Note: Descriptions are shown in the official language in which they were submitted.
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
PSEUDOMYCIN PHOSPHATE PRODRUGS
FIELD OF THE INVENTION
The present invention relates to pseudomycin compounds,
in particular, phosphate prodrugs of pseudomycin compounds.
BACKGROUND OF THE INVENTION
Pseudomycins are natural products isolated from liquid
cultures of Pseudomonas syringae (plant-associated
bacterium) and have been shown to have antifungal
activities. (see i.e., Harrison, L., et al., "Pseudomycins,
a family of novel peptides from Pseudomonas syringae
possessing broad-spectrum antifungal activity," J. Gen.
Microbiology, 137(12), 2857-65 (1991) and US Patent Nos.
5,576,298 and 5,837,685) Unlike the previously described
antimycotics from P. syringae (e. g., syringomycins,
syringotoxins and syringostatins), pseudomycins A-C contain
hydroxyaspartic acid, aspartic acid, serine,
dehydroaminobutyric acid, lysine and diaminobutyric acid.
The peptide moiety for pseudomycins A, A', B, B', C, C'
corresponds to L-Ser-D-Dab-L-Asp-L-Lys-L-Dab-L-aThr-Z-Dhb-L-
Asp(3-OH)-L-Thr(4-C1) with the terminal carboxyl group
closing a macrocyclic ring on the OH group of the N-terminal
Ser. The analogs are distinguished by the N-acyl side
chain, i.e., pseudomycin A is N-acylated by
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
3,4-dihydroxytetradecanoate, pseudomycin A' by
3,4-dihydroxypentadecanoate, pseudomycin B by
3-hydroxytetradecanoate, pseudomycin B' by
3-hydroxydodecanoate, pseudomycin C by
3,4-dihydroxyhexadecanoate and pseudomycin C' by
3-hydroxyhexadecanoate. (see i.e., Ballio, A., et al.,
"Novel bioactive lipodepsipeptides from Pseudomonas
syringae: the pseudomycins," FEES Letters, 355(1), 96-100,
(1994) and Coiro, V.M., et al., "Solution conformation of
the Pseudomonas syringae MSU 16H phytotoxic lipodepsipeptide
Pseudomycin A determined by computer simulations using
distance geometry and molecular dynamics from NMR data,"
Eur. J. Biochem., 257(2), 449-456 (1998).)
Pseudomycins are known to have certain adverse
biological effects. For example, destruction of the
endothelium of the vein, destruction of tissue,
inflammation, and local toxicity to host tissues have been
observed when pseudomycin is administered intraveneously.
Therefore, there is a need to identify compounds within this
class that are useful for treating fungal infections without
the currently observed adverse side effects.
2
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
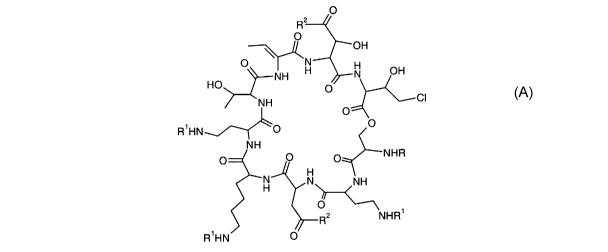
BRIEF SUMMARY OF THE INVENTION
The present invention provides a pseudomycin prodrug
represented by the following structure which is useful as an
antifungal agent.
O
R3
O
OH
O~ N H
NH H N OH
HO O
NH SCI
O
O O
R'HN O
NH N R
O O O H
N NH
O
R2 NHR'
R~HN O
wherein R is
Rb b. Rd
Rr
Ra Ra
~c ~e
R R
where
Ra and Ra~ are independently hydrogen or methyl, or
either Ra or Ra~ is alkyl amino, taken together with Rb
or Rb~ forms a six-membered cycloalkyl ring, a six-
membered aromatic ring or a double bond, or taken
together with R° forms a six-membered aromatic ring;
3
CA 02396513 2002-06-13
wo o»~s~e petNSOw~oi6i
Rb triad Rb' are independc~atly hydrogen , halogets, flr
methyl, or either Rb or R°' is amino, alkylantino, a-
8cetoaCetate, methoxy, er hydroxy:
R~ is hydrogen, hydroxy. C1-C, alko~qr, hydroxy Cl-
C~ nlkoxy, or taken together with R' foams s 6-me~nbered
aromatic ring or cs-c, cycloe11q~1 ring
R° is ~ydxogen~
R' is hydrogen, or taken together with At is a
aix-taembexed aromatic ring, C5-C~, alkoxy substituted
to . six-ine~nbered aromatic ring, or CS~Cla alkyl substituted
six-meatbersd aromatic ~rinQ, au&
R= is Ce-Ci, alkyl, C5-Cil elkoxy, or biphenyl;
or R is
R~
O
' where
R° is hydrogea. or Ci-Ci~ alkyl, and
Rh is Cl-Cls alkyl, C~-C~~ alkoxy, lCl-Clo
al7cyl ) phe~syl . - (CH=) p-axyl, or - ~CH~ ) n- (C6-Cs.
cycloal~l) , where n ; '1 or 21 or
R is
0
avaaTZTOTB EaxET
Eac fang z AM Newly filed
voVV3Ulbl
- . , , - '" '~~~' ~~ ~ 31? 2?6 1294 TO 9011a9gg239g4q65 P.09~?xt
' WO OtM 153 P'CtNS00~30~ 6~
R~
m
~her~
Ri ie a k~y8rogen, hsloger~, or C5-C~ alkoxy, aad
m ie 2, 2 ox 3;
ar R is
H
r R7 i
vdhere
Rj is CS-Cl~ alkoxy or C~-Cl, a17pr1, eisd n ~ 0, 1 or
2t
or R is
\ Px
whore
Rk is CS--Cl~ nlkoxy: ar
R is - (Cxy ? -NR°'- ( Ci~-Cls alkyl > , whore R"' is 13. -CHj or
, -C(0)CIi~;
R~' 1s in0ependently hydrogen pr a group represented by
formula 2fel, i(b)~ or 1(cl
S
$08aT=TOE 8~T
Eno far AM Newly filed . .
