Note: Descriptions are shown in the official language in which they were submitted.
CA 02390501 2002-05-07
WO 01/34202 PCT/US00/31068
DEPLETION OF CELLULAR COENZYME-A LEVELS AS A MEANS
TO SELECTIVELY KILL CANCER CELLS
Review of Related Art
A number of studies have demonstrated surprisingly high levels of
fatty acid synthase expression (FAS, E.C. 2.3.1.85) in virulent human breast
cancer
(Alo, P. L., Visca, P., Marci, A., Mangoni, A., Botti, C., and Di Tondo, U.
Expression of fatty acid synthase (FASO as a predictor of recurrence in stage
I breast
carcinoma patients., Cancer. 77: 474-482, 1996; Jensen, V., Ladekarl, M., Holm-
Nielsen, P., Melsen, F., and Soerensen, F. B. The prognostic value of
oncogenic
antigen 519 (OA-519) expression and proliferative activity detected by
antibody
MIB-1 in node-negative breast cancer., Journal of Pathology. 17G: 343-352,
1995),
as well as other cancers (Rashid, A., Pfizer, E. S., Moga, M., Milgraum, L.
Z.,
Zahurak, M., Pasternack, G. R., Kuhajda, F. P., and Hamilton, S. R. Elevated
expression of fatty acid synthase and fatty acid synthetic activity in
colorectal
neoplasia., American Journal of Pathology. 150: 201=2U8, 1997; Pfizer, E.,
Lax, S.,
Kuhajda, F., Pasternack, G., and Kurman, R. Fatty acid synthase expression in
endometrial carcinoma: correlation with cell proliferation and hormone
receptors.,
Cancer. 83: 528-537, 1998). FAS expression has also been identified in
intraductal
and lobular in situ breast carcinoma; lesions associated with increased risk
for the
development of infiltrating breast cancer (Milgraum, L. Z., Witters, L. A.,
Pastcrnack, G. R., and Kuhajda, F. P. Enzymes of the fatty acid synthesis
pathway
are highly expressed in in situ breast carcinoma., Clinical Cancer Research.
3: 21 1 ~-
2120, 1997). FAS is the principal synthetic enzyme of fatty acid synthesis (FA
synthesis) which catalyzes the NADPH dependent condensation of malonyl-CoA
and acetyl-CoA to produce predominantly the 16-carbon saturated free fatty
acid,
palmitate (Wakil, S. Fatty acid synthase, a proficient multifunctional
enzyme.,
Biochemistry. 28: 4523-4530, 1989). Ex vivo measurements in tumor tissue have
revealed high levels of both FAS and FA synthesis indicating that the entire
genetic
program is highly active consisting of some 25 enzymes from hexokinase to FAS
(Rashid, et al., 1997).
WO 01/34202 CA 02390501 2002-05-07 PCT/US00/31068
2
Cultured human cancer cells treated with inhibitors of FAS, including
the fungal product, cerulenin, and the novel compound, C75, demonstrated a
rapid
decline in FA synthesis, with subsequent reduction of DNA synthesis and cell
cycle
arrest, culminating in apoptosis (Pfizer, E. S., Jackisch, C., Wood, F. D.,
Pastemack,
G. R., Davidson, N. E., and Kuhajda, F. Inhibition of fatty acid synthesis
induces
programmed cell death in human breast cancer cells., Cancer Research. 56: 2745-
2747, 1996, Pfizer, E. S., Chrest, F. J., DiGiuseppe, J. A., and Han, W. F.
Pharmacological inhibitors of mammalian fatty acid synthase suppress DNA
replication and induce apoptosis in tumor cell lines., Cancer Research. 58:
4611-
461 S, 1998). Pharmacological inhibition of mammalian fatty acid synthase
activity
lead to inhibition of DNA replication within about 90 minutes of drug
application.
These findings suggested a vital biochemical link between FA synthesis and
cancer
cell growth. While generating a great deal of interest, the question of how
inhibition
of fatty acid synthase triggered this phenomenon remained unknown.
Importantly,
these effects occurred despite the presence of exogenous fatty acids in the
culture
medium derived from fetal bovine serum. While it has been possible to rescue
the
cytotoxic effect of cerulenin on certain cells in fatty acid-free culture
conditions by
the addition of exogenous palmitate, most cancer cells were not rescued from
FA
synthesis inhibition by the pathway endproduct (data not shown) (Pfizer, E.
S.,
Wood, F. D.. Pasternack, G. R., and Kuhajda, F. P. Fatty acid synthase (FAS):
A
target for cvtotoxic antimetabolities in HL60 promyelocytic leukemia cells.,
Cancer
Research. 1996: 745-751, 1996). Thus, it has been unresolved whether the
cytotoxic
effect of FA synthesis inhibition on most cancer cells resulted from end
product
starvation, or from some other biochemical mechanism.
Summary of the lnvention
This invention describes a method to inhibit growth or kill cancer
cells by acute depletion of free cellular Coenzyme A (CoA). This invention
encompasses: any method to selectively decrease CoA in cancer cells by
increasing
the utilization of CoA and/or reducing its synthesis.
This therapeutic strategy will lead to novel chemotherapeutic agents
for a wide variety of human cancers. In addition, as this is a novel pathway
leading
CA 02390501 2002-05-07
WO 01/34202 PCT/US00/31068
3
to apoptosis not shared by other cancer drugs, it may be anticipated that this
therapeutic strategy may potentiate other commonly utilized cancer therapeutic
agents.
