Note: Descriptions are shown in the official language in which they were submitted.
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
A METHOD OF IDENTIFYING LIGANDS OF BIOLOGICAL TARGET
MOLECULES
FIELD OF INVENTION
The present invention relates to a novel method useful for identifying small
organic molecule ligands (in the following also denoted "compounds") for
binding
to specific sites on biological target molecules such as proteins, nucleic
acids,
carbohydrates, nucleoproteins, glycoproteins and glycolipids. The compounds
are
capable of interacting with the biological target molecule, in particular with
a
protein, in such a way as to modify the biological activity thereof.
The invention further relates to methods of identifying compounds acting as
ligands of biological target molecules such as, e.g., proteins involving the
introduction of metal ion binding sites into the biological target molecules,
including a method of identifying compounds that bind to orphan receptors.
Small organic ligands identified according to the methods of the present
invention
find use, for example, as novel therapeutic drug compounds or drug lead
compounds, enzyme inhibitors, labelling compounds, diagnostic reagents,
affinity
reagents e.g. for protein purification etc.
INTRODUCTION
The initial phase in developing novel biologically active compounds such as,
e.g., therapeutically or propylactically active drug compounds is to identify
and
characterize one or more binding ligand(s) for a given biological target. Many
molecular techniques have been developed and are currently being employed for
identifying novel ligands or compounds that bind to the biological target. In
the
following proteins are used as an example on a biological target molecule.
Proteins as dru _~ tar gets
Most drug compounds act by binding to and altering the function of proteins.
These can be intracellular proteins such as, for example enzymes and
transcription
factors, or they can be extracellular proteins, for example enzymes, or they
can be
membrane proteins. Membrane proteins constitute a numerous and varied group
whose function is either structural, for example being involved in cell
adhesion
processes, or the membrane proteins are involved in intercellular
communication
and communication between the cell exterior and the interior by transducing
chemical signals across cell membranes, or they facilitate or mediate
transport of
compounds across the lipid membrane. Membrane proteins are for instance
receptors and ion channels to which specific chemical messengers termed
ligands
bind resulting in the generation of a signal, which gives rise to a specific
CONFIRMATION COPY
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
2
intracellular response (this process is known as signal transduction).
Membrane
proteins can, for example also be enzymes which are associated to the membrane
for
functional purposes, e.g. proximity to their substrates. Most membrane
proteins are
anchored in the cell membrane by a sequence of amino acid residues, which are
predominantly hydrophobic to form hydrophobic interactions with the lipid
bilayer
of the cell membrane. Such membrane proteins are also known as integral
membrane proteins. In most cases, the integral membrane proteins extend
through
the cell membrane into the interior of the cell, thus comprising an
extracellular
domain, one or more transmembrane domains and an intracellular domain.
A large fraction of current drugs act on membrane proteins and among these the
majority are targeted towards the G protein coupled receptors (GPCR) with
their
seven transmembrane segments, also called 7TM receptors.
Identification of lead compounds in drug discoverx
Drug discovery traditionally involves a process where a lead compound first is
identified and then subsequently chemically optimised for high affinity and
selectivity for the protein target (or another biological target molecule) and
optimised for other drug-like properties such as lack of toxic effects and
desirable
pharmacokinetics.
Recent drug development has focused on screening of large libraries of
chemical compounds in order to identify lead compounds, which are capable of
either upregulating (called agonists) or downregulating the activity of the
protein
target (called antagonists), as required. Screening has usually been performed
in a
"shot-gun" fashion by setting up an assay for screening large numbers of
compounds, e.g. Iarge files of compounds or compounds in combinatorial
libraries,
in order to identify compounds with the desired activity. The subsequent
chemical
optimization of the lead compounds obtained from such screening procedures has
been performed very much in a trial-and-error fashion and has been quite
cumbersome and resource-demanding, involving procedures such as described by
E.
Sun and F.E. Cohen, Gene 1993 137(1),127-32, or J. Kuhlmann, I~tJClin
Pha~macol Then. 1999 37(12), 575-83. A major disadvantage of the drug
discovery
process is.that it is difficult to identify active compounds with sufficient
selectivity
and specificity for a given target protein or in many cases it is even
difficult at all to
identify suitable Iead compounds, for example for interfering with protein-
protein
interactions.
Optimization of lead compounds to high affinity ligands
Through the generation of chemical analogs of the lead compound and testing
of these for binding or activity on the biological target molecules such as a
protein
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
3
target, the lead compound is gradually improved in affinity for the target.
Also this
process in to a large degree done by trial-and-error, although the medicinal
chemist
usually is guided by a gradually increasing knowledge in the structure
activity
relationship (SAR) of the compounds, i.e. the observation of which
modification at
which site in the compound that increase or decrease the activity of the
compound.
The SAR can provide a great deal of information regarding the nature of ligand-
receptor interactions, but no detailed information about the location and
actual
chemical nature of the binding site in the target protein is provided. A
number of
closely related chemical structures are used to direct the orientation of the
ligand
within the putative binding cavity and to determine what part of the Iigand is
involved in binding to the receptor. This technique has its limitations due to
the fact
that changing the structure of the ligand may result in a actual change in the
binding
site of the receptor (Mattos et al. St~uct. Biol., 1995 1:55-58) , a fact
which
obviously still would be un-know to the medicinal chemist. Thus, in most cases
the
lack of knowledge of the precise molecular interaction with the receptor of
the lead
compounds found by chemical screening has prevented a rational chemical
approach to the optimisation of the lead compound.
Identification of li~and binding sites
Determination of the three-dimensional structure of the target protein either
alone or even better in complex with the ligand by X-ray crystallography
provides
high-resolution and very high quality information about the molecular
recognition
of the compound in the target protein structure. In the case, where the target
is a
soluble protein it is often possible to perform rationalized lead compound
optimization through crystallisation of the lead compound in complex with the
target protein, analyse the molecular interactions and identify possible ways
of
improving these interactions and on this basis new compounds with improved
affinity are synthesised. Subsequent X-ray analysis of complexes of these
improved
compounds and the target protein can then lead to the synthesis of a new
series of
further improved compounds, new compound-target crystalisations and so on
until
the desired affinity has been obtained.
However, these methods of structure based lead compound optimization or
"rational drug discovery" can only be applied to soluble proteins, which are
relatively easy to crystallise. For example, membrane proteins which
constitute a
majority of drug targets are very difficult or in most cases still impossible
to
crystallise. A variety of methods have been employed in order to characterize
ligand-receptor interactions in proteins where three-dimensional structures
cannot
be obtained. For example, site-directed mutagenesis is used to eliminate a
ligand
binding site or part of a ligand binding site by substitution of selected
amino acid
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
4
residues with other residues, e.g. ala.nine. Only a few cases have been
presented
where ligand binding sites have been thoroughly investigated by an extensive
and
systematic mutational analysis of alI possible residues in a given area and
with
combination of both mutational analysis of the receptor and chemical analysis
of the
ligand (e.g. the ~i-adrenergic receptor, Strader et al., FASEB J. 3, 1989, pp.
1825
1832; Strader et al, J. Biol. Chem. 266(1), 1991, pp. 5-8; Schambye et al.
Mol.
Pha~m., 1995 47:425-431).
A general problem of the site-directed mutagenesis method is that it is not
clear whether the substitution of a residue affects the binding of a ligand
directly
(i.e. the residue is directly involved in ligand binding) or indirectly (i.e.
the residue
is only involved in the structure of the receptor). Another problem of Ala
substitution is false negative results because the procedure basically creates
another
"hole" in the presumed binding pocket through removal of the side chain on the
residue replaced by Ala. The effect of Ala substitution is highly dependent on
the
relative contribution to the binding energy of the replaced residue. An
alternative to
Ala substitution is steric hindrance mutagenesis where for example a larger
side
chain, e.g. Trp, are introduced in a presumed binding pocket as described by
Holst
et al., Mol Pha~macol. 53(1), 1998, pp. 166-175.
Methods such as photoaffinity labelling has also been proven to be a useful
tool in identifying domains of receptors involved in ligand binding (Dohlman
et al.,
Ann. Rev. Biochem. 60, 1991, pp. 653-688). A photoreactive group is attached
or
built into the ligand. After binding, the ligand-receptor complex is exposed
to UV
light, resulting in crosslinking of the ligand to the receptor. Finally the
complex is
digested with proteases and the ligand-binding part of the receptor can be
identified.
It should be noted however, that except for proteins where crystal- or NMR-
structures can be made, it is only in a few cases where binding pockets for
ligands in
fact have been identified with a reasonable degree of accuracy. This is
especially the
case for membrane proteins. In even fewer cases have the actual pattern of
chemical
recognition been determined well in these proteins, i.e. identification of
which
chemical moiety of the ligand interacts with which side-chain or with which
part of
the backbone in the target (Schwartz et al. Current Opiu. Biotechnol., 1994
4:434-444). In the very few cases of for example membrane proteins where some
information is available concerning the presumed binding pocket or perhaps
even
about actual chemical interactions, this is only the case for final, high-
affinity
optimized drugs. No information along these lines are today known for lead
compounds found by chemical screening in for example membrane proteins. Even
in the case where an X-ray structure is known for a complex between a compound
or a drug and its target protein, it is often not possible to predict the
binding mode of
close analogs of this since modification of the compound may seriously alter
the
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
overall binding mode involving also parts of the compound which have not been
chemically modified (Mattos et al. St~uct. Biol, 1995 1:55-58). Thus, a
chemical
"anchor", i.e. a well identified binding point between a chemical moiety in
the
compound and a particular site in the target protein, would be highly
beneficial in
5 order to efficiently apply structure based drug discovery techniques to both
proteins
with known three dimensional structures and to protein targets for which
meaningful molecular models can be built based on homology to known protein
structures.
The present invention deals with methods involving a chemical "anchor" by
making use of a metal binding site in the target biological molecule as well a
metal
binding site in a chemical compound. The metal binding site in the biological
target
molecule such as, e.g., a target protein may be a natural metal-ion binding
site or it
may be a metal-ion binding site that has been introduced into the protein by
artificial
means such as, e.g., engineering means.
BACKGROUND OF THE INVENTION
Natural metal-ion sites in proteins
Many proteins contain metal-ion binding sites. These metal-ion sites serve
either structural purposes, for example stabilizing the three-dimensional
structure of
the protein, or they serve functional purposes, where the metal-ion may for
example
be part of the active site of an enzyme. It is well known that also several
integral
membrane proteins include binding sites for metal ions. The coordination of
metal
ions to metal ion binding sites is well characterized in numerous high-
resolution X-
ray and NMR structures of soluble proteins; for example, distances from the
chelating atoms to the metal ion as well as the preferred conformation of the
chelating side chains are known (e.g. J.P. Glusker, Adv. Protein Chem. 42,
1991, pp.
3-76; P. Chakrabarty, P~oteih Eng. 4, 1990, pp. 57-63; R. Jerigan et al., Cur.
Opin.
St~uct. Biol. 4, 1994, pp. 256-263). Thus, metal-ion binding in proteins is
one of the
most well characterised forms of ligand-protein interactions known. Hence,
characterising a metal ion-binding site in a membrane protein using, for
example,
molecular models and site directed mutagenesis can yield information about the
structure of the membrane protein and importantly where the "ligand" (metal
ion)
binds (e.g. Elling et al. Fold. Des. 2(4), 1997, pp. S76-80).
Metal-ion site en ing_ eerin,~ in proteins
Engineering of artificial metal ion binding sites into membrane proteins has
been employed to explore the structure and function of these proteins. Thus,
C.E.
Elling et al., Nature 374, 1995, pp. 74-77, have reported how the binding site
fox a
proto-type antagonists for the tachykinin NK-I receptor could be converted
into a
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
6
metal ion-binding site by systematic substitution of residues in the binding
pocket
with His residues. If side chains of amino acid residues participating in
metal ion
binding are known, it imposes a distance constraint on the protein structure
which
can be used in the interpretation of unknown protein structures (C.E. Elling
and
T.W. Schwartz, EMBO J. 15(22), 1996, pp. d2I3-6219; C.E. Elling et aL, Fold.
Des.
2(4), 1997, pp. S76-80). Recently the generation of an activating metal ion
binding
site has been reported for the ~i2-adrenergic receptor, where the binding site
for the
normal catecholamine ligands was exchanged with a metal-ion site through
specific
substitutions in the binding pocket for the agonists (C.E. Elling et al, PNAS
96,
1999, pp. 12322-12327). This metal-ion binding site could be addressed also
with
metal-ions in complex with metal-ion chelators, i.e. small organic compounds
binding metal-ions.
However, none of the above-mentioned documents address the concept of
using a chemical "anchor" in the drug discovery process.
SUMMARY OF THE INVENTION
The present invention provides a molecular approach for rapidly and
selectively identifying small organic molecule ligands, i.e. compounds, that
are
capable of interacting with and binding to specific sites on biological target
molecules. The methods described herein make it possible to construct and
screen
libraries of compounds specifically directed against predetermined epitopes on
the
biological target molecules. The compounds are initially constructed to be bi-
functional, i.e. having both a metal-ion binding moiety, which conveys them
with
the ability to bind to either a natural or an artificially constructed metal-
ion binding
site as well as a variable moiety, which is varied chemically to probe for
interactions
with specific parts of the biological target molecule located spatially
adjacent to the
metal-ion binding site. Compounds may subsequently be further modified to bind
to
the unmodified biological target molecule without help of the bridging metal-
ion.
The methods according to the invention may be performed easily and quickly and
lead to unambiguous results. The compounds identified by the methods described
herein may themselves be employed for various applications or may be further
derivatised or modified to provide novel compounds
The methods of the present invention are applicable to any biological target
molecule that has or can be manipulated to have a metal-ion binding site.
However,
in the following proteins are used as examples of biological target molecules.
Parts of the present invention utilise the finding that many proteins in their
natural form posses a metal-ion binding site, which may or may not have been
recognized previously. However, in order to obtain a general applicability of
the
technology to a broad range of biological target molecules, the invention
especially
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
7
utilizes the possibility to mutate proteins, for example a receptor, an enzyme
or a
transcriptional regulator in such a way, that they comprise a metal ion
binding site.
The metal-ion site is then used as an anchor-point for the initial parts of
the
medicinal chemistry drug-discovery process, during which test compounds can be
synthesized, which due to their specific interaction with the metal-ion
binding site
can be deliberately directed towards interaction with specific, functionally
interesting parts of the biological target molecule.. The test compounds are
subsequently structurally optimised for interaction with spatially
neighbouring parts
of the proteins (that is, interaction with the side chains or backbone of one
or more
neighbouring amino acid residues). These compounds can then be utilized as
leads
or starting points for the construction of ligands binding to the wild-type
protein. In
this way it is possible to predetermine the binding site of a compound to a
particular
location in a protein structure and thereby target the optimised compounds to
sites
where binding of the compound will alter the biological activity of the
protein in a
desired way, for example to increase or decrease its biological activity. By
selecting
the binding site for a test compound at will and thereby selecting the binding
site for
the optimised compound (such as a drug candidate) in a protein, it is for
example
possible to
1) speed up the process of development of high affinity drug candidates or
other compounds with biological activity because a more efficient structure-
based
compound optimisation process can be applied; .
2) obtain high selectivity for a given member of a protein family by targeting
the compound to a site in the protein which differs between different members
of
the protein family;
3) obtain new functionalities of compounds by targeting them to allosteric
modulatory sites in proteins.
These constitute some of the advantages of the present invention.
In the course of research leading to the present invention, the inventors have
found that certain small organic compounds which bind metal ions (i.e. metal
ion
chelators) are also able to bind to metal ion binding sites in various
proteins,
including membrane proteins for example receptors, in such a way that the
metal ion
acts as a bridge between the small organic compound and the protein.
Importantly,
the present invention has made it possible to predetermine or identify and
localise
the exact binding site and binding mode of such metal ion chelates used as
test
compounds, contrary to what has been known in the art for test compounds in
general. Based on the identification or confirmation of the binding site of
the test
compounds, using for example site-directed mutagenesis, three-dimensional
structure determination by for example X-ray crystallography or NMR or
molecular
models of the protein and techniques such as those described above, a rational
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
g
approach may be taken to the chemical optimisation of the test compounds.
Thus,
relatively small chemical libraries may be made, the compounds in which may be
designed to interact with specific amino acid residues of the protein in
question.
Compounds that exhibit a high affinity binding to the protein and affect the
biological activity of the protein in a desired way may then be selected for
further
optimisation.
The metal-ion binding portion of the test compounds may subsequently be
removed or altered to no longer posses metal-ion binding properties, and the
test
compounds, as well as chemical derivatives thereof may be constructed to
interact
with side chains of other amino acids in the vicinity of the artificial metal
ion
binding site, and tested for binding to the wild-type protein which does not
include a
metal ion binding site. Accordingly, relatively small chemical libraries may
be
made, the compounds in which may be designed to interact with the specific
amino
acid residues found in the wild-type protein at or spatially surrounding the
location
where the metal ion site had initially been engineered.
Thus, the present invention is based on the general principle, applicable to
any
biological target molecule including a protein, of introducing metal ion
binding sites
at any position in e.g. the protein where a test compound binding to the
protein is
likely to exert an effect on the biological activity of the protein. This may
for
example be 1) at a site where the test compound will interfere with the
binding to
another protein, for example a regulatory protein, or to a domain of the same
protein; 2) at a site where the binding of the test compound will interfere
with the
cellulaa- targeting of the protein; 3) at a site where the binding of the test
compound
will directly or indirectly interfere with the binding of substrate or the
binding of an
allosteric modulatory factor for the protein; 4) at a site where the binding
of the test
compound may interfere with the intra-molecular interaction of domains within
the
protein, for example the interaction of a regulatory domain with a catalytic
domain;
5) at a site where binding of the test compound will interfere with the
folding of the
protein, for example the folding of the protein into its active conformation;
or 6) at a
site which will interfere with the activity of the protein, for example by an
allosteric
mechanism. Subsequent to identifying test compounds that bind to the
artificial
metal ion binding site of the protein, information may be acquired of the
structure of
the binding site and of amino acid residues in its immediate vicinity. Such
information may be used in the design of compounds with improved binding
affinity
to the proteins resulting from interaction with one or more amino acid
residues in
the vicinity of the metal ion binding site. Such compounds may, in turn, be
used in
the design of potential drug candidates or other compounds with a desired
activity
on the corresponding wild-type, non-mutated protein.
