC13, 300 MHz) 1.42 (s, 9H), 2.36 (s, 3H),
3.14 (s,
2H), 3.38 (d, 2H), 6.47 (t, 1H), 7.26-7.44 (m, SH), 7.77 (s, 1H), 7.86 (s,
2H).
CA 02387660 2002-04-15
WO 01/32602 - 24 - PCT/US00/30074
6-10: N-(3,5-biphenyl-2-penten-4-yn-1-yl)-sarcosine, 'butyl ester, (G).
Prepared in a similar fashion from iodobenzene to provide 46 mg (33%) of a
yellow oil.
'H NMR (CDCl3, 300 MHz) 1.42 (s, 9H), 2.36 (s, 3H), 3.13 (s, 2H), 3.36 (d,
2H), 6.38 (t,
1 H), 7.26-7.46 (m, 1 OH).
6-11: N-(3-Phenyl-5-(4-thiomethylphenyl)-2-penten-4-yn-1-yl)-sarcosine, 'butyl
ester, (G).
Prepared in a similar fashion from 4-thiomethyliodobenzene to provide 30.0 mg
(70%)
of a yellow oil.
6-12: N-(3-Phenyl-5-(4-methylphenyl)-2-penten-4-yn-1-yl)-sarcosine, 'butyl
ester,
(G)
Prepared in a similar fashion from 4-methyliodobenzene to provide 33.0 mg
(85%) of a
yellow oil.
6-13: N-(5-(3-Thiophene)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, 'butyl ester,
(G).
Prepared in a similar fashion from 3-iodothiophene to provide 30.0 mg (78%) of
a brown
oil.
6-14: N-(3-Phenyl-5-(4-tbutylphenyl)-2-penten-4-yn-1-yl)-sarcosine, 'butyl
ester,
(G).
Prepared in a similar fashion from 4-t-butyliodobenzene to provide 38.0 mg
(86%) of a
yellow oil.
6-15: N-(5-(4-Methoxyphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, 'butyl
ester,
(G).
Prepared in a similar fashion from 4-methoxyiodobenzene to provide 31.0 mg
(73%) of a
yellow oil.
6-16: N-(5-(2-Isopropylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, 'butyl
ester,
(G).
Prepared in a similar fashion from 2-isopropyliodobenzene to provide 27.0 mg
(64%) of
an amber oil.
CA 02387660 2002-04-15
WO 01/32602 - 25 - PCT/US00/30074
6-17: N-(5-(4-diphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, tbutyl ester,
(G).
Prepared in a similar fashion from 4-biphenyliodobenzene to provide 260 mg
(85%) of a
yellow oil.
6-18: N-(5-(4-trifluoromethylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine,
butyl
ester, (G).
Prepared in a similar fashion from 4-trifluoromethyliodobenzene to provide 240
mg
(80%) of a yellow oil.
6-19: N-(5-(4-benzylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, 'butyl
ester,
(G).
Prepared in a similar fashion from 4-benzyliodobenzene to provide 240 mg (80%)
of a
light yellow oil.
6-20: N-(5-(4-ethylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, tbutyl
ester, (G).
Prepared in a similar fashion from 4-ethyliodobenzene to provide 240 mg (88%)
of
product. 6-20: 'H NMR (CDCI3, 300 MHz) 1.22 (t, 3H),1.43 (s, 9H), 2.36 (s,
3H),
2.63(q, 2H), 3.13 (s, 2H), 3.36 (d, 2H), 6.37 (t, 1H), 7.13 (d, 2H), 7.26-7.43
(m, 7H).
6-21: N-(5-(4-npropylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, tbutyl
ester,
(G).
Prepared in a similar fashion from 4-n-propyliodobenzene to provide 240 mg
(85%) of
product. 6-21: 1H NMR (CDC13, 300 MHz) 0.93 (t, 3H),1.43 (s, 9H), 1.57
(sextet, 2H),
2.36 (s, 3H), 2.57 (t, 2H), 3.14 (s, 2H), 3.37 (d, 2H), 6.37 (t, 1H), 7.12 (d,
2H), 7.24-7.43
(m, 7H).
6-22: N-(5-(4-nbutylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, tbutyl
ester,
(G).
Prepared in a similar fashion from 4-n-butyliodobenzene to provide 260 mg
(89%) of a
yellow oil.
CA 02387660 2002-04-15
WO 01/32602 - 26 - PCT/US00/30074
6-23: N-(5-(4-npentylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, tbutyl
ester,
(G).
Prepared in a similar fashion from 4-n-pentyliodobenzene to provide 240 mg
(79%) of a
yellow oil.
6-24: N-(5-(4-phenoxyphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, tbutyl
ester,
(G).
Prepared in a similar fashion from 4-phenoxyiodobenzene to provide 34.7 mg
(56%) of a
yellow film.
6-25: N-(5-(1-naphthyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, tbutyl ester,
(G).
Prepared in a similar fashion from 1-iodonaphthalene to provide 35.8 mg
(63.5%) of
product. 6-25: 'H NMR (CDC13, 300 MHz) 1.43 (s, 9H), 2.40 (s, 3H), 3.17 (s,
2H), 3.42
(d, 2H), 6.53 (t, 1H), 7.33-7.57 (m, 8H), 7.67 (d, 1H), 7.75-7.85 (m, 2H),
8.30 (d, 1H)
6-26: N-(5-(4-methyphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, tbutyl
ester,
(G).
Prepared in a similar fashion from 4-methyliodobenzene to provide 34.7 mg
(88%) of a
light yellow oil.
6-27: N-(5-(3-isopropylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, tbutyl
ester,
(G).
Prepared in a similar fashion from 3-isopropyliodobenzene to provide 17.6 mg
(42%) of
product. 6-27: 'H NMR (CDC13, 300 MHz) 1.23 (d, 6H),1.42 (s, 9H), 2.36 (s,
3H), 2.87
(septtet, 1H), 3.13 (s, 2H), 3.36 (d, 2H), 6.38 (t, 1H), 7.15-7.42 (m, 8H),
7.70-7.71 (m,
1 H)
6-28: N-(5-(2-naphthyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, 'butyl ester,
(G).
Prepared in a similar fashion from 2-iodonaphthalene to provide 30.0 mg (69%)
of a
colourless oil.
