Note: Descriptions are shown in the official language in which they were submitted.
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
HETEROCYCLIC CYTOTOXIC AGENTS
Government Funding
The invention described herein was made with U.S. Government
S support under grant number CA39662 awarded by the National Cancer Institute.
The United States Government has certain rights in the invention.
Background of the Invention
DNA-topoisomerases are enzymes which are present in the nuclei of
cells where they catalyze the breaking and rejoining of DNA strands, which
control
the topological state of DNA. Recent studies also suggest that topoisomerases
are
also involved in regulating template supercoiling during RNA transcription.
There
are two major classes of mammalian topoisomerases. DNA-topoisomerase-I
catalyzes changes in the topological state of duplex DNA by performing
transient
single-strand breakage-union cycles. In contrast, mammalian topoisomerase II
alters
the topology of DNA by causing a transient enzyme bridged double-strand break,
followed by strand passing and resealing. Mammalian topoisomerase II has been
further classified as Type II a and Type II (3. The antitumor activity
associated with
agents which are topoisomerase poisons is associated with their ability to
stabilize
the enzyme-DNA cleavable complex. This drug-induced stabilization of the
enzyme-DNA cleavable complex effectively converts the enzyme into a cellular
poison.
Several antitumor agents in clinical use have potent activity as
mammalian topoisomerase II poisons. These include adriamycin, actinomycin D,
daunomycin, VP-16, and VM-26 (teniposide or epipodophyllotoxin). In contrast
to
the number of clinical and experimental drugs which act as topoisomerase II
poisons, there are currently only a limited number of agents which have been
identified as topoisomerase I poisons. Camptothecin and its structurally-
related
analogs are among the most extensively studied topoisomerase I poisons.
Recently,
bi- and terbenzimidazoles (Chen et al., Cancer Res. 1993, 53, 1332-1335; Sun
et
al., J. Med. Chem. 1995, 38, 3638-3644; Kim et al., J. Med. Chem. 1996, 39,
992-
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
2
998), certain benzo[c]phenanthridine and protoberberine alkaloids and their
synthetic analogs (Makhey et al., Med. Chem. Res. 1995, S, 1-12; Janin et al.,
J.
Med. Chem 1975, 18, 708-713; Makhey et al., Bioorg. & Med. Chem. 1996, 4, 781-
791), as well as the fungal metabolites, bulgarein (Fujii et al., J. Biol.
Chem. 1993,
268, 13160-13165) and saintopin (Yamashita et al., Biochemistry 1991, 30, 5838-
5845) and indolocarbazoles (Yamashita et al., Biochemistry 1992, 31, 12069-
12075)
have been identified as topoisomerase I poisons.
The exceptional topoisomerase poisoning observed with coralyne,
nitidine, 5,6-dihydro-8-desmethylcoralyne and related analogs prompted several
recent studies on those structural features which are associated with their
ability to
act specifically as poisons of topoisomerase I or topoisomerase II (Gatto et
al.,
Cancer Res. 1996, 56, 2795-2800; Wang et al., Chem. Res. Toxicol. 1996, 9, 75-
83;
Wang et al., Chem. Res. Toxicol., 1993, 6, 813-818). A common feature
associated
with all three of these agents is the presence of a 3-phenylisoquinolinium
moiety
within their structure.
Despite the observation that several of these compounds had similar
potency to camptothecin as a topoisomerase I poison or similar potency to VM-
26 as
a topoisomerase II poison, they possessed only modest cytotoxic activity. The
absence of a more direct correlation with their potency as topoisomerase
poisons
was attributed, in part, to the likelihood that these agents are not likely to
be
absorbed as effectively into cells as either camptothecin or VM-26. The
presence of
the quaternary ammonium group most likely impedes cellular uptake. It has been
speculated that agents such as coralyne and nitidine may need to undergo
hydrolysis
to permit effective transport, with subsequent dehydration or cyclodehydration
to
reform the quaternary ammonium parent compound. This may explain the
relatively
poor antitumor activity observed in vivo with agents such as coralyne or
nitidine.
Presently, a need exists for additional agents that are useful for
treating cancer.
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
3
Summary of the Invention
Applicant has discovered compounds that show inhibitory activity
against topoisomerase I andlor topoisomerase II, and compounds that are
effective
cytotoxic agents against cancer cells, including drug-resistant cancer cells.
S Accordingly, the invention provides a compound of the invention which is a
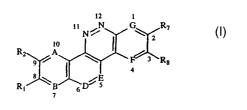
compound of formula I:
12 1
1 N~ N \ R~
I 2
Rz 9 iA \ / 3 Ra
4
~E
R, B D 5
7 6
wherein:
A is N or CR3;
B is N or CRS;
10 D is NRe or CR~Rb;
E is NRf or CR~Rd;
F is N or CR,;
G is N or CR6;
R,, Rz and R~ are each individually hydrogen, (C,-C6)alkyl, (C3-
C6)cycloalkyl, (C,-C6)alkoxy, vitro, hydroxy, NRgRh, COORk, ORm, or halo; or
R,
and RZ taken together are methylenedioxy and R~ is hydrogen, (C,-C6)alkyl, (C3-
C6)cycloalkyl, (C,-C6)alkoxy, vitro, hydroxy, NR~R,,, COORk, ORm, or halo; or
RZ
and R3 taken together are methylenedioxy and R, is hydrogen, (C,-C6)alkyl, (C3-
C~)cycloalkyl, (C,-C6)alkoxy, vitro, hydroxy, NRgR,,, COORk, ORm, or halo;
R6, R-, and R8 are each individually hydrogen, (C,-C6)alkyl, (C3
C6)cycloalkyl, (C,-C6)alkoxy, vitro, hydroxy, NRgR,,, COORk, ORm, or halo; or
R6
and R~ taken together are methylenedioxy and Rg is hydrogen, (C,-C6)alkyl, (C3-
C6)cycloalkyl, (C,-C6)alkoxy, vitro, hydroxy, NRgR,,, COORk, ORm, or halo; or
R~
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
4
and R8 taken together are methylenedioxy and R6 is hydrogen, (C,-C6)alkyl, (C3-
C6)cycloalkyl, (C,-C6)alkoxy, vitro, hydroxy, NRgR,,, C(=O)Rk, COORk, OR~,, or
halo;
each bond represented by ----- is individually present or absent;
R~ and Rb are each independently hydrogen or (C,-C6)alkyl if the bond
between the 11- and 12-positions represented by ----- is absent; or Ra is
hydrogen or
(C,-C6)alkyl and Rb is absent if the bond between the 11- and 12-positions
represented by ----- is present;
R~ and Rd are each independently hydrogen or (C,-C6)alkyl if the bond
between the 11- and 12-positions represented by ----- is absent; or R~ is
hydrogen or
(C,-C6)alkyl and Rd is absent if the bond between the 11- and 12-positions
represented by ----- is present;
Re is hydrogen or (C,-C6)alkyl if the bond between the 5- and
6-positions represented by ----- is absent; or Re is absent if the bond
between the 5-
1 S and 6-positions represented by ----- is present;
Rf is hydrogen or (C,-C~)alkyl if the bond between the 5- and
6-positions represented by ----- is absent; or Rf is absent if the bond
between the 5-
and 6-positions represented by ----- is present;
each R~ and R,, is independently hydrogen, (C,-C6)alkyl, (C3-
C6)cycloalkyl, (C,-C6)alkoxy, (C,-C~,)alkanoyl, aryl, aryl(C,-C~)alkyl,
aryloxy, or
aryl(C,-C6)alkoxy; or R~ and R,, together with the nitrogen to which they are
attached
are pyrrolidino, piperidino, morpholino, or thiomorpholino;
each Rk is independently hydrogen, or (C,-C6)alkyl; and
each Rm is independently (C,-C6)alkanoyl, aryl, or aryl(C,-C6)alkyl;
each RS and R~ is independently hydrogen, methyl, vitro, hydroxy,
amino, or halo;
wherein any (C,-C6)alkyl, (C3-C~)cycloalkyl, or (C,-C6)alkoxy of R',
R2, R3, R6, R', Rg, or Rk is optionally substituted on carbon with 1, 2, or 3
substituents independently selected from hydroxy, halo, NR"RP, (C3-
C6)cycloalkyl,
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
or (C,-Cb)alkoxy; wherein each R~ and RP is independently hydrogen, (C1-
C6)alkyl,
(C3 C6)cycloalkyl, (C,-C6)alkoxy, or (C,-C6)alkanoyl; or R~ and Rp together
with the
nitrogen to which they are attached are pyrrolidino, piperidino, morpholino,
or
thiomorpholino;
$ wherein any aryl is optionally be substituted with 1, 2, or 3 substituents
independently selected from hydroxy, halo, vitro, trifluoromethyl,
trifluoromethoxy,
carboxy, amino, (C,-C6)alkyl, (C3-C6)cycloalkyl, and (C,-C6)alkoxy;
provided no more than two of A-G comprise nitrogen;
or a pharmaceutically acceptable salt thereof.
The invention also provides a pharmaceutical composition comprising
a effective amount of a compound of formula I, or a pharmaceutically
acceptable
salt thereof, in combination with a pharmaceutically acceptable diluent or
carrier.
The invention also provides a method of inhibiting cancer cell growth,
comprising administering to a mammal afflicted with cancer, an amount of a
compound of formula (I), effective to inhibit the growth of said cancer cells.
The invention also provides a method comprising inhibiting cancer cell
growth by contacting said cancer cell in vitro or in vivo with an amount of a
compound of claim l, effective to inhibit the growth of said cancer cell.
The invention also provides a compound of formula I for use in
medical therapy (preferably for use in treating cancer, e.g. solid tumors), as
well as
the use of a compound of formula I for the manufacture of a medicament useful
for
the treatment of cancer, e.g. solid tumors.
The invention also provides processes and novel intermediates
disclosed herein which are useful for preparing compounds of the invention.
Some
of the compounds of formula I are useful to prepare other compounds of formula
I.
K.W. Gopinath et al., Indian J. Chem., 1958, 504-509, disclose the
preparation of 2,3,8,9-tetramethoxy-5,6-diazachrysene and 2,3-8,9-
bismethylenedioxy-5,6-diazacrysene. Accordingly, the compounds of the
invention
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
6
may preferably exclude the compounds 2,3,8,9-tetramethoxy-5,6-diazachrysene
and
2,3-8,9-bismethylenedioxy-5,6-diazacrysene.
The compounds of the invention may also preferably exclude
compounds of formula (I) wherein D is NRe; when A CR3; B is CRS; E is CR~Rd; F
is CR,; and G is CR6.
The compounds of the invention may also preferably exclude
compounds wherein R,-R~ and R~ R8 are each hydrogen.
The compounds of the invention may also preferably exclude
9-hydroxy-2,3,8-trimethoxydibenzo[c,h]cinnoline.
Preferably, for a compound of formula I one of RZ and R8 is hydrogen,
methyl, vitro, hydroxy, amino, fluoro or chloro; or at least one of RZ and Rg
forms
part of a methylenedioxy.
Brief Description of the Figures
Figures 1-5: illustrate the synthesis of compounds of the invention.
Figure 6: illustrates specific compounds of Formula I.
Figures 7-10: illustrate the synthesis of compounds of the invention.
Figure 11: shows the structure of reference compounds tested
hereinbelow.
Detailed DescriRtion
The following definitions are used, unless otherwise described: halo is
fluoro, chloro, bromo, or iodo. Alkyl, alkoxy, alkenyl, alkynyl, etc. denote
both
straight and branched groups; but reference to an individual radical such as
"propyl"
embraces only the straight chain radical, a branched chain isomer such as
"isopropyl" being specifically referred to. Aryl denotes a phenyl radical or
an ortho-
fused bicyclic carbocyclic radical having about nine to ten ring atoms in
which at
least one ring is aromatic.
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
7
Specific values listed below for radicals, substituents, and ranges, are
for illustration only; they do not exclude other defined values or other
values within
defined ranges for the radicals and substituents
Specifically, (C1-C6)alkyl can be methyl, ethyl, propyl, isopropyl,
butyl, iso-butyl, sec-butyl, pentyl, 3-pentyl, or hexyl; (C3-C6)cycloalkyl can
be
cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl; (C,-C6)alkoxy can be
methoxy,
ethoxy, propoxy, isopropoxy, butoxy, iso-butoxy, sec-butoxy, pentoxy, 3-
pentoxy,
or hexyloxy; (C,-C6)alkanoyl can be acetyl, propanoyl, butanoyl, pentanoyl, or
hexanoyl; and aryl can be phenyl, indenyl, or naphthyl;.
Specifically, Rz or R~ can be hydroxy, methoxy, benzyloxy, amino,
hydroxymethyl, aminomethyl, aminocarbonyl, methoxycarbonyl, trifluoromethyl, 3-
aminopropoxycarbonyl, or 2-hydroxyethyl.
Specifically, R3 can be hydrogen.
Specifically, RS and R, are each hydrogen.
