Note: Descriptions are shown in the official language in which they were submitted.
07-01-2002 _2p02 hlON 03:35 PM F~ HQ, US00300f
CA 02387988 2002-04-04 t, w
1
METHOD r"OR MAKING SULFUR TRIOXiD~,
SULI~UtZIC AC1D, AND OLEUM lrROM SULFUR DIO'7fIDR
rlELD Or THE INVENTION
This invention relates to a novel process for preparing sulfiu trioxide (50J)
by
oxidizing sulfur dioxide (S02). This invention also relates to apracess for
preparing
..
' liquid sulfuric acid (H~SO~ and/or oleutn from SOj by the contact process,
wherein
S0~ is oxidized to i-orm SO~, which, in torn, is contacted wish water or a
solution of
sulFuric acid to produce additional sulfitric acid andlor oleutn, This
invention further
relates to recovering high grade energy from the heat produced.during such a
contact
process.
' B~ACKGROUNf) (~F THI:1N~~EN'rIOI~I ~ - - -
Sulfwic acid is the highest volume chemical mattttfactured in the world.
Much of the sulfuric acid is used to produce phosphoric acid in integrated
fertilirxr
complexes. Sulfuric acid is also used, for example, in dyes and pigntents,
industcill
explosives, etching applications, alkylalion catalysis, electroplating baths,
and
. . .
t nonfewotts metallurgy. Current worldwide production is reported to be
about.570,000
tons per day, with about 30% being Produced in the United States.
The contact pTOCC55 has been one of the most popularmethods for making
sulfuric acid and otcum ("oleum" is a solution ofSO, in sulfuric acid, and
also is
known as "fuming sulfuric acid" or "HzS~O?"). This process generally comprises
3
. . steps: (1) forming SOz from a sulfur-containing raw material, (2)
catalytically
vxidizins the 50~ to form S03 and (3) contacting the SO~ with water or
concentrated
sulfuric acid to hydrate the SO~ and form sulfuric acid andlor oleum.
Awide variety of sulfur-containing raw materials have been used in the
contact process to form SOZ. Most sulfuric acid plants, for examplo, form S02
by
oxidizing an oxidizable sulfur-containing material (e.g., elemental sulftrc or
motel ores
' containing sulfides) in a thermal combustion gone. A significant number of
other
AMENDED SHEET
Ernpf.zPit:07/Olmn.r~ i'r:u4 F~f nr ~~A~ p nna
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
2
plants (e.g., sulfuric acid regeneration plants), in contrast, burn a
carbonaceous
material (i.e., a fuel) in the presence of a decomposable sulfate to provide
the heat
necessary to decompose the sulfate into SOZ and various byproducts.
After being formed, the SOZ is normally oxidized to S03 by contacting it with
a catalyst (e.g., a vanadium pentoxide (V205) catalyst) at a temperature
effective for
catalytic oxidation of SOZ (e.g., at least about 410 to about 420°C for
a V205 catalyst)
in the presence of molecular oxygen. This reaction is often conducted in a
catalytic
converter which comprises a plurality of catalyst beds in series
(conventionally, 4 or
more catalyst beds). One of the difficulties with this reaction stems from the
fact that
it is a highly exothermic reaction. This requires that the reaction conditions
be
controlled so that the heat evolved from the oxidation reaction does not
overheat the
catalyst to a temperature which may lead to thermal damage and premature
deactivation of the catalyst and/or adversely affect the reaction equilibrium.
The
oxidation reaction can be controlled, for example, by limiting the
concentration of
SOZ or oxygen fed into the catalytic converter, or by using a converter
comprising a
tube-in-shell device such as that disclosed by Daley et al. in U.S. Patent No.
4,643,887 wherein the catalyst is cooled by indirect heat exchange with a
cooling
medium(e.g., air or molten salts). In processes using a V205 catalyst, for
example, the
reaction conditions are typically controlled so that the temperature of the
catalyst
beds) is maintained at less than about 650°C, and more typically less
than about
630°C.
The formation of sulfuric acid and/or oleum is normally conducted in an
absorption zone within an S03 absorption tower, in which the conversion gas
containing the S03 is contacted with water, or, more typically, a concentrated
solution
of sulfuric acid (e.g., a solution containing about 98.5 weight% sulfuric
acid) to form
sulfuric acid and/or oleum. Water is normally less preferred because it tends
to form
an acid mist of HZS04 that is difficult to condense.
While the reaction of S03 with the concentrated HzS04 is rapid and virtually
complete, the oxidation of SOZ to S03 is typically less complete. Thus, the
tail gas
leaving the S03 absorption tower will typically contain residual SO2. In most
countries, HzS04 plants are limited by the amount of SOz that they are allowed
to emit
into the atmosphere. The U.S. Environmental Protection Agency, for example,
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
currently limits SOZ emissions to 4 pounds per short ton (2 kg per metric ton)
of
HZS04 produced. This is equivalent to a minimum SOZ to S03 conversion of 99.7%
in
the catalytic oxidation step (i.e., no greater than 0.3% of the entering SOZ
may exit the
system in the S03 absorber tail gas).
Increasing the concentration of SOZ in the gas fed to the catalytic converter
generally tends to reduce the efficiency of the reaction. This, in turn, leads
to more
SOZ remaining in the tail gas discharged from the plant. Consequently, as a
sulfuric
acid plant operator seeks to increase production by increasing the
concentration of
SOZ in the gas fed to the converter, the SOZ emissions from the plant will
tend to
increase. As a result, sulfuric acid plants have generally been forced to
limit their rate
of production or risk non-compliance with environmental regulations.
In some instances, sulfuric acid plants have been able to increase their
production by using tail-gas scrubbers to remove SOz before it is emitted to
the
atmosphere. Tail-gas scrubbers have been particularly useful in conjunction
with
low-conversion, single-stage S03 absorption plants. A number of SOZ tail-gas
scrubbing processes are available, many of which use non-regenerable scrubbing
mediums such as ammonia, sodium hydroxide, or hydrogen peroxide. Such
techniques, however, have various disadvantages. For example, they require
expensive equipment (e.g., a separate scrubbing tower). Such equipment takes
up
valuable space and produces an additional pressure drop in the overall gas
system,
which decreases the gas handling capacity of the system. In addition, the
scrubbing
processes using a base often produce a by-product which must be properly
disposed of
(e.g., when ammonia is used to scrub the tail gas, a side stream of ammonium
sulfate
is produced; and when sodium hydroxide is used, a side stream of sodium
sulfate is
produced). And the use of ammonium salt scrubbing solutions, in particular,
typically
results in the formation of submicron aerosol fumes which must be removed
using
sophisticated and expensive mist eliminators.
Sulfuric acid plants have also controlled SOZ emissions by using a dual S03
absorption process. In such a process, an S03 absorption tower containing an
intermediate S03 absorption zone is positioned between two of the catalyst
beds of the
converter. For example, in many conventional systems using a 4 bed catalytic
converter, gas exiting the second or third catalyst bed is passed through an
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
4
intermediate 503 absorption zone wherein the gas is contacted with a
concentrated
solution of HZS04to form product acid. Gas exiting the intermediate S03
absorption
zone is returned to the next bed of the converter. Because the oxidation of
SOz to S03
is an equilibrium-controlled reaction, removal of S03 in the intermediate
absorption
zone helps drive the reaction forward in the succeeding beds of the converter
to
achieve higher conversions and thereby reduce SOz emissions in the tail gas
exiting
the final S03 absorption tower. Such a process is disadvantageous, however,
because
the intermediate absorption zone contributes substantially to the capital and
operating
costs of the system. In addition, even with the dual absorption, plant
capacity may
still be limited to assure high conversions and low SOZ emissions.
In addition to the goal of increasing capacity while controlling SOZ
emissions,
another goal related to sulfuric acid contact plants has been to maximize the
recovery
of useable energy from the heat produced during the exothermic steps of the
contact
process. Until recently, only from about SS to about 60% of the heat generated
in the
contact sulfuric acid process was recovered in useful form. A major
improvement in
energy recovery, however, was provided by McAlister et al. in U.S. Patent Nos.
5,503,821; 5,118,490; 4,670,242; and 4,576,813. These patents describe
processes
which can, for example, recover heat of S03 absorption in the form of medium
pressure steam. In each process, an 503 absorption tower is operated at high
temperature, and heat is transferred from the absorption acid to produce
steam. By
maintaining the acid concentration in the range of 99 to 100%, alloy heat
exchangers
may be used for recovery of the absorption heat. These processes allow the
process
heat energy recovery capability to be increased to greater than 90%.
SUMMARY OF THE INVENTION
This invention provides for an improved process for making S03 which
comprises oxidizing SOz in a catalytic converter. More particularly, this
invention
provides for a process for making S03 which can be implemented with relatively
low
capital and operating costs; a process for making 503 which allows for a
minimal
volume of gas to be handled upstream of the catalytic converter, thus allowing
for
smaller equipment (i.e., equipment having lower capital and operating costs)
to be
used upstream of the converter; a process for making 503 from an SOz source
gas that
07-01-2002 2002 tlON 03:36 PM FAX N0,
US0030
CA 02387988 2002-04-04
has a nlativcly low SOi gas strength; a pmcess for making S03 wherein the
catalytic
converter can be operated without the use of an extraneous energy source to
bring the
SO= converter feed gas to the activation temperature of ihc S02 oxidation
catalyst (i.e.,
a process wherein the catalytic converter operates "autothermally'~; a process
for
making S03 wherein the catalytic converter may be operated aatothermally even
when
a weak source gas (e.g., a source gas having an SOZ concentration of less
th~.n-about S
mole%) is used; a process for making S03 from spent sulfuric acid; a pmcass
for
making SOs from sulfidic metal oxidation oi'fgascs; and a pmcess for the
production
of sulfuric acid andJor oleum wherein the recovery of heat energy-is enhanced:
.
14 Briefly, therefore, the present invention is directed to a process for
snaking
S03 from a source gas comprising 50=. In one cmbodunent, ihe,process comprises
contacting the source gas with a liquid SOZ absorption, solvent u1 an SOz
absorption
zone to selectively transfer SOz fmni the source gas to the SOZ absorption
solccnt and
form an SOi-depleted gas and an SOz-enriched solvent. Sulfuric dioxide is then
IS stripped from the 50i-enriched solvent in art SOZ stripping zone to form an
50=-
dcpleted absorption solvent and an SO~-enriched stripper gas having an SOZ gas
strength greater than the S02 gas strength of the source gas. A reaction gas
comprising a first portion of the SO~-cnriclied stripper gas is then formed.
~n
oxidation product gas (comprising S03 ~tnd residual SO~), in turn, is Formed
by a
20 process comprising passing the reaction gas through a plurality oCeatalyst
beds in
series (this plurality comprises at least 2 and no greaser than 4 catalyst
beds urhich
contain a catalyst etTcctive for oxidizing SOZ into 503). In-this embodiment,
a second
portion of the SOZ-enriched gas is introduced into at least one catalyst bed
downstream of the most upstream catalyst bed to increase the amount of SOZ
being
25 fed info the dvwnstrearrt bed.
In another embodiment far making SOj from a source gas comprlsinb SOz, the
procc;ss comprises contacting the sotuce gas with a liquid SOZ absorption
solvent in an
S02 absorption zone to selectively transfer S0~ frorn the source gas to the
S02
absorption solvent and form an SOi-depleted gas and an SOz-enriched solvent.
Sulfur
30 dioridc is than stripped from the SO2-enriched solvent in an SOz stripping
zone to
fonn an SOZ-depleted absorption solvent and an SO~-enriched stripper gas
having an
SOZ gas strengtli ~,reater than the SOZ gas strength ofthe source gas. A
raactiott gas is
AMENDED SHEET
Fm~f -oit'n?mwnn~ ~~AA ~_...,i __ .ono n nni
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
6
then formed which comprises a first portion of the SOZ-enriched stripper gas
(this first
portion comprises at least about 30% of the SOZ in the SOZ-enriched stripper
gas).
Afterward, an oxidation product gas (comprising S03 and residual SOz) is
formed by a
process comprising passing the reaction gas through a plurality of catalyst
beds in
series (this plurality comprises at least 2 catalyst beds which contain a
catalyst
effective for oxidizing SOz into S03). In this embodiment, a second portion of
the
SOZ-enriched gas is introduced into at least one catalyst bed downstream of
the most
upstream catalyst bed to increase the amount of SOZ being fed into the
downstream
bed.
In another embodiment for making S03 from a source gas comprising SOz, the
process comprises contacting the source gas with a liquid SOz absorption
solvent in an
SOZ absorption zone to selectively transfer SOz from the source gas to the SOZ
absorption solvent and form an SOZ-depleted gas and an SOZ-enriched solvent.
Sulfur
dioxide is then stripped from the SOZ-enriched solvent in an SOZ stripping
zone to
form an SOZ depleted absorption solvent and an SOZ enriched stripper gas
having an
SOz gas strength greater than the SOz gas strength of the source gas. A
reaction gas is
then formed which comprises a first portion of the SOZ-enriched stripper gas.