CA 02387660 2002-04-15
WO 01/32602 - 27 - PCT/LTS00/30074
6-29: N-(5-(3,4-dimethylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, tbutyl
ester, (G).
Prepared in a similar fashion from 3,4-dimethyliodobenzene to provide 40.0 mg
(98%)
of a yellow film.
10
6-30: N-(5-(2-isopropylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, tbutyl
ester,
(G).
Prepared in a similar fashion from 2-isopropyliodobenzene to provide 27.0 mg
(64%) of
an amber oil.
6-31: N-(5-(3,4-methylenedioxyphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine,
tbutyl ester, (G).
Prepared in a similar fashion from 3,4-methylenedioxyiodobenzene to provide
40.0 mg
94%) of a yellow oil.
20
6-32: N-(5-(4-pyrrolylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, butyl
ester,
(G).
Prepared in a similar fashion from 4-pyrrolyliodobenzene to provide 41.0 mg
(92%) of a
light yellow oil.
6-33: N-(5-(4-trifluoromethoxyphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine,
tbutyl ester, (G).
Prepared in a similar fashion from 4-trifluoromethoxyiodobenzene to provide
28.5 mg
(61%) of product. 6-33: 1H NMR (CDC13, 300 MHz) 1.42 (s, 9H), 2.35 (s, 3H),
3.13 (s,
2H), 3.36 (d, 2H), 6.39 (t, 1H), 7.15 (d, 2H), 7.26-7.39 (d, 2H), 7.46 (d, 2H)
6-34: N-(5-(3,4-dimethoxyphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine,
tbutyl
ester, (G).
Prepared in a similar fashion from 3,4-dimethoxyiodoenzene to provide 35.0 mg
(80%)
of a colourless oil.
In a similar fashion the following compounds are prepared from intermediate F
and 1.3
equivalents of the corresponding aryliodide treated under the conditions
described above:
CA 02387660 2002-04-15
WO 01/32602 - 2g - PCT/US00/30074
6-35: N-(5-(2-quinoline)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, 'butyl ester,
(G).
Prepare in a similar fashion from 2-iodoquinolene.
6-36: N-(5-(indanyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, 'butyl ester,
(G).
Prepare in a similar fashion from iodoindane.
Example 7-1: N-(5-(4-Fluorophenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine,
(H).
A solution of t-butyl ester 6-1 (51 mg, 0.135 mmol) in 96% formic acid was
heated at 40
C for 16 hours. The reaction mixture was cooled and concentrated. The residue
was
taken up into dichloromethane and passed through a 2g solid phase extraction
tube,
eluting with dichloromethane, then ethyl acetate, then methanol. The methanol
fraction
was concentrated to provide amino acid 7-1 (39 mg, 90%) as a colourless foam :
1H
NMR (CDCl3, 300 MHz) 2.72 (s, 3H), 3.49 (s, 2H), 3.92 (d, 2H), 6.38 (t, 1H),
6.98 (dd,
2H), 7.26-7.42 (m, 7H). HRMS calc 324.1400, found 324.1386.
In a similar fashion the following compounds were prepared from the
corresponding
intermediate treated under the conditions described above
7-2: N-(5-(2-Fluorophenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, (H).
Prepared in a similar fashion from intermediate 6-2 to provide 31 mg (81 %) of
a
colourless foam. 1H NMR (CDC13, 300 MHz) 2.68 (s, 3H), 3.47 (s, 2H), 3.90 (s,
2H),
6.43 (s, 1 H), 7.05 (dd, 2H), 7.23-7.42 (m, 7H). HRMS calc 324.1400, found
324.1408.
7-3: N-(5-(2,4-Difluorophenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, (H).
Prepared in a similar fashion from intermediate 6-3 to provide 34 mg (82%) of
a
colourless foam. 'H NMR (CDC13, 300 MHz) 2.70 (s, 3H), 3.48 (s, 2H), 3.91 (s,
2H),
6.42 (s, 1H), 6.78 (dd, 2H), 7.26-7.38 (m, 6H). HRMS calc 342.1306, found
342.1333.
7-4: N-(5-(3-Nitrophenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, (H).
Prepared in a similar fashion from intermediate 6-4 to provide 42 mg (68%) of
a
colourless foam. 'H NMR (CDC13, 300 MHz) 2.72 (s, 3H), 3.50 (s, 2H), 3.94 (d,
2H),
CA 02387660 2002-04-15
WO 01/32602 - 29 - PCT/US00/30074
6.50 (t, 1 H), 7.26-7.48 (m, 6H), 7.70 (d, 1 H), 8.12 (d, 1 H), 8.22 (s, 1 H).
HRMS calc
351.1345, found 351.1353.
7-5: N-(5-(4-Nitrophenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, (H).
Prepared in a similar fashion from intermediate 6-5 to provide 21 mg (80%) of
a
colourless foam. 1H NMR (CDC13, 300 MHz) 2.66 (s, 3H), 3.43 (s, 2H), 3.85 (d,
2H),
6.51 (s, 1H), 7.26-7.53 (m, SH), 7.54 (d, 2H), 8.14 (d, 2H).
7-6: N-(3-Phenyl-5-(2-thiomethylphenyl)-2-penten-4-yn-1-yl)-sarcosine, (H).
Prepared in a similar fashion from intermediate 6-6 to provide 14 mg (87%) of
a
colourless foam. 1H NMR (CDC13, 300 MHz) 2.42 (s, 3H), 2.66 (s, 3H), 3.48 (s,
2H),
3.88 (s, 2H), 6.40 (s, 1 H), 7.12-7.68 (m, 9H).
7-7: N-(5-(4-Chlorophenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, (H).
Prepared in a similar fashion from intermediate 6-7 to provide 40 mg (90%) of
a
colourless foam. 'H NMR (CDCl3, 300 MHz) 2.68 (s, 3H), 3.48 (s, 2H), 3.87 (s,
2H),
6.39 (s, 1H), 7.24-7.37 (m, 9H). HRMS calc 340.1104, found 340.1097.
7-8: N-(5-(4-Isopropylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, (H).
Prepared in a similar fashion from intermediate 6-8 to provide 32 mg (99%) of
a
colourless foam. 1H NMR (CDCl3, 300 MHz) 1.21 (d, 6H), 2.65 (s, 3H), 2.86
(hept,
1H), 3.43 (s, 2H), 3.86 (d, 2H), 6.36 (t, 1H), 7.14 (d, 2H), 7.26-7.36 (m,
7H). HRMS
calc 348.1964, found 348.1998.
