Note: Descriptions are shown in the official language in which they were submitted.
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
COMPOSITIONS AND METHODS FOR REGULATING ENDOGENOUS
INHIBITOR OF ATP SYNTHASE, INCLUDING TREATMENT FOR DIABETES
CROSS REFERENCE TO RELATED APPLICATION
This application claims the benefit of U.S. Provisional Patent
Application No. 60/164,622 filed November 10, 1999, which is incorporated
herein by
reference in its entirety.
TECHNICAL FIELD
The present invention relates generally to diabetes mellitus, and in
particular to compositions and methods for the diagnosis, prognosis and
treatment of
type 2 diabetes. More specifically, the invention relates to compositions and
methods
for the direct delivery of extracellular polypeptides to organelles, to the
use of increased
mitochondria) A'TP production to treat diabetes, to methods of using IF 1 (the
inhibitory
factor of mitochondria) A'TPase) and derivatives thereof as therapeutic agents
for
diabetes mellitus, and to methods of using IF 1 and derivatives thereof as
reagents for
assays designed to identify agents that either (i) cause or contribute to, or
(ii) ameliorate
or treat, diabetes mellitus.
BACKGROUND OF THE INVENTION
Type 2 diabetes mellitus, or ''late onset" diabetes, is a common,
degenerative disease affecting 5 to 10 percent of the population in developed
countries.
The propensity for developing type 2 diabetes mellitus ("type 2 DM") is
reportedly
maternally inherited, suggesting a mitochondria) genetic involvement.
(Alcolado, J.C.
and Alcolado, R., Br. Med. J. 302:1178-1180 (1991); Reny, S.L., International
J.
Epidem. 23:886-890 (1994)). Diabetes is a heterogeneous disorder with a strong
genetic component; monozygotic twins are highly concordant and there is a high
incidence of the disease among first degree relatives of affected individuals.
Current pharmacological therapies for type 2 DM include injected
insulin, and oral agents that are designed to lower blood glucose levels.
Currently
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
available oral agents include (i) the sulfonylureas, which act by enhancing
the
sensitivity of the pancreatic beta cell to glucose, thereby increasing insulin
secretion in
response to a given glucose load; (ii) the biguanides, which improve glucose
disposal
rates and inhibit hepatic glucose output; (iii) the thiazolidinediones, which
improve
peripheral insulin sensitivity through interaction with nuclear peroxisome
proliferator-
activated receptors (PPAR, see, e.g., Spiegelman, 1998 Diabetes 47:507-514;
Schoonjans et al., 1997 Curr. Opin. Lipidol. 8:159-166; Staels et al., 1997
Biochimie
79:95-99), (iv) repaglinide, which enhances insulin secretion through
interaction with
ATP-dependent potassium channels; and (v) acarbose, which decreases intestinal
absorption of carbohydrates.
At the cellular level, the degenerative phenotype that . may be
characteristic of late onset diabetes mellitus includes indicators of altered
mitochondrial
respiratory function, for example impaired insulin secretion, decreased ATP
synthesis
and increased levels of reactive oxygen species. Studies have shown that type
2 DM
may be preceded by or associated with certain related disorders. For example,
it is
estimated that forty million individuals in the U.S. suffer from impaired
glucose
tolerance (IGT'). Following a glucose load, ciruculating glucose
concentrations in IGT
patients rise to higher levels, and return to baseline levels more slowly,
than in
unaffected individuals. A small percentage of IGT individuals (5-10%) progress
to
non-insulin dependent diabetes (NIDDM) each year. This form of diabetes
mellitus,
type 2 DM, is associated with decreased release of insulin by pancreatic beta
cells and a
decreased end-organ response to insulin. Other symptoms of diabetes mellitus
and
conditions that precede or are associated with diabetes mellitus include
obesity, vascular
pathologies, peripheral and sensory neuropathies and blindness.
It is clear that none of the current pharmacological therapies corrects the
underlying biochemical defect in type 2 DM. Neither do any of these currently
available treatments improve all of the physiological abnormalities in type 2
DM such
as impaired insulin secretion, insulin resistance and/or excessive hepatic
glucose output.
In addition, treatment failures are common with these agents, such that multi-
drug
therapy is frequently necessary.
2
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
Due to the strong genetic component of diabetes mellitus, the nuclear
genome has been the main focus of the search for causative genetic mutations.
However, despite intense effort, nuclear genes that segregate with diabetes
mellitus are
rare and include, for example, mutations in the insulin gene, the insulin
receptor gene
and the glucokinase gene. By comparison, although a number of altered
mitochondria)
genes that segregate with diabetes mellitus have been reported (see generally
e.g.,
PCT/I1S95/04063), relationships amongst mitochondria) and extramitochondrial
factors
that contribute to cellular respiratory and/or metabolic activities as they
pertain to
diabetes remain poorly understood.
Mitochondria are the main energy source in cells of higher organisms,
and provide direct and indirect biochemical regulation of a wide array of
cellular
respiratory, oxidative and metabolic processes. Such processes include
electron
transport chain (ETC) activity, which drives oxidative phosphorylation to
produce
metabolic energy in the form of adenosine triphosphate (ATP), and which
controls
mitochondria) regulation of intracellular and intramitochondrial calcium
homeostasis.
Mitochondria) ultrastructural characterization reveals the presence of an
outer mitochondria) membrane that serves as an interface between the organelle
and the
cytosol, a highly folded inner mitochondria) membrane that appears to form
attachments
to the outer membrane at multiple sites, and an intermembrane space between
the two
mitochondria) membranes. The subcompartment within the inner mitochondria)
membrane is commonly referred to as the mitochondria) matrix. (For a review,
see,
e.g., Ernster et al., 1981 J. Cell Biol. 91:227s.) The cristae, originally
postulated to
occur as infoldings of the inner mitochondria) membrane, have recently been
characterized using three-dimensional electron tomography as also including
tube-like
conduits that may form networks, and that can be connected to the inner
membrane
and/or the intermembrane space by open, circular (30 nm diameter) junctions
(Perkins
et al., 1997, Journal of Structural Biology 119:260-272). While the outer
membrane is
freely permeable to ionic and non-ionic solutes having molecular weights less
than
about ten kilodaltons, the inner mitochondria) membrane exhibits selective and
3
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
regulated permeability for many small molecules, including certain canons, and
is
impermeable to large (> ~10 kDa) molecules.
Four of the five multi-subunit protein complexes (Complexes I, III, I V
and V) that mediate ETC activity are localized to the inner mitochondria)
membrane.
The remaining ETC complex (Complex II) is situated in the matrix. In at least
three
distinct chemical reactions known to take place within the ETC, protons are
moved
from the mitochondria) matrix, across the inner membrane, to the intermembrane
space.
This disequilibrium of charged species creates an electrochemical membrane
potential
of approximately 220 mV referred to as the "protonmotive force" (PMF). The
PMF,
which is often represented by the notation Op, corresponds to the sum of the
electric
potential (~~fm) and the pH differential (OpH) across the inner membrane
according to
the equation
Op = 0'hm - ZOpH
wherein Z stands for -2.303 RT/F. The value of Z is -59 at 25°C when 0p
and O~I'm are
expressed in mV and ~pH is expressed in pH units (see, e.g., Ernster et al.,
J. Cell Biol.
91:227s, 1981 and references cited therein).
~'~'m provides the energy for phosphorylation of adenosine diphosphate
(ADP) to yield ATP by ETC Complex V, a process that is coupled
stoichiometrically
with transport of a proton into the matrix. D~fm is also the driving force for
the influx
of cytosolic Ca'+ into the mitochondrion. Even fundamental biological
processes. such
as translation of mRNA molecules to produce polypeptides, may be dependent on
O~I'm
(Cote et al., J. Biol. Chem. 265:7532-7538, 1990). Under normal metabolic
conditions,
the inner membrane is largely impermeable to proton movement from the
intermembrane space into the matrix, leaving ETC Complex V as the primary
means
whereby protons can return to the matrix. When, however, the integrity of the
inner
mitochondria) membrane is compromised, as occurs during mitochondria)
permeability
transition (MPT) that accompanies certain diseases associated with altered
mitochondria) function, protons are able to bypass the conduit of Complex V
without
generating ATP, thereby uncoupling respiration from ATP production. During
MPT,
4
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
D~I'm collapses and mitochondria) membranes lose the ability to selectively
regulate
permeability to solutes both small (e.g., ionic Caz', Na+, K+ and H+) and
large (e.g.,
proteins). Loss of mitochondria) potential also appears to be a critical event
in the
progression of diseases associated with altered mitochondria) function such as
diabetes
mellitus, and also including degenerative diseases such as Alzheimer's
Disease;
Parkinson's Disease; Huntington's disease; dystonia; Leber's hereditary optic
neuropathy; schizophrenia; mitochondria) encephalopathy, lactic acidosis, and
stroke
(MELAS); cancer; psoriasis; hyperproliferative disorders; mitochondria)
diabetes and
deafness (MIDD) and myoclonic epilepsy ragged red fiber syndrome.
As noted above, biochemical energy is produced by mitochondria)
oxidative phosphorylation, whereby electrons are transported along the ETC
from donor
NADH and ultimately transferred to acceptor oxygen in a process coupled to ATP
synthesis. In a manner that is dependent on the electrochemical proton
gradient across
the inner mitochondria) membrane as described above, ETC Complex V (ATP
synthase,
also referred to as FoF,ATPase or ATPase herein) is capable of reversibly
interconverting the reactants ADP plus energy into ATP, in response to
cellular energy
demand. ATP synthase occurs as a multi-component complex of at least 16
differenct
polypeptides (Walker et al., 1994 FEBS Lett. 346:39), including the
transmembrane Fo
portion in which resides proton pump activity, and the F, extramembrane
portion having
catalytic (e.g., ATP synthesis or ATP hydrolysis) activity. The globular
catalytic F,
ATP synthase portion comprises six polypeptides (subunits a, (3, x, 8, s and
the ATP
synthase inhibitor protein IF,), which are encoded by nuclear genes and are
imported
into the mitochondria during mitochdrial biogenesis. Enzyme complexes similar
to
mammalian ATP synthase are found in all cell types and in chloroplast and
bacterial
membranes.
Regulation of ATP production is mediated in part by IF1 (also notated
IF,), which inhibits catalytic activity of the ATP synthase F, portion (see,
e.g., Pullman
et al., 1963 J. Biol. Chem. 238:3762; Tuena et al., 1988 Biochem. Cell Biol.
66:677;
Walker et al., 1987 Biochem. 26:8613; Higuti et al., 1993 Biochim. Biophys.
Acta
1172:311; U.S. Patent No. 5,906,923; and references cited therein). Mature IF1
protein
5
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
is approximately 84 amino acids in length (9.6 kDa) and is synthesized as an
approximately 105 amino acid precursor protein from which the N-terminal
signal
sequence is cleaved after importation into mitochondria. IF1 features pH-
sensitive,
primarily alpha-helical structure that is highly conserved in eukaryotes such
as yeast
and mammals (Lebowitz et al. 1993 Arch. Biochem. Biophys. 301:64). In the
alpha
helix conformation IF 1 is inactive as an ATP synthase inhibitor, but at
pH<6.7 IF 1 loses
its helical structure and is activated to bind to the catalytic portion ~i
(and possibly a)
subunit and inhibit ATP synthase (Jackson et al., 1988 FEBS Lett. 229:224;
Mimura et
al., 1993 J. Biochem. 113:350). IF1 inhibition of ATPase activity may also be
influenced by mitochondria) membrane potential and/or by IF 1 interactions
with
phospholipids (see, e.g., Solaini et al., 1997 Biochem J. 327:443 and
references cited
therein). IF 1 and related proteins are described, for example, in W098/33909
and
references cited therein.
Mitochondria) energy production is related to glucose homeostasis
primarily through the regulation of glucose stimulated insulin secretion
(GSIS).
According to the generally accepted paradigm of glucose-mediated insulin
secretion, the
initial step is uptake of glucose into the (3-cells via glut 2 glucose
transporters (Figure
2). Uptake significantly exceeds glucose utilization and is therefore not rate-
limiting
for the sequence of events that triggers insulin release (Matschinsky,
Diabetes, 45:223-
241 (1996); Newgard and McGarry, Artnu Rev Biochem, 64:689-719 (1995)).
Rather, it
is the subsequent phosphorylation of glucose to glucose-6-phosphate (G6P) that
appears
to define the setpoint at which secretion is initiated. Pancreatic islets of
Langerhans
("islets") contain both a low Km (hexokinase I) and a high Km (glucokinase =
hexokinase IV) glucose phosphorylating activity. The Km of glucokinase is 6-11
mM,
while that of hexokinase I is 10-100 ~M. Furthermore, hexokinase I is
inhibited by its
product, G6P, while glucokinase is not. The majority of glucose
phosphorylating
activity in (3-cells is accounted for by the high Km glucokinase. The low Km
hexokinase I is believed to be inactive in the islets due to inhibition by
G6P. As a
result, insulin secretion normally occurs when the blood glucose begins to
rise above
the physiological level of ~5.5 mM.
6
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
Glucokinase is bound to the outer surface of the mitochondria in beta
cells through its interaction with the mitochondria) membrane protein porin
(also called
VDAC or voltage-dependent anion channel) (Malaisse-Lagae and Malaisse, Biochem
Med Metab Biol, 39:80-89, (1988); Sener et al., Arch Biochem Biophys, 251:61-
67
(1986); Muller et al., Arch Biochem Biophys, 308:8-23 (1994)). This situation
is
analogous to the interaction of hexokinase II with mitochondria in liver and
skeletal
muscle, and hexokinase I with mitochondria in liver (Gerbitz et al., Diabetes
45:113-
126 (1996); Weiler et al., Biochem Med, 33:223-235 (1985); Adams et al.,
Biochim
Biophys Acta 932:195-205 (1988)). Because porin is associated with the
mitochondria)
adenine nucleotide translocator (ANT), binding of GK to the pore may
facilitate
delivery of oxidatively produced ATP to the enzyme, which preferentially uses
ATP
produced by the mitochondria (Rasschaert and Malaisse, Biochim Biophys Acta,
1015:353-360 (1990)). Delivery of ADP back to the mitochondria for resynthesis
of
ATP by complex V may also be a function of this association (Laterveer et al.,
In
Gnaiger E, Gellerich FN, and Wyss M (eds.): Modern Trends in Biothermokinetics
3,
New York: Plenum, pp.186-190 (1994)). The importance of glucokinase as the
glucose
sensor is illustrated by the development of diabetes in individuals who have
mutations
of the glucokinase gene (Froguel et al., N Engl J Med, 328:697-702 ( 1993);
Bell et al.,
Annu Rev Physiol, 58:171-186 (1996)). Maturity Onset Diabetes of the Young
(MODY) is a form of diabetes mellitus that resembles NIDDM clinically, but has
its
onset before the age of 25, is generally milder, and has an autosomal dominant
mode of
transmission. At least three distinct mutations have been identified in MODY
families
(Bell et al., Annu Rev Physiol, 58:171-186 (1996)). MODY 2 is characterized by
mutations of the glucokinase gene, resulting in a predicted 50-100% decrease
in
glucokinase activity and impaired insulin secretion (Froguel et al., N Engl J
Med,
328:697-702 (1993); Hattersley et al., Lancet, 339:1307-1310 (1992)). The
occurrence
of diabetes in heterozygous individuals who have some residual glucokinase
activity
underscores the role of glucokinase as the rate limiting glucose sensor of the
beta cell.
Following glucose phosphorylation, the subsequent fate of G6P is almost
entirely via metabolism through the glycolytic pathway, because the pentose
phosphate
7
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
shunt is relatively inactive in pancreatic (3-cells (Asheroft et al., Biochem
J, 126:525-
532 (1972)), and glycogen synthesis accounts for no more than 7% of glucose
flux
(Meglasson and Matschinsky, Diabetes Metab Rev, 2:163-214 ( 1986)). The
glycolytic
pathway distal to G6P appears particularly important for insulin secretion
with regards
to the production of NADH (Dukes et al., J Biol Chem, 269:10979-10982 (1994);
MacDonald and Fahien, Arch Biochem Biophys, 279:104-108 (1990)), which is
efficiently shuttled from the cytosol to the mitochondria. There, it enters
the electron
transport chain at complex 1 and fuels oxidative production of ATP. The next
clear-cut
correlation between cellular metabolism and insulin secretion is the rise in
the
intracellular ATP:ADP ratio (Erecinska et al., Biochim Biophys Acta, 1101:271-
295
( 1992); Longo et al., J Biol Chem, 266:9314-9319 ( 1991 ); Ashcroft et al.,
Biochem J,
132:223-231 ( 1973)), which triggers closure of the ATP-sensitive K+ channel
at the
(3-cell plasma membrane, resulting in depolarization of the cell. The increase
in
ATP:ADP following a glucose load is believed to be due to a rise in ATP of
predominantly oxidative origin (Malaisse, Int J Biochem, 5:593-701 (1992)). In
fact,
using a series of glycolytic inhibitors, Dukes et al. (J .Biol Chem, 269:10979-
10982
(1994)) demonstrated that only oxidatively derived ATP could trigger closure
of the K+
channel in ~3-cells. This membrane depolarization leads to opening of Caz+
channels
with influx of calcium to the cytosol. It is the rise in intracellular calcium
that
ultimately causes the exocytosis of insulin. Using (3-cells in culture (HIT),
Civelek and
coworkers (Civelek et al, Biochem J, 318:615-621 ( 1996a); Civelek et al.,
Biochem J,
315:1015-1019 (1996b)) have confirmed the proposed temporal relationship,
establishing that glucose phosphorylation precedes the rise in ATP:ADP, which
precedes the rise in intracellular calcium.
According to this model of glucose-mediated insulin secretion, the ~i-cell
mitochondria play an important role in insulin release. In cellular studies,
manipulation
of mitochondrial function can alter normal glucose homeostasis. Thus, for
instance,
glucose-stimulated insulin secretion can be abrogated at the cellular level by
a variety of
metabolic inhibitors, including oligomycin, azide, antimycin A, rotenone,
cyanide, and
the uncouples FCCP (MacDonald and Fahien, Arch Biochem Biophys, 279:104-108
8
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
(1990); Detimary et al., Biochem J, 297:455-461 (1994); Dukes et al., J Biol
Chem,
269:10979-10982 (1994); Kiranadi et al., FEBS Lett, 283:93-96 (1991)). Using
oligomycin to inhibit ATP synthase, the catalytic activity of ATP synthase has
been
shown to be tightly coupled to insulin secretion, such that even a minor
defect in the
activity of the enzyme is predicted to cause a similar impairment of glucose-
stimulated
insulin secretion (Anderson, Drug Development Research, 46(1):47-67 (1999)).
Similarly, decreasing all mitochondrial-encoded enzyme activities by
depleting mtDNA eliminates glucose stimulated insulin secretion. Soejima and
coworkers (J Biol Chem, 271:26194-26199 (1996)) used bis-4-piperidyl
dichloride to
deplete mtDNA from the mouse pancreatic (3-cell line MIN6. These cells
expressed no
detectable mitochondrially encoded proteins, no cytochrome oxidase activity,
and no
glucose-stimulated insulin secretion. Tsuruzoe and coworkers (Diabetes, 47:621-
631
(1998)) performed a similar series of experiments using MIN6 cells that were
depleted
of mtDNA with ethidium bromide. Their cell line also lost the ability to
secrete insulin
or to increase intracellular ATP in response to glucose, but retained the
ability to secrete
insulin in response to sulfonylurea or KCI. Kennedy and colleagues (Int J
Diabetes,
pp.l-11 (1998)) treated the rat-derived INS-1 cell line with ethidium bromide
to deplete
the majority of mtDNA, with a similar loss of ATP and insulin responses to
glucose. A
significantly different approach, using the antiviral compound dideoxycytidine
(ddC) to
deplete mtDNA from INS-1 cells, has also been described (Anderson et al.,
Diabetes,
47 Suppl 1:A260 (1998)). Like the cells constructed by Kennedy and coworkers
(Int J
Diabetes, pp.l-11 (1998)), p° INS-1 cells produced using ddC retained
normal basal
insulin secretion, but failed to increase insulin secretion in response to a
glucose
challenge. In contrast, a normal insulin secretory response to KCI was
observed in
these cells, suggesting that the insulin secretory machinery distal to the
mitochondria
was intact. Similarly, intracellular ATP levels did not change in response to
glucose in
these p° INS-1 cells (Anderson, Drug Development Research, 46:57-67
(1999)). The
shift from oxidative to glycolytic ATP production in this p° cell line
was also
demonstrated by an increase in lactate production by the p° cells as
compared to the
parental INS-1 cells.
9
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
Thus, there is little doubt about the importance of normal mitochondria)
function in glucose-stimulated insulin secretion. However, the role of
mitochondria in
glucose utilization, the other key component of glucose homeostasis, is not
well
understood. The occurrence of mild to moderate insulin resistance in some
cases of
mitochondria) diabetes suggests that mitochondria) function may be involved in
insulin
sensitivity (Sue et al., Lancet 341:437-438 (1993); van den Ouweland et al.,
Nature
Genetics, 1:368-371 (1992); Kishimota et al., Diabetologia, 38:193-200
(1995)).
Moreover, two separate studies have shown an increased incidence of mtDNA
alterations in populations of patients with NIDDM or impaired glucose
tolerance (which
is characterized by insulin resistance) as compared to individuals with normal
glucose
tolerance (Poulton et al., Diabetologia, 41:54-48 (1998); Liang et al.,
Diabetes, 46:920-
923 (1997)). The role of mitochondria in peripheral insulin sensitivity may
relate to the
interaction of hexokinase with the mitochondria) protein porin. As noted
above,
hexokinase associates with the mitochondria in skeletal muscle, resulting in
activation
of the enzyme (De Vos et al., Biochem Int, 24:117-121 (1991); Adams, Bioehim
Biophys Acta, 932:195-205 (1988); Weiler, Biochem Med, 33:223-235 (1985)), and
facilitating delivery of ATP to the enzyme. The specific effects of
mitochondria)
mutations and mitochondria) dysfunction on the activity of hexokinase remain
to be
determined, but may contribute to impaired insulin-mediated glucose
utilization. In
individuals with the common form of NIDDM, hexokinase activity in skeletal
muscle
was reported to be low (Vestergaard et al., J Clin Invest, 96:2639-2645
(1995);
Kruszynska et al., Diabetes, 47:A66 (1998)), and failed to increase normally
in during a
hyperinsulinemic clamp study (Kruszynska et al., Diabetes, 47:A66 (1998)).
Although
Simoneau and Kelley (J Appl Physiol, 83:166-171 (1997)) observed a slight
increase
rather than a decrease in hexokinase activity in NIDDM skeletal muscle, they
documented an overall decline in oxidative enzyme activities relative to
glycolytic
activities. While it is not yet clear whether such alterations in metabolism
in NIDDM
are primary or secondary events, these observations further illustrate a
potential role for
mitochondria) metabolism in peripheral glucose utilization.
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
In addition to their role in energy production in growing cells,
mitochondria (or, at least, mitochondria) components) participate in
programmed cell
death (PCD), also known as apoptosis (Newmeyer et al., Cell 79:353-364, 1994;
Liu et
al., Cell 86:147-157, 1996). Apoptosis is apparently required for normal
development
of the nervous system and for proper functioning of the immune system.
Moreover,
some disease states are thought to be associated with either insufficient or
excessive
levels of apoptosis (e.g., cancer, autoimmune diseases and possibly certain
forms of
diabetes in the first instance, and stroke damage and neurodegeneration in
Alzheimer's
disease in the latter case). For general reviews of apoptosis, and the role of
mitochondria therein, see, e.g., Green and Reed (Science 281:1309-1312, 1998),
Green
(Cell 94:695-698, 1998) and Kroemer (Nature Medicine 3:614-620, 1997).
Clearly there is a need for improved diagnostic methods for early
detection of a risk for developing type 2 DM, and for better therapeutics that
are
targeted to correct biochemical and/or metabolic defects responsible for this
disease,
regardless of whether such a defect underlying altered mitochondria) function
may have
mitochondria) or extramitochondrial origins. The present invention provides
compositions and methods related to exploiting the regulation of glucose-
stimulated
insulin secretion by mitochondria) energy production to at least partially
overcome the
inadequate GSIS in type 2 DM, and offers other related advantages.
SUMMARY OF THE INVENTION
It is an aspect of the present invention to provide a method for
identifying an agent that alters mitochondria) ATP production, comprising:
comparing
(i) a level of binding of an endogenous inhibitor of ATP synthase to an ATP
synthase
subunit in the presence of a candidate agent to (ii) the level of binding of
an endogenous
inhibitor of ATP synthase to an ATP synthase subunit in the absence of the
candidate
agent, wherein an altered level of binding indicates that the agent alters
mitochondria)
ATP production. In certain embodiments the endogenous inhibitor of ATP
synthase is
an IFI, in certain further embodiments the IF1 is a mammalian IF1, and in
certain
11
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
further embodiments the mammalian IF1 is a mouse IF1, .a rat IF1, a rabbit
IF1, a
bovine IF 1, a canine IF 1, a non-human primate IF 1 or a human IF 1.
In another embodiment the invention provides a method for identifying
an agent that alters mitochondria) ATP production, comprising contacting, in
the
absence and presence of a candidate agent, an isolated IF 1 polypeptide and an
isolated
mitochondria) ATP synthase, wherein the ATP synthase is capable of ATP
synthesis,
under conditions and for a time sufficient for ATP production to occur; and
comparing a
level of ATP production by the ATP synthase in the presence of the candidate
agent to a
level of ATP production in the absence of the candidate agent, and therefrom
identifying an agent that alters mitochondria) ATP production.
In another embodiment the invention provides a method of treating
diabetes comprising administering to a patient in need thereof an effective
amount of a
compound that (a) increases the synthesis of mitochondria) ATP in cells, (b)
decreases
the hydrolosis of mitochondria) ATP in cells, or (c) does both (a) and (b). In
certain
embodiments the compound is a composition that inhibits one or more activities
of IF 1
or a composition that mimics IF 1. In another embodiment the invention
provides a
method of identifying agents useful for treating diabetes, comprising
contacting a
sample comprising mitochondria with a candidate agent and determining an
effect of
the compound on the amount of ATP in the sample, wherein a compound that
results in
increased ATP in the sample is identified as an agent useful for treating
diabetes.
In another embodiment there is provided by the present invention a
method of identifying an agent useful for treating diabetes, comprising
contacting a
sample comprising mitochondria with a candidate agent and determining an
effect of
the compound on the amount of ATP in the mitochondria, wherein a compound that
results in increased ATP in the mitochondria is identified as an agent useful
for treating
diabetes.
It is another aspect of the invention to provide an organelle-targeted
fusion protein comprising: (a) a first polypeptide portion comprising an
organelle
targeting sequence, wherein the organelle targeting sequence is capable of
promoting
the localization of a protein to a selected organelle; (b) a second
polypeptide portion
12
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
comprising a tat sequence; and (c) a third polypeptide portion having an amino
acid
sequence distinct from the first or the second polypeptide portion, wherein
the
organelle-targeted fusion protein is taken up by cells upon contact and is
preferentially
localized to the selected organelle.
In other embodiments, there is provided an organelle-targeted compound
comprising: (a) a first polypeptide portion comprising an organelle targeting
sequence,
wherein the organelle targeting sequence is capable of promoting the
localization of a
protein to a selected organelle; (b) a second polypeptide portion comprising a
"tat
sequence"; and (c) a nucleic acid portion, wherein the organelle-targeted
compound is
taken up by cells upon contact and is preferentially localized to the selected
organelle.
In certain further embodiments, the nucleic acid portion is a DNA, an RNA, an
oligonucleotide, a ribozyme, an expression cassette, an expression construct,
or a
peptide nucleic acid.
In certain other embodiments the organelle-targeted compound further
comprises a detectable label. In certain embodiments the selected organelle is
a
mitochondrion, and the organelle targeted sequence is a mitochondria)
targeting
sequence, which in certain further embodiments is SEQ ID NO: 10, SEQ ID NO:11
or
SEQ ID N0:14. In certain other embodiments, the selected organelle is a Golgi
apparatus, and the organelle targeted sequence is a Golgi targeting sequence.
In certain
other embodiments the selected organelle is a nucleus, and the organelle
targeted
sequence is a nuclear targeting sequence. In certain other embodiments the
selected
organelle is a chloroplast, and the organelle targeted sequence is a
chloroplast targeting
sequence. In certain other embodiments the selected organelle is the
endoplasmic
reticulum, and the organelle targeted sequence is an ER targeting sequence. In
another
embodiment the present invention provides a method of delivering an organelle-
targeted
fusion protein to an organelle in a cell comprising contacting the cell with
the organelle-
targeted fusion protein as just described.
In another embodiment the invention provides an expression construct
encoding an organelle-targeted fusion protein, wherein the organelle-targeted
fusion
protein comprises: (a) a first polypeptide portion comprising an organelle
targeted
13
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
sequence wherein the organelle targeted sequence is capable of promoting the
localization of a protein to a selected organelle; (b) a second polypeptide
portion
comprising a tat sequence; and (c) a third polypeptide portion having an amino
acid
sequence distinct from the first or the second polypeptide portion, wherein
the
organelle-targeted fusion protein is taken up by cells upon contact and is
preferentially
localized to the selected organelle. In another embodiment the invention
provides a
host cell comprising the expression construct as just described. In certain
other
embodiments the invention provides a method of producing an organelle-targeted
fusion
protein comprising culturing the host cell just described, and recovering the
organelle-
targeted fusion protein therefrom.
These and other aspects of the present invention will become evident
upon reference to the following detailed description. In addition, various
references are
set forth herein which describe in more detail certain aspects of this
invention, and are
therefore incorporated by reference in their entireties.
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1 shows the loss over time of mitochondrial DNA (mtDNA) from
INS-1 cells treated with ddC (panel 1A) and the secretion of insulin by these
cells and
the parent INS-1 cells in response to glucose treatment (panel 1B).
Figure 2 shows the results of experiments in which INS-1 cells and
mtDNA-depleted INS-1 cells are treated with glucose and measured for their
ability to
produce ATP (panel 2A) or lactate (panel 2B).
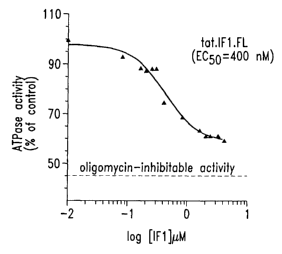
Figure 3 shows the inhibition of purified F1-ATPase by Aurovertin-B.
Figure 4 shows the inhibition of purified F1-ATPase by partially purified
bovine cardiac IF 1.
Figure 5 shows the amino acid sequence of rat IF 1 (SEQ ID N0:13) and
results from experiments in which two different synthetic polypeptides derived
from rat
IF 1 were tested for their ability to inhibit purified F 1-ATPase or FO-F 1-
ATPase.
14
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
Figure 6 shows the results of an experiment in which a synthetic
polypeptide corresponding to amino acids 42-58 of rat IF1 was tested for its
ability to
inhibit FO-F1-ATPase in rat alkaline submitochondrial particles.
Figure 7 shows gel electrophoretic (Coomassie stain) and western blot
characterization of recombinant IF 1 fusion proteins.
Figure 8 shows inhibition of ATP hydrolase activity in rat liver
submitochondrial particles by a recombinant IF 1 fusion protein.
Figure 9 shows enhancement of glucose-stimulated insulin secretion
(GSIS) by a recombinant IF 1 fusion protein.
DETAILED DESCRIPTION OF THE INVENTION
The present invention relates to compositions and methods for treating
type 2 DM, including methods for identifying an agent that alters
mitochondria) ATP
production. The invention is therefore directed in pertinent part to the
unexpected
observation that regulation of glucose-stimulated insulin secretion (GSIS) by
mitochondria) energy production can be manipulated in a manner that permits
restoration of some or all of the inadequate GSIS present in type 2 DM.