A specific group of compounds are compounds of formula I:
12 I
II N G R~
Ni~ ~ \ 2
R2 9 ~A \ F 3 Rx
4
8 \ ~ .i E
R~ B D
'7 6
wherein:
A is N or CR3;
B is N or CRS;
D is NRe or CR~RE,;
E is NRf or CR~Rd;
F is N or CRS;
G is N or CR6;
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
8
R,, RZ and R3 are each individually hydrogen, (C,-C6)alkyl, (C3
C6)cycloalkyl, (C,-C6)alkoxy, vitro, hydroxy, NRgR," COORk, ORm, or halo; or
R,
and RZ taken together are methylenedioxy and R3 is hydrogen, (C,-C6)alkyl, (C3
C6)cycloalkyl, (C,-C6)alkoxy, vitro, hydroxy, NRgR," COORk, ORm, or halo; or
Rz
and R3 taken together are methylenedioxy and R, is hydrogen, (C,-C6)alkyl, (C3-
C6)cycloalkyl, (C,-C6)alkoxy, vitro, hydroxy, NR~R," COORk, ORm, or halo;
R6, R~ and Rg are each individually hydrogen, (C,-C6)alkyl, (C3-
C6)cycloalkyl, (C,-C6)alkoxy, vitro, hydroxy, NRgR," COORk, ORm, or halo; or
R6
and R~ taken together are methylenedioxy and Rg is hydrogen, (C,-C6)alkyl, (C3-
C~)cycloalkyl, (C,-C6)alkoxy, vitro, hydroxy, NRfiR,,, COORk, ORm, or halo; or
R~
and Rg taken together are methylenedioxy and R6 is hydrogen, (C,-C6)alkyl, (C3
C6)cycloalkyl, (C,-C6)alkoxy, vitro, hydroxy, NRgR,,, C(=O)Rk, COORk, ORm, or
halo;
each bond represented by ----- is individually present or absent;
Ra and R,, are each independently hydrogen or (C,-C6)alkyl if the bond
between the 11- and 12-positions represented by ----- is absent; or R~ is
hydrogen or
(C,-C6)alkyl and Rb is absent if the bond between the I I- and 12-positions
represented by ----- is present;
R~ and R~ are each independently hydrogen or (C,-C6)alkyl if the bond
between the I 1- and 12-positions represented by ----- is absent; or R~ is
hydrogen or
(C1-C6)alkyl and Rd is absent if the bond between the I 1- and 12-positions
represented by ----- is present;
Re is hydrogen or (C,-C6)alkyl if the bond between the 5- and
6-positions represented by ----- is absent; or Re is absent if the bond
between the 5-
and 6-positions represented by ----- is present;
Rf is hydrogen or (C,-C6)alkyl if the bond between the 5- and
6-positions represented by ----- is absent; or Rf is absent if the bond
between the S-
and 6-positions represented by ----- is present;
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
9
each Rg and R,, is independently hydrogen, (C,-C6)alkyl, (C3-
C6)cycloalkyl, (C1-C6)alkoxy, (C,-C6)alkanoyl, aryl, aryl(C,-C6)alkyl,
aryloxy, or
aryl(C,-C6)alkoxy; or Rg and Rh together with the nitrogen to which they are
attached
are pyrrolidino, piperidino, morpholino, or thiomorpholino;
each Rk is independently hydrogen, or (C,-C6)alkyl; and
each Rm is independently (C,-C6)alkanoyl, aryl, or aryl(C,-C6)alkyl;
each RS and R~ is independently hydrogen, methyl, nitro, hydroxy,
amino, or halo;
wherein any (C,-C6)alkyl, (C3-C6)cycloalkyl, or (C,-C6)alkoxy of R~,
RZ, R3, R6, R', Rg, or Rk is optionally substituted on carbon with 1, 2, or 3
substituents independently selected from hydroxy, halo, NRnRp, (C~-
C~)cycloalkyl,
or (C,-C6)alkoxy; wherein each R" and RP is independently hydrogen, (C,-
C6)alkyl,
(C~-C6)cycloalkyl, (C,-C6)alkoxy, or (C,-C6)alkanoyl; or R" and R~, together
with the
nitrogen to which they are attached are pyrrolidino, piperidino, morpholino,
or
thiomorpholino;
wherein any aryl is optionally be substituted with 1, 2, or 3 substituents
independently selected from hydroxy, halo, vitro, trifluoromethyl,
trifluoromethoxy,
carboxy, amino, (C,-C6)alkyl, (C3-C~)cycloalkyl, and (C,-C~)alkoxy;
provided no more than two of A-G comprise nitrogen; and
provided at least one of RZ and R~ is hydrogen, methyl, vitro, hydroxy,
amino, fluoro or chloro; or at least one of RZ and R~ forms part of a
methylenedioxy;
or a pharmaceutically acceptable salt thereof. Preferably, within this
specific group of compounds the compound of formula I is not 2,3-8,9-
bismethylenedioxy-5,6-diazacrysene; and R,-R3 and R6 R$ are not each hydrogen.
A specific group of compounds are compounds of forn~ula I wherein
R,, R~ and R3 are each individually hydrogen, or (C,-C6)alkoxy; or R, and RZ
taken
together are methylenedioxy (-OCH~O-) and R3 is hydrogen or (C,-C6)alkoxy; or
a
pharmaceutically acceptable salt thereof.
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
Another specific group of compounds are compounds of formula I
wherein R~ or R8 is (C,-C6)alkoxy; or R~ and Rg taken together are
methylenedioxy;
or a pharmaceutically acceptable salt thereof.
Another specific group of compounds are compounds of formula I
S wherein R-, and R8 taken together are methylenedioxy; or a pharmaceutically
acceptable salt thereof.
Another specific group of compounds are compounds of formula I
wherein the bonds represented by ----- are both present; or a pharmaceutically
acceptable salt thereof.
10 Another specific group of compounds are compounds of formula I
wherein the bond between the 5- and the 6-positions that is represented by ----
- is
absent; or a pharmaceutically acceptable salt thereof.
Another specific group of compounds are compounds of formula I
wherein the bond between the I 1- and the 12-positions that is represented by -
---- is
absent; or a pharmaceutically acceptable salt thereof.
Another specific group of compounds are compounds of formula I
wherein the bonds represented by ----- are both absent; or a pharmaceutically
acceptable salt thereof.
A specific compound of formula I is a compound of formula II, III, N,
V, VI, VII, VIII, IX or X (Figure 6) wherein R,-R~, R~-R, have any of the
values,
specific values or preferred values described herein for a compound of formula
I.
Compounds of formulae II-X can be prepared from available starting materials
using
procedures known in the art, or using procedures analogous to those described
herein.
A compound of formula I can be prepared by subjecting an
intermediate of formula XX (wherein R,-R8 and A-G have any of the values,
specific values, or preferred values described herein for a corresponding
substituent
in a compound of formula I):
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
I1
HZN G R~
2
w
R ~ A ~ \ F~ R8
,E
R, B D
to conditions suitable for formation of the tetracyclic ring system.
Conditions suitable for formation of the tetracyclic ring system are well
known to
the art. For example, see Example I hereinbelow.
An intermediate of formula XX can be prepared from readily available
S starting materials using procedures that are known in the art, or can be
prepared
using the procedures described hereinbelow, which are illustrated in the
figures.
As illustrated in Figure 1 and as shown in Example I, reduction of 6,7-
dimethoxy-I-oxo-1,2,3,4-tetrahydronaphthlene, provides an alcohol 1, which can
be
dehydrated to provide dihydronaphthlene 2. Coupling with an 2-iodo-
nitrobenzene
provides 3 which can be oxidized to provide 4. Reduction of the vitro group
provides amine 5, which is a compound of formula XX.
As illustrated in Figures 2, 3, and 4, and as shown in Example 2,
nitration of 4-bromoveratrole under standard conditions provides vitro
compound 7,
which can be converted to stannane 8 under standard conditions. Coupling of
stannane 8 with triflate 9 provides 11, which can be oxidized to provide 12.
Alternatively, stannane 8 can be coupled with triflate 10 to provide 12.
Reduction
of the vitro group in 12 under standard conditions, provides an intermediate
of
formula XX. As illustrated in Figure 4, triflate 9 can be prepared from 6,7-
dimethoxy-2-oxo -1,2,3,4-tetrahydronaphthlene by formation of the
eneoltriflate,
under standard conditions. Triflate 10 can be prepared from 9 by oxidation
under
standard conditions.
As illustrated in Figures 4 and 5, and as shown in Example 3, an
intermediate 16 can be prepared by nitration of readily available 3,4-
dimethoxybromobenzene under standard conditions, followed by formation of the
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
12
corresponding stannane 16. Coupling of triflate 10 and stannane 16 under
standard
conditions, provides nitro compound 17 which can be reduced to provide an
intermediate of formula XX.
Other intermediates of formula XX can be prepared using procedures
S similar to those described herein by selecting appropriate starting
materials to
provide the desired intermediate of formula XX.
In cases where compounds are sufficiently basic or acidic to form
stable nontoxic acid or base salts, administration of the compounds as salts
may be
appropriate. Examples of pharmaceutically acceptable salts are organic acid
addition salts formed with acids which form a physiological acceptable anion,
for
example, tosylate, methanesulfonate, acetate, citrate, malonate, tartarate,
succinate,
benzoate, ascorbate, a-ketoglutarate, and a-glycerophosphate. Suitable
inorganic
salts may also be formed, including hydrochloride, sulfate, nitrate,
bicarbonate, and
carbonate salts.
Pharmaceutically acceptable salts may be obtained using standard
procedures well known in the art, for example by reacting a sufficiently basic
compound such as an amine with a suitable acid affording a physiologically
acceptable anion. Alkali metal (for example, sodium, potassium or lithium) or
alkaline earth metal (for example calcium) salts of carboxylic acids can also
be
made.
The compounds of formula I can be formulated as pharmaceutical
compositions and administered to a mammalian host, such as a human patient in
a
variety of forms adapted to the chosen route of administration, i.e., orally
or
parenterally, by intravenous, intramuscular, topical or subcutaneous routes.
Thus, the present compounds may be systemically administered, e.g.,
orally, in combination with a pharmaceutically acceptable vehicle such as an
inert
diluent or an assimilable edible carrier. They may be enclosed in hard or soft
shell
gelatin capsules, may be compressed into tablets, or may be incorporated
directly
with the food of the patient's diet. For oral therapeutic administration, the
active
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
13
compound may be combined with one or more excipients and used in the form of
ingestible tablets, buccal tablets, troches, capsules, elixirs, suspensions,
syrups,
wafers, and the like. Such compositions and preparations should contain at
least
0.1 % of active compound. The percentage of the compositions and preparations
may, of course, be varied and may conveniently be between about 2 to about 60%
of
the weight of a given unit dosage form. The amount of active compound in such
therapeutically useful compositions is such that an effective dosage level
will be
obtained.
The tablets, troches, pills, capsules, and the like may also contain the
following: binders such as gum tragacanth, acacia, corn starch or gelatin;
excipients
such as dicalcium phosphate; a disintegrating agent such as corn starch,
potato
starch, alginic acid and the like; a lubricant such as magnesium stearate; and
a
sweetening agent such as sucrose, fructose, lactose or aspartame or a
flavoring agent
such as peppermint, oil of wintergreen, or cherry flavoring may be added. When
the
unit dosage form is a capsule, it may contain, in addition to materials of the
above
type, a liquid carrier, such as a vegetable oil or a polyethylene glycol.
Various other
materials may be present as coatings or to otherwise modify the physical form
of the
solid unit dosage form. For instance, tablets, pills, or capsules may be
coated with
gelatin, wax, shellac or sugar and the like. A syrup or elixir may contain the
active
compound, sucrose or fructose as a sweetening agent, methyl and propylparabens
as
preservatives, a dye and flavoring such as cherry or orange flavor. Of course,
any
material used in preparing any unit dosage form should be pharmaceutically
acceptable and substantially non-toxic in the amounts employed. In addition,
the
active compound may be incorporated into sustained-release preparations and
devices.
The active compound may also be administered intravenously or
intraperitoneally by infusion or injection. Solutions of the active compound
or its
salts can be prepared in water, optionally mixed with a nontoxic surfactant.
Dispersions can also be prepared in glycerol, liquid polyethylene glycols,
triacetin,
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
14
and mixtures thereof and in oils. Under ordinary conditions of storage and
use,
these preparations contain a preservative to prevent the growth of
microorganisms.
The pharmaceutical dosage forms suitable for injection or infusion can
include sterile aqueous solutions or dispersions or sterile powders comprising
the
active ingredient which are adapted for the extemporaneous preparation of
sterile
injectable or infusible solutions or dispersions, optionally encapsulated in
liposomes. In all cases, the ultimate dosage form must be sterile, fluid and
stable
under the conditions of manufacture and storage. The liquid carrier or vehicle
can
be a solvent or liquid dispersion medium comprising, for example, water,
ethanol, a
polyol (for example, glycerol, propylene glycol, liquid polyethylene glycols,
and the
like), vegetable oils, nontoxic glyceryl esters, and suitable mixtures
thereof. The
proper fluidity can be maintained, for example, by the formation of liposomes,
by
the maintenance of the required particle size in the case of dispersions or by
the use
of surfactants. The prevention of the action of microorganisms can be brought
about
by various antibacterial and antifungal agents, for example, parabens,
chlorobutanol,
phenol, sorbic acid, thimerosal, and the like. In many cases, it will be
preferable to
include isotonic agents, for example, sugars, buffers or sodium chloride.
Prolonged
absorption of the injectable compositions can be brought about by the use in
the
compositions of agents delaying absorption, for example, aluminum monostearate
and gelatin.
Sterile injectable solutions are prepared by incorporating the active
compound in the required amount in the appropriate solvent with various of the
other ingredients enumerated above, as required, followed by filter
sterilization. In
the case of sterile powders for the preparation of sterile injectable
solutions, the
preferred methods of preparation are vacuum drying and the freeze drying
techniques, which yield a powder of the active ingredient plus any additional
desired
ingredient present in the previously sterile-filtered solutions.
For topical administration, the present compounds may be applied in
pure form, i.e., when they are liquids. However, it will generally be
desirable to
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
administer them to the skin as compositions or formulations, in combination
with a
dermatologically acceptable carrier, which may be a solid or a liquid.