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
9
Accordingly, the present invention relates to a drug discovery process for
identification of a small organic compound that is able to bind to a
biological target
molecule, the process comprising mutating a biological target molecule in such
a
way that at least one amino acid residue capable of binding a metal ion is
introduced
into the biological target molecule so as to obtain a metal ion binding site
as an
anchor point in the mutated biological target molecule.
The mutated biological target molecule may furthermore be contacted with a
test compound which comprises a moiety including at Ieast two heteroatoms for
chelating a metal ion, under conditions permitting non-covalent binding of the
test
compound to the introduced metal ion binding site of the mutated biological
target
molecule, and then followed by detection of any change in the activity of the
mutated biological target molecule or determation of the binding affinity of
the test
compound to the mutated biological target molecule.
The present invention relates also to a drug discovery process for
identification of a small organic compound that is able to bind to a
biological target
molecule which has at least one metal ion binding site, the process comprising
(a) contacting the biological target molecule with a test compound which
comprises a moiety including at least two heteroatoms for chelating a metal
ion,
under conditions permitting non-covalent binding of the test compound to the
metal
ion binding site of the biological target molecule, and
(b) detecting any change in the activity of the biological target molecule or
determining the binding affinity of the test compound to the biological target
molecule.
A very important class of biological target molecules amenable to testing
according to the present invention are proteins such as membrane proteins
which
includes proteins that are involved in intercellular communication and other
biological processes of profound importance for cellular activity. Thus, in
another
aspect, the present invention relates to a method of identifying a metal ion
binding
site in a protein, the method comprising
(a) selecting a nucleotide sequence suspected of coding for a protein and
deducing the amino acid sequence thereof,
(b) expressing said nucleotide sequence in a suitable host cell,
(c) contacting said cell or a portion thereof including the expressed
protein with a test compound which comprises a moiety including at least two
heteroatoms for chelating a metal ion, under conditions permitting non-
covalent
binding of the test compound to the protein, and detecting any change in the
activity
of the protein or determining the binding affinity of the test compound to the
protein, and
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
(c) determining, based on the generic three-dimensional model of the class o90
proteins to which the protein or suspected protein belongs, at least one metal
ion
binding amino acid residue located in said protein to locate the metal ion
binding
site of said protein.
5 In a still further aspect the invention relates to a method of mapping a
metal
ion binding site of a protein, the method comprising
(a) contacting the protein with a test compound which comprises a moiety
including at least two heteroatoms for chelating a metal ion, under conditions
permitting non-covalent binding of the test compound to the protein, and
detecting
10 any change in the activity of the protein or determining the binding
affinity of the
test compound to the protein, and
(b) determining, based on the primary structure of the specific protein in
question and the generic three-dimensional model of the class of proteins to
which
the specific protein of step (a) belongs, at least one metal ion binding amino
acid
residue located in the membrane protein to identify the metal ion binding site
of said
membrane protein.
In a further aspect the invention relates to chemical libraries comprising
test
compounds in chelated or non-chelated form and to a chemical library
comprising
metal ions suitable for chelating test compounds. The metal ions are generally
presented in salt form or in the form of complexes or solvates.
In still further aspects the invention relates to the use of test compounds as
tracers in binding assays for orphan receptors and in pharmacological l~nock-
out
experiments.
Further aspects of the invention as well as preferred embodiments of the
invention appear from the appended claims.
The details and particulars described for e.g. the drug discovery process
aspect
apply mutatis mutandis - whenever relevant - to all other aspects of the
invention.
DETAILED DESCRIPTION OF THE INVENTION
Essential parts of the present invention relates to methods of identifying
compounds that are capable of binding to specific sites on biological target
molecules. Much of the detailed description of the invention is dealt with in
the
description of the examples presented in "EXPERIMENTAL". In a typical form of
this
process the following steps are involved:
(1) Ide~ttification o~ engineering of ~r2etal-ion bi~di~cg sites to be
exploited as
anclzo~ ports fog lead compounds - In one embodiment of the invention, the
biological target molecule already has a suitable metal-ion site, which may or
may
not previously have been recognized. In another more broadly applicable form
of
the invention such metal-ion sites are introduced, for example through
mutagenesis,
SUBSTITUTE SHEET (RULE 26)
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
11
at specific sites in the biological target molecule expected to be useful as
anchor
points for the development of compounds affecting the function of the target
molecule in a desired way. In one form of the invention a number of such sites
are
introduced and one or more axe selected for further use.
(2) Selection of lead compound fi°om library of metal ion chelating
compounds
- Basic libraries of metal-ion chelators exposing a systematic range of
chemical
moieties differing in potential chemical interaction-mode with the surrounding
parts
of the biological target molecule are screened for lead or test compounds
which will
bind to the metal-ion site in the biological target molecule and affect its
function in
a desired way.
(3) Chemical optimisation of lead compound fog secohda~y iute~actioh points
in the biological target molecule - Based on the selected lead compound,
libraries of
basic bi-functional compounds are being constructed in which the compounds
have
both a anchoring metal-iou binding moiety, which conveys them with the ability
to
bind to the metal-ion binding site in the biological target molecule, as well
as a
variable moiety, which is varied chemically to probe for improved interactions
with
specific parts of the biological target molecule located spatially adjacent to
the
metal-ion binding site. In one preferred form of the invention these libraries
are
constructed based on structural knowledge of the chemical target moiety in the
biological target molecule. In another form a more broad screening of larger
libraries of compounds is performed without detailed knowledge of the
structure of
the biological target molecule surrounding the anchoring metal-ion site.
(4) Chemical optimisation of lead compound fog high a~hity inte~~action with
wild
type biological target molecule - exchange of metal ion anchor with "o~diha~y"
chemical ihte~action - When a compound has been developed having a suitable,
detectable affinity also on the wild-type form of the biological target
molecule
usually without metal-ion present, then this compound is further optimized for
high
affinity binding and effect on the wild-type molecule. In one form of the
invention
structure-based construction of chemical libraries will be performed in order
to take
advantage of the possibility to directly exchange the metal-ion bridge with
other
types of chemical interactions with the amino acid residues found in the wild
type
molecule.
The present invention is directed to methods directly or indirectly involved
in
the above-mentioned drug discovery process. Furthermore, it is directed to the
use
of chemical libraries and to a method for selecting a chemical compound from a
library.
The following detailed description of the invention is mainly concerned with
methods of identifying compounds interacting with proteins such as, e.g.,
membrane
proteins. It should be understood, however, that the discussion of the
detailed
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
12
method steps apply equally to other biological target molecules like nucleic
acids,
carbohydrates, nucleoproteins, glycoproteins and glycolipids.
In the following some definitions are first dealt with following by a detailed
description of the invention according to the four main steps of the drug
discovery
process:
DEFII~TITIONS
Throughout the text including the claims, the following terms shall be defined
as indicated below.
A "test compound" is intended to indicate a small organic molecule ligand or a
small organic compound which is capable of interacting with a biological
target
molecule, in particular with a protein, in such a way as to modify the
biological
activity thereof. The term includes in its meaning metal ion chelates of the
formulas
shown below. Furthermore, the term includes in its meaning metal ion chelates
of
the formulas shown below as well as chemical derivatives thereof constructed
to
interact with other parts) of the biological target molecule than the metal
ion
binding site. In proteins such an interaction may take place with side chains
of
amino acids or amino acid residues in the vicinity of the natural or
artificial metal
ion binding site. A test compound may also be an organic compound which in its
structure includes a metal atom via a covalent binding. Such test compounds
will
generally contain at least one heteroatom such as, e.g., N, O, S, Se and/or P.
A "metal ion chelator" is intended to indicate a compound capable of forming
a complex with a metal atom or ion. Such a compound will generally contain a
heteroatom such as N, O, S, Se or P with which the metal atom or ion is
capable of
forming a complex.
A "metal ion chelate" is intended to indicate a complex of a metal ion
chelator
and a metal atom or ion.
A "metal ion binding site" is intended to indicate a part of a biological
target
molecule which comprises an atom or atoms capable of complexing with a metal
atom or ion. Such an atom will typically be a heteroatom, in particular N, O,
S, Se
or P. With respect to proteins a metal ion binding site is typically an amino
acid
residue of the protein which comprises an atom capable of complexing with a
metal
ion. These amino acid residues are typically but nor respricted to histidine,
cysteine,
and aspartate.
A "ligand" is intended to include any substance that either inhibits or
stimulates the activity of the membrane protein or that competes for the
receptor in a
binding assay. An "agonist" is defined as a ligand increasing the functional
activity
of a membrane protein (e.g. signal transduction through a receptor). An
"antagonist"
is defined as a ligand decreasing the functional activity of a membrane
protein either
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
13
by inhibiting the action of an agonist or by its own intrinsic activity. An
"inverse
agonist" (also termed "negative antagonist") is defined as a ligand decreasing
the
basal functional activity of a membrane protein.
A "biological target molecule" is intended to include proteins such as, e.g.,
membrane proteins, nucleic acids, carbohydrates, nucleoproteins, glycoproteins
and
glycolipids. In the present context the biological target molecule contains or
has
been manipulated to contain a metal ion binding site.
A 'protein" is intended to include any protein, polypeptide or oligopeptide
with a discernible biological activity in any unicellular or multicellular
organism,
including bacteria, fungi, plants, insects, animals or mammals, including
humans.
Thus, the protein may suitably be a drug target, i.e. any protein which
activity is
important for the development or amelioration of a disease state, or any
protein
which level of activity may be altered (i.e. up- or down-regulated) due to the
influence of a biologically active substance such as a small organic chemical
compound.
A "memb~ahe p~oteih" is intended to include but is not limited to any protein
anchored in a cell membrane and mediating cellular signalling from the cell
exterior
to the cell interior. Important classes of membrane proteins include receptors
such
as tyrosine kinase receptors, G-protein coupled receptors, adhesion molecules,
ligand- or voltage-gated ion channels, or enzymes. The term is intended to
include
membrane proteins whose function is not known, such as orphan receptors. In
recent
years, largely as part of the human genome project, large numbers of receptor-
like
proteins have been cloned and sequenced, but their function is as yet not
known.
The present invention may be of use in elucidating the function of the
presumed
receptor proteins by making it possible to develop methods of identifying
ligand for
orphan receptors based on compounds developed from metal ion chelates that
bind
to mutated orphan receptors into which artificial metal ion binding sites have
been
introduced.
"Signal t~ahsductio~c" is defined as the process by which extracellular
information is communicated to a cell by a pathway initiated by binding of a
ligand
to a membrane protein, leading to a series of conformational changes resulting
in a
physiological change in the cell in the form of a cellular signal.
A 'fuhctiohal group" is intended to indicate any chemical entity which is a
component part of the test compound and which is capable of interacting with
an
amino acid residue or a side chain of an amino acid residue of the membrane
protein. A functional group is also intended to indicate any chemical entity
which is
a component part of the biological target molecule and which is capable of
interacting with other parts of the biological target molecule or with a part
of the test
compound. Examples of such functional groups include, but~are not limited to,
ionic
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
14
groups involved in ionic interactions such as e.g. the ammonium ion or
carboxylate;
ion; hydrogen bond donor or acceptor groups such as amino, amide, carboxy,
sulphonate, etc.; and hydrophobic groups involved in hydrophobic interactions,
pi-
stacking and the like.
A "wild type"membrane protein is understood to be a membrane protein in its
native, non-mutated form, in this case not comprising an introduced metal ion
binding site
The term "ih the vicinity of is intended to include an amino acid residue
located in the area defining the binding site of the metal ion chelate and at
such a
distance from the metal ion binding amino acid residue that it is possible, by
attaching suitable functional groups to the test compound, to generate an
interaction
between said functional group or groups and said amino acid residue.
IDENTIFICATION OR ENGINEERING OF METAL-ION BINDING SITES 1N
BIOLOGICAL TARGET MOLECULES TO BE EXPLOITED AS ANCHOR
POINTS FOR LEAD COMPOUNDS
NATURE OF THE BIOLOGICAL TARGET MOLECULES
The biological target molecules include but are not restricted to proteins,
nucleoproteins, glycoproteins, nucleic acids, carbohydrates, and glycolipids.
In the
present context the biological target molecule contains or has been
manipulated to
contain a metal ion binding site. In preferred embodiments the biological
target
molecule is a protein, which may be for example a membrane receptor, a protein
involved in signal transduction, a scaffolding protein, a nuclear receptor, a
steroid
receptor, a transciption factor, an enzyme, and an allosteric regulator
protein, or it
may be a growth factor, a hormone, a neuropeptide or an immunoglobulin.
In particularly preferred embodiments the biological target molecule is a
membrane protein which suitably is an integral membrane protein, which is to
say a
membrane protein anchored in the cell membrane. The membrane protein is
preferably of a type comprising at least one transmembrane domain. Interesting
membrane proteins for the present purpose are mainly found in classes
comprising
1-14 transmembrane domains.
1 TM - membrane proteins of interest comprising one transmembrane domain
include but are not restricted to receptors such as tyrosine kinase receptors,
e.g. a
growth factor receptor such as the growth hormone, insulin, epidermal growth
factor, transforming growth factor, erythropoietin, colony-stimulating factor,
platelet-derived growth factor receptor or nerve growth factor receptor (TrkA
or
TrkB).
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
2TM - membrane proteins of interest comprising two transmembrane domains
include but are not restricted to, e.g., purinergic ion channels.
3, 4, STM - membrane proteins of interest comprising 3, 4 or 5 transmembrane
domains includes but are not restricted to e.g. ligand-gated ion channels,
such as
5 nicotinic acetylcholine receptors, GABA receptors, or glutamate receptors
(NMDA
or AMPA).
6TM - membrane proteins of interest comprising 6 transmembrane domains
include but are not restricted to e.g., voltage-gated ion channels, such as
potassium,
sodium, chloride or calcium channels.
10 7TM - membrane proteins of interest comprising 7 transmembrane domains
include but are not restricted to G-protein coupled receptors, such as
receptors for:
acetylcholine, adenosine, norepinephrin and epinephrine, anaphylatoxin
chemotactic
factor, angiotensin, bombesin (neuromedin), bradykinin, calcitonin, calcitonin
gene
related peptide, conopressin, corticotropin releasing factor, amylin,
adrenomedullin,
15 calcium, cannabinoid, CC-chemokines, CXC-chemokines, cholecystokinin,
conopressin, corticotropin-releasing factor, dopamine, eicosanoid, endothelin,
fMLP, GABAB, galanin, gastrin, gastric inhibitory peptide, glucagon, glucagon-
like
peptide I and II, glutamate, glycoprotein hormone (e.g. FSH, LSH, TSH, LH),
gonadotropin releasing hormone, growth hormone releasing hormone, growth
hormone releasing peptide (Ghrelin), histamine, 5-hydroxytryptamine,
leukotriene,
lysophospholipid, melanocortin, melanin concentrating hormone, melatonin,
motilin, neuropeptide Y, neurotensin, nocioceptin, odor components , opiods,
retinal, orexin, oxytocin, parathyroid hormone/parathyroid hormone-related
peptide,
pheromones, platelet-activating factor, prostanoids, secretin, somatostatin,
tachykinin, thrombin and other proteases acting through 7TM receptor,
thyrotropin-
releasing hormone, pituitary adenylate activating peptide, vasopressin,
vasoactive
intestinal peptide and virally encoded receptors; in pa~~ticula~: adenosin,
galanin,
CC-chemokines, CXC-chemokines, melanocortin, bombesin, cannabinoid,
lysophospholipid, fIVILP, neuropeptide Y, tachykinin, dopamine, histamine, 5-
hydroxytryptamine, histamine, mas-proto-oncogene, acetylcholine, oxytocin,
human
herpes virus encoded receptors, Epstein Barr virus induced receptors,
cytomegalovirus encoded receptors and bradykinin receptors; preferably the
galanin
receptor type 1, leukotriene B4 receptor, CCRl, CCR2, CCR3, CCR4, CCRS,
CCR6, CCR7, CCRB, CCR9, CCR10, CXCRl, CXCR2, CXCR3, CXCR4, CXCRS,
CXCR6, CX3CR1, melanocortin-1 receptor, melanocortin-3 receptor, melanocortin-
4 receptor, melanocortin-5 receptor, bombesin receptor subtype 3, cannabinoid
xeceptor 1, cannabinoid receptor 2, EDG-2, EDG-4, FMLP-related receptor I,
FMLP-related receptor II, NPY Y6 receptor, NPY YS receptor, NPY Y4 receptor,
NK-1 receptor, NK-3 receptor, D2 receptor (short), D2 receptor (long), duffy
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
16
antigen; US27, US28, UL33 and U78 from human cytomegalovirus; U12 and US1
from human herpes virus 6 or 7, ORF74 from human herpes virus 8, and histamine
H1 receptor, MAS proto-oncogene, muscarinic Ml receptor, muscarinic M2
receptor, muscarinic M3 receptor, muscarinic MS receptor, oxytocin receptor,
S XCRl receptor, EBI2 receptor, RDC 1 receptor, GPRI2 receptor or GPR3
receptor.
8, 9, 10, 1 l, 12, 13, I4TM - Membrane proteins of interest comprising 8 to 14
transmembrane domains include but are not restricted to e.g., transporter
proteins,
such as a GABA, monoamine or nucleoside transporter.
The membrane protein may also be a multidrug resistance protein, e.g. a P-
glycoprotein, multidrug resistance associated protein, drug resistance
associated
protein, lung resistance related protein, breast cancer resistance protein,
adenosine
triphosphate-binding cassette protein, Bmx, QacA or EmrAB/ToIC pump.
The membrane protein may also be a cell adhesion molecule, including but not
restricted to for example NCAM, VCAM, ICAM or LFA-1.
1 S Furthermore, the membrane protein may be an enzyme such as adenylyl
cyclase.
In a particularly preferred embodiment of the invention, the biological target
molecules are 7 transmembrane domain receptors (7TM receptors) also known as
G-protein coupled receptors (GPCRs).
