Note: Descriptions are shown in the official language in which they were submitted.
CA 02398362 2002-07-19
WO 01/53526 PCT/1B01/00077
1
HOMOGENOUS ASSAY OF DUPLEX OR TRIPLEX
HYBRIDIZATION BY MEANS OF MULTIPLE
MEASUREMENTS UNDER VARIED CONDITIONS
SPECIFICATION
BACKGROUND OF THE INVENTION
1. Field of Invention
The invention relates to methods of sequencing or
assaying nucleic acids, and more particularly to methods
of accurately assaying triplex and duplex nucleic acid
hybridization complexes.
2. Description of Related Art
It has been understood for a number of years that
biological molecules can be isolated and characterized
through the application of an electric field to a sample.
Electrophoresis is perhaps_ the most well-known
example of an isolation and characterization technique
based on the influence of electric fields on biological
molecules. In gel electrophoresis, a uniform matrix or
gel is formed of, for example, polyacrylamide, to which an
electric field is applied. Mixtures applied to one end of
the gel will migrate through the gel according to their
size and interaction with the electric field. Mobility is
dependent upon the unique characteristics of the substance
such as conformation, size and charge. Mobilities can be
influenced by altering pore sizes of the gel, such as by
formation of a concentration or pH gradient, or by
altering the composition of the buffer (pH, SDS, DOC,
glycine, salt). One- and two-dimensional gel
electrophoresis are fairly routine procedures in most
research laboratories. Target substances can be purified
by passage through and/or physical extraction from the
gel.
SUBSTITUTE SHEET (RULE 26)
CA 02398362 2002-07-19
WO 01/53526 PCT/1B01/00077
2
A more recently developed process in which an
electric field is applied to a biological sample is
disclosed in U.S. Patent No. 5,824,477 to Stanley. The
Stanley patent discloses a process for detecting the
presence or absence of a predetermined nucleic acid
sequence in a sample. The process comprises: (a)
denaturing a sample double-stranded nucleic acid by means
of a voltage applied to the sample in a solution by means
of an electrode; (b) hybridizing the denatured nucleic
acid with an oligonucleotide probe for the sequence; and
(c) determining whether the hybridization has occurred.
The Stanley patent discloses the application of an
electric field to the sample to be assayed for the limited
purpose of denaturing the target sequence.
A more well-known type of hybridization assay is
based on the use of fluorescent marking agents. In their
most basic form, fluorescent intensity-based assays have
typically comprised contacting a target with a
fluorophore-containing probe, removing any unbound probe
from bound probe, and detecting fluorescence in the washed
sample. Homogeneous assays improve upon such basic
assays, in that the former do not require a washing step
or the provision of a non-liquid phase support.
Some assays have employed intercalating fluorophores
to detect nucleic acid hybridization, based on the ability
of such fluorophores to bind between strands of nucleic
acid in a hybridization complex.
For example, U.S. Patent No. 5,824,557 to Burke et
al. discloses a method and kit for detecting and
quantitating nucleic acid molecules. A preferred
embodiment relies on the intercalation of a dye into a
double-stranded nucleic acid helix or single-stranded
nucleic acid. The dye fluoresces after intercalation and
CA 02398362 2007-11-07
3
the intensity is a direct measurement of the amount of
nucleic acid present in the sample. While the method of
Burke et al. is purported to be useful for measuring the
amount of nucleic acid in a sample, the non-specific
binding between intercalator and nucleic acid upon which
the method is based renders the method impractical for
detecting specific binding, particularly under conditions
where non-target nucleic acid duplexes are present.
U.S. Patent No. 5,814,447 to Ishiguro et al.
discloses an assay which is purported to improve upon
assays that rely on non-specific interaction between
intercalating agents and nucleic acid duplexes, such as
Burke et al. and an earlier assay described by Ishiguro et
al. in Japanese Patent Public Disclosure No. 237000/1993.
The earlier development comprised adding an intercalating
fluorochrome having a tendency to exhibit- increased
intensity of fluorescence when intercalated to a sample
solution before a specific region of a target nucleic acid
was amplified by PCR, and measuring the intensity of
fluorescence from the reaction solution at given time
intervals to detect and quantitate the target nucleic acid
before amplification. The 1447 patent attetnpted to
improve upon the earlier development by providing an assay
having improved specificity, characterized in that the
probe is a single-stranded oligonucleotide labeled with an
intercalating fluorochrome which is to be intercalated
into a complementary binding portion between a target
nucleic acid and a single-stranded oligonucleotide probe.
In the ongoing search for more sensitive, accurate
and rapid assay techniques, one research group developed
an assay comprising analyzing the effects of an electric
field on the fluorescent intensity of nucleic acid
hybridization duplexes. See U.S. Patent No. 6,060,242
CA 02398362 2007-11-07
4
issued May 9, 2000. The researchers indicated that the
fluorescent intensity of a one base-pair mismatched duplex
differed from that of a perfectly matched duplex. Thus,
the applications purport to disclose a method for
detecting a nucleotide sequence, wherein an electric field
is applied to a liquid meditim prior to or concurrently
with a detecting step, and a change in an intensity of a
fluorescent emission as a function of the electric field
is detected as an indication of whether the probe is
hybridized to a completely complementary nucleotide
sequence or an incompletely complementary nucleotide
sequence.
Despite the foregoing developments, a need has
continued to exist in the art for a simple, highly
sensitive, effective and rapid method for analyzing
interaction between nucleic acids and/or nucleic acid
analogs.
SUMMARY OF THE INVENTION
The invention provides a method for assaying
sequence-specific hybridization, said method comprising:
providing a target comprising at least one nucleic
acid sequence;
providing a probe comprising a nucleic acid or
nucleic acid analog sequence;
adding said probe and said target to a hybridization
medium to provide a test sample;
applying a first stimulus to said test sample to
provide a first stimulated test sample;
detecting a first signal from said first stimulated
test sample, wherein said first signal is
CA 02398362 2002-07-19
WO 01/53526 PCT/1B01/00077
correlated with a binding affinity between said
probe and said target;
calibrating said first signal against a reference
signal exhibited by a reference sample
5 comprising at least one reference probe combined
with said target, wherein relative to said
target, each of said probe and said at least one
reference probe is a different member selected
from the group consisting of a perfect match, a
one-base mismatch, a two-base mismatch, a
three-base mismatch, a one-base deletion, a two-
base deletion and a three-base deletion; and
determining from said calibrating a first
determination of an extent of matching between
said probe and said target;
applying a second stimulus to said first stimulated
test sample to provide a second stimulated test
sample; and
detecting a second signal from said second stimulated
test sample, wherein said second signal is
correlated with said binding affinity between
said probe and said target;
determining from said detecting of said second signal
a second determination of said extent of
matching between said probe and said target; and
comparing said first determination and said second
determination.
Also provided is another method for assaying
hybridization, said method comprising:
providing a target comprising at least one nucleic
acid sequence;
providing a probe comprising a nucleic acid or
nucleic acid analog sequence;
CA 02398362 2002-07-19
WO 01/53526 PCT/1B01/00077
6
adding said probe and said target to a hybridization
medium to provide a test sample;
measuring a first signal of a first condition of said
test sample to provide a primary determination
regarding hybridization between said probe and
said target, wherein said first signal is
correlated with hybridization between said probe
and said target;
measuring a second signal of a second condition of
said test sample to provide a secondary
determination regarding hybridization between
said probe and said target, wherein said second
signal is correlated with hybridization between
said probe and said target, provided that when
said first condition and said second condition
are alike, a stimulus is applied to said test
sample after measuring said first signal and
before measuring said second signal, wherein
said stimulus significantly affects imperfectly
complementary hybridization between said probe
and said target and does not significantly
affect perfectly complementary hybridization
between said probe and said target; and
comparing said primary determination and said
secondary determination to evaluate whether any
inconsistency therebetween warrants retesting.