Afterward, an oxidation product gas (comprising S03 and residual SOZ) is
formed by a
process comprising passing the reaction gas through a plurality of catalyst
beds in
series (this plurality comprises at least two catalyst beds which comprise a
catalyst
effective for oxidizing SOZ into S03). In this embodiment, a second portion of
the
SOZ-enriched gas is introduced into at least one catalyst bed downstream of
the most
upstream catalyst bed to increase the amount of SOZ being fed into the
downstream
bed. In addition, the molar ratio of OZ to SOZ is greater than about 0.2:1 in
the gas
entering each of the catalyst beds in the plurality.
In another embodiment for making S03 from a source gas comprising SO2, the
process comprises contacting the source gas with a liquid SOz absorption
solvent in an
SOz absorption zone to selectively transfer SOZ from the source gas to the SOZ
absorption solvent and form an SOZ-depleted gas and an SOZ-enriched solvent.
Sulfur
dioxide is then stripped from the SOZ-enriched solvent in an SOz stripping
zone to
form an SOz-depleted absorption solvent and an SOZ-enriched stripper gas. A
converter feed gas is formed which comprises a first portion of the SOz-
enriched
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
7
stripper gas. This converter feed gas is divided into a first portion and a
second
portion. A first partial conversion gas and a second partial conversion gas
(both
comprising S03 and residual SOz) are then formed by passing the first portion
of the
converter feed gas through a catalyst bed, and passing the second portion
through a
different catalyst bed in parallel with the catalyst bed through which the
first portion
of the converter feed gas is passed (both catalyst beds comprise an oxidation
catalyst
effective for oxidizing SOz to S03). A first portion of the remainder of the
SOz-
enriched stripper gas is then combined with the first partial conversion gas
to fortify
the SOZ gas strength of the first partial conversion gas. Likewise, a second
portion of
the remainder of the SOZ-enriched stripper gas is combined with the second
partial
conversion gas to fortify the SOz gas strength of the second partial
conversion gas.
The fortified first partial conversion gas and the fortified second partial
conversion
gas are then passed through at least one fiu-ther catalyst bed (also
comprising an
oxidation catalyst effective for oxidizing SOZ to S03), thereby oxidizing
additional
SOZ to S03 and forming a conversion gas comprising S03 and SO2.
This invention also provides for an improved process for making sulfuric acid
and/or oleum. More particularly, this invention provides for a process for
making
sulfizric acid and/or oleum which meets SOZ emissions standards; a process for
making sulfiiric acid and/or oleum having greater SOZ oxidation capacity than
typical
conventional sulfuric acid plants without having greater SOZ emissions; a
process for
making sulfuric acid and/or oleum in which SOZ emissions are confined to a
single
purge stream for simple control and monitoring; a process for making sulfuric
acid
and/or oleum which achieves at least about 99.7% recovery of SO2, even at low
single
pass SOz conversions (e.g., SOz single-pass conversions of as low as about 75%
or
lower); a process for making sulfizric acid and/or oleum which can be
implemented
with relatively low capital and operating costs; a process for making sulfiuic
acid
and/or oleum which allows for a lesser volume of gas to be handled upstream of
the
catalytic converter than typical conventional sulfuric acid contact plants,
thus
allowing for smaller equipment to be used upstream of the converter; a process
for
making sulfizric acid and/or oleum which achieves low SOz emissions without
requiring the installation of separate SOz non-regenerable tail gas scrubbing
treatments and/or an S03 intermediate absorption zone (i.e., low emissions may
be
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
achieved using a single S03 absorber); a process for making sulfuric acid
and/or
oleum from an SOz source stream that has an H20/SOZ molar ratio greater than
the
desired HZO/S03 molar ratio in the product acid stream; a process for making
sulfuric
acid and/or oleum from an SOZ source gas that has a relatively low SOZ gas
strength; a
process for making sulfuric acid and/or oleum wherein the catalytic converter
operates
autothermally; a process for making sulfuric acid and/or oleum wherein the
catalytic
converter can be operated autothermally even when a weak source gas is used; a
process for making sulfuric acid and/or oleum from spent sulfuric acid; a
process for
making sulfuric acid and/or oleum from sulfidic metal oxidation off gases; a
process
for making sulfuric acid and/or oleum in which process energy is recovered in
high
grade form; a process for making sulfuric acid and/or oleum from a wet source
gas; a
process for making sulfuric acid and/or oleum from a wet source gas without
first
requiring the source gas to be passed through a drying tower; and a process
for
making sulfuric acid and/or oleum from a wet source gas in which heat
generated by
vapor phase formation of sulfuric acid (i.e., sulfuric acid formation from
water vapor
in the source gas reacting with S03 in the conversion gas) is recovered.
Briefly, therefore, the present invention is directed to a process for making
sulfuric acid and/or oleum from a source gas comprising SOZ. In one
embodiment,
the process comprises contacting at least a portion of the source gas with a
liquid SOz
absorption solvent in an SOz absorption zone to selectively transfer SOz from
the
portion of the source gas to the SOZ absorption solvent and form an SOZ-
depleted gas
and an SOZ-enriched solvent. Sulfur dioxide is then stripped from the SOZ-
enriched
solvent in an SOz stripping zone to form an SOZ-depleted absorption solvent
and an
SOZ enriched stripper gas having an SOZ gas strength greater than the SOZ gas
strength
of the source gas. An oxidation product gas (comprising S03 and residual SOZ)
is
then formed by a process comprising passing the SOZ-enriched stripper gas
through a
plurality of catalyst beds in series (each comprising an oxidation catalyst
effective for
oxidizing SOZ to S03). The oxidation product gas, in turn, is combined with
water
vapor to form an acid product gas comprising: (a) sulfuric acid formed by a
gas phase
reaction between S03 from the oxidation product gas and water vapor, thereby
generating the heat of formation of sulfuric acid in the gas phase; (b) S03;
and (c)
SO2. Heat energy from the gas phase heat of formation of sulfuric acid is
recovered
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
9
by transfer of heat from the acid product gas to steam or feed water in an
indirect heat
exchanger. The cooled acid product gas is then contacted with liquid sulfuric
acid in
an S03 absorption zone to form additional sulfuric acid and/or oleum and an
S03-
depleted gas comprising SO2.
In another embodiment for making sulfuric acid and/or oleum from a source
gas comprising SOz, the process comprises forming an oxidation product gas
(comprising S03 and residual SOZ) by a process comprising passing a first
portion of
the source gas through a plurality of catalyst beds in series ( this plurality
comprises at
least 2 catalyst beds which contain a catalyst effective for oxidizing SOZ
into S03).
Here, a second portion of the source gas is introduced into at least one
catalyst bed
downstream of the most upstream catalyst bed to increase the amount of SOz
being
fed into the downstream bed. The oxidation product gas, in turn, is combined
with
water vapor to form an acid product gas comprising: (a) sulfuric acid formed
by a gas
phase reaction between S03 from the oxidation product gas and water vapor,
thereby
generating the heat of formation of sulfuric acid in the gas phase; (b) 503;
and (c)
SOz. Heat energy from the gas phase heat of formation of sulfuric acid is
recovered
by transfer of heat from the acid product gas to steam or feed water in an
indirect heat
exchanger. The cooled acid product gas is then contacted with liquid sulfuric
acid in
an S03 absorption zone to form additional sulfuric acid and/or oleum and an
503-
depleted gas comprising SO2.
Another embodiment of this invention is directed to an improved process for
making sulfuric acid and/or oleum from a source gas comprising SOZ and water
vapor.
This process comprises forming a reaction gas comprising SO2, and then forming
an
oxidation product gas (comprising 503 and residual SOZ) by a process
comprising
passing the reaction gas through a plurality of catalyst beds in series (each
catalyst bed
comprises an oxidation catalyst effective for oxidizing SOZ into S03). The
oxidation
product gas, in turn, is combined with water vapor to form an acid product gas
comprising: (a) sulfuric acid formed by a gas phase reaction between 503 from
the
oxidation product gas and water vapor, thereby generating the heat of
formation of
sulfuric acid in the gas phase; (b) 503; and (c) SO2. Heat energy is recovered
from the
gas phase heat of formation of sulfuric acid by transferring heat from the
acid product
gas to steam or feed water in an indirect heat exchanger. Afterward, the
cooled acid
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
product gas is contacted with a solution comprising sulfuric acid in an S03
absorption
zone to form additional sulfuric acid and/or oleum and an S03-depleted gas
comprising SO2. The improvement in this process comprises combining at least a
portion of the source gas with the oxidation product gas to form the acid
product gas,
5 and forming the reaction gas from the S03-depleted gas.
Other objects and features of this invention will be in part apparent and in
part
pointed out hereinafter.
BRIEF DESCRIPTION OF THE FIGURES
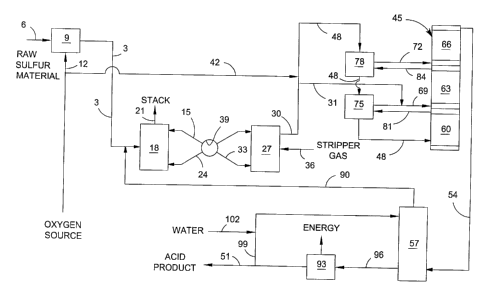
Fig. 1 is a schematic flow sheet illustrating various features of one
10 embodiment of the process of the present invention.
Fig. 2 is a schematic flow sheet showing a 4 bed catalytic converter of a
contact sulfuric acid plant modified in accordance with the present invention.
Fig. 3 is a schematic flow sheet illustrating various features of another
embodiment of the process of the present invention for use with a wet SOZ
source gas.
Fig. 4 is a schematic flow sheet illustrating an embodiment of the process of
the present invention described in the Example below.
DESCRIPTION OF THE PREFERRED EMBODIMENTS
A. Formation of the Source Gas Containing Sulfur Dioxide
Refernng to Fig. l, a source gas 3 containing SOZ is formed from a sulfur-
containing raw material 6. A wide variety of sulfur-containing raw materials
may be
used. For example, a decomposable sulfate is often suitable. Such a sulfate
may
include, for example, calcium sulfate, ammonium sulfate, or spent HZS04 (i.e.,
contaminated or diluted HZS04). To form SOZ, the sulfate is typically injected
as a
liquid spray into a combustion zone 9, along with a carbonaceous material
(i.e., a fuel)
and an oxygen source 12 (normally air). This mixture is then burned to provide
the
heat necessary to evaporate water and decompose the sulfate. For example, in a
spent
acid recovery plant where spent HZS04 is used as the raw material 6, a gas 3
is formed
which typically contains sulfurous acid (HzS03), SO2, O2, CO2, N2, and water
vapor.
In a particularly preferred embodiment, the sulfur containing raw material 6
is
an oxidizable material, such as elemental sulfur, hydrogen sulfide (HzS), or
iron pyrite
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
11
(FeSz) or another sulfide-containing metal ore. In this embodiment, the
sulfur-containing material 6 is typically burned with an oxygen source 12 in a
kiln or
other suitable thermal combustion zone 9 to produce a source gas 3 containing
SO2.
The most economically practical oxygen source 12 is normally air, which, when
burned with the oxidizable sulfur material 6, produces a source gas 3
containing SOZ,
Oz, and Nz (and water vapor if, for example, the air and/or the raw sulfur
material
contains water, or the sulfur-containing raw material is HzS).
It should be recognized that the process of this invention may be practiced
with a wide range of SOZ concentration in the source gas 3 (i.e., the source
gas 3 may
contain from about 0.1 to about 100 mole% SOZ). In some embodiments, for
example, the process is used in conjunction with other manufacturing processes
which
either need to reduce or eliminate the sulfur content in a particular
material, or need to
reduce or eliminate a sulfur-containing material in a waste stream. As
suggested
above, this process provides, for example, a practical way to utilize the SOz
which is
produced as an off gas when a metal ore is roasted or smelted during a metal
recovery
operation. This process also, for example, provides a practical way to utilize
spent
HzS04. In most of these SOZ salvage processes, the SOZ concentration in the
source
gas 3 is typically less than about 11 mole%, and more typically from about 0.1
to
about S mole%.
Because there are greater operational and capital costs associated with larger
process equipment, it is often preferable to minimize the volume of the SOZ
source
gas 3, while also increasing the concentration of SOZ in the source gas 3. In
a
particularly preferred embodiment, this is achieved by using elemental sulfur
as the
raw material 6. When elemental sulfur is burned in air, for example, SOZ
concentrations of from about 11 to about 21 mole% (and more typically, from
about
15 to about 20 mole%) may be obtained.
Regardless of the content of the oxygen source 12, it is also preferable to
minimize the volume of the source gas 3 by burning the elemental sulfur in the
least
amount of OZ necessary to allow substantially complete conversion of the
elemental
sulfur. In other words, the amount of the oxygen source 12 fed into the
combustion
zone 9 of the sulfur burner preferably is the amount necessary to maintain the
molar
ratio of OZ to elemental sulfur at slightly greater than about 1.0, more
preferably from
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
12
about 1.05 to about 1.3; and most preferably about 1.05 to about 1.1. In most
embodiments, it is preferred for the Oz concentration in the source gas 3 to
be from
about 0.5 to about 5 mole%, more preferably from about 0.5 to about 3 mole%,
and
most preferably from about 0.5 to about 2 mole%.
