Note: Descriptions are shown in the official language in which they were submitted.
CA 02388834 2002-06-25
- 1 -
Method for detecting and evaluating a potentially aberrantly methylated
DNA region on the X chromosome. or the clonality
There is provided a method for detecting and evaluating a potentially
aberrantly methylated DNS
region on the X chromosome, or the clonality, the use of such a method as well
as a kit for carry-
ing out said method.
DNA, the Garner of genetic information, is present in the nuclei of eukaryotes
tightly packed in
structural units, the chromosomes. Changes in the numbers or even structures
of the chromo-
somes as well as alterations of the DNA itself without inducing microscopic
alterations on the
chromosomes usually result in serious syndromes in men. Diseases can also be
caused by epige-
netic alterations such as by an aberrant DNA methylation. DNA methylation
occurs by the cova-
lent binding of methyl groups to nucleotides and constitutes an important
regulation mechanism:
In the case of the mammalian genome, active genes or their regulatory units
are frequently un-
denmethylated, while inactive DNA sections are frequently characterized by
strong methylation.
Therein, primarily the cytosine residues in CpG dinucleotides are methylated
in 5'. Accordingly,
in methylated DNA, methyl cytosine (mC) is preferably found as mCpG in the CpG
islets of
DNA. In most cases, these are to be found in regulatory DNA units and
originally unmethylated.
Physiologically, unmethylated active DNA and methylated inactive DNA sections
are usually
found in the diploid mammalian genome mCpG in imprinted genes - as a function
of the parental
origin of the respective gene or chromosome. Thus, imprinted genes are
expressed by just one of
the two homologous chromosomes. A similar ratio is found in cells of female
individuals for the
dose compensation of genes that are localized on X chromosomes: since female
cells (XX), un-
like male cells (XY), carry two X chromosomes, one of the two X chromosomes is
inactivated.
This inactive X chromosome, or its DNA (in the form of mCpG), is methylated
except for a few
regions (XIC ... X inactivation center, pseudo-autosomal regions). This X
inactivation occurs
randomly in the course of the development of an individual (random X
inactivation), whereupon
the entirety of cells consists of a population of cells with inactive paternal
and active maternal X
chromosomes as well as an equally large population of cells with active
paternal and inactive
maternal X chromosomes. Hence results a ratio of active to inactive (Xa/Xi)
alleles of Xa/Xi =
50/50 in respect to their parental origin. In many cases, also a deviation
from this 50/50 ratio
may, however, occur in female individuals such that, although the ratio of
methylated to unmeth-
ylated, or active to inactive, X chromosomes (per cell and also per overall
population) of 1:1 is
maintained, the size of the cell population with the active/inactive
chromosome does not equal
50/50 in respect to the parental origin of the same. This means that the cell
population (clone)
with the active X chromosome of one parent (El ) is larger and that with the
active X chromo-
some of the other parent (E2) is smaller, or the clone with the inactive X
chromosome of E1 is
smaller and that with the inactive X chromosome of E2 is larger. This
phenomenon of a "shifted"
clone size or clonality is described by the term "skewed X inactivation",
which can be observed
even in normal females with increasing age.
CA 02388834 2002-06-25
Experimentally, the methylation of DNA can be detected, for instance, by
cutting with methyla-
tion-sensitive enzymes, by means of mC-specific antibodies, by genomically
sequencing bisul-
fite-deaminated DNA, by the selective hydrazine cleavage of unmethylated DNA,
etc.
If a DNA section comprises an incorrect (aberrant) methylation (i.e. a
hypermethylation of
physiologically unmethylated DNA or a hypomethylation of physiologically
methylated DNA),
this will in most cases lead to a deregulation of the physiological function
of this DNA section
and can result in a disease. The determination of the phenotype is frequently
insufficient to diag-
nose a particular disease. In order to be able to determine the disease for
sure and to diagnose also
the variant or disease, an analysis of the hereditary substance of the
patient, in particular the
methylation of a given DNA section, are usually required.
A number of diseases are caused by an alteration of the DNA on an X
chromosome. A DNA re-
gion may, for instance, be methylated on the X chromosome so as to inactivate
this gene. In ad-
dition, the gene function can be disturbed and additionally methylated, for
instance, by an
alteration (mutation) of the DNA, for instance by the expansion of a repeat
region, (FraX-A,
FraX-E, FraX-F) (cf. Carrel et al., American Journal of Medical Genetics 64:27-
30 (1996);
Hirst et al., Hum. Molec. Genet. 2: 197-200, 1993; Parrish et al., Nature
Genet. 8 : 229-235,
1994 ; Ritchie et al., Hum. Molec. Genet. 3 : 2115-2121, 1994 ; Sutherland et
al., Hum. Molec.
Gent. 1 : 111-113, 1992). In other cases, diseases may go back to alterations
of DNA sections or
whole chromosomes (regions) by duplication, insertion, translocation. If such
an alteration affects
the X chromosome already in the early development stages of an individual, one
will have to
speak of X-chromosomal diseases in many cases. Also with sporadic diseases, as
is the case with
many tumor diseases, X-chromosomal alterations could be proved (Gale et al.,
Blood, Vol. 83,
No. 10 ( 1994), 2899-2905). Consequently, different variants of the X
chromosome are present
concurrently, or also different cell populations (clones) besides the normal,
healthy clone. These
populations (clones) may differentiate both in the allele pattern and in the
methylation pattern of
the alleles or the altered genetic material, respectively (cf. Cammarata et
al., Am. J. Med. Genet.,
1999; 85(1): 86-7; ISSN: 0148-7299; Dierlamm et al., Br. J. Haematol., 1995;
91 (4): 885-91;
ISSN: 0007-1048; James et al., Ann. Hum. Genet., 1997; 61(6): 485-90; ISSN:
0003-4800; Leal
et al., Hum. Genet., 1994; 94(4): 423-6; ISSN: 0340-6717; MacDonald et al.,
Hum. Mol. Genet.,
19,94; 3(8): 1365-71; ISSN: 0964-6906; and Martinez-Pasarell et al., Horm.
Res., October
1999; 51(5): 248-252; ISSN: 0301-0163).
In genetic assays the affected gene, or the affected gene locus, is assayed by
cytogenetic, South-
ern blot, PCR, reverse transcription PCR (RT-PCR) or immunologic analyses.
Those assays
serve to analyze the sequence, the expression, the extent of methylation and
the like.
The fragile X syndrome (FX or FraX-A syndrome) is an example of a hereditary
disease which
CA 02388834 2002-06-25
- 3 -
affects primarily males. The clinical phenotype of the FX syndrome displays
moderate to severe
mental retardation, large ears, a long face, hyperactivity, autistic traits,
etc. The FX syndrome re-
sults from a functional failure or a suppressed expression of the FMR 1 gene,
which is located in
the telomeric region of the long arm of the X chromosome, X (q27). In the vast
majority of cases,
the gene transcription is impaired by an unphysiologic expansion of a
polymorphous CGG-triplet
repeat region on the 5' end of the FMR 1 gene. In normal individuals, the FMR
1 gene repeat re-
gion comprises between 6 and (48-) 54 repeat units with an occurrence maximum
of the allele
frequency having a repeat length of approximately 30 units. Individuals with
alleles in the so-
called premutation comprise between (48-) 54 and 200 repeat units and do not
show any clinical
failures, either. There is a grey zone in the range of between 48 and 54
repeat units, i.e., patients
having a repeat region length of 48-54 units will have to be classified as
premutations with fur-
ther FX patients in the family. Individuals with premutations have increased
risks to pass on full
mutations to the subsequent generation, such full mutations consisting of more
than 200 triplet
repeat units. The risk to transmit full mutations to subsequent generations
depends on the size of
the premutation, on the sex of the transmitting parent and on the position of
the parent in the
family tree. Individuals with larger premutation alleles bear higher risks to
transmit full mutations
than those having shorter premutations. However, this phenomenon only relates
to females such
that children from mothers with premutations can be afflicted by full
mutations. On the other
hand, children from fathers having a premutation allele will only inherit the
premutation.
Moreover, other rare mutations such as, e.g., small deletions of the FMR 1
gene region or point
mutations within the coding sequence, can induce a lack of, or the production
of a functionally
incapable, FMR 1 protein.
It could be demonstrated that the FMR 1 gene region with a normal or
premutation repeat number
is not methylated, while, in the event of full mutations, cytosines in CpG
dinucleotides are meth-
ylated in the expanded repeat region and in the surrounding S' promoter region
of the FMR 1
gene.