In one embodiment, this invention provides a method for inhibiting
growth of tumor cells in an organism comprising administering to the organism
a
composition which causes acute depletion of intracellular free Coenzyme A in
cancer cells in said organism. In particular, upon administration of the
composition,
intracellular malonyl CoA in cells of the organism rises abruptly, preferably
within 3
hours of the administration. It is expected that intracellular malonyl CoA
rises prior
to growth inhibition of the cells, and preferably, the rise in intracellular
malonyl
CoA is correlated with reduced consumption of malonyl CoA. More preferably,
the
rise in intracellular malonyl CoA occurs prior 1o any increase in rate of
consumption
of malonyl CoA. In one mode, the rise in intracellular malonyl CoA is
correlated
with reduced intracellular activity of malonyl CoA decarboxylase (MCD) or
reduced
1 S intracellular activity of fatty acid synthase, and the composition may
comprises an
inhibitor of MCD. In another mode, the rise in intracellular malonyl CoA is
correlated with increased synthesis of malonyl CoA.
In another embodiment, this invention provides a method for
inhibiting growth of tumor cells in an organism comprising administering to
the
organism a composition which causes acute depletion of intracellular free
Coenzyme
A in cancer cells in said organism and the rise in intracellular malonyl CoA
is
correlated with increased intracellular activity of acetyl-CoA carboxylase
(ACCj.
Alternatively, the composition may comprises an activator of ACC, an activator
of
citrate synthase, an inhibitor of 5'-AMP-activated protein kinase (AMPK),
andior an
inhibitor of acyl CoA synthase. In a preferred mode, a second chemotherapeutic
agent is also administered to the organism.
In the method of this invention, intracellular malonyl CoA level prior
to administration of said composition is preferably at least 2-fold above
normal
malonyl CoA level in non-malignant cells. Generally, intracellular level of
malonyl
CoA is elevated and intracellular level of acetyl CoA and free CoA are reduced
relative to pre-treatment levels. Preferably, fatty acid synthesis rate in
some cells of
WO 01/34202 CA 02390501 2002-05-07 PCT/USO~/31068
4
the organism is at least 2-fold above normal prior to administration of the
composition, and administration of the composition is cytotoxic to the cells.
The
fall in intracellular free Coenzyme A level may be expected to be correlated
with
appoptosis of cells having decreased Coenzyme A.
S In a preferred embodiment of the method of this invention, the
composition comprises an inhibitor of Pantothenate kinase, an inhibitor of
Phosphopantothenoylcysteine synthetase, an inhibitor of
Phosphopantothencylcysteine decarboxylase, and/or an inhibitor of
Phosphopantotheine adenylyltransferase. In another preferred mode, the
composition comprises a substrate capable of esterification to CoA. Typically,
the
organism treated according to this invention comprises tumor cells having
elevated
fatty acid synthesis rates and cell number of said tumor cells is reduced
subsequent
to administration of said composition.
In another embodiment, this invention provides a screening method
to assist in detecting compositions which are selectively cytotoxic to tumor
cells
comprising administering a target composition to a target cell, monitoring
intracellular levels of free and/or derivatized Coenzyme A in said cell
subsequent to
said administration, wherein an abrupt decrease in intracellular free Coenzyme
A is
indicative of selective cytotoxicity.
In yet another embodiment, this invention provides a screening
method to assist in classifying compositions which are selectively cvtotoxic
to tumor
cells comprising administering a target composition to a target cell, in the
absence,
and in parallel, in the presence of sufficient ACC inhibitor to limit the
production of
malonyl-CoA, wherein a difference in cytotoxicity is indicative of a cytotoxic
activity derived from an effect on intracellular levels of free and/or
derivatized
Coenzyme A.
Brief Description of the Figures
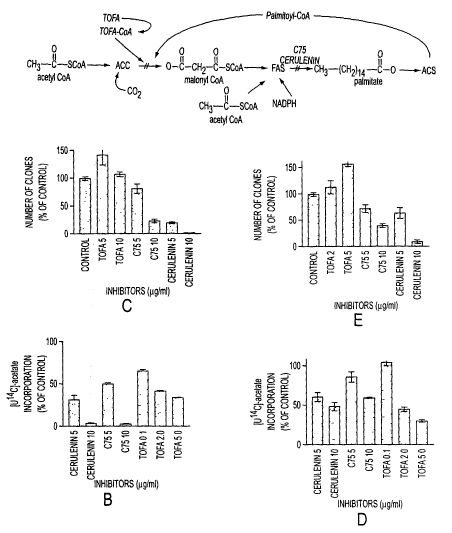
Figure 1 shows the fatty acid synthesis pathway, and the effect of
various fatty acid synthase inhibitors on fatty acid synthesis and tumor cell
growth.
Figure 2 shows malonyl CoA levels under various conditions.
CA 02390501 2002-05-07
WO 01/34202 PCT/US00/31068
Figure 3 show the results of clonogenic assays and apoptosis assays
on breast cancer cells treated with various inhibitors.
Figure 4 shows various parameters in tumor cells and liver cells.
Figure 5 shows malonyl CoA levels in tumor cells and liver cells.
5 Detailed Description of the Embodiments
If fatty acid starvation mediated the cytotoxic effects of cerulenin and
C75, then any other FA synthesis inhibitor of similar potency should produce
similar
effects. To test this idea, we compared the effects on cancer cells of
inhibition of
acetyl-CoA carboxylase (ACC, E.C. 6.4.1.?), the rate limiting enzyme of fatty
acid
synthesis, with the effects of FAS inhibitors. The inventors have now
demonstrated
that inhibition of FAS leads to high levels of malonyl-CoA which occurs within
30
minutes of C75 treatment. These superphysiological levels of malonyl-CoA, not
low levels of endogenously synthesized fatty acids, are responsible for breast
cancer
cell apoptosis. In addition, this is a novel pathway which leads to selective
apoptosis of cancer cells.