'------,--~
CA 02396513 2002-06-13
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
O
O ~ ~~ORta
O O~ ~ORta
O ~ O 1~
~O
~P~ORta
O/ \ORta
1 (a) 1 (b)
O O
~OR'b
P
O~O~ ~OR~b
1 (c)
where
Rla is hydrogen, C1-C6 alkyl (e. g., methyl, ethyl,
n-propyl, i-propyl, n-butyl, i-butyl, s-butyl, t-butyl,
etc. ) , benzyl, or -CHZCHZSi (CH3) 3, and
Rlb is hydrogen or C1-C6 alkyl,
provided that at least one R1 is a group represented by
formula 1(a), 1(b) or 1(c);
R2 and R3 are independently -ORZa, or -N (R2b) (R2~) ,
where
RZa and R2b are independently hydrogen, C1-Clo alkyl
(e.g., methyl, ethyl, n-propyl, i-propyl, n-butyl, i-
butyl, s-butyl, t-butyl, etc.), C3_C6 cycloalkyl (e. g.,
cyclopropyl, cyclobutyl, cyclopentyl,
cyclopentylmethylene, methylcyclopentyl, cyclohexyl,
etc . ) hydroxy ( Cl-Clo ) alkyl , alkoxy ( C1-Clo ) alkyl ( a . g,
2 0 methoxyethyl ) , or C2-Clo alkenyl , amino ( C1-C1o ) alkyl ,
6
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
mono- or di-alkylamino (C1-Clo) alkyl, aryl (C1-Clo) alkyl
(e.g., benzyl), heteroaryl(Cl-Clo)alkyl (e.g., 3-
pyridylmethyl, 4-pyridylmethyl), or
cycloheteroalkyl(C1-Clo)alkyl (e. g., N-tetrahydro-1,4-
oxazinylethyl and N-piperazinylethyl), or
Rzb is an alkyl carboxylate residue of an
aminoacid alkyl ester ( a . g . , -CH2COZCH3 ,
-CH ( COzCH3 ) CH ( CH3 ) z , -CH ( COZCH3 ) CH ( phenyl ) ,
-CH ( COZCH3 ) CHZOH, -CH ( C02CH3 ) CHZ (p-hydroxyphenyl ) ,
-CH ( COZCH3 ) CH2 SH , -CH ( COzCH3 ) CH2 ( CHz ) 3NH2 ,
-CH ( C02CH3 ) CH2 ( 4- or 5-imidazole ) , -CH ( C02CH3 ) CHZCOzCH3,
-CH ( C02CH3 ) CHZCOZNH2 , and the like ) , and
R2~ is hydrogen or C1-C6 alkyl; and pharmaceutically
acceptable salts and solvates thereof. In a preferred
embodiment, R is represented by the structure
Rb Rb~ Ra
Rf
Ra Ra
~c ~e
R R
where Rb~ is hydroxy, Ra, Ra~ , Rb, R~, Rd, and Re are all
hydrogen, and Rf is n-octyl.
In another embodiment of the present invention, a
pharmaceutical formulation is provided which includes the
pseudomycin prodrug described above and a pharmaceutically
acceptable carrier.
7
CA 02396513 2002-06-13
. w0 ~I11133i PCtNS0013p167
Iz~ yet another embodiment of the pzesent invention, a
method is provide8 for treating a fungal infection in an
animal in need thereof, which comprises ad,>nihictering to the
anisaal the pscudomycib pzodrue desczibed above. The use of
the pseudomycin prodrug descri)~ed above in the manufacture
of a medicament for uQe in troating a fungal irrfectioa in an
animal ie also provided,
Dsrfir~it~one
As used herein, the term "alkyl" refers to a
hydrocarbon zadical of the general, f ortaula C"H=~.~ containing
fr~n 1 to 30 carboy atoms unless othe~ise indicateB, 'The
alk~ne radical may be straight (o, g. methyl, ethyl, propyl,
butyl, etc.), braz~ched (e. g., isopropyl, isobutyl, terti~.ry
butyl, nevpentyl, etc.), cyclic (e.9., cyclopropyl,
cyclobutyl, cyclopentyl, methylcyclope~ntyl, cyclohexyl,
ate.), or muzti-cyclic (e.g " bicyelo(2.2.17hegtane,
Bpiro [2.27p~tane, etc . ) , ?'he alkar~m rsdical may b0
substituted or unsubetituted. Similarly, the alkyl yortion
of axe alkoxy group, alknhoyl, ox alkanc~ate have the save
definition as above.
The torm 'alkenyl' refers to an acyclic hydrocarbon
concnining at least one carbon carbon double bond. the
alkene radical asay be straight, branched, cyclic, or >nulti-
cyclic. The alkene ra6ical away be substituted er
a
soas~x~ e~s
E ~ o i a n 6 s; AM Newly filed
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
unsubstituted. The alkenyl portion of an alkenoxy, alkenoyl
or alkenoate group has the same definition as above.
The term "aryl" refers to aromatic moieties having
single (e. g., phenyl) or fused ring systems (e. g.,
naphthalene, anthracene, phenanthrene, etc.). The aryl
groups may be substituted or unsubstituted.
Within the field of organic chemistry and particularly
within the field of organic biochemistry, it is widely
understood that significant substitution of compounds is
tolerated or even useful. In the present invention, for
example, the term alkyl group allows for substitutents which
is a classic alkyl, such as methyl, ethyl, propyl, hexyl,
isooctyl, dodecyl, stearyl, etc. The term "group"
specifically envisions and allows for substitutions on
alkyls which are common in the art, such as hydroxy,
halogen, alkoxy, carbonyl, keto, ester, carbamato, etc., as
well as including the unsubstituted alkyl moiety. However,
it is generally understood by those skilled in the art that
the substituents should be selected so as to not adversely
affect the pharmacological characteristics of the compound
or adversely interfere with the use of the medicament.
Suitable substituents for any of the groups defined above
include alkyl, alkenyl, alkynyl, aryl, halo, hydroxy,
alkoxy, aryloxy, mercapto, alkylthio, arylthio, mono- and
di-alkyl amino, quaternary ammonium salts, aminoalkoxy,
9
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
hydroxyalkylamino, aminoalkylthio, carbamyl, carbonyl,
carboxy, glycolyl, glycyl, hydrazino, guanyl, and
combinations thereof.
The term "prodrug" refers to a class of drugs which
result in pharmacological action due to conversion by
metabolic processes within the body (i.e.,
biotransformation). In the present invention, the
pseudomycin prodrug compounds contain linkers that can be
cleaved by esterases in the plasma to produce the active
drug.