Nevertheless, FMR 1 alleles with a vast number of repeat units (300-800) can
be reactivated in
vitro by treatment with demethylating agents such as S-azadeoxycytidine. In
addition, males
were found who, despite repeat numbers of above 200 (full mutation), which
were, however, un-
methylated, showed normal intelligence and almost normal FMR 1 protein
concentrations. From
this, it can be concluded that the de novo methylation of the FMR1 promoter is
more likely to
impair the gene function than the length of the expanded repeat region.
Different degrees of
methylation can result in different degrees of the disease.
Current methods for evaluating the FX syndrome comprise cytogenetic, Southern
blot, PCR, re-
verse transcription PCR (RT-PCR) and immunohistochemical analyses. These
assays, as a rule,
analyze the size of the expanded repeat region, the de novo methylation of
this gene region or the
CA 02388834 2002-06-25
- 4 -
gene expression.
In WO 92/14840, the detection of the fragile X syndrome by the aid of
restriction enzymes is de-
scribed, wherein the restriction enzymes, for instance, cut only those sites
which include unmeth-
ylated cytosines. Unlike unmethylated DNA, methylated DNA is, thus, digested
and the digestion
products are detectable.
WO 91/09140 relates to an oligonucleotide having a defined sequence and
binding to a given site,
M54, of the region of the fragile gene. Patients with fragile X syndrome can
be detected by in situ
hybridization with the labeled oligonucleotide.
WO 92/20825 likewise relates to a method for diagnosing the fragile X
syndrome, wherein either
the amount of mDNA is determined, for instance, by RT-PCR or the amount of
protein is deter-
mined, for instance, by means of immunological methods of the FMR 1 gene.
Another method
for detecting the fragile X syndrome consists in determining the length of the
repeat region. This
is done, for instance, by the digestion of the FMR 1 gene with restrictions
enzymes or by PCR
methods with primers that are specific for the repeat region of the FMR 1
gene, and subsequent
gel electrophoresis.
Also the method according to US 5 658 764 for detecting the fragile X syndrome
comprises size
measurements of the GC-rich sequence by PCR methods.
The drawbacks of the above-described methods consist in that the analysis of
but one gene re-
gion does not allow an unambiguous assertion as to the variant of the disease.
The conventional
PCR of a single gene region, for instance, does not yield sufficiently clear
results, because in the
case of the fragile X syndrome full mutations can, for instance, not be
amplified by PCR on ac-
count of their high CG content and their repetitive nature. Since negative
results might, thus, also
suggest full mutations, a Southern blot assay would have to be carried out
subsequently. Such
Southern blot assays yield information on the length of the repeat region as
well as on the methy-
lation. The drawbacks of Southern blotting reside in that it is time-
consuming, that relatively
large quantities of DNA are necessary, which involves difficulties
particularly in prenatal analy-
ses, and also the usually required use of radioisotopes is disadvantageous to
the laboratory per-
sonnel. A disadvantage of PCR consists in that it cannot distinguish between a
full mutation and
a premutation, in particular in the event of a borderline full
mutation/premutation, i.e., the length
of the repeat region is about 200 units and the sequence is partially
methylated and partially un-
methylated. Furthermore, the distinction between a full mutation and a mosaic
(i.e., e.g., a full
mutation on one allele and a premutation or an expansion in the normal range
on the other allele)
is problematic or impossible. Moreover, those known methods are very time-
consuming, requir-
ing several method steps. In females, those methods also involve high "false-
negative" rates.
CA 02388834 2002-06-25
- 5 -
The publication of Das et al. "Methylation Analysis of the Fragile X Syndrome
by PCR" (Genetic
Testing, Vol. 1, No. 3, 1997/98) relates to a method for detecting the fragile
X snydrom by meth-
ylation-specific PCR (MS-PCR). Thereby, the unmethylated cytosine residues are
converted
into uracil by the addition of an agent, while the methylated cytosine
residues are not converted.
In DNA replication, uracil is replaced with thymidine in the modified DNA. By
constructing
specific primers that hybridize with the modified DNA sequence, either the
methylated or the un-
methylated sequence is amplified. If primers resulting in varying product
lengths are selected, it
is feasible to determine as a function of the PCR product whether a given site
of the FMR 1 gene
is methylated or not. Due to the PCR amplification of the unmethylated
sequence, MS-PCR dis-
tinguishes normal and premutated alleles from full mutation and mosaic
sequences - amplifica-
tion of the methylated sequence. After this, normal and premutated sequences
must be
distinguished by conventional PCR (p. 1 S2, col. 2, 2"d paragraph). In
conventional PCR, only
normal sequences (S-SO repeat units) but no premutated sequences (SO-200
repeat units) are am-
plified (cf. Fig. 2B). Furthermore, MS-PCR cannot differentiate between
individuals with full
mutations and those having full mutation/premutation mosaics. Such a
determination must be
carried out subsequently by Southern blot analysis (cf. p. 1 S3, col. 2 below
to p. 1 S4, col. 1 on
top). It is, moreover, impossible to test female syndromes by said MS-PCR,
because the inactive
X chromosome is always methylated and hence a product will be obtained in any
event. Afflicted
female patients consequently can be correctly diagnosed only by conventional
Southern blot
analysis (cf. p. 1SS, last-but-one paragraph).
Even when carrying out the above-mentioned methods in combination, the
determination of the
clone size, i.e., the clonality, is impossible in the event of a mosaic, since
in that case only meth-
ylation or non-methylation is qualitatively assessed. This can only be
realized by analyzing an
additional informative gene section (i.e., two distinguishable alleles),
whereby the extent of a
skewed X inactivation must be performed on the X chromosome by the methylation
analysis of
such an informative region. In addition, this region has to be chosen in a
manner that the physiol-
ogic methylation pattern is known beforehand. A gene Locus frequently used to
this end is the
HUMARA (human androgen receptor) gene (cf. Pardini et aL, The New England
Journal of
Medicine, Vol. 338, No. S (1998), 291-295; Kubota et al., Hum. Gent. (1999)
104: 49-SS).
These problems are faced in the diagnostics of all X-chromosomal diseases
whose pertinent gene
regions are differently methylated by X inactivation and, in particular, in
the detection of the
variant of the disease (normal mutation/premutation/full mutation) and the
clonality or clone size.
Examples encompass FraX-A, FraX-E, FraX-F and various X-chromosomal diseases:
As already pointed out in connection with the fragile X syndrome (FraX-A), the
ratios with
FraX-E and FraX-F are similar, the disease likewise developing by repeat
expansion and con-
current methylation.
CA 02388834 2002-06-25
In FraX-E-positive individuals, a GCC repeat expansion present at Xq28
adjacent a CpG islet,
comprises more than 200 GCC units as compared to 6-25 GCC units in normal
individuals. Be-
sides, the CpG islet is methylated in FraX-E-positive individuals. Those
patients show mental
retardations (cf. Knight et al., Am. J. Hum. Gent. 53:A79, 1993; Knight et
al., Cell 74: 127-134,
1993). A gene, FMR2, which is transcribed distally from the CpG islet with
FraX-E and down-
regulated by repeat expansion and methylation was identified (Gu et al.,
Nature Gent. 13: 109-
113).
An expansion of a sequence (GCCGTC)"(GCC)m can be found in FraX-F-positive
patients,
wherein m can be more than 900 and the adjacent CpG islet is methylated. In
normal persons, m
is 12 to 26 and n is 3 (Ritchie et al., Hum. Molec. Gent. 3: 2115-2121, 1994).
The drawbacks of the known assaying techniques reside in that several
different method steps are
frequently necessary to diagnose the diseases and the disease variants. In
many cases, an unambi-
guous assertion as to the syndrome is not possible, because the applied
assaying techniques only
enable assertions about specific gene regions.
It is, therefore, an object of the present invention to provide a method for
diagnosing a potentially
aberrantly methylated DNA region on the X chromosome, or the clonality, which
can be realized
rapidly and readily by as few methods steps as possible with distinction being
made between the
various variants, and which method can be applied in both male and female
patients to distin-
guish between different genotypes.