B. Sulfur Dioxide Gas StrenEthening
The SOZ-containing source gas 3 is preferably introduced into an SOZ
absorption/stripping zone to remove and recover SOZ in the form of an SOz
enriched
gas (i.e., a gas having an increased SOZ content relative to the source gas
3).
If the source gas 3 is at an elevated temperature (i. e., greater than about
50°C)
and/or contains entrained particulate impurities, it is generally preferred to
first
condition the source gas 3 to cool the gas 3 and remove particulates from the
gas 3
before introducing it into the SOz absorption/desorption zone. There are a
variety of
well-known techniques which may be used to condition the source gas 3. For
example, if the source gas 3 is a combustion gas exiting a sulfur burner, its
temperature is typically from about 900 to about 1600°C, and more
typically from
about 1050 to about 1600°C. This gas 3 may, for example, be cooled by:
(a) passing
the gas 3 through an indirect heat exchanger where heat from the gas 3 is
used, for
example, to the preheat the oxygen source 12 (e.g., air) being used in the
combustion
chamber 9, thereby reducing fuel costs in heating the oxygen source 12 with an
external source; (b) by passing the gas 3 through a waste heat boiler where it
is cooled
by generation of high pressure steam (i.e., steam having a pressure of at
least about 27
bar (gauge)); and/or (c) passing the gas 3 through a humidifying tower and one
or
more indirect heat exchangers, where it is further cooled with, for example,
cooling
tower water. Particularly where the source gas 3 is formed from spent sulfuric
acid or
is the off gas from a metal roasting or smelting operation, an electrostatic
precipitator
is often used to remove particulates from the gas after it is cooled.
Alternatively, such
a gas 3 may be conditioned by passing the gas 3 through one or more reverse
jet
scrubbers of the type, for example, sold by Monsanto Enviro-Chem Systems, Inc.
(St.
Louis, MO, USA) under the trademark "DYNAWAVE". It should be noted that a
portion (e.g., 5-10%) of the conditioned source gas 3 may be recycled back to
the
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
13
combustion zone 9 (particularly a sulfur burner) to control the temperature in
the
combustion zone 9 below a desired maximum temperature.
Preferably, in the first step of the SOZ absorption/desorption process, the
SOZ
containing source gas 3 is contacted with a liquid SOZ absorption solvent 15
in an SOZ
absorption zone 18. The liquid SOZ absorption solvent 15 selectively absorbs
SOz
from the source gas 3, thereby transfernng SOz from the source gas 3 to the
SOZ
absorption solvent 15 and producing an SOZ depleted exhaust stripper gas 21
(from
which the SOz has been substantially removed) and an SOZ enriched absorption
solvent 24. The SOZ-enriched absorption solvent 24, in turn, is stripped of
SOZ in an
SOZ stripper zone 27 to yield an SOZ-enriched stripper gas 30 and an SOZ-
depleted
solvent 33 (which preferably is subsequently recycled back to the SOZ
absorption zone
18 for further selective absorption of SOz from the source gas 3).
The liquid SOz absorption solvent 15 may be either a physical or a chemical
solvent. Physical solvents, however, are generally more preferred. Suitable
absorbents include various organic absorbents (e.g., tetraethylene glycol
dimethyl
ether), and aqueous solutions of alkali metals (e.g., a sodium
sulfite/bisulfite solution).
An example of a suitable physical sulfur dioxide absorption solvent is one
comprising tetra ethylene glycol diethel ether such as that disclosed and
utilized in the
sulfur dioxide recovery processes described in U.S. Patent No. 4,659,553
(Line) and
U.S. Patent No. 4,795,553 (Hensel et al.), the entire disclosures of which are
incorporated herein by reference. The liquid sulfur dioxide absorbent
preferably
contains more than 50% by weight tetra ethylene glycol diethel ether. Such a
liquid
sulfur dioxide absorbent suitably comprises, on a dry weight basis, from about
60% to
about 80% tetra ethylene glycol diethel ether, from about 15% to about 25%
triethylene glycol diethel ether, from about 2.5% to about 7.5% pentaethylene
glycol
diethel ether and from about 2.5% to about 7.5% mono ethers. The circulating
tetra
ethylene glycol diethel ether-containing absorbent may contain water, for
example, up
to about 10% by weight. Use of sulfur dioxide absorbents based on tetra
ethylene
glycol diethel ether in the absorption and stripping stages of a sulfur
dioxide recovery
system, including the process equipment and operating conditions employed, is
described in U.S. Patent Nos. 4,659,553 (Line) and U.S. Patent No. 4,795,553
(Hensel
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
14
et al.) and may be applied by one skilled in the art in the practice of the
present
invention.
Another example of suitable SOz absorption solvents include aqueous
solutions of various amines. Exemplary amine absorbing agents include, for
example,
aniline derivatives (e.g., dimethylaniline), alkanolamines (e.g.,
diethanolamine,
triethanolamine, tripropanolamine, and tributanolamine),
tetrahydroxyethylalkylenediamines (e.g., tetrahydroxymethylenediamine,
tetrahydroxyethylethylenediamine, tetrahydroxyethyl-1,3-propylenediamine,
tetrahydroxyethyl-1,2-propylenediamine, tetrahydroxyethyl-1,5-
pentylpentylenediamine), and heterocyclic diamines (e.g., piperazine;
dimethylpiperazine; N,N'-bis(2-hydroxyethyl)piperazine; -methylpyrrolidone;
and
sulfonate as disclosed in U.S. Patent No. 3,764,665, Groenendael et al. the
entire
disclosure which is herein incorporated by reference).
An even more preferred traditionally used absorbing agent is a half salt of a
diamine having the following formula (I):
R~ R3
NAN
Rzi ~Rq
(I),
wherein A is alkylene having 2 or 3 carbon atoms; R', R2, R3, and R4 may be
the same
or different, and can be hydrogen, alkyl (preferably having from 1 to about 8
carbon
atoms, and including cycloalkyls), hydroxyalkyl (preferably having from 2 to
about 8
carbon atoms), aralkyl (preferably having from about 7 to about 20 carbon
atoms),
aryl (preferably monocyclic or bicyclic), or alkylaryl (preferably having from
about 7
to about 20 carbon atoms). It should be noted that any of R', R2, R3, and R4
may
together form cyclic structures. The free nitrogen of the half salt preferably
has a pKa
of from about 4.5 to about 7.3. Examples of particularly preferred diamines
include
the sulfite half salts of N,N',N'-(trimethyl)-N(2-
hydroxyethyl)ethylenediamine;
N,N,N',N'-tetramethylethylenediamine; N,N,N',N'-tetrakis(2-
hydroxyethyl)ethylenediamine; N-(2-hydroxyethyl)ethylenediamine; N,N'-
dimethylpiperazine; N,N,N',N'-tetrakis(2-hydroxyethyl)-1,3-diaminopropane; and
N,N'-dimethyl-N,N-bis(2-hydroxyethyl)ethylenediamine. These half salt diamine
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
absorbents are described by Hakka in U.S. Patent No. 5,019,361 (incorporated
herein
by reference).
In the embodiments using an aqueous solution comprising an amine or an
amine salt, the absorption solvent 15 preferably comprises an aqueous solution
S containing from about 20 to about 40 weight% of the absorbing agent on an
amine
(rather than an amine salt) basis. The SOz-enriched absorption solvent 24, in
turn,
preferably has an SOz/amine-absorbing-agent weight ratio of from about 0.1:1
to
about 0.25:1.
The above-listed traditional SOZ absorbents are often hampered by one or
10 more shortcomings. These shortcomings include, for example, relatively low
SOZ
absorption capacity and the tendency to absorb substantial quantities of water
vapor
from the source gas 3. Absorption of substantial quantities of water, in turn,
can lead
to a significant reduction in the SOZ absorption capacity of the SOZ
absorption solvent
15, thereby requiring a greater flow of the SOZ absorption solvent 15. Such
water
15 absorption can also lead to excessive corrosion of the equipment used in
the SOZ
absorption/stripping process. Further, such absorption requires energy and
capital
input for the water to be separated from the SOZ-depleted solvent 33 so that
the
solvent 33 may be recycled back to the SOz absorption zone 18 and used for
further
SOZ absorption.
In a particularly preferred embodiment, the SOZ absorption solvent 15
comprises an organic phosphorous compound, as described in U.S. Patent No.
5,851,265 (Burmaster et al.) which the entire disclosure is herein
incorporated by
reference). In this embodiment, the SOz absorption solvent 15 preferably
comprises a
phosphate triester, phosphonate diester, phosphinate monoester, or a mixture
thereof.
The substituents bonded to the phosphorous atom, as well as the organic
radicals of
the ester functionality, in the compounds are preferably independently aryl or
Cl to C8
alkyl (i.e., an alkyl group containing from 1 to 8 carbon atoms). Examples of
suitable
phosphate triesters include: tributyl phosphate, tripentyl phosphate, trihexyl
phosphate, and triphenyl phosphate. Examples of suitable phosphinate
monoesters
include: butyl dibutyl phosphinate, pentyl dipentyl phosphinate, hexyl dihexyl
phosphinate, and phenyl diphenyl phosphinate.
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
16
In accordance with an even more preferred embodiment of the present
invention, the SOZ absorption solvent 15 comprises at least one substantially
water-
immiscible organic phosphonate diester having formula (II)
0
i1
Rs0- I -R~
o R 2 (II),
wherein R', R2, and R3 are independently aryl or C~ to Cg alkyl, with R', Rz,
and R3
being selected such that (1) the organic phosphonate diester has a vapor
pressure of
less than about 1 Pa at 25 ° C, and (2) the solubility of water in the
organic
phosphonate diester is less than about 10 weight% at 25 ° C.
Preferably, the organic
phosphonate is a dialkyl alkyl phosphonate, and R', RZ, and R3 are
independently C,
to C6 alkyl. More preferably, to simplify preparation and reduce the
manufacturing
costs of the phosphonate deters solvent, R', RZ, and R3 are identical, with
each
containing at least 4 carbon atoms. Examples of suitable organic phosphonate
deters
for use in the practice of the present invention include dibutyl butyl
phosphonate,
dipentyl pantile phosphonate, dihexyl hassle phosphonate and diphenyl phenyl
1 S phosphonate. In accordance with an especially preferred embodiment of the
present
invention, the SOZ absorption solvent 15 comprises dibutyl butyl phosphonate.
Dibutyl butyl phosphonate is a neutral diester of phosphonic acid, and is a
clear,
colorless liquid with a relatively low viscosity and very mild odor. Dibutyl
butyl
phosphonate has a molecular weight of 250.3 and a vapor pressure of about 0.1
Pa at
25 ° C: The solubility of water in dibutyl butyl phosphonate is about
5.5 weight% at
° C.
An SOZ absorption solvent comprising at least one organic phosphonate diester
as defined above tends to be more preferred because such a solvent typically
possesses a combination of characteristics which renders it particularly
useful in an
25 SOZ absorption/desorption process, including: (1) increased SOZ solubility,
especially
at low partial pressures of SOZ in the source gas 3; (2) high heats of
solution, which
reduce the amount of energy required for stripping SOZ from the SOZ-enriched
absorption solvent 24; (3) low melting points, so that the solvent 15 will
remain a
liquid over a wide range of process temperatures; (4) low viscosity, which
allows the
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
17
size of both thermal and absorption/stripping equipment to be reduced; (S) low
vapor
pressure, which reduces solvent 15 losses; (6) decreased tendency to react
with water
and undergo hydrolysis; and (7) being substantially water immiscible (i.e.,
non-
hygroscopic) such that the solubility of water in the solvent 15 is decreased.
The fact
that the organic phosphonate diesters are substantially water immiscible is
particularly
advantageous in the practice of the present invention. This characteristic
provides an
SOZ absorption solvent 15 which does not absorb excessive amounts of water
from the
SOZ-containing source gas 3.
The SOz absorption zone 18 preferably comprises a means for promoting mass
transfer between the gas and liquid phases, and more preferably comprises a
bed of
random packings such as saddles or rings in a vertical tower. Preferably, the
source
gas 3 is contacted countercurrently with the SOz absorption solvent 15. In
such an
embodiment, the source gas 3 is preferably introduced through an inlet near
the
bottom of the SOz absorption zone 18, and the SOZ absorption solvent 15 is
introduced through an inlet near the top of the SOZ absorption zone 18 and
distributed
over the packing. The SOZ enriched absorption solvent 24 is then withdrawn
from an
outlet near the bottom of the SOZ absorption zone 18, and the exhaust gas
substantially free of SOZ (i.e., the SOZ-depleted gas 21) is removed from an
outlet near
the top of the SOZ absorption zone 18. Although the SOZ absorption zone 18 may
comprise a conventional, randomly packed tower, those skilled in the art will
appreciate that other configurations may be suitably used as well. For
example, the
tower may contain structured packing or comprise a tray tower, in either of
which the
process streams preferably flow countercurrently.
