Note: Descriptions are shown in the official language in which they were submitted.
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
HMG-COA REDUCTASE INHIBITOR EXTENDED
RELEASE FORMULATION
BACKGROUND OF THE INVENTION
The use of HMG-COA reductase inhibitors for the reduction of serum cholesterol
levels is well known. These compounds include alkyl esters of hydroxy
substituted
naphthalenes which are orally effective in the reduction of serum cholesterol
levels.
Examples of these compounds include mevastatin which is described in U.S.
3,671,523;
lovastatin which is described in U.S. 4,231,938; pravastatin which is
described in U.S.
4,346,227; and simvastatin which is described in U.S. 4,444,784. All of these
patents are
incorporated by reference.
Lovastatin is a metabolite which is produced by the natural fermentation of an
fungus
of the Aspergillus genus. Lovastatin acts systemically to lower blood serum
cholesterol
levels by disrupting the biosynthesis of cholesterol in the liver, where 70%
to 80% of body
cholesterol is produced. Specifically lovastatin interrupts a step in the
endogenous
production of cholesterol by inhibiting the HMG coenzyme A reductase from
combining with
bile acids in the digestive tract such that the bile acids are excreted from
the body without
reabsorption. With synthesis in the liver thusly inhibited, the liver cells
must take cholesterol
from the bloodstream, and they do so by increasing their production of cell
surface receptors
for LDL cholesterol. Lovastatin formulations are generally capable of lowering
the blood
serum cholesterol level by about 30-40%. The other compounds of this class are
derived
from natural or synthetic sources using well known procedures and have similar
mechanisms
of activity.
However, it is desirable to enhance the activity of these compounds to achieve
even
greater reductions of blood serum cholesterol levels in connection with the
treatment of
hypercholesterolemia and other maladies. Accordingly, the present invention
provides a novel
controlled release formulation of a compound which is an alkyl ester of a
hydroxy substituted
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
naphthalene derivative which provides for a gradual release of the compound.
This formulation
has been prepared to provide a slow controlled release of these compounds in
order to provide
a more constant level of bioavailability in order to provide an enhanced
effect that cannot be
achieved by conventional immediate release dosing. The use of a controlled
release form of is
believed to be specially useful for those who have meals at irregular times or
those who
frequently eat snacks between meals. These subjects include night shift
workers, airline
personnel and travelers, and those individuals with blood sugar problems who
eat frequent small
meals. In addition, it is believed that the human body synthesizes high
amounts of cholesterol
during the hours of sleep and it is desirable in certain cases to provide
therapeutic level of these
compounds during periods of sleep.
Controlled release formulations have been described in U. S. 4,615,698 which
have been
based on an osmotic dosage form which is designed to collapse and cause the
faced surfaces to
come into a close contacting arrangement as the drug is delivered through a
passageway in the
semi-permeable wall of the dosage form. In addition, U.S. 4,503,030 discloses
an osmotic
dosage form which has a passageway and a semi-permeable membrane consisting of
a particular
cellulose polymer and a pH-sensitive material which could be an enteric
coating material. This
patent describes the use of 1:1 mixtures of a pH sensitive material and
cellulose polymer which
are applied at a level of about 70% by weight based on the total weight of the
osmotic core tablet
and coating material.
In the parent application, the applicants have discovered that a ratio of
0.75:1, and lower,
of pH sensitive material to cellulose polymer may be used to provide a stable
membrane around
an osmotic core tablet at a coating level of 1-4% by weight based on the total
weight of the
osmotic core tablet and coating material. These osmotic tablets will
substantially, completely
deliver the compound without the need to provide a passageway in the tablet
according to the
teachings of the prior art. In addition the osmotic tablet of the invention
will provide higher
bioavailability and lower peak plasma drug concentrations than are provided by
the same weight
of the alkyl ester of a hydroxy substituted naphthalene derivative in a
conventional immediate
release dosage form.
2
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
SUMMARY OF THE INVENTION
It is an object of the present invention to provide oral compositions which
enhance the
activity of antilipemic agents, specifically HMG-CoA reductase inhibitors, to
achieve even
greater reductions of blood serum cholesterol levels in connection with the
treatment of
hypercholesterolemia and other maladies.
It is an object of the present invention to provide a controlled release form
of an alkyl
ester of a hydroxy substituted naphthalene derivative.
It is an object of the present invention to provide a controlled release form
of an alkyl
ester of a hydroxy substituted naphthalene derivative which provides effective
yet novel
plasma concentration profiles of the drug.
It is a further object of the present invention to provide methods of treating
human
patients who have high serum cholesterol levels.
It is a further object of the present invention to provide methods of
enhancing the
activity of antilipemic agents, specifically HMG-CoA reductase inhibitors, to
achieve even
greater reductions of blood serum cholesterol levels in connection with the
treatment of
hypercholesterolemia and other maladies.
It is also an object of the present invention to provide a controlled release
dosage
formulation of an alkyl ester of a hydroxy substituted naphthalene derivative
which
substantially completely releases said alkyl ester in about 4 to 30 hours in
vitro in a Type 2
USP 23 dissolution apparatus in 2% sodium lauryl sulfate, pH buffer to 7.0 at
37°C and
SOrpm.
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
In accordance with the above-mentioned objects and others, the present
invention
provides a novel controlled release formulation of a compound which is an
alkyl ester of a
hydroxy substituted naphthalene derivative which provides for a gradual
release of the
compound. This formulation has been prepared to provide a slow controlled
release of these
compounds in order to provide a more constant level of bioavailability in
order to provide an
enhanced effect that cannot be achieved by conventional immediate release
dosing.
In view of the above objects and others, the invention is in part directed to
a
controlled release oral solid dosage form for the reduction of serum
cholesterol levels in
humans comprising a drug comprising an alkyl ester of hydroxy substituted
naphthalenes
(e.g., lovastatin) and a controlled release carrier in an amount effective to
provide a controlled
release of the drug, the dosage form providing a mean time to maximum plasma
concentration (TmaX) of the drug which occurs at about 10 to about 32 hours
after oral
administration to human patients, the dosage form providing a reduction in
serum cholesterol
levels when administered to human patients on a once-a-day basis.
The invention is further directed to a controlled release oral solid dosage
form for the
reduction of serum cholesterol levels in humans, comprising a drug comprising
an alkyl ester
of hydroxy substituted naphthalenes, and a controlled release Garner for the
drug, the dosage
form providing a substantially complete release of the drug in about 4 to 30
hours in vitro in a
Type 2 USP 23 dissolution apparatus in 2% sodium lauryl sulfate, pH buffer to
7.0 at 37°C
and SOrpm, the dosage form providing a mean time to maximum plasma
concentration (TmaX)
which occurs at about 10 to about 32 hours after oral administration to human
patients, the
dosage form achieving a reduction in serum cholesterol levels when
administered to human
patients on a once-a-day basis.
Further embodiments of the invention relate to a controlled release oral solid
dosage
form for the reduction of serum cholesterol levels in humans, comprising a
drug comprising
an alkyl ester of hydroxy substituted naphthalenes, and a controlled release
carrier for the
drug, the controlled release dosage form providing a dissolution of from about
0% to about
4
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
25% drug released after 2 hours; from about 40% to about 85% drug released
after 6 hours;
and not less than about 75% drug released after 16 hours, when measured in
vitro in a Type 2
USP 23 dissolution apparatus in 2% sodium lauryl sulfate, pH buffer to 7.0 at
37°C and
50rpm, and which preferably provides a time to maximum plasma concentration
(Tm~) of the
drug which occurs at about 10 to about 32 hours after oral administration to
human patients,
the dosage form achieving a reduction in serum cholesterol levels when
administered to
human patients on a once-a-day basis.
In certain preferred embodiments, the controlled release dosage form provides
a
dissolution of from about 0% to about 20% drug released after 2 hours; from
about 50% to
about 80% drug released after 6 hours; and not less than about 80% drug
released after 16
hours, when measured in vitro in a Type 2 USP 23 dissolution apparatus in 2%
sodium lauryl
sulfate, pH buffer to 7.0 at 37°C and 50rpm. In certain further
preferred embodiments, the
controlled release dosage form provides a dissolution of from about 10% to
about 15% drug
released after 2 hours; from about 65% to about 75% drug released after 6
hours; and not less
than about 79% drug released after 16 hours, when measured in vitro in a Type
2 USP 23
dissolution apparatus in 2% sodium lauryl sulfate, pH buffer to 7.0 at
37°C and 50rpm.
In certain embodiments, the mean time to maximum plasma concentration of the
drug
preferably occurs at about 14 to about 24 hours after oral administration.
In certain preferred embodiments of the invention, the mean time to maximum
plasma
concentration of total HMG-CoA Reductase Inhibitors preferably occurs at about
13 to about
21 hours after oral administration. Preferably, the controlled release
formulations of the
invention provide a mean time to maximum plasma concentration of active HMG-
CoA
Reductase Inhibitors which preferably occurs at about 6.2 to about 20.1 hours,
and more
preferably from about 9.5 to about 15.2 hours after oral administration.
Preferably, the
controlled release formulations in accordance with the invention provide a
mean maximum
plasma concentration (CmaX) of total HMG-CoA Reductase Inhibitors from about
4.7 ng/ml to
about 25.4 ng/ml, preferably from about 10.5 ng/ml to about 17.3 ng/ml, and a
mean
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
maximum plasma concentration (Cmax) of active HMG-CoA Reductase Inhibitors
preferably
from about 2.1 ng/ml to about 22.5 ng/ml, preferably 6.4 ng/ml to about 13.4
ng/ml (e.g.,
based on a 40 mg dose of lovastatin).
In certain preferred embodiments of the invention, the drug is selected from
the group
consisting of lovastatin, a derivative of lovastatin, an active metabolite of
lovastatin,
mevastatin, pravastatin, simvastatin, and mixtures thereof.
In certain preferred embodiments of the invention, the controlled release oral
solid
dosage form includes lovastatin in an amount of from about 10 to about 80 mg
(e.g., 10, 20,
40 or 80 mg). In embodiments in which the drug is lovastatin, the formulations
preferably
provide a maximum plasma concentration (Cmax) of lovastatin from about 1 ng/ml
to about 8
ng/ml, preferably from about 1.5 ng/ml to about 7.1 ng/ml, and more preferably
from about 3
ng/ml to about 4 ng/ml, based on a 40 mg dose of lovastatin (the plasma levels
of lovastatin
preferably being dose proportional, as described herein in the appended
Examples). When
the drug is lovastatin, the controlled release formulations of the invention
preferably provide
a mean AUCo~Bhr of lovastatin from about 15 to about 90 ng~hr/ml, more
preferably from
about 34 to about 77 ng~hr/ml. In certain preferred embodiments of the
invention where the
drug is lovastatin, the dosage form preferably provides a mean time to maximum
plasma
concentration of lovastatin acid at about 5.3 to about 28.7 hours after oral
administration, and
more preferably at about 13.0 to about 20.9 hours, after oral administration.
In further
preferred embodiments where the drug is lovastatin, the dosage form provides a
mean
maximum plasma concentration (Cr"~) of lovastatin acid from about 1.05 ng/ml
to about 7.22
ng/ml, preferably from about 2.50 ng/ml to about 4.90 ng/ml, based on a 40 mg
dose of
lovastatin. In such embodiments, the dosage from may provide a mean AUCo~B~ of
lovastatin acid from about 9.96 to about 132.54 ng~hr/ml, preferably from
about 47.5 to
about 91.2 ng~hr/ml.