The method according to the invention of the initially defined kind is
characterized in that
a) either the extent of methylation of the potentially aberrantly methylated
DNA region and/or the
extent of methylation in a definitely physiologically methylated DNA region of
the X chromo-
some is determined in a sample and the presence and optionally the variant and
the extent of the
potential aberration are subsequently diagnosed from the comparison of the
methylation determi-
nations, or
b) the extent of methylation of the potentially aberrantly methylated DNA
region and/or the ex-
tent of methylation as well as optionally the sequence variant of a
polymorphous DNA region of
the X chromosome, which may either be physiologically or aberrantly
methylated, are determined
in a sample, and the presence and optionally the variant and the extent of the
potential aberration,
or the clonality, are subsequently diagnosed from the comparison of the
methylation detenmina-
tions and optionally the sequence variant, or
c) the extent of methylation of the potentially aberrantly methylated DNA
region, the extent of
methylation in a definitely physiologically methylated DNA region of the X
chromosome, and
CA 02388834 2002-06-25
_ 7
the extent of methylation as well as the sequence variant of a polymorphous
DNA region of the X
chromosome, which may either be physiologically or aberrantly methylated, are
determined in a
sample, and the presence and optionally the variant and the extent of the
potential aberration are
subsequently diagnosed from the comparison of the methylation determinations
and the sequence
variant.
Within the context of the present invention, extent of methylation is meant to
denote the ratio of
methylated and unmethylated alleles in a chromosome, which means that the
methylated and the
unmethylated DNA are simultaneously determined in a reaction. Said
simultaneous determination
allows a precise and quantitative assertion about the ratio of methylation to
non-methylation of a
specific sequence. It is, thereby, feasible to diagnose the extent and variant
of the disease in ques-
tion.
Within the context of the present invention, potentially aberrantly methylated
DNA region is
meant to denote a DNA region that is methylated or unmethylated. If this DNA
region is affected
by a methylation, an aberrant methylation can induce a disease.
It could be demonstrated that a quantitative methylation analysis of the two
alleles of a methy-
lated DNA region and/or the determination of the clone size via the
methylation analysis of a
polymorphous DNA region give an unambiguous and a clear solution as to the
methylation pat-
tern and hence the syndrome. The different genotypes of both male and female
patients can thus
be distinguished, because the result exhibits a specific pattern as a function
of the respective
genotype.
A quantitative methylation analysis which yields sound results in the
determination of the clonal-
ity (clone size) is suitable for the clarification of all X-chromosomal
diseases, and also those dis-
eases which are based on a deviation from the normal number of X chromosomes
(Klinefelter,
Turner, Multiple X syndromes), or also those diseases whose affected cell
population (clone) ex-
hibits X aneuploidy. This is found very frequently in various tumor diseases
and hematologic
neoplasms. The detection and determination of the clone size/clonality of
these neoplastic dis-
eases is an important parameter in diagnosing and monitoring. In the event of
autosomal diseases
relating to imprinted genes, conditions are similar.
Thereby, also the extent of methylation in a definitely physiologically
methylated DNA region of
the X chromosome can be determined. The definitely physiologically methylated
DNA region
merely relates to one allele of the X chromosome, while the other allele is
not methylated.
If the extent of the definitely physiologically methylated DNA region known,
this additional
control serves as an internal standard.
CA 02388834 2002-06-25
This additional analysis will be of diagnostic value if the potentially
aberrantly methylated DNA
region comprises deletions such that it cannot be detected irrespective of
whether it comprises
methylations or not. The detection of the potentially aberrantly methylated
DNA region would
then be missing in the result, yet the definitely physiologically methylated
DNA region would be
detected. From this, a deletion can be concluded. Without such an additional
control, it would not
be possible to tell whether the DNA region does comprise deletions, and was
therefore not de-
tected, or v~ihether the detection has failed.
According to point b), however, another option for a possible additional
determination resides in
the determination of the extent of methylation as well as optionally the
sequence variant of a
polymorphous DNA region of the X chromosome. A syndrome is, for instance,
determined by the
evaluation of the potentially aberrantly methylated DNA region and the
polymorphous DNA re-
gion.
In the context of the present invention, the term polymorphous DNA region
serves to denote the
variability of a DNA sequence and, for instance, a DNA section which comprises
different alleles
in a population, such as the length of a given DNA section in the event of
microsatellite repeats
or nucleotide and amino acid polymorphisms. The simultaneous determination of
the extent of
methylation of the sequence variant along with the determination of the extent
of methylation of
the potentially aberrantly methylated DNA region enables a perfect statement
as to the variant or
clone size of the respective disease.
According to point c), these three different determinations can also be
carried out at one and the
same time, which will result in the maximum clarification of the syndrome,
since the variant of
the aberrantly methylated DNA region can be precisely assigned. The method can
be carried out
rapidly and the result is faultless and easy to interpret. As opposed to
conventional analytical
procedures, this method enables the examination of both male and female
patients. This analysis
yields unambiguous results even in borderline cases.
The extent of methylation can be determined by various methods, e.g., by
methylation-specific
restriction enzymes, oligonucleotides specifically recognizing and hybridizing
methylation-spe-
cifically altered DNA sections, etc. Also the sequence variant of the
polymorphous DNA region
can be determined in various ways such as, for instance, by PCR or by the aid
of restriction en-
zyrnes.
For a rapid analysis, it is particularly advantageous if the determination of
the extent of methyla-
tion as well as optionally the determination of the sequence variant are
performed by a methyla-
tion-specific PCR (MS-PCR), using primers by which a methylated or
unmethylated DNA
region is each amplified in a methylation-specific manner. The MS-PCR
technique is a simple
method to distinguish between methylated sequences and unmethylated sequences
and is used to
CA 02388834 2002-06-25
_ g _
diagnose patients, because methylation or non-methylation frequently is an
indication for a ge-
netically caused disease, or also gives rise to the same.
MS-PCR is based on the principle of using primers which are specific for a
methylated or un-
methylated sequence such that the respective ones of the primers will each
hybridize with the se-
quence as a function of whether the latter is methylated or not. The PCR
products will
subsequently suggest a methylation or non-methylation. The primers may, for
instance, be cho-
sen such that the PCR product for the methylated sequence will have a certain
length and the
PCR product for the unmethylated sequence will have another length. The PCR
products, thus,
will give information about the methylation of amplified DNA sections on
account of their sizes.
A common method for obtaining MS primers consists in specifically altering the
methylated
and/or unmethylated sequence and constructing appropriate primers that are
specific for the al-
tered sequences. An option to alter DNA in a methylation-specific manner
involves treatment of
the DNA with a deaminating agent such as, e.g., sodium bisulfite. Sodium
bisulfite converts the
unmethylated cytosine into uracil, which is replaced with thymidine in the
subsequent DNA am-
plification. This method, thus, produces different DNA sequences, departing
from originally ho-
mologous, yet differently methylated alleles. The primers that hybridize with
the unmethylated
sequence comprise thyrnidine instead of cytosine. On the other hand, the
primers that are specific
for the methylated sequence continue to comprise cytosine on the sites of
methylated Cs.
In the instant case, it is advantageous for an unambiguous result if the
primers are chosen such
that the PCR product of the methylated sequence will always have another
length than the PCR
product of the unmethylated sequence - irrespective of the length of the
repeat region.
The determination by means of MS-PCR accordingly guarantees a particularly
time-saving and
efficient method, requiring little DNA and involving less work. The results of
PCRs are obtained
simultaneously. Nor are any additional method steps like Southern blotting,
hybridization meth-
ods, etc. required.
It is particularly beneficial, for instance in the event of the CGG repeat of
the FMR 1 gene, if the
MS-PCR primers are realized on an antisense strand or on antisense strands.
The melting tem-
perature will thereby be reduced from 95°C to a melting temperature of
75°C, which enables the
PCR to be carried out at moderate annealing temperatures, thus improving the
process course.
These temperature conditions are inoffensive to the reaction components and
DNA polymerase.
Preferably, the MS-PCRs are carned out in duplex reactions. This means that
the PCRs for the
amplification of a given methylated sequence and the same given unmethylated
sequence are car-
ried out in one reaction, e.g., the PCRs for the amplification of the
polymorphous methylated and
unmethylated DNA regions. In this manner it is safeguarded that the reaction
products obtained
CA 02388834 2002-06-25
- 10 -
will be unambiguously assigned in the subsequent analysis on account of their
sizes, thus render-
ing the result clearly interpretable.
Advantageously, the MS-PCR for the determination of the potentially aberrantly
methylated
DNA region and the MS-PCR for the determination of the definitely
physiologically methylated
DNA region are carned out in a joint multiplex reaction. Since the MS-PCRs, as
a rule, concern
two different genes and hence two different sequences, the MS-PCRs will not be
mutually influ-
enced. The determination of the polymorphous DNA region would be carned out in
a separate
duplex reaction so as to enable clear determination of the polymorphism, for
instance, in order to
prevent the same having the size of variable PCR products from overlaying with
other PCR
products in the subsequent evaluation.