7-9: N-(5-(3,5-Bis(trifluoromethyl)phenyl)-3-phenyl-2-penten-4-yn-1-yl)-
sarcosine,
(H).
Prepared in a similar fashion from intermediate 6-9 to provide 26 mg (76%) of
a
colourless foam. 'H NMR (CDCl3, 300 MHz) 2.67 (s, 3H), 3.46 (s, 2H), 3.87 (d,
2H),
6.52 (t, 1H), 7.26-7.40 (m, SH), 7.77 (s, 1H), 7.83 (s, 2H). HRMS calc
442.1242, found
442.1173.
CA 02387660 2002-04-15
WO 01/32602 - 30 - PCT/US00/30074
7-10: N-(3,5-biphenyl-2-penten-4-yn-1-yl)-sarcosine, (H).
Prepared in a similar fashion from intermediate 6-10 to provide 18 mg (46%) of
a
colourless foam. 1H NMR (CDC13, 300 MHz) 2.69 (s, 3H), 3.48 (s, 2H), 3.89 (d,
2H),
6.40 (t, 1H), 7.26-7.44 (m, 10H). HRMS calc 306.1494, found 306.1432.
7-11: N-(5-(4-Biphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, (H).
Prepared in a similar fashion from intermediate 6-17 to provide 220.0 mg (97%)
of a
yellow solid.
7-12: N-(5-(4-trifluoromethylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine,
(H).
Prepared in a similar fashion from intermediate 6-18 to provide 200.0 mg (96%)
of a
yellow film.
7-13: N-(5-(4-benzylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, (H).
Prepared in a similar fashion from intermediate 6-19 to provide 190.0 mg (87%)
of a
light yellow solid.
7-14: N-(5-(4-ethylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, (H).
Prepared in a similar fashion from intermediate 6-20, to provide 176.1 mg
(86%) of a
green-grey solid.
7-15: N-(5-(4-"propylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, (H).
Prepared in a similar fashion from intermediate 6-21 to provide 190.9 mg (93%)
of an
orange-white solid.
7-16: N-(5-(4-"butylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, (H).
Prepared in a similar fashion from intermediate 6-22 to provide 206.0 mg (91
%) of a
yellow solid.
7-17: N-(5-(4-"pentylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, (H).
Prepared in a similar fashion from intermediate 6-23 to provide 204.4 mg (98%)
of a
yellow solid.
CA 02387660 2002-04-15
WO 01/32602 - 31 - PCT/US00/30074
7-18: N-(5-(4-phenoxyphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, (H).
Prepared in a similar fashion from intermediate 6-24 to provide 33.0 mg (100%)
of a
light yellow solid.
7-19: N-(5-(1-naphthyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, (H).
Prepared in a similar fashion from intermediate 6-25 to provide 25.4 mg (82%)
of a
yellow oil.
7-20: N-(5-(4-methyphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, (H).
Prepared in a similar fashion from intermediate 6-26 to provide 12.6 mg (55%)
of a
yellow solid.
7-21: N-(5-(3-isopropylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, (H).
Prepared in a similar fashion from intermediate 6-27 to provide 12.6 mg (83%)
of a
green-brown oil.
7-22: N-(5-(2-naphthyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, (H).
Prepared in a similar fashion from intermediate 6-28 to provide 25.1 mg (97%)
of a
yellow solid.
7-23: N-(5-(3,4-dimethylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, (H).
Prepared in a similar fashion from intermediate 6-29 to provide 33.2 mg (97%)
of a light
yellow solid.
7-24: N-(5-(2-isopropylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, (H).
Prepared in a similar fashion from intermediate 6-30 to provide 15.2 mg (66%)
of a flaky
yellow solid.
7-25: N-(5-(3,4-methylenedioxyphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine,
(H).
Prepared in a similar fashion from intermediate 6-31 to provide 9.Smg (31%) of
an off
white solid.
CA 02387660 2002-04-15
WO 01/32602 - 32 - PCT/US00/30074
7-26: N-(5-(4-pyrrolylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, (H).
Prepared in a similar fashion from intermediate 6-32 to provide 24.1 mg (68%)
of a
yellow solid.
7-27: N-(5-(4-trifluoromethoxyphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine,
(H).
Prepared in a similar fashion from intermediate 6-33 to provide 23.0 mg (92%)
of a
yellow solid.
7-28: N-(5-(3,4-dimethoxyphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, (H).
Prepared in a similar fashion from intermediate 6-34 to provide 25.7 mg (86%)
of a
yellow solid.
In a similar fashion the following compounds are prepared from the
corresponding
intermediate treated under the conditions described above:
7-29: N-(5-(2-quinoline)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine,
Prepare in a similar fashion from 6-35.
7-30: N-(5-(indanyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine,
Prepare in a similar fashion from 6-36.
Example 8-1: N-(3-Phenyl-5-(4-thiomethylphenyl)-2-penten-4-yn-1-yl)-sarcosine,
(H).
A solution of 'butyl ester 6-11 G(vi) (30.0 mg, 0.0736 mmol) in 96% formic
acid was
heated at 50 C for 3 hours. The reaction mixture was cooled and concentrated.
The
residue was taken up in dichloromethane and passed through a 2 g solid phase
extraction
tube, eluting with dichloromethane, then ethyl acetate, then methanol. The
methanol
fraction was concentrated to provide amino acid 8-1 (14.9 mg, 58%) as a light
yellow
solid.
In a similar fashion the following compounds were prepared from the
corresponding
intermediate under the conditions described above:
CA 02387660 2002-04-15
WO 01/32602 - 33 - PCT/US00/30074
8-2: N-(5-(4-Methylphenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine, (H).
Prepared in a similar fashion from intermediate 6-12 to provide 30.0 mg (91%)
of a light
yellow solid.
8-3: N-(3-Phenyl-5-(3-thiophene)-2-penten-4-yn-1-yl)-sarcosine, (H).
Prepared in a similar fashion from intermediate 6-13 to provide 22.0 mg (87%)
of a
brown solid foam.
8-4: N-(3-Phenyl-5-(4-tbutylphenyl)-2-penten-4-yn-1-yl)-sarcosine, (H).
Prepared in a similar fashion from intermediate 6-14 to provide 22.9 mg (66%)
of a light
yellow solid.