The present invention further relates to a method for reducing serum
cholesterol levels
in humans, comprising orally administering a drug comprising an alkyl ester of
hydroxy
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
substituted naphthalenes in a controlled release oral solid dosage form which
provides a mean
time to maximum plasma concentration (TmaX) of the drug which occurs at about
10 to about
32 hours after oral administration of said dosage form to human patients, and
which achieves
an effective reduction in serum cholesterol levels when administered to human
patients on a
once-a-day basis. In certain preferred embodiments where the drug is
lovastatin, it is
preferred that after administration the dosage form provides a mean maximum
plasma
concentration (Cmax) of lovastatin from about 1.5 ng/ml to about 7.1 ng/ml,
based on a 40 mg
dose of lovastatin (e.g., where the plasma levels and CmaX of lovastatin is
dose proportional),
after administration of a single dose to human patients. In certain preferred
embodiments,
orally administering lovastatin in a controlled release oral solid dosage form
provides a mean
time to maximum plasma concentration (TmaX) which occurs at about 14 to about
24 hours
after oral administration of said dosage form to human patients, and a maximum
plasma
concentration (Cmax) of the drug of from about 3 ng/ml to about 4 ng/ml (based
on a 40 mg
dose of lovastatin), such that the dosage form achieves a reduction in serum
cholesterol levels
when administered to human patients on a once-a-day basis.
The present invention further relates to a method for reducing serum
cholesterol levels
in humans, comprising orally administering a drug comprising an alkyl ester of
hydroxy
substituted naphthalenes in a controlled release oral solid dosage form to
human patients in
the morning (e.g., after administration of a single dose of lovastatin), which
dosage form
provides a mean time to maximum plasma concentration (TmaX) at about 11 to
about 32 hours
after oral administration to human patients, the dosage form achieving a
reduction in serum
cholesterol levels when administered to human patients on a once-a-day basis.
In this
embodiment, when the drug is lovastatin, the dosage form preferably provides a
mean
maximum plasma concentration (CmaX) of said drug from about 1.5 ng/ml to about
6.9 ng/ml,
based on a 40 mg dose of lovastatin. In this embodiment, where the human
patients are
administered the dosage form after breakfast in the fed state, it is preferred
that the dosage
form provides a mean time to maximum plasma concentration (TmaX) which occurs
at about
16 to about 32 hours after oral administration of a single dose, more
preferably at about 22 to
about 26 hours after oral administration, and that it further preferably
provides a mean
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
maximum plasma concentration (Cmax) of the drug from about 1.5 ng/ml to about
4.5 ng/ml,
based on a 40 mg dose of lovastatin, after oral administration of a single
dose. When the
human patients are adminstered the dosage form in the morning in the fasted
state, it is
preferred that the dosage form provides a mean time to maximum plasma
concentration (TmaX)
which occurs at about 5.3 to about 17 hours after oral administration of a
single dose, more
preferably at about 9 to about 13 hours after oral administration, and
preferably provides a
mean maximum plasma concentration (Cmax) of the drug from about 2.9 ng/ml to
about 6.9
ng/ml, based on a 40 mg dose of lovastatin, after oral administration of a
single dose.
The present invention further relates to a method for reducing serum
cholesterol levels
in humans, comprising orally administering a drug comprising an alkyl ester of
hydroxy
substituted naphthalenes in a controlled release oral solid dosage form to
human patients at
dinner time, which dosage form provides a mean time to maximum plasma
concentration
(T",~) at about 10.4 to about 20.6 hours after oral administration (e.g.,
after a single dose), the
dosage form achieving a reduction in serum cholesterol levels when
administered to human
patients on a once-a-day basis. Preferably, where the drug is lovastatin, the
dosage form
provides a mean maximum plasma concentration (Cmax) of said drug from about
1.9 ng/ml to
about 4.4 ng/ml, based on a 40 mg dose of lovastatin. In such embodiments, it
is preferred
that the mean time to maximum plasma concentration (Tmax) is at about 13.5 to
about 17.5
hours after oral administration, and more preferably at about 15.5 hours after
oral
administration. It is further preferred the dosage form provides a mean
maximum plasma
concentration (Cmax) of said drug of about 3 ng/ml, based on a 40 mg dose of
lovastatin.
The present invention further relates to a method for reducing serum
cholesterol levels
in humans, comprising orally administering a drug comprising an alkyl ester of
hydroxy
substituted naphthalenes in a controlled release oral solid dosage form to
human patients at
bedtime, which dosage form provides a mean time to maximum plasma
concentration (TmaX)
which occurs at about 10 to about 23.2 hours after oral administration (e.g.,
of a single dose)
to a population of human patients, the dosage 'form achieving a reduction in
serum
cholesterol levels when administered to human patients on a once-a-day basis.
Preferably,
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
where the drug is lovastatin, the dosage form provides a mean maximum plasma
concentration (C~"~) of said drug from about 1 ng/ml to about 8 ng/ml, based
on a 40 mg dose
of lovastatin. In this embodiment, where the human patients are administered
the dosage
form at bedtime, it is preferred that the dosage form provides a mean time to
maximum
S plasma concentration (TmaX) which occurs at about 14.2 to about 16.9 hours
after oral
administration of a single dose, and preferably provides a mean maximum plasma
concentration (Cmax) of the drug of about 4 ng/ml, based on a 40 mg dose of
lovastatin, after
oral administration of a single dose. When administered at bedtime, the dosage
forms of the
invention preferably provide a mean time to maximum plasma concentration
(Tmax) which
occurs at about 10 to about 22 hours after oral administration to human
patients at steady-
state, preferably at about 12 to about 16 hours, and more preferably at about
14 hours after
oral administration. Preferably, the dosage form provides a mean maximum
plasma
concentration (CmaX) of said drug from about 3 ng/ml to about 8 ng/ml, more
preferably about
5.5 ng/ml, based on a 40 mg dose of lovastatin at steady-state.
The invention also relates to a method for improving the dose-response
relationship
achieved via the administration of a statin drug orally administered in
immediate release
form, comprising orally administering the statin in a controlled release
dosage form which
provides a mean time to maximum plasma concentration (TmaX) of the statin drug
which
occurs at about 10 to about 32 hours after oral administration to human
patients.
In certain preferred embodiments of the present invention, administration of
the
controlled release oral solid dosage forms of the invention achieves an
accumulation of
lovastatin and its latent and active metabolites at steady-state conditions of
about 1.4 to about
2 fold the levels attained by immediate release lovastatin administered once
daily. In certain
preferred embodiments, the dosage forms of the invention provide increased
systemic
bioavailability of lovastatin, but at the same time do not provide increased
bioavailability of
lovastatin acid, active or total inhibitors.
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
The invention is further related to a controlled release oral solid dosage
form,
comprising a therapeutically effective amount of lovastatin, and a controlled
release carrier
providing delivery of said lovastatin when said dosage form is orally
administered to human
patients, such that a mean maximum plasma concentration (Cmax) of lovastatin
from about 1
ng/ml to about 5.5 ng/ml and preferably from about 3 ng/ml to about 5.5 ng/ml
is attained,
after administration of a single dose or at steady-state in a population of
human patients in
need of such therapy, per 40 mg dose of lovastatin.
The invention is further related to a method for reducing serum cholesterol
levels in
humans, comprising orally administering a controlled release oral solid dosage
form
containing a therapeutically effective amount of lovastatin which provides a
mean maximum
plasma concentration (Cmax) of lovastatin from about 1 ng/ml to about 5.5
ng/ml, preferably
from about 3 ng/ml to about 5.5 ng/ml, after administration of a single dose
or at steady-state
in a population of human patients in need of such therapy, per 40 mg dose of
lovastatin.
The invention is further related to a method for providing increased systemic
bioavailability of lovastatin, while at the same time not increasing the
bioavailability of
lovastatin acid, active or total inhibitors compared to an immediate release
reference standard
form of lovastatin, comprising preparing a controlled release oral solid
dosage form of
lovastatin which comprises a therapeutically effective amount of lovastatin
and a sufficient
amount of a controlled release Garner such that the controlled release dosage
form provides a
dissolution of from about 0% to about 25% lovastatin released after 2 hours;
from about 40%
to about 85% lovastatin released after 6 hours; and not less than about 75%
lovastatin
released after 16 hours, when measured in vitro in a Type 2 USP 23 dissolution
apparatus in
2% sodium lauryl sulfate, pH buffer to 7.0 at 37°C and SOrpm, and such
that the dosage form
provides a mean time to maximum plasma concentration (TmaX) of lovastatin from
about 10
to about 32 hours after oral administration to human patients. The dosage form
is
administered to human patients on a once-a-day basis.
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
For purposes of this disclosure, the term "statin" encompasses alkyl esters of
hydroxy
substituted naphthalenes which are orally effective in the reduction of serum
cholesterol
levels, and includes but is not limited to examples of these compounds
described in U.S.
3,671,523 (include mevastatin); compounds described in U.S. 4,231,938
(including
lovastatin); compounds described in U.S. 4,346,227 (including pravastatin);
and compounds
is described in U.S. 4,444,784 (including simvastatin).
The term "steady state" means that the blood plasma concentration curve for a
given
drug has been substantially repeated from dose to dose of the drug
formulation. The term
"single dose" means that the human patient has received a single dose of the
drug formulation
and the drug plasma concentration has not achieved steady state. The term
"multiple dose"
means that the human patient has received at least two doses of the drug
formulation in
accordance with the dosing interval for that formulation (e.g, on a once a day
basis). Patients
who have received multiple doses of the controlled release formulations of the
invention may
1 S but not necessarily have attained steady state drug plasma levels, as the
term multiple dose is
defined herein.
The term "morning" as it is used herein with respect to the dosing of the
controlled
release formulations of the invention means that the controlled release
formulation is orally
administered early in the day after the patient has awakened from overnight
sleep, generally
between about 6 a.m. and 11 a.m. (regardless of whether breakfast is eaten at
that time, unless
so specified herein). The term "dinnertime" as it is used herein with respect
to the dosing of
the controlled release formulations of the invention means that the controlled
release
formulation is orally administered at a time when dinner is normally eaten
(regardless of
whether a meal is actually eaten at that time, unless so specified herein),
generally between
about 4 p.m. and 8 p.m. The term "bedtime" as it is used herein with respect
to the dosing of
the controlled release formulations of the invention means that the controlled
release
formulation is orally administered before the patient goes to bed in the
evening, generally
between about 8 p.m. and 12 p.m.
n
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
The phrase "therapeutically effective reduction" when used herein is meant to
signify
that serum cholesterol levels are reduced by approximately the same amount as
an immediate
release reference standard (e.g., Mevacor~ ) or more, when the controlled
release dosage form
is orally administered to a human patient on a once-a-day basis.
BRIEF DESCRIPTION OF THE DRAWINGS
FIG.1 is a graph of in vitro dissolution data which shows the dissolution
profile of the
formulation of Example 1 in 2% sodium lauryl sulfate at pH 7.0 in NaHZP04
buffer in a USP
XXII Type II dissolution apparatus at 50 rpm at 37°C.