When the above-described solvents comprising an organic phosphorus
compound are used, the SOZ absorption zone 18 preferably is operated at an
average
temperature of from about 10 to about 60°C (more preferably from about
10 to about
50°C, and most preferably from about 30 to about 40°C), and a
pressure of from
about SO to about 150 kPa (absolute). It should be recognized that although
pressure
increases the amount of SOZ that the SOZ absorption solvent 15 can absorb, the
absorption can alternatively be carried out at a relatively low pressure,
thereby
reducing equipment costs.
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
18
Condensation of water vapor from the source gas 3 in the SOz absorption zone
18 may lead to formation of a separate water phase, which could increase the
corrosion rate of metallic process equipment and complicate later removal of
the
absorbed SOZ in the subsequent solvent regeneration step. To avoid such
condensation, the temperature of the solvent 15 introduced into the absorption
zone 18
preferably is above the dew point temperature of the source gas 3 fed into the
absorption zone 18.
The mass flow rate ratio (L/G) of the SOZ absorption solvent 15 and the source
gas 3 necessary to achieve substantial transfer of SOz from the source gas 3
to the SOZ
absorption solvent 15 in the absorption zone 18 may be determined by
conventional
design practice. Preferably, the SOZ absorption zone 18 is designed and
operated such
that the SOz content of the SOZ-depleted gas 21 is less than about 400 ppmv,
more
preferably less than about 200 ppmv, and most preferably less than about 150
ppmv.
This trace amount of SO2, along with most of the Oz, inert gases (e.g., NZ),
and water
vapor contained in the source gas 3, are eliminated from the system as part of
the SOZ-
depleted gas 21 vented from the top of the SOz absorption zone 18. If
necessary to
achieve satisfactory emission standards, the SOZ-depleted gas 21 may be passed
through a mist eliminator for recovery of entrained liquid before being
discharged
through a stack.
Use of the highly efficient organic phosphorous solvents discussed above
allows the concentration of the SOz in the SOZ-enriched stripper gas 30
exiting the
stripper zone 27 to be significantly greater than the concentration of the SOZ
in the
source gas 3 fed to the system. For example, for source gases containing from
about
0.1 to about 5 percent by volume SOz, the process of the present invention may
be
operated such that the ratio of the SOz molar concentration in the in the SOZ-
enriched
stripper gas 30 to the SOZ molar concentration in the source gas 3 is greater
than about
1.1:1, preferably at least about 2.75:1, more preferably at least about 4:1,
even more
preferably at least about 7:1, and most preferably at least about 10:1. It
should be
recognized that even greater ratios may often be achieved, depending on the
SOZ
concentration of the source gas 3. Generally, it is preferred that at least 67
mole%
(more preferably at least about 75 mole%, still more preferably at least about
85
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
19
mole%, and most preferably at least about 90 mole%) of the SOZ-enriched
stripper gas
30 consist of SOz.
Various methods for stripping SOZ from the SOZ-enriched absorption solvent
24 may be used. For example, SOz may be stripped by contacting the SOZ-
enriched
absorption solvent 24 with a non-condensable, oxygen-containing stripping gas
36
such that SOZ is transferred from the SOZ-enriched absorption solvent 24 to
the
stripping gas 36 to produce the SOz-enriched stripper gas 30 and the SOZ-
depleted
absorption solvent 33. Preferably, the non-condensable, oxygen-containing
stripping
gas 36 comprises air. It should be recognized that one of the advantages
provided by
the above-described solvents comprising organic phosphorous compounds (and
especially solvents comprising phosphonate diesters) is their inherent flame
retarding
property and resistance to oxidation. Thus, unlike some organic solvents used
in
conventional SOZ absorption/desorption cycles (e.g., tetraethylene glycol
dimethyl
ether), the organic solvents utilized in the present invention can be readily
stripped of
SOZ using an oxygen-containing stripping gas with minimal risk of solvent
degradation or explosion.
The SOz stripper zone 27 preferably comprises a means for promoting mass
transfer between the gas and liquid phases. Like the SOZ absorption zone 18,
the SOZ
stripper zone 27 preferably comprises a bed of conventional random packing in
a
vertical tower. To maximize transfer of SO2, the SOZ-enriched absorption
solvent 24
is preferably contacted countercurrently with the SOZ stripping gas 36. In
this
embodiment, a non-condensable, oxygen-containing SOZ stripping gas 36
preferably
is introduced through an inlet near the bottom of the SOZ stripper zone 27,
and the
SOz-enriched absorption solvent 24 is introduced through a liquid inlet near
the top of
the SOZ stripper zone 27 and distributed over the packing material. The SOZ-
depleted
absorption solvent 33 is then preferably withdrawn from an outlet near the
bottom of
the SOZ stripper zone 27, and the SOZ-enriched stripper gas 30 is removed from
an
outlet near the top of the SOz stripper zone 27. In a particularly preferred
embodiment, the SOZ-depleted absorption solvent 33 is recycled back to the
solvent
inlet near the top of the SOZ absorption zone 18, thereby serving as the SOz
absorption
solvent 15 for further absorption of SOz from the source gas 3. Although a
conventional packed tower is typically preferred, those skilled in the art
will
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
appreciate that the SOZ stripper zone 27, like the SOz absorption zone 18, may
have
other suitable configurations, including structured packing or a tray tower.
The mass flow rate ratio (L/G) of the SOz-enriched absorption solvent 24 to
the stripping gas 36 necessary to achieve substantial transfer of SOZ from the
SOZ
5 enriched absorption solvent 24 to the stripper gas 36 may be determined by
conventional design practice. Preferably, essentially all (i.e., at least
about 90%, and
more preferably at least about 95%) of the SOZ contained in the SOZ-enriched
absorption solvent 24 is transferred to the stripper gas 36.
The SOZ-enriched stripper gas 30 exiting the top of the SOZ stripper zone 27
is
10 preferably passed to an overhead condenser, and a portion of any water
vapor
contained in the SOZ-enriched stripper gas 30 is condensed by transfer of heat
in the
SOZ-enriched stripper gas 30 to cooling water. This condensate and the
remainder of
the SOz-enriched stripper gas 30 are then preferably transferred to liquid/gas
phase
separator. In this instance, the cooled SOZ enriched stripper gas 30 exits the
separator
15 and a liquid stream comprising the condensate is refluxed and introduced
into an
upper section of the tower containing the SOZ stripper zone 27 over a second
bed of
packing material. Solvent that may have been vaporized in the SOZ stripper
zone 27
may also be condensed in the overhead condenser and form part of the refluxed
condensate. However, to avoid formation of two liquid phases in the separator,
it is
20 preferred to operate the condenser such that the condensate refluxed to the
stripper
consists essentially of water vapor condensed from the SOZ-enriched stripper
gas 30.
Alternatively, the SOz-enriched absorption solvent 24 can be stripped by steam
distillation (i.e., contacting the SOz-enriched absorption solvent 24 with
live steam
introduced into the bottom of the SOz stripper zone 27) to recover the SOZ
from the
SOz-enriched absorption solvent 24. Regardless of how the SOZ
stripping/solvent
regeneration step is conducted, the SOZ preferably is stripped from the SOZ-
enriched
absorption solvent 24 under non-reducing conditions.
To promote desorption of SOZ and avoid thermal degradation of the SOz
absorption solvent 15, the SOZ stripper zone 27 preferably is operated at an
average
temperature of from about 80 to about 120°C, and more preferably from
about 90 to
about 110 ° C. When air stripping is employed, the preferred operating
pressure in the
SOZ stripper zone 27 is from about 20 to about 150 kPa (absolute).
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
21
Temperature control within the SOz absorption zone 18 and SOZ stripper zone
27 may be achieved by controlling the temperature of the various process
streams fed
to these apparatus. Preferably, the temperature in the SOZ stripper zone 27 is
maintained within the desired range by controlling only the temperature of the
SOZ-
enriched absorption solvent 24, while air is introduced at from about 20 to
about
120°C as the non-condensable, oxygen-containing stripping gas 36. As
noted above,
the SOZ-enriched absorption solvent 24 exiting the SOZ absorption zone 18
preferably
is at a temperature of from about 10 to about 60°C, more preferably
from about 10 to
about 50°C, and most preferably from about 30 to about 40°C.
This SOz-enriched
absorption solvent 24 is preferably passed through a solvent heat interchanges
39
where it is preheated by indirect transfer of heat from the SOZ-depleted
solvent 33
being recycled from the SOZ stripper zone 27 to the SOZ absorption zone 18
(this, in
turn, cools the SOz-depleted solvent 33 exiting the SOZ stripper zone 27,
which is
typically at a temperature from about 80 to about 120°C). If further
heating is
required to achieve the desired temperature in the SOz stripper zone 27, the
preheated
SOZ-enriched absorption solvent 24 leaving the interchanges 39 may be passed
through a solvent heater, where it is further heated by indirect heat exchange
with
steam. If further cooling of the SOZ-depleted solvent 33 is required to
maintain the
desired temperature in the SOZ absorption zone 18, the SOZ-depleted solvent 33
leaving the interchanges 39 may be passed through a solvent cooler where it is
further
cooled by indirect heat exchange with cooling tower water. It should be
recognized
that the use of a solvent interchanges 39 reduces the energy demands of the
solvent
heater, and reduces the cooling water required in the solvent cooler.
During the course of operation, inorganic salts and strong acids may
accumulate in the solvent circulated between the SOZ absorption zone 18 and
the S0z
stripper zone 27. When this occurs, a purge stream may be periodically or
continuously removed from the SOz-depleted solvent 33 and directed to a
solvent
purification vessel. An aqueous wash stream, such as water or a mildly
alkaline
aqueous solution (e.g., a sodium bicarbonate solution), is also introduced
into the
purification vessel and contacted with the purge stream. The resulting two-
phase
mixture is then decanted to separate the aqueous phase containing the
inorganic salt
contaminants from the organic phase comprising SOZ-depleted solvent 33 having
a
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
22
reduced contaminant concentration. A waste stream comprising the aqueous waste
is
discharged from the purification vessel, while a liquid stream comprising the
purified
SOZ absorption solvent is returned to the remaining SOz-depleted solvent 33
routed
back to the SOZ absorption zone 18. The quantity of solvent 33 treated in this
manner
preferably is sufficient to maintain the contaminant concentration in the
circulating
solvent 33 at a level low enough to provide low process equipment corrosion
rates and
not materially compromise SOZ absorption efficiency. It should be understood
that
the washing of the SOz-depleted solvent 33 may be carried out in a batch or a
continuous fashion. If the SOZ-depleted solvent 33 is washed continuously, a
suitable
liquid-liquid phase separator (e.g., a centrifugal contactor) may be used to
separate the
aqueous waste and purified organic phases.
It should be recognized that the SOZ absorption/stripping zones are
particularly
useful when the source gas 3 has a relatively weak SOz concentration (i.e.,
from about
0.1 to about 11 mole%, and even more so at from about 0.1 to about S mole%)
because they can be used to remove the inert gases (most notably, NZ) from the
source
gas 3 and thereby significantly increase the SOZ concentration. One advantage
of
having a greater SOZ concentration is that it allows for a smaller volume of
gas to be
handled during the process, thereby permitting the use of smaller equipment
(which
has cheaper capital and operational costs). Also, by removing inert gases
during the
SOZ absorption/stripping process and then combining the SOZ-enriched stripper
gas 30
with a fresh oxygen source 42 (and/or providing oxygen by way of the stripper
gas 36
itself), the oxygen concentration in the gas 30 can be increased without
necessarily
increasing the total volume of the SOz-containing gas. This process also
provides a
mechanism for delaying the introduction of the oxygen needed for the SOZ
oxidation
until the oxygen is actually needed (i.e., in the catalytic converter 45).
This is
particularly advantageous because, under such a scheme, only the amount of
oxygen
needed for producing the SOZ has to be introduced into combustion zone 9.
Thus, the
combustion zone 9 and other equipment upstream of the converter 45 does not
have to
be sized to handle the oxygen-containing gas which is required for the SOZ
oxidation.
Because smaller equipment can be used upstream of the catalytic converter 45,
significant capital and operational expenses can be avoided.
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
23
Because the SOZ absorption/stripping zones may be used to remove water
from the source gas 3, they are particularly useful in embodiments where it is
desirable to remove water vapor from the source gas 3 so that the SOZ
containing gas
fed to the catalytic converter 45 contains essentially no water vapor. Such
S embodiments include, for example, embodiments where the converter 45 and/or
equipment downstream of the converter 45 are made of material which is
vulnerable
to corrosion caused by sulfuric acid formed by the vapor phase reaction of
water
vapor with S03. The SOZ absorption/stripping zones are also particularly
useful in
embodiments where the H20/SOZ molar ratio in the source gas 3 is greater than
the
molar ratio of Hz0/S03 in the desired acid product 51 (this situation may
especially
occur when the source gas 3 is prepared from spent acid, the off gas of a
metal
roasting or smelting operation, or HZS). For example, if the desired product
acid
concentration is 98.5 weight%, the H20/S03 molar ratio in the conversion gas
54 fed
to the S03 absorption zone 57 cannot exceed about 1.08. Consequently, if there
is no
water removal in the system between the source gas 3 and the S03 absorption
zone 57,
the H20/SOZ molar ratio in the source gas 3 also preferably does not exceed
about
1.08. The SOZ absorption/stripping zone may be used (alone or together with,
for
example, a drying tower and/or a cooling towers) which condenses liquid out of
the
source gas 3) to ensure that the H20/S03 molar ratio is maintained below this
value.