The term "animal" refers to humans, companion animals
(e. g., dogs, cats and horses), food-source animals (e. g.,
cows, pigs, sheep and poultry), zoo animals, marine animals,
birds and other similar animal species.
DETAILED DESCRIPTION OF THE INVENTION
Applicants have discovered that a prodrug derivative of
the pseudomycin natural or semi-synthetic products provide
less adverse side effects than the corresponding natural
products and maintains in vivo efficacy against C. albican,
C. neoformans, and A. fumigatus. The prodrug is produced by
acylating at least one of the pendant amino groups attached
to the lysine or 2,4-diaminobutyric acid peptide units in
the pseudomycin cyclopeptide ring system to form an acyl
phosphate substituent(s). The phosphate acylating agent (or
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
linker) is generally a compound having one of following
formulae 1a-L, 1b-L or 1c-L
O
~~OR~a
O O~ ~~R~a
L O ~ O
L O
~~OR~a
\OR~a
1a-L 1b-L
~~OR~b
~ P
L~O
oR
1 n-T.
where L is a suitable leaving group such that a carbamate
linkage with the pendant amino group on the pseudomycin
structure can be formed. Suitable leaving groups are well
known to those skilled in the art and include groups such as
p-nitrophenoxy and N-oxysuccinimide. R1a and Rlb are as
defined above.
The benzyl phosphate linkers (1a-L) may be synthesized
using the synthetic route shown in scheme I below. For
illustrative purposes, a specific para-phenylmethylene
substituted acylating compound having a p-nitrophenoxy
leaving group is depicted. However, it will be understood
by those skilled in the art that one could synthesize a
variety of derivatives (including the ortho substituted
derivative (1b-L)) using the same basic synthetic method.
11
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
P-ORIa EtPr2N/DMAP ~ O-P-ORIa
HO OH + H/ 'ORIa Cc~ N HO ~ORia
4
O ~ ~ O
p-N02PhOC(O)Ct O-P~ RRaa
pyridine Q2 ~ ~ Q O O
C' .-. 1-. .-..... .-. T
For a more detailed description of the synthetic procedures,
see the preparation section of the Examples below.
The phosphate linker (1a-L or 1b-L) where Rla is
-CHzCH2Si(CH3)3 may be prepared by oxidative coupling of bis-
teocphosphite with 4-hydroxybenzyl alcohol in carbon
tetrachloride as described in Li, J., et al., Bioorg. Med.
Chem. Lett., 8, 3159-3164 (1998). Bis-teocphosphite may be
prepared from 2-2-trimethylsilylethanol and PC13 as
described in McCombie, et al., J. Chem. Soc., 381 (1945).
The methylene phosphate linker (1c-L) may be
synthesized using the synthetic route shown in scheme II
below. For illustrative purposes, a specific acylating
compound with an N-succinimide leaving group is depicted.
However, it will be understood by those skilled in the art
that one could synthesize a variety of derivatives using the
same basic synthetic method.
12
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
O ~ O O
P-OR~b ~ ~~ 1b
P-OR
EtS O I n'Bu4N+ O~ ~OR'b EtS O~O~ ~OR'b
O O O
S02C12 ~ ~ ~p~ OR'b
N-hydroxysuccinimide N-O O O OR'b
O
Scheme II
For a more detailed description of the synthetic procedures,
see the preparation section of the Examples below. The
phosphate triesters (methyleneoxy and benzyloxy derivatives)
may be converted to their phosphate monoester derivatives
via hydrogenation (e. g., hydrogen over Pd/C in methanol).
As discussed earlier, pseudomycins are natural products
isolated from the bacterium Pseudomonas syringae that have
been characterized as lipodepsinonapetpides containing a
cyclic peptide portion closed by a lactone bond and
including the unusual amino acids 4-chlorothreonine (ClThr),
3-hydroxyaspartic acid (HOAsp), 2,3-dehydro-2-aminobutyric
acid (Dhb), and 2,4-diaminobutyric acid (Dab). Methods for
growth of various strains of P. syringae to produce the
different pseudomycin analogs (A, A', B, B', C, and C') are
described below and described in more detail in PCT Patent
Application Serial No. PCT/US00/08728 filed by Hilton, et
13
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
al. on April 14, 2000 entitled "Pseudomycin Production by
Pseudomonas Syringae," incorporated herein by reference, PCT
Patent Application Serial No. PCT/US00/08727 filed by
Kulanthaivel, et al. on April 14, 2000 entitled "Pseudomycin
Natural Products," incorporated herein by reference, and
U.S. Patent Nos. 5,576,298 and 5,837,685, each of which are
incorporated herein by reference.
Isolated strains of P. syringae that produce one or
more pseudomycins are known in the art. Wild type strain
MSU 174 and a mutant of this strain generated by transposon
mutagenesis, MSU 16H (ATCC 67028) are described in U.S.
Patent Nos. 5,576,298 and 5,837,685; Harrison, et al.,
"Pseudomycins, a family of novel peptides from Pseudomonas
syringae possessing broad-spectrum antifungal activity," J.
Gen. Microbiology, 137, 2857-2865 (1991); and Lamb et al.,
"Transposon mutagenesis and tagging of fluorescent
pseudomonas: Antimycotic production is necessary for control
of Dutch elm disease," Proc. Natl. Acad. Sci. USA, 84, 6447-
6451 (1987).
A strain of P. syringae that is suitable for production
of one or more pseudomycins can be isolated from
environmental sources including plants (e. g., barley plants,
citrus plants, and lilac plants) as well as, sources such as
soil, water, air, and dust. A preferred stain is isolated
14
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
from plants. Strains of P. syringae that are isolated from
environmental sources can be referred to as wild type. As
used herein, "wild type" refers to a dominant genotype which
naturally occurs in the normal population of P. syringae
(e.g., strains or isolates of P. syringae that are found in
nature and not produced by laboratory manipulation). Like
most organisms, the characteristics of the pseudomycin-
producing cultures employed (P. syringae strains such as MSU
174, MSU 16H, MSU 206, 25-B1, 7H9-1) are subject to
variation. Hence, progeny of these strains (e. g.,
recombinants, mutants and variants) may be obtained by
methods known in the art.