FIG. 2 is a graph of in vitro dissolution data which shows the dissolution
profiles of
the formulations of Examples 2, 3 and 4 in 2% sodium lauryl sulfate at pH 7.0
in NaHZP04
buffer in a USP XXII Type II dissolution apparatus at 50 rpm at 37°C.
FIG. 3 is a graph of the in-vitro dissolution data which shows the dissolution
profiles
of Examples 2 and 5-7 under similar conditions as set forth above with respect
to Fig. 2.
FIG. 4 is a graph of the in-vitro dissolution data which shows the dissolution
profiles
of Examples 8 under similar conditions as set forth above with respect to Fig.
3.
FIG. 5 is a graph of the in-vitro dissolution data which shows the dissolution
profiles
of Examples 9 under similar conditions as set forth above with respect to Fig.
3.
FIG. 6 is a graph of comparative data which shows mean (~SD) plasma
concentration
time profiles of lovastatin in healthy subjects (n=8) following a single oral
dose of a
conventional immediate release dose of 40mg of lovastatin and an extended
release 40mg
dose of lovastatin according to the invention (Example 5).
12
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
FIG. 7 is a graph of comparative data which shows mean (~SD) plasma
concentration
time profiles of lovastatin acid in healthy subjects (n=8) following a single
oral dose of a
conventional immediate release dose of 40mg of lovastatin and an extended
release 40mg
dose of lovastatin according to the invention (Example 5).
FIG. 8 is a graph of comparative data which shows mean (~SD) plasma
concentration
time profiles of lovastatin in healthy subjects (n=9) following a single oral
dose in the
morning of a conventional immediate release dose of 40mg of lovastatin with
breakfast and
an extended release 40mg dose of lovastatin according to the invention
(Example 5) with and
without breakfast.
FIG. 9 is a graph of comparative data which shows mean (~SD) plasma
concentration
time profiles of lovastatin acid in healthy subjects (n=9) following a single
oral dose in the
morning of a conventional immediate release dose of 40mg of lovastatin with
breakfast and
an extended release 40mg dose of lovastatin according to the invention
(Example 5) with and
without breakfast.
FIG. 10 is a graph of the mean plasma concentration-time profiles of
lovastatin and
lovastatin acid in patients (n=12) after multiple-dose administration of 40mg
Lovastatin XL
and a conventional 40mg immediate release dose of lovastatin (Study No. 5, Day
1 and Day
28).
FIG. 11 is a graph of the mean plasma concentration-time profiles of total and
active
inhibitors of HMG-CoA Reductase in patients (n=12) after multiple-dose
administration of
40mg Lovastatin XL and a conventional 40mg immediate release dose of
lovastatin (Study
No. 5, Day 1 and Day 28).
FIG. 12 is a graph of a regression line depicting CmaX plotted against dose
for Study
No. 5.
13
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
FIG. 13 is a graph of a regression line depicting AUCo_ash~ plotted against
dose for
Study No. 5.
DETAILED DESCRIPTION OF THE INVENTION
The controlled release dosage form is preferably prepared by combining
mevastatin,
pravastatin, simvastatin or lovastatin with a pharmaceutically acceptable,
water swellable
polymer and an osmotic agent into a compressed tablet core having an optional
first coating
for sealing and protection and a second coating comprising a pH sensitive
agent water
insoluble polymer. Mevastatin, pravastatin, simvastatin and lovastatin are
well known
compounds that are described in the prior art including the particular patents
which have been
cited herein. It is also within the scope of the invention to use mixtures of
different alkyl
esters of hydroxy substituted naphthalenes.
Specifically, a pharmaceutically acceptable, water swellable polymer and an
osmotic
agent are combined with the drug which may be micronized or unmicronized or
amorphous
or crystalline and compressed to form the tablet core. The osmotic agent is
any nontoxic
pharmaceutically acceptable water soluble compound which will dissolve
sufficiently in
water and increase the osmotic pressure inside the core of the tablet. The
osmotic agents
include the simple sugars and salts such as sodium chloride, potassium
chloride, magnesium
sulfate, magnesium sulfate, magnesium chloride, sodium sulfate, lithium
sulfate, urea,
inositol, sucrose, lactose, glucose, sorbitol, fructose, mannitol, dextrose,
magnesium
succinate, potassium acid phosphate and the like. The preferred osmotic agent
for the tablet
core is a simple sugar such as anhydrous lactose in the range of 0-50% by
weight, based on
the weight of the compressed, uncoated tablet.
The pharmaceutically acceptable, water swellable polymer may be any
pharmaceutically acceptable polymer which swells and expands in the presence
of water to
slowly release the lovastatin. These polymers include polyethylene oxide,
methylcellulose,
hydroxypropylcellulose, hydroxypropylmethylcellulose and the like. In a
preferred
14
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
embodiment, the water swellable polymer will be polyethylene oxide (obtained
from Union
Carbide Corporation under the trade name Polyox WSR Coagulant or Polyox WSR N
80)
These materials form a viscous gel in water or other solvent system at a
sufficient
concentration to control the release of the lovastatin. This will generally
require a
concentration of the pharmaceutically acceptable, water swellable polymer of
about 0-50% by
weight of the compressed, uncoated tablet.
Binder may be employed in a sufficient amount so that when it is combined with
a
suitable solvent, mixed with the water soluble osmotic agent and agitated,
granules will be
formed which may be compressed into a tablet core. Prior to compressing the
granules, the
conventional solid pharmaceutical diluents such as microcrystalline cellulose,
lactose,
dextrose and the like may be added to the granule forming mixture in amounts
from about 0
to 51 % weight based on the weight of the compressed, uncoated tablet. In the
present case,
the above mentioned osmotic agent, lactose, may function as a binder in the
tablet
compression step.
In the preparation of the tablets of the invention, various solvents may be
used to
prepare the granules. In addition, various other diluents, excipients,
lubricants, dyes,
pigments, dispersants, emulsifiers, etc. may be used to optimize the
formulations of the
invention.
Additionally, a surfactant is used in the preferred embodiment. The surfactant
may be
any ionic or non-ionic water soluble surfactant which may be employed in the
range of 0-50%
by weight or preferably 1-5% by weight. The preferred surfactant for the
present formulation
is sodium lauryl sulfate but other surfactants such as polysorbate 20, 60 or
80; polyoxl
stearate and the like.
Furthermore, the preferred embodiment may comprise a lubricant. Ideally, the
lubricant will be in the range of 0.5 to 2.5% by weight of the compressed,
uncoated tablet.
15
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
After the above described tablet~ore is formed, it is coated with: 1) an
optional
protective first coating on the tablet core and/or an optional pH sensitive
coating; 2) an outer
coating comprising a pH sensitive agent and a water insoluble polymer.
Specifically, a protective first coating may be used at a level in the range
of 0-10% by
weight which may be applied from a coating system such as opadry Clear sold by
Colorcon
Corporation. In an especially preferred embodiment, the Opadry Clear will be
2.83% by
weight and will be combined with an osmotic agent in the range of 0-10% by
weight. While
the osmotic agent may be any salt, low molecular weight molecule or water
soluble polymers,
the preferred agent is sodium chloride. The osmotic agent is added to the
coating system
when the coating system is being dispersed into purified water. The coating
system which
contains the osmotic agent may then be sprayed onto the tablets to form a
protective coating
layer. As mentioned above, this protective first coating is optional.
1 S An optional inner or over coat over the outer coat may also be applied
which
comprises a pH sensitive polymer which functions as an enteric polymer in that
it does not
begin to dissolve until pH conditions in excess of the stomach region are
encountered.
Generally, the pH sensitive materials do not dissolve and begin to release the
active drug until
a pH above 3.0 and preferably above 5.5. Materials such as such as Eudragit L
(copolymer of
poly(methacrylic acid, methylmethacrylate), 1:1 ratio; MW (No. Av. 135,000 USP
Type A)
or Eudragit S (copolymer of poly(methacrylic acid, methylmethacrylate, 1:2
ratio MW (No.
Av. 135,000 - USP Type B) hydroxypropyl methyl cellulose phthalate, cellulose
acetate
phthalate, polyvinyl acetate phthalate and the like may be used in the range
of 0-30% by
weight and preferably 2 to 4 percent by weight of the combined weight of the
compressed,
uncoated tablet and the inner coating of the pH sensitive polymer.
The outer coating comprises a pH sensitive polymer which functions as an
enteric
polymer in that it does not begin to dissolve until pH conditions in excess of
the pH of the
stomach region are encountered and a water insoluble polymer which provide
controlled
release properties to the coating formulation. The pH sensitive polymer is the
same type of
16
CA 02390301 2002-05-07
WO 01/34123 PCTNS00/30415
material that is described above as the 6ptional inner coating layer. The
water insoluble
polymer may be a cellulosic polymer such as ethylcellulose, cellulose acylate,
cellulose
mono-, di- or triacetate. The pH sensitive polymer and the insoluble
cellulosic polymer are
used at a weight ratio of about 0.1:1 to 0.75:1, preferably 0.25:1 to 0.5:1 of
pH sensitive
polymer to water insoluble cellulosic polymer. A combined coating weight of
about 0.5-5%
by weight and preferably 1 to 4% by weight and especially preferred is 1 to 3%
by weight of
the gained weight based on the weight of the coated tablet core. Cellulose
acetate is the
preferred water insoluble polymer and the outer coating is preferably applied
as a suspension
in acetone.
Furthermore, a plasticizer or combination of plasticizers may be added to the
inner,
outer or over coating to provide elasticity and shape to the coating. while
the plasticizer or
combination of plasticizers may be any water soluble or water insoluble
formulation in the
range of 0-10% by weight and preferably 0.5 to 5% by weight of the outer
coating
composition. Acetyltributyl citrate is the preferred plasticizer but materials
such as acetyl
triethyl citrate, dibutyl phthalate, triacetin, diethyl phthalate,
polyethylene glycol, propylene
glycol and the like may be utilized.
An antioxidant such as BHA or BHT may be added to the tablet core as a
stabilizer at
a level of 0.001 to 0.01 % by weight of the tablet core.
Lastly, a channelling agent is mixed with the aforementioned components of the
outer
coating. A channelling agent may be employed to increase the porosity of the
film coating in
order- to increase the amount of the fluids that penetrate the tablet core and
increase the rate
of hydration. This allows the release of the lovastatin after the outer film
coat ruptures.
Generally, channelling agents may be any salts, surfactants, or short-chain
water soluble
polymers in a water channel forming effective amount i.e. 1 to S% by weight,
based on the
total weight of the core and all coating components. The channeling agents
include any
pharmaceutically acceptable water soluble salt, surfactant, or short chain
water soluble
17
CA 02390301 2002-05-07
WO 01/34123 PCT1US00/30415
polymer such as sodium chloride, potassium chloride, sucrose, polysorbate-80,
hydroxypropyl
cellulose, hydroxyethyl cellulose and the like.
Also, the preferred embodiment of the inner or over coating is supplied with
an anti-
sticking agent such as talc to overcome any tablet to tablet stickiness during
the coating
process. The amount of anti-sticking agent is an amount which prevents
sticking which may
be in the range of 0-6% by weight based on the weight of the tablets and the
coating materials
on a dry weight basis.