Useful solid carriers include finely divided solids such as talc, clay,
microcrystalline cellulose, silica, alumina and the like. Useful liquid
carriers
S include water, alcohols or glycols or water-alcohol/glycol blends, in which
the
present compounds can be dissolved or dispersed at effective levels,
optionally with
the aid of non-toxic surfactants. Adjuvants such as fragrances and additional
antimicrobial agents can be added to optimize the properties for a given use.
The
resultant liquid compositions can be applied from absorbent pads, used to
10 impregnate bandages and other dressings, or sprayed onto the affected area
using
pump-type or aerosol sprayers.
Thickeners such as synthetic polymers, fatty acids, fatty acid salts and
esters, fatty alcohols, modified celluloses or modified mineral materials can
also be
employed with liquid carriers to form spreadable pastes, gels, ointments,
soaps, and
15 the like, for application directly to the skin of the user.
Examples of useful dermatological compositions which can be used to
deliver the compounds of formula I to the skin are known to the art; for
example, see
Jacquet et al. (U.S. Pat. No. 4,608,392), Geria (U.S. Pat. No. 4,992,478),
Smith et al.
(U.S. Pat. No. 4,559,157) and Wortzman (U.S. Pat. No. 4,820,508).
Useful dosages of the compounds of formula I can be determined by
comparing their in vitro activity, and in vivo activity in animal models.
Methods for
the extrapolation of effective dosages in mice, and other animals, to humans
are
known to the art; for example, see U.S. Pat. No. 4,938,949.
Generally, the concentration of the compounds) of formula I in a
liquid composition, such as a lotion, will be from about 0.1-25 wt-%,
preferably
from about 0.5-10 wt-%. The concentration in a semi-solid or solid composition
such as a gel or a powder will be about 0.1-5 wt-%, preferably about 0.5-2.5
wt-%.
The amount of the compound, or an active salt or derivative thereof,
required for use in treatment will vary not only with the particular salt
selected but
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
16
also with the route of administration, the nature of the condition being
treated and
the age and condition of the patient and will be ultimately at the discretion
of the
attendant physician or clinician.
In general, however, a suitable dose will be in the range of from about
0.5 to about 100 mg/kg, e.g., from about 10 to about 75 mg/kg of body weight
per
day, such as 3 to about SO mg per kilogram body weight of the recipient per
day,
preferably in the range of 6 to 90 mg/kg/day, most preferably in the range of
15 to
60 mg/kg/day.
The compound may conveniently be administered in unit dosage form;
for example, containing 5 to 1000 mg, conveniently 10 to 750 mg, most
conveniently, 50 to S00 mg of active ingredient per unit dosage form.
Ideally, the active ingredient should be administered to achieve peak
plasma concentrations of the active compound of from about 0.5 to about 75 pM,
preferably, about I to 50 pM, most preferably, about 2 to about 30 NM. This
may be
achieved, for example, by the intravenous injection of a 0.05 to 5% solution
of the
active ingredient, optionally in saline, or orally administered as a bolus
containing
about 1-100 mg of the active ingredient. Desirable blood levels may be
maintained
by continuous infusion to provide about 0.01-5.0 mg/kg/hr or by intermittent
infusions containing about 0.4-15 mg/kg of the active ingredient(s).
The desired dose may conveniently be presented in a single dose or as
divided doses administered at appropriate intervals, for example, as two,
three, four
or more sub-doses per day. The sub-dose itself may be further divided, e.g.,
into a
number of discrete loosely spaced administrations; such as multiple
inhalations from
an insufflator or by application of a plurality of drops into the eye.
The ability of a compound of the invention to effect topoisomerase I or
II mediated DNA cleavage can be determined using pharmacological models that
are
well known to the art, for example, using a model like Test A described below.
Test A.Topoisomerase I-mediated DNA cleavage assay
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
17
Human topoisomerase I was expressed in E. Coli and isolated as
a recombinant fusion protein using a T7 expression system as described
previously (Makhey, D. et al., Bioorg. Med. Chem., 2000, 8, 1-11). DNA
S topoisomerase I was purified from calf thymus gland as reported previously
(Maniatis, T., et al., J. Molecular Cloning, a Laboratory Manual, Cold Spring
Harbor Laboratory, Cold Spring Harbor, New York, 149-185). Plasmid YepG
was also purified by the alkali lysis method followed by phenol deproteination
and CsCI/ethidium isopycnic centrifugation method as described (Maniatis,
T.; Fritsch, E. F.; Sambrook, J. Molecular Cloning, a Laboratory Manual;
Cold Spring Harbor Laboratory: Cold Spring Harbor, NY 1982; pp 149-
185).'°2 The end-labeling of the plasmid was accomplished by digestion
with
a restriction enzyme followed by end-filling with Klenow polymerase as
previously described (Liu, L. F.; Rowe, T. C.; Yang, L.; Tewey, K. M.; Chen,
G. L. "Cleavage of DNA by mammalian topoisomerase II," J. Biol. Chem.
1983, 258, 15365).'°3 Cleavage assays were performed as previously
reported
(B. Gatto et al. Cancer Res., 1996, 56, 2795-2800)." The drug and the DNA
in presence of topoisomerase I was incubated for 30 minutes at 37 °C.
After
development of the gels, typically 24-hour exposure was used to obtain
autoradiograms outlining the extent of DNA fragmentation. Topoisomerase I-
mediated DNA cleavage values are reported as REC, Relative Effective
Concentration, i.e. concentrations relative to 2,3-dimethoxy-8,9-
methylenedioxybenzo[i]phenanthridine, whose value is arbitrarily assumed as
1.0, that are able to produce the same cleavage on the plasmid DNA in the
presence of human topoisomerase I. Relative potency was based upon the
relative amount of drug needed to induce approximately 10% DNA
fragmentation. Assays were performed under the direction of Dr. L. F. Liu,
Department of Pharmacology, The University of Medicine and Dentistry of
New Jersey, Robert Wood Johnson Medical School, Piscataway, New Jersey.
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
18
Data from Test A for representative compounds of the invention is shown in
Table 1.
Table 1
S Compound Topoisomerase I-mediated DNA cleavage
6 5
14 0.01
19 >100
60 2
61 10
The cytotoxic effects of a compound of the invention can be
determined using pharmacological models that are well known to the art, for
example, using a model like Test B described below.
Test B. Inhibition of Cell Growth: MTT-microtiter plate tetrazolinium
cytotoxicity assay (RPMI 8402, CPT-K5, U937, U937/CR Cells)
The cytotoxicity was determined using the MTT-microtiter plate
tetrazolinium cytotoxicity assay (MTA) (See Chen A.Y. et al. Cancer Res. 1993,
53,
1332; Mosmann, T. J., J. Immuuol. Methods 1983, 65, 55; and Carmichael, J. et
al.
Cancer Res. 1987, 47, 936). The human lymphoblast RPMI 8402 and its
camptothecin-resistant variant cell line, CPT-KS were provided by Dr. Toshiwo
Andoh (Anchi Cancer Research Institute, Nagoya, Japan) (see Andoh, T.; Okada,
K.
"Drug resistance mechanisms of topoisomerase I drugs," Adv. in Pharmacology
1994, 29B, 93). Human U-937 myeloid leukemia cells and U-937/CR cells were
described by Rubin et al., J. Biol. Chem., 269, 2433-2439 (1994). The
cytotoxicity
assay was performed by using 96-well microtiter plates using 2000 cells/well,
in
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
19
200 mL of growth medium. Cells were grown in suspension at 37 °C in 5%
COZ
and maintained by regular passage in RPMI medium supplemented with 10% heat-
inactivated fetal bovine serum, L-glutamine (2 mM), penicillin ( 100U/mL), and
streptomycin (0.1 mg/mL). For determination of ICSO, cells were exposed
continuously for 3-4 days to varying concentrations of drug, and MTT assays
were
performed at the end of the fourth day. Each assay was performed with a
control
that did not contain any drug. All assays were performed at least twice in 6
replicate wells. All assays were performed under the direction of Dr. L. F.
Liu,
Department of Pharmacology, The University of Medicine and Dentistry of New
Jersey, Robert Wood Johnson Medical School, Piscataway, New Jersey.
Representative data is shown in Tables 2 and 3.
Table 2
[ ] pM KB3- KBV-I KBH1.0+ HELA HCT116 ZR-75-1
I V
(oral)(mdr-1) (H033342 (cervical(colon)(breast)
r.) )
CPT* 0.006 0.006 0.007 0.004 0.003 0.004
VBS+ 0.003 0.4 0.0022 0.002 0.003 0.003
H033342* 0.25 3 >10 0.6 0.22 0.06
BZ-III-26*0.3 0.2 0.15 0.15 0.25 0.2
DL-II-91 0.2 0.18 0.12 0. I 0.2 0.2
* 2
6 6 4 5 6 3.2 4
14 0.07 0.08 0.06 0.05 0.035 0.04
Table 3
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
[ ] pM RPMI 8402 CPT-KS U937 U937/CR
Bz-III-26* 0.5 0.3 0.09 0.06
DL-II-91 0.3 0.13 0.03 0.025
*
6 3 10 3 3
5 14 0.06 6 0.06 3
19 10 >10 10 20
60 2 4 1.9 2.5
61 4 31 3 6
34 >100 >100 >100 >100
10 42 38 61 >100 40
43 >100 52 38 30
44 0.5 I .3 43 0.9
*See Figure I I +VBS = Vinblastine
The data in Tables 2 and 3 demonstrates that representative
15 compounds of the invention function as cytotoxic agents against tumor cell
lines,
including multidrug resistant tumor cell lines. Thus, the compounds are useful
to
treat cancer and can be used to treat tumors that are resistant to other
specific
chemotherapeutic agents.
Topoisomerase inhibitors are also known to possess antibacterial,
20 antifungal, antiprotozoal, antihelmetic, and antiviral activity.
Accordingly, the
topoisomerase inhibitors of the invention may also be useful as antibacterial,
antifungal, antiprotozoal, antihelmetic, or antiviral agents. In particular,
compounds
of the invention that demonstrate little or no activity as mammalian
topoisomerase I
poisons, because of the possibility of similar molecular mechanism of action,
could
be highly active and selective antibacterial, antifungal, antiprotozoal,
antihelmetic,
or antiviral agents. Thus, certain compounds of the invention may be
particularly
useful as systemic antibacterial, antifungal, antiprotozoal, antihelmetic, or
antiviral
agents in mammals. The invention also provides the use of a compound of the
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
21
invention for the manufacture of a medicament useful for producing an .
antibacterial, antifungal, antiprotozoal, antihelmetic, or antiviral effect in
a
mammal.
As used herein, the term "solid mammalian tumors" include cancers of the
head and neck, lung, mesothelioma, mediastinum, esophagus, stomach, pancreas,
hepatobiliary system, small intestine, colon, rectum, anus, kidney, ureter,
bladder,
prostate, urethra, penis, testis, gynecological organs, ovarian, breast,
endocrine
system, skin central nervous system; sarcomas of the soft tissue and bone; and
melanoma of cutaneous and intraocular origin. The term "hematological
malignancies" includes childhood leukemia and lymphomas, Hodgkin's disease,
lymphomas of lymphocytic and cutaneous origin, acute and chronic leukemia,
plasma cell neoplasm and cancers associated with AmS. The preferred mammalian
species for treatment are humans and domesticated animals.
The invention will now be illustrated by the following non-limiting
Examples, wherein unless otherwise stated: melting points were determined with
a
Thomas-Hoover Unimelt capillary melting point apparatus; column chromatography
refers to flash chromatography conducted on SiliTech 32-63 m, (ICN
Biomedicals,
Eschwegge, Ger.) using the solvent systems indicated; radial chromatography
refers
to the use of a Model 8924 chromatotron (Harrison Research, CA); infrared
spectral
data (IR) were obtained on a Perkin-Elmer 1600 Fourier transform
spectrophotometer and are reported in cm '; proton ('H NMR) and carbon ('3C
NMR) nuclear magnetic resonance were recorded on a Varian Gemini-200 Fourier
Transform spectrometer; NMR spectra (200 MHz'H and 50 MHz'~C) were
recorded in the deuterated solvent indicated with chemical shifts reported in
units
downfield from tetramethylsilane (TMS); coupling constants are reported in
hertz
(Hz), a few drops of CF3COOH improved "C NMR spectra by allowing for
increased solubility and formation of the protonated form of the
terbenzimidazoles,
thereby eliminating tautomeric differences among carbon atoms; mass spectra
were
obtained from Washington University Resource for Biomedical and Bio-organic
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
22
Mass Spectrometry within the Department of Chemistry at Washington University,
St. Louis, MO; combustion analyses were performed by Atlantic Microlabs, Inc.,
Norcross, GA, and were within 0.4% of the theoretical value; compounds 7 and
15
were prepared by nitration of 4-bromoveratrole and 4-bromo-1,2-
(methylenedioxy)benzene as previously described (Pettit, G.R.; Piatak, D.M. J.
Org. Chem., 25, 1960, 721; Dallacker, F.; Wagner, A. Z. Naturforsch., 1984,
39b,
936).
Example 1 2,3-Dimethoxy-dibenzo[c,h]cinnoline (6).
6-(2-Aminophenyl)-2,3-dimethoxynaphthalene (5, 70 mg, 0.25 mmol) was
dissolved in 48% hydrobromic acid (4.25 mL), cooled in ice-salt bath, and
treated
dropwise with stirring with sodium nitrite (0.13 g) in water (2.2 mL).