Figure 1 A outlines the portion of the FA synthesis pathway
containing the target enzymes of the inhibitors used in this study. TOFA (5-
(tetradecyloxy)-2-furoic acid) is an allosteric inhibitor of acetyl-CoA
carboxylase
(ACC, E.C. 6.4.1.?), blocking the carboxylation of acetyl-CoA to malonyl-CoA.
Once esterified to coenzyme-A, TOFA-CoA allosterically inhibits ACC with a
mechanism similar to long chain acyl-CoA's, the physiological end-product
inhibitors of ACC (Halvorson, D. L. and McCune, S. A. Inhibition of fatty acid
synthesis in isolated adipocytes by S-(tetradecyloxy)-2-furoic acid., Lipids.
19: 851-
856, 1984). Both cerulenin (Funabashi, H., Kawaguchi, A., Tomoda, H., Omura,
S.,
Okuda, S., and lwasaki, S. Binding site of cerulenin in fatty acid
synthetase., J.
Biochem. 105: 751-755, 1989) and C75 (Pfizer, et al., 1998) are inhibitors of
FAS,
preventing the condensation of malonyl-CoA and acetyl-CoA into fatty acids.
Cerulenin is a suicide inhibitor, forming a covalent adduct with FAS (Moche,
M.,
Schneider, G., Edwards, P., Dehesh, K., and Lindqvist, Y. Structure of the
complex
WO 01/34202 CA 02390501 2002-05-07 PCT/US00/31068
6
between the antibiotic cerulenin and its target, beta-ketoacyl carrier protein
synthase., J Biol Chem. 274: 6031-6034, 1999), while C75 is likely a slow-
binding
inhibitor (Kuhajda FP, Pizer ES, Mani NS, Pinn ML, Han WF, Chrest FJ, and CA,
T. Synthesis and anti-tumor activity of a novel inhibitor of fatty acid
synthase.,
S Proceeding of the American Association for Cancer Research. 40: 121, 1999).
Using TOFA, the inventors have achieved FA synthesis inhibition in human
breast
cancer cell lines comparable to inhibition by cerulenin or C75. Surprisingly,
however, TOFA was essentially non-toxic to human breast cancer cells. These
data
indicate that fatty acid starvation is not a major source of cytotoxicity to
cancer cells
in serum supplemented culture. An alternative effect of FAS inhibition,
production
of high levels of the substrate, malonyl-CoA, resulting specifically from
inhibition
of FAS, appears to mediate cytotoxicity of cerulenin and C75.
Malonyl-CoA, the enzymatic product of acetyl-CoA carboxylase
(ACC, E.C. 6.4.1.2), is a key regulatory molecule_ in cellular metabolism. In
addition to its role as a substrate in fatty acid synthesis, malonyl-CoA
re~~ulates (3
oxidation of fatty acids through its interaction with carnitine
palmitoyltransferase-I
(CPT-I ) at the outer membrane of the mitochondria. CPT-1 regulates (3-
oxidation of
fatty acids in the mitochondrion by controlling the passage of long-chain acyl-
CoA
derivatives such as palmitoyl-CoA through the outer mitochondrial membrane.
Physiologically, cytoplasmic malonyl-CoA levels are higher during fatty acid
synthesis. The higher steady state level of malonvl-CoA blocks entry of lone-
chain
acyl-CoA's into the mitochondrion thus preventing the futile cycle of
oxidizing
endogenously synthesized fatty acids.
Coenzyme-A is a vital cofactor for cellular processes involved in
energy generation, lipid biosynthesis, and energy regulation. For example,
acetyl
CoA and malonyl-CoA are substrates for fatty acid and cholesterol synthesis.
All
fatty acids must be esterified to CoA before they can be incorporated into
cellular
structures, or oxidized in the mitochondria for energy. Succinyl-CoA is an
intermediate of the TCA cycle. Thus, maintenance of an adequate supply of CoA
is
vital for cell survival.
CA 02390501 2002-05-07
WO 01/34202 PCT/US00/31068
7
Many types of cancer cells have high levels of fatty acid synthesis.
As expected, cells with high levels of fatty acid synthesis have high steady
state
levels of malonyl-CoA, at least six times the levels in normal cells (see
Example 6).
To deplete the available supply of free CoA, intracellular malonyl CoA levels
can be
selectively and abruptly raised to superphysiological levels in tumor cells by
treating
them with inhibitors of FAS. This maneuver raises malonyl-CoA levels by both
blocking utilization of malonyl-CoA as a substrate in fatty acid synthesis and
concomitantly stimulating malonyl-CoA synthesis by relieving fatty acyl-CoA
inhibition of ACC (Figure 1A). Since FAS is preferentially expressed in cancer
cells, the malonvl-CoA elevation is largely restricted to tumors cells. This
leads to
cancer cell apoptosis and sparing of norn~al tissues as occurs in human cancer
Ycno~rafts treated with FAS inhibitors (Sec Example ~). By abruptly incrcasin~
malonyl-CoA levels, adequate free CoA is not available for other cellular
processes,
leading to cell death. _
I S Free CoA levels may be manipulated using a variety of methods and
target enzymes. The Examples demonstrate reduction of free CoA in conjunction
with elevation of malonyl-CoA levels through reduced utilization and
simultaneous
enhanced production of malonyl CoA. Evidence utilizing metabolic labeling with
[U-~'~C] acetate documents the high levels of fatty acid synthesis in human
cancer
cells (Kuhajda, F. P., Jenner, K., Wood, F. D., Hcnnigar, R. A., Jacobs, L.