In addition, the invention provides still another
hybridization assay method comprising:
providing a target comprising at least one nucleic
acid sequence;
providing a probe comprising a nucleic acid or
nucleic acid analog sequence;
CA 02398362 2002-07-19
WO 01/53526 PCT/1B01/00077
7
adding said probe and said target to a hybridization
medium to provide a test sample;
measuring a pre-electrif ication fluorescent intensity
of said test sample to provide a primary
determination regarding hybridization between
said probe and said target, wherein said
pre-electrification fluorescent intensity is
correlated with hybridization between said probe
and said target;
applying a voltage to said test sample;
measuring a post-electrification fluorescent
intensity of said test sample, during or after
said voltage applying, to provide a secondary
determination regarding hybridization between
said probe and said target, wherein said
post-electrification fluorescent intensity is
correlated with hybridization between said probe
and said target; and
comparing said primary determination and said
secondary determination to evaluate whether any
inconsistency therebetween warrants retesting.
Also provided is a method for assaying
sequence-specific hybridization, said method comprising:
providing a target comprising at least one nucleic
acid sequence;
providing a probe comprising a nucleic acid or
nucleic acid analog sequence;
adding said probe and said target to a hybridization
medium to provide a test sample;
applying an electrical voltage to-said test sample;
detecting a signal of said test sample during or
after said applying of said electrical voltage,
CA 02398362 2002-07-19
WO 01/53526 PCT/1B01/00077
g
wherein said signal is correlated with a binding
affinity between said probe and said target;
calibrating said signal against a reference signal
exhibited by a reference sample comprising at
least one reference probe combined with said
target, wherein relative to said target, each of
said probe and said at least one reference probe
is a different member selected from the group
consisting of a perfect match, a one-base
mismatch, a two-base mismatch, a three-base
mismatch, a one-base deletion, a two-base
deletion and a three-base deletion; and
determining from said calibrating an extent of
matching between said probe and said target.
BRIEF DESCRIPTION OF THE DRAWINGS
The invention will be described in conjunction with
the following drawings in which like reference numerals
designate like elements and wherein:
Figs. 1A and 1B are graphs of current as a function
of time and complementarity;
Figs. 1C and 1D are graphs of current as a function
of temperature and complementarity;
Figs. 2A, 2B, 2C, 3A and 3B are graphs of current as
a function of temperature, complementarity and additional
factors;
Fig. 4 is a graph of current as a function of time
and complementarity; and
Figs. 5A, 5B, 5C and 6 are fluorescent intensity
spectra.
DETAILED DESCRIPTION OF PREFERRED EMBODIMENTS
The invention provides a rapid, sensitive,
environmentally friendly, and safe method for assaying
binding between a target and a probe, wherein the target
CA 02398362 2002-07-19
WO 01/53526 PCT/1B01/00077
9
comprises a nucleic acid sequence or a nucleic acid analog
sequence and the probe comprises a nucleic acid sequence
or a nucleic acid analog sequence.
Unlike certain prior art assays, the invention not
only detects the presence of hybridization, but also
provides qualitative and quantitative information
regarding the nature of hybridization between a probe and
target. Thus, the invention enables the practitioner to
distinguish among a perfect match, a one base pair
mismatch, a two base pair mismatch, a three base pair
mismatch, a one base pair deletion, a two base pair
deletion and a three base pair deletion.
Embodiments of the invention comprise calibrating the
measured signal (e.g., electric current and/or fluorescent
intensity) for a first probe-target mixture against the
same type of signal exhibited by other probes combined
with the same target, wherein each of the other probes
differs from the first probe by at least one base.
In certain embodiments, a low voltage is applied to
the sample prior to or concurrent with measuring said
signal. Generally, the voltage is selected such that it
is high enough to destabilize imperfectly matched
hybridization partners but not so high as to destabilize
perfectly matched hybridization partners. In certain
preferred embodiments, the voltage is about 1V to about
20V.
A calibration curve can be generated,' wherein the
magnitude of the measured signal (e.g., electric current
and/or fluorescent intensity) is a function of the binding
affinity between the target and probe. As the binding
affinity between the target and a plurality of different
probes varies with the number of mismatched bases, the
nature of the mismatch (A-G vs. A-C vs. T-G vs. T-C,
CA 02398362 2007-11-07
etc.), the location of the mismatch(es) within the
hybridization complex, etc., the assay of the invention
can be used to sequence the target.
The signal measured can be, e.g., electrical
5 conductance. In such embodiments, the binding affinity
between the probe and target is directly correlated with
the magnitude of the signal. That is, the electrical
conductance increases along with the extent of matching
between the probe and target, preferably over a range
10 inclusive of 0-2 mismatches and/or deletions, more
preferably over a range inclusive of 0-3 mismatches and/or
deletions.
In other embodiments, the signal measured can be the
fluorescent intensity.c3f a fluorophore included in the
test sample. In such embodiments, the binding affinity
between the probe and target can be directly or inversely
correlated with the intensity, depending on whether the
fluorophore signals hybridization through signal quenching
or signal amplification. Thus, the fluorescent intensity
generated by intercalating agents is directly correlated
with probe-target binding affinity, whereas the intensity
of embodiments employing non-intercalating fluorophores
covalently bound to the probe is inversely correlated with
probe-target binding affinity. The fluorescent intensity
increases (or decreases for non-intercalators) along with
the extent of matching between the probe and target,
preferably over a range inclusive of 0-2 mismatches and/or
deletions, more preferably over a range inclusive of 0-3
mismatches and/or deletions.
Although the inventors have previously disclosed the
advantages of fluorescent intensity assays for
hybridization (see U.S. Patent No. 6,403,313,
issued June 11, 2002), the application of an electric
CA 02398362 2002-07-19
WO 01/53526 PCT/1B01/00077
I1
field to the sample appears to increase the resolution of
the assay, as shown in Example 6 below.
Moreover, in particularly preferred embodiments of
the invention, the assay comprises measuring at least two
signals of the sample. The first signal is preferably
fluorescent intensity and the second signal is preferably
selected from several electrical conductance measurements
(or vice versa) .
In the preferred multiple measurement embodiments,
the first signal can be the same as or different from the
second signal. When the first and second signals measured
are the same, the second signal can be calibrated against
the first signal and/or against the same reference
signal(s) used to calibrate the first signal. In
addition, a condition-altering stimulus is preferably
applied to the test sample after the first signal is
measured and before the second signal is measured. The
stimulus is preferably sufficient to significantly affect
imperfectly complementary hybridization between the probe
and the target and insufficient to significantly affect
perfectly complementary hybridization between the probe
and the target.
For example, in a particularly preferred embodiment
of the invention, the first signal measured is
pre-electrif ication fluorescent intensity (i.e., intensity
measured before a condition-altering voltage is applied to
the test sample) and the second signal measured is
post-electrification fluorescent intensity (i.e.,
intensity measured during or after the condition-altering
voltage has been applied to the test sample).