7TM overview - This family of receptors constitutes the largest super-family
of proteins in the human body and a large number of current drugs are directed
towards 7TM receptors, for example: antihistamines (for allergy and gastric
ulcer),
beta-blockers (for cardiovascular diseases), opioids (for pain), and
angiotensin
antagonists (for hypertension). These current drugs are directed against
relatively
2S few receptors, which have been known for many years. To date, several
hundred
7TMs have been cloned and characterized, and the total number of different
types of
7TMs in humans is presumed to be between 1 and 2.000. The spectrum of ligands
acting through 7TMs includes a wide variety of chemical messengers such as
ions
(e.g. calcium ions), amino acids (glutamate, y-amino butyric acid), monoamines
(serotonin, histamine, dopamine, adrenalin, noradrenalin, acetylcholine,
cathecolamine, etc.), lipid messengers (prostaglandins, thromboxane,
anandamide,
etc.), purines (adenosine, ATP), neuropeptides (tachykinin, neuropeptide Y,
enkephalins, cholecystokinin, vasoactive intestinal polypeptide, etc.),
peptide
hormones (angiotensin, bradykinin, glucagon, calcitonin, parathyroid hormone,
3S etc.), chemokines (interleukin-8, RANTES, etc.), glycoprotein hormones (LH,
FSH,
TSH, choriogonadotropin, etc.) and proteases (thrombin). It is expected that a
large
number of the members of the 7TM superfamily of receptors will be suitable as
drug
targets. This notion is based on the fact that these receptors axe involved in
controlling major parts of the chemical transmission of signals between cells
both in
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
17
the endocrine and the paracrine system in the body as well as within the
nervous
system.
7TM receptor sigr~allihg - In 7TMs, binding of the chemical. messenger to the
receptor leads to the association of an intracellular G-protein, which in turn
is linked
to a secondary messenger pathway. The G-protein consists of three subunits, an
a-
subunit that binds and hydrolyses GTP, and a ~3y-subunit. When GDP is bound,
the
a subunit associates with the ~iy subunit to form an inactive heterotrimer
that binds
to the receptor. When the receptor is activated, a signal is transduced by a
changed
receptor conformation that activates the G-protein. This leads to the exchange
of
GDP for GTP on the a subunit, which subsequently dissociates from the receptor
and the (3y dimer, and activates downstream second messenger systems (e.g.
adenylyl cyclase). The a subunit will activate the effector system until its
intrinsic
GTPase activity hydrolyses the bound GTP to GDP, thereby inactivating the a
subunit. The ~iy subunit increases the affinity of the a subunit for GDP but
may also
be directly involved in intracellular signalling events. - 7TMligand bihdihg
sites
Mutational analysis of 7TMs has demonstrated that functionally similar but
chemically very different types of ligands can apparently bind in several
different
ways and still lead to the same function. Thus monoamine agonists appear to
bind in
a pocket relatively deep between TM-III, TM-V and TM-VI, while peptide
agonists
mainly appear to bind to the exterior parts of the receptors and the
extracellular ends
ofthe TMs (Strader et al., (1991) J. Biol. Chem. 266: 5-8; Strader et al.,
(1994) Ann.
Rev. Biochem. 63: 101-132; Schwartz et al. Curr. Pharmaceut. Design. (1995),
1:
325-342). Moreover, ligands can be developed independent on the chemical
nature
of the endogenous ligand, for example non-peptide agonists or antagonists for
peptide receptors. Such non-peptide antagonists for peptide receptors often
bind at
different sites from the peptide agonists of the receptors. For instance, non-
peptide
antagonists may bind in the pocket between TM-III, TM-V, TM-VI and TM-VII
corresponding to the site where agonists and antagonists for monoamine
receptors
bind. It has been found that in the substance P receptor, when the binding
site for a
non-peptide antagonist has been exchanged for a metal ion binding site through
introduction of His residues, no effect on agonist binding was observed
(Elling et
al., (1995) Nature 374: 74-77; Elling et al. (1996) EMBO J. 15: 6213-6219). It
is
believed that the non-peptide antagonist and the zinc ions act as antagonists
by
selecting and stabilizing an inactive conformation of the receptor that
prevents the
binding and action of the agonist. This illustrates that drugs can be
developed totally
independent on knowledge of the endogenous ligand, since there need not be any
overlap in their binding sites.
Gehe~ic v~umbe~ivcg system fog 7TMs - a useful tool in the identification and
engineering of metal-ion sites is the generic numbering system for residue of
7TM
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
18
receptors. The largest family of 7TM receptors is composed of the rhodopsin-
like
receptors, which are named after the light-sensing molecule from our eye.
Within
the many hundred members of the rhodopsin-like receptor family, a number of
residues especially within each of the transmembrane segments are highly but
not
totally conserved. However, due to differences in the length of especially the
N-
terminal segment, residues located at corresponding positions in different 7TM
receptors are numbered differently in different receptors. However, based on
the
conserved key residues in each TM, a generic numbering system has been
suggested
(JM Baldwin, EMBO J. 12(4), 1993, pp. 1693-1703; TW Schwartz, Curr. Opin.
Biotech. 5, 1994, pp. 434-444). In Fig. IV a schematic depiction of the
structure of
rhodopsin-like 7TMs is shown with one or two conserved, key residues
highlighted
in each TM: AsnI:lB; AspII:lO; CysIII:01 and ArgIII:26; TrpIV:lO; ProV:l6;
ProVI:15; ProVII:17. In relation to the present invention it is important that
residues
involved in for example metal ion binding sites can be described in this
generic
numbering system. For example, a tri-dentate metal ion site constructed in the
tachy-
kinin NKl receptor (Elling et al., (1995) Nature 374, 74-77) and subsequently
trans-
ferred to the kappa-opioid receptor (Thirstrup et al., (1996) J. Biol. Chem.
271,
7875-7878) and to the viral chemolcine receptor ORF74 (Rosenkilde et al., J.
Biol.
Chem. 1999 Jan 8; 274(2), 956-61) can be described to be located between
residues
V:01, V:05, and VI:24 in all of these receptors although the specific
numbering of
the residues is very different in each of the receptors. It is only in the
rhodopsin-like
receptor family that a generic numbering system has been established; however,
it
should be noted that although the sequence identity between the different
families of
7TM receptors is very low, it is believed that they may share a more-or-less
conunon seven helical bundle structure. Thus, all the techniques described in
the
present invention can be applied to the other families of 7TM receptors with
minor
modifications. This generic numbering system together with general knowledge
of
the 3D structure of the 7TM receptors and knowledge from systematic metal-ion
site
engineering makes it possible to predict or identify the presence of metal-ion
sites
based on the DNA sequence coding for the 7TM receptor (see examples).
Orphan 7TMrecepto~s - one embodiment of the invention is directed to a
method of developing assay for orphan 7TM receptors by the introduction of
metal-
ion sites in the orphan receptor. During the cloning of 7TM receptors many
"extra"
receptors were discovered for which no ligand was known, the so-called orphan
receptors. Today there are several hundreds of such orphan 7TM receptors.
Based
on characterization of their expression pattern in different tissues or
expression
during development or under particular physiological or patho-physiological
conditions and based on the fact that the orphan receptors sequence-wise
appear to
belong to either established sub-families of 7TM receptors or together with
other
SUBSTITUTE SHEET (RULE 26)
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
19
orphans in new families, it is believed that the majority of the orphan
receptors are
in fact important entities. As stated by representatives from the big
pharmaceutical
companies: Orphan 7TMs are "the next generation of drug targets" or "A
neglected
opportunity for pioneer drug discovery" (Wilson et al. Br.J.Pharmacol. (1998)
125:
1387-92; Stadel et al. Trends Pharmacol.Sci. (1997) 18: 430-37). Over the
years
ligands have been discovered for some of the orphan 7TM receptors, which then
immediately have been recognized as "real" drug targets, for example:
nocioceptin
(for pain) (Reinscheid et al. Science (1995) 270: 792-94), orexin (for
appetite
regulation and regulation of energy homeostasis) (Sakurai et al. Cell (1998)
92: 573-
85), melanin-concentrating hormone (for appetite regulation) (Chambers et al.
Nature (1999) 400: 261-65),~and cysteinyl leukotrienes (inflammation,
especially
asthma) (Sarau et al. Mol. Pharmacol. (1999) 56: 657-63). In the latter case,
a
number of drugs (for example pranlukast, zafirlukast, montelukast, pobilukast)
had
in fact been developed in recent years against the receptor as a physiological
entity
without having access to the cloned receptor - which turned out to be a "well
known" orphan receptor. The problem is that it is very difficult to
characterize
orphan receptors and find their endogenous ligands, since no assays are
available for
these receptors due to the lack of specific ligands - a "catch 22" situation.
The
present invention is aimed at eliminating this problem. By introducing metal
ion
binding sites in orphan receptors at locations where it is known from previous
work
on multiple other 7TM receptors with known ligands and with binding and
functional assays that binding of metal ions and metal ion chelates will act
as either
agonists or more common as antagonists, then it will be possible to establish
binding
assays and functional assays for the orphan receptors. Binding of metal ion
chelates
can be monitored either through functional assays in cases where agonistic
metal ion
sites are created, or through ligand binding assays. For example, many
aromatic
metal ion chelators are by themselves fluorescent and can therefore directly
be used
as tracers in binding assays. Or, radioactive or other measurable indicators
can be
incorporated into the metal ion chelator. By establishing a metal ion chelator
based
receptor analysis for the orphan receptors, it will become possible to search
for the
elusive endogenous ligands or it will be possible to use the orphan receptors
in
various forms for drug discovery technology, for example high throughput .
screening. It should be noted that due to the initial lack of knowledge of the
endogenous ligand and therefore also lack of knowledge of the binding site for
this
3 5 ligand in the 7TM receptor, there is a certain danger that the introduced
metal ion
binding site can interfere with ligand binding or signal transduction.
However,
based on metal ion site engineering in multiple 7TM receptors and on
mutational
mapping of binding sites in multiple 7TM receptors, it will be possible to
introduce
such metal ion sites at different locations in the receptor in an attempt to
eliminate
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
2a
this problem. Moreover, an artificial binding site and binding analysis, which
may
interfere with the binding of the natural ligand, may still .be useful for
screening for
receptor ligands, for example antagonists.
SOURCE OF THE BIOLOGICAL TARGET MOLECULES -
The biological target molecules of interest may be obtained in a useful form
by
different ways including but not limited to recombinantly, synthetically or
commercially.
Clohihg ahd exp~essioh - In a preferred embodiment the biological target
molecule being a protein is obtained recombinantly. This can be achieved
through
cloning of the gene for the protein from genomic or cDNA libraries generally
by the
use of PCR techniques in accordance with standard techniques (eg. Sambrook et
al.
Molecular Cloning: A laboratory manual, 2. Ed. Cold Spring Harbor Laboratory,
New York 1989) and expression of the gene in a suitable cell. The nucleotide
sequence encoding the target protein - and mutant versions thereof (see below)
-
may be inserted into a suitable expression vector for the purpose of
expression and
analysis in a host organism. Thus regulatory element ensuring either
constitutive or
inducible expression of the protein of interest should be present in the
vector,
including promoter elements. The host organism into which the nucleotide
sequence
is introduced may be any cell type or cell line, which is capable of producing
the
target molecule in a suitable form for the test to be performed including but
not
restricted to. eg. yeast cells and higher eukaryotic cells such as eg. insect
or
mammalian cells. Transformation of the cell line of choice may be performed by
standard techniques routinely employed in the field as described eg. in Wigler
et al.
Cell (1978) 14: 725 and in accordance with standard techniques (Sambrook et
al.
Molecular Cloning: A Laboratory Manual, 2. ed. Cold Spring Harbor Laboratory,
Cold Spring Harbor, New York, 1989). In a particularly preferred embodiment
the
biological target molecule being a membrane protein is expressed and tested in
mammalian cells usually within the membrane and usually in whole cells or in
isolated membrane preparations, which is dealt with and described further in
the
examples presented in "EXPERIMENTAL". Examples of suitable mammalian cell
lines are the COS (ATCC CRL 1650 and 1651), BHK (ATCC CRL 1632, ATCC
CCL 10), CHL (ATCC CCL39), CHO (ATCC CCL 61), HEK293 (ATCC CRL
1573) and NIH/3T3 (ATCC CRL 1658) cell lines.
Isolation and purification - In the case where the biological target molecules
is
a soluble protein, for example an enzyme, a preferred source may be
recombinantly
produced protein, which subsequently is isolated and purified to a suitable
purity
and in a form suited for functional testing by various standard protein
chemistry
methods well known to those skilled in the art.
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
21
Functional testing of the biolo ical target molecules
As part of the drug discovery process of the current invention, the biological
target molecule comprising a natural or an engineered metal-ion binding site
is
contacted with a test compound for example consisting of a metal-ion in
complex
with a metal-ion chelator and any change in the biological activity of the
biological
target molecule is detected or the binding affinity of the test compound is
determined.
Due to the diversity of biological target molecules, a wide variety of
functional
test can be performed depending on the individual target molecule and its
functions.
For example, for a soluble enzyme a suitable enzymatic analysis could be used
on
the purified enzyme (as described fox Factor VIIa in the examples). For
certain
transcription factors a suitable gene-expression reporter assay could, for
example be
performed in a whole cell preparation. In a preferred embodiment of the
invention
the biological target molecule is a membrane protein and the effect of test
compounds is monitored on the signal transduction process of the receptor,
i.e. its
ability to influence intracellular levels of for example cAMP, inositol
phosphates,
calcium mobilization etc. in response to the natural ligand (as described in
"EXPERIMENTAL"). For instance, in the case of a 7TM receptor, this may entail
the
effect on signalling mediated through the intracellular G-protein. In this
way, the
testing may reveal whether the binding of a metal-ion (complex) may affect the
activity of the target in for instance an antagonistic or an agonistic
fashion. For the
most part tests are performed as dose-response analysis in which a range of
concentration of metal-ion chelator complexes axe exposed to the biological
target
molecule.
When appropriate, the binding affinity of the test compound to the biological
target molecule is determined, for example in competition binding experiments
against a suitable radioactively labelled ligand for the protein target (as
described in
"EXPERIMENTAL"). Or, the affinity of the test compound can in some cases be
determined by use of a chelating agent which is in itself is detectable or
which can
be labelled with a detectable labelling agent.
STRUCTURAL TESTING OF THE BIOLOGICAL TARGET MOLECULES
In a preferred embodiment of the invention, the 3D structure of the test
compound in complex with the biological target molecule is determined, for
example by techniques such as X-ray analysis of crystals of the ligand-protein
complex or, for example by nuclear magnetic resonance (NMR) spectroscopic
analysis of complexes in solution - all known to those skilled in the art. In
this way
the amino acid residues located in the vicinity of the metal-ion site and the
chemical
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
22
interaction of the bifunctional test compound with specific residues in the
biological
target molecule can be determined as control and as basis for the structure-
based
design of further modifications of the Lead test compound and design of new
libraries of compounds. Further, the effect of the test compound on the
structure of
the biological target protein, domains of this and or effect on the
interaction of the
target protein with other proteins can be determined.
IDENTIFICATION OF METAL-ION SITES IN BIOLOGICAL TARGET MOLECULES
In a preferred embodiment of the present invention, naturally occurring metal-
ion sites axe used as initial attachment sites for metal-ion chelating test
compounds
in the drug discovery process. In general, such natural metal-ion sites can be
identified functionally by studying the effect of either free metal-ions or by
the
effect of a library of metal-ion chelator complexes on any function of the
biological
target molecule. Metal-ion sites can also be identified or confirmed by
structural
means as described above and location of the site can also be identified by
careful,
controlled mutagenesis, i.e. exchanging of the residues involved in metal-ion
binding with residues not having this property. Natural metal-ion sites are
interesting drug targets since binding of a drug at or close by a natural
metal-ion site
often will act as an allosteric agent, i.e. affecting the structure and
fixnction of the
biological target molecule at a site different from the usual active site,
where most
ligands will bind and act (see below).
Natural metal-ion sites ire p~oteihs i~ general - Metal-ion sites are known to
occur in many biological target molecules including but not restricted to, for
example proteins, glycoproteins, RNA, etc. These sites can serve either
structural or
functional purposes. Some metal-ion sites are known to occur solely from
functional
data, for example Zn(II)-sites in ligand gated ion channels. Or previously
unknown
metal-ion sites axe discovered in the crystal structure of the protein, as for
example
Zn(II) sites in rhodopsin. Independent on the physiological purpose of the
naturally
occurring metal-ion site they may be targeted by the technology of the present
invention, where they are addressed not only by a metal-ion, but by a metal-
ion in
complex with a metal-ion chelator, which can affect the protein structure and
function differently than the free metal-ion.
Natural metal iorc sites in 7TM~ecepto~s - naturally occurring metal ion sites
have been described in two 7TM receptors: the tachykinin NK3 receptor
(Rosenkilde et al. (1998) FEBS Lett. 439: 35-40) and the galanin receptor
(Kask et
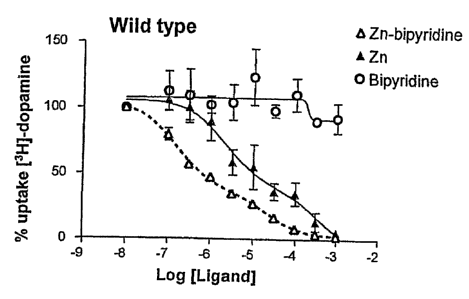
al. (1996) EMBO J. 15: 236-240). In the NK3 receptor Zn(II) was shown to act
as
an enhancer (positive modulator) for agonist binding and action without itself
being
an agonist. Through mutagenesis the metal ion binding site was mapped to
residues
V:01 and V:OS at the extra-cellular end of TM-V. In the galanin receptor
Zn(II) was
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
23
shown to act as an antagonist for galanin binding, but the site was not
characterized
further (see "EXPERIMENTALS"). However, based on knowledge from metal-ion site
engineering in 7TM receptors in general (see below) it is possible based on
sequence analysis and molecular models to find previously unnoticed and often
physiologically silent metal-ion sites in 7TM receptors. Some of these sites,
for
example the known one in the NK3 receptor, may be affected physiologically by
free metal ions, for example when a receptor is expressed in brain regions
where
extra-cellular zinc concentrations may vary around 10-5 molar. However, many
of
the previously unnoticed metal ion sites may just be a reflection of the fact
that
polar, metal-ion binding amino acid residues (for example: His, Cys, Asp etc.)
frequently are found in the water-exposed main ligand-binding crevice of 7TM
receptors. In one embodiment of the present invention, these residues are used
as
initial attachment sites for metal ion chelating test compounds, i.e. lead
compounds
in the drug discovery process (see for example the LTB4 receptor in
"EXPERIMENTAL"~.