B., Dick,
J. D., and Pasternack, G. R. Fatty acid synthesis: a potential selective
tartlet for
antineoplastic therapy., Proceedings of National Academy of Science. ~l: 6379-
6383, 1994; Rashid, A., Pizer, E. S., Moga, M., Milgraum, L. Z., Zahurak, M.,
Pasternack, G. R., Kuhajda. F. P., and Hamilton, S. R. Elevated expression of
fatty
acid synthase and fatty acid synthetic activity in colorectal neoplasia.,
American
Journal of Pathology. I~O: 201-208, 1997). This high level of fatty acid
synthesis in
human cancer cells allows for the selective manipulation of malonyl-CoA levels
to
induce apoptosis. Acute increase in malonyl-CoA levels leads to the selective
destruction of cancer cells via apoptosis, leaving norn~al cells unaffected.
This
therapeutic strategy identifies potential new targets and strategies for
cancer
chemotherapy based upon alteration of malonyl-CoA levels.
WO 01/34202 CA 02390501 2002-05-07 PCT/US00/31068
8
Preferably, manipulation of free Coenzyme A levels according to this
invention is accomplished by administering a composition (or multiple
compositions) to an organism in need thereof. The composition administered to
the
organism may contain an agent having a biological effect of reducing the
available
S supply of free CoA. Agents which interfere with biosynthesis of CoA, or
agents that
are incorporated into CoA-esters, reducing the pool of free CoA, may be used
alone
or together with other agents of this invention. Preferred agents have the
effect, at
least in part, of raising intracellular malonyl-CoA levels. Typically, the
organism
will be a mammal, such as a mouse, rat, rabbit, guinea pig, cat dog, horse,
cow,
sheep, goat, pig. or a primate, such as a chimpanzee, baboon, or preferably a
human.
Usually, the organism will contain neoplastic (malignant) cells. The method of
this
invention is directed to selectively affecting malignant cells, and having
less effect
(or more preferably no effect) on normal (non-malignant) cells.
The agent in the composition administered to the organism will
preferably raise intracellular malonyl-CoA levels in at least a portion of the
malignant cells in the organism. Preferably the malonyl CoA level will be
raised at
least 2-fold, more preferably at least 5-fold. Preferably, the agent will
raise the
intracellular malonyl-CoA concentration in the malignant cells to a level
higher than
the level in surrounding normal cells. Suitable agents may raise the malonyl
CoA
level by any of a number of methods (see alternative mechanisms listed below).
In
some embodiments, two or more agents are administered, and some or all of
these
agents may affect malonyl CoA level by a different mechanism. Agents acting by
any of the modes of the following list may be used in compositions of this
invention.
Assays for the following activities are available in the literature, and
determination
of whether a particular agent exhibits one of these activities is within the
skill in the
art.
Methods to acutely decrease free CoA for cancer treatment.
Acute (i.e., abrupt or preciptous) decrease in free CoA levels leads to the
selective
destruction of cancer cells via apoptosis. This therapeutic strategy
identifies
potential new targets and strategies for cancer chemotherapy based upon
alteration
WO 01/34202 CA 02390501 2002-05-07 pCT/US00/31068
9
of malonyl-CoA levels that occur selectively in cancer cells, with coordinate
changes in free CoA levels.
Agents for increasing malonyl-CoA production:
Acetyl-CoA carboxylase (ACC)effectors: Agents which increase ACC activity,
S reduce ACC inhibition, or increase the mass of active ACC enzyme will lead
to
increased levels of malonyl-CoA.
5' c-AMP protein kinase effectors: 5' c-AMP protein kinase inhibits ACC by
phosphorylation leading to acute reduction of malonyl-CoA. Inhibitors of this
kinase would lead to acutely increased levels of malonyl-CoA by releasing
inhibition of ACC.
Citrate synthase effeetors: Increasing mitochondria) citrate would provide
substrate for fatty acid synthesis and citrate also acts as a "feed-forward"
activator of
ACC causing increase malonyl-CoA synthesis.
Acyl-CoA svnthase effectors: Inhibition of acyl-CoA synthase would reduce
cellular fatty acyl-CoA concentration releasing inhibition of ACC. This would
result in increased ACC activity and malonyl-CoA levels.
Agents to decrease malonyl-CoA utilization:
Malonyl-CoA decarboxylase (MCD) effectors: This enzyme catalyzes an ATP
dependent dccarboxylation of malonyl-CoA back to acetyl-CoA. Inhibition of MCD
would acutely raise malonyl-CoA levels.
Simultaneously decreased malonyl-CoA utilization and increased production:
Fatty acid synthase (FAS) effectors: Inhibition of FAS leads to decreased
utilization of malonyl-CoA by blocking its incorporation into fatty acids. FAS
inhibition also leads to reduced fatty acyl-CoA levels which will activate
ACC.
Exemplary FAS inhibitors may be obtained as described in U.S. Patent Nos.
5,759.837 and 5,981,575, incorporated herein by reference.
These strategies for acutely increasing malonyl-CoA levels may be
used together or in concert with other drugs to enhance apoptosis of cancer
cells.
Preferably, at least one agent in the compositions of this invention raises
the level of
malonyl-CoA by a mechanism other than inhibiting FAS.
WO 01/34202 CA 02390501 2002-05-07 PCT/US00/31068
Decreasing CoA synthesis:
Pantothenate kinase (PanK) effectors: this enzyme catalyses , an ATP
dependent phosphorylation of pantothenic acid, the first step in Coenzyme A
synthesis, and reduction in its activity may be expected to reduce the total
amount of
S CoA with the consequent lowering of available free CoA.
Phosphopantothenoylcysteine synthetase effectors: this enzyme catalyses the
ATP dependent addition of cysteine to 4-phosphopantothenic acid to form 4
phosphopantothenoyl-L-cysteine, the second step in Coenzyme A synthesis, and
reduction in its activity may be expected to reduce the total amount of CoA
with the
10 consequent lowering of available free CoA.