The additional measurements in the foregoing
embodiments increase the reliability of the assay and
enable immediately retesting suspect results.
CA 02398362 2002-07-19
WO 01/53526 PCT/1B01/00077
12
Inconsistent results achieved by the at least two
measurements will typically warrant retesting.
The invention enables quantifying the binding
affinity between probe and target. Such information can
be valuable for a variety of uses, including designing
antisense drugs with optimized binding characteristics.
Unlike prior art methods, the assay of the invention
is preferably homogeneous. The assay can be conducted
without separating the probe-target complex from the free
probe and target prior to detecting the magnitude of the
measured signal. The assay does not require a gel
separation step, thereby allowing a great increase in
testing throughput. Quantitative analyses are simple and
accurate. Consequently the binding assay saves a lot of
time and expense, and can be easily automated.
Furthermore, it enables binding variables such as buffer,
pH, ionic concentration, temperature, incubation time,
relative concentrations of probe and target sequences,
intercalator concentration, length of target sequences,
length of probe sequences, and possible cofactor
requirements to be rapidly determined.
The assay can be conducted in e. g., a solution within
a well, on an impermeable surface or on a biochip.
Moreover, the inventive assay is preferably conducted
without providing a signal quenching agent on the target
or on the probe.
Preferred embodiments of the invention specifically
detect triplex hybridization between the probe and the
double-stranded target, thus obviating the need to
denature the target. While PNA probes have been known to
form triplexes with certain classes of targets (see, e.g.,
Egholm et al., 365 Nature 566 (1993), and Tomac et al.,
118 J.Am.Chem.Soc. 5544 (1996)), the inventors were
CA 02398362 2002-07-19
WO 01/53526 PCT/1B01/00077
13
surprised that they were able to specifically assay
triplexes formed between single-stranded nucleic acid
(e.g., ssDNA and RNA) probes and double-stranded nucleic
acid (e.g., dsDNA) targets. Triplex formation and/or
stabilization is enhanced by the presence of an
intercalating agent in the sample being tested.
Suitable probes for use in the inventive assay
include, e.g., ssDNA, RNA, PNA and other nucleic acid
analogs having uncharged or partially-charged backbones.
Although antiparallel probes are preferred in certain
embodiments, PNA probes can also be parallel. Probe
sequences having any length from 8 to 20 bases are
preferred since this is the range within which the
smallest unique DNA sequences of prokaryotes and
eukaryotes are found. Probes of 12 to 18 bases are
particularly preferred since this is the length of the
smallest unique sequences in the human genome. In
embodiments, probes of 6 to 30 bases are most preferred.
However, a plurality of shorter probes can be used to
detect a nucleotide sequence having a plurality of
non-unique target sequences therein, which combine to
uniquely identify the nucleotide sequence. The length of
the probe can be selected to match the length of the
target.
The invention does not require the use of radioactive
probes, which are hazardous, tedious and time-consuming to
use, and need to be constantly regenerated. Probes of the
invention are preferably safe to use and stable for years.
Accordingly, probes can be made or ordered in large
quantities and stored.
It is preferred that the probe and target be
unlabeled, but in alternative embodiments, there is an
intercalating agent covalently bound to the probe. In
CA 02398362 2002-07-19
WO 01/53526 PCT/1B01/00077
14
such embodiments, the intercalating agent is preferably
bound to the probe at either end.
In other embodiments, the intercalating agent is not
covalently bound to the probe, although it can insert
itself between the probe and target during the assay, in
a sense bonding to the probe in a non-covalent fashion.
Preferred intercalating agents for use in the
invention include, e.g., YOYO-1, TOTO-l, ethidium bromide,
ethidium homodimer-1, ethidium homodimer-2 and acridine.
In general, the intercalating agent is a moiety that is
able to intercalate between strands of a duplex and/or a
triplex nucleic acid complex. In preferred embodiments,
the intercalating agent (or a component thereof) is
essentially non-fluorescent in the absence of nucleic
acids and fluoresces when intercalated and excited by
radiation of an appropriate wavelength, exhibiting a 100-
fold to 10,000-fold enhancement of fluorescence when
intercalated within a duplex or triplex nucleic acid
complex.
In alternative embodiments, the intercalating agent
may exhibit a shift in fluorescent wavelength upon
intercalation and excitation by radiation of an
appropriate wavelength. The exact fluorescent wavelength
may depend on the structure of the nucleic acid that is
intercalated, for example, DNA vs. RNA, duplex vs.
triplex, etc.
The excitation wavelength is selected (by routine
experimentation and/or conventional knowledge) to
correspond to this excitation maximum for the fluorophore
being used, and is preferably 200 to 1000 nm.
Intercalating agents are preferably selected to have an
emission wavelength of 200 to 1000 nm. In preferred
embodiments, an argon ion laser is used to irradiate the
CA 02398362 2002-07-19
WO 01/53526 PCT/1B01/00077
fluorophore with light having a wavelength in a range of
400 to 540 nm, and fluorescent emission is detected in a
range of 500 to 750 nm.
The assay of the invention can be performed over a
5 wide variety of temperatures, such as, e.g., from 5 to
85 C. Certain prior art assays require elevated
temperatures, adding cost and delay to the assay. On the
other hand, the invention can be conducted at room
temperature or below (e.g., at a temperature below 25 C).
10 The inventive assay is extremely sensitive, thereby
obviating the need to conduct PCR amplification of the
target. For example, in at least the fluorescent
intensity embodiments, it is possible to assay a test
sample having a volume of about 20 microliters, which
15 contains about 10 femtomoles of target and about 10
femtomoles of probe. Embodiments of the invention are
sensitive enough to assay targets at a concentration of 5
X 10"9M, preferably at a concentration of not more than 5
x 10-10 M. Embodiments of the invention are sensitive
enough to employ probes at a concentration of 5 X 10-9M,
preferably at a concentration of not more than 5 x 10-10 M.
Conductivity measurements can distinguish samples
having as little as about 1 pmole of probe and 1 pmole of
target in 40 microliters. Decreasing the sample volume
would permit the use of even smaller amounts of probe and
target.
It should go without saying that the foregoing values
are not intended to suggest that the method cannot detect
higher concentrations.
A wide range of intercalator concentrations are
tolerated at each concentration of probe and target
tested. For example, when 5 X 10-10 M probe and 5 X 10-10
M target are hybridized, the optimal concentration of the
CA 02398362 2002-07-19
WO 01/53526 PCT/1B01/00077
16
intercalator YOYO-1 ranges from 25 nM to 2.5 nM. At a 5
X 10-6 M concentration of both probe and target, the
preferred YOYO-1 concentration range is 1000 nM to 100 nM.
The assay is sufficiently sensitive to distinguish a
one base-pair mismatched probe-target complex from a two
base-pair mismatched probe-target complex, and preferably
a two base-pair mismatched probe-target complex from a
three base-pair mismatched probe-target complex. Of
course, the assay is sufficiently sensitive to distinguish
a perfectly matched probe-target complex from any of the
above mismatched complexes.
The hybridization medium can be any conventional
medium known to be suitable for preserving nucleotides.
See, e.g., Sambrook et al., "Molecular Cloning: A Lab
Manual," Vol. 2 (1989). For example, the liquid medium
can comprise nucleotides, water, buffers and standard salt
concentrations.