P. syringae MSU 16H is publicly available from the
American Type Culture Collection, Parklawn Drive, Rockville,
MD, USA as Accession No. ATCC 67028. P. syringae strains
25-B1, 7H9-1, and 67 H1 were deposited with the American
Type Culture Collection on March 23, 2000 and were assigned
the following Accession Nos.:
25-B1 Accession No. PTA-1622
7H9-1 Accession No. PTA-1623
67 H1 Accession No. PTA-1621
Mutant strains of P. syringae are also suitable for
production of one or more pseudomycins. As used herein,
"mutant" refers to a sudden heritable change in the
phenotype of a strain, which can be spontaneous or induced
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
by known mutagenic agents, such as radiation (e. g.,
ultraviolet radiation or x-rays), chemical mutagens (e. g.,
ethyl methanesulfonate (EMS), diepoxyoctane, N-methyl-N-
nitro-N'-nitrosoguanine (NTG), and nitrous acid), site-
specific mutagenesis, and transposon mediated mutagenesis.
Pseudomycin-producing mutants of P. syringae can be produced
by treating the bacteria with an amount of a mutagenic agent
effective to produce mutants that overproduce one or more
pseudomycins, that produce one pseudomycin (e. g.,
pseudomycin B) in excess over other pseudomycins, or that
produce one or more pseudomycins under advantageous growth
conditions. While the type and amount of mutagenic agent to
be used can vary, a preferred method is to serially dilute
NTG to levels ranging from 1 to 100 ~,g/ml. Preferred
mutants are those that overproduce pseudomycin B and grow in
minimal defined media.
Environmental isolates, mutant strains, and other
desirable strains of P. syringae can be subjected to
selection for desirable traits of growth habit, growth
medium nutrient source, carbon source, growth conditions,
amino acid requirements, and the like. Preferably, a
pseudomycin producing strain of P. syringae is selected for
growth on minimal defined medium such as N21 medium and/or
for production of one or more pseudomycins at levels greater
16
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
than about 10 ~g/ml. Preferred strains exhibit the
characteristic of producing one or more pseudomycins when
grown on a medium including three or fewer amino acids and
optionally, either a lipid, a potato product or combination
thereof.
Recombinant strains can be developed by transforming
the P. syringae strains, using procedures known in the art.
Through the use of recombinant DNA technology, the P.
syringae strains can be transformed to express a variety of
gene products in addition to the antibiotics these strains
produce. For example, one can modify the strains to
introduce multiple copies of the endogenous pseudomycin-
biosynthesis genes to achieve greater pseudomycin yield.
To produce one or more pseudomycins from a wild type or
mutant strain of P. syringae, the organism is cultured with
agitation in an aqueous nutrient medium including an
effective amount of three or fewer amino acids, preferably
glutamic acid, glycine, histidine, or a combination thereof.
Alternatively, glycine is combined with one or more of a
potato product and a lipid. Culturing is conducted under
conditions effective for growth of P. syringae and
production of the desired pseudomycin or pseudomycins.
Effective conditions include temperatures from about 22°-C to
about 27-°C, and a duration of about 36 hours to about 96
17
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
hours. Controlling the concentration of oxygen in the
medium during culturing of P. syringae is advantageous for
production of a pseudomycin. Preferably, oxygen levels are
maintained at about 5 to 50o saturation, more preferably
about 30o saturation. Sparging with air, pure oxygen, or
gas mixtures including oxygen can regulate the concentration
of oxygen in the medium.
Controlling the pH of the medium during culturing of P.
syringae is also advantageous. Pseudomycins are labile at
basic pH, and significant degradation can occur if the pH of
the culture medium is above about 6 for more than about 12
hours. Preferably, the pH of the culture medium is
maintained between 6 and 4. P. syringae can produce one or
more pseudomycins when grown in batch culture. However,
fed-bath or semi-continuous feed of glucose and optionally,
an acid or base (e.g., ammonium hydroxide) to control pH,
enhances production. Pseudomycin production can be further
enhanced by using continuous culture methods in which
glucose and ammonium hydroxide are fed automatically.
Choice of P. syringae strain can affect the amount and
distribution of pseudomycin or pseudomycins produced. For
example, strains MSU 16H and 67 H1 each produce
predominantly pseudomycin A, but also produce pseudomycin B
and C, typically in ratios of 4:2:1. Strain 67 H1 typically
produces levels of pseudomycins about three to five fold
18
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
larger than are produced by strain MSU 16H. Compared to
strains MSU 16H and 67 H1, strain 25-B1 produces more
pseudomycin B and less pseudomycin C. Strain 7H9-1 are
distinctive in producing predominantly pseudomycin B and
larger amount of pseudomycin B than other strains. For
example, this strain can produce pseudomycin B in at least a
ten fold excess over either pseudomycin A or C.
Alternatively, the prodrug can be formed from an N-acyl
semi-synthetic compound. Semi-synthetic pseudomycin
compounds may be synthesized by exchanging the N-acyl group
on the L-serine unit. Examples of various N-aryl
derivatives are described in PCT Application No.
PCT/US00/15017 filed by Chen, et al. on June 8, 2000
entitled "Pseudomycin N-Acyl Side-Chain Analogs" and
incorporated herein by reference. In general, four
synthetic steps are used to produce the semi-synthetic
compounds from naturally occurring pseudomycin compounds:
(1) selective amino protection; (2) chemical or enzymatic
deacylation of the N-aryl side-chain; (3) reacylation with a
different side-chain; and (4) deprotection of the amino
groups.
The pendant amino groups at positions 2, 4 and 5 may be
protected using any standard means known to those skilled in
the art for amino protection. The exact genus and species
of amino protecting group employed is not critical so long
19
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
as the derivatized amino group is stable to the condition of
subsequent reactions) on other positions of the
intermediate molecule and the protecting group can be
selectively removed at the appropriate point without
disrupting the remainder of the molecule including any other
amino protecting group(s). Suitable amino-protecting groups
include benzyloxycarbonyl, p-nitrobenzyloxycarbonyl,
p-bromobenzyloxycarbonyl, p-methoxybenxyloxycarbonyl,
p-methoxyphenylazobenzyloxycarbonyl,
p-phenylazobenzyloxycarbonyl, t-butyloxycarbonyl,
cyclopentyloxycarbonyl, and phthalimido. Preferred amino
protecting groups are t-butoxycarbonyl (t-Boc),
allyloxycarbonyl (Alloc), phthalimido, and benzyloxycarbonyl
(CbZ or CBZ). Further examples of suitable protecting
groups are described in T.W. Greene, "Protective Groups in
Organic Synthesis," John Wiley and Sons, New York, N.Y.,
(2nd ed., 1991), at chapter 7.