Accordingly, in certain preferred embodiments of the present invention,
mitochondria) function may be altered (e.g., increased or decreased in a
statistically
significant manner relative to an appropriate control, and in certain highly
preferred
embodiments, increased) by alteration of interactions between IF 1 and ATP
synthase as
described herein. In certain embodiments IF 1 interactions with ATP synthase
may be
altered, for example, by altering the binding of IF 1 to ATP synthase, and in
certain
embodiments the ability of IF1 to alter or regulate ATP synthase catalytic
activity,
which may include ATP synthase activity and/or ATP hydrolysis activity, may be
altered. By way of illustration and not limitation, the invention therefore
contemplates
compositions and methods for altering the association of IF 1 with at least
one ATP
synthase subunit, or for altering the expression level of IF1, or for altering
the activity
of IF 1.
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
For instance, the invention contemplates agents, and screening assays to
identify them, that interfere with IF 1 binding to ATP synthase subunits in a
manner that
prevents IF 1 inhibition of ATP synthase catalytic synthesis of ATP; the
invention also
contemplates agents, and screening assays to identify them, that interfere
with IF1-
encoding gene expression; the invention also contemplates mutant IF1 that is
altered in
its ability to interact with ATP synthase. Also, as noted above, IF 1 may bind
to sites on
ATP synthase Fl a and/or (3 subunits, such that the invention also
contemplates mutant
ATP synthase subunits which, by virtue of their mutation(s), are altered in
their ability
to functionally interact with IF 1.
The present invention is also directed in part to organellar-targeted
fusion proteins, and in particular embodiments, to IF 1 fusion proteins
comprising
organelle-selective or organelle-specific targeting sequences (OTS). Examples
of
organelles for which polypeptide targeting domains are known in the art are
briefly
described here. Based on the disclosure herein and as known in the art, a
person having
ordinary skill in the art may employ these or other polypeptide sequences (or
nucleic
acid sequences encoding them) and determine the appropriate structure and
delivery of
IF 1 fusion proteins to the desired organelles) by routine methods and without
undue
experimentation.
Mitochondria: As described above, mitochondria are the main energy
source in cells of higher organisms, and provide direct and indirect
biochemical
regulation of a wide array of cellular respiratory, oxidative and metabolic
processes,
including oxidative phosphorylation to produce ATP, intracellular calcium
homeostasis
and apoptosis. Thus, for instance, agents including mitochondrially targeted
fusion
proteins comprising mitochondrial targeting sequences, which fusion proteins
further
comprise polypeptide domains able to interact with and/or influence
mitochondrial
components, might have a variety of remedial, therapeutic, palliative,
rehabilitative,
preventative, prophylactic or disease-impeditive uses.
By way of example and not limitation, green fluorescent protein (GFP)
fusion protein derivatives have been targeted to the mitochondrial matrix
using
cytochrome c oxidase subunit IV protein sequences sequences (Llopis et al.,
Proc. Natl.
16
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
Acad. Sci. U.S.A. 95:6803-6808, 1993), to the mitochondria) intermembrane
space using
cytochrome c protein sequences (Mahajan et al., Nature Biotech. 16:547-552,
1998),
and to the outer membrane of mitochondria using hexokinase (Sui et al., Arch.
Biochem.
Biophys. 345:111-125, 1997), Bcl-2 or Bax (Mahajan et al., Nature Biotech.
16:547-
552, 1998) protein sequences. GFP fusion proteins have also been targeted to
mitochondria using 3-oxoacyl-CoA thiolase (Zhang et al., Biochem. Biophys.
Res.
Commun. 242:390-395, 1998), OSCP (Prescott et al., FEBS Letts. 411:97-101,
1997)
and BNIP3 (Yasuda et al., J. Biol. Chem. 273:12415-12421, 1998) protein
sequences.
Aequorin fusion protein derivatives have been targeted to mitochondria using
cytochrome c oxidase protein sequences (Proton et al., Biofactors 8:243-253,
1998;
Rizzuto et al., Nature 358:325-327, 1992). Other fusion proteins have been
described
that target mitochondria) sites using protein sequences from mitochondria) (or
bacterial)
thiolases (Arakawa et al., J. Biochem., Tokyo, 107:160-164, 1990), Fo-ATPase
subunit 9
(J. Biol. Chem. 271:25208-25212, 1996), manganese superoxide dismutase (Balzan
et
al., Proc. Nat). Acad. Sci. U.S.A. 92:4219-4223, 1995), and P-450(SCC)
(Kumamoto et
al., J. Biochem., Tokyo, 105:72-78, 1989).
Chloroplasts: The chloroplast is an organelle found in plant cells
wherein photosynthesis takes place. Photosynthesis, in addition to being an
integral
part of a plant cell's metabolism, is an important process that impacts many
other living
organisms as well. The reason for this is twofold: photosynthesis "fixes"
atmospheric
COZ into biologically usable carbohydrate (CHO)~ molecules and also produces
Oz
which is required by all aerobic organisms. Like mitochondria, chloroplasts
have a
double (outer and inner) membrane, contain their own DNA and have translation
factors
(ribosomes, tRNAs, etc.) that are distinct from those found in the cytoplasm.
Electron
microscopy demonstrates that, like mitochondria, chloroplasts have a highly
organized
internal ultrastructure which includes flattened membranous bodies known as
lamellae
or thykaloid discs. Chloroplasts are, however, typically much larger than
mitochondria;
in higher plants they are generally cylindrical in shape and range from about
5 to 10 ~
in length and from 0.5 to 2 ~ in diameter. Like mitochondria, which are
present in
greater numbers in certain tissues (e.g., liver) than others, chloroplasts
have greater
17
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
copy numbers in some tissues than others. For example, mature leaves contain
many
chloroplasts and the total amount of chloroplast DNA in such leaves is about
twice that
of nuclear DNA (Dope et al., J. Cell. Biol. 79:631-636, 1978).
By way of illustration and not limitation, fusion proteins have been
targeted to the chloroplast outer membrane by use of the SCE70 heat shock
protein
targeting sequence (Wu et al., J. Biol. Chem. 268:19384-19391, 1993). Other
targeting
sequences, such as those from the Rieske iron-sulfiur protein (Madueno et al.,
J. Biol.
Chem. 269:17458-17463, 1994), direct fusion proteins across the chloroplast
thylakoid
membrane. In certain embodiments wherein the invention contemplates fusion
proteins
capable of dual targeting to mitochondria and to chloroplasts, fusion proteins
comprising dual targeting polypeptide sequences may be employed as described
(Creissen et al., Plant J. 8:167-175, 1995; Huang et al., Plant Cell 2:1249-
1260, 1990).
Conversely,. when plant cells are being used and targeting to only
mitochondria or
chloroplasts is desired, care must be taken to ensure that a dual targeting
sequence is not
employed.
The Nucleus: The nucleus is the organelle that comprises most (from the
standpoint of information, if not mass) of a cell's DNA in the form of several
chromosomes (Mitochondria and chloroplasts have their own DNA molecules that
are
typically much smaller than the nuclear genomes, and thus encode fewer
functions;
however, as a cell contains only one nucleus and may contain many mitochondria
and/or chloroplasts, the total mass of the DNA molecules in these organelles
may
approach that of the nuclear DNA.) The nucleus is bounded by two membranes
collectively called the nuclear envelope (the membranes are known as the inner
and
outer nuclear membranes). Macromolecules, most particulary RNA molecules, are
conveyed to or from the cytosol through openings in the nuclear envelope
called nuclear
pores. In the case of the nucleus, by way of example and not limitation,
aequorin fusion
protein derivatives have been targeted to the nucleus using nucleoplasmin
protein
sequences (Badminton et al., J. Biol. Chem. 271:31210-31214, 1997).
Endoplasmic Reticulum: The endoplasmic reticulum (ER) is composed
of a series of flattened sheets, tubes and sacs that enclose a large
intracellular space.
18
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
The membrane of the ER is in structural continuity with the outer nuclear
membrane
and extends throughout the cytoplasm. Some functions of the ER include the
synthesis
and transport of membrane proteins and lipids. Generally speaking, two types
of ERs
may exist in a cell. Smooth ER are generally tubular in shape and are
typically devoid
of attached ribosomes; one major function of smooth ER is lipid metabolism.
Rough
ER typically occurs as flattened sheets, the cytosolic side of which is
usually associated
with many active (protein-synthesizing) ribosomes. As a non-limiting example,
aequorin fusion protein derivatives have been targeted to the endoplasmic
reticulum
using calreticulin protein sequences (Kendall et al., Biochem. Biophys. Res.
Commun.
189:1008-1016, 1992).
The Gobi Apparatus: The Golgi apparatus is a system of stacked,
flattened and membrane-enclosed sacs and is generally thought to be involved
in the
modification, sorting and packaging of macromolecules for secretion or for
delivery to
other subcellular compartments. Numerous small (> ~50 nM) membrane-enclosed
vesicles which are thought to comprise macromolecules in order to carry out
the
transport thereof between the Golgi apparatus and other subcellular
compartments.
Aequorin fusion protein derivatives, for example, have been targeted to
the Golgi membrane using galactosyltransferase, SNAP-25, connexin and 5-HT~~
receptor protein sequences (Burton et al., Mol. Cell. Biol. 7:419-434, 1996;
Marsault et
al., EMBO J. 16:1575-1581, 1997; Daguzan et al., Int. J. Dev. Biol. 39:653-
657, 1995).
GFP fusion proteins have been targeted to the Golgi apparatus using
galactosyltransferase protein sequences (Llopis et al., Proc. Natl. Acad. Sci.
U.S.A.
95:6803-6808, 1993).
In general, the organelle-targeted molecules of the invention have the
following structures:
( OET ) - ( CTS ) - ( OTS ) - ( MOI ) (Structure 1 ),
( OET ) - ( OTS ) - ( CTS ) - ( MOI ) (Structure 2),
( OET ) - ( CTS ) - ( MOI ) - ( OTS ) (Structure 3),
( OET ) - ( OTS ) - ( MOI ) - ( CTS ) (Structure 4),
19
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
( OET ) - ( MOI ) - ( OTS ) - ( CTS ) (Structure 5),
( OET ) - ( MOI ) - ( CTS ) - ( OTS ) (Structure 6),
( OTS ) - ( CTS ) - ( OET ) - ( MOI ) (Structure 7),
( OTS ) - ( OET ) - ( CTS ) - ( MOI ) (Structure 8),
( OTS ) - ( CTS ( MOI ( OET (Structure
) - ) - ) 9),
( OTS ) - ( OET ( MOI - ( CTS (Structure
) - ) ) 10),
( CTS ) - ( OTS ( MOI ( OET (Structure
) - ) - ) 11),
( CTS ) - ( OTS ( OET ( MOI (Structure
) - ) - ) 12),
( MOI ) - ( - ( - ( CTS (Structure
OET OTS ) 13),
) )
( MOI ) - ( - ( - ( OTS (Structure
OET CTS ) 14),
) )
and the like, wherein:
"OET" indicates an optional epitope tag, for example, a His tag (SEQ ID
NO:1), a FLAG~ epitope (SEQ ID N0:2), an AU1 epitope (SEQ ID N0:3), an AUS
epitope (SEQ ID N0:4), a c-myc epitope (SEQ ID NO:S), a Glu-Glu epitope (SEQ
ID
N0:6), an HA.11 epitope (SEQ ID N0:7), an IRS eptiope (SEQ ID N0:8), or a KT3
epitope (SEQ ID N0:9);
"CTS" indicates a cellular transport sequence, a preferred sequence
being that described as "TAT CTS" (SEQ ID NOS:10, 11 and 27) herein;
"OTS" indicates an organellar targeting sequence; and
"MOI" indicates a molecule of interest that one desires to target to a
specific organelle, for example, a polypeptide or a nucleic acid. In certain
preferred
embodiments, the MOI is an IF 1 polypeptide and in certain other preferred
embodiments the MOI is a nucleic acid sequence encoding an IF 1 polypeptide or
a
fragment, derivative, mutant or variant thereof as provided herein. In certain
embodiments, the MOI may be an antisense IF 1 nucleic acid, for example, the
reverse
complement of a portion of an IF1 encoding nucleic acid sequence (e.g.,
containing the
reverse complement of the ATG sequence encoding initiating methionine). In
other
embodiments, the MOI may be a mutated IF 1 polypeptide (or a nucleic acid
encoding
such a mutated IF1) selected for its failure to bind to ATP synthase. In other
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
embodiments, the MOI may be a mutated IF 1 polypeptide (or a nucleic acid
encoding
such a mutated IF 1 ) selected for its ability to activate ATP synthase
catalytic activity.
As shown above, these elements are arranged from the amino (N-)
terminal end on the left to the carboxy (C-) terminal end on the right. It
will be
appreciated by those skilled in the art that the order of these elements can
be altered,
and additional elements can be added to the organelle-targeted molecules so
long as the
functionality of the various elements is retained and delivery to the desired
organelles is
not impaired.
The CTS (cellular transport sequence) is a polypeptide capable of
delivering a covalently attached molecule into a target cell. A preferred CTS
is a HIV-1
tat protein or a HIV-1 tat-derived polypeptide, such as are described herein
or in U.S.
Patents 5,670,617; 5,674,980; 5,747,641; or 5,804,604. Tat proteins from other
viruses,
such as HIV-2 (Guyader et al., Nature 326:662-669, 1987), equine infectious
anemia
virus (Carroll et al., J. Virol. 65:3460-3467,1991 ), and simian
immunodeficiency virus
(Chakrabarti et al., Nature 328:543-547, 1987); Arya et al., Nature 328:548-
550, 1987)
are known. It should be understood that TAT polypeptides derived from those
tat
proteins fall within the scope of the present invention. TAT polypeptides
comprising
the region that mediates entry and uptake into cells can be defined using
known
techniques (see, e.g., Frankel et al., Proc. Nat). Acad. Sci. U.S.A. 86:7397-
7401, 1989)
and the present disclosure as a guide.
The present invention is directed in part to compositions and methods for
the modulation of mitochondria) energy production and GSIS for the treatment
of type 2
diabetes mellitus (type 2 DM). Certain useful embodiments of the invention
include:
(1) a method for treating diabetes that includes increasing
mitochondria) ATP synthesis or decreasing hydrolysis of mitochondria) ATP (or
both) in cells of an individual in need thereof;
(2) a method of screening for or identifying an agent that alters (e.g.,
increases or decreases) the binding interaction of at least one IF 1 protein
and at
least one ATP synthase subunit that comprises comparing the level of IF 1
binding to an ATP synthase subunit after contacting at least one IF1 protein
and
21
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
at least one ATP synthase subunit in the presence or absence of one or more
candidate agents or test compounds under conditions and for a time sufficient
to
permit IF 1 binding to the ATP synthase subunit(s);
(3) a method of screening for or identifying an agent that alters the
effect of an IF 1 protein on an ATP synthase catalytic activity that comprises
comparing the level of ATP synthase catalytic activity (e.g., synthase
activity
and/or hydrolase activity) in the presence of a candidate agent to the level
of
activity in the absence of the agent by contacting an IF 1 protein and a
catalytically competent ATP synthase under conditions and for a time
sufficient
to detect ATP synthase catalytic activity in the presence or absence of one or
more candidate agents or test compounds, and determining the activity of the
ATP synthase;
(4) a method of screening for or identifying an agent that influences
the activity of an IF1 protein, comprising contacting at least one cell
comprising
an IF 1 protein with one or more candidate agents or test compounds, and
measuring at least one mitochondrial activity; and
(5) a method of screening for or identifying an agent that alters the
expression of a nucleic acid that encodes an IF 1 protein that comprises
contacting at least one cell comprising a nucleic acid that encodes an IF 1
protein
with one or more candidate agents or test compounds and measuring expression
of an IF I protein.
DEFINITIONS AND GENERAL METHODS
Unless defined otherwise, all technical and scientific terms used herein
have the same meaning as commonly understood by one of ordinary skill in the
art to
which this invention is directed. Generally, the nomenclature used herein and
the
laboratory procedures in cell biology, chemistry, microbiology, molecular
biology, cell
science, cell culture and tissue culture described below are well known and
commonly
employed in the art. Conventional methods are used for these procedures, such
as those
provided in the art and various general references (Sambrook et al., Molecular
Cloning.
22
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
A Laboratory Manual, 2nd edition, Cold Spring Harbor Press, Cold Spring
Harbor,
N.Y. (1989)). Where a term is provided in the singular, the inventors also
contemplate
the plural of that term. The nomenclature used herein and the laboratory
procedures
described below are those well known and commonly employed in the art.
DEFINITIONS
"Membrane permeant derivative" refers to a chemical derivative of a
compound that increases membrane permeability of the compound. These
derivatives
are made better able to cross cell membranes because hydrophilic groups are
masked to
provide more hydrophobic derivatives. Also, the making groups can be designed
to be
cleaved from the compound within a cell to make the compound more hydrophilic
once
within the cell. Because the substrate is more hydrophilic than the membrane
permeant
derivative, it preferentially localizes within the cell (U.S. Patent No.
5,741,657 to Tsien
et al., issued April 21, 1998).
"Isolated polynucleotide" refers to a polynucleotide of genomic, cDNA,
PCR or synthetic origin, or some combination thereof, which by virtue of its
origin, the
isolated polynucleotide ( 1 ) is not associated with the cell in which the
isolated
polynucleotide is found in nature, or (2) is operably linked to a
polynucleotide that it is
not linked to in nature. The isolated polynucleotide can optionally be linked
to
promoters, enhancers, or other regulatory sequences.
"Isolated protein" refers to a protein of cDNA, recombinant RNA, or
synthetic origin, or some combination thereof, which by virtue of its origin
the isolated
protein ( 1 ) is not associated with proteins normally found within nature, or
(2) is
isolated from the cell in which it normally occurs, or (3) is isolated free of
other
proteins from the same cellular source, for example, free of cellular
proteins), or (4) is
expressed by a cell from a different species, or (5) does not occur in nature.
"Polypeptide" is used herein as a generic term to refer to native protein,
fragments, or analogs of a polypeptide sequence.
23
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
"Active fragment" refers to a fragment of a parent molecule, such as an
organic molecule, nucleic acid molecule, or protein or polypeptide, or
combinations
thereof, that retains at least one activity of the parent molecule.
"Naturally occurring" refers to the fact that an object can be found in
nature. For example, a polypeptide or polynucleotide sequence that is present
in an
organism, including viruses, that can be isolated from a source in nature and
which has
not been intentionally modified by man in the laboratory is naturally
occurring.
"Operably linked" refers to a juxtaposition wherein the components so
described are in a relationship permitting them to function in their intended
manner. A
control sequence operably linked to a coding sequence is ligated in such a way
that
expression of the coding sequence is achieved under conditions compatible with
the
control sequences.
"Control sequences" refer to polynucleotide sequences that effect the
expression of coding and non-coding sequences to which they are ligated. The
nature of
such control sequences differs depending upon the host organism; in
prokaryotes, such
control sequences generally include promoter, ribosomal biding site, and
transcription
termination sequences; in eukaryotes, generally, such control sequences
include
promoters and transcription termination sequences. The term control sequences
is
intended to include components whose presence can influence expression, and
can also
include additional components whose presence is advantageous, for example,
leader
sequences and fusion partner sequences.
"Polynucleotide" refers to a polymeric form of nucleotides of a least ten
bases in length, either ribonucleotides or deoxyribonucleotides or a modified
from of
either type of nucleotide. The term includes single and double stranded forms
of DNA
or RNA.
"Genomic polynucleotide" refers to a portion of the nuclear genome.
"Mitochondrial genomic polynucleotide" refers to a portion of the
mitochondria genome.
24
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
"Active genomic polynucleotide" or active portion of a genome" refer to
regions of a genome (nuclear or mitrochondrial) that can be up regulated, down
regulated or both, either directly or indirectly, by a biological process.
"Ribozyme" means enzymatic RNA molecules capable of catalyzing the
specific cleavage of RNA. The mechanism of ribozyme action involves sequence-
specific hybridization of the ribozyme molecule to complementary target RNA,
followed by endonucleolytic cleavage. Specific ribozyme cleavage sites within
any
potential RNA target are initially identified by scanning the target RNA
target for
ribozyme cleavage sites which include the sequences GUA, GUU and GUC. Once
identified, short RNA sequences between 15 and 20 ribonucleotides
corresponding to
the region of the target gene containing the cleavage site can be evaluated
for secondary
structural features which may render the oligonucleotide inoperable. The
suitability of
candidate targets can also be evaluated by testing accessibility to
hybridization with
complementary oligonucleotides using ribonuclease protection assays.
"Directly" in the context of a biological process or processes, refers to
direct causation of a process that does not require intermediate steps,
usually caused by
one molecule contacting or binding to another molecule (the same type or
different type
of molecule). For example, molecule A contacts molecule B, which causes
molecule B
to exert effect X that is part of a biological process.
"Indirectly" in the context of a biological process or precesses, refers to
indirect causation that requires intermediate steps, usually caused by two or
more direct
steps. For example, molecule A contacts molecule B to exert effect X which in
turn
causes effect Y.
"Sequence identity" refers to the proportion of base matches between
two nucleic acid sequences or the proportion of amino acid matches between two
amino
acid sequences. When sequence identity is expressed as a percentage, for
example 50%,
the percentage denotes the proportion of matches of the length of sequences
from a
desired sequence that is compared to some other sequence. Gaps (in either of
the two
sequences) are permitted to maximize matching; gap lengths of 15 bases or less
are
usually used, 6 bases or less are preferred with 2 bases or less more
preferred. When
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
using oligonuleotides as probes, the sequence identity between the target
nucleic acid
and the oligonucleotide sequence is preferably not less than 10 target base
matches out
of 20 (50% identity) and more preferably not less than about 60% identity, 70%
identity, 80% identity or 90% identity), and most preferably not less than 95%
identity.
S "Selectively hybridize" refers to detectably and specifically bind.
Polynucleotides, oligonucleotides and fragments thereof selectively hybridize
to target
nucleic acid strands, under hybridization and wash conditions that minimize
appreciable
amounts of detectable binding to nonspecific nucleic acids. High stringency
conditions
can be used to achieve selective hybridization conditions as known in the art.
Generally, the nucleic acid sequence identity between the polynucleotides,
oligonucleotides, and fragments thereof and a nucleic acid sequence of
interest will be
at least 30%, and more typically and preferably of at least 40%, 50%, 60%,
70%, 80%
or 90%.
Hybridization and washing conditions are typically performed at high
stringency according to conventional hybridization procedures. Positive clones
are
isolated and sequenced. For example, a full length polynucleotide sequence can
be
labeled and used as a hybridization probe to isolate genomic clones from an
appropriate
target library as they are known in the art. Typical hybridization conditions
and
methods for screening plaque lifts and other purposes are known in the art
(Benton and
Davis, Science 196:180 (1978); Sambrook et al., supra, (1989)).
Two amino acid sequences have share identity if there is a partial or
complete identity between their sequences. For example, 85% identity means
that 85%
of the amino acids are identical when the two sequences are aligned for
maximum
matching. Gaps (in either of the two sequences being matched) are allowed in
maximizing matching; gap lengths of S or less are preferred with 2 or less
being more
preferred. Alternatively and preferably, two protein sequences (or polypeptide
sequences derived from them of at least 30 amino acids in length) share
identity, as this
term is used herein, if they have an alignment score of at least 5 (in
standard deviation
units) using the program ALIGN with the mutation data matrix and a gap penalty
of 6
26
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
or greater (Dayhoff, in Atlas of Protein Sequence and Structure, National
Biomedical
Research Foundation, volume 5, pp. 101-110 (1972) and Supplement 2, pp. 1-10).
"Corresponds to" refers to a polynucleotide sequence that shares identity
(for example is identical) to all or a portion of a reference polynucleotide
sequence, or
that a polypeptide sequence is identical to all or a portion of a reference
polypeptide
sequence. In contradistinction, the term "complementary to" is used herein to
mean that
the complementary sequence is homologous to all or a portion of a reference
polynucleotide sequence. For illustration, the nucleotide sequence TATAC
corresponds
to a reference sequence TATAC and is complementary to a reference sequence
GTATA.
The following terms are used to describe the sequence relationships
between two or more polynucleotides: "reference sequence," "comparison
window,"
"sequence identity," "percentage of sequence identity," and "substantial
identity." A
reference sequence is a defined sequence used as a basis for a sequence
comparison; a
reference sequence can be a subset of a larger sequence, for example, as a
segment of a
full length cDNA or gene sequence given in a sequence listing, or may comprise
a
complete cDNA or gene sequence. Generally, a reference sequence is at least 20
nucleotides in length, frequently at least 25 nucleotides in length, and often
at least 50
nucleotides in length. Since two polynucleotides can each ( 1 ) comprise a
sequence (for
example a portion of the complete polynucleotide sequence) that is similar
between the
two polynucleotides, and (2) may further comprise a sequence that is divergent
between
the two polynucleotides, sequence comparisons between two (or more)
polynucleotides
are typically performed by comparing sequences of the two polynucleotides over
a
"comparison window" to identify and compare local regions of sequence
similarity. A
comparison window, as used herein, refers to a conceptual segment of at least
20
contiguous nucleotide positions wherein a polynucleotide sequence may be
compared to
a reference sequence of at least 20 contiguous nucleotides and wherein the
portion of
the polynucleotide sequence in the comparison window can comprise additions
and
deletions (for example, gaps) of 20 percent or less as compared to the
reference
sequence (which would not comprise additions or deletions) for optimal
alignment of
27
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
the two sequences. Optimal alignment of sequences for aligning a comparison
window
can be conducted by the local identity algorithm (Smith and Waterman, Adv.
Appl.
Math. 2:482 (1981)), by the identity alignment algorithm (Needleman and
Wunsch, J.
Mol. Bio., 48:443 (1970)), by the search for similarity method (Pearson and
Lipman,
Proc. Natl. Acid. Sci. U.S.A. 85:2444 (1988)), by the computerized
implementations of
these algorithms such as GAP, BESTFIT, FASTA and TFASTA (Wisconsin Genetics
Software Page Release 7.0, Genetics Computer Group, Madison, WI), or by
inspection.
Preferably, the best alignment (for example, the result having the highest
percentage of
identity over the comparison window) generated by the various methods is
selected.
"Complete sequence identity" means that two polynucleotide sequences
are identical (for example, on a nucleotide-by-nucleotide basis) over the
window of
comparison.
"Percentage of sequence identity" is calculated by comparing two
optimally aligned sequences over the window of comparison, determining the
number
of positions at which the identical nucleic acid base occurs in both sequences
to yield
the number of matched positions, dividing the number of matched positions by
the total
number of positions in the window of comparison (for example, the window
size), and
multiplying the result by 100 to yield the percentage of sequence identity.
"Substantial identity" as used herein denotes a characteristic of a
polynucleotide sequence, wherein the polynucleotide comprises a sequence that
has at
least 30 percent sequence identity, preferably at least 50 to 60 percent
sequence, more
usually at least 60 percent sequence identity as compared to a reference
sequence over a
comparison window of at least 20 nucleotide positions, frequently over a
window of at
least 25 to SO nucleotides, wherein the percentage of sequence identity is
calculated by
comparing the reference sequence to the polynucleotide sequence that may
include
deletions or addition which total 20 percent or less of the reference sequence
over the
window of comparison.
"Substantial identity" as applied to polypeptides herein means that two
peptide sequences, when optimally aligned, such as by the programs GAP or
BESTFIT
using default gap weights, share at least 30 percent sequence identity,
preferably at least
28
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
40 percent sequence identity, and more preferably at least 50 percent sequence
identity,
and most preferably at lest 60 percent sequence identity. Preferably, residue
positions,
which are not identical, differ by conservative amino acid substitutions.
"Conservative amino acid substitutions" refer to the interchangeability of
residues having similar side chains. For example, a group of amino acids
having
aliphatic side chains is glycine, alanine, valine, leucine, and isoleucine; a
group of
amino acids having aliphatic-hydroxyl side chains is serine and threonine; a
group of
amino acids having amide-containing side chains is asparagine and glutamine; a
group
of amino acids having aromatic side chains is phenylalanine, tyrosine and
tryptophan; a
group of amino acids having basic side chains is lysine, arginine and
histidine; and a
group of amino acids having sulfur-containing side chan is cystein and
methionine.
Preferred conservative amino acid substitution groups are:' valine-leucine-
isoleucine;
phenylalanine-tyrosine; lysine-arginine; alanine-valine; glutamic-aspartic;
and
asparagine-glutamine.
"Modulation" refers to the capacity to either enhance or inhibit a
functional property of a biological activity or process, for example, enzyme
activity or
receptor binding. Such enhancement or inhibition may be contingent on the
occurrence
of a specific event, such as activation of a signal transduction pathway
and/or may be
manifest only in particular cell types.
"Modulator" refers to a chemical (naturally occurring or non-naturally
occurring), such as a biological macromolecule (for example, nucleic acid,
protein, non-
peptide or organic molecule) or an extract made from biological materials,
such as
prokaryotes, bacteria, eukaryotes, plants, fungi, multicellular organisms or
animals,
invertebrates, vertebrates, mammals and humans, including, where appropriate,
extracts
of: whole organisms or portions of organisms, cells, organs, tissues, fluids,
whole
cultures or portions of cultures, or environmental samples or portions
thereof.
Modulators are typically evaluated for potential activity as inhibitors or
activators
(directly or indirectly) of a biological process or processes (for example,
agonist, partial
antagonist, partial agonist, antagonist, antineoplastic, cytotoxic, inhibitors
of neoplastic
transformation or cell proliferation, cell proliferation promoting agents,
antiviral agents,
29
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
antimicrobial agents, antibacterial agents, antibiotics, and the like) by
inclusion in
assays described herein. The activity of a modulator may be known, unknown or
partially known.
"Test chemical" refers to a chemical or extract, including an agent or
compound such as a "test compound", to be tested by at least one method of the
present
invention to be a putative modulator. A test chemical is usually not known to
bind to
the target of interest. "Control test chemical" refers to a chemical known to
bind to the
target (for example, a known agonist, antagonist, partial agonist or inverse
agonist).
Test chemical does not typically include a chemical added to a mixture as a
control
condition that alters the function of the target to determine signal
specificity in an assay.
Such control chemicals or conditions include chemicals that (1) non-
specifically or
substantially disrupt protein structure (for example denaturing agents such as
urea or
guandium, sulfhydryl reagents such as dithiotritol and beta-mercaptoethanol),
(2)
generally inhibit cell metabolism (for example mitochondrial uncouples) and
(3) non-
specifically disrupt electrostatic or hydrophobic interactions of a protein
(for example,
high salt concentrations or detergents at concentrations sufficient to non-
specifically
disrupt hydrophobic or electrostatic interactions). The term test chemical
also does not
typically include chemicals known to be unsuitable for a therapeutic use for a
particular
indication due to toxicity of the subject. Usually, various predetermined
concentrations
of test chemicals are used for determining their activity. If the molecular
weight of a
test chemical is known, the following ranges of concentrations can be used:
between
about 0.001 micromolar and about 10 millimolar, preferably between about 0.01
micromolar and about 1 millimolar, more preferably between about 0.1
micromolar and
about 100 micromolar. When extracts are uses a test chemicals, the
concentration of
test chemical used can be expressed on a weight to volume basis. Under these
circumstances, the following ranges of concentrations can be used: between
about 0.001
micrograms/ml and about 100 milligram/ml, preferably between about 0.01
micrograms/ml and about 10 milligrams/ml, and more preferably between about
0.1
micrograms/ml and about 1 milligrams/ml or between about 1 microgram/ml and
about
100 micrograms/ml.
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
"Target" refers to a biochemical entity involved in a biological process.
Targets are typically proteins that play a useful role in the physiology or
biology of an
organism. A therapeutic chemical typically binds to a target to alter or
modulate its
function. As used herein, targets can include, but not be limited to, cell
surface
receptors, G-proteins, G-protein coupled receptors, kinases, phosphatases, ion
channels,
lipases, phosholipases, nuclear receptors, intracellular structures, tubules,
tubulin, and
the like.
"Label" or "labeled" refers to incorporation of a detectable marker, for
example by incorporation of a radiolabled compound or attachment to a
polypeptide of
moieties such as biotin that can be detected by the binding of a section
moiety, such as
marked avidin. Various methods of labeling polypeptide, nucleic acids,
carbohydrates,
and other biological or organic molecules are known in the art. Such labels
can have a
variety of readouts, such as radioactivity, fluorescence, color,
chemiluminescence or
other readouts known in the art or later developed. The readouts can be based
on
enzymatic activity, such as beta-galactosidase, beta-lactamase, horseradish
peroxidase,
alkaline phosphatase, luciferase; radioisotopes such as 3H, '4C, 355, 'zsI or
'3'I);
fluorescent proteins, such as green fluorescent proteins; or other fluorescent
labels, such
as FITC, rhodamine, and lanthanides. Where appropriate, these labels can be
the
product of the expression of reporter genes, as that term is understood in the
art.