Although the applicants do not wish to be bound by any theory by which the
invention
operates, it is believed that the tablets of the invention release the
lovastatin by osmotic
pressure. Water is drawn into the tablet and it expands to the point where the
outer coating
fails in one particular area to form a constricted opening which releases the
internal contents
of the tablet which contain the drug. Thereafter, the aqueous medium of the
tablet shell will
continue to release the drug as it dissolves until the osmotic pressure inside
the tablet shell
equals that of the surrounding environment. At the late stages of the in vivo
release of
lovastatin, it is believed that the tablet shell will collapse and/or
disintegrate completely to
substantially completely release the remaining drug. The water insoluble
coating is not
absorbed in the gastrointestinal tract and is eliminated in the feces.
The tablets of the invention may be made in a smooth faced tablet die.
Thereafter the
tablet is provided with the outer coating which, because of surface tension,
will result in a
thinner coating layer over the corners of the tablet which will provide an
area in the outer
coating which will form a channel to is allow intestinal fluid to reach the
core of the tablet.
18
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
In certain preferred embodiments, the tablets of the invention will have the
following
general formula:
TABLE 1
INGREDIENTS POSSIBLE RANGE, wt%
Tablet Core
Alkyl ester of a substituted naphthalene3-20
Water Swellable Polymer 10-40
Antioxidant 0.001-0.01
Osmotic Agents 20-80
Surfactant 0-5
Lubricant 0-5
Coatings:
Seal Coating 0-10
Osmotic Agents 0-10
Inner Coating
Enteric Polymer 0-30
Anti-sticking Agent 0-6
Plasticizer 0-6
Channelling Agents 0-6
Outer Coating
Blend of Enteric Polymer and Water-insoluble0.5-5
Polymer
Plasticizer(s) 0-1
Channelling Agents 0.2.5
Overcoat
Enteric Polymer 0-30
Anti-sticking Agent 0-6
Plasticizer 0-6
Channelling Agents 0-6
TOTAL 100
Other controlled release technologies known to those skilled in the art can be
used in
order to achieve the controlled release formulations of the present invention,
i.e.,
formulations which provide a mean Tmax of the drug (i.e., a HMG-CoA reductase
inhibitor)
at the desired time after oral administration, e.g., in general, at about 10
to about 32 hours
19
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
after oral administration to a population of human patients, and which
preferably provide
other pharmacokinetic parameters described herein when orally administered to
human
patients. Such formulations can be manufactured as a controlled oral
formulation in a
suitable tablet or multiparticulate formulation known to those skilled in the
art. In either case,
the controlled release dosage form may optionally include a controlled release
carrier which
is incorporated into a matrix along with the drug (e.g., HMG-COA reductase
inhibitors), or
which is applied as a controlled release coating.
An oral dosage form according to the invention may be provided as, for
example,
granules, spheroids, beads, pellets (hereinafter collectively referred to as
"multiparticulates")
and/or particles. An amount of the multiparticulates which is effective to
provide the desired
dose of opioid over time may be placed in a capsule or may be incorporated in
any other suit-
able oral solid form. In one preferred embodiment of the present invention,
the controlled
release dosage form comprises such particles containing or comprising the
active ingredient,
wherein the particles have diameter from about 0.1 mm to about 2.5 mm,
preferably from
about 0.5 mm to about 2 mm.
Examples of suitable multiparticulate formulations are those in which the
particles
comprise inert beads which are coated with the drug. Thereafter, a coating
comprising the
controlled release Garner is applied onto the beads. Alternatively, a
spheronizing agent,
together with the drug can be spheronized to form spheroids. Microcrystalline
cellulose is
preferred. A suitable microcrystalline cellulose is, for example, the material
sold as Avicel
PH 101 (Trade Mark, FMC Corporation). In such embodiments, in addition to drug
and
spheronizing agent, the spheroids may also contain a binder. Suitable binders,
such as low
viscosity, water soluble polymers, will be well known to those skilled in the
pharmaceutical
art. However, water soluble hydroxy lower alkyl cellulose, such as
hydroxypropylcellulose,
are preferred. Additionally (or alternatively) the spheroids may contain a
water insoluble
polymer, especially an acrylic polymer, an acrylic copolymer, such as a
methacrylic acid-ethyl
acrylate copolymer, or ethyl cellulose.
20
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
In certain embodiments, the particles comprise normal release matrixes
containing the
drug. These particles are then coated with the controlled release carrier.
The controlled release coatings useful in the formulations of the present
invention
should be capable of producing a strong, continuous film that is smooth and
elegant, capable
of supporting pigments and other coating additives, non-toxic, inert, and tack-
free. In one
embodiment, coatings are provided to permit either pH-dependent or pH-
independent release,
e.g., when exposed to gastrointestinal fluid. A pH-dependent coating serves to
release the
opioid in desired areas of the gastro-intestinal (GI) tract, e.g., the stomach
or small intestine,
such that an absorption profile is provided which is capable of providing at
least about twelve
hour and preferably up to twenty-four hour analgesia to a patient. When a pH-
independent
coating is desired, the coating is designed to achieve optimal release
regardless of pH-
changes in the environmental fluid, e.g., the GI tract. It is also possible to
formulate
compositions which release a portion of the dose in one desired area of the GI
tract, e.g., the
stomach, and release the remainder of the dose in another area of the GI
tract, e.g., the small
intestine.
Formulations according to the invention that utilize pH-dependent coatings to
obtain
formulations may also impart a repeat-action effect whereby unprotected drug
is coated over
the enteric coat and is released in the stomach, while the remainder, being
protected by the
enteric coating, is released further down the gastrointestinal tract. Coatings
which are pH-
dependent may be used in accordance with the present invention include
shellac, cellulose
acetate phthalate (CAP), polyvinyl acetate phthalate (PVAP),
hydroxypropylmethylcellulose
phthalate, and methacrylic acid ester copolymers, and the like.
In certain preferred embodiments, the tablet core or multiparticulates
containing the
drug are coated with a hydrophobic material selected from (i) an
alkylcellulose; (ii) an acrylic
polymer; or (iii) mixtures thereof. The coating may be applied in the form of
an organic or
aqueous solution or dispersion. The coating may be applied to obtain a weight
gain from
about 2 to about 25% of the substrate in order to obtain a desired sustained
release profile.
21
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
Such formulations are described, e.g., in detail in U.S. Patent Nos. 5,273,760
and 5,286,493,
hereby incorporated by reference. Other examples of sustained release
formulations and
coatings which may be used in accordance with the present invention include
U.S. Patent
Nos. 5,324,351; 5,356,467, and 5,472,712, hereby incorporated by reference in
their entirety.
The sustained release coatings of the present invention may also include an
exit means
comprising at least one passageway, orifice, or the like. The passageway may
be formed by
such methods as those disclosed in U.S. Patent Nos. 3,845,770; 3,916,889;
4,063,064; and
4,088,864 (all of which are hereby incorporated by reference). The passageway
can have any
shape such as round, triangular, square, elliptical, irregular, etc.
In other embodiments of the present invention, the desired controlled release
of the
formulation is achieved via a matrix (either normal or controlled release)
having a controlled
release coating as set forth above.
1 S The present invention may also utilize a controlled release matrix that
affords in-vitro
dissolution rates of the drug within the preferred ranges and that releases
the drug in a pH-
dependent or pH-independent manner. The materials suitable for inclusion in a
controlled
release matrix will depend on the method used to form the matrix. The
controlled release
material which may be included in a matrix in addition to the drug includes
hydrophilic
and/or hydrophobic materials, such as gums, alkylcelluloses, cellulose ethers,
acrylic resins,
and protein derived materials. This list is not meant to be exclusive, and any
pharmaceutically acceptable hydrophobic material or hydrophilic material which
is capable of
imparting controlled release of the active agent may be used in accordance
with the present
invention.
The hydrophobic material is preferably selected from the group consisting of
alkylcelluloses, cellulose ethers, acrylic and methacrylic acid polymers and
copolymers,
hydrogenated castor oil, hydrogenated vegetable oil, or mixtures thereof. In
certain preferred
embodiments of the present invention, the hydrophobic material is a
pharmaceutically
acceptable acrylic polymer, including but not limited to acrylic acid and
methacrylic acid
22
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
copolymers, methyl methacrylate, methyl methacrylate copolymers, ethoxyethyl
methacrylates, cynaoethyl methacrylate, aminoalkyl methacrylate copolymer,
poly(acrylic
acid), poly(methacrylic acid), methacrylic acid alkylamine copolymer,
poly(methyl
methacrylate), poly(methacrylic acid)(anhydride), polymethacrylate,
polyacrylamide,
poly(methacrylic acid anhydride), and glycidyl methacrylate copolymers. In
other
embodiments, the hydrophobic material is selected from materials such as
ethylcellulose.
In yet other alternative embodiments, a spheronizing agent, together with the
active
ingredient can be spheronized to form spheroids. Microcrystalline cellulose is
preferred. A
suitable microcrystalline cellulose is, for example, the material sold as
Avicel PH 101 (Trade
Mark, FMC Corporation). In such embodiments, in addition to the active
ingredient and
spheronizing agent, the spheroids may also contain a binder. Suitable binders,
such as low
viscosity, water soluble polymers, will be well known to those skilled in the
pharmaceutical
art. However, water soluble hydroxyalkylcelluloses (such as
hydroxypropylcellulose) are pre-
ferred. Additionally (or alternatively) the spheroids may contain a water
insoluble polymer,
especially an acrylic polymer, an acrylic copolymer, such as a methacrylic
acid-ethyl acrylate
copolymer, or ethyl cellulose.
In addition to the above ingredients, a controlled release matrix may also
contain
suitable quantities of other materials, e.g. diluents, lubricants, binders,
granulating aids,
colorants, flavorants and glidants that are conventional in the pharmaceutical
art. The
quantities of these additional materials will be sufficient to provide the
desired effect to the
desired formulation. Specific examples of pharmaceutically acceptable Garners
and
excipients that may be used to formulate oral dosage forms are described in
the Handbook of
Pharmaceutical Excipients, American Pharmaceutical Association (1986),
incorporated by
reference herein.
If necessary to achieve the desired plasma curve (e.g., Tmax), a portion of
the drug
may be included in the formulations of the invention in immediate release
form. For
example, the drug may be included in separate normal release matrix particles,
or may be co-
23
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
administered in a different immediate release composition which is either
enveloped within a
gelatin capsule or is administered separately. In other embodiments, the
particles comprise
inert beads which are coated with the drug. Thereafter, a coating comprising
the controlled
release carrier is applied onto the beads, and then an immediate release drug
layer is applied
on top of the controlled release coating as an overcoat. Alternatively, in
controlled release
tablet formulations in which the controlled release Garner is included in a
matrix with the
drug or the controlled release Garner is applied in a coating on the surface
of the tablet, a
portion of the drug may be applied on the surface of the tablet as an
immediate release drug
layer (as an overcoat if the tablet has a controlled release coating).
Further specific controlled release technologies which may be used in
conjunction
with the present invention include the Assignee's U.S. Patent Nos. 5,837,379;
5,34,023;
5,830,503; 5,736,159; 5,728,402; 5,654,005; 5,567,441; 5,558,879; 5,532,275;
5,508,040;
5,472,708; 5,458,888; 5,458,887; and 5,419,917, all of which are hereby
incorporated by
reference.