Example 9: N-(5-(4-Bromophenyl)-3-phenyl-2-penten-4yn-1-yl)sarcosine tButyl
ester, (S)
To a solution of terminal acetylene F (3.25 g, 11.4 mmol) in Et3N (75 mL) was
added 4-
bromoiodobenzene (4.19 g, 14.8 mmol), Pd(PPh3)4 (1.32 g, 1.14 mmol), and CuI
(0.65 g,
3.42 mmol). The mixture was stirred overnight, and concentrated. Column
chromatography (10% EtOAc/hexanes) provided bromide S (3.84 g, 76%) as a
yellow
oil.
Example 10-1: N-(5-(4-(3furyl)phenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine
Butyl ester, (G').
To a solution of bromide S (3.84 g, 8.72 mmol) in DME (25 mL) was added 3-
furanboronic acid (1.47 g, 13.1 mmol), Pd(PPh3)4 (1.01 g, 0.872 mmol), and 2M
Na2C03
(25 mL). The mixture was refluxed for 1 hour, cooled, and partitioned between
EtOAc
and water. The organic phase was washed with brine, dried (MgS04), filtered,
and
concentrated. Column chromatography (10-12.5% EtOAc/hexanes) provided ester G'
(2.62 g, 78%) as a yellow oil.
In a similar fashion the following compounds were made from the corresponding
boronic
acid under the conditions described above:
CA 02387660 2002-04-15
WO 01/32602 - 34 - PCT/US00/30074
10-2: N-(5-(4-(3-thiophene)phenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine
Butyl
ester, (G').
Prepared in a similar fashion from 3-thiopheneboronic acid to provide 21.0 mg
(46%) of
a colourless film.
In a similar fashion the following compounds are made from the corresponding
boronic
acid under the conditions described above:
10-3: N-(5-(4-(4Methyl-3-thiophene)phenyl)-3-phenyl-2-penten-4yn-1-yl)-
sarcosine
t-Butyl ester,
Prepare in a similar fashion from S and 4-methyl-3-thiopheneboronic acrd.
10-4: N-(5-(4-(4Methyl-3-furyl)phenyl)-3-phenyl-2-penten-4yn-1-yl)-sarcosine
t-Butyl ester,
Prepare in a similar fashion from S and 4-methyl-3-furanboronic acid.
10-5: N-(5-(4-(cyclohexyl)-phenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine
tButyl
Ester,
Prepare in a similar fashion from S and cyclohexylboronic acid.
10-6: N-(5-(4-(cyclopentyl)-phenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine
tButyl
Ester,
Prepare in a similar fashion from S and cyclopentylboronic acid.
Example 11-1: N-(5-(4-(3furyl)phenyl)-3-phenyl-2-penten-4yn-1-yl)-sarcosine,
(H').
The ester G' (2.62 g, 6.13 mmol) was dissolved in 96% formic acid (26 mL). The
solution was warmed at 40 °C overnight, then concentrated. Column
chromatography
(0-8% MeOH/CHZCl2) provided a pale yellow solid. Trituration with MeOH
provided
pure H' (0.78 g, 34%) as a white solid. Conversion of 11-1 to the
corresponding sodium
salt was achieved by suspending 11-1 in methanol and adding 1 equivalent of 6M
sodium hydroxide. The solution was then concentrated and the residue was
triturated
with isopropanol to provide a white solid.
CA 02387660 2002-04-15
WO 01/32602 - 35 - PCT/US00/30074
In a similar fashion the following compounds were made from the corresponding
intermediate treated under the conditions described above:
11-2: N-(5-(4-(3-thiophene)phenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine,
(H').
Prepared in a similar fashion from intermediate 10-2 to provide 11.7 mg (59%)
of an off
white solid.
11-3: N-(5-(4-(4-Methyl-3-thiophene)phenyl)-3-phenyl-2-penten-4-yn-1-yl)-
sarcosme,
Prepare in a similar fashion from 10-3.
11-4: N-(5-(4-(4-Methyl-3-furyl)phenyl)-3-phenyl-2-penten-4-yn-1-yl)-
sarcosine,
Prepare in a similar fashion from 10-4.
11-5: N-(5-(4-(cyclohexyl)-phenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine,
Prepare in a similar fashion from 10-5.
10-6: N-(5-(4-(cyclopentyl)-phenyl)-3-phenyl-2-penten-4-yn-1-yl)-sarcosine,
Prepare in a similar fashion from 10-6.
Example 12-1: Ethyl 4-(trifluoromethyl)phenylpropiolate (A).
To a solution of 4-iodobenzotrifluoride (256 mg, 0.941 mmol) in triethylamine
(2.5 mL)
was added ethyl propiolate (0.124 mL, 120 mg, 1.22 mmol), Pd(PPh3)4 (109 mg,
0.0941
mmol), and CuI (54 mg, 0.282 mmol). After 24 hours the reaction mixture was
concentrated. Column chromatography (10% EtOAc/hexanes) provided 12-1 (149 mg,
65%) as a colourless oil.
In a similar fashion the following compounds were prepared from the
corresponding
aryliodide and 1.3 equivalents of ethylpropiolate treated under the conditions
described
above:
CA 02387660 2002-04-15
WO 01/32602 - 36 - PCT/US00/30074
12-2: Ethyl 4-fluorophenylpropiolate (A).
Prepared in a similar fashion from 4-fluoroiodobenzene to provide 33 mg (4%)
of a
colourless solid.
S 12-3: Ethyl 2-fluorophenylpropiolate (A).
Prepared in a similar fashion from 2-fluoroiodobenzene to provide 3.46 g (93%)
of a
colourless oil.
12-4: Ethyl 4-chlorophenylpropiolate (A).
Prepared in a similar fashion from 4-chloroiodobenzene to provide 4.60 g
(100%) of a
colourless solid.
12-5: Ethyl Z-chlorophenylpropiolate (A).
Prepared in a similar fashion from 2-chloroiodobenzene to provide 7.64 g
(100%) of a
yellow liquid.
12-6: Ethyl 3-thienylpropiolate (A).
Prepared in a similar fashion from 3-thienyliodobenzene to provide 90 mg (53%)
of a
yellow solid.
12-7: Ethyl 4-methoxyphenylpropiolate (A).
Prepared in a similar fashion from 4-methoxyiodobenzene to provide 117 mg
(13%) of a
colourless oil.