In a particularly preferred manner, a method is provided, wherein the
polymorphous DNA region
is a DNA region with a repeat polymorphism, which is preferably connected with
the potentially
aberrantly methylated DNA region, whereby the length of the repeat region is
determined as a
sequence variant and subsequently both the presence and the extent of the
potential aberration are
determined.
In the context of the present invention, repeat polymorphism is meant to
denote a region which
comprises several repetitions of a repeat unit, i.e., a certain sequence of
several nucleotides. The
number of repetitions, or so-called repeat units, differs as a function of the
gene or disease, the
extent of the disease, as a rule, being related to the length of the repeat
region. In most cases, a
repeat region having a particularly unnatural length is also methylated at the
same time if this
DNA region is normally unrnethylated. Mostly, in that case, also the, e.g.,
adjacent or potentially
aberrantly methylated region is accordingly strongly methylated such that the
determination of
the potentially aberrantly methylated DNA region and the determination of the
polymorphous
DNA region yield complementary results and can, thus, be regarded as
additional controls.
In a particularly preferred manner, the polymorphous DNA region is the CGG
trinucleotide re-
peat region of the FMR I gene. The determination in this region yields a
repeatable and unambi-
guous result. If the determination is, for instance, carried out by an MS-PCR,
both the length of
the repeat region and the extent of methylation can be determined in a single
method step if the
primers are chosen such that they encompass the whole repeat region. As
already pointed out in
the introductory part of the specification, the CGG trinucleotide repeat
region of the FRM 1 gene
is a sequence region that gives information about diseases and, in particular,
the fragile X syn-
drome, since the length of the repeat region is characteristic of the variant
of the disease.
It is particularly beneficial if the polymorphous DNA region is in the first
untranslated exon of
the FMR 1 gene. This is a region which is advantageous for the determination,
yielding a fault-
less result.
CA 02388834 2002-06-25
- 11 -
Preferably, the potentially aberrantly methylated DNA region is the FMR 1 gene
or a part of the
same. This guarantees a facilitated procedure, since only the FMR 1 gene
region is required for at
least part of the analysis. It could be used for the analysis upon isolation
from the remaining
DNA, or the analysis could also be carried out on isolated X chromosomes.
Preferably, the potentially aberrantly methylated DNA region is the 5'-
untranslated region of the
FMR 1 gene, i.e., the FMR 1 promoter, or a part of the same. On the one hand,
this yields reliable
results and, on the other hand, the potentially aberrantly methylated DNA
region is, thus, close to
the CGG repeat region, which is located in the first untranslated exon. In
this manner, it is not
necessary to use the whole gene for the analysis, which might possibly
disintegrate during de--
amination. If the potentially aberrantly methylated DNA region is located in
the FMR 1 pro-
moter, a shorter DNA section can be used for the assay.
In this case, it is advantageous if the definitely physiologically methylated
DNA region is a DNA
region that is definitely methylated on the active X chromosome. Its
methylation pattern is,
therefore, contrary to that of the FMR 1 gene which is methylated on the
inactive X chromosome.
This additional determination, thus, provides a novel and inventive method
which also allows the
determination of the degree of the respective disease in females in a
particularly simple and un-
ambiguous manner. To this end, the ratio of the methylated to the unmethylated
DNA regions of
the one gene (preferably the FMR 1 gene) is compared to the ratio of the
reciprocally methylated
DNA region (methylated : unmethylated) as well as the ratio of the methylated
repeat polymor-
phism to the unmethylated repeat polymorphism (preferably of the FMR 1 gene).
Preferably, the definitely physiologically methylated DNA region is a region
that comprises the
XIST gene or parts of the same. The XIST gene is methylated on the active
chromosome X such
that it comprises a methylation pattern reciprocal to that of the FMR 1 gene.
The determination
by the aid of the XIST gene as an internal standard constitutes a reliable,
unambiguous and sim-
ple method for detecting and evaluating a potentially aberrantly methylated
DNA region on the X
chromosome, in particular in females.
It is particularly favorable if the definitely physiologically methylated DNA
region is a region in
the XIST gene promoter. It turned out that the methylation of the promoter of
the XIST gene is
easy to determine and leads to reliable results.
For a particularly advantageous method it is provided that the potentially
aberrantly methylated
DNA region and the polymorphous DNA region are two sequences that do not
overlap each
other. It is, thus, ensured that, even if both DNA regions are on the FMR 1
gene, two separate,
independent DNA regions are analyzed in a manner that the results of the
investigations into both
regions complement each other, thus enabling the premature recognition of
possible confusion
CA 02388834 2002-06-25
- 12 -
errors.
Preferably, two or more potentially aberrantly methylated DNA regions are
simultaneously de-
tected on the X chromosome and evaluated. It is, for instance, feasible to
simultaneously deter-
mine in one reaction the syndromes relating to FraX-A, FraX-E and FraX-F,
whereby an exact
and reliable result will be obtained for any syndrome without the results of
the different syn-
dromes influencing one another.
A further aspect of the present invention is the use of an above-described
method according to
the invention for the detection and evaluation of X-chromosomal diseases. Such
diseases, which
have already been described above, are diseases induced by an aberrant
methylation on at least
one site in the X chromosome. Depending on the disease to be detected and
evaluated, a specific
potentially aberrantly methylated DNA region and optionally a definitely
physiologically methy-
lated DNA region of the X chromosome and/or a specific polymorphous DNA region
of the X
chromosome are analyzed.
In doing so, it is particularly advantageous if the method is used, in
particular, for the detection
and evaluation of the FraX-A, FraX-E and FraX-F syndromes, the clonality and X-
ane-
uploidies.
In the event of the fragile X syndrome, it is particularly favorable if the
FMR 1 promoter is de-
termined as a potentially aberrantly methylated DNA region and/or the XIST
gene or a part of the
same is determined as a definitely physiologically methylated DNA region
and/or the CGG tri-
nucleotide region of the FMR I gene is determined as a polymorphous DNA
region.
In the event of FraX-E, it is advantageous to determine the CpG islet at Sq28
as a potentially
aberrantly methylated DNA region and/or the GCC trinucleotide region as a
polymorphous DNA
region and/or the XIST gene or a part of the same as a definitely
physiologically methylated
DNA region.
In the event of FraX-F, its is advantageous to determine the (GCCGTC)"(GCC)m
region as a
polymorphous DNA region and/or the adjacent CpG islet as a potentially
aberrantly methylated
DNA region and/or the XIST gene or a part of the same as a definitely
physiologically methy-
lated DNA region.
Concerning the clone size, the CGG nucleotide repeat region of the FMRI gene
is preferably de-
termined as a polymorphous DNA region.
As already pointed out in the introductory part of the specification, normal
sequences comprise a
repeat region of S-(48) 54 CGG units, premutated sequences comprise (48-) 54-
200 CGG units
CA 02388834 2002-06-25
- 13 -
and full mutations comprise over 200 CGG units, in the event of FraX-A.
In order to completely ascertain the syndrome in the event of the fragile X
syndrome (and, of
course, also with, e.g., FraX-E and FraX-F), an unambiguous and rapid
distinction between the
different genotypes of the fragile X syndrome can be made by the analysis
according to the in-
vention even with female patients in which methylation analyses do not yield
clear results, since
- concerning the repeat region - the latter is competed in a PCR, particularly
at a length of more
than 200 units, by a repeat of normal length. By determining the extent of
methylation of the po-
tentially aberrantly methylated DNA region, the sample can be clearly assigned
to a certain geno-
type, since in the normal and premutation cases this DNA region is not
methylated, whereas it is
methylated in the event of full mutations. In male pre-/full mutation mosaics,
conditions are
similar to those already described for female patients. No clear diagnosis can
be obtained from a
conventional analysis in which merely the repeat region is determined, in
particular in borderline
cases, without additional method steps such as, e.g., Southern blotting. Even
if Southern blotting
is applied, it only is feasible to detect mosaics to a certain extent because
of the limited sensitiv-
ity. By contrast, an improved assessment ofthe syndrome can be obtained at a
reduced demand of
patient DNA by the higher-sensitivity PCR methods mentioned.
If, furthermore, besides the determination of a potentially aberrantly
methylated DNA region also
a definitely physiologically methylated DNA region is analyzed, the latter can
be employed as an
internal standard. If the potentially aberrantly methylated DNA region cannot
be detected on ac-
count of deletions, the definitely methylated DNA region (e.g., the XIST gene)
will be detected in
any event. It is, thus, guaranteed that the method did function and the
potentially aberrantly
methylated DNA region is possibly missing at least partially, i.e., deleted
(FMR 1 gene, for in-
stance).