DESCRIPTION OF THE PREFERRED EMBODIMENTS
The following examples illustrate various aspects of the present invention.
They are
not to be construed to limit the claims in any manner whatsoever.
EXAMPLE 1
A tablet having the following formula was prepared:
lovastatin 11.99wt% 40.Omg
Polyox WSR Coagulant, NF* 4.50wt% lS.Omg
Polyox WSR N 80, NF** 17.98wt% 60.Omg
lactose (anhydrous) 50.65wt% 169.Omg
sodium lauryl sulfate 3.OOwt% lO.Omg
silicon dioxide Fumed USP/NF 0.45wt% l.5mg
24
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
Myvaplex 600P*** - 1.80wt% 6.Omg
* polyethylene oxide Mw No av 5,000,000
** polyethylene oxide Mw No av 200,000
*** glyceryl monostearate
Seal Coating:
Opadry Clear* * * * 2. 81 wt% 9.4mg
sodium chloride 0.93wt% 3.lmg
**** mixture containing hydroxypropyl methyl cellulose and polyethylene glycol
Inner Coating:
hydroxypropylmethylcell. phtha1.55 2.27wt% 7.58mg
talc 0.78wt% 2.6mg
acetyl tributyl citrate 0.22wt% 0.75mg
sugar, confectioners 6X micronized 0.62wt% 2.08mg
Outer Coating:
cellulose acetate 1.00wt% 3.32mg
Eudragit S 100, 0.34wt% 1.13mg
Triacetin 0.08wt% 0.27mg
polyethylene glycol 400 0.08wt% 0.27mg
sugar, confectioners 6X micronized 0.50wt% 1.66m~
1 OO.Owt%333.66mg
' Eudragit S 100 (poly(methacrylic acid,
methylmethacrylate, 1:2 ratio MW (No.
Av.
135,000 - USP Type B)
The following describes the process of making the above described dosage form:
STEP 1, THE TABLET CORE
(a) Granulation
25
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
1. Pass Polyox WSR N80, sodiurrrlauryl sulfate and anhydrous lactose through a
30 mesh
stainless steel screen.
2. Charge the screened materials and lovastatin (micronized) into a vertical
granulator.
3. Dissolve butylated hydroxy anisole in ethanol.
4. Mix ethanol, and purified water.
5. Pre-mix the powder mixture for 5 minutes.
6. Blend the powder mixture again, add the butylated hydroxyanisole solution
and then
the ethanol/water mixture.
7. Dry the granules at 45-50°C until the moisture content is lower than
l.8wt%.
8. Pass the granules through a 1575 mesh using a Comil.
Tabletting
1. Mix Cab-O-Sil and Polyox WSR N80.
2. Pass the mixture of Cab-o-Sil and Polyox WSR N80 through a 24 mesh
stainless steel
screen with the Polyox WSR Coagulant.
3. Blend the screen materials with lovastatin granules for 15 minutes.
4. Pass Myvaplex through a 30 mesh stainless steel screen and combine with the
other
screen materials.
5. Blend for five minutes.
6. Compress the blend into tablets (300mg, round, standard concave, 11/32")
which
contain 40mg of lovastatin.
Seal Coating: Opadry Clear
1. Dissolve sodium chloride in purified water.
2. Disperse Opadry Clear into the sodium chloride solution.
3. Spray lovastatin tablets with the aqueous coating suspension using a
c6ater.
Inner Coating: Hydroxypropyl methylcellulose phthalate 55
26
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
1. Dissolve hydroxypropyl methylcellulose phthalate SS in acetone using a
homogenizer.
2. Add acetyl tributyl citrate to the acetone solution and mix it with a
homogenizer until
a homogenized dispersion is obtained.
3. Add talc and sugar to the solution and mix it with a homogenizer until a
homogenized
S dispersion is obtained.
4. Replace the homogenizer with a magnetic mixer and stir the coating mixture
throughout the coating process.
5. Spray the Opadry Clear coated lovastatin tablets with the coating
dispersion in a
coater.
Outer Coating: cellulose acetate
1. Dissolve cellulose acetate and Eudragit S 100 in acetone using a
homogenizer.
2. Add polyethylene glycol 400, triactein and sugar to the solution and mix
until a
homogeneous dispersion is obtained.
3. Spray the coating suspension onto the tablets in a water.
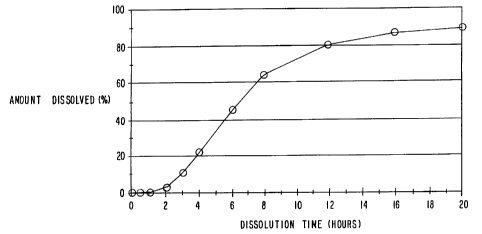
Release in the above described manner will result in the dissolution profile
shown in
FIG. l, which is a graph of in vitro dissolution data which shows the
dissolution profile of the
formulation of Example 1 in 2% sodium lauryl sulfate at pH 7.0 in NaH2P04
buffer in a USP
XXII Type II dissolution apparatus at 50 rpm at 37°C.
It is believed that administration of the above described micronized
Lovastatin in
these amounts will be particularly effective in inhibiting the biosynthesis of
cholesterol in the
liver through interruption of HMG coenzyme A reductase. The dosage of
lovastatin should
be individualized depending on the desired and/or degree of serum cholesterol
that is desired.
Generally 10 to 80mg of lovastatin per day should be administered by mouth
depending on
the response and the degree of reduction in serum cholesterol level that is
indicated.
27
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
EXAMPLE 2
A tablet having the following formula was prepared:
lovastatin l2.llwt% 40.Omg
Polyox WSR Coagulant, NF* 4.54wt% lS.Omg
Polyox WSR N 80, NF** 17.71wt% 58.Smg
lactose (anhydrous) 51.13wt% 168.9mg
sodium lauryl sulfate 3.03t% lO.Omg
silicon dioxide Fumed USP/NF 0.45wt% l .5mg
butylated hydroxy anisole 0.03wt% 0.1
Omg
Myvaplex 600P*** 1.82wt% 6.Omg
* polyethylene oxide Mw No av 5,000,000
** polyethylene oxide Mw No av 200,000
*** glyceryl monostearate
Seal Coating:
Opadry Clear**** 2.85wt% 9.4mg
sodium chloride 0.94wt% 3.lmg
**** mixture containing hydroxypropyl methyl cellulose and polyethylene glycol
Inner Coating:
hydroxypropylmethylcell. phtha1.55 2.29wt% 7.58mg
talc 0.79wt% 2.6mg
acetyl tributyl citrate 0.23wt% 0.75mg
sugar, confectioners 6X micronized 0.08wt% 0.27mg
28
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
Outer Coating: -
cellulose acetate l.OOwt% 3.32mg
Eudragit S 100 0.34wt% 1.13mg
triacetin 0.08wt% 0.27mg
polyethylene glycol 400 0.08wt% 0.27mg
sugar, confectioners 6X micronized O.SOwt% 1.66mg
100.Owt% 330.35mg
' Eudragit S 100 (poly(methacrylicacid,
methylmethacrylate, 1:2 ratio MW
(No. Av. 135,000
- USP Type B)
Coated tablets were prepared using the general procedure of Example 1, except
that an
indentation was made on the tablet surface.
EXAMPLE 3
A tablet having the following formula was prepared:
lovastatin 12.14wt% 20.Omg
Polyox WSR Coagulant, NF* 4.SSwt% 7.Smg
Polyox WSR N 80, NF** 17.76wt% 29.25mg
lactose (anhydrous) 51.30wt% 84.Smg
sodium lauryl sulfate 3.04wt% S.Omg
silicon dioxide Fumed USP/NF 0.46wt% 0.75mg
butylated hydroxy anisole 0.03wt% O.OSmg
Myvaplex 600P*** 1.82wt% 3.Omg
* polyethylene oxide Mw No av 5,000,000
** polyethylene oxide Mw No av 200,000
*** glyceryl monostearate
29
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
Seal Coating: -
Opadry Clear**** 3.42wt% 5.63mg
sodium chloride 1.14wt% 1.88mg
**** mixture containing hydroxypropyl methyl cellulose and polyethylene glycol
Outer Coating:
cellulose acetate 1.43wt% 2.36mg
Eudragit S 100' 0.49wt% 0.80mg
triacetin O.llwt% 0.19mg
polyethylene glycol 400 O.llwt% 0.19mg
sugar, confectioners 6X micronized 0.72wt% 1.18mg
Overcoat:
hydroxypropylmethylcell. phtha1.55 0.77wt% 1.27mg
talc 0.30wt% 0.49mg
triacetin 0.12wt% 0.20mg
sugar, confectioners 6X micronized0.30wt% 0.49mg
100.Owt% 146.73mg
' Eudragit S 100 (poly(methacrylic acid, methylmethacrylate, 1:2 ratio MW (No.
Av.
135,000 - USP Type B)
The following describes the process of making the above described dosage form:
STEP 1, THE TABLET CORE
(a) Granulation
30
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
1. Pass Polyox WSR N80, sodium tauryl sulfate and anhydrous lactose through a
30
mesh stainless steel screen.
2. Charge the screened materials and lovastatin (micronized) into a vertical
granulator.
3. Dissolve butylated hydroxy anisole in ethanol.
4. Mix ethanol and purified water.
5. Pre-mix the powder mixture for 5 minutes.
6. Blend the powder mixture again, add the butylated hydroxyanisole solution
and then
the ethanol/water mixture.
7. Dry the granules at 45-50°C until the moisture content is lower than
l.8wt%.
8. Pass the granules through a 1575 mesh using a Comil.
Tabletting
1. Mix Cab-O-Sil and Polyox WSR NBO.
2. Pass the mixture of Cab-O-Sil and Polyox WSR N80 through a 24 mesh
stainless
steel screen with the Polyox WSR Coagulant'.
3. Blend the screen materials with lovastatin granules for 15 minutes.
4. Pass Myvaplex through a 30 mesh stainless steel screen and combine with the
other
screen materials.
5. Blend for five minutes.
6. Compress the blend into tablets (164.72mg, round, standard concave, 17/6411
dia.)
which contain 20mg of lovastatin.
Seal Coating: Opadry Clear
1. Dissolve sodium chloride in purified water.
2. Disperse Opadry Clear into the sodium chloride solution.
3. Spray lovastatin tablets with the aqueous coating suspension using a
coater.
Inner Coating: None
31
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
Outer Coating: cellulose acetate -
1. Dissolve cellulose acetate and Eudragit S 100 in acetone using a
homogenizer.
2. Add polyethylene glycol 400, triactein and sugar to the solution and mix
until a
homogeneous dispersion is obtained.
3. Spray the coating suspension onto the tablets in a coater.
Overcoating: Hydroxypropyl methylcellulose phthalate 55
1. Dissolve hydroxypropyl methylcellulose phthalate 55 in acetone using a
homogenizer.
2. Add acetyl tributyl citrate to the acetone solution and mix it with a
homogenizer until
a homogenized dispersion is obtained.
3. Add talc and sugar to the solution and mix it with a homogenizer until a
homogenized
dispersion is obtained.
4. Replace the homogenizer with a magnetic mixer and stir the coating mixture
throughout the coating process.