ENGINEERING OF METAL-ION SITES IN BIOLOGICAL TARGET MOLECULES
It is generally known that metal-ion sites can be built into proteins by
introduction of metal-ion chelating residues at appropriate sites. In a
particularly
preferred embodiment of the invention such sites are constructed at strategic
sites in
the biological target molecule with the purpose to serve as anchor sites for
test
compounds in a drug discovery process and thereby target the medicinal
chemistry
part of the process towards particularly interesting epitopes on the target
molecule.
Mutagercesis - the nucleotide sequence encoding the target protein of interest
may be subjected to site-directed mutagenesis in order to introduce the amino
acid
residue, which includes the metal-ion bindning site. Site-directed mutagenesis
may
be performed according to well-known techniques. Eg. as desribed in Ho et al.
Gene
(1989) 77: 51-59. In a specific, non-limiting example the mutation is
introduced into
the coding sequence of the target molecule by the use of a set of overlapping
oligonucleotide primer both of which encode the mutation of choice and through
polymerisation using a high-fidelity DNA polymerise such as eg. Pfu Polymerise
(Stratagene) according to manufacturers specifications. The presence of the
site-
directed mutation event is subsequently confirmed through DNA sequence
analysis
throughout the genetic segment generated by PCR. In order to generate a metal-
ion
binding site this may involve the introduction of one or more amino acid
residues
capable of binding metal-ions including but not restricted to, for example
His, Asp,
Cys or Glu residues.
Generally the mutated target molecule will initially be tested with respect to
the ability to still constitute a functional, although altered, molecule
through the use
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
24
of an activity assay suitable in the specific case. It should be noted that
although
mutations in proteins may obviously occasionally alter the structure and
affect the
function of the protein, this is by far always the case. For example, only a
very small
fraction (less than ten) of the many hundred Cys mutations performed in
rhodopsin
as the basis for site directed spin-labelling experiments and in for example
the
dopamine and other 7TM receptors as the basis for Cys accessibility scanning
experiments have impaired the function of these molecules. Similarly, in the
bacterial transport protein Lac-permease almost all residues have been mutated
and
only a few of these substitutions directly affect the function of the protein.
Mutations will often also be performed in the biological target molecule to
confirm
or probe for the chemical interaction of test compounds with other residues in
the
vicinity of the natural or the engineered metal-ion site as an often
integrated part of
the general drug discovery method of the invention.
Metal-ion site engineering in protein targets in general - The method of the
invention may suitably include a step of determining the location of, fox
example the
metal ion binding amino acid residues) in a mutated protein and determining
the
location of at least one other amino acid residue in the vicinity of the metal
ion
binding amino acid residue, based on either the actual three-dimensional
structure of
the specific biological target molecule in question (e.g. by conventional X-
ray
crystallographic or NMR methods) or based on molecular models based on the
primary structure of the specific molecule together with the three-dimensional
structure of the class of molecules to which the specific molecule belongs
(e.g.
established by sequence homology searches in DNA or amino acid sequence
databases).
In the biological target molecule, the metal-ion binding site may suitably be
introduced to serve as an anchoring, primary binding site for the test
compound,
which can thereby be targeted to affect a site in the biological target
molecule
having one or more of the following properties (the metal-ion site may be
placed
either within or close to this site):
a site where the biological target molecule binds to another biological target
molecule, for example a regulatory protein.
a site which will control the activity of the biological target molecule in a
positive or negative fashion (i.e. up-regulating or down regulating the
activity of the
biological target molecule), for example by an allosteric mechanism.
a site where the binding of the test compound will directly or indirectly
interfere with the binding of the substrate or natural ligand or the binding
of an
allosteric modulatory factor for the biological target molecule.
SUBSTITUTE SHEET (RULE 26)
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
a site where the binding of the test compound may interfere with the intra-
molecular interaction of domains within the biological target molecule, for
example
the interaction of a regulatory domain with a catalytic domain.
a site where binding of the test compound will interfere with the folding of
the
5 biological target molecule, for example the folding of a protein into its
active
conformation.
a site where the binding of the test compound will interfere with the cellular
targeting of the biological target molecule.
a site where the binding of the test compound will stabilise a conformation of
10 the biological target molecule, which presents an epitope normally involved
in
protein-protein interactions in a non-functional form.
This list of properties is by no means exhaustive and only serves to give some
examples of the possibilities which can be obtained by targeting the test
compound
and thereby the final drug candidate to specific epitopes in the biological
target
15 molecule through the drug discovery process of the present invention.
This will potentially provide the ligand with other pharmacological properties
than agents normally acting at the active site. It is for example likely that
compounds binding at allosteric sites will be more efficacious in interfering
with for
example protein-protein interactions, which notoriously have been diff cult as
drug
20 targets. Allosteric agents will, for example have the possibility of
stabilising a
conformation of the biological target molecule where major parts of the
protein-
protein interface are vastly different from the one enabling the normal
interaction.
Metal-ion site et~ginee~ivcg in 7TMp~oteins - in a preferred embodiment of the
invention metalsites are introduced in 7TM receptors as part of the drug
discovery
25 process. Much experience has been obtained in building artificial metal-ion
sites in
7TM receptors in general (Elling et al. Nature (1995) 374: 74-7; Elling et al.
EMBO
J. (1996) 15: 6213-9; Elling et al. Fold Des. (1997) 2: S76-80; Elling et al.,
Proc Natl
Acad Sci USA (1999) 96:12322-7; Sheikh et al. Nature. (1996) 383: 347-50).
Based
on this protein engineering work and on mutational analysis of ligand-binding
sites
as such at multiple locations in a number of wild-type 7TM receptors
(Schwartz,
T.W. (1994) Curr. Opin. Biotech. 5: 434-444, Schwartz et al. Curr. Pharmaceut.
Design (1995) 1: 325-342). However, in the present context such metal-ion
sites are
introduced in 7TM receptors as anchor points for lead compounds with the
purpose
of improving these compounds for high affinity binding and particular
pharmacological profiles depending on their molecular interactions with the
target
molecule. The introduction of the sites is helped by molecular models of the
7TM
receptors established on the basis of, e.g., X-ray crystallographic data of a
membrane protein of the same family, electron density maps of the membrane
protein generated by cryo-electronmicroscopic analysis of two-dimensional
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
26
membrane crystals (Baldwin, EMBO J. 12(4), 1993, pp. 1693-1703; Baldwin, Curr.
Opinion. Cell. Biol. 6, 1997, pp. 180-190; Herzyk et al. J. Mol. Biol. 281(4),
1998,
pp. 741-754).
SELECTION OF LEAD COMPOUND FROM LIBRARY OF METAL ION
CHELATING COMPOUNDS
TEST COMPOUNDS
Test compounds which have been found suitable for use in the present
methods are any compound which is capable of forming a complex with a metal
ion.
All of the groups of a test compound which is attached directly to the metal
atom or
metal ion (central metal or coordinated metal) - whether ions or molecules -
are the
coordinating groups or ligands. A ligand attached directly through only one
coordinating atom (or using only one coordination site on the metal) is called
a
monodentate ligand. A ligand that may be attached through more than one atom
is
multidentate, the number of actual coordinating sites being indicated by the
terms
bidentate, tridentate, tetradentate and so forth. Multidentate ligands
attached to a
central metal by more than one coordinating atom are called chelating ligands.
A
test compound for use in the present context is at least bidentate, i.e. it is
a so-called
metal ion chelator.
In the present context useful metal ion chelators generally have a log K value
in a range of from about 3 to about 18 such as, e.g. from about 3 to about 15,
from
about 3 to about 12, from about 4 to about 10, from about 4 to about 8, from
about
4.5 to about 7, from about 5 to about 6.5 such as from about 5.5 to about 6.5.
K is an
individual complex constant (also denoted equilibrium or stability constant).
The
constant's subscript 1, 2, 3 etc. indicates which coordination step the
constant is
valid for, i.e. K1 is the complex constant for the coordination of the first
ligand, K2
is for the second ligand and so forth. log K can be determined as described in
W.A.E. McBryde, "A Critical Review of Equilibrium Data for Protons and Metal
Complexes of 1,10-Phenanthroline, 2,2'-bipyridyl and related Compounds."
Pergamon Press, Oxford, 1978.
In general, metal ion chelators can form complexes with different metal ions.
In such cases it suffices for the purpose of the present invention that only
one of the
log K values for a given metal ion chelator is within the ranges specified
above.
Metal atoms or ions of particular relevance are: Co, Cu, Ni, Pt and Zn
including the
various oxidation steps such as, e.g., Co (II), Co (III), Cu (I), Cu (II), Ni
(II), Ni
(III), Pt (II), Pt (IV) and Zn (II).
More specifically, a test compound for use in a method according to the
invention has at Ieast two heteroatoms, similar or different, selected from
the group
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
consisting of nitrogen (N), oxygen (O),sulfur (S), selenium (Se) and
phosphorous
(p),
Test compounds which have been found to be useful in the present methods
are typically compounds comprising a heteroalkyl, heteroalkenyl, heteroalkynyl
moiety or a heterocyclyl moiety for chelating the metal ion. The term
"heteroalkyl"
is understood to indicate a branched or straight-chain chemical entity of 1-15
carbon
atoms containing at least one heteroatom. The term "heteroalkenyl" is intended
to
indicate a branched or straight-chain chemical entity of 2-15 carbon atoms
containing at least one double bond and at least one heteroatom. The term
"heteroalkynyl" is intended to indicate a branched or straight-chain chemical
entity
of 2-15 carbon atoms containing at least one triple bond and at least one
heteroatom.
The term "heterocyclyl" is intended to indicate a cyclic unsaturated
(heteroalkenyl),
aromatic ("heteroaryl") or saturated ("heterocycloalkyl") group comprising at
least
one heteroatom. Preferred "heterocyclyl" groups comprise 5- or 6-membered
rings
with 1-4 heteroatoms or fused 5- or 6-membered rings comprising 1-4
heteroatoms.
The heteroatom is typically N, O, S, Se or P, normally N, O or S. The
heteroatom is
either an integrated part of the cyclic, branched or straight-chain chemical
entity or
it may be present as a substituent on the chemical entity such as, e.g., a
thiophenol,
phenol, hydroxyl, thiol, amine, carboxy, etc. Examples of heteroaryl groups
are
indolyl, dihydroindolyl, furanyl, benzofuranyl, pyridinyl, pyrimidinyl,
quinolinyl,
triazolyl, imidazolyl, thiazolyl, tetrazolyl and benzimidazolyl. The
heterocycloalkyl
group generally includes 3-20 carbon atoms, and 1-4 heteroatoms.
Particularly useful test compounds are those having at least two heteroatoms
of
general formula I
R1 F ~X)vG-R2
i
~Y)m
wherein F is N, O, S, Se or P; and G is N, O, S, Se or P;
at least one of (X)n and (Y)m is present and if n is 0, then -(X)n - is
absent,
and if m is 0, then -(Y)m- is absent, and both n and m are not 0;
Rl and R2, which are the same or different, are radicals preferably selected
from the group consisting of: hydrogen, a C 1-C 15 alkyl, C~-C 15 alkenyl, C2-
C 15
alkynyl, aryl, cycloalkyl, alkoxy, ester, -OCOR', -COOR', heteroalkyl,
heteroalkenyl, heteroalkynyl, heterocycloalkyl, heterocycloalkenyl,
heterocycloalkynyl or heteroaryl group, an amine, imine, nitro, cyano,
hydroxyl,
alkoxy, ketone, aldelhyde, carboxylic acid, thiol, amide, sulfonate, sulfonic
acid,
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
28
sulfonamide, phosphonate, phosphonic acid group or a combination thereof,
optionally substituted with one or more substituents selected from the same
group as
R1 and/or a halogen such as F, Cl, Br or I;
R' is hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl,
alkynyl,
substituted alkynyl, aryl, substituted aryl, arylalkyl, substituted arylalkyl,
heteroalkyl, substituted heteroalkyl, heteroalkenyl, substituted
heteroalkenyl,
heteroalkynyl, heteroaryl, substituted heteroaryl, cycloalkyl, substituted
cycloalkyl,
cycloalkenyl, substituted cycloalkenyl, heterocycloalkyl, substituted
heterocycloalkyl, heterocycloalkenyl or substituted heterocycloalkenyl;
Rl and/or R2 optionally forming a fused ring together with any of F, (X)n or a
part of (X)n G, (Y)m or a part of (Y)m or R1 and R2 themselves forming a fused
ring;
X and Y are the same or different and have the same meaning as R' such as,
e.g., -CH2-, CH2-CHI-, -CH2-S-CH2-, -CH2-N-CH2-, -CH=CH-CH=CH-,
-(CH2)d- (Z)e-(V) f (W)g-(CH2)h-, -CH2-O-CH2-, wherein
each of Z and W are independently C, S, O, N, Se or P and
V is -CH- or -CH2-;
(X)n and/or (Y)m optionally being substituted with one or more substituents
selected from the same group as R1 and/or a halogen such as F, Cl, Br or I;
n is 0 or an integer of 1-5,
m is 0 or an integer of 1-5,
a and/or g are an integer of 1-3,
d, f and/or h are an integer of I-7.
As mentioned above m and n are not 0 at the same time. When m = 0, the
formula I is
~°~X~'l
R1-F G-R2
In the present context, the term "alkyl" is intended to indicate a branched or
straight-chain, saturated chemical group containing 1-15 such as, e.g. 1-10,
preferably 1-8, in particular 1-6 carbon atoms, such as methyl, ethyl, propyl,
isopropyl, butyl, sec. butyl, tart. butyl, pentyl, isopentyl, hexyl, isohexyl,
heptyl etc.
The term "alkenyl" is intended to indicate an unsaturated alkyl group having
one or more double bonds between two adjacent carbon atoms.
The term "alkynyl" is intended to indicate an unsaturated alkyl group having
one or more triple bonds between two adjacent carbon atoms.
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
29
The term "cycloalkyl" is intended to denote a cyclic, saturated alkyl group of
3-7 carbon atoms.
The term "cycloalkenyl" is intended to denote a cyclic, unsaturated alkyl
group of 3-7 carbon atoms having one or more double bonds between two adjacent
carbon atoms.
The term "aryl" is intended to denote an aromatic (unsaturated), typically 5-
or
6-membered, ring, which may be a single ring (e.g. phenyl) or fused with other
5- or
6-membered rings (e.g. naphthyl or anthracyl).
The term "alkoxy" is intended to indicate the group alkyl-O-.
The term "amino" is intended to indicate the group -NR'R" where R' and R",
which are the same or different, have the same meaning as R~ in Formula I. In
a
primary amine group, both R' and R" are hydrogen, whereas in a secondary amino
group, either but not both R' and R" is hydrogen. R' and R" may also be fused
to
form a ring.
The term "ester" is intended to indicate the group COO-R', where R' is as
indicated above except hydrogen, -OCOR", or a sulfonic acid ester or a
phosphonic
acid ester.
Examples of halogen include fluorine, chlorine, bromine and iodine.
In the formula I above it is contemplated that if the valency of the
heteroatoms
F and/or G is more than 2 then further Rl and/or R2 groups are present
adjacent to
the F and/or G groups.
For the purpose of the present invention, other particular useful test
compounds are those having the general formula II below
R3 R4
~~) rr-(~)m
R ~ Fi y-R2
II
In the above formula II F, G, R1 and R2 have the same meaning as above. R3
and R4 have the same meaning as Rl and/or R2, and A and B have independently
the same meaning as X and Y in formula I. n and m have the same meaning as in
formula I except that n and m may be 0 at the same time and then the basic
structure
is Rl-F-G-R2 and when n or m are 0, respectively, then the basic structures of
formula II axe
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
R3 R4
~A)n ~B)m
R~-F ~G-R2 R~-F ~ ~G-R2
In formulas II (A) and (B) above the radicals R3 and R4 may be situated
anywhere on A and B, respectively, or anywhere on (A)n and (B)m, respectively.
5 For repeating units of e.g. A (or B) the group R3 (or R4) may be
independently
chosen in each of the repeating units.
Examples of interesting structures contained in test compounds for use in
methods according to the present invention are given below.
The following formulas are based on the formula II above and F and/or G are
10 nitrogen (N) or oxygen (O). T and Q are heteroatoms, and q and s
independently are
0 or an integer of from 1 to 4. The meanings of q and s for q and/or s being 0
are the
same as in Formula II for n and m. As an example, if q is 0 in Formula IIIA
then the
heterocyclic ring containing N is present, but the ring system does not
contain any
T. A circle indicates a fused alkyl, alkenyl, aryl, heteroalkyl,
heteroalkenyl,
15 heteroalkynyl or heteroaryl ring having from 3-7 atoms in the ring. RS has
the same
meaning as Rl and/or R2. In Formulas III C-G, IV C and V C-D, T and/or Q may
be placed anywhere in the cyclic system. This means for example that when q is
1,
then one heteroatom T is present in the ring system and the position of the
heteroatom is in principle freely chosen (of course the heteroatom F is also
present,
20 i.e. a total of two heteroatoms in the ring, when q is 1).
In the formulas below, the structure of the compounds are given in different
structure levels. Firstly, a in very general form and then in more and more
specif c
forms. Furthermore, all Formulas III are based on the same F-G-structure. The
same
applies for Formulas IV, V, VI and VII, respectively.
Rs
Ra
/~N~~N_R2
III A
Rs
Z~~-~~~4
9
N N-RZ
3 0 III B
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
31
R4 4
(~ ~ RS(T)
\-N N-Rz 'N N-Rz
R6 5
u 6
(~ ~ Q) (T~ ~ (=~)S 6 5_(T~~~~)s
~=N N~ 5 ~~J R R 'N N J R
N
III C-G
Rs
Ra
(~ ( ~m
N N-Rz
5 IV A
Ra
R5
Ra,
(r)q
N~ Rz,
Rz
IV B
R4
R5
Ra
(T q~~
~N N~ Rz
R
IV C
R5
( q B 4
\N-Rz
OH
VA
R5
4
(T). "
[ '~~~N-Rz
'OH
V$
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
32
s a Rs Rs
(~ ~ 2 (T~ ~ J Q)P
N-R N
OH OH
VC VD
R3
Rz,
R~-Ni 'NvRz
VI A
R~
R~'N N Rz
(A)ri (g)m
R3 Ra
VII A
R~
NH HN
VII B
For the purpose of the present invention test compounds having a structure
based on Formula III are suitable for use. Such compounds may comprise a
heterocyclic moiety of the general formula VIII.