C. Oxidation of Sulfur Dioxide to Sulfur Trioxide
The SOZ-enriched stripper gas 30 is preferably combined with a source of
molecular oxygen 42 to form a converter feed gas 48, which is then passed
through a
catalytic converter 45 to oxidize the SOZ to form a conversion gas 54
containing 503.
The oxygen source 42 may be any oxygen-containing gas. As used herein, an
"oxygen-containing gas" is a gas comprising molecular oxygen (Oz), which
optionally
may also comprise one or more diluents which are non-reactive with Oz, SOz,
S03,
and sulfuric acid under the reaction conditions. Examples of such gases are
air, pure
molecular oxygen, or molecular oxygen diluted with nitrogen and/or another
inert
gas(es). For economic reasons, the oxygen source 42 preferably is air or
essentially
pure molecular oxygen, with air being most preferred. It should be recognized
that
the stripper gas 36 advantageously may provide part (or, in some instances,
all) of the
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
24
oxygen required in the converter feed gas 48 if the stripper gas 36 is air or
another OZ-
containing gas.
In a particularly preferred embodiment, the converter feed gas 48 contains
essentially no water vapor, thereby reducing the risk of corrosion to process
equipment downstream. Here, if the SOZ-enriched stripper gas 30 is wet, it
preferably
is dried, such as by being contacted with concentrated sulfuric acid in a
drying tower
before being introduced into the catalytic converter 45. If the SOz absorption
solvent
is an organic phosphorous solvent. as described above and dry air is used to
strip
the SOZ from the SOZ-enriched absorption solvent 24, the SOZ enriched stripper
gas 30
10 often does not need to be dried before being routed to the converter 45.
The catalytic converter typically comprises at least two catalyst beds in
series
through which the converter feed gas 48 passes. The catalyst in each of the
catalyst
beds may generally be any material which catalyzes the oxidation reaction of
SOZ to
S03. Conventionally used catalysts include, for example, various vanadium
15 compounds, platinum compounds (e.g., platinized asbestos), silver
compounds, fernc
oxide, chromium oxide, etc. In a particularly preferred embodiment, the
catalyst
comprises vanadium or a combination of vanadium and cesium. In the most
preferred
embodiment, the catalyst comprises vanadium pentoxide (V205).
As noted above, in the more preferred embodiments of this invention, the SOZ-
enriched stripper gas 30 is normally at a temperature of no greater than about
120°C
upon exiting the SOZ stripper zone 27. And this temperature is typically
decreased
when the SOz-enriched stripper gas 30 is combined with the oxygen source 42,
which
is often near ambient temperature. The more preferred oxidation catalysts,
however,
have an activation temperature which is significantly greater than
120°C. Thus, the
converter feed gas 48 is often preferably heated before being introduced into
the first
catalyst bed 60 of the converter 45. On the other hand, because the oxidation
of SOz
to S03 is an exothermic reaction, the reaction is also preferably controlled
so that the
temperature of the catalyst bed 60 does not increase so much as to deactivate
the
catalyst and/or shift the reaction equilibrium to favor the reverse reaction.
When, for example, a vanadium-containing catalyst (e.g., V205) is used, it is
typically preferred for the converter feed gas 48 and partial conversion gas
69 and 72
entering catalyst beds 60, 63 and 66, respectively, to have a temperature of
from
07-01-2002 -2002 MOH 03:36 PM F~ N0, US00300~
CA 02387988 2002-04-04 -~ --
about 410 to about 450°C (even more preferably from about 415 to about
435°C), and
then to control the temperattu~e in each bed so that the gas ienzpetature
approaches, but
does not exceed, about 650°C (more preCECably about 630"C), Temperature
control in
the converter45 is preferably accornplishedby maintaining the SOZ strength
(i.e., the
5 S0~ concentration) in the converter feed gas 48 and partial conversion gas
69 and 72
i~lroduced into catalyst beds 60, G3 and 66, respectively, at no greaser stout
about 15
mole%, nzoro preferably no greater than about 13.5 mole%, and still mere
preferably
no greater than about 12 mole%. Tt is also preferred that lhc amount of the
oxygen
source 4Z combined with the SOi-e~trichcd stripper gas 30 be such that the
molar ratio
10 of Oi to SO~ in the converter Feed gas 48 and partial conversion gas 69 and
72
intmdttced into catalyst beds 60, G3 smd 6G, respectively, be greater than
about 0.2:I,
snore preferably at least about O,S:I, even more preferably at least about
U.7:1, still
even more profcrably from about 0.7:1 to about 1.4:1, and most preferably from
about
0.9:1 to about 1.2;I.
15 l3ccause the SO= oxidation reaction is exvthe2mic, it is oiien advantageous
to
use an indirect heat exchanrer(s) 75 and 78 to heat the converter feed gas 4g
v~~iih the
partial conversion gas 81 and 84 exitiltg the catalyst beds 60 and 63 of the
catalytic ,
coxtvertcr 45. Generally, if the convener feed gas 48 contains at least about
5 mole%
5Ui (and particularly at least about 8 mole%) and an excess amount of Oi, the
20 oxidation reaction can evolve sufficient heat for inereasa~g the
temperature of tho
converter feed gas 48 to the activRtiou ten~pcrature of tlic oxidation
catalyst,. thus
avoiding thQ need for any cxtraiieotts heat source for heating the converter
feed gas 48
after starhtp (i.E., making U,e converter 45 energy set f sustaining or
"autolhcrmaT~.
Thus, the converter feed gas 48 preferably has an SOi concentration of from
about 7
25 to about 15 mole%, more preferably from aboua7 to about 13.5 mole%, even
more
preferably from about 7 to about 12 mole%, still even more preferably from
about 10
to about 12 mole%, and most preferably about l 1.5 male%. The converter feed
bas
48 preferably is preheated using two indirect heat exchangers in series;
first, a cold
heat exchanger 78 is which the converter feed gas 48 is preheated by transfer
of heat
from the partial conversion gas 84 leaving the second bed 63 of the converter
45; and,
second, a hot heat exchanger 75 in which the convener feed gas 48 is further
heated
by transfer of heat from the partial conversion gas Si leaving the first
catalyst bets GO
AMENDED SHEET
EtfIPf .ZE?1t:~7inlltU~.JZ Lt:4~ I-mof nr ~'~'II'~ D fXl~
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
26
of the converter 45. In the embodiments where the source gas 3 is a hot gas
exiting
from a combustion chamber 9, the converter feed gas 48 may also (or
alternatively),
for example, be heated by passing it through an indirect heat exchanger to
transfer
heat from the source gas 3 to the converter feed gas 48.
In a particularly preferred embodiment, the SOZ-enriched stripper gas 30 is
split into at least two streams. Preferably, a portion, preferably at least
about 30%
(more preferably at least about 40%, and even more preferably at least about
50%) of
the SOZ enriched stripper gas 30 is combined with the oxygen source 42 (either
before
or after being preheated, and preferably before) to form the converter feed
gas 48,
which, in turn, is introduced into the first catalyst bed 60 of the converter
45 wherein
a portion of the SOZ content of the gas 48 is oxidized to S03 to form a
partial
conversion gas 81 containing S03 and residual SO2. The cooled partial
conversion
gas exiting indirect heat exchanger 75 is then combined with a second portion
31 of
the SOZ enriched stripper gas 30 to fortify the SOZ concentration in the
partial
conversion gas. The fortified partial conversion gas 69 is then passed through
at least
one additional catalyst bed (63 and 66 in Fig. 1) to oxidize further SOZ in
the gas 69.
Fortifying the SOZ gas strength of the partial conversion gas is advantageous
because
it significantly increases the capacity of the converter 45. As noted above,
the
maximum SOz concentration of the gas fed into the first catalyst bed 60 is
normally
limited (in the presence of excess oxygen) to about 15 mole% (more typically
about
13.5 mole%, and even more typically about 12 mole%) because greater SOZ
concentrations will typically cause too much heat to be released during the
oxidation
reaction, thereby causing the catalyst to deactivate and/or the reaction
equilibrium to
shift unfavorably. However, by adding additional SOZ to the partial conversion
gas
fed into the second catalyst bed 63 (and, in some embodiments, a subsequent
catalyst
bed as well), that additional SOz may be oxidized without causing the
temperature in
any bed to increase to an undesirable level (as long as the amount of SOZ
added does
not cause the SOZ concentration in the fortified partial conversion gas 69 to
be greater
than about 15 mole%). Preferably, the amount of SOZ added to the partial
conversion
gas increases the SOZ concentration to no greater about 15 mole%, more
preferably
from about 7 to about 13.5 mole%, even more preferably from about 7 to about
12
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
27
mole%, still even more preferably from about 10 to about 12 mole%, and most
preferably about 11.5 mole%.
Although it is especially preferred for the partial conversion gas to be
fortified
with the entire portion of the SOZ-enriched stripper gas 31 which is not fed
into the
first catalyst bed 60, it should be recognized that this invention also
encompasses
embodiments wherein the SOZ-enriched stripper gas 30 is split into more than 2
portions and subsequently used to fortify the feed gas to more than one
catalyst bed of
the converter. Thus, where the catalytic converter has 4 catalyst beds in
series, the
SOZ-enriched stripper gas may, for example, be split into three portions. To
illustrate,
in one such embodiment, the first portion of the SOz-enriched stripper gas is
combined with the oxygen source to form the converter feed gas, which, in
turn, is
introduced into the first catalyst bed of the converter where SOZ in the gas
is oxidized
to form a partial conversion gas. This partial conversion gas is then combined
with
the second portion of the SOZ enriched stripper gas to fortify the SOZ
strength in the
partial conversion gas. The fortified partial conversion gas is then passed
through the
second catalyst bed to oxidize further SOZ and form a second partial
conversion gas.
This second partial conversion gas is then combined with the third portion of
the SOz-
enriched stripper gas to fortify the SOz strength in the second partial
conversion gas.
This fortified second partial conversion gas is then passed through the third
catalyst
bed to oxidize still further SOZ and form a third partial conversion gas. This
third
partial conversion gas is then passed through the fourth (i.e., the final)
catalyst bed to
oxidize at least a portion of any remaining SO2.
D. Production of Sulfuric Acid and/or Oleum from Sulfur Trioxide
The conversion gas 54 exiting the catalytic converter 45 preferably is
contacted with water or, more preferably, concentrated sulfuric acid 87
(preferably an
aqueous solution containing from about 96 to about 99.5 weight% HZS04, more
preferably from about 98.5 to about 99.5 weight%, and most preferably from
about 99
to about 99.5 weight%) in an S03 absorption zone 57 to absorb S03 from the
conversion gas 54, thereby forming an S03-depleted gas 90 and additional
sulfuric
acid and/or oleum 51. There is preferably also a heat recovery zone 93
associated
with the S03 absorption zone 57. This heat recovery zone 93 preferably
recovers
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
28
energy from the heat of absorption of the S03 in the S03 absorption zone 57.
Sulfur
trioxide absorption zones and heat recovery zones suitable for use in
accordance with
this invention are well-known in the art. See, e.g., McAlister et al., U.S.
Patent Nos.
4,670,242 and 4,576,813 (both incorporated herein by reference).
In a particularly preferred embodiment employing a heat recovery zone 93 in
association with an S03 absorption zone 57, the conversion gas 54 is cooled in
an
economizer to a temperature which is above the dew point of the conversion gas
54,
and then introduced into the lower portion of a vertical tower comprising the
S03
absorption zone 57. The S03 absorption zone 57 preferably comprises a bed of
random packing (although the S03 absorption zone 57 may alternatively comprise
another gas-liquid contacting device, such as a tray tower). Preferably, the
cooled
conversion gas 54 flows upward through the S03 absorption zone 57. At the same
time, hot, concentrated liquid sulfuric acid 87 is sprayed from the top of the
absorption zone 57 and flows downward through the packing. As the concentrated
sulfuric acid 87 and S03 countercurrently contact each other, the S03 is
absorbed into
the concentrated sulfuric acid 87. This concentrated sulfuric acid 87
preferably has a
temperature of greater than about 120°C. Such conditions tend to reduce
sulfuric acid
corrosiveness to alloys used in many conventional absorption towers, while
providing
a high degree of S03 absorption.