The deacylation of a N-acyl group having a gamma or
delta hydroxylated side chain (e. g., 3,4-dihydroxytetra-
deconoate) may be accomplished by treating the amino-
protected pseudomycin compound with acid in an aqueous
solvent. Suitable acids include acetic acid and
trifluoroacetic acid. A preferred acid is trifluoroacetic
acid. If trifluoroacetic acid is used, the reaction may be
accomplished at or near room temperature. However, when
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
acetic acid is used the reaction is generally ran at about
40°C. Suitable aqueous solvent systems include
acetonitrile, water, and mixtures thereof. Organic solvents
accelerate the reaction; however, the addition of an organic
solvent may lead to other by-products. Pseudomycin
compounds lacking a delta or gamma hydroxy group on the side
chain (e. g., Pseudomycin B and C') may be deacylated
enzymatically. Suitable deacylase enzymes include Polymyxin
Acylase (164-16081 Fatty Acylase (crude) or 161-16091 Fatty
Acylase (pure) available from Wako Pure Chemical Industries,
Ltd.), or ECB deacylase. The enzymatic deacylation may be
accomplished using standard deacylation procedures well
known to those skilled in the art. For example, general
procedures for using polymyxin acylase may be found in
Yasuda, N., et al, Agric. Biol. Chem., 53, 3245 (1989) and
Kimura, Y., et al., Agric. Biol. Chem., 53, 497 (1989).
The deacylated product (also known as the pseudomycin
nucleus) is reacylated using the corresponding acid of the
desired acyl group in the presence of a carbonyl activating
agent. "Carbonyl activating group" refers to a substituent
of a carbonyl that promotes nucleophilic addition reactions
at that carbonyl. Suitable activating substituents are
those which have a net electron withdrawing effect on the
carbonyl. Such groups include, but are not limited to,
alkoxy, aryloxy, nitrogen containing aromatic heterocycles,
21
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
or amino groups (e. g., oxybenzotriazole, imidazolyl,
nitrophenoxy, pentachlorophenoxy, N-oxysuccinimide, N,N'-
dicyclohexylisoure-0-yl, and N-hydroxy-N-methoxyamino);
acetates; formates; sulfonates (e. g., methanesulfonate,
ethanesulfonate, benzenesulfonate, and p-tolylsulfonate);
and halides (e. g., chloride, bromide, and iodide).
A variety of acids may be used in the acylation
process. Suitable acids include aliphatic acids containing
one or more pendant aryl, alkyl, amino(including primary,
secondary and tertiary amines), hydroxy, alkoxy, and amido
groups; aliphatic acids containing nitrogen or oxygen within
the aliphatic chain; aromatic acids substituted with alkyl,
hydroxy, alkoxy and/or alkyl amino groups; and
heteroaromatic acids substituted with alkyl, hydroxy, alkoxy
and/or alkyl amino groups.
Alternatively, a solid phase synthesis may be used
where a hydroxybenzotriazole-resin (HOBt-resin) serves as
the coupling agent for the acylation reaction.
Once the amino group is deacylated and reacylated
(described above), then the amino protecting groups (at
positions 2, 4 and 5) can be removed by hydrogenation in the
presence of a hydrogenation catalyst (e. g., 10% Pd/C).
When the amino protecting group is allyloxycarbonyl, then
the protecting group can be removed using tributyltinhydride
and triphenylphosphine palladium dichloride. This
22
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
particular protection/deprotection scheme has the advantage
of reducing the potential for hydrogenating the vinyl group
of the Z-Dhb unit of the pseudomycin structure.
The prodrug is then produced by acylating at least one
of the pendant amino groups attached to the lysine or 2,4-
diaminobutyric acid peptide units of the N-acyl modified
semi-synthetic pseudomycin compound to form the desired
carbamate linkage.
Other modified prodrug pseudomycin compounds may be
synthesized by amidation or esterification of the pendant
carboxylic acid group of the aspartic acid and/or
hydroxyaspartic acid units of the pseudomycin ring.
Examples of various acid-modified derivatives are described
in PCT Application No. PCT/US00/15021 filed by Chen, et al.
on June 8, 2000 entitled "Pseudomycin Amide & Ester Analogs"
and incorporated herein by reference. The acid-modified
derivatives may be formed by condensing any of the
previously described prodrugs with the appropriate alcohol
or amine to produce the respective ester or amide.
Formation of the ester groups may be accomplished using
standard esterification procedures well-known to those
skilled in the art. Esterification under acidic conditions
typically includes dissolving or suspending the pseudomycin
compound in the appropriate alcohol in the presence of a
protic acid (e. g., HCl, TFA, etc.). Under basic conditions,
23
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
the pseudomycin compound is generally reacted with the
appropriate alkyl halide in the presence of a weak base
(e. g., sodium bicarbonate and potassium carbonate).
Formation of the amide groups may be accomplished using
standard amidation procedures well-known to those skilled in
the art. However, the choice of coupling agents provides
selective modification of the acid groups. For example, the
use of benzotriazol-1-yloxy-tripyrrolidinophosphonium
hexafluorophosphate(PyBOP) as the coupling agent allows one
to isolate pure mono-amides at residue 8 and (in some cases)
pure bis amides simultaneously. Whereas, the use of o-
benzotriazol-1-yl-N,N,N',N'-tetramethyluronium
tetrafluoroborate (TBTU) as the coupling agent favors
formation of monoamides at residue 3.
The pseudomycin prodrug may be isolated and used per se
or in the form of its pharmaceutically acceptable salt or
solvate. The prodrug is prepared by forming at least one
phosphate carbamate linkage as described earlier. The term
"pharmaceutically acceptable salt" refers to non-toxic acid
addition salts derived from inorganic and organic acids.
Suitable salt derivatives include halides, thiocyanates,
sulfates, bisulfates, sulfites, bisulfites, arylsulfonates,
alkylsulfates, phosphonates, monohydrogen-phosphates,
dihydrogenphosphates, metaphosphates, pyrophosphonates,
alkanoates, cycloalkylalkanoates, arylalkonates, adipates,
24
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
alginates, aspartates, benzoates, fumarates,
glucoheptanoates, glycerophosphates, lactates, maleates,
nicotinates, oxalates, palmitates, pectinates, picrates,
pivalates, succinates, tartarates, citrates, camphorates,
camphorsulfonates, digluconates, trifluoroacetates, and the
like.