(P)~
(Z)~ '(W)p
-r)
~N N~
VIII
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
33
wherein R3, R4, Z, W and P are as defined herein before, a and/or b are an
integer of 1-7 and c is 0 or an integer of 1-7, and each of Q and T is
independently -
CH- or -CH2-, s is an integer of 1-7, and t is an integer of 1-7, are believed
to be
particularly suitable. When c is 0 in the above Formula VIII then -(P)~ is
absent,
i.e. there is no bond between (Z)a and (W)b.
Test compounds in which the heterocyclyl moiety has the general formula IX .
R3 (P)~ R4
(X)n
N N
IX
wherein R3, R4, P, X and n are as indicated above, and c is 0 or an integer of
1
3, are also believed to be useful fox the use in the present invention. When c
is 0
then -(P)~ is absent.
Other suitable test compounds are those in which the structure corresponds to
Formula VII. More specifically, the heterocyclyl moiety may have the general
formula X
([F(CHR)n]p-[G(CH2)m]r)u
X
wherein F is N,O or S and G is N,O or S,
n is an integer from 1 to 5,
m is 0 or an integer from 1 to 5,
p and/or r are 0 or an integer from 1 to 8,
a is an integer from 1 to 8, and
R has the same meaning as Rl in Formula I.
As an example of the meaning of p, r and/or a in the above formula the
following applies: When r is 0 in the above Formula X then the Formula is
([F(CHR)n]p)u
XI
SUBSTITUTE SHEET (RULE 26)
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
34
In analogy, the same meaning applies for p equal to 0, respectively, i.e. when
p
is 0 then the Formula is
(~G(CH2)m~r)u
XII
In all of the formulas given herein it is contemplated that when the valency
of
the heteroatoms F and/or G is more than 2 then, whenever relevant, further R1
and/or R2 groups are present adjacent to the F and/or G atoms.
Further useful test compounds are those in which the heterocyclic moiety is
selected from a compound of formula XIIIa, XIIIb or XIIIc.
R4 n
/ \ / \~ RAN N/R4.
N N N N
N N
XIIIa XIIIb XIIIc
wherein R3 and R4 are as indicated above in formula I.
In Formulas VII, VIII, IX, X and XI the groups R3 and R4 only indicate that
the rings) may be substituted with a group similar to R3 and/or R'~. R1 and R2
in
the meaning of formula I are included in the structures given above.
Furthermore, it
is understood that more than one R or substituent may be present whenever
relevant
and any R may also be substituted, cf. the meaning of e.g. R1 given under
Formula
I.
Examples of test compounds may be those in which the heterocyclic moiety is
selected from a compound shown in Table 1:
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
0
\ \ / ~ r ~
r r
o~
~a
/ \
\ / N /
/ 1 / \
'N N'
I~
/\
0
. ~ w _
r l ~ ~ i
r
r
~\ w
I r N
N I w
\ N~~
I
N~
Table I
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
36
In the following Table II is given further examples of useful test compounds.
The number given refers to an internal numbering system applied in the
experiments
performed.
OH - / \ Hz
\ i v / / \ '=N N-OH
\ ~ 2os
121 OH ~~ 132
_ ~ / \ H2~
_ \ ~ \ N N-O
/ \ ' H OH 210
-N / 133
\ / \'77' \/ 122 ~\ %
OH
H H 151
~O ~ O /
\
O \ N ' / N 134
/ \ N ~ / \ 124 I iN NHZ
79 184
/ \
\
N
C13C 135 CCI~
\ /~~+ / u
/ \ ~N~ - H f~
OH
O \ / 125 \ ' / \ 198
84 ..
OH H 172 ~
\ ~ / I II
OH f~
O 160 205
/ \ ~ N / \ \ 7 / \
86 N 176 H ~O
/ \' N 1O~'
180 / \ 201
/ \ ~ ~ \ / HZ H H
89 - - / \
\ H \ / \ N / \ HS SH
137
186 183
118
\ ~ ~ / HZN NHZ
185 166
ONa N 154 N ~ p~0
O _ _ ~N'H H~N
COOH CNH N' I % C
/ \ N119 / I t~ COOH Jl N
149 59 128
Table II
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
37
Metal atoms or ions forming the complex with the heteroalkyl or heterocyclyl
moiety in the test compounds may advantageously be selected from metal atoms
or
ions which have been tested for or are used for pharmaceutical purposes.
Such metal atoms or ions belongs to the groups denoted light metals,
transition
metals, posttransition metals or semi-metals (according to the periodic
system).
Thus the metal ion is selected from the group consisting of aluminium,
antimony, arsenic, astatine, barium, beryllium, bismuth, boron, cadmium,
calcium,
cerium, cesium, chromium, cobalt, copper, dysprosium, erbium, europium,
gadolinium, gallium, germanium, gold, hafnium, holmium, indium, iridium, iron,
lanthanum, lead, lutetium, magnesium, manganese, mercury, molybdenum,
neodymium, nickel, niobium, osmium, palladium, platinum, polonium,
praseodymium, promethium, rhenium, rhodium, rubidium, ruthenium, samarium,
scandium, selenium, silicon, silver, strontium, tantalum, technetium,
tellurium,
terbium, thallium, thorium, thulium, tin, titanium, tungsten, vanadium,
ytterbium,
yttrium, zinc, zirconium, and oxidation states and isotopes thereof; in
particular
aluminium, antimony, barium, bismuth, calcium, chromium, cobalt, copper,
europium, gadolinium, gallium, germanium, gold, indium, iron, lutetium,
manganese, magnesium, nickel, osmium, palladium, platinum, rhenium, rhodium,
rubidium, ruthenium, samarium, silver, strontium, technetium, terbium,
thallium,
thorium, tin, yttrium, zinc, and oxidation states or isotopes thereof; in
particular
cobalt, copper, nickel, platinum, ruthenium, and zink, and oxidation states
and
isotopes thereof, preferably calcium (II), cobalt (II) and (III), copper (I)
and (II),
europium (III), iron (II) and (III), magnesium (II), manganese (II), nickel
(II) and
(III), palladium (II), platinum (II) and (V), ruthenium (II), (III), (IV),
(VI) and
(VIII), samarium (III), terbium (III), zinc (II),- or isotopes thereof,
preferably cobalt
(II) and (III), copper (I) and (II), nickel (II) and (III), zink (II) and
platinum (II) and
(V), or isotopes thereof.
For the present purpose, a particularly favourable test compound is a chelate
between any of the test compounds of the formulas mentioned above and any of
the
metal atoms or ions mentioned above. In particular chelates between any of the
test
compounds and any of atom or ion of Co, Cu, Ni, Zn, Rn and Pt are of interest
in
methods of the present invention. Especially, chelates like e.g. metal ion-
phenanthroline complex, metal ion-bipyridyl complex and metal ion-1,4,~,11-
tetraazacyclotetradecane complex are suitable for use in methods of the
present
invention such as, e. g., a Cu2+-phenanthroline complex, a Zn2+-phenanthroline
complex, a Cud+-bipyridyl complex, a Zn2+-bipyridyl complex, a Ca2+-bipyridyl
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
38
complex, aCu2~-1,4,8,11-tetraazacyclotetradecane, aZn2+-1,4,8,11-
tetraazacyclotetradecane.
LIBRARIES
The invention also relates to chemical libraries of test compounds and their
use
in drug discovery processes. More specifically, a chemical libery is claimed
comprising test compounds according to the above-mentioned formula I and
wherein the test compound is or is not in chelated form with any of the metal
ions
mentioned above. A chemical library of salt, solvates or complexes of the
above-
mentioned metal ions is also claimed. Besides the chemical structure, the test
compounds contained in the libraries must fulfil certain criteria with respect
to
molecular weight (at the most 2000 such as, e.g., at the most 1500, at the
most 1000,
at the most 750, at the most 500), lipophilicity (log P at the most 7 such as,
e.g., at
the most 6 or at the most 5), number of hydrogen bond donors (at the most 15
such
as, e.g. at the most 13, 12, 11, 10, 8, 7, 6 or at the host 5) and number of
hydrogen
bond acceptors (at the most 15 such as, e.g. at the most 13, 12, 11, 10, 8, 7,
6 or at
the most 5).
Libraries of test compounds or of salts, solvates or complexes of the above-
mentioned metal ions which find use herein will generally comprise at least 2
compounds, often at least about 25 different compounds such as, e.g., at least
about
100 different compounds, at least about 500 different compounds, at least
about
1000 different compounds or at least about 1000 different compounds. The
method
by which the population of compounds are prepared are not critical to the
invention
and a person spilled in the field of chemistry will be able to select suitable
synthetic
methods for the preparation of the compounds.
CHEMICAL OPTIMIZATION OF LEAD COMPOUND FOR SECONDARY
INTERACTION POINTS IN THE BIOLOGICAL TARGET MOLECULE
IDENTIFICATION OF CHEMICAL INTERACTIONS
The chemical optimization of the test compound can be guided by detailed
l~nowledge of the 3D structures) of the biological target molecule,
preferentially
determined in complex initially with the un-substituted metal-ion chelator and
3 S subsequently in complex with the chemically modified metal-ion chelator in
which
attempts have been made to establish first one secondary interaction and
subsequently further secondary or tertiary interactions. For some biological
target
molecules such as soluble proteins this can be achieved through for example
SUBSTITUTE SHEET (RULE 26)
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
39
crystallization and standard X-ray analysis procedures or through, for example
NMR analysis of the complex in solution again using standard procedures.
For membrane proteins high resolution structures are in general not available.
However determination of chemical interactions may be performed using a
generic
three-dimensional model of the membrane protein showing the spatial
arrangement
of the amino acid residues defining the area of the metal ion binding site.
Such a
determination is then performed using site-directed mutagenesis of a least one
amino acid residue potentially involved in interaction with said functional
group of
the test compound other than the metal ion. Followed by expression of the
mutated
membrane protein in a suitable cell, contacting said cell or a portion thereof
including the mutated membrane protein with the test compound, and determining
any effect on binding in a competitive binding assay using a labelled ligand
of the
membrane protein, detection of any changes in signal transduction from the
membrane protein or using a chelating agent which is in itself detectable or
labelled
1 S with a detectable labelling agent. If an amino acid residue involved in
interaction
with such a functional group of the test compound is mutated to one, which is
not -
this may be detected as a decrease in binding or other activity
GENERATION OF NEW SPECIFIC INTERACTIONS
During the chemical optimisation of the test compound methods developed for
structure-based drug discovery in general can be utilized, as knowledge of the
3D
structure of the target epitopes makes it possible to apply classical
structure-based
approaches such as structure-based library design for the establishment of
secondary
and tertiary interaction sites for the lead compound in the target molecule.
However,
it should be noted, that a major advantage and difference of the present
method is,
that the lead compound is anchored to a particular site and thereby to a
certain
degree in a particular conformation in the biological target molecule through
binding to the bridging metal-ion site while the compound is being optimized
for
chemical recognition with the target molecule.
In the case of membrane proteins suitable X-ray structures are generally not
available. However, the molecular models are often rather detailed and in the
case
of the 7TM receptors they are in fact rather precise and correspond well with
the X-
ray structure of rhodopsin which was recently published. Thus the combination
of
relatively good molecular models (which have allowed for the construction of
interhelical metal-ion sites) and the present method does to a certain degree
compensate for the lack of detailed knowledge of the 3D structure of the
target
molecule because the Iead compound is anchored and thereby create a fix-point
for
the subsequent medicinal chemical optimization point guided by the molecular
models.
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
By using relatively flexible spacers in between,the metal-ion chelating moiety
and the variable chemical moiety of the test compound it becomes possible to
probe
for interaction or binding to structurally and functionally interesting
epitopes of the
biological target molecule with chemical moieties, which due to their
intrinsic low
5 affinity would normally not be detectable in the analytical systems on their
own.
Due to the local high concentration of the chemical moieties, which is created
by the
tethering to the metal-ion chelating moiety bound to the metal-ion site, these
compounds can now be detected.
1 O USE OF TEST COMPOUNDS IN VIVO TARGET VALIDATION
In an embodiment of the invention the method will be used to increase the
affinity and specificity of metal-ion chelator compounds to be used in
pharmacological knock-out applications for in vivo target validation; i.e. to
determine the effect of a specific agonist or antagonist for a biological
target
15 molecule. Here, the compounds will be used as metal-ion chelator complexes.
This
procedure has in principle been described previously (Elling et al. (1999)
Proc.Natl.Acad.Sci.USA 96:12322-12327); however only for basic metal-ion
chelating agents. The technology is based on the introduction of a silent
metal-ion
site in a potential drug target, i.e. creation of a metal-ion site in which
the mutations
20 do not affect the binding and action of the endogenous ligand for the
receptor. When
such a metal-ion site engineered receptor is introduced into an animal by
classical
gene-replacement technology, i.e. exchange of the endogenous receptor with the
metal-ion site engineered receptor, then the animals will develop normally
without
any compensatory mechanisms, which otherwise frequently impair the
25 interpretations of the phenotypes of the animals in classical gene knock-
out
technology. In the adult animals or whenever it is found appropriate the
animals are
then treated with an appropriate metal-ion-chelating agent which then will act
as an
antagonist (or agonist) and turn off (or on) the function of the metal-ion
site
engineered receptor. Currently, this approach is impaired by the fact, that
the
30 generally available metal-ion chelating agents only will bind with at best
~,M
affinity to the metal-ion site engineered biological target molecule, which
will give
similar ~,M or lower antagonistic potencies. These relatively low potencies
and the
relative low specificity of the basic test compounds impairs the general
applicability
of the technology due to simple pharmacolcinetic and toxicology problems. With
the
35 technology presented in the present invention above it will be possible to
increase
the affinity of metal-ion chelators significantly, which will make it
considerably
more easy to reach therapeutic, efficient antagonistic concentrations of the
metal-ion
chelator in the animals and also to increase the "therapeutic window" due to
the
higher degree of selectivity of the compounds caused by the establislnnent of
more
SUBSTITUTE SHEET (RULE 26)
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
41
than one molecular interaction point. Establishment of just a single suitable
charge-
charge interaction will increase the affinity of the metal-ion chelator by 10
to 100-
fold or more.
CHEMICAL OPTIMIZATION OF LEAD COMPOUND FOR HIGH AFFINITY
INTERACTION WITH WILD TYPE BIOLOGICAL TARGET MOLECULE
EXCHANGE OF METAL ION ANCHOR WITH "ORDINARY" CHEMICAL INTERACTION
In the case, where the initial binding of the metal-ion chelator was obtained
through mutational introduction of an anchoring metal-ion site in the
biological
target molecule, a final step of optimization will have to be performed to
obtain high
affinity binding or potency on the wild-type target molecule without the metal-
ion
bridge. Through the methods described in the previous experiments, the metal-
ion
chelator lead compound will gradually be optimized for interactions with
chemical
groups in the biological target molecule spatially surrounding the metal-ion
site - i.e.
interactions with chemical groups found also in the wild-type target molecule.
Thus,
the test compound will gradually increase its affinity not only for the metal-
ion site
engineered molecule but also for the wild-type biological target molecule.
When
two to three secondary interaction points have been established, the affinity
of the
test compound for the wild-type target molecule, which is being tested in
parallel
with the metal-ion site engineered molecule, will have reached micro-molar
affinities, i.e. a lead compound on the wild-type target molecule has been
created.
At this point one or more of the following three approaches will be followed:
1)
structure-based further chemical optimization of the compound in general
aiming at
improving recognition at various known chemical moieties of the target
molecule;
2) structure-based further chemical optimization of the compound at which the
"metal-ion site bridge" is exchanged by a more classical type of chemical
interaction
with the residues) which had been modified to create the metal-ion site in the
biological target molecule. Here advantage can be taken of the fact that the
geometry of the metal-ion site anchor is well known in general and, that
relatively
limited structure-based libraries can be established to create a new type of
interaction; 3) further chemical optimization of the compound through.more-or-
less
random generation of chemical diversity in general in the compound.