Phosphopantothenoylcysteine decarboxylase effectors: this enzyme catalyses
the removal of the alpha-carboxyl group of cysteine from 4-phosphopantothenoyl-
L
cysteine to form 4-phosphopantotheine, the. third step in Coenzyme A
synthesis, and
reduction in its activity may be expected to reduce the total amount of CoA
with the
consequent lowering of available free CoA.
Phosphopantotheine adenylyltransferase (also called dephospho-CoA
pyrophosphorylase) effectors: this enzyme catalyses the addition of adenine to
4-
phosphopantotheine, consuming ATP and producing dephospho-CoA and
pyrophosphate, the fourth step in Coenzyme A synthesis. The final step in CoA
synthesis, ATP dependent phosphorylation of dephospho-CoA to CoA, is
performed by dephospho-CoA kinase, which is probably an additional catalytic
activity of the phosphopantotheine adenylyltransferase enzyme. Inhibition of
PanK,
or of phosphopantothenoylcysteine synthetase, or of
phosphopantothenoylcysteine
decarboxylase, or of phosphopantotheine adenylyltransferase would acutely
inhibit
CoA synthesis, and would decrease free CoA.
Sequestration of cellular CoA in the form of stable CoA-esters:
Certain synthetic agents are taken up by cells and esterified with CoA by
various cellular enzymes to form stable CoA-esters. A direct effect of such
agents is
to decrease free CoA by the amount of CoA that is incorporated into stable CoA-
esters. These CoA-esters may or may not have additional biological activities
within
the cell. Two examples of such synthetic agents are TOFA and etomoxir.
WO 01/34202 CA 02390501 2002-05-07 PCT/US00/31068
11
Administration of a sufficiently large dose of such an agent to a tumor cell
would
sequester enough CoA in the form of its stable CoA-ester to decrease free CoA
by a
functionally significant amount.
ADMINISTRATION OF THE COMPONENTS
Therapeutic agents according to this invention are preferably
formulated in pharmaceutical compositions containing the agent and a
pharmaceutically acceptable carrier. The pharmaceutical composition may
contain
other components so long as the other components do not reduce the
effectiveness of
the agent according to this invention so much that the therapy is ne~~ated.
Pharmaceutically acceptable carriers are well known, and one skilled in the
pharmaceutical art can easily select carriers suitable for particular routes
of
administration (Remington's Pharmaceutical Sciences, Mack Publishing Co.,
Easton,
PA, 1985).
The pharmaceutical compositions containing any of the a~.:ents of this
invention may be administered by parenteral (subcutaneously, intramuscularly,
intravenously, intraperitoneally, intraplcurally, intravesicularly or
intrathecally),
topical, oral, rectal, or nasal route, as necessitated by choice of drug. The
concentrations of the active agent in pharmaceutically acceptable carriers may
range
from 0.01 mM to 1 M or higher, so lOllg as the concentration does not exceed
an
acceptable level of toxicity at the point of administration.
Dose and duration of therapy will depend on a variety of factors.
including the therapeutic index of the drugs, disease type, patient aye,
patient
weight, and tolerance of toxicity. Dose will generally be chosen to achieve
serum
concentrations from about 0.1 pg/ml to about 100 pg/ml. Preferably, initial
dose
levels will be selected based on their ability to achieve ambient
concentrations
shown to be effective in in-vitro models, such as those described herein. and
in-vivo
models and in clinical trials, up to maximum tolerated levels. Standard
clinical
procedure prefers that chemotherapy be tailored to the individual patient and
the
systemic concentration of the chemotherapeutic agent be monitored regularly.
The
dose of a particular drug and duration of therapy for a particular patient can
be
determined by the skilled clinician using standard pharmacological approaches
in
CA 02390501 2002-05-07
WO 01/34202 PCT/US00/31068
12
view of the above factors. The response to treatment may be monitored by
analysis
of blood or body fluid levels of the agent according to this invention,
measurement
of activity of the agent or its levels in relevant tissues or monitoring
disease state in
the patient. The skilled clinician will adjust the dose and duration of
therapy based
on the response to treatment revealed by these measurements.
EXAMPLES
In order to facilitate a more complete understanding of the invention,
a number of Examples are provided below. However, the scope of the invention
is
not limited to specific embodiments disclosed in these Examples, which are for
purposes of illustration only.
Example 1. Inhibition of FAS in cells iir vitro
TOFA, Cerulenin, and C7~ all inhibited fatty acid synthesis in human
breast cancer cells. The human breast cancer cell lines, SKBR3 and MCF7 were
maintained in RPMI with 10% fetal bovine serum. Cells were screened
periodically
for Mycoplasma contamination (Gen-probe). All inhibitors were added as stock S
mg/ml solutions in DMSO. For fatty acid synthesis activity determinations,
SxIO'~
cells/well in 24 well plates were pulse labeled with [LI-'°C~-acetate
after exposure to
drug, and lipids were extracted and quantified as described previously
(Pfizer, et al.,
1988). For MCF7 cells, pathway activity was detcnnined after 2 hours of
inhibitor
exposure. SKBR3 cells demonstrated slower response to FAS inhibitors, possible
because of their extremely hi~7h FAS content. so pathway activity was
detenninecl
after 6 hours of inhibitor exposure.
In standard pulse labeling experiments in which breast cancer cell
lines, SKBR3 and MCF7 were labeled for 2 hours after exposure to FA synthesis
inhibitors, TOFA, C75, and cerulenin all inhibited [U'°C-acetates
incorporation into
lipids to a similar extent (Figure 1 B and D). In numerous similar experiments
(not
shown), TOFA maximally inhibited FA synthesis in the 1 to Sug/ml dose range in
all cell lines tested, and cerulenin and C75 maximally inhibited FA synthesis
in the
range of l Op.g/ml.