Example 13-1: 1-Ethoxycarbonyl-2-(4-(trifluoromethyl)phenyl)-4-trimethylsilyl-
1-
buten-4-yne (C).
To a solution of Pd(OAc)2 (2.6 mg, 0.0115 mmol) in PhMe (2 mL) was added
tris(2,6-
dimethoxyphenyl)phosphine (5.1 mg, 0.0115 mmol). After 15 minutes a solution
of 12-
1 (117 mg, 0.573 mmol) in PhMe (3 mL) was added. After 5 minutes
(trimethylsilyl)acetylene (0.081 mL, 56 mg, 0.573 mmol) was added. After 21
hours the
reaction mixture was concentrated. Column chromatography ( 10% EtOAc/hexanes)
provided 13-1 (144 mg, 83%) as a yellow oil.
CA 02387660 2002-04-15
WO 01/32602 - 37 - PCT/US00/30074
In a similar fashion the following compounds were prepared from the
corresponding
propiolate intermediate treated by the conditions described above:
13-2: 1-Ethoxycarbonyl-2-(4-fluorophenyl)-4-trimethylsilyl-1-buten-4-yne (C).
Prepared in a similar fashion from intermediate 12-2 to provide 29 mg (58%) of
a yellow
oil.
13-3: 1-Ethoxycarbonyl-2-(2-fluorophenyl)-4-trimethylsilyl-1-buten-4-yne (C).
Prepared in a similar fashion from intermediate 12-3 to provide 4.19 g (80%)
of a yellow
oil.
13-4: 1-Ethoxycarbonyl-2-(4-chlorophenyl)-4-trimethylsilyl-1-buten-4-yne (C).
Prepared in a similar fashion from intermediate 12-4 to provide 4.04 g (60%)
of a brown
oil.
13-5: 1-Ethoxycarbonyl-2-(2-chlorophenyl)-4-trimethylsilyl-1-buten-4-yne (C).
Prepared in a similar fashion from intermediate 12-5 to provide 10.4 g (93%)
of a brown
oil.
13-6: 1-Ethoxycarbonyl-2-(3-fluorophenyl)-4-trimethylsilyl-1-buten-4-yne (C).
Prepared in a similar fashion from the commercially available intermediate
Ethyl 3-
fluorophenylpropiolate to provide 0.73 g (85%) of a yellow oil.
13-7: 1-Ethoxycarbonyl-2-(3-thienyl)-4-trimethylsilyl-1-buten-4-yne (C).
Prepared in a similar fashion from intermediate 12-6 to provide 123 mg (90%)
of a
yellow oil.
13-8: 1-Ethoxycarbonyl-2-(4-methoxyphenyl)-4-trimethylsilyl-1-buten-4-yne (C).
Prepared in a similar fashion from intermediate 12-7 to provide 144 mg (83%)
of a
yellow oil.
CA 02387660 2002-04-15
WO 01/32602 - 38 - PCT/US00/30074
Example 14-1: 1-Hydroxy-3-(4-(trifluoromethyl)phenyl)-5-trimethylsilyl-2-
penten-4-yne.
A solution of 13-1 (144 mg, 0.476 mmol) in anhydrous PhMe (2 mL) was chilled
in a
dry-ice/acetone bath. A 1.0 M solution of DIBAL-H in PhMe (1.2 mL, 1.19 mmol)
was
added dropwise. After 5 minutes the chilling bath was removed. After an
additional 15
minutes the reaction mixture was chilled in an ice bath and Celite and
Na2S04~1 OH20
were added to quench the reaction. The reaction mixture was filtered through
Celite.
The filtrate was concentrated. Column chromatography (20% EtOAc/hexanes)
provided
14-1 (114 mg, 92%) as a yellow oil.
In a similar fashion the following compounds were prepared form the
corresponding
ester intermediates under the conditions described above:
14-2: 1-Hydroxy-3-(4-fluorophenyl)-5-trimethylsilyl-2-penten-4-yne.
Prepared in a similar fashion from intermediate 13-2 to provide 19 mg (80%) of
a
colourless oil.
14-3: 1-Hydroxy-3-(2-fluorophenyl)-5-trimethylsilyl-2-penten-4-yne.
Prepared in a similar fashion from intermediate 13-3 to provide 2.65 g (74%)
of a yellow
oil.
14-4: 1-Hydroxy-3-(4-chlorophenyl)-5-trimethylsilyl-2-penten-4-yne.
Prepared in a similar fashion from intermediate 13-4 to provide 2.16 g (62%)
of a yellow
oil.
14-5: 1-Hydroxy-3-(2-chlorophenyl)-5-trimethylsilyl-2-penten-4-yne.
Prepared in a similar fashion from intermediate 13-5 to provide 4.86 g (54%)
of a yellow
oil.
14-6: 1-Hydroxy-3-(3-fluorophenyl)-5-trimethylsilyl-2-penten-4-yne.
Prepared in a similar fashion from intermediate 13-6 to provide 0.47 g (74%)
of a pale
yellow oil.
CA 02387660 2002-04-15
WO 01/32602 - 39 - PCT/US00/30074
14-7: 1-Hydroxy-3-(3-thienyl)-5-trimethylsilyl-2-penten-4-yne.
Prepared in a similar fashion from intermediate 13-7 to provide 56 mg (77%) of
a yellow
oil.
14-8: 1-Hydroxy-3-(4-methoxyphenyl)-5-trimethylsilyl-2-penten-4-yne.
Prepared in a similar fashion from intermediate 13-8 to provide 114 mg (92%)
of a
yellow oil.
Example 15-1: N-(3-(4-(Trifluoromethyl)phenyl)-5-(trimethylsilyl)-2-penten-4-
yn-1-
yl)-sarcosine, tbutyl ester (E).
A solution of 14-1 (11~ mg, 0.385 mmol) in anhydrous CHZC12 (4 mL) was chilled
in a
dry-ice/acetonitrile bath. PPh3 (152 mg, 0.578 mmol) and NBS (103 mg, 0.578
mmol)
were added. After 40 minutes saturated NaHC03 was added. The reaction mixture
was
partitioned between CHZC12 and saturated NaHC03. The organic phase was washed
with
brine, dried (Na2S04), filtered, and concentrated to provide crude
intermediate D (1-
Bromo-3-(4-(trifluoromethyl)phenyl)-5-trimethylsilyl-2-penten-4-yne) used
directly
in the next step.