5. Spray the Opadry Clear coated lovastatin tablets with the coating
dispersion in a
coater.
EXAMPLE 4
A tablet having the following formula was prepared:
lovastatin 12.20wt% 20.Omg
Polyox WSR Coagulant, NF* 4.57wt% 7.Smg
Polyox WSR N 80, NF** 17.84wt% 29.25mg
lactose (anhydrous) 51.53wt% 84.Smg
sodium lauryl sulfate 3.OSwt% S.Omg
silicon dioxide Fumed USP/NF 0.46% 0.75mg
butylated hydroxy anisole 0.03wt% O.OSmg
Myvaplex 600P*** 1.83wt% 3.Omg
32
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
* polyethylene oxide Mw No av S,OOO,OflO
** polyethylene oxide Mw No av 200,000
*** glyceryl monostearate
Seal Coating:
Opadry Clear**** 3.43wt% 5.63mg
sodium chloride 1.15wt% 1.88mg
**** mixture containing hydroxypropyl methyl cellulose and polyethylene glycol
Inner Coating: None
Outer Coating:
cellulose acetate 1.96wt% 3.21mg
Eudragit S 100' 0.66wt% ~ 1.09mg
acetyl tributyl citrate 0.32wt% 0.52mg
sugar, confectioners 6X micronized 0.98wt% 1.61mg
100.00wt% 163.99mg
Coated tablets were prepared using the general procedure of Example 3 except
that no inner
coating was applied and an outer enteric coating was applied as an overcoat
over the outer
layer.
A comparison of Examples 2, 3 and 4 shows that the following was the weight of
the
coatings that were applied:
Example 2 Inner Coating 4wt%
Outer Coating 2wt%
Over Coating 0%
Example 3 Inner Coating 0%
33
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
Outer Coating 3wt%
Over Coating 2.5%
Example 4 Inner Coating Owt%
Outer Coating 4wt%
Over Coating Owt%
FIG. 2 is a graph of in vitro dissolution data which shows the dissolution
profiles of
the formulations of Examples 2, 3 and 4 in 2% sodium lauryl sulfate at pH 7.0
in NaH2P04
buffer in a USP XXII Type II dissolution apparatus at 50 rpm at 37°C.
It is believed that administration of the above described micronized
Lovastatin in
these amounts will be particularly effective in inhibiting the biosynthesis of
cholesterol in the
liver through interruption of HMG coenzyme A reductase. The dosage of
lovastatin should
be individualized depending on the desired and/or degree of serum cholesterol
that is desired.
Generally 10 to 80mg of lovastatin per day should be administered by mouth
depending on
the response and the degree of reduction in serum cholesterol level that is
indicated.
EXAMPLES 5-7
In Examples 5-7, 40mg lovastatin tablets were prepared using the general
procedure
of Example 1. The ingredients of Examples 5-7 are set forth in Table 2 below.
TABLE 2
Summary of Lovastatin
Formulations
Weight Percent
Ingredient
Example 5 Example 6 Example 7
Lovastatin (strength, 40 40 40
mg)
34
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
Tablet Core
1. Lovastatin 12.11 12.28 12.28
2. Lactose (Anhydrous) 51.13 51.8 51.8
3. Polyoxo WSR Coagulant 4.54 4.6 4.6
4. Polyoxo WSR N80 17.71 17.94 17.94
5. Sodium Lauryl Sulfate 3.03 3.06 3.06
6. Glyceryl Monostearate 1.82 1.84 1.84
7. Silicon Dioxide 0.45 0.46 0.46
8. Butylated Hydroxyanisole0.03 0.02 0.02
Seal Coat
1. Opadry Clear 2.85 2.88 2.88
2. Sodium Chloride Powder 0.94 0.96 0.96
Inner Coat
1. HPMCP 55 2.29 1.61 1.61
2. Talc, USP 0.79 0.55 0.55
3. Acetyltributyl Citrate 0.23 0.16 0.16
4. Sugar, Micronized 0:64 0.44 0.44
Outer Coat
1. Cellulose Acetate 1 0.7 0.7
2. Eudragit S 100 0.34 0.24 0.24
3. Triacetin 0.08 0.06 0.06
4. Polyethylene Glycol 0.08 0.6 0.6
400
5. Acetyltributyl Citrate - - -
6. Sugar, Micronized 0.5 0.35 0.35
Overcoat
1. HPMCP 55 - - -
2. Talc, USP - - -
3. Triacetin - - -
4. Su ar, Micronized - - -
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
5. Opadry Yellow - - - -
6. Opadry Pink - - -
Total Tablet Weight, % 100 100 100
S
As can be ascertained from Table 2, Example 5 has the same composition as
Example
2. However, the tablets of Example 5 do not contain the indentation in the
tablet surface that
was present in Example 2. Preliminary results of a single-dose study in
healthy humans
indicated that the indentation in the lovastatin formulation of Example 2 did
not improve its
pharmacokinetics. Because Example S provided the desired safety, efficacy and
pharmacokinetic profiles in Studies 1-3 described in detail herein, two more
clinical lots were
made based on the composition and process of Example 5. Examples 6 and 7 are
the two
clinical lots made based on the composition of Example 5. A different coater
(Glatt coater
GPCG3) was used for the manufacturing of these two lots. Since the new coater
had a better
coating efficiency, less coating mixtures was required to achieve the same
dissolution as that
of Example 5. Considering the similarity of the dissolution profiles and the
identical coating
mixture composition, the "actual composition" of Examples 6 and 7 should be
very similar to
that of Example 5.
EXAMPLES 8 - 9
In Example 8, 20mg lovastatin tablets were prepared. In Example 9, l Omg
lovastatin
tablets were prepared. Both formulations were prepared using the general
procedure of
Example 1.
The composition of Example 9 is the same as Example 8, except that l Omg of
lactose
has been used to replace l Omg of drug substance (lovastatin).
The ingredients of Examples 8-9 are set forth in Table 3 below:
36
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
TABLE 3
Summary of Lovastatin Formulations
Weight Percent
Ingredients
Example 8 Example 9
Lovastatin (strength, mg) 20 10
Tablet Core
1. Lovastatin 11.69 5.84
2. Lactose (Anhydrous) 49.32 55.18
3. Polyox~ WSR Coagulant 4.38 4.38
4. Polyoxo WSR N80 18.08 17.09
S. Sodium Lauryl Sulfate 2.92 2.92
6. Blyceryl Monostearate 1.75 1.75
7. Silicon Dioxide 0.44 0.44
8. Butylated Hydroxyanisole0.02 0.01
37
CA 02390301 2002-05-07
WO 01!34123 PCT/US00/30415
Seal Coat
1. Opadry Clear 2.74 2.74
2. Sodium Chloride Powder 0.91 0.91
Inner Coat
1. HPMCP 55 2.21 2.21
2. Talc, USP 0.76 0.76
3. Acetyltributyl Citrate 0.22 0.22
4. Sugar, Micronized 0.61 0.61
Outer Coat
1. Cellulose Acetate 0:97 0.97
2. Eudragit S 100 0.33 0.33
3. Triacetin 0.08 0.08
4. Polyethylene Glycol 0.08 0.08
400
5. Acetyltributyl Citrate - -
6. Sugar, Micronized 0.49 0.49
Overcoat
1. HPMCP SO ' - -
2. Talc - -
3. Triacetin - -
4. Sugar, Micronized - -
5. Opadry Yellow 3 3
6. Opadry Pink - -
Total Tablet Weight % 100 100
Each of Examples 5-9 were evaluated by dissolution testing under the following
conditions: USP apparatus 2 (paddles); medium: 2% SLS/sodium phosphate buffer
(0.01M),
pH 7.0, stir speed SO rpm, and temperature 37°C. The dissolution
profile for Examples 2 and
5-7 are illustrated in Figure 3. The dissolution profiles for Examples 3,4 and
8 are illustrated
in Figure 4. The dissolution profile for Example 9 is illustrated in Figure S.
The dissolution
38
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
profiles of Examples 5-7 and 2 are further provided in Table 4 below. The
dissolution
profiles of Examples 3,4,8 and 9 are further provided in Table 5 below.
TABLE 4
Time (Hours) Ex. 5 Ex. 6 Ex. 7 Ex. 2
0 0 0 0 0
0.5 0 0 0 0
1 1 1 1 0
2 12 14 13 1
3 29 32 30 11
4 44 48 46 27
6 67 72 72 55
8 80 82 83 72
12 86 86 88 84
16 90 86 89 87
20 90 85 89 87
TABLE 5
Time (Hours) Ex. 3 Ex. 4 Ex. 8 Ex. 9
0 0 0 0 0
0.5 5 0 0 0
1 15 0 0 1
2 36 4 4 6
4 70 18 33 39
6 86 37 65 67
4
5
8 90 54 82 80
12 92 78 89 86
16 99 85 92 88
20 99 87 92 89
39
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
CLINICAL STUDIES
Oral Pharmacokinetics
Studies were conducted to evaluate the formulations of Examples 5-9
(hereinafter
collectively referred to as "Lovastatin XLT"~"). In certain of these studies,
an immediate
release tablet of lovastatin commercially available for more than 10 years
(Mevacor°, Merck
& Co., Inc.) was used as a reference standard.
Study No. 1 was a pharmacokinetics, safety and tolerability open-label, single
dose,
two-period crossover study of 40 mg Lovastatin XL tablets (Example 5) in
comparison to
Mevacor. There were 8 healthy male volunteers. The dose of Lovastatin XL and
Mevacor
was administered at 6:30 p.m., immediately after dinner.
Study No. 2 was a safety, pharmacokinetics and effect of food open-label,
single dose,
three-period crossover study comparing Lovastatin XL tablets (Example 5) to
Mevacor.
There were 9 healthy male volunteers. The dose of Lovastatin XL was
administered at 8:00
a.m. (fasting), 8:00 a.m. immediately after breakfast (fed conditions), in
comparison to
Mevacor administered at 8:00 a.m., immediately after breakfast.
Study No. 3 examined the safety and pharmacokinetics of Lovastatin XL tablets
(Example 5 and Example 2) in an open-label, single-dose, two-period crossover.
There were
6 healthy male volunteers. The doses of Lovastatin XL were administered at 8
a.m.,
immediately after breakfast.
Study No. 4 was a multiple-dose, safety, tolerability, efficacy,
pharmacodynamics and
pharmacokinetics single-blind, 4-week active treatment, 2-period cross-over
study with a 4-
week diet/placebo run-in period in which 40 mg Lovastatin XL tablets (Example
5) were
compared to Mevacor 40 mg tablets. Patients had a diet/placebo run-in period
of 4 weeks
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
prior to randomization to the active treatment. A total of 24 patients were
randomized to
receive 40 mg/day of Lovastatin XL, or 40 mg/day of Mevacor once daily in
active treatment
Period I and was switched to the alternate treatment drug in Period II. The
Lovastatin XL
tablets were administered on a once-daily basis for 4 weeks at about 10:00
p.m. The Mevacor
tablets were administered once daily for 4 weeks at about 6 p.m., immediately
after dinner.
There was a two week placebo washout period between treatments. Of the 24
patients with
hypercholesterolemia, 12 were male and 12 were female, and 13 of the subjects
participated
in the pharmacokinetic substudy. The results are summarized in Table 6.