Hybridization between complementary bases occurs
under a wide variety of conditions having variations in
temperature, salt concentration, electrostatic strength,
and buffer composition. Examples of these conditions and
methods for applying them are known in the art.
It is preferred that hybridization complexes be
formed at a temperature of about 15 C to about 25 C for
about 1 minute to about 5 minutes. Longer reaction times
are not required, but incubation for several hours will
not adversely affect the hybridization complexes.
It is possible (although unnecessary, particularly
for embodiments containing an intercalating agent) to
facilitate hybridization in solution by using certain
reagents. Preferred examples of these reagents include
single stranded binding proteins such as Rec A protein, T4
gene 32 protein, E. coli single stranded binding protein,
CA 02398362 2002-07-19
WO 01/53526 PCT/1B01/00077
17
major or minor nucleic acid groove binding proteins,
divalent ions, polyvalent ions, viologen and intercalating
substances such as ethidium bromide, actinomycin D,
psoralen, and angelicin. Such facilitating reagents may
prove useful in extreme operating conditions, for example,
under abnormal pH levels or extremely high temperatures.
The inventive assay can be used to, e.g., identify
accessible regions in folded nucleotide sequences, to
determine the number of mismatched base pairs in a
hybridization complex, and to map genomes.
In embodiments wherein fluorescent intensity is
detected using an intercalating agent, intensity increases
with increasing binding affinity between the probe and
target. In embodiments wherein fluorescent intensity is
detected using a non-intercalating fluorophore, intensity
decreases as binding affinity increases between the probe
and target. Regardless of whether the fluorophore
intercalates or not, the instant method does not require
the measurement of the polarization of fluorescence,
unlike fluorescent anisotropy methods.
The invention will be illustrated in more detail with
reference to the following Examples, but it should be
understood that the present invention is not deemed to be
limited thereto.
EXAMPLES
Example 1
Sense and antisense 50-mer ssDNA target sequences,
derived from exon 10 of the human cystic fibrosis gene
(Nature 380, 207 (1996)) were synthesized on a DNA
synthesizer (Expedite 8909, PerSeptive Biosystems) and
purified by HPLC. Equimolar amounts of complementary
oligonucleotides were denatured at 95 C for 10 min and
allowed to anneal gradually as the temperature cooled to
CA 02398362 2002-07-19
WO 01/53526 PCT/1B01/00077
18
21 C over 1.5 hours. Double stranded DNA (dsDNA)
oligonucleotides were dissolved in ddHzO at a
concentration of 1 pmole/,ul.
Sequence for the sense strand of the wild-type target
DNA (SEQ ID NO:1): 51 -TGG CAC CAT TAA AGA AAA TAT
CAT CTT TGG TGT TTC CTA TGA TGA ATA TA-3'.
Sequence for the antisense strand of the wild-type
target DNA (SEQ ID NO:1): 5'-TAT ATT CAT CAT AGG AAA
CAC CAA AGA TGA TAT TTT CTT TAA TGG TGC CA-3'.
The predicted melting temperature (Tm) of dsDNA (SEQ
ID NO:1) is 65.2 C.
SEQ ID NO:2 was a 50-mer mutant dsDNA target sequence
identical to wild-type target DNA (SEQ ID NO:1) except for
a one base pair mutation (underlined) at amino acid
position 507 at which the wild-type sequence CAT was
changed to CGT.
Sequence for the sense strand of SEQ ID NO:2: 5'-TGG
CAC CAT TAA AGA AAA TAT CGT CTT TGG TGT TTC CTA
TGA TGA ATA TA-3'.
Sequence for the antisense strand of SEQ ID NO: 2: 5' -
TAT ATT CAT CAT AGG AAA CAC CAA AGA CGA TAT TTT
CTT TAA TGG TGC CA-3'.
The predicted melting temperature (Tn,) of dsDNA (SEQ
ID NO:2) is 66.0 C.
SEQ ID NO:3 was a 50-mer mutant dsDNA target sequence
identical to wild-type target DNA (SEQ ID NO:1) except for
a consecutive two base pair mutation (underlined) at amino
acid positions 506 and 507 at which the wild-type sequence
CAT was changed to ACT.
Sequence for the sense strand of SEQ ID NO:3: 5'-TGG
CAC CAT TAA AGA AAA TAT ACT CTT TGG TGT TTC CTA
TGA TGA ATA TA-31.
CA 02398362 2002-07-19
WO 01/53526 PCT/1B01/00077
19
Sequence for the antisense strand of SEQ ID N0: 3: 5' -
TAT ATT CAT CAT AGG AAA CAC CAA AGA GTA TAT TTT
CTT TAA TGG TGC CA-31.
The predicted melting temperature (Tm) of dsDNA (SEQ
ID NO:3) is 65.2 C.
The PNA probes used in the Examples were synthesized,
HPLC purified and confirmed by mass spectroscopy by
Commonwealth Biotechnologies, Inc. (Richmond, VA, USA).
PNA probes were first dissolved in 0.1% TFA
(trifluoroacetic acid) to a concentration of 10 mg/ml, and
then diluted to 1 mg/ml by the addition of ddH2O. Final
PNA stock solutions were prepared in ddH2O at a
concentration of 1 pmole/,ul.
Probe No. 1 was a 15-mer antiparallel PNA probe
designed to be completely complementary to a 15 nucleotide
segment of the sense strand of the 50-mer wild-type target
DNA (SEQ ID N0:1), overlapping amino acid positions 505 to
510 (Nature 380, 207 (1996) ). The probe had the following
structure (SEQ ID NO:8) :
5'-H-CAC CAA AGA TGA TAT-Lys-CONH2-3'
The hybridization reaction mixture (80,ul) contained
the following: 2 pmoles of target dsDNA, 2 pmoles of PNA
probe, 0.5X TBE and 250 nM of the DNA intercalator YOYO-1
(Molecular Probes, Eugene, OR, USA). Samples were placed
into a 3 mm quartz cuvette and were subjected to 1 or 5
volts DC (V) electrification for 15 seconds. The
amperometric assay consisted of the monitoring of current
while the voltage was being applied to the solution. A
temperature probe was placed in each solution to measure
temperature at the time of amperometric assessment. At 1
volt, a current peak was observed during the first 2
seconds of electrification. The current declined sharply
over the following 13 seconds. Experiments applying 5
CA 02398362 2002-07-19
WO 01/53526 PCT/1B01/00077
volts gave rise to currents that remained relatively
stable over the entire electrification period (15
seconds).