To a solution of the crude bromide D (139 mg, 0.385 mmol) in anhydrous MeCN (4
mL)
was added t-butyl sarcosine hydrochloride (77 mg, 0.424 mmol), K~C03 (532 mg,
3.85
mmol), and KI (320 mg, 1.92 mmol). After 24 hours the reaction mixture was
poured
into water and extracted with EtOAc. The organic phase was washed with water
and
brine, dried (Na2S04), filtered, and concentrated. Column chromatography (15%
EtOAc/hexanes) provided 15-1 (62 mg, 38% over 2 steps) as a colourless oil.
In a similar fashion the following compounds were prepared from the
corresponding
crude bromide treated under the conditions described above:
15-2: N-(3-(4-fluorophenyl)-5-(trimethylsilyl)-2-penten-4-yn-1-yl)-sarcosine,
'butyl
ester (E).
Prepared in a similar fashion from intermediate 14-2 to provide 18 mg (63%
over 2
steps) of a colourless oil.
CA 02387660 2002-04-15
WO 01/32602 - 40 - PCT/US00/30074
15-3: N-(3-(2-fluorophenyl)-5-(trimethylsilyl)-2-penten-4-yn-1-yl)-sarcosine,
'butyl
ester (E).
Prepared in a similar fashion from intermediate 14-3 to provide 3.24 g (81 %
over 2
steps) of a yellow oil.
15-4: N-(3-(4-chlorophenyl)-5-(trimethylsilyl)-2-penten-4-yn-1-yl)-sarcosine,
'butyl
ester (E).
Prepared in a similar fashion from intermediate 14-4 to provide 1.55 g (49%)
of a yellow
oil.
15-5: N-(3-(2-chlorophenyl)-5-(trimethylsilyl)-2-penten-4-yn-1-yl)-sarcosine,
'butyl
ester (E).
Prepared in a similar fashion from intermediate 14-5 to provide 5.39 g (75%)
of a pale
yellow oil.
15-6: N-(3-(3-fluorophenyl)-5-(trimethylsilyl)-2-penten-4-yn-1-yl)-sarcosine,
'butyl
ester (E).
Prepared in a similar fashion from intermediate 14-6 to provide 0.63 g (89%)
of a yellow
oil.
15-7: N-(3-(3-thienyl)-5-(trimethylsilyl)-2-penten-4-yn-1-yl)-sarcosine,
'butyl ester
(E).
Prepared in a similar fashion from intermediate 14-7 to provide 61 mg (71 %)
of a yellow
oil.
15-8: N-(3-(4-methoxyphenyl)-5-(trimethylsilyl)-2-penten-4-yn-1-yl)-sarcosine,
'butyl ester (E).
Prepared in a similar fashion from intermediate 14-8 to provide 14 mg ( 10 %)
of a
yellow oil.
CA 02387660 2002-04-15
WO 01/32602 - 41 - PCT/L1S00/30074
Example 16-1: N-(3-(4-(Trifluoromethyl)phenyl)-2-penten-4-yn-1-yl)sarcosine,
butyl ester (F).
To a solution of 15-1 (62 mg, 0.146 mmol) in MeOH (2 mL) was added K2C03 (101
mg,
0.730 mmol). After 15 minutes the reaction mixture was poured into water and
extracted
with EtOAc. The organic phase was washed with brine, dried (MgS04), filtered,
and
concentrated to provide 16-1 (36 mg, 71%) as a yellow oil.
In a similar fashion the following compounds were prepared from the
corresponding
trimethylsilyl intermediates under the conditions described above:
16-2: N-(3-(4-fluorophenyl)-2-penten-4-yn-1-yl)sarcosine, tbutyl ester (F).
Prepared in a similar fashion from intermediate 15-2 to provide 13 mg (93%) of
a yellow
oil.
16-3: N-(3-(2-fluorophenyl)-2-penten-4-yn-1-yl)sarcosine, tbutyl ester (F).
Prepared in a similar fashion from intermediate 15-3 to provide 2.22 g (85%)
of a
colourless oil.
16-4: N-(3-(4-chlorophenyl)-2-penten-4-yn-1-yl)sarcosine, 'butyl ester (F).
Prepared in a similar fashion from intermediate 15-4 to provide 0.80 g (76%)
of a yellow
oil.
16-5: N-(3-(2-chlorophenyl)-2-penten-4-yn-1-yl)sarcosine, tbutyl ester (F).
Prepared in a similar fashion from intermediate 15-5 to provide 3.72 g (85%)
of a yellow
oil.
16-6: N-(3-(3-fluorophenyl)-2-penten-4-yn-1-yl)sarcosine, butyl ester (F).
Prepared in a similar fashion from intermediate 15-6 to provide 0.42 g (83%)
of a pale
yellow solid.
16-7: N-(3-(3-thienyl)-2-penten-4-yn-1-yl)sarcosine, tbutyl ester (F).
Prepared in a similar fashion from intermediate 15-7 to provide 46 mg (96%) of
a yellow
solid.
CA 02387660 2002-04-15
WO 01/32602 - 42 - PCT/LTS00/30074
16-8: N-(3-(4-methoxyphenyl)-2-penten-4-yn-1-yl)sarcosine, tbutyl ester (F).
Prepared in a similar fashion from intermediate 15-8 to provide 15 mg (136%)
of a
yellow oil.
S
Example 17-1: N-(5-(4-Isopropylphenyl)-3-(4-(trifluoromethyl)phenyl)-2-penten-
4-
yn-1-yl)-sarcosine, tbutyl ester (G).
To a solution of 16-1 (35 mg, 0.099 mmol) in triethylamine (2 mL) was added 4-
iodoisopropylbenzene (32 mg, 0.129 mmol), Pd(PPh3)4 ( 11 mg, 0.0099 mmol), and
CuI
(5.5 mg, 0.029 mmol). After 18 hours the reaction mixture was concentrated.
Column
chromatography (10% EtOAc/hexanes) provided 17-1 (40 mg, 86%) as a colourless
oil.
In a similar fashion the following compounds were prepared from 1.3
equivalents of the
appropriate aryliodide with the corresponding alkyne intermediate according to
the
conditions described above:
17-2: N-(5-(4-Isopropylphenyl)-3-(4-fluorophenyl)-2-penten-4-yn-1-yl)-
sarcosine,
tbutyl ester (G).