In the event that the FMR 1 repeat region is determined by means of MS-PCR,
the number of
normal and premutation repeat units can be readily calculated from the length
of the MS-PCR
products. Thus, a method is provided, in which the degree of methylation and
the length of the
repeat regions are determined at one and the same time. The degree of the
fragile X syndrome is,
thus, determined in a single method step (either a single MS-PCR reaction or
several MS-PCR
reactions concurrently). MS-PCR is a rapid and reliable method which is cost-
effective and can
be carried out in any laboratory. Moreover, only a small quantity of DNA is
required for this
analysis. Furthermore, no additional, frequently time-consuming and expensive
methods such as,
for instance, immunologic or Southern blot methods need be carried out. Unlike
conventional
methods for determining the fragile X syndrome, which comprise PCR and an
additional South-
ern blot in most cases, no radioactive substances are employed, which is
advantageous, in par-
ticular for the laboratory personnel. In addition, a method is provided which
saves cumbersome
operating steps and is suitable, in particular, for routine diagnoses and
screening assays. Moreo-
ver, this analysis is suitable for DNA extracted from Guthrie maps and for
assays of small tissue
CA 02388834 2002-06-25
- 14 -
samples taken from the body by the aid of fine needles.
If a triple MS-PCR analysis is carried out for the detection and evaluation of
the fragile X syn-
drome, the following results will be obtained:
- Normal male individuals comprise an unmethylated repeat region (as a
polymorphous DNA re-
gion) of up to (48-) 54 units as well as an unmethylated FMR 1 DNA region,
e.g., the pro-
moter (as a potentially aberrantly methylated DNA region).
- Male patients afflicted with a premutation exhibit the same pattern, yet
with approximately
(48-) 54 to 200 repeat units.
- Male patients afflicted with a full mutation comprise a methylated repeat
region with more than
200 units (which is usually not amplified) as well as a methylated FMR 1
promoter region (as a
potentially aberrantly methylated DNA region). In full mutation/premutation
borderline cases, it
is decisive whether the methylated or unmethylated FMR 1 promoter region is
amplified. Even if
the methylated repeat region of a full mutation is as long so as not to be
methylated, the full mu-
tation can be detected because of the amplification of the methylated FMR 1
promoter region.
- Mosaic full mutation/premutation: the repeat region of the allele with the
full mutation is not
amplified, the unmethylated repeat region of the premutation allele is
amplified, comprising
(48-) 54 to 200 units. The methylated FMR 1 promoter region is amplified.
- On account of the random X inactivation in normal homozygous females, the
ratio of the un-
methylated to the methylated FMR 1 DNA region as well as of the unmethylated
to the methy-
lated XIST gene is 1 : 1. Since the two homologs comprise identical repeat
region lengths, only
one unmethylated and one methylated FMR 1 repeat region is each visible.
- In heterozygous females having different repeat region lengths, the same
pattern as described
above is to be observed, except that two unmethylated and two methylated FMR 1
repeat regions
having different lengths are to be seen.
- Females afflicted by a premutation exhibit similar patterns as heterozygous
females, the differ-
ence being that one methylated and one unmethylated FMR 1 repeat region of an
allele have
lengths of (48-) 54 to 200 units.
- Females afflicted by a full mutation always comprise a methylated FMR 1
promoter region on
the expanded allele irrespective of whether this is on the active or inactive
chromosome. In the
event of a random X inactivation, the methylation ratio of the FMR 1 DNA
region promoter re-
gion is 3 : 1 (methylation : non-methylation), while the methylation ratio of
the XIST gene is still
1:1.
- Females afflicted by a full mutation and either a skewed X inactivation or a
mosaic exhibit the
same pattern as respective females with a random X inactivation. A skewed X
inactivation and
mosaics can be identified by the quantitative assessment of the methylation
ratio of the FMR 1
promoter region with that of the XIST gene and the FMR 1 repeat region. The
methylation ratio
of the XIST gene will remain 1 : 1 in all cases, since every cell continues to
contain an active and
an inactive chromosome. The methylation ratio of the section of the further
gene (XIST), thus,
CA 02388834 2002-06-25
- 15 -
constitutes a standard. By contrast, the methylation ratio of the FMR 1
promoter region and that
of the repeat region will shift towards non-methylation or methylation,
respectively, as a function
of whether the normal chromosome or that with the expanded allele comprises a
skewed X inac-
tivation. A similar pattern can be observed in mosaic women having a normal
cell population and
one with a full mutation.
According to another aspect, the present invention relates to a kit of the
initially defined kind,
which comprises primer sets for the specific amplification of the respective
one of the methylated
and unmethylated DNA variants of a potentially aberrantly methylated DNA
region and/or a
definitely physiologically methylated DNA region of the X chromosome and/or a
polymorphous
DNA region of the X chromosome, which may either be physiologically or
aberrantly methy-
lated. This kit serves to carry out the above-described method according to
the invention.
Said kit preferably comprises, in addition to the primer sets, further
substances that are necessary
to carry out an MS-PCR: a substance for the conversion of the methylated or
unmethylated se-
quence (e.g., sodium bisulfite for the conversion of unmethylated cytosine
residues), the neces-
sary enzymes, buffers, etc. In this manner, a kit is provided which only
requires addition of the
DNA to be assayed in order to carry out the method according to the invention.
By the aid of this
kit, routine diagnoses can be performed in any laboratory.
In a particularly advantageous manner, said kit can be employed for the
detection and evaluation
of X-chromosomal diseases, in which case it comprises primer sets for the
methylation-specific
amplification of a DNA region with a repeat polymorphism for the determination
of the poly-
morphous DNA region. These primer sets serve to determine the extent of
methylation and the
length of the repeat polymorphism in order to assay the degree of the disease
and the clonality of
the same. Such chromosomal diseases include, for instance, FraX-A, FraX-E,
FraX-F, X-chro-
mosomal retardations, clonality, etc.
Preferably, the kit comprises primer sets for the amplification of a
polymorphous DNA region of
the CGG nucleotide repeat region in the first exon of the FMR 1 gene. Since
the repeat region of
the FMR 1 gene guarantees a safe and clear assertion as to the degree of
disease of the fragile X
syndrome or the clonicity, respectively, as already described above, a rapid
and reliable analytical
method can, thus, be provided by said kit.
Lt is, furthermore, beneficial if the kit comprises primer sets for the
amplification of the XIST
gene or parts of the same in order to determine the definitely physiologically
methylated DNA
region. As already described above, the XIST gene has a methylation pattern
reciprocal to that of
the FMR 1 gene. The methylation pattern in the result allows for a precise
interpretation in re-
spect to the variant of the fragile X syndrome.
CA 02388834 2002-06-25
- 16 -
It is, furthermore, advantageous if the kit comprises primer sets for the
amplification of the FMR
1 gene or a part of the same in order to determine the potentially aberrantly
methylated DNA re-
gion. In a favorable manner, the kit comprises primer sets for the
amplification of the FMR 1
gene promoter or a part of the same. With that kit, the above-described
preferred analytical
method can be carned out.
In doing so, it is advantageous if the primer sets are each provided as duplex
sets in a spatially
separated manner. It is thereby ensured, as described above, that clear and
readily interpretable
results will be obtained.
It is particularly favorable if the primer sets for the amplification of the
definitely physiologically
methylated DNA region and the potentially aberrantly methylated DNA region are
provided to-
gether in a multiplex set or a 4-plex set. Thus, the methylation-specific PCR
reactions of two
DNA regions are carried out simultaneously in one reaction mixture, which is
why the method
requires even less time. This is feasible, because the two genes (FMR 1 and
XIST) comprise dif-
ferent sequences such that the two PCR reactions will not influence each
other.
In the following, the present invention will be explained in more detail by
way of examples and
drawing figures to which it is, however, not to be limited, wherein:
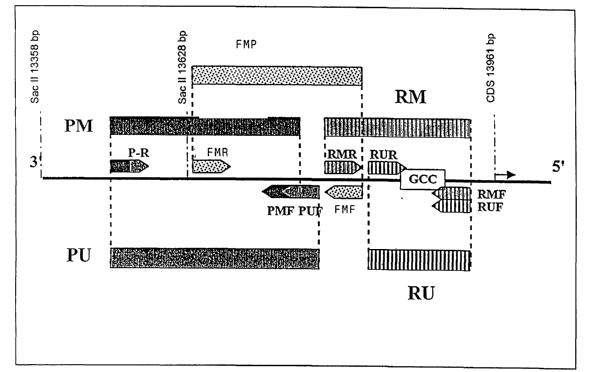
Fig. 1 illustrates the position of the primers for the FMR 1 gene;
Fig. 2 schematically illustrates the respective methylation patterns of the
FMR l and XIST genes
as well as the size and methylation of the repeat region;
Figs. 3a and 3b depict gel electrophoreses for the separation of the MS-PCR
products;
Fig. 4 shows the results of a multiplex PCR at the HUMAR.A locus;
Figs. 5 and 6 show the results of MS-PCRs.