Examples of reporter genes are beta-lactamase (U.S. Patent No. 5,741,657 to
Tsien et
al., issued April 21, 1998) and green fluorescent protein (U.S. Patent No.
5,777,079 to
Tsien et al., issued July 7, 1998; U.S. Patent No. 5,804,387 to Cormack et
al., issued
September 8, 1998).
"Substantially pure" refers to an object species or activity that is the
predominant species or activity present (for example on a molar basis it is
more
abundant than any other individual species or activities in the composition)
and
preferably a substantially purified fraction is a composition wherein the
object species
or activity comprises at least about 50 percent (on a molar, weight or
activity basis) of
all macromolecules or activities present. Generally, a substantially pure
composition
will comprise more than about 80 percent of all macromolecular species or
activities
31
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
present in a composition, more preferably more than about 85%, 90%, 95% and
99%.
Most preferably, the object species or activity is purified to essential
homogeneity,
wherein contaminant species or activities cannot be detected by conventional
detection
methods) wherein the composition consists essentially of a single
macromolecular
species or activity. The inventors recognize that an activity may be caused,
directly or
indirectly, by a single species or a plurality of species within a
composition, particularly
with extracts.
"Pharmaceutical agent or drug" refers to a chemical, composition or
activity capable of inducing a desired therapeutic effect when property
administered by
an appropriate dose, regime, route of administration, time and delivery
modality.
"Pharmaceutical agent or drug" refers to a chemical, composition or
activity capable of inducing a desired therapeutic effect when property
administered by
an appropriate dose, regime, route of administration, time and delivery
modality.
A "bioactive compound" refers to a compound that exhibits at least one
bioactivity.
A "bioactivity" refers to a composition that exhibits at least one activity
that modulates a biological process, cellular process or disease state.
Preferred
bioactivities include, but are not limited to activities that modulate at
least one
mitochondria) activity or mitochondria) function as provided herein (such as
the
production of ATP) or mitochondria) mass, such as by an increase
(mitochondria)
biogenesis) or decrease in the number of mitochondria or the amount of
mitochondria)
DNA. Another preferred bioactivity includes an activity that modulates a
cellular
process, such as the production or secretion of insulin. A further preferred
bioactivity
includes an activity that modulates a disease states such as diabetes type I
(type 1 DM)
or diabetes type II (type 2 DM).
A "mitochondria) biogenesis activity" is an activity that modulates the
production of active, inactive or defective mitochondria, preferably active
mitochondria.
A "mitoclastic activity" is an activity that modulates the destruction of
mitochondria.
32
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
An "anti-diabetic activity" is an activity that modulates the disease state
of diabetes, including diabetes type I and diabetes type II. Preferably, an
anti-diabetic
activity is also, directly or indirectly, a mitochondria) biogenesis activity.
A "bioactive derivative" refers to a modification of a bioactive
compound or bioactivity that retains at least one characteristic activity of
the parent
compound.
A "bioactive precursor" refers to a precursor of a bioactive compound or
bioactivity that exhibits at least one characteristic activity of the
resulting bioactive
compound or bioactivity.
A "patient" or "subject" refers a whole organism in need of treatment,
such as a farm animal, companion animal or human. An animal refers to any non-
human animal.
An "indicator of mitochondria) function" is any parameter that is
indicative of mitochondria) function that can be measured by one skilled in
the art. In
certain embodiments, the indicator of mitochondria) function is a
mitochondria) electron
transport chain enzyme, a Krebs cycle enzyme, a mitochondria) matrix
component, a
mitochondria) membrane component or an ATP biosynthesis factor. In other
embodiments, the indicator of mitochondria) function is mitochondria) number
per cell
or mitochondria) mass per cell. In other embodiments, the indicator of
mitochondria)
function is an ATP biosynthesis factor. In other embodiments, the indicator of
mitochondria) function is the amount of ATP per mitochondrion, the amount of
ATP
per unit mitochondria) mass, the amount of ATP per unit protein or the amount
of ATP
per unit mitochondria) protein. In other embodiments, the indicator of
mitochondria)
function comprises free radical production. In other embodiments, the
indicator of
mitochondria) function comprises a cellular response to elevated intracellular
calcium.
In other embodiments, the indicator of mitochondria) function is the activity
of a
mitochondria) enzyme such as, by way of non-limiting example, citrate
synthase,
hexokinase II, cytochrome c oxidase, phosphofructokinase, glyceraldehyde
phosphate
dehydrogenase, glycogen phosphorylase, creatine kinase, NADH dehydrogenase,
glycerol 3-phosphate dehydrogenase, triose phosphate dehydrogenase or malate
33
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
dehydrogenase. In other embodiments, the indicator of mitochondria) function
is the
realtive or absolute amount of mitochondria) DNA per cell in the patient.
"Improving mitochondria) function" may refer to (a) substantially
restoring to a normal level at least one indicator of glucose responsiveness
in cells
having reduced glucose responsiveness and reduced mitochondria) mass and/or
impaired mitochondria) function; or (b) substantially restoring to a normal
level, or
increasing to a level above and beyond normal levels, at least one indicator
of
mitochondria) function in cells having impaired mitochondria) function or in
cells
having normal mitochondria) function, respectively. Improved mitochondria)
function
may result from changes in extramitochondrial structures or events, as well as
from
mitochondria) structures or events, in direct interactions between
mitochondria) and
extramitochondrial genes and/or their gene products, or in structural or
functional
changes that occur as the result of interactions between intermediates that
may be
formed as the result of such interactions, including metabolites, catabolites,
substrates,
precursors, cofactors and the like.
"Impaired mitochondria) function" may include impairments in the level
and/or rate of any respiratory, metabolic or other biochemical or biophysical
activity in
some or all cells of a biological source. As non-limiting examples, markedly
impaired
ETC activity may be related to impaired mitochondria) function, as may be
generation
of increased ROS or defective oxidative phosphorylation. As further examples,
altered
mitochondria) membrane potential, induction of apoptotic pathways and
formation of
atypical chemical and biochemical crosslinked species within a cell, whether
by
enzymatic or non-enzymatic mechanisms, may all be regarded as indicative of
mitochondria) function. These and other non-limiting examples of impaired
mitochondria) function are described in greater detail below.
Other technical terms used herein have their ordinary meaning in the art
that they are used, as exemplified by a variety of technical dictionaries,
such as the
McGraw-Hill Dictionary of Chemical Terms and the Stedman's Medical Dictionary.
ASSAYS OF MITOCHONDRIAL FUNCTION
34
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
According to certain embodiments within any of the above aspects of the
invention, certain assays of mitochondria) function may be practiced, which in
preferred
embodiments may pertain to determination of ATP biosynthesis or of an ATP
biosynthesis factor as provided herein. Thus, in certain embodiments of any of
the
above aspects of the invention, mitochondria) function is determined as the
amount of
ATP per cell, per unit protein or per mitochondrion in a sample, and in
certain
embodiments the rate of ATP synthesis in the sample is determined. In certain
embodiments an ATP biosynthesis factor as provided herein is determined.
An "ATP biosynthesis factor" refers to any naturally occurring cellular
component that contributes to the efficiency of ATP production in
mitochondria. Such
a cellular component may be a protein, polypeptide, peptide, amino acid, or
derivative
thereof; a lipid, fatty acid or the like, or derivative thereof; a
carbohydrate, saccharide or
the like or derivative thereof, a nucleic acid, nucleotide, nucleoside,
purine, pyrimidine
or related molecule, or derivative thereof, or the like. An ATP biosynthesis
factor
includes at least the components of the ETC and of the Krebs cycle (see,
e.g., Lehninger, Biochemistry, 1975 Worth Publishers, New York; Voet and Voet,
Biochemistry, 1990 John Wiley & Sons, New York; Mathews and van Holde,
Biochemistry, 1990 Benjamin Cummings, Menlo Park, California) and any protein,
enzyme or other cellular component that participates in ATP synthesis,
regardless of
whether such ATP biosynthesis factor is the product of a nuclear gene or of an
extranuclear gene (e.g., a mitochondria) gene). Participation in ATP synthesis
may
include, but need not be limited to, catalysis of any reaction related to ATP
synthesis,
transmembrane import and/or export of ATP or of an enzyme cofactor,
transcription of
a gene encoding a mitochondria) enzyme and/or translation of such a gene
transcript.
Compositions and methods for determining whether a cellular
component is an ATP biosynthesis factor are well known in the art, and include
methods for determining ATP production (including determination of the rate of
ATP
production in a sample) and methods for quantifying ATP itself. The
contribution of an
ATP biosynthesis factor to ATP production can be determined, for example,
using an
isolated ATP biosynthesis factor that is added to cells or to a cell-free
system. The ATP
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
biosynthesis factor may directly or indirectly mediate a step or steps in a
biosynthetic
pathway that influences ATP production. For example, an ATP biosynthesis
factor may
be an enzyme that catalyzes a particular chemical reaction leading to ATP
production.
As another example, an ATP biosynthesis factor may be a cofactor that enhances
the
efficiency of such an enzyme. As another example, an ATP biosynthesis factor
may be
an exogenous genetic element introduced into a cell or a cell-free system that
directly or
indirectly affects an ATP biosynthetic pathway. Those having ordinary skill in
the art
are readily able to compare ATP production by an ATP biosynthetic pathway in
the
presence and absence of a candidate ATP biosynthesis factor. Routine
determination of
ATP production may be accomplished using any known method for quantitative ATP
detection, for example by way of illustration and not limitation, by
differential
extraction from a sample optionally including chromatographic isolation; by
spectrophotometry; by quantification of labeled ATP recovered from a sample
contacted with a suitable form of a detectably labeled ATP precursor molecule
such as,
for example, 32P; by quantification of an enzyme activity associated with ATP
synthesis
or degradation; or by other techniques that are known in the art. Accordingly,
in certain
embodiments of the present invention, the amount of ATP in a biological sample
or the
production of ATP (including the rate of ATP production) in a biological
sample may
be an indicator of mitochondrial function. In one embodiment, for instance,
ATP~may
be quantified by measuring luminescence of luciferase catalyzed oxidation of D-
luciferin, an ATP dependent process.
"Enzyme catalytic activity" refers to any function performed by a
particular enzyme or category of enzymes that is directed to one or more
particular
cellular function(s). For example, "ATP biosynthesis factor catalytic
activity" refers to
any function performed by an ATP biosynthesis factor as provided herein that
contributes to the production of ATP. Typically, enzyme catalytic activity is
manifested as facilitation of a chemical reaction by a particular enzyme, for
instance an
enzyme that is an ATP biosynthesis factor, wherein at least one enzyme
substrate or
reactant is covalently modified to form a product. For example, enzyme
catalytic
activity may result in a substrate or reactant being modified by formation or
cleavage of
36
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
a covalent chemical bond, but the invention need not be so limited. Various
methods of
measuring enzyme catalytic activity are known to those having ordinary skill
in the art
and depend on the particular activity to be determined.
For many enzymes, including mitochondrial enzymes or enzymes that
are ATP biosynthesis factors as provided herein, quantitative criteria for
enzyme
catalytic activity are well established. These criteria include, for example,
activity that
may be defined by international units (IU), by enzyme turnover number, by
catalytic
rate constant (K~a~), by Michaelis-Menten constant (K",), by specific activity
or by any
other enzymological method known in the art for measuring a level of at least
one
enzyme catalytic activity. Specific activity of a mitochondrial enzyme, such
as an ATP
biosynthesis factor, may be expressed as units of substrate detectably
converted to
product per unit time and, optionally, further per unit sample mass (e.g., per
unit protein
or per unit mitochondrial mass).
In certain preferred embodiments of the invention, enzyme catalytic
activity may be expressed as units of substrate detectably converted by an
enzyme to a
product per unit time per unit total protein in a sample. In certain
particularly preferred
embodiments, enzyme catalytic activity may be expressed' as units of substrate
detectably converted by an enzyme to product per unit time per unit
mitochondria) mass
in a sample. In certain highly preferred embodiments, enzyme catalytic
activity may be
expressed as units of substrate detectably converted by an enzyme to product
per unit
time per unit mitochondria) protein mass in a sample. Products of enzyme
catalytic
activity may be detected by suitable methods that will depend on the quantity
and
physicochemical properties of the particular product. Thus, detection may be,
for
example by way of illustration and not limitation, by radiometric,
colorimetric,
spectrophotometric, fluorimetric, immunometric or mass spectrometric
procedures, or
by other suitable means that will be readily apparent to a person having
ordinary skill in
the art.
In certain embodiments of the invention, detection of a product of
enzyme catalytic activity may be accomplished directly, and in certain other
embodiments detection of a product may be accomplished by introduction of a
37
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
detectable reporter moiety or label into a substrate or reactant such as a
marker enzyme,
dye, radionuclide, luminescent group, fluorescent group or biotin, or the
like. The
amount of such a label that is present as unreacted substrate and/or as
reaction product,
following a reaction to assay enzyme catalytic activity, is then determined
using a
method appropriate for the specific detectable reporter moiety or label. For
radioactive
groups, radionuclide decay monitoring, scintillation counting, scintillation
proximity
assays (SPA) or autoradiographic methods are generally appropriate. For
immunometric measurements, suitably labeled antibodies may be prepared
including,
for example, those labeled with radionuclides, with fluorophores, with
affinity tags,
with biotin or biotin mimetic sequences or those prepared as antibody-enzyme
conjugates (see, e.g., Weir, D.M., Handbook of Experimental Immunology, 1986,
Blackwell Scientific, Boston; Scouten, W.H., Methods in Enrymology 135:30-65,
1987;
Harlow and Lane, Antibodies: A Laboratory Manual, Cold Spring Harbor
Laboratory,
1988; Haugland, 1996 Handbook of Fluorescent Probes and Research Chemicals-
Sixth
Ed., Molecular Probes, Eugene, Oregon; Scopes, R.K., Protein Purification:
Principles
and Practice, 1987, Springer-Verlag, New York; Hermanson, G.T. et al.,
Immobilized
Affinity Ligand Techniques, 1992, Academic Press, Inc., NY; Luo et al., 1998
J.
Biotechnol. 65:225 and references cited therein). Spectroscopic methods may be
used
to detect dyes (including, for example, colorimetric products of enzyme
reactions),
luminescent groups and fluorescent groups. Biotin may be detected using avidin
or
streptavidin, coupled to a different reporter group (commonly a radioactive or
fluorescent group or an enzyme). Enzyme reporter groups may generally be
detected by
the addition of substrate (generally for a specific period of time), followed
by
spectroscopic, spectrophotometric or other analysis of the reaction products.
Standards
and standard additions may be used to determine the level of enzyme catalytic
activity
in a sample, using well known techniques.
As noted above, enzyme catalytic activity of an ATP biosynthesis factor
may further include other functional activities that lead to ATP production,
beyond
those involving covalent alteration of a substrate or reactant. For example by
way of
illustration and not limitation, an ATP biosynthesis factor that is an enzyme
may refer
38
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
to a transmembrane transporter molecule that, through its enzyme catalytic
activity,
facilitates the movement of metabolites between cellular compartments. Such
metabolites may be ATP or other cellular components involved in ATP synthesis,
such
as gene products and their downstream intermediates, including metabolites,
catabolites,
substrates, precursors, cofactors and the like. As another non-limiting
example, an ATP
biosynthesis factor that is an enzyme may, through its enzyme catalytic
activity,
transiently bind to a cellular component involved in ATP synthesis in a manner
that
promotes ATP synthesis. Such a binding event may, for instance, deliver the
cellular
component to another enzyme involved in ATP synthesis and/or may alter the
conformation of the cellular component in a manner that promotes ATP
synthesis.
Further to this example, such conformational alteration may be part of a
signal
transduction pathway, an allosteric activation pathway, a transcriptional
activation
pathway or the like, where an interaction between cellular components leads to
ATP
production.
Thus, according to the present invention, an ATP biosynthesis factor
may include, for example, a mitochondria) membrane protein. Suitable
mitochondria)
membrane proteins include such mitochondria) components as the adenine
nucleotide
transporter (ANT; e.g., Fiore et al., 1998 Biochimie 80:137; Klingenberg 1985
Ann.
N.Y.Acad. Sci. 456:279), the voltage dependent anion channel (VDAC, also
referred to
as porin; e.g., Manella, 1997 J. Bioenergetics Biomembr. 29:525), the malate-
aspartate
shuttle, the mitochondria) calcium uniporter (e.g., Litsky et al., 1997
Biochem.
36:7071), uncoupling proteins (UCP-1, -2, -3; see e.g., Jezek et al., 1998
Int. J.
Biochem. Cell Biol. 30:1163), a hexokinase, a peripheral benzodiazepine
receptor, a
mitochondria) intermembrane creatine kinase, cyclophilin D, a Bcl-2 gene
family
encoded polypeptide, the tricarboxylate carrier (e.g., Iocobazzi et al., 1996
Biochim.
Biophys. Acta 1284:9; Bisaccia et al., 1990 Biochim. Biophys. Acta 1019:250)
and the
dicarboxylate carrier (e.g., Fiermonte et al., 1998 J. Biol. Chem. 273:24754;
Indiveri et
al., 1993 Biochim. Biophys. Acta 1143:310; for a general review of
mitochondria)
membrane transporters, see, e.g., Zonatti et al., 1994 J. Bioenergetics
Biomembr. 26:543
and references cited therein).
39
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
Affinity techniques are particularly useful in the context of isolating an
enzyme or an ATP biosynthesis factor protein or polypeptide for use according
to the
methods of the present invention, and may include any method that exploits a
specific
binding interaction involving an enzyme or an ATP biosynthesis factor to
effect a
separation. For example, because an enzyme or an ATP biosynthesis factor
protein or
polypeptide may contain covalently attached oligosaccharide moieties, an
affinity
technique such as binding of the enzyme (or ATP biosynthesis factor) to a
suitable
immobilized lectin under conditions that permit carbohydrate binding by the
lectin may
be a particularly useful affinity technique.
Other useful affinity techniques include immunological and other
biochemical affinity techniques for isolating and/or detecting a specific
protein or
polypeptide antigen (e.g., an enzyme or ATP biosynthesis factor) and/or a
specific
binding interaction between biomolecules such as proteins, which techniques
rely on
specific binding interaction between antibody combining sites for antigen and
antigenic
determinants present on the factor, or between protein-protein binding sites,
ligand-
receptor binding sites, receptor-counterreceptor binding sites or the like.
Binding of an
antibody or other affinity reagent to an antigen or other cognate ligand,
receptor or
counterreceptor is "specific" where the binding interaction involves a Ka of
greater than
or equal to about 104 M-1, preferably of greater than or equal to about 105 M-
1, more
preferably of greater than or equal to about 106 M-1 and still more preferably
of greater
than or equal to about 107 M-1 . Affinities of binding partners or antibodies
can be
readily determined using conventional techniques, for example those described
by
Scatchard et al., Ann. N. Y. Acad. Sci. 51:660 ( 1949).
Immunological techniques include, but need not be limited to,
immunoaffinity chromatography, immunoprecipitation, solid phase
immunoadsorption
or other immunoaffinity methods. For these and other useful affinity
techniques, see,
for example, Scopes, R.K., Protein Purification: Principles and Practice,
1987,
Springer-Verlag, New York; Weir, D.M., Handbook of Experimental Immunology,
1986, Blackwell Scientific, Boston; and Hermanson, G.T. et al., Immobilized
Amity
Ligand Technigues, 1992, Academic Press, Inc., California; which are hereby
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
incorporated by reference in their entireties, for details regarding
techniques for
isolating and characterizing complexes, including affinity techniques.
SAMPLES
Samples of cells for the present invention can be provided as cells in
culture or from a subject, such as a tissue, fluid or organ or a portion of
any of the
foregoing. For example, cells can preferably be from tissues that are involved
in
glucose metabolism, such as pancreatic cells, islates of Langerhans,
pancreatic beta
cells, muscle cells, liver cells or other appropriate cells. Preferably, cells
are provided
in culture and can be a primary cell line or a continuous cell line and can be
provided as
a clonal population of cells or a mixed population of cells. Preferably, the
cells are
insulin producing (and more preferably insulin secreting) cells in that they
naturally
produce and optionally secrete insulin or have been engineered to produce and
optionally secrete insulin under appropriate stimuli, such as in the presence
of glucose.
Preferred cells include, but are not limited to, a glucose-responsive,
insulin-producing cell line such as the rat-derived INS-1 cell line; cells
(particularly
beta cells) derived from Zucker diabetic fatty rat (ZDF) or cells
(particularly beta cells)
from Zucker lean control rates (ZLC) ) (Shafrir et al., J. Basic Clin.
Physiol.
Pharmacol. 9:347-385, 1988). Other preferred cells include derivatives of the
above
cell lines that have been depleted of their mitochondrial DNA (mtDNA); such
cells are
commonly referred to as "p°" ("rho-zero"). Other preferred cells
include cybrid cells,
i. e., derivatives of the above cell lines in which the endogenous mtDNA has
been
replaced by mtDNA from an individual suffering from diabetes or another
mitochondrial disease of interest. General methods for preparing, using and
assaying
the mitochondrial functions of rho-zero and cybrid cells are described in U.S.
Patent
No. 5,888,438, published PCT applications WO 95/26973 and WO 98/17826, King
and
Attardi (Science 246:500-503, 1989), Chomyn et al. (Mol. Cell. Biol. 11:2236-
2244,
1991), Miller et al. (J. Neurochem. 67:1897-1907, 1996), Swerdlow et al.
(Annals of
Neurology 40:663-671, 1996), Cassarino et al. (Biochim. Biophys. Acta 1362:77-
86,
41
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
1997), Swerdlow et al. (Neurology 49:918-925, 1997), Sheehan et al. (J.
Neurochem.
68:1221-1233, 1997), and Sheehan et al. (J. Neurosci. 17:4612-4622, 1997).
Cybrid
cells comprising mitochondria dervied from diabetic individuals are described
in
published PCT applications WO 95/26973 and WO 98/17826.
Cybrid cells can be made using mitochondria from healthy subjects or
from subjects that may have mitochondria) defects. Briefly, a host cell line
is treated
with ethidium bromide, or an antiviral agent (as described in copending U.S.
patent
applications 09/069,489 and 09/237,999) such as ddC, to substantially deplete
cells of
mitochondria) DNA (mtDNA). Platelets, or other sources of mitochondria, are
fused
with the mitochondria depleted cells to form a hybrid cell that includes the
nuclear
genome of the host cell and the mitochondria (and thus mitochondria) genome)
of the
subject.
In the beta cells of ZDF rats, increased ceramide synthesis and nitric
oxide increases beta cell apoptosis. Ceramide (particularly C2 ceramide, but
not C2
dihydroceramide) and nitric oxide are stimulated by FAA (oleate:palmitate).
Also, C6
ceramide can induce caspase 3 activation in INS-1 cells. Furthermore, sodium
nitroprusside (SNP) can induce INS-1 cell death.
Biological samples may comprise any tissue or cell preparation in which
at least one mitochondria) function can be detected (and which in certain
preferred
embodiments pertains to mitochondria) ATP production and/or an ATP
biosynthesis
factor as provided herein), and may vary in nature accordingly, depending on
the
particular indicators) to be compared. Biological samples may be provided by
obtaining a blood sample, biopsy specimen, tissue explant, organ culture or
any other
tissue or cell preparation from a subject or a biological source. The subject
or
biological source may be a human or non-human animal, a primary cell culture
or
culture adapted cell line including but not limited to genetically engineered
cell lines
that may contain chromosomally integrated or episomal recombinant nucleic acid
sequences, immortalized or immortalizable cell lines, somatic cell hybrid or
cytoplasmic hybrid "cybrid" cell lines, differentiated or differentiatable
cell lines,
transformed cell lines and the like. In certain preferred embodiments of the
invention,
42
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
the subject or biological source may be suspected of having or being at risk
for having
type 2 diabetes mellitus, and in certain preferred embodiments of the
invention the
subject or biological source may be known to be free of a risk or presence of
such as
disease.
In certain other preferred embodiments where it is desirable to determine
whether or not a subject or biological source falls within clinical parameters
indicative
of type 2 diabetes mellitus, signs and symptoms of type 2 diabetes that are
accepted by
those skilled in the art may be used to so designate a subject or biological
source, for
example clinical signs referred to in Gavin et al. (Diabetes Care 22(suppl.
1):SS-S19,
1999, American Diabetes Association Expert Committee on the Diagnosis and
Classification of Diabetes Mellitus) and references cited therein, or other
means known
in the art for diagnosing type 2 diabetes. In certain aspects of the
invention, biological
samples may be obtained from the subject or biological source before and after
contacting the subject or biological source with a candidate agent, for
example to
identify a candidate agent capable of effecting a change in the level of a
mitochondria)
function, relative to the level before exposure of the subject or biological
source to the
agent.
Candidate agents for use in screening assay methods provided by the
present invention, such as methods for identifying an agent that alters
mitochondria)
ATP production or methods for identifying an agent for treating diabetes, may
be
provided as "libraries" or collections of compounds, compositions or
molecules. Such
molecules typically include compounds known in the art as "small molecules"
and
having molecular weights less than 105 daltons, preferably less than 104
daltons and still
more preferably less than 103 daltons. For example, members of a library of
test
compounds can be administered to a plurality of samples, and then assayed for
their
ability to increase or decrease the level of at least one indicator of
mitochondria)
function.
Candidate agents further may be provided as members of a combinatorial
library, which preferably includes synthetic agents prepared according to a
plurality of
predetermined chemical reactions performed in a plurality of reaction vessels.
For
43
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
example, various starting compounds may be prepared employing one or more of
solid-
phase synthesis, recorded random mix methodologies and recorded reaction split
techniques that permit a given constituent to traceably undergo a plurality of
permutations and/or combinations of reaction conditions. The resulting
products
comprise a library that can be screened followed by iterative selection and
synthesis
procedures, such as a synthetic combinatorial library of peptides (see e.g.,
PCT/US91/08694, PCT/L1S91/04666, which are hereby incorporated by reference in
their entireties) or other compositions that may include small molecules as
provided
herein (see e.g., PCT/LJS94/08542, EP 0774464, U.S. 5,798,035, U.S. 5,789,172,
U.S.
5,751,629, which are hereby incorporated by reference in their entireties).
Those
having ordinary skill in the art will appreciate that a diverse assortment of
such libraries
may be prepared according to established procedures, and tested for their
influence on
an indicator of mitochondria) function, according to the present disclosure.
In certain other embodiments, the invention provides a method of
treating a patient having type 2 DM by administering to the patient an agent
that
substantially increases mitochondria) ATP synthesis in cells, and/or that
substantially
decreases mitochondria) ATP hydrolysis in cells, and/or that restores at least
one
mitochondria) function to a level found in control or normal subjects. In one
preferred
embodiment the restored or increased mitochondria) function is the amount of
ATP
produced. In a most preferred embodiment, an agent that substantially restores
or alters
(e.g., increases or decreases in a statistically significant fashion)
mitochondria) ATP
biosynthetic and/or mitochondria) ATP hydrolytic function to a normal level
effects the
return of the level of ATP to a level found in control subjects. In another
preferred
embodiment, the agent that substantially restores such mitochondria) function
confers a
clinically beneficial effect on the subject. In another embodiment, the agent
that
substantially restores such mitochondria) function promotes a statistically
significant
change the mitochondria) function. As noted herein, those having ordinary
skill in the
art can readily determine whether a change in the level of a particular
mitochondria)
function brings that level closer to a normal value and/or clinically benefits
the subject.
Thus, an agent that substantially restores at least one mitochondria) function
to a normal
44
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
level may include an agent capable of fully or partially restoring such level.
For
example, and according to non-limiting theory, a preferred agent according to
certain
embodiments of the present invention comprises a composition that inhibits
(i.e.,
impairs, hinders or otherwise down-regulates) one or more activities of IF 1.
In certain
other embodiments, a preferred agent may comprise a composition that mimics
IF1,
which relates to a composition that structurally and/or functionally
resembles, imitates,
supplants, supplements, augments, enhances, substitutes or otherwise replaces
all or a
portion of a native IF 1 molecule, for example by possessing a three-
dimensional
structure capable of binding interactions with binding partners to which IF 1
binds, or as
another example, by exhibiting greater stability and/or specificity under
physiological
conditions than might be expected of IF 1.
EXPRESSION SYSTEMS
In order to produce a gene product of interest in sufficient quantities for
further embodiments of the invention, the nucleotide sequence of interest,
such as a
nucleotide sequence encoding an IF 1, or functional equivalents thereof, is
inserted into
an appropriate "expression vector," i. e., a genetic element, often capable of
autonomous
replication, which contains the necessary elements for the transcription and,
in instances
where the gene product is a protein, translation of the inserted nucleotide
sequence. A
genetic element that comprises an expression vector and a nucleic acid of
interest in an
arrangement appropriate for expression of a gene product of interest is
referred to herein
as an "expression construct."
Methods which are well known to those skilled in the art can be used to
prepare expression constructs containing a nucleotide sequence of interest and
appropriate transcriptional and translational controls. These methods include
in vitro
recombinant DNA techniques, synthetic techniques and in vivo recombination or
genetic recombination. Such techniques are known in the art (see, e.g.,
Sambrook et al.,
Molecular Cloning: A Laboratory Manual, Second Edition, Cold Spring Harbor
Press,
Plainview N.Y., 1989; Ausubel et al. , eds., Short Protocols in Molecular
Biology,
Second Edition, John Wiley & Sons, New York N.Y., 1992).
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
A variety of expression vector/host systems may be utilized to contain
and express a nucleotide sequence of interest. These include but are not
limited to
microorganisms such as bacteria transformed with recombinant bacteriophage,
plasmid
or cosmid DNA expression vectors; yeast transformed with yeast expression
vectors;
insect cell systems infected with virus expression vectors (e.g.,
baculovirus); plant cell
systems transfected with virus expression vectors (e.g., cauliflower mosaic
virus,
CaMV; tobacco mosaic virus, TMV) or transformed with bacterial expression
vectors
(e.g., Ti or pBR322-based plasmids); or animal cell systems.
The "control elements" or "regulatory sequences" of these systems,
which may vary in their strength and specificities, are those nontranslated
regions of the
vector, enhancers, promoters, and S' and 3' untranslated regions, which
interact with
host cellular proteins to carry out transcription and, where the gene product
of interest is
a protein, translation. Depending on the vector system and host utilized, any
number of
suitable transcription and translation elements, including constitutive and
inducible
promoters, may be used. For example, when cloning in bacterial systems,
inducible
promoters, including hybrid promoters, such as lacZ promoter of the
BluescriptTM
phagemid (Stratagene, La Jolla, CA.) or pSportl (Life Technologies, Inc.,
Rockville,
MD) and ptrp-lac hybrids and the like may be used. In insect cells, the
baculovirus
polyhedrin promoter may be used. Promoters and/or enhancers derived from the
genomes of plant cells (e.g., heat shock, RUBISCO; and storage protein gene
promoters) or from plant viruses or pathogens (e.g., viral or Agrobacterium-
based
promoters or leader sequences) may be cloned into the vector. In mammalian
cell
systems, promoters from mammalian genes or from mammalian viruses are
appropriate.
If it is necessary to generate a cell line that contains multiple copies of
the nucleotide
sequence of interest, vectors based on SV40 or EBV may be used with an
appropriate
selectable marker.
In bacterial systems, a number of expression vectors may be selected
depending upon the use intended for expressed gene product of interest. For
example,
when large quantities of a protein of interest are needed for the induction of
antibodies,
46
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
vectors which direct high level expression of the protein of interest, or
fusion proteins
derived therefrom that are more readily assayed and/or purified, may be
desirable.
Such vectors include, but are not limited to, Escherica coli cloning and
expression vectors such as pET (Stratagene, La Jolla, California), pRSET
(Invitrogen,
Carlsbad, California) or pGEMEXTM (Promega, Madison, WI) vectors, in which the
sequence encoding a protein of interest is ligated downstream from a
bacteriophage T7
promoter and ribosome binding site so that, when the expression construct is
transformed into E. coli expressing the T7 RNA polymerase, large levels of the
polypeptide of interest are produced; pGEMTM vectors (Promega), in which
inserts into
sequences encoding the lacZ a-peptide may be detected using colorimetric
screening;
and the like. For polypeptides that are relatively insoluble, it may be
desirable to
produce thioredoxin fusion proteins using, for example, pBAD/Thio-TOPO vectors
(Invitrogen).