Prepared in a similar fashion from intermediate 16-2 and 4-
isopropyliodobenzene to
provide 14 mg (76%) of a colourless oil.
17-3: N-(5-(4-Isopropylphenyl)-3-(2-fluorophenyl)-2-penten-4-yn-1-yl)-
sarcosine,
'butyl ester (G).
Prepared in a similar fashion from intermediate 16-3 and 4-
isopropyliodobenzene to
provide 440 mg (79%) of a yellow oil.
17-4: N-(5-(4-t-Butylphenyl)-3-(2-fluorophenyl)-2-penten-4-yn-1-yl)-sarcosine,
tbutyl ester (G).
Prepared in a similar fashion from intermediate 16-3 and 4-t-butyliodobenzene
to
provide 500 mg (87%) of a yellow oil.
CA 02387660 2002-04-15
WO 01/32602 - 43 - PCT/US00/30074
17-5: N-(5-(4-Isopropylphenyl)-3-(4-chlorophenyl)-2-penten-4-yn-1-yl)-
sarcosine,
tbutyl ester (G).
Prepared in a similar fashion from intermediate 16-4 and 4-
isopropyliodobenzene to
provide 0.50 g (88%) of a pale yellow oil.
17-6: N-(5-(4-t-Butylphenyl)-3-(4-chlorophenyl)-2-penten-4-yn-1-yl)-sarcosine,
butyl ester (G).
Prepared in a similar fashion from intermediate 16-4 and 4-tbutyliodobenzene
to provide
514 mg (83%) of a pale yellow oil.
17-7: N-(5-(4-Isopropylphenyl)-3-(2-chlorophenyl)-2-penten-4-yn-1-yl)-
sarcosine,
tbutyl ester (G).
Prepared in a similar fashion from intermediate 16-5 and 4-
isopropyliodobenzene to
provide 0.53 g (97%) of a yellow oil.
17-8: N-(5-(4-t-Butylphenyl)-3-(2-chlorophenyl)-2-penten-4-yn-1-yl)-sarcosine,
tbutyl ester (G).
Prepared in a similar fashion from intermediate 16-5 and 4-t-butyliodobenzene
to
provide 0.52 g (92%) of a yellow oil.
17-9: N-(5-(4-Isopropylphenyl)-3-(3-fluorophenyl)-2-penten-4-yn-1-yl)-
sarcosine,
'butyl ester (G).
Prepared in a similar fashion from intermediate 16-6 and 4-
isopropyliodobenzene to
provide 0.16 g (103%) of a yellow oil.
17-10: N-(5-(4-Isopropylphenyl)-3-(3-thienyl)-2-penten-4-yn-1-yl)-sarcosine,
tbutyl
ester (G).
Prepared in a similar fashion from intermediate 16-7 and 4-
isopropyliodobenzene to
provide 54 mg (86%) of a yellow oil.
CA 02387660 2002-04-15
WO 01/32602 - 44 - PCT/US00/30074
17-11: N-(5-(4-Isopropylphenyl)-3-(4-methoxyphenyl)-2-penten-4-yn-1-yl)-
sarcosine, tbutyl ester (G).
Prepared in a similar fashion from intermediate 16-8 and 4-
isopropyliodobenzene to
provide 21 mg (129%) of a colourless oil.
17-12: N-(5-(3,4-Methylenedioxyphenyl)-3-(3-fluorophenyl)-2-penten-4-yn-1-yl)-
sarcosine, butyl ester (G).
Prepared in a similar fashion from intermediate 16-6 and 3,4-
methylenedioxyiodobenzene to provide 74.1 mg (106%) of a brown oil.
17-13: N-(5-(4-Ethylphenyl)-3-(2-chlorophenyl)-2-penten-4-yn-1-yl)-sarcosine,
tbutyl ester (G).
Prepared in a similar fashion from intermediate 16-5 and 4-ethyliodobenzene to
provide
44.0 mg (110%) of a light yellow oil.
17-4: N-(5-(4-Propylphenyl)-3-(2-chlorophenyl)-2-penten-4-yn-1-yl)-sarcosine,
tbutyl ester (G).
Prepared in a similar fashion from intermediate 16-5 and 4-propyliodobenzene
to provide
39.5 mg (96%) of a light yellow oil.
Example 18-1: N-(5-(4-Isopropylphenyl)-3-(4-(trifluoromethyl)phenyl)-2-penten-
4-
yn-1-yl)-sarcosine (H).
A solution of 17-1 (40 mg, 0.0849 mmol) in formic acid (2 mL) was warmed at 40
°C for
18 hours. The reaction mixture was concentrated. Column chromatography (0-100%
MeOH/CH2C12) provided 18-1 (36 mg, 99%) as a yellow oil.
In a similar fashion the following compounds were prepared from the
corresponding t-
butyl ester intermediate under the conditions described above:
18-2: N-(5-(4-Isopropylphenyl)-3-(4-fluorophenyl)-2-penten-4-yn-1-yl)-
sarcosine
(H).
Prepared in a similar fashion from intermediate 17-2 to provide 13 mg (107%)
of a
colourless oil.
CA 02387660 2002-04-15
WO 01/32602 - 45 - PCT/US00/30074
18-3: N-(5-(4-Isopropylphenyl)-3-(2-fluorophenyl)-2-penten-4-yn-1-yl)-
sarcosine
(H).
Prepared in a similar fashion from intermediate 17-3 to provide 379 mg (99%)
of a pale
yellow oil.
18-4: N-(5-(4-t-Butylphenyl)-3-(2-fluorophenyl)-2-penten-4-yn-1-yl)-sarcosine
(H).
Prepared in a similar fashion from intermediate 17-4 to provide 434 mg (100%)
of a
yellow oil.
18-5: N-(5-(4-Isopropylphenyl)-3-(4-chlorophenyl)-2-penten-4-yn-1-yl)-
sarcosine
(H).
Prepared in a similar fashion from intermediate 17-5 to provide 436 mg (96%)
of a beige
solid.
18-6: N-(5-(4-t-Butylphenyl)-3-(4-chlorophenyl)-2-penten-4-yn-1-yl)-sarcosine
(H).
Prepared in a similar fashion from intermediate 17-6 to provide 408 mg (88%)
of a beige
solid.
18-7: N-(5-(4-Isopropylphenyl)-3-(2-chlorophenyl)-2-penten-4-yn-1-yl)-
sarcosine
(H).