The term "solvate" refers to an aggregate that
comprises one or more molecules of the solute (i.e.,
pseudomycin prodrug compound) with one or more molecules of
a pharmaceutical solvent, such as water, ethanol, and the
like. When the solvent is water, then the aggregate is
referred to as a hydrate. Solvates are generally formed by
dissolving the prodrug in the appropriate solvent with heat
and slowing cooling to generate an amorphous or crystalline
solvate form.
Each pseudomycin, semi-synthetic pseudomycin,
pseudomycin prodrug and mixtures can be detected,
determined, isolated, and/or purified by any variety of
methods known to those skilled in the art. For example, the
level of pseudomycin or pseudomycin prodrug activity in a
broth or in an isolate or purified composition can be
determined by antifungal action against a fungus such as
Candida and can be isolated and purified by high performance
liquid chromatography.
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
The active ingredient (i.e., pseudomycin derivative) is
typically formulated into pharmaceutical dosage forms to
provide an easily controllable dosage of the drug and to
give the physician, patient, or veterinarian an elegant and
easily handleable product. Formulations may comprise from
0.1% to 99.9% by weight of active ingredient, more generally
from about 10% to about 30% by weight.
As used herein, the term "unit dose" or "unit dosage"
refers to physically discrete units that contain a
predetermined quantity of active ingredient calculated to
produce a desired therapeutic effect. When a unit dose is
administered orally or parenterally, it is typically
provided in the form of a tablet, capsule, pill, powder
packet, topical composition, suppository, wafer, measured
units in ampoules or in multidose containers, etc.
Alternatively, a unit dose may be administered in the form
of a dry or liquid aerosol which may be inhaled or sprayed.
The dosage to be administered may vary depending upon
the physical characteristics of the animal, the severity of
the animal's symptoms, and the means used to administer the
drug. The specific dose for a given animal is usually set
by the judgment of the attending physician or veterinarian.
Suitable carriers, diluents and excipients are well
known to those skilled in the art and include materials such
as carbohydrates, waxes, water soluble and/or swellable
26
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
polymers, hydrophilic or hydrophobic materials, gelatin,
oils, solvents, water, and the like. The particular
carrier, diluent or excipient used will depend upon the
means and purpose for which the active ingredient is being
applied. The formulations may also include wetting agents,
lubricating agents, surfactants, buffers, tonicity agents,
bulking agents, stabilizers, emulsifiers, suspending agents,
preservatives, sweeteners, perfuming agents, flavoring
agents and combinations thereof.
A pharmaceutical composition may be administered using
a variety of methods. Suitable methods include topical
(e. g., ointments or sprays), oral, injection and inhalation.
The particular treatment method used will depend upon the
type of infection being addressed.
In parenteral iv applications, the formulations are
typically diluted or reconstituted (if freeze-dried) and
further diluted if necessary, prior to administration. An
example of reconstitution instructions for the freeze-dried
product are to add ten ml of water for injection (WFI) to
the vial and gently agitate to dissolve. Typical
reconstitution times are less than one minute. The
resulting solution is then further diluted in an infusion
solution such as dextrose 5o in water (D5W), prior to
administration.
27
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
Pseudomycin compounds have been shown to exhibit
antifungal activity such as growth inhibition of various
infectious fungi including Candida spp. (i.e., C. Albicans,
C. Parapsilosis, C. Krusei, C. Glabrata, C. Tropicalis, or
C. Lusitaniaw); Torulopus spp.(i.e., T. Glabrata);
Aspergillus spp. (i.e., A. Fumigates); Histoplasma spp.
(i.e., H. Capsulatum); Cryptococcus spp. (i.e., C.
Neoformans); Blastomyces spp. (i.e., B. Dermatitidis);
Fusarium spp.; Trichophyton spp., Pseudallescheria boydii,
Coccidioides immits, Sporothrix schenckii, etc.
Consequently, the compounds and formulations of the
present invention are useful in the preparation of
medicaments for use in combating either systemic fungal
infections or fungal skin infections. Accordingly, a method
is provided for inhibiting fungal activity comprising
contacting the pseudomycin prodrug of the present invention
with a fungus. A preferred method includes inhibiting
Candida albicans or Aspergillus fumigates activity. The
term "contacting" includes a union or junction, or apparent
touching or mutual tangency of a compound of the invention
with a fungus. The term does not imply any further
limitations to the process, such as by mechanism of
inhibition. The methods are defined to encompass the
inhibition of parasitic and fungal activity by the action of
the compounds and their inherent antifungal properties.
28
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
A method for treating a fungal infection which
comprises administering an effective amount of a
pharmaceutical formulation of the present invention to a
host in need of such treatment is also provided. A
preferred method includes treating a Candida albicans,
Cryptococcus neoformans, or Aspergillus fumigatus infection.
The term "effective amount" refers to an amount of active
compound which is capable of inhibiting fungal activity.
The dose administered will vary depending on such factors as
the nature and severity of the infection, the age and
general health of the host and the tolerance of the host to
the antifungal agent. The particular dose regimen likewise
may vary according to these factors. The medicament may be
given in a single daily dose or in multiple doses during the
day. The regimen may last from about 2-3 days to about 2-3
weeks or longer. A typical daily dose (administered in
single or divided doses) contains a dosage level between
about 0.01 mg/kg to 100 mg/kg of body weight of an active
compound. Preferred daily doses are generally between about
0.1 mg/kg to 60 mg/kg and more preferably between about 2.5
mg/kg to 40 mg/kg. The host may be any animal including
humans, companion animals (e. g., dogs, cats and horses),
food-source animals (e. g., cows, pigs, sheep and poultry),
zoo animals, marine animals, birds and the like.
29
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
EXAMPLES
Unless indicated otherwise, all chemicals can be
acquired from Aldrich Chemical (Milwaukee, WI).