Stirring was
continued for 0.5 h., and to the cold solution was then added with stirring
freshly
precipitated copper (0.5 g). The mixture was allowed to rise slowly to room
temperature and left overnight. The solid was filtered off and washed with hot
chloroform. The chloroform solution was washed with diluted sodium hydroxide
solution, then with water, dried (anhydrous NazS04) and rotaevaporated to give
the
crude product. Chromatography on silica gel using 50:50 hexanes:ethyl acetate
afforded the title compound ( 13 mg) in I 8% yield; 'H NMR (CDCI~) d 4.11 (3H,
s),
4.24(3H, s), 7.37( 1 H, s), 7.897.94 (2H, m), 8.14( 1 H, d, J=8.9), 8.41 ( 1
H, d, J=8.8),
8.61--8.66( I H, m), 8.75-8.80( I H, m), 9.24( I H, s); '~C NMR d 56.1, 56.4,
104.0,
107.3, 112.3, 116.5, 118.5, 121.7, 126.7, 128.6, 128.8, 131.1, 131.2, 131.9,
141.5,
146.3, 150.9, 151Ø
The intermediate 6-(2-aminophenyl)-2,3-dimethoxynaphthalene (5) was
prepared as follows.
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
23
a. 6-(2-Nitrophenyl)-2,3-dimethoxy-7,8-dihydronaphthalene (3).
Pd(PPh3)ZCIZ (840 mg, 1.2 mmol) and sodium acetate (200 mg, 2.4 mmol) were
added to a solution of 6,7-dimethoxy-3,4-dihydronaphthalene (2, 700 mg, 3.7
mmol) and 1-iodo-2-nitrobenzene (925 mg, 3.7 mmol) in dimethylacetamide (50
mL). The mixture was stirred under nitrogen at 140 °C overnight, and
then
concentrated in vacuo. Ethyl acetate (60 mL) was added to the residue and
washed
with distilled water (SO mL). The organic layer was separated and passed
through a
celite bed. The organic layer was then washed with brine, dried (anhydrous
Na2S04)
and concentrated irz vacuo. The residue was chromatographed to give compound 3
(330 mg) in 29% yield; 'H NMR (CDC13) d 2.51 (2H, t, J=8.1 ), 2.92(2H, t,
J=8.1 ),
3.87(3H, s), 3.90(3H, s), 6.45( 1 H, s), 6.67( 1 H, s), 6.73( 1 H, s),
7.38~7.45(2H, m),
7.54--7.62( 1 H, m), 7.88--7.93( 1 H, m); '~C NMR d 28.5, 28.5, 56.6, 111.0,
111.7,
124.9, 127.0, 127.1, 128.1, 128.3, 131.3, 133.3, 135.7, 138.7, 147.9, 148.9.
b. 6-(2-Nitrophenyl)-2,3-dimethoxynaphthalene (4). 6-(2-Nitrophenyl)-
2,3-dimethoxy-7,8-dihydronaphthalene ( 100 mg, 0.32 mmol) was refluxed
overnight in toluene (20 mL) with DDQ (109 mg, 0.48 mmol). Cooled down to
room temperature and filtered through celite bed. The filtrate was
rotaevaporated to
dryness to give the crude product. Chromatography on silica gel using 80:20
hexanes:ethyl acetate afforded compound 4 (90 mg) in 91 % yield; 'H NMR
(CDCI~)
d 4.00(3H, s), 4.01(3H, s), 7.12(1H, s), 7.14(1H, s), 7.27(1H, dd, J=8.4,
J=1.7),
7.46--7.62(3H, m), 7.66( 1 H, s), 7.72( 1 H, d, J=8.4), 7.86( 1 H, d, J=8.1 );
'3C NMR d
56.4, 106.6, 107.1, 124.5, 124.6, 125.9, 127.3, 128.4, 129.3, 129.6, 132.7,
132.7,
133.6, 137.0, 149.9, 150.5, 150.6.
c. 6-(2-Aminophenyl)-2,3-dimethoxynaphthalene (S). 6-(2-Nitrophenyl)-
2,3-dimethoxynaphthalene (70 mg, 0.23 mmol) was hydrogenated overnight in
ethyl
acetate (45 mL) at 40-45 Ib./sq. in. under catalysis of palladium (10 wt% on
activated carbon, 20 mg). The solution was passeded through a celite bed and
the
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
24
catalyst was washed with ethyl acetate (3x 10 mL). Concentration in vacuo gave
compound 5 (60 mg) in 99% yield;'H NMR (CDC13) d 4.02(3H, s), 4.04(3H, s),
6.82( 1 H, d, J=8.0), 6.85--6.93 ( 1 H, m), 7.16( 1 H, s), 7.18( 1 H, s),
7.20~7.26(2H, m),
7.47( 1 H, dd, J=8.3, J=1.6), 7.78( 1 H, d, J=8.8), 7.80( 1 H, s); '3C NMR d
56.4, 106.6,
106.9, 116.1, 119.2, 126.1, 126.8, 127.3, 128.3, 128.7, 128.9, 129.9, 131.2,
135.8,
144.3, 150.2, 150.3.
Compound 2 was prepared as illustrated in Figure 1, from readily available
starting materials, using standard procedures.
Example 2 2,3-Dimethoxy-8,9-methylenedioxy-dibenzo[c,h]cinnoline (14).
6-(2-Amino-4,5-methylenedioxyphenyl)-2,3-dimethoxy-naphthalene (13,
40 mg, 0.13 mmol) in acetic acid (2 mL) and concentrated hydrochloric acid
(0.3
mL) was cooled to 0 °C and diazotised with a solution of sodium nitrite
(0.09 g in
1.5 mL water). The diazonium solution was allowed to rise slowly to room
temperature and left overnight. Water (50 mL) was added to the red solution
with
some precipitate. The resulting mixture was extracted with ethyl acetate,
washed
with diluted sodium hydroxide solution, then with water, dried (anhydrous
NazS04)
and rotaevaporated to give the crude product. Chromatography on silica gel
using
40:60 hexanes:ethyl acetate afforded the title compound (20 mg) in 50% yield;
'H
NMR (CDCl3) d 4.09(3H, s), 4.22(3H, s), 6.24(2H, s), 7.31(1H, s), 7.80(1H, s),
7.95( 1 H, s), 8.00( 1 H, d, J=9.2), 8.13( I H, d, J=8.9), 9.14( I H, s); '3C
NMR d 56.5,
56.9, 98.1, 102.9, 104.6, 107.5, 107.9, 117.1, 119.6, 120.6, 126.8, 128.5,
131.7,
141.9, 145.6, 150.2, 151.2, 152.1.
The intermediate 6-(2-Amino-4,5-methylenedioxyphenyl)-2,3-
dimethoxynaphthalene (13) was prepared as follows.
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
a. 6,7-Dimethoxy-3,4-dihydro-2-naphthalenetritlate (9). A solution of
6,7-dimethoxy-2-tetralone (250 mg, 1.2 mmol) in THF (5 mL) was added to a
suspension of sodium hydride (60 wt%, 75 mg, 1.9 mmol) in THF (10 mL) cooled
by ice bath and stirred 0.5 h. A solution of N-phenyltrifluoromethane-
sulfonimide
5 (500 mg, 1.4 mmol) in THF (5 mL) was then added, and the reaction was
stirred at
0 °C for 9 hours. After concentration in vacuo, the residue was
chromatographed
using 80.20 hexanes:ethyl acetate to give compound 9 (300 mg) in 73% yield;'H
NMR (CDC13) d 2.66(2H, t, J=8.5), 3.00(2H, t, J=8.4), 3.86(3H, s), 3.88(3H,
s),
6.40( 1 H, s), 6.62( 1 H, s), 6.68( 1 H, s); '3C NMR d 27.1, 28.9, 56.5, I I
1.3, 111.7,
10 115.9, 118.7, 122.3, 124.0, 126.0, 148.2, 148.9, 149.3.
b. 6,7-Dimethoxy-2-naphthalenetriflate (10). 6,7-Dimethoxy-3,4-dihydro-
2-naphthalenetriflate (200 mg, 0.59 mmol) was refluxed overnight in toluene
(30
mL) with DDQ ( 166 mg, 0.73 mmol), cooled to room temperature, and filtered
15 through celite bed. The filtrate was concentrated in vacuo to give the
crude product.
Chromatography on silica gel using a 80:20 hexanes:ethyl acetate afforded
compound 10 (190 mg) in 95% yield;'H NMR (CDC13) d 4.00(6H, s), 7.10(1H, s),
7.12( 1 H, s), 7.21 ( 1 H, dd, J=8.9, J=2.5), 7.58 ( 1 H, d, J=2.5), 7.71 ( 1
H, d, J=8.9); "C
NMR d 56.4, 106.6, 106.6, 109.7, 116.1, I 17.9, 118.2, 122.5, 128.9, 129.0,
129.8,
20 146.6, 150.9, 151.3.
c. 6-(4,5-Methylenedioxy-2-nitrophenyl)-2,3-dimethoxy-
naphthalene (12). Tetrakis(triphenylphosphine)palladium(0) (40 mg) and cuprous
bromide (8 mg) were added to a solution of 6,7-dimethoxy-2-naphthalenetriflate
25 ( 160 mg, 0.48 mmol) and trimethyl(3,4-methylenedioxy-6-
nitrophenyl)stannane (8,
187 mg, 0.57 mmol) in THF (20 mL). The mixture was stirred at room temperature
for 0.5 h., and then refluxed under nitrogen for 18 h. After Cooling, THF was
rotaevaporated and ethyl acetate (50 ml) was added to the residue. the
solution was
washed with water (30 mL). The organic layer was separated and passed through
a
WO 01/32631 CA 02389312 2002-04-29 pCT~S00/29583
26
celite bed to remove suspended particles. The organic layer was then washed
with
brine, dried (anhydrous Na2S04) and concentrated in vacuo. The residue was
chromatographed on silica gel using 70:30 hexanes:ethyl acetate to give a
mixture
of two compounds with same Rf value. The mixture can be separated after the
hydrogenation step.
d. 6-(2-Amino-4,5-methylenedioxyphenyl)-2,3-dimethoxy-
naphthalene (13). 6-(4,S-Methylenedioxy-2-nitrophenyl)-2,3-
dimethoxynaphthalene (2S mg, 0.071 mmol) was hydrogenated overnight in ethyl
acetate (40 mL) at 4045 lb./sq. in. under catalysis of palladium (10 wt% on
activated carbon, 20 mg). The solution was passed through celite bed and the
catalyst was washed with ethyl acetate (3x 10 ml). Concentration in vacuo gave
the
crude product. Chromatography using 60:40 hexanes:ethyl acetate gave compound
13 ( I S mg) in 66% yield; 'H NMR (CDCI~) d 4.01 (3H, s), 4.02(3H, s), 5.91
(2H, s),
1 S 6.40( 1 H, s), 6.73( 1 H, s), 7.13( 1 H, s), 7.1 S( 1 H, s), 7.39( 1 H,
dd, J=8.2, J=1.8),
7.72( 1 H, s), 7.74( 1 H, d, J=8.S); "C NMR d 56.4, 98.3, 101.2, 106.6, 106.8,
110.7,
120.5, 126.3, 127.0,127.3, 128.5, 129.9, 135.7, 138.9, 141.1, 148.0, 1 S0.1,
150.3.
The intermediate trimethyl(3,4- methylenedioxy-6-nitrophenyl)-
stannane (8) in sub-part c above was prepared as follows.
e. Trimethyl (3,4- methylenedioxy-6-nitrophenyl)stannane (8). A mixture
of hexmethylditin (3 g, 9.2 mmol), 4-bromoveratrole 7 (Pettit, G.R.; Piatak,
D.M.
J. Org. Chem., 25, 1960, 1.6 g, 6.1 mmol) and Pd(PPh~)4 (200 mg) in anhydrous
2S THF (30 ml) was heated to reflux under nitrogen for 10 h. After cooling to
room
temperature, THF was evaporated and methylene chloride (30 mL) was added to
the
residue. To this mixture, aqueous potassium fluoride (7.0M, 2 mL) was added
dropwise with vigorous stirring. The mixture was passed through a celite bed
and
the filtrate was washed with brine. The methylene chloride layer was dried
(anhyd.
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
27
NazS04), filtered and evaporated in vacuo. The residue was chromatographed
over
100 g of silica gel using 1:6 ethyl acetate:hexanes to give 8 in 65% yield; 'H
NMR
(CDC13) d 0.32 (9H, s), 6.12 (2H, s), 7.04 ( 1 H, s), 7.82 ( 1 H, s); '3C NMR
(CDC13) d
-6.8, 103.3, 105.8, 114.5, 137.2, 147.9, 149.4, 153.4; HRMS calcd for
C,oH,3N04Sn-CH3: 315.9632; found: 315.9638.
Example 3 2,3,8,9-Tetramethoxy-dibenzo[c,h]cinnoline (19).
6-(2-Amino-4,5-dimethoxyphenyl)-2,3-dimethoxynaphthalene (18)
( 11 mg, 0.033 mmol) in acetic acid (0.6 mL) and concentrated hydrochloric
acid
(0.06 mL) was cooled to 0 °C and diazotised with a solution of sodium
nitrite (0.026
g in 0.5 mL water). The diazonium solution was allowed to rise slowly to room
temperature and left overnight. Water (30 mL) was added to the red solution
with
some precipitate. The resulting mixture was extracted with ethyl acetate,
washed
with diluted sodium hydroxide solution, then with water, dried (anhydrous
NazS04)
and rotaevaporated to give the crude product. Chromatography on silica gel
using
40:60 chloroform:ethyl acetate afforded the title compound (5 mg) in 44%
yield; 'H
NMR (CDC1~) d 4.09(3H, s), 4.18(6H, s), 4.23(3H, s), 7.31 ( 1 H, s), 7.74( 1
H, s),
8.00( 1 H, s), 8.01 ( I H, d, J=8.5), 8.20( I H, d, J=8.9), 9. I 5( 1 H, s); '
~C NMR d 56.5,
56.9, 99.9, 104.5, 107.9, 109.5, 116.9, 118.5, 118.9, 127.0, 128.4, 131.6,
141.8,
144.6, I 51.1, 151.2, 152.0, 153.9.