Prepared in a similar fashion from intermediate 17-7 to provide 438 mg (95%)
of an off
white foam.
18-8: N-(5-(4-t-Butylphenyl)-3-(2-chlorophenyl)-2-penten-4-yn-1-yl)-sarcosine
(H).
Prepared in a similar fashion from intermediate 17-8 to provide 448 mg (97%)
of a
colourless foam.
18-9: N-(5-(4-Isopropylphenyl)-3-(3-fluorophenyl)-2-penten-4-yn-1-yl)-
sarcosine
(H).
Prepared in a similar fashion from intermediate 17-9 to provide 0.12 g (93%)
of a
colourless oil.
CA 02387660 2002-04-15
WO 01/32602 - 46 - PCT/US00/30074
18-10: N-(5-(4-Isopropylphenyl)-3-(3-thienyl)-2-penten-4-yn-1-yl)-sarcosine
(H).
Prepared in a similar fashion from intermediate 17-10 to provide 34 mg (72%)
of a
yellow solid.
18-11: N-(5-(4-Isopropylphenyl)-3-(4-methoxyphenyl)-2-penten-4-yn-1-yl)-
sarcosine
(H).
Prepared in a similar fashion from intermediate 17-11 to provide 14 mg (76%)
of a
colourless oil.
18-12: N-(5-(3,4-Methylenedioxyphenyl)-3-(3-fluorophenyl)-2-penten-4-yn-1-yl)-
sarcosine (H).
Prepared in a similar fashion from intermediate 17-12 to provide 64.1 mg (88%)
of a
orange-brown oil.
18-13:N-(5-(4-Ethylphenyl)-3-(2-chlorophenyl)-2-penten-4-yn-1-yl)-sarcosine
(H).
Prepared in a similar fashion from intermediate 17-13 to provide 30.1 mg (79%)
of a
colourless oil.
18-14: N-(5-(4-Propylphenyl)-3-(2-chlorophenyl)-2-penten-4-yn-1-yl)-sarcosine
(H).
Prepared in a similar fashion from intermediate 17-14 to provide 13.9 mg (40%)
of a
light yellow oil.
Example 20 - Assay of Transport via GIyT-1
This example illustrates a method for the measurement of glycine uptake by
transfected
cultured cells.
Cells stably transfected with GIyT-1 C (see Kim, et al., Molecular
Pharmacology, 45,
1994:608-617) were washed twice with HEPES buffered saline (HBS). The cells
were
then incubated 10 minutes at 37 C, after which a solution was added containing
50 nM
[3H]glycine (17.5 Ci/mmol) and either (a) no potential competitor, (b) 10 mM
nonradioactive glycine or (c) a concentration of a candidate drug. A range of
concentrations of the candidate drug was used to generate data for calculating
the
concentration resulting in 50% of the effect (e.g., the ICsos, which are the
concentrations
CA 02387660 2002-04-15
WO 01/32602 - 47 - PCT/US00/30074
of drug inhibiting glycine uptake by 50%). The cells were then incubated
another 10
minutes at 37°C, after which the cells were aspirated and washed three
times with ice-
cold HBS. The cells were harvested, scintillant was added to the cells, the
cells were
shaken for 30 minutes, and the radioactivity in the cells was counted using a
scintillation
counter. Data were compared between the same cells contacted or not contacted
by a
candidate agent, depending on the assay being conducted.
The compounds of the present invention were active as GIyT-1 inhibitors.
Example 21 - Assay of Binding to NMDA Receptors
This example illustrates binding assays to measure interaction of compounds
with the
glycine site on the NMDA receptor.
Direct binding of [3H]glycine to the NMDA-glycine site was performed according
to the
method of Grimwood et al., Molecular Pharmacology, 41, 923-930 (1992); Yoneda
et
al., J. Neurochem, 62, 102-112 (1994).
The binding test was performed in eppendorf tubes containing 150 pg of
membrane
protein and 50 nM [3H]glycine in a volume of 0.5 ml. Non-specific binding was
determined with 1 mM glycine. Drugs were dissolved in assay buffer (50 mM Tris-
acetate, pH 7.4) or DMSO (final concentration of 0.1%). Membranes were
incubated on
ice for 30 minutes and bound radioligand was separated from free radioligand
by
filtration on Whatman GF/B glass fiber filters or by centrifugation (18,000 x
g, 20 min).
Filters or pellet was washed three times quickly with ice-cold 5 mM Tris-
acetate buffer.
Filters were dried and placed in scintillation tubes and counted. Pellets were
dissolved in
deoxycholate/NaOH (0.1 N) solution overnight, neutralized and radioactivity
was
determined by scintillation counting.
A second binding test for the NMDA-glycine site used [3H]dichlorokynurenic
acid
(DCKA) and membranes prepared as above. See, Yoneda et al., J. Neurochem.,
60,634-
645 (1993). The binding assay was performed as described for [3H]glycine above
except
that [3H]DCKA was used to label the glycine site. The final concentration of
[3H]DCKA
was 10 nM, and the assay was performed for 10 minutes on ice.
CA 02387660 2002-04-15
WO 01/32602 - 48 - PCT/US00/30074
A third binding test used for the NMDA-glycine site used indirect assessment
of affinity
of ligands for the site by measuring the binding of [3H]MK-801 (dizocilpine).
See,
Palmer and Burns, J. Neurochem., 62, 187-196 (1994). Preparation of membranes
for
the test was the same as above. The binding assay allowed separate detection
of
antagonists and agonists.
The third binding test was operated to identify antagonists as follows: 100 pg
of
membranes were added to wells of a 96-well plate, along with glutamate (10 pM)
and
glycine (200 nM) and various concentrations of the ligand to be tested. The
assay was
started by the addition of 5 nM [3H]MK-801 (23.9 Ci/mmol), which binds to the
ion
channel associated with NMDA receptors. The final volume of the assay was 200
p1.
The assay was performed for 1 hour at room temperature. Bound radioactivity
was
separated from free by filtration, using a TOMTEC harvester. Antagonist
activity was
indicated by decreasing radioactivity associated with the NMDA receptor with
increasing concentration of the tested ligand.
The third binding test was operated to identify agonists by performing the
test as above,
except that the concentration of glycine was 200 nM. Agonist activity was
indicated by
increasing radioactivity associated with the NMDA receptor with increasing
concentration of the tested ligand.