Biological Samples
P. syringae MSU 16H is publicly available from the
American Type Culture Collection, Parklawn Drive, Rockville,
MD, USA as Accession No. ATCC 67028. P. syringae strains
25-B1, 7H9-1, and 67 H1 were deposited with the American
Type Culture Collection on March 23, 2000 and were assigned
the following Accession Nos.:
25-B1 Accession No. PTA-1622
7H9-1 Accession No. PTA-1623
67 H1 Accession No. PTA-1621
Chemical Abbreviations
The following abbreviations are used through out the
examples to represent the respective listed materials:
ACN - acetonitrile
TFA - trifluoroacetic acid
DMAP - 4-dimethylaminopyridine
DMF - dimethylformamide
EDCI - 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide
hydrochloride
BOC = t-butoxycarbonyl, (CH3)3C-O-C(O)-
CBZ = benzyloxycarbonyl, C6HSCH2-O-C (0) -
PyBOP = benzotriazol-1-yloxy-tripyrrolidinophosphonium
hexafluorophosphate
TBTU = o-Benzotriazol-1-yl-N,N,N',N'-tetramethyluronium
tetrafluoroborate
CA 02396513 2002-06-13 - - -- ~ .. wVVJV 1 O1
WO 0114!534 , PCTIUS00l3Qlff
DZF.A ~ N,rt-diisopxopyle~thy7:arl~ne
Tha fallvwing st~cy.ctuze 2I will bs used Zo deecriba the
products observed in Exaatpl~s I and 2.
R~wNN
S =Z
Detertjon and pvantifjcatjon of Au tifun4al Act~vit
~.atifungal activity in the following exasaples were
Qetermined in vitro by obtaining the minimum inhibitoxy
concentration (~C~ of the confound ueiRg a standard agar
1o dilution test or a disc-diffusion teat. A typical fungus
employed in testing antifungal act~.vity is Canc3.tda albicans.
Iustifungal e.ctivity is conaldexed significant whets the tBSt
sam~Ie (50 ~r11 causes 10~12 mm diameter zones of inhibition
on ,C, aIbicans x657 needed agar ple~tes.
~s
Ta.~ I Ve in TO~ci ri ty:
31
av$ast~rv~rs ~~
Ert, P f a~n a s . AM Newly tiled
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
Mice were treated intravenously (IV) through the
lateral tail vein with 0.1 ml of testing compound (20 mg/kg)
at 0, 24, 48 and 72 hours. Two mice were included in each
group. Compounds were formulated in 5.0o dextrose and
sterile water for injection. The mice were monitored for 7
days following the first treatment and observed closely for
signs of irritation including erythema, swelling,
discoloration, necrosis, tail loss and any other signs of
adverse effects indicating toxicity.
The mice used in the study were outbred, male ICR mice
having an average weight between 18-20 g (available from
Harlan Sprangue Dawley, Indianapolis, IN).
In TTivo Testing Procedures:
The examples below were evaluated in a Mouse Model of
Disseminated Candidiasis. Outbred, male ICR mice (average
weight, 18-20 g; Harlan Sprangue Dawley, Indianapolis, IN)
were used in a disseminated candidiasis EDSO survival study.
Candida albicans A26 was cultured on Sabouraud dextrose agar
(SDA; DIFCO Laboratories; Detroit, MI) slants at 35 °C
overnight. Blastoconidia were washed from the surface of
the slant in sterile saline and quantitated using a
hemacytometer. Mice were x-irradiated with 400 r 24 hr
prior to infection with a Gamacell 40 (Atomatic Energy of
Canada Limited Commercial Products, Ottawa, Canada). Mice
32
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
were infected by an intravenous (IV) injection of 0.1 mL
(containing 2 x 106 blastoconidia per mouse) in the lateral
tail vein. Untreated controls were moribund within 3-4 days
post-infection. Mice were dosed four times at 0, 4, 24 and
48 hr post-infection with 0.2 mL of testing compounds which
were given at 20, 10 and 5 mg/kg. Compounds were formulated
in 4.0% hydroxypropyl cyclodextrin and sodium acetate, pH
7.0 buffer and 1.750 dextrose. Infected sham-treated mice
(10 animals) were dosed with vehicle alone. Morbidity and
mortality were recorded for 7 days. The 50o effective doses
(EDso) were determined using the method of Reed and Muench.
Statistical differences in treated groups compared to
untreated infection controls were determined using the
Student's t test.
Preparations
Preparation of (1a-1: Rla - benzyl):
O
O-P~ ORaa
HO OR
1a-1
To a cooled (-20°C) acetonitrile solution (31.5 mL) of
4-hydroxybenzyl alcohol (657 mg, 5.30 mmol) was added CC14
(2.5 mL, 26.5 mmol). The resulting milky solution was
treated with EtPr2N (1.9 mL, 11.13 mmol) and DMAP (65 mg,
33
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
0.53 mmol). After 10 min, the reaction mixture was treated
with dibenzyl phosphite (1.39 g, 5.30 mmol) at -20°C. Upon
completion, the reaction mixture was quenched with 0.5 N
KHZP04. The organic layer was separated and saved. The
aqueous layer was extracted with EtOAc (3 x 10 mL). The
combined organic layers were washed with brine and dried and
concentrated in vacuo to give 2.27 g of the crude product
1a-1 in ~100o yield.
Compounds where Rla - -CH3 ( la-2 ) , Rla - -CH2CH3 ( 1a-3 )
and Rla - -CH2CH2Si ( CH3 ) 3 ( 1a-4 ) were also prepared using the
same general procedures described above with the appropriate
starting materials.
Preparation of (1b-1: Rla - benzyl):
O
O-P~ ORaa
O ~ ~ O O O R'
2
1b-1
To a dichloromethane solution (12 mL) of the crude 1a-1
(2.27 g, 5.3 mmol) was added at 0°C p-nitrophenyl
chloroformate (1.18 g, 5.8 mmol). This was followed by
pyridine (0.5 mL, 6.1 mmol). The reaction was stirred at
0°C for 2 hr and quenched with saturated solution of NaHC03.
The reaction mixture was extracted with EtOAc and the
34
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
extract was washed with water and brine. The organic layer
was dried and concentrated in vacuo to give an oily residue,
which was purified with silica gel chromatography to afford
1.55 g (53%) of the desired product 1b-1.
Compounds where Rla - -CH3 ( 1b-2 ) , Rla - -CH2CH3 ( 1b-3 )
and Rla - -CHzCHzSi (CH3 ) 3 ( 1b-4 ) were also prepared using the
same general procedures described above with the appropriate
starting materials.
Preparation of (2a-1: R1b - benzyl)
O
_ 1b
n-Bu N+ -O~P' ~Rb
a OR
2a-1
7.51 g (27 mmol) of dibenzyl phosphate was mixed with
17.5g (27 mmol) of 40% tetrabutyl ammonium hydroxide and the
mixture was lyophilized to form an oil.
Compounds where Rlb - -CHZCH3 ( 2a-2 ) and Rlb - -CH2CH2CH3
(2a-3) were also prepared using the same general procedures
described above with the appropriate starting materials.