A series of experiments were carried out where the
5 conductance values were observed when no DNA or PNA was
present (control), or when wild-type SEQ ID NO:1, mutant
SEQ ID NO:2 or mutant SEQ ID NO:3 were reacted with
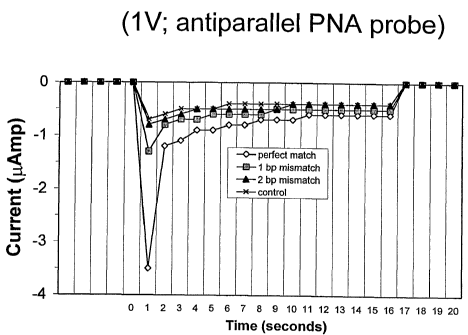
antiparallel PNA Probe No. 1. Figures 1A and 1B plot the
data obtained for conductance in the individual
10 experiments. Figure lA displays the results of the
application of 1V electrification and Figure 1B the
application of 5V. Double stranded DNA:PNA hybrid
triplexes consisting of perfectly complementary sequences
(SEQ ID NO:1 + Probe No. 1) allowed maximum intercalation
15 of YOYO-1, yielding the highest conductance values
(depicted on the figures as negative current values)
throughout the entire 15 seconds of 1V application. The
normalized peak conductance for the triplex hybridization
of the antiparallel PNA probe with a 1 bp mismatched dsDNA
20 (SEQ ID NO:2 + Probe No. 1) and with the 2 bp mismatched
dsDNA (SEQ ID NO:3 + Probe No. 1) were respectively 790
and 960 lower than that observed with the perfectly
matched dsDNA:PNA triplex hybrid (SEQ ID NO:1 + Probe No.
1) during the first second of voltage application (Fig.
1A). Similar percent decreases in conductance between
perfectly complementary triplexes and triplexes containing
base pair mismatches were obtained when the conductance
values over the entire 15 seconds of voltage application
were averaged. In Fig. 1A the 1 bp and 2 bp mismatched
dsDNA:PNA hybrids resulted in average conductance values
that were 65% and 91% lower, respectively, than those for
the perfectly matched dsDNA:PNA hybrid. All experiments
expressed in Figure lA were carried out at room
CA 02398362 2002-07-19
WO 01/53526 PCT/1B01/00077
21
temperature (23 C). As the degree of mismatch between the
probe and the double stranded target increased', the level
of intercalation by YOYO-1 diminished and the level of
conductance decreased. These relationships were also
observed when the experiments referred to above were
repeated and a higher voltage (5V) was applied. During
the 5V application the normalized average conductance
values for the 1 bp mismatched dsDNA:PNA triplex (SEQ ID
NO:2 + Probe No. 1) and the 2 bp mismatched dsDNA:PNA
triplex (SEQ ID NO:3 + Probe No. 1) were respectively 52%
and 67% lower than that observed for the perfectly matched
dsDNA:PNA triplex (SEQ ID NO:3 + Probe No. 1) (Fig. 1B).
Experiments expressed in Figure 1B were performed at room
temperature (23 C).
When the experiments were repeated with the
temperature increased to 50 C and 65 C, similar
amperometric values were observed. At 50 C, the
application of 1V for 15 seconds to the perfectly matched
dsDNA:PNA triplex (SEQ ID NO:1 + Probe No. 1) produced an
average current of -0.25 ,uAmp as compared to values of -
0.15 ,uAmp (a 40% reduction) and -0.06 ,uAmp (a 76%
reduction) for the 1 bp mismatched dsDNA:PNA triplex (SEQ
ID NO:2 + Probe No. 1) and the 2 bp mismatched dsDNA:PNA
triplex (SEQ ID NO:3 + Probe No. 1), respectively (Fig.
1C). At 65 C, similar observations were noted when 1V of
electricity was applied for 15 seconds. Perfectly matched
nucleic acid hybrids produced an average current of -0.37
,uAmp compared with -0.16 ,uAmp (a 57% reduction) and -0.01
,uAmp (a 97% reduction) for 1 bp and 2 bp mismatched
hybrids, respectively (Fig. 1C). The application of 5
volts at high temperatures produced analogous results.
While experiments performed at 50 C generated average
currents of -0.27 mAmp, -0.13 mAmp (a 52% reduction), and
CA 02398362 2002-07-19
WO 01/53526 PCT/1B01/00077
22
-0.08 mAmp (a 70% reduction), for perfectly matched
hybrids, 1 bp mismatched hybrids, and 2 bp mismatched
hybrids, respectively, experiments performed at 65 C
resulted in average current values of -0.31 mAmp, -0.14
mAmp (a 55% reduction), and -0.10 mAmp (a 68% reduction)
for the same three respective groups (Fig. 1D). For all
of the foregoing experiments, dsDNA was not denatured
prior to triplex hybridization with the antiparallel PNA
Probe No. 1.
Similar experiments were done at varying temperatures
after the hybridization mixes had been heated to 65 C and
immediately allowed to cool. After cooling to room
temperature (23 C), applying 1V for 15 seconds to the
perfectly matched sample (SEQ ID NO:1 + Probe No. 1)
produced an average current of -0.18 ,uAmp. By comparison,
values of -0.06 gAmp (a 67% reduction) and -0.05 gAmp (a
72% reduction) for the 1 bp mismatched dsDNA:PNA triplex
hybrid (SEQ ID NO:2 + Probe No. 1) and the 2 bp mismatched
dsDNA:PNA triplex hybrid (SEQ ID NO:3 + Probe No. 1), were
respectively observed (data not shown). When the samples
were cooled from 65 C to 50 C, similar observations were
noted when 1V was subsequently applied for 15 seconds.
The perfectly matched sample (SEQ ID NO:1 + Probe No. 1)
produced an average current of -0.23 ,uAmp compared with
-0.11 ,uAmp (a 52% reduction) and -0.01 Amp (a 96%
reduction) observed for the 1 bp and 2 bp mismatched
samples, respectively (data not shown) When 5V was
applied after cooling to 23 C or 50 C, the average current
generated in the perfectly matched triplex hybrid (SEQ ID
NO:l + Probe No. 1), the 1 bp mismatched triplex hybrid
(SEQ ID NO:2 + Probe No. 1), and the 2 bp mismatched
triplex hybrid (SEQ ID NO:3 + Probe No. 1) were: -0.15
mAmp, -0.09 mAmp (a 40% reduction), and -0.07 mAmp (a 53%
CA 02398362 2002-07-19
WO 01/53526 PCT/1B01/00077
23
reduction) , respectively at 23 C, and -0.23 mAmp, -0.09
mAmp (a 61% reduction), and -0.09 mAmp (a 61% reduction),
respectively at 50 C (data not shown).
Pretreatment of hybridization mixes at 65 C (the Tm
of the 50-mer dsDNA sequences) followed by cooling did not
significantly affect the difference in conductance
observed between perfectly complementary dsDNA:PNA
triplexes and those containing 1 or 2 bp mismatches when
measured directly at 23 C or 50 C (without preheating at
65 C) when an antiparallel PNA probe was used. Clearly,
the antiparallel PNA probe in the presence of the DNA
intercalator YOYO-l was able to form triplex structures
with the dsDNA targets. Application of low levels of
electricity (such as 1V or 5V) allowed the perfectly
matched dsDNA:PNA triplex sequences to be distinguished
from those containing 1 bp or 2 bp mutations, without
prior denaturation of sequences.
Example 2
Figure 2 demonstrates that the amperometric assay of
the invention can also discriminate between perfectly
matched dsDNA:PNA triplex hybrids and those containing 1
bp or 2 bp mismatches when the PNA probe used is in a
parallel orientation with respect to the target DNA
sequence. Probe No. 2 was a 15-mer PNA probe identical in
sequence to Probe No. 1, but was synthesized to match the
parallel orientation of the target DNA, instead of the
conventional anti-parallel orientation. Probe No. 2 had
the following structure (SEQ ID NO:9) :
5'-H-TAT AGT AGA AAC CAC-Lys-CONH2-3'
Experiments with assay conditions identical to those
described in Example 1 were carried out with the sole
difference that Probe No. 2 was used in place of Probe No.