After passing through the absorption zone 57, the sulfuric acid concentration
in the sulfuric acid solution 96 is preferably greater than about 98 weight%
(more
preferably greater than about 98.5 weight%, even more preferably greater than
about
99 weight%, and most preferably from about 99 to about 100 weight%). It should
be
recognized that these preferred concentrations can be greater if the S03
absorption
zone 57 is operated at pressure significantly greater than atmospheric
pressure.
Because the absorption of S03 into the concentrated liquid sulfuric acid is an
exothermic process, the temperature of the liquid sulfuric acid increases as
the liquid
sulfuric acid becomes more concentrated while passing through the absorption
zone
57. In fact, while passing through the absorption zone 57, the temperature of
the
concentrated sulfuric acid preferably increases to a temperature of up to
about 250°C
(this preferred maximum temperature is greater at absorber pressures greater
than
atmospheric pressure). Consequently, the liquid sulfuric acid 96 preferably is
passed
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
29
through a heat recovery zone 93 (which may either be physically inside or
outside of
the absorption zone 57, and most preferably comprises an indirect heat
exchanger
outside the absorption zone 57) to remove the heat of absorption of the 503.
This heat
may, in turn, be used, for example, to generate low to medium pressure steam
(typically up to about 10.5 bar (gauge)) for use within the manufacturing
complex
surrounding the sulfuric acid plant or to generate electricity.
To minimize corrosion of the heat exchanger in the heat recovery zone 93, the
liquid sulfuric acid concentration preferably is at least about 99 weight%
throughout
the course of the heat transfer. It is also preferred that the temperature of
the liquid
sulfuric acid 96 throughout the heat exchanger be greater than about
130°C (more
preferably greater than about 140°C, and most preferably greater than
about 150°C)
where low pressure steam is desired (i.e., up to about 3.5 bar (gauge)), and
be greater
than about 150°C (more preferably greater than about 175°C, and
most preferably
greater than about 200°C) where medium pressure steam is desired (i.e.,
from about
6.5 to about 10.5 bar (gauge)). A portion of the sulfuric acid stream 96
preferably is
recovered as product 51. The remainder 99 preferably is diluted with water 102
(in
either liquid or vapor form) or dilute sulfuric acid, and reticulated to the
top of the
S03 absorption zone 57 to again be passed through the S03 absorption zone 57.
After the S03-depleted gas 90 exits from the top of the S03 absorption zone
57, the gas 90 may optionally be passed through a second S03 absorption zone
which
may be a second stage of the tower containing the first S03 absorption zone
57, or
may be located in a separate tower. The purpose of such a second stage or
tower is to
remove any residual S03 that remains in the S03-depleted gas 90. It should be
recognized, however, that in many instances, essentially all the S03 is
absorbed in the
primary S03 absorption zone 57, rendering a second stage or a second tower
unnecessary. And, even if a second stage or tower is used, it is typically not
economically productive to incorporate a heat exchanger to recover energy from
the
heat of absorption of the S03 in the second stage or second tower, given the
small
amount (if any) of S03 being absorbed there. Use of a second S03 absorption
zone is
described, for example, by McAlister, et al. in U.S. Patent No. 4,996,038
(incorporated herein by reference).
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
It should be recognized that the process of this invention may comprise more
than one S03 absorption zone such that partial conversion exiting an
intermediate
catalyst bed of the converter is contacted with water or a liquid comprising
sulfuric
acid to absorb S03 from the gas before the gas is passed through one or more
5 subsequent catalyst beds of the converter (i.e., the process may be used
with a system
comprising an intermediate S03 absorber). For example, where a catalytic
converter
comprising 4 catalyst beds is used, the partial conversion gas leaving the
second or
third bed may be passed through an intermediate S03 absorption zone (i. e., an
interpass absorption zone) for removal of S03 in the form of product acid
and/or
10 oleum. Gas exiting the intermediate absorption zone is then returned to the
next
downstream catalyst bed of the converter. Because the conversion of SOZ to S03
is an
equilibrium reaction, removal of S03 in the interpass absorption zone helps
drive the
reaction forward in the succeeding bed or beds of the converter to achieve
higher
conversions. Use of an intermediate S03 absorption zone, however, is normally
less
15 preferred in the practice of the present invention because it substantially
adds to the
capital and operating costs.
E. Recvclin~ the Tail Gas
In a particularly preferred embodiment of this invention, at least a portion
of
the S03-depleted gas 90 exiting the S03 absorption zone 57 (i.e., the tail
gas) is
20 recycled back to the SOZ absorption zone 18 and contacted with the SOZ
absorption
solvent 15 along with the source gas 3. In this manner, unconverted SOZ in the
tail
gas 90 is thereby recaptured in the S03-enriched absorption solvent 24 exiting
the SOZ
absorption zone 18, stripped from the S03-enriched absorption solvent 24 in
the SOz
stripper zone 27, and returned to the catalytic converter 45 as part of the
SOz-enriched
25 stripper gas 30 for ultimate recovery as product acid 51. In such an
embodiment, at
least a substantial portion of the inert gases and excess OZ in the recycled
tail gas 90
will be purged from the process in the SOz-depleted gas 21 exiting the SOz
absorption
zone 18.
Those skilled in the art will recognize that, depending on the efficiency of
the
30 converter 45, emission standards may be met by recycling less than all of
the tail gas
90 from the S03 absorption zone 57. In fact, depending on local prevailing
emission
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
31
standards, target emissions may be met by recycling 90%, 75%, or even SO% of
the
tail gas 90, with some resultant savings in energy costs for gas compression.
It is
ordinarily preferred, however, that substantially all the tail gas 90 be
recycled. While
non-condensable gases separated from the process gas in both the SOZ and S03
absorption zones must be purged to the atmosphere, emissions are confined to a
single
location (i.e., the SOz-depleted gas from the SOZ absorption zone) when the
entire tail
gas 90 is recycled back to the SOZ absorption zone 18. This facilitates both
monitoring and control of SOZ emissions. Moreover, by recycling all the tail
gas 90 to
the SOZ absorption zone 18, 99.7 percent or more of the SOz in the source gas
3 fed to
the SOz absorption zone 18 may ultimately be recovered as product acid 51,
even
where single-pass conversion efficiencies in the sulfuric acid plant are
relatively low.
In other words, by recycling all the tail gas 90 to the SOZ absorption zone
18, SOZ
emissions from the contact sulfuric acid plant may be essentially eliminated.
And,
recycle of the entire tail gas 90 allows the acid plant to be operated with a
single S03
absorption zone 57, entirely eliminating the interpass S03 absorption step
that has
become standard throughout much of the sulfuric acid industry as a means of
controlling SOZ emissions. And, even with single rather than dual absorption,
the
converter 45 may be operated at a single-pass efficiency of less than 98
percent.
Where the tail gas 90 is recycled, it is typically preferred that the SOz
conversion per
single pass through the entire converter 45 (i.e., the total amount of SOz
consumed
during a single pass through the entire converter = total amount of SOZ fed
into the
converter x 100%) be at least 75%, more preferably at least about 85%, even
more
preferably at least about 90%, and most preferably at least about 95%.
In one embodiment, instead recycling all the tail gas 90 to the SOZ absorption
zone 18, only a portion (e.g., from about 80 to about 90%) of the tail gas 90
is
recycled to the SOZ absorption zone 18, while another portion (preferably the
entire
remainder of the tail gas 90) is routed directly to the converter feed gas 48,
and
thereby fed back into the converter 45. This allows for a smaller SOZ
absorption zone
18 to be used.
Advantageously, the process of this invention may be implemented using only
two (or, more preferably, three (as shown in Fig. 1)) catalyst beds in the
catalytic
converter 45. It should be recognized, however, that this process may also be
07-01-2002 -2002 »0N 03:36 PM FAX N0, US003009
CA 02387988 2002-04-04 ~~ --
32
implomcnted using a double S03 absorption plant aadlor 4 or more catalyst beds
in
the catalytic converter. For example, an existing contact acid plant (having,
for
example, a 4-catalyst bed converter) can be retrofitted unth the features of
this
invention to operate al greater than design throughput without exceeding
emission
limits.
In another embodiment of the present invention, an already-existing contact
sulfuric acid production plant including a catalytic converter with 4 catalyst
beds in
series and at least two associated indirect heat exchangers for cooling the
partial
conversion gas passing bctvvee~i catalyse beds is modified (i.e., retrofitted)
so that the
converter comprises 2 parallel sets of 2 catalyst beds in series. The flow
scheme for
such a retnofitled catalytic converter is schematically illustrated in Fig. Z,
Tn the
retrofitted converter 45A, the parallel sets of catalyst heds.aie typically
contained
within the single vessel which housed the serial catalyst beds of the original
convertor.
FTowcver, it should be understood that the parallel sets of catalyst beds
could be
hoascd in separate vessels. 1n the mvdired flow schane, the SOz-auiched
stripper
Sas 30 is preferably ultimately divided into 4 portions. . A first portion of
the SOZ-
enrichcd stripper gas 3D is combined with an oxygen source 42 to form a
converter
feed gas 48, which is subsequently divided to form a first converter feed gas
48A and
a second converter feed gas 48B. 7.'he First converter feed gas 48A is heated
in .
indirect heat exchanger 75 and passed through the Crst catalyst bed 6D of the
first set
of catalyst beds to form a first partial eonversivwgas 81A, and the second
converter
feed gas 48'fi is simultaneously heated in indirect heat exchanger 78 Rnd
passed
through the first catalyst bed 65 of the second set of catalyst beds to form a
second
partial conversion gas 84A. The remainder of the SOi-enriched stripper gas 31
is
divided and a first portion 31A is combined with the cooled first partial
conversion
gas exiting indirect heat exchanger 75 la fortify the SOz concentration in the
first
partial conversion gas artdproduce a fortified first partial conversion gas
69~~. The
second portion 31B of the remainder afthe SOZ-enriched stripper gas 31 is
lilcewise
combined with the cooled second partial conversion ias exiting indirect heat
exchanger 78 to fareify the SO, concentration in the second partial conversion
gas and
produce a ford lied second partial conversion gas 7ZA. The first fortified
partial
conversion gas 69A is passed through the second catalyst bad 63 of the first
set of
AMENDED SHEET
Efid~f.G~31t.~7~(~1/LIAJ~ 1L.4~ I-mDf nr 'vA'~ D fYl~
07-01-2002 -2002 LION 03.37 PhI F~ ~, US00300~
CA 02387988 2002-04-04
. 33
catalyst beds to form a first conversion gas 54A, and the second fortiCcd
partial
conversion gas 72A is passed through the second catalyst bed 66 of the second
set of
catalyst beds to form a second conversion gas 54B. The first and second
conversion
gases 54A and 54B may rhea be combined iv fonn conversion gas acid intcaduced
into a single SO~ absorption zonQ. Where the existing contact suifaric acid
plaint is a
dual SOj absorption plant, however, the first conversion gas 54A preferably is
introduced into one of the S03 absorption zones, while the second conversion
gas 54B
is introduced into tbc other SO, absorption zone (i.e., tho two S03 absorption
cones
are operated in parallel). jn either case, it is particularly prcfetred to
recycle the S03-
depleted tail gas exiting the SO, absorption tone (or ioncs) to the SOZ
absorption
TJana.
F. Particulars Preferred )gmbodi~ents far Aiah Grade Energy Recover
Water vapor may be introduced into the conversion gas 54 exiting tho catalytic
converter 45. Ia such embodiments, upon mixing, the water vapor reacts wikh
the SOz
. 15 ' in tho conversion gas 54 to produce gaseous sulfuric acid. A portion of
the energy
fmm the heat of formation of the gaseous sulfuric acid may, in turn, be
recovered by,
fur example, passing the resulting gas through a heax exchanger.. Substantial
additi oval energy may bE recovered by also (or alternatively) passisag lhc
gas lhr ough
~ CDlldetl5111~' economizer. . .
The source of the water vapor may, for example, be low pressure steam (i.E~.,
f
' ' up to about 6.5 box (gauge), more preferably up io about 3.5 bar (gauge),
and most
proferably from about Q.Z to about I bar (gauge)). '>,his low pressure steam
shay be
obtained from a varioty of sources at a sulfuric plant, such as, for
ex~.rnple, a low
pressure pore on a steam turbine for an electrical generator, steam generated
from low
temperature sulfucic acid, ctc.
- 1n aparticularly preferred embodiment, a wet SOz source gas 3 is used, and
at
least a portion (often prefcmbly all) of the source gas 3 is combined with the
conversion gas 54 to supply at least a portion (preferably all) of flue water
vapor. An
example oFsuch an embodiment is illustrated in hig. 3. In this embodiment,
thawator
3U vapor in the wet source gas 1003 reacts with the S03 in the conversion ors
I006 to
pmduce gaseous sulfuric acid. 1'he vapor phase formation of gaseous sulfiiric
acid
AMENDED SHEET
Ern~f.zPit:07~O1~~u~~~ c~.4~ ~-~t_~r ~_~,a~ p non
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
34
generates heat which preferably is recovered as energy by, for example,
transferring
the heat to steam or feed water in an indirect heat exchanger 1012. In
addition, more
energy is preferably recovered by condensing at least a portion of the gaseous
sulfuric
acid into liquid sulfuric acid in a condensing economizer 1015. The gas 1018
exiting
S the condensing economizer 1015 is then preferably passed through an S03
absorption
zone 1021 (which preferably is associated with a heat recovery means 1024
which
recovers the energy from the heat of absorption produced in the S03 absorption
zone
1021) where S03, water vapor, and any additional gaseous sulfuric acid is
separated
from the gas 1018 to form a dry S03-depleted gas 1066. This dry S03-depleted
gas
1066, in turn, is a/the source of SOZ for the converter feed gas 1030.