Preparation of (2b-1: Rlb - benzyl):
O O
~P; OR~b
EtS O O OR'b
2b-1
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
Compound 2a-1 (2.34 g, 4.3 mmol) was dissolved in 25 ml
THF. S-ethyl-iodomethylthiocarbonate (1.14 g, 4.7 mmol)
(prepared according to Folkmann, et al., Synthesis, 1159
(1990)) was added to the above solution at room temperature.
The reaction was stirred at room temperature overnight. The
mixture was filtered and the filtrates were concentrated in
vacuo to give a brown oily residue, which was purified by
column chromatography using 40o ethyl acetate in hexane to
provide 0.86 g (510) of 2b-1 as oil.
Compounds where R1b - -CHzCH3 ( 2b-2 ) and R1b - -CH2CH2CH3
(2b-3) were also prepared using the same general procedures
described above with the appropriate starting materials.
Preparation of (2c-1: Rlb - benzyl)
O O O
I I
~P~ OR'b
N-O O O OR'b
O
2c-1
Compound 2b-1 (0.46 g, 1.1 mmol) was dissolved in 1 ml
dichloromethane and cooled to -78°C. Sulfonylchloride (0.12
ml, 1.49 mmol) was added slowly. The mixture was stirred at
-78°C for 15 minutes and warmed slowly to room temperature.
Stirring was continued at room temperature for three hours.
36
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
The mixture was concentrated in vacuo to give the requisite
chloroformate intermediate. The crude phosphate bearing
chloroformate thus obtained was dissolved in 3 ml
dichloromethane and cooled to 0°C. Ethyldipropylamine (0.3
ml) and 134 mg hydroxyl succinimide was added. The mixture
was stirred at room temperature for 30 minutes to provide
the crude 2c-1, which was used directly in Example 2.
Compounds where Rlb - -CH2CH3 ( 2c-2 ) and Rlb - -CHZCHzCH3
(2c-3) were also prepared using the same general procedures
described above with the appropriate starting materials.
Example Z
The following example demonstrates the formation of
mono-, di- and tri-substituted phosphate benzyloxycarbamate
prodrugs of pseudomycin B (n = 10, R2 and R3 - -OH).
-ORIa
'OR~a
Rl, ~ Rl, , ~ Rl, , , - ~ Rla - benzyl
RZ - -OH
R3 - -OH
1A-1
To a DMF solution (100 mL) of pseudomycin B (604 mg,
0.50 mmol) was treated with 1b-1 (0.909 g, 1.65 mmol) at
room temperature. After stirring at room temperature for 24
hours, the reaction mixture was concentrated in vacuo, the
o / ~ O_
37
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
resulting reaction mixture (containing mono-, di- and the
desired triprodrug) was purified by reverse phase
chromatography to afford 418 mg (340) of the desired
triprodrug 1A-1.
Compounds where Rla - - CH3 ( 1A-2 ) , Rla - -CHZCH3 ( 1A-3 )
and Rla - -CHzCH2Si ( CH3 ) 3 ( 1A-4 ) were also prepared using the
same general procedures described above with the appropriate
starting materials.
The phosphate triester prodrug 1A-1 may be converted to
the phosphoric acid monoester prodrug 1B via hydrogenation.
O
i O~ O-P~ ORaa
O OR
R1 ~ and R1, , - ~ Ria - -H
R1" , - -H
R2 - -OH
R3 - -OH
1B
To a MeOH solution (10 mL) of 1A-1 (110.6 mg, 0.045
mmol) was added 10% Pd/C (60 mg) under nitrogen with
caution. The reaction mixture was subjected to standard
hydrogenation under 1.5 atm H2. After 30 min, the reaction
was stopped and the catalyst was filtered off. The
filtrates were concentrated and diluted with 1:1 CH3CN/H20
38
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
and lyophilized to give 67.6 mg (89%) of product (mainly as
diprodrug 1B as judged by its mass spectrum).
The ortho benzyl derivative (e.g., R1 - 1(b) below) was
synthesized using the same general procedures described
above with the appropriate starting materials.
0
IP-OR~a
O O~ ~ORia
~O
1(b)
Example 2
The following examples illustrates the formation of
mono-, di- and tri-substituted phosphate and phosphoric acid
monoester methyleneoxycarbamate prodrugs of pseudomycin B (n
- 10 , R2 and R3 - -OH ) .
O O
IP~OR'b
Rl' ~ R1, , ' Rl, , , - O~O/ \OR~b ~ Rlb - benZyl
R2 - -OH
R3 - -OH
2A-1
The crude 2c-1 was added to a solution of 300 mg (0.25
mmol) Pseudomycin B in 100 ml DMF. The mixture was stirred
39
CA 02396513 2002-06-13
WO 01/41534 PCT/US00/30167
at room temperature overnight. The mixture was concentrated
and purified by reverse phase preparative HPLC. 50 mg of
2A-1 (19%) was obtained.
Compounds where Rlb - -CH2CH3 ( 2A-2 ) and Rlb - -CHzCH2CH3
(2A-3) were also prepared using the same general procedures
described above with the appropriate starting materials.
The phosphate triester prodrug 2A-1 may be converted to
the phosphoric acid monoester prodrug 2B via hydrogenation.
O O
~~OR'b
P
p~ ~OR~b
Rl' ~ Rl , , and Rl , , , - ~ Rib - -H
RZ - -OH
R3 - -OH
2B
The purified product 2A-1 was dissolved 5 ml methanol.
50 mg of 10o Pd/C was added under nitrogen. Hydrogen was
applied and the reaction mixture was stirred for one hour at
room temperature. The mixture was filtered and the solvent
was removed. The product was dissolved in
water/acetonitrile (1/1) and lyophilized to give 32 mg (90%
yield) of the desired product 2B.
An improvement in tail vein toxicity was observed in
the phosphoric acid triester and monoester
benxyloxycarbamate prodrugs (1A-1, 1A-2, 1A-3, 1A-4 and 1B)
CA 02396513 2002-06-13
WO 01/41534 PCT/LJS00/30167
and the phosphoric acid triester and monoester
methyleneoxycarbamate prodrugs (2A-1, 2A-2, 2A-3 and 2B) of
pseudomycin B in comparison to pseudomycin B. Although both
the para and ortho phosphoric acid monoester
benzyloxycarbamate prodrugs showed in vivo activity, the
best in vivo activity was observed with Compound 1B.
41