Example 1: DNA Preparation
EDTA-anticoagulated, peripheral blood samples from healthy patients as well as
patients af-
flicted by the fragile X syndrome were stored at -20°C in 500 p1
aliquots until DNA extraction.
For DNA deamination and subsequent MS-PCR analysis, DNA was extracted from 80
u1 blood
with DNAzoI (Vienna Lab, Vienna, Austria) and resuspended in 30 p1 sterile
water.
0.5 pg DNA was deaminated according to the protocols of Zeschnigk et al., "A
single-tube PCR
test for the diagnosis of Angelman and Prader-Willi syndrome based on allelic
methylation dif-
ferences at the SNRPN locus", Eur. J. Hum. Genet. S, 94-8 ( 1997); "Imprinted
segments in the
human genome: different DNA methylation patterns in the Prader-Willi/Angelman
syndrome re-
gion as determined by the genomic sequencing method", Hum. Mol. Genet. 6, 387-
95 (1997),
said deamination having been carried out at 55°C for two hours with the
addition of 8 N,1 poly-
acrylic carrier to shorten DNA precipitation to 10 minutes at -20°C.
The deaminated DNA was
CA 02388834 2002-06-25
- 17 -
dissolved in 20 ~,l sterile water.
Example 2: Methylation-Specific PCR
13 primers (cf. SEQ. ID. No. 1-13) were synthesized in accordance with the
deaminated DNA
sequence of the respective unmethylated or methylated gene regions (cf. Table
l and Fig. 1).
CA 02388834 2002-06-25
- 18 -
ro ~ ~
M M 01 e~i
V
w" ~° "
a H
V v
O H
O 1!
~ a
N
r1 N M 01 V If1 b l~
M M
V M V ~ ~ ~
M M ~ M
~ E ~ ~ ~
E ~
m
n
E
V A
i~ n. yn n ;~ '~
in n
0
O
H ~ N ~ ~ ~ N
o a o ec
w ,Q w ,q w ~c~ w .ct
b b
0
o W-~1N aotH ~w ~H
v ~ u° ~
o ~ o x m
CA 02388834 2002-06-25
- 19 -
,~ a ~ a
., . .
.. + + M
n
a,
a
x ~ ~
H o +~
c~
" ...,
~
a ~ , . . .
a a ~ a
o H
..
0 0
a a
x '
a
~a
'
M
~ ' ' '
' f~l M Irf
V
E
0
s ~ : : '
n i n n n n
n
H vi
o ~
m
b
N ~d ~d
b ~d ~d ~
a ~ ~ ~ H.
a
w w ~ w a
a
a b o
~
H ~~ b ~ a ~ o
0
a
CA 02388834 2002-06-25
- 20 -
To a multiplex PCR mixture were added two forward primers (PUF, PMF) specific
for unmethy-
lated and methylated DNA and a mutual reverse primer (P-R) specific for the
deaminated un-
methylated and methylated FMR 1 promoters, as well as two forward primers
(XUF, XMF) and
two reverse primers (XUR, XMR) specific for the deaminated unmethylated and
methylated
XIST promoters. The duplex PCR mix comprises two forward primers (RUF, RMF)
and two re-
verse primers (RUR, RMR) which are specific for the deaminated unmethylated
and methylated
triplet repeat regions.
An additional primer pair detects a further methylated sequence section in the
FMR 1 promoter
(FMF, FMR), yet cannot be combined with the other primers.
For the design of these primers, the sequence of the antisense strand of the
FMR 1 gene region
(Acc. No. L29074, L38501, U80460) was used as the target sequence to be
amplified in order to
reduce the melting temperature from Tm = 95°C to Tm = 75°C.
The PCR was carried out in a reaction volume of 25 p1 under oil, whereby 1
E.iI of the 20 N,I de-
aminated DNA was each used from patients and normal controls. For the
multiplex PCR, the am-
plification buffer F-511 (10 mM Tris, pH 8.8, 50 mM KCI, 1.5 mM MgCl2, 0.1%
Triton-X-100;
Finnzymes Oy, Espoo, Finland) (DYN) was used; optimized buffer EXT (50 mM
Tris, 15 mM
NH4C1, 1.5 mM MgCl2, 0.1% Triton-X-100, pH 9.0) was used for the duplex
reaction with 4%
DMSO and 60 mM TMAC and for the FMP amplification without amplifier (DMSO,
TMAC).
The dNTP concentrations were 200 NM of each nucleotide. Table 1 lists the
optimum primer
concentrations. The amplifications were performed on a Biometra TrioBlock
(Biometra, Goettin-
gen, Germany) and initiated with one unit of Dynazyme SOl L (Finnzymes Oy,
Espoo, Finland),
with a first denaturation step at 95°C for 5 minutes. The multiplex PCR
profiles were 33 cycles at
95°C/30s [Program 1], 60°C/20s, 72°C/40s [Program 2].
Duplex and FMP profiles included 35
cycles at 95°C/45s, 63°C/lmin and 72°C/lmin. Finally,
incubation took place at 72°C for 7 min-
utes in all cases.
PCR products (5 p1) were separated on NOVEX-TBE gels (Novex, San Diego,
California, USA)
in O.Sx TBE buffer (90 mM Tris, 90 mM borate, 2 mM EDTA, pH 8.0). The bands
were detected
by staining with ethidium bromide (EtBr). Densitometric analyses were
performed with
KODAK-1D'~"''2Ø2 software package (Kodak, New Haven, CT, USA).
Repeat units in the normal and premutation ranges, but no full mutations could
be amplified. The
number of repeat units could be calculated from the lengths of the PCR
products in normal indi-
viduals and premutation carriers.
Table 2 lists the results to be expected, wherein "-" means no PCR product,
"+" means a PCR
product and "2+" means two products having different lengths. In Fig. 2, the
results to be ex-
CA 02388834 2002-06-25
- 21 -
pected are schematically illustrated using the same numbering as in Table 2.
Table 2
Target gene sequence ~lt~plw Duplex 8CR
PCR
PCR product ~1 XIST FEZ
Praaaoter Praaaoter Repeat
PIT 8M XU --- RU Rllt
~d
1 Normal males + - - + + -
2 'Males with premutatioa + - - + + -
3 Males with full mutation - + - + - -
4 Mosaic males with full mutation- + - + +
Males with deletion - - - + - -
6 Normal females with identical 1s1 IsI + +
repeat-lengths on both alleles
Normal females with different 1:1 1:1 2+ 2+
repeat-lengths on both alleles
8 Females with presnutatioa 1s1 1:1 2+ 2+
9 Females with full mutation 1s3 1s1 + +
Females with full mutation (>)1s3 isi a D
and
as elevated amount of cells
with active normal alleles
11 Fe~xaales with full mutation (<)1s3 1:1
and
D
as elevated amauat of cells
with actively affected alleles
12 Mosaic females with premutation(>)1s3 a p
or 1 s it ar
(<)1s3
D
a v
13 Mosaic females with full mutatio(>)1s3
or 1s1 or
(<)1s3 D a
CA 02388834 2002-06-25
- 22 -
( 1 ) Normal male individuals exhibit an unmethylated repeat region from up to
(48-) 54 triplets
as well as an unmethylated FMR 1 promoter.
(2) Male patients afflicted by a premutation exhibit the same pattern, yet
with about (48-) 54 to
200 repeat units.
(3) Male patients afflicted by a full mutation exhibit in the result a
methylated repeat region with
more than 200 triplets (which in most cases is not amplified in the MS-PCR) as
well as a methy-
lated FMR 1 promoter.
(4) Male patients with a full mutation/premutation mosaic exhibit a methylated
repeat region with
more than 200 units (which is not amplified) as well as an unmethylated repeat
region of (48-) 54
to 200 units and, in addition, an unmethylated and a methylated FMR 1
promoter. By the detec-
tion of the specific product of the XIST gene, a clear distinction between
female and male pa-
tients is feasible in all cases.
(5) With deletions in the FMR 1 gene region to be amplified, merely the
methylated PCR product
of the XIST gene promoter is visible.