Plasmids such as pGEX vectors (Amersham Pharmacia Biotech,
Piscataway, NJ) may be used to express polypeptides of interest as fusion
proteins.
Such vectors comprise a promoter operably linked to a glutathione S-
transferase (GS'T)
gene from Schistosoma japonicum (Smith et al., 1988, Gene 67:31-40), the
coding
sequence of which has been modified to comprise a thrombin cleavage site-
encoding
nucleotide sequence immediately 5' from a multiple cloning site. GST fusion
proteins
can be detected by Western blots with anti-GST or by using a colorimetric
assay; the
latter assay utilizes glutathione and 1-chloro-2-4-dinitrobenzene (CDNB) as
substrates
for GST and yields a yellow product detectable at 340 nm (Habig et al., 1974,
J. Biol.
Chem. 249:7130-7139). GST fusion proteins produced from expression constructs
derived from this expression vector can be purified by, e.g., adsorption to
glutathione-
agarose beads followed by elution in the presence of free glutathione. Another
series of
expression vectors of this type are the pBAD/His vectors (Guzman et al., J.
Bact.
177:4121-4130, 1997; Invitrogen, Carlsbad, CA), which contains the following
elements operably linked in a 5' to 3' orientation: the inducible, but tightly
regulatable,
araBAD promoter; optimized E coli translation initiation signals; an amino
terminal
polyhistidine(6xHis)-encoding sequence (also referred to as a "His-tag"); an
XPRESS~r"'
47
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
epitope-encoding sequence; an enterokinase cleavage site which can be used to
remove
the preceding N-terminal amino acids following protein purification, if so
desired; a
multiple cloning site; and an in-frame termination codon. Fusion proteins made
from
pBAD/His expression constructs can be purified using substrates or antibodies
that
specifically bind to the His-tag, and assayed by Western analysis using the
Anti-
XpressTM antibody. Proteins made in such systems are designed to include
heparin,
thrombin, enterokinase, factor XA or other protease cleavage sites so that the
cloned
polypeptide of interest can be released from the GST moiety by treatment with
the
appropriate protease.
Expression vectors derived from bacteriophage, including cosmids and
phagemids, may also be used to express nucleic acids of interest in bacterial
cells. Such
vectors include, but are not limited to, ZAP ExpressTM, Lambda ZAPTM, and
Lambda
gtll bacteriophage vectors, pBluescriptTM phagemids, (all available from
Stratagene)
and the pSL 1180 Superlinker Phagemid (Amersham Pharmacia Biotech).
In yeast such as Saccharomyces cerevisiae or Pichia pastoris, a number
of vectors containing constitutive or inducible promoters such as those for
mating factor
alpha, GALL, TEFL AOXI or GAP may be used. Appropriate expression vectors
include various pYES, pYD and pTEF derivatives (Invitrogen) (see, for example,
Grant
et al., Methods in Enzymology 153:516-544, 1987; Lundblad et al., Units 13.4
to 13.7 of
Chapter 13 in: Short Protocols in Molecular Biology, 2nd Ed., Ausubel et al.,
eds., John
Wiley & Sons, New York, New York, 1992, pages 13-19 to 13-33).
In cases where plant expression vectors are used, the expression of a
nucleotide sequence of interest may be driven by any of a number of promoters.
For
example, viral promoters such as the 35S and 19S promoters of CaMV (Brisson et
al.,
Nature 310:511-514, 1984) may be used alone or in combination with the omega
leader
sequence from TMV (Takamatsu et al., EMBO J. 6:307-311, 1987). Alternatively,
plant promoters such as the promoter of the gene encoding the small subunit of
RUBISCO (Coruzzi et al.., EMBO J. 3:1671-1680, 1984; Broglie et al., Science
224:838-843, 1984); or heat shock promoters (Winter and Sinibaldi, Results
Probl. Cell.
Differ. 17:85-105, 1991) may be used. These constructs can be introduced into
plant
48
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
cells by direct DNA transformation or pathogen-mediated transfection. For
reviews of
such techniques, see Gossen et al. (Curr. Opin. Biotechnol. 5:516-520, 1994),
Porta and
Lomonossoff (Mol. Biotechnol. 3:209-221, 1996) and Turner and Foster (Mol.
Biotechnol. 3:225-36, 1995)..
Another expression system which may be used to express a gene product
of interest is an insect system. In one such system, Autographa californica
nuclear
polyhedrosis virus (AcNPV) is used as a vector to express foreign genes in
Spodoptera
frugiperda cells or in Trichoplusia larvae. The nucleotide sequence of
interest may be
cloned into a nonessential region of the virus, such as the polyhedrin gene,
and placed
under control of the polyhedrin promoter. Successful insertion of the sequence
of
interest will render the polyhedrin gene inactive and produce recombinant
virus lacking
coat protein. The recombinant viruses are then used to infect S. frugiperda
cells or
Trichoplusia larvae in which the gene product of interest is expressed (see
"Piwnica-
Worms, Expression of Proteins in Insect Cells Using Baculovirus Vectors,"
Section II
of Chapter 16 in: Short Protocols in Molecular Biology, 2nd Ed., Ausubel et
al., eds.,
John Wiley & Sons, New York, New York, 1992, pages 16-32 to 16-48; Lopez-
Ferber
et al., Chapter 2 in: Baculovirus Expression Protocols, Methods in Molecular
Biology,
Vol. 39, C.R. Richardson, Ed., Humana Press,1'otawa, New Jersey, 1995, pages
25-63).
S. frugiperda cells (Sf~, Sf21 or High FiveTM cells) and appropriate
baculovirus transfer
vectors are commercially available from, e.g., Invitrogen. Expression systems
utilizing
Drosophila S2 cells (also available from Invitrogen) may also be utilized.
Expression constructs for expressing nucleic acids of interest in
mammalian cells are prepared in a step-wise process. First, expression
cassettes that
comprise a promoter (and associated regulatory sequences) operably linked to a
nucleic
acid of interest are constructed in bacterial plasmid-based systems; these
expression
cassette-comprising constructs are evaluated and optimized for their ability
to produce
the gene product of interest in mammalian cells that are transiently
transfected
therewith. Second, these expression cassettes are transferred to viral systems
that
produce recombinant proteins during lytic growth of the virus (e.g., SV40,
BPV, EBV,
49
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
adenovirus; see below) or from a virus that can stably integrate into and
transduce a
mammalian cellular genome (e.g., a retroviral expression construct).
With regard to the first step, commercially available "shuttle"
(i.e., capable of replication in both E. coli and mammalian cells) vectors
that comprise
S promoters that function in mammalian cells and can be operably linked to a
nucleic acid
of interest include, but are not limited to, SV40 late promoter expression
vectors
(e.g., pSVL, Amersham Pharmacia Biotech), glucocorticoid-inducible promoter
expression vectors (e.g., pMSG, Amersham Pharmacia Biotech), Rous sarcoma
enhancer-promoter expression vectors (e.g., pRc/RSV, Invitrogen) and CMV
immediate
early promoter expression vectors, including derivatives thereof having
selectable
markers to agents such as Neomycin, Hygromycin or ZEOCINr"'' (e.g., pRc/CMV2,
pCDMB, pcDNA 1.1, pcDNA 1.1 /Amp, pcDNA3. l , pcDNA3. l /Zeo and
pcDNA3.l/Hygro, Invitrogen). In general, preferred shuttle vectors for nucleic
acids of
interest are those having selectable markers (for ease of isolation and
maintenance of
I S transformed cells) and inducible, and thus regulatable, promoters as
overexpression of a
gene product of interest may have toxic effects.
Methods for transfecting mammalian cells are known in the art (see,
Kingston et al., "Transfection of DNA into Eukaryotic Cells," Section I of
Chapter 9 in:
Short Protocols in Molecular Biology, 2nd Ed., Ausubel et al., eds., John
Wiley &
Sons, New York, New York, 1992, pages 9-3 to 9-16). A control plasmid, such as
pCH 110 (Pharmacia), may be cotransfected with the expression construct being
examined so that levels of the gene product of interest can be normalized to a
gene
product expressed from the control plasmid. Preferred expression cassettes,
consisting
essentially of a promoter and associated regulatory sequences operably linked
to a
nucleic acid of interest, are identified by the ability of cells transiently
transformed with
a vector comprising a given expression cassette to express high levels of the
gene
product of interest, or a fusion protein derived therefrom, when induced to do
so.
Expression may be monitored by Northern or Western analysis or, in the case of
fusion
proteins, by a reporter moiety such as an enzyme or epitope. Effective
expression
cassettes are then incorporated into viral expression vectors.
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
Nucleic acids, preferably DNA, comprising preferred expression
cassettes are isolated from the transient expression constructs in which they
were
prepared, characterized and optimized. A preferred method of isolating such
expression
cassettes is by amplification by PCR, although other methods (e.g., digestion
with
appropriate restriction enzymes) can be used. Preferred expression cassettes
are
introduced into viral expression vectors, preferably retroviral expression
vectors, in the
following manner.
A DNA molecule comprising a preferred expression cassette is
introduced into a retroviral transfer vector by ligation. Two types of
retroviral transfer
vectors are known in the art: replication-incompetent and replication-
competent.
Replication-incompetent vectors lack viral genes necessary to produce
infectious
particles but retain cis-acting viral sequences necessary for viral
transmission. Such cis-
acting sequences include the 'Y packaging sequence, signals for reverse
transcription
and integration, and viral promoter, enhancer, polyadenylation and other
regulatory
sequences. Replication-competent vectors retain all these elements as well as
genes
encoding virion structural proteins (typically, those encoded by genes
designated gag,
pol and env) and can thus infectious particles. In contrast, these functions
are supplied
in trans to replication-incompetent vectors in a packaging cell line, i.e., a
cell line that
produces mRNAs encoding gag, pol and env genes but lacking the ~I' packaging
sequence. See, generally, Cepko, Unit 9.10 of Chapter 9 in: Short Protocols in
Molecular Biology, 2nd Ed., Ausubel et al., eds., John Wiley & Sons, New York,
New
York, 1992, pages 9-30 to 9-35.
A retroviral construct comprising an expression cassette comprising a
nucleic acid of interest produces RNA molecules comprising the cassette
sequences and
the'I' packaging sequence. These RNA molecules correspond to viral genomes
that are
encapsidated by viral structural proteins in an appropriate cell line (by
"appropriate" it
is meant that, for example, a packaging cell line must be used for constructs
based on
replication-incompetent retroviral vectors). Infectious viral particles are
then produced,
and released into the culture supernatant, by budding from the cellular
membrane. The
infectious particles, which comprise a viral RNA genome that includes the
expression
51
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
cassette for the gene product of interest, are prepared and concentrated
according to
known methods. It may be desirable to monitor undesirable helper virus, i. e.,
viral
particles which do not comprise the expression cassette for the gene product
of interest.
See, generally, Cepko, Units 9.11, 9.12 and 9.13 of Chapter 9 in: Short
Protocols in
Molecular Biology, 2nd Ed., Ausubel et al., eds., John Wiley & Sons, New York,
New
York, 1992, pages 9-36 to 9-45.
Viral particles comprising an expression cassette for the gene product of
interest are used to infect in vitro (e.g., cultured cells) or in vivo (e.g.,
cells of a rodent,
or of an avian species, which are part of a whole animal). Tissue explants or
cultured
embryos may also be infected according to methods known in the art. See,
generally,
Cepko, Unit 9.14 of Chapter 9 in: Short Protocols in Molecular Biology, 2nd
Ed.,
Ausubel et al., eds., John Wiley & Sons, New York, New York, 1992, pages 9-45
to 9-
48. Regardless of the type of cell used, production of the gene product of
interest is
directed by the recombinant viral genome.
In eukaryotic expression systems, host cells may be chosen for its ability
to modulate the expression of the inserted sequences or, when the gene product
of
interest is a protein, to process the protein of interest in the desired
fashion. Such
modifications of proteins include, but are not limited to, acetylation,
carboxylation,
glycosylation, phosphorylation, lipidation and acylation. Post-translational
processing
which cleaves a "prepro" form of the protein of interest may also be important
for its
correct intracellular localization, folding and/or function. Different host
cells such as
CHO, HeLa, MDCK, HEK293, WI38, etc. have specific cellular machinery and
characteristic mechanisms for such post-translational activities and may be
chosen to
ensure the correct modification and processing of a protein of interest.
Expression systems of the invention also include the few systems in
which a nucleic acid of interest is expressed from an organellar genome. Means
for the
genetic manipulation of the mitochondrial genome of Saccharomyces cerevisiae
(Steele
et al., Proc. Natl. Acad. Sci. U.S.A. 93:5253-5257, 1996) and systems for the
genetic
manipulation of plant chlorplasts (U.5. Patent No. 5,693,507; Daniell et al.,
Nature
Biotechnology 16:345-348, 1998) have been described. Naturally, nucleic acids
that
52
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
encode polypeptide sequences may have to be altered in organellar expression
systems
in order to reflect the differences in the genetic codes of organelles (see,
e.g., Table 1).
NUCLEIC ACIDS AND NUCLEOTIDE SEQUENCES
Once a nucleic of interest has been identified, it can be used to generate
other useful nucleic acids having related sequences, including without
limitation
deoxyribonucleic acids (DNA). In a preferred embodiment, an RNA of interest is
used
to generate a cDNA molecule that can be used to detect nucleic acids having
the
sequence of interest, or to produce a polypeptide encoded by the sequence of
the RNA
of interest.
For example, it is known in the art to isolate mRNAs of interest and have
them reverse-transcribed. Reverse transcription is a process by which a
reverse
complementary DNA (cDNA) is produced from an RNA molecule which acts as a
template. The RNA portion of the resultant (RNA:DNA) hybrid may then be
displaced
or enzymatically degraded, after which the single-stranded DNA (ssDNA) is used
as a
template for one or more rounds of DNA polymerization, the product of which is
a
double-stranded DNA (dsDNA) molecule. The dsDNA molecule includes the sequence
of the RNA of interest (except that uridine residues in the RNA are replaced
by
thymidine residues in the DNA). The nucleotide sequence of the dsDNA is then
determined and analyzed; additionally or alternatively, the dsDNA is cloned,
i.e.,
incorporated into a vector DNA that is capable of replication in an
appropriate host cell.
If the dsDNA molecule includes a sequence that encodes a polypeptide, a
preferred
vector is an expression vector.
A DNA molecule prepared according to the methods of the invention can
be a full-length cDNA, i.e., one comprising a nucleotide sequence that encodes
an entire
protein. At a minimum, a full-length cDNA will encompass a "start"
(translation
initiating) codon, a "stop" (translation terminating) codon, and all the
polypeptide-
encoding sequences in-between.
Alternatively, a DNA molecule prepared according to the methods of the
invention can be an Expressed Sequence Tag (EST), i.e., one which does not
comprise a
53
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
complete full length cDNA but which does comprise a nucleotide sequence that
is a
portion of an full length cDNA or of a mRNA comprising a full length cDNA. An
EST
is useful in and of itself as, e.g., a probe in methods for detecting a mRNA
of interest.
Because a full-length cDNA is required for, e.g., recombinant DNA expression
of a
protein encoded by a mRNA interest, it may also be desirable to use an EST as
a tool to
isolate a full-length cDNA according to a variety of methods. For example, a
nucleic
acid comprising an EST sequence of interest can be labeled and used to probe
preparations of cellular DNA, cDNA or RNA for hybridizing sequences, and such
hybridizing sequences can be isolated, amplified and cloned according to known
methods. As another example, the sequence of an EST can be used to prepare
primers
for inverse PCR, a process by which sequences flanking an EST of interest can
be
determined (see, e.g., Benkel and Fong, Genet. Anal. 13:123-127, 1996;
Silverman,
Methods Mol. Biol. 54:145-155, 1996; Pang and Knecht, BioTeehnigues 22:1046-
1048,
1997; Huang, Methods Mol. Biol. 69:89-96, 1997; Huang, Methods Mol. Biol.
67:287-
294, 1997; and Offringa and van der Lee, Methods ..Mol. Biol. 49:181-195,
1996; all of
which are hereby incorporated by reference).
In methods of cloning full-length cDNAs from ESTs, and as a useful
method in its own right, it is desirable to screen mRNA or cDNA libraries
prepared
from various cells and tissues in order to identify cells and tissues that
express relatively
high levels of a nucleic acid of interest. For example, a nucleic acid of
interest can be
used to examine tissue- or temporal-specific patterns of expression of a
nucleic acid of
interest in a variety of methods known in the art. The nucleic acid of
interest can be
detectably labeled and used to probe (i) an immobilized collection of mRNA
molecules
(e.g., RNA Master BlotsTM or Multiple Tissue Northern, MTNTM, Blots from
Clontech)
or (ii) a cDNA library (prepared according to methods known in the art or
available
from, e.g., Clontech or from depositories such as the American Type Culture
Collection, ATCC, Manassas, VA). Alternatively or additionally, a sequence of
interest
can be used to design specific PCR primers that can be used in amplification
reactions
in 96-well plates wherein each well comprises first strand cDNAs from a
particular
tissue (such as, e.g., the Rapid-ScanTM gene expression panel from OriGene
54
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
Technologies, Inc., Rockville, MD). In this embodiment, automated, semi-
automated
or robotic means may be used to carry out such assays.
Mammalian tissues that may be examined include but are not limited to
brain (including, by way of example but not limitation, whole brain and
subsections
thereof, e.g., amygdala, caudate nucleus, cerebellum, cerebral cortex, frontal
lobe,
hippocampus, medulla oblongata, occipital lobe, putamen, substantia nigra,
temporal
lobe, thalamus, acumens, subthalamic nucleus, inferio temporal cortex, medial
frontal
cortex, occipital pole), heart, kidney, spleen, liver, colon, lung, small
intestine, stomach,
skeletal muscle, smooth muscle, testis, uterus, bladder, lymph nodes, spinal
cord, dorsal
root ganglia, trachea, bone marrow, placenta, salivary glands, thyroid glands,
thymus,
adrenal glands, pancreas, ovary, uterus, prostate, skin, bone marrow, pancreas
or
portions thereof such as beta cells, fetal brain and fetal liver.
In order to identify tissues or cells from which a cDNA corresponding to
an EST of interest can optimally be prepared, mRNA or cDNA libraries or arrays
derived from the organism from which the EST of interest was isolated are
probed.
Tissues or cells having a high level of expression of the nucleic acid of
interest are
preferably used as sources for full-length nucleic acids, i.e., nucleic acids
containing all
the genetic information required to express a complete gene product of
interest. The
full-length nucleic acids are used, e.g., to express the gene product (i.e.,
RNA or
protein) of interest or to prepare manipulated cells or transgenic animals in
which the
level of expression or activity, or tissue- or temporal-specific patterns of
expression, of
the gene product of interest is altered relative to the wildtype condition.
Another utility of ESTs and full-length cDNAs is to search in silico for
corresponding protein sequences, in order to identify proteins of interest
encoded
thereby and to prepare antibodies thereto. For example, the nucleotide
sequence of an
EST or cDNA of interest is translated in silico in all six potential reading
frames (three
reading frames on each strand of a dsDNA), and the resulting amino acid
sequences are
used as probes to search protein databases for a match to a portion of a
protein having a
known amino acid sequence. In the case of mitochondrial proteins, it is
desirable to
perform such in silico translations using both the "universal" genetic code
and the
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
somewhat different genetic code utilized in mitochondria (TABLE 1 ), as
different
amino acid sequences will result in each case.
TABLE l:
S DIFFERENCES BETWEEN THE "UNIVERSAL" AND MITOCHONDRIAL GENETIC CODES
"Universal" Yeast Mitochondria)Mammalian Mitochondria)
Codon Genetic Code Genetic Code Genetic Code
AGA Arg Arg (stop)
AGG Arg Arg (stop)
AUA Ile Met Met
CUA Leu Thr Leu
UGA (stop) Trp Trp
Nucleic acids having or comprising a sequence of interest can be
prepared by a variety of methods known in the art. For example, such nucleic
acids can
be made using molecular biology or synthetic techniques (Sambrook et al.,
Molecular
Cloning, A Laboratory Manual, Cold Spring Harbor Press (1989)). Many
equivalent
bases, both naturally occurring and synthetic, in nucleotide sequences are
known in the
art. For example, thymine (T) residues in DNA are transcribed into uracil (U)
residues
in RNA molecules but, because both T and U specifically pair with adenine (A)
1 S residues, these changes do not impact hybridization specificity. Nucleic
acids
comprising such equivalent substitutions are within the scope of the
disclosure. In
addition, nucleic acids of the invention may have one or more non-nucleotide
moieties.
These non-nucleotides and their use in ribozymes are described in U.S. Patent
No.
5,891,683 and includes polyethers, polyamines, polyamides, polyhydrocarbons
and
abasic nucleotides.
As another example, such nucleic acids can be oligonucleotides,
including oligodeoxyribonucleotides and oligoribonucleotides synthesized in
vitro by,
for example, the phosphotriester, phosphoramidite or H-phosphanate
methodologies
S6
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
(see, respectively, Christodoulou, "Oligonucleotide Synthesis: Phosphotriester
Approach," Chapter 2 In: Protocols for Oligonucleotides and Analogs: Synthesis
and
Properties, Agrawal, ed., Methods in Molecular Biology Vol. 20, Humana Press,
Totowa, NJ (1993); Beaucage, "Oligodeoxyribonucleotide Synthesis:
Phosphoramidite
Approach," Chapter 3, Id.; and Froehler, "Oligodeoxynucleotide Synthesis: H-
phosphonate Approach," , Chapter 4, Id., all of which are hereby incorporated
by
reference).
The length of a nucleic acid according to the present invention can be
chosen by one skilled in the art depending on the particular purpose for which
the
nucleic acid is intended. For PCR primers and antisense oligonucleotides, the
length of
the nucleic acid is preferably from about 10 to about 100 base nucleotides
(nt), more
preferably from about 12 nt to about 60 nt, and most preferably from about 15
nt to
' about 30 rt. For ribozymes, the length of the nucleic acid is preferably
from about 20 nt
to about 200 nt, more preferably from about 30 nt to about 100 nt, and most
preferably
from about 40 nt to about 80 rt. For probes, the length of the nucleic acid is
preferably
from about 10 nt to about 5,000 nt, more preferably from about 15 to about
1,000 nt,
and most preferably from about 20 nt to about S00 rt.
Appropriate chemical modifications to nucleic acids of the invention are
also readily chosen by one skilled in the art. Such modifications may include,
for
example, means by which the nucleic acid is detectably labeled for use as a
probe.
Typical detectable labels include radioactive moieties and reporter groups
such as, e.g.,
enzymes and fluorescent or luminescent moieties. Other chemical modifications
appropriate for particular uses, such as antisense applications, as explained
herein.
Detectably labeled nucleic acids are preferred for diagnostic, prognostic
and pharmacogenetic methods of the invention. Whether labeled or unlabeled,
nucleic
acids of the invention can be provided in kit form, e.g., in a single or
separate container,
along with other reagents, buffers, enzymes or materials to be used in
practicing at least
one method of the invention. The kit can be provided in a container that can
optionally
include instructions or software for performing a method of the invention.
Such
57
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
instructions or software can be provided in any language or human- or machine-
readable format.
DETECTING NUCLEIC ACIDS. INCLUDING DIFFERENTIALLY EXPRESSED
NUCLEIC ACIDS
A variety of methods for detecting nucleic acids, including differentially
expressed nucleic acids, may be used in the methods of the invention. Such
methods
include, without limitation, the following methodologies. It should be noted
that,
regardless of which method is used to identify candidate differentially
expressed genes,
a second independent method should be used to verify the results obtained from
the first
method. Preferably, in the present invention, cells that do not express an IF
1 are used
as a first cell and cells that express the IF 1 are used as the second cell
such that
differential display of the first cell and the second cell is determined. In
the present
invention, the first cell and the second cell can be the same cell, however,
the second
cell has been induced to express a particular IF 1 by an appropriate inducer,
such as
tetracycline, in a construct such as that described in FIG. 1.
Subtractive Hybridization: In a typical procedure for applying the
technique of subtraction hybridization (Hedriek et al., Nature 308:149-153,
1984) to
investigate differences in the expression of genes of a certain sample of test
or target
cells, e.g. from tumor tissues or tissues in a disease state, such as tissues
affected by
diabetes, as compared with the expression of genes of a sample of reference
cells, e.g.
cells from corresponding normal tissue, total cell mRNA is extracted (using
any
preferred method) from both samples of cells. The mRNA in the extract from the
test
or target cells is then used in a conventional manner to synthesize
corresponding single
stranded cDNA using an appropriate primer and a reverse transcriptase in the
presence
of the necessary deoxynucleoside triphosphates, and the template mRNA is
subsequently degraded by alkaline hydrolysis or RNase H to leave only the
single
stranded cDNA. The single stranded cDNA thus derived from the mRNA expressed
by
the test or target cells is then mixed under hybridizing conditions with an
excess
quantity of the mRNA extract from the reference (normal) cells; this mRNA is
58
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
generally termed the subtraction hybridization "driver" since it is this mRNA
or other
single stranded nucleic acid present in excess which "drives" the subtraction
process.
As a result, cDNA strands having common complementary sequences anneal with
the
mRNA strands to form mRNA/cDNA duplexes and are thus subtracted from the
single
stranded species present. The only single stranded DNA remaining is then the
unique
cDNA that is derived specifically from the mRNA produced by genes which are
expressed solely by the test or target cells. Alternatively, the reference
cells may be
used as a source of single-stranded DNA, and the test or target cells may be
used as a
source of driver RNA. In this case the remaining single-stranded DNA is
derived from
mRNA produced by genes expressed in the reference cells but not in the target
cells.
To complete the subtraction process, it is generally desirable to
physically to separate out the common mRNA/cDNA duplexes, using for example
hydroxyapatite (HAP) or (strept)avidin-biotin in a chromatographic separation
method.
One or more repeat rounds of the subtraction hybridization may be carried out
to
improve the degree of removal of commonly expressed sequences, although other
means may be employed (see, e.g., U.S. Patent No. 5,589,339). It is generally
desirable
to clone the sequences isolated by subtractive hybridization, such that they
may be
amplified and to facilitate identification. The single-stranded cDNA may be
converted
to double-stranded DNA by methods or means know in the art. For example,
multiple
copies of a single nucleotide, for example deoxycytidine may be added, onto
the 3' end
of the single-stranded DNA molecules using an enzyme such as terminal
transferase,
and then an oligonucleotide of complementary sequence, e.g. poly G to prime
synthesis
of the complementary strand using any of a number of commercially available
DNA
polymerases can be used. The cDNA sequences obtained from subtractive
hybridization may be used to produce labeled probes that may perhaps then be
used for
detecting or identifying corresponding cloned copies in a cDNA clone colony or
cDNA
library (labeling of such probes is frequently introduced by using labeled
deoxynucleoside triphosphates in synthesis of the cDNA),
High Density Array: Multiple sample nucleic acid hybridization
analysis can be carried out on micro-formatted multiplex or matrix devices
(e.g., DNA
59
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
or RNA chips, filters and microarrays) (see, e.g., Bains, BiolTechnology
10:757-758,
1992). These hybridization formats are micro-scale versions of the
conventional "dot
blot" and "sandwich" hybridization systems. In these methods, specific DNA
sequences are typically attached to, or synthesized on, very small specific
areas of a
solid support, allowing large numbers of different DNA sequences to be placed
in a
small area. The high density arrays comprise target elements, i.e., target
nucleic acid
molecules bound to a solid support. The nucleic acids for both the target
elements and
the probes may be, for example, RNA, DNA, or cDNA. In one type of array,
target
elements comprising nucleic acid elements that are short synthetic
oligonucleotides
derived from mRNA, eDNA or EST sequences are used to carry out serial analysis
of
gene expression (SAGE; U.S. Patent No. 5,866,330).
In methods for comparing two nucleic acid collections, nucleic acid
molecules in the test and control collections (which may be, e.g., mRNA
preparations
from a diseased and undiseased human) are detectably labeled. The first and
second
I S labeled probes thus formed are each contacted to an identical high density
array
comprising a plurality of target elements under conditions such that nucleic
acid
hybridization to the target elements can occur.
After contacting the probes to the target elements the amount of binding
to each target element in each of the two arrays is measured, and the binding
ratio (i.e.,
amount bound in the disease sample / amount bound in the control sample) is
determined for each target element. A binding ratio > 1 indicates that nucleic
acids
hybridizing to the particular target element are "up-regulated" in the nucleic
acid
collection prepared from the diseased patient relative to the nucleic acid
prepared from
the control individual, whereas a binding ratio <1 indicates that nucleic
acids
hybridizing to the particular target element are "down-regulated" in the
diseased patient.
High density cDNA arrays that may be used in the invention include but
are not limited to GeneChipTM arrays comprising synthetic oligonucleotides
(Affymetrix, Inc., Santa Clara, CA); GeneFiltersTM yeast or human cDNA arrays
(Research Genetics, Huntsville, AL); ATLAST"' cDNA arrays (Clontech); and
GEMTM
and Gene Display Arrays (GDA) cDNA arrays (Genome Systems, Inc., St. Louis,
MO).
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
Furthermore, one method for building a microarrayer (a machine that produces
microarrays) is available on-line at http://cmgm.stanford.edu/pbrown/mguide/
index.html.
One type of high density array uses electronic hybridization, i.e., a
method that directs sample DNA molecules to, and concentrates them at, test
sites on a
microchip that can be electronically activated by a positive charge. Because
DNA
molecules in solution have strong negative charges, they are attracted to
activated sites.
The electronic hybridization of sample DNA molecules at each test site
promotes rapid
hybridization of the sample DNAs with the nucleic acids of the target
elements.
Materials for electronic hybridization are available from Nanogen (San Diego,
CA) and
the method is described in U.S. Patent No. 5,849,486.
Differential Display: To investigate differences in the expression of
genes of a certain sample of test or target cells, such as tissues affected by
diabetes, as
compared with the expression of genes of a sample of reference cells, e.g.
cells from
corresponding normal brain tissue, the RNA may be reverse transcribed and
amplified
with specific primer sets, and the resulting amplification products from the
two samples
compared (Hipfel R, et al. (1998) J. Biochem Biophys. Methods 37: 131-135;
Bosch
TC and Lohmann JU (1998) Mthods Mol Biol 86: 153-160). Total cell RNA is
extracted (using any preferred method) from both samples of cells. The RNA
from both
samples is reverse transcribed using a set of twelve primers containing a
sequences of
poly (T) terminating in one of either AA, AC, AG, AT, CA, CC, CG, CT, GA,GC,
GG,
or GT. The single stranded cDNAs of the resulting cDNA/mRNA hybrids are then
amplified in separate reactions, with each reaction using one of the set of
twelve "3' "
primers used in the reverse transcription reaction and one of a set of "5'
"primers.
Typically a set of about twenty 5' primers is used, each with a different
arbitrary
sequence. The resulting amplification products are labeled, preferably by
using primers
which have incorporated a fluorescent dye, but other labeling methods and
other labels
may be used, and electrophoresed such as on gels. The products resulting from
reverse
transcription and amplification of RNA from two different samples with the
same
primer sets are compared. Bands which are overexpressed or underexpressed in
one
61
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
sample when compared with another sample may be excised from the gel,
reamplified,
cloned, and sequenced to identify genes with different levels of expression in
the two
samples.
GENETIC MODULATION OF NUCLEIC ACIDS AND GENE PRODUCTS
Various antisense-based methodologies may be used to modulate (reduce
or eliminate) the expression of a nucleic acid of interest, and the
corresponding gene
product, in organelles, cells, tissues, organs and organisms. Such antisense
modulation
may be used to validate the role of a gene of interest in a disease or
disorder or, when
the causes or symptoms of a disease or disorder result from the over-
expression of a
nucleic acid of interest, as therapeutic agents. In the case of the present
invention, the
expression of IF 1 can be increased by interfering with the transcription or
translation of
inhibitors of IF 1 transcription or translation. Alternatively, the expression
of IF 1 can be
decreased by interfering with the transcription or translation of activators
of IF 1
transcription or translation or by interfering with the transcription or
translation of IF 1
itself.