1. When 1 volt was applied, the average current for a 1
CA 02398362 2002-07-19
WO 01/53526 PCT/1B01/00077
24
bp mismatched dsDNA:PNA triplex (SEQ ID NO:2 + Probe No.
2), and a consecutive 2 bp mismatched dsDNA:PNA triplex
(SEQ ID NO:3 + Probe No. 2), were respectively 25% and 32%
lower at 23 C, respectively 30% and 23% lower at 50 C, and
respectively 28% and 53% lower at 65 C than that observed
with the perfectly matched dsDNA:PNA triplex (SEQ ID NO:1
+ Probe No. 2) at matching temperatures (Fig. 2A).
Similar results were obtained when 5V (instead of 1V)
was applied for 15 seconds. Perfectly matched dsDNA:PNA
hybrids at 23 C, 50 C and 65 C generated average currents
of -0.15 mAmp, -0.24 mAmp and -0.17 mAmp, respectively
(Fig. 2B). Incompletely complementary triplexes with a 1
bp mismatch and a 2 bp mismatch produced average currents
that were 27% less (-0.11 mAmp) and 53% less (-0.07 mAmp),
respectively at 23 C, 21% less (-0.19 mAmp) and 46% less
(-0.13 mAmp), respectively at 50 C, and unchanged
(-0.17mAmp) and 18% less (-0.14 mAmp), respectively at
65 C, than that achieved by the perfectly matched hybrid
samples (Fig. 2B).
The results illustrated in Figures 2A and 2B
indicated that when the parallel PNA Probe No. 2 was used,
the differences in conductivity obtained between perfectly
matched dsDNA:PNA triplexes and those containing 1 bp or
2 bp mismatches were less dramatic than that achieved with
the antiparallel PNA Probe No. 1 (Fig. 1).
However, experiments involving parallel Probe No. 2
and the application of 5V after the samples have been
heated to 65 C and immediately allowed to cool disclosed
amperometric measurements which demonstrated enhanced
signaling differences between perfectly matched dsDNA:PNA
triplexes and the 1 bp or 2 bp mismatched dsDNA:PNA
triplexes (Fig. 2C). The perfectly matched hybrids (SEQ
ID NO:1 + Probe No. 2), the 1 bp mismatched hybrids (SEQ
CA 02398362 2002-07-19
WO 01/53526 PCT/1B01/00077
ID NO:2 + Probe No. 2) and the 2 bp mismatched hybrids
(SEQ ID NO:3 + Probe No. 2) yielded average conductance
values of -0.19 mAmps, -0.08 mAmps and -0.06 mAmps,
respectively at 23 C, -0.17 mAmps, -0.09 mAmps and -0.07
5 mAmps, respectively at 50 C, and -0.23 mAmps, -0.13 mAmps
and -0.08 mAmps, respectively at 65 C. This translated to
reductions in conductivity of 58% and 68% at 23 C, 47% and
59% at 50 C, and 43% and 65% at 65 C for the 1 bp and 2 bp
mismatched samples, respectively, when compared to the
10 values achieved by the perfectly complementary samples
(Fig. 2C).
Therefore, both antiparallel and parallel PNA probes
in the amperometric assay are capable of discriminating
between perfectly complementary dsDNA targets and
15 incompletely complementary dsDNA targets containing 1 bp
or 2 bp mutations.
Example 3
Probe No. 3 was a 15-mer ssDNA probe identical in
sequence and orientation to the 15-mer antiparallel PNA
20 Probe No. l(SEQ ID NO:8). Probe No. 3 had the following
structure:
5'-CAC CAA AGA TGA TAT-3'
The specificity of the amperometric assay was further
investigated by reacting ssDNA Probe No. 3 with the 50-mer
25 wild-type and mutant dsDNA target sequences in the absence
of prior denaturation. The assay conditions were identical
to that described in Example 1.
Enhanced by the DNA intercalator YOYO-1, dsDNA:ssDNA
triplexes were formed between 30 C and 65 C. Upon 1 volt
treatment, the perfectly matched DNA triplex, consisting
of SEQ ID N0:1 + Probe No. 3, yielded the highest
conductivity values (Fig. 3A) . In contrast, incompletely
complementary probe and target combinations generating a
CA 02398362 2002-07-19
WO 01/53526 PCT/1B01/00077
26
1 bp mismatch (SEQ ID NO:2 + Probe No. 3), and a
consecutive 2 bp mismatch (SEQ ID NO:3 + Probe No. 3),
resulted in average conductance values that were 14% and
64% lower at 23 C, 30% and 70% lower at 50 C, and 25% and
72% lower at 65 C, respectively, than that observed with
the perfectly complementary sequences at matching
temperatures (Fig. 3A). The application of a higher
voltage (5V) to these samples resulted in greater
amperometric differences observed between perfectly
matched and mismatched samples, than that obtained at 1V,
particularly at lower temperatures. After a 5V treatment
for 15 seconds, the average currents for the 1 bp
mismatched DNA triplex and the 2 bp mismatched DNA triplex
were 54% and 78% lower, respectively at 23 C, 68% and 70%
lower, respectively at 50 C, and 33% and 61% lower,
respectively at 65 C, than that observed with the
perfectly matched DNA triplex at matching temperatures
(Fig. 3B).
In similar electricity experiments, the hybridization
mixes were heated to 65 C and were either maintained at
this temperature or immediately allowed to cool to 50 C or
23 C prior to application of 1V or 5V. A 1V treatment for
15 seconds to the perfectly matched DNA triplex sequences
(SEQ ID NO:1 + Probe No. 3) produced the highest
conductance values at 23 C, 50 C and 65 C (Fig. 3A). The
DNA triplexes containing a 1 bp mismatch (SEQ ID NO:2 +
Probe No. 3) or a 2 bp mismatch (SEQ ID NO:3 + Probe No.
3) were less conductive by 21% and 63%, respectively at
23 C, by 18% and 74%, respectively at 50 C, and by 12% and
106%, respectively at 65 C (Fig. 3A). Similarly, when 5V
were applied for 15 seconds to pre-heated samples, the
average conductance values for the 1 bp mismatched DNA
triplexes and the 2 bp mismatched DNA triplexes were
CA 02398362 2002-07-19
WO 01/53526 PCT/1B01/00077
27
reduced by 24% and 104%, respectively at 23 C, by 42% and
44%, respectively at 50 C, and by 38% and 102%,
respectively at 65 C, when compared to the average
conductance values generated by the perfectly matched DNA
triplexes (Fig. 3B).
The observation that the antiparallel PNA probe (Fig.
1) and ssDNA probe (Fig. 3) behaved in a similar fashion
in the amperometric assay, suggested that the backbone of
the nucleic acid entity used as the probe was not
particularly important. The presence of YOYO-l allowed
the dsDNA targets and the ssDNA probe to form a triple
helix conformation capable of generating different
electrical charges depending on the level of sequence
complementarity between the target and the probe in
solution. As the degree of mismatch between the probe and
the target increased, the level of conductance decreased,
proving the reliability of the amperometric assay when a
natural DNA probe was used in the absence of prior
denaturation.
Example 4
In the amperometric assays illustrated in Examples 1
to 3, the DNA intercalator YOYO-l was added to the
solution containing the hybridization mixes.