(6) In normal homozygous females, the ratio of unmethylated to methylated FMR
1 promoter and
that of the XIST gene promoter is about 1 : 1. Since both homologs comprise
identical repeat re-
gion lengths, only one unmethylated and one methylated FMR 1 repeat region is
each visible.
(7) In heterozygous females exhibiting different repeat region lengths, the
same pattern as de-
scribed above can be observed, only two unmethylated and two methylated FMR 1
repeat regions
having different lengths being visible.
(8) Females afflicted by a premutation exhibit a similar pattern as
heterozygous females with the
difference that one methylated and one unmethylated FMR 1 repeat region have a
length of be-
tween (48-) 54 and 200 units each.
(9) Females afflicted by a full mutation always exhibit a methylated FMR 1
promoter on the ex-
panded allele irrespective of whether this is located on the active or
inactive chromosome. In the
event of a random X inactivation, the methylation ratio of the FMR 1 promoter
is 1 : 3 (non-
methylation : methylation), while the methylation ratio of the XIST gene
promoter continues to
bell.
( 10) - ( 13) Females with a full mutation and either a skewed X inactivation
or a mosaic exhibit
the same pattern as afflicted females with a random X inactivation, yet with
the methylation ratio
in the FMR 1 gene being shifted in one or the other direction. A skewed X
inactivation and mo-
saic can be identified by the semi-quantitative comparison of the methylation
ratio of the FMR 1
promoter and the FMR 1 repeat region with that of the gene section of the XIST
gene. The meth-
ylation ratio of the XIST gene remains 1 : 1 in all cases, since every cell
continues to carry 1 ac-
tive and 1 inactive chromosome. By contrast, the methylation ratio of the FMR
1 promoter and
that of the repeat region shift towards non-methylation or methylation,
respectively depending on
whether the normal chromosome or that with the expanded allele comprises a
skewed X inactiva-
tion. A similar pattern can be observed in mosaic females with a normal cell
population and one
with a full mutation.
CA 02388834 2002-06-25
- 23 -
Thus, all possible variants can be detected to diagnose the fragile X syndrome
and similar dis-
eases such as FraX-E, FraX-F and other X-chromosomal diseases, but also the
clonality and X-
aneuploids.
Figs. 3a and 3b depict gel electrophoreses of the MS-PCR products, Fig. 3a
illustrating the prod-
ucts of the multiplex reaction (FMR 1 promoter, XIST promoter) and Fig. 3b
showing the prod-
ucts of the duplex reaction (FMR 1 repeat expansion) with the numbering of
Table 2 having been
retained and n indicating the number of repeat units.
( 1 ) Normal male patient (n = 34)
(2) Male patient with a premutation (n = 130)
(3) Male patient with a full mutation (n > 200)
(4) Male patient with a mosaic full mutation (n > 200 and n = 100)
(5) Male patient with a deletion in the FMR 1 gene promoter as well as in the
repeat region
(6) Normal homozygous female (n = 32)
(7) Normal heterozygous female (n = 23+30)
(8) Female with a premutation (n = 33+66)
(9) Female with a full mutation (n > 200 and n = 23)
( 10) Female with a full mutation and an elevated amount of cells with normal
alleles due to a
skewed X inactivation (n > 200 and n = 30; elevated amount of the RU product)
( I 1 ) Female with an elevated amount of cells with full mutations due to a
skewed X inactiva-
tion (n > 200 and n = 46; elevated amount of the RM product)
( 12) Negative control (native placenta DNA which was not deaminated prior to
amplification)
(s) Size standard.
Table 3 illustrates the results from a densitometric analysis of the multiplex
PCR products of fe-
male patients. It shows the net intensities, the calculated ratios of
unmethylated and methylated
products as well as the standardized ratio based on the intergenic XIST
standard ratio (XM per
XU), the columns being numbered in accordance with Figs. l and 3. The ratio of
unmethylated
and methylated XIST products specific for the inactive and active X chromose,
respectively, re-
mained stable within all female samples (mean value (XM/XU) ) = 1.033 + 0.21).
In afflicted fe-
males (9 and 11 ), the ratios of the FMR 1 methylation based on the XIST
standard ratio were
outside the 95% confidence range (ratio (PU/PM) / (XM/XU) = 0.32 + 0.06). In
the event of a
skewed X inactivation, which prefers the full mutation allele (col. 10), the
ratio remains within
the normal range and, on account of the different band intensities of the RU
and RM products
specific for afflicted females, the patient was diagnosed as suffering from
the fragile X syndrome
(col. 10, cf. also Fig. 3 below).
Table 3
CA 02388834 2002-06-25
- 24 -
Product - Saa~ple.(net
intensity)
(bpl 6 7 8 9 10 11
PU I3181 1206 2585 1122 1244 916 3i8
PM L2881 3812 5317 3863 6512 3278 2977
1Q3 [2411 1177 2476 1972 3529 1854 1698
XU (1981 1157 1862 1700 3143 2363 2155
Ratio intragenic
ratios
(pM/PU) 0:32 0.49 0.29 0.19 0.28 0.11
(~i/XU) 1.02 1.33 1.16 1.12 0.78 0.79 .
standardized
intergenic
ratios
(PM/PU)
0.31 0.37 0.25 0.17 0:36 0.14
Example 3: Evaluation of the Clonality at the Humara Gene Locus
In order to detect a skewed X inactivation, a PCR was carried out for the
amplification of a se-
quence in the Humara locus (Human Androgen Receptor). Three primers were used:
hum-A (um): SEQ. ID. No. 18
hum-B2 (m): SEQ. ID. No. 19
hum-C (com): SEQ. ID. No. 20
wherein hum-A hybridizes with the unmethylated sequence, hum-B2 with the
methylated se-
quence and hum-C in both cases. 25 NI hum-A [20 pmol/l.il], 37.5 w1 hum-B2 [20
pmol/~.ll], 50 p1
hurn-C [20 pmol/~,l], 672 ~,1 AD, 100 ~.il DYN and 100 p1 dNTPs [2 mM] were
used per batch.
The PCR profiles comprised 33 cycles 95°C/20s, 54°C/40s,
72°C/40s [Program 3].
Table 4 illustrates the results of a densitometric analysis of the multiplex
PCR products, the result
being visible in the gel illustrated in Fig. 4. Numbering of the bands in the
gel of Fig. 4 corre-
sponds with the numbering of the columns in Table 4. The net intensities are
calculated from the
ratios of unmethylated and methylated products, the mean value (A + A')/2
deviating the more
from 0.5 (i.e., 50% of the overall cell population) the more intensively
skewing occurs. Patients
15, 16 and 19 exhibit intensive skewing.
CA 02388834 2002-06-25
- 25 -
o M M ~ w
n n
~~~" ~, o0 0
N
~ M M p p
m
~ m M p 0 0 0
M p
I
l1
O
N
~ it P w M 111
m M o ~'
H o0
p p
m 0 0
0 1 e! M 0
~'
of M Q 1 ~~ ~ M
p
CM0 M ~ 0 00
p
f l Q
a a I
aa
M ~ w IMlf~ N~ n
I~fl
10
M 0o p
M 0
a
00 p C1
- m n n
M H 00' ~ !!IfI Hla.t l'
0 00
~
N
n
1 0
m
ri
n.n w "~ 'o a ran
o Y w M p t ! i t
t N
H e w a M 0 0
r~ r l
n
m oo ~ ~r~ N ~ o
o
~ ~ ~ ~ o 0 0
n er of a~ H ~ o
soc
a m ~ w0 e-
0 0 0
n t~r 10 0 0
u 1
t
m o ~ a a o n
o , ~ In Ml o r v Ino
" d n o d
-
y 7 m H ~t H
~ i o
H
n n ~ Dw p1 M n
01w Ip w
'
~ M p 0 0 d o
x V f N ~
0
,
.. .,
~ w M M H N
rt
'a _ .. y y ~ r
H 5
C
~ M ~ m
~
U La~ H M a m cti s' a m
e!
CA 02388834 2002-06-25
- 26 -
It is, thus, apparent that in a multiplex reaction the intensity of skewing
and hence the extent of
the clonality in the patient can be precisely evaluated by the method
according to the invention.
Example 4: Detection of FraX-A, FraX-E and FraX-F Patients in an MS-PCR
Multiplex MS-PCRs are carried out on patients in order to detect and evaluate
in a single reac-
tion the occurrence and extent of a possible aberrant methylation of the
promoter regions of
FraX-A, FraX-E and FraX-F. Table 5 indicates the primers employed [20
pmol/~tl], the amounts
of the substances used for the MS-PCRs and the product sizes to be expected,
the concentration
of the dNTPs being 2 mM. Fig. 5 shows the results (in the form of a gel
illustration) of these
multiplex MS-PCRs [Program 1 ], wherein A is the respective size standard, B
refers to normal
females, C refers to normal males, D represents male patients suf~'ering from
the FraX-E disease
and E refers to native non-deaminated DNA. It is apparent that, compared to
normal males, the
afflicted male patients exhibit bands in the order of 248 bp, yet no Fra.X-F
bands (191 bp) occur.