The term "antisense" refers to nucleic acids that comprise one or more
sequences that are the reverse complement of the "sense" strand of a gene,
i.e., the
strand that is transcribed and, in the case of protein-encoding sequences,
translated.
Because antisense nucleic acids bind with high specificity to their targeted
nucleic
acids, selectivity is high and toxic side effects resulting from misdirection
of the
compounds can be minimal.
In general, antisense compositions are of two types: (i) synthetic
antisense oligonucleotides, including enzymatic ones such as, e.g., ribozymes;
and
(ii) antisense expression constructs. One skilled in the art will be able to
utilize either
modality as is appropriate to the given situation.
Synthetic antisense oligonucleotides are prepared from the reverse
complement of a nucleic acid of interest. An antisense oligonucleotide
consists of
nucleic acid sequences corresponding to the reverse complement of a
differentially
expressed RNA. When introduced into cells expressing the RNA of interest, the
62
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
antisense oligonucleotides specifically bind to the RNA molecules and
interfere with
their function by preventing secondary structures from forming or blocking the
binding
of regulatory or RNA-stabilizing factors. In addition, in the case of protein-
encoding
RNA species, oligonucleotides can inhibit RNA splicing, polyadenylation or
protein
translation, thus limiting or preventing the amount of protein made from such
mRNAs.
Additionally or alternatively, such oligoncuelotides can bind to double-
stranded DNA
molecules and form triplexes therewith, and thus interfere with the
transcription of such
sequences.
In instances where it is desired to target antisense oligonucleotides to
RNAs produced from organellar genomes, peptide nucleic acids (PNAs) are
preferred
synthetic oligonucleotides. In PNAs, the sugar-phosphate backbone of
biological
nucleic acids has been replaced with a polypeptide-like chain. Targeting
sequences that
direct proteins to organelles can be conjugated to the backbone of antisense
PNAs, with
the result being that such conjugates are preferentially delivered to the
targeted
organelle (see, for example, published PCT applications WO 97/41150 and WO
99/05302.
Antisense oligonucleotides may be inherently enzymatic in nature, that
is, capable of degrading the RNA molecule towards which they are targeted;
such
molecules are generally referred to as "ribozymes." A variety of increasingly
short
synthetic ribozyr~e frameworks that can be modified to comprise a nucleic acid
sequence of interest have been described (Couture and Stinchcomb, Trends
Genet.
12:510-515, 1996), including but not limited to hairpin ribozymes (Hampel,
Prog.
Nucleic Acid Res. Mol. Biol. 58:1-39, 1998), hammerhead ribozymes (Birikh et
al., Eur.
J. Biochem. 245:1-16, 1997) and minizymes (Kuwabara et al., Nature
Biotechnology
16:961-965, 1998).
In the case of non-catalytic antisense nucleic acids and ribozymes
antisense modulation of gene expression in a cell can also be achieved by
expression
constructs that direct the transcription of the reverse complement of a
nucleotide
sequence of interest in vivo. For example, in order to express non-catalytic
antisense
transcripts in mammalian or plant cells, all that may be required is the
"flipping"
63
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
(i.e., reversing the orientation) of a nucleic acid of interest that has been
cloned into a
mammalian or plant expression vector, respectively. It is not necessary to
maintain the
proper relationship of elements such as translation signals and the like, as
the minimum
requirement for an antisense expression construct of this type is a promoter
operably
S linked to the reverse complement of a nucleic acid of interest. It is also
possible to
design expression constructs that express ribozymes in cells. Antisense and
ribozyme
expression constructs are also used to produce transgenic animals in which the
level of
expression of a gene of interest can be modulated in a temporal- or tissue-
specific
manner (see Sokol and Murray, Transgenic Res. 5:363-371, 1996, for a review).
Nucleic acid sequences derived according to the present invention may
also be used to design "RNA decoys," i.e., short RNA molecules corresponding
to cis-
acting regulatory sequences that bind trans-acting regulatory factors. When
overexpressed in a cell or administered in excess thereto, such RNA decoys
competitively inhibit the binding and thus action of the traps-acting
regulatory factors,
and thus limit or prevent the ability of such factors to carry out processes
that stabilize
(or destabilize) the RNA of interest, or enhance (or decrease) the
polyadenylation,
splicing nuclear transport, or translation of the RNA (Sullenger et al., J.
Virol. 65:6811-
6816, 1991 ). Expression of the RNA of interest may thus be either enhanced or
decreased for therapeutic purposes.
POLYPEPTIDES AND PROTEINS
The nucleic acids of interest identified according to the methods of the
invention may encode amino acid sequences. Such amino acid sequences may
correspond to a full-length protein or to a polypeptide portion thereof. The
present
invention also includes polypeptides that are derivatives of IF1, or
polypeptides that
have at least one activity of IFl. For example, as discussed above, certain
polypeptides
according to the present invention may comprise IF 1 mutant, variant,
derivative, analog,
fusion or fragment polypeptides or the like, which are unable to bind to an
ATP
synthase subunit or which, upon binding to ATP synthase, activate rather than
inhibit
ATP synthase activity. Identification, construction, expression, detection and
functional
64
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
assays of such polypeptides are readily performed by the person having
ordinary skill in
the art based upon the present disclosure.
In instances wherein a full-length protein is encoded by a nucleic acid of
interest, the protein may be a known protein that is commercially available or
one to
which antibodies are known and can be used to isolate the protein from
appropriate
biological samples. If a full-length protein of the invention has not
previously been
described, it may be produced via recombinant DNA methodologies for example,
using
the expression systems described previously, or prepared from biological
samples using
known biochemical techniques. Short (i.e., having less than about 30 amino
acids)
polypeptides that are encoded by short (i.e., having less than about 100
nucleotides)
nucleic acids of the invention or derived from the amino acid sequences
encoded by
longer nucleic acids or from full-length proteins can be synthesized in vitro
by methods
known in the art. Fusion proteins comprising amino acid sequences of interest
may also
be prepared and are included within the scope of the polypeptides and proteins
of the
invention.
Regardless of the means by which they are prepared, the polypeptides
and proteins of the invention have a variety of applications. They may be used
to
generate antibodies or to screen for ligands that may serve as therapeutic
agents, or may
themselves be used as therapeutic agents. Full-length proteins of the
invention may
have the activity of the wildtype protein and may thus be used to treat
conditions
resulting from a loss of such activity. Polypeptides of the invention may also
have such
activities, or may competitively inhibit a protein of interest in vivo by
binding a ligand
of the protein. If the ligand is an activator of the protein, such
polypeptides may be
used to treat conditions resulting from the over-expression or over-activation
of the
protein in vivo. If the ligand is a toxin or activator of cell death
(apoptosis or necrosis),
administration of a protein or polypeptide that binds such a ligand to a
patient in need
thereof will have the beneficial effect of competitively inhibiting the action
of the toxin
or cell death activator.
ANTIBODIES
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
Antibodies to a protein or polypeptide of interest are prepared according
to a variety of methods known in the art. In particular, antibodies that bind
with IF 1 or
a label sequence, such as FLAG, can be used to detect IF 1 or a label
sequence,
particularly in a cell, using labeled antibodies that bind with such
polypeptides. In
general, such antibodies may be polyclonal, monoclonal or monospecific
antibodies.
Primary antibodies of the invention bind specifically to a particular protein
or
polypeptide of interest and are thus used in assays to detect and quantitate
such proteins
and polypeptides. The invention also includes active fragments or active
portions that
exhibit the binding specificity or the substantial binding specificity of the
intact
antibody they were derived from. In such assays, generally referred to in the
art as
immunoassays, a primary antibody of the invention is detectably labeled or is
specifically recognized and monitored by a detectably labeled secondary
antibody or a
combination of a secondary antibody and a tertiary molecule (which may also be
an
antibody) that is detectably labeled. Regardless of the specific format, the
primary
antibody of the invention provides a means by which a protein or polypeptide
of interest
is specifically bound and subsequently detected. One preferred assay format is
the
Enzyme-Linked Immunosorbent Assay (ELISA) format.
A nucleic acid of interest may encode a known protein or a portion
thereof, or a polypeptide sequence that is homologous to a known protein. In
such
instances, antisera to the known protein, or the known protein itself, may be
commercially available. In the latter instance, or when the nucleic acid of
interest can
be used to produce a protein of interest (or a polypeptide portion thereof
greater than
about 30 amino acids in length) via recombinant DNA expression techniques, the
known or recombinantly-produced protein can be used to immunize a mammal of
choice (e.g., a rabbit, mouse or rat) in order to produce antisera from which
polyclonal
antibodies can be prepared (see, e.g., Cooper and Paterson, Units 11.12 and
11.13 in
Chapter 11 in: Short Protocols in Molecular Biology, 2nd Ed., Ausubel et al.,
eds., John
Wiley & Sons, New York, New York, 1992, pages 11-37 to 11-41).
In the event that a nucleic acid sequence of interest encodes a
polypeptide sequence for which no complete protein (or homolog thereof) is
known, is
66
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
too short to encode more than about 30 amino acids (i.e., the nucleic acid of
interest is
less than about 100 nucleotides in length), or encodes more than one
polypeptide
sequence of potential interest, such candidate amino acid sequences can be
used to
synthesize one or more polypeptide molecules, each of which has a defined
amino acid
sequence. Such synthetic polypeptides can then be used to immunize animals
(e.g., rabbits) according to methods known in the art (Collawn and Paterson,
Units
11.14 and 11.1 S in Chapter 11 in: Short Protocols in Molecular Biology, 2nd
Ed.,
Ausubel et al., eds., John Wiley & Sons, New York, New York, 1992, pages 11-42
to
11-46; Cooper and Paterson, Units 11.12 and 11.13 in Chapter 11 in: Short
Protocols in
Molecular Biology, 2nd Ed., Ausubel et al., eds., John Wiley & Sons, New York,
New
York, 1992, pages 11-37 to 11-41). The resulting antisera, sometimes referred
to as
"monospecific," may then be used to probe cells from which the nucleic acid of
interest
was isolated. A positive response to a given antiserum indicates that the
candidate
reading frame from which the synthetic polypeptide used to raise the antiserum
was
derived is a reading frame used to encode at least one protein in the cells)
so examined.
Moreover, such an antiserum can be used to identify proteins of interest in
the cells
from which the nucleic acid of interest was isolated.
Because of their high degree of specificity and homogeneity, monoclonal
antibodies are often the preferred type of antibody for a variety of
applications.
Methods for producing and preparing monoclonal antibodies are known in the art
(see,
e.g., Fuller et al., Units 11.4 to 11.11 in Chapter 11 in: Short Protocols in
Molecular
Biology, 2nd Ed., Ausubel et al., eds., John Wiley & Sons, New York, New York,
1992,
pages 11-22 to 11-36). Murine monoclonal antibodies may be "humanized" to
reduce
their antigenicity in humans and used as therapeutic agents (see, e.g., Gussow
and
Seemann, Methods in Enzymology 203:99-121, 1991; Vaughan et al., Nature
Biotechnology 16:535-539, 1998).
Antibodies to proteins and polypeptides of interest are used to detect
such proteins and polypeptides in a variety of assay formats. Such
immunoassays may
useful in diagnostic, prognostic or pharmacogenetic methods of the invention,
or in
methods in which various cell types, tissues or organs are probed for the
presence of a
67
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
protein of interest. Monoclonal antibodies are generally preferred for such
methods due
to their high degree of specificity and homogeneity.
DIAGNOSTIC. PROGNOSTIC AND PHARMACOGENETIC METHODS
Assays for or utilizing one or more of the antibodies, polypeptides and
proteins, ligands therefor and nucleic acids of the invention are used in
diagnostic,
prognostic and pharmacogenetic methods of the invention. The term "diagnostic"
refers
to assays that provide results which can be used by one skilled in the art,
typically in
combination with results from other assays, to determine if an individual is
suffering
from a disease or disorder of interest such as diabetes, including type I and
type II,
whereas the term "prognostic" refers to the use of such assays to evaluate the
response
of an individual having such a disease or disorder to therapeutic or
prophylactic
treatment. The term "pharmacogenetic" refers to the use of assays to predict
which
individual patients in a group will best respond to a particular therapeutic
or
prophylactic composition or treatment.
The terms "disease" and "disorder" refers to diabetes, either type I or
type II.
In diagnostic and prognostic applications of the invention, samples from
individuals are assayed with regard to the relative or absolute amounts of a
"marker,"
i.e., a nucleic acid or protein of interest, or an endogenous ligand of or
antibody to a
nucleic acid or protein of interest. An increased or decreased level of a
marker relative
to control levels indicates that the individual from which the sample was
taken has, has
had, or is likely to develop the disease or disorder of interest. The term
"control level"
refers to the level of marker present in samples taken from one or more
individuals
known to not have the disease or disorder of interest, or to the level of
marker present in
a sample taken from the individual in question before or after the diagnostic
sample.
Additionally or alternatively, a number of individuals known to not have the
disease or
disorder of interest are tested for levels of the marker, and an absolute
amount or
concentration corresponding to a normal level of the marker is established; in
this
embodiment, affected individuals are identified as those having a level of
marker that is
68
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
significantly lower or higher than the normal value. In addition, nucleic
acids of the
invention may be used to screen for single nucleotide polymorphisms (SNPs) and
other
mutations such as gene deletions or insertions, by hybridization methods
(Sapolsky RJ
et al. Genet. Anal. (1999) 14: 187-192), or other methods as they are known or
later
S developed in the art.
In pharmacogenetic applications of the invention, patients suffering from
a disease or disorder of interest are stratified with regards to desirable or
undesirable
responses to a potential treatment using one or more assays of the invention.
A
therapeutic composition and/or treatment known to be more effective, or which
produces fewer side-effects, in some patients as compared to others is
administered a
group of patients suffering from a disease or disorder of interest. A method
of
identifying which patients having the disease are more likely to respond to a
therapeutic
composition and/or treatment comprises providing samples from a group of
patients
having said disease; measuring the amount or molecular attribute of a protein
or
polypeptide of interest, or of a nucleic acid of interest, or a ligand
therefor or antibody
thereto, or any combination thereof present in said samples; providing the
therapeutic
composition and/or treatment to the patients; measuring the degree, frequency,
rate or
extent of responses of the patients to the therapeutic composition and/or
treatment; and
determining if a correlation exists between the amount or molecular attributes
of a
nucleic acid of interest, or the amount or molecular attributes of a protein
or polypeptide
of interest, or a ligand therefor or antibody thereto present in said samples
and the
degree, frequency, rate or extent of such responses.
The resulting correlations are used to stratify patients in the following
manner. If such a correlation is a positive correlation, the presence of such
correlation
indicates that patients yielding samples having an increased or decreased
amount,
relative to the established normal range, of the protein or polypeptide of
interest, or the
ligand or antibodies therefor, or nucleic acid molecules, or an increase or
decreased
amount, relative to the established normal range, of the nucleic acid of
interest, are more
likely to respond to said treatment. In contrast, if the correlation is a
negative
correlation, the presence of said correlation indicates that patients yielding
samples
69
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
having an increased amount of the protein or polypeptide of interest, or the
ligand
therefor, or of the nucleic acid of interest are less likely to respond to
said treatment.
Additionally, molecular attributes of nucleic acids and/or polypeptides of the
invention
may correlate positively or negatively with patients' responses to therapeutic
compositions and treatments, and methods to screen for the relevant molecular
attributes to stratify patients to determine optimal therapeutic courses are
also part of
the invention.
The responses) that are measured in these methods can be desirable
response(s), in which case it is preferred to provide the therapeutic
composition and/or
treatment to patients having a relatively high level of the protein or
polypeptide of
interest, or the ligand therefor, or of the nucleic acid of interest present.
Alternatively,
the responses) that are measured in these methods can be undesirable
response(s), in
which case it is preferred to avoid providing the therapeutic composition
and/or
treatment to patients having a relatively high level of the protein or
polypeptide of
interest, or the ligand therefor, or of the nucleic acid of interest.
The assays for the preceding methods may be performed at a laboratory
to which patient-derived samples or delivered, or at the site of patient
treatment. In the
latter instance, kits for performing one or more assays of the invention are
preferred.
Antibodies, polypeptides and proteins, ligands therefor and nucleic acid
probes and
primers of the invention can be provided in kit form, e.g., in a single or
separate
container, along with other reagents, buffers, enzymes or materials to be used
in
practicing at least one method of the invention. Such kits can be provided in
a container
that can optionally include instructions or software for performing a method
of the
invention. Such instructions or software can be provided in any language or
human- or
machine-readable format.
COMPOUND SCREENING, INCLUDING HIGH-THROUGHPUT ASSAYS
The nucleic acids, proteins, polypeptides, antibodies and transgenic
animals of the invention may be used to validate the role of a gene product of
interest in
a particular disease, disorder or undesirable response, and to screen for
conditions or
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
compounds that can be used to treat such diseases, disorders and undesirable
responses,
preferably using high-throughput screening methods such as they are known in
the art
or later developed. Such treatment can be remedial, therapeutic, palliative,
rehabilitative, preventative, impeditive or prophylactic in nature. Diseases
and
disorders to which the invention may be applied include diabetes, including
type I and
type II.
The term "undesirable response" refers to a biological or biochemical
response by one or more cells of an organism to one or more physical
conditions,
chemical agents, or combinations thereof that leads to an undesirable
consequence. An
undesirable response can occur at the organellar level (e.g., loss of Dyr in
mitochondria),
the cellular level (e.g., cell death such as apoptosis or necrosis), in
tissues
(e.g., ischemia), in organs (e.g., ischemic heart disease) or to the organism
as a whole
(e.g., death; loss of reproductive capacity or cognitive processes).
Physical conditions that may produce an undesirable response include,
without limitation, hypothermia, hyperthermia, dehydration, exposure to
ultraviolet and
other types of radiation, micro-gravity, physical trauma, tensile stress, and
exposure to
electrical or magnetic fields. Chemical agents that may produce an undesirable
response include without limitation reactive oxygen species (ROS), apoptogens,
and the
like.
Nucleic acids of the invention are used to screen for conditions or
compounds that can be used to treat disease states and undesirable responses
in the
following manner. Treatment of cells with antisense molecules, including
ribozymes,
or introduction thereinto of antisense constructs specific for a given gene
product of
interest, should result in such cells demonstrating at least one of the
biochemical or
biological defects associated with the disease or disorder for which the gene
product is
being validated. In like fashion, transgenic animals comprising constructs
directing the
over-expression of a gene of interest, or an antisense or ribozyme expression
construct,
or animals to which antisense, ribozyme or molecular decoy oligonucleotides
are
administered, will demonstrate at least one of the biochemical or biological
defects
associated with the disease or disorder of interest if the nucleic acid
encodes a gene
71
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
product that is a valid target for the disease or disorder. In addition, SNPs
or mutant
forms of the gene identified by the invention and correlated with diseases or
disorders
may be introduced into cells or animals by homologous recombination. Such
cells or
animals or cells derived from such animals, may be used to assess responses to
conditions or compounds that can be used to treat disease states by any of a
variety of
assays or physiological assessments/measurements.
Similarly, for polypeptides of interest that may be targets for therapeutic
intervention, cells may be contacted with one or more antibodies specific for
the
polypeptide, and the presentation of responses associated with the disease or
disorder
will be seen with valid targets. Polypeptides and proteins of the invention
are also used
to screen for conditions or compounds that can be used to treat disease states
and
undesirable responses. In one type of screen, the protein of interest, or a
polypeptide
derived therefrom having at least one activity of the protein of interest, is
produced by
recombinant DNA methods or in vitro synthetic techniques. The protein or
I S polypepeptide, which may be attached to a solid support, is contacted with
a detectably
labeled ligand (including, for example, an antibody). A compound is then
introduced to
the reaction vessel, and active compounds are identified as those that cause
the release
of the detectably labeled ligand.
Assays involving nucleic acids, polypeptides, or antibodies of the
invention may be automated for rapid screening of multiple compounds. The
invention
includes high throughput screens that may be developed as having particular
applicability to the nucleic acids, polypeptides, antibodies, and genetically
manipulated
cells of the invention, and also high throughput screens as they are currently
known in
the art (for example, Stockwell, BR et al. ( 1999) Chem. Biol. 6: 71-83;
McDonald, OB
et al. (1999) Anal. Biochem. 268: 318-329; Sapolsky, RJ et al. Genet. Anal.
(1999) 14:
187-192; Swartzmann, EE et al. (1999) Anal. Biochem. 271: 143-151; Gonzalez,
JE and
Neglescu PA (1998) Curr. Opin. Biotech. 624-631), and as may be adapted for
the
purposes of the invention.
As noted above, the present invention exploits the binding interaction
between IFI and ATP synthase as described herein to provide a method of
identifying
72
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
an agent which alters the interactions between IFl and ATP synthase. The
binding
interaction between an IF 1 and at least one ATP synthase subunit and or an
ATP
synthase mufti-subunit complex (e.g., the Fl portion) may result in the
formation of a
complex, which refers to a specific intermolecular association that results
from an
affinity interaction between an IF 1 and ATP synthase. as provided herein.
An IF 1 and ATP synthase complex may be identified by any of a variety
of techniques known in the art for demonstrating an intermolecular interaction
between
two polypeptides, for example, co-purification, co-precipitation, co-
immunoprecipitation, radiometric or fluorimetric assays, western immunoblot
analyses,
affinity capture including affinity techniques such as solid-phase ligand-
counterligand
sorbent techniques, affinity chromatography and surface affinity plasmon
resonance,
and the like. Determination of the presence of a complex may employ
antibodies,
including monoclonal, polyclonal, chimeric and single-chain antibodies, and
the like,
that specifically bind to the IF 1 as provided herein and/or to the ATP
synthase.
Labeled IF 1 polypeptides as provided herein and/or one or more labeled
ATP synthase subunits can also be used to detect the presence of a complex.
These
proteins can be labeled by covalently or non-covalently attaching a suitable
reporter
molecule or moiety, for example any of various enzymes, fluorescent materials,
luminescent materials and radioactive materials. Examples of suitable enzymes
include,
but are not limited to, horseradish peroxidase, biotin, alkaline phosphatase,
[~-
galactosidase and acetylcholinesterase. Examples of suitable fluorescent
materials
include, but are not limited to, umbelliferone, fluorescein, fluorescein
isothiocyanate,
rhodamine, dichlorotriazinylamine fluorescein, dansyl chloride and
phycoerythrin.
Appropriate luminescent materials include luminol, and suitable radioactive
materials
include radioactive phosphorus [;ZP], iodine ['Z'I or'3'I] or tritium [3H].
According to the subject invention, at least one ATP synthase subunit
polypeptide as provided herein is combined with at least one detectably
labeled IFl
polypeptide (e.g., an epitope tagged IF1 fusion protein) which interacts with
the ATP
synthase subunit under conditions and for a time sufficient to permit
formation of an
intermolecular complex.. Suitable conditions for formation of such complexes
are
73
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
known in the art and can be readily determined based on teachings provided
herein,
including solution conditions and methods for detecting the presence of a
complex
and/or for detecting free substrate in solution. According to the present
invention, there
is provided a method for identifying an agent that alters the interaction
between an IF 1
and an ATP synthase, comprising comparing the level of IFl-ATP synthase
binding in
the presence of a candidate agent to the level of binding in the absence of
such agent.
Such binding assays may employ direct or indirect detection of the presence of
a
binding complex, and are readily adaptable to high throughput screening
formats with
which those having ordinary skill in the art will be familiar. In certain
other related
embodiments, ATP synthase catalytic activity (instead of binding interactions
between
IF 1 and ATP synthase) may be determined according to established methods,
such that
the influence of a candidate agent on the IF 1 effect upon ATP synthase
catalytic activity
may be observed.
THERAPEUTIC APPLICATIONS
Therapeutic agents derived therefrom according to the above
embodiments can be employed in combination with conventional excipients, i.
e.,
pharmaceutically acceptable organic or inorganic carrier substances suitable
for
parenteral application which do not deleteriously react with the active
compound.
Suitable pharmaceutically acceptable carriers include, but are not limited to,
water, salt
solutions, alcohol, vegetable oils, polyethylene glycols, gelatin, lactose,
amylose,
magnesium stearate, talc, silicic acid, viscous paraffin, perfume oil, fatty
acid
monoglycerides and diglycerides, petroethral fatty acid esters,
hydroxymethylcellulose,
polyvinylpyrrolidone, etc. The pharmaceutical preparations can be sterilized
and if
desired, mixed with auxiliary agents, e.g., lubricants, preservatives,
stabilizers, wetting
agents, emulsifiers, salts for influencing osmotic pressure, buffers,
colorings, flavoring
and/or aromatic substances and the like which do not deleteriously react with
the active
compounds. For parenteral application, particularly suitable vehicles consist
of
solutions, preferably oily or aqueous solutions, as well as suspensions,
emulsions, or
implants. Aqueous suspensions may contain substances which increase the
viscosity of
74
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
the suspension and include, for example, sodium carboxymethyl cellulose,
sorbitol,
and/or dextran. Optionally, the suspension may also contain stabilizers (see
generally
WO 98/13353 to Whitney, published April 2, 1998).
The term "therapeutically effective amount," for the purposes of the
invention, refers to the amount of a therapeutic agent which is effective to
achieve its
intended purpose. While individual needs vary, determination of optimal ranges
for
effective amounts of a therapeutic agent is within the skill of the art. Human
doses can
be extrapolated from animal studies (Fingle and Woodbury, Chapter 1 in Goodman
and
Gilman's The Pharmacological Basis of Therapeutics, 5th Ed., MacMillan
Publishing
Co., New York (1975), pages 1-46). Generally, the dosage required to provide
an
effective amount of the composition, and which can be adjusted by one of
ordinary skill
in the art will vary, depending on the age, health physical condition, weight,
extent of
disease of the recipient, frequency of treatment and the nature and scope of
the desired
effect.
Therapeutic agents of the invention can be delivered to mammals via
intermittent or continuous intravenous injection of one or more these
compositions or of
a liposome (Rahman and Schein, in Liposomes as Drug Carriers, Gregoriadis,
ed., John
Wiley, New York (1988), pages 381-400; Gabizon, A., in Drug Carrier Systems,
Vol.
9, Roerdink et al., eds., John Wiley, New York, 1989, pp. 185-212)
microparticle (Tice
et al., U.S. Patent 4,542,025), or a formulation comprising one or more of
these
compositions; via subdermal implantation of drug-polymer conjugates (Duncan,
Anti-
cancer Drugs 3:175-210, 1992; via microparticle bombardment (Sanford et al.,
U.S.
Patent 4,945,050); via infusion pumps (Blackshear and Rohde, in: Drug Carrier
Systems, Vol. 9, Roerdink et al., eds., John Wiley, New York, 1989, pp. 293-
310) or by
other appropriate methods known in the art (see, generally, Remington's
Pharmaceutical Sciences, 18th Ed., Gennaro, ed., Mack Publishing Co., Easton,
PA,
1990).
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
TRANSGENIC ANIMALS
Transgenic animals, modified with regards to a nucleic acid of interest,
may be prepared. Such animals are useful for developing animal models of human
disease and for evaluating the safety and effectiveness of therapeutic agents
of the
invention. In general, such transgenic animals are of four types: (i)
"transgenic knock-
outs," in which the animal's homologs of a gene of interest are disrupted or
removed,
with a resulting loss of function of the corresponding gene product; (ii)
"constitutive
transgenics," in which the gene of interest in operably linked to a
constitutive promoter,
(iii) "regulatable transgenics," in which the gene of interest is operably
linked to an
inducible promoter; and (iv) "replacement transgenics," in which the animal's
homolog
of the gene of interest has been replaced with the human gene of interest, or
with an
alternate form, for example a mutated form, of the gene of interest, which may
be
expressed from an endogenous or inducible promoter.
The non-human transgenic animals of the invention comprise any animal
that can be genetically manipulated to produce one or more of the above-
described
classes of transgenic animals. Such non-human animals include vertebrates such
as
rodents, non-human primates, sheep, dog, cow, amphibians, reptiles, etc.
Preferred
non-human animals are selected from non-human mammalian species of animals,
including without limitation animals from the rodent family including but not
limited to
rats and mice, most preferably mice (see, e.g., U.S. Patents 5,675,060 and
5,850,001).
Other non-human transgenic animals that may be prepared include without
limitation
rabbits (U.5. Patent No. 5,792,902), pigs (U.S. Patent No. 5,573,933), bovine
species
(U.S. Patents 5,633,076 and 5,741,957) and ovine species such as goats and
sheep (U.5.
Patents 5,827690; 5,831,141; and 5,849,992).
In one aspect of the present invention, animals, such as mice or rats, that
have identified IFl genes can be engineered such that the animal IF1 is
"knocked out"
and replaced with the human version. Such mice can be made using homologous
recombination. These animals can be compared to their non-engineered
counterparts to
evaluate the activity of the human IF 1.
76
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
The transgenic animals of the invention are animals into which has been
introduced by nonnatural means (i.e., by human manipulation), one or more
genes that
do not occur naturally in the animal, e.g., foreign genes, genetically
engineered
endogenous genes, etc. The nonnaturally introduced genes, known as transgenes,
may
be from the same species as the animal but not naturally found in the animal
in the
configuration and/or at the chromosomal locus conferred by the transgene, or
they may
be from a different species. Transgenes may comprise foreign DNA sequences,
i.e.,
sequences not normally found in the genome of the host animal. Alternatively
or
additionally, transgenes may comprise endogenous DNA sequences that are
abnormal
in that they have been rearranged or mutated in vitro in order to alter the
normal in vivo
pattern of expression of the gene, or to alter or eliminate the biological
activity of an
endogenous gene product encoded by the gene. (Watson et al., in Recombinant
DNA,
2d Ed., W.H. Freeman & Co., New York, 1992), pages 255-272; Gordon, Intl. Rev.
Cytol. 115:171-229, 1989; Jaenisch, Science 240:1468-1474, 1989; Rossant,
Neuron
1 ~ 2:323-334, 1990). Transgenes may be introduced into the genome by
homologous
recombination, whereby the transgene replaces the endogenous copy of the gene
in the
recipient animal's genome. Methods of generating and screening targeted gene
replacements and the generation of transgenic animals carrying targeted gene
replacements are described in U.S. Patent No. 5,814,300.
The transgenic non-human animals of the invention are produced by
introducing transgenic constructs comprising sequences of interest, or the
host animal's
homologs thereof, into the germline of the non-human animal. Embryonic target
cells
at various developmental stages are used to introduce the transgenes of the
invention.
Different methods are used depending on the stage of development of the
embryonic
target cell(s).
Microinjection of zygotes is the preferred method for incorporating
transgenes into animal genomes in the course of practicing the invention. A
zygote, a
fertilized ovum that has not undergone pronuclei fusion or subsequent cell
division, is
the preferred target cell for microinjection of transgenic DNA sequences. The
murine
male pronucleus reaches a size of approximately 20 micrometers in diameter, a
feature
77
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
which allows for the reproducible injection of 1-2 picoliters of a solution
containing
transgenic DNA sequences. The use of a zygote for introduction of transgenes
has the
advantage that, in most cases, the injected transgenic DNA sequences will be
incorporated into the host animal's genome before the first cell division
(Brinster et al.,
Proc. Natl. Acad. Sci. U.S.A. 82:4438-4442, 1985). As a consequence, all cells
of the
resultant transgenic animals (founder animals) stably carry an incorporated
transgene at
a particular genetic locus, referred to as a transgenic allele. The transgenic
allele
demonstrates Mendelian inheritance: half of the offspring resulting from the
cross of a
transgenic animal with a non-transgenic animal will inherit the transgenic
allele, in
accordance with Mendel's rules of random assortment.
Viral integration can also be used to introduce the transgenes of the
invention into an animal. The developing embryos are cultured in vitro to the
developmental stage known as a blastocyte. At this time, the blastomeres may
be
infected with appropriate retroviruses (Jaenisch, Proc. Natl. Sci. U.S.A.
73:1260-1264,
1976; Soriano and Jaenisch, Cell 46:19-29, 1986). Infection of the blastomeres
is
enhanced by enzymatic removal of the zona pellucida (Hogan, et al., in
Manipulating
the Mouse Embryo, Cold Spring Harbor Press, Cold Spring Harbor, N.Y., 1986).