Intercalation by YOYO-l facilitated the formation of the
dsDNA:PNA triplexes and dsDNA:ssDNA triplexes. The
possibility of utilizing an intercalator moiety covalently
tethered to a ssDNA probe in the amperometric assay was
evaluated in Example 4.
Acridine is an alternative dsDNA intercalator, that
also possesses the ability to intercalate into triplex
nucleic acid structures, thereby stabilizing the triple
helix formation. See, e.g., Kukreti et al., "Extension of
the range of DNA sequences available for triple helix
CA 02398362 2002-07-19
WO 01/53526 PCT/1B01/00077
28
formation: stabilization of mismatched triplexes by
acridine-containing oligonucleotides." 25 Nucleic Acids
Research 4264-4270 (1997). A ssDNA probe containing an
acridine molecule (Glen Research, Sterling, VA, USA)
covalently attached at the 31 end was synthesized on a DNA
synthesizer (Expedite 8909, PerSeptive Biosystems) and
purified by HPLC.
Probe No. 4 was a 15-mer ssDNA probe identical in
sequence and orientation to the 15-mer Probe No. 3 (and
thus also identical in sequence and orientation to the 15-
mer antiparallel PNA Probe No. 1 (SEQ ID NO:8)) but with
the addition of an acridine moiety at the 3' position.
The probe had the following structure:
5'-CAC CAA AGA TGA TAT-acridine-3'
The hybridization reaction mixture (80 ,ul) contained
the following: 2 pmoles of target dsDNA, 2 pmoles of
ssDNA Probe No. 4 and 0.5X TBE. Samples were placed into
a 3 mm quartz cuvette and were subjected to 5V DC
electrification for 11 seconds at 23 C. The current and
temperature were monitored as described in Example 1.
As shown in Fig. 4, the ssDNA Probe No. 4 was able to
hybridize with the 50-mer perfectly matched dsDNA target
(SEQ ID NO:1) as a result of the stable intercalation of
the covalently tethered acridine moiety, generating an
average current of -0.53 mAmp. By comparison, the less
stable DNA triplexes containing a 1 bp mismatch (SEQ ID
NO:2 + Probe No. 4) or a 2 bp mismatch (SEQ ID NO:3 +
Probe No. 4) produced average currents that were 52% and
66% lower, respectively, than that achieved by the
perfectly matched DNA triplex, when normalized against the
control (Probe No. 4 without target DNA) (Fig. 4).
Therefore, the acridine attached to a ssDNA probe was
equally as efficient as untethered YOYO-l in forming
CA 02398362 2002-07-19
WO 01/53526 PCT/1B01/00077
29
triple DNA helices that generated different electrical
currents depending on the level of sequence
complementarity between the target and the probe in the
amperometric assay.
Example 5
Sense and antisense 15-mer ssDNA target sequences,
derived from exon 10 of the human cystic fibrosis gene,
were synthesized, purified and annealed as described in
Example 1. DsDNA oligonucleotides were dissolved in ddH2O
at a concentration of 1 pmole/,ul.
SEQ ID NO:4 was a 15-mer dsDNA target sequence
derived from SEQ ID NO:1, designed to be completely
complementary to Probe No. 1.
Sequence for the sense strand of the wild-type target
DNA (SEQ ID NO:4): 5'-ATA TCA TCT TTG GTG-3'.
Sequence for the antisense strand of the wild-type
target DNA (SEQ ID NO:4): 51-CAC CAA AGA TGA TAT-3'.
The predicted melting temperature (Tm) of dsDNA (SEQ
ID NO:4) is 40.0 C.
SEQ ID NO:5 was a 15-mer mutant dsDNA target sequence
identical to wild-type target DNA (SEQ ID NO:4) except for
a one base pair mutation (underlined), at which the
sequence TTT was changed to TAT.
Sequence for the sense strand of the mutant target
DNA (SEQ ID NO:.5):
5'-ATA TCA TCT ATG GTG-3'.
Sequence for the antisense strand of the mutant
target DNA (SEQ ID NO:5):
5'-CAC CAT AGA TGA TAT-3'.
The predicted melting temperature (Tm) of dsDNA (SEQ
ID NO:5) is 40.0 C.
SEQ ID NO:6 was a 15-mer mutant dsDNA target sequence
identical to wild-type target DNA (SEQ ID NO:4) except for
CA 02398362 2002-07-19
WO 01/53526 PCT/1B01/00077
a consecutive two base pair mutation (underlined), at
which the sequence ATC was changed to GGC.
Sequence for the sense strand of the mutant target
DNA (SEQ ID NO:6):
5 5'-ATA TCG GCT TTG GTG-3'.
Sequence for the antisense strand of the mutant
target DNA (SEQ ID NO : 6):
5'-CAC CAA AGC CGA TAT-3'.
The predicted melting temperature (Tm) of dsDNA (SEQ
10 ID NO:6) is 44.0 C.
SEQ ID NO:7 was a 15-mer mutant dsDNA target sequence
identical to wild-type target DNA (SEQ ID NO:4) except for
a separated three base pair mutation (underlined), wherein
three 1 bp mutations were separated by 3 base pairs each.
15 The sequences ATC, TCT and TGG were changed to ACC, TAT
and TAG, respectively.
Sequence for the sense strand of the mutant target
DNA (SEQ ID NO:7): 5'-ATA CCA TAT TTA GTG-3'.
Sequence for the antisense strand of the mutant
20 target DNA (SEQ ID NO:7): 5'-CAC TAA ATA TGG TAT-3'.
The predicted melting temperature (Tm) of dsDNA (SEQ
ID NO:7) is 38.0 C.
The hybridization reaction mixture (80 ,ul) contained
the following: 2 pmoles of target dsDNA, 2 pmoles of
25 parallel PNA Probe No. 2, 0.5X TBE and 250 nM of the DNA
intercalator YOYO-1. The reaction mixtures were incubated
at 95 C for 5 - 10 minutes to allow denaturation, and then
maintained at 65 C until assayed. Samples were placed
into a quartz cuvette, irradiated with an argon ion laser
30 beam having a wavelength of 488 nm and monitored for
fluorescent emission at 65 C. Concurrent temperature
measurements were achieved by a software-controlled
temperature probe placed directly into each sample. The
CA 02398362 2002-07-19
WO 01/53526 PCT/1B01/00077
31
maximum fluorescent intensity occurred at a wavelength of
536 nm, indicative of intercalation of YOYO-1 in the
PNA:DNA hybrids. As a second assay, following the initial
laser irradiation of each sample, the same samples were
subjected to 1V DC electrification for 4 seconds. During
the final second of electrification the samples were
irradiated a second time with the argon ion laser and
monitored for fluorescent emission at 65 C. Fluorescent
intensities were plotted as a function of wavelength for
each sample analyzed.
SsDNA:PNA hybrids consisting of perfectly
complementary sequences (SEQ ID NO:4 + Probe No. 2)
allowed maximum intercalation of YOYO-1, yielding the
highest fluorescent intensities (Fig. 5A). The
fluorescent intensities for a 1 bp mismatched ssDNA:PNA
hybrid (SEQ ID NO:5 + Probe No. 2), a consecutive 2 bp
mismatched ssDNA:PNA hybrid (SEQ ID NO:6 + Probe No. 2),
and a separated 3 bp mismatched ssDNA:PNA hybrid (SEQ ID
NO:7 + Probe No. 2) were all lower than that observed with
the perfectly matched asDNA : PNA hybrid at 650C ( Fig . 5 and
data not shown). As the degree of mismatch between the
probe and the target increased, the level of intercalation
by YOYO-l diminished and hence the level of fluorescent
intensity decreased. Only background levels of
fluorescence were observed when no DNA or PNA was present
(YOYO-1 alone) (Fig. 5A).