Suitable methods for recovering energy using an indirect heat exchanger, a
condensing economizer, and/or a heat-recovery/S03-absorption tower are
described,
for example, by McAlister et al. in U.S. Patent Nos. 5,503,821; 5,130,112; and
5,118,490 (all incorporated herein by reference).
The condensing economizer 1015 preferably comprises an indirect heat
exchanger in which heat is transferred to a heat transfer fluid (e.g., boiler
feed water).
This indirect heat exchanger preferably comprises heat transfer wall means
(e.g., the
tubes of a shell and tube type heat exchanger), preferably constructed of an
alloy (e.g.,
an Fe/Cr or Fe/Cr/Ni alloy) which is resistant to corrosion by condensing
sulfuric
acid. Preferably, at least a portion of the wall means on the gas stream side
of the
exchanger is at a temperature which is less than the dew point of the gas
stream in the
exchanger. Thus, sulfuric acid condenses on the heat transfer wall, and the
heat of
formation of the condensing acid is transferred to the boiler feed water.
The condensing economizer 1015 may be operated to condense as sulfuric
acid as much as from about 5 to about 20% of the S03 generated in the
catalytic
converter 1009. Table 1 shows the heat evolved when S03 and water react to
form
sulfuric acid under various phase conditions.
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
Table 1
Sulfuric Acid
Heat of Reaction
from Standard
Heat of Formation
(25C)
No. Reaction Conditions Heat of Reaction
1) S03 (g) + H20 (1) ---> -31.7 kcal/mole
HZS04 (1)
5 2) S03 (g) + HZO (g) ---> -23.3 kcal/mole
HzS04 (g)
3) S03 (g) + H20 (g) ---> -42.2 kcal/mole
HZS04 (1)
The gas phase reaction (Equation 2) produces 74% of the heat produced by the
normal
liquid phase reaction (Equation 1). Transfernng the heat from condensing
sulfuric
acid to boiler feed water results in the ultimate recovery of both the heat of
formation
10 and heat of condensation of the sulfuric acid. The boiler feed water, in
turn, may be
further heated with the source gas 1003 as the source gas 1003 exits the SOz-
producing combustion zone 1002 to form high grade energy, i.e., steam at a
pressure
of at least about 30 bar (gauge), and more preferably from about 40 to about
60 bar
(gauge). This steam may be further heated by, for example, the conversion-
15 gas/source-gas mixture 1039 in the indirect heat exchanger 1012.
The conversion of S03 to sulfuric acid in the vapor phase increases as the
temperature of the vapor phase decreases. Thus, it is advantageous to decrease
the
temperature in the condensing economizer 1015 to the maximum extent compatible
with effective operation of the S03 absorber 1021 downstream. Not only is the
20 reaction forced to the maximum degree of completion and generation of the
heat of
formation, but the maximum proportion of the heat of formation and
condensation of
sulfuric acid is recovered in high grade form by transfer to high pressure
boiler feed
water for the waste heat boiler. Fortuitously, the condensing economizer 1015
can be
operated to extract a maximum amount of the vapor phase energy of formation of
25 sulfuric acid without the necessity for close control of the fluid flow
rates or wall
temperatures within the economizer 1015. The concentration of acid in the
condensate 1033 varies only slightly with the H20/S03 molar ratio in the gas
1036,
and consequently does not vary significantly with either the temperature to
which the
gas 1036 is cooled or the wall temperature of the condensing economizer 1015.
Thus,
30 it is not necessary to closely control the operation of the condensing
economizer 1015
to avoid corrosive conditions therein. And, variations in inlet air humidity,
or
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
36
excursions in sulfur flow rate, do not materially affect the concentration of
the acid
condensate 1033 on the tube walls of the condensing economizer 1015. As much
as
140% of the stoichiometric amount of water vapor may be present in the gas
1036
without reducing the concentration of the condensing acid condensate 1033 to
less
than 98%.
The energy equivalent of from about 40 to about 70% (most typically about
60%) of the heat of formation of sulfuric acid vapor may be recovered by
cooling the
gas 1039 before it enters the S03 absorption zone 1021. Where both an initial
heat
exchanger 1012 and a condensing economizer 1015 are used, typically from about
70% to about 90% (and more typically about 75%) of the recovered heat of
formation
is recovered in the condensing economizer 1015.
Preferably, the boiler feed water enters the condensing economizer at a
temperature of from about 110 to about 180°C, and the gas 1036 enters
the
condensing economizer 1015 at a temperature of from about 320 to about
470°C, and
with an H20/S03 mole ratio of from about 0.2 to about 1.05. The gas 1018
leaving
the condensing economizer 1015, on the other hand, preferably has a
temperature of
from about 240 to about 300°C.
It should be understood that a substantial portion of the vapor phase heat of
formation of sulfuric acid can be extracted without condensation in the
economizer
1015. In some circumstances, for example, it may be desirable to operate the
economizer 1015 under conditions which preclude condensation because this
allows
the economizer 1015 to be constructed of carbon steel instead a more costly
material
(e.g., a Fe/Cr or Fe/Cr/Ni alloy} which is resistant to sulfuric acid
corrosion. Thus, for
example, recovery of a substantial fraction of the heat of formation may be
achieved
without condensation by transfernng heat from the gas 1036 to boiler feed
water in a
co-current heat exchanger. Nevertheless, in most instances, it is preferred
that an
alloy exchanger be used and that the tube walls be operated at a temperature
low
enough to cause condensation thereon (though not so low as to cause nucleation
and
mist formation within the bulk gas). By such means, a substantial portion of
the heat
of formation and a significant portion of the heat of condensation may be
recovered in
the form of high pressure steam.
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
37
The wet gas 1018 leaving the condensing economizer 1015 preferably is
directed to an S03 absorption zone 1021 where it is contacted countercurrently
with a
concentrated solution of sulfuric acid 1048. Preferably, the S03 absorption
zone 1021
comprises a means in a vertical tower for promoting mass transfer and heat
transfer
between the gas and liquid phases within the tower (preferably a bed of random
packings such as saddles or rings, although it should be understood that other
gas
liquid contacting devices, e.g., a countercurrent tray tower or a co-current
venturi
absorber, may be used in lieu of random packing). The inlet gas 1018 to the
absorption zone 1021 comprises S03 and sulfuric acid vapor. Contact of the gas
1018
with the liquid sulfuric acid 1048 causes absorption of 503, condensation and
absorption of any water vapor, and condensation and absorption of sulfuric
acid vapor
into the sulfuric acid solution. It should be understood that, within the
context of this
disclosure, the terms "heat of absorption" and "energy of absorption" include
all these
various heat effects, and may also include energy of formation of sulfuric
acid in the
vapor phase that has not been recovered in condensing economizer 1015.
The use of hot acid for S03 absorption provides at least two advantages.
First,
the heat of absorption is generated at relatively high temperature which
allows
subsequent recovery of this energy at high temperature. Additionally, the use
of high
temperature acid avoids shock cooling of the gas 1018 and consequently
minimizes
the formation of acid mist in the wet gas. Preferably, the temperature of the
acid 1051
at the exit of the absorption zone 1021 is no greater than about 40°C
less than (and
more preferably no greater than about 20°C less than) the dew point of
the inlet gas
1018. The gas 1018 can typically be at a temperature of up to about
300°C as it enters
the S03 absorption zone 1021, thereby allowing recovery of the maximum amount
of
the energy of vapor phase formation and condensation of sulfuric acid in the
form of
high pressure steam as a result of the transfer of this heat to the high
pressure boiler
feed water for waste heat boiler.
In a particularly preferred embodiment, the concentrated sulfuric acid contact
solution 1048 is introduced at an inlet near the top of the S03 absorption
zone 1021,
while the gas 1018 is introduced at an inlet near the lower end of the S03
absorption
zone 1021. The acid solution 1048 at the acid inlet preferably has a
temperature of
from about 170 to about 220°C, and a sulfuric acid concentration of
from about 98.5
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
38
to about 99.5%, and more preferably from about 99 to about 99.5%. The gas 1018
at
the gas inlet, on the other hand, preferably has a temperature of from about
240 to
about 300°C, and an H20/S03 molar ratio which preferably is less than
the HZO/S03
molar ratio in the acid solution 1048 , and equals from about 0.2 to about
1.05 (more
preferably from about 0.7 to about 1.0). If the water vapor concentration in
the source
gas 1003 is so great that the HZO/S03 molar ratio in the gas 1018 entering the
S03
absorption zone 1021 exceeds the H20/S03 molar ratio in the concentrated
sulfuric
acid contact solution 1048 when the entire source gas 1003 is combined with
the
conversion gas 1006, the H20/S03 molar ratio in the gas 1018 entering the S03
absorption zone1021 preferably is reduced by either partially drying the
source gas
1003 in a drying tower before it is combined with the conversion gas 1006; or
by only
combining a portion 1042 of the source gas 1003 with the conversion gas 1006,
and
routing the remaining portion 1045 directly to the SOZ absorption/stripper
zones (and,
optionally a drying tower, if the SOz absorption/stripper zone is unable to
remove
essentially all the water content).
The acid solution 1051 preferably is discharged from the S03 absorption zone
1021 at a temperature of at least about 190°C, more preferably from
about 190 to
about 250°C, and even more preferably from about 210 to about
250°C. At least a
major portion of this solution preferably flows to a circulating pump, and
passed
through an indirect heat exchanger 1024 where the energy of absorption is
recovered
by transfer of heat to another fluid. Preferably, the indirect heat exchanger
1024
comprises a heat recovery system boiler, and the heat energy is ultimately
recovered
in the form of low to medium pressure (i.e., up to about 10.5 bar (gauge)).
The acid solution 1054 from the indirect heat exchanger 1024 is preferably
recirculated back to the S03 absorption zone 1021. To recover the acid
product, a
portion 1057 of the acid solution 1054 preferably is removed as product before
the
acid solution 1054 is recirculated (additional heat energy may be recovered
from this
acid product by, for example, passing it through one or more additional
indirect heat
exchangers). An equal amount of water 1060 is then added to the remaining
sulfuric
acid solution 1063. This water 1060 may, for example, be added in liquid or
vapor
form, or in the form of diluted sulfuric acid.
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
39
As a result of the high temperature operation of the S03 absorption zone 1021,
the S03-depleted gas 1066 exiting the top of this zone 1021 is relatively hot.
This, in
turn, often results in the stripping of sulfuric acid from the acid stream
into the gas
stream. In other words, although the absorption efficiency of the S03
absorption zone
1021 is at least about 90%, high temperature operation of the absorption zone
1021
also typically results in some unabsorbed S03 passing through the absorption
zone
1021. Gas 1066 exiting the top of the S03 absorption zone 1021 is therefore
preferably directed to a condensing stage for absorption of residual S03 and
condensation of sulfuric acid vapor. This condensing stage preferably contains
means
for promoting gas/liquid contact and mass transfer and heat transfer. For
example, in
one embodiment, this stage comprises a countercurrent packed section wherein
relatively cool acid having a concentration of about 98.5% is fed to the top
of this
stage and gas 1066 leaving the main S03 absorption zone 1021 (which is
typically at a
temperature of from about 170 to about 230°C) enters the bottom of the
condensing
stage. In this embodiment, the acid entering the condensing stage preferably
is at a
temperature of less than about 120°C, most preferably from about 60 to
about 80°C.
On passage through the condensing stage, the gas 1066 preferably is cooled to
a
temperature of from about 75 to about 140°C, and more preferably from
about 80 to
about 120°C. Gas leaving the condensing stage is then preferably passed
through a
mist eliminator. The acid flow rate in the condensing stage preferably is
maintained
at a rate low enough so that the acid leaves the stage at a temperature which
approaches the temperature of the acid entering the main S03 absorption packed
bed.
In this wet gas embodiment, the gas 1066 exiting the S03 absorption zone 1021
(i.e., the S03-depleted gas), along with any portion 1045 of the source gas
1003 that is
not combined with the conversion gas 1006, is preferably used to form the
converter
feed gas 1030. More specifically, the S03-depleted gas 1066 (along with any
portion
1045 of the source gas 1003 which is not combined with the conversion gas
1006) is
first passed through the SOZ absorption/stripper zones described previously.