Transgenes are introduced via viral vectors which are typically replication-
defective but
which remain competent for integration of viral-associated DNA sequences,
including
transgenic DNA sequences linked to such viral sequences, into the host
animal's
genome (Jahner et al., Proc. Natl. Acad. Sci. U.SA. 82:6927-6931, 1985; Van
der
Putten et al., Proc. Natl. Acad. Sci. U.S.A. 82:6148-6152, 1985). Transfection
is easily
and efficiently obtained by culture of blastomeres on a mono-layer of cells
producing
the transgene-containing viral vector (Van der Putten et al., Proc. Natl.
Acad. Sci.
U.S.A. 82:6148-6152, 1985; Stewart, et al., EMBO J. 6:383-388, 1987).
Alternatively,
infection may be performed at a later stage, such as a blastocoele (Jahneret
al., Nature
298:623-628, 1982). In any event, most transgenic founder animals produced by
viral
integration will be mosaics for the transgenic allele; that is, the transgene
is
incorporated into only a subset of all the cells that form the transgenic
founder animal.
Moreover, multiple viral integration events may occur in a single founder
animal,
78
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
generating multiple transgenic alleles which will segregate in future
generations of
offspring. Introduction of transgenes into germline cells by this method is
possible but
probably occurs at a low frequency (Jahner et al., Nature 298:623-628, 1982).
However, once a transgene has been introduced into germline cells by this
method,
offspring may be produced in which the transgenic allele is present in all of
the animal's
cells, i.e., in both somatic and germline cells.
Embryonic stem (ES) cells can also serve as target cells for introduction
of the transgenes of the invention into animals. ES cells are obtained from
pre-
implantation embryos that are cultured in vitro (Evans et al., Nature 292:154-
156,
1981; Bradley et al., Nature 309:255-258, 1984; Gossler et al., Proc. Natl.
Acad. Sci.
U.S.A. 83:9065-9069, 1986; Robertson et al., Nature 322:445-448, 1986;
Robertson,
E.J., in Teratocarcinomas and Embryonic Stem Cells: A Practical Approach,
Robertson, E.J., ed., IRL Press, Oxford, 1987, pp. 71-112). ES cells, which
are
commercially available (from, e.g., Genome Systems, Inc., St. Louis, MO), can
be
transformed with one or more transgenes by established methods (Lovell-Badge,
R.H.,
in Teratocarcinomas and Embryonic Stem Cells: A Practical Approach, Robertson,
E.J., ed., IRL Press, Oxford, 1987, pp. 153-182). Transformed ES cells can be
combined with an animal blastocyst, whereafter the ES cells colonize the
embryo and
contribute to the germline of the resulting animal, which is a chimera
(composed of
cells derived from two or more animals) (Jaenisch, Science 240:1468-1474,
1988;
Bradley in: Teratocarcinomas and Embryonic Stem Cells: A Practical Approach,
Robertson, E.J., ed., IRL Press, Oxford 1987, pp. 113-151 ). Again, once a
transgene
has been introduced into germline cells by this method, offspring may be
produced in
which the transgenic allele is present in all of the animal's cells, i.e., in
both somatic and
germline cells.
However it occurs, the initial introduction of a transgene is a non-
Mendelian event. However, the transgenes of the invention may be stably
integrated
into germ line cells and transmitted to offspring of the transgenic animal as
Mendelian
loci. In mosaic transgenic animals, some cells carry the transgenes and other
cells do
not. In mosaic transgenic animals in which germ line cells do not carry the
transgenes,
79
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
transmission of the transgenes to offspring does not occur. Nevertheless,
mosaic
transgenic animals are capable of demonstrating phenotypes associated with the
transgenes.
Offspring that have inherited the transgenes of the invention are
distinguished from littermates that have not inherited transgenes by analysis
of genetic
material from the offspring for the presence of biomolecules that comprise
unique
sequences corresponding to sequences of, or encoded by, the transgenes of the
invention. For example, biological fluids that contain polypeptides uniquely
encoded
by the transgenes of the invention may be immunoassayed for the presence of
the
polypeptides. A more simple and reliable means of identifying transgenic
offspring
comprises obtaining a tissue sample from an extremity of an animal, e.g., a
tail, and
analyzing the sample for the presence of nucleic acid sequences corresponding
to the
DNA sequence of a unique portion or portions of the transgenes of the
invention. The
presence of such nucleic acid sequences may be determined by, e.g.,
hybridization
("Southern") analysis with DNA sequences corresponding to unique portions of
the
transgene, analysis of the products of PCR reactions using DNA sequences in a
sample
as substrates and oligonucleotides derived from the transgene's DNA sequence,
etc.
Cloned animals, transgenic and otherwise, of the invention may also be
prepared (for a review of mammalian cloning techniques, see Wolf et al., J.
Assist.
Reprod. Genet. 15:235-239, 1998). Such cloned animals include, without
limitation,
ovine species such as sheep (Campbell et al., Nature 380:64-66, 1996; Wells et
al., Biol.
Reprocl. 57:385-393, 1997) rodents such as mice (Wakayama et al., Nature
394:369-
374, 1998) and non-human primates such as rhesus monkeys (Meng et al., Biol.
Reprod.
57:454-459, 1997).
The transgenic and cloned animals of the invention may be used as
animal models of human disease states and to evaluate potential therapies for
such
disease states. For example, in such methods, a first transgenic animal having
a disease
state (or one or more symptomatic components thereof) is given a known dose of
a
candidate therapeutic composition or exposed to a candidate therapeutic
treatment, and
a second (control) transgenic animal is given a placebo or not exposed to the
candidate
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
therapeutic treatment. Symptoms and/or clinical end-points relevant to the
disease state
are measured in both animals over appropriate intervals of time, and the
results are
compared. Therapeutic (desirable) compositions and treatments are identified
as those
which ameriolate, delay the onset of or eliminate such symptoms and end-points
in the
treated animal relative to the control animal. In like fashion, undesirable
compositions
and treatments that aggravate or accelerate the disease state are identified
as those
which enhance the degree of such symptoms and end-points and/or hasten their
onset.
Because of their high degree of genetic identity, cloned transgenic animals
are preferred
in such methods.
EMBODIMENTS OF THE INVENTION
A. Methods for Increasing at Least One Mitochondria) Function in a Cell.
In certain embodiments the present invention provides a method to
increase at least one mitochondria) function in cells, particularly ex vivo or
in vivo. The
present invention is not limited to any particular cell type, disease or
disorder.
Preferably, the present invention increases at least one mitochondria)
function in
diabetic or prediabetic cells or subjects (diabetes type I or diabetes type
II), particularly
in insulin producing cells or glucose responsive cells. Such increase in at
least one
mitochondria) function can preferably be accomplished by regulating the
transcription,
translation or activity of IF 1.
Thus, the invention provides a method for treating diabetes that includes
improving at least one mitochondria) function in cells in a subject in need
thereof. This
method can be accomplished in any number of ways, including providing
appropriate
stimuli, compounds or compositions, including small molecules, polypeptides,
nucleic
acid molecules, gene therapy constructs or organic molecules, compounds or
compositions identified using a method of the present invention or
combinations
thereof.
Increasing or improving at least one mitochondria) function in a cell can
be accomplished in any manner. Preferably, the mitochondria) function being
improved
81
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
is in functional (i. e., not uncoupled) mitochondria such that ATP production
within the
cell is increased. However, mitochondria may according to certain embodiments
be
uncoupled to some degree, for example by uncoupling factors such as UCP's (Wu
et al.,
Cell 98:115-124 (1999)). Alternatively, mitochondria) function in increased
such that
ATP production within the cell is increased. Not wanting to be limited to
theory, the
increase in ATP production related to the increase in mitochondria) function
in insulin
producing cells results in an increase in insulin production and/or insulin
secretion.
Alternatively, the increase in ATP production can increase the sensitivity of
insulin
sensitive cells to insulin.
The cells can be any cells within the subject, preferably insulin
producing cells or insulin sensitive cells. Preferred insulin producing cells
are
pancreatic cells, such as within the islets of Langerhans, preferably the beta
cells.
Preferred insulin sensitive cells are those cells involved in glucose
metabolism,
homeostasis and/or storage, such as liver cells and/or muscle cells. One
additional
l5 benefit to increasing mitochondria) function in liver cells is that the
activity of the liver
can increase such that these cells can perform detoxification functions, such
as for
reducing the toxicity or increasing the solubility of compounds, including
therapeutics
such as antiviral compounds and antisense compounds. In addition, subjects
that have
liver diseases or disorders, such as hepatitis, cirrhosis, toxic intake of
compounds, can
have their liver function increased using the methods of the present
invention.
In certain embodiments of the present invention, the subject and/or the
cells are treated with at least one agent that enhances at least one activity
of an IF1 gene
or polypeptide. Agents that increase the activity of an IF 1 gene are those
that can
directly or indirectly increase the transcription of such gene, modulate post-
transcriptional modification or mRNA half life. Examples of such compounds can
include cold and caloric intake. Alternatively, the cell or subject can
include a nucleic
acid molecule that can be induced to increase the transcription of endogenous
or
exogenous IF 1 genes. For example, such constructs can include an IF 1 gene
operably
linked to an inducible or constitutive promoter such that IF 1 transcription
can be
increase in a regulated or non-regulated fashion.
82
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
IF 1 can be any IF 1, such as a wildtype or mutated rat, mouse or human
IF 1. An IF 1 can have at least one activity of an IF 1, preferably binding to
a subunit of
an ATP synthase but without an inhibitory effect on ATP production, which can
then
lead to increased ATP synthesis. Wildtype IFIs that bind to and inhibit ATP
synthase
catalytic activity are also useful according to the present invention, for
example in
screening assays for agents that interfere with these functional activities.
Various IF 1
nucleic acid sequences and amino acid sequences from a variety of biological
sources
are provided in SEQ ID NOs:l2-16. These sequences or portions thereof or
related
sequences as described herein that include at least one activity of an IF 1
can be used in
the present invention.
B. Methods for Screening for Test Compounds that Increase Mitochondria)
Function.
As provided herein, according to certain embodiments the present
invention provides a method to screen for compounds that increase
mitochondria)
1 S function, particularly ex vivo or in vivo. The present invention is not
limited to a
particular mechanism cell type, disease state or disorder. Preferably,
mitochondria)
function is increased in cells that are prediabetic or diabetic in nature,
particularly
insulin producing cells, including glucose responsive cells (diabetes type I
or diabetes
type II). Such increase in mitochondria) function can be accomplished by
regulating the
transcription, translation or activity of IF 1.
One embodiment of the present invention is a method for screening for
identifying test compounds that influence the expression of a nucleic acid
that encodes
an IF 1 protein, that includes contacting at least one cell that includes a
nucleic acid
molecule that encodes an IF 1 protein with one or more test compounds; and
measuring
the expression of an IF1 protein.
The cell used in the methods of the present invention can be any cell
including, preferably a cell that is insulin producing or insulin sensitive,
preferably cells
in culture, such as continuous cell lines. In addition, cells from whole
organisms,
including cells in suspension or from a tissue or organ or fluid from an
organism, such
83
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
as Zucker diabetic fatty rats (ZDF's), preferably pancreatic cells such as
beta cells can
be used. For insulin producing cells, the rat cell line INS 1 is preferred.
Other preferred
cells include SYSY cells, HEK293 cells, G7/V79 cells, rho° 3T3-L1, and
rho° INS-1
cells, and 293 cells. For insulin sensitive cells, muscle cells or liver cells
are preferred
as they are known in the art, such as HEPG2 cells.
The nucleic acid molecules that encode an IF 1 can be endogenous to the
genome of the cell or can be engineered into the genome such as by homologous
recombination or by random integration (Whitney et al., W098/13353, published
April
2, 1998, Smith et al., WO 94/24301, published October 27, 1994). When
endogenous,
the expression of the IF 1 can be enhanced using stimuli or compounds known or
expected to enhance such expression. When randomly integrated, such nucleic
acid
molecules can be operably linked to an endogenous regulatory element or an
exogenous
regulatory element that can be modulated in the presence of an inducer or
repressor,
such as 2XTet0,. Optionally, the IF 1 gene can be operably linked to a
reporter gene,
such as green fluorescent protein, beta-lactamase or luciferase, for example,
or tag, such
as FLAG, such that the expression of the IF 1 gene can be monitored by
measuring the
expression of the reporter gene or tag.
In the case of exogenous IF 1 genes, the genes can be operably linked to a
regulatory element to form a construct that is extra-chromosomal, such as a
plasmid.
The expression of the IF1 gene can be modulated by a repressor or inducer of
the
regulatory element. Optionally, the IF 1 gene can be operably linked to a
reporter gene,
such as green fluorescent protein, beta-lactamase or luciferase, for example,
or a tag,
such as FLAG, such that the expression of the IF 1 gene can be monitored by
measuring
the expression of the reporter gene or tag in vitro, ex vivo or in vivo. The
IF 1 gene and,
optionally, a second gene of interest, when provided together in the same
cell, can be on
the same or different extra-chromosomal elements. Such general technology is
known
in the art (U.S. Patent NO. 5,298,429 to Evans issued March 29, 1994).
As discussed above, the expression of IF1 can be measured using a
variety of methods (in vitro, ex vivo or in vivo), including reporter genes or
tags, such as
immunological tags. In addition, other detection methods, such as Northern
blots or
84
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
Southern blots can be used. Furthermore, nucleic acid amplification methods,
such as
PCR, such as quantitative PCR or RT-PCR can be used. Also, in situ
hybridization
methods or immunohistochemical or other receptor-ligand reactions can be used.
Alternatively, the activity of an IF 1 can be directly measured, such as
IF1 binding to at least one ATP synthase subunit or to an IF1-specific
antibody, or by
other methods known in the art. Compounds that alter or modulate IFl activity
can also
presumptively influence mitochondria) biogenesis, ATP synthesis, insulin
production or
insulin secretion, among others.
The cells of the present invention can be contacted with one or more test
chemicals. The expression of IF 1 in the cells can be monitored and test
compounds that
increase such expression can be identified. Alternatively, test compounds that
increase
the production of ATP, decrease the hydrolysis of ATP, increase the synthesis
or
secretion of insulin or increase the insulin sensitivity of the cell can be
monitored using
methods known in the art. Test compounds having such activity can be
identified and
screened for other activities described herein.
GENERAL MATERIALS AND METHODS
1. EXPRESSION CONSTRUCTS AND CELLS
Nucleic acid molecules of the present invention can be provided as part
of an expression construct. An expression construct is a nucleic acid molecule
that
includes expression control sequences, such as promoters, appropriate for the
expression of a nucleic acid molecule in an appropriate expression system.
Preferably,
a nucleic acid molecule of the present invention is operably linked to an
expression
control sequence, such as a promoter, that is appropriate for a particular
expression
system, such as an in vitro expression system or a host cell, such as a
bacterial or
eukaryotic cell.
"Operably linked" refers to a juxtaposition wherein the components so
described are in a relationship permitting them to function in their intended
manner. A
control sequence operably linked to a coding sequence is ligated in such a way
that
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
expression of the coding sequence is achieved under conditions compatible with
the
control sequences.
"Control sequences" refer to polynucleotide sequences that effect the
expression of coding and non-coding sequences to which they are ligated. The
nature of
such control sequences differs depending upon the host organism; in
prokaryotes, such
control sequences generally include promoter, ribosomal binding site, and
transcription
termination sequences; in eukaryotes, generally, such control sequences
include
promoters and transcription termination sequences. The term control sequences
is
intended to include components whose presence can influence expression, and
can also
include additional components whose presence is advantageous, for example,
leader
sequences and fusion partner sequences.
A nucleic acid molecule can be engineered into an expression construct,
such as a plasmid or viral vector, using methods known in the art (Sambrook et
al.,
supra, 1989). The nucleic acid molecule is preferably inserted in-frame and in
the
proper orientation in the expression construct such that a polypeptide of
appropriate
amino acid sequence relative to the native polypeptide coded by the nucleic
acid is
produced upon expression thereof. Such in-frame insertions can be inferred
from the
nucleotide sequence of a nucleic acid molecule and be confirmed using a
variety of
methods, including computer anlaysis of predicted amino acid sequences and the
folding thereof, or by binding with antibodies that specifically bind with
identified or
orphan proteins, such as unidentified proteins or portions of proteins that do
not have an
identified function.
The nucleic acid molecules of the invention, preferably in an expression
construct, can be inserted into a host cell, such as a prokaryotic cell (such
as a bacterium
such as E. coli) or a eukaryotic cell (such as a HeLa cell) using methods
known in the
art, such as electroporation or treatment with cold calcium solutions. The
expression
construct is preferably configured such that an expression control element,
such as a
promoter, is operably linked to a nucleic acid molecule of the present
invention in-
frame and in the proper orientation such that the native amino acid sequence
encoded by
the nucleic acid molecule of the present invention are expressed by the
expression
86
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
construct. Expression constructs can be chosen such that the nucleic acid
molecule of
the present invention is expressed efficiently in a chosen host cell. The
products of the
expressed nucleic acid of the present invention, including RNA transcripts and
at least
one polypeptide, can be collected and identified using methods known in the
art. "RNA
transcripts" are RNA molecules that are synthesized ("transcribed") by RNA
polymerase using DNA as a template.
2. GENE THERAPY CONSTRUCTS
Another aspect of the present invention is a gene therapy construct that
includes an expression vector that includes a promoter operably linked to at
least one
nucleic acid of the present invention. Preferably, the nucleic acid of the
present
invention is selected from a) a substantially pure nucleic acid molecule
including at
least one of SEQ ID NO:1 through SEQ ID N0:12 and reverse complements thereof,
a
cDNA molecule prepared by a method of the present invention and reverse
complements thereof.
The gene therapy construct is preferably a viral vector, such as a
retrovirus, adenovirus, adenoassociated virus, papilloma virus or other type
of virus
vector used in gene therapy systems or genetic manipulation of cells.
Preferred gene
therapy constructs include those that can target insulin producing cells or
insulin
sensitive cells. Coxsakievisus, particular Coxsackievirus B and Coxsackievirus
B4,
Echoviruses, such as Echo 1 l, certain adenoviral vectors and certain
retroviruses, such
as C-type retroviruses, can target pancreatic cells, such as beta cells
(Ramsingh et al.,
Bioessays 19:793-800 (1997), Hyoty et al., Clin. Diagn. Virol. 9:77-84 (1998),
Jenson
et al., Lancet, 2(8190):354-358 (1980), Luppi et al., J. Biol. Regul. Homeost.
Agents
13:14-24 ( 1999), Tsumura et al., Lab. Anim. 32:86-94 ( 1998), Frisk et al.,
virus Res.
33:229-240 (1994), Giannoukakis et al. Diabetes 48:1730-1736 (1999)). In
addition,
liposomes and lipid preparations can also be used as vectors. A variety of
these types of
vectors are known in the art (see, for example: U.S. Patent No. 5,399,346 to
Anderson
et al., issued March 21, 1995; Bandara et al., DNA and Cell Biology, 11:227-
231
(1992); Berkner, Biotechniques 6:616-629 (1989); U.S. Patent No. 5,240,846 to
Collins
87
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
et al., issued August 31, 1993; Culver and Blaese, TIG 5:171-178 (1994);
Goldman et
al., Gene Therapy 3:811-818 (1996); Hamada et al., Gynecologic Oncology 63:219-
227
( 1996); Holmberg et al., J. Liposome Res. 1:393-406 ( 1990); Hurford et al.,
Nature
Genetics 10:430-435 (1995); Karlsson et al., The EMBO J. 5:2377-2385 (1986);
Kleinerman et al., Cancer Res. 55:2831-2836 (1995); Krul et al., Cancer
Immunol.
Immunother. 43:44-48 (1996); U.1S. Patent No. 5,532,220 to Lee et al., issued
July 2,
1996; Liu et al., Nature Biotechnology 15:167-173 (1997); Mathiowitz et al.,
Nature
386:410- (1997); Nabel et al. Proc. Natl. Acad. Sci. USA 90:11307-11311
(1993);
Nabel et al., Science, 14 Sep:1285-1288 (1990); Ram et al., Cancer Res. 53:83-
88
(1993); Rosenfeld et al., Cell 68:143-155 (1992); I1.S. Patent No. 5,580,859
to Felgner
et al., issued December 30, 1997 WO 98/13353 to Whitney et al., published
April 2,
1998; U.S. Patent No. 5,298,429 to Evans et al., issued March 29, 1994; U.S.
Patent No.
5,514,561 to Quante et al., issued May 7, 1996; WO 96/24301 to The University
of
Edinburgh, published October 27, 1994; WO 96/30540 to The Regents of the
University of California, published October 3, 1996; Larrick and Burck, Gene
Therapy,
Application of Molecular Biology, Elsevier, New York (1991); and Pinkert,
Tansgenic
Animal Technology, a Laboratory Handbook, Academic Press, Inc., San Diego (
1994)).
Appropriate viral vectors can be selected based on the route of
administration and the target cell type or population. For example,
retroviruses are
preferred if the target cell type or population is actively proliferating and
other viruses,
such as lentivirus, adeno associated virus, adenoviruses, are preferred if the
target cell
type or population is not actively proliferating (see, for example, Larrick et
al, Gene
Therapy, Elsevier, New York ( 1991 )). Different viruses have different
specificity for
different cell types and populations. Thus, viruses that infect a targeted
cell type of
population of cells can be selected. The viral vector can be provided as a
pharmaceutical composition in an appropriate pharmaceutically acceptable
carrier, such
as an exciptient, at an appropriate dose for an appropriate route of
administration and
regime.
The gene therapy construct can also be a naked DNA construct such as
plasmids that are useful in a gene therapy treatment system (see, for example,
U.S.
88
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
Patent No. 5,580,859 to Felgner et al., issued December 3, 1996; U.S. Patent
No.
5,703,055 to Felgner et al., issued December 30, 1997; U.S. Patent No.
5,846,946 to
Huebner et al., issued December 8, 1998; and U.S. Patent No. 5,910,488 to
Nabel et al.,
issued June 8, 1999). A particular vector can be made with a particular target
tissue,
cell type or population of cells in mind. For example, particular regulatory
elements,
such as control elements and promoters, can be chosen based on the target
cells such
that the regulatory elements are operable in the target cells. The vector is
preferably
introduced into a subject via direct injection into the pathological location,
such as the
brain, but other methods of delivery, such as systemic or intra-tissue or
organ
administration distal from the pathological location, such as the muscle, may
also be
used. These types of vectors can be provided as a pharmaceutical composition
in an
appropriate pharmaceutically acceptable carrier, such as an exipient, at an
appropriate
dose for an appropriate route of administration and regime.
3. SCREENING METHODS
The present invention also includes a variety of methods to identify
biologically active agents that can modulate the activity of at least one
function of a
polypeptide of the present invention. The functions can be in vitro (outside
of a whole
cell), ex vivo (within or on a cell but not in a whole organism such as
samples from a
whole organism or cells in culture) or in vivo (within a whole organism). The
present
invention also includes biologically active agents identified by these
methods.
Organism refers to a subject, such as a non-human animal (such as a test
animal or
transgenic animal) or a human.
The term "modulation" refers to the capacity to either enhance or inhibit
a functional property of a biological activity or process, for example, enzyme
activity or
receptor binding. Such enhancement or inhibition may be contingent on the
occurrence
of a specific event, such as activation of a signal transduction pathway
and/or may be
manifest only in particular cell types.
The term "modulator" refers to a chemical (naturally occurring or non-
naturally occurring), such as a biological macromolecule (for example, nucleic
acid,
89
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
protein, non-peptide or organic molecule) or an extract made from biological
materials,
such as prokaryotes, bacteria, eukaryotes, plants, fungi, multicellular
organisms or
animals, invertebrates, vertebrates, mammals and humans, including, where
appropriate,
extracts of: whole organisms or portions of organisms, cells, organs, tissues,
fluids,
whole cultures or portions of cultures, or environmental samples or portions
thereof.
Modulators are typically evaluated for potential activity as inhibitors or
activators
(directly or indirectly) of a biological process or processes (for example,
agonists,
partial antagonists, partial agonists, antagonists, antineoplastic agents,
cytotoxic agents,
inhibitors of neoplastic transformation or cell proliferation, cell
proliferation promoting
agents, antiviral agents, antimicrobial agents, antibacterial agents,
antibiotics, and the
like) by inclusion in assays described herein. The activity of a modulator may
be
known, unknown or partially known.
The terms "test compound" or "test chemical" refers to a chemical,
compound, composition or extract to be tested by at least one method of the
present
invention to be a putative modulator. A test compound or test chemical
identified by
the present invention is a "biologically active agent." Test compounds can
include
small molecules, such as drugs, proteins or peptides or active fragments
thereof, such as
antibodies, nucleic acid molecules such as DNA, RNA or combinations thereof,
antisense molecules or ribozymes, or other organic or inorganic molecules,
such as
lipids, carboydrates, or any combinations thereof. Test compounds that include
nucleic
acid molecules can be provided in a vector, such as a viral vector, such as a
retrovirus,
adenovirus or adeno-associated virus, a liposome, a plasmid or with a
lipofection agent.
Test compounds, once identified, can be agonists, antagonists, partial
agonists or
inverse agonists of a target. A test compound is usually not known to bind to
the target
of interest. "Control test compound" refers to a compound known to bind to the
target
(for example, a known agonist, antagonist, partial agonist or inverse
agonist). Test
compound does not typically include a compound added to a mixture as a control
condition that alters the function of the target to determine signal
specificity in an assay.
Such control compounds or conditions include chemicals that (1) non-
specifically or
substantially disrupt protein structure (for example denaturing agents such as
urea or
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
guandium, sulfhydryl reagents such as dithiotritol and beta-mercaptoethanol),
(2)
generally inhibit cell metabolism (for example mitochondrial uncouplers) or
(3) non-
specifically disrupt electrostatic or hydrophobic interactions of a protein
(for example,
high salt concentrations or detergents at concentrations sufficient to non-
specifically
disrupt hydrophobic or electrostatic interactions). The term test compound
also does
not typically include compounds known to be unsuitable for a therapeutic use
for a
particular indication due to toxicity to the subject. Usually, various
predetermined
concentrations of test compounds are used for determining their activity. If
the
molecular weight of a test chemical is known, the following ranges of
concentrations
can be used: between about 0.001 micromolar and about 10 millimolar,
preferably
between about 0.01 micromolar and about 1 millimolar, more preferably between
about
0.1 micromolar and about 100 micromolar. When extracts are uses a test
compounds,
the concentration of test chemical used can be expressed on a weight to volume
basis.
Under these circumstances, the following ranges of concentrations can be used:
between about 0.001 micrograms/ml and about 1 milligram/ml, preferably between
about 0.01 micrograms/ml and about 100 micrograms/ml, and more preferably
between
about 0.1 micrograms/ml and about 10 micrograms/ml.
Test compounds that modulate the activity of the at least one in vitro or
ex vivo function of a polypeptide of the present invention have presumptive
therapeutic
activity in modulating the activity of that in vivo function in a subject,
including a
human. The present invention includes biologically active agents identified by
a
method of the present invention. Such biologically active agents can be
provided as a
pharmaceutical, such as with an excipient.
4. IN VITRO FUNCTION
Another aspect of the invention involves a method for identifying
biologically active agents, including: providing a sample that includes at
least one
polypeptide of the present invention; contacting the sample with at least one
test
chemical; detecting at least one in vitro function of the polypeptide; and
identifying at
least one test chemical that modulates (such as enhances or inhibits) the at
least one in
91
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
vitro function of the polypeptide. Preferably, this method is practiced in a
high
throughput format and device, such as described in WO 98/52047 to Stylli et
al.,
published November 19, 1998.
In operation, a polypeptide of the present invention having at least one in
vitro function that is detectable using a compound that provides a readout of
the at least
one in vitro function, such as an enzymatic substrate that changes at least
one property,
such as, for example, colormetric, spectrographic or fluorescent properties,
upon the
action of the at least one in vitro function upon the enzymatic substrate is
provided.
Such enzymatic substrates are known in the art for a variety of activities,
such as, for
example, proteases and kinases (see, for example, WO 97/28261 to Tsien et al.,
published August 7, 1997; WO 98/02571 to Tsien et al., published January 22,
1998;
and The Sigma Catalogue, Sigma Chemical Company, St. Louis, MO (1999)).
The polypeptide of the present invention having at least one in vitro
function is contacted with a test chemical before or contemporaneously with
being
contacted with the compound that provides a readout for the at least one in
vitro
function. The at least one in vitro function is monitored by monitoring the
readout of
that activity. The results of these studies can be compared to an appropriate
control to
determine the ability of a test chemical to modulate the activity of the at
least one in
vitro function. Appropriate controls are known in the art, such as performing
the test in
the absence of the test chemical. The control can be performed at the same
time as the
test, but can also be performed at a time and place distant from the test. For
example,
standard curves or values can be obtained and provided for a particular test
which can
be used in the comparison.
5. EX VIVO FUNCTION
Another aspect of the invention involves a method for identifying
biologically active agents, including: providing a sample that includes at
least one cell
that includes at least one polypeptide of the present invention; contacting
the sample
with at least one test chemical; detecting at least one ex vivo function of
the
polypeptide; and identifying at least one test chemical that modulates (such
as enhances
92
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
or inhibits) the at least one ex vivo function of the polypeptide. The
polypeptide of the
present invention is preferably within or associated with a cell and the test
chemical is
contacted with the cell. Preferably, this method is practiced in a high
throughput format
and device, such as described in WO 98/52047 to Stylli et al., published
November 19,
1998. The at least one cell can be from a sample from a subject, such as a
test animal,
transgenic animal, or human, or can be a cell in culture.
In operation, a polypeptide of the present invention having at least one ex
vivo function that is detectable using a compound that provides a readout of
the at least
one in vitro function, such as an enzymatic substrate that changes at least
one property,
such as, for example, colormetric, spectrographic or fluorescent properties,
upon the
action of the at least one ex vivo function upon the enzymatic substrate is
provided.
Such enzymatic substrates are known in the art for a variety of activities,
such as, for
example, proteases, kinases (see, for example, WO 97/28261 to Tsien et al.,
published
August 7, 1997; WO 98/02571 to Tsien et al., published January 22, 1998; and
The
Sigma Catalogue, Sigma Chemical Company, St. Louis, MO (1999)).
The cell that includes at least one polypeptide of the present invention
having at least one ex vivo function is contacted with a test chemical before
or
contemporaneously with being contacted with the compound that provides a
readout for
the at least one ex vivo function. The at least one ex vivo function is
monitored by
monitoring the readout of that activity. The results of these studies can be
compared to
an appropriate control to determine the ability of a test chemical to modulate
the activity
of the at least one ex vivo function. Appropriate controls are known in the
art, such as
performing the test in the absence of the test chemical. The control can be
performed at
the same time as the test, but can also be performed at a time and place
distant from the
test. For example, standard curves or values can be obtained and provided for
a
particular test which can be used in the comparison.
6. IN VIVO FUNCTION
Another aspect of the invention involves a method for identifying
biologically active agents, including: providing at least one subject that
includes at least
93
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
one polypeptide of the present invention; contacting the at least one subject
with a test
chemical; detecting at least one in vivo function of the polypeptide; and
identifying at
least one test chemical that modulates (such as enhances or inhibits) the at
least one in
vivo function of the polypeptide.
In operation, a polypeptide of the present invention having at least one in
vivo function that is detectable using a compound that provides a readout of
the at least
one in vitro function, such as an enzymatic substrate that changes at least
one property,
such as, for example, colorimetric, spectrographic or fluorescent properties,
upon the
action of the at least one in vivo function upon the enzymatic substrate is
provided.
Such enzymatic substrates are known in the art for a variety of activities,
such as, for
example, proteases, kinases (see, for example, WO 97/28261 to Tsien et al.,
published
August 7, 1997; WO 98/02571 to Tsien et al., published January 22, 1998; and
The
Sigma Catalogue, Sigma Chemical Company, St. Louis, MO (1999)).