The intermediate 6-(2-amino-4,5-dimethoxyphenyl)-2,3-
dimethoxynaphthalene (18) was prepared as follows.
a. Trimethyl(3,4-dimethoxy-6-nitrophenyl)stannane (16). A mixture of
hexmethylditin (3 g, 9.2 mmol), 4-bromo-1,2-(methylenedioxy)benzene 15
(Dallacker, F.; Wagner, A. Z. Naturforsch., 1984, 39b, 936, 1.6 g, 6.1 mmol)
and
Pd(PPh~)4 (200 mg) in anhydrous THF (30 ml) was heated to reflux under
nitrogen
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
28
for 10 h. After cooling to room temperature, THF was evaporated and methylene
chloride (30 mL) was added to the residue. To this mixture, aqueous potassium
fluoride (7.0M, 2 mL) was added dropwise with vigorous stirring. The mixture
was
passed through a celite bed and the filtrate was washed with brine. The
methylene
chloride layer was dried (anhyd. Na2S04), filtered and evaporated in vacuo.
The
residue was chromatographed over 100 g of silica gel using 1:6 ethyl
acetate:hexanes to give 16 in 70% yield; mp 115-117 °C;'H NMR (CDCl3) d
0.32
(9H, s), 3.94 (3H, s), 3.99 (3H, s), 7.03 ( 1 H, s), 7.88 ( 1 H, s); '~C NMR
(CDCl3) d -
7.2, 56.7, 107.7, 117.3, 134.0, 146.8, 149.8, 154.1; HRMS calcd for
C"H,~N04Sn-CH3: 329.9937; found: 329.9939.
b. 6-(4,5-Dimethoxy-2-nitrophenyl)-2,3-dimethoxynaphthalene (17).
Tetrakis(triphenylphosphine)palladium(0) (80 mg) and cuprous bromide (16 mg)
were added to a solution of 6,7-dimethoxy-2-naphthalenetriflate (10, 220 mg,
0.655
mmol) and trimethyl(3,4-dimethoxy-6-nitrophenyl)stannane (16, 220 mg, 0.64
mmol) in THF (25 mL). The mixture was stirred at room temperature for 0.5 h.,
and
then refluxed under nitrogen for 32 hr. After Cooling, THF was rotaevaporated
and
ethyl acetate (50 ml) was added to the residue. the solution was washed with
water
(30 mL). The organic layer was separated and passed through a celite bed to
remove
suspended particles. The organic layer was then washed with brine, dried
(anhydrous Na2S04) and concentrated in vacuo. The residue was chromatographed
on silica gel using 60:40 hexanes:ethyl acetate to give compound 17; 'H NMR
(CDCI~) d 3.95(3H, s), 3.99(6H, s), 4.01 (3H, s), 6.86( 1 H, s), 7.12( 1 H,
s), 7.14( 1 H,
s), 7.23(1H, dd, J=8.4, J=1.8), 7.56(1H, s), 7.61(1H, d, J=1.7), 7.70(1H, d,
J=8.4);
'3C NMR d 56.4, 56.9, 106.7, 107.0, 108.3, 114.4, 124.9, 125.7, 127.0, 129.0,
129.5, 132.2, 134.6, 141.6, 148.4, 150.4, 152.7.
c. 6-(2-Amino-4,5-dimethoxyphenyl)-2,3-dimethoxynaphthalene (18). 6-
(4,5-Dimethoxy-2-nitrophenyl)-2,3-dimethoxynaphthalene ( 12 mg, 0.03 mmol) was
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
29
hydrogenated overnight in ethyl acetate (20 mL) at 40-45 Ib./sq. in. under
catalysis
of palladium ( 10 wt% on activated carbon, 10 mg). The solution was passed
through
celite bed and the catalyst was washed with ethyl acetate (3x 10 mL).
Concentration
in vacuo gave the crude product. Chromatography using 65:35 hexanes:ethyl
acetate
gave compound 18 ( 1 I mg) in nearly 100% yield; 'H NMR (CDCI3) d 3.84(3H, s),
3.89(3H, s), 4.01 (3H, s), 4.02(3H, s), 6.41 ( 1 H, s), 6.80( 1 H, s), 7. I 4(
1 H, s), 7.15( 1 H,
s), 7.43(1H, dd, J=8.4, J=1.6), 7.75(1H, d, J=1.5), 7.75(1H, d, J=8.4);'3C NMR
d
56.4, 57.2, 101.3, 106.6, 106.8, 115.2, 119.9, 126.2, 126.8, 127.3, 128.5,
129.9,
135.7, 138.0, 142.7, 149.8, 150.1, 150.3.
Example 4 2,3,8-Trimethoxydibenzo[c,h]cinnoline (60)
6-(2-Amino-4-methoxyphenyl)-2,3-dimethoxynaphthalene (32) (12
mg, 0.039 mmol) was dissolved in acetic acid (0.6 mL) and concentrated
hydrochloric acid (0.06 mL). The solution was cooled in an ice bath and
diazotized
by the dropwise addition of a solution of sodium nitrite (0.026 g in 0.5 mL
water).
The resulting diazonium solution was allowed to rise to room temperature
slowly
and left overnight. To the resulting red solution which contained some
precipitate
was added 30 mL water and the mixture was extracted with ethyl acetate (30 mL
x
3). The organic layers were combined and washed with diluted sodium hydroxide
solution first, then with water and brine. The ethyl acetate extracts were
dried with
anhydrous sodium sulfate and evaporated in vacuo. The crude product was
purified
by column chromatography on silica gel using 40:60 hexanes:ethyl acetate to
give
the pure 4 (5 mg) in 40% yield; mp 244-246 °C; 1R (KBr) 2919, 1619,
1507, 1388,
1292, 1277, 1204 cm'; UV (MeOH) 292, 266, 216 nm; 'H NMR (CDCl3) 8 4.10
(6H, s), 4.22 (3H, s), 7.32 (1H, s), 7.54 (1H, dd, J,=9.1, JZ=2.6), 8.04-8.08
(2H, m),
8.28 ( 1 H, d, J=8.9), 8.49 ( I H, d, J=9.1 ), 9.16 ( 1 H, s); '~C NMR b
56.33, 56.52,
56.92, 104.36, 107.93, 108.92, 116.88, 117.08, 119.48, 123.51, 124.54, 127.14,
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
128.42, 132.46, 141.77, 148.52, 151.12, 151.40, 160.45; HRMS (EI) calcd for
C19H16N2O3 ~z: 320.1161; found: 320.0384.
The intermediate compound 32 was prepared as follows.
5
a. 6-(4-Methoxy-2-nitrophenyl)-2,3-dimethoxynaphthalene (29).
Tetrakis(triphenylphosphine)palladium (0) (60 mg) and cuprous bromide (10 mg)
were added to a solution of 6,7-dimethoxy-2-trifluoromethanesulfonyloxy-
naphthalene 10 (150 mg, 0.45 mmol) and trimethylnitroarylstannane 26 (140 mg,
10 0.45 mmol) in THF (20 mL) at room temperature and stirred for 0.5 h. The
mixture
was then refluxed under NZ for 36 h. After cooling, THF was evaporated and
ethyl
acetate (30 mL) was added to the residue. The solution was washed with water.
The organic layer was separated and passed through a Celite bed to remove
suspended particles. The organic layer was then washed with brine, dried
15 (anhydrous Na2S0,~), and evaporated in vacuo. The residue was
chromatographed
using a 70:30 mixture of hexanes:ethyl acetate to give 29 (60 mg) in 43%
yield; 'H
NMR (CDCI~) 8 3.92 (3H, s), 4.01 (3H, s), 4.02 (3H, s), 7.12-7.26 (4H, m),
7.39-
7.46 (2H, m), 7.61 ( I H, d, J=1.7), 7.71 ( 1 H, d, J=8.4); '~C NMR (CDCI~) b
56.42,
106.64, 106.98, 109.50, 119.23, 124.83, 125.89, 127.20, 129.02, 129.38,
129.60,
20 133.53, 150.25, 150.42, 159.46; HRMS (EI) calcd for C,9H,~N05 m/z:
339.1107;
found: 339.1108.
b. 6-(2-Amino-4-methoxyphenyl)-2,3-dimethoxynaphthalene (32). 6-(4-
Methoxy-2-nitrophenyl)-2,3-dimethoxynaphthalene 29 ( 18 mg, 0.053 mmol) was
25 hydrogenated overnight in ethyl acetate (20 mL) at 4045 lb./sq. in. using
10%
palladium on carbon ( 10 mg) as catalyst. The reaction solution was passed
through
a Celite bed and the catalyst was washed with ethyl acetate (10 mL x 3).
Concentration in vacuo gave the crude product. The residue was chromatographed
using a 60:40 mixture of hexanes:ethyl acetate to give 32 (14 mg) in 85%
yield;'H
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
31
NMR (CDC13) b 3.83 (3H, s), 4.01 (3H, s), 4.03 (3H, s), 6.37 (1H, d, J=2.5),
6.45
(1H, dd, J,=8.3, JZ=2.4), 7.12-7.16 (3H, m), 7.42 (1H, dd, J,=8.4, JZ=1.7),
7.72-7.77
(2H, m); '3C NMR 8 55.72, 56.40, 101.56, 104.79, 106.59, 106.78, 121.49,
126.36,
126.81, 127.24, 128.47, 129.89, 131.98, 135.55, 145.28, 150.01, 150.25,
160.49;
HRMS (E1) calcd for C,9H,9N03 m/z: 309.1365; found: 309.1355.
The intermediate compound 26 used in sub-part a above was prepared
as follows.
c. Trimethyl(4-methoxy-2-nitrophenyl)stannane (2G). A mixture of
hexamethylditin (1.0 g, 3.1 mmol), 4-methoxy-2-nitrobromobenzene 23 (0.5 g,
2.16
mmol) and Pd(PPh3)4 (60 mg) in anhydrous THF (20 mL) was heated to reflux
under nitrogen until thin layer chromatography no longer showed the presence
of
starting material. After cooling to room temperature, THF was evaporated and
methylene chloride was added to the residue. To this mixture, aqueous
potassium
fluoride (7.0 M, 1.0 mL) was added dropwise with vigorous stirring. The
mixture
was passed through a Celite bed and the filtrate washed with brine. The
methylene.
chloride layer was dried (anhydrous NazSO,r), filtered and the solution
concentrated
in vacuo. The residue was chromatographed using a 95:5 mixture of
hexanes:ethyl
acetate to give 26 (260 mg) in 38% yield; mp 93-5°C;'H NMR (CDCI~) 8
0.32 (9H,
s), 3.89 (3H, s), 7.21 ( 1 H, dd, JI=8.0, JZ=2.6), 7.57 ( 1 H, d, J=8.0), 7.86
( 1 H, d,
J=2.6);'~C NMR (CDC13) b -7.1, 56.7, 107.7, 117.3, 133.9, 146.8, 149.6, 154.1;
HRMS (EI) calcd for C,oH,sNO~Sn-CH3 m/z: 301.9839; found: 301.9832.
The starting 4-Bromo-3-nitroanisole (23) was purchased from Aldrich
Chemical Company (Milwaukee, WI) [5344-78-5].
Example S 2,3,9-Trimethoxydibenzo[c,h~cinnoline (61)
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
32
6-(2-Amino-5-methoxyphenyl)-2,3-dimethoxynaphthalene (33) (60
mg, 0.20 mmol) was dissolved in acetic acid (1.5 mL) and concentrated
hydrochloric acid (0.3 mL). The solution was cooled in an ice bath and
diazotized
by dropwise addition of a solution of sodium nitrite (0.12 g in 1.2 mL water).
The
resulting diazonium solution was allowed to rise to room temperature slowly
and
left overnight. To the resulting red solution with some precipitate, 50 mL
water was
added and then extracted with ethyl acetate (40 mL x 3). The organic layers
were
combined and washed with diluted sodium hydroxide solution first, then with
water
and brine. Dried with anhydrous sodium sulfate and evaporated in vacuo. The
crude product was purified by column chromatography on silica gel using 35:65
hexanes:ethyl acetate to give the pure S (16 mg) in 26% yield; mp 215-217
°C; IR
(KBr) 2987, 1617, 1504, 1486, 1394, 1277, 1231, 1167 cm-';UV (MeOH) 288, 262,
232 nm (log E = 4.71, 4.66, 4.58); 'H NMR (CDC13) ~ 4.07 (3H, s), 4.09 (3H,
s),
4.22 ( 1 H, s), 7.29 ( 1 H, s), 7.46 ( 1 H, dd, J'=9.1, JZ=2.6), 7.72 ( 1 H,
d, J=2.5), 7.99
( 1 H, d, J=8.9), 8.20 ( 1 H, d, J=9.0), 8.60 ( 1 H, d, J=9.1 ), 9.17 ( 1 H,
s); '~C NMR b
56.31, 56.51, 56.90, 100.27, 104.61, 107.73, 117.00, 118.66, I 21.31, 124.25,
126.98, 129.09, 131.40, 133.34, 141.76, 143.70, 151.23, 151.31, 161.95; HRMS
(El) calcd for C,9H,6N20~ m/z: 320.1161; found: 320.1144.