All patients display the control (unmethylated promoter, PU).
r
CA 02388834 2002-06-25
- 27 -
x°
A N .-1 eh ,a-1 e~i .~-1 ~~-1
H
a
w
m
e~ 0D sw 01
M N N ri
<A
i~
N N
w W
cn a
m a ~ ~ a
~ ~ ~ ~
a a t~
is n ~ a a
a a ~ u~
U H
H
x x x x x x ~ x ~ x
O O a~ t0 O N N tD t0 OO
O O N ~-I N e-1 e-I e-1 t-1 c~1
~i e-1 d~
..
H
t1a 1 1 1 I
w w ~ ~
A
CA 02388834 2002-06-25
- 28 -
In the case of females, the net intensities of the bands in the gel are
measured, whereby conclu-
sions as to the extent of the disease can be drawn from the mutual intensities
of the individual
bands. If, for instance, one of the bands specific for the methylated promoter
(PM, FraX-E AB,
FraX-F AB) is amplified relative to the other bands at a ratio of 3:2, this is
a sign of a disease. In
the instant gel of Fig. 5, all females show ratios of 2:2, from which it can
be taken that these fe-
males are healthy (suffering neither from FraX-A or FraX-E nor from FraX-F).
Example 5: Combination of a Duplex PCR with a Potentially Methylated DNA
Region
A PCR [Program 2] is carried out for the amplification of the polymorphous
gene region (in the
instant case the FX repeat (RU, RM)) along with the potentially aberrantly
methylated gene re-
gion (in the example, the methylated FMR 1 promoter (FX-JK)). From the
evaluation of the band
patterns and the band intensities as illustrated in Fig. 6, a disease can be
concluded and, if de-
sired, the extent of the affection can be determined.
Table 6 indicates the different constellations, "XY" representing male and
"XX" representing fe-
male patients.
CA 02388834 2002-06-25
- 29 -
..
N
v
F1 e~ e-1
ra
W
x
N N
. v v
i~
N e-I e-I
ri
N N
~1
N O
O
O
G',N N N
e-I I
e-I
_
I
~O ri
H
~
1 1 ei
~1
a
I 1 -1
0
a
,, .. ..
m
b
..
b
~
0
a
0
~
H
P~ m
pr N
b
~
,-
l
r
~ m
.b .
CA 02388834 2002-06-25
- 30 -
Table 7 indicates the primers used and the PCR products to be expected.
Table 7
Primer Concentrations (20 pmol/~,l)
Primer 1 Prod. size;
RUF 30
RUR 30 RU:
64+3x(CGG)n
RMF 20
RMR 20 RM:
120+3x(CGG)n
FX-K 20
FX-J 20 FX-JK:
302bp
TMAC (1,5M) 40
DMSO 40
EXT 100
dNTPs 100
AD 468
FX-K: 5'-GGAAGTGAAATCGAAACGGAGTTGAGC-3' (SEQ ID NO 21)
FX-J:. 5'-AACGTTCTAACCCTCGCGAAACAATACG-3' (SEQ ID NO 22)
Fig. 6 depicts the separation of the PCR products, S being the standard, 21 a
normal male, 22 a
male patient with premutation, 23 a male patient with full mutation, 24 a
normal female (homo-
zygous repeat), 25 a normal female (heterozygous repeat), 26 a female patient
with premutation,
27 a female patient with full mutation, 28 native non-deaminated DNA as a
negative control.
In a normal male only the unmethylated repeat is amplified. In male a patient
afflicted by a per-
mutation also the unmethylated expanded repeat is amplified.
In a male patient afflicted by a full mutation the methylated promoter is
amplified, and optionally
also the methylated repeat (as already indicated, the repeat is not amplified
if it exceeds a certain
size).
CA 02388834 2002-06-25
- 31 -
Normal females exhibit an intensity ratio of 2 : 2 : 2 (methylated promoter :
methylated repeat
unmethylated repeat), homozygous females having two identical alleles
(identical repeat num-
ber), heterozygous females, however, having different repeat lengths such that
two different al-
leles with different repeat lengths (2 x 1 ) are to be observed.
Females with permutations exhibit a ratio of 2 : 1 : 1 (the expanded repeat
cannot be amplified
from a certain size; however, if it is amplified, the normal and expanded
repeats are to be ob-
served).
Females with full mutations exhibit a ratio of 3 : 1 : 1 (so far, an expanded
methylated full muta-
tion repeat could not be amplified).
It is apparent that this embodiment of the method according to the invention
guarantees an exact
evaluation of the syndrome both in male and in female patients.
CA 02388834 2002-06-25
- 32 -
SEQUENCE LISTING
<110> St. Anna-Kinderspital
<120> Method for detecting and evaluating
a potentially aberrant methylated DNA region on
the X-Chromosome
<130> FMR1
<140>
<141>
<160> 22
<170> PatentIn Ver. 2.1
<210> 1
<211> 25
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of the artificial sequence: Primer
<400> 1
gtgtttgatt gaggttgaat ttttg 25
<210> 2
<211> 28
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of the artificial sequence: Primer
<400> 2
atttaatttc ccacrccact aaatacac 28
<210> 3
CA 02388834 2002-06-25
- 33 -
<211> 27
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of the artificial sequence: Primer
<400> 3
gttgcgggtg taaatattga aattacg 27
<210> 4
<211> 26
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of the artificial sequence: Primer
<400> 4
aattaaagta ggtattcgcg gtttcg 26
<210> 5
<211> 26
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of the artificial sequence: Primer
<400> 5
tttttcctta acccatcgaa atatcg 26
<210> 6
<211> 27
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of the artificial sequence: Primer
CA 02388834 2002-06-25
- 34 -
<400> 6
aaaagtggtt gttattttag atttgtt 2~
<210> 7
<211> 25
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of the artificial sequence: Primer
<400> 7
ctacctccca atacaacaat cacac 25
<210> 8
<211> 29
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of the artificial sequence: Primer
<400> 8
tttgagaggt gggttgtggg tgtttgagg 29
<210> 9
<211> 34
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of the artificial sequence: Primer
<400> 9
aacaccacta ccaaaaaaca tacaacaaca caac 34
<210> 10
<211> 30
<212> DNA
<213> Artificial Sequence
CA 02388834 2002-06-25
- 35 -
<220>
<223> Description of the artificial sequence: Primer
<400> 10
ccgcctctaa acgaacgacg aaccgacgac 30
<210> 11
<211> 29
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of the artificial sequence: Primer
<400> 11
tttcgagagg tgggttgcgg gcgttcgag 29
<210> 12
<211> 29
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of the artificial sequence: Primer
<400> 12
cgtcgtcggt tcgtcgttcg tttagaggc 29
<210> 13
<211> 28
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of the artificial sequence: Primer
<400> 13
ccgaccgatt cccaacaacg cgcatacg 28
CA 02388834 2002-06-25
- 36 -
<210> 14
<211> 21
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of the artificial sequence: Primer
<400> 14
ttcgtcgtcg ttgtcgtcgt c 21
<210> 15
<211> 27
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of the artificial sequence: Primer
<400> 15
aactaaaaat atccgaaccg catcgac 27
<210> 16
<211> 21
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of the artificial sequence: Primer
<400> 16
agttcgtagc gcggattttc g 21
<210> 17
<211> 25
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of the artificial sequence: Primer
CA 02388834 2002-06-25
- 37 -
<400> 17
aacgtaaacg cgactaacgc taacg 25
<210> 18
<211> 22
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of the artificial sequence: Primer
<400> 18
aatttgtttt agagtgtgtg tg 22
<210> 19
<211> 21
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of the artificial sequence: Primer
<400> 19
gcgagcgtag tatttttcgg c 21
<210> 20
<211> 21
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of the artificial sequence: Primer
<400> 20
ctactaccta aaactaatct c 21
<210> 21
<211> 27
CA 02388834 2002-06-25
- 38 -
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of the artificial sequence: Primer
<400> 21
ggaagtgaaa tcgaaacgga gttgagc 2~
<210> 22
<211> 28
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of the artificial sequence: Primer
<.400> 22
aacgttctaa ccctcgcgaa acaatacg 28