Study No. 5 examined the oral pharmacokinetics, pharmacodynamics and safety of
10
mg, 20 mg and 40 mg Lovastatin XL tablets (Examples 9, 8 and S, respectively).
The study
design was single-dose, 3-period cross-over, with 8 healthy male volunteers.
Each of these
dosages were administered at bedtime (about 10:00 p.m.).
Tables 6 and 7 provide mean pharmacokinetic values (for lovastatin and
lovastatin
acid, respectively) for both Lovastatin XL and Mevacor for Study Nos. 1-5.
Table 8 provides
mean AUC and CmaX ratios for the 40mg tablets in Study Nos. 1, 2 and 4. Table
9 provides
pharmacokinetic data (mean AUC and Cy"aX ratios) for Lovastatin XL 40mg doses
for Study
Nos. 1, 2 and 5. FIG. 6 is a graph of Study 1 of comparative data which shows
mean (~SD)
plasma concentration time profiles of lovastatin in healthy subjects (n=8)
following a single
oral dose of a conventional immediate release dose of 40mg of lovastatin and
an extended
release 40mg dose of lovastatin according to the invention (Example 5). FIG. 7
is a graph of
Study 1 of comparative data which shows mean (~SD) plasma concentration time
profiles of
lovastatin acid in healthy subjects (n=8) following a single oral dose of a
conventional
immediate release dose of 40mg of lovastatin and an extended release 40mg dose
of
lovastatin according to the invention (Example 5). FIG. 8 is a graph of Study
2 comparative
data which shows mean (~SD) plasma concentration time profiles of lovastatin
in healthy
subjects (n=9) following a single oral dose in the morning of a conventional
immediate
release dose of 40mg of lovastatin with breakfast and an extended release 40mg
dose of
lovastatin according to the invention (Example 5) with and without breakfast.
FIG. 9 is a
41
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
graph of Study 2 of comparative data which shows mean (~SD) plasma
concentration time
profiles of lovastatin acid in healthy subjects (n=9) following a single oral
dose in the
morning of a conventional immediate release dose of 40mg of lovastatin with
breakfast and
an extended release 40mg dose of lovastatin according to the invention
(Example S) with and
without breakfast.
The mean AUC values for lovastatin acid, active and total inhibitors at steady
state
were similar for both Lovastatin XL and Mevacor (Tables 7 and 8). From the
results of these
studies, it was found that when compared to Mevacor and regardless of dosing
time,
Lovastatin XL had higher bioavailability of inactive prodrug (lovastatin), as
reflected by
mean AUC values and by geometric mean AUC ratios of lovastatin which were
greater than
unity, after administration of a single dose (Table 6). Geometric AUC ratios
for the acid
were close to unity (Table 7).
As can be ascertained from the results provided in Table 10, ingestion of a
high-fat
breakfast prior to the administration of Lovastatin XL reduced the AUCo~B~ and
CmaX values
of lovastatin and lovastatin acid by approximately 40-50%. Thus, a high-fat
meal decreased
the rate and extent of absorption of lovastatin and lovastatin acid after
administration of
Lovastatin XL.
Efficacy
Study No. 4 was designed to evaluate the safety, efficacy, pharmadynamics,
pharmacokinetics, and tolerability of Lovastatin XL relative to MEVACOR after
multiple
dose treatment in patients with fasting plasma LDL-cholesterol levels between
130 and 250
mg/dl and triglyceride levels below 350 mg/dl. This study had a single-center,
single-blind,
randomized, two-way crossover design. Patients had a diet/placebo run-in
period of 4 weeks
prior to randomization to the active treatment. A total of 24 patients were
randomized to
receive 40 mg/day of lovastatin to the alternate treatment drug in Period II.
During the active
treatment periods, patients were instructed to take Lovastatin XL daily in the
evening at
42
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
approximately 10:00 p.m. for 4 weeks, or MEVACOR daily with the evening meal
(about
6:00 p.m.) for 4 weeks. There was a two-week placebo washout period between
treatments.
The multiple-dose pharmacokinetics of Lovastatin XL studied in patients in
Study No.
4 are depicted in Figures 10 and 11 and in Table 11. FIG. 10 is a graph of the
mean
plasma concentration-time profiles of lovastatin and lovastatin acid in
patients (n=12) after
multiple-dose administration of 40mg Lovastatin XL and a conventional 40mg
immediate
release dose of lovastatin (Study No. 4, Day l and Day 28). FIG. 11 is a graph
of the mean
plasma concentration-time profiles of total and active inhibitors of HMG-CoA
Reductase in
patients (n=12) after multiple-dose administration of 40mg Lovastatin XL and a
conventional
40mg immediate release dose of lovastatin (Study No. 4, Day 1 and Day 28).
The mean plasma concentration-time profiles of lovastatin, lovastatin acid,
and total
and active inhibitors of HMG-CoA reductase following administration of 40
mg/day of
Lovastatin XL for four weeks exhibited extended release characteristics, as
depicted in
Figures 10 and 1 l and in Table 11. Table 12 provides the Least Squares Means -
% change in
LDL-Cholesterol, HDL-Cholesterol, Total Cholesterol and Triglycerides of Study
No. 4. The
increased systemic bioavailability of lovastatin when administered as the XL
formulation was
not accompanied by increased bioavailability of lovastatin acid, active or
total inhibitors. The
mean plasma concentration-time profiles of these compounds following
administration of 40
mg/day of Mevacor for four weeks exhibited immediate-release characteristics.
No
accumulation was observed with Mevacor. As can be ascertained from these
results, the
accumulation of lovastatin and its latent and active metabolites after chronic
once-daily
administration of Lovastatin XL was approximately 1.4- to 2-fold. The primary
efficacy
endpoints defined by the protocol were the changes from baseline of blood
lipid values at
weeks 3 and 4 (combined) of treatment. Values were also calculated for Weeks 3
and 4
individually and were not combined if the treatment-by-time interaction was
significant.
Results of this study demonstrated that Lovastatin XL lowered LDL-cholesterol
4.4
percentage points more (p = 0.0605) than MEVACOR at Week 3, 3.6 percentage
points more
(p = 0.0737) at Week 4, and 3.9 percentage points more (p = 0.0435) when Weeks
3 and 4 are
43
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
combined (Table 9). When compared with MEVACOR, mean Lovastatin XL HDL-
cholesterol values increased 2.7 percentage points more (p = 0.2588) at Week 3
and 3.0
percentage points more (p = 0.2741) at Week 4 of each treatment period, and
3.0 percentage
points more (p = 0.1402) with Week 3 and 4 combined. Mean Lovastatin XL total
cholesterol values decreased 3.4 percentage points more (p = 0.0245) at Week
3, 1.7
percentage points more (p = 0.3422) at Week 4, and 2.5 percentage points more
(p = 0.0721)
with Week 3 and 4 combined. Mean Lovastatin XL triglyceride values decreased
101.2
percentage points more (p = 0.1067) at Week 3, but increased 3.1 percentage
points more (p =
0.5297) at Week 4. Lovastatin XL, at 40 mg daily, produced 41 percent lowering
in mean
LDL-cholesterol. The magnitude of this reduction was 3.9 percentage points
greater (p =
0.0435, based on combined Weeks 3 and 4 data) than was produced by an equal
dose of
MEVACOR. Based on the well-recognized dose-response relationship that doubling
the dose
of a statin produces approximately a 6 to 7 percentage point further decline
in LDL-
cholesterol, the 3.9 percentage point differential observed in this trial
would, if reproducible,
translate to the response expected from a dosage equivalent of MEVACOR of more
than 60
mg.
44
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
O V' O\ V1 N
~ I i i '
I I
i
> _ ~ ~ I i
i M i
W N N M M
01 V M ~V V-700
00
I I I ~I i MI ~ I
~I ~ NI ~ I
~
' v~ N V vp ~ ~ M N
O
O .--.
N N
I '
i
N i i ~I i i i
I
b~
U N '
~ ~ d O ~ O Vr ~Y
O~
~ N 'r'o 0
~
+I +I + ~ l I I
+I I +
I ;
I
O ~ ~ O _ O ~
~
o O ~ ~' p p p
0
M M ~ d' N
~
E" ~
'b
~_
RH 3E ~ # jF
Q O V N
U U ~ .-
_ .
'
U
' ~I
~I
d
M
N M
O
m i.--~
V'
c~o ~ ~ I~ 01
O
~ ~
o
U >~ "~ t t ~,~ ~ v-,
+I ~I ~I+I ' +I +I I ~I
+ I
I
v ~
-, ~
+ ~ l dvo oo, ~ 'r'
I
V1 l~~ ~ M
01
N ~
00 O~ WO~
O ~ ~ N
~" o0 00 00
,~
O
o L1 f~o N ~ v N
O
~ ~ O ~ ~
O
pp _ ii
N
_y
~
~
ate-~ ~ ~ N
~"
bn ~
w w ~ ~ ~ sue.
~
N N N
~1 oa 0.~ l~ ~
~ f~
b ~ b y b b b
~ ~ ~ ~
~
3 3 3 ~ c~ pa as
a 3 ~
3
c
N M ~ ~ '~
-H
o ~n o
'-' ""'' N
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
O O 01 N O
O I i n i I
~I ~I MIn
M N v~ u1
H
00 00 O~~!1M I ~' I l~
OO O1 I~~ V~ OO O
M O ~Y00 M
O
O O o0 O~ N
I I I O
y ~I +I i
'b
.U
Q
E
U ~ O~O~ M 0~1V~ 00
o .~+I +I +I~+I+I . d+1. +I
~l ~ ~ o "N , omo
N ~hN N ~ M
E
H
b ~ ~ ~ ~ #
c~C O M --' M
V'1i i i i i
H i M
~ ~I I I
~ t ~ 00 oNo
pp
w
O
N U ~C~ V \O01 ~ N N
~n OvN ,_.'
Q ~ m.INI~I +I I ~I
~ Ov
I a
N
D1 ~
U
O ~ U ~ ~ O O
.4: ,~ ~ N
~ .--.
O N O
O ~ C/~~ V7 N .,-.,r~
~ .~b~A~.
N w ~'w ~ ~ ~ ~,'
w
~ U ~ N v
N
.v~ ,~ ~ .~~ b ,.Yb .~b
O
C7 ~3 ~3 w ~3 as ~30.1~30.'1
b -n N M ~' \p
O
z
46
~n o ~n p
N
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
O ,.,- o O
d . NI ~ +I ~ ~ NI~ NI
I~ 01 > ~ N
(7~ M M (3~ d'
r~M
ate.MI i NIi ~ ~ NI i +~ i
D,
H
_O
l~ O V, N
U a, U
Q . ~~ . +I ~ . ~~. +I
N > Vr
.b
M ~ ~ N N
b~
U ' X oo ... ~ ~C ,-.