WO 01/34202 CA 02390501 2002-05-07 PCT/US00/31068
13
Example 2. Effect of the same inhibitors on cell growth
TOFA, Cerulenin, and C75 all inhibited fatty acid synthesis in human
breast cancer cells, but showed differential cytotoxicity. Cells and
inhibitors were as
described for Example 1. For clonogenic assays, 4x 1 OS cells were plated in
25 cmz
flasks with inhibitors added for 6 hours in concentrations listed. Equal
numbers of
treated cells and controls were plated in 60 mm dishes. Clones were stained
and
counted after 7 to I 0 days.
Although all inhibitors reduced FA synthesis to a similar degree,
TOFA was non-toxic or stimulatory to the cancer cell growth in the dose range
for
ACC inhibition, as measured by clonogenic assays, while cerulenin and C75 were
significantly cvtotoxic in the dose range for FAS inhibition (Figure 1 C and
E). The
profound difference between the cytotoxic effects of ACC and FAS inhibition
demonstrate that the acute reduction of fatty acid production per se is not
the major
source of cell injury after FAS inhibition.
1 S Example 3. Measurement of malonvl-CoA.
The most obvious difference in the expected results of inhibiting
these two enzymes was that malonyl-CoA levels should fall after ACC
inhibition,
but should increase after FAS inhibition. Although not previously investigated
in
eukarvotcs, recent data in E. coli have demonstrated elevated levels of
malonyl-CoA
resulting from exposure to cerulenin (Chohnan, et al., 1997, "Changes in the
size
and composition of intracellular pools of non-esterified coenzyme A and
coenzyme
A thioesters in aerobic and facultatively anaerobi bacteria," Applied and
Environmental Microbiology, 63:555-560). Malonyl-CoA levels were measured in
cells subjected to FAS inhibition and to inhibition by TOFA under conditions
described in Example 2.
Malonyl-CoA levels were measured in MCF-7 cells using the HPLC
method of Corkey, et al ("Analysis of acyl-coenzyme A esters in biological
samples,"Methods in Enzvmologt:~, 166:55-70). Briefly, 2.5 x 105 cells/well in
24
well plates were subjected to 1.2 ml of 10% TCA at 4° C after various
drug
treatments. The pellet mass was recorded and the supernatant was washed 6
times
with 1.2 ml of ether and reduced to dryness using vacuum centrifugation at
25° C.
WO 01/34202 CA 02390501 2002-05-07 PCT/IIS00/31068
14
Coenzyme-A esters were separated and quantitated using reversed phase HPLC on
a
p Supelco C18 column with a Waters HPLC system running Millenium'~ software
monitoring 254 nm as the maximum absorbance for coenzyme-A. The following
gradients and buffers were utilized: Buffer A: 0.1 M potassium phosphate, pH
5.0,
5 Buffer B: 0.1 M potassium phosphate, pH 5.0, with 40% acetonitrile.
Following a
20 min. isocratic run with 92% A, 8% B at 0.4 ml/min, flow was increased to
0.8
ml/min over one minute whereupon a linear gradient to 10% B was run until 24
min.
then held at 10% B until 50 min. where a linear gradient was run to 100% B at
55
min., completing at 60 min. The following coenzyme-A esters (Sigma) were run
as
standards: malonyl-CoA, acetyl-CoA, glutathione-CoA, succinyl-CoA, HMG-CoA,
and free CoA. Samples and standards were dissolved in 50 ~1 of buffer A.
Coenzyme-A esters eluted sequentially as follows: malonyl-CoA, glutathione-
CoA,
free CoA, succinyl-CoA, HMG-CoA, and acetyl-CoA. Quantitation of coenzyme-A
esters was performed by the Millenium3'' software.
I S Direct measurement of coenzyme-A derivatives in MCF-7 cells by
reversed phase HPLC of acid soluble extracts from drug treated cells confirmed
that
both cerulenin and C75 caused a rapid increase in malonyl-CoA levels while
TOFA
reduced malonyl-CoA levels. Figure 2A is a representative chromatograph
demonstrating the separation and identification of coenzyme-.A derivatives
important in cellular metabolism. Malonyl-CoA is the first of these to elute,
with a
column retention time of 19-22 minutes. The overlay of chromatographs in
Figurc
2B shows that cerulenin treatment lead to a marked increase in malonyl-CoA
over
the control while TOFA caused a significant reduction. The chemical identity
of the
malonyl-CoA was independently confirmed by spiking samples with standards (not
shown).
Malonyl-CoA levels were markedly increased with FAS inhibition
and reduced by TOFA. Analysis of multiple experiments in Figure 2C
demonstrated
that following a 1 hour exposure to cerulenin or C75 at 10 ~g/ml, malonyl-CoA
levels increased by 930% and 370% respectively, over controls, while TOFA
treatment (20 pg/ml) led to a 60% reduction of malonyl-CoA levels. The
concentration of TOFA required for maximal reduction of malonyl-CoA levels was
WO 01/34202 CA 02390501 2002-05-07 pCT/US00/31068
4 fold higher than the dose for pathway inhibition in Figure 1 B and D.
However,
optimal cultures for extraction of CoA derivatives had S fold higher cell
density than
the cultures used in the other biochemical and viability assays presented.