The intermediate compound 33 was prepared as follows.
a. 6-(S-Methoxy-2-nitrophenyl)-2,3-dirnethoxynaphthalene (30).
Tetrakis(triphenylphosphine)palladium (0) (80 mg) and cuprous bromide (6 mg)
were added to a solution of 6,7-dimethoxy-2-trifluoromethanesulfonyloxy-
naphthalene 10 (200 mg, 0.60 mmol) and trimethylnitroarylstannane 27 (200 mg,
0.64 mmol) in THF (25 mL) at room temperature and stirred for 0.5 h. The
mixture
was then refluxed under NZ overnight. After cooling, THF was evaporated and
ethyl acetate (30 mL) was added to the residue. The solution was washed with
water. The organic layer was separated and passed through a Celite bed to
remove
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
33
suspended particles. The organic layer was then washed with brine, dried
(anhydrous NaZS04), and evaporated in vacuo. The residue was chromatographed
using a 75:25 mixture of hexanes:ethyl acetate to give a mixture of two
compounds
with similar Rfvalues. This mixture was used for next step without further
purification.
b. 6-(2-Amino-5-methoxyphenyl)-2,3-dimethoxynaphthalene (33). Crude 6-
(5-methoxy-2-nitrophenyl)-2,3-dimethoxynaphthalene 30 (100 mg, approximately
90% pure) was hydrogenated overnight in ethyl acetate (40 mL) at 4045 lb./sq.
in.
using 10% palladium on carbon (30 mg) as catalyst. The reaction solution was
passed through a Celite bed and the catalyst was washed with ethyl acetate (10
mL x
3). Concentration in vacuo gave the crude product. The residue was
chromatographed using a 50:50 mixture of hexanes:ethyl acetate to give 33 (66
mg);
mp 158-160 °C; IR (KBr) 3408, 3354, 2936, 1633, 1499, 1249, 1 166
crri';'H NMR
(CDCI~) b 3.57 (2H, s), 3.79 (3H, s), 4.01 (3H, s), 4.03 (3H, s), 6.77-6.84
(3H, m),
7.15 ( 1 H, s), 7.16 ( 1 H, s), 7.45 ( 1 H, dd, J,=8.3, JZ=I .8), 7.75-7.79
(2H, m); '3C
NMR 8 56.34, 56.42, 106.59, 106.85, 114.80, 116.35, 117.41, 125.99, 126.81,
127.32, 128.73, 129.42, 129.82, 135.75, 137.89, 150.19, 150.32, 153.25; HRMS
(EI) calcd for C,9H,9N03 m/z: 309.1365; found: 309.1375.
The intermediate compound 27 was perpared as follows.
c. 3-Methoxy-6-nitrobromobenzene (24). Nitric acid (70%, 5 mL) was
placed in a 25 mL round-bottomed flask. Concentrated sulphuric acid (4 mL) was
then added dropwise with stirring. The mixture was kept cool during the
addition
by immersing the flask in an ice bath. 3-Methoxybromobenzene (4 g, 21.5 mmol)
was then introduced dropwise. The reaction mixture was then heated to 50
°C and
stirred for 5 h. After cooling, the mixture was poured into 100 mL of cold
water
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
34
and extracted with ethyl acetate (30 mL x 3). The organic layers were combined
and washed with water (50 mL x 4) and brine. The ethyl acetate layer was dried
with anhydrous sodium sulfate and concentrated in vacuo. The crude product was
purified by column chromatography on silica gel using 95:5 hexanes:ethyl
acetate.
The first compound that eluted from the column was 3-methoxy-4-
nitrobromobenzene ( 1.2 g) in 24% yield; 'H NMR (CDCI3) 8 3.96(3H, s), 7.17 (
1 H,
dd, J,=8.6, Jz=1.9), 7.24 ( 1 H, d, J=1.9), 7.75 ( 1 H, d, J=8.6); "C NMR 8
57.34,
117.58, 124.00, 127.41, 129.05, 142.26, 154.02. The second compound eluting
from the column was 24 (1.5 g, 30% yield); 43-45 °C;'H NMR (CDCl3) 8
3.89
(3H, s), 6.91 ( 1 H, dd, J,=9.1, JZ=2.7), 7.21 ( 1 H, d, J=2.7), 7.98 ( 1 H,
d, J=9.1 ); ' ~C
NMR b 56.68, 114.02, 117.29, 120.61, 128.46, 163.23.
d. Trimethyl(3-methoxy-6-nitrophenyl)stannane (27). A mixture of
hexamethylditin (2 g, 6.13 mmol), 3-methoxy-6-nitrobromobenzene 24 (0.70 g,
3.0
mmol) and Pd(PPh3)4 (100 mg) in anhydrous THF (20 mL) was heated to reflux
under nitrogen until thin layer chromatography no longer showed the presence
of
starting material. After cooling to room temperature, THF was evaporated and
methylene chloride was added to the residue. To this mixture, aqueous
potassium
fluoride (7.0 M, 1.5 mL) was added dropwise with vigorous stirring. The
mixture
was passed through a Celite bed and the filtrate washed with brine. The
methylene
chloride layer was dried (anhydrous NazSO~), filtered and the solution
concentrated
in vacuo. The residue was chromatographed using a 500:8 mixture of
hexanes:ethyl
acetate to give 27 (200 mg) in 21 % yield; 'H NMR (CDCl3) 8 0.34 (9H, s), 3.91
(3H, s), 6.92 (1H, dd, J,=9.1, J2=2.7), 7.13 (1H, d, J=2.8), 8.33 (1H, d,
J=9.1);'~C
NMR 8 -7.09, 56.23, 114.22, 122.38, 127.03, 143.62, 146.84, 164.05.
Example 6 9-Hydroxy-2,3,8-trimethoxydibenzo[c,h]cinnoline (34)
CA 02389312 2002-04-29
WO 01/32631 PCT/IJS00/29583
9-Benzyloxy-2,3,8-trimethoxydibenzo[c,hJcinnoline, 42, (5 mg, 0.012
mmol) was hydrogenated overnight in ethyl acetate (25 mL) at 26 Ib./sq. in.
using
10% palladium on carbon (1.5 mg). The solution was passed through a Celite bed
and the catalyst was washed with ethyl acetate ( 10 mL x 3). Concentration of
the
5 ethyl acetate solution in vacuo gave the crude product. Chromatography using
a
50:45:5 mixture of hexanes:ethyl acetate:methanol as eluting solvent gave
compound 34 (3 mg) in 76% yield; 'H NMR (DMSO-db) 8 4.00 (3H, s), 4.10 (3H,
s), 4. I 2 (3H, s), 7.66 ( 1 H, s), 8.02 (2H, s), 8.21 ( 1 H, d, J=8.4), 8.38
( 1 H, d, J=8.9),
8.96 ( 1 H, s); '3C NMR 8 55.9, 56.4, 103.1, I 03.8, 108.5, 109.1, 117.4,
117.8, 118.0,
10 125.7, 128.1, 131.3, 140.4, 143.8, 150.5, 150.7, 151.3, 152.4; HRMS (El)
calcd for
C'9H'6NzO4 m/z.336.111; found: 336.1109.
Example 7 9-Benzyloxy-2,3,8-trimethoxydibenzo[c,h]cinnoline (42)
I S 6-(2-Amino-5-benzyloxy-4-methoxyphenyl)-2,3-dimethoxy-
naphthalene 41 (35 mg, 0.084 mmol) was dissolved in acetic acid (0.65 mL) and
concentrated hydrochloric acid (0.13 mL). The solution was cooled in an ice
bath
and diazotized by the dropwise addition of a solution of sodium nitrite (0.052
g in
0.52 mL water). The reaction mixture was allowed to warm slowly to room
20 temperature and left for 1 day. To the resulting red solution containing
some
precipitate was added 50 mL water and the mixture was extracted with ethyl
acetate
(30 mL x 3). The organic layers were combined and washed with diluted sodium
hydroxide solution first, then with water and brine. The organic layer was
dried
using anhydrous sodium sulfate and evaporated in vacuo. The crude product was
25 purified by column chromatography on silica gel using 20:80 hexanes:ethyl
acetate
to give the pure 42 (24 mg) in 67% yield; mp 244-246 °C; IR (KBr) 2935,
1621,
1507, 1466, 1307, 1269, 1234, 1206, 1168 cm-'; 'H NMR (CDCI~) b 4.07 (3H, s),
4.14 (3H, s), 4.21 (3H, s), 5.40 (2H, s), 7.25 (1H, s), 7.37-7.60 (5H, m),
7.91 (1H, d,
J=9.0), 7.97 ( 1 H, s), 8.01 ( I H, d, J=9.0), 9.11 ( 1 H, s); '~C NMR 8
56.48, 56.86,
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
36
56.91, 71.64, 101.69, 104.43, 107.82, 109.58, 116.84, 118.19, 118.81, 126.95,
127.97, 128.34, 128.90, 129.34, 131.51, 136.31, 141.61, 144.52, 151.00,
151.14,
152.31, 152.93; HRMS (E~ calcd for CZ6Hz2Nz04 mlz: 426.1580; found: 426.1577.
$ The intermediate compound 41 was prepared as follows.
a. 5-Bromo-2-methoxyphenol (35). To a solution of 5-bromo-2-
methoxybenzaldehyde (2.4 g, 11.2 mmol) in 50 mL CHZC12, m-chloroperbenzoic
acid (70-75%, 7 g, 28.4 mmol pure m-CPBA,) was added and the mixture was
stirred at ambient temperature for 2 days. The reaction was quenched with
aqueous
saturated NaHC03 solution and extracted with ethyl acetate (50 mL x 3). The
organic extract was dried with anhydrous sodium sulfate and filtered through a
silica gel bed. Evaporation of the solvent gave compound 35 (2.1 g) in 92%
yield;
mp 62-64 °C; 'H NMR (CDCI3) 8 3.87 (3H, s), 6.7 I ( I H, d, J=8.6),
6.97 ( 1 H, dd,
J,=8.6, JZ=2.4), 7.07 ( 1 H, d, J=2.4); '~C NMR 8 56.59, I 12.36, I 13.75,
118.33,
123.29, 146.37, 146.98.
b. 3-Benzyloxy-1-bromo-4-methoxybenzene (36). A solution of 5-bromo-2-
methoxyphenol, 35, (2.0 g, 10 mmol) and a-bromotoluene (2.6 g, 15.3 mmol) in
CH~CN (30 mL) and acetone (25 mL) was treated with potassium carbonate (2.1 g,
15.2 mmol). The resulting mixture was heated to reflux under nitrogen for 18
h.
After cooling to room temperature, the reaction mixture was filtered through a
Celite bed. The acetone was removed in vacuo and 50 mL ethyl acetate was added
to the residue. The ethyl acetate solution was washed with water, brine, dried
with
anhydrous NaZSO~, and then evaporated ifz vacuo. The residue was
chromatographed
using a 90:10 mixture of hexanes:ethyl acetate to give compound 36 (2.77 g) in
96% yield; mp 70-71 °C;'H NMR (CDCI~) 8 3.86 (3H, s), 5.12 (2H. s),
7.77 (1H,
J=9.2), 7.04-7.08 (2H, m), 7.33-7.48 (5H, m); '~C NMR b 56.66, 71.68, 113.04,
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
37
113.54, 117.71, 124.47, 127.91, 128.58, 129.13, 136.91, 149.45, 149.50; HRMS
(E>7 calcd for C~4H,302Br m/z: 292.0099; found: 292.0085.
c. 3-Benzyloxy-4-methoxy-6-nitrobromobenzene (37). 3-Benzyloxy-1-
bromo-4-methoxybenzene, 36, (1 g, 3.4 mmol) was dissolved in 50 mL acetic acid
in a 100 mL round-bottomed flask and cooled to 0 °C using an ice bath.
2.5 mL
nitric acid (70%) in 6 mL acetic acid was added dropwise. The reaction mixture
was allowed to slowly rise to room temperature. After 3 h no starting material
was
detected by thin layer chromatography. Evaporation of acetic acid gave the
crude
product, which was filtered through a short silica gel column using a 80:20
mixture
of hexanes:ethyl acetate to give 3-benzyloxy-4-methoxy-6-nitrobromobenzene
1.15 g) in quantitative yield; mp 134-135 °C; IR (KBr) 2946, 1577,
1518, 1468,
1382, 1329, 1266, 1211 crri';UV (MeOH) 246, 212 nm (log E = 3.91, 4.13);'H
NMR (CDCI~) 8 3.93 (3H, s), 5.19 (2H. s), 7.17 ( I H, s), 7.38-7.45 (5H, m),
7.57
( 1 H, s); '~C NMR b 56.99, 71.98, 107.69, 109.74, 118.73, 127.98, I 29.11,
129.36,
135.50, 142.42, 149.18, 152.44; HRMS (EI) calcd for C,4H,~N04Br m/z: 336.9950;
found: 336.9941.
d. Trimethyl(3-benzyloxy-4-methoxy-6-nitrophenyl)stannane (38). A
mixture of hexamethylditin (2 g, 6.13 mmol), 3-benzyloxy-4-methoxy-6-nitro-
bromobenzene 37 ( 1.4 g, 4.14 mmol) and Pd(PPh3)4 (200 mg) in anhydrous THF
(40 mL) was heated to reflux under nitrogen for 2 days. After cooling to room
temperature, THF was evaporated and methylene chloride was added to the
residue.