3 S APPLICATIONS
The small organic molecular ligands (compounds) identified according to the
methods of the present invention will find use as e.g. drug compounds with
abortifacient, acromegalic, alcohol deterrent, amebicidic, anabolic,
analeptic,
analgesic, anesthetic, antiacne, antiallergic, ophthalmic, anti-Alzheimer's
disease,
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
42
antianginal, antiarrhythmic, antiarthritic, antiasthmatic, antibacterial,
antibiotic,
anticancer, anticholelithogenic, anticoagulant, anticonvulsant,
antidepressant,
antidiabetic, antidiarrheal, antiemetic, antiepileptic, antiestrogen,
antifungal,
antiglaucoma, antihistamine, antihypertensive, antiinflammatory,
antilipidemic,
antimalarial, antimigraine, antinauseant, antineoplastic, antiobesity,
antiparasitic,
antiparkinsonian, antiperistaltic, antiprogestogen, antiprolactin,
antiprostatic
hypertrophy, antipsoriatic, antipsychotic, antirheumatic, antisecretory,
antiseptic,
antispasmodic, antithrombotic, antitussive, antiulcer, antiviral, anxiolytic,
bronchodilator, calcium regulator, cardioprotective, cardiostimulant,
cardiotonic,
cephalosporin, cerebral vasodilator, chelator, choleretic chrysotherapeutic,
cognition
enhancer, congestive heart failure, coronary vasodilator, cystic fibrosis,
cytoprotective, dependence treatment, diuretic, dyslipidemia, enzyme,
expectorant,
fertility enhancer, fibrinolytic, gastroprokinetic, Gaucher's disease, growth
hormone, growth hormone insensitivity, haemophilia, heart failure,
hematologic,
hematopoetic, hemostatic, hepatroprotective, hormone, hyperphenylalaninemia,
hyperprolactinemia, hypertensive, hypnotic, hypoammonnuric, hypocalciuric,
hypocholesterolemic, hypoglycemia, hypolipaemic, hypolipidemic, idiopathic
hypersomnia, immunomodulator, immunostimulant, immunosuppressant, beta-
lactamase inhibitor, leukopenia, lung surfactant, mucolytic, muscle relaxant,
multiple sclerosis, muscle relaxant, narcotic antagonist, nasal decongestant,
neuroleptic, neuromuscular blocker, neuroprotective blocker, neuroprotective,
nootropic, non-steroid antiinflammatory disease disease (NSAID), osteoporosis,
Paget's disease, platelet aggregation inhibitor, platelet antiaggregant,
pneumonia,
precocious puberty progestogen, protease inhibitor, psychostimulant, 5-alpha-
reductase inhibitor, respiratory surfactant, subarachnoid hemorrhage,
thrombolytic,
ulcerative colitis, urolithiasis, urologic, vasoprotective, vulnerary and
wound
healing properties. Important proteins for the present purpose are proteins,
which
may be stabilised in an active or inactive conformation by a biologically
active
substance. In this way, it may be possible to obtain an effect of a test
compound of
the type described herein irrespective of whether the active site of the
protein is
known, or whether the structure of the active site has been resolved (e.g. by
X-ray
crystallisation). Examples of such proteins are enzymes, receptors, hormones
and
other signalling molecules, transcriptional factors and regulators, intra- or
extracellulax structural proteins, in particular actins; adaptins; antibodies;
ATPases;
cyclins; dehydrogenases; GTP-binding proteins; GTP/GDP-exchange factors;
GTPase activating proteins; GTP/GDP dissociation inhibitors; chaperones;
histones;
histone acetyltransferases & deacetyltransferases; hormones and other
signalling
proteins and peptides; kinases; lipases; major facilitator superfamily
proteins;
motorproteins; nucleases; polymerases; isomerases; proteases; protease
inhibitors;
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
43
phosphatases; ubiquitin-system proteins; membrane proteins including
receptors,
transporters and channels; transcription factors and tubulins; preferably
membrane
receptors; nuclear receptors, zinc finger proteins; proteases, tyrosine
kinases and
matrix proteins. Other important proteins for the present purpose are proteins
whose
biological activity is regulated by their cellular targeting and whose
biological
activity therefore can be modulated by drugs, which alter their cellular
targeting
with or without altering their actual intrinsic activity.
The invention is further illustrated in the following non-limiting examples.
LEGEND TO FIGURES
Fig L1
Identification of naturally occurring metal-ion binding site in the 7TM
leukotriene LT$4 receptor
Whole cell competition binding experiment with COS-7 cells expressing the
wild type and mutant variants of the leukotriene LTB4 receptor using [3H]-LTB4
as
the radioligand.
Panel A. Affinity of Cu(II), 2,2'-bipyridine and the complex therof in the
wild
type LTB4 receptor.
Panel B. Affnity of Cu(biprydine) in mutant forms of the LTB4 receptor in
which the metal-ion binding is severely imparired.
Panel C.Helical wheel diagram illustrating the transmembrane segments of the
LTB4 receptor. The two cysteine residues within the transmembrane segment III
which have been identif ed as critical for metal-ion chelator complex binding,
Cys93 and Cys97 are indicated in dark gray.
Fig L2
Identification of naturally occurring metal-ion binding site in the 7TM
galanin
receptor
Whole cell competition binding experiment with COS-7 cells expressing the
wild type and mutant forms of the galanin receptor using [1251]-galanin as
radioligand.
Panel A. Affinity of the free copper metal-ion, the free chelator and the
phenanthroline complex on the wild-type galanin receptor.
Panel B. Affinity of the copper-phenanthroline copmplex on two mutant forms
of the galanin receptor, in which the binding of the metal-ion complex is
impaired.
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
44
Fig L3
Identification of naturally occurring metal-ion binding site in the 12TM
protein, the dopamine transporter.
Competition analysis of uptake of [3H]-dopamine in whole COS-7 cells
expressing the dopamine transporter.
Panel A. Uptake of [3H]-dopamine by the wild-type dopamine transporter in
the presence of free metal zinc-ion and zinc in complex with the chelator 2,2'-
bipyridine.
Panel B. Dopamine uptake analysis in a mutant form of the dopamine
transporter, [H193K], in which binding of the metal-ion complex has been
eliminated (Noregaard et al. EMBO J. (1998) I7: 4266-4273).
Panel C. Effect of metal-ion complex formation on the ability to inhibit [3H]-
dopamine uptake in the wild-type and [H193K] mutant dopamine transporter. (For
compounds, 209 and 210, see list of compounds in Appendix).
Fig IL1
Binding of various metal-ion complexes to a library of inter-helical metal-ion
sites engineered into the tachykinin NKl receptor.
COS-7 cells expressing various engineered forms of the NK1 receptor were
analyzed by competition binding using [1251]-Substance P as radioligand.
Panel A. IC50 values for the zinc and copper metal-ions and complexes
thereof with the chelators, 2,2'-bipyridine and phenanthroline are presented
in the
table. N indicated the number of experiments performed.
Panel B. Data obtained using the chelator cyclam are presented for the NK1
mutant in which an inter-helical metal-ion site has been generated through the
introduction of the HisV:05;HisVI:24 exchanges.
Panel C. A helical diagram representing the four sets of inter-helical metal-
ion
sites which appear in Panel A are indicated.
Fig IL2
Re-engineering of a metal-ion chelator binding site in the 12TM dopamine
transporter.
Dopamine uptake was analysed in COS-7 cells expressing the wild type and
mutant forms of the dopamine transporter in competition with the metal-ion
chelator
complex, zinc(II)-2,2'-bipyridine. The two panels show two forms of re-
engineered
dopamine transporters in which the ability to bind the metal-ion chelator
complexes have been reconstituted following the elimination of the His193
interaction point.
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
Fig IIT.1
Structure-activity relationship of antagonist metal-ion complexes in the
galanin
and the leukotriene LTB4 receptors.
Panel A. Competition binding analysis in COS-7 cells expressing the galanin
5 receptor. Binding of [1251]_galanin was analysed in the presence of various
copper-
ion chelator complexes.
Panel B. Competition binding analysis in COS-7 cells expressing the LTB4
receptor. Binding of [3H]-LTB4 was analysed in the presence of various copper-
ion
chelator complexes.
10 For structures of the chelators employed in both panels, see Appendix.
Fig. ITL2
Structure-activity relationship of antagonist metal-ion complexes in the metal-
ion site engineered tachykinin NK1 receptor
15 Binding of [1251]-Substance P was analysed in COS-7 cells expressing NI~1
receptor which have been engineered to bind the zinc metal-ion. Ligand binding
is
presented in competiton with the zinc metal-ion, the zinc-1,10-phenanthroline
complex and with other zinc-chelator xomplexes as indicated. For structures of
the
chelators, see Appendix.
Fig. IIL3
Structure-activity relation ship of agonistic metal-ion complexes in the metal
ion Site Beta2-adrenergic receptor.
The effect of Cu(II) and copper-chelator complexes on stimulation of
accumulation of intracellular cAMP was analyzed in COS-7 cells expressing the
beta2-adrenoceptor.
Panel A. Washing experiment demonstrating the reversibility of the
stimulatory action of the metal-ion complexes.
Panel B. The effect of copper and complexes in the wild-type beta2-AR and in
engineered forms of the receptor.
Panel C. Dosis-response analysis of selected copper-chelator complexes on the
[F289C;N312C] beta2-AR.
Fig. TIL4
Structure-activity relationship of antagonistic metal-ion complexes in a
soluble
protein, the enzyme FVIIa.
A comparison of selected metal-ion complexes on the binding of [3H]-LTB4
and the inhibition of the enzymatic activity of the active form of Factor VII
(FVIIa)
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
46
in COS-7 cells expressing respectively the LTB4 receptor (Panel B) and the
FVIIa
(Panles A and C). For stucture of the chelators see the Appendix.
Fig. IILS
Structure-based optimization of metal-ion chelators for secondary interactions
in the CXCR4 receptor and other biological targets.
Helical wheel diagram for the CXCR4 receptor. The Asp 171 residue present in
the transmembrane segment IV, and which is considered a major attachment site
for
the binding of the cyclam chelator is shown in white on black. Positions which
in
combination are proposed to constitute putative metal-ion binding sites are
high-
lighted in pairs and in black on dark gray.
Fig. IV
Schematic depiction of the structure of rhodopsin-like 7TMs with one or two
conserved, key residues highlighted in each TM: Asn1:18; AspII:lO; CysIII:01
and ArgIII:26; TrpIV:lO; ProV:lS; ProVII:l7.
Fig. V
A table of test compounds wherein log K values are given.
EXAMPLES
The examples presented encompass naturally occurring as well as specifically
engineered metal-ion binding sites in a number of different proteins
representing
several different classes of membrane proteins: 7TM proteins (examples being
various G-protein coupled receptors), and 12TM proteins (example - the
dopamine
transporter) as well as an example comprising a soluble protein, Factor VIIa,
the
active form of the FVII protease.
The examples are chosen with the intent of illustrating the sequential and
rational process through which small organic compounds, the metal-ion
chelators,
may be identified as ligands and subsequently optimized with respect to the
affinity
by which they recognize the protein targets.
Overall, the examples serve to illustrate how the activity of potential drug
targets may be affected through interaction with small metal-ion chelators and
importantly how the present technology provides the opportunity to aim the
active
drug candidates towards functionally significant domains of the target.
Throughout
this section, 'the affinity' of the metal-ion chelator complexes refers to the
ability of
the complex to displace the binding of a radioligand and the potency of the
metal-
ion chelator complexes refers to the ability of the substances to activate or
inactivate
the drug targets.
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
47
I. Binding Of Metal-Ions and Metal-Ion Complexes To Various Drug
Targets With Natural Metal-Ion Sites
The examples compiled in this section illustrate how metal-ion binding sites
may be identified in the native forms of various drug targets, and how these
sites
may be addressed by metal-ions in complex with certain chelators, as observed
either through an effect on the binding affinity of a radioactive ligand or
through a
direct effect on activation or inactivation of the target.
Examble L 1- Identification of a naturally occurring metal-ion chelator
binding=site
in the 7TM leukotriene LTB4 receptor
The present example illustrates how the presence of a previously unnoticed,
naturally occurring metal-ion binding site within a transmembrane segment of a
7TM receptor may be predicted through analysis of the nucleotide sequence of
the
gene coding for the protein and how it can subsequently be experimentally
identified. Briefly, molecular models of 7TM receptors can be built based on
the
deduced amino acid sequence and identification of the seven transmembrane
segments (eg.Unger at al. (1997) Nature 389: 203-206). In these molecular
models,
illustrated in the helical wheel diagram shown in Fig. L 1B, potential metal-
ion sites
can be identified by the presence of metal-ion binding residues, for example
histidine, cysteine, or aspartate residues located in suitable relative
positions, for
example in an i and i + 4 arrangement (i.e. with three residues in between) on
a
helical face within the so-called main ligand-binding crevice of the receptor
between TM-II, III, IV, V, VI, and VII (Schwartz et al, (1996) Trends
Pharmacol.
Sci.17:213-216).
Methods - The leukotriene LTB4 receptor cDNA was cloned by PCR from a
leukocyte cDNA library, built into an eukaryotic expression vector and
introduced
into COS-7 cells by a standard calcium phosphate transfection method. One day
after transfection the cells were transferred and seeded in mufti-well plates
fox
assay. The number of cells plated per well was chosen so as to obtain 5 to 10%
binding of the radioligand added. Two days after transfection the cells were
assayed
for the presence of [3H]-LTB4 binding activity. Radioligand was bound in a
buffer
composed of 50 mM Tris-HCl (pH 7.4), 3 mM MgCl2, 0.1 % BSA, 100 mg/ml
Bacitracin and displaced in a dose dependent manner by unlabelled LTB4 ligand.
The assay was performed in duplicate for 3 hours at 4 °C, and stopped
by washing
twice in buffer. Cell associated, receptor bound radioligand was determined by
the
addition of lysis buffer (48% urea, 2% NP-40 in 3M acetic acid). The
concentration
of radioligand in the assay corresponds to a final concentration of 45 pM. The
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
48
metal-ion chelating complex, 2,2'-bipyridine was added in a two-fold molar
excess
in order to ensure that no free metal-ion was present.
Results and discussion - As shown in the helical wheel diagram of the
leukotriene LTB4 receptor (Fig. L 1 C), two Cys residues are located on the
face of
TM-III pointing inwards, i.e. towards the main ligand-binding pocket of the
receptor
(positions III:04, Cys93 and III:08, Cys97). Theoretically these residues
could
constitute a metal-ion binding site. The actual presence of a naturally
occurring
metal-ion binding site in the leukotriene LTB4 receptor is demonstrated by the
fact,
that binding of the radioligand, [3H]-LTB4 to the receptor expressed in COS-7
cells
could be displaced by Cu(II), IC50 = 70 ~.M (Fig. I. 1A). In agreement with
the fact
that the proposed metal-ion site is located in the main ligand-binding pocket
of the
receptor, i.e. with amble space towards the center of the receptor, the
complex
between the metal-ion and the chelator, 2,2'-bipyridine bound equally well as
the
free metal-ion, i.e. the 2,2'-bipyridine did neither impair nor improve the
binding
affinity (Fig. L 1A). As shown in Fig. L 1 B, Ala-substitution of Cys93
severely
impaired the effect of the metal-ion chelator complex on LTB4 binding. Ala-
substitution of Cys97 also clearly impaired the effect of the metal-ion
complex. The
combined substitution of both Cys residues totally eliminated the metal-ion
chelator
effect (Fig. L 1B) demonstrating that these two residues on the central face
of TM-III
are involved in the binding of the metal-ion chelator complex. Thus, the two
residues represent a naturally occurring intra-helical 'bis-Cys-site', which
can be
addressed with for example Cu(II) in complex with bipyridine..
Example L2 - Identification of naturally occurring metal-ion chelator bindin_
site
in the 7TM ~alanin receptor
Whereas the naturally occurring metal-ion site in the LTB4 receptor is located
within a transmembrane helix, the metal-ion site in the receptor for the
neuropeptide
galanin exemplifies the identification of an inter-helical metal ion site in a
7TM
receptor. Furthermore, this is an example in which the metal-ion chelator
positively
contributes to the affinity of the metal-ion.
Methods - The galanin receptor cDNA was introduced into COS-7 cells by the
standard calcium phosphate transfection method. The cells were transferred and
seeded in mufti-well plates for assay one day following the transfection and
the
number of cells plated per well was adjusted for each individual (wild type
and
mutant) construct aiming at the binding of 5 to 10% of the radioligand present
in the
assay. Two days post-transfection the cells were assayed for the presence of
[1251]-
Galanin binding activity. Radioligand was bound in buffer composed of 25 mM
Hepes (pH 7.4), 2.5 mM MgCl2, 100 mg/ml Bacitracin and displaced in a dose
dependent manner by unlabelled ligand. The assay was performed in triplicate
for 3
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
49
hours at 4 °C, and terminated by the addition of lysis buffer (48%
urea, 2% NP-40 in
3M acetic acid). The concentration of radioligand in the assay corresponds to
a final
concentration of 20 pM.
Results and discussion - Binding of [lasl]-galanin to the galanin Rl receptor
expressed in COS-7 cells is displaced by Cu(II) with an ICS° of 28 ~.M
and a Hill
coefficient of -4; whereas the I,10-phenanthroline alone has no effect on
[1251]-
galanin binding (Fig. L2A). 1,10'-Phenanthroline binds Cu(II) with very high
affinity, 7.6 x 10-1° M, and importantly, the Cu(II)-phenanthroline
complex inhibits
galanin binding with an affinity of 2 ~.M, i.e. 14-fold better than the free
metal-ion.
Mutational substitution identified residues CysBg (II:17) and Cys29o (VII:07)
as being
essential for the binding of the metal-ion complex (Fig. L2B).
These experiments demonstrate, that a naturally occurring inter-helical metal-
ion site in a 7TM receptor can be addressed by a metal-ion chelator complex
with
even higher affinity than by the free metal-ion.
Example L3 - Identification of naturally occurring metal-ion chelator bindin
site in
the I2TM dopamine trans~outer.
In the literature, a naturally occurring allosteric metal-ion binding site has
been
demonstrated in the dopamine transporter, a membrane protein having supposedly
12 transmembrane spanning segments, 12TM (Norregaard et al EMBO J. 17: 4266-
4273 (1998)). Here Zn(II) binds in a two-component fashion to a tridentate
metal-
ion site composed of residues His193, HiS375, and G1u396 and thereby blocks
dopamine transport. This effect of Zn(II) can be eliminated by mutational
exchange
of any of the three residues with a non-chelating residue.
Methods - The dopamine transporter cDNA was introduced into COS-7 by the
standard calcium phosphate transfection method. Two days post-transfection the
cells were assayed for [3H]-Dopamine uptake activity. The uptake assays was
performed in 251nM Hepes pH 7.4, 120 mM NaCI, 5 mM ICI, 1.2 mM CaCl2, 1.2
1nM MgS04, I mM ascorbic acid and 5 mM D-glucose and in the presence of
various concentrations of unlabelled dopamine as indicated in the figures. The
assay
was performed in triplicate at 37°C for 10 minutes, and terminated by
washing with
buffer twice and the addition of lysis buffer (48% urea, 2% NP-40 in 3M acetic
acid).
Results and discussion - As shown in Fig. L3A, 2,2'-bipyridine in complex
with Zn(II) inhibits the transport of [3H]dopamine by the dopamine
transporter,
transiently expressed in COS-7 cells, in a two component fashion, i.e. with
ICSo
values of 0.16 and 20 ~.M, corresponding to a slightly higher potency than the
free
metal-ion, which similarly acts in a two component fashion, i.e. with ICso
values of
2.2 and 338 ~.M,. Importantly, the chelator bipyridine had no effect on the
dopamine
SUBSTITUTE SHEET (RULE 26)
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
transport without being on complex with the metal-ion (Fig. L3A). That the
metal-
ion chelator complex acts through the same site as the free metal-ion was
demonstrated by the mutational exchange of residue His193 (Fig. L3B). Dopamine
transport could be inhibited also by a structurally distinct class of metal-
ion .