The remarkable increase in malonyl-CoA after FAS inhibition can be
5 attributed in part to the release of long-chain fatty acyl-CoA inhibition of
ACC
leading to an increase in ACC activity (Figure 1 A). Moreover, the cerulenin-
induced increase in malonyl-CoA levels occurred within 30 minutes of treatment
(930 +/-15% increase over control, not shown), within the time frame of FA
synthesis inhibition, and well before the onset of DNA synthesis inhibition or
early
10 apoptotic events (Pfizer, et al., 1998). Thus, high levels of malonyl-CoA
were a
characteristic effect of FAS inhibitors and temporally preceded the other
cellular
responses, including apoptosis.
The levels of cerulenin or C7~ which induce high levels of malonyl-
CoA _are cytotoxic to human breast cancer cells as measured by clonogenic
assays
15 and flow-cytometric analysis of apoptosis using merocyanin 450 staining
(Pfizer, et
al., 1998). FAS inhibition causes high malonyl-CoA levels by inhibiting its
consumption through FAS inhibition, with concomitant stimulation of synthesis
by
relieving the inhibitory effect of long-chain acyl-CoA's upon ACC activity
(Figure
2).
Example 4. TOFA rescue of FAS inhibition
TOFA rescue of FAS inhibition demonstrates that high Icvcls of
malonyl-CoA are responsible for cancer cell cytotoxicity. If the elevated
levels of
malonyl-CoA resulting from FAS inhibition were responsible for cvtotoxicity,
then
it should be possible to rescue cells from FAS inhibition by reducing malonyl-
CoA
accumulation with TOFA. Co-administration of TOFA and cerulenin to SKBR3
cells (Figure 3A) abrogated the cytotoxic effect of cerulenin alone in
clonogenic
assays performed as described in Example 2. In MCF7 cells (Figure 3C), TOFA
produced a rescue of both cerulenin and C75 under similar experimental
conditions.
Representative flow cytometric analyses of SKBR3 cells (Figure 3B)
and MCF7 (Figure 3D) substantiated these findings, since TOFA rescued cells
from
cerulenin induced apoptosis. Apoptosis was measured by multiparameter flow
CA 02390501 2002-05-07
WO 01/34202 PCT/US00/31068
16
cytometry using a FACStarY~°S flow cytometer equipped with argon and
krypton
lasers (Becton Dickinson). Apoptosis was quantified using merocyanine 540
staining (Sigma), which detects altered plasma membrane phospholipid packing
that
occurs early in apoptosis, added directly to cells from culture (Pfizer, et
al., 1998;
Mower, et al., 1994, "Decreased membrane pospholipid packing and decreased
cell
size precede DNA cleavage in mature mouse B cell apoptosis, J. Inrmunol.,
152:4832-4842). In some experiments, chromatin conformational changes of
apoptosis were simultaneously measured as decreased staining with LDS-751
(Exciton) (Frey, et al., 1995, "Nucleic acid dyes for detection of apoptosis
in live
cells," Cvtonuety, 21:265-274). Merocyanine 540 [10~g/ml] was added as a
I mg/ml stock in water. Cells were stained with LDS-751 at a final
concentration of
100nM from a 1mM stock in DMSO. The mcrocyanine _540-positive cells were
marked by an increase in red fluorescence, collected at 575 +/- 20 nm, 0.5 to
2 logs
over merocyaninc 540-negative cells. Similarly, the LDS-751 dim cells
demonstrated a reduction in fluorescence of 0.5 to I.5 logs relative to normal
cells,
collected at 660 nm with a DF20 band pass filter. Data were collected and
analyzed
using CellQuest software (Becton Dickinson).
In these experiments, all LDS-751 dim cells were merocyanine 540
bright, however a population of merocyanine 540 brigin cells were detected
that
were not yet LDS-751 dim. All merocyaninc 540 bri~~ht cells were classified as
apoptotic. These experiments also confimed the differemial cvtotoxicitv
between
TOFA (<5% increase in apoptosis; no reduction in clonogenicity) compared to
cerulenin (>85% apoptosis; 70% reduction in clonogenicity). Taken together,
these
studies show that high malonyl-CoA levels play a role in the cytotoxic effect
of FAS
inhibitors on cancer cells.
Example 5. Effect of FAS inhibitors on tumor cell growth in vivo
To determine if the effects of FAS inhibition seen in vitro would
translate to an in vivo setting requiring systemic activity, C75 was tested
against
subcutaneous MCF-7 xenografts in athymic nude mice, to quantitate effects on
FA
synthesis and the growth of established solid tumor. Previous studies have
demonstrated local efficacy of cerulenin against a human cancer xenograft
(Pfizer, et
CA 02390501 2002-05-07
WO 01/34202 PCT/US00/31068
17
al., 1996, "Inhibition of fatty acid synthesis delays disease progression in a
xenograft
model of ovarian cancer," Cancer Res., 56: 1189-1193), but were limited by the
failure of cerulenin to act systemically. The similar responses of breast
cancer cells
to cerulenin and C75 in vitro suggested that C75 might be effective in vivo
against
xenografted breast cancer cells.
Subcutaneous flank xenografts of the human breast cancer ccll line,
MCF-7 in nu/nu female mice (Harlan) were used to study the anti-tumor effects
of
C75 in vivo. All animal experiments complied with institutional animal care
guidelines. All mice received a 90-day slow-release subcutaneous estrogen
pellet
(Innovative Research) in the anterior flank 7 days before tumor inoculation.
10'
MCF-7 cells were xenografted from culture in DMEM supplemented with 10% FBS
and insulin 10 ~tg/ml.
Treatment began when measurable tumors developed about 10 days
after inoculation. Eleven mice (divided among two separate experiments of 5
and 6
mice each) were treated intraperitoneally with weekly doses of C75 at 30 mg/kg
in
0.1 ml RPMI. Dosing was based on a single dose LD,~ determination of 40 mg/kg
in BALB/c mice; 30 mg/kg has been well tolerated in outbred nude mice. Eleven
control mice (divided in the same way as the treatment groups) received RPMI
alone. Tumor volume was measured with calipers in three dimensions. Experiment
was terminated when controls reached the surrogate endpoint.