The subject that includes at least one polypeptide of the present
1 S invention having at least one in vivo function is contacted with a test
chemical before or
contemporaneously with being contacted with the compound that provides a
readout for
the at least one in vivo function. The at least one in vivo function is
monitored by
monitoring the readout of that activity. The results of these studies can be
compared to
an appropriate control to determine the ability of a test chemical to modulate
the activity
of the at least one in vivo function. Appropriate controls are known in the
art, such as
performing the test in the absence of the test chemical. The control can be
performed at
the same time as the test, but can also be performed at a time and place
distant from the
test. For example, standard curves or values can be obtained and provided for
a
particular test which can be used in the comparison.
In the case of diabetes, a preferred animal model is the non-obese
diabetic (NOD) mouse. The successful use of this animal model in diabetic drug
discovery is reported in the literature (Yang et al., J. Autoimmun. 10:257-260
(1997),
Akashi et al., Int. Immunol. 9:1159-1164 (1997), Suri and Katz, Immunol. Rev.
169:55-
65 (1999), Pak et al., Autoimmunity 20:19-24 (1995), Toyoda and Formby,
Bioessays
20:750-757 (1998), Cohen, Res. Immunol. 148:286-291 (1997), Baxter and Cooke,
94
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
Diabetes Metal. Rev. 11:315-335 (1995), McDuffie, Curr. Opin. Immunol. 10:704-
709
(1998), Shieh et al. Autoimmunity 15:123-135 (1993), Anderson et al.,
Autoimmunity
15:113-122 (1993)). According to certain embodiments of the present invention
as
provided herein using the NOD mouse, or in another suitable animal model,
there is
contemplated the testing of the ability of a candidate agent, for example an
agent
identified using one or more of the in vitro screening assays described
herein, to
regulate (and preferably lower) blood glucose in the test animal.
7. PHARMACOLOGY AND TOXICITY OF TEST COMPOUNDS
The structure of a test compound can be determined or confirmed by
methods known in the art, such as mass spectroscopy. For test compounds stored
for
extended periods of time under a variety of conditions, the structure,
activity and
potency thereof can be confirmed.
Identified test compounds can be evaluated for a particular activity using
recognized methods and those disclosed herein. For example, if an identified
test
compound is found to have anticancer cell activity in vitro, then the test
compound
would have presumptive pharmacological properties as a chemotherapeutic to
treat
cancer. Such nexuses are known in the art for several disease states, and more
are
expected to be discovered over time. Based on such nexuses, appropriate
confirmatory
in vitro and in vivo models of pharmacological activity, and toxicology, can
be selected
and performed. The methods described herein can also be used to assess
pharmacological selectivity and specificity, and toxicity.
Identified test compounds can be evaluated for toxicological effects
using known methods (see, Lu, Basic Toxicology, Fundamentals, Target Organs,
and
Risk Assessment, Hemisphere Publishing Corp., Washington (1985); U.S. Patent
Nos;
5,196,313 to Culbreth (issued March 23, 1993) and 5,567,952 to Benet (issued
October
22, 1996)). For example, toxicology of a test compound can be established by
determining in vitro toxicity towards a cell line, such as a mammalian, for
example a
human cell line. Test compounds can be treated with, for example, tissue
extracts, such
as preparations of liver, such as microsomal preparations, to determine
increased or
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
decreased toxicological properties of the test compound after being
metabolized by a
whole organism. The results of these types of studies are predictive of
toxicological
properties of chemical's in animals, such as mammals, including humans.
Alternatively, or in addition to these in vitro studies, the toxicological
properties of a test compound in an animal model, such as mice, rats, rabbits,
dogs or
monkeys, can be determined using established methods (see, Lu, supra (1985);
and
Creasey, Drug Disposition in Humans, The Basis of Clinical Pharmacology,
Oxford
University Press, Oxford ( 1979)). Depending on the toxicity, target organ,
tissue, locus
and presumptive mechanism of the test compound, the skilled artisan would not
be
burdened to determine appropriate doses, LDSO values, routes of administration
and
regimes that would be appropriate to determine the toxicological properties of
the test
compound. In addition to animal models, human clinical trials can be performed
following established procedures, such as those set forth by the United States
Food and
Drug Administration (USFDA) or equivalents of other governments. These
toxicity
studies provide the basis for determining the efficacy of a test compound in
vivo.
8. EFFICACY OF TEST COMPOUNDS
Efficacy of a test compound can be established using several art
recognized methods, such as in vitro methods, animal models or human clinical
trials
(see, Creasey, supra (1979)). Recognized in vitro models exist for several
diseases or
conditions. For example, the ability of a test compound to extend the life-
span of HIV-
infected cells in vitro is recognized as an acceptable model to identify
chemicals
expected to be efficacious to treat HIV infection or AIDS (see, Daluge et al.,
Antimicro.
Agents Chemother. 41:1082-1093 (1995)). Furthermore, the ability of
cyclosporin A
(CsA) to prevent proliferation of T-cells in vitro has been established as an
acceptable
model to identify chemicals expected to be efficacious as immunosuppressants
(see,
Suthanthiran et al., supra (1996)). For nearly every class of therapeutic
agent, disease
or condition, an acceptable in vitro or animal model is available. The skilled
artisan is
armed with a wide variety of such models as they are available in the
literature or from
the USFDA or the National Institutes of Health (NIH). In addition, these in
vitro
96
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
methods can use tissue extracts, such as preparations of liver, such as.
microsomal
preparations, to provide a reliable indication of the effects of metabolism on
a test
compound. Similarly, acceptable animal models can be used to establish
efficacy of
test compounds to treat various diseases or conditions. For example, the
rabbit knee is
an accepted model for testing agents for efficacy in treating arthritis (see,
Shaw and
Lacy, J. Bone Joint Surg. (Br.) 55:197-205 (1973)). Hydrocortisone, which is
approved
for use in humans to treat arthritis, is efficacious in this model which
confirms the
validity of this model (see, McDonough, Phys. Ther. 62:835-839 (1982)). When
choosing an appropriate model to determine efficacy of test compounds, the
skilled
artisan can be guided by the state of the art, the USFDA or the NIH to choose
an
appropriate model, dose and route of administration, regime and endpoint and
as such
would not be unduly burdened.
In addition to animal models, human clinical trials can be used to
determine the efficacy of test compounds. The USFDA, or equivalent
governmental
agencies, have established procedures for such studies.
9. SELECTIVITY OF TEST COMPOUNDS
The in vitro and in vivo methods described above also establish the
selectivity of a candidate modulator. It is recognized that chemicals can
modulate a
wide variety of biological processes or may be selective. Panels of cells as
they are
known in the art can be used to determine the specificity of the a test
compound (WO
98/13353 to Whitney et al., published April 2, 1998). Selectivity is evident,
for
example, in the field of chemotherapy, where the selectivity of a chemical to
be toxic
towards cancerous cells, but not towards non-cancerous cells, is obviously
desirable.
Selective modulators are preferable because they have fewer side effects in
the clinical
setting. The selectivity of a test compound can be established in vitro by
testing the
toxicity and effect of a test compound on a plurality of cell lines that
exhibit a variety of
cellular pathways and sensitivities. The data obtained form these in vitro
toxicity
studies can be extended to animal model studies, including human clinical
trials, to
determine toxicity, efficacy and selectivity of a test compound.
97
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
The selectivity, specificity and toxicology, as well as the general
pharmacology, of a test compound can be often improved by generating
additional test
compounds based on the structure/property relationship of a test compound
originally
identified as having activity. Test compounds can be modified to improve
various
properties, such as affinity, life-time in blood, toxicology, specificity and
membrane
permeability. Such refined test compounds can be subjected to additional
assays as they
are known in the art or described herein. Methods for generating and analyzing
such
compounds or compositions are known in the art, such as U.S. Patent No.
5,574,656 to
Agrafiotis et al.
10. PHARMACEUTICAL COMPOSITIONS
The present invention also encompasses a test compound in a
pharmaceutical composition comprising a pharmaceutically acceptable carrier
prepared
for storage and preferably subsequent administration, which has a
pharmaceutically
effective amount of the test compound in a pharmaceutically acceptable carrier
or
diluent. Acceptable carriers or diluents for therapeutic use are well known in
the
pharmaceutical art, and are described, for example, in Remington's
Pharmaceutical
Sciences, Mack Publishing Co., (A.R. Gennaro edit. (1985)). Preservatives,
stabilizers,
dyes and even flavoring agents can be provided in the pharmaceutical
composition. For
example, sodium benzoate, sorbic acid and esters of p-hydroxybenzoic acid can
be
added as preservatives. In addition, antioxidants and suspending agents can be
used.
The test compounds of the present invention can be formulated and used
as tablets, capsules or elixirs for oral administration; suppositories for
rectal
administration; sterile solutions or suspensions or injectable administration;
and the
like. Injectables can be prepared in conventional forms either as liquid
solutions or
suspensions, solid forms suitable for solution or suspension in liquid prior
to injection,
or as emulsions. Suitable excipients are, for example, water, saline,
dextrose, mannitol,
lactose, lecithin, albumin, sodium glutamate, cysteine hydrochloride and the
like. In
addition, if desired, the injectable pharmaceutical compositions can contain
minor
amounts of nontoxic auxiliary substances, such as wetting agents, pH buffering
agents
98
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
and the like. If desired, absorption enhancing preparations, such as
liposomes, can be
used.
The pharmaceutically effective amount of a test compound required as a
dose will depend on the route of administration, the type of animal or patient
being
treated, and the physical characteristics of the specific animal under
consideration. The
dose can be tailored to achieve a desired effect, but will depend on such
factors as
weight, diet, concurrent medication and other factors which those skilled in
the medical
arts will recognize. In practicing the methods of the present invention, the
pharmaceutical compositions can be used alone or in combination with one
another, or
in combination with other therapeutic or diagnostic agents. These products can
be
utilized in vivo, preferably in a mammalian patient, preferably in a human, or
in vitro.
In employing them in vivo, the pharmaceutical compositions can be administered
to the
patient in a variety of ways, including parenterally, intravenously,
subcutaneously,
intramuscularly, colonically, rectally, nasally or intraperiotoneally,
employing a variety
of dosage forms. Such methods can also be used in testing the activity of test
compounds in vivo.
As will be readily apparent to one skilled in the art, the useful in vivo
dosage to be administered and the particular mode of administration will vary
depending upon the age, weight and type of patient being treated, the
particular
pharmaceutical composition employed, and the specific use for which the
pharmaceutical composition is employed. The determination of effective dosage
levels,
that is the dose levels necessary to achieve the desired result, can be
accomplished by
one skilled in the art using routine methods as discussed above, and can be
guided by
agencies such as the USFDA or NIH. Typically, human clinical applications of
products are commenced at lower dosage levels, with dosage level being
increased until
the desired effect is achieved. Alternatively, acceptable in vitro studies can
be used to
establish useful doses and routes of administration of the test compounds.
In non-human animal studies, applications of the pharmaceutical
compositions are commenced at higher dose levels, with the dosage being
decreased
until the desired effect is no longer achieved or adverse side effects are
reduced of
99
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
disappear. The dosage for the test compounds of the present invention can
range
broadly depending upon the desired affects, the therapeutic indication, route
of
administration and purity and activity of the test compound. Typically,
dosages can be
between about 1 ng/kg and about 10 mg/kg, preferably between about 10 ng/kg
and
about 1 mg/kg, more preferably between about 100 ng/kg and about 100
micrograms/kg, and most preferably between about 1 microgram/kg and about 10
micrograms/kg.
The exact formulation, route of administration and dosage can be chosen
by the individual physician in view of the patient's condition (see, Fingle et
al., in The
Pharmacological Basis of Therapeutics (1975)). It should be noted that the
attending
physician would know how to and when to terminate, interrupt or adjust
administration
due to toxicity, organ dysfunction or other adverse effects. Conversely, the
attending
physician would also know to adjust treatment to higher levels if the clinical
response
were not adequate. The magnitude of an administrated does in the management of
the
I S disorder of interest will vary with the severity of the condition to be
treated and to the
route of administration. The severity of the condition may, for example, be
evaluated,
in part, by standard prognostic evaluation methods. Further, the dose and
perhaps dose
frequency, will also vary according to the age, body weight and response of
the
individual patient, including those for veterinary applications.
Depending on the specific conditions being treated, such pharmaceutical
compositions can be formulated and administered systemically or locally.
Techniques
for formation and administration can be found in Remington's Pharmaceutical
Sciences,
18th Ed., Mack Publishing Co., Easton, PA (1990). Suitable routes of
administration
can include oral, rectal, transdermal, otic, ocular, vaginal, transmucosal or
intestinal
administration; parenteral delivery, including intramuscular, subcutaneous,
intramedullary injections, as well as intrathecal, direct intraventricular,
intravenous,
intraperitoneal, intranasal, or intraocular injections.
For injection, the pharmaceutical compositions of the present invention
can be formulated in aqueous solutions, preferably in physiologically
compatible
buffers such as Hanks' solution, Ringer's solution or physiological saline
buffer. For
100
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
such transmucosal administration, penetrans appropriate to the barrier to be
permeated
are used in the formulation. Such penetrans are generally known in the art.
Use of
pharmaceutically acceptable carriers to formulate the pharmaceutical
compositions
herein disclosed for the practice of the invention into dosages suitable for
systemic
administration is within the scope of the invention. With proper choice of
carrier and
suitable manufacturing practice, the compositions of the present invention, in
particular,
those formulation as solutions, can be administered parenterally, such as by
intravenous
injection. The pharmaceutical compositions can be formulated readily using
pharmaceutically acceptable carriers well known in the art into dosages
suitable for oral
administrations. Such carriers enable the test compounds of the invention to
be
formulated as tables, pills, capsules, liquids, gels, syrups, slurries,
suspensions and the
like, for oral ingestion by a patient to be treated.
Agents intended to be administered intracellularly may be administered
using techniques well known to those of ordinary skill in the art. For
example, such
agents may be encapsulated into liposomes, then administered as described
above.
Substantially all molecules present in an aqueous solution at the time of
liposome
formation are incorporated into or within the liposomes thus formed. The
liposomal
contents are both protected from the external micro-environment and, because
liposomes fuse will cell membranes, are efficiently delivered into the cell
cytoplasm.
Additionally, due to their hydrophobicity, small organic molecules can be
directly
administered intracellularly.
Pharmaceutical compositions suitable for use in the present invention
include compositions wherein the active ingredients are contained in an
effective
amount to achieve its intended purpose. Determination of the effective amount
of a
pharmaceutical composition is well within the capability of those skilled in
the art,
especially in light of the detailed disclosure provided herein. In addition to
the active
ingredients, these pharmaceutical compositions can contain suitable
pharmaceutically
acceptable carriers comprising excipients and auxiliaries which facilitate
processing of
the active chemicals into preparations which can be used pharmaceutically. The
preparations formulated for oral administration may be in the form of tables,
dragees,
101
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
capsules or solutions. The pharmaceutical compositions of the present
invention can be
manufactured in a manner that is itself known, for example by means of
conventional
mixing, dissolving, granulating, dragee-making, emulsifying, encapsulating,
entrapping
or lyophilizing processes. Pharmaceutical formulations for parenteral
administration
include aqueous solutions of active chemicals in water-soluble form.
Additionally, suspensions of the active chemicals may be prepared as
appropriate oily injection suspensions. Suitable lipophilic solvents or
vehicles include
fatty oils such as sesame oil, or synthetic fatty acid esters, such as ethyl
oleate or
triglycerides or liposomes. Aqueous injection suspensions may contain
substances what
increase the viscosity of the suspension, such as sodium carboxymethyl
cellulose,
sorbitol or dextran. Optionally, the suspension can also contain suitable
stabilizers or
agents that increase the solubility of the chemicals to allow for the
preparation of highly
concentrated solutions.
Pharmaceutical compositions for oral use can be obtained by combining
the active chemicals with solid excipient, optionally grinding a resulting
mixture, and
processing the mixture of granules, after adding suitable auxiliaries, if
desired, to obtain
tables or dragee cores. Suitable excipients are, in particular, fillers such
as sugars,
including lactose, sucrose, mannitol or sorbitol; cellulose preparations such
as, for
example, maize starch, wheat starch, rice starch, potato starch, gelatin, gum
tragacanth,
methyl cellulose, hydroxypropylmethyl-cellulose, sodium carboxymethylcellulose
and/or polyvinylpyrrolidone. If desired, disintegrating agents can be added,
such as the
cross-linked polyvinyl pyrolidone, agar, alginic acid or a salt thereof such
as sodium
alginate. Dragee cores can be provided with suitable coatings. Dyes or
pigments can be
added to the tablets or dragee coatings for identification or to characterize
different
combinations of active doses.
The test compounds of the present invention, and pharmaceutical
compositions that include such test compounds are useful for treating a
variety of
ailments in a patient, including a human. A patient in need of such treatment
can be
provided a test compound of the present invention, preferably in a
pharmacological
composition in an effective amount to reduce the symptoms, pathology or rate
of
102
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
progression of a disease or disorder in a patient. The amount, dosage, route
of
administration, regime and endpoint can all be determined using the procedures
described herein or by appropriate government agencies, such as the United
Stated Food
and Drug Administration.
11. TREATING DIABETES USING IDENTIFIED COMPOUNDS
Another aspect of the invention involves a method of treating diabetes by
administering an effective amount of pharmaceutical composition of the present
invention to a subject, such as a human patient, in need of treatment of
diabetes. The
pharmaceutical composition is administered to the subject in an amount, route
of
administration and regime sufficient to have a therapeutic, palliative,
prophylactic,
impeditive effect to ameliorate the effects, reversing the course of, delaying
the onset of
or preventing diabetes. The subject preferably is suspected of having or being
at risk of
developing diabetes.
An "effective amount" is the amount of a therapeutic reagent that when
administered to a subject by an appropriate dose and regime results the
desired result.
A "subject in need of treatment of diabetes" is a subject diagnosed with
diabetes or is suspected of having diabetes.
A "therapeutic effect" is the reduction or elimination of a disease state or
pathological condition.
A "palliative effect" is the alleviation of symptoms associated with a
disease or pathological condition.
A "prophylactic effect" is the prevention of a disease state or
pathological condition.
An "impeditive effect" is the reduction of the rate of progression of a
disease state or pathological condition.
To "ameriolate the effects" of refers to the reduction of the severity of
the symptoms of a disease state or pathological condition.
To "reverse the course of diabetes disease" refers to the restoration or
improvement of glucose metabolism in a subject.
103
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
1. NUCLEIC ACID MOLECULES
Therapeutic composition. The therapeutic composition of the present
invention includes at least one nucleic acid molecule of the present
invention,
preferably a nucleic. The nucleic acids may be covalently or noncovalently
conjugated
or bound to other molecules, such as, but not limited to, proteins that may
facilitate their
delivery to the target tissue or tissues. Small molecules such as folate may
be
conjugated to nucleic acid molecules to enhance transport across the blood-
brain barrier
(Wu, D. et al. (1999) Pharm. Res. 16: 415-19.)
The nucleic acid molecules can be complexed with cationic lipids,
packaged within liposomes, incorporated into hydrogels, cyclodextrins,
biodegradable
nanocapsules, or bioadhesive microspheres. The pharmaceutical compostition may
include carriers, thickeners, diluents, buffers, preservatives, surface active
agents, and
the like in addition to oligonucleotides. Pharmaceutical compositions can also
include
one or more active ingredients such as antimicrobial agents, antiinflammatory
agents,
anesthetics, and the like in addition to oligonucleotides. If administration
is by
injection or infusion, the nucleic acid molecules cann be delivered directly
or in the
aforementioned compositions in sterile solution, which may also contain
buffers,
diluents, and other suitable additives. Formulations for topical
administration may
include ointments, lotions, creams, gels, drops, suppositories, sprays,
liquids, and
powders. Conventional pharmaceutical carriers, aqueous, powder or oily bases,
thickeners and the like can be necessary or desirable.
Nasal inhalation may be particularly effective for delivery of
pharmaceutical compositions to the brain (Wang, Y. et al. (1998) Biopharm Drug
Dispos. 19: 571-5) and/or cerebrospinal fluid (Sakane T. (1991) J. Pharm.
Pharmacol.
43: 449-51 ). Pharmaceutical compositions that include nucleic acid molecules
can also
include compounds that enhance absorption by nasal epithelial cells such as
cationic
compounds (Natsume, H. et al. (1999) Int. J. Pharm. 185: 1-12), cyclodextrins
(Martin,
et al., J. Drug Target. , 6: 17-36), or other compounds that are known or may
be later
discovered to enhance nasal absorption. Solutions containing nucleic acids for
nasal
delivery may be supplied in spray containers for aerosol inhalation.
. 104
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
Compositions for oral delivery include powders or granules, suspensions
or solutions in water or nonaqueous media, capsules, sachets, or tablets.
Thickeners,
flavorings, diluents, emulsifiers, dispersing aids or binders may be
desirable.
Dose. Optimum doses of pharmaceutical compositions that include
nucleic acid molecules depends on a variety of factors, including the severity
of the
condition to be treated, the toxicity of the nucleic acid molecules being
delivered, the
route of administration, and the individual patient's response to the
treatment. The
skilled practitioner is able to determine the appropriate dose based on these
factors and
the effective dose derived from animal and clinical studies. In general,
dosage is from
0.01 micrograms to 100 g per kg of body weight, and may be given once or more
daily,
weekly, monthly, or yearly, or even once every 2 to 20 years. Persons of
ordinary skill
in the art can estimate repetition rates for dosing based on measured
residence times and
concentrations of the drug in bodily fluids or tissues. It may be desirable to
have the
patient undergo maintenance therapy to prevent the recurrence of the disease
state,
wherein the nucleic acids are administered in maintenance doses, ranging from
0.01
microgram to 100 g per kg of body weight once or more daily to once every 20
years.
Route of Administration. Nucleic acid molecules may be administered
by any appropriate route of administration, such as, for example, parenteral
or
intravenous injection. Nucleic acids may also be delivered intravenously
through
pump, stmt, or drip. Nucleic acid molecules may be introduced into the
cerebrospinal
fluid by injection into the spinal column. For delivery into the brain,
injection may be
into the brain cavity via a canula. Other routes of delivery include oral
delivery and
topical application. Nasal inhalation of aerosols may be particularly
effective for
administering the nucleic acids of the invention and their formulations to the
brain.
Nucleic acids may also be encased in or applied to a polymer, solid support or
fabric, or
gel which is delivered locally. Such solid supports, fabrics, polymers, or
gels may be
biodegradable.
Re_eime. The dose regime is determined experimentally based on animal
studies and clinical trials. Doses may be given once or more daily, weekly,
monthly, or
yearly, or even once every 2 to 20 years. Persons of ordinary skill in the art
can
105
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
estimate repetition rates based on measured residence times and concentrations
of the
drug in bodily fluids or tissues. Following successful treatment, it may be
desirable to
have the patient under maintenance therapy to prevent the recurrence of the
disease
state, wherein the oligonucliotide is administered in maintenance doses,
ranging from
0.01 micorgrams to 100 grams per kg of body weight, once or more daily, to
once every
20 years.
Monitoring Progress. The progress of treatment for diabetes, either type I
or type II, can be measured using methods known in the art. For example, blood
glucose, urine glucose or blood or serum insulin levels can be monitored using
established methods. These measurements can be taken at appropriate intervals,
including before, during and after feeding or fasting. In this instance, the
caloric intake
and type of caloric intake, such as carbohydrates, should be noted.
2. GENE THERAPY CONSTRUCTS
Gene therapy constructs contain nucleic acids comprising a nucleic acid
molecule of the present invention optionally operably linked to gene
regulatory
elements. The nucleic acid molecule and gene regulatory elements may be in a
plasmid
or may be incorporated into a vector, such as, but not limited to, a
retroviral vector, an
adenoviral vector, an adeno-associated viral vector, a vaccinia viral vector,
a herpes
viral vector, or other vectors as they are known or later developed in the
art. The gene
therapy constructs may be administered as DNA, as viral particles, or in
cells.
Therapeutic composition. Gene therapy constructs that consist of nucleic
acid molecules not incorporated into vectors such as viruses may be delivered
as free
nucleic acids, or may be delivered covalently or noncovalently conjugated or
bound to
other molecules, such as, but not limited to, molecules that enhance their
transport
across the blood-brain barrier or that may facilitate their delivery to the
target tissue or
tissues. Other DNA sequences, such as adenovirus VA genes can be included in
the
administration medium and be co-transfected with the gene of interest. The
presence of
genes coding for the adenovirus VA gene product may significantly enhance the
translation of mRNA transcribed from the plasmid. Gene therapy constructs that
are
106
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
packaged in viruses may have proteins or other molecules or compounds, such
as, but
not limited to lipids, proteins, or polymers incorporated into or associated
with the virus
to enhance delivery into cells. The gene therapy constructs, whether naked DNA
or
packaged vector constructs, may be complexed with cationic lipids, packaged
within
liposomes, incorporated into hydrogels, cyclodextrins, biodegradable
nanocapsules, or
bioadhesive microspheres. The pharmaceutical composition may include carriers,
thickeners, diluents, buffers, preservatives, surface active agents, and the
like in
addition to oligonucleotides. Pharmaceutical compositions may also include one
or
more active ingredients such as antimicrobial agents, antiinflammatory agents,
anesthetics, and the like in addition to oligonucleotides. If administration
is by
injection or infusion, the gene therapy constructs may be delivered directly
or in the
aforementioned compositions in sterile solution, which may also contain
buffers,
diluents, and other suitable additives. Formulations for topical
administration may
include ointments, lotions, creams, gels, drops, suppositories, sprays,
liquids, and
powders. Conventional pharmaceutical carriers, aqueous, powder or oily bases,
thickeners and the like may be necessary or desirable.
Nasal inhalation may be particularly effective for delivery of
pharmaceutical compositions to the brain (Wang, Y. et al. (1998) Biopharm Drug
Dispos. 19: 571-5) and/or cerebrospinal fluid (Sakane T. (1991) J. Pharm.
Pharmacol.
43: 449-51 ). Pharmaceutical compositions that include gene therapy constructs
may
also include compounds that enhance absorption by nasal epithelial cells such
as
cationic compounds (Natsume, H. et al. (1999) Int. J. Pharm. 185: 1-12),
cyclodextrins
(Martin, et al., J. Drug Target. 6: 17-36), or other compounds that are known
or may be
later discovered to enhance nasal absorption. Solutions containing gene
therapy
constructs may be supplied in spray containers for aerosol inhalation.
Compositions for oral delivery include powders or granules, suspensions
or solutions in water or nonaqueous media, capsules, sachets, or tablets.
Thickeners,
flavorings, diluents, emulsifiers, dispersing aids or binders may be
desirable. Nucleic
acids may also be encased in or applied to a polymer, solid support or fabric,
or gel
107
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
which is delivered locally. Such solid supports, fabrics, polymers, or gels
may be
biodegradable.
Gene therapy constructsJmay also be delivered in cells. Cells containing
gene therapy constructs may be derived from the patient, another human being,
or even
an animal of another species. Gene therapy constructs may be introduced into
the cells
ex vivo by viral transfection, electroporation, membrane fusion with
liposomes, high
velocity bombardment with DNA coated microprojectiles, incubation with calcium-
phosphate-DNA precipitate, tansfection with DEAE-dextran, direct
microinjection, or
other methods known or later developed in the art. The cells are then
delivered to the
patient by any of a variety of means, including implantation or injection. The
cells may
express the gene therapy construct in vivo to obtain the therapeutic effect in
the patient.
Alternatively, after introduction into the patient, the cells containing the
gene therapy
construct may replicate and/or package the gene therapy construct such that
endogenous
cells in the patient may be infected, transformed, or transfected with the
gene therapy
construct and thereby express it. Cells containing gene therapy constructs may
be
enclosed in structures composed of polymers or other materials to retain them
at the
instillation site or to protect them from the patient's cellular immunity
mechanisms.
Dose. Optimum doses depend on the severity of the condition to be
treated, the toxicity of the gene therapy construct being delivered, the route
of
administration, and the individual patient's response to the treatment. The
skilled
practitioner is able to determine the appropriate dose based on these factors
and the
effective dose derived from animal and clinical studies. In general, for naked
DNA
gene therapy constructs, the dosage is from 0.01 micrograms to 100 g per kg of
body
weight. For viral gene therapy constructs, an appropriate dose is in the range
of 0.1 to
50 ml of 106 to 10" particle forming units per ml viral expression vectors..
For cells
containing viral expression constructs, about 105 to about 10g cells may be
delivered to
an appropriate site.
Route of Administration. Naked DNA gene therapy constructs and viral
gene therapy constructs may be delivered by intravenous or intraperitoneal
injection,
intratracheally, intrathecally parenterally, intraarticularly,
intramuscularly, or introduced
108
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
into the brain by injection via a cannula or injected into the spinal column
for
distribution within the cerebrospinal fluid. Gene therapy constructs may be
administered intravenously, by injection, catheter, pump, or drip.
Alternatively, Cells
containing gene therapy constructs may be implanted surgically into the brain,
or they
may be delivered to another site in the body. This may be convenient if the
protein or
nucleic acid molecules expressed from the gene therapy construct is targeted
to the
brain or, if the cells are packaging cells, the virus produced by the
introduced cells may
be targeted to the brain or other relevant tissue. Cells may be administered
topically,
intraocularly, parenterally, intranasally, intratracheally, intrabronchially,
intramuscularly, subcutaneously, or by any other means.
Regime. The dose regime is determined experimentally based on animal
studies and clinical trials. Doses may be given once or more daily, weekly,
monthly, or
yearly, or even once every 2 to 20 years. Persons of ordinary skill in the art
can
estimate repetition rates based on measured residence times and concentrations
of the
gene product of the gene therapy vector in bodily fluids or tissues. Following
successful treatment, it may be desirable to have the patient receive
additional doses of
the gene therapy vector if it is determined that levels of the gene product
have declined
below a level necessary to prevent disease progression, or if there are
symptoms of
disease progression. The gene therapy construct or cells containing the gene
therapy
construct may be administered in maintenance doses, where the dose has been
determined based on animal and clinical studies, and may be monitored by
measuring
the expression product of the gene therapy construct in the patient's bodily
fluids.
Monitorin~gress. The progress of treatment for diabetes, either type I
or type II, can be measured using methods known in the art. For example, blood
glucose, urine glucose or blood or serum insulin levels can be monitored using
established methods. These measurements can be taken at appropriate intervals,
including before, during and after feeding or fasting. In this instance, the
caloric intake
and type of caloric intake, such as carbohydrates, should be noted.
3. BIOLOGICALLY ACTIVE AGENTS.
109
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
Therapeutic com osp ition. A therapeutic composition of the present
invention can include at least one biologically active agent of the present
invention. At
least one biologically active agent of the present invention can optionally be
covalently
or noncovalently conjugated or bound to other molecules, such as, but not
limited to,
proteins that may facilitate their delivery to the target tissue or tissues.
Small molecules
such as folate may be conjugated to the biologically active agents of the
invention to
enhance transport across the blood-brain barrier (Wu, D. et al. (1999) Pharm.
Res. 16:
41 S-19.). The pharmaceutical composition may comprise a pharmaceutically
acceptable carrier prepared for storage and preferably subsequent
administration, which
has a pharmaceutically effective amount of the biologically active agent in a
pharmaceutically acceptable carrier or diluent. Acceptable carriers or
diluents for
therapeutic use are well known in the pharmaceutical art, and are described,
for
example, in Remington's Pharmaceutical Sciences, Mack Publishing Co., (A.R.
Gennaro edit. (1985)). Preservatives, stabilizers, dyes and even flavoring
agents can be
provided in the pharmaceutical composition. For example, sodium benzoate,
sorbic
acid and esters of p-hydroxybenzoic acid can be added as preservatives. In
addition,
antioxidants and suspending agents can be used.
The biologically active agents of the present invention can be formulated
and used as tablets, capsules or elixirs for oral administration;
suppositories for rectal
administration; sterile solutions or suspensions for injectable
administration; and the
like. Injectables can be prepared in conventional forms either as liquid
solutions or
suspensions, solid forms suitable for solution or suspension in liquid prior
to injection,
or as emulsions. Suitable excipients are, for example, water, saline,
dextrose, mannitol,
lactose, lecithin, albumin, sodium glutamate, cysteine hydrochloride and the
like. In
addition, if desired, the injectable pharmaceutical compositions can contain
minor
amounts of nontoxic auxiliary substances, such as wetting agents, pH buffering
agents
and the like. If desired, absorption enhancing preparations, such as
liposomes, can be
used. The pharmaceutical composition may also include carriers, thickeners,
diluents,
buffers, preservatives, surface active agents, and the like in addition to one
or more
biologically active agents. Pharmaceutical compositions may also include one
or more
110
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
active ingredients such as antimicrobial agents, antiinflammatory agents,
anesthetics,
and the like in addition to the biologically active agents of the invention.