To this mixture, aqueous potassium fluoride (7.0 M, 1.5 mL) was added dropwise
with vigorous stirring. The mixture was passed through a Celite bed and the
filtrate
washed with brine. The methylene chloride layer was dried (anhydrous Na2S04),
filtered and evaporated in vacuo. The residue was chromatographed using a
90:10
mixture of hexanes:ethyl acetate to give 38 (1.16 g) in 66% yield; mp 81-83
°C; IR
(KBr) 2908, 1569, 1518, 1454, 1318, 1275, 1215 crri'; UV (MeOH) 248, 214 nm
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
38
(log E = 4.08, 4.21); 'H NMR (CDC13) ~ 0.27 (9H, s), 3.97 (3H, s), 5.29 (2H.
s),
7.04 ( 1 H, J=9.2), 7.36-7.44 (5H, m), 7.91 ( I H, s); '3C NMR 8 -7.17, 56.75,
71.59,
108.01, 119.55, 127.80, 128.85, 129.31, 123.64, 136.42, 146.89, 150.18,
153.25;
HRMS (En calcd for C,.,HZ,N04Sn m/z: 408.0258; found: 408.0243.
e. 6-(5-Benzyloxy-4-methoxy-2-nitrophenyl)-2,3-dimethoxy-
naphthalene (39). Tetrakis(triphenylphosphine)palladium (0) (200 mg) and
cuprous bromide (20 mg) were added to a solution of 6,7-dimethoxy-2-
trifluoromethanesulfonyloxy-naphthalene 10 (500 mg, 1.49 mmol) and
trimethylnitroarylstannane 38 (950 mg, 2.25 mmol) in THF (40 mL) at room
temperature and stirred for 0.5 h. The mixture was then refluxed under NZ for
2
days. After cooling, THF was evaporated and ethyl acetate (30 mL) was added to
the residue. The solution was washed with water. The organic layer was
separated
and passed through a Celite bed to remove suspended particles. The organic
layer
was then washed with brine, dried (anhydrous Na2S04), and evaporated in vacuo.
The residue was chromatographed using a 70:30 mixture of hexanes:ethyl acetate
to
give 39 (230 mg) in 35% yield; mp 151-153 °C; IR (KBr) 2962, 1608,
1573, 1508,
1416, 1330, 1275, 1254 cm-'; 'H NMR (CDCI~) 8 3.98 (3H, s), 3.99 (3H, s), 4.01
(3H, s), 5.20 (2H. s), 6.94 ( 1 H, s), 7.10 ( I H, s), 7.14 ( 1 H, s), 7.18 (
1 H, dd, J,=8.4,
JZ=1.8), 7.36-7.43 (5H, m), 7.54 ( 1 H, d, J= I .5), 7.56 ( 1 H, s), 7.68 ( 1
H, d, J=8.3);
'~C NMR b 56.37, 56.95, 71.69, 106.69, 106.99, 108.67, 116.35, 124.91, 125.78,
127.00, 128.00, 128.87, 129.00, 129.23, 129.53, 131.81, 134.49, 136.15,
141.89,
148.95, 150.41, 151.85; HRMS (EI) calcd for Cz6HZ~N06 m/z: 445.1525; found:
445.1355.
f. 6-(2-Amino-5-henzyloxy-4-methoxyphenyl)-2,3-dimethoxy-
naphthalene (41). Compound 39 (50 mg, 0.112 mmol) was hydrogenated in ethyl
acetate (40 mL) at 30 lb./sq. in. using 10% palladium on carbon (15 mg) as
catalyst
for 16 hours. The solution was passed through a Celite bed and the catalyst
was
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
39
washed with ethyl acetate ( 10 mL x 3). Concentration of the ethyl acetate
solution
in vacuo gave a crude product. Column chromatography was performed using a
35:65 mixture of hexanes:ethyl acetate as eluting solvent to give two
compounds.
The compound having the higher Rf material on thin layer chromatography was
isolated as compound 41 (34 mg, 73%); IR (KBr) 3446, 2932, 1610, 1509, 1461,
1421, 1254 crri';'H NMR (CDCI~) 8 3.90 (3H, s), 4.01 (3H, s), 4.02 (3H, s),
5.08
(2H. s), 6.42 ( 1 H, s), 6.87 ( 1 H, s), 7.12 ( 1 H, s), 7.15 ( 1 H, s), 7.33-
7.48 (6H, m),
7.71-7.75 (2H, m); '~C NMR 8 56.40, 56.42, 56.48, 73.07, 101.48, 106.59,
106.78,
119.09, 119.96, 126.24, 126.78, 127.29, 128.16, 128.20, 128.45, 128.90,
129.86,
135.56, 138.19, 138.88, 141.70, 150.07, 150.30, 150.92; HRMS (En calcd for
C26H25N04 ~z: 415.1784; found: 415.1775.
The compound having the lower Rf was isolated as 6-(2-Amino-5-
hydroxy-4-methoxyphenyl)-2,3-dimethoxynaphthalene (40) (6 mg, 16%); IR
(KBr) 3432, 2937, 2364, 1625, 1508, 1459, 1252, 1232 cm-'; UV (MeOH) 238, 208
nm;'H NMR (CDCI~) b 3.88 (3H, s), 4.01 (3H, s), 4.02 (3H, s), 6.40 (1H, s),
6.85
(/H, s), 7.12 (/H, s), 7.15 (/H, s), 7.41 (1H, dd, J,=8.4, JZ=1.7); 7.73 (/H,
d, J=1.5),
7.74 ( I H, d, J=8.3); '3C NMR b 56.40, 100.47, 106.59, 106.82, 116.90,
121.07,
126.29, 126.83, 127.25, 128.47, 129.85, 135.50, 137.17, 139.05, 147.11,
150.06,
150.27.
Example 8 2-Methoxy-8,9-methylenedioxydibenzo[c,h]cinnoline (43)
6-(2-Amino-4,5-methylenedioxyphenyl)-2-methoxynaphthalene 46
(170 mg, 0.58 mmol) was dissolved in acetic acid (4.5 mL) and concentrated
hydrochloric acid (0.9 mL). The solution was cooled in an ice bath and
diazotized
by the dropwise addition of a solution of sodium nitrite (0.36 g in 3.6 mL
water).
The resulting diazonium solution was allowed to warm slowly to room
temperature
and left for I day. To the resulting red solution containing some precipitate
was
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
added 50 mL water and the mixture was extracted with ethyl acetate (30 mL x
3).
The organic layers were combined and washed with diluted sodium hydroxide
solution first, then with water and brine. The organic layer was dried over
anhydrous sodium sulfate and evaporated in vacuo. The crude product was
purified
S by column chromatography on silica gel using 50:50 hexanes:ethyl acetate to
give
the pure 43 (20 mg) in 11% yield; mp 258-260 °C; IR (KBr) 2922, 1611,
1497,
1465, 1414, 1370, 1272, 1201 cm'; UV (MeOH) 286, 228 nm (log E = 4.72, 4.41);
'H NMR (CDCl3) b 4.01 (3H, s), 6.23 (2H, s), 7.30 (1H, J=2.6), 7.48 (1H, dd,
J,=9.1, JZ=2.6), 7.75 ( 1 H, s), 7.95 ( 1 H, s), 8.00 ( 1 H, d, J=9.2), 8. I 9
( 1 H, d, J=9.1 ),
10 9.62 ( 1 H, d, J=9.2); ' ~C NMR b 56.01, 97.95, 102.88, 107.48, 108.31,
119.09,
1 I 9.61, 119.71, 120.57, 125.89, 126.56, 132.26, 134.59, 142.52, 145.88,
150.21,
152.14, 160.16; HRMS (EI) calcd for C,xH12N20~ m/z: 304.0848; found: 304.0843.
The intermediate compound 46 was prepared as follows.
a. 6-(4,5-Methylenedioxy-2-nitrophenyl)-2-methoxynaphthalene (45).
Tetrakis(triphenylphosphine)palladium (0) ( 120 mg) and cuprous bromide (20
mg)
were added to a solution of 2-bromo-6-methoxynaphthalene (0.3 g, 1.27 mmol)
and
trimethyl(3,4-methylenedioxy-6-nitrophenyl)stannane, 62, (0.45 g, 1.37 mmol)
in
THF (30 mL) at room temperature and stirred for O.Sh. The mixture was then
refluxed under N2 for 16 h. After cooling, THF was evaporated and 50 mL ethyl
acetate was added to the residue. The solution was washed with water. The
organic
layer was separated and passed through a Celite bed to remove suspended
particles.
The organic layer was then washed with brine, dried (anhydrous NazS04), and
evaporated in vacuo. The residue was chromatographed using a 80:20 mixture of
hexanes:ethyl acetate to give the desired product 45 (0.29 g) in 71 % yield;
mp 165-
167 °C; IR (KBr) 291 l, 1609, 1520, 1482, 1429, 1393, 1344, 1257, 1199
crri'; 1H
NMR (CDCl3) b 3.94 (3H, s), 6.14 (2H, s), 6.88 (1H, s), 7.16-7.21 (2H, m),
7.31
(1H, dd, J,=8.5, JZ=1.9), 7.47 (1H, s), 7.67-7.77 (3H, m); 13C NMR b 55.89,
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
41
103.45, 105.92, 106.19, 111.69, 119.86, 126.90, 127.02, 127.55, 129.23,
130.09,
133.76, 133.85, 134.48, 143.38, 147.52, 151.47, 158.62; HRMS (El) calcd for
C,8H,3N05 m/z: 323.0794; found: 323.0788.
b. 6-(2-Amino-4,S-methylenedioxyphenyl)-2-methoxynaphthalene (46).
6-(4,5-Methylenedioxy-2-nitrophenyl)-2-methoxynaphthalene 45 (260 mg, 0.81
mmol) was hydrogenated overnight in ethyl acetate (35 mL) at 4045 lb./sq. in.
using 10% palladium on carbon (70 mg) as catalyst. The reaction solution was
passed through a Celite bed and the catalyst was washed with ethyl acetate (10
mL x
3). Concentration of the ethyl acetate solution in vacuo gave the crude
product.
The residue was chromatographed using a 75:25 mixture of hexanes:ethyl acetate
to
give 46 (180 mg) in 76% yield; mp 130-132 °C; IR (KBr) 3463, 3372,
2874, 1631,
1494, 1441, 1389, 1260, 1187 crri';UV (MeOH) 234 nm (log E =4.73); 1H NMR
(CDC13) 8 3.56 (2H, s), 3.95 (3H, s), 5.92 (2H, s), 6.40 ( I H, s), 6.75 ( 1
H, s), 7.17-
7.22 (2H, m), 7.51 (1H, dd, J,=8.5, Jz=1.6), 7.73-7.82 (3H, m); 13C NMR b
55.84,
98.33, 101.25, 106.1 1, 110.71, I 19.66, 120.30, 127.79, 128.27, 128.61,
129.60,
129.91, 133.95, 135.1 1, 138.95, 141.17, 148.05, 158.29; HRMS (EI) calcd for
C,8H,5N0~ m/z: 293.1052; found: 293.1051.
The intermediate compound 62 was prepared as follows.
c. Trimethyl(3,4-methylenedioxy-6-nitrophenyl)stannane (62). A mixture
of hexamethylditin ( 1 g, 3.1 mmol), compound 16 (0.7 g, 2.9 mmol) and
tetrakis(triphenylphosphine)palladium ( 100 mg) in anhydrous THF (20 ml) was
heated to reflux under nitrogen for 10 h. After cooling to room temperature,
THF
was evaporated and methylene chloride (30 mL) was added to the residue. To
this
mixture, aqueous potassium fluoride (7.0M, 1 mL) was added dropwise with
vigorous stirring. The mixture was passed through a Celite bed and the
filtrate was
washed with brine. The methylene chloride layer was dried (anhydrous Na2S04),
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
42
filtered and evaporated in vacuo. The residue was chromatographed using 95:5
hexanes:ethyl acetate to give 62 (0.S g) in S4% yield; 'H NMR (CDCl3) 8 0.32
(9H,
s), 6.12 (2H, s), 7.04 ( 1 H, s), 7.82 ( 1 H, s); '3C NMR (CDCl3) b -6.94,
103.27,
10S.82, 114.76, 137.19, 147.90, 149.36, 153.36; HRMS (En calcd for
S C,oH,~N04Sn-CH3 m/z: 315.9632; found: 315.9638.
Example 9 3-Methoxy-8,9-methylenedioxydibenzo[c,h]cinnoline (44)
7-(2-Amino-4,S-methylenedioxyphenyl)-2-methoxynaphthalene 49
(70 mg, 0.24 mmol) was dissolved in acetic acid (2.0 mL) and concentrated
hydrochloric acid (0.4 mL). The solution was cooled in an ice bath and
diazotized
by the dropwise addition of a solution of sodium nitrite (0.16 g in 1.6 mL
water).
The resulting diazonium solution was allowed to warm slowly to room
temperature
and left overnight. To the resulting red solution containing some precipitate
was
1 S added SO mL water and the reaction mixture was extracted with ethyl
acetate (30
mL x 3). The organic layers were combined and washed with diluted sodium
hydroxide solution first, then with water and brine. The organic layer was
dried
over anhydrous sodium sulfate and evaporated in vacuo. The crude product was
purified by column chromatography on silica gel using SS:4S hexanes:ethyl
acetate
to give compound 44 (60 mg 83%); mp 259-261 °C; IR (KBr) 2923, 1612,
1498,
1468, 1234, 1199 cm '; UV (MeOH) 270, 250, 228 nm (log E = 4.68, 4.37, 4.44);
'H
NMR (CDCI~) 8 4.12 (3H, s), 6.24 (2H, s), 7.37 (1H, dd, J,=8.8, JZ=2.7), 7.80
(1H,
s), 7.88 ( 1 H, d, J=8.8), 7.96 ( 1 H, s), 8.03 ( 1 H, d, J=9.1 ), 8.09 ( 1 H,
d, J=9.0), 9.1 S
(1H, d, J=2.7);'~C NMR 8 56.32, 98.31, 102.93, 104.03, 107.49, 116.36, 120.40,
2S 120.50, 121.09, 127.96, 130.00, 132.54, 133.34, 141.99, 146.10, 150.53,
152.09,
160.28; HRMS (EI) calcd for C,BH,~N~O~ m/z: 304.0848; found: 304.0852.