In a parallel experiment to determine fatty acid synthesis activity in
treated and control tumors, a group of MCF-7 xenografted mice were treated
with
C75 or vehicle at above doses and sacrificed after 3 hours. Tumor and liver
tissue
were ex oivo labeled with [U~4~C], lipids were extracted and counted as
described
(Rashid, et al., 1997).
In an additional parallel experiment to histologically examine treated
and control tumors, 6 C75 treated and 6 vehicle control mice were sacrificed 6
hours
after treatment. Tumor and normal tissues were fixed in neutral-buffered
formalin,
processed for routine histology, and immunohistochemistry for FAS was
performed.
Immunohistochemistry for FAS was performed on the MCF-7 xenografts using a
CA 02390501 2002-05-07
WO 01/34202 PCT/US00131068
18
mouse monoclonal anti-FAS antibody (Alo, et al., 1996) at 1:2000 on the Dako
Immunostainer using the LSAB2 detection kit.
Fatty acid synthesis pathway activity in tissues of xenografted mice
was determined by ex vivo pulse labeling with [U~4C]-acetate. The tumor
xenografts
had 10-fold higher FA synthesis activity than liver, highlighting the
difference in
pathway activity between benign and malignant tissues (Figure 4A). FAS
expression
in the MCF-7 xenograft paralleled the high level of FA synthesis activity
(Figure
4B). Intraperitoneal injections of C75 at 30 mg/kg reduced fatty acid
synthesis in ex
vivo labeled liver by 76% and in the MCF-7 xenografts by 70% within 3 hours
(Figure 4A). These changes in FA synthesis preceded histological evidence of
cytotoxicity in the xenograft, which became evident 6 hours after treatment
(Figures
4 C and 4D). The C75 treated xenografts showed numerous apoptotic bodies
throughout the tumor tissue, which were not seen in vehicle treated tumors.
Histological analysis of liver and other host tissues following C75 treatment
showed
no evidence of any short or long term toxicity (not shown).
C75 treatment of the xenografts leads to cytotoxicity and reduction in
tumor growth without injury to normal tissues. Tumor histology 6 hours
following a
30 mg/kg dose of C75 demonstrates significant cytotoxicity compared to control
tumor (Figures 4 C and 4D, attached preprint). Note the evidence of apoptotic
bodies in the C75 treated xenograft while examination of liver and other
organs
show no evidence of tissue injury (data not shown). Weekly intrapcritoneal C75
treatment retarded the growth of established subcutaneous MCF-7 tumors
compared
to vehicle controls, demonstrating a systemic anti-tumor effect (Figure 4E).
After
32 days of weekly treatments, there was a greater than eight-fold difference
in tumor
growth in the treatment group compared to vehicle controls. Similar to
cerulenin,
transient reversible weight loss was the only toxicity noted (Pfizer, et al.,
1996).
The systemic pharmacologic activity of C75 provided the first
analysis of the outcome of systemic FAS inhibitor treatment. The significant
anti-
tumor effect of C75 on a human breast cancer xenograft in the setting of
physiological levels of ambient fatty acids was similar to the in vitro result
in serum
WO 01/34202 CA 02390501 2002-05-07 pCT/US00/31068
19
supplemented culture, and was consistent with a cytotoxic mechanism
independent
of fatty acid starvation.
Example 6. Human cancer cells have high steady state levels of malonyl-CoA
lIi VIVO.
S The result in Example 5 suggested that malonyl-CoA accumulation
may not be a significant problem in normal tissues, possibly because FA
synthesis
pathway activity is normally low, even in lipogenic organs such as the liver.
It is of
further interest that, while malonyl-CoA was the predominant low molecular
weight
CoA conjugate detected in breast cancer cells in these experiments, other
studies
have reported predominantly succinyl-CoA and acetyl-CoA in cultured
hcpatocytes
(Corkey, 1988). The high level of malonyl-CoA in the tumor tissues reflects
the
high level of fatty acid synthesis in the tumor cells compared to
liver(Rashid. et al.,
1997).
Using the MCF7 human breast cancer xenograft model of Example 5,
malonyl-CoA levels were measured in the tumor xenog~raft and liver from the
same
animal using high-performance liquid chromatography. Figure 3 below shows high
levels of malonyl-CoA in the tumor tissue compared to the liver. In addition,
the
distribution of other CoA derivatives are markedly altered. For example. while
liver
has about 10 fold less malonyl-CoA compared to the xenograft, it has about 10
fold
higher levels of acetyl-CoA, and higher levels of other CoA derivatives,
particularly
succinyl-CoA. Differences in CoA derivative profiles may be indicative of
larger
differences in energy metabolism between cancer cells and hepatocytes.
For purposes of clarity of understanding, the foregoing invention has
been described in some detail by way of illustration and example in
conjunction with
specific embodiments, although other aspects, advantages and modifications
will be
apparent to those skilled in the art to which the invention pertains. The
foregoing
description and examples are intended to illustrate, but not limit the scope
of the
invention. Modifications of the above-described modes for carrying out the
invention that are apparent to persons of skill in medicine, immunology,
hybridoma
technology, pharmacology, and/or related fields are intended to be within the
scope
of the invention, which is limited only by the appended claims.
CA 02390501 2002-05-07
WO 01/34202 PCT/US00/31068
All publications and patent applications mentioned in this specification
are indicative of the level of skill of those skilled in the art to which this
invention
pertains. All publications and patent applications are herein incorporated by
reference to the same extent as if each individual publication or patent
application
5 was specifically and individually indicated to be incorporated by reference.