(,~O U ~ ~i i oon ~ ~ M a\ i
r~.i Y ~! MI .~ p\4 a.~.~~+I y,l
I C',
c~ ~ ~ ~ cC ~ M
~ J O
a U
Q '
O O ~ y O ~ O
0 ~ U ~ U
d ~ Q W ~ --~Q W ~ ~n
E'~O O ~ ~ N N O
U
C7
c x ' x ~'? ~ x on x ~
i i i , i
~ U ~ ~ v~ j ~ ~ ~ N
U M~ ~I .U ~ Y ~+I +I
CG\O d o ~ M M
U ~ ~ ~ U > M
a N Q
Q
w
O ~ ~ ~ ,N-,N ~ N N N N
U
c~
+ N G7
~
N O N O
O ~ ~ O
N ~] O ~ U (~O U
Q N O U
O ~ b~~D~.~ O ~ ~ ~. ~
a ~ ~ ..~~ a
b ~ b ~ .~ b
~1 as ~3 ca~3 ~ ~ ~3~ ~3
z ~ z
47
~n o ~n o
~-' ~-y N
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
~.
0
~ i
V
.
0 0
~.
0 i i N
O t
.fl
"' O O
;'d ~O l~ M M
V l~ l~ N
7 O O O O
d
,n O
'b
C
O
N .
y
O
b ~ > 0 0 0 0 0
O
d
:, >
.~
0
r
>s
d' oo ~
o 0 U
d ~'. ~
,~
E
0
II
.o
~.
U is m o
v o . . ~? av
~
N 0 0
,~
U '"'
d
a~
y
U N
7 .~ ~ O O
d
O O
c
N
O y
Y
V1
\_ \_
_~
d4 i. O ~ ~ t-.
bD w i
a ~ Cl
N
. b
O O
... ~ u,
~r~ . ~H~~ ~~~~~ ~~~~~
~
~~~
48
o ~n o ~n o
N N M
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
a, a\
+I +I ~~ +I +I
E-~ o ' [~ ~ o,
'. .~
0
N ~ N
.d 'O ~ O
I I 1 ~ ~ I + I i ,''"'.s,
,~
- N N N
O
O
W
J
O
W N ~ O~ oo N ~n
4-V '~ 01 d. ~ M d'
p ~ ~
ff + +
I
p~ ~ U ~ o U o x
.b , o o
o
o ~ 00 0 ~ a, v,
>~ ~ ~ M 'd N s.r.
~
.fl
O
N
I~
W
P.
~r Y
O _~
O
O
O
O ~ O t~ ~ v0 Vr
v O (~ o O~ v~ O
o ~1 M ~' '
~ I y..,I
I I
W ~-'7-O ~ N Gy,
~ i M
O O
O ~t - O
d o, "-, d o, ~,
v v
o ~ o
.3 ~ ~ .~ .3
X x ~ ~ x x
>~ C".~. ~ L~7 .~.~. Cue.'
. . U
. .
. .
.
N ~ Y ~ N
~ W ~ Y BFI
Y ~I-H ~ ~I--1
~
(~ ~" C f~ C~ '~ CCS
G x ~
~ O > ~ ~ O
49
~n o ~n o ~n o
N N M
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
Dose Proportionality
Study No. 5 was conducted in healthy volunteers receiving separately single
oral
doses of 10, 20 and 40 mg of Lovastatin XL. Results of this study indicated
that, as the dose
of Lovastatin XL increased from 10 to 40 mg, the AUCo_asnr and CmaX values of
lovastatin
appeared to increase linearly (see results for Study No. 5 depicted in Table
6). Figure 12 is a
graph of a regression line depicting CmaX plotted against dose for Study No.
5. Figure 13 is a
graph of a regression line depicting AUCo_a$hr plotted against dose for Study
No. 5.
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
.x.~ _
o c~ o
ri
I + I
l
N c~ ~! o M,
p
b0 ~ U1 ~n t~ M
cct
V7
C'
00 O
"t7 O '~ M
~~ ~I ~ ~I
~
U ' ' o
~ ~ ~~
.-~, ~ N ~ 'O ''"
a
N
O
O-0CC$
~r ~ rv
U
W
a
,~ o
d
o
Ei n
0
0
N O N v:7 ~ ~ N
~~ I y M N
: ~
~
Q. ~ ri c.i .-.
l~ 00
N
t/y"
CC cti
f~ .- ~n ~ cn t~ -~
,.~ M
I o ~ N M
I
I I M I
y ~ f~ a, t~ M
- 01 00
- M ~ 00
~r
C~
N
4~
O c,..,
v~ O
t
.
"O O y.,
O
O N ~ O Q)
w, U
.
o
CG c0 ~ U ? C~JU
~ Y b ~U ~ b
a ~. N x ~ d x
~
51
~n o ~n o ~n
~' '-" N N
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
TABLE
12
Least
Squares
Means
Change
in LDL-Cholesterol,
HDL-Cholesterol,
Total
Cholesterol,
and
Triglycerides
[Study
No. 4]
ParameterWeek n LovastatinMEVACOR Differencep-value
XL
LDL-C 3 24 -40.60 -36.19 -4.41 .0605
4 22 -42.48 -38.86 -3.62 .0737
3&4 24 -41.32 -37.45 -3.87 .0435*
HDL-C 3 24 +7.73 +5.06 +2.67 .2588
4 22 +8.81 +5.77 +3.04 .2741
3&4 24 +8.19 +5.18 +3.01 .1402
3 24 -27.80 -24.44 -3.36 .0245*
Total-C 4 22 -29.27 -27.60 -1.67 .3422
3&4 24 -28.48 -25.99 -2.48 .0721
Trigylcerides3 24 -20.53 -10.33 -10.20 .1067
4 22 -21.02 -24.14 +3.12 .5297
*Significant at p<0.05
Analytical Methods
Concentrations of lovastatin and lovastatin acid in plasma samples from the
first
single-dose, safety and pharmacokinetic study [Study No. 1 ] were determined
by a
LC/MS/MS method. The internal standards (mevastatin and mevastatin 13-
hydroxyacid) were
52
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
added to each plasma sample (1 ml). Each sample was extracted on a SPEC-Plus
C18 disc,
which was previously conditioned with methanol and water. The disc was then
washed with
water and formic acid. The analytes were eluted with 70:30 (v:v)
methanol:water and 75:25
(v:v) acetonitrile:ethyl acetate. The eluate was then evaporated to dryness
and reconstituted
S in SO,uI of mobile phase (sodium acetate in acetonitrile/water). The sample
was injected onto
a SCIEX API III-Plus LC/MS/MS equipped with a short C18 HPLC column. The peak
area
of the m/z 421.3319.3 product ion of lovastatin acid was measured against the
m/z
407.6--305.0 product ion of the internal standard (mevastatin l3-hydroxyacid)
using negative
ion MRM mode. The peak area of the m/z 427.4-325.0 product ion of lovastatin
was
measured against the m/z 413,2311.0 product ion of the internal standard
(mevastatin) using
positive ion MRM mode. Quantitation was performed using a weighted
(1/concentration2)
linear least squares regression line generated from plasma calibration
standards. The standard
lines for lovastatin and lovastatin acid were linear over the concentration
range of 0.1-50
ng/ml. The interday precision and accuracy values were 3.7-24.3% relative
standard
deviation and within 13%, respectively.
Concentrations of lovastatin and lovastatin acid in plasma samples from Study
Nos. 2-
5 were determined by a LC/MS/MS method. The pH of each plasma sample was
adjusted
with ammonium formate buffer. Lovastatin, lovastatin acid and their
corresponding internal
standards (d5-lovastatin and d5-lovastatin acid) were extracted using SPE
cartridges and
eluted with 75% methanol followed by acetonitrile. The extract was dried under
nitrogen,
reconstituted and injected onto a LC/MS/MS. The peak area of the m/z
423.2303.2 product
ion of lovastatin acid was measured against the m/z 428.2303.2 product ion of
the internal
standard (ds-lovastatin acid) using MRM mode. The peak area of the m/z
405.2285.2
product ion of lovastatin was measured against the m/z 410.2- 285.2 product
ion of the
internal standard (d5-lovastatin) using MRM mode. Quantitation was performed
using a
weighted (1/concentration) linear least squares regression line generated from
plasma
calibration standards. The standard lines for lovastatin and lovastatin acid
were linear over
the concentration range of 0.1-20 ng/ml. The interday precision and accuracy
values were
6.4-9.3% relative standard deviation and within 7%, respectively.
53
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
An enzymatic assay method was used to determine concentrations of active
inhibitors
(lovastatin acid + active metabolites of lovastatin) and total inhibitors
(lovastatin + lovastatin
acid + latent and active metabolites of lovastatin) of HMG-CoA reductase
inhibitors in
plasma samples from the multiple-dose study [Study No. 4]: An aliquot of each
sample was
subjected to alkaline hydrolysis to determine the concentration of total
inhibitors.
Concentrations of active inhibitors were determined in unhydrolyzed samples.
The inhibitory
activity was measured against lovastatin acid as a standard. Lovastatin acid
was used in its
ammonium salt form and all results were expressed as nanogram equivalent per
milliliter.
The standard curves for total and active inhibitors were over the
concentration range of 0.5-
100 ng eq/ml. The interday precision and accuracy values were 2.64-9.84%
relative standard
deviation and within 12.5%, respectively.
Conclusions
The bioavailability of Lovastatin XL relative to MEVACOR, in terms of
AUCo_Zanr or
AUCo_a8hr ratio of lovastatin, is greater than 100% after single-dose
administration.
The same is true at steady state. However, the relative bioavailability of
Lovastatin
XL to MEVACOR at steady state, in terms of AUCo_z4hr ratio of lovastatin acid
or
active or total inhibitors of HMG-CoA reductase, is close to or less than
100%.
2. Together with low CmaX values, the relative bioavailability of Lovastatin
XL indicates
that the systemic exposure of patients to active drug and metabolites of
lovastatin as
well as total inhibitors of HMG-CoA reductase after administration of
Lovastatin XL
is not higher than that after administration of MEVACOR.
Administration of Lovastatin XL following a high-fat breakfast decreases the
bioavailability of lovastatin acid.
54
CA 02390301 2002-05-07
WO 01/34123 PCT/US00/30415
4. As the dose of Lovastatin XL increases from 10 to 40mg, the plasma profiles
(including the AUCo_4snr and C",a~ values) of lovastatin appear to increase
linearly.
5. Accumulation of lovastatin and its latent and active metabolites after
chronic once-
daily administration of Lovastatin XL is approximately 1.4- to 2-fold.
6. When compared to MEVACOR, the increased benefit on lipid levels produced by
Lovastatin XL is achieved with similar systemic exposure to active (and total)
inhibitors of HMC-CoA reductase. No evidence of unfavorable safety and
tolerability
characteristics is observed.
The foregoing description of a preferred embodiment of the invention has been
presented for purposes of illustration and description. It is not intended to
be exhaustive or to
limit the invention precise form disclosed. In certain further preferred
emodiments, the
controlled release oral lovastatin formulations of the invention may be
characterized by other
pharmacokinetic values which are set forth in the data provided in the
appended examples,
which data can be readily gleaned by one of ordinary skill in the art
reviewing the appended
Tables and Figures. Such pharmacokinetic values may be derived in part based
on
parameters such as Cmax (ng/mL); Cmin (ng/mL); Tmax (hr); fluctuation
(%)(expressed as
the difference between Cmax and Cmin expressed as a percentage of Cmin); the
area under
the curve (AUC); and any combination thereof. Obvious modifications or
variations are
possible in light of the above 40 teachings. All such obvious modifications
and variations are
intended to be within the scope of the appended claims.