The intermediate compound 49 was prepared as follows.
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
43
a. 7-Methoxy-2-trifluoromethanesulfonyloxynaphthalene (47). A solution
of 7-methoxy-2-naphthol (0.75 g, 4.3 mmol) in THF (10 mL) was added to a
suspension of sodium hydride (60 wt%, 205 mg, 5.1 mmol) in THF (10 mL) cooled
by ice bath and stirred for 1.5 h. A solution of N-
phenyltrifluoromethanesulfonimide (1.55 g, 4.34 mmol) in THF (10 mL) was then
added, and the reaction mixture was stirred for 9 h. After evaporation of the
solvent
in vacuo, the residue was mixed with silica gel (4 g) and then chromatographed
using 500:18 hexanes:ethyl acetate to give pure 47 (1.19 g) in 90% yield; mp
34 °C
(lit'°° 34 °C);'H NMR (CDC13) 3.93 (3H, s), 7.13-7.25
(3H, m), 7.65 (1H, d,
J=2.5), 7.77 ( I H, d, J=9.1 ), 7.83 ( 1 H, d, J=8.8); 13C NMR b 55.88,
106.25, 116.13,
117.49, 118.52, 120.75, 122.51, 128.34, 129.87, 130.72, 135.41, 148.29,
159.39;
HRMS (EI) calcd for C,ZHyS04F3 m/z: 306.0174; found: 306.0176.
b. 7-(4,5-Methylenedioxy-2-nitrophenyl)-2-methoxynaphthalene (48).
Tetrakis(triphenylphosphine)palladium (0) ( 120 mg) and cuprous bromide (20
mg)
were added to a solution of 7-Methoxy-2-trifluoromethanesulfonyloxynaphthalene
47 (336 mg, 1.1 mmol) and trimethylnitroarylstannane 62 (300 mg, 0.92 mmol) in
THF (30 mL) at room temperature and stirred for 0.5 h. The mixture was then
refluxed under NZ overnight. After cooling, THF was evaporated in vacuo and
ethyl
acetate (30 mL) was added to the residue. The solution was washed with water.
The organic layer was separated and passed through a Celite bed to remove
suspended particles. The organic layer was then washed with brine, dried
(anhydrous NaZSO,~), and evaporated in vacuo. The residue was chromatographed
using a 80:20 mixture of hexanes:ethyl acetate to give 48 (100 mg) in 34%
yield; IR
(KBr) 2915, 1627, 1509, 1481, 1425, 1333, 1262, 1218 crri';1H NMR (CDC13) b
3.92 (3H, s), 6.15 (2H, s), 6.88 (1H, s), 7.13-7.23 (3H, m), 7.48 (1H, s),
7.64 (1H,
s), 7.74-7.81 (2H, m); 13C NMR 8 55.81, 103.47, 105.90, 106.46, 111.60,
119.84,
124.13, 126.07, 128.50, 128.72, 129.75, 133.89, 134.97, 136.60, 143.42,
147.63,
151.45, 158.61; HRMS (El) calcd for C,~H,3N05 m/z: 323.0794; found: 323.0787.
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
44
c. 7-(2-Amino-4,5-methylenedioxyphenyl)-2-methoxynaphthalene (49).
7-(4,5-Methylenedioxy-2-nitrophenyl)-2-methoxynaphthalene 48 (100 mg, 0.31
mmol) was hydrogenated overnight in ethyl acetate (35 mL) at 4045 lb./sq. in.
using 10% palladium on carbon (30 mg) as catalyst. The reaction solution was
passed through a Celite bed and the catalyst was washed with ethyl acetate (10
mL x
3). The ethyl acetate solution was concentrated in vacuo gave the crude
product.
The residue was chromatographed using a 75:25 mixture of hexanes:ethyl acetate
to
give 49 (75 mg) in 83% yield; IR (KBr) 3426, 3366, 2364, 2339, 1629, 1503,
1487,
1467, 1233, 1215, 1187 cm 1; 1H NMR (CDCI~) 8 3.64 (2H, s), 3.94 (3H, s), 5.92
(2H, s), 6.40 ( 1 H, s), 6.76 ( 1 H, s), 7.15-7.20 (2H, m), 7.40 ( 1 H, dd,
J,=8.3, Jz=1.7),
7.75-7.85 (3H, m); 13C NMR b 55.83, 98.35, 101.27, 106.25, 110.65, 119.35,
120.29, 125.82, 127.35, 128.31, 128.72, 129.67, 135.34, 137.99, 138.99,
141.17,
148.16, 158.49; HRMS (EI) calcd for C,~H,SN03 m/z: 293.1052; found: 293.1052.
Example 10 3-Methoxy-8,9-methylenedioxydihenzo[c,h]cinnoline (44)
The compound of Example 9 (compound 44) was also prepare as
follows. Lithium aluminum hydride (46 mg, 1.2 mmol) was added to a stirred
solution of compound 54 (74 mg, 0.2 mmol) in diethyl ether (10 mL) and benzene
( 10 mL). The mixture was stirred under reflux for 1 h. After cooling to room
temperature, the excess hydride was decomposed with 0.05 mL water, 0.05 mL 15%
NaOH and 0.15 mL water, and the reaction mixture filtered through a Celite
bed.
Evaporation of solvent in vacrso gave the crude product, which was purified by
column chromatography using 50:50 hexanes:ethyl acetate mixture as eluting
solvent to provide compound 44 (46 mg,75%).
The intermediate compound 54 was prepared as follows.
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
a. 4-Methyl-2,3,4,5-tetrabromophenol (50). p-Cresol (5 g, 46 mmol) was
added dropwise to 15 mL (0.29 mol) of bromine containing 0.25 g Fe filings at
room temperature. During the addition of p-cresol, small portions of
chloroform
were added from time to time to facilitate stirring. After 6 h, HBr evolution
5 subsided. The residue was dissolved in hot chloroform, washed with aqueous
NazS203, NaHC03. dried with anhydrous sodium sulfate and evaporated in vacuo.
The crude product was purified by column chromatography using a 95:5 mixture
of
hexanes and ethyl acetate to give 3.57 g of 50 (92% yield); mp 195-196
°C (1it93 196
°C); 1H NMR (acetone-d6) 8 2.71 (3H, s); 13C NMR b 28.10, 115.20,
127.66,
10 133.24, 152.19.
b. 4-Methyl-4-nitro-2,3,5,6-tetrabromo-2,5-cyctohexadien-1-one (51). A
solution containing 1.6 mL of nitric acid (d=1.52, 70°10) in 10 mL of
acetic acid was
added over a 10 minute period to a solution of compound 50 (3.2 g, 7.6 mmol)
in 25
15 mL of pure acetic acid at about 10 °C: The reaction mixture was
stirred for 4 h and
30 mL of water was then added. The precipitates were filtered and washed with
water and heptane and dried in vacuum to give 2.9 g of pure 51 (82% yield); 1H
NMR (CDCl3) 8 2.26 (3H, s); IR (KBr) 1680 (C=O) (1it93).
20 c. 2-Hydroxy-7-methoxy-1-nitronaphthalene (52). 7-Methoxy-2-naphthol
(871 mg, 5 mmol) was dissolved in 40 mL of dry ether. To this solution was
added
51 (2.33 g, 5 mmol) as a solid. The color of the solution slowly became red,
and
eventually dark red with some dark precipitate adhering to the inside surface
of the
flask. The reaction continued for 2.5 h at room temperature. Evaporation of
the
25 solvent gave the crude product. To the residue was added 20 mL of
methanol/water
(80/20). The reaction mixture was filtered and washed with methanol/water
(80/20). The filtrate was then evaporated under vacuum and purified using
column.
A 90:10 mixture of hexanes and ethyl acetate was used as the eluting solvent.
The
yield was of 380 mg 52 (35%); mp 130-131 °C (1it93 130 °C); 1H
NMR (CDC13) 8
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
46
3.96 (3H, s), 7.04 ( 1 H, d, J=9.0), 7.10 ( 1 H, dd, J=9.0, J=2.6), 7.67 ( 1
H, d, J=8.9),
7.87 ( 1 H, d, J=8.9), 8.37 ( 1 H, d, J=2.5); 13C NMR b 56.03, 104.30, 116.80,
117.28, 124.22, 129.35, 131.52, 139.59, 160.41, 162.75.
d. 7-Methoxy-1-nitro-2-trifluoromethanesulfonyloxynaphthalene (53). A
solution of compound 52 (380 mg, 2.24 mmol) in THF (15 mL) was added to a
suspension of sodium hydride (60 wt% in mineral oil, 90 mg, 2.25 mmol) in THF
( 10 mL) cooled in an ice bath and stirred for 0.5 h. A solution of N-
phenyltrifluoromethanesulfonimide (800 mg, 2.24 mmol) in THF ( 10 mL) was then
added, and the reaction stirred at 0 °C for 8 h. After concentration in
vacuo, the
residue was chromatographed using 85:15 hexanes:ethyl acetate to give triflate
53
(526 mg) containing approximately 10% N-phenyltrifluoromethanesulfonamide.
e. 6-(4,5-Methylenedioxy-2-nitrophenyl)-2,3-dimethoxy-5-
IS nitronaphthalene (54). Tetrakis(triphenylphosphine)palladium (0) (100 mg)
and
cuprous bromide (20 mg) was added to a solution of 7-Methoxy-I-nitro-2-
trifluoromethanesulfonyloxy-naphthalene 53 (366 mg, I .04 mmol) and
trimethylnitroarylstannane 62 (500 mg, 1.52 mmol) in THF (30 mL) at room
temperature and stirred for 0.5 h. The mixture was then refluxed under NZ
overnight. After cooling, THF was evaporated and ethyl acetate (30 mL) was
added
to the residue. The solution was washed with water. The organic layer was
separated and passed through a Celite bed to remove suspended particles. The
organic layer was then washed with brine, dried (anhydrous NazS04), and
evaporated in vacaso. The residue was chromatographed using a 70:30 mixture of
hexanes:ethyl acetate to give 54 (160 mg) in 42% yield; mp 187-189 °C;
IR (KBr)
2925, 1628, 1526, 1487, 1364, 1332, 1265, 1230 cm'; 1H NMR (CDC13) b 3.92
(3H, s), 6.19 (2H, d), 6.76 ( 1 H, s), 7.12 ( 1 H, d, J=2.5), 7.18 ( I H, d,
J=8.3), 7.28
( 1 H, dd, J,=9.0, J~=2.3), 7.70 ( 1 H, s), 7.85 ( 1 H, d, J=9.2), 7.93 ( 1 H,
d, J=8.4); 13C
CA 02389312 2002-04-29
WO 01/32631 PCT/LTS00/29583
47
NMR 8 56.08, 100.59, 103.93, 106.31, 110.70, 121.54, 124.07, 126.47, 128.71,
129.55, 130.33, 130.99, 131.23, 142.83, 148.92, 152.07, 160.68.
Example 10 The following illustrate representative pharmaceutical dosage
forms,
containing a compound of formula I ('Compound X'), for therapeutic or
prophylactic
use in humans.
(i) Tablet 1 m~/tablet
'Compound X' 100.0
10Lactose 77.5
Povidone 15.0
Croscarmellose sodium 12.0
Microcrystalline cellulose92.5
Magnesium stearate 3.0
300.0
(ii) Tablet 2 m /t~ ablet
'Compound X' 20.0
Microcrystalline cellulose410.0
Starch 50.0
20Sodium starch glycolate15.0
Magnesium stearate 5.0
500.0
(iii) Capsule m~/capsule
25'Compound X' 10.0
Colloidal silicon dioxide1.5
Lactose 465.5
Pregelatinized starch 120.0
Magnesium stearate 3.0
30 600.0
(iv) Injection 1 (1 mg/ml) m./g-ml
'Compound X' (free acid 1.0
form)
Dibasic sodium phosphate 12.0
35 Monobasic sodium phosphate0.7
Sodium chloride 4.5
1.0 N Sodium hydroxide solution
(pH adjustment to 7.0-7.5; q.s.
Water for injection q.s. ad
1 mL
40
CA 02389312 2002-04-29
WO 01/32631 PCT/US00/29583
48
(v) Infection 2 (10 m~/ml) mg/ml
'Compound X' (free acid 10.0
form)
Monobasic sodium phosphate0.3
Dibasic sodium phosphate 1.1
Polyethylene glycol 400 200.0
O1 N Sodium hydroxide solution
(pH adjustment to 7.0-7.5) q.s.
Water for injection q.s. ad
I mL
(vi) Aerosol m /
'Compound X' 20.0
Oleic acid 10.0
Trichloromonofluoromethane 5,000.0
Dichlorodifluoromethane 10,000.0
Dichlorotetrafluoroethane 5,000.0
The above formulations may be obtained by conventional procedures well known
in
the pharmaceutical art.
All publications, patents, and patent documents are incorporated by
reference herein, as though individually incorporated by reference. The
invention
has been described with reference to various specific and preferred
embodiments
and techniques. However, it should be understood that many variations and
modifications may be made while remaining within the spirit and scope of the
invention.