Formulations
for topical administration may include ointments, lotions, creams, gels,
drops,
suppositories, sprays, liquids, and powders. Conventional pharmaceutical
carriers,
aqueous, powder or oily bases, thickeners and the like may be necessary or
desirable.
Agents intended to be administered intracellularly may be administered
using techniques well known to those of ordinary skill in the art. For
example, such
agents may be encapsulated into liposomes, then administered as described
above.
Substantially all organic molecules present in an aqueous solution at the time
of
liposome formation are incorporated into or within the liposomes thus formed.
The
liposomal contents are both protected from the external micro-environment and,
because liposomes fuse with cell membranes, are efficiently delivered into the
cell
cytoplasm. Additionally, due to their hydrophobicity, small organic molecules
can be
directly administered intracellularly.
Nasal inhalation may be particularly effective for delivery of
pharmaceutical compositions to the brain (Wang, Y. et al. (1998) Biopharm Drug
Dispos. 19: 571-5) and/or cerebrospinal fluid (Sakane T. (1991) J. Pharm.
Pharmacol.
43: 449-51). Pharmaceutical compositions that include biologically active
agents may
also include compounds that enhance absorption by nasal epithelial cells such
as
cationic compounds (Natsume, H. et al. (1999) Int. J. Pharm. 185: 1-12),
cyclodextrins
(Martin, et al., J. Drug Target. 6: 17-36), or other compounds that are known
or may be
later discovered to enhance nasal absorption. Solutions containing
biologically active
agents for nasal delivery may be supplied in spray containers for aerosol
inhalation.
Dose. The pharmaceutically effective amount of a biologically active
agent of the present invention required as a dose will depend on the route of
administration and the physical characteristics of the specific animal under
consideration. The dose can be tailored to achieve a desired effect, but will
depend on
such factors as weight, diet, concurrent medication and other factors which
those skilled
in the medical arts will recognize. In practicing the methods of the present
invention,
the pharmaceutical compositions can be used alone or in combination with one
another,
111
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
or in combination with other therapeutic or diagnostic agents. The skilled
practitioner
is able to determine the appropriate dose based on these factors and the
effective dose
derived from animal and clinical studies.. The determination of effective
dosage levels,
that is the dose levels necessary to achieve the desired result, can be
accomplished by
one skilled in the art using routine methods. Typically, human clinical
applications of
products are commenced at lower dosage levels, with dosage level being
increased until
the desired effect is achieved. Alternatively, acceptable in vitro studies can
be used to
establish useful doses and routes of administration of the bioactive compounds
and
bioactivities.
Route of Administration. In employing them in vivo, the pharmaceutical
compositions containing at least one biologically active agent of the present
invention
can be administered to the patient in a variety of ways, including, for
example,
parenterally, intravenously, subcutaneously, intramuscularly, colonically,
rectally,
nasally or intraperiotoneally, employing a variety of dosage forms.
Biologically active
agents may be introduced into the cerebrospinal fluid by injection into the
spinal
column. For delivery into the brain, injection may be into the brain via
cannula. Other
routes of delivery include oral delivery and topical application. Nasal
inhalation of
aerosols may be particularly effective for administering the biologically
active agents of
the invention and their formulations to the brain.
Regime. It will be recognized by one of skill in the art that the optimal
quantity and spacing of individual dosages of a biologically active agent of
the present
invention will be determined by the nature and extent of the condition being
treated, the
form, route and site of administration, and the particular patient being
treated, and that
such optimums can be determined by conventional techniques. It will also be
appreciated by one of skill in the art that the optimal course of treatment,
i.e., the
number of doses of biologically active agent of the invention given per day
for a
defined number of days, can be ascertained by those skilled in the art using
conventional course of treatment determination tests. Persons of ordinary
skill in the art
can estimate repetition rates based on measured residence times and
concentrations of
the biologically active agent in bodily fluids or tissues. Following
successful treatment,
112
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
it may be desirable to have the patient receive maintenance doses of the
biologically
active agent, where the maintenance dose has been determined based on animal
and
clinical studies..
Monitoring Progress. The progress of treatment for diabetes, either type I
or type II, can be measured using methods known in the art. For example, blood
glucose, urine glucose or blood or serum insulin levels can be monitored using
established methods. These measurements can be taken at appropriate intervals,
including before, during and after feeding or fasting. In this instance, the
caloric intake
and type of caloric intake, such as carbohydrates, should be noted.
EXAMPLES
The following examples illustrate the invention and are not intended to
limit the same. Those skilled in the art will recognize, or be able to
ascertain through
routine experimentation, numerous equivalents to the specific substances and
procedures described herein. Such equivalents are considered to be within the
scope of
the present invention.
EXAMPLE 1:
GLUCOSE RESPONSIVENESS IS LINKED TO MITOCHONDRIAL DNA CONTENT
In order to determine if a correlation exists between mitochondria) mass
and/or function, the following experiments were carried out.
Generation of INS-1 Cells Depleted of Mitochondria) DNA
INS-1 rat insulinoma cells were provided by Prof. Claes Wollheim,
University Medical Centre, Geneva, Switzerland, and cultured at 37°C in
a humidified
5% COZ environment in RPMI cell culture media (Gibco BRL, Gaithersburg, MD)
supplemented with 10% fetal bovine serum (Irvine Scientific), 2 mM L-
glutamine, 100
U/ml penicillin, 100 pg/ml streptomycin, 10 mM HEPES, 1 mM sodium pyruvate and
50 pM (3-mercaptoethanol.
INS-1 cells were cultured for 3-60 days under conditions as described
above except media were additionally supplemented with 50 pg/ml uridine and
113
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
nucleoside analogs 2'3'-dideoxycytidine [ddC], 2'3'-dideoxyinosine [ddI] or
2'3'-didehydro-3-deoxythymidine [d4T] (all from Sigma) at varying
concentrations
(1-500 ~M) diluted from IOOX stock in PBS or a comparable dilution of PBS
without.
Media were replenished every two days. Cells were harvested at periodic
intervals and
assayed for insulin secretion and mtDNA content.
Total DNA was prepared from rat liver (for probing rat-derived cells) or
the murine cell line 3T3 Ll (for probing mouse-derived cells; see Green et
al., Cell
3:127-133, 1974 and Cell 5:19-27, 1975) using DNAzoITM reagents (Molecular
Research Center, Inc., Cincinnati, OH) and method essentially according to the
manufacturer's instructions. The template DNAs were examined by agarose gel
electrophoresis and ethidium bromide staining and found to be roughly
equivalent.
Each template DNA was used in separate polymerase chain reaction (PCR)
reactions to
prepare DNA molecules having 1,207 base pairs and corresponding to either
nucleotides 5342 to 6549 of the rat (Rattus norvegicus) mitochondrial genome
(GenBank Accession No. X14848, Anderson et al., Nature 260:497-516, 1981) or
nucleotides 5361 to 6568 of the murine (Mus musculus) mitochondrial genome
(GenBank Accession No. V00711, Bibb et al., Cell 26:167-180, 1981). The same
pair
of oligonucleotide primers, specific for the mitochondrially encoded
cytochrome c
oxidase subunit I (COX-I) gene, were used for reactions for either rat or
mouse
templates. The pair of primers consisted of forward and reverse
oligonucleotides
having the following sequences:
Forward: S'-CACAAAGATATCGGAACCCTCTA
(SEQ ID NO: 17)
Reverse: 5'-AAGTGGGCTTTTGCTCATGTGTCAT (SEQ ID NO: 18)
The PCR reactions contained appropriate amounts of template DNA,
primers, MgCl2, all four dNTPs, reaction buffer, and Taq polymerase, brought
up to a
volume of 50 u1 using sterile water. The reactions were incubated at
95°C for 10
seconds, followed by 30 cycles of 95°C for 1 minute, 60°C for 1
minute and 72°C for 1
114
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
minute, after which the reactions were incubated at 72°C for 4 minutes
and then cooled
to 4°C.
The PCR reactions mixes were extracted with phenol:chloroform and,
along with a series of molecular weight markers, electrophoresed on an agarose
gel that
was stained with ethidium bromide and visualized with ultraviolet light. For
both
reactions, a single band of the predicted size (i. e., about 1.2 kilobases)
was observed.
The rat probe was radiolabeled with 32P using a Prime-a-Gene~ random priming
kit
(Promega, Madison, WI) essentially according to the manufacturer's
instructions.
To quantify mitochondrial DNA by slot blotting, INS-1 cells, or p°
INS-
1 cells generated using ddC as described above, were seeded into 12-well
plates
containing RPMI media supplemented as described above at 0.4 x 106 cells/well
and
cultured at 37°C, 5% COz for 2 days. Cells (0.7 x 106 cells/well) were
rinsed with PBS
and total cellular DNA was extracted using DNAzoI (Molecular Research Center,
Inc.,
Cincinnati, Ohio) according to the manufacturer's instructions. One hundred ng
DNA
from each cell preparation was slot-blotted onto a Zeta-Probe membrane (Bio-
Rad,
Hercules, California) and crosslinked at 125 joules using a BioRad GS
GeneLinker
irradiation/energy source.
The membranes were rinsed in hybridization buffer (5X SSC, 0.1 % N-
laurylsarcosine, 0.02% SDS, 1% blocking solution, Boehringer Mannheim,
Indianapolis, Indiana) and hybridized overnight in the same buffer at
42°C with the
[3zP]-labeled rat COX I probe. Following hybridization, membranes were washed
twice
with 2X SSC/0.1% SDS and twice with O.1X SSC/0.1% SDS and exposed to X-ray
film. Mitochondrial DNA was quantified by densitometric scanning of the
resulting
autoradiographs.
Incubation of INS-1 cells with ddC, ddI or d4T for seven days decreased
mtDNA content in a dose-dependent fashion. The relative mtDNA content (mean
COX-I hybridization signal + SEM) of the cells, normalized to total cellular
DNA, is
plotted as a function of nucleoside analog concentration in Figure 1A. The
ICS° for ddC
was approximately 50 pM. In INS-1 cells incubated with 25 ~M ddC for up to 40
days,
115
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
the decline in mtDNA content was time-dependent, with a t"2 of approximately
three
days; mtDNA was undetectable in these cells after 21 days.
Glucose-Responsive Insulin Production by INS-1 Cells Depleted of Mitochondrial
DNA
INS-1 cells, or p° INS-1 cells generated using ddC as described
above,
were seeded into 12-well plates containing RPMI media supplemented as
described at
0.5 x 106 cells/well and cultured at 37°C, 5% COz for 2 days. Cells
(0.7 x 106 cells/well
were rinsed with glucose-free KRH buffer (134 mM NaCI, 4.7 mM KCI, 1.2 mM
KHZP04, 1.2 mM MgS04, 1.0 mM CaCIZ, 10 mM HEPES, 10 mM NaHC03, 0.5%
BSA), then incubated in the same buffer for 1 hr at 37°C in a
humidified 5% C02 /95%
air atmosphere. Fresh KRH buffer containing 0.5 mM isobutylmethyl xanthine and
the
following secretagogues was added: 5 mM glucose, 10 mM glucose, 20 mM glucose,
5
mM KCl or 20 mM KCI. After an additional 1 hr at 37°C, 5% COZ the
culture
supernatants were collected. Insulin concentrations in the supernatants were
measured
1 S and normalized to cell number using an insulin-specific radioimmunoassay
kit (ICN
Biochemicals, Irvine, CA) according to the manufacturer's instructions.
As expected, untreated (mitochondrially proficient) INS-1 cells begin to
exhibit glucose-mediated insulin secretion at concentrations of glucose
starting at 5 mM
(Figure 1B, "parental INS-1"). In contrast, in cells treated with ddC (10 p.M)
for over
20 days, at which time mtDNA was significantly reduced, glucose stimulated
insulin
secretion was not observed at any glucose level tested (Figure 1 B, "mtDNA-
depleted
IN S-1 ").
Other Glucose-Mediated Responses are Blunted in INS-1 Cells Depleted of
Mitochondrial DNA
The ability of mitochondrially proficient and INS-1 cells that have been
treated with ddC, and thus depleted of mtDNA, to respond to glucose in other
ways was
examined.
116
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
Intracellular ATP levels were determined using an ATP bioluminescent
assay kit (Sigma) for both types of cells in response to various doses of
glucose. The
results (Figure 2A) show that untreated INS-1 cells produce increasing amounts
of ATP
in response to increasing amounts of glucose. In contrast, INS-1 cells that
have been
substantially depleted of mtDNA, although able to maintain a basal level of
ATP, do
not show any substantial response to stimulation by glucose.
Lactate production was also determined for both types of cells in
response to various doses of glucose. Cells were grown in 35 mm dishes with
various
concentrations of glucose. Media were replenished about 16 hr before assay
with
normal culture media containing various amounts of glucose. The media were
then
collected, and lactate measured using a commercially available kit, in which
lactate
dehydrogenase is used to produce a fluorescent compound (Sigma, St. Louis,
MO),
essentially according to the manufacturer's instructions.
The results (Figure 2B) show that untreated INS-1 cells maintain abasal
level of lactate and produce only slightly increasing amounts of ATP in
response to
increasing amounts of glucose. In contrast, INS-1 cells that have been
substantially
depleted of mtDNA show any substantial response to stimulation by glucose.
These results indicate that, at a minimum, functioning mitochondria
promote glucose-responsiveness in insulin-secreting cells, and suggest that
functioning
mitochondria are required for a robust production of insulin, ATP and lactate
in
response to glucose in such cells.
EXAMPLE 2:
CONSTRUCTION OF IF1 FUSION PROTEIN EXPRESSION CONSTRUCTS
Two IF1-derived fusion proteins were constructed (see Example 3 for
details) having structures that may be diagrammed as follows:
TAT.IF 1.Ma: ( His tag ~( TAT ~( IF 1 )
TAT.IF 1.FL: ( His tag ~--( TAT ~( mtOTS ~(IF 1 ),
117
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
wherein:
"His tag" denotes 6 histidine amino acid residues in contiguous order
(SEQ ID NO:1);
"TAT" denotes a cellular targeting sequence (CTS) derived from HIV-1
(SEQ ID NO:10);
"mtOTS" denotes a mitochondrial targeting sequence (SEQ ID N0:14)
derived from Rattus norvegicus IF 1; and
"IF 1" denotes the IF 1 polypeptide derived from Rattus norvegicus (SEQ
ID N0:13).
Expression constructs designed to encode and direct the production of
these IF1-derived fusion proteins were constructed as follows.
Rat Heart cDNA Library:
A cDNA library derived from total cellular RNA rat heart was prepared
according to methods known in the art. In brief, rat hearts were dissected
away from
associated tissues and gently minced in a buffer containing 40 mM Trsi-HCI, pH
7.0,
with surgical instruments that had been treated to remove any RNase or
contaminating
RNAs. The cells were lysed and RNA was purified from the lysate and clarified
by
treatment with RNase-free DNase I (Roche Molecular Biochemicals, formerly
Boehringer Mannheim Biochemicals, Indianapolis, IN) using 1 u1 of DNase I (10
u/ul)
in a buffer containing 40 mM Trsi-HCI, pH 7.0, 6 mM magnesium chloride and 2
mM
calcium chloride for 30 minutes at 37°C. This treatment was followed by
two
phenol/chloroform extractions, one chloroform extraction and an ethanol
precipitation
in the presence of sodium acetate. The RNA pellet was collected by
centrifugation,
washed with 70% ethanol, air dried, and resuspended in RNase-free sterile
water. The
RNA was reverse transcribed to generate cDNA using RNase H-deficient Reverse
Transcriptase (SUPERSCRIPTTM; Life Technologies, Rockville, MD).
TAT.IF1.FL insert:
118
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
Rat IF 1 cDNAs were amplified from the rat heart cDNA library by
polymerase chain reactions (PCR) in a thermal cycler using the following
primers,
AMPLITAQTM DNA Polymerase (Perkin-Elmer), and reagents and buffers supplied in
a GENEAMPTM PCR Reagent Kit (Perkin-Elmer), according to the manufacturer's
instructions. In the following representations of the PCR primers, underlined
nucleotides indicate sequences complementary to the 5'-ends and 3'-ends of the
rat IF1
cDNAs, double-underlined nucleotides indicate recognition sequences for the
restriction
enzymes SacI (recognition sequence: 5'-GAGCTC) and HindIII (recognition
sequence:
5'-AAGCTT), and the rat IF 1 start codon (ATG) and the reverse complement of
the
stop codon (TGA, having the reverse complement TCA) are emboldened.
For TAT.IF l .FL, in which the full length rat IF 1 (SEQ ID NOS:12 and
13) is linked to_.a TAT sequence, primers having the following nucleotide
sequence
were used:
Forward-FL (sense):
5'-TGAGCTCAGATATGGCAGGAAGAAGCGGAGACAGAGAGGAATGGCAG
SEQ ID NO: 19,
and
Reverse (antisense):
5'-ATATAAGCTTTCAATGCTCACTATTCTTTAGGTA
SEQ ID NO: 20.
In the Forward-FL primer, the sequence between the SacI restriction
enzyme site and the start codon for rat IF 1 encodes a Tat-derived cellular
targeting
sequence, underlined in the following representation thereof:
AGATATGGCAGGAAGAAGCGGAGACAGAGAGGA SEQID N0:21
ArgTyrGlyArgLysLysArgArgGlnAraGly SEQID NO:22
119
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
The PCR products were digested with SacI and HindIII (both enzymes
from Roche Molecular Biochemicals) essentially according to the manufacturer's
recommendations using manufacturer-supplied reaction buffers. The restriction
enzyme
digested DNAs were purified by horizontal agarose gel electrophoresis and band
extraction using the UltraCleanTM GelSpin kit (Mo Bio Laboratories, Inc.,
Solana
Beach, CA).
TAT.IF 1.Ma insert:
Rat IF 1 cDNAs were amplified from the rat heart cDNA library by PCR
as in the preceding section, with the exception that the following primers
were used. In
the following representations of the PCR primers, underlined nucleotides
indicate
sequences complementary to the 5'-ends and 3'-ends of the rat IF1 cDNAs,
double-
underlined nucleotides indicate recognition sequences for the restriction
enzymes SacI
(recognition sequence: 5'-GAGCTC) and HindIII (recognition sequence: 5'-
AAGCTT),
and the reverse complement of the rat IF 1 stop codon (TGA, having the reverse
complement TCA) is emboldened.
For TAT.IF l .FL, in which rat IF 1 lacking its natural mitochondrial
targeting sequence (SEQ ID N0:14) is linked to a TAT sequence, primers having
the
following nucleotide sequence were used:
Forward-Ma (sense):
5'-TGAGCTCAGGATATGGCAGGAAGAAGCGGAGACAGAGAGGAGGCTCGG
SEQ ID NO: 23,
and
Reverse (antisense):
5'-ATATAAGCTTTCAATGCTCACTATTCTTTAGGTA
SEQ ID NO: 24.
As in the Forward-FL primer, the sequence between the SacI restriction
enzyme site and the start codon for rat IF 1 encodes a Tat-derived cellular
targeting
sequence. The PCR products were digested with SacI and HindIII (Roche) and
purified
120
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
by horizontal agarose gel electrophoresis and band extraction using the
UltraCleanTM
GelSpin kit (Mo Bio Laboratories).
Preparation of Expression Vector and Ligation:
The expression vector pBAD/His (Invitrogen, Carlsbad, CA) was used.
This vector contains the following elements operably linked in a S' to 3'
orientation: the
inducible, but tightly regulatable, araBAD promoter; optimized E. coli
translation
initiation signals; an amino terminal polyhistidine (6xHis)-encoding sequence
(also
referred to as a "His-Tag"); an XPRESSTM epitope-encoding sequence; an
enterokinase
cleavage site which can be used to remove the preceding N-terminal amino acids
following protein purification, if so desired; a multiple cloning site; and an
in-frame
termination codon.
Plasmid pBAD/His DNA was prepared by digestion with the restriction
endonucleases SacI and HindIII essentially according to the manufacturer's
instructions
and subjected to horizontal agarose gel electrophoresis and band extraction
using the
UltraCIeanTM GelSpin kit (Mo Bio Laboratories). Restricted and purified IF.FL
or
IF.Ma DNAs were ligated with restricted expression vector DNA using T4 DNA
ligase
(New England Biolabs, Beverly, MA) using the manufacturer's reaction buffer
and
following the manufacturer's instructions. Competent recAl hsdR endAlE. coli
cells
(strain TOP10F'; Invitrogen) were transformed with ligation mixtures
containing the
prokaryotic vector construct according to the manufacturer's instructions.
Single
colonies were selected and grown in 3-5 ml of LB broth (Sambrook, J., Fritsch,
E.F.,
and ' Maniatis, T., Molecular Cloning.' A Laboratory Manual, Cold Spring
Harbor
Laboratory Press, Cold Spring Harbor, NY, 1989) containing 50 ~g/ml ampicillin
(Roche Molecular Biochemicals). Plasmid DNA was isolated from the bacterial
cultures using the WIZARDTM Plus Series 9600 Miniprep Reagents System
(Promega,
Madison, WI). A few candidate isolates of "pBAD/His.TAT.IF 1.FL" and
"pBAD/His.TAT.IFl.Ma" expression constructs were restriction mapped to confirm
their structures. One isolate of each expression construct having the
predicted
restriction map was selected for further experiments.
121
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
EXAMPLE 3
EXPRESSION AND PURIFICATION OF IFI FUSION PROTEINS
Inducible expression of the TAT.IFI.ma and TAT.IFI.ma fusion
proteins was examined as follows. Following overnight culture and dilution
into fresh
media, E. coli cells harboring pBAD/His.TAT.IFl.FL or pBAD/His.TAT.IFI.Ma were
induced by treatment with L-arabinose at a concentration of 0.02% for about 4
hours.
Cells were harvested, lysed and sonicated to prepare protein extracts. The
protein
content of the extracts was determined and equivalent amounts of protein were
subject
to Western analysis.
The results show that a specific band of the predicted molecular weight
(His-Tag + enterokinase site + epitope + full-length rat IF 1 ) was observed
in the
arabinose induced Ecoli that were transformed with the pBAD/His.TAT.IF1.FL
expression construct, but was absent in the non-induced control culture.
Similarly, a
band corresponding to the predicted molecular weight of TAT.IFI.Ma (His-Tag +
enterokinase site + epitope + rat IF 1 - rat IF 1 mitochondria) targeting
sequence) was
observed in induced pBAD/His.TAT.IFI.Ma-transformed E. coli but was not
present in
the uninduced cultures. The His-tagged proteins were purified from induced
cells using
the ProBondTM Nickel-chelating resin (Invitrogen) essentially according to the
manufacturer's instructions. Nickel-affinity column purified IF 1 fusion
proteins are
shown in Figure 7, which depicts a Coomassie blue stained electrophoretogram
of the
expressed products of the indicated constructs, and which also shows western
blot
analysis of IF 1 fusion proteins detected using an antibody specific for the
XpressTM
epitope tag as described above.
EXAMPLE 4
122
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
DIRECT DELIVERY OF IF 1 FUSION PROTEINS TO MITOCHONDRIA
In order to examine the ability of the two IF 1-derived fusion proteins to
enter cells and be delivered directly to mitochondria, the following
experiments were
carried out.
The purified IF 1 fusion protein derivatives described in the preceding
Example were labeled by attaching a fluorescent moiety, Oregon GreenTM, using
the
Oregon GreenTM FluoReporter Protein Labeling Kit (Molecular Probes, Eugene,
OR).
INS-1 cells were cultured as in Example 4, and purified TAT.IFl.Ma and
TAT.IF1.FL
polypeptides were added to separate sets of cells to a final concentration of
100 ug/ml.
The cells were visually examined by fluorescent microscopy. Control
INS-1 cells, to which unlabeled TAT.IF1.FL polypeptide was added, exhibited a
slight
diffuse fluroescence, as the cells naturally fluoresce to some degree,
producing some
background signal. Similarly, cells to which the labeled TAT.IFl.Ma
polypeptide was
added exhibited a diffuse pattern of fluorescence, which was not more intense
than the
background signal. In contrast, cells to which the labeled TAT.IF1.FL
polypeptide was
added demonstrated a punctate pattern of fluorescence, indicating organellar
delivery
thereof.
EXAMPLE 5
PREPARATION OF REAGENTS FOR ASSAYS OF ATPASE AND IF 1 ACTIVITY
Structure and Preparation of IF 1 PolYpeptide Derivatives:
Synthetic polypeptides corresponding to portions of rat IF 1 were
prepared using Fmoc chemistry according to methods known in the art. After
synthesis
was completed, protecting groups were removed and the polypeptide chains were
cleaved from the resin in order to achieve their release therefrom.
Two polypeptides were prepared. IF 1 fizz-a6~ consists of amino acids 22
46 of the mature form of rat IF 1 (i. e., the protein remaining after cleavage
and removal
of the mitochondria) targeting sequence) and has the sequence:
123
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
FGKREKAEEDRYFREKTREQLAALK (SEQ ID NO: 25 )
IF 1 ~42_5g~ consists of amino acids 42-58 of the mature form of rat IF 1 and
has the
sequence:
LAALKKHHEDEIDHHSK (SEQ ID NO: 26 )
Preparation of FO-F 1 ATPase and F 1 ATPase:
The complete mitochondria) ATP synthase complex is thought to
comprise a membrane-bound portion (FO) and a "lollipop-shaped" portion (F1)
that
projects into the matrix. F1-ATPase is an active, water-soluble subcomplex of
ATPase
that represents the portion of the mitochondria) ATPase that faces into the
mitochondria) matrix. F I ATPase can be isolated from F0, and retains some
enzymatic
activities as an isolated subcomplex. Isolated Fl ATPase can be reassociated
with the-
FO subcomplex to reform the original membrane-bound structure (Kagawa and
Racker,
J. Biol. Chem. 241:2467, 1966).
FO-F1 ATPase complexes and Fl ATPase subcomplexes were isolated
from bovine cardiac samples essentially according to the method of Walker et
al.
(Methods in Enzymology 260:163-190, 1995), with the exception that, instead of
preparing SMPs (submitochondrial particles) at neutral pH, ASMPs (alkaline
submitochondrial particles; see Rouslin and Broge, Anal. Biochem. 222:68-75,
1994)
were prepared from bovine heart by sonication in a basic solution. The ASMPs
were
extracted with chloroform and used to prepare FO-F 1 ATPase and F 1 ATPase
according
to the method of Walker et al. (Methods in Enzymology 260:163-190, 1995).
Preparation of Bovine Cardiac IF 1:
IF 1 was isolated from bovine cardiac samples essentially according to
the method of Rouslin and Broge (Anal. Biochem. 222:68-75, 1994) with the
following
exceptions. Following their preparation, SMPs heated in a solution having a pH
of
124
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
about 5Ø The filtrate was poured over a Dowex 50 column which was washed
with
ammonium sulfate, H20 and 7 M urea. Proteins were eluted by applying a
solution of 7
M urea, 100 mM Tris-S04, pH 7.3, to the column. Eluted fractions were pooled
and
IFl proteins were concentrated using a Centricon~ concentrator (Millipore,
Bedford,
MA). .
Preparation of Rat ASMPs:
ASMPs (alkaline submitochondrial particles) were prepared from rat
cardiac samples essentially according to the method of Rouslin and Broge
(Anal.
Biochem. 222:68-75, 1994).
EXAMPLE 6
METHODS FOR EVALUATING ATPASE ACTIVITY AND THE INHIBITION THEREOF
One assay for ATPase activity involves measuring ATP hydrolysis
according to methods known in the art (Walker et al., Methods in Enzymology
260:.163-
190, 1995; Rouslin and Broge, Anal. Biochem. 222:68-75, 1994). Such assays can
be
performed using purified FO-F 1 ATPase complexes or F 1 ATPase subcomplexes,
on
using alkaline submitochondrial particles (ASMPs).
In an initial experiment to validate the reagents and assay system,
Aurovertin-B was used as a positive control. This antibiotic binds to and
inhibits the
activity of bacterial and mitochondria) ATPases (van Raaij et al., Proc. Nat).
Acad. Sci.
U.S.A. 93:14 6913-14 6917, 1996). The purified F1-ATPase described in the
preceding
Example was treated with varying concentrations of Aurovertin-B, and ATP
hydrolyis
was measured. As shown in Figure 3, Aurovertin-B inhibited ATP hydrolysis by
purified F1-ATPase in a dose-dependent fashion (IC50 = 0.85 uM).
In a similar fashion, the partially purified IF 1 of the preceding Example
was tested for its ability to inhibit the ATP hydrolytic activity of the
purified F1-
ATPase. As shown in Figure 4, pooled fractions 12-16 of the IF1 preparation
(filled
125
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
circles) inhibited ATP hydrolysis by purified F 1-ATPase in a manner that was
dependent upon the amount (volume) of the pooled fractions. In contrast,
pooled
fractions 8-11 (open circles in Figure 4) had essentially no effect on the F1-
ATPase,
indicating that these fractions contained insubstantial amounts of IF 1.
In like fashion, the two IF 1 polypeptide derivatives were tested for their
ability to inhibit the purified Fl-ATPase and FO-F1-ATPase. The results
(Figure 5)
indicate that both synthetic polypeptides inhibited the F 1 ATPase. The IF 1
~ZZ-46>
polypeptide had an IC50 of approximately 1.39 uM, whereas the IF1~42-ss>
polypeptide
had an IC50 of approximately 0.87 uM. In contrast, only the IF1~4Z_SS~
polypeptide
inhibited the FO-F1-ATPase, with an IC50 of approximately 0.18 uM; the IF1~22-
a6~
polypeptide had no effect ("N.E." in Figure 5). Without wishing to be bound by
theory,
this result suggests that there may be two modes of action of IF 1. The
results even raise
the possibility that there may be two separable ATPase binding sites in IF1, a
first site
that is contained in amino acids 22-46 of the mature IF 1 protein which is
blocked by the
FO subunit, and a second site that is present in amino acids 42-58 of the
mature IF1
protein and which is not impacted by the presence of FO in ATPase complexes.
The IF1~42-ss> polypeptide was also tested for its ability to inhibit the FO-
F1-ATPase in rat ASMPs (alkaline submitochondrial particles) prepared as in
the
preceding Example. The results (Figure 6) show that the IF 1 ~4z-sad
Polypeptide inhibits
the FO-F1-ATPase in rat ASMPs in a dose-dependent fashion. Although the IC50
of the
IF 1 X42-ss~ polypeptide in this experiment (about 2.5 uM) was somewhat
different than
that seen with the purified bovine FO-F1-ATPase (0.18 uM, Figure 5), this may
reflect a
species difference between the bovine and rat FO-F1-ATPases. Figure 8 shows an
inhibition curve that was generated when the indicated concentration of
recombinant
tat.IFl.fl (described above) was incubated with rat liver submitochondrial
particles and
ATP hydrolase activity was measured. ATP hydrolase activity was expressed as
the
percent of detectable activity in the absence of added IF1-containing fusion
protein, and
the level of oligomycin-inhibitable activity was also determined (Fig. 8).
The tat.IFl.fl fusion protein also enhanced GSIS in INS-1 cells using the
assay conditions described above in Example 1 (Figure 9). Briefly, INS-1 cells
were
126
CA 02390646 2002-05-08
WO 01/34833 PCT/US00/30862
incubated in the presence of varying amounts of tat.IFl.fl or vehicle control
in RPMI at
37°C for 1 hr. Glucose-free RPMI was then added for 1 hr with the
continued presence
of the fusion protein (or vehicle control), media were collected and insulin
concentrations were determined by ELISA. Fig. 9 shows insulin secretion
expressed as
the percentage of insulin secretion determined in the absence of glucose and
IF 1 fusion
protein.
From the foregoing it will be appreciated that, although specific
embodiments of the invention have been described herein for purposes of
illustration,
various modifications may be made without deviating from the spirit and scope
of the
invention. Accordingly, the invention is not limited except as by the appended
claims.
127