This
removes the excess inert gases, and can be used to enhance the SOZ
concentration in
the S03-depleted gas 1066 where the SOZ concentration in the S03-depleted gas
1066
is less than the desired concentration. The gas exiting the SOZ
absorption/stripper
zones (i.e., the SOz-enriched stripper gas 1069) is then preferably combined
with a dry
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
oxygen source 1072 if the stripper gas 1075 does not supply the desired level
of
oxygen. It should be recognized that the SOZ-enriched stripper gas 1069 may
also be
divided into 2 or more portions in the same manner as described above wherein
one
portion of the SOZ-enriched stripper gas 1069 is combined with the dry oxygen
source
5 1072 and fed into the first catalyst bed 1078 of converter 1009, and a
second portion
1070 is used to fortify the SOZ concentration of the partial conversion gas
1081
exiting the first catalyst bed 1078.
It is especially preferred for the gas passing through the converter 1009 to
be
essentially free of water vapor. By passing essentially moisture free gas
through the
10 converter 1009, the risk of corrosion (or the added cost of using corrosion-
resistant
material) in the converter 1009 (and any process equipment between the
catalyst beds
of the converter 1009) caused by sulfuric acid formed by the vapor phase
reaction of
S03 and water vapor is generally avoided. To ensure that the gas 1030 being
fed into
the converter 1009 is essentially free of water vapor, any oxygen source 1072
15 combined with the SOz-enriched stripper gas 1069 preferably is dried
beforehand. It
is also preferred that the SOZ absorption solvent 1084 consist essentially of
a
composition that transfers little or no water to the SOZ enriched stripper gas
1069.
The organic phosphorus solvents discussed above are generally suitable for
this
purpose, particularly where the stripper gas 1075 is dry air.
20 The wet-source-gas embodiment described above is advantageous because it
produces a dry SOZ gas 1030 for the converter 1009 without having to first
pass the
entire source gas 1003 through a drying tower, thereby avoiding the capital
and
operational expenses associated with such a tower (and associated equipment,
e.g., a
pump, piping, a pump tank, and a cooler) as to the portion 1042 of the source
gas
25 1003 that is combined with the conversion gas 1006 (as noted above, it is
most often
preferred that this portion 1042 be the entire source gas 1003). In addition,
this
process is advantageous because it produces heat (i.e., the heat of formation
of
gaseous sulfuric acid, the heat of condensation of gaseous sulfuric acid, and
the heat
of condensation of water vapor) which may be transferred and used elsewhere as
30 energy.
Although the above discussion focuses on heat recovery in the particularly
preferred embodiment where a wet source gas is combined with the conversion
gas to
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
41
supply all the water vapor, it should be understood that the general heat
recovery
principles discussed above also apply to embodiments where a different source
of
water vapor is used (e.g., low pressure steam), or where a wet source gas and
a
different source of water vapor are both combined with the conversion gas to
supply
the water vapor.
G. Preferred Equipment for Handling Gases Containing Sulfuric Acid
Wet S03-containing gas can be handled in carbon steel equipment, although the
gas temperature in such equipment preferably is kept above the dew point to
avoid the
condensation of gaseous sulfuric acid formed from the water vapor and 503. In
the
more preferred embodiments of the present invention, however, the dew point is
generally high and much of the equipment (particularly the condensing
economizer) is
operated at a temperature below the dew point. This equipment, therefore,
preferably
is made of a material that is resistant to sulfuric acid corrosion under the
conditions of
this invention. There are a number of conventionally used materials,
particularly
stainless steel and nickel alloys, that can be used in for this purpose. Alloy
performance may be characterized by a corrosion index (CI), which is defined
in
terms of alloy composition by the following relationship:
CI = 0.4[Cr] - 0.05 [Ni] - 0.1 [Mo] - 0.1 [Ni] x [Mo]
wherein [Cr] is the weight percent of chromium in the alloy, [Ni] is the
weight percent
of nickel in the alloy, and [Mo] is the weight percent of molybdenum in the
alloy.
Alloys which work best in high temperature strong sulfuric acid service have
been
found to have a corrosion index of greater than 7, and particularly greater
than 8.
The alloys most likely to exhibit low corrosion rates are those with the
highest
corrosion index. As indicated by the corrosion index formula, high chromium is
desirable, and it is preferable to avoid alloys which have both high nickel
and high
molybdenum. It should be recognized, however, that alloys which contain high
nickel
and very low molybdenum, or low nickel and moderate amounts of molybdenum, are
often acceptable. Particular alloys found suitable for use in contact with
liquid phase
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
42
sulfuric acid at high temperature include, for example, those having UNS
designations
S30403, 530908, 531008, S44627, 532304, and 544800.
EXAMPLE
This example further illustrates and explains the invention. The invention,
however, should not be considered to be limited to any of the details in this
example.
Using a computer model, the performance of the system shown in Fig. 4 was
assessed. A source gas 2003 containing about 19 mole% SOz, about 2 mole % O2,
and about 79 mole% NZ is formed in a sulfur burner 2006 by burning sulfur 2009
in
the presence of dry air 2012. This source gas 2003 (initially at a temperature
of about
1538°C upon exiting the sulfur burner 2006) is cooled to about
548°C in a waste heat
boiler 2002. The source gas 2004 is further cooled to about 337°C in an
indirect heat
exchanger 2015 (i.e., MonplexTM, Monsanto Environ-Chem Systems, Inc., St.
Louis,
MO, USA) by transfernng heat from the source gas 2004 to the gas 2018 being
fed
into the SOz oxidation catalytic converter 2021. Finally, the source gas 2024
is cooled
1 S even further to about 204°C in yet another indirect heat exchanger
2023 which uses
heat in the source gas 2024 to form steam.
The cooled source gas 2025 is split into two portions: one portion 2026 (being
about 6.6 volume% of the cooled source gas 2025) is fed back into the sulfur
burner
2006 to maintain the desired temperature in the burner 2006, and the remaining
portion 2028 (being about 94% of the cooled source gas 2025) is introduced
into the
SOZ absorption/stripping zones (i.e., a Claus MasterTM, Monsanto Environ-Chem
Systems, Inc., St. Louis, MO, USA). Here, the source gas 2028 is passed
through a
packed SOZ absorption column 2027, where it is contacted with a liquid SOZ
absorption solvent comprising dibutyl butyl phosphonate 2030 flowing
countercurrently to the source gas 2028. The dibutyl butyl phosphonate 2030
selectively absorbs SOZ to form an SOZ-enriched absorption solvent 2033 and an
SOZ
depleted gas 2036 (the SOZ-depleted gas 2036 containing substantially all the
residual
OZ and inert gases (mostly Nz) from the source gas 2028).
The SOZ-depleted gas 2036 is discharged from the system, and the SOZ-enriched
absorption solvent 2033 is introduced into a packed SOz stripper column 2039,
where
the SOZ-enriched absorption solvent 2033 is contacted with a countercurrent
flow of
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
43
dry air 2042 (the dry stripper air 2042 entering the column 2039 has a
temperature of
about 110°C) to form an SOZ-enriched stripping gas 2045 (containing
about 90
mole% SO2, with the remaining being air) and an SOz-depleted absorption
solvent
2048 (which is recycled back to the SOz absorption column 2027 to be used
again as
the SOz absorption solvent 2030). Both the SOZ absorption column 2027 and the
SOZ
stripper column 2039 are operated at nearly atmospheric pressure.
The SOz-enriched stripping gas 2045 is divided into two portions: one portion
(i.e., the primary SOZ gas 2051) being about 54 volume% of the SOZ-enriched
stripping gas 2045, and the other portion (i.e., the bypass SOZ gas 2054)
being about
46 volume% SOZ-enriched stripping gas 2045 (both portions having the same
composition, i.e., 90 mole% SO2, with the remaining being air). The primary
SOZ gas
2051 is combined with dry air 2057 (the dry air 2057 having a temperature of
about
66°C) to form a converter feed gas 2018 containing about 12 mole% SOZ
and having a
temperature of about 130°C. This converter feed gas 2018 is heated to a
temperature
of about 410°C by the gas 2004 coming from the sulfur burner 2006 using
the
MonplexTM indirect heat exchanger 2015. After being heated, the converter feed
gas
2060 is passed through a first catalyst bed 2063 containing V205 which
converts (i.e.,
oxidizes) about 67% of the SOZ in the converter feed gas 2060 into 503,
thereby
forming a partial conversion gas 2066 having a temperature of about
637°C, and
containing about 4 mole% SOZ and about 8 mole% S03. The V205 catalyst in the
first
catalyst bed 2063 is a potassium-promoted catalyst coated on a silica support,
and is
in the shape of rings having an outer diameter of 12.5 mm, an inner diameter
of 5 mm,
and an average length of 14 mm (Cat. No. LP-120, Monsanto Environ-Chem
Systems,
Inc., St. Louis, MO, USA). The first catalyst bed 2063 has a diameter of about
26.25
feet and contains about 50,000 liters of the catalyst. The total flowrate of
the
converter feed gas 2060 into the first catalyst bed 2063 is about 50,767 scfin
(i.e.,
standard cubic feet per minute (defined at 70°F and 1 atm)).
The partial conversion gas 2066 exiting the first catalyst bed 2063 is cooled
to
about 420°C by transfernng heat to feed water in an indirect heat
exchanger 2069.
The cooled partial conversion gas 2072 is then combined with the bypass SOZ
gas
2054 to increase the SOZ concentration in the partial conversion gas 2072 to
about 13
mole%. The SOz-fortified partial conversion gas 2075 (having a temperature of
about
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
44
423°C) is then passed through a second catalyst bed 2078 containing
Vz05 to oxidize
more SOZ to form a second partial conversion gas 2081 having a temperature of
about
607°C, and containing about 15.4 mole% S03 and about 6.1 mole% un-
oxidized SOz.
The second catalyst bed 2078 has the same dimensions, the same Vz05 catalyst,
and
the same volume of catalyst as the first catalyst bed 2063. The total flowrate
of gas
entering the second catalyst bed 2078 is about 54,366 scfin.
The second partial conversion gas 2081 is cooled to about 420°C by
transfernng
heat to feed water in a second indirect heat exchanger 2084, and then passed
through a
third catalyst bed 2087 containing V205 to oxidize still more SOZ and form a
final
conversion gas 2090 having a temperature of about 519°C, and containing
about 20.0
mole% S03 and about 2.1 mole% un-oxidized residual SO2. The V205 catalyst in
the
third catalyst bed 2087 is a potassium-promoted catalyst coated on a silica
support and
is in the shape of rings having an outer diameter of 9.5 mm, an inner diameter
of 4
mm, and an average length of 13 mm (Cat. No. LP-110, Monsanto Environ-Chem
1 S Systems, Inc., St. Louis, MO, USA). The third catalyst bed 2087 has a
diameter of
about 26.25 feet and contains about 80,000 liters of the catalyst. The total
flowrate of
gas entering the third catalyst bed 2087 is about 52,406 scfm.
The final conversion gas 2090 is cooled to a temperature of about
166°C in an
indirect heat exchanger 2091, and then contacted in a packed S03 absorption
column
2093 (having a diameter of about 12 feet and a height of about 40 feet) with a
countercurrent flow of an aqueous solution 2096 containing about 98.5 weight%
HZS04 to form a more concentrated sulfuric acid solution 2097 having a
sulfuric acid
concentration of about 99.5 weight%. The flowrate of the conversion gas 2090
is
about 51,349 scfin, while the flowrate of the aqueous sulfuric acid solution
2096 is
about 1,700 gallons per minute. The temperature of the aqueous sulfuric acid
solution
2096 entering the column 2093 is about 82°C, and the temperature of the
sulfuric acid
solution 2097 exiting the S03 absorption column 2093 is about 110°C.
The gas 2102 exiting the S03 absorption column 2093 (i.e., "the S03-depleted
gas" or "tail gas") is split into 2 portions: one portion 2103 (being about 80
volume%
of the S03-depleted gas 2102) is combined with the source gas stream 2028, and
thereby routed to the SOZ absorption column 2027. The other portion 2104
(being
about 20 volume% of the S03-depleted gas 2102) is combined with the dry air
2057
CA 02387988 2002-04-04
WO 01/36324 PCT/US00/30095
being combined with the primary SOZ gas 2051, thereby maintaining a smaller
volume of total gas being fed into the SOZ absorption column 2027. Thus, both
portions of the S03-depleted gas 2102 are ultimately recycled back to the
converter
2021 so that substantially all the residual SOz in the S03-depleted gas 2102
can
eventually be converted into sulfuric acid.
The single pass SOZ conversion for the whole converter 2021 is about 90.4%.
The overall conversion of the SOZ in the source gas 2003 is about 99.87%.
*********
The above description of the preferred embodiments and accompanying figures
10 are intended only to acquaint others skilled in the art with the invention,
its principles,
and its practical application, so that others skilled in the art may adapt and
apply the
invention in its numerous forms, as may be best suited to the requirements of
a
particular use. The present invention, therefore, is not limited to the above
embodiments, and may be variously modified.
15 With reference to the use of the words) "comprise" or "comprises" or
"comprising" in the above description and/or in the following claims,
applicant notes
that unless the context requires otherwise, those words are used on the basis
and clear
understanding that they are to be interpreted inclusively, rather than
exclusively, and
that applicant intends each of those words to be so interpreted in construing
the above
20 description and/or the following claims.