When the perfectly matched ssDNA:PNA hybrids were
subjected to 1V of electricity for 4 seconds at 65 C, the
fluorescent intensity remained relatively constant,
decreasing by only 20 (Fig. 5A). In contrast, application
of 1V to the incompletely complementary duplexes
containing a 1 bp mismatch (Fig. 5B), a 2 bp mismatch
(Fig. 5C) and a 3 bp mismatch (data not shown) produced
CA 02398362 2002-07-19
WO 01/53526 PCT/1B01/00077
32
fluorescent intensities that were 18%, 39% and 71% lower,
respectively, than that achieved with the same samples
irradiated in the absence of electricity. Treatment with
low levels of electricity (such as 1V) further diminished
the stability of the ssDNA:PNA hybrids containing bp
mismatches. As the degree of sequence complementarity
between the probe and the target decreased, the level of
fluorescent intensity diminished dramatically in the
presence of electricity, providing a highly reliable and
accurate second assay to differentiate between perfectly
matched sequences and those containing 1 bp, 2 bp or 3 bp
mutations.
Example 6
The hybridization assay in Example 5 was performed
after denaturation of the dsDNA target sequences and
measured ssDNA:PNA hybrid formation at a temperature above
the melting point (Tm) of the dsDNA targets. Example 6
will demonstrate the reliability of the fluorescent
intensity assay in the absence and presence of applied
electricity to differentiate between perfect matches and
base pair mismatches without the requirement for prior
denaturation.
The hybridization reaction mixture (80,ul) contained
the following: 4 pmoles of target dsDNA, 4 pmoles of
antiparallel PNA Probe No. 1, 0.5X TEE and 250 nM of the
DNA intercalator YOYO-1. Samples were placed into a
quartz cuvette, irradiated with an argon ion laser beam
having a wavelength of 488 nm for 80 msec and monitored
for fluorescent emission at 23 C. Concurrent temperature
measurements were achieved by a software-controlled
temperature probe placed directly into each sample. The
maximum fluorescent intensity occurred at a wavelength of
536 nm, indicative of intercalation of YOYO-l in the
CA 02398362 2002-07-19
WO 01/53526 PCT/1B01/00077
33
PNA:DNA hybrids. As a second assay, following the initial
laser irradiation of each sample, the same samples were
subjected to 20V DC electrification for 4 seconds.
Immediately after 3 seconds of electrification the samples
were irradiated a second time with the argon ion laser for
80 msec and monitored for fluorescent emission at 23 C.
Fluorescent intensities were plotted as a function of
wavelength for each sample analyzed.
Enhanced by the intercalator YOYO-1, dsDNA:PNA
triplexes were formed at 23 C. The highest fluorescent
intensity was achieved when the wild-type 50-mer dsDNA
target sequence (SEQ ID N0:1) was hybridized with the 15-
mer antiparallel PNA Probe No. 1 (Fig. 6). By comparison,
the fluorescent intensities for a 1 bp mismatched
dsDNA:PNA triplex (SEQ ID NO:2 + Probe No. 1) and a
consecutive 2 bp mismatched dsDNA:PNA triplex (SEQ ID NO:3
+ Probe No. 1) were 60% and 91% lower, respectively, than
that observed with the perfectly matched dsDNA:PNA triplex
at 23 C (Fig. 6). When no DNA or PNA was present in the
reaction mixture containing YOYO-1, only background levels
of fluorescence were observed.
The difference in fluorescent intensities obtained by
the perfectly complementary triplexes and those containing
1 bp or 2 bp mismatches were significantly greater than
that achieved between perfectly matched duplexes and
incompletely complementary duplexes (compare Figs. 5 and
6). Clearly the fluorescent intensity assay of triplex
formation possessed enhanced discriminatory ability to
detect base pair mismatches.
Moreover, even further discrimination between wild-
type and mutated sequences was possible with the secondary
application of electricity. A 20V treatment for 3 seconds
to the perfectly matched dsDNA: PNA triplexes produced a
CA 02398362 2002-07-19
WO 01/53526 PCT/1B01/00077
34
fluorescent intensity spectrum virtually identical to that
achieved by the same sample not subjected to electricity
(Fig. 6). However, application of 20V for 3 seconds to
the incompletely complementary triplexes containing a 1 bp
mismatch and a 2 bp mismatch produced fluorescent
intensities that were 23% and 71% lower, respectively,
than that obtained with the same samples irradiated in the
absence of electricity (Fig. 6) . The 20V treatment of
electricity did not affect the stability of the perfectly
complementary triplexes, but weakened the stability of the
dsDNA:PNA triplexes containing base pair mismatches at a
level dependent on the degree of sequence complementarity
between the probe and the target. Therefore, the
application of electricity to the fluorescent intensity
assay provided an even more highly reliable assay to
distinguish between wild-type sequences and those
containing 1 bp or 2 bp mutations, without prior
denaturation of sequences.
While the invention has been described in detail and
with reference to specific examples thereof, it will be
apparent to one skilled in the art that various changes
and modifications can be made therein without departing
from the spirit and scope thereof.
CA 02398362 2002-09-26
1
SEQUENCE LISTING
<110> Ingeneus Corporation
<120> HOMOGENOUS ASSAY OF DUPLEX OR TRIPLEX HYBRIDIZATION BY MEANS OF
MULTIPLE MEASUREMENTS UNDER VARIED CONDITIONS
<130> 08-895359CA
<140> Not Yet Known
<141> 2001-01-23
<150> US 09/490,273
<151> 2000-01-24
<160> 9
<170> PatentIn Ver. 2.1
<210> 1
<211> 50
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: derived from
exon 10 of the human cystic fibrosis gene
<400> 1
tggcaccatt aaagaaaata tcatctttgg tgtttcctat gatgaatata 50
<210> 2
<211> 50
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: derived from
exon 10 of the human cystic fibrosis gene
<400> 2
tggcaccatt aaagaaaata tcgtctttgg tgtttcctat gatgaatata 50
<210> 3
<211> 50
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: derived from
exon 10 of the human cystic fibrosis gene
<400> 3
CA 02398362 2002-09-26
2
tggcaccatt aaagaaaata tactctttgg tgtttcctat gatgaatata 50
<210> 4
<211> 15
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: derived from
exon 10 of the human cystic fibrosis gene
<400> 4
atatcatctt tggtg 15
<210> 5
<211> 15
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: derived from
exon 10 of the human cystic fibrosis gene
<400> 5
atatcatcta tggtg 15
<210> 6
<211> 15
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: derived from
exon 10 of the human cystic fibrosis gene
<400> 6
atatcggctt tggtg 15
<210> 7
<211> 15
<212> DNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: derived from
exon 10 of the human cystic fibrosis gene
<400> 7
CA 02398362 2002-09-26
3
ataccatatt tagtg 15
<210> 8
<211> 15
<212> PNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: ssPNA Probe
<400> 8
caccaaagat gatat 15
<210> 9
<211> 15
<212> PNA
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence: ssPNA Probe
<400> 9
tatagtagaa accac 15