5 chelators, exemplified by 2-pyridylamidoxime,0-acetyl (compound 210), which
like
2,2'-bipyridine does not affect dopamine transport by itself, but blocks
dopamine
transport with a potency approx. 10-fold higher than free Zn(II) and
interestingly
acts in a mono-component fashion (Fig. L3C). This effect of the metal-ion
chelator
complex was eliminated by mutational substitution of His193 known to be
involved
10 in metal-ion binding (Fig. L3C). This substitution is known not to affect
the
transport of catecholamine (Norregaard et al (1998) EMBO J. 17: 4266-4273)
indicating that the effect of the metal-ion chelator complexes is mediated
through
the binding to a site (i.e. the endogenous metal-ion site), which is different
from the
catecholamine binding site. Thus, the metal-ion chelator complexes act as
blockers
15 of transport through a novel allosteric molecular mechanism and could
therefore
serve as Iead compounds in the development of a new type of transport
blockers. It
should be noted that the affinity of, for example 2-pyridylamidoxime,0-acetyl
(compound 210) corresponds to even a very good lead compound found by simple
screening.
20 The experiments presented in this section demonstrate that metal-ion
chelator
complexes of very different chemical structures can act as allosteric blockers
of
function - in these cases of either 7TM receptors or 12TM transporter proteins
-
through binding to naturally occurring metal-ion sites. Furthermore, it is
shown that
these compounds can bind with affinities similar to that of lead compounds
found
25 by conventional drug screening techniques. Thus, these metal-ion chelators
can
function as lead compounds in a chemical optimization process to obtain high
affinity compounds acting as drug candidates.
II. Binding Of Metal-Ion Complexes In Engineered Metal-Ion Sites In Various
30 Potential Drug Targets
Natural metal-ion sites are only found in a subset of potential drug targets.
However, through mutagenesis it is possible to introduce metal-ion binding
sites in
proteins by introduction of metal-ion binding residues such as His, Cys, or
Asp. The
examples in the present section demonstrate how metal-ion complexes can bind
to
35 and affect the function of proteins after mutational engineering of metal-
ion sites
into the proteins.
Example IL 1- Binding of various metal-ion complexes to a library of inter-
helical
metal-ion sites en~;ineered into the tachykinin NK1 receptor.
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
51
This example illustrates that different epitopes of a target protein - here a
NK1
receptor - can be addressed by metal-ion chelator complexes, i.e. potential
lead
compounds for antagonists, after systematic mutational engineering of metal-
ion
sites into these different epitopes. Previously, a series of metal-ion sites
have been
built into the tachykinin NK1 receptor to probe helix-helix interactions, i.e.
providing distance constraints in molecular models of the receptor (Elling et
al.
(1995) Nature 374: 74-77, Elling et al. (1996) EMBO J. 15: 6213-6219; Holst et
al.
(2000) Mol.Pharmacol. 58: 263-270). Here, such metal-ion sites are used as
anchor
points for potential lead compounds - i.e. metal-ion chelator complexes - for
the
development of receptor antagonists with different molecular mechanisms of
actions.
Methods - The tachykinin NK1 receptor cDNA was expressed in COS-7 cells.
Two days after transfection whole cells were assayed with respect to binding
of
radioactively labeled substance P ([lasl]-Bolton Hunter labeled Substance P),
in
displacement with substance P, ZnClz, CuCl2 or various chelator complexes
thereof
present in a three fold molar ratio with respect to the metal-ion
concentration. The
zinc(cyclam) complex was prepared by co-incubation at 60 °C for one
hour followed
by overnight incubation at 37 °C. The assay was typically performed in
12 or 24
well plates. On the day of assay, the cells were washed with binding buffer
(50 mM
Tris-HCl (pH 7.4), 150 mM NaCI, 5 mM MnCl2, 0.1 % BSA, 0.1 % and Bacitracin
(100 mg/ml). Unlabelled competitor ligand and radioligand (20,000 cpm-
approximately 20 pM) was added to the cells in binding buffer and incubation
continued for 3 hours at 4 0C. The assay was terminated by washing of the
cells and
lysis. The assay was performed in duplicate.
Results and discussion - Four different inter-helical metal-ion sites located
between respectively TM-II and -III, TM-III and -V, TM-III and -VII, and TM-V
and VI (Fig. IL 1, Table in Panel A) were here probed with metal-ion chelator
complexes in competition binding experiments against [lasl]-substance P in COS-
7
cells transiently transfected with the NK1 receptor. An increase in affinity
from
approx. 10-fold to around 50-fold was observed in the metal-ion site
engineered
receptors as opposed to the wild-type NKl receptor for free Zn(II) as well as
for
Zn(IT) in complex with either 1,10-phenanthroline or in complex with 2,2'-
bipyridine (Fig. IL 1A). Thus, single to double digit ~,M affinities were
obtained for
the metal-ion chelator complexes in these metal-ion site engineered receptors,
corresponding to affinities observed for lead compounds in general found by
conventional chemical screening. In the sites between TM-II and -III and
between
TM-III and -VII a similar increase in affinity was found for Cu(II) and Cu(II)
in
complex with the chelators as observed with the zinc-ions. However in the
sites
between TM-TII and -V and between TM-V and -VI no increase or just a marginal
SUBSTITUTE SHEET (RULE 26)
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
52
increase in affinity was observed for copper and the copper-chelator
complexes.
Thus, different metal-ions can be exploited in different sites. In Fig.ILB is
demonstrated that an inter-helical bis-His site, in this case constructed
between TM-
V and TM-VI, can also be addressed by a metal-ion chelator complex where the
ion,
in this case Zn(II), is bound in a circular chelator, here cyclam. Cyclam
binds Zn(II)
with a very high affinity, 3.2 x 10-16 M, which can be noted by the fact that
the
Zn(II)-cyclam complex has no effect on the wild-type NK1 receptor even at 10-3
M
conc. i.e. an even smaller effect than the free metal-ion. Thus, the effect of
the
metal-ion chelator complex on the metal-ion site engineered receptor cannot be
caused by the presence of free metal-ions.
The present example demonstrates that metal-ion chelator complexes can bind
with suitable affinity, i.e. corresponding to ordinary lead compounds, in
different
parts of the main ligand-binding crevice of a 7TM receptor. This can be
utilized, for
example to target the lead compound and thereby subsequently the chemically
optimized compound, i.e. the drug candidate, to bind and interact with
different
parts of the target molecule. In the present case, the metal-ion site between
TM-II
and -III can be used as anchor point for lead compounds addressing chemical
interactions with wild-type residues located in the pocket between TM-I, -II, -
III,
and VII; whereas the metal-ion sites located between TM-III and -V and TM-V
and
-VI can be used as anchor points for chelating lead-compounds addressing
residues
in the pocket between TM-III, -IV, -V, -VI and -VII (see helical wheel diagram
in
Fig. IL 1 C). The metal-ion site located between TM-III and -VII may in
principle be
used to address either of these pockets. This approach can be used to
deliberately
direct the chemical optimization process, i.e. the molecular recognition
towards
specifically interesting parts of the target protein in order to obtain for
example
selectivity for a certain receptor subtype or a certain member of a family of
related
proteins. For example, families of monoamine and adenosine 7TM receptors are
generally very highly - if not totally - conserved in the binding pocket for
the natural
ligand, i.e. the pocket between TM-III, -IV, -V, -VI, and -VII; however, they
differ
more in the pocket between TM-I, -II, -III, and VII. Conventional drug
discovery
methods are for various reasons highly biased towards the binding pocket for
the
natural ligand. The present approach allows for deliberate targeting of the
lead
compound and thereby also the final drug candidate for allosteric sites, i.e.
pockets
or epitopes distinct from the one used by the natural ligand.
Example IL2 - Re-en 'neerin~ of a metal-ion chelator binding site in the 12TM
dopamine transporter.
In example L3 , it was shown that the Zn(II)-bipyridine inhibited dopamine
transport in a two-component fashion. This complicated type of interaction
could
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
53
hamper a subsequent further medicinal chemistry optimization of the chelator
for
high affinity interaction. In this example, the naturally occurring metal-ion
site was
re-engineered by elimination of one part of the metal-ion binding site and by
introduction a new metal-ion binding residue.
Methods - as in example L3.
Results and discussion - Re-engineering of the metal-ion site in the dopamine
transporter was done by eliminating His 193, i.e. the residue found in the
proposed
extra-cellular loop 1, by substitution with a Lys residue and by introduction
of an
alternative metal-ion chelating His residue either in exchange for GIu396
located at
the extra-cellular end of TM-8 or in exchange for Va1377 located in TM-7. Both
of
these introduced His residues are located in a potentially favorable
configuration for
participating in metal-ion binding with His375 in TM 7. As shown in Fig. IL2,
in
both cases - [H193K;E396H] and [H193K;V377H] - more mono-component
interaction curves were obtained for the metal-ion chelator complex in the re-
engineered transporter mutants as compared to the wild-type transporter
protein.
This example demonstrates that a natural metal-ion site can successfully be re-
engineered to create a less complex molecular or pharmacological phenotype. In
a
subsequent medicinal chemical optimization process such re-engineered metal-
ion
sites will be used in parallel with the natural site during the screening of
chemical
Libraries.
In biological target molecules in general, more than one version of an
engineered metal-ion site can in a similar fashion be used in parallel in the
screening
process in order to exploit the chemical libraries more efficiently. This
approach
enables each compound to contact, for example the same amino acid side chain
located on an opposing transmembrane helix in more than one configuration.
The experiments presented in this section demonstrate that metal-ion chelator
complexes can act as blockers of the function of biological target molecules -
in
these cases of either 7TM receptors or 12TM transporter proteins - through
binding
to metal-ion sites introduced by mutagenesis. Furthermore, these compounds can
bind with similar affinity as lead compounds found by conventional drug
screening
techniques. Thus, these metal-ion chelators can function as lead compounds in
a
chemical optimization process to obtain high affinity compounds acting as drug
candidates.
III. Increasing The Affinity / Potency Of The Metal-Ion Chelator Complexes
Through Chemical Modifications Of The Chelator Molecule
In the present collection of examples, the metal-ion chelators are considered
as
being bi-functional compounds, i.e., being composed of a metal-ion chelating
moiety and a variable chemical moiety which interacts positively or negatively
-
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
54
depending on the chemical recognition - with spatially surrounding parts of
the
biological target molecule to which the chelator binds through either a
natural or an
engineered metal-ion site.
Example IIL 1- Structure-activity relationship of antagonist metal-ion
complexes in
the ~alanin Rl and the leukotriene LTB4 7TM receptors.
As discussed in examples L l and L2, the human galanin receptor possesses a
natural, antagonistic metal-ion site located between Cysgg in TM-II and Cys290
in
TM-VII, whereas the human leukotriene LTB4 receptor has a metal-ion site
located
between Cys93 and Cys97, both located in TM-III.
Methods - as in examples L 1 and L2.
Results and discussion - A small library of commercially available 1,10-
phenanthroline analogs in complex with Cu(II) were tested in competition for
binding against [125I~-galanin to the galanin Rl receptor expressed in COS-7
cells.
This demonstrated, that many metal-ion chelator complexes bound with a similar
affinity as the basic chelator compound, i.e. 1,10-phenanthroline, indicating
that the
modifications of the variable chemical moiety of the metal-ion chelator
neither
increased nor decreased the binding affinity (in Fig. III.1 is shown 5-phenyl-
1,10-
phenanthroline, compound 134, as an example). However, some chemical
modifications clearly decrease the affinity of the metal-ion chelator complex,
for
example 2,9-dimethanol-1,10-phenanthroline (compound 133), or-importantly -
some chemical modifications increase the binding affinity, for example 5-
methyl-
1,10-phenanthroline (compound 176) (Fig. III.1). In the LTB4 receptor similar
results were obtained, however here different phenanthroline analogs yield
different
results. For example, the 5-phenyl substitution of phenanthroline (compound
134),
which had no effect on the binding affinity in the galanin R1 receptor,
entirely
eliminated the binding of the metal-ion chelator complex in the LTB4 receptor
(Fig.
IIL 1 ).
Example IIL2 - Structure-activity relationship of antagonistic metal-ion
complexes
in the metal-ion site en~Lneered tachykinin NKl 7TM receptor
The tachykinin NK1 receptor, which currently in the industry is a major
putative target for the development of anxiolytic, antidepressive, as well as
anti-
emetic drugs, is here used as an example of a biological target molecule, in
which
an engineered metal-ion site can be used as an anchor point for the discovery
and
development of antagonistic drug candidates. As demonstrated in example IL 1 a
number of metal-ion sites could be built into the NK1 receptor and addressed
by
metal-ion chelator complexes competing for binding against radioactive
substance P
through interactions at different sites in the main ligand-binding pocket of
the
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
receptor, depending on the location of the metal-ion. Here, structure-activity
relationships are demonstrated for a series of chelator analogs in two of
these sites,
i.e. the site between V:OS and VI:24 and the site between III:08 and VII:06.
Methods - as in example IL l .
5 Results and discussion - As observed in for example the galanin Rl receptor,
many of the chemical variations of the variable part of the chelator were
tolerated in
the structure of the NK1 receptor when bound to the engineered metal-ion sites
in
complex with Zn(II). However, as demonstrated in Fig IIL2, clear differences
were
observed for some of the analogs in the two selected sites. Thus, 2,9-
10 bis(trichloromethyl)-1,10-phenanthroline (compound 135) and 1,10-
phenanthroline-
5,6-dione (compound 175) bound 6- and 10-fold better than 1,10-phenanthroline
in
the [HisV:OS,HisVI:24] site, but almost similar to 1,10'-phenanthroline in the
[HisIII:08;CysVII:06] site - all in complex with Zn(II). In contrast the 5-
phenyl-
1,10-phenanthroline (compound 134) was 7-fold more potent in the
15 [HisIII:08;CysVII:06] site than phenanthroline but only slightly more
potent in the
[HisV:OS,HisVI:24] site - again all in complex with Zn(II). It should be noted
here,
that 5-phenyl-1,10-phenanthroline (compound I34) bound like 1,10'-
phenanthroline
in the galanin receptor, but was totally inactive in the leukotrien LTB4
receptor (see
Fig. IIL 1.).
20 This example together with the previous example demonstrate, that
relatively
minor chemical modification of the variable, "non-metal binding" part of the
chelator molecule can alter the recognition and antagonistic property of the
metal-
ion chelator complex both in biological target molecules having naturally
occurring
metal-ion sites as well as in molecules into which metal-ion sites have
deliberately
25 been engineered. Importantly, increases in affinities are observed
demonstrating that
the metal-ion chelators can be utilized as Iead compounds in a drug discovery
process towards high affinity compounds.
Example IIL3 - Structure-activity relationship of agohist metal-ion complexes
in
30 the metal-ion site engineered beta_2-adrenergic 7TM receptor.
It is generally known in the field that while it is possible to find
a~tagohistic
lead compounds and optimize these for high affinity through medicinal
chemistry
efforts in many biological target molecules, it is generally much more
difficult to
find and develop agohist compounds, that is compounds, drug candidates, which
35 activate the biological target molecule. The present example demonstrates
how an
engineered agonistic metal-ion site can be used as anchor-point for the
development
of agonists in a 7TM receptor.
Methods - Mutations were created in the beta2-AR cDNA by the PCR
directed overlap-extension method (Ho et al. (1989) Gene 77: 51-59). The beta2
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
56
AR cDNA was expressed by transient transfection into COS-7 cells. Two days
after
transfection the cells were assayed for intracellular levels of basal and
ligand-
induced cyclic AMP. The assay employed is essentially as described in Solomon
et
al (Anal.Biochem. (1974) 58: 541). Labelled adenine (2 ~,Ci, [3H]adenine,
Amersham TRK311) was added to cells seeded in 6-well culture dishes. The
following day the cells were washed twice with HBS buffer [25 mM Hepes, 0.75
mM NaH2P04, 140 mM NaCI (pH 7.2)] and incubated in buffer supplemented with
1 mM 3-isobutyl-1-methylxanthine (Sigma I-5879). Agonists were added and the
cells were incubated for 30 min at 37 °C. The assay was terminated by
placing the
cells on ice and aspiration of the buffer followed by addition of ice-cold 5%
trichloroacetic acid containing 0.1 mM unlabelled camp (Sigma A-9062) and ATP
(Sigma A-9501). Cyclic AMP was then isolated by application of the supernatant
to
a SOW-X4 resin (BioRad) and subsequently an alumina resin (A-9003; Sigma)
eluting the cyclic AMP with 0.1 M imidazole (Sigma I-0125). Determinations
were
done in duplicate.
Results and discussion - The inventors have previously demonstrated that
Cys-substitution of Asn3ia (AsnVII:06) in TMVII in the beta2-adrenergic
receptor
creates a bi-dentate metal-ion binding site with AspIII:08 at which metal-ion
chelator complexes such as 1,10-phenanthroline and 2,2'-bipyridine in complex
with either Zn(II) or Cu(II) can bind and act as agonists for the receptor
(Elling et al.
PNAS 1999, 15: 6213-6219). As shown in Fig. IIL3B an extended version of this
site including also a substituted residue, Phe2g9 (PheVI:16) located in the
important
TM-VI, metal-ion chelator complexes, in this case Cu(II)-1,10-phenanthroline
and
Cu(II)-bipyridine display higher agonistic efficacy than in the TM-III to TM-
VII
site. The free metal-ion or the chelator by itself has no stimulatory effect
in the
metal-ion-site engineered receptor (Fig. IIL3B). That the agonistic effect of
the
metal-ion chelator complex is not caused by some kind of covalent modification
of
the receptor - for example oxidation - is shown in Fig. IIL3, where a simple
washing experiment demonstrates how the stimulatory effect quickly disappears,
when the metal-ion chelator is removed, while the stimulation continues if the
metal-ion chelator complex is re-added. When a library of bipyridine analogs
were
tested for agonistic activity in this site, many were found not to be active
(data not
shown), while some were shown to be as potent as bipyridine itself (Fig .
IIL3C).
Importantly, a compounds such as 2,2'-di(4-(benzimidazol-2-yl)-
quinoline),(compound 85) was found to stimulate signal transduction as
determined
in CAMP accumulation in the metal-ion site engineered receptor with an x-fold
improved potency, i.e. ECso = 470 nM.
This example demonstrates, that the variable, non-metal-ion binding part of
the
chelators can be modified to create nanomolar affinity agonists in metal-ion
site
SUBSTITUTE SHEET (RULE 26)
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
57
engineered biological target molecules. Such a compound could serve as an
intermediate "chemical stepping-stone" in the process of developing high
affinity
agonists for the metal-ion site engineered receptor. And, similarly agonistic
metal-
ion sites can be engineered into other 7TM receptors and other biological
target
molecules in general to serve as anchor points for the initial identification
as well as
the initial optimization process for agonist leads for such target molecules.
Example IIL4 - Structure-activit~relationship of antagonistic metal-ion
complexes
in a soluble protein, the enzyme FVIIa
The previously presented examples have all represented membrane proteins,
which obviously constitute a very large group of biological target molecules
for
medical drugs. In the present example, Factor VIIa, i.e. the active form of
the FVII
protease involved in the coagulation cascade is used to demonstrate that metal-
ion
chelator complexes can modulate the function of a soluble protein, in this
case an
enzyme which is known to possess an appropriate, allosteric metal-ion site
(Dennis
et al. Nature (2000) 404: 465-470).
Method - The amidolytic activity of Factor VIIa (FVIIa) was measured by the
incubation of 2.5 ~,1 FVIIa (100 nM final concentration, obtained from
American
Diagnostica), 2.5 ~,l ligand and 4 ~,1 substrate (10 mM, 52288 obtained from
Chromogenix) in 42.5 ~.l buffer (50 mM Hepes pH 7.4, 1 1nM CaCl2, 100 mM
NaCI, 0,02% Tween 20). The assay was performed in 96-well plates (Costar).
Incubation was performed at room temperature for five hours with absorbance
read
every 10 minutes.
Results and discussion - As shown in Fig. IIL4A, 2,2'-bipyridine without
metal-ions has no effect on the activity of FVIIa; however in complex with
Zn(II),
2,2'-bipyridine inhibits the enzymatic activity with a 100 ~,M affinity. Many
bipyridine analogs act with a similar potency as the basic chelator, however
for
example Zn(II)-4,4'-di-terbutyl-2,2'-dipyridyl (compound 180) inhibits FVIIa
enzyme activity with an 8.5-fold increased potency as compared to Zn(II)-
bipyridine (Fig. IIL4A). In contrast Zn(II)- 4,4'-di-terbutyl-2,2'-dipyridyl
(compound 180) inhibits LTB4 binding to the LTB4 receptor with a potency which
is 10-fold lowef° than Zn(II)-bipyridine alone (Fig. IIL4B). As shown
in Fig. IIL4C,
1,10-phenanthroline had no effect on FVIIa activity by itself, however in
complex
with Zn(II) 1,10-phenanthroline inhibits the enzyme activity with a potency of
110
~,M. As with 2,2'-bipyridine, many phenanthroline analogs act with a potency
similar to or lower than 1,10'-phenanthroline itself (data not shown);
however, for
example 2,9-bis(trichloromethyl)-1,10-phenanthroline in complex with Zn(II)
inhibits FVIIa activity with increased potency as compared to Zn(II)-1,10'-
phenanthroline (Fig. IIL4C).
SUBSTITUTE SHEET (RULE 26)
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
58
Most enzyme inhibitors act by binding at - or near by - the active site of the
target molecule. However, as recently demonstrated for FVIIa, very efficient
inhibition can be obtained also by binding instead at exosites or allosteric
sites
located far away from the active site in the biological target molecule
(Dennis et al.
Nature (2000) 404: 465-470). The method described here can be utilized to
specifically target the lead compound and thereby the final drug candidate to
act at
allosteric sites in the target molecule, as the binding site is determined by
the site at
which the anchoring metal-ion site is engineered. Inhibition of enzymes and
proteins
in general at allosteric sites is particularly interesting since the active
site often is
relatively similar in enzymes belonging to a particular protein family, for
example
kinases or phosphatases, which means that it can be difficult to obtain
selectivity of
drugs acting at the active site. This is not the case with drugs acting at
allosteric
sites.
Example IILS - Structure-based optimization of metal-ion chelators for
secondarX
interactions in the CXCR4 receptor and other biolog-'lcal target molecules.
The previous examples in this session have demonstrated, that it is possible
to
obtain both decreased, but importantly, also increased affinity by modifying
the
variable, non-metal binding part of metal-ion chelators, which in various
biological
target molecules bind to either natural or engineered metal-ion sites. These
examples were gathered mainly from screenings of commercially available, small
libraries of chelator analogs. In the present example it is described how the
process
of increasing the affinity or potency of the metal-ion chelator can be
performed in a
deliberate structure based fashion in this case through the establishment of a
charge-
charge interaction. The metal-ion-mediated binding of the metal-ion chelator
is here
considered as being the "primary interaction point" or the anchor point, while
the
subsequent establishment of other chemical interactions is considered to be
"secondary interaction points".
Methods -The cDNA coding for, for example the CXCR4 chemokine receptor
can be expressed in COS-7 cells as described for other 7TM and 12TM proteins
previously. Metal-ion sites may be engineered through PCCR-directed
mutagenesis
and the functional activity of the receptor be tested for instance by
(established)
binding experiments employing the radiolabelled ligand, [125I]-SDF 1 a.
Results and discussion - The inventors have demonstrated that Asp171
(AspIV:20) located at the extracellular end of TM-IV on the face pointing
inwards,
towards the main ligand binding crevice of the CXCR4 receptor is exposed and
can
be used as attachment site for the positively charged cyclam ring of non-
peptide
bicyclam antagonists for this receptor. Metal-ion binding sites will be
introduced in
the CXCR4 receptor in the spatial vicinity of AspIV:20 by introduction of a
His
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
59
residue at position V:01 which will form a bis-His metal-ion binding site with
the
naturally occurring HisIII:05 in the CXCR4 receptor - as previously
demonstrated in
the NI~l receptor (Elling et al. EMBO J. (1996) 15: 6213-6219). Similarly an
intra-
helical bis-His site will be introduced between residues V:O1 and V:OS through
introduction of two His residues at these positions and between III:OS and
IV:24
through His substitution at position IV:24. Thus three metal-ion sites will be
constructed all within few A's of AspIV:20 (see helical wheel diagram in Fig.
IILS).
A small library of 1,10-phenanthroline analogs will be obtained or synthesized
in
which amino-methyl, amino-ethyl, amino-propyl, and aminobutyl will be placed
in
either the 2, 3, 4, or 5 positions and a similar small library where the same
substituents will be placed in either the 3, 4, or 5 position of bipyridine
will
similarly be constructed. In a typical experiment, these libraries of amino-
substituted
chelators will be tested in complex with either Zn(II) or Cu(II) in the metal-
ion-site
engineered CXCR4 receptors, and the compounds ability to inhibit the binding
of
lasl-SDF 1 or the binding of [l2sl]-1265 monoclonal antibody or the ability of
the
compounds to inhibit the signal transduction mechanism induced by SDF-la will
be
tested as performed for metal-ion chelators in the previous examples described
above. Due to the spatial proximity as well as the relative conformational
flexibility
of the system, several of these compounds will in several of the sites have
the
opportunity of forming a salt-bridge between the amino function of the amino-
substituted metal-ion chelator and the carboxylic acid function of Asplm
(AspIV:20). This formation of a secondary interaction will be quantified as an
increased affinity or an increased potency of the metal-ion complex of the
amino-
substituted chelator in comparison to the corresponding metal-ion complex of
the
non-substituted phenanthroline or dipyridine. Due to the relatively high
energy in
the charge-charge-interaction a considerable increase in affinity or potency
will be
observed. The molecular interaction mode of the amino-substituted chelator(s)
will
be confirmed through mutational substitutions of Aspl~l with Asn, Ala and
other
residues. Depending on the structure of the most optimal amino-substituted
analogs) a second and third round of analogs will be synthesized which
conceiveably will present an appropriate basic moiety in a more
conformationally
constrained fashion.
These mini-libraries of amino-substituted metal-ion chelators can be utilized
in
several biological target molecules, which present Asp or Glu residues in an
appropriate fashion. For example, in the CXCR4 receptor Asp26a (AspVI:23) is
equally available as Asp171 for interaction as previously described (Gerlach
et al.).
Similarly AspIII:08 is conserved among monoamine receptors and, for example
opioid and somatostatin receptors and this residue is a known interaction
point for
amine functions (Strader et al (1991) 266: 5-8). These and other acidic,
potential
SUBSTITUTE SHEET (RULE 26)
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
secondary interaction points for amino-substituted metal-ion chelators can be
addressed through construction of a small number of metal-ion sites placed in
their
spatial vicinity - as described above for Aspl~l (AspIV:20). Similarly amino-
functions in a biological target molecule - for example, epsilon amino groups
of Lys
5 residues -can be addressed by, for example mini-libraries of tetrazol
substituted
metal-ion chelators. As described, charge-charge interactions will initially
be
pursued for establishing secondary interactions for the metal-ion chelator
lead
compounds. However, other types of weaker interactions such as hydrogen-bonds,
amino-aromatic interactions, aromatic-aromatic interactions, aliphatic
hydrophobic
10 interactions, van der Walls interactions etc. will also be exploited in a
similar,
systematic fashion as described above for the charge-charge interactions.
In the present section, a 7TM receptor is for convenience used as an example
of a biological target molecule. In this system, very useful molecular models
are
available, which have been refined and have allowed for, for example the
15 construction of intra- and especially inter-helical metal-ion sites.
However, due to
lack of, for example an array of suitable X-ray structures of this or similar
targets in
complex with agonists and antagonists it is not possible to apply classical
structure-
based drug design methodology in full. Nevertheless, for example in these
membrane proteins the present method does to a certain degree compensate for
the
20 lack of knowledge of the detailed 3D structure of the target molecule by
anchoring
the lead compound and thereby creating a fix-point for the subsequent
medicinal
chemical optimization point guided by the molecular models.
The approach described above could be further helped and guided by detailed
knowledge of the 3D structures) of the biological target molecule,
preferentially
25 determined in complex initially with the un-substituted metal-ion chelator
and
subsequently in complex with the chemically modified metal-ion chelator in
which
attempts have been made to establish first one secondary interaction and
subsequently further secondary or tertiary interactions. For some biological
target
molecules such as soluble proteins this can be achieved through for example
30 crystallization and standard X-ray analysis procedures or through, for
example
NMR analysis of the complex in solution again using standard procedures. Here,
the
method can take advantage of methods developed for structure-based drug
discovery in general. This would make it possible to apply classical structure-
based
approaches such as structure-based library design for the establishment of
secondary
35 and tertiary interaction sites for the lead compound in the target
molecule. However,
it should be noted, that a major advantage and difference of the present
method is,
that the lead compound is anchored to a particular site and thereby to a
certain
degree in a particular conformation in the biological target molecule through
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
61
binding to the bridging metal-ion site while the compound is being optimized
for
chemical recognition with the target molecule.
Also it should be noted that through the application of a more-or-less
flexible
spacer in between the metal-ion chelating moiety and the so-called variable
chemical moiety of the test compound it becomes possible to probe for
interaction
or binding to structurally and functionally interesting epitopes of the
biological
target molecule with variable chemical moieties, which due to their intrinsic
low
affinity would not be detectable in the analytical systems on their own; but,
which -
due to the local high concentration of these created by the binding of the
tethering
metal-ion chelating moiety to the metal-ion site - now are detected.
Example IIL6 - Structure-based optimization of metal-ion chelators to use as
antagonists in "pharmacological knock-out" experiments
The approach described in the previous examples will be used as (a) steps) in
the drug development process in general to increase the affinity of lead
compounds
for the biological target molecule through establislunent of chemical
recognition
between the ligand and structural elements found in the wild-type target
molecule,
i.e. in the unmodified vicinity of the engineered metal-ion site. However, the
method will also be used for example to increase the affinity and specificity
of
metal-ion chelator compounds to be used in pharmacological knock-out
applications. This procedure has in principle been described previously
(Elling et al.
(1999) Proc.Natl.Acad.Sci.USA 96: 12322-12327); however only for basic metal-
ion chelating agents. Briefly, the method is based on the introduction of a
silent
metal-ion site in a potential drug target, i.e. creation of a metal-ion site
in which the
mutations do not affect the binding and action of the endogenous ligand for
the
receptor. When such a metal-ion site engineered receptor is introduced into an
animal by classical gene-replacement technology, i.e. exchange of the
endogenous
receptor with the metal-ion site engineered receptor, then the animals will
develop
normally without any development of compensatory mechanisms, which otherwise
frequently impair the interpretations of the phenotypes in classical gene
knock-out
technology. In the adult animals or whenever it is found appropriate the
animals are
then treated with an appropriate metal-ion-chelating agent which then will act
as an
antagonist and turn off the function of the metal-ion site engineered
receptor.
Currently, this approach is impaired by the fact, that the generally available
metal-
ion chelating agents only will bind with at best ~,M affinity to the metal-ion
site
engineered biological target molecule, which will give similar ~,M or lower
antagonistic potencies. These relatively low potencies and the relative low
specificity of the basic test compounds impairs the general applicability of
the
technology due to simple pharmacokinetic and toxicology problems.
SUBSTITUTE SHEET (RULE 26)
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
62
By applying the technology described in the previous example and in the
previous examples in general, it will be possible to increase the affinity of
metal-ion
chelators significantly, which will make it considerably more easy to reach
therapeutic, efficient antagonistic concentrations of the metal-ion chelator
in the
animals and also to increase the "therapeutic window" due to the higher degree
of
selectivity of the compounds caused by the establishment of more than one
molecular interaction point. Establishment of just a single suitable charge-
charge
interaction will increase the affinity of the metal-ion chelator by 10 to 100-
fold or
more. This will be performed as an example in the so-called RASSL a modified
kappa-opioid receptor, which previously has been used in gene-knock out
experiments (Redfern et al. Nat. Biotechnol. (1999) 17:165-169). By
introduction of
metal-ion sites, for example between TM-V and TM-VI or between TM-VI and
TM-VII or between TM-II and TM-III or between TM-III and TM-VII in a kappa-
opiod RASSL molecule and through screening of, for example the mini-library of
amino-substituted metal-ion chelators it will be possible to select a nano-
molar
affinity antagonist because of the formation of a secondary charge-charge
interaction with AspIII:08, i.e. the Asp in TM-III corresponding to the amine-
binding Asp in monoamine receptors.
IV. Optimization Of Compounds On The Wild-Type Biological Target
Molecule
In the case, where the initial binding of the metal-ion chelator was obtained
through mutational introduction of an anchoring metal-ion site in the
biological
target molecule, a final step of optimization will have to be performed to
obtain high
affinity binding or potency on the wild-type target molecule without the metal-
ion
bridge. Through the methods described in the previous experiments, the metal-
ion
chelator lead compound will gradually be optimized for interactions with
chemical
groups in the biological target molecule spatially surrounding the metal-ion
site - i.e.
interactions with chemical groups found also in the wild-type target molecule.
Thus,
the test compound will gradually increase its affinity not only for the metal-
ion site
engineered molecule but also for the wild-type biological target molecule.
When
two to three secondary interaction points have been established, the affnity
of the
test compound for the wild-type target molecule, which is being tested in
parallel
with the metal-ion site engineered molecule, will have reached micro-molar
affinities, i.e. a lead compound on the wild-type target molecule has been
created.
At this point one or more of the following three approaches will be followed;
1)
structure-based further chemical optimization of the compound in general
aiming at
improving recognition at various known chemical moieties of the target
molecule;
2) structure-based further chemical optimization of the compound at which the
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
63
"metal-ion site bridge" is exchanged by a more classical type of chemical
interaction
with the residues) which had been modified to create the metal-ion site in the
biological target molecule. Here advantage can be taken of the fact that the
geometry of the metal-ion site anchor is well known in general and, that
relatively
limited structure-based libraries can be established to create a new type of
interaction; 3) further chemical optimization of the compound through more-or-
less
random generation of chemical diversity in general in the compound.
The above-given examples describe specific methods that can be employed to
practice the present invention. Based on the details given a person skilled in
the art
will be able to devise alternative methods at arriving in the same information
using
the concept of the invention. However, the examples are not to be construed to
limit
the invention in any way.
CA 02395999 2002-06-28
WO 01/50127 PCT/EP00/13389
64
Patent EXAMPLES - Figures
I. BINDING OF METAL-IONS AND METAL-ION COMPLEXES TO VARIOUS
DRUG TARGETS WITH NATURAL METAL-ION SITES
L1 Identification of naturally occurring metal-ion chelator binding site in
the 7TM leukotriene LTB4
receptor
L2 Identification of naturally occurring metal-ion chelator binding site in
the
7TM galanin receptor
L3 Identification of naturally occurring metal-ion chelator binding site in a
12TM protein, the dopamine transporter.
II. BINDING OF METAL-ION COMPLEXES IN ENGINEERED
METAL-ION SITES IN VARIOUS POTENTIAL DRUG TARGETS.
II:1 Binding of various metal-ion complexes to a library of inter-helical
metal-ion sites engineered into
the tachykinin NKl receptor
IL2 Re-engineering of a metal-ion chelator binding site in the 12TM dopamine
transporter.
III. INCREASING THE AFFNITY/POTENCY OF THE METAL-ION CHELATOR
COMPLEXES THROUGH CHEMICAL MODIFICATIONS OF THE
CHELATOR MOLECULE.
IIL1 Structure-activity relationship of antagonist metal-ion complexes in the
galanin and the leukotrien
LTB4 receptors.
IIL2 Structure-activity relationship of antagonistic metal-ion complexes in
the metal-ion site
engineered tachykinin ~NKl receptor. '
IIL3 Structure-activity relationship of agonist metal-ion complexes in the
metal-ion site engineered
beta2-adrenergic 7TM receptor.
IIL4 Structure-activity relationship of antagonistic metal-ion complexes in a
soluble protein,
the enzyme FVIIa.
IILS Structure-based optimisation of metal-ion chelators for secondary
interactions in the CXCR4 receptor
and other biological target molecules.
IIL6 Structure-based optimisation of metal-ion chelators to use as antagonists
in 'pharmacological knock-out'
experiments. No figure !
1V. OPTIMIZATION OF COMPOUNDS ON THE WILD-TYPE BIOLOGICAL TARGET MOLECULE
No ftgures !
APPENDIX - List of compounds